

Abstract
A central question in cell biology is how cells respond to stress signals and biochemically regulate apoptosis. One critical pathway involves the change of mitochondrial function and release of cytochrome c to initiate apoptosis. In response to apoptotic stimuli, we found that maspin—a noninhibitory member of the serine protease inhibitor superfamily—translocates from the cytosol to mitochondria and binds to cardiolipin in the inner mitochondrial membrane. Biolayer interferometry assay revealed that recombinant maspin binds cardiolipin with an apparent Kd,of ∼15.8 μM and competes with cytochrome c (apparent Kd of ∼1.31 μM) for binding to cardiolipin-enriched membranes. A hydrophobic, lysine-rich domain in maspin consists of 27 aa, is located at position 268–294, and is responsible for the interaction of this protein with cardiolipin. Depletion of cardiolipin in cells significantly prevents maspin binding to the inner mitochondrial membrane and decreases cytochrome c release and apoptosis. Alteration to maspin’s cardiolipin binding domain changes its ability to bind cardiolipin, and tumor cells expressing this mutant have a low frequency of apoptosis. We propose a model of apoptosis in which maspin binds to cardiolipin, displaces cytochrome c from the membrane, and facilitates its release to the cytoplasm.—Mahajan, N., Hoover, B., Rajendram, M., Shi, H. Y., Kawasaki, K., Weibel, D. B., Zhang, M. Maspin binds to cardiolipin in mitochondria and triggers apoptosis.
Keywords: diphosphatidylglycerol, cytochrome c, mitochondrial membranes, anionic phospholipids
Many signals lead to programmed cell death; however, there is a single biochemical pathway for this process, which occurs in the mitochondria and makes this organelle the central executioner of apoptosis (1). Several major steps are involved in mitochondria-mediated programmed cell death: 1) the permeability of the outer mitochondrial membrane to molecular transport is increased; 2) cytochrome c is released from the outer mitochondrial membrane; and 3) the caspase family of cysteine-aspartic proteases is activated and degrades cellular organelles (2, 3). The molecular mechanism or mechanisms involved in regulating the release of cytochrome c from mitochondrial membranes remain poorly understood, and consequently the biochemistry of programmed cell death is still emerging.
Maspin is a noninhibitory member of the serine protease inhibitor superfamily and has been characterized as a tumor suppressor gene in various cancer types (1, 4). Since its discovery 2 decades ago (5), maspin has been established as a participant in different cellular processes, including apoptosis (6, 7), embryonic development (8), tumor suppression (6, 9, 10), and oxidative stress (11, 12). Previously, we demonstrated that under a variety of apoptosis-stimulating conditions, maspin translocates from the cytosol to mitochondria and is accompanied by dissipation of the transmembrane potential, release of cytochrome c, and caspase activation or modulation of expression of B-cell lymphoma 2 (Bcl-2) family members (7, 13, 14). Suppression of maspin overexpression by RNA interference in mammary tumor cells that had been modified to overexpress maspin desensitized cells to apoptosis (7). Subcellular fractionation experiments demonstrated that maspin migrates to the inner mitochondrial membrane (IMM) after the induction of apoptosis (7). However, the biochemical role of maspin in the mitochondria remains unknown.
Our search for maspin binding partners among the mitochondrial proteome using a yeast 2-hybrid screen was unsuccessful (unpublished results). We instead turned our attention to its interaction with mitochondrial phospholipids, which participate in a broad range of processes (15, 16). For example, the dianionic phospholipid cardiolipin (CL) is essential in mitochondria and uniquely found only in mitochondrial and bacterial membranes (17). CL is concentrated in the IMM (∼10−20% of total phospholipids) (18), is required for the formation of the cristae structure of the IMM, maintains the activity of several essential IMM proteins, and plays a central role in the execution of apoptosis and modulation of the proapoptotic action of caspase-truncated Bcl-2 homology domain 3 interacting-domain death agonist (Bid) with Bcl-2–associated X protein or Bcl-2 homologous antagonist-killer (19). Cytochrome c is bound to CL through electrostatic interactions (20), and the disruption of this complex is hypothesized to form an “apoptotic trigger” in which release of cytochrome c activates downstream biochemical processes that lead to programmed cell death (21).
In the present study, we demonstrate that maspin binds CL located in the mitochondria through a CL binding domain (CLBD), competes with cytochrome c for CL binding in the mitochondria, and activates apoptosis. Maspin is more abundant than cytochrome c in some maspin-expressing mammary tumor cells, thereby enabling it to compete effectively with cytochrome c for binding to CL, despite its lower affinity. The results lay the foundation for unraveling the mechanism of maspin actions in mitochondria and apoptosis and offer the basis for developing novel therapy targeting maspin-mediated apoptosis in cancer and a variety of other cells.
MATERIALS AND METHODS
Reagents
Phosphatidylethanolamine (MilliporeSigma, Burlington, MA, USA), phosphatidylcholine (PC) (MilliporeSigma), lysophosphatidylcholine (Avanti Polar Lipids, Alabaster, AL, USA), CL (Avanti Polar Lipids), and phosphatidic acid (MilliporeSigma) were purchased and used without further purification. Antibodies used for Western blot analysis were as follows: rabbit polyclonal anti-maspin antibody (9), anti–glutathione S-transferase (GST) antibody (Santa Cruz Biotechnology, Dallas, TX, USA), anti-actin antibody (MilliporeSigma), anti-cytochrome c (Cell Signaling Technology, Danvers, MA, USA), anti–cytochrome c oxidase subunit IV (COX IV) (Cell Signaling Technology), and anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (MilliporeSigma). Horseradish peroxidase–labeled goat anti-rabbit pAb or anti-mouse antibody was used as secondary antibody as required and purchased from MilliporeSigma. Proteasomal inhibitors [i.e., MG132 (MilliporeSigma), lactacystin (Cayman Chemicals, Ann Arbor, MI, USA)] and lysosomal inhibitors, such as NH4Cl (MilliporeSigma) and chloroquine (MilliporeSigma), were purchased and used without additional purification.
Plasmid constructs and cell culture
A GST-containing fusion protein to full-length, wild-type maspin (MpWT), Arg-Ala point mutation (Mp*340), maspin mutants containing truncations in aa 1–139 (Mp-N) and aa 1–225 (Mp-BglII), and His-tagged maspin have been previously described (9). R340A is a mutation of the P1 position of the reactive center loop that has differentially been described to be involved in various functions of maspin (12, 22). To generate a mutant version of maspin (MpPS) in which the lysine-rich region is swapped with the corresponding region in plasminogen activator inhibitor 1 (PAI-1; PS comes from PAI-1 swap), we introduced a HindIII site in maspin cDNA by site-directed mutagenesis at position C323 as previously described (11). A PCR amplicon was amplified from PAI-1 cDNA using the following primers: sense: 5′-GGGGCCATGGCCAGGCTGCCCCGCCT-3′; antisense: 5′-GGGGAAGCTTACTTTCTGCAGCGC-3′. The PCR amplicon was ligated in between NcoI and HindIII in maspin, swapping the region from MpWT with the region from PAI-1. The fusion proteins were induced with isopropyl β-d-1-thiogalactopyranoside (1 mM) and purified using glutathione agarose (MilliporeSigma). The fusion proteins were eluted using 50 mM Tris buffer (pH 7.5) containing 10 mM reduced glutathione. The size and purity of proteins were confirmed by SDS-PAGE and Western blot analysis.
We used a temperature-sensitive Chinese hamster ovary (CHO-K1) cell line (named PGS-S) that exhibits diminished phosphatidylglycerophosphate synthase (PGS) activity, which is a key enzyme in CL synthesis. Incubation of PGS-S cells at 40°C for 4–5 d resulted in a 2–3-fold reduction in CL without affecting the concentration of other major phospholipids (23). We used cell line PGS-S/cPGS1, which is stably transfected with a cDNA encoding PGS as a control (24). PGS-S and PGS-S/cPGS1 cells were transfected with pEF-IRIS-puro-h.maspin or empty vector (puro, used to clone human maspin) using Effectene (Qiagen, Germantown, MD, USA). To avoid clonal selection–caused bias, we chose to pool stable transfectants and selected them with puromycin medium (10 μg/ml) for 14 d. We then confirmed overexpression of maspin in these cells by Western blot analysis. Unless otherwise indicated, all CHO-K1 cell mutants and transfectants were grown in Ham’s F-12 medium supplemented with 10% (v/v) bovine serum, penicillin G (100 units/ml), and streptomycin sulfate (100 mg/ml), under 5% CO2 atmosphere at 100% humidity at either 40, 37, or 33°C.
Murine mammary tumor TM40D cells were used and maintained as described previously (25). TM40D cells were transfected with pEF-IRIS-neo-h.maspin wild type (MpWT), pEF-IRIS-neo-h.maspin–Pai-1 swap (MpPS), or control vector alone (Neo) using Effectene (Qiagen). The stable transfectants were pooled together to avoid clonal bias and selected with G418 medium (600 μg/ml) for 14 d, and expression of maspin was determined by Western blot analysis.
Staurosporine (STS) (LC Laboratories, Woburn, MA, USA) is a potent inhibitor of a wide range of protein kinases—such as PKC and tyrosine protein kinases—and was used to induce apoptosis as described previously (7, 26).
Western blot analysis
Cell lysates were prepared in RIPA buffer with protease cocktail inhibitor (Thermo Fisher Scientific, Waltham, MA, USA). Cellular debris was cleared from lysates by centrifugation, and protein concentrations were determined using a Bicinchoninic Acid Protein Assay Kit (Pierce, Rockford, IL, USA). Samples were separated on SDS-PAGE and transferred to a membrane (Hybond-P; GE Healthcare, Waukesha, WI, USA), and proteins were visualized using ECL (Pierce). Quantification of Western blots was performed using ImageJ v.64 software (National Institutes of Health, Bethesda, MD, USA). Levels were calculated by normalizing the band intensities of the test gene to that of loading control and presented as arbitrary units. GAPDH and COX IV were used as loading controls for cytosolic and mitochondrial fractions, respectively.
Protein lipid overlay assay
To assess the lipid binding properties of GST-fused proteins, a protein lipid overlay assay was performed as previously described (27). Lipids were dissolved in a solution of methanol:chloroform:water (2:1:0.8) and were spotted on Hybond-P membrane (GE Healthcare) at a concentration of 20 nmol per spot and dried at 25°C for 1 h, or stored at 4°C and used within 10 d. The membrane was blocked with 3% fatty acid free bovine serum albumin (MilliporeSigma) in Tris–buffered saline (20 mM Tris, 150 mM NaCl, pH 8.0) containing 0.1% Tween 20 for 1 h at 25°C followed by overnight incubation with indicated protein (5 μg/ml) at 4°C. The next day, we washed the membrane 5 times for 10 min each in Tris-buffered saline containing Tween 20 and incubated with either maspin or GST antibody for 3 h at 25°C. The membrane was washed as before, and then incubated with horseradish peroxidase–conjugated antibody for 1 h at 25°C. Finally, proteins bound to the membrane—through interaction with lipids—were detected by ECL.
Modified gel mobility shift assay
To evaluate maspin binding to lipids in solution, we used another approach based on the principle that tight binding to lipids may induce changes in the electrophoretic mobility of proteins under nondenaturing conditions. To test this hypothesis, we performed PAGE without SDS followed by standard Western blotting (PAGE-Western) (28, 29) Protein (20 ng) and lipids (2 mM) were mixed in PBS containing 1 mM DTT to a final volume of 20 μl. Samples were incubated for 1 h at 25°C, and the reaction was terminated by addition of sample buffer (20% glycerol, 0.1 M Tris-HCl at pH 6.8, and 0.01% bromophenol blue). Samples were separated on 8% native gels, and Western blot analysis was performed.
Preparation of CL liposomes
We used lipids from Avanti Polar Lipids, including 1,2-di-(9Z-octadecenoyl)-sn-glycero-d-phosphocholine (DOPC; herein referred to as PC) and 1,1′,2,2′-tetra-(9Z-octadecenoyl) (herein referred to as CL). To create liposomes, we dried aliquots of the mixtures of the lipids dissolved in chloroform in a glass vial under argon and removed traces of solvent by incubating in a vacuum chamber for 12–15 h at 25°C. We resuspended the dried lipid fractions in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) at pH 7.4 (Thermo Fisher Scientific) and sonicated (Branson 2510 bath sonicator) the suspensions for 8 min. We immersed the vials containing the lipids in liquid nitrogen for ∼2 min. We repeated the sonication and liquid nitrogen step 5 times. To prepare liposomes of the lipid mixtures, we extruded sonicated suspensions of lipids (Avanti Polar Lipid Mini Extruder) through a membrane containing 0.1-μm-diameter pores 21 times on a hot plate heated to 60°C. To confirm the homogeneity of our liposome preparations, we used dynamic light scattering (Zetasizer Systems, Malvern Panalytical, Malvern, United Kingdom) to quantify their size and dispersity (Supplemental Fig. S1).
Biolayer interferometry assays of maspin and cytochrome binding to bilayer membranes consisting of CL and PC
We used an Octet Red96 biolayer interferometry (BLI) instrument (ForteBio, Pall, Port Washington, NY, USA) operating in kinetics mode to monitor the competitive binding of maspin and cytochrome c to PC and CL liposomes. All wash steps, baseline determination, and loading of liposomes and protein were carried out in 25 mM HEPES pH 7.4 in Costar 96-well black polystyrene plates at 30°C. After prewetting aminopropylsilane (APS)–coated biosensors (Pall) for 10 min, we established a baseline by dipping tips into wells containing buffer for 300 s. Biosensors were equilibrated against PC:CL liposomes (50:50 M concentration) at a concentration of 200 μM until saturation was reached (7500 s). Tips were washed in buffer and equilibrated for 300 s in a solution of 0.1% bovine serum albumin (MilliporeSigma) to block unbound hydrophobic sites (30). Following a 120-s wash in buffer, we established a second baseline for 300 s. We performed 2 types of experiments to measure the competitive protein association step. In the first experiment, we fixed the total protein concentration at 5 μM in each well and monitored the competitive binding of cytochrome c and maspin at a range of molar ratios for 9000 s. In the second experiment, 5 μM cytochrome c (MilliporeSigma) was initially equilibrated with surface immobilized vesicles for 2000 s before dipping biosensors into wells containing 5 μM cytochrome c with varying molar ratios of purified maspin for 9000 s. We recorded the dissociation of protein from the liposomes for 9000 s after returning the tips to buffer. The protein association and dissociation steps were maintained at a plate agitation speed of 1000 rpm, and the loading of liposomes was maintained at 100 rpm. For all other steps, the agitation speed was fixed at 600 rpm.
Octet data acquisition and analysis software v.8.01 was used as previously described (31). Acquired raw data were analyzed in MatLab (MathWorks, Natick, MA, USA). To determine the apparent association constant Ka for cytochrome c and maspin, the same program template and buffers as the competitive binding protein assay were used. For cytochrome c, the protein concentration was varied until saturation of the equilibrium wavelength shift was reached (∼100 μM). For maspin, we were unable to reach saturation because of the large quantity of protein required. We provide a measurement for the apparent Ka for cytochrome c and an estimate for maspin fitting with a 1:1 binding model (Eq. 1).
 |
Req represents the equilibrium wavelength shift, Rmax the maximal value of wavelength shift, and [P] the total concentration of protein in solution.
We determined the dissociation of protein from liposomes by fitting the data to Eq. 2.
In this equation, y is the wavelength shift, A and B are integers, t is time, koff is the dissociation rate constant, and C is a baseline coefficient.
Translocation of maspin to mitochondria and release of cytochrome c
STS has previously been shown to induce apoptosis in CHO-K1 cells at a concentration of 1 μm for 4 h (26). Mitochondrial and cytosolic fractions were isolated from ∼5 × 107 cells after STS treatment using a Mitochondrial-Cytosolic Factionation Kit (BioVision, Milpitas, CA, USA). We used a Bicinchoninic Acid Protein Assay Kit (Pierce) to determine the concentration of total protein. Release of cytochrome c to the cytosol and translocation of maspin to mitochondria was determined by Western blot analysis. GAPDH and COX IV were used as loading controls for cytosolic and mitochondrial fractions, respectively.
Measurements of caspase 3 activity
One hundred micrograms of maspin protein were used to measure the caspase 3 activity in the absence or presence of STS (1 μM) for 4 h in cells using a commercially available Colorimetric Assay Kit (BioVision).
Import assay to determine binding of maspin to CL and release of CL-bound cytochrome c
Mitochondria from PGS-S and PGS-S/cPGS1 cells grown at 40°C for 4–5 d were isolated as described above, and an import assay was performed as previously described (7). Atractyloside (ATR, 5 mM) (MilliporeSigma) was added to open the permeability transition pore in the membrane. Isolated mitochondria (200 μg) were incubated with ATR (5 mM) in 250 μl of PBS for 15 min. Following this step, we added 100 ng of GST fused with MpWT (GST.MpWT) (from a 100 ng/μl stock) to the reaction mixture and incubated for 30 min. Mitochondria were washed twice with 1 ml of cold PBS to remove unbound maspin. The pellet containing mitochondria were lysed in mitochondria extraction buffer provided with the kit (BioVision), and Western blot analysis was performed for maspin and COX IV (as loading control). To determine whether the binding of maspin to CL in the IMM is correlated to the release of CL-bound cytochrome c, we isolated mitochondria from mouse liver (32) and added 500 μg of ATR-treated mitochondria (as previously described) with an equal amount of GST or GST.MpWT (Supplemental Fig. S4). Release of CL-bound cytochrome c was determined by Western blot analysis; treatment with ATR led to the release of most of the cytochrome c (∼80% of total cytochrome c in mitochondria) present in the intermitochondrial membrane space (Supplemental Fig. S4).
Protein Abundance Database
The Protein Abundance Database (PaxDb; https://pax-db.org/) is used to compare the abundance of maspin and cytochrome c in human epithelial-derived cells in this study.
Statistical analysis
Quantification of Western blots was performed using ImageJ. Statistical analysis (2-tailed Student’s t test or ANOVA) was based on a minimum of 3 replicates using Prism 5 for Mac statistical software (v.5; Graphpad Software, La Jolla, CA, USA). Results were shown as means ± sd. The difference is considered significant if P < 0.05.
RESULTS
Maspin binds to CL tightly
We initially used 2 different in vitro approaches to determine the interaction of maspin with phospholipids: protein lipid overlay assays and protein mobility shift assays. The protein lipid overlay assay revealed that recombinant maspin interacted with CL but not with the other phospholipids, including the anionic phospholipid phosphatidic acid (Fig. 1A); we did not assess other anionic phospholipids such as phosphatidylglycerol because of their low abundance (∼1% of total phospholipids) compared with the more predominate phospholipids (PC and CL, ∼40 and 20% of total phospholipids respectively) in mitochondrial membranes (33, 34). To confirm this interaction, we performed gel mobility shift assays and found that incubating recombinant maspin with CL retarded its mobility in PAGE (Fig. 1B). As a negative control, we tested the binding of GST and found that it did not bind to any phospholipids in these assays. We confirmed these results by quantitatively measuring the interaction of cytochrome c and maspin to phospholipid bilayers containing a 50:50 M ratio of CL to PC using BLI. We selected a high CL concentration to ensure saturation of the available binding sites on cytochrome c. Kinetic assays have demonstrated that a higher anionic phospholipid content (from increasing CL) correlates with tighter cytochrome c binding (31). BLI is a biophysical method for measuring the binding kinetics of proteins to the surface of fiber optic biosensors that are modified to display specific molecules (in this case, phospholipid bilayers); absorption of protein on the surface changes its optical thickness and can be measured spectrometrically (31). We immobilized PC:CL liposomes (containing 50:50 PC:CL) on the surface of APS-terminated biosensor surfaces. We analyzed the homogeneity and dispersity of PC:CL liposome preparations using DLS and found that liposome quality and homogeneity is preserved at high molar concentrations of CL (see Supplemental Fig. S1). We then measured the interaction between cytochrome c and CL-containing bilayers by quantifying the equilibrium wavelength shift, Req (Fig. 1C). These measurements yielded a Ka value of ∼7 × 105 M−1 and koff value of ∼3.31 × 10−4 s−1 that we used to calculate an apparent dissociation constant, Kd, of ∼1.31 ± 0.4 μM for cytochrome c binding to CL, which is in agreement with values measured using surface plasmon resonance and BLI-based techniques (see Supplemental Table S1) (31, 35, 36). Using a similar approach to analyze the interaction of maspin with CL-containing membranes, we estimated a Ka of 6.3 104 M−1 and a koff of 1.08 × 10−5 s−1, which yields an apparent Kd of ∼15.8 μM. These data suggest that the binding affinity of maspin for CL is ∼1 order of magnitude weaker than for cytochrome c, making it reasonable that these 2 proteins compete for binding to CL in the IMM.
Figure 1.

Maspin binds to CL. A) A protein-lipid overlay assay demonstrating the interaction between maspin and various lipids [phospholipids (PL), phosphatidylethanolamine (PE), PC, CL, phosphatidic acid (PA), BSA (Mock)]. Wild-type maspin (GST.MpWT) binds specifically to CL and not to the other lipids tested. GST protein (as control) did not bind to the lipids. B) Binding of maspin to CL was determined by retardation of protein mobility in a PAGE–Western blot (WB). T20, Tween 20. Incubation of GST.MpWT with CL reduced the mobility of the protein in contrast to the other lipids. GST was used as a control. C) Dependence of the equilibrium and maximal wavelength shift (Req and Rmax) on protein concentration. Liposomes (50:50 PC:CL by molar concentration) were immobilized on the surface of APS biosensor tips. The interaction between vesicles, cytochrome c (solid line), and maspin (dotted line) was monitored until equilibrium was reached in 25 mM HEPES pH 7.4. Fit to Eq. 1 yielded the apparent association constants (Ka) of 6.9 × 105 M−1 and 6.3 × 104 M−1 for cytochrome c and maspin, respectively. All data points represent means ± sd (n = 3). Representative figures are shown of experiments completed a minimum of 6 times.
Maspin contains a CLBD
CL binding proteins, including caspase-truncated Bid (37), p53 (38), cytochrome c (20), and β2 glycoprotein I (39), share a common structural feature: the presence of positively charged, Lys-rich regions that are also hydrophobic and thus attractive to anionic phospholipids such as CL; these regions have been referred to as CLBDs (14). We closely examined the protein sequence of maspin and discovered 3 putative CLBDs that are positively charged, Lys-rich regions of the protein (Fig. 2A). To identify the protein domain responsible for the binding of maspin to CL, we created various maspin mutants with truncations highlighted in Fig. 2A and tested their ability to bind to phospholipids in a protein lipid overlay assay. Maspin mutants containing aa 1–139 and 1–225 lost their ability to bind CL (Fig. 2B).
Figure 2.

Maspin binds CL through a CLBD. A) A schematic representing 6 different putative CLBDs (highlighted with blue, yellow, and red bars) in full-length maspin and truncated mutants containing protein sequences rich in hydrophobic amino acids and Lys residues. B) Comparison of the binding of different constructs of maspin to CL using a protein-lipid overlay assay. Only MpWT bound to CL. C) A schematic depicting a maspin construct in which the 7 Lys residues in the CLBD (region highlighted in red) were swapped with the corresponding sequence from the serpin family member, PAI-1 (pink box). A sequence alignment of maspin (AAA18957.1) and PAI-1 (AAA60009.1) demonstrates the homology of these 2 domains in which 7 Lys residues are present in this region of maspin (red box, aa 268–294) and 2 Lys residues in PAI-1 (pink box, aa 298–324). D) Three-dimensional structure of maspin (Protein Data Bank ID: 1XU8) highlighting the CLBD (depicted in black) and Lys residues (depicted in purple) using Internal Coordinate Mechanics (ICM) Pro v.3.48 software. E) A comparison of binding of GST.MpWT and GST.MpPS with various lipids [phospholipids (PL), phosphatidylethanolamine (PE), PC, CL, phosphatidic acid (PA), BSA (mock)] demonstrates the importance of the CLBD in maspin for binding CL. F) PAGE-Western blots demonstrate that CL causes a gel shift of wild-type maspin (GST.MpWT). Representative figures are shown of experiments completed a minimum of 6 times.
Our deletion analysis suggested that the region located after aa 226 in maspin might represent a CLBD. This region of aa 268–294 has a noticeable concentration of hydrophobic residues and contains 7 Lys residues. An analysis of the structure of maspin (Protein Data Bank ID: 1XU8; https://www.wwpdb.org/) indicates that all of these Lys residues are located in a groove or along a face of the protein that may be suitable for binding to CL-rich membranes (Fig. 2C, D). To confirm the role of this CLBD, we swapped this region of maspin with a region from PAI-1, a protein that is functionally homologous to maspin, has not been reported to bind to membranes, and does not contain a characteristically hydrophobic and positively charged region (Fig. 2C). We tested the resulting mutant protein, GST fused with MpPS (GST.MpPS) in the protein lipid overlay assay and protein mobility shift assay and found that the mutant was unable to bind CL in both assays, confirming this region as a CLBD (Fig. 2E, F).
Maspin binds CL, enhances cytochrome c release from mitochondria, and increases cell apoptosis
Guided by these in vitro experiments, we studied the maspin-CL interaction in live mammalian cells. We first manipulated the CL content in cells and analyzed its impact on maspin binding and the induction of apoptosis. For these experiments, we used the CL-deficient cell line PGS-S, which is a Chinese hamster ovary–derived mutant cell line that synthesizes 2–3-fold less CL than wild-type cells at a nonpermissive temperature, and PGS-S/cPGS1 cell line, which is stably transfected with a cDNA encoding PGS as a control (24). We observed that PGS-S and PGS-S/cPGS1 cells express a low level of maspin; therefore, we transfected a maspin expression plasmid into these cells and established stable clones overexpressing maspin (PGS-SMp and PGS-S/cPGS1Mp) (Supplemental Fig. S2). To measure the translocation of maspin from the cytosol to mitochondria in PGS-SMp cells, we treated cells with STS to broadly inhibit protein kinases and induce apoptosis, and we subsequently fractionated their mitochondria and cytosol. We observed that maspin translocated from the cytosol to mitochondria in both PGS-S/cPGS1Mp cells and PGS-S mutant cells (Fig. 3A), which indicates that maspin translocation is not obviously dependent on the concentration of CL in the membrane. Because maspin does not contain a mitochondrial-targeting peptide sequence, these data suggest that its mitochondrial translocation might be facilitated by interactions with other chaperon proteins, as reported for p53 transportation to mitochondria (40).
Figure 3.

The cellular CL concentration does not affect maspin translocation into mitochondria; however, it affects cytochrome c release and the activation of caspase 3. A) A plot comparing maspin translocated into mitochondria in PGS-S/cPGS1Mp and PGS-SMp cells after STS treatment. The amount of CL does not obviously increase the translocation of maspin into cells. COX IV was used as mitochondrial loading control. B) A plot comparing caspase 3 activity [optical density at 405 nm (OD405)] in PGS-S/cPGS1Mp and PGS-SMp cell lysates in the absence and presence of STS using a colorimetric assay. The data indicate that a 2–3-fold reduction in cellular CL reduces the caspase activity by ∼50%. C) A plot comparing cytochrome c (Cyt C) release in PGS-S/cPGS1Mp and PGS-SMp cells after apoptosis induction indicates that reduction in CL leads to a large decrease in cytochrome c in the cytosol.
Release of cytochrome c from mitochondria to the cytosol activates caspase 3 and induces cell apoptosis (41, 42). We next determined whether translocated maspin affects the activation of apoptosis in cells treated with STS by quantifying caspase 3 activity. We observed a significant reduction of caspase 3 activity in PGS-SMp cells that have 2–3-fold less CL compared with the activity present in the wild-type PGS-S/cPGS1Mp cells (Fig. 3B). We also found a significant decrease in the amount of cytochrome c released from mitochondria to the cytosol in PGS-SMp cells compared with PGS-S/cPGS1Mp control cells (Fig. 3C). Following the translocation of maspin into mitochondria in response to an apoptotic stimulus, its influence on apoptosis and cytochrome c release in PGS-SMp cells was attenuated because of a reduced concentration of CL.
Maspin binds CL in the mitochondria and competes with cytochrome c
We performed a mitochondrial import assay to provide further support for the binding of maspin to CL after its translocation into the mitochondria. We harvested mitochondria from wild-type PGS-S/cPGS1 cells and CL-deficient PGS-S cells. Previously, we demonstrated that maspin translocated into mitochondria and associated with the IMM using a mitochondria import assay and by detecting maspin in different mitochondria fractions (7). We performed an import assay using isolated mitochondria that were pretreated with ATR to open the permeability transition pore in the membrane, considering the size of the maspin (43, 44). We then incubated ATR-treated mitochondria with recombinant maspin proteins. After washing the organelles thoroughly, we found a high level of recombinant GST-tagged maspin (GST.MpWT) retained in mitochondria isolated from wild-type PGS-S/cPGS1 cells and an ∼50% lower amount of GST.MpWT from mitochondria of CL-deficient PGS-S cells (Fig. 4A). Critically, the maspin mutant GST.MpPS lacking the CLBD was not retained in the mitochondria of wild-type PGS-S/cPGS1 cells (Supplemental Fig. S3), which we hypothesize is due to the absence of its ability to bind CL tightly. These results confirm the interaction of maspin with CL in the mitochondria.
Figure 4.

Maspin binds CL in the mitochondria and displaces CL-bound cytochrome c (Cyt C). A) A plot comparing the amount of maspin retained in mitochondria isolated from PGS-S/cPGS1 and PGS-S cells; COX IV was used as a mitochondrial loading control. The data indicate that a reduction in cellular CL reduces the concentration of mitochondrial maspin. B) A plot comparing cytochrome c (from the intermitochondrial membrane space) released after ATR treatment of mitochondria isolated from mouse liver. The supernatants were concentrated in a MicroVac and loaded on SDS-PAGE to determine the relative concentration of cytochrome c. ATR treatment led to a large increase in cytochrome c. C) A plot of the amount of cytochrome c released from ATR-treated mitochondria incubated with GST.MpWT compared with GST. The supernatants were concentrated in a MicroVac and loaded on SDS-PAGE to determine the relative concentration of cytochrome c; GST was used as a negative control. D) A plot comparing IMM-bound cytochrome c in mitochondrial pellets after incubating organelles with either GST or GST.MpWT.
Cytochrome c is present in both the intermitochondrial membrane space and in the IMM. At least 15–20% of the cytochrome c in the IMM is bound to CL under normal cellular conditions (45, 46). Our hypothesis is that during the induction of apoptosis, maspin is translocated into mitochondria and competes with cytochrome c for binding to CL-rich regions of the IMM. To test this hypothesis, we isolated mitochondria from mouse liver, treated them with ATR, incubated the organelles with GST.MpWT or GST (negative control), and measured cytochrome c release from the IMM (Supplemental Fig. S4A, B). Mitochondria treated with ATR released cytochrome c from the IMM (Fig. 4B). Incubation of recombinant maspin (GST.MpWT) with ATR-treated mitochondria released significantly more cytochrome c from the IMM than the GST protein control (Fig. 4C). Consequently, we observed a significant reduction in the concentration of cytochrome c in the IMM of mitochondria treated with GST.MpWT compared with the GST-treated organelles (Fig. 4D). These experiments are consistent with the binding of maspin to CL in the mitochondria, which competes for binding sites with cytochrome c and displaces it from the membrane.
Maspin competes with cytochrome c for binding to CL in vitro
To confirm that the competition of maspin for CL reduces the interaction of cytochrome c with the IMM, we designed a competitive binding assay using BLI. We immobilized liposomes containing a mixture of 50:50 PC:CL on the surface of APS-terminated BLI biosensors and monitored the equilibrium wavelength shift, Req, for the binding of protein. To monitor the direct competition between recombinant maspin and cytochrome c binding to CL-containing membranes, we dipped biosensors into wells containing a fixed concentration of total protein (set at 5 μM) and different molar ratios of cytochrome c and maspin. Figure 5A demonstrates the slower binding of maspin to CL-containing membranes compared to cytochrome c. We found that increasing the molar ratio of maspin to cytochrome c reduces Req monotonically (e.g., adding ∼1% maspin was sufficient to reduce Req by ∼25%). At a threshold concentration of ∼2:1 cytochrome c to maspin, we observed kinetic profiles for protein binding to CL-containing membranes that resembled the binding of maspin, and large values of Req at time scales that were longer than for cytochrome c alone, suggesting a slower approach to equilibrium binding. These results were consistent with the kon values (The rate constant for the protein binding to the membranes) that we measured from the apparent Kd and koff values of the individual proteins. Maspin has a slower kon of ∼0.68 M/s compared with a rate of ∼252.6 M/s for cytochrome c. These kinetic values suggest that cytochrome c binds CL faster and more tightly than maspin, which is consistent with our observation that cytochrome c reaches saturation binding to membranes more rapidly than maspin.
Figure 5.
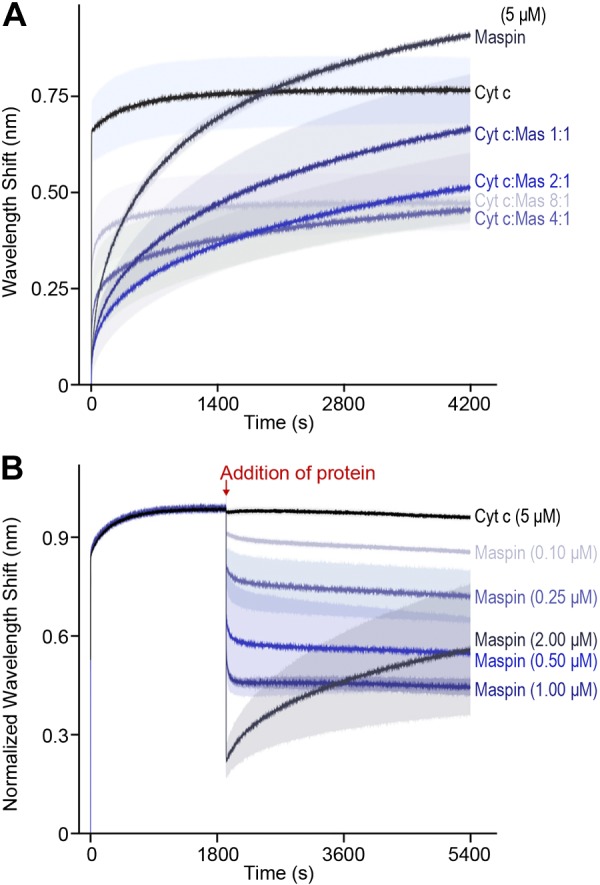
Maspin (mas) occludes and displaces cytochrome c (Cyt c) from binding to PC and CL liposomes. A) To measure the competition of cytochrome c and maspin at membranes, we dipped APS-coated BLI biosensors containing adsorbed PC:CL liposomes (50:50 by molar concentration) into wells containing varying ratios of cytochrome c and maspin and a total protein concentration of 5 μM. We measured the competition of protein for the membrane surface for 4200 s. Cytochrome c and maspin (5 μM) are shown in black and dark blue, respectively. Decreasing concentrations of maspin (cytochrome c:maspin molar ratios of 1:1, 2:1, 4:1, and 8:1) are indicated by the color gradient (dark to light blue). Lines indicate the mean wavelength shift values (based on n = 3 measurements), and the shading indicates the the mean ± sd. B) To measure the displacement of cytochrome c by maspin, we equilibrated APS-coated BLI biosensors containing adsorbed PC:CL liposomes (50:50 by molar concentration) into wells containing cytochrome c (5 μM) for 2000 s, then dipped into wells containing 5 μM cytochrome and different concentrations of maspin. The black curve depicts data for cytochrome c (5 μM) dipped into a well containing 5 μM cytochrome c (as a control). Increasing concentrations of maspin are indicated by the color gradient (from light to dark blue). The competition of maspin and cytochrome c was monitored for 7000 s before returning tips to baseline wells to monitor dissociation (unpublished results). Lines indicate the means of 6 experiments, and shading indicates the mean ± sd (A, B).
The significance of the 2:1 cytochrome c to maspin stoichiometry that we observed may be connected to the in vivo concentration of maspin required to displace cytochrome c from the membrane and inhibit its reassociation. Strikingly, the results from the competitive binding assay diverge from what would be expected based on a comparison of the apparent Kd. The apparent Kd for maspin estimated from the binding isotherm suggests that it is a weaker binder by a factor of 10 compared with cytochrome c. Consequently, it is surprising that maspin can significantly compete for binding and displace cytochrome c from CL-containing membranes at a concentration as low as 1% of the total protein. However, the data from our competition experiments between maspin and cytochrome c indicate that maspin monotonically decreases Req and inhibits the fast reassociation of cytochrome c with CL in the membrane. To interpret this reduction in Req with increasing maspin concentration, we estimated the volume occupied by a molecule of cytochrome c and maspin at the biosensor surface by assuming spherical geometry. We used the reported value of the Stokes radius of cytochrome c of 17 Å (47) to estimate a volume of ∼20 nm3. Although hydrodynamic data are not available for maspin, we estimated the protein to have a minimum molecular volume of ∼50 nm3 based on its MW; we expect that its volume in solution is likely larger because of hydration and irregularities in the protein surface (48). This simple model estimates that maspin occupies ∼2.5 times more volume at a membrane compared with cytochrome c, and each maspin molecule occludes ∼3 molecules of cytochrome, which is compatible with the data we observed (Fig. 5A). An alternative explanation that is also consistent with our results is that maspin binds to CL cooperatively and decreases the affinity of cytochrome c for the membrane. We were unable to measure cooperativity because of the large quantity of protein required to reach saturation with this assay, and this hypothesis requires further investigation to test how the cooperativity of maspin may be connected to apoptotic signaling.
To study maspin competing directly with cytochrome c for binding to CL in the membrane, we adsorbed liposomes (50:50 PC:CL) on BLI biosensors and incubated them in a solution of 5 μM cytochrome c until we observed binding reach equilibrium (Fig. 5B). We subsequently transferred the sensors containing cytochrome c bound to bilayer membranes into wells containing different relative concentrations of maspin and cytochrome c. We hypothesized that introducing maspin to membrane-bound cytochrome c would decrease Req because of a reduction in the amount of protein on the membrane. To test this hypothesis, we transferred biosensors equilibrated with 5 μM cytochrome c into wells containing cytochrome c and increasing concentrations of maspin. When cytochrome c–bound sensors were placed in wells containing 5 μM cytochrome c and 2 μM maspin, we observed a sharp decrease in sensor response and reduced binding of cytochrome c that correlated to an increase in maspin concentration (Fig. 5B). The results of our in vitro experiments indicate that maspin binds CL, competes with cytochrome c for binding to the membrane, and displaces it from the membrane, thereby corroborating our in vivo results.
A search of PaxDb enabled us to compare the abundance of maspin and cytochrome c in human epithelial-derived tumor cells, including those from mammary, prostate, and Hela cell lines (49, 50). As shown in Supplemental Table S2, the concentration of maspin and cytochrome c is similar in these tumor cells. In this current study and our previous reports (7, 25), we used maspin-overexpressing TM40D (TM40DMp) cells in which maspin expression level is elevated at least 10-fold higher than in a parental tumor cell line. Consequently, we estimate that the concentration of maspin in our TM40DMp tumor cells is likely 10 times higher than cytochrome c in these tumor cells, which offsets its ∼1 order of magnitude lower binding affinity for CL in the mitochondria.
Tumor cells expressing a mutant maspin with reduced CL-binding deficiency have a low frequency of apoptosis
Cancer cells are defective in their response to apoptosis. In contrast to normal cells, defects in the ability of tumor cells to activate the death-signaling pathway reduces their frequency of apoptosis (3). Overexpression of maspin in TM40D murine cancer cells induced higher rates of tumor cell apoptosis in mammary tumors (6, 51). With these observations in mind, we selected a mouse mammary cell line (TM40D) as a model system to determine the effect of the maspin-CL interaction on cancer cell apoptosis. We transfected cells with constructs expressing MpWT or MpPS. We generated stable expression clones from cells transfected with MpWT (TM40DMp) and mutant MpPS (TM40DPS) and confirmed the expression level of maspin by Western blot analysis (Supplemental Fig. S5A).
We stimulated TM40DMp and TM40DPS cells with STS and observed that maspin translocates into the mitochondria (Fig. 6A and Supplemental Fig. S5B, C). Although the mutant maspin (TM40DPS) is still able to translocate to mitochondria, it clearly loses its ability to affect cell apoptosis. These data also suggest that maspin’s translocation to mitochondria is not dependent on its CLBD domain. TM40DPS cells showed an attenuated level of caspase 3 activity compared with TM40DMp cells (Fig. 6B), indicating that reduced maspin binding to CL decreased the cellular apoptotic response. Accordingly, the release of cytochrome c was attenuated in TM40DPS cells compared with TM40DMp cells (Fig. 6C). These observations confirm that maspin binding to CL in the mitochondria plays a role in tumor cell apoptosis.
Figure 6.

TM40DPS cells expressing mutant maspin lacking the CLBD translocate to mitochondria and display a low apoptosis efficiency. A) A plot comparing maspin translocated to mitochondria in TM40DMp (MpWT) and TM40DPS cells (containing the corresponding sequence from PAI-1 swapped with the CLBD in maspin) after STS treatment; COX IV was used as a mitochondrial loading control. B) A plot comparing caspase 3 activity [optical density at 405 nm (OD405)] in TM40DMp and TM40DPS cell lysates in the absence and presence of STS using a Colorimetric Assay Kit. C) A plot comparing cytochrome c (Cyt C) released into the cytosol was determined in TM40DMp and TM40DPS cells after apoptosis induction. GAPDH was as used as a cytosolic loading control.
DISCUSSION
Our study addresses the molecular mechanism that underlies the release of cytochrome c from mitochondria and induction of apoptosis. Based on the results of our current and previous studies (7), we propose a model for the action of maspin in apoptosis (Fig. 7). Upon induction of apoptosis, maspin translocates to the IMM in a step that is independent of the amount of CL in mitochondria and binds to CL through a specific CLBD that contains a segment of 27 aa enriched in hydrophobic and Lys residues. Although this CLBD region in maspin is necessary for CL binding and apoptosis induction, simply incubating a peptide containing the 27 aa with mammary tumor cells does not increase tumor cell apoptosis (unpublished results). This is likely because peptide alone could not hold the structure required for CL binding. The interaction of maspin and CL competes for membrane-binding sites in the IMM and leads to the displacement of cytochrome c from the membrane in vivo and in vitro, enhances caspase 3 activity in cells, and increases the frequency of cell apoptosis. A significant rearrangement of the membrane takes place during apoptosis (52–58) that may also contribute to the interaction of maspin with the IMM and its relationship to membrane-bound cytochrome c. Results of this study provide a biochemical and biophysical basis for the interaction of maspin with the IMM and its effect on cytochrome c anchoring in the membrane, which has a direct consequence on the mediation of apoptosis by mitochondria.
Figure 7.

Proposed model for maspin-CL binding in the IMM and release of cytochrome c into the cytoplasm A) Cytochrome c (Cyt C) is bound to CL (depicted in red) in the IMM. Maspin (Mp) is in the cytosol. B) Apoptosis is induced and maspin translocated from the cytosol into mitochondria. C) Maspin competes with cytochrome c for binding to CL in the IMM, and cytochrome c is released from mitochondria and triggers apoptosis.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Dr. M. Geiger (Medical University of Vienna, Vienna, Austria) for stimulating discussions and for providing the protein-lipid overlay assay protocol. This work was supported by the U.S. National Institutes of Health (NIH) National Cancer Institute (Grant CA079736 to M.Z.), NIH Office of the Director (Grant 1DP2OD008735-01 to D.B.W.) and the U.S. National Science Foundation (Grant DMR-1121288; University of Wisconsin–Madison). M.R. was supported by the Dr. James Chieh-Hsia Mao Wisconsin Distinguished Graduate Fellowship, B.H. by an NIH Molecular Biophysics Predoctoral Traineeship (T32 GM08293), and H.Y.S by the Northwestern University Zell Foundation. The authors declare no conflicts of interest.
Glossary
- ATR
atractyloside
- APS
aminopropylsilane
- Bcl-2
B-cell lymphoma 2
- Bid
Bcl-2 homology domain 3 interacting-domain death agonist
- BLI
biolayer interferometry
- CHO-K1
temperature-sensitive Chinese hamster ovary
- CL
cardiolipin
- CLBD
CL binding domain
- COX IV
cytochrome c oxidase subunit IV
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GST
glutathione S-transferase
- GST.MpPS
GST fused with MpPS
- GST.MpWT
GST fused with MpWT
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IMM
inner mitochondrial membrane
- MpPS
maspin–PAI-1 swap
- MpWT
wild-type maspin
- PC
phosphatidylcholine
- PAI-1
plasminogen activator inhibitor 1
- PaxDb
Protein Abundance Database
- PGS
phosphatidylglycerophosphate synthase
- STS
staurosporine
- TM40DMp
maspin-overexpressing TM40D
- TM40DPS
MpPS-transfected TM40D
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
N. Mahajan, H. Y. Shi, and M. Zhang designed the research, performed the experiments, analyzed the data, and wrote the manuscript; B. Hoover, M. Rajendram, and D. B. Weibel designed the research, the experiments, analyzed the data, and wrote the section for the maspin-cardiolipin binding assays; and K. Kawasaki proposed the apoptosis assay involving PGS-S and PGS-S/cPGS1 cells and contributed the reagents.
REFERENCES
- 1.Galluzzi L., Kepp O., Kroemer G. (2012) Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788 [DOI] [PubMed] [Google Scholar]
- 2.De Bruin E. C., Medema J. P. (2008) Apoptosis and non-apoptotic deaths in cancer development and treatment response. Cancer Treat. Rev. 34, 737–749 [DOI] [PubMed] [Google Scholar]
- 3.Estaquier J., Vallette F., Vayssiere J. L., Mignotte B. (2012) The mitochondrial pathways of apoptosis. Adv. Exp. Med. Biol. 942, 157–183 [DOI] [PubMed] [Google Scholar]
- 4.Bailey C. M., Khalkhali-Ellis Z., Seftor E. A., Hendrix M. J. (2006) Biological functions of maspin. J. Cell. Physiol. 209, 617–624 [DOI] [PubMed] [Google Scholar]
- 5.Zou Z., Anisowicz A., Hendrix M. J., Thor A., Neveu M., Sheng S., Rafidi K., Seftor E., Sager R. (1994) Maspin, a serpin with tumor-suppressing activity in human mammary epithelial cells. Science 263, 526–529 [DOI] [PubMed] [Google Scholar]
- 6.Jiang N., Meng Y., Zhang S., Mensah-Osman E., Sheng S. (2002) Maspin sensitizes breast carcinoma cells to induced apoptosis. Oncogene 21, 4089–4098 [DOI] [PubMed] [Google Scholar]
- 7.Latha K., Zhang W., Cella N., Shi H. Y., Zhang M. (2005) Maspin mediates increased tumor cell apoptosis upon induction of the mitochondrial permeability transition. Mol. Cell. Biol. 25, 1737–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao F., Shi H. Y., Daughty C., Cella N., Zhang M. (2004) Maspin plays an essential role in early embryonic development. Development 131, 1479–1489 [DOI] [PubMed] [Google Scholar]
- 9.Cella N., Contreras A., Latha K., Rosen J. M., Zhang M. (2006) Maspin is physically associated with [beta]1 integrin regulating cell adhesion in mammary epithelial cells. FASEB J. 20, 1510–1512 [DOI] [PubMed] [Google Scholar]
- 10.Li J. J., Colburn N. H., Oberley L. W. (1998) Maspin gene expression in tumor suppression induced by overexpressing manganese-containing superoxide dismutase cDNA in human breast cancer cells. Carcinogenesis 19, 833–839 [DOI] [PubMed] [Google Scholar]
- 11.Mahajan N., Shi H. Y., Lukas T. J., Zhang M. (2013) Tumor-suppressive maspin functions as a reactive oxygen species scavenger: importance of cysteine residues. J. Biol. Chem. 288, 11611–11620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin S., Li X., Meng Y., Finley R. L., Jr., Sakr W., Yang H., Reddy N., Sheng S. (2005) Tumor-suppressive maspin regulates cell response to oxidative stress by direct interaction with glutathione S-transferase. J. Biol. Chem. 280, 34985–34996 [DOI] [PubMed] [Google Scholar]
- 13.Sheng S. (2006) A role of novel serpin maspin in tumor progression: the divergence revealed through efforts to converge. J. Cell. Physiol. 209, 631–635 [DOI] [PubMed] [Google Scholar]
- 14.Liu J., Yin S., Reddy N., Spencer C., Sheng S. (2004) Bax mediates the apoptosis-sensitizing effect of maspin. Cancer Res. 64, 1703–1711 [DOI] [PubMed] [Google Scholar]
- 15.Gonzalvez F., Gottlieb E. (2007) Cardiolipin: setting the beat of apoptosis. Apoptosis 12, 877–885 [DOI] [PubMed] [Google Scholar]
- 16.Schug Z. T., Gottlieb E. (2009) Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta 1788, 2022–2031 [DOI] [PubMed] [Google Scholar]
- 17.Schlame M. (2008) Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 49, 1607–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Claypool S. M., Koehler C. M. (2012) The complexity of cardiolipin in health and disease. Trends Biochem. Sci. 37, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terrones O., Antonsson B., Yamaguchi H., Wang H. G., Liu J., Lee R. M., Herrmann A., Basañez G. (2004) Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J. Biol. Chem. 279, 30081–30091 [DOI] [PubMed] [Google Scholar]
- 20.Nicholls P. (1974) Cytochrome c binding to enzymes and membranes. Biochim. Biophys. Acta 346, 261–310 [DOI] [PubMed] [Google Scholar]
- 21.O’Brien E. S., Nucci N. V., Fuglestad B., Tommos C., Wand A. J. (2015) Defining the apoptotic trigger: the interaction of cytochrome c and cardiolipin. J. Biol. Chem. 290, 30879–30887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravenhill L., Wagstaff L., Edwards D. R., Ellis V., Bass R. (2010) G-helix of maspin mediates effects on cell migration and adhesion. J. Biol. Chem. 285, 36285–36292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohtsuka T., Nishijima M., Akamatsu Y. (1993) A somatic cell mutant defective in phosphatidylglycerophosphate synthase, with impaired phosphatidylglycerol and cardiolipin biosynthesis. J. Biol. Chem. 268, 22908–22913 [PubMed] [Google Scholar]
- 24.Kawasaki K., Kuge O., Chang S. C., Heacock P. N., Rho M., Suzuki K., Nishijima M., Dowhan W. (1999) Isolation of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylglycerophosphate (PGP) synthase, expression of which corrects the mitochondrial abnormalities of a PGP synthase-defective mutant of CHO-K1 cells. J. Biol. Chem. 274, 1828–1834 [DOI] [PubMed] [Google Scholar]
- 25.Zhang W., Shi H. Y., Zhang M. (2005) Maspin overexpression modulates tumor cell apoptosis through the regulation of Bcl-2 family proteins. BMC Cancer 5, 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang G., Yan G., Gurtu V., Spencer C., Kain S. R. (1998) Caspase inhibition prevents staurosporine-induced apoptosis in CHO-K1 cells. Apoptosis 3, 27–33 [DOI] [PubMed] [Google Scholar]
- 27.Deak M., Casamayor A., Currie R. A., Downes C. P., Alessi D. R. (1999) Characterisation of a plant 3-phosphoinositide-dependent protein kinase-1 homologue which contains a pleckstrin homology domain. FEBS Lett. 451, 220–226 [DOI] [PubMed] [Google Scholar]
- 28.Esposti M. D., Cristea I. M., Gaskell S. J., Nakao Y., Dive C. (2003) Proapoptotic Bid binds to monolysocardiolipin, a new molecular connection between mitochondrial membranes and cell death. Cell Death Differ. 10, 1300–1309 [DOI] [PubMed] [Google Scholar]
- 29.Arnold M., Ringler P., Brisson A. (1995) A quantitative electrophoretic migration shift assay for analyzing the specific binding of proteins to lipid ligands in vesicles or micelles. Biochim. Biophys. Acta 1233, 198–204 [DOI] [PubMed] [Google Scholar]
- 30.Dennison S. M., Anasti K. M., Jaeger F. H., Stewart S. M., Pollara J., Liu P., Kunz E. L., Zhang R., Vandergrift N., Permar S., Ferrari G., Tomaras G. D., Bonsignori M., Michael N. L., Kim J. H., Kaewkungwal J., Nitayaphan S., Pitisuttithum P., Rerks-Ngarm S., Liao H. X., Haynes B. F., Alam S. M. (2014) Vaccine-induced HIV-1 envelope gp120 constant region 1-specific antibodies expose a CD4-inducible epitope and block the interaction of HIV-1 gp140 with galactosylceramide. J. Virol. 88, 9406–9417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong Y., Muenzner J., Grimm S. K., Pletneva E. V. (2012) Origin of the conformational heterogeneity of cardiolipin-bound cytochrome C. J. Am. Chem. Soc. 134, 18713–18723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frezza C., Cipolat S., Scorrano L. (2007) Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2, 287–295 [DOI] [PubMed] [Google Scholar]
- 33.Daum G. (1985) Lipids of mitochondria. Biochim. Biophys. Acta 822, 1–42 [DOI] [PubMed] [Google Scholar]
- 34.Khalifat N., Puff N., Bonneau S., Fournier J. B., Angelova M. I. (2008) Membrane deformation under local pH gradient: mimicking mitochondrial cristae dynamics. Biophys. J. 95, 4924–4933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stepanov G., Gnedenko O., Mol’nar A., Ivanov A., Vladimirov Y., Osipov A. (2009) Evaluation of cytochrome c affinity to anionic phospholipids by means of surface plasmon resonance. FEBS Lett. 583, 97–100 [DOI] [PubMed] [Google Scholar]
- 36.Domanov Y. A., Molotkovsky J. G., Gorbenko G. P. (2005) Coverage-dependent changes of cytochrome c transverse location in phospholipid membranes revealed by FRET. Biochim. Biophys. Acta 1716, 49–58 [DOI] [PubMed] [Google Scholar]
- 37.Lutter M., Fang M., Luo X., Nishijima M., Xie X., Wang X. (2000) Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat. Cell Biol. 2, 754–761 [DOI] [PubMed] [Google Scholar]
- 38.Li C. H., Cheng Y. W., Liao P. L., Kang J. J. (2010) Translocation of p53 to mitochondria is regulated by its lipid binding property to anionic phospholipids and it participates in cell death control. Neoplasia 12, 150–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kertesz Z., Yu B. B., Steinkasserer A., Haupt H., Benham A., Sim R. B. (1995) Characterization of binding of human beta 2-glycoprotein I to cardiolipin. Biochem. J. 310, 315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merrick B. A., He C., Witcher L. L., Patterson R. M., Reid J. J., Pence-Pawlowski P. M., Selkirk J. K. (1996) HSP binding and mitochondrial localization of p53 protein in human HT1080 and mouse C3H10T1/2 cell lines. Biochim. Biophys. Acta 1297, 57–68 [DOI] [PubMed] [Google Scholar]
- 41.Ott M., Robertson J. D., Gogvadze V., Zhivotovsky B., Orrenius S. (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 99, 1259–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J., Yuan J. (2008) Caspases in apoptosis and beyond. Oncogene 27, 6194–6206 [DOI] [PubMed] [Google Scholar]
- 43.Bruey J. M., Ducasse C., Bonniaud P., Ravagnan L., Susin S. A., Diaz-Latoud C., Gurbuxani S., Arrigo A. P., Kroemer G., Solary E., Garrido C. (2000) Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2, 645–652 [DOI] [PubMed] [Google Scholar]
- 44.Joshi A., Bondada V., Geddes J. W. (2009) Mitochondrial micro-calpain is not involved in the processing of apoptosis-inducing factor. Exp. Neurol. 218, 221–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ow Y. P., Green D. R., Hao Z., Mak T. W. (2008) Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542 [DOI] [PubMed] [Google Scholar]
- 46.Garrido C., Galluzzi L., Brunet M., Puig P. E., Didelot C., Kroemer G. (2006) Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 13, 1423–1433 [DOI] [PubMed] [Google Scholar]
- 47.Talmard C., Guilloreau L., Coppel Y., Mazarguil H., Faller P. (2007) Amyloid-beta peptide forms monomeric complexes with Cu(II) and Zn(II) prior to aggregation. Chembiochem 8, 163–165 [DOI] [PubMed] [Google Scholar]
- 48.Erickson H. P. (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 11, 32–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geiger T., Wehner A., Schaab C., Cox J., Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell Proteomics 11, M111.014050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Pääbo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 7, 548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi H. Y., Zhang W., Liang R., Abraham S., Kittrell F. S., Medina D., Zhang M. (2001) Blocking tumor growth, invasion, and metastasis by maspin in a syngeneic breast cancer model. Cancer Res. 61, 6945–6951 [PubMed] [Google Scholar]
- 52.Fadok V. A., Voelker D. R., Campbell P. A., Cohen J. J., Bratton D. L., Henson P. M. (1992) Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216 [PubMed] [Google Scholar]
- 53.Ashman R. F., Peckham D., Alhasan S., Stunz L. L. (1995) Membrane unpacking and the rapid disposal of apoptotic cells. Immunol. Lett. 48, 159–166 [DOI] [PubMed] [Google Scholar]
- 54.Verhoven B., Schlegel R. A., Williamson P. (1995) Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J. Exp. Med. 182, 1597–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlegel R. A., Callahan M., Krahling S., Pradhan D., Williamson P. (1996) Mechanisms for recognition and phagocytosis of apoptotic lymphocytes by macrophages. Adv. Exp. Med. Biol. 406, 21–28 [DOI] [PubMed] [Google Scholar]
- 56.Krahling S., Callahan M. K., Williamson P., Schlegel R. A. (1999) Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 6, 183–189 [DOI] [PubMed] [Google Scholar]
- 57.Callahan M. K., Williamson P., Schlegel R. A. (2000) Surface expression of phosphatidylserine on macrophages is required for phagocytosis of apoptotic thymocytes. Cell Death Differ. 7, 645–653 [DOI] [PubMed] [Google Scholar]
- 58.Fadeel B., Orrenius S. (2005) Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J. Intern. Med. 258, 479–517 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.