

Abstract
Background
People experiencing acute psychotic illnesses, especially those associated with agitated or violent behaviour, may require urgent pharmacological tranquillisation or sedation. Droperidol, a butyrophenone antipsychotic, has been used for this purpose in several countries.
Objectives
To estimate the effects of droperidol, including its cost‐effectiveness, when compared to placebo, other 'standard' or 'non‐standard' treatments, or other forms of management of psychotic illness, in controlling acutely disturbed behaviour and reducing psychotic symptoms in people with schizophrenia‐like illnesses.
Search methods
We updated previous searches by searching the Cochrane Schizophrenia Group Register (18 December 2015). We searched references of all identified studies for further trial citations and contacted authors of trials. We supplemented these electronic searches by handsearching reference lists and contacting both the pharmaceutical industry and relevant authors.
Selection criteria
We included all randomised controlled trials (RCTs) with useable data that compared droperidol to any other treatment for people acutely ill with suspected acute psychotic illnesses, including schizophrenia, schizoaffective disorder, mixed affective disorders, the manic phase of bipolar disorder or a brief psychotic episode.
Data collection and analysis
For included studies, we assessed quality, risk of bias and extracted data. We excluded data when more than 50% of participants were lost to follow‐up. For binary outcomes, we calculated standard estimates of risk ratio (RR) and the corresponding 95% confidence intervals (CI). We created a 'Summary of findings' table using GRADE.
Main results
We identified four relevant trials from the update search (previous version of this review included only two trials). When droperidol was compared with placebo, for the outcome of tranquillisation or asleep by 30 minutes we found evidence of a clear difference (1 RCT, N = 227, RR 1.18, 95% CI 1.05 to 1.31, high‐quality evidence). There was a clear demonstration of reduced risk of needing additional medication after 60 minutes for the droperidol group (1 RCT, N = 227, RR 0.55, 95% CI 0.36 to 0.85, high‐quality evidence). There was no evidence that droperidol caused more cardiovascular arrhythmia (1 RCT, N = 227, RR 0.34, 95% CI 0.01 to 8.31, moderate‐quality evidence) and respiratory airway obstruction (1 RCT, N = 227, RR 0.62, 95% CI 0.15 to 2.52, low‐quality evidence) than placebo. For 'being ready for discharge', there was no clear difference between groups (1 RCT, N = 227, RR 1.16, 95% CI 0.90 to 1.48, high‐quality evidence). There were no data for mental state and costs.
Similarly, when droperidol was compared to haloperidol, for the outcome of tranquillisation or asleep by 30 minutes we found evidence of a clear difference (1 RCT, N = 228, RR 1.01, 95% CI 0.93 to 1.09, high‐quality evidence). There was a clear demonstration of reduced risk of needing additional medication after 60 minutes for participants in the droperidol group (2 RCTs, N = 255, RR 0.37, 95% CI 0.16 to 0.90, high‐quality evidence). There was no evidence that droperidol caused more cardiovascular hypotension (1 RCT, N = 228, RR 2.80, 95% CI 0.30 to 26.49,moderate‐quality evidence) and cardiovascular hypotension/desaturation (1 RCT, N = 228, RR 2.80, 95% CI 0.12 to 67.98, low‐quality evidence) than haloperidol. There was no suggestion that use of droperidol was unsafe. For mental state, there was no evidence of clear difference between the efficacy of droperidol compared to haloperidol (Scale for Quantification of Psychotic Symptom Severity, 1 RCT, N = 40, mean difference (MD) 0.11, 95% CI ‐0.07 to 0.29, low‐quality evidence). There were no data for service use and costs.
Whereas, when droperidol was compared with midazolam, for the outcome of tranquillisation or asleep by 30 minutes we found droperidol to be less acutely tranquillising than midazolam (1 RCT, N = 153, RR 0.96, 95% CI 0.72 to 1.28, high‐quality evidence). As regards the 'need for additional medication by 60 minutes after initial adequate sedation, we found an effect (1 RCT, N = 153, RR 0.54, 95% CI 0.24 to 1.20, moderate‐quality evidence). In terms of adverse effects, we found no statistically significant differences between the two drugs for either airway obstruction (1 RCT, N = 153, RR 0.13, 95% CI 0.01 to 2.55, low‐quality evidence) or respiratory hypoxia (1 RCT, N = 153, RR 0.70, 95% CI 0.16 to 3.03, moderate‐quality evidence) ‐ but use of midazolam did result in three people (out of around 70) needing some sort of 'airway management' with no such events in the droperidol group. There were no data for mental state, service use and costs.
Furthermore, when droperidol was compared to olanzapine, for the outcome of tranquillisation or asleep by any time point, we found no clear differences between the older drug (droperidol) and olanzapine (e.g. at 30 minutes: 1 RCT, N = 221, RR 1.02, 95% CI 0.94 to 1.11, high‐quality evidence). There was a suggestion that participants allocated droperidol needed less additional medication after 60 minutes than people given the olanzapine (1 RCT, N = 221, RR 0.56, 95% CI 0.36 to 0.87, high‐quality evidence). There was no evidence that droperidol caused more cardiovascular arrhythmia (1 RCT, N = 221, RR 0.32, 95% CI 0.01 to 7.88, moderate‐quality evidence) and respiratory airway obstruction (1 RCT, N = 221, RR 0.97, 95% CI 0.20 to 4.72, low‐quality evidence) than olanzapine. For 'being ready for discharge', there was no difference between groups (1 RCT, N = 221, RR 1.06, 95% CI 0.83 to 1.34, high‐quality evidence). There were no data for mental state and costs.
Authors' conclusions
Previously, the use of droperidol was justified based on experience rather than evidence from well‐conducted and reported randomised trials. However, this update found high‐quality evidence with minimal risk of bias to support the use of droperidol for acute psychosis. Also, we found no evidence to suggest that droperidol should not be a treatment option for people acutely ill and disturbed because of serious mental illnesses.
Plain language summary
Droperidol for psychosis‐induced aggression or agitation
Is droperidol effective for managing people who are aggressive or agitated due to psychosis?
Background
People with psychosis can experience symptoms such as hallucinations (seeing or hearing things that are not there) or delusions (belief in things that are bizarre or obviously not true). These symptoms are often disturbing and frightening, and can lead to people with psychosis becoming very disturbed, violent or agitated. Droperidol is one of the medicines normally used to help calm (tranquillise) people in this situation. Previously, the use of this drug was based on results from small clinical trials with no firm conclusion regarding its effects. Larger trials were needed.
Searching
In 2015, the Information Specialist of the Cochrane Schizophrenia Group updated previous searches of their specialised register of studies. The review authors identified and screened 21 records.
Description of studies
Six randomised controlled studies are now included in the review. All the studies randomised people who were aggressive or agitated due to psychosis to receive either droperidol or placebo (a pretend medicine), haloperidol, olanzapine or midazolam. The size of the studies ranged from 40 to 221 participants. All took place in within a hospital. Four of the six studies were under two hours of duration.
Main results
Compared to placebo, droperidol was more effective at tranquillising agitated participants 30 minutes after taking it. Similar results were found for tranquillisation when droperidol was compared with haloperidol but this effect was less clear, and not evident when droperidol was compared to midazolam or olanzapine. Droperidol did not cause more side effects than the other drugs in the studies. The studies did not look at costs.
Review authors' conclusions
Although we could only include six studies, they provided high‐quality evidence suggesting that droperidol is effective and can be used to control people with very disturbed and aggressive behaviours caused by psychosis.
Summary of findings
Background
Description of the condition
Violent or acutely disturbed people pose a risk to themselves and to others, as well as a diagnostic dilemma (Thomas 1992). The actual prevalence of violent behaviour is high although percentages differ according to setting, definition, client group and measure (Latalova 2014). For people presenting with first episode of illness, serious violence has been reported in anything between 2% and nearly 30% (Latalova 2014). Violent behaviour may be more prevalent at this point in a person's illness, when their symptoms may have gone unnoticed for some time, and they are more vigorous than later on in life (Winsper 2013).
Ideally, to ensure a safe and therapeutic environment, attempts should be made to calm the person either through verbal de‐escalation or intensive nursing techniques. Behaviour may frequently be too disturbed or agitated for 'verbal tranquillisation' to be effective, and further action, in the form of rapid tranquillisation, may be necessary.
Description of the intervention
Various drug regimens are used in such emergency situations, and clinical practice differs. One survey from the USA found that the medical directors of 20 emergency rooms preferred drug management for aggressive people to be a haloperidol‐lorazepam mixture (Table 5) (Binder 1999). In 1993, a similar survey of clinicians' preferences in the UK found that chlorpromazine was the most common choice (Cunnane 1994). Another survey of emergency rooms in Rio de Janeiro found that a haloperidol‐promethazine mixture was commonly used for emergency intramuscular (IM) sedation of severely agitated/aggressive people (70 to 100 people with suspected psychotic illness per week per 3.5 million; Table 6) (Huf 2002). A survey of frequency of emergency prescribing in a general psychiatric hospital in South London (UK) showed that rapid medical tranquillisation was required 102 times in 160 days (Pilowsky 1992). Eight different drugs were used, amongst which diazepam, haloperidol and droperidol were used most often (Table 7).
1. Survey of 20 medical directors of emergency departments in the USA.
| Favoured drug | Number |
| Haloperidol + lorazepam ± benztropine | 11 |
| Droperidol | 4 |
| Benzodiazepine (unspecified) alone | 3 |
| Droperidol + lorazepam + diphenhydramine | 1 |
| Haloperidol + benztropine | 1 |
2. Preferred medication for rapid tranquillisation in Rio de Janeiro.
| Drug of choice | Mean dose (mg) | Frequency of use |
| Haloperidol + promethazine | 5 (2.5 to 10) + 50 (25 to 100) | 61% |
| Haloperidol + promethazine + diazepam | 5 (2.5 to 10) + 50 (25 to 100) +10 | 15% |
| Diazepam | 10 | 9% |
| Haloperidol + promethazine + chlorpromazine | 5 + 50 + 25 | 7% |
| Chlorpromazine + diazepam + promethazine | 25 + 10 + 50 | 1% |
| Chlorpromazine + promethazine | 25 + 50 | 1% |
| Chlorpromazine | 25 | 1% |
| Diazepam + promethazine | 10 + 50 | 1% |
| Haloperidol + diazepam | 5 + 10 | 1% |
| Promethazine | 50 | 1% |
3. Drugs for rapid tranquillisation in London survey.
| Drug of choice | Mean dose (mg) |
| Diazepam* | 27 (10 to 80) |
| Haloperidol | 22 (10 to 60) |
| Chlorpromazine | 162 (50 to 400) |
| Droperidol | 14 (10 to 20) |
| Paraldehyde | U/K |
| Amytal | U/K |
| Lorazepam | U/K |
| Nitrazepam** | U/K |
* most frequent; **least frequent; U/K: unknown.
Droperidol (marketed as Dehydrobenzperidol, Dridol, Droleptan, Droperidols, Inapsin, Inapsine, Leptanal comp, Leptofen, Paxical or Sintodian) has been widely used in Europe by psychiatrists since the 1960s for treating acute or chronic psychoses (Cocito 1970; Resnick 1984). It inhibits the effects of dopamine. In the USA, it is used primarily in conjunction with anaesthetics because of its sedative and antiemetic properties (Resnick 1984). Reported advantages of droperidol over haloperidol (another inhibitor of the effects of dopamine) include: a faster onset of action when given IM, swifter elimination from the body and fewer adverse effects (Richards 1998). The most commonly reported adverse effects for droperidol include hypotension (abnormally low blood pressure) and tachycardia (above normal heart rate). Other adverse effects include restlessness, hyperactivity, anxiety and dysphoria (feeling ill at ease). The frequency of adverse effects involving movement disorders is reported to be 20% to 40% (Cocito 1970). Droperidol has been associated only rarely with serious adverse effects such as neuroleptic malignant syndrome (altered consciousness, muscle rigidity and autonomic instability) and sudden death. Sudden death has been reported to be associated with high doses of droperidol (25 mg or more) in people at risk for cardiac dysrhythmia, such as people with severe electrolyte disturbances or alcohol withdrawal (RxList 2000). Droperidol should not be given to people with severe depression as it may aggravate their symptoms (Martindale 1982).
Following an extensive risk‐benefit assessment requested by the Medicines Control Agency, Janssen‐Cilag, the pharmaceutical company who market droperidol, concluded that the oral form of droperidol should be discontinued and that the injectable form would no longer be commercially viable. The Medical Director of Janssen‐Cilag told PharmaTimes (www.pharmatimes.co.uk/) that the decision had been taken because many people who receive droperidol also receive other medications that extend QT prolongation, and are more likely to have background illnesses that may exacerbate the problem. He added that the company intended to implement a world‐wide withdrawal of droperidol, and supplies would stop entering the distribution chain at the end of March 2001. This seems not to have happened and droperidol has been used for this and other purposes (Furyk 2015; Storrar 2014), and research has continued. Some authoritative findings are not supportive of the original decision regarding QT prolongation (Calver 2015), and call into question the original Food and Drugs Authority decision and their decision‐making process (Newman 2015).
How the intervention might work
Droperidol, 1‐(1‐3‐(p‐fluorobenzoyl)propyl‐1,2,3,6‐tetrahydro‐4‐pyridyl)‐2‐benzimidazolinone, is a butyrophenone neuroleptic drug (Figure 1). Butyrophenones inhibit the effects of dopamine and resemble phenothiazines such as trifluoperazine. They have fewer sedative and antimuscarinic effects than other phenothiazine derived antipsychotic drugs, but exhibit more pronounced adverse effects upon the extrapyramidal nerve system. Back in March 2000, the cost of medication with droperidol (Droleptan) in Great Britain was GBP0.90 for a 2 mL amp injection, or GBP0.25 for a 10 mg tablet (BNF 2000). Droperidol may be taken orally (5 mg to 20 mg repeated every four to six hours, as necessary) or as an IM or intravenous (IV) injection (dosages: up to 10 mg repeated every four to six hours for IM; and 5 mg to 15 mg repeated every four to six hours for IV). The onset of action from injection is 3 to 10 minutes, although the peak effect may not be apparent for 30 minutes. The duration of sedation and tranquillisation may last for two to four hours, although alteration of alertness may persist for up to 12 hours (RxList 2000).
1.
Droperidol structure.
Why it is important to do this review
Droperidol is still in use. We think it is still being used in Australia, Belgium, Brazil, Czech Republic, Denmark, Finland, Greece, India, Italy, Netherlands, New Zealand, South Africa, Spain, Sweden, Thailand, and the USA. It is of interest to researchers and clinicians in the area of management of acute aggression. Previous versions of this review are out of date (Cure 2001; Cure 2004), and this review forms one of a family of related work (Table 8).
4. Other relevant Cochrane reviews.
| Focus of review | Reference |
| Completed and maintained reviews | |
| 'As required' medication regimens for seriously mentally ill people in hospital | Chakrabarti 2007 |
| Benzodiazepines for psychosis‐induced aggression or agitation | Gillies 2005 |
| Chlorpromazine for psychosis‐induced aggression or agitation | Ahmed 2010 |
| Clotiapine for acute psychotic illnesses | Berk 2004 |
| Containment strategies for people with serious mental illness | Muralidharan 2006 |
| Droperidol for acute psychosis | This review |
| Haloperidol for psychosis‐induced aggression or agitation (rapid tranquillisation) | Powney 2012 |
| Haloperidol + promethazine for psychosis‐induced aggression | Huf 2009 |
| Olanzapine IM or olanzapine orodispersible tablet for acutely disturbed/agitated people with suspected serious mental illnesses | Belgamwar 2005 |
| Seclusion and restraint for serious mental illnesses | Sailas 2000 |
| Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses | Gibson 2004 |
| Reviews in the process of being completed or updated | |
| Risperidone for psychosis‐induced aggression or agitation | Ahmed 2011 |
| Haloperidol for long‐term aggression in psychosis | Khushu 2012 |
| Loxapine inhaler for psychosis‐induced aggression | Vangala 2012 |
| Clozapine for people with schizophrenia and recurrent physical aggression (Title) | Toal 2012 |
| Quetiapine for psychosis‐induced aggression | Wilkie 2012 |
| De‐escalation techniques for psychosis‐induced aggression | Rao 2012 |
IM: intramuscular.
Objectives
To estimate the effects of droperidol, including its cost‐effectiveness, when compared to placebo, other 'standard' or 'non‐standard' treatments, or other forms of management of psychotic illness, in controlling acutely disturbed behaviour and reducing psychotic symptoms in people with schizophrenia‐like illnesses.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs). If a trial had been described as 'double‐blind' but implied randomisation, we would have included such trials in a sensitivity analysis (see Sensitivity analysis). We excluded quasi‐randomised studies, such as those allocating by alternate days of the week.
Types of participants
Any people with acutely disturbed/aggressive/agitated behaviour secondary to psychotic illnesses such as schizophrenia, schizoaffective disorder, mixed affective disorders, the manic phase of bipolar disorder or a brief psychotic episode, irrespective of age and sex. The definition of 'acute' adopted for the purposes of this review was determined by the statements and implications made by the authors of the trials that the behavioural disturbances of the participants were of sudden onset or extreme in nature, or both. If trial participants were included who were not clearly acutely disturbed, we analysed data together and separately to see what effect the results had on the summated outcome. If there were differences, we presented data separately. We only included trials of people with organic illnesses or people abusing substances if participants were exhibiting disturbed behaviour resulting from a psychotic episode, and we analysed these data separately. For the 2015 update, we decided to slightly widen our inclusion criteria by including studies where the majority of people in the study had some form of mental illness that was thought to be fuelling their aggression/agitation ‐ even if their data were 'contaminated' by data relating to people who were aggressive for reasons thought to not be because of mental illness.
Types of interventions
1 Droperidol
Any dose, given orally, or by IM or IV injection
Compared with:
a. Standard medication
Drug treatments that fit with normal 'custom and practice': this may have involved increasing the dose of standard medication or addition of another 'standard' psychotropic drug, such as an antipsychotic, an anxiolytic (benzodiazepine or other) or a mood stabiliser. We proposed to report the effects of separate preparations distinctly.
b. Non‐standard medication
Drug treatments that were evaluated as a new type of intervention. We proposed to report the effects of separate preparations distinctly.
c. Placebo
d. Any other means of management
Types of outcome measures
We planned to divide outcomes into immediate (within two hours), short term (longer than two hours to 24 hours), medium term (longer than 24 hours to two weeks) and long term (beyond two weeks).
Primary outcomes
1. Tranquilisation or asleep: tranquillised/sleep ‐ by up to 30 minutes
2. Specific behaviours: aggression ‐ another episode of aggression by 24 hours
3. Adverse effect: specific and serious adverse effects by 24 hours
Secondary outcomes
1. Tranquillisation or asleep
1.1 Tranquil/asleep ‐ after 30 minutes 1.2 Time to tranquillisation/sleep.
2. Specific behaviours
2.1 Self‐harm, including suicide. 2.2 Injury to others. 2.3 Aggression. 2.3.1 Clinically important change in aggression. 2.3.2 Any change in aggression. 2.3.3 Average endpoint aggression score. 2.3.4 Average change in aggression scores.
3. Global state
3.1 Overall improvement. 3.2 Use of additional medication. 3.3 Use of restraints/seclusion. 3.4 Relapse ‐ as defined by each study. 3.5 Recurrence of violent incidents. 3.6 Needing extra visits from the doctor. 3.7 Refusing oral medication. 3.8 Accepting treatment. 3.9 Average endpoint acceptance score. 3.10 Average change in acceptance score.
4. Adverse effects
4.1 Death. 4.2 Other clinically important general adverse effects. 4.3 Any general adverse effects. 4.4 Any serious, specific adverse effects ‐ after 24 hours. 4.5 Average endpoint general adverse effect score. 4.6 Average change in general adverse effect scores. 4.7 Clinically important change in specific adverse effects. 4.8 Any change in specific adverse effects. 4.9 Average endpoint‐specific adverse effects. 4.10 Average change in specific adverse effects.
5. Service outcomes
5.1 Duration of hospital stay. 5.2 Re‐admission. 5.3 No clinically important engagement with services. 5.4 Not any engagement with services. 5.5 Average endpoint engagement score. 5.6 Average change in engagement scores.
6. Mental state
6.1 Clinically important change in general mental state. 6.2 Any change in general mental state. 6.3 Average endpoint general mental state score. 6.4 Average change in general mental state scores.
7. Leaving the study early
7.1 For specific reasons. 7.2 For general reasons.
8. Satisfaction with treatment
8.1 Recipient of treatment not satisfied with treatment. 8.2 Recipient of treatment average satisfaction score. 8.3 Recipient of treatment average change in satisfaction scores. 8.4 Informal treatment provider not satisfied with treatment. 8.5 Informal treatment providers' average satisfaction score. 8.6 Informal treatment providers' average change in satisfaction scores. 8.7 Professional providers not satisfied with treatment. 8.8 Professional providers' average satisfaction score. 8.9 Professional providers' average change in satisfaction scores.
9. Acceptance of treatment
9.1 Accepting treatment. 9.2 Average endpoint acceptance score. 9.3 Average change in acceptance score.
10. Quality of life
10.1 Clinically important change in quality of life. 10.2 Any change in quality of life. 10.3 Average endpoint quality of life score. 10.4 Average change in quality of life scores. 10.5 Clinically important change in specific aspects of quality of life. 10.6 Any change in specific aspects of quality of life. 10.7 Average endpoint‐specific aspects of quality of life. 10.8 Average change in specific aspects of quality of life.
11. Economic outcomes
11.1 Direct costs. 11.2 Indirect costs.
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2008), and GRADE profiler (GRADEpro) to import data from Review Manager 5 (Review Manager) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined and the sum of available data on all outcomes that we rated as important to patient care and decision making. We aimed to select the following main outcomes for inclusion in the 'Summary of findings' tables.
Tranquillisation or asleep: tranquillised/sleep ‐ by up to 30 minutes.
Specific behaviours: aggression ‐ another episode of aggression ‐ by 24 hours.
Adverse effect ‐ specific and serious adverse effects by 24 hours (not death).
Adverse effect ‐ specific and serious adverse effects (death).
Service outcome ‐ satisfaction with treatment (not discharged).
Mental state ‐ improvement.
Economic outcomes ‐ direct costs.
Search methods for identification of studies
Electronic searches
On 18 December 2015, we searched the Cochrane Schizophrenia Group's Register of Trials using the following search string:
*Droperidol* in Intervention Field of STUDY
In such a study‐based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
The Cochrane Schizophrenia Group's Register of Trials is compiled by systematic searches of major resources (including AMED, BIOSIS, CINAHL, Embase, MEDLINE, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature and conference proceedings (see Group's Module). The register as no language, date, document type or publication status limitations for inclusion of records.
For previous searches, see Appendix 1.
Searching other resources
1 Reference searching
We inspected references of all included studies for further relevant studies.
2 Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
Data collection and analysis
For previous data collection and analysis see Appendix 2.
Selection of studies
Review authors (MAK) and CEA (see Acknowledgements) independently inspected citations from the 2015 search and identified relevant abstracts. We compared findings to ensure reliability. In case of disputes, we would have acquired the full report for more detailed scrutiny. One review author (MAK) obtained and inspected full reports of the abstracts meeting the review criteria, which CEA re‐inspected to ensure a reliable selection. We did not disagree on selection. In future versions, if it is not possible to resolve disagreements by discussion, we will attempt to contact the study authors for clarification.
Data extraction and management
1 Extraction
Review author (MAK) independently extracted data from all included studies and CA independently extracted data from a random 20% sample. We discussed any disagreements and documented decisions; if necessary, we contacted authors of studies for clarification. We extracted data presented only in graphs and figures whenever possible, but included these data in the review only if two review authors independently had the same result. We attempted to contact authors through an open‐ended request to obtain missing information or for clarification whenever necessary. If studies were multicentre, we would have extracted data relevant to each component centre separately. Where possible, we reported total end‐scale measures, as opposed to subscale measures.
2 Management
2.1 Forms
We extracted data onto simple standard forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if:
the psychometric properties of the measuring instrument were described in a peer‐reviewed journal (Marshall 2000); and
the measuring instrument had not been written or modified by one of the trialists for that trial.
Ideally, the measuring instrument should have been either a self‐report or completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; in Description of studies we noted if this was the case.
2.3 Endpoint versus change data
Both endpoint and change data have advantages. Change data can remove a component of between‐person variability from the analysis. However, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided to use primarily endpoint data, and only use change data if the former were not available. We combined endpoint and change data in the analysis, as we preferred to use mean differences (MD) rather than standardised mean differences throughout (Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to all data before inclusion.
For change data
We entered change data, as when continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. We presented and entered change data into statistical analyses.
For endpoint data
When a scale started from the finite number 0, we subtracted the lowest possible value from the mean and divided this by the standard deviation (SD). If this value was lower than 1, it strongly suggested a skew, and we would have excluded the study. If this ratio was higher than 1 but below 2, there was suggestion of skew. We would have entered the study and tested whether its inclusion or exclusion would have changed the results substantially. Finally, if the ratio was larger than 2, we would have included the study, because skew was less likely (Altman 1996; Higgins 2011).
If a scale started from a positive value (such as the Positive and Negative Syndrome Scale, which can have values from 30 to 210) (Kay 1986), we would have modified the calculation described above to take into account the scale starting point. In such cases, skew is present if 2 SD > (S ‐ Smin), where S is the mean score and Smin is the minimum score.
(Please note, irrespective of the above rules, we would enter endpoint data from studies of at least 200 participants in the analysis because skewed data pose less of a problem in large studies.)
2.5 Common measure
To facilitate comparison between trials, we intended to convert variables that could be reported in different metrics, such as days in hospital (e.g. mean days per year, per week or per month) to a common metric (e.g. mean days per month).
2.6 Conversion of continuous to binary data
Where possible, we attempted to convert outcome measures to dichotomous data. We did this by identifying cutoff points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS), in Overall 1962, or the Positive and Negative Syndrome Scale, in Kay 1986, this could be considered to be a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cutoff presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for droperidol intervention. Where keeping to this made it impossible to avoid outcome titles with clumsy double‐negatives (e.g. 'not un‐improved'), we reported data where the left of the line indicated an unfavourable outcome. We noted this in the relevant graphs.
Assessment of risk of bias in included studies
One review author (MAK) assessed risk of bias by using criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where a study provided inadequate details of randomisation and other characteristics of the trial, she attempted to contact the study authors to obtain further information.
We noted the level of risk of bias in both the text of the review and in the 'Risk of bias' table within the Characteristics of included studies table; Table 1; Table 2; Table 3; and Table 4.
Summary of findings for the main comparison. Droperidol versus placebo.
| Droperidol versus placebo | ||||||
|
Patient or population: acute psychosis Setting: inpatient Intervention: droperidol Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with droperidol | |||||
| Tranquillisation or asleep: tranquillised/sleep ‐ by around 30 minutes | Moderate | RR 1.18 (1.05 to 1.31) | 227 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 800 per 1000 | 944 per 1000 (840 to 1000) | |||||
| Global state: use of additional medication ‐ by 60 minutes after initial adequate sedation until ED discharge (various psychotropic drugs) | Moderate | RR 0.55 (0.36 to 0.85) | 227 (1 RCT) | ⊕⊕⊕⊕ High 1,2 | 'Moderate' control risk approximately that of trial population. | |
| 400 per 1000 | 220 per 1000 (144 to 340) | |||||
| Adverse effects ‐ cardiovascular ‐ arrhythmia | Moderate | RR 0.34 (0.01 to 8.31) | 227 (1 RCT) | ⊕⊕⊕⊝ Moderate 1,3 | 'Moderate' control risk approximately that of trial population. | |
| 10 per 1000 | 3 per 1000 (0 to 83) | |||||
| Adverse effects ‐ respiratory ‐ airway obstruction | Moderate | RR 0.62 (0.15 to 2.52) | 227 (1 RCT) | ⊕⊕⊝⊝ Low 3,4 | 'Moderate' control risk approximately that of trial population. | |
| 40 per 1000 | 25 per 1000 (6 to 101) | |||||
| Service use: person able to be discharged home | Moderate | RR 1.16 (0.90 to 1.48) | 227 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 500 per 1000 | 580 per 1000 (450 to 740) | |||||
| Mental state ‐ improvement | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| Economic: direct costs | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ED: emergency department; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: rated 'not serious' (no downgrade) ‐ clear reporting of good methods.
2 Indirectness: rated 'not serious' (no downgrade) ‐ but proxy outcome for 'Another episode of aggression by 24 hours'.
3 Imprecision: rated 'serious' (downgraded by 1) ‐ few events, wide confidence intervals.
4 Indirectness: rated 'serious' (downgraded by 1) ‐ respiratory obstruction proxy measure ‐ not 'death'.
Summary of findings 2. Droperidol versus haloperidol.
| Droperidol versus haloperidol | ||||||
|
Patient or population: acute psychosis Setting: inpatient Intervention: droperidol Comparison: haloperidol | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with haloperidol | Risk with droperidol | |||||
| Tranquillisation or asleep: tranquillised/sleep ‐ by around 30 minutes | Moderate | RR 1.01 (0.93 to 1.09) | 228 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 920 per 1000 | 929 per 1000 (856 to 1000) | |||||
| Global state: use of additional medication ‐ by 60 minutes after initial adequate sedation until ED discharge (various psychotropic drugs) | Moderate | RR 0.37 (0.16 to 0.90) | 255 (2 RCTs) | ⊕⊕⊕⊕ High 1,2 | 'Moderate' control risk approximately that of trial population. | |
| 160 per 1000 | 59 per 1000 (26 to 144) | |||||
| Adverse effects ‐ cardiovascular ‐ hypotension | Moderate | RR 2.80 (0.30 to 26.49) | 228 (1 RCT) | ⊕⊕⊕⊝ Moderate 1,3 | 'Moderate' control risk approximately that of trial population. | |
| 10 per 1000 | 28 per 1000 (3 to 265) | |||||
| Adverse effects ‐ cardiovascular ‐ hypotension/desaturation | Study population | RR 2.80 (0.12 to 67.98) | 228 (1 RCT) | ⊕⊕⊝⊝ Low 1,3,4 | 'Moderate' control risk approximately that of trial population. | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Service use: person able to be discharged home | Study population | Not pooled | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| Mental state: mean score by 13 days (Scale for Quantification of Psychotic Symptom Severity, high = poor) | The mean mental state: mean score by 13 days (Scale for Quantification of Psychotic Symptom Severity, high = poor) was 0 | The mean mental state: mean score by 13 days (Scale for Quantification of Psychotic Symptom Severity, high = poor) in the intervention group was 0.11 undefined more (0.07 fewer to 0.29 more) | MD 0.11 CI ‐0.07 to 0.29 |
40 (1 RCT) | ⊕⊕⊝⊝ Low 1,3,4 | 'Moderate' control risk approximately that of trial population. |
| Economic: direct costs | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ED: emergency department; MD: mean difference; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: rated 'not serious' (no downgrade) ‐ clear reporting of good methods.
2 Indirectness: rated 'not serious' (no downgrade) ‐ but proxy outcome for 'Another episode of aggression by 24 hours'.
3 Imprecision: rated 'serious' (downgraded by 1) ‐ few events, wide confidence intervals.
4 Indirectness: rated 'serious' (downgraded by 1) ‐ hypotension/desaturation proxy measure ‐ not 'death'.
Summary of findings 3. Droperidol versus midazolam.
| Droperidol versus midazolam | ||||||
|
Patient or population: acute psychosis Setting: inpatient Intervention: droperidol Comparison: midazolam | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with midazolam | Risk with droperidol | |||||
| Tranquillisation or asleep: tranquillised/asleep ‐ by 30 minutes (at 10 minutes) | Moderate | RR 0.96 (0.72 to 1.28) | 153 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 550 per 1000 | 528 per 1000 (396 to 704) | |||||
| Global state: use of additional medication ‐ by 60 minutes after initial adequate sedation until ED discharge (various psychotropic drugs) | Moderate | RR 0.54 (0.24 to 1.20) | 153 (1 RCT) | ⊕⊕⊕⊝ Moderate 1,2 | 'Moderate' control risk approximately that of trial population. | |
| 190 per 1000 | 101 per 1000 (42 to 224) | |||||
| Adverse effects ‐ respiratory ‐ airway obstruction | Moderate | RR 0.13 (0.01 to 2.55) | 153 (1 RCT) | ⊕⊕⊝⊝ Low 1,2,3 | 'Moderate' control risk approximately that of trial population. | |
| 40 per 1000 | 5 per 1000 (0 to 102) | |||||
| Adverse effects ‐ respiratory ‐ hypoxia | Moderate | RR 0.70 (0.16 to 3.03) | 153 (1 RCT) | ⊕⊕⊕⊝ Moderate 1,2 | 'Moderate' control risk approximately that of trial population. | |
| 50 per 1000 | 35 per 1000 (8 to 143) | |||||
| Service use: person able to be discharged home | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| Mental state ‐ improvement | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| Economic: direct costs | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ED: emergency department; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: rated 'not serious' (no downgrade) ‐ clear reporting of good methods.
2 Imprecision: rated 'serious' (downgraded by 1) ‐ few events, wide confidence intervals.
3 Indirectness: rated 'serious' (downgraded by 1) ‐ respiratory obstruction proxy measure ‐ not 'death'.
Summary of findings 4. Droperidol versus olanzapine.
| Droperidol versus olanzapine | ||||||
|
Patient or population: acute psychosis Setting: inpatient Intervention: droperidol Comparison: olanzapine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with olanzapine | Risk with droperidol | |||||
| Tranquillisation or asleep: tranquillised/asleep ‐ by around 30 minutes | Moderate | RR 1.02 (0.94 to 1.11) | 221 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 900 per 1000 | 918 per 1000 (846 to 999) | |||||
| Global state: use of additional medication ‐ by 60 minutes after initial adequate sedation until ED discharge (various psychotropic drugs) | Moderate | RR 0.56 (0.36 to 0.87) | 221 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 370 per 1000 | 207 per 1000 (133 to 322) | |||||
| Adverse effects ‐ cardiovascular ‐ arrhythmia | Moderate | RR 0.32 (0.01 to 7.88) | 221 (1 RCT) | ⊕⊕⊕⊝ Moderate 1,2 | 'Moderate' control risk approximately that of trial population. | |
| 10 per 1000 | 3 per 1000 (0 to 79) | |||||
| Adverse effects ‐ respiratory ‐ airway obstruction | Moderate | RR 0.97 (0.20 to 4.72) | 221 (1 RCT) | ⊕⊕⊝⊝ Low 2,3 | 'Moderate' control risk approximately that of trial population. | |
| 30 per 1000 | 29 per 1000 (6 to 142) | |||||
| Service use: person able to be discharged home | Moderate | RR 1.06 (0.83 to 1.34) | 221 (1 RCT) | ⊕⊕⊕⊕ High 1 | 'Moderate' control risk approximately that of trial population. | |
| 530 per 1000 | 562 per 1000 (440 to 710) | |||||
| Mental state ‐ improvement | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| Economic: direct costs | Study population | Not estimable | (0 studies) | ‐ | No trial reported this important outcome. | |
| Not pooled | Not pooled | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; ED: emergency department; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: rated 'not serious' (no downgrade) ‐ clear reporting of good methods.
2 Imprecision: rated 'serious' (downgraded by 1) ‐ few events, wide confidence intervals.
3 Indirectness: rated 'serious' (downgraded by 1) ‐ respiratory obstruction proxy measure ‐ not 'death'.
Measures of treatment effect
1 Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive than odds ratios, and that odds ratios tend to be interpreted as RR by clinicians (Boissel 1999; Deeks 2000). The number needed to treat for an additional beneficial outcome/number needed to treat for an additional harmful outcome statistic with its CIs is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and its interpretation (Hutton 2009). For binary data presented in the Table 1, where possible, we calculated illustrative comparative risks.
2 Continuous data
For continuous outcomes, we estimated MD between groups with 95% CI. We preferred not to calculate effect size measures (standardised mean difference). However, if scales of very considerable similarity had been used, we presumed there was a small difference in measurement, and we calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1 Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data pose problems. Authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, CIs unduly narrow, and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
If clustering had not been accounted for in primary studies, we would have presented data in a table, with an asterisk symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intraclass correlation coefficients (ICCs) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). If clustering was incorporated into the analysis of primary studies, we would have presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We sought statistical advice and were advised that the binary data as presented in a report should be divided by a 'design effect'. We calculated this using the mean number of participants per cluster (m) and the ICC (design effect = 1 + (m ‐ 1) × ICC) (Donner 2002). If the ICC was not reported, we would have assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies had been appropriately analysed taking into account ICCs and relevant data documented in the report, we would have synthesised these with other studies using the generic inverse‐variance technique.
2 Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase, the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we had planned to use only the data of the first phase of cross‐over studies.
3 Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary, we simply added these and combined them within the two‐by‐two table. If data were continuous, we combined data following the formula in Section 7.7.3.8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Where the additional treatment arms were not relevant, we did not use these data.
Dealing with missing data
1 Overall loss of credibility
At some degree of loss of follow‐up data must lose credibility (Xia 2009). We chose that, for any outcome, should more than 50% of the data be unaccounted for, we would not reproduce these data or use them within analyses (except for the outcome 'leaving the study early'). However, if more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we would have marked such data with an asterisk to indicate that such a result may well be prone to bias.
2 Binary
In the case where attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). We assumed all participants leaving the study early to have the same rates of negative outcome as participants who completed, except for the outcomes of death and adverse effects. For these outcomes, we used the rate of participants who stayed in the study ‐ in that arm of the trial ‐ for participants who did not. We undertook a sensitivity analysis to test how prone the primary outcomes were to change when data only from people who completed the study to that point were compared to the intention‐to‐treat analysis using the above assumptions.
3 Continuous
3.1 Attrition
In the case where attrition for a continuous outcome was between 0% and 50%, and data only from people who completed the study to that point were reported, we used these data.
3.2 Standard deviations
If in future updates SDs are not reported, we will first try to obtain the missing values from the authors. If these are not available, where measures of variance for continuous data are missing, but an exact standard error (SE) and CIs are available for group means, and either P value or t value is available for differences in mean, we can calculate them according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). When only the SE is reported, SDs can be calculated by the formula SD = SE × square root (n). Sections 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions present detailed formulae for estimating SDs from P values, t or F values, CIs, ranges or other statistics (Higgins 2011). If these formulae do not apply, we will calculate the SDs according to a validated imputation method that is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. We nevertheless will examine the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data were used in the trial, if less than 50% of the data were assumed, we presented and used these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
1 Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations that we had not predicted would arise. If such situations or participant groups arose, we would have fully discussed these.
2 Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods that we had not predicted would arise. If such methodological outliers had been present, we would have fully discussed these.
3 Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of the I2 statistic depends on the magnitude and direction of effects and the strength of evidence for heterogeneity (e.g. P value from Chi2 test, or a CI for the I2 statistic). We interpreted an I2 statistic estimate of 50% or greater accompanied by a statistically significant Chi2 statistic as evidence of substantial levels of heterogeneity (Higgins 2011). When we found substantial levels of heterogeneity in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1 Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in Section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of included RCTs. If the protocol was available, we compared outcomes in the protocol with those in the published report. If the protocol was not available, we compared outcomes listed in the methods section of the trial report with reported results.
2 Funnel plot
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Chapter 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar size. In future updates of this review, if funnel plots are possible, we will seek statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seemed to be true to us, and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. However, there is a disadvantage to the random‐effects model, in that it puts added weight on to small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed‐effect model for all analyses.
Subgroup analysis and investigation of heterogeneity
1 Subgroup analyses
1.1 Primary outcomes
We did not anticipate a need for any subgroup analysis.
1.2 Clinical state, stage or problem
We proposed to undertake this review as part of a family of similar reviews that will provide an overview of the effects of droperidol for people with psychosis induced aggression or agitation in general. In addition, we aimed to report data on subgroups of people in the same clinical state, stage and with similar problems.
2 Investigation of heterogeneity
If inconsistency was high, we reported this. We first investigated whether data had been entered correctly. Second, if data were correct, we visually inspected the graph and successively removed outlying studies to see if homogeneity was restored. For this review, we decided that should this occur with data contributing to the summary finding of no more than around 10% of the total weighting, we would present the data. If not, we would not pool the data and we would discuss these issues. We know of no supporting research for this 10% cutoff, but we used prediction intervals as an alternative to this unsatisfactory state.
If in future updates of this review unanticipated clinical or methodological heterogeneity is obvious, we will simply state hypotheses regarding these. We do not anticipate undertaking analyses relating to such situations.
Sensitivity analysis
1 Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in such a way as to imply randomisation. For the primary outcomes, we would have included these studies, and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we would have employed all data from these studies.
2 Assumptions for lost binary data
Where we had to make assumptions regarding people lost to follow‐up (see Dealing with missing data), we compared the findings of the primary outcomes when we used our assumption(s) and when we used data only from people who completed the study to that point. If there was a substantial difference, we would have reported results and discussed them but continued to employ our assumption.
If we had needed to make assumptions regarding missing SDs data (see Dealing with missing data), we would have compared the findings of the primary outcomes when we used our assumption(s) and when we used data only from people who completed the study to that point. We would have undertaken a sensitivity analysis testing how prone results were to change when completer‐only data only were compared to imputed data using the above assumption. If there was a substantial difference, we would have reported results and discussed them but continued to employ our assumption.
3 Risk of bias
For the primary outcome, we analysed the effects of excluding trials that we judged to be at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting. If the exclusion of trials at high risk of bias had substantially altered the direction of effect or the precision of the effect estimates, then we would not have included data from these trials in the analysis.
4 Imputed values
We had intended to undertake a sensitivity analysis to assess, if necessary, the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials.
If we had noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but would have presented them separately.
5 Fixed effect and random effects
We synthesised all data using a fixed‐effect model, however we also aimed to synthesise data for the primary outcome using a random‐effects model to evaluate whether this altered the significance of the results. If the significance of results changed, we would have noted this in the text.
Results
Description of studies
See Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
We included six studies in this review. In the update search, we undertook for this review, we found 14 records that were potentially relevant. We identified no duplicates. We screened these 14 records and removed two records. We assessed 12 full‐text articles for eligibility and excluded six from the review with reasons. Three of these studies were already included in the previous version of the review and we added three new studies. The PRISMA table shows results of our search (Figure 2).
2.
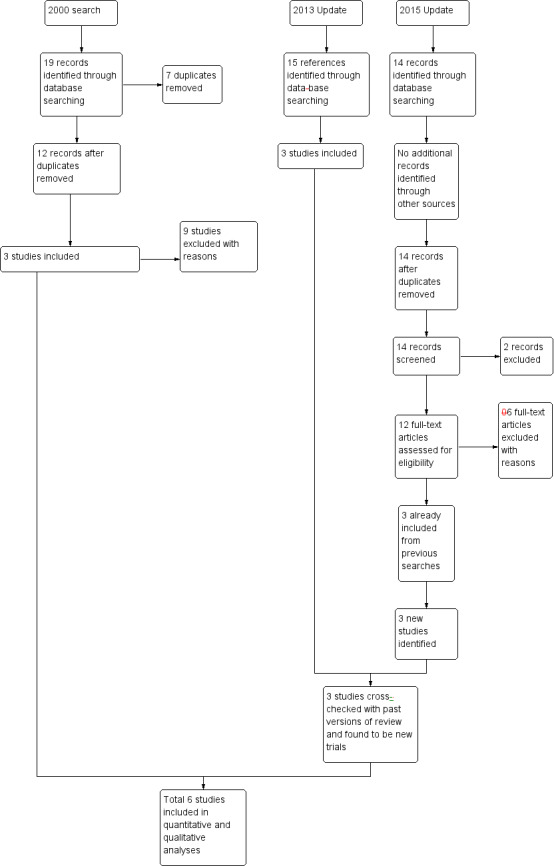
2015 study flow diagram.
Included studies
1 Methods
All the included trials were randomised including one study that employed block randomisation (Calver 2015). Five out of six included trials were double‐blind (Chan 2013; Cocchi 1971; Knott 2006; Resnick 1984; Van Leeuwen 1977), while Resnick 1984 gave no clear details of blinding. In an effort to minimise bias, three of the included studies stated that the outcome assessor was blind to group allocation (Calver 2015; Chan 2013; Knott 2006), one study reported no detail of blinding the outcome assessor (Van Leeuwen 1977).
2 Length of trials
The overall duration of the included trials varied in length from immediate (within two hours), short term (more than two hours to 24 hours) to long term (beyond 2 weeks) as listed in Table 9.
5. Length of included studies.
| Study | Immediate (< 2 hours) | Short term (> 2 hours to 24 hours) | Medium term (> 24 hours to 2 weeks) | Long term (> 2 weeks) |
| Van Leeuwen 1977 | ✓ (3 and 30 min) | ‐ | ‐ | ‐ |
| Chan 2013 | ✓ (5, 10 and 60 min) | ‐ | ‐ | ‐ |
| Calver 2015 | ✓ (10 min) | ✓ (120 min) | ‐ | ‐ |
| Knott 2006 | ✓ (within 60 min) | ✓ (2 hours) | ‐ | ‐ |
| Resnick 1984 | ✓ (15 and 30 min) | ✓ (24 hours) | ‐ | ‐ |
| Cocchi 1971 | ‐ | ‐ | ‐ | ✓ (30 days) |
min: minute.
3 Participants
A total of 733 people participated in the six studies. Three of the included studies included more than 100 participants (Calver 2015; Chan 2013; Knott 2006); the remaining studies included 40 (Cocchi 1971), 27 (Resnick 1984), and 41 (Van Leeuwen 1977) participants. Only three studies specified inclusion of both male and female participants (Calver 2015; Cocchi 1971; Van Leeuwen 1977).
All trials included people with psychoses. Resnick 1984 did not specify beyond stating that participants were admitted involuntarily to the emergency department of a psychiatric unit. Van Leeuwen 1977 included people with schizophrenia, manic depression or in a 'confusional state'; however, 10 participants had no specific diagnosis. Cocchi 1971 stated that all participants had schizophrenia.
All studies included people with acutely disturbed/aggressive/agitated behaviour secondary to psychotic illnesses such as schizophrenia, schizoaffective disorder, mixed affective disorders, the manic phase of bipolar disorder or a brief psychotic episode, irrespective of age and sex. For the 2015 update, we widened the criteria to include studies where the majority of people in the study had some form of mental illness that was thought to be fuelling their aggression/agitation. We included these studies even if their data were 'contaminated' by data relating to people who were aggressive for reasons other than mental illness. Therefore, we included Knott 2006 (60% of participants had mental illness) in the review. However, none of the studies employed diagnostic criteria; it is unknown whether this influenced the validity of findings.
Five out of six trials referred to the current clinical state of participants: agitation or aggression (Calver 2015; Chan 2013); schizophrenic ‐ acutely exacerbated (Cocchi 1971); marked agitation requiring chemical restraint (Knott 2006); unspecified psychosis (Resnick 1984); and a combination of schizophrenia, mania, confusional state and miscellaneous disorders (Van Leeuwen 1977).
4 Setting
Two trials took place in large metropolitan emergency departments (Chan 2013; Knott 2006), and Calver 2015 was in a psychiatric intensive care unit of a large tertiary specialist mental health facility in Australia. One trial was conducted in an Emergency Department and Psychiatric crisis unit, Oregon Health Sciences University, Portland (Resnick 1984). The setting of Van Leeuwen 1977 was unclear and Cocchi 1971 stated the trial took place in a hospital setting.
5 Interventions
| Trial drug | 5 mg IM | 5 mg IV | 10 mg IM | 10 mg IV |
| Droperidol | ✓ (Resnick 1984) | ✓ (Knott 2006; Chan 2013) | ✓ (Calver 2015) | ✓ (Van Leeuwen 1977) |
| Haloperidol | ✓ (Resnick 1984) | ‐ | ✓ (Calver 2015) | ‐ |
| Olanzapine | ‐ | ✓ (Chan 2013) | ‐ | ‐ |
| Midazolam | ‐ | ✓ (Knott 2006) | ‐ | ‐ |
| Placebo | ‐ | ✓ (Chan 2013) | ‐ | ✓ (Van Leeuwen 1977) |
| IM: intramuscular; IV: intravenous. | ||||
6 Outcomes
6.1 Overall
The outcomes for which we could obtain useable data were: tranquillisation or asleep, global state, service use, mental state and Adverse effects.
6.2 Outcome scales
The scales used by trials that provided useable data are described below.
6.1.1 Mental state
i. Scale for Quantification of Psychotic Symptom Severity, high = poor) (Goodrich 1953)
A research rating scale for use by hospital psychiatrists to express quantitatively the severity of 'incapacitation' due to psychotic symptoms. A rating of from 1.0 to 2.0, extreme behaviour disorganisation requiring vigilance by hospital staff; from 2.0 to 3.0, severity requiring "security ward" care; from 3.0 to 3.7, severity requiring open convalescent ward care; and from 3.8 to 4.0, not requiring hospitalisation, or person ready for discharge.
ii. Glasgow Coma Scale (Teasdale 1974)
The Glasgow Coma Scale (GCS) is a scoring system used to describe the level of consciousness in a person following a traumatic brain injury. The test is simple, reliable, correlates well with outcome and is an objective way of recording the initial and subsequent level of consciousness in a person after a brain injury. It is used by trained staff at the site of an injury (e.g. at a car crash or sports injury), and in the emergency department and intensive care units. Clinicians use this scale to rate the best eye opening response, the best verbal response and the best motor response aperson makes. Generally, brain injury is classified as 'severe' (GCS 3 to 8, cannot score lower than 3), 'moderate' (GCS 9 to 12) and mild (GCS 13 to 15).
iii. Brief Psychiatric Rating Scale (Overall 1962)
The BPRS is used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The original scale has 16 items, although a revised 18‐item scale is commonly used. Each item is defined on a 7‐point scale varying from 'not present' to 'extremely severe', scoring from 0 to 6 or 1 to 7. Total scores can range from 0 to 126, with high scores indicating more severe symptoms.
6.3 Missing outcomes
No trial reported outcomes directly relevant to satisfaction with treatment, acceptance of treatment, quality of life or economics.
Excluded studies
See Characteristics of excluded studies table for details of excluded studies and Table 10 for details of randomised excluded studies which are potentially relevant to other reviews.
6. Randomised excluded studies relevant to other reviews.
| Excluded study | Participants | Suggested comparison | Existing review | |
| People with serious mental illness | People without mental illness | |||
| Hu 2014 | Acute agitation and schizophrenia. | ‐ | Haloperidol vs. ziprasidone for acute agitation and schizophrenia. | Powney 2012 |
| Cocito 1970 | With psychosis in hospital, not acutely ill. | Droperidol for (non‐acute) psychosis. | ‐ | |
| Isbister 2010; Rosen 1997 | ‐ | People with violent and acute behavioural disturbance. No mention of any underlying psychiatric illness. | Droperidol for acute non‐psychiatric disturbance. | ‐ |
| Mostly people with trauma and medical reasons for their disturbance (total 46), 1 with 'psychiatric' diagnosis. | ||||
|
Richards 1997; Richards 1998; Thomas 1992 |
People with drug‐induced toxicity, not people with severe mental illnesses. | Droperidol for drug‐induced toxicity. | ‐ | |
| Foster 1995 | Healthy women attending day hospital for minor surgery. | Droperidol for minor surgery. | ‐ | |
We excluded 14 studies, four of which were not randomised. Girard 1972 and Lilburn 1977 were case‐control studies, and Weiser 1973 was a case series. After emails from Dr Hooper it was clear that his study also had to be excluded, as allocation to groups had not been random, with participants being alternately allocated to either the treatment or the control intervention (Hooper 1983). Most of the remaining trials were excluded because participants were not clearly experiencing psychotic illnesses. Foster 1995 included female participants undergoing minor gynaecological surgery. Richards 1998 and Thomas 1992 both included predominantly 'intoxicated' people. Thomas 1992 also included people experiencing trauma, an underlying medical condition or who were undiagnosed, as did Rosen 1997. Both Richards 1998 and Rosen 1997 included some people with a 'psychiatric' diagnosis but their studies had to be excluded because outcomes for these participants, a small minority of the total, were not separately analysed. Cocito 1970 included only people with psychosis, but not necessarily with acute illnesses, while Isbister 2010, although randomised, did not mention any underlying psychiatric illness. Weiser 1975 would have been included, except for the addition of five people to replace those who left the study early. It is not clear to which group(s) those leaving early belonged, so the remaining data were rendered of little value once data from the non‐random replacements had been incorporated.
Awaiting assessment
No studies are currently awaiting assessment.
Ongoing studies
We are not aware of any ongoing studies.
Risk of bias in included studies
See also 'Risk of bias' tables in the Characteristics of included studies table and Figure 3.
3.
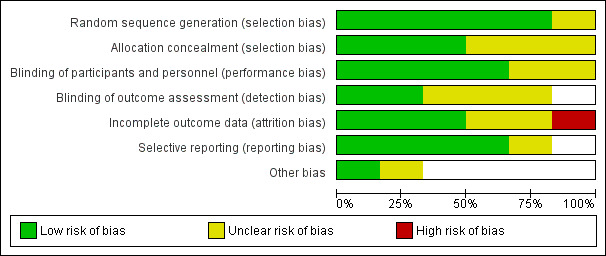
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
All the included trials were randomised including one study (Calver 2015) that employed block randomisation. Van Leeuwen 1977 specified that treatment was "randomly assigned" with participants listed in chronological order and assigned individually numbered vials. Therefore, it was unclear whether those randomising could have ascertained the order of prescribing. Resnick 1984 did not specify the explicit means of allocation, although he stated that participants received treatment on a 'randomised basis', and that the codes identifying the packages of medication were "kept in the pharmacy until the conclusion of the study". Cocchi 1971 specified only that the study was randomised, with no details regarding the means of allocation.
Blinding
Five out of six included trials were double‐blind (Chan 2013; Cocchi 1971; Knott 2006; Resnick 1984; Van Leeuwen 1977). To minimise bias, three of the included studies stated that the outcome assessor was blinded to group allocation (Calver 2015; Chan 2013; Knott 2006). One study reported no detail of blinding the outcome assessor (Van Leeuwen 1977), while Resnick 1984 gave no clear details of blinding.
Incomplete outcome data
We rated only three studies at low risk bias with regard to attrition bias, as all participants were continued to follow‐up (Calver 2015;Chan 2013;Resnick 1984), and only one study with high risk of attrition bias as it did not include all randomised participants in the final analysis (Knott 2006). We rated two studies as having unclear risk of bias (Cocchi 1971; Van Leeuwen 1977).
Selective reporting
All studies reported data for all outcomes listed. We rated five studies at low risk of bias and one study at unclear risk of reporting bias (Knott 2006).
Other potential sources of bias
We identified no other potential sources of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
There are four comparisons: droperidol compared with placebo, droperidol compared with haloperidol, droperidol compared with midazolam and droperidol compared with olanzapine. The studies reported outcomes for intervals within the 'immediate' time frame as defined in Criteria for considering studies for this review. We reported these immediate outcomes individually.
1 Comparison 1: Droperidol versus placebo
Two studies provided data for the comparison of droperidol versus placebo (Chan 2013, N = 227; Van Leeuwen 1977, N = 41).
1.1 Tranquillisation or asleep: 1. tranquilised/asleep (minutes)
One study provided data for tranquillisation or asleep (Chan 2013).
1.1.1 By five minutes
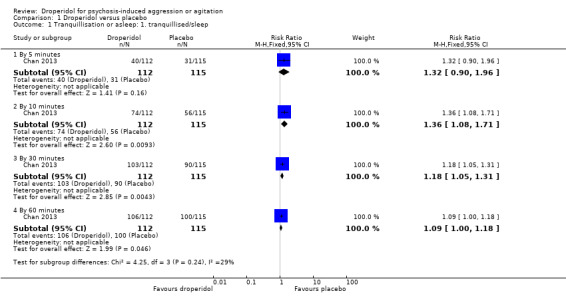
There was no clear difference between droperidol and placebo for by five minutes (1 RCT, N = 227, RR 1.32, 95% CI 0.90 to 1.96; Analysis 1.1).
1.1. Analysis.
Comparison 1 Droperidol versus placebo, Outcome 1 Tranquillisation or asleep: 1. tranquillised/sleep.
1.1.2 By 10 minutes
By 10 minutes, there was evidence that droperidol was clearly different in its effects compared with placebo (1 RCT, N = 227, RR 1.36, 95% CI 1.08 to 1.71; Analysis 1.1).
1.1.3 By 30 minutes
By 30 minutes, we found evidence of a clear difference between droperidol and placebo (1 RCT, N = 227, RR 1.18, 95% CI 1.05 to 1.31; Analysis 1.1).
1.1.4 By 60 minutes
By 60 minutes, we found evidence of a clear difference between droperidol and placebo (1 RCT, N = 227, RR 1.09, 95% CI 1.00 to 1.18; Analysis 1.1).
1.2 Tranquillisation or asleep: 2. difficulty in achieving tranquillisation/sleep
Chan 2013 provided data for difficulty in achieving tranquillisation/sleep. There was no clear difference between droperidol and placebo (1 RCT, N = 227, RR 0.51, 95% CI 0.10 to 2.75; Analysis 1.2).
1.2. Analysis.
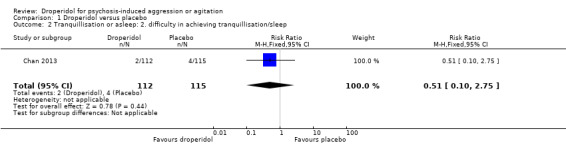
Comparison 1 Droperidol versus placebo, Outcome 2 Tranquillisation or asleep: 2. difficulty in achieving tranquillisation/sleep.
1.3 Tranquillisation or asleep: 3. time to tranquillisation/sleep
Chan 2013 provided data for time to tranquillisation/sleep. We found evidence of a clear difference between droperidol and placebo in the mean time (in minutes) taken to become tranquil or asleep (1 RCT, N = 227, MD ‐46.50, 95% CI ‐86.83 to ‐6.17; Analysis 1.3).
1.3. Analysis.
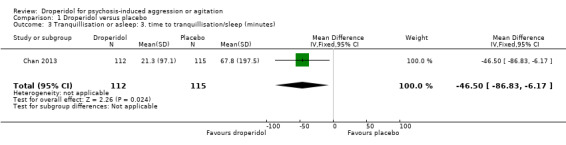
Comparison 1 Droperidol versus placebo, Outcome 3 Tranquillisation or asleep: 3. time to tranquillisation/sleep (minutes).
1.4 Global state: use of additional medication
Two studies provided data on use of additional medication (Chan 2013; Van Leeuwen 1977).
1.4.1 "To reach initial adequate sedation"
Chan 2013 provided data for numbers needing additional medication to reach initial adequate sedation. We found evidence that droperidol was clearly different in its effects compared with placebo (1 RCT, N = 227, RR 0.50, 95% CI 0.28 to 0.89; Analysis 1.4).
1.4. Analysis.
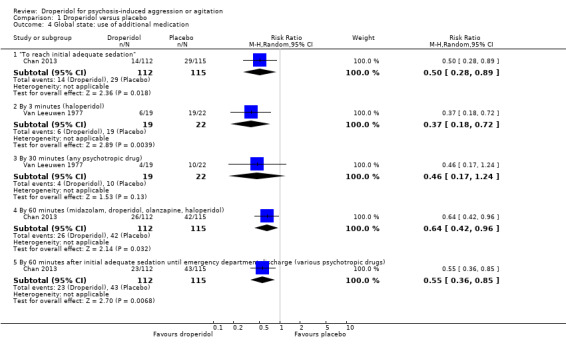
Comparison 1 Droperidol versus placebo, Outcome 4 Global state: use of additional medication.
1.4.2 By three minutes (haloperidol)
Van Leeuwen 1977 provided data for numbers needing additional haloperidol by three minutes. We found evidence of a clear difference between droperidol and placebo for use of additional medication (1 RCT, N = 41, RR 0.37, 95% CI 0.18 to 0.72; Analysis 1.4).
1.4.3 By 30 minutes (any psychotropic drug)
Van Leeuwen 1977 provided data for numbers needing any additional psychotropic drug by 30 minutes. We found no evidence of a clear difference between droperidol and placebo (1 RCT, N = 41, RR 0.46, 95% CI 0.17 to 1.24; Analysis 1.4).
1.4.4 By 60 minutes (midazolam, droperidol, olanzapine, haloperidol)
Chan 2013 provided data for use of additional medication. By 60 minutes, there was evidence of a clear difference favouring droperidol (1 RCT, N = 227, RR 0.64, 95% CI 0.42 to 0.96; Analysis 1.4).
1.4.5 From 60 minutes after initial adequate sedation until emergency department discharge (various psychotropic drugs)
We found evidence of a clear difference for use of additional medication favouring droperidol (1 RCT, N = 227, RR 0.55, 95% CI 0.36 to 0.85; Analysis 1.4).
1.5 Adverse effects
Two studies provided adverse effect data (Chan 2013; Van Leeuwen 1977).
1.5.1 Cardiovascular ‐ arrhythmia
Chan 2013 provided data for arrhythmia. There was no clear difference between droperidol and placebo (1 RCT, N = 227, RR 0.34, 95% CI 0.01 to 8.31; Analysis 1.5).
1.5. Analysis.
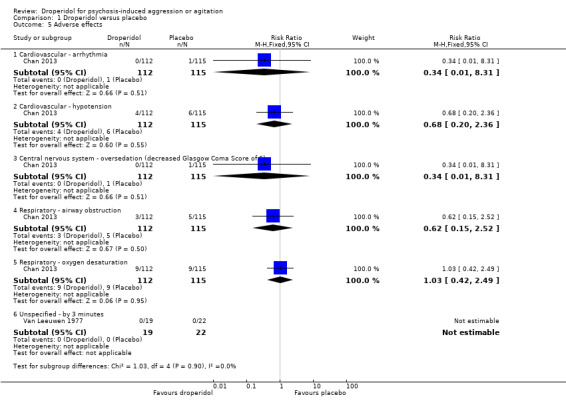
Comparison 1 Droperidol versus placebo, Outcome 5 Adverse effects.
1.5.2 Cardiovascular ‐ hypotension
Chan 2013 provided data for hypotension. We found no evidence of a clear difference between droperidol and placebo (1 RCT, N = 227, RR 0.68, 95% CI 0.20 to 2.36; Analysis 1.5).
1.5.3 Central nervous system ‐ oversedation (decreased Glasgow Coma Score of 6)
Chan 2013 provided data for oversedation. We found no evidence of a clear difference between droperidol and placebo (1 RCT, N = 227, RR 0.34, 95% CI 0.01 to 8.31; Analysis 1.5).
1.5.4 Respiratory ‐ airway obstruction
Chan 2013 provided data for airway obstruction. There was noclear difference between droperidol and placebo (1 RCT, N = 227, RR 0.62, 95% CI 0.15 to 2.52; Analysis 1.5).
1.5.5 Respiratory ‐ oxygen desaturation
Chan 2013 provided data for oxygen desaturation. We found no evidence of a clear difference between droperidol and placebo (1 RCT, N = 227, RR 1.03, 95% CI 0.42 to 2.49; Analysis 1.5).
1.5.6 Unspecified ‐ by three minutes
One trial provided data for unspecified Adverse effects by three minutes (Van Leeuwen 1977, N = 41). There were no events in either the droperidol or placebo group (Analysis 1.5).
1.6 Service use: person able to be discharged home
Chan 2013 provided data for discharge. There was no clear difference between droperidol and placebo (N = 227, RR 1.16, 95% CI 0.90 to 1.48; Analysis 1.6).
1.6. Analysis.
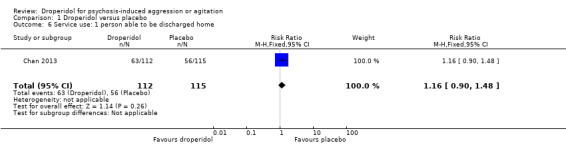
Comparison 1 Droperidol versus placebo, Outcome 6 Service use: 1 person able to be discharged home.
1.7 Service use: emergency department length of stay
Chan 2013 (N = 227) provided data for length of stay in emergency department. Data were skewed and can be viewed in Analysis 1.7). There was no suggestion of a difference between droperidol and placebo (median stay was around 10 hours for both groups).
1.7. Analysis.
Comparison 1 Droperidol versus placebo, Outcome 7 Service use: 2 emergency department length of stay.
| Service use: 2 emergency department length of stay | ||||
|---|---|---|---|---|
| Study | Intervention | number of participants | Median (hours) | Interquartile range |
| Chan 2013 | Droperidol | 112 | 10.0 | 6.7 to 13.2 |
| Chan 2013 | Placebo | 115 | 9.7 | 5.7 to 14.7 |
2 Comparison 2: Droperidol versus haloperidol
Three studies provided data for the comparison of droperidol versus haloperidol (Calver 2015, N = 228; Cocchi 1971, N = 40; Resnick 1984, N = 27).
2.1 Tranquillisation or asleep: 1. tranquillised/sleep within 120 minutes
Calver 2015 provided useable data for tranquillised/sleep within 120 minutes. We found no evidence of a clear difference between droperidol and haloperidol(1 RCT, N = 228, RR 1.01, 95% CI 0.93 to 1.09).
2.2 Tranquillisation or asleep: 2. time to tranquillisation/sleep
Calver 2015 provided data for time to tranquillisation/sleep, we have presented them in Analysis 2.2. There was no suggestion of clear difference between droperidol and haloperidol (median time was around 25 minutes for both groups).
2.2. Analysis.
Comparison 2 Droperidol versus haloperidol, Outcome 2 Tranquillisation or asleep: 2. time to tranquillisation/sleep.
| Tranquillisation or asleep: 2. time to tranquillisation/sleep | ||||
|---|---|---|---|---|
| Study | Intervention | Number of participants | Median (minutes) | Interquartile range |
| Calver 2015 | Droperidol | 118 | 25 | 15 to 30 |
| Calver 2015 | Haloperidol | 110 | 20 | 15 to 30 |
2.3 Global state: use of additional medication
2.3.1 Midazolam administered initially
Calver 2015 provided data that showed no clear difference between droperidol and haloperidol for use of additional medication (1 RCT, N = 228, RR 3.26, 95% CI 0.69 to 15.37; Analysis 2.3).
2.3. Analysis.
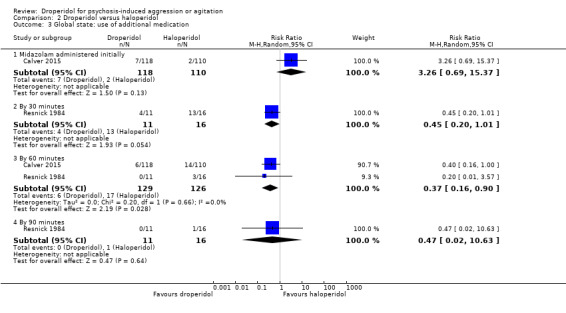
Comparison 2 Droperidol versus haloperidol, Outcome 3 Global state: use of additional medication.
2.3.2 By 30 minutes
Resnick 1984 provided data for use of additional medication by 30 minutes. We found no clear difference between droperidol and haloperidol (1 RCT, N = 27, RR 0.45, 95% CI 0.20 to 1.01; Analysis 2.3).
2.3.3 By 60 minutes
Two trials provided data for use of additional medication by 60 minutes (Calver 2015; Resnick 1984). There was evidence of a clear effect, favouring droperidol for this outcome. (2 RCTs, N = 255, RR 0.37, 95% CI 0.16 to 0.9; Analysis 2.3).
2.3.4 By 90 minute
Resnick 1984 provided data for use of additional medication by 90 minutes. There was no evidence of a clear difference between droperidol and haloperidol(1 RCT, N = 27, RR 0.47, 95% CI 0.02 to 10.63; Analysis 2.3).
2.4 Global state: no overall improvement ‐ by 30 days
Cocchi 1971 provided data for overall improvement. There was no clear difference between droperidol and haloperidol (1 RCT, N = 40, RR 0.67, 95% CI 0.29 to 1.52; Analysis 2.4).
2.4. Analysis.
Comparison 2 Droperidol versus haloperidol, Outcome 4 Global state: no overall improvement ‐ by 30 days.
2.5 Adverse effects
One study provided adverse effect data (Calver 2015, N = 228).
2.5.1 Cardiovascular ‐ hypotension
There was no clear difference between droperidol and haloperidol for hypotension (RR 2.80, 95% CI 0.30 to 26.49; Analysis 2.5).
2.5. Analysis.
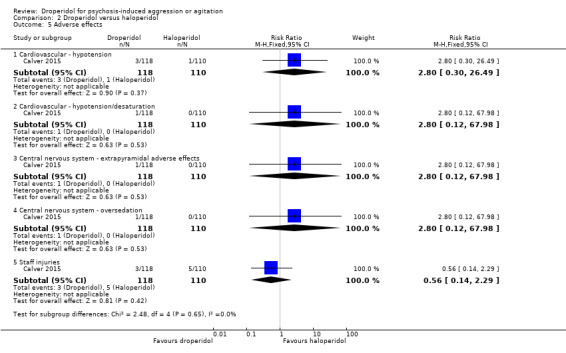
Comparison 2 Droperidol versus haloperidol, Outcome 5 Adverse effects.
2.5.2 Cardiovascular ‐ hypotension/desaturation
We found no evidence of a clear difference between the two treatments for hypotension/desaturation (RR 2.80, 95% CI 0.12 to 67.98; Analysis 2.5).
2.5.3 Central nervous system ‐ extrapyramidal adverse effects
There was no clear difference in extrapyramidal adverse effects between droperidol and haloperidol (RR 2.80, 95% CI 0.12 to 67.98; Analysis 2.5).
2.5.4 Central nervous system ‐ oversedation
There was no clear difference between droperidol and haloperidol for oversedation (RR 2.80, 95% CI 0.12 to 67.98; Analysis 2.5).
2.5.5 Staff injuries
There was no evidence of a clear difference in staff injuries between droperidol and haloperidol (RR 0.56, 95% CI 0.14 to 2.29; Analysis 2.5).
2.6 Mental state: mean score by 13 days (Scale for Quantification of Psychotic Symptom Severity, high = poor)
Cocchi 1971, N = 40 provided mental state data and we found no evidence of a clear difference between droperidol and haloperidol (MD 0.11, 95% CI ‐0.07 to 0.29; Analysis 2.6).
2.6. Analysis.
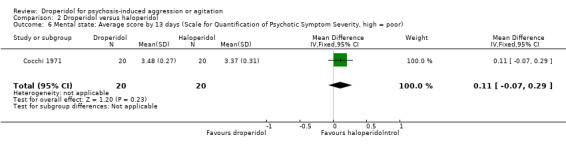
Comparison 2 Droperidol versus haloperidol, Outcome 6 Mental state: Average score by 13 days (Scale for Quantification of Psychotic Symptom Severity, high = poor).
3 Comparison 3: Droperidol versus midazolam
One study provided data for the comparison of droperidol versus midazolam (Knott 2006, N = 153).
3.1 Tranquillisation or asleep: 1. tranquillised/asleep
We identified one study relevant to this outcome and categorised data into two subsets: by 5 minutes and by 10 minutes.
3.2.1 By five minutes
We found evidence of a clear difference between droperidol and midazolam by five minutes (RR 0.37, 95% CI 0.21 to 0.64; Analysis 3.1).
3.1. Analysis.
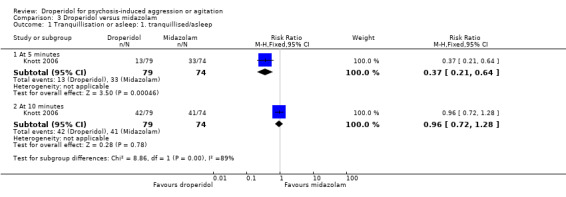
Comparison 3 Droperidol versus midazolam, Outcome 1 Tranquillisation or asleep: 1. tranquillised/asleep.
3.2.2 By 10 minutes
There was no clear difference between droperidol and midazolam by 10 minutes (RR 0.96, 95% CI 0.72 to 1.28; Analysis 3.1).
3.2 Tranquillisation or asleep: 2. time to tranquillisation/sleep
There was no suggestion of a clear difference between droperidol and midazolam (median time was around 10 minutes for both groups; Analysis 3.2).
3.2. Analysis.
Comparison 3 Droperidol versus midazolam, Outcome 2 Tranquillisation or asleep: 2 time to tranquillisation/sleep.
| Tranquillisation or asleep: 2 time to tranquillisation/sleep | |||
|---|---|---|---|
| Study | Intervention | Number of participants | Median (minutes) |
| Knott 2006 | Droperidol | 79 | 8 |
| Knott 2006 | Midazolam | 74 | 6.5 |
3.3 Global state: use of additional medication
We found one study reporting data on use of additional medication and categorised data into one subset involving 153 participants (Knott 2006).
3.3.1 By 60 minutes
There was no clear difference between droperidol and midazolam by 60 minutes (RR 0.54, 95% CI 0.24 to 1.20; Analysis 3.3).
3.3. Analysis.
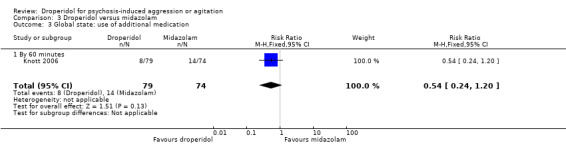
Comparison 3 Droperidol versus midazolam, Outcome 3 Global state: use of additional medication.
3.4 Adverse effects
One study provided adverse effect data (Knott 2006, N = 153).
3.4.1 Cardiovascular ‐ arrhythmia (bradycardia)
There was no evidence of a clear difference between droperidol and midazolam for arrhythmia (RR 2.81, 95% CI 0.12 to 67.98; Analysis 3.4).
3.4. Analysis.
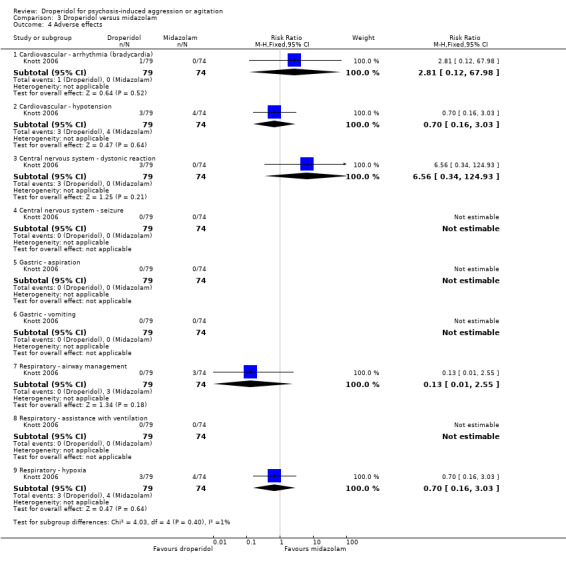
Comparison 3 Droperidol versus midazolam, Outcome 4 Adverse effects.
3.4.2 Cardiovascular ‐ hypotension
There was no clear difference between droperidol and midazolam for hypotension (RR 0.70, 95% CI 0.16 to 3.03; Analysis 3.4).
3.4.3 Central nervous system ‐ dystonic reaction
There was no evidence of a clear difference between droperidol and midazolam for dystonic reaction (RR 6.56, 95% CI 0.34 to 124.93; Analysis 3.4).
3.4.4 Central nervous system ‐ seizure
There were no seizures in either the droperidol or midazolam group (Analysis 3.4).
3.4.5 Gastric ‐ aspiration
There were no aspirations in either the droperidol or midazolam group (Analysis 3.4).
3.4.6 Gastric ‐ vomiting
There were no vomiting episodes in either the droperidol or midazolam group (Analysis 3.4).
3.4.7 Respiratory ‐ airway management
There was no evidence of a clear difference between droperidol and midazolam (RR 0.13, 95% CI 0.01 to 2.55; Analysis 3.4).
3.4.8 Respiratory ‐ assistance with ventilation
There was no need for assistance with ventilation with either droperidol or midazolam (Analysis 3.4).
3.4.9 Respiratory ‐ hypoxia
There was no clear difference between droperidol and midazolam for hypoxia (RR 0.70, 95% CI 0.16 to 3.03; Analysis 3.4)
4 Comparison 4: Droperidol versus olanzapine
One study provided data for the comparison of droperidol versus olanzapine (Chan 2013). In this comparison, there were seven outcomes.
4.3 Tranquillisation or asleep: 3. tranquillised/asleep
We divided the data into four subsets, with a total of 884 people. There was no clear difference between droperidol and olanzapine (RR 1.00, 95% CI 0.93 to 1.07).
4.3.1 At five minutes
There was no clear difference between droperidol and olanzapine at five minutes (RR 1.00, 95% CI 0.70 to 1.42; Analysis 4.1).
4.1. Analysis.
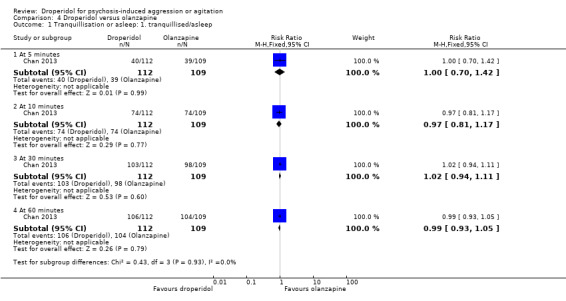
Comparison 4 Droperidol versus olanzapine, Outcome 1 Tranquillisation or asleep: 1. tranquillised/asleep.
4.3.2 At 10 minutes
There was no evidence of a clear difference between droperidol and olanzapine at 10 minutes (RR 0.97, 95% CI 0.81 to 1.17; Analysis 4.1).
4.3.3 At 30 minutes
There was no clear difference between droperidol and olanzapine at 30 minutes (RR 1.02, 95% CI 0.94 to 1.11; Analysis 4.1).
4.3.4 At 60 minutes
There was no clear difference between droperidol and olanzapine at 60 minutes (RR 0.99, 95% CI 0.93 to 1.05; Analysis 4.1).
4.2 Tranquillisation or asleep: 2. difficulty in achieving tranquillisation/sleep
We found no evidence of a clear difference between droperidol and olanzapine for difficulty in achieving tranquillisation/sleep (RR 0.65, 95% CI 0.11 to 3.81; Analysis 4.2).
4.2. Analysis.
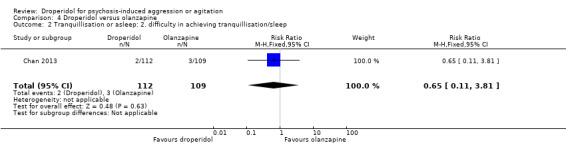
Comparison 4 Droperidol versus olanzapine, Outcome 2 Tranquillisation or asleep: 2. difficulty in achieving tranquillisation/sleep.
4.3 Tranquillisation or asleep: 3. time to tranquillisation/sleep
There was no clear difference between droperidol and olanzapine for time to tranquillisation/sleep (in minutes) (1 RCT, N = 221, MD 7.3 95% CI ‐11.74 to 26.34; Analysis 4.3).
4.3. Analysis.
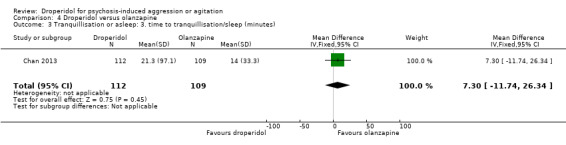
Comparison 4 Droperidol versus olanzapine, Outcome 3 Tranquillisation or asleep: 3. time to tranquillisation/sleep (minutes).
4.4 Global state: use of additional medication
We identified one study reporting use of additional medication and categorised data into three subsets (Chan 2013, N = 221).
4.4.1 "To reach initial adequate sedation"
There was no clear difference between droperidol and olanzapine for use of additional medication "to reach initial adequate sedation" (RR 0.68, 95% CI 0.36 to 1.28; Analysis 4.4).
4.4. Analysis.
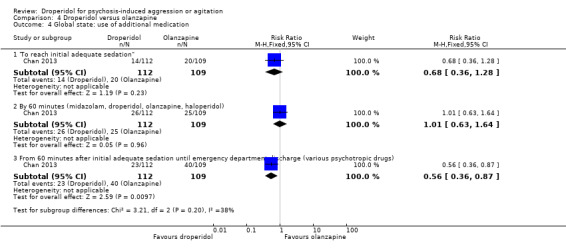
Comparison 4 Droperidol versus olanzapine, Outcome 4 Global state: use of additional medication.
4.4.2 By 60 minutes (midazolam, droperidol, olanzapine, haloperidol)
We found no evidence of a clear difference between droperidol and olanzapine for use of additional medication by 60 minutes (RR 1.01, 95% CI 0.63 to 1.64; Analysis 4.4).
4.4.3 From 60 minutes after initial adequate sedation until emergency department discharge (various psychotropic drugs)
We found evidence of a clear difference, favouring droperidol for use of additional medication from 60 minutes after initial adequate sedation until emergency department discharge (RR 0.56, 95% CI 0.36 to 0.87; Analysis 4.4).
4.5 Adverse effects
One study provided adverse effect data (Chan 2013, N = 221). We divided the data into five subsets.
4.5.1 Cardiovascular ‐ arrhythmia
There was no clear difference between droperidol and olanzapine for arrhythmia (RR 0.32, 95% CI 0.01 to 7.88; Analysis 4.5).
4.5. Analysis.
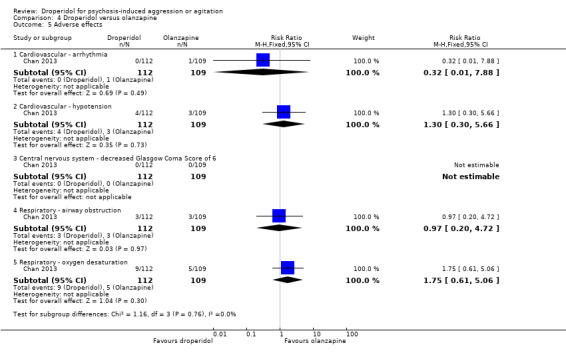
Comparison 4 Droperidol versus olanzapine, Outcome 5 Adverse effects.
4.5.2 Cardiovascular ‐ hypotension
There was no clear difference between droperidol and olanzapine for hypotension (RR 1.30, 95% CI 0.30 to 5.66; Analysis 4.5).
4.5.3 Central nervous system ‐ decreased Glasgow Coma Score (score of 6)
There were no reports of decreased GCS (score of 6) in either the droperidol or olanzapine group (Analysis 4.5).
4.5.4 Respiratory ‐ airway obstruction
We found no evidence of a clear difference between droperidol and olanzapine (RR 0.97, 95% CI 0.20 to 4.72; Analysis 4.5).
4.5.5 Respiratory ‐ oxygen desaturation
We found no evidence of a clear difference between droperidol and olanzapine (RR 1.75, 95% CI 0.61 to 5.06; Analysis 4.5).
4.6 Service use: 1. person able to be discharged home
One study provided data for discharge (Chan 2013, N = 221). There are no subsets in this outcome. We found no evidence of a clear difference between droperidol and olanzapine (RR 1.06, 95% CI 0.83 to 1.34; Analysis 4.6).
4.6. Analysis.
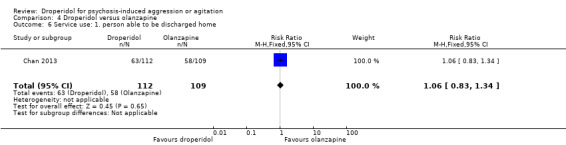
Comparison 4 Droperidol versus olanzapine, Outcome 6 Service use: 1. person able to be discharged home.
4.7 Service use: 2. emergency department length of stay
One study provided data for length of stay in emergency department (N = 221). There was no suggestion of clear difference between droperidol and olanzapine (median stay was around 11 hours for both groups; Analysis 4.7).
4.7. Analysis.
Comparison 4 Droperidol versus olanzapine, Outcome 7 Service use: 2. emergency department length of stay.
| Service use: 2. emergency department length of stay | ||||
|---|---|---|---|---|
| Study | Intervention | Number of participants | Median (hours) | Interquartile range |
| Chan 2013 | Droperidol | 112 | 10.0 | 6.7 to 13.2 |
| Chan 2013 | Olanzapine | 109 | 11.0 | 7.2 to 14.7 |
5 Missing outcomes
We found no data for satisfaction with treatment, acceptance of treamtent, quality of life or economic outcomes.
Discussion
Summary of main results
1 Comparison 1: droperidol versus placebo
1.1 Tranquillisation or asleep: tranquillised/sleep/global state/service use
By 30 minutes, data that we categorise as being of 'high quality' derived from a single trial of over 200 people suggested that droperidol was more acutely tranquillising than placebo (Analysis 1.1). This would fit with clinical experience. This finding also fits with the clear demonstration within the same study of reduced risk of needing additional medication (Analysis 1.4). For the outcome of being ready for discharge, there was no clear difference between groups (RR 1.16, 95% CI 0.9 to 1.48).
1.2 Adverse effects
The one relevant trial (N = 227) found no evidence that droperidol caused more cardiovascular arrhythmia and respiratory airway obstruction than placebo. Droperidol has become less accessible because it has been reported that people who receive droperidol are at higher risk of QT prolongation (Wooltorton 2002). We found no evidence for concern in these short trials for people with acute aggressive behaviour.
1.3 Missing outcomes
It seems worth noting that the one study we found did not report any key outcomes for mental state and none for costs. The global state reported were very useful and, perhaps, trialists considered them to be adequate for the purposes of this question. However, some type of economic consideration of the outcomes is always important and omission of this from a trial conducted in 2012 to 2013 leaves managers and policy makers less informed than they could have been.
2 Comparison 2: droperidol versus haloperidol
2.1 Tranquillisation or asleep: tranquillised/sleep/global state/mental state
By 30 minutes, data that we categorised as being of 'high quality' from a single trial of over 200 people suggested that droperidol was more acutely tranquillising than haloperidol (Analysis 2.1). This finding also fits with the clear demonstration within the same and one other study of reduced risk of needing additional medication (Analysis 2.3). For mental state, there was no evidence of clear difference between the efficacy of droperidol and haloperidol (Scale for Quantification of Psychotic Symptom Severity: MD 0.11, 95% CI ‐0.07 to 0.29) in terms of a reduced mean score by 13 days (Analysis 2.6). This is probably no surprise as key effects of importance in this acutely aggressive situation are measured in hours and by nearly two weeks it would seem unlikely that there should be a discernible difference.
2.1. Analysis.
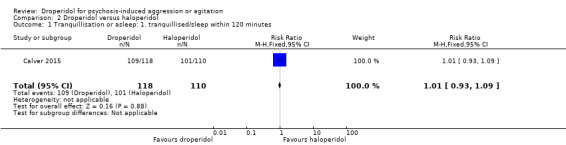
Comparison 2 Droperidol versus haloperidol, Outcome 1 Tranquillisation or asleep: 1. tranquillised/sleep within 120 minutes.
2.2 Adverse effects
The one relevant trial (N = 228) found no evidence that droperidol caused more cardiovascular hypotension and cardiovascular hypotension/desaturation than haloperidol (Analysis 2.5). There was no suggestion that use of droperidol was unsafe.
2.3 Missing outcomes
The one included trial did not report outcomes of service use and costs. Acknowledging their importance, global and mental state were adequately reported. However, economic consideration and service use were omitted from the study which leaves managers and policy makers less informed regarding the cost effectiveness of droperidol over haloperidol.
3 Comparison 3: droperidol versus midazolam
3.1 Tranquillisation or asleep: tranquillised/asleep/global state
By 30 minutes, data that we categorised as being of 'high quality' derived from a single trial of more than 200 people suggested that droperidol was less acutely tranquillising than midazolam in the first few minutes (Analysis 3.1). This would fit with what is known about midazolam from other studies. This finding does not fit with the suggestion within the same study of reduced risk of needing additional medication (Analysis 3.3).
3.2 Adverse effects
The one relevant trial (N = 153) reported no statistically significant differences between droperidol and midazolam ‐ but use of midazolam did result in three people (out of around 70) needing some type of 'airway management' with no such events in the droperidol group. Respiratory depression remains a known concern with midazolam (TREC 2003). It is entirely reversible with the use of flumazenil but even these small trials involving midazolam suggested that use of this effective compound should continue to be in units skilled in recognition of respiratory problems and their management (Analysis 3.4).
3.3 Missing outcomes
The one relevant study did not report service use, mental state and economic costs. Omitting mental state from a study leaves the clinicians less informed of the relative efficacy of droperidol compared to midazolam. Likewise, not reporting important outcome of costs leaves managers and policy makers less informed regarding the cost effectiveness of droperidol over haloperidol.
4 Comparison 4: droperidol versus olanzapine
4.1 Tranquillisation or asleep: tranquillised/asleep/global state/service use
By any time point, we found no clear differences between the older drug (droperidol) and olanzapine (Analysis 4.1). There also was a suggestion that participants allocated to droperidol needed less additional medication than people given the olanzapine (Analysis 4.4). This would fit with clinical experience and other studies of a similar nature (Raveendran 2007).
4.2 Adverse effects
The one relevant trial (N = 221) found no evidence that droperidol caused more cardiovascular arrhythmia and respiratory airway obstruction than olanzapine. The concern regarding droperidol and QT prolongation is not obviously supported by the data we found. We found no evidence for concern in this short trial for people with acute aggressive behaviour.
4.3 Missing outcomes
We found no economic data. However, droperidol should be cheaper than the olanzapine preparation which may in itself increase the risk of further administration of medications. It would seem that droperidol could be both as or more effective and more cost effective.
Overall completeness and applicability of evidence
1 Completeness
Evidence was certainly relevant, but overall data were too sparse to extensively address the objectives of this review. The search strategy identified six trials involving 733 participants comparing droperidol to placebo, non‐standard medication and standard medication. The included studies addressed most of the outcomes being investigated for this review. However, there still were a few outcomes which were not catered for by the trials. These missing outcomes, such as costs, are of prime importance when estimating the cost effectiveness of droperidol when compared with placebo, other 'standard' treatments or 'non‐standard' treatments.
2 Applicability
The included trials were set in psychiatric hospitals, emergency departments and psychiatric crisis units. All trials included people with psychoses. Resnick 1984 did not specify beyond stating that participants were admitted involuntarily to the emergency department of a psychiatric unit. Van Leeuwen 1977 included people with schizophrenia, manic depression or in a 'confusional state'; however, 10 participants had no specific diagnosis. Cocchi 1971 stated that all participants had schizophrenia. These inclusion criteria should make any findings applicable to the acute management of disturbed people thought to experience serious mental illnesses. It is noteworthy that along with inclusion of need for repeat injection in the included studies, outcomes such as further aggressive episodes, tranquillisation, sedation and mental state were also included. However, it is desirable to include outcomes such as quality of life, carer satisfaction, economic costs and loss to follow‐up.
Quality of the evidence
See also Risk of bias in included studies and Table 1; Table 2; Table 3; and Table 4.
Overall the quality of the six include trials was moderate to high based on GRADE. One of the fundamental prerequisites of a randomised trial methodology is random sequence allocation which all trials employed. Calver 2015 used block randomisation. Van Leeuwen 1977 specified that treatment was "randomly assigned" with participants listed in chronological order and assigned individually numbered vials. Therefore, it is unclear whether the people randomising participants could have ascertained the order of prescribing. Resnick 1984 did not specify the explicit means of allocation, although they stated that participants received treatment on a "randomised basis", and that the codes identifying the packages of medication were "kept in the pharmacy until the conclusion of the study". Cocchi 1971 specified only that the study was randomised, with no details regarding the means of allocation. In effort to minimise bias, most of the included trials were double‐blind. All studies reported data for all outcomes listed and were therefore rated at low risk of bias with the exception of one study (Knott 2006), which we rated as having unclear risk of reporting bias. Therefore, we considered the quality of evidence high.
Potential biases in the review process
The search criteria on the Cochrane Schizophrenia Group Trials Register (December 2015) are sufficiently robust to detect relevant studies. However, it is possible that we have failed to identify small studies but we think it unlikely that we would have missed large trials. Studies published in languages other than English, and those with equivocal results, are often difficult to find (Egger 1997). Our search was biased by use of English phrases. However, given that the Cochrane Schizophrenia Group's Register covers many languages but is indexed in English we feel that this would not have missed many studies within the register. For example, the search uncovered two studies for which the title was only available in Chinese characters. A Chinese‐speaking colleague (Jun Xia) checked these for relevance and neither were relevant to this review.
Furthermore, we were not blinded to the names of the authors, institutions or journal of publication which may have introduced some type of bias in the review process.
Agreements and disagreements with other studies or reviews
A previous version of this review did not identify many studies that met the inclusion criteria (Cure 2004). However, this updated version of the review found three more relevant studies. At this point, we are unaware of any other similar reviews or studies.
Authors' conclusions
Implications for practice.
1 For people with psychotic illness
Acute psychotic illness, especially with agitated, aggressive or violent behaviours, may require rapid tranquillisation or sedation. Droperidol, a butyrophenone neuroleptic that is no longer commercially promoted or manufactured, remains a viable option for this purpose based on more evidence from randomised controlled trials in this 2016 update.
2 For clinicians
Intramuscular droperidol was once a popular choice for the acute management of very psychotic aggressive and agitated people. The first version of this review thought that evidence relating to use of droperidol was of historical interest only (Cure 2004). This seems to be untrue. Droperidol is still used. There remains compelling evidence that droperidol has a place in short‐term management of psychotic aggressive people. The evidence presented in this review allows conclusions to be drawn about its comparative efficacy to haloperidol, midazolam and olanzapine that concur with the impressions of the effectiveness of these other compounds from other sources. Clinicians could help evaluate these different approaches by supporting clinically relevant randomised controlled trials. We found no evidence of reasons for any more concern over cardiac problems than with other approaches, and less for respiratory difficulties than midazolam.
3 For managers/policy makers
Currently it seems that people in Australia, Belgium, Brazil, Czech Republic, Denmark, Finland, Greece, India, Italy, Netherlands, New Zealand, South Africa, Spain, Sweden, Thailand, and the USA have droperidol as one option for treatment of aggression thought due to psychosis. Much evidence for other compounds or approaches is no stronger than for this old drug.
Implications for research.
1 General
As with all similar studies, public registration of a study before randomisation commences would ensure that participants could be confident that people would know that the study had at least taken place. Better reporting of data would have allowed us to determine the effects of this compound in emergency situations. Newer trials tended to comply with CONSORT making it much easier to understand the methods of the studies. We hope that trials in the next version of this review will go that one last and important step and allow full access to all data (AllTrials; OpenTrials).
2 Specific
2.1 Reviews
Several of the excluded studies in this review would be relevant for inclusion in related Cochrane reviews (Table 10).
2.2 Trials
This review highlights the urgent need for more good quality controlled trials of other compounds for management of acute psychosis that address outcomes of major importance such as quality of life, economic costs and satisfaction of carers. We realise that design of such studies takes time and a great deal of thought and commitment. However, we have given this area some thought and suggest the broad outline of a trial in Table 11.
7. Design of a future study.
| Methods | Allocation: randomised (clearly described). Blinding: single blind (outcomes assessor). Duration: up to 2 weeks. Design: parallel. Setting: emergency settings. |
| Participants | Diagnosis: people whose aggressive behaviour is thought due to psychotic illness. N = 300. Age: > 18 years. Sex: not applicable. Inclusion criteria: other measures failed. Exclusion criteria: specific contraindication to evaluated treatments. |
| Interventions | 1. Droperidol. N = 150. 2. Drug intervention of choice. N = 150. Both drugs should be known to be effective, but the comparative effectiveness be unclear. |
| Outcomes | Tranquil/asleep: binary outcomes, time. Behaviour: need for additional medication, additional aggressive episode. Adverse effects. Acceptability of treatment. Costs: cost of services, cost of care. Quality of life. Service outcomes: days in hospital, discharged, transfer to secure unit. |
| Notes | Study should comply with CONSORT and AllTrials. |
Feedback
Error in publication date of van Leeuwen study, 14 May 2008
Summary
I would like to advise of an error in the date of one of the studies included. Reference by van Leeuwen was published in 1977 not 1997? (1997 was used in various sites of the paper).
Reply
We thanked the contributor for alerting the review authors to this error and have now made the necessary amendments to the review.
Contributors
Feedback submitted by Esther Chan.
Reply submitted by Bethany York, Review Group Co‐ordinator, Cochrane Schizophrenia Group.
What's new
| Date | Event | Description |
|---|---|---|
| 1 June 2016 | New citation required but conclusions have not changed | New evidence added to review but conclusions unchanged |
| 13 January 2016 | New search has been performed | Major update. Two new trials added. Conclusions unchanged but strengthened. |
History
Protocol first published: Issue 4, 2000 Review first published: Issue 2, 2001
| Date | Event | Description |
|---|---|---|
| 13 November 2013 | Amended | Eight new references from updated search (August 12, 2013) were added to 'Pending classification references' section of the review. |
| 3 May 2012 | Amended | Additional table linked to text |
| 13 April 2011 | Amended | Contact details updated. |
| 20 April 2009 | Feedback has been incorporated | Further to submitted feedback the publication date of the van Leeuwen study has been corrected from 1997 to 1977. |
| 24 April 2008 | Amended | Converted to new review format. |
| 18 September 2003 | New citation required and conclusions have changed | Substantive amendment |
Notes
None.
Acknowledgements
We would like to thank Sharon Cure and Simone Carpenter for their contributions to earlier versions of this review. The review authors would like to acknowledge the help of Jo Wood of Janssen‐Cilag Pharmaceuticals, England (in 2000), for her help identifying trials relevant to droperidol; Brian Devine and Mike Musker of the Ashworth Hospital, for their initial support; and Nicola Howson and Nancy Owens from the Cochrane Schizophrenia Group (Oxford) for their help and support for the 2004 version of this review. Thanks now to Clive E Adams (CEA) and the editorial team at the Nottingham University Cochrane Schizophrenia Group for their unwavering support in the writing of this review. The Cochrane Schizophrenia Group Editorial Base in Nottingham produces and maintains standard text for use in the Methods sections of their reviews. We have used this text as the basis of what appears here and adapted it as required. Thanks also to Jun Xia who examined the Chinese language titles returned by our search. We would also like to thank Ji Xu and Vivek Agarwal for peer reviewing this version of the review.
We would also like to acknowledge Linda Gowing who submitted a comment just before publication of this update. Her comment was correct, the review was out of date and refers to injectable forms of droperidol being withdrawn by the manufacturer and worldwide stocks of intramuscular droperidol running low ‐ which is no longer true. We have addressed this issue in our current background by updating this statement, indicating the withdrawal of droperidol has not happened.
Appendices
Appendix 1. Previous searches
1.1 Searches in 1998 to 2000
This review is part of a larger project attempting to identify all randomised trials relevant to the management of aggressive or violent people. The searches are therefore more general than would be expected for such a review (see 2.6).
1.1.1 Electronic searches
1.1.1.1 AMED (1983 to December 1998)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat* or "ANGER‐" in SH or "AGGRESSION‐" in SH or explode "CRIME" or "EXHIBITIONISM‐" in SH or "JUVENILE‐DELINQUENCY" in SH or "PRISONS‐" in SH or "PRISONERS‐" in SH or explode "VIOLENCE")
1.1.1.2 ASSIA (1987 to December 1998)
randomi* or random* and allocat* or random* and assign* or singl* blind* or doubl* blind* or tripl* blind* or trebl* blind* The resulting records were then searched in ProCite using the phrase: homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or assault* or assail* or attacker* or ((physical or spouse or elder or sexual* or child or partner) and abus*) or rape* or rapist* or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* or hostil* or cruel* or delinquen* or threaten* or disorderly or affray* or felon* or unlawful* or penal* or penol* or crim* or offen* or prison* or inmate* or correctional* or firearm* or weapon* or gun* or (intent and (kill or harm)) or (bodily and harm) or (child and neglect) or masturbat* or (breach* and peace) or (dangerous and (behav* or histor* or conduct*)) or (disrupt* and (behav* or histor* or conduct*)) or ((destruct* and not self‐destruc*) and (behav* or histor* or conduct*)) or ((anti‐social or antisocial) and (behav* or histor* or conduct*)) or agitat*
1.1.1.3 Biological Abstracts (1993 to September 1999) and BA on CD (1982 to 1985)
They were searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat*)
1.1.1.4 Brainwave (Pharmaceutical Newsletters, Pharmaceutical Research, Pharmaceutical Industry News, Social Sciences, Medical Research)
It was searched on the internet (http://www.brainwave.telebase.com [Searched on May 5, 2000]) using the phrase: ((random* or doubl*) and aggress*) or ((random* or doubl*) and violen*) within the title field
1.1.1.5 British Nursing Index/RCN Journals (1988 to September 1999)
They were searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat*)
1.1.1.6 Cambridge Scientific Abstracts (1982 to January 2000)
It was searched using the phrase: (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or assault* or assail* or attacker* or ((physical or spouse or elder or sexual* or child or partner) and abus*) or rape* or rapist* or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* or hostil* or cruel* or delinquen* or threaten* or disorderly or affray* or felon* or unlawful* or penal* or penol* or crim* or offen* or prison* or inmate* or correctional* or firearm* or weapon* or gun* or (intent and (kill or harm)) or (bodily and harm) or (child and neglect) or masturbat* or (breach* and peace) or (dangerous and (behav* or histor* or conduct*)) or (disrupt* and (behav* or histor* or conduct*)) or ((destruct* and not self‐destruc*) and (behav* or histor* or conduct*)) or ((anti‐social or antisocial) and (behav* or histor* or conduct*))) or agitat* and (randomi* or (random* and alloc*) or (random* and assign*) or (doubl* and blind))
1.1.1.7 CINAHL (1982 to October 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat* or "Anal‐Intercourse"/ all topical subheadings / all age subheadings or explode "Aggression"/ all topical subheadings / all age subheadings or explode "Public‐Offenders"/ all topical subheadings / all age subheadings or explode "Risk‐for‐Violence‐Self‐Directed‐or‐Directed‐at‐Others‐(NANDA)"/ all topical subheadings / all age subheadings or explode "Crime"/ all topical subheadings / all age subheadingsv or "Masturbation"/ all topical subheadings / all age subheadings or "Anger"/ all topical subheadings / all age subheadings or explode "Weapons"/ all topical subheadings / all age subheadings or "Prisoners"/ all topical subheadings / all age subheadings or "Correctional‐Facilities"/ all topical subheadings / all age subheadings)
1.1.1.8 The Cochrane Controlled Trials Register (1999, Issue 4)
It was searched using the phrase: (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near (kill or harm) or bodily near harm or assault* or assail* or attacker* or physical near abus* or spouse near abus* or partner near abus* or child:ti near neglect:ti or child:ab near neglect:ab or child:ti near abus*:ti child:ab near abus*:ab or elder near abus* or rape*:ti rape*:ab or rapist* or sexual*:ti near abus*:ti or sexual*:ab near abus*:ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic*:ti malic*:ab or hostil* or (dangerous or disrupt*) near (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near (behav* or histor* or conduct*) or (antisocial or anti‐social) near (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near peace or felon* or unlawful* or penal* or penol* or crim*:ti crim*:ab or offen*:ti offen*:ab or prison* or inmate* or correctional* or firearm* or weapon* or gun*:ti gun*:ab or agitat*) NOT (Cancer or carcinoma)
1.1.1.9 The Cochrane Schizophrenia Group's Database of Conference Abstracts (1971 to December 1999)
It was searched using the phrase: homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or assault* or assail* or attacker* or ((physical or spouse or elder or sexual* or child or partner) and abus*) or rape* or rapist* or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* or hostil* or cruel* or delinquen* or threaten* or disorderly or affray* or felon* or unlawful* or penal* or penol* or crim* or offen* or prison* or inmate* or correctional* or firearm* or weapon* or gun* or (intent and (kill or harm)) or (bodily and harm) or (child and neglect) or masturbat* or (breach* and peace) or (dangerous and (behav* or histor* or conduct*)) or (disrupt* and (behav* or histor* or conduct*)) or ((destruct* and not self‐destruc*) and (behav* or histor* or conduct*)) or ((anti‐social or antisocial) and (behav* or histor* or conduct*)) or agitat*
1.1.1.10 The Cochrane Schizophrenia Group Register (updated June 2000)
It was searched using the phrase: homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or "intent to kill" or "intent to harm" or "bodily harm" or assault* or assail* or attacker* or "physical abus*" or "spouse abus*" or "partner abus*" or "child neglect" or "child abus*" or "elder abus*" or rape* or rapist* or "sexual* abus*" or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* or hostil* or "(dangerous or disrupt*) near (behav* or histor* or conduct*)"or "dangerousness or (destruct* not self‐destruct*) near (behav* or histor* or conduct*)"or "(antisocial or anti‐social) near (behav* or histor* or conduct*)" or cruel* or delinquen* or threaten* or disorderly or affray* or "breach* of the peace" or felon* or unlawful* or penal* or penol* or crim* or offen* or prison* or inmate* or correctional* or firearm* or weapon* or gun* or agitat*
1.1.1.11 Criminal Justice Abstracts (January to September 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson)
1.1.1.12 Dissertations Abstracts (1861 to December 1999)
It was searched using the phrase: (Homicid? or murder? or manslaughter? or infanticid? or parricid? or tortur? or (intent and (kill or harm)) or (bodily and harm) or assault? or assail? or attacker? or physical abus? or spouse abus? or partner abus? or child neglect or child abus? or elder abus? or rape? or rapist? or (sexual? and abus?) or bugger? or sodom? or molest? or pedophil? or paedophil? or indecen? or masturbat? or exhibitionis? or lewd? or sadis? or sadomasochis? or aggress? or violen? or anger or malic? or hostil? or cruel? or delinquen? or threaten? or disorderly or affray? or (breach? and peace) or felon? or unlawful? or penal? or penol? or crim? or offen? or ((dangerous or disrupt?) or dangerousness or (destruct? and not self‐destruct?) or (antisocial or anti‐social) or abduct? or kidnap? or prison? or inmate? or correctional? or firearm? or weapon? or gun?) or agitat*) and (randomi? or (random? and assign?) or (random? and allocat?) or blind?)
1.1.1.13 Embase (1980 to October 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials (see Group Module) combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat* or explode "aggression"/ all subheadings or explode "antisocial‐behavior"/ all subheadings or "pedophilia"/ all subheadings or "offender"/ all subheadings or "masturbation"/ all subheadings or "exhibitionism"/ all subheadings or "prison"/ all subheadings or "prisoner"/ all subheadings or "sadism"/ all subheadings or explode "crime"/ all subheadings or explode "weapon"/ all subheadings)
1.1.1.14 Health CD (1994 to December 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat*)
1.1.1.15 Index to Scientific and Technical Proceedings (1990 to March 2000)
It was searched using the phrase: randomised or randomized or randomly & allocat* or randomly & assign* or doubl* & blind*
1.1.1.16 International Bibliography of the Social Sciences (1951 to January 2000)
It was searched using the phrase: (random* alloc*) or randomi* or (random* assign*) or (double blind)
1.1.1.17 International Pharmaceutical Abstracts (1970 to December 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson)
1.1.1.18 MEDLINE (1966 to December 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat* or explode "Aggression"/ all subheadings or "Sadism"/ all subheadings or "Firearms"/ all subheadings or explode "Anger"/ all subheadings or "Dangerous‐Behavior" or explode "Homicide"/ all subheadings or explode "Sex‐Offenses"/ all subheadings or explode "Violence"/ all subheadings or "Pedophilia"/ all subheadings or "Masturbation"/ all subheadings or "Exhibitionism"/ all subheadings or "Prisoners"/ all subheadings or explode "Prisons"/ all subheadings" or Juvenile‐Delinquency"/ all subheadings or "Hostility")
1.1.1.19 NCCAN
It was searched on the internet (http://www.CALIB.com/NCCANCH/ [Searched on December 14, 1999) using the phrase: random or randomize or randomised or randomly
1.1.1.20 NCJRS
It was searched on the internet (http://www.NCJRS.org [Searched December 9, 1999] using the phrase: randomi$ or (random$ and (alloc$ or assign$))
1.1.1.21 PAIS (1972 to October 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat*)
1.1.1.22 PASCAL (1984 to January 2000)
It was searched using the phrase: (aggress* & randomi*) or (aggress* & randomly) or (violen* & randomi*) or (violen* & randomly) or (abus* & randomi*) or (abus* & randomly)
1.1.1.23 PsycLIT (1897 to September 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat* or explode "Antisocial‐Behavior" or explode "Sadomasochism" or "Weapons‐" in DE or "Prisoners‐" in DE or explode "Anger" or "Penology‐" in DE or "Exhibitionism‐" in DE or "Aggressive‐Behavior" in DE or "Aggressiveness‐" in DE or "Dangerousness‐" in DE or explode "Correctional‐Institutions" or explode "Criminals" or explode "Homicide" or "Pedophilia‐" in DE or "Masturbation‐" in DE)
1.1.1.24 Sociological Abstracts (1963 to September 1999)
It was searched using the Cochrane Schizophrenia Group's phrase for randomised controlled trials combined with the phrase: and (homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or intent near2 (kill or harm) or bodily near1 harm or assault* or assail* or attacker* or physical abus* or spouse abus* or partner abus* or child neglect in ti or child neglect in ab or child abus* in ti or child abus* in ab or elder abus* or rape* in ti or rape* in ab or rapist* or sexual* abus* in ti or sexual* abus* in ab or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or masturbat* near2 public or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* in ti or malic* in ab or hostil* or (dangerous or disrupt*) near2 (behav* or histor* or conduct*) or dangerousness or (destruct* not self‐destruct*) near2 (behav* or histor* or conduct*) or (antisocial or anti‐social) near2 (behav* or histor* or conduct*) or cruel* or delinquen* or threaten* or disorderly or affray* or breach* near3 peace or felon* or unlawful* or penal* or penol* or crim* in ti or crim* in ab or offen* in ti or offen* in ab or prison* or inmate* or correctional* or firearm* or weapon* or (gun* in ti or gun* in ab) not gunderson or agitat* or "Aggression‐" in DE or "Violence‐" in DE or "Family‐Violence" in DE or explode "Homicide" or "Kidnapping‐" in DE or "Torture‐" in DE or explode "Assault" or "Attack‐" in DE or explode "Child‐Abuse" or explode "Elder‐Abuse" or explode "Sexual‐Abuse" or explode "Spouse‐Abuse" or "Abuse‐" in DE or "Child‐Neglect" in DE or explode "Offenders" or "Masturbation‐" in DE or explode "Sexual‐Deviation" or explode "Anger" or "Deviant‐Behavior" in DE or "Threat‐" in DE or explode "Criminality" or "Correctional‐System" in DE or explode "Crime" or "Prisoners‐" in DE or "Imprisonment‐" in DE or "Juvenile‐Correctional‐Institutions" in DE or "Firearms‐" in DE)
1.1.1.25 SPECTR (ERIC, 1966 to 1998; Criminal Justice Abstracts, 1968 to 1998; Sociological Abstracts, 1974 to 1996)
It was searched using the phrase: homicid* or murder* or manslaughter* or infanticid* or parricid* or tortur* or assault* or assail* or attacker* or ((physical or spouse or elder or sexual* or child or partner) and abus*) or rape* or rapist* or bugger* or sodom* or molest* or pedophil* or paedophil* or indecen* or exhibitionis* or lewd* or sadis* or sadomasochis* or abduct* or kidnap* or aggress* or violen* or anger or malic* or hostil* or cruel* or delinquen* or threaten* or disorderly or affray* or felon* or unlawful* or penal* or penol* or crim* or offen* or prison* or inmate* or correctional* or firearm* or weapon* or gun* or (intent and (kill or harm)) or (bodily and harm) or (child and neglect) or masturbat* or (breach* and peace) or (dangerous and (behav* or histor* or conduct*)) or (disrupt* and (behav* or histor* or conduct*)) or ((destruct* and not self‐destruc*) and (behav* or histor* or conduct*)) or ((anti‐social or antisocial) and (behav* or histor* or conduct*)) or agitat*
1.1.1.26 The Composite Aggression/Violence Trials Database
Searches 1.1 to 1.25 identified some 22,000 references of which 2,200 appeared to be randomised controlled trials relevant to the management of aggressive or violent people. This database was searched using the following phrase: Droperidol* OR Inapsin* OR Droleptan OR dehydrobenzperidol OR Dridol OR Sintodian OR Paxical OR (Leptanal AND comp*) OR Leptofen OR r04749*
1.1.2 Searching other resources
1.1.2.1 Reference searching
We inspected references of all included studies for further relevant studies.
1.1.2.2 Personal contact
We contacted the first author of each included study for information regarding unpublished trials.
1.2 Search in 2013
1.2.1 Electronic searches
1.2.1.1 Cochrane Schizophrenia Group's Register
We searched Cochrane Schizophrenia Group's Register (August 12, 2013) using the phrase: (droperidol* or *4749* or Dehydrobenzperidol* or Dridol* or Droleptan* or Inapsin* or Leptanal* or Leptofen* or Paxical* or Sintodian* or Thalamonal* in title) or (*droperidol* or *4749* or *Dehydrobenzperidol* or *Dridol* or *Droleptan* or *Inapsin* or *Leptanal* or *Leptofen* or *Paxical* or *Sintodian* or *Thalamonal* in abstract, index or title terms of REFERENCE) or droperidol* or *4749* or Dehydrobenzperidol* or Dridol* or Droleptan* or Inapsin* or Leptanal* or Leptofen* or Paxical* or Sintodian* or Thalamonal* in interventions of STUDY }
The Schizophrenia Group's trials register is based on regular searches of BIOSIS Inside, CENTRAL, CAJ, CINAHL, EMBASE, MEDLINE and PsycINFO; the hand searching of relevant journals and conference proceedings, and searches of several key grey literature sources. A full description is given in the Group's module.
Appendix 2. Previous data collection and analysis
We (MM and AL) searched The Cochrane Schizophrenia Group's register. Working independently we examined the papers identified from the search strategy. We discarded obviously irrelevant publications and retained only those in which some form of early intervention had been compared against a control treatment, and obtained copies of papers relating to relevant trials. Once we had obtained these papers, we decided whether the trials were eligible. We resolved any disagreements by discussion. For the 2006 update we (MM and JR) independently inspected citations. Where disagreement occurred, we sought to resolve this by discussion, or where doubt remained, we acquired the full article for further inspection. Once we had obtained the full articles, we independently decided whether they met the review criteria. We resolved any disagreements that occurred by discussion, and when this was not possible we added trials to the list of those awaiting assessment until we acquired further information. For the 2009 update we (MM and JR) inspected all study citations identified by the searches, and obtained full reports of the studies of agreed relevance.
Data extraction and management
1. Extraction
We (MM, AL) independently extracted and entered trial data into Review Manager (RevMan) twice, cross‐checking for consistency (RevMan 2008). An initial analysis included all trials meeting inclusion criteria, whilst a second sensitivity analysis excluded all but the highest quality trials (Category A and B). For the 2006 and 2010 update, we (MM and JR) independently extracted and entered data into RevMan, cross‐checking again for consistency. Where disputes arose, we attempted to resolve these by discussion. When this was not possible and further information was needed to resolve the dilemma, we did not enter the data, and added this outcome of the trial to the list of those awaiting assessment.
2. Management
2.1 Forms
We extracted the data onto standard, simple forms.
2.2 Direction of graphs
Where possible, we entered data into RevMan in such a way that the area to the left of the 'line of no effect' indicates a 'favourable' outcome for early intervention. Where this was not possible, (for example, scales that calculate higher scores=improvement) we inserted a minus sign into the data tables to reverse the graphical display in RevMan analyses so that the direction of effect was clear.
2.3 Scale‐derived data
Unpublished scales are known to be subject to bias in trials of treatments for schizophrenia (Marshall 2000). Therefore we only included continuous data from rating scales were if the measuring instrument had been described in a peer‐reviewed journal.
2.4 Skewed data
Continuous data on outcomes in trials relevant to mental health issues are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data we applied the following standards to continuous final value endpoint data before inclusion: (a) standard deviations and means were reported in the paper or were obtainable from the authors; (b) when a scale started from zero, the standard deviation, when multiplied by two, should be less than the mean (otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution ‐ Altman 1996); in cases with data that are greater than the mean we entered them into the 'Other data' table as skewed data. Where the skewed data are derived from a trial with ≧ 200 participants, the skewed data pose less of a problem when looking at means if the sample size is large and were entered into syntheses.
If a scale starts from a positive value (such as PANSS, which can have values from 30 to 210) the calculation described above in (b) should be modified to take the scale starting point into account. In these cases skewness is present if 2SD>(S‐Smin), where S is the mean score and Smin is the minimum score. We reported non‐normally distributed data (skewed) in the 'other data types' tables.
For change data (mean change from baseline on a rating scale) it is impossible to tell whether data are non‐normally distributed (skewed) or not, unless individual patient data are available. After consulting the ALLSTAT electronic statistics mailing list, we entered change data in RevMan analyses and reported the finding in the text to summarise available information. In doing this, we assumed either that data were not skewed or that the analysis could cope with the unknown degree of skew.
2.5 Final endpoint value versus change data
Where both final endpoint data and change data were available for the same outcome category, only final endpoint data were presented. We acknowledge that by doing this much of the published change data may be excluded, but argue that endpoint data is more clinically relevant and that if change data were to be presented along with endpoint data, it would be given undeserved equal prominence. Where studies reported only change data we contacted authors for endpoint figures.
2.6 Common measure
To facilitate comparison between trials, we converted variables (such as days in hospital) that could be reported in different metrics (mean days per year, per week or per month) to a common metric (for example, mean days per month).
2.7 Conversion of continuous to binary
Where possible, efforts were made to convert outcome measures to dichotomous data. This could be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the Positive and Negative Syndrome Scale (PANSS, Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.8 Summary of findings table
For the 2011 version of the review we had available to us the possibility of producing Summary of Findings tables. These should be considered before being biased by the results of analyses, but for us this is impossible. We have chosen to present two ‐ but this choice is post hoc. We chose to present data from PACE‐Australia and OPUS‐Scandinavia as these are benchmark trials in this area and outcomes from these trials that we think to be clinically important.
Progression to psychosis
Compliance with treatment ‐ treatment stopped in spite of need
Leaving the study early
Service use: 1. Average mean number of days per month in hospital
Service use: 2. Not hospitalised
Social outcomes: 1. Not living independently
Social outcomes: 2. Not working or in education
Assessment of risk of bias in included studies
Again working independently, we assessed risk of bias using the tool described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008). This tool encourages consideration of how the sequence was generated, how allocation was concealed, the integrity of blinding at outcome, the completeness of outcome data, selective reporting and other biases. We would not have included studies where sequence generation was at high risk of bias or where allocation was clearly not concealed.
The categories are defined below.
‐ YES ‐ low risk of bias ‐ NO ‐ high risk of bias ‐ UNCLEAR ‐ uncertain risk of bias
If disputes arose as to which category we should allocate a trial, again, we achieved resolution by discussion, after working with a third reviewer.
Earlier versions of this review used a different, less well‐developed, means of categorising risk of bias (see Appendix 2).
Measures of treatment effect
1. Binary data
For binary outcomes we calculated an estimate of the risk ratio (RR) and its 95% (fixed‐effect) confidence intervals (CI). RR is more intuitive (Boissel 1999) than odds ratios and odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). This misinterpretation then leads to an overestimate of the impression of the effect. When the overall results were significant we calculated the number needed to treat/harm (NNT/NNH) using Visual Rx.
2. Continuous data
For continuous outcomes we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference SMD). However, had scales of very considerable similarity been used, we would have presumed there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ cluster randomisation (such as randomisation by clinician or practice), but analysis and pooling of clustered data pose problems. Firstly, authors often fail to account for intra‐class correlation in clustered studies, leading to a unit‐of‐analysis error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes Type I errors (Bland 1997; Gulliford 1999).
Where clustering had not been accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation co‐efficients (ICCs) of their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a design effect. This is calculated using the mean number of participants per cluster (M) and the ICC (Design effect=1+ (M ‐1)*ICC) (Donner 2002). If the ICC is not reported we assumed it to be 0.1 (Ukoumunne 1999). If cluster studies had been appropriately analysed taking into account ICCs and relevant data documented in the report, we synthesised these¬with other studies using the generic inverse variance technique.
2. Cross‐over design
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (for example, pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state, despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia,¬we will only use¬data¬of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
We presented studies involving more than two treatment arms, if relevant, in comparisons.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss to follow‐up data must lose credibility (Xia 2007). We are forced to make a judgment where this is for the trials likely to be included in this review. Should more than 50% of data be unaccounted for by eight weeks, we did not reproduce these data or use them within analyses.
2. Intention to treat analysis
2.1 Binary data
We excluded data from studies where more than 50% of participants in any group were lost to follow‐up (this did not include the outcome of 'leaving the study early'). In studies with less than 50% dropout rate, people leaving early were considered to have had the negative outcome, For example, those lost to follow‐up for the outcome of relapse were treated in the analysis as having relapsed. Suicide was treated as relapse.
2.2 Continuous data
2.2.1 Attrition
In the case where attrition for a continuous outcome is between 0% and 50% and completer‐only data were reported, we have reproduced these.
2.2.2 Standard deviations
We first tried to obtain the missing values from the authors. If not available, where there were missing measures of variance for continuous data but an exact standard error and confidence interval were available for group means, and either P value or T value were available for differences in mean, we noted these, and in future versions will calculate them according to the rules described in the Handbook (Higgins 2008): When only the standard error (SE) is reported, standard deviations (SDs) can be calculated by the formula SD=SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Handbook (Higgins 2008) present detailed formula for estimating SDs from P values, T or F values, confidence intervals, ranges or other statistics. If these formula do not apply, we, in the future will calculate SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Some of these imputation strategies can introduce error. The alternative would be to exclude a given study’s outcome and thus to lose information. We will examine the validity of the imputations in a sensitivity analysis excluding imputed values.
2.2.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results. Therefore, where LOCF data have been used in the trial, if less than 50% of the data had been assumed, we reproduced these data and indicated that they are the product of LOCF assumptions.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying situations or people which we had not predicted would arise. When such situations or participant groups arose, we would have fully discussed these.
2. Methodological heterogeneity
We considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. Should such methodological outliers arise we would have fully discussed these.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 method alongside the Chi2 P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on i. magnitude and direction of effects and ii. strength of evidence for heterogeneity (e.g. P value from Chi2¬test, or a confidence interval for I2). We interpreted I2 estimate greater than or equal to 50% accompanied by a statistically significant Chi2 statistic as evidence of substantial levels of heterogeneity (Section 9.5.2 ‐ Higgins 2008). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. These are described in section 10.1 of the Handbook (Higgins 2006). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects (Egger 1997). We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
Where possible we employed a fixed‐effect model for analyses. We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us; however, random‐effects does put added weight onto the smaller of the studies ‐ those trials that are most vulnerable to bias. For this reason we favour using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We did not anticipate subgroup analyses.
2. Investigation of heterogeneity
If inconsistency was high, we have reported this. First we investigated whether data had been entered correctly. Second, if data had been correct, we visually inspected the graph and successively removed studies outside of the company of the rest to see if homogeneity was restored. Should this occur with no more than 10% of the data being excluded, we have presented data. If not, we have not pooled data and have discussed relevant issues.
Should unanticipated clinical or methodological heterogeneity be obvious we simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
For the 2011 version of this review we did not anticipate undertaking any additional sensitivity analyses.
Appendix 3. Previous effects of interventions
1. Comparison 1. droperidol versus placebo
The search identified only one small (N = 41) randomised trial comparing droperidol (10 mg IV) with placebo (IV) Van Leeuwen 1977a.
1.1 Global impression: needing additional injections within the first 90 minutes. Those allocated to droperidol were significantly less likely to need additional haloperidol injections within the first few minutes (N = 41, RR 0.37, 95% CI 0.2 to 0.7, NNTB 2, 95% CI 1 to 10) than those given an injection of placebo. By 90 minutes, this difference was still evident but not statistically significant (RR 0.46, 95% CI 0.2 to 1.2).
1.2 Adverse effects The Van Leeuwen 1977a trial reported that no adverse effects were apparent for the 41 people randomised to droperidol or placebo. However, this observation referred only to the first three minutes after the initial injection had been given. After three minutes haloperidol was administered to 25/41 trial participants and some adverse effects may have been caused by that drug. However, any that occurred after the first three minutes were not reported.
2. Comparison 2. droperidol versus haloperidol
The search identified only two small trials. Resnick 1984 (N = 27) was clearly relevant to the acute management of disturbed people. In this study, participants were randomised to 5 mg of IM droperidol or 5 mg of IM haloperidol. Cocchi 1971, however, was also identified by the search and is less clearly relevant. Although this study does deal with acutely disturbed or relapsing people, it allocated them to oral droperidol or haloperidol, suggesting that these trial participants were not as disturbed as in the other studies. Outcomes were also measured after 30 days rather than after a few hours, which makes this trial less relevant to the emergency situation investigated by this review.
2.1 Global impression: needing additional injections within the first 90 minutes Those allocated to droperidol were less likely to need additional injections by 30 minutes than those given haloperidol, but this result did not quite reach conventional levels of statistical significance (N = 27, 1 RCT, RR 0.45, 95% CI 0.2 to 1.0). Resnick 1984 reported the need for additional repeat injections up to 90 minutes. Not one of those allocated to droperidol required repeat injections, but three in the haloperidol group were given another injection at 60 minutes (N = 27, RR 0.20, 95% CI 0.01 to 3.6), and one unfortunate person had to be medicated yet again at 90 minutes (N = 27, 1 RCT, RR 0.47, 95% CI 0.02 to 10.6).
2.2 Global impression: no clear improvement by 30 days Cocchi 1971 reported that oral droperidol was no more likely to afford improvement in acutely ill people than oral haloperidol at 30 days (N = 40, RR 0.67, 95% CI 0.3 to 1.5).
2.3 Mental state: mean score on the Rating Scale for Quantification of Psychotic Symptoms The study on less acutely disturbed people in the non‐emergency situation found no difference between oral droperidol and oral haloperidol on ratings of this scale (N = 40, mean difference 0.11, 95% CI ‐0.1 to 0.3) (Cocchi 1971).
2.4 Adverse effects: mild dystonia Resnick 1984 reported that one person experienced a mild dystonic reaction when given haloperidol IM (N = 27, RR 0.47, 95% CI 0.02 to 10.6).
Appendix 4. Previous discussion
1. Droperidol discontinued Janssen‐Cilag Limited produced droperidol (www. janssen‐cilag.co.uk) until March 2001, when production of all formulations of its branded form (Droleptan) were discontinued. The Medicines Control Agency in the UK (www.mca.gov.uk/) had raised concerns about the potential effects of long‐term droperidol on the electrical conduction of the heart (cardiac Q‐T interval prolongation) and requested a risk‐benefit assessment. The company concluded that the oral formulations should be discontinued to prevent use. The authors have asked Janssen‐Cilag UK whether or not the injectable form for rapid tranquillisation was withdrawn because of concerns about safety. The company have informed us that the injectable form of droperidol also carries significant risks (Lawrence 1997) and has been associated with prolonged Q‐T intervals (Guy 1991; Lischke 1994) as well as the rare, but potentially fatal, cardiac arrhythmia, 'Torsade de pointes' (Guy 1991; Michalets 1998). Although the benefits of its continued use in the acute situation may have outweighed these risks, careful monitoring ‐ including ECGs and electrolyte assays ‐ would have been necessary. This would have compromised the cost effective production of injectable droperidol, and so it was discontinued along with the oral preparations.
2. Small number of studies Acute psychosis is difficult to study and co‐operation from the study population is rare. This may be one of the reasons for the scarcity of controlled clinical trials using droperidol solely for this indication. Droperidol appears to have been widely used in emergency room situations for people who are agitated or acutely disturbed but who have not, at point of medication, been diagnosed (Binder 1999; Pilowsky 1992). Several papers identified by our searches concerned the use of droperidol for people who were later diagnosed as having either trauma, an underlying organic condition, or who were intoxicated. As these diagnoses fell outside our remit for types of participants, the trials were excluded from this review. Nevertheless, acute disturbance due to suspected mental illness is so common (Huf 2002), and management of such situations so important, that there is little excuse not to have good evidence for the use of droperidol. However, a total of only 69 people seem to have been randomised into trials of droperidol versus placebo or haloperidol that are relevant to the emergency control of disturbance thought to be due to mental illnesses.
3. Quality No trial reported adequate methods of random sequence generation, and only Van Leeuwen 1977a included any description of the method of randomisation. No trial included in this review would have rated highly with respect to the CONSORT statement (Begg 1996; Moher 2001) and the inclusion of bias is likely.
4. Publication bias With such small studies publication bias is also likely, and for both truly relevant trials (Resnick 1984; Van Leeuwen 1977) droperidol was the experimental intervention, so any publication bias would favour droperidol.
5. Applicability of findings The included trials were set in a psychiatric hospital, an emergency department, and a psychiatric crisis unit. The truly relevant studies (Resnick 1984; Van Leeuwen 1977) reported outcomes for the very short term, i. e. those of value in the crisis situation. Van Leeuwen 1977a included people with schizophrenia, manic‐depression and confusional states, with seven of the 41 participants having no diagnosis recorded. Resnick 1984 included participants who were involuntarily hospitalised and had 'underlying psychoses'. This trial specifically excluded patients who were intoxicated (the only trial to specifically mention exclusion criteria). These inclusion criteria should make any findings applicable to the acute management of disturbed people thought to suffer from serious mental illnesses. However, although need for repeat injection is of importance, it would also have been desirable to have had outcomes such as further aggressive episodes, tranquillisation, sedation and carer satisfaction.
5. Comparison 1. droperidol versus placebo
The small amount of data available (N = 41) suggests that droperidol is superior to placebo three minutes after injection (fewer repeat injections needed). However, the design of this study precluded good information about adverse effects, and, while such limited data could generate hypotheses, it does not provide conclusive evidence.
6. Comparison 2. droperidol versus haloperidol
Although the search identified two trials, only Resnick 1984 (N = 27) was clearly relevant to the acute management of disturbed people. Cocchi 1971 did not assess the immediate effects of droperidol given as emergency medication, but compared it to haloperidol in a study of 30 days' duration. Droperidol was not statistically different to haloperidol for the proportion of participants needing additional injections at 30 minutes, but this difference would be clinically relevant if sustained in larger studies (˜40% droperidol versus ˜60% haloperidol). Haloperidol did cause a mild dystonic reaction in one person. The results of Resnick 1984 indicate that droperidol is a valuable drug in the acute situation, but, were it still being used, all findings would need to be replicated.
7. Heterogeneity
This review is a re‐presentation of the findings of trials rather than a meta‐analysis in which heterogeneity could operate.
8. Sensitivity analysis
It had been hoped to conduct sensitivity analyses comparing results when trials with high attrition with those with only competer data. All three trials reported on loss to follow up and this was not undertaken.
Data and analyses
Comparison 1. Droperidol versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep: 1. tranquillised/sleep | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 By 5 minutes | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.90, 1.96] |
| 1.2 By 10 minutes | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.36 [1.08, 1.71] |
| 1.3 By 30 minutes | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [1.05, 1.31] |
| 1.4 By 60 minutes | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [1.00, 1.18] |
| 2 Tranquillisation or asleep: 2. difficulty in achieving tranquillisation/sleep | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.10, 2.75] |
| 3 Tranquillisation or asleep: 3. time to tranquillisation/sleep (minutes) | 1 | 227 | Mean Difference (IV, Fixed, 95% CI) | ‐46.5 [‐86.83, ‐6.17] |
| 4 Global state: use of additional medication | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 "To reach initial adequate sedation" | 1 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.28, 0.89] |
| 4.2 By 3 minutes (haloperidol) | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.18, 0.72] |
| 4.3 By 30 minutes (any psychotropic drug) | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.46 [0.17, 1.24] |
| 4.4 By 60 minutes (midazolam, droperidol, olanzapine, haloperidol) | 1 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 0.64 [0.42, 0.96] |
| 4.5 By 60 minutes after initial adequate sedation until emergency department discharge (various psychotropic drugs) | 1 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 0.55 [0.36, 0.85] |
| 5 Adverse effects | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Cardiovascular ‐ arrhythmia | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.31] |
| 5.2 Cardiovascular ‐ hypotension | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.20, 2.36] |
| 5.3 Central nervous system ‐ oversedation (decreased Glasgow Coma Score of 6) | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.31] |
| 5.4 Respiratory ‐ airway obstruction | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.15, 2.52] |
| 5.5 Respiratory ‐ oxygen desaturation | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.42, 2.49] |
| 5.6 Unspecified ‐ by 3 minutes | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6 Service use: 1 person able to be discharged home | 1 | 227 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.90, 1.48] |
| 7 Service use: 2 emergency department length of stay | Other data | No numeric data |
Comparison 2. Droperidol versus haloperidol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep: 1. tranquillised/sleep within 120 minutes | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.93, 1.09] |
| 2 Tranquillisation or asleep: 2. time to tranquillisation/sleep | Other data | No numeric data | ||
| 3 Global state: use of additional medication | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Midazolam administered initially | 1 | 228 | Risk Ratio (M‐H, Random, 95% CI) | 3.26 [0.69, 15.37] |
| 3.2 By 30 minutes | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.20, 1.01] |
| 3.3 By 60 minutes | 2 | 255 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.16, 0.90] |
| 3.4 By 90 minutes | 1 | 27 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.02, 10.63] |
| 4 Global state: no overall improvement ‐ by 30 days | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.29, 1.52] |
| 5 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Cardiovascular ‐ hypotension | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.80 [0.30, 26.49] |
| 5.2 Cardiovascular ‐ hypotension/desaturation | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.80 [0.12, 67.98] |
| 5.3 Central nervous system ‐ extrapyramidal adverse effects | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.80 [0.12, 67.98] |
| 5.4 Central nervous system ‐ oversedation | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.80 [0.12, 67.98] |
| 5.5 Staff injuries | 1 | 228 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.14, 2.29] |
| 6 Mental state: Average score by 13 days (Scale for Quantification of Psychotic Symptom Severity, high = poor) | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | 0.11 [‐0.07, 0.29] |
Comparison 3. Droperidol versus midazolam.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep: 1. tranquillised/asleep | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 At 5 minutes | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.21, 0.64] |
| 1.2 At 10 minutes | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.72, 1.28] |
| 2 Tranquillisation or asleep: 2 time to tranquillisation/sleep | Other data | No numeric data | ||
| 3 Global state: use of additional medication | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.24, 1.20] |
| 3.1 By 60 minutes | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.24, 1.20] |
| 4 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Cardiovascular ‐ arrhythmia (bradycardia) | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.81 [0.12, 67.98] |
| 4.2 Cardiovascular ‐ hypotension | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.16, 3.03] |
| 4.3 Central nervous system ‐ dystonic reaction | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.56 [0.34, 124.93] |
| 4.4 Central nervous system ‐ seizure | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.5 Gastric ‐ aspiration | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.6 Gastric ‐ vomiting | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.7 Respiratory ‐ airway management | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.01, 2.55] |
| 4.8 Respiratory ‐ assistance with ventilation | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.9 Respiratory ‐ hypoxia | 1 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.16, 3.03] |
Comparison 4. Droperidol versus olanzapine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tranquillisation or asleep: 1. tranquillised/asleep | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 At 5 minutes | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.70, 1.42] |
| 1.2 At 10 minutes | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.81, 1.17] |
| 1.3 At 30 minutes | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.94, 1.11] |
| 1.4 At 60 minutes | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.93, 1.05] |
| 2 Tranquillisation or asleep: 2. difficulty in achieving tranquillisation/sleep | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.11, 3.81] |
| 3 Tranquillisation or asleep: 3. time to tranquillisation/sleep (minutes) | 1 | 221 | Mean Difference (IV, Fixed, 95% CI) | 7.30 [‐11.74, 26.34] |
| 4 Global state: use of additional medication | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 'To reach initial adequate sedation" | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.68 [0.36, 1.28] |
| 4.2 By 60 minutes (midazolam, droperidol, olanzapine, haloperidol) | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.63, 1.64] |
| 4.3 From 60 minutes after initial adequate sedation until emergency department discharge (various psychotropic drugs) | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.36, 0.87] |
| 5 Adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 Cardiovascular ‐ arrhythmia | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.88] |
| 5.2 Cardiovascular ‐ hypotension | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [0.30, 5.66] |
| 5.3 Central nervous system ‐ decreased Glasgow Coma Score of 6 | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Respiratory ‐ airway obstruction | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.20, 4.72] |
| 5.5 Respiratory ‐ oxygen desaturation | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.75 [0.61, 5.06] |
| 6 Service use: 1. person able to be discharged home | 1 | 221 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.83, 1.34] |
| 7 Service use: 2. emergency department length of stay | Other data | No numeric data |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Calver 2015.
| Methods | Allocation: randomised. Blinding: 'masked study'. Duration: 120 minutes. Settings: psychiatric intensive care unit of a large tertiary specialist mental health facility, Australia. |
|
| Participants | Diagnosis: people with agitation or aggression admitted involuntarily to psychiatric intensive care unit from the psychiatric emergency care centre. N = 228. Age: ≥ 18 years. Sex: men and women. History: adults (> 18 years of age) with acute behavioural disturbance requiring parenteral medication for sedation and in whom verbal de‐escalation or oral medication (or both) had failed. Excluded: people < 18 years old and willing to take oral medication for sedation without physical restraint or seclusion. |
|
| Interventions | 1. Droperidol 10 mg IM. N = 118. 2. Haloperidol 10 mg IM. N = 110. |
|
| Outcomes | Global state: time to sedation, failed sedation, use of additional sedation, successful sedation. Adverse drug effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Block randomisation was used". Response: randomisation probably done. |
| Allocation concealment (selection bias) | Low risk | Quote: "Microsoft Excel was used to randomly create blocks of four (ABAB, AABB, etc.) or six (ABABAB, AAABBB, etc.). The use of different block sizes meant that it was impossible to predict the next treatment". Response: allocation concealment done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The list of study codes with allocations was generated by a research assistant and supplied to the Calvary Mater Newcastle pharmacy, so that the investigators and treating staff remained unaware of the allocations". Response: both participants and personnel were blind to the allocations. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The pharmacy re labelled the vials of haloperidol or droperidol with study numbers based on the list of allocations". Response: blinding of outcome assessment done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "There were 584 sedation episodes during the 23‐month study period and of these 356 were not included in the analysis because the treating clinician elected to give labelled parenteral sedation... an initial SAT score was recorded which was similar to those of the study participants". Response: participants initially recruited were included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported. |
| Other bias | Low risk | |
Chan 2013.
| Methods | Allocation: randomised. Design: multicentre, randomised, double‐blind, placebo‐controlled, double‐dummy, clinical trial. Duration: initially at 5 minutes followed by at 10, 30 and 60 minutes. Settings: trial undertaken in 3 large metropolitan EDs. |
|
| Participants | Diagnosis: people with agitation or aggression. N = 227. Age: 18 to 65 years. Sex: not specified. History: highly agitated people aged 18 to 65 years, requiring parenteral drug sedation for acute agitation, as determined by a registrar (senior resident) or consultant emergency physician. Excluded: people with known hypersensitivity or contraindication to midazolam, droperidol or olanzapine; obvious reversible cause for agitation (e.g. hypotension, hypoxia, hypoglycaemia); known pregnancy; acute alcohol withdrawal; received (within the previous 12 hours) oral or parenteral sedative drug(s) either as usual or out‐of‐hospital acute agitation treatment. |
|
| Interventions | 1. Droperidol 5 mg IV + placebo‐olanzapine. N = 112. 2. Olanzapine 5 mg IV + placebo‐droperidol. N = 109. 3. Control group: placebo‐droperidol, placebo‐olanzapine. N = 115. |
|
| Outcomes | Global state: time to achieve adequate sedation for first time, need for additional parenteral sedative drugs, need for repeat sedation within 60 minutes of initial sedation, total midazolam dose administered in the 60 minutes after initial adequate sedation and from 60 minutes after initial adequate sedation until ED discharge, proportion adequately sedated at 5 and 10 minutes after study drug administration. Service use: ED length of stay. Adverse effects: corrected QT interval (QTc), need for airway management or assisted ventilation, oxygen desaturation (90%). Physiological: systolic blood pressure < 90 mmHg, dystonic reactions, seizures, vomiting or aspiration, and movement disorders. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Computerized block randomisation (blocks of 6), stratified by study site, was performed by an independent pharmacist". Response: randomisation probably done. |
| Allocation concealment (selection bias) | Low risk | Quote: "After enrolment, patients were assigned to the next study pack in the allocated sequence". Response: low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All patients, ED staff, and study personnel remained blinded to group allocation until data entry and analyses were completed". Response: low risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "The appearance of the drug vials and the dosage instructions for the placebo and active study drugs were identical". Response: low risk of detection bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of 457 patients screened for eligibility, 121 were excluded and 336 were enrolled. All groups had similar baseline characteristics. Patients with minor protocol violations (mainly delays in initial midazolam administration) were included in the analysis. The nature of the violations did not differ substantially between the groups". Response: incomplete outcome data addressed. |
| Selective reporting (reporting bias) | Low risk | All the outcomes were adequately reported. |
Cocchi 1971.
| Methods | Allocation: Allocation: randomised ‐ no further details. Blinding: double (drugs packaging was indistinguishable; assessors were external doctors. However, they discussed every clinical case with doctors involved in patients' care). Design: randomised (with same drug schedule ‐ dose and duration ‐ for almost all randomised participants. 4 people in droperidol group did not receive the plateau dose due to EPS). Duration: 30 days. Settings: hospital. |
|
| Participants | Diagnosis: schizophrenia ‐ acute exacerbation. N = 40. Age: range 17 to 51 years, median 25 years. Sex: female 16, male 24. History: psychiatric inpatients. Excluded: no exclusions mentioned. |
|
| Interventions | 1. Droperidol 2 mg to 10 mg orally. N = 20. 2. Haloperidol 2 mg to 10 mg orally. N = 20. |
|
| Outcomes | Global state: clinical improvement. Mental state: Rating Scale for Quantification of Psychotic Symptom Severity. Mental state: Specific Symptoms Scale (unpublished scale, specific symptoms ‐ no standard deviation) ‐ unable to use. Adverse effects: EPS (no data) ‐ unable to use. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised ‐ no details on how random sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | No information provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study described as "double‐blind", but external assessors and doctors involved in patient care discussed every case in order to get a global evaluation of them. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information about data analysis (it seems that rating scales were administered 4 times to all participants, but no details reported). |
| Selective reporting (reporting bias) | Unclear risk | Data reported for outcomes listed in the methods. |
Knott 2006.
| Methods | Allocation: randomised. Blinding: double‐blind. Duration: 2 hours. Settings: conducted in the ED of a large Australian metropolitan university hospital. |
|
| Participants | Diagnosis: marked agitation, required chemical restraint (about 66% 'mental illness'). N = 170. Age: range 18 to 65 years. Sex: unspecified. History: aged judged to be 18 to 65 years (inclusive), exhibited marked agitation that required chemical restraint (decision of consultant (attending) emergency physician or a senior accredited resident of the Australasian College for Emergency Medicine). Excluded: people with known hypersensitivity to either drug, known pregnancy or readily reversible causes for the agitation (systolic blood pressure < 90 mmHg, hypoxia, hypoglycaemia). If treating physician believed agitation was due to acute alcohol withdrawal, participant excluded because this condition is particularly amenable to treatment with benzodiazepines. |
|
| Interventions | 1. Droperidol5 mg IV. N = 86. 2. Midazolam5 mg IV. N = 84. |
|
| Outcomes | Global state: time to sedation, need for subsequent sedation within 60 minutes of initial (adequate) sedation. Adverse effects: ECG, corrected QT (QTc) interval on a 12‐lead ECG. Physiological: pulse rate, blood pressure, oxygen saturation. Loss to follow‐up. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was determined from random‐number tables". Response: randomisation probably done. |
| Allocation concealment (selection bias) | Low risk | Quote: "These solutions were packaged in identical vials and randomly assigned to serially numbered study packs". Response: low risk of selection bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "patients and staff remained blinded to which drug was used throughout each patient’s stay". Response: blinding probably practiced. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "and the codes remained with pharmacy until the study was complete". Response: blinding of the outcome assessment probably done. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "One hundred seventy patients were enrolled by study‐pack allocation. Of these, 17 packs were lost so that data on 153 patients were available for analysis". Response: 17 study packs were lost. It is unknown whether these were selected and discarded unused or used for sedation, with all documentation subsequently lost. |
Resnick 1984.
| Methods | Allocation: unclear ‐ 'code in pharmacy', probably randomised. Design: unspecified. Blinding: double ‐ identical vials. Duration: 24 hours (re‐evaluated at 15 minutes after the initial injection and at 30 minute intervals for 3 hours). Settings: ED and Psychiatric Crisis Unit, Oregon Health Sciences University, Portland. |
|
| Participants | Diagnosis: psychotic ‐ unspecified. N = 27. Age: range 18 to 65 years. Sex: unspecified. History: admitted to ED of psychiatric unit with symptoms of acute agitation and achieved a score of ≥ 17 on a subset of 6 categories on the BPRS. Excluded: people who were intoxicated; had known sensitivity to droperidol or haloperidol; or showed evidence of active renal, hepatic or cardiac disease. |
|
| Interventions | 1. Droperidol5 mg IM. N = 11. 2. Haloperidol5 mg IM. N = 16. |
|
| Outcomes | Global state: needing additional injection, time to control, BPRS. Adverse effects. Adverse effects: EPS. Vital signs: blood pressure, pulse, respiration (no data) ‐ unable to use. |
|
| Notes | Predefined levels of BPRS (subset ‐ anxiety, tension, mannerisms and posturing, hostility, unco‐operativeness, excitement) to a score of > 15 used to instigate reinjection. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...haloperidol on a randomised basis from identical appearing vials..." Response: probably randomised. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Packages of medication were identified only by a code which was kept in the pharmacy until the conclusion of the study." Response: unclear whether and how allocation concealment was assured. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "a double‐blind clinical comparison of droperidol and haloperidol was undertaken". Response: personnel and participants were probably blind to the interventions. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear if blinding of outcome assessment was carried out. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Of the 16 patients in the haloperidol group, 3 required a single injection and 13 required two or more injections... In droperidol group, 7 of the 11 patients required one injection and 4 required two injection". Response: low risk of attrition bias as the number of people initially randomised were all included in the final analysis. |
| Selective reporting (reporting bias) | Low risk | All the stated outcomes were reported. |
| Other bias | Unclear risk | Unclear. |
Van Leeuwen 1977.
| Methods | Allocation: randomly assigned ‐ participants listed in chronological order assigned individually numbered vials. Blinding: double ‐ no further details. Design: double‐blind placebo‐controlled study. Duration: 30 minutes. Settings: not clear. |
|
| Participants | Diagnosis: schizophrenia (N = 20); mania/manic depression (N = 9), confusional state (N = 2), miscellaneous (N = 3), not recorded (N = 7). N = 41. Age: range 14 to 78 years, median 33.5 years. Sex: female 18, male 23. History: acutely agitated, about 50% already taking maintenance psychotropic drugs, about 20% already received inadequate treatment for agitation. Excluded: no exclusions mentioned. |
|
| Interventions | 1. Droperidol 10 mg IV. N = 19. 2. Placebo 10 mg IV. N = 22. |
|
| Outcomes | Global state: needing additional injection, time to control (no measure/scale given). Adverse effects. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The vials containing this solution were individually numbered and their content (droperidol or placebo) was randomly assigned". Response: probably randomised. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "On entering the trial, the patients were chronologically numbered and this number indicated the vial which was to be used". Response: unclear method of concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the study was strictly double blinded..." Response: participants and personnel were probably blind to the intervention. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No mention of blinding of outcome assessment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "breaking the code revealed that 19 patients had been treated with droperidol and 22 patients with placebo". Response: all participants who were initially included were analysed. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported. |
BPRS: Brief Psychiatric Rating Scale; ECG: electrocardiogram; ED: emergency department; EPS: extrapyramidal adverse effects; IM: intramuscular; IV: intravenous; SAT: Social Attribution Task.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Cocito 1970 | Allocation: randomised. Participants: people with psychosis in hospital, not acutely ill. |
| Fang 2014 | Allocation: no information regarding randomisation. Interventions: no mention of droperidol. |
| Foster 1995 | Allocation: randomised. Participants: healthy women attending day hospital for minor surgery. |
| Girard 1972 | Allocation: not randomised, case‐control study. |
| Hooper 1983 | Allocation: unclear; Dr Hooper kindly responded to email ‐ allocation was quasi‐randomised, "every other patient". |
| Hu 2014 | Allocation: quasi‐randomised. |
| Isbister 2010 | Allocation: randomised. Participants: people with violent and acute behavioural disturbance. No mention of any underlying psychiatric illness. |
| Lilburn 1977 | Allocation: not randomised. |
| Richards 1997 | Allocation: randomised. Participants: people with methamphetamine toxicity, not people with severe mental illnesses. |
| Richards 1998 | Allocation: randomised. Participants: mostly people with drug‐induced toxicity (total 202), 20 with 'psychiatric illness'. Interventions: droperidol vs. lorazepam. Outcomes: sedation, re‐admission, adverse effects, additional drugs, time in emergency department, vital signs ‐ no data exclusively for 20 people with 'psychiatric illness'. |
| Rosen 1997 | Allocation: randomised. Participants: mostly people with trauma and medical reasons for their disturbance (total 46), 1 with 'psychiatric' diagnosis. Interventions: droperidol vs. placebo. Outcomes: sedation, re‐admission, adverse effects, additional drugs, time in emergency department, vital signs ‐ no data exclusively for the person with 'psychiatric diagnosis'. |
| Thomas 1992 | Allocation: randomised. Participants: mostly people who were intoxicated or had some form of underlying illness (trauma), no mention of psychoses or psychiatric illness. |
| Weiser 1973 | Allocation: not randomised, case series. |
| Weiser 1975 | Allocation: randomly assigned. Participants: people with schizophrenia ‐ acute/subacute (N = 50 but 5 added during study). Interventions: droperidol 100 mg vs. droperidol 150 mg vs. droperidol 200 mg vs. clopenthixol vs. clozapine. Outcomes: behaviour, mental state, length of stay in hospital, leaving the study early ‐ but not presented free of data from 5 non‐random additional participants. |
Differences between protocol and review
We added further details in the background information slightly to reflect more recent literature.
We amended some of the outcomes between the protocol and this update to reflect Cochrane Schizophrenia Group presentation and wording of outcomes, the type of outcomes remains the same.We have grouped primary outcomes under three main outcomes: tranquil or asleep by up to 30 minutes, another episode of aggression by 24 hours, and specific and serious adverse effects by 24 hours. The secondary outcomes are under 11 main headings: tranquillisation or asleep, specific behaviours, global state, service outcomes, mental state, adverse effects, leaving the study early, satisfaction with treatment, acceptance of treatment, quality of life and economic outcomes. We felt in retrospect that these outcomes were important given the persistent and all‐encompassing nature of schizophrenia. As no relapse data were available, we did not present 'relapse' data in the 'Summary of findings' tables, presenting 'leaving the study early' data instead.
We have also slightly widened our inclusion criteria by including studies where the majority of people in the study had some form of mental illness that was thought to be fuelling their aggression/agitation ‐ even if their data were 'contaminated' by data relating to people who were aggressive for reasons thought to not be because of mental illness.
We have updated the methods section with the latest template provided by the Cochrane Schizophrenia Group (see Acknowledgements).
Contributions of authors
Mariam A. Khokhar (update 2016): primary review author, results and discussion writing.
John Rathbone (2011): study selection, data extraction, writing review.
Sources of support
Internal sources
Academic Unit of Psychiatry, University of Leeds, UK.
Said Business School, University of Oxford, UK.
External sources
NHS National R&D Programme on Forensic Mental Health, UK.
Declarations of interest
None known.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Calver 2015 {published data only}
- Calver L, Drinkwater V, Gupta R, Page CB, Isbister GK. Droperidol v. haloperidol for sedation of aggressive behaviour in acute mental health: randomized controlled trial. British Journal of Psychiatry 2015;206(3):223‐8. [DOI] [PubMed] [Google Scholar]
Chan 2013 {published data only}
- Chan E, Taylor D, Kong D, Knott J, Phillips G, Castle D. IV droperidol or olanzapine as adjuncts to midazolam for the acutely agitated patient: a multi‐centre, randomised, double‐blind, placebo‐controlled, clinical trial. Schizophrenia Research 2012;136:S259. [DOI] [PubMed] [Google Scholar]
- Chan EW, Taylor DM, Knott JC, Phillips GA, Castle DJ, Kong DCM. Intravenous droperidol or olanzapine as an adjunct to midazolam for the acutely agitated patient: a multicenter, randomized, double‐blind, placebo‐controlled clinical trial. Annals of Emergency Medicine 2013;61(1):72‐81. [DOI] [PubMed] [Google Scholar]
Cocchi 1971 {published data only}
- Cocchi A, Fonda P, Perosino N. Droperidol: a double‐blind clinical study [Droperidol: studio clinico in doppio cieco]. Rivista Sperimentale di Freniatria e Medicina Legale Delle Alienazioni Mentali 1971;95(6):1109‐25. [PubMed] [Google Scholar]
Knott 2006 {published data only}
Resnick 1984 {published data only}
- Resnick M, Burton BT. Droperidol versus haloperidol in the initial management of acutely agitated patients. Journal of Clinical Psychiatry 1984;45(7):298‐9. [PubMed] [Google Scholar]
Van Leeuwen 1977 {published data only}
- Leeuwen AMH, Molders J, Sterkmans P, Mielants P, Martens C, Toussaint C, et al. Brief communication: droperidol in acutely agitated patients. A double blind placebo‐controlled study. Journal of Nervous and Mental Disease 1977;164(4):280‐3. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Cocito 1970 {published data only}
- Cocito E, Ambrosini G, Arata A, Bevilacqua P, Tortora E. Clinical evaluation of 112 psychiatric patients of a butyrophenone neuroleptic, dehydrobenzperidol (R 4749). Arzneimittelforschung 1970;20(8):1119‐25. [PubMed] [Google Scholar]
Fang 2014 {published data only}
- 方政华, 周剑, 曾宪祥, 左静, 刘芳, 漆靖. Clinical acute schizophrenia ziprasidone different dosing regimens in treatment [Google Translate] [齐拉西酮片治疗精神分裂症急性期 不同给药方案的临床观察]. 延边医学 2014;6(125):52‐4. [Google Scholar]
Foster 1995 {published data only}
- Foster PN, Stickle BR, Dale M, Laurence AS. Akathisia after low‐dose droperidol. British Journal of Anaesthesia 1995;74:477P. [Google Scholar]
Girard 1972 {published data only}
- Girard R, Blondel F. Clinical evaluation of oral droperidol in psychiatry [Interet du droperidol en psychiatrie]. Psychologie Medicale 1972;4(6):1169‐75. [Google Scholar]
Hooper 1983 {published data only}
- Hooper JF, Minter G. Droperidol in the management of psychiatric emergencies. Journal of Clinical Psychopharmacology 1983;3(4):262‐3. [DOI] [PubMed] [Google Scholar]
Hu 2014 {published data only}
- 胡光涛, 李志宏, 王国威, 黄一, 贺英. Mesylate injection of acute agitation in schizophrenia symptoms. Efficacy of ziprasidone treatment [Google translate] [注射用甲磺酸齐拉西酮治疗精神分裂症急性激越症状疗效观察]. 中国新药与临床杂志 2014;33(1):57‐60. [Google Scholar]
Isbister 2010 {published data only}
- Isbister GK, Calver LA, Page CB, Stokes B, Bryant JL, Downes MA. Randomized controlled trial of intramuscular droperidol versus midazolam for violence and acute behavioral disturbance: the DORM study. Annals of Emergency Medicine 2010;56(4):392‐401. [DOI] [PubMed] [Google Scholar]
Lilburn 1977 {published data only}
Richards 1997 {published data only}
- Richards JR, Derlet RW, Duncan DR. Metamphetamine toxicity: treatment with a benzodiazepine versus a butyrophenone. European Journal of Emergency Medicine 1997;4:130‐5. [DOI] [PubMed] [Google Scholar]
Richards 1998 {published data only}
- Richards JR, Derlet RW, Duncan DR. Chemical restraint for the agitated patient in the emergency department: lorazepam versus droperidol. Journal of Emergency Medicine 1998;16(4):567‐73. [DOI] [PubMed] [Google Scholar]
Rosen 1997 {published data only}
- Rosen CL, Ratliff AF, Wolfe RE, Branney SW, Roe EJ, Pons PT. The efficacy of intravenous droperidol in the prehospital setting. Journal of Emergency Medicine 1997;15(1):13‐7. [DOI] [PubMed] [Google Scholar]
Thomas 1992 {published data only}
- Thomas H Jr, Schwartz E, Petrilli R. Droperidol versus haloperidol for chemical restraint of agitated and combative patients. Annals of Emergency Medicine 1992;21(4):407‐13. [DOI] [PubMed] [Google Scholar]
Weiser 1973 {published data only}
- Weiser G. Initial treatment of schizophrenia using droperidol [Initialbehandlung der Schizophrenie mit Droperidol]. Wiener Zeitschrift fur Nervenheilkunde und deren Grenzgebiete 1973;31:176‐88. [PubMed] [Google Scholar]
Weiser 1975 {published data only}
- Weiser G, Tahedl A, Reisecker F, Meyer H. Advantages of the initial therapy of acute schizophrenia with large doses of droperidol: a comparative study [Vorteile der Initialbehandlung akuter Schizophrenien mit hochdosiertem Droperidol. Eine Vergleichsstudie]. Arzneimittel Forschung 1975;25(11):1845‐8. [PubMed] [Google Scholar]
Additional references
Ahmed 2010
- Ahmed U, Jones H, Adams CE. Chlorpromazine for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2010, Issue 4. [DOI: 10.1002/14651858.CD007445.pub2; PUBMED: 20393959] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahmed 2011
- Ahmed U, Rehman F, Jones H, Adams CE. Risperidone for psychosis induced aggression or agitation. Cochrane Database of Systematic Reviews 2011, Issue 11. [DOI: 10.1002/14651858.CD009412; CD009412] [DOI] [PMC free article] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT Statement. JAMA 1996;276(8):637‐9. [DOI] [PubMed] [Google Scholar]
Belgamwar 2005
- Belgamwar RB, Fenton M. Olanzapine IM or velotab for acutely disturbed/agitated people with suspected serious mental illnesses. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD003729.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Berk 2004
- Berk M, Rathbone J, Mandriota‐Carpenter SL. Clotiapine for acute psychotic illnesses. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD002304.pub2; CD002304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Binder 1999
- Binder RL, McNiel DE. Contemporary practices in managing acute violent patients in 20 psychiatric emergency rooms. Psychiatric Services 1999;50(12):1553‐4. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM, Kerry SM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
BNF 2000
- British Medical Association. British National Formulary. 9th Edition. London: British Medical Association, 2000. [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Chakrabarti 2007
- Chakrabarti A, Whicher EV, Morrison M, Douglas‐Hall P. 'As required' medication regimens for seriously mentally ill people in hospital. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD003441.pub2] [DOI] [PubMed] [Google Scholar]
Cunnane 1994
- Cunnane JG. Drug management of disturbed behaviour by psychiatrists. Psychiatric Bulletin 1994;18:138‐9. [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. 8th International Cochrane Colloquium, 2000 Oct 25‐28; Cape Town: The Cochrane Collaboration. 2000.
Divine 1992
- Divine GW, Brown JT, Frazer LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7:623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Furyk 2015
- Furyk JS, Meek RA, Egerton‐Warburton D. Drugs for the treatment of nausea and vomiting in adults in the emergency department setting. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD010106.pub2; PUBMED: 26411330] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gibson 2004
- Gibson RC, Fenton M, Coutinho ES, Campbell C. Zuclopenthixol acetate for acute schizophrenia and similar serious mental illnesses. Cochrane Database of Systematic Reviews 2004, Issue 3. [DOI: 10.1002/14651858.CD000525.pub2] [DOI] [PubMed] [Google Scholar]
Gillies 2005
- Gillies D, Beck A, McCloud A, Rathbone J. Benzodiazepines for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD003079.pub2] [DOI] [PubMed] [Google Scholar]
Goodrich 1953
- Goodrich DW. Quantification of the severity of overt psychotic symptoms. American Journal of Psychiatry 1953;110:334. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1991
- Guy JM, Andre‐Fouet X, Porte J, Bertrand M, Lamaud M, Verneyre H. Torsade de pointe and prolongation of the duration of the QT interval after injection of droperidol. Annales de Cardilogie et d'Angeiologie 1991;40(9):541‐5. [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Huf 2002
- Huf G, Silva Freire Coutinho E, Fagundes HM Jr, Oliveira ES, Lopez JR, Gewandszajder M, et al. Current practices in managing acutely disturbed patients at three hospitals in Rio de Janeiro‐Brazil: a prevalence study. BMC Psychiatry 2002;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huf 2009
- Huf G, Alexander J, Allen Michael H, Raveendran Nirmal S. Haloperidol plus promethazine for psychosis‐induced aggression. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD005146.pub2; CD005146] [DOI] [PubMed] [Google Scholar]
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British Journal of Haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Khushu 2012
- Khushu A, Powney MJ, Adams CE. Haloperidol for long‐term aggression in psychosis. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD009830] [DOI] [PMC free article] [PubMed] [Google Scholar]
Latalova 2014
- Latalova K. Violence and duration of untreated psychosis in first‐episode patients. International Journal of Clinical Practice 2014;68(3):330‐5. [PUBMED: 24471741] [DOI] [PubMed] [Google Scholar]
Lawrence 1997
- Lawrence KR, Nasrawy SA. Conduction disturbances associated with administration of butyrophenone antipsychotics in the critically ill: a review of the literature. Pharmacotherapy 1997;17(3):531‐7. [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lischke 1994
- Lischke V, Behne M, Doelken P, Schledt U, Probst S, Vettermann J. Droperidol causes a dose‐dependent prolongation of the QT interval. Anaesthesia & Analgesia 1994;79:983‐6. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Martindale 1982
- Martindale W, Pharmaceutical Society of Great Britain. Martindale: The Extra Pharmacopoeia. 28th Edition. London: Pharmaceutical Press, 1982. [Google Scholar]
Michalets 1998
- Michalets EL, Smith LK, Tassel ED. Torsade de pointes resulting from the addition of droperidol to an existing cytochrome P450 drug interaction. Annals of Pharmacotherapy 1998;32:761‐5. [DOI] [PubMed] [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D, CONSORT Group. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomized trials. JAMA 2001;285:1987‐91. [DOI] [PubMed] [Google Scholar]
Muralidharan 2006
- Muralidharan S, Fenton M. Containment strategies for people with serious mental illness. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD002084.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Newman 2015
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Pilowsky 1992
- Pilowsky LS, Ring H, Shine PJ, Battersby M, Lader M. Rapid tranquillisation. A survey of emergency prescribing in a general psychiatric hospital. British Journal of Psychiatry 1992;160:831‐5. [DOI] [PubMed] [Google Scholar]
Powney 2012
- Powney MJ, Adams CE, Jones H. Haloperidol for psychosis‐induced aggression or agitation (rapid tranquillisation). Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD009377.pub2; PUBMED: 23152276] [DOI] [PubMed] [Google Scholar]
Rao 2012
- Rao H, Yeung WL, Jayaram MB. De‐escalation techniques for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD009922] [DOI] [PMC free article] [PubMed] [Google Scholar]
Raveendran 2007
- Raveendran NS, Tharyan P, Alexander J, Adams CE, Trec‐India II Collaborative Group. Rapid tranquillisation in psychiatric emergency settings in India: a pragmatic randomised controlled trial of intramuscular olanzapine versus intramuscular haloperidol plus promethazine. BMJ 2007;335(7625):865. [DOI] [PMC free article] [PubMed] [Google Scholar]
RxList 2000
- RxList.com, Inc. RxList ‐ Internet Drug Index. www.rxlist.com Accessed 26 July 2000.
Sailas 2000
- Sailas E, Fenton M. Seclusion and restraint for people with serious mental illnesses. Cochrane Database of Systematic Reviews 2000, Issue 2. [DOI: 10.1002/14651858.CD001163] [DOI] [PMC free article] [PubMed] [Google Scholar]
Storrar 2014
- Storrar J, Hitchens M, Platt T, Dorman S. Droperidol for treatment of nausea and vomiting in palliative care patients. Cochrane Database of Systematic Reviews 2014, Issue 11. [DOI: 10.1002/14651858.CD006938.pub3; CD006938] [DOI] [PMC free article] [PubMed] [Google Scholar]
Teasdale 1974
- Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet (London, England) 1974;2(7872):81‐4. [PUBMED: 4136544] [DOI] [PubMed] [Google Scholar]
Toal 2012
- Toal F, Roberts K. Clozapine for people with schizophrenia and recurrent physical aggression. Cochrane Database of Systematic Reviews: Registered title.
TREC 2003
- TREC Collaborative Group. Rapid tranquillisation for agitated patients in emergency psychiatric rooms: a randomised trial of midazolam versus haloperidol plus promethazine. BMJ 2003;327(7417):708‐13. [EMBASE: 2003210869] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii‐92. [MEDLINE: ] [PubMed] [Google Scholar]
Vangala 2012
- Vangala R, Ahmed U, Ahmed R. Loxapine inhaler for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD010190] [DOI] [Google Scholar]
Wilkie 2012
- Wilkie F, Fenton M. Quetiapine for psychosis‐induced aggression or agitation. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD009801] [DOI] [Google Scholar]
Winsper 2013
- Winsper C, Singh SP, Marwaha S, Amos T, Lester H, Everard L, et al. Pathways to violent behavior during first‐episode psychosis: a report from the UK National EDEN Study. JAMA Psychiatry 2013;70(12):1287‐93. [PUBMED: 24089149] [DOI] [PubMed] [Google Scholar]
Wooltorton 2002
- Eric Wooltorton. Droperidol: cardiovascular toxicity and deaths [Droperidol: cardiovascular toxicity and deaths]. Canadian Medical Association Journal 2002; Vol. 166, issue 7. [PMC free article] [PubMed]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
References to other published versions of this review
Cure 2001
- Cure S, Carpenter S. Droperidol for acute psychosis. Cochrane Database of Systematic Reviews 2001, Issue 2. [DOI: 10.1002/14651858.CD002830; PUBMED: 11406047] [DOI] [PubMed] [Google Scholar]
Cure 2004
- Cure S, Rathbone J, Carpenter S. Droperidol for acute psychosis. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD002830.pub2; PUBMED: 15495037] [DOI] [PubMed] [Google Scholar]
Rathbone 2004
- Rathbone J, Mandriota‐Carpenter SL, Cure SJ. Droperidol for acute psychosis. Cochrane Database of Systematic Reviews 2004, Issue 1. [DOI: 10.1002/14651858.CD002830.pub2; CD002830] [DOI] [PubMed] [Google Scholar]