

Notes
Editorial note
This review has been superseded by a new review with an expanded scope:
Foong WC, Loh CK, Ho JJ, Lau DSC. Foetal haemoglobin inducers for reducing blood transfusion in non‐transfusion‐dependent beta‐thalassaemias. Cochrane Database of Systematic Reviews 2023, Issue 1. Art. No.: CD013767. DOI: 10.1002/14651858.CD013767.pub2.
Abstract
Background
Non‐transfusion dependent beta thalassaemia is a subset of inherited haemoglobin disorders characterised by reduced production of the beta globin chain of the haemoglobin molecule leading to anaemia of varying severity. Although blood transfusion is not a necessity for survival, it is required when episodes of chronic anaemia occur. This chronic anaemia can impair growth and affect quality of life. People with non‐transfusion dependent beta thalassaemia suffer from iron overload due to their body's increased capability of absorbing iron from food sources. Iron overload becomes more pronounced in those requiring blood transfusion. People with a higher foetal haemoglobin level have been found to require fewer blood transfusions. Hydroxyurea has been used to increase foetal haemoglobin level; however, its efficacy in reducing transfusion, chronic anaemia complications and its safety need to be established.
Objectives
To assess the effectiveness, safety and appropriate dose regimen of hydroxyurea in people with non‐transfusion dependent beta thalassaemia (haemoglobin E combined with beta thalassaemia and beta thalassaemia intermedia).
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register, compiled from electronic database searches and handsearching of relevant journals. We also searched ongoing trials registries and the reference lists of relevant articles and reviews.
Date of last search: 30 April 2016.
Selection criteria
Randomised or quasi‐randomised controlled trials of hydroxyurea in people with non‐transfusion dependent beta thalassaemia comparing hydroxyurea with placebo or standard treatment or comparing different doses of hydroxyurea.
Data collection and analysis
Two authors independently applied the inclusion criteria in order to select trials for inclusion. Both authors assessed the risk of bias of trials and extracted the data. A third author verified these assessments.
Main results
No trials comparing hydroxyurea with placebo or standard care were found. However, we included one randomised controlled trial (n = 61) comparing 20 mg/kg/day with 10 mg/kg/day of hydroxyurea for 24 weeks.
Both haemoglobin and foetal haemoglobin levels were lower at 24 weeks in the 20 mg group compared with the 10 mg group, mean difference ‐2.39 (95% confidence interval ‐ 2.8 to ‐1.98) and mean difference ‐1.5 (95% confidence interval ‐1.83 to ‐1.17), respectively. Major adverse effects were significantly more common in the 20 mg group, for neutropenia risk ratio 9.93 (95% confidence interval 1.34 to 73.97) and for thrombocytopenia risk ratio 3.68 (95% confidence interval 1.13 to 12.07). No difference was reported for minor adverse effects (gastrointestinal disturbances and raised liver enzymes). The effect of hydroxyurea on transfusion frequency was not reported.
The overall quality for the outcomes reported was graded as very low mainly because the outcomes were derived from only one small study with an unclear method of allocation concealment.
Authors' conclusions
There is no evidence from randomised controlled trials to show whether hydroxyurea has any effect compared with controls on the need for blood transfusion. Administration of 10 mg/kg/day compared to 20 mg/kg/day of hydroxyurea resulted in higher haemoglobin levels and seems safer with fewer adverse effects. It has not been reported whether hydroxyurea is capable of reducing the need for blood transfusion. Large well‐designed randomised controlled trials with sufficient duration of follow up are recommended.
Keywords: Humans, beta-Thalassemia, beta-Thalassemia/blood, beta-Thalassemia/drug therapy, Blood Transfusion, Blood Transfusion/statistics & numerical data, Hematinics, Hematinics/administration & dosage, Hemoglobin A, Hemoglobin A/analysis, Hydroxyurea, Hydroxyurea/administration & dosage, Randomized Controlled Trials as Topic
Plain language summary
Hydroxyurea for reducing blood transfusion in non‐transfusion dependent beta thalassaemias
Review question
We wanted to find out if giving hydroxyurea to people with non‐transfusion dependent beta thalassaemia would reduce the need for blood transfusion.
Background
Thalassaemia is a genetic blood disorder causing defective adult haemoglobin (the oxygen carrying component of red blood cells). This causes anaemia with different degrees of severity. People with non‐transfusion dependent beta thalassaemia do not depend on regular transfusions for survival, but may require blood transfusion from time to time. Persistent anaemia affects growth, may delay puberty and reduce quality of life. However, transfusion should be avoided, if possible, because it leads to excess iron being deposited in various organs affecting how they function.
People with non‐transfusion dependent beta thalassaemia have higher levels of foetal haemoglobin (the main form of haemoglobin found during the development of a baby before birth). After birth, foetal haemoglobin gradually disappears and is replaced by the defective adult haemoglobin. A small amount of foetal haemoglobin remains after birth and is often present in people with non‐transfusion dependent beta thalassaemia. The higher the level of foetal haemoglobin the less transfusion could be needed.
Hydroxyurea is an anti‐cancer treatment which increases the level of foetal haemoglobin. Therefore, it might reduce the need for blood transfusion in people with non‐transfusion dependent beta thalassaemia. However, it is not known whether hydroxyurea is effective and safe and if so, which is the best dose and at which age treatment should start.
Search date
The evidence is current to 30 April 2016.
Study characteristics
We did not find any randomised controlled trials (where people taking part in the trial have equal chances of being in the treatment or the control group) comparing hydroxyurea with a placebo (a dummy drug) or usual care. However, we found one randomised controlled trial comparing two different doses of hydroxyurea (10 mg/kg/day versus 20 mg/kg/day given for 24 weeks) and included it in this review. A total of 61 people took part in this trial.
Key results
The lower dose of hydroxyurea appeared to increase levels of foetal haemoglobin, but the higher dose did not. We found some evidence that the higher dose was harmful, particularly to the bone marrow. The trial did not look at whether blood transfusions could be given less often or whether the effects of the anaemia were reduced. In the short term, the lower dose does not appear to have any side effects. The trial duration was very short and we need to know what might happen if treatment with hydroxyurea is continued for a longer period of time.
Quality of the evidence
We graded the quality of the evidence as very low. This was because our key results are based on only one small trial. In addition we can not be sure whether the trial methods were of high quality because the authors have not completely described them.
Summary of findings
Summary of findings 1. Hydroxyurea versus placebo or standard care for non‐transfusion dependent beta thalassaemia.
| Hydroxyurea compared with placebo for people with non‐transfusion dependent beta thalassaemia | ||||||
|
Patient or population: people with non‐transfusion dependent beta thalassaemia Intervention: hydroxyurea Comparison: placebo or standard care | ||||||
| Outcomes | Illustrative comparative risks (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Hydroxyurea | |||||
| Frequency of transfusion | No trials found. | |||||
| Major adverse effects | No trials found. | |||||
| Quality of life | No trials found. | |||||
| Mean Hb F (g/dL) | No trials found. | |||||
| CI: confidence interval; Hb F: foetal haemoglobin. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
Summary of findings 2. Different doses or dose regimens of hydroxyurea compared.
| Different doses or dose regimens of hydroxyurea compared | ||||||
| Patient or population: people with non‐transfusion dependent beta thalassaemia Settings: specialised outpatient clinic Intervention: 20 mg/kg/day hydroxyurea versus 10 mg/kg/day | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| 10 mg/kg/day | 20 mg/kg/day | |||||
| Frequency of transfusion | Outcome not reported. | |||||
| Major adverse effects ‐ Neutropenia | Study population | RR 9.93 (1.34 to 73.67) | 61 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Not downgraded for lack of blinding because outcome considered to be objective. | |
| 31 per 1000 | 310 per 1000 (42 to 1000) | |||||
| Major adverse effects ‐ Thrombocytopenia | Study population | RR 3.68 (1.12 to 12.07) | 61 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 | Not downgraded for lack of blinding because outcome considered to be objective. | |
| 94 per 1000 | 345 per 1000 (105 to 1000) | |||||
| Quality of life | Outcome not reported | |||||
| Mean Hb F at 24 weeks (g/dL) | The mean Hb F at 24 weeks (g/dL) in the intervention group was 1.5 lower (1.83 to 1.17 lower) | 61 (1 trial) | ⊕⊕⊝⊝ low1,2 | Not downgraded for lack of blinding because outcome considered to be objective. | ||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; Hb F: foetal haemoglobin; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | ||||||
1 Downgraded one level for unclear allocation concealment 2 Downgraded one level because conclusions based on one small trial 3 Downgraded one level for extremely wide CI
Background
Description of the condition
Non‐transfusion dependent thalassaemia (NTDT) refers to a group of thalassaemias not needing regular blood transfusion for survival, but where the anaemia is severe enough to cause clinical manifestations, whereas the carrier state (or trait) is asymptomatic (Camaschella 1995; Weatherall 2001). The types of thalassaemia that present as NTDT include beta (ß) thalassaemia intermedia, haemoglobin E (Hb E) combined with ß thalassaemia (Hb E/ß thalassaemia) and haemoglobin H (Hb H) disease. In Hb E/ß thalassaemia and ß thalassaemia intermedia, the ß haemoglobin chain is affected; whereas Hb H disease affects the alpha (α) haemoglobin chain. Collectively, NTDT affects nearly 70,000 children annually, about one quarter of the total thalassaemia burden (Weatherall 2012). It is an inherited disease found mainly in populations originating from Africa, Asia and the Mediterranean, but which has spread globally due to population migration (Kohne 2010; Weatherall 2012).
In this review, ß thalassaemia intermedia and Hb E/ß thalassaemia will be collectively referred to as non‐transfusion dependent ß thalassaemia (NTDßT) as these are major contributors to NTDT. People with NTDßT have an extremely diverse clinical spectrum of chronic anaemia. This diversity arises from multiple genetic differences which affect the amount of haemoglobin produced, its functional properties and its stability which determines the lifespan of the haemoglobin molecule. Together, these factors ameliorate or worsen the chronic anaemia (Kohne 2011; Weatherall 2001). Another factor is the persistence of foetal haemoglobin (Hb F) which has some functional properties in postnatal life. If the function of Hb F is greater than the other haemoglobins produced by an individual with NTDßT, then its persistence might create a milder presentation (Musallam 2012a; Pourfarzad 2013; Weatherall 2001). However, the proportion of Hb F produced in any individual with NTDßT varies even within one genetic group (Italia 2009). The reasons for this are not clearly understood, but it may be due to other independent genetic factors (Menzel 2007; Pandey 2012; Panigrahi 2005; Sankaran 2008). All these factors contribute to the severity of the clinical picture manifesting as poor growth, bone deformities, extramedullary haemopoietic pseudo‐tumour, pulmonary hypertension and thrombotic hypercoagulable episodes presenting mainly as leg ulcers and pulmonary hypertension (Aessopos 2001; Matta 2013; Taher 2006).
The iron deposition pathophysiology in NTDßT differs from that seen in thalassaemia major. In thalassaemia major, which is a condition where the individual is dependant on transfusion for survival, iron deposition occurs as a result of frequent transfusions. In NTDßT iron deposition may occur if frequent transfusion is required, but there is also greater gut iron absorption and greater iron release by the reticuloendothelial system, the normal iron storage sites (Pippard 1979). The deposited iron tends to accumulate in the organs causing endocrinopathies, liver cirrhosis, hepatocarcinoma and cardiac siderosis, just as is seen in thalassaemia major (Taher 2006; Weatherall 2001).
Treatment of non‐transfusion dependent beta thalassaemia
Transfusion of red cells remains the mainstay of treatment to minimize chronic anaemia and its complications (Musallam 2012a). The OPTIMAL CARE study found that transfusion reduced the complications caused by the anaemia (Taher 2010). However, while transfusions are beneficial in reducing the complications of chronic anaemia, they will exacerbate the existing problem of iron deposition. Chelating agents can be used to remove excessive iron deposition, but they in themselves bring further additional problems (Fisher 2013). The decision to initiate chelation therapy depends on the degree of overload, the rate of further iron accumulation and on the availability of resources to detect body iron concentration. The body iron is best measured by magnetic resonance imaging (MRI) using either R2 (1/T2) or R2* (1/T2*) pulse sequences on the liver. This has been validated against liver biopsy (St Pierre 2005). Alternatively serum ferritin can be used but is less accurate (Baynes 1986; Musallam 2012; Musallam 2012c; Origa 2007). Suggested values derived from retrospective studies suggest initiation of chelators at liver iron concentrations of five to seven mg Fe / dry weight or serum ferritin reaches 500 to 800 µg/l or above (Musallam 2011; Taher 2009). Multiple transfusion carries risks of its own, including exposure to serious blood‐transmitted infections, red cell antibody formation and hypersensitivity reactions; and this needs to be taken into consideration in the treatment of these individuals. In children, the frequency of transfusion is determined by clinical criteria such as poor growth, poor school performance, and clinical evidence of extramedullary erythropoiesis. Adults are transfused if they undergo transient stressful conditions such as pregnancy or are at a high risk of complications from chronic anaemia (Taher 2010; TIF 2013a; TIF 2013b; TIF 2013c).
Splenectomy has been used in an attempt to reduce transfusion frequency by reducing the removal of abnormal red cells. However, it has a known risk of infection and is also associated with an increase in thrombotic events (Borgna‐Pignatti 2007; de Montalembert 1990; Musallam 2012b). A further suggested treatment option is the use of drugs to increase the production of Hb F; hydroxyurea is one such option.
Description of the intervention
Hydroxyurea (or hydroxycarbamide) is a drug that has been identified to have Hb F‐inducing properties. Its usage has been widely studied in people with sickle cell disease, but this is a relatively new therapy for people with NTDßT.
A Cochrane systematic review has found that hydroxyurea appears to be safe and effective in reducing transfusion in sickle cell disease (Jones 2001). In both sickle cell disease and NTDßT the dose used varies widely and is guided both by the therapeutic response and the onset of toxicity, which are both dose‐dependant. Therefore, the initial dose is incrementally increased to achieve the greatest possible increase in Hb F production without adverse effects. A response should be evident within three to six months of treatment (Italia 2010; Strouse 2012). Monitoring is continued every six to 12 months to ensure the response is maintained and to look for adverse effects. At present, there is little information on how long hydroxyurea needs to be continued in people with NTDßT(Kinney 1999; Ware 2011; Zimmerman 2004). It has been reported that when hydroxyurea is taken for more than 12 months, a decline in its effectiveness may occur (Mancuso 2006; Rigano 2010). It has been postulated that this could be because of the myelotoxic effect of hydroxyurea (de Paula 2003; Fucharoen 1996). Due to this possible decline in efficacy, combining hydroxyurea with other co‐interventions has been suggested, which might sustain or induce further production of foetal haemoglobin. The co‐interventions used with hydroxyurea which have been found in the literature are L‐carnitine (Karimi 2010), sodium hydroxybutyrate (Hoppe 1999; Singer 2005a; Singer 2005b) and erythropoietin (Elalfy 2013).
The most important adverse effect reported in the literature is neutropenia, but other reported adverse effects include thrombocytopenia, oligospermia, skin rashes and nail changes (Zimmerman 2004). These adverse effects are reversible (Strouse 2008). A study by Taher (who observed 584 participants from six thalassaemia care centres where 34% of the participants were on hydroxyurea), reported that hydroxyurea was associated with an increased risk of hypogonadism but protective for extramedullary haemopoiesis, pulmonary hypertension, leg ulcers, hypothyroidism and osteoporosis (Taher 2010). No long‐term adverse effects were reported in a 20‐years follow up of individuals with sickle cell disease (Steinberg 2010; Ware 2002).
The dose of hydroxyurea used for NTDßT is generally lower than that used for sickle cell disease and ranges from 3 mg/kg/day to 20 mg/kg/day. It reaches its peak plasma level within one to four hours after ingestion and is mainly metabolized by the liver. It has been reported to have been used in children with NTDßT as young as four years of age (Bradai 2007); however, there are no data to suggest the optimum age to start treatment with hydroxyurea.
Other factors influencing the response to hydroxyurea include age, pre‐treatment Hb F level, ß globin genotype and haplotype, co‐inheritance with α thalassaemia and the degree of iron overload (Musallam 2013).
How the intervention might work
Hydroxyurea has the capability of increasing the level of Hb F (Bauer 2012; Borgna‐Pignatti 2007). Its exact mechanism of action remains unclear, but could be due to stress erythropoiesis (Pourfarzad 2013). Since Hb F has some functional properties, an increase in its concentration might reduce the long‐term effects of chronic anaemia and the requirement for transfusion (Italia 2010). If transfusion is reduced then iron deposition may be less severe and the transfusion‐related risks might also be reduced. Several small studies and individual case reports have reported reduced extramedullary haemopoiesis (Meo 2008). Response to hydroxyurea is a complex matter. Laboratory work by Pourfarzad suggested that individuals with a low baseline Hb F level respond better to hydroxyurea; but the Hb F which is induced, although greater in quantity, is less functional and hence results in a lower overall rise in haemoglobin (Pourfarzad 2013).
Why it is important to do this review
An evaluation of the benefits and harms of hydroxyurea and its appropriate dosage are urgently needed since it is already being used for people with NTDßT. Reports show that it is widely used in India and the Middle East (Borgna‐Pignatti 2007; Kosaryan 2009; Kosaryan 2014). In addition, its use has been recommended for people with NTDßT in a guideline produced by the Thalassaemia International Federation (TIF 2013a; TIF 2013b; TIF 2013c). If hydroxyurea was found to be safe and effective in reducing transfusion requirements, it could bring benefits at little cost to those with NTDßT; even a small reduction in transfusion requirement might be beneficial.
Objectives
To assess the effectiveness, safety and appropriate dose regimen of hydroxyurea in people with NTDßT (Hb E/ß thalassaemia and ß thalassaemia intermedia).
Methods
Criteria for considering studies for this review
Types of studies
Randomised and quasi‐randomised controlled trials.
Types of participants
People of any age with thalassaemia who have been defined as having NTDßT, either ß thalassaemia intermedia or Hb E/ß thalassaemia.
Since there is a wide range of transfusion dependency ranging from occasional to regular transfusion during certain circumstances, NTDßT is difficult to define. For this review, we have considered non‐transfusion dependence to be present when an individual has one of these conditions and transfusion is based on circumstances such as poor growth, excessive extramedullary haemopoiesis and pregnancy.
We specified the ages of the included participants.
Types of interventions
The use of hydroxyurea in any dose or duration longer than three months compared with a standard management protocol or placebo.
One dose or dose regimen of hydroxyurea compared with another different dose or dose regimen.
At the review stage we made a post hoc decision to include studies comparing different doses of hydroxyurea.
Types of outcome measures
Primary outcomes
Frequency of blood transfusion measured by the volume transfused per kg per year (units transfused per kg per year (if sufficient data available) or other similar measures)
Transfusion‐free interval (in months)
Haemoglobin level (in g/dl or equivalent at intervals after commencing the intervention (e.g. every three to six months)
Secondary outcomes
-
Thalassaemia‐ and transfusion‐related complications
liver and spleen size
pubertal stage (for boys: testicular size in ml, for girls: breast enlargement according to Tanner staging) and growth velocity in cm per year
paraspinal extramedullary haemopoiesis, pulmonary hypertension, cardiovascular complications, leg ulcers and thrombotic events
iron deposition (in the heart and liver measured by quantifiable methods or serum ferritin in ng/mL or pmol/L)
endocrine dysfunction (e.g. male and female reproduction organ dysfunction (subfertility), diabetes)
-
Adverse effects or toxicity
major (e.g. neutropenia due to myelosuppression)
minor (e.g. gastrointestinal disturbances)
Hb F level (in per cent of total haemoglobin or equivalent at intervals after commencing the intervention (e.g. every three to six months)
Quality of life (QoL) (measured by validated instruments or scales such as health‐related QoL tool (HRQoL) or health outcome rating scales, or self‐reported satisfaction or dissatisfaction)
Search methods for identification of studies
Electronic searches
We identified relevant studies from the Cystic Fibrosis and Genetic Disorders Group's Haemoglobinopathies Trials Register using the terms: (thalassaemia OR (haemoglobinopathies AND general) AND hydroxyurea).
The Haemoglobinopathies Trials Register is compiled from electronic searches of the Cochrane Central Register of Controlled Trials (CENTRAL) (updated each new issue of The Cochrane Library) and weekly searches of MEDLINE. Unpublished work is identified by searching the abstract books of five major conferences: the European Haematology Association conference; the American Society of Hematology conference; the British Society for Haematology Annual Scientific Meeting; the Caribbean Health Research Council Meetings; and the National Sickle Cell Disease Program Annual Meeting. For full details of all searching activities for the register, please see the relevant section of the Cochrane Cystic Fibrosis and Genetic Disorders Group Module.
Date of most recent search: 06 July 2016.
We additionally searched PubMed from 1966 and Embase from 1980 using the search strategies detailed in the appendices (Appendix 1; Appendix 2). No language or publication status restrictions were applied. If we had encountered non‐English language studies, we planned to attempt to source a translation to see any possibility of including these studies in the review.
Date of most recent search: 30 April 2016.
To capture ongoing or unpublished trials, we used clinicaltrials.gov (www.clinicaltrials.gov) and WHO ICTRP (www.who.int/ictrp) using the keywords: hydroxyurea AND (thalassaemia OR thalassemia).
Date of most recent search: 4 May 2016.
Searching other resources
We scanned the reference lists for relevant articles retrieved by the electronic searches to assess any identified trials for possible inclusion. If we had found any ongoing trials, we planned to contact the principal investigators for trial information. We contacted the authors of the RCTs we identified in our search for information on any other potential trials for inclusion in the review.
Data collection and analysis
Selection of studies
Two authors (WCF, CKL) independently screened the results (titles and abstracts) of the literature search for potentially relevant trials. We retrieved full reports of the potentially relevant trials and independently determined if they met the review's predefined inclusion criteria using a pre‐tested eligibility form. If we were unable to ascertain the relevance, we resolved contentious issues by discussion. We resolved any further disagreements through discussion and consensus with a third review author (JJH or VV). We consulted an editor within the Cochrane Cystic Fibrosis and Genetic Disorders Group with regards to the change in inclusion criteria for the review (seeDifferences between protocol and review below), which resolved the issue of trial eligibility for the only included trial. If required, we would have contacted the trial authors for further information if trial eligibility was unclear; however it was not necessary to do this for the only included trial. We have listed all excluded trials and documented the reason for excluding them (Higgins 2011a; Higgins 2011b).
Data extraction and management
We extracted data from the included trial onto a specially designed data collection form. Two authors (WCF and CKL) independently collected trial details and outcome data. The authors then entered the information into the agreed form, compared results and resolved any disagreements by discussion, and by consultation with a third review author (JJH). Where available we extracted the following details:
general information (title, authors, source, country, year of publication, setting);
trial characteristics (design, risk of bias);
participant characteristics (inclusion and exclusion criteria, type of NTDßT, severity of anaemia, indication for hydroxyurea, ongoing standard management protocol including the indication for blood transfusion, sample size, losses to follow up, baseline characteristics such as age, ethnic group, pre‐existing co‐morbidities, duration of transfusion, prior splenectomy and iron chelation);
intervention (data regarding the usage of hydroxyurea, e.g. amount, frequency of delivery, timing from diagnosis, length of treatment, participant responsiveness);
comparison (data regarding standard management protocol with or without a placebo; in trials using placebo, we collected data on amount and frequency of delivery);
outcomes (as specified under Types of outcome measures); and
additional data (adverse events, QoL measurements and outcomes not described above).
If we had found it necessary, we planned to contact the original investigators in an effort to obtain any data needing further clarification or that were not reported.
We planned to group outcome data into those measured at up to six months, up to one year, up to three years, up to five years and up to 10 years. If outcomes were measured at other time periods, we would have considered whether to include these as well. For the one included trial, we reported outcomes at time points up to 24 weeks.
Assessment of risk of bias in included studies
Two review authors (WCF, CKL) independently assessed the risk of bias of each included trial using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). We resolved any disagreements by discussion with a third review author (JJH or VV) or by correspondence with the trial authors.
For each trial included, we assessed the following criteria.
1. Sequence generation
We described the method used to generate the allocation sequence in terms of sufficient details and appropriateness. We assessed the method as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital or clinic record number); or
unclear risk of bias.
2. Allocation concealment
We described the method used to conceal allocation to interventions prior to assignment to judge whether the intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment. We assessed the method as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes, alternation; date of birth);
unclear risk of bias.
3. Blinding of clinician (person delivering the treatment), participant and outcome assessors to treatment allocation
We described the methods used, if any, to blind participants and personnel from knowledge of which intervention a participant received. We made a judgement on whether the blinding was adequate. We considered that trials are at low risk of bias if they were blinded, or if we judged that the lack of blinding would be unlikely to affect results. We assessed blinding separately for different outcomes or classes of outcomes. We assessed the methods as:
low, high or unclear risk of bias for participants;
low, high or unclear risk of bias for personnel;
low, high or unclear risk of bias for outcome assessors.
4. Completeness of the outcome data, checking for possible attrition bias through withdrawals, loss to follow up and protocol violations
We described whether there are significant missing outcome data based on the number of participants recruited or randomised and the number analysed for each outcome at the end of the trial period. If the percentage loss to follow up had exceeded 10%, we would have made a decision about using appropriate sensitivity analysis. We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; 'as treated' analysis done with substantial departure of intervention received from that assigned at randomisation);
unclear risk of bias.
5. Selective reporting bias, checking that all of a trial’s pre‐specified outcomes and all expected outcomes of interest to the review have been reported
We attempted to describe how we investigated the possibility of selective outcome reporting bias and what we found including non‐significant results that were not reported adequately or missed important outcome variables. We assessed the methods as:
low risk of bias (where it is clear that all of the trial's pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
high risk of bias (where not all the trial's pre‐specified outcomes have been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest were reported incompletely and so cannot be used; trial failed to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
6. Other sources of bias in the included trials
We described any important concerns we have about other possible sources of bias. If we had found cross‐over trials, we planned to assess the risk of bias related to any possible carry‐over effect as well as assess the effect of the duration of the intervention and follow up on outcomes. We assessed whether each trial was free of other problems that could put it at risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there is risk of other bias.
7. An overall risk of bias assessment was made based on the items above
We made explicit judgements about whether trials are at high risk of bias, according to the criteria given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011c). With reference to (1) to (6) above, we assessed the likely magnitude and direction of the bias and whether we considered it is likely to impact on the findings. We explored the impact of the level of bias through undertaking sensitivity analysis.
Measures of treatment effect
We anticipated that one of our primary outcomes (frequency of transfusion) may be reported as rates. If we had encountered this, we planned to calculate rate ratios with 95% confidence intervals (CI). We calculated risk ratios (RR) and their associated 95% CI for all dichotomous outcomes. For continuous outcomes, where outcomes were measured in the same way between trials, we planned to report the mean relative change from baseline or the mean post‐intervention value as well as the difference in means between treatment groups or by combining the baseline and post‐intervention data, and their 95% CIs. Where publications present the mean difference (MD) and standard error (SE), we planned to calculate the standard deviation (SD). Where outcomes were reported on different scales, such as QoL scales, and we judged them to be reporting the same thing, we planned to use standardised mean difference (SMD).
Unit of analysis issues
We treated the participant as the unit of analysis.
For this intervention cross‐over trials are possible. Cross‐over trials might be useful for short‐term effects such as some adverse effects. If in future we encounter cross‐over trials, we will include them and analyse them separately by following the methods in the Cochrane Handbook for Systematic Reviews of Interventions when analysing and incorporating the trials for meta‐analysis (Higgins 2011d). First we will carefully consider the washout period.The half life of hydroxyurea is between two to four hours. Studies done on people with sickle cell disease have shown a recovery of marrow effects such as blood counts within one to two weeks after withholding hydroxyurea (Strouse 2012; Ware 2011). If individual patient data is available or the cross‐over trials are analysed appropriately using a paired analysis and the SD for participant‐specific differences is present or can be obtained, we will incorporate such trials into the meta‐analysis. If not, we will use the data from the first period if available. We will not attempt to impute missing SDs. Besides, the clinical status of NTDßT is not stable over time, e.g., pre‐pubertal children require less transfusion than pubertal individuals or pregnant females and trials recruiting pre‐pubertal children might continue from pre‐puberty into puberty.
We did not encounter cluster RCTs. If in the future we do, we plan to follow the methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b). We will use an estimate of the intra‐cluster correlation co‐efficient (ICC) derived from the trial (if possible), or from another source. If we use ICCs from other sources, we will report this and conduct sensitivity analyses to investigate the effect of variation in the ICC.
Dealing with missing data
If we had encountered missing or unclear data, we planned to contact the primary investigators for up to three attempts after which the data will be determined missing or unclear data. We planned to assess data on the number of participants who entered the trial and the availability of data at the time of follow up, and those who completed the trial for each outcome. If there had been a difference, we planned to calculate the percentage change and report it. We also attempted to establish whether the primary study used an intention‐to‐treat analysis, such as the participants were analysed in the groups to which they were randomised. We have contacted the trial investigator (Bohara 2014) for more information on the number of participants in each group at each time point. We have tried twice and have to date not received a response. In future, if we encounter trials in abstract format we will contact the authors for any full reports (Higgins 2011d).
Assessment of heterogeneity
If in future we are able to include more than one trial, we plan to test for heterogeneity between trials using the I2 statistic and the Chi2 test (both of which are automatically calculated in the Review Manager software (RevMan 2014)). We will regard heterogeneity as substantial if an I2 value is greater than 80%. We will note a low P value (less than 0.10) in the Chi2 test and interpret it in the light of the number of included trials. We will explore any heterogeneity by subgroup analysis.
Assessment of reporting biases
We attempted to minimize publication and reporting bias by conducting a comprehensive search. If we had obtained sufficient number of trials (at least 10), we planned to assess publication bias by constructing a funnel plot and reviewing its symmetry. If we had found any funnel plot asymmetry, we planned to consider whether this could be due to publication bias (Sterne 2011).
Data synthesis
We have presented the data from the single included trial. If no meta‐analysis had been possible, or if we had encountered substantial heterogeneity (as defined above), we would have given a descriptive, qualitative critical appraisal of the outcome information from the included trials (Deeks 2011). We analysed the data reported in the included trial which are relevant to the primary and secondary outcomes of this review using the Review Manager software (RevMan 2014).
If we are able to include more trials in the future and they are clinically and methodologically comparable, we plan to carry out meta‐analyses initially using a random‐effects model.
Subgroup analysis and investigation of heterogeneity
If we had identified substantial heterogeneity, we planned to investigate it using subgroup analyses. We would have considered whether an overall summary was meaningful, and if it was, we would have used a random‐effects analysis to produce it. We planned to only carry out subgroup analysis for the primary outcome, considering the following subgroup analyses:
dose of hydroxyurea used (for trials included in Comparison 1, hydroxyurea versus placebo or standard care);
type of NTDßT (Hb E/ß thalassaemia, ß thalassaemia intermedia, other) (for both comparisons);
baseline Hb F (high or low) (for both comparisons).
Sensitivity analysis
We planned to conduct sensitivity analyses to assess the robustness of our review results by repeating the analysis with the following adjustments, exclusion of trials with:
unclear or high risk of bias regarding allocation concealment;
unclear or high risk of bias regarding blinding of outcomes assessment;
unclear or high risk of bias regarding completeness of follow up.
We also planned to use a sensitivity analysis to test the robustness of our choice of model used for meta‐analysis (Deeks 2011).
Summary of findings table
We used GRADE to reflect the quality of evidence and used GRADEPro software (GRADE 2015) to create a summary of findings (SoF) table. The SoF table had the following components:
summarised key findings (participants, comparison and baseline information, outcome) (Schünemann 2011);
summarised statistical results;
summarised quality of evidence, magnitude of the effect including the source of any external information used in the ‘Assumed risk’ column or any departures from the standard methods, and reasons behind the decisions.
We included the following outcomes in our SoF table:
frequency of transfusion;
major adverse effects;
QoL;
foetal haemoglobin (Hb F) level.
Results
Description of studies
Results of the search
All searches combined identified 461 citations, including 74 duplicates. After title and abstract screening, we retrieved the full‐text of 53 records. Among them were six randomised controlled trials (seven records), but only one trial could be included in this review (Bohara 2014) (Figure 1).
1.
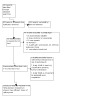
Study flow diagram.
Included studies
We did not find any randomised controlled trials comparing hydroxyurea with a placebo or standard care. The only included trial was a randomised controlled trial comparing x10 mg/kg/day with 20 mg/kg/day of hydroxyurea at a single centre in India (Bohara 2014). Treatment was stopped if participants developed adverse effects or if they failed to respond to the allocated treatment, but they remained in the trial until trial completion at 24 weeks. The participants (n = 61) had NTDßT of thalassaemia intermedia phenotype. The primary outcome was to assess the difference in responses to the different doses of hydroxyurea. The secondary outcomes were assessment of tolerability and safety of hydroxyurea at different doses.
Excluded studies
We excluded three trials (three unique records) (Biswas 2014; Elalfy 2013; Karimi 2010). Two trials compared hydroxyurea alone with hydroxyurea and another foetal haemoglobin inducer as a co‐intervention (Elalfy 2013; Karimi 2010). The third trial, available only in abstract format, was excluded because the participants had severe transfusion dependent Hb E/ß thalassaemia (mean transfusion about 13 to 14 per year) (Biswas 2014). Details of these excluded trials can be found in the tables (Characteristics of excluded studies).
Studies awaiting classification
One identified trial is awaiting classification (Huang 2016). The full paper is published only in Chinese with a short abstract in English; we will assess the trial once we have obtained a translation. The abstract describes a prospective trial with two groups of participants with ß‐thalassaemia intermedia, but does not mention if the trial was randomised. The intervention is either hydroxyurea or no hydroxyurea. Further details can be found in the tables (Characteristics of studies awaiting classification).
Ongoing studies
One trial is still ongoing, but has completed recruitment of participants (Haghpanah 2015). It is comparing a herbal product (resveratrol) to hydroxyurea and placebo or to resveratrol and placebo in people with in non‐transfusion dependent ß‐thalassaemia intermedia. Further details can be found in the tables (Characteristics of ongoing studies).
Risk of bias in included studies
Risk of bias of the included study is summarised in Figure 2.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Sequence generation
The Bohara trial generated a random sequence by using a random number table (Bohara 2014). We judged this trial to have a low risk of bias for this criterion.
Allocation concealment
Allocation concealment was not reported in the trial; therefore, we judged it to have unclear risk of bias for this criterion (Bohara 2014).
Blinding
The included trial did not report of any blinding of the participants, treating physicians and outcome assessors (Bohara 2014). The authors did not describe whether there was blinding of the intervention and control. Therefore, we judge there to be an unclear risk of bias for this criterion.
Incomplete outcome data
The participants were analysed in their allocated group irrespective of the treatment received. All participants were accounted for at the end of the trial and all participants were included in the final analysis (Bohara 2014). Therefore, we judge there to be a low risk of bias for this criterion.
Selective reporting
Although the 'Methods' section stated that participants were interviewed regarding their sense of well being and state of energy, these results were not reported. This could be selective outcome reporting. Therefore, this trial was judged to have an unclear risk of reporting bias. The trial was not registered prospectively on a clinical trials register (Bohara 2014).
Other potential sources of bias
The baseline characteristics between the two groups in the included trial showed no significant difference in age, gender, ß genotype, reticulocytes and mean haemoglobin, Hb F and Hb E (Bohara 2014). However, there was a significant difference in the mean of the mean corpuscular volume (MCV) levels, which was said to be non‐influential to hydroxyurea response. Therefore, this trial was judged to have low risk for any potential sources of bias.
Effects of interventions
Hydroxyurea versus placebo or standard care
No trials were included.
10 mg/kg/day hydroxyurea versus 20 mg/kg/day hydroxyurea
One trial involving a total of 61 participants was included (Bohara 2014).
Primary outcomes
1. Frequency of blood transfusion
Frequency of blood transfusion was not reported (Bohara 2014).
2. Transfusion‐free interval
Transfusion‐free interval was not reported (Bohara 2014).
3. Haemoglobin level
The trial authors reported that mean haemoglobin was significantly lower in the 20 mg/kg/day group at six, 12 and 24 weeks; our analysis shows a MD at 24 weeks ‐2.39 g/dL (95% CI ‐2.80 to ‐1.98) (Analysis 1.1).
1.1. Analysis.

Comparison 1: 20 mg/kg/day versus 10 mg/kg/day hydroxyurea, Outcome 1: Mean haemoglobin (g/dL)
Secondary outcomes
1. Thalassaemia‐ and transfusion‐related complications
None of our prespecified complications were reported in the included trial (Bohara 2014).
2. Adverse effects or toxicity
The major adverse effects reported were neutropenia (defined by the authors as absolute neutrophil count below 0.5 x 103/μL), leukopenia (white cell count below 3 x 103/μL) and thrombocytopenia (platelet count below 50 x 100/μL) (Bohara 2014). The 20 mg/kg/day group had a higher risk of the pre‐specified major adverse effects: neutropenia, RR 9.93 (95% CI 1.34 to 73.67); and thrombocytopenia, RR 3.68 (95% CI 1.12 to 12.07) (Analysis 1.2).
1.2. Analysis.

Comparison 1: 20 mg/kg/day versus 10 mg/kg/day hydroxyurea, Outcome 2: Major adverse effects (at 24 weeks)
There was no significant difference between the two groups for minor adverse effects (gastrointestinal disturbances and raised liver enzymes) (Analysis 1.3).
1.3. Analysis.

Comparison 1: 20 mg/kg/day versus 10 mg/kg/day hydroxyurea, Outcome 3: Minor adverse effects (at 24 weeks)
3. Foetal haemoglobin (Hb F) level
At 24 weeks, the mean foetal haemoglobin was significantly lower in the 20 mg/kg/day group, MD ‐1.50 g/dL (95% CI ‐1.83 to ‐1.17) (Analysis 1.4).
1.4. Analysis.

Comparison 1: 20 mg/kg/day versus 10 mg/kg/day hydroxyurea, Outcome 4: Mean foetal haemoglobin (g/dL)
4. Quality of life
No data on quality of life were reported (Bohara 2014).
Summary of findings for the main comparisons
We have summarised the findings for the main comparisons in relevant tables (Table 1; Table 2).
Discussion
Summary of main results
We did not find any trials comparing hydroxyurea with a control (placebo or standard care), so we are not able to answer the question about whether hydroxyurea has an effect on our primary outcomes. However, the single included trial comparing two doses of hydroxyurea found that haemoglobin was significantly higher in the group that received the lower hydroxyurea dose (10 mg/kg/day) at 24 weeks (and also at six and 12 weeks, as reported in the original trial). The higher dose group (20 mg/kg/day) did not experience any change from baseline in haemoglobin and Hb F at 24 weeks, whereas the lower dose experienced a significant increase in both compared to baseline (P < 0.000). In addition to the poorer response to treatment, there were more major and minor adverse events in the 20 mg/kg/day group.
Results from this single trial suggest that a dose of 10 mg/kg/day of hydroxyurea is safer and more effective than a higher dose.
Overall completeness and applicability of evidence
The major weakness of this review is that it does not include any trials comparing hydroxyurea with placebo or no treatment. Although the only included trial had relevant participants, interventions and outcomes, all participants received hydroxyurea and therefore lacked a usual care control group (Bohara 2014). It was observed that better outcomes were found with the lower dose; the poorer outcome with the higher dose may be due to the known adverse effects that hydroxyurea has on the bone marrow (de Paula 2003; Fucharoen 1996). We do not know whether even better outcomes and fewer adverse effects might be achieved with doses lower than 10 mg or even without hydroxyurea. There are no data from randomised controlled trials on the frequency of transfusion and the duration of transfusion free intervals. It should be noted that any observed improvement in haemoglobin or foetal haemoglobin (Hb F) may not result in a reduction in transfusion requirement.
Quality of the evidence
The single included trial has a small sample size (61 participants) and we judged allocation concealment to be unclear (Bohara 2014 ). Our analysis showed extremely wide confidence intervals for adverse events. Therefore, data from two of the three laboratory outcomes were judged to be from very low quality evidence and the data for mean Hb F was judged to be low quality.
Potential biases in the review process
At protocol stage, we had intended to include either randomised or quasi‐randomised trials comparing the efficacy and safety of hydroxyurea compared to placebo or no treatment for people with non‐transfusion dependent beta thalassaemia (NTDßT). During our search we found one trial comparing two doses of hydroxyurea. We noted that this trial met our objectives, so we made a post hoc change to our inclusion criteria by adding a second comparison. We do not think this post hoc change weakens the findings of this review. We also excluded two other randomised controlled trials comparing hydroxyurea alone with hydroxyurea in combination with another foetal haemoglobin enhancer, L‐carnitine (Karimi 2010) and erythropoietin (Elalfy 2013).
Agreements and disagreements with other studies or reviews
As stated previously, a Cochrane review found that hydroxyurea was effective in reducing transfusions in sickle cell disease (Jones 2001). The dose used in the two included studies in that review was high (15 mg/kg/day and 20 mg/kg/day) and the investigators continued to increase the dose beyond this until there was evidence of bone marrow depression. The only included trial in this review suggested that doses in this range were less effective than a low dose (10 mg/kg/day) (Bohara 2014). During our search we found a number of observational studies with a dose of 10 mg/kg/day reporting significant increases in haemoglobin (Amoozgar 2011; Dixit 2005; El‐Beshlawy 2014; Fucharoen 1996; Karimi 2005; Karimi 2010a) and foetal haemoglobin levels (Amoozgar 2011). An improvement in the haemoglobin concentrations after hydroxyurea treatment was also reported as a modest but significant increase in a meta‐analysis using before and after studies, and case reports. The authors of that review reported that they did not find any RCTs (Kosaryan 2014a).
While the trial included in this review showed unfavourable outcomes with 20 mg/kg/day hydroxyurea treatment (Bohara 2014), observational studies successfully used a dose of 20 mg/kg/day without any adverse effects (Ansari 2011; Bradai 2007; de Paula 2003; Dixit 2005; Fucharoen 1996). However, this observational data should be treated cautiously.
Haematological adverse effects, namely neutropenia, leukopenia and thrombocytopenia, were the main major adverse effects noted especially with the 20 mg/kg/day hydroxyurea in the included trial (Bohara 2014. This corresponds to the observational studies (Bradai 2007; Dixit 2005; Fucharoen 1996). One study observed 143 participants with a hydroxyurea dose of 8 mg/kg/day to 12 mg/kg/day for a median of 5.7 years did not encounter bone marrow depression (Karimi 2010a). Other adverse effects, hepatotoxicity and gastrointestinal disturbances found in this review have been reported in other observational studies (Dixit 2005; Karimi 2010a; Taher 2010).
Authors' conclusions
Implications for practice.
There is no evidence from randomised controlled trials to show whether hydroxyurea has any effect compared with controls on the need for blood transfusion. Therefore, we cannot currently recommend its use outside of the setting of a randomised controlled trial. We are also unable to conclude whether 10 mg/kg/day hydroxyurea is the appropriate dose, but could caution users to be aware of adverse effects at a higher dose. There is no evidence from randomised controlled trials as to whether hydroxyurea is capable of reducing the need for blood transfusion through a rise in the haemoglobin and Hb F levels.
Implications for research.
Hydroxyurea appears to have found an established role in the treatment of NTDßT without adequate evaluation. Therefore, there is an urgent need for large well‐designed randomised controlled trials comparing hydroxyurea with placebo or standard care. Such trials should have a sufficiently long follow‐up period to study the effect of hydroxyurea on the complications of the disease and from blood transfusion. Trials should also closely monitor adverse events from the drug and complications of both the disease and blood transfusion. Different doses and dosing schedules need to be compared. Research is also required to test whether there is any benefit of combining hydroxyurea with other Hb F enhancers.
What's new
| Date | Event | Description |
|---|---|---|
| 24 March 2023 | Amended | Notification of new review that has superseded this review (see Published Notes). |
History
Protocol first published: Issue 3, 2015 Review first published: Issue 10, 2016
Acknowledgements
We would like to thank Jill Fitzpatrick and Natalie Hall for their contribution and support in the process of searching for trials.
Appendices
Appendix 1. PubMed search strategy
#1 hydroxyurea
#2 hydroxycarbamide
#3 oncocarbide
#4 hydrea
#5 droxia
#6 #1 OR #2 OR #3 OR #4 OR #5
#7 beta‐thalassemia[tw]
#8 beta‐thalassaemia[tw]
#9 b‐thalassemia[tw]
#10 b‐thalassaemia[tw]
#11 "Thalassemia"[Mesh]
#12 #7 OR #8 OR #9 OR #10 OR #11
#13 randomised controlled trial[pt]
#14 controlled clinical trial[pt]
#15 randomised[ab]
#16 placebo[ab]
#17 drug therapy[sh]
#18 randomly[ab]
#19 trial[ab]
#20 groups[ab]
#21 #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20
#22 animals[mh] NOT humans[mh]
#23 #21 NOT #22
#24 #6 AND #12 AND #23
Appendix 2. EMBASE search strategy
#1 beta‐thalassemia: ab,ti #2 beta‐thalassaemia: ab,ti #3 b‐thalassemia: ab,ti #4 b‐thalassaemia: ab,ti #5 THALASSEMIA/exp #6 #1 OR #2 OR #3 OR #4 OR #5
#7 (hydroxyurea/exp OR hydroxyurea OR hydroxycarbamide/exp OR hydroxycarbamide OR oncocarbide/exp OR oncocarbide OR hydrea/exp OR hydrea OR droxia/exp OR droxia).ti,ab,tn
#8 hydroxyurea:tn,ab,ti
#9 hydroxycarbamide:tn,ab,ti
#10 oncocarbide:tn,ab,ti
#11 hydrea:tn,ab,ti
#12 droxia:tn,ab,ti
#13 #8 OR #9 OR #10 OR #11 OR #12
#14 hydroxyurea/exp
#15 #13 OR #14
#16 #7 OR #14
#17 cross over procedure/exp
#18 cross over clinical trial:ab,ti
#19 allocat*:ti,ab
#20 doubl* ADJ blind*:ti,ab
#21 trial*:ti
#22 placebo*:ti,ab
#23 random*:ti,ab
#24 double blind procedure/exp
#25 crossover*:ti,ab
#26 single blind procedure/exp
#27 randomised controlled trial/exp
#28 #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27
#29 #6 AND #15 AND #16 AND #28
Data and analyses
Comparison 1. 20 mg/kg/day versus 10 mg/kg/day hydroxyurea.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Mean haemoglobin (g/dL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1.1 At 24 weeks | 1 | 61 | Mean Difference (IV, Fixed, 95% CI) | ‐2.39 [‐2.80, ‐1.98] |
| 1.2 Major adverse effects (at 24 weeks) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.2.1 Neutropenia | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.93 [1.34, 73.67] |
| 1.2.2 Thrombocytopenia | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.68 [1.12, 12.07] |
| 1.3 Minor adverse effects (at 24 weeks) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.3.1 Gastrointestinal disturbances | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.31 [0.99, 11.06] |
| 1.3.2 Raised liver enzymes | 1 | 61 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.76 [0.58, 13.14] |
| 1.4 Mean foetal haemoglobin (g/dL) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.4.1 At 24 weeks | 1 | 61 | Mean Difference (IV, Fixed, 95% CI) | ‐1.50 [‐1.83, ‐1.17] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bohara 2014.
| Study characteristics | ||
| Methods | Randomised controlled trial. Single centre in India. |
|
| Participants | 61 participants aged 8 years and above with Hb E/ß thalassaemia with the phenotype of ß thalassaemia intermedia. Gender split: 31 males and 30 females. 10 mg/kg/day hydroxyurea group (n = 32) mean (SD) age 16.68 (6.05) years. 20 mg/kg/day hydroxyurea group (n = 29) mean (SD) age 16.17 (5.75) years. |
|
| Interventions | 10 mg/kg/day hydroxyurea (designated control in this review) versus 20 mg/kg/day hydroxyurea. There were no participants on either placebo or standard care in the study. If any participant presented with adverse events (e.g. a fall in haemoglobin > 0.5 g/dL which led to a need for transfusion, ANC < 1.5 x 103/mL, platelet count < 50 x 103/mL, significant hepatic or renal dysfunction) hydroxyurea therapy was withheld for 1 week. Therapy (at 5 mg/kg/day lower than the dose at which the toxicity had occurred) to be restarted after the resolution of the toxicity. These participants then "resumed the therapy as per study protocol". For any participant deemed to be non‐responsive (when hydroxyurea caused a rise in haemoglobin of < 0.5 g/dl in 12 weeks or a drop in haemoglobin from pre‐hydroxyurea value), therapy was permanently withheld. This was the case for 5 participants from the 10 mg/kg/day group and 12 participants in 20 mg/kg/day group. The number of participants remaining on hydroxyurea on each arm declined markedly as the study progressed. At the end of the study, there were 25 participants still receiving the lower dose of hydroxyurea while 13 participants were still on the higher dose. |
|
| Outcomes | Haemoglobin and Hb F were measured at baseline, 6, 12 and 24 weeks. Adverse effects (neutropenia, thrombocytopenia, gastrointestinal disturbances and raised liver enzymes) were reported. Response rates (good response, intermediate response and no response) based on arbitrary criteria were also reported. |
|
| Notes | No funding statement. The trial was not registered prospectively on a clinical trials register. Available as published abstract and full text publication. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomised ... using a random number table." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not reported. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding of participants or trial personnel was not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding of outcome assessors was not reported. Note: The primary outcomes were laboratory assessments and it is possible that they were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Although not all participants received the intervention that they were assigned for the full duration of the trial, all enrolled participants were analysed in the group they were assigned to "irrespective of how long they received hydroxyurea therapy". |
| Selective reporting (reporting bias) | Unclear risk | In the methods section of the published report, participants were reported to have undergone an interview regarding their sense of well being and state of energy and this was not reported in the results. Comment: the authors were unsure whether this was evidence of reporting bias since this outcome, although related to QoL, was not one of our pre‐specified outcomes. No protocol was available and other evidence of selective reporting was detected. |
| Other bias | Low risk | The baseline characteristics between the 2 groups had no significant difference in age, gender, ß genotype, reticulocytes and mean haemoglobin, Hb F and Hb E. However, there was a significant difference in the mean of the MCV levels, judged to not influence response to hydroxyurea. |
ANC: absolute neutrophil count Hb E: haemoglobin E Hb F: foetal haemoglobin MCV: mean corpuscular volume QoL: quality of life SD: standard deviation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Biswas 2014 | This is randomised study, available in abstract only, which evaluated the efficacy of hydroxyurea against valproic acid among transfusion dependent Hb E/ß thalassaemia patients. |
| Elalfy 2013 | This 15‐month trial randomised young people with ß thalassaemia into 2 groups, either hydroxyurea alone or hydroxyurea in combination with erythropoietin. Erythropoietin induces production of red blood cells. |
| Karimi 2010 | This is a randomised controlled trial carried out over 6 months to assess the effect of combination therapy of hydroxyurea with L‐carnitine and magnesium chloride. The participants received either hydroxyurea alone, hydroxyurea and L‐carnitine, hydroxyurea and magnesium chloride, or a combination of all 3 drugs. Both hydroxyurea and L‐carnitine are foetal haemoglobin enhancers, while magnesium chloride is known to stabilize erythrocytes' membrane and morphology. |
Characteristics of studies awaiting classification [ordered by study ID]
Huang 2016.
| Methods | Prospective study with two groups of participants. Allocation and randomisation not mentioned. |
| Participants | Participants with beta‐thalassaemia intermedia. |
| Interventions | Hydroxyurea or no hydroxyurea. No further details available. |
| Outcomes | Haemoglobin levels, reticulocytes percentage, serum ferritin, blood transfusion dependency and adverse effects. |
| Notes | Full paper only available in Chinese (awaiting translation). Information above obtained from English language abstract. |
Characteristics of ongoing studies [ordered by study ID]
Haghpanah 2015.
| Study name | Comparison of the efficacy and safety of a herbal product (resveratrol) with hydroxyurea in non‐transfusion dependent ß thalassemia intermedia. |
| Methods | Convenience sampling randomisation using block randomisation. |
| Participants | Participants with ß‐thalassaemia intermedia who were not transfused and did not receive Hb F inducer for 6 months prior to the study. Those with abnormal liver or kidney function tests were excluded. |
| Interventions | The participants will be receiving either hydroxyurea and herbal product (resveratrol), hydroxyurea and placebo or resveratrol and placebo. |
| Outcomes | The primary outcomes are haemoglobin and Hb F levels. The secondary outcomes are safety and adverse effects of either resveratrol or hydroxyurea or both. |
| Starting date | 23 August 2015. |
| Contact information | Sezaneh Haghpanah (haghpanah@sums.ac.ir). |
| Notes | An ongoing study which has completed recruitment of participants. |
Differences between protocol and review
At the stage of conducting our search we made a post hoc adjustment to the sections 'Objectives' and 'Types of interventions' to allow an additional comparison for different doses or dose regimens of hydroxyurea.
Contributions of authors
Title development: WCF and VV.
Title registration: WCF drafted the proposal with substantial input from JJH and CKL along with some comments from VV.
Protocol: WCF drafted the protocol with substantial input from JJH and CKL.
| Roles and responsibilities | |
| TASK | WHO WILL UNDERTAKE THE TASK? |
| Protocol stage: draft the protocol | WCF with input from all authors |
| Review stage: select which trials to include (2 people) | WCF, CKL |
| Review stage: extract data from trials (2 people) | WCF, CKL |
| Review stage: enter data into RevMan | WCF |
| Review stage: carry out the analysis | WCF, CKL, JJH |
| Review stage: interpret the analysis | All authors |
| Review stage: draft the final review | WCF with input from all authors |
| Update stage: update the review | All authors |
Sources of support
Internal sources
Penang Medical College, Malaysia
Universiti Kebangsaan Malaysia Medical Centre, Malaysia
Mahidol University, Bangkok, Thailand
External sources
-
National Institute for Health Research, UK
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
WCF is involved in the management of people with non‐transfusion dependent thalassaemia. This author has received small pharmaceutical grants to attend conferences.
JJH has no known conflict of interest and has nothing to declare.
CKL is involved in the management of people with non‐transfusion dependent thalassaemia. This author has received small pharmaceutical grants to attend conferences.
VV is involved in the management of people with non‐transfusion dependent thalassaemia. This author has received honoraria and travel support from companies that manufacture iron chelators to attend conferences. All of these were unrelated to hydroxyurea.
Edited (no change to conclusions)
References
References to studies included in this review
Bohara 2014 {published data only}
- Bohara VV, Raut L, Bardarkhe G, Pujari G, Chakrabarti P, Ray S, Nath U, Chaudhuri U. Optimizing the dose of hydroxyurea in thalassemia intermedia (Conference abstract). Indian Journal Hematology Blood Transfusion 2012;28(4):191-256. [Google Scholar]
- Bohara VV, Ray S, Chakrabarti P, Ray SS, Nath UK, Chaudhuri U. Optimizing the dose of hydroxyurea therapy for patients with beta-thalassemia intermedia (Hb E-beta-thalassemia): a single center study from Eastern India. Hemoglobin 2014;38(1):44-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Biswas 2014 {published data only}
- Biswas S, Chaudhuri S, Kumar M, Sen A, Bhattyacharyya M, Ghosh K. The role of hydroxyurea and valproic acid in the management of severe HbE-ß thalassaemia (Conference abstract). Indian Journal of Hematology & Blood Transfusion 2014;30(Suppl 2):S448-546. [Google Scholar]
Elalfy 2013 {published data only}
- Elalfy MS, Adly AA, Ismail EA, Elhenawy YI, Elghamry IR. Therapeutic superiority and safety of combined hydroxyurea with recombinant human erythropoietin over hydroxyurea in young beta-thalassemia intermedia patients. European Journal of Haematology 2013;91(6):522-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
Karimi 2010 {published data only}
- Karimi M, Mohammadi F, Behmanesh F, Samani SM, Borzouee M, Amoozgar H, et al. Effect of combination therapy of hydroxyurea with l-carnitine and magnesium chloride on hematologic parameters and cardiac function of patients with beta-thalassemia intermedia. European Journal of Haematology 2010;84(1):52-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Huang 2016 {published data only}
- Huang L, Yao HX. [Curative effects of hydroxyurea on the patients with beta-thalassaemia Intermedia]. Zhongguo Shi Yan Xue Ye Xue za Zhi [Journal of Experimental Hematology] 2016;24(3):806-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Haghpanah 2015 {published data only}
- Haghpanah S, Karimi M. Comparison of the efficacy and safety of herbal product (Resveratrol) with Hydroxyurea (HU) in non- transfusion- dependent B-thalassemia-intermedia. http://www.irct.ir/searchresult.php?id=20051&number=2.
Additional references
Aessopos 2001
- Aessopos A, Farmakis D, Karagiorga M, Voskaridou E, Loutradi A, Hatziliami A, et al. Cardiac involvement in thalassemia intermedia: a multicenter study. Blood 2001;97(11):3411-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Amoozgar 2011
- Amoozgar H, Farhani N, Khodadadi N, Karimi M, Cheriki S. Comparative study of pulmonary circulation and myocardial function in patients with beta-thalassemia intermedia with and without hydroxyurea, a case-control study. European Journal of Haematology 2011;87(1):61-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ansari 2011
- Ansari SH, Shamsi TS, Ashraf M, Perveen K, Farzana T, Bohray M, et al. Efficacy of hydroxyurea in providing transfusion independence in beta-thalassemia. Journal of Pediatric Hematology/Oncology 2011;33(5):339-43. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bauer 2012
- Bauer DE, Kamran SC, Orkin SH. Reawakening fetal hemoglobin: prospects for new therapies for the beta-globin disorders. Blood 2012;120(15):2945-53. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Baynes 1986
- Baynes R, Bezwoda W, Bothwell T, Khan Q, Mansoor N. The non-immune inflammatory response: serial changes in plasma iron, iron-binding capacity, lactoferrin, ferritin and C-reactive protein. Scandinavian journal of clinical and laboratory investigation 1986;46(7):695-704. [PMID: ] [DOI] [PubMed] [Google Scholar]
Borgna‐Pignatti 2007
- Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. British Journal of Haematology 2007;138(3):291-304. [PMID: ] [DOI] [PubMed] [Google Scholar]
Bradai 2007
- Bradai M, Pissard S, Abad MT, Dechartres A, Ribeil JA, Landais P, et al. Decreased transfusion needs associated with hydroxyurea therapy in Algerian patients with thalassemia major or intermedia. Transfusion 2007;47(10):1830-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Camaschella 1995
- Camaschella C, Cappellini MD. Thalassemia intermedia. Haematologica 1995;80(1):58-68. [PMID: ] [PubMed] [Google Scholar]
de Montalembert 1990
- Montalembert M, Girot R, Revillon Y, Jan D, Adjrad L, Ardjoun FZ, et al. Partial splenectomy in homozygous beta thalassaemia. Archives of Disease in Childhood 1990;65(3):304-7. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
de Paula 2003
- Paula EV, Lima CS, Arruda VR, Alberto FL, Saad ST, Costa FF. Long-term hydroxyurea therapy in beta-thalassaemia patients. European Journal of Haematology 2003;70(3):151-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Deeks 2011
- Deeks JJ, Higgins JPT, Altman DG on behalf of the Cochrane Statistical Methods Group. Chapter 9 Analysing data and undertaking meta-analysis. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Dixit 2005
- Dixit A, Chatterjee TC, Mishra P, Choudhry DR, Mahapatra M, Tyagi S, et al. Hydroxyurea in thalassemia intermedia--a promising therapy. Annals of Hematology 2005;84(7):441-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
El‐Beshlawy 2014
- El-Beshlawy A, El-Ghamrawy M, EL-Ela MA, Said F, Adolf S, Abdel-Razek AR, et al. Response to hydroxycarbamide in pediatric beta-thalassemia intermedia: 8 years' follow-up in Egypt. Annals of Hematology 2014;93(12):2045-50. [PMID: ] [DOI] [PubMed] [Google Scholar]
Fisher 2013
- Fisher SA, Brunskill SJ, Doree C, Gooding S, Chowdhury O, Roberts DJ. Desferrioxamine mesylate for managing transfusional iron overload in people with transfusion-dependent thalassaemia. Cochrane Database of Systematic Reviews 2013, Issue 8. Art. No: CD004450. [DOI: 10.1002/14651858.CD004450.pub3] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fucharoen 1996
- Fucharoen S, Siritanaratkul N, Winichagoon P, Chowthaworn J, Siriboon W, Muangsup W, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood 1996;87(3):887-92. [PMID: ] [PubMed] [Google Scholar]
GRADE 2015 [Computer program]
- GRADEpro GDT: GRADEpro Guideline Development Tool. McMaster University, 2015 (developed by Evidence Prime, Inc.), 2015. Available from gradepro.org.
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration 2011. Available from www.cochrane-handbook.org.
Higgins 2011b
- Higgins JPT, Deeks JJ (editors). Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Higgins 2011c
- Higgins JPT, Altman DG, Sterne JAC on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Higgins 2011d
- Higgins JPT, Deeks JJ, Altman DG on behalf of the Cochrane Statistical Methods Group (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors). Cochrane Handbook of Systematic Reviews of Interventions. Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Hoppe 1999
- Hoppe C, Vichinsky E, Lewis B, Foote D, Styles L. Hydroxyurea and sodium phenylbutyrate therapy in thalassemia intermedia. American Journal of Hematology 1999;62(4):221-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Italia 2009
- Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni AH, Sawant PM, et al. Response to hydroxyurea in beta thalassemia major and intermedia: experience in Western India. Clinica Chimica Acta; International Journal of Clinical Chemistry 2009;407(1-2):10-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Italia 2010
- Italia KY, Jijina FF, Merchant R, Panjwani S, Nadkarni AH, Sawant PM, et al. Effect of hydroxyurea on the transfusion requirements in patients with severe HbE-beta-thalassaemia: a genotypic and phenotypic study. Journal of Clinical Pathology 2010;63(2):147-50. [PMID: ] [DOI] [PubMed] [Google Scholar]
Jones 2001
- Jones AP, Davies SC, Olujohungbe A. Hydroxyurea for sickle cell disease. Cochrane Database of Systematic Reviews 2001, Issue 2. Art. No: CD002202. [DOI: 10.1002/14651858.CD002202] [DOI] [PubMed] [Google Scholar]
Karimi 2005
- Karimi M, Darzi H, Yavarian M. Hematologic and clinical responses of thalassemia intermedia patients to hydroxyurea during 6 years of therapy in Iran. Journal of Pediatric Hematology/Oncology 2005;27(7):380-5. [PMID: ] [DOI] [PubMed] [Google Scholar]
Karimi 2010a
- Karimi M, Cohan N, Mousavizadeh K, Falahi MJ, Haghpanah S. Adverse effects of hydroxyurea in beta-thalassemia intermedia patients: 10 years' experience. Pediatric Hematology and Oncology 2010;27(3):205-11. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kinney 1999
- Kinney TR, Helms RW, O'Branski EE, Ohene-Frempong K, Wang W, Daeschner C, et al. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Pediatric Hydroxyurea Group. Blood 1999;94(5):1550-4. [PMID: ] [PubMed] [Google Scholar]
Kohne 2010
- Kohne E, Kleihauer E. Hemoglobinopathies: a longitudinal study over four decades. Deutsches Arzteblatt International 2010;107(5):65-71. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kohne 2011
- Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Deutsches Arzteblatt International 2011;108(31-2):532-40. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kosaryan 2009
- Kosaryan M, Vahidshahi K, Karami H, Ehteshami S. Effect of hydroxyurea on thalassemia major and thalassemia intermedia in Iranian patients. Pakistan Journal of Medical Sciences Quarterly 2009;25(1):74-8. [Google Scholar]
Kosaryan 2014
- Kosaryan M, Karami H, Zafari M, Yaghobi N. Report on patients with non transfusion-dependent beta-thalassemia major being treated with hydroxyurea attending the Thalassemia Research Center, Sari, Mazandaran Province, Islamic Republic of Iran in 2013. Hemoglobin 2014;38(2):115-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kosaryan 2014a
- Kosaryan M, Zafari M, Alipur A, Hedayatizadeh-Omran A. The effect and side effect of hydroxyurea therapy on patients with beta-thalassemia: a systematic review to December 2012. Hemoglobin 2014;38(4):262-71. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mancuso 2006
- Mancuso A, Maggio A, Renda D, Di Marzo R, Rigano P. Treatment with hydroxycarbamide for intermedia thalassaemia: decrease of efficacy in some patients during long-term follow up. British Journal of Haematology 2006;133(1):105-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Matta 2013
Menzel 2007
- Menzel S, Jiang J, Silver N, Gallagher J, Cunningham J, Surdulescu G, et al. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood 2007;110(10):3624-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Meo 2008
- Meo A, Cassinerio E, Castelli R, Bignamini D, Perego L, Cappellini MD. Effect of hydroxyurea on extramedullary haematopoiesis in thalassaemia intermedia: case reports and literature review. International Journal of Laboratory Hematology 2008;30(5):425-31. [PMID: ] [DOI] [PubMed] [Google Scholar]
Musallam 2011
- Musallam KM, Cappellini MD, Wood JC, Motta I, Graziadei G, Tamim H, et al. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica 2011;96(11):1605-12. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Musallam 2012
- Musallam KM, Cappellini MD, Wood JC, Taher AT. Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood reviews 2012;26 Suppl 1:S16-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Musallam 2012a
- Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood 2012;119(2):364-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Musallam 2012b
- Musallam KM, Taher AT, Rachmilewitz EA. Beta-thalassemia intermedia: a clinical perspective. Cold Spring Harbor Perspectives in Medicine 2012;2(7):a013482. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Musallam 2012c
- Musallam KM, Cappellini MD, Daar S, Karimi M, El-Beshlawy A and Taher AT. Serum Ferritin Levels and Morbidity in β-Thalassemia Intermedia: A 10-Year Cohort Study. In: 54th ASH Annual Meeting and Exposition. 2012.
Musallam 2013
- Musallam KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassaemia. Blood 2013;121(12):2199-212. [DOI: 10.1182/blood-2012-10-408021] [DOI] [PubMed] [Google Scholar]
Origa 2007
- Origa R, Galanello R, Ganz T, Giagu N, Maccioni L, Faa G, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica 2007;92(5):583-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pandey 2012
- Pandey S, Pandey S, Mishra RM, Saxena R. Modulating effect of the -158 gamma (C-->T) Xmn1 polymorphism in Indian sickle cell patients. Mediterranean Journal of Hematology and Infectious Diseases 2012;4(1):e2012001. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Panigrahi 2005
- Panigrahi I, Agarwal S, Gupta T, Singhal P, Pradhan M. Hemoglobin E-beta thalassemia: factors affecting phenotype. Indian Pediatrics 2005;42(4):357-62. [PMID: ] [PubMed] [Google Scholar]
Pippard 1979
- Pippard MJ, Callender ST, Warner GT, Weatherall DJ. Iron absorption and loading in beta-thalassaemia intermedia. Lancet 1979;2(8147):819-21. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pourfarzad 2013
- Pourfarzad F, Lindern M, Azarkeivan A, Hou J, Kia SK, Esteghamat F, et al. Hydroxyurea responsiveness in beta-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica 2013;98(5):696-704. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rigano 2010
- Rigano P, Pecoraro A, Calzolari R, Troia A, Acuto S, Renda D, et al. Desensitization to hydroxycarbamide following long-term treatment of thalassaemia intermedia as observed in vivo and in primary erythroid cultures from treated patients. British Journal of Haematology 2010;151(5):509-15. [PMID: ] [DOI] [PubMed] [Google Scholar]
Sankaran 2008
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science (New York, N.Y.) 2008;322(5909):1839-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH on behalf of the Cochrane Applicability and Recommendations Methods Group and the Cochrane Statistical Methods Group. Chapter 11: Presenting results and ‘Summary of findings’ tables. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Singer 2005a
- Singer ST, Kuypers FA, Olivieri NF, Weatherall DJ, Mignacca R, Coates TD, et al. Fetal haemoglobin augmentation in E/beta(0) thalassaemia: clinical and haematological outcome. British Journal of Haematology 2005;131(3):378-88. [PMID: ] [DOI] [PubMed] [Google Scholar]
Singer 2005b
- Singer ST, Kuypers FA, Olivieri NF, Weatherall DJ, Mignacca R, Coates TD, et al. Single and combination drug therapy for fetal hemoglobin augmentation in hemoglobin E-beta 0-thalassemia: Considerations for treatment. Annals of the New York Academy of Sciences 2005;1054:250-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
St Pierre 2005
- St Pierre TG, Clark PR, Chua-anusorn W, Fleming AJ, Jeffrey GP, Olynyk JK, et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood 2005;105(2):855-61. [PMID: ] [DOI] [PubMed] [Google Scholar]
Steinberg 2010
- Steinberg MH, McCarthy, William F. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. American Journal of Hematology June 2010;85(6):401-2. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D on behalf of the Cochrane Bias Methods Group (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S (editors). Cochrane Handbook forSystematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Strouse 2008
- Strouse JJ, Lanzkron S, Beach MC, Haywood C, Park H, Witkop C, et al. Hydroxyurea for sickle cell disease: a systematic review for efficacy and toxicity in children. Pediatrics 2008;122(6):1332-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Strouse 2012
- Strouse JJ, Heeney MM. Hydroxyurea for the treatment of sickle cell disease: efficacy, barriers, toxicity, and management in children. Pediatric Blood & Cancer 2012;59(2):365-71. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Taher 2006
- Taher A, Isma'eel H, Cappellini MD. Thalassemia intermedia: revisited. Blood Cells, Molecules & Diseases 2006;37(1):12-20. [PMID: ] [DOI] [PubMed] [Google Scholar]
Taher 2009
- Taher A, Hershko C, Cappellini MD. Iron overload in thalassaemia intermedia: reassessment of iron chelation strategies. British Journal of Haematology 2009;147(5):634-40. [PMID: ] [DOI] [PubMed] [Google Scholar]
Taher 2010
- Taher AT, Musallam KM, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood 2010;115(10):1886-92. [PMID: ] [DOI] [PubMed] [Google Scholar]
TIF 2013a
- Taher A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V. Chapter 01: Introduction. In: Guidelines for The Management of Non-transfusion Dependent Thalassaemia (NTDT). Nicosia, Cyprus: Thalassaemia International Federation, 2013:1-11. [PubMed] [Google Scholar]
TIF 2013b
- Taher A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V. Chapter 02: Blood Transfusion. In: Guidelines for The Management of Non-transfusion Dependent Thalassaemia (NTDT). Nicosia, Cyprus: Thalassaemia International Federation, 2013:12-8. [PubMed] [Google Scholar]
TIF 2013c
- Taher A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V. Chapter 04: Fetal Hemoglobin Induction. In: Guidelines for The Management of Non-transfusion Dependent Thalassaemia (NTDT). Nicosia, Cyprus: Thalassaemia International Federation, 2013:27-34. [PubMed] [Google Scholar]
Ware 2002
- Ware RE, Eggleston B, Redding-Lallinger R, Wang WC, Smith-Whitley K, Daeschner C, et al. Predictors of fetal hemoglobin response in children with sickle cell anemia receiving hydroxyurea therapy. Blood 2002;99(1):10-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Ware 2011
- Ware RE, Despotovic JM, Mortier NA, Flanagan JM, He J, Smeltzer MP, et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood 2011;118(18):4985-91. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weatherall 2001
- Weatherall DJ. The Thalassaemia Syndrome. 4th edition. Blackwell Science, 2001. [Google Scholar]
Weatherall 2012
- Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood reviews 2012;26 Suppl 1:S3-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Zimmerman 2004
- Zimmerman SA, Schultz WH, Davis JS, Pickens CV, Mortier NA, Howard TA, et al. Sustained long-term hematologic efficacy of hydroxyurea at maximum tolerated dose in children with sickle cell disease. Blood 2004;103(6):2039-45. [PMID: ] [DOI] [PubMed] [Google Scholar]