

Abstract
A novel cycloartane triterpenoid alkaloid, kleinhospitine E (1), six new cycloartane triterpenoids (2−7), three known cycloartane triterpenoids (8−10), and taraxerone (11) were isolated from a methanol extract of Kleinhovia hospita. Their structures were elucidated by 1D-and 2D-NMR spectroscopy as well as HRMS analysis. The absolute configurations of all isolated compounds were determined from their ECD spectra by comparison with theoretical values. Kleinhospitine E (1) is the first cycloartane alkaloid possessing an unusual γ-lactam with an oxopropylidene side chain. Compounds 2, 3, and 6 were assigned as cycloartane triterpenoids with a 9α,10α-cyclopropyl ring, which is found rarely among naturally occurring compounds, while 4 and 5 were established as isomers of compound 3 containing a 21,23-diacetal side chain. Biological evaluation revealed that compounds 4 and 9 exhibited more potent antiproliferative activities against a multidrug-resistant tumor cell line compared with its parent chemosensitive cell line. Furthermore, compound 6 exhibited submicromolar anti-HIV activity.
Graphical Abstract
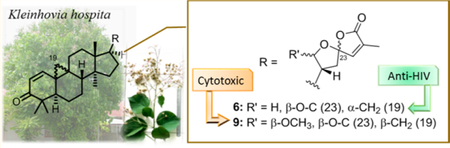
Graphical Abstract
The Indonesian plant Kleinhovia hospita L. (Malvaceae) is locally called “tahongai” (Kalimantan Timur), “timoho” (Java), and “paliasa” (Makassar, South Sulawesi), although two additional plant species, Melochia umbellata var. degrabrata and M. umbellata var. visenia, are also known as “paliasa”.1 In Indonesia, some tribes have used the leaves of the plant for the treatment of various diseases, such as hepatitis, liver cancer (Makassar and Bugis, South Sulawesi), gastritis (Moronene, Southeast Sulawesi), hypertension (Wawoni, Southeast Sulawesi), and high cholesterol levels (Lombok, North Lombok). In Papua New Guinea and the Solomon Islands, the cambium of this plant has been used to treat pneumonia, while the leaves and bark are used to kill ectoparasites, such as lice.2 Preliminary screening of the cytotoxic activity of some selected medicinal plants collected from South Sulawesi, Indonesia, showed that a crude methanol extract of K. hospita displayed potent cytotoxic activity against the A549, MDA-MB-231, MCF-7, KB, and KB-VIN cell lines with an IC50 value of <20 μg/mL. Despite the interesting biological activities mentioned above, not many detailed phytochemical studies have been reported so far on K. hospita. To date, cycloartane triterpenoids, the alkaloids kleinhospitines A−D, and several flavonoids as well as coumarins were isolated from the stem bark3 and leaves.4–7 Cycloartane triterpenoids are found widely in plants and algae and are associated with the biosynthesis of steroidal compounds.8 They exhibit numerous biological activities, including having cytotoxic,9–22 antimicrobial,23,24 anti-HIV,25 antituberculosis,26 hepatoprotective,5,6 and antifeedant effects.19 In most cases, the cyclopropyl moiety on the skeleton has a β-orientation, produced biosynthetically through the chair-boat-chair-boat conformation of 1,3-oxidosqualene. However, cycloartanes with an α-cyclopropyl ring are rare in Nature; examples include kleinhospitines C and D found in K. hospita as a mixture of C-23 epimers,6 as well as four compounds from Neoboutonia melleri.22 As part of our continuing phytochemical research on tropical rainforest plants,27–31 herein are reported the isolation and structure elucidation of a novel cycloartane triterpenoid alkaloid (1) and cycloartane triterpenoids with both α- and β-cyclopropyl rings (2−10) from Indonesian K. hospita. All isolated compounds were evaluated for antiproliferative effects against five human tumor cell lines, including a multidrug-resistant (MDR) subline.
RESULTS AND DISCUSSION
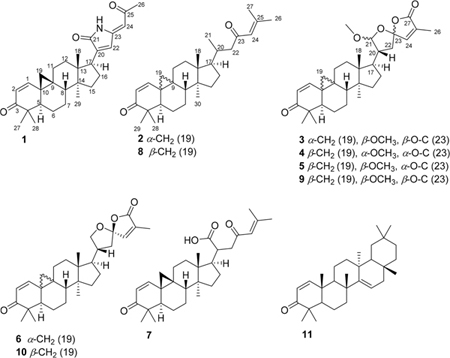
A crude methanol extract of K. hospita was suspended in MeOH−H2O (9:1) and partitioned with n-hexane. The MeOH−H2O layer was evaporated and further partitioned between H2O and EtOAc. The n-hexane and EtOAc layers were separately fractionated by a combination of various chromatographic techniques to provide seven new (1−7) and four known compounds, with the latter identified as cycloartan-1,24-diene-3,23-dione (8), (21S,23R)-2½3,23/27-diepoxy-21-methoxycycloartan-1,24-diene-3,27-dione (9),5 (23R)-21,23:23,27-diepoxycycloarta-1,24-diene-3,27-dione (10), and taraxerone (11),7 by comparison with their previously reported spectroscopic data.
Compound 1 was isolated as a pale yellow, amorphous powder and exhibited a positive HRMS protonated molecular ion peak at m/z 448.2854 [M + H]+ (calcd 448.2852), consistent with the molecular formula C29H38NO3. This compound also showed a positive response to Dragendorff reagent for an alkaloid. The 1H NMR spectrum (Table 1) displayed five methyl signals at δH 0.83 (3H, s), 0.96 (3H, s), 1.01 (3H, s), 1.09 (3H, s), and 2.28 (3H, s). A cyclopropyl ring was characterized by a proton signal at δH 0.68 (1H, d, J = 5.0 Hz) and 1.32 (1H, d, J = 5.0 Hz) conjugated with an α,β-unsaturated ketone at δH 6.79 (1H, d, J = 10.1 Hz) and 5.97 conjugated with a NH group was postulated from the signal at δC 172.5. The complete assignments of the overall structure were based on the HMBC data obtained (Figure 1). The correlations of H3-27/H3-28 with C-4, H-1/H3-27 with C-3, H-2 with C-4/C-10, and H-5 with C-10/C-28 revealed the structure of the A ring, while those of H2-19 with C-1/C-5/C-8, H-1 with C-9, H2-6 with C-10, and H2-7 with C-9 indicated the structures of the 9,19-cyclopropyl and B rings. Similarly, the structures of rings C and D were developed through a combination of HMQC, 1H−1H COSY, and HMBC correlations, such as H3-18 with C-12/C-13/C-14/C-17 and H3-29 with C-8/C-13/C-14/C-15, as shown in Figure 1. The key correlations of δH 9.58 (NH) with δC 144.3 (C-20), 133.1 (C-22) and 147.9 (C-23), as well as δH 6.64 (H-22) with δC 172.5 (CO-21) and 144.3 (C-20), indicated the presence of a side chain containing a 1,5-dihydro-2H-pyrrol-2-one substructure. In addition, the correlations between the proton signal at δH 5.65 (H-24) and δC 133.1 (C-22), 147.9 (C-23), and 199.2 (CO-25) indicated a 2-oxopropylidene group to be linked to C-23. The relative configuration of 1 was established from the NOESY data (Figure 2). Key correlations between H2-19 and H3-27/H-8/H3-18 suggested β-orientations for these four groups. The correlations of H-5 with H3-28/H3-29 and of H-29 with H-17 revealed α-orientations of the protons involved. The absolute configuration of 1 was determined by the electronic circular dichroism (ECD) according to the reported theory of the related cycloartanes6,7 (Figure 3). In the (1H, d, J = 10.1 Hz). Two additional olefinic protons that were observed at δH 6.64 (1H, s) and 5.65 (1H, s) were most likely due to double bonds in the side chain. In addition, a proton signal at δH 9.58 (1H, s) was assigned to a NH group. The 13C NMR (Table 2) and DEPT spectra identified 29 signals, classified as five methyls, seven methylenes, seven methines, seven quaternary carbons, and three carbonyls. The presence of two α,β-unsaturated ketone groups was based on carbon signals observed at δC 205.2 and 199.2, while a keto-carbonyl 200−400 nm region, which is attributable to the configuration of the cyclopropane and ring A, the spectrum of 1 showed a first negative Cotton effect at around 340 nm, consistent with configurations of (9S,10S). Accordingly, the structure of compound 1 (kleinhospitine E) was determined as (9S,10S)-21,23-lactamcycloartan-1,24-diene-3,25-dione. The unusual side chain present, 1,5-dihydro-2H-pyrrol-2-one, has been found in Nature previously and occurred in a 3,4-seco triterpenoid without a cyclopropyl ring from the bark of Entandrophragma congoënse (Meliaceae)32 as well as a spiro γ-lactamlactone unit in kleinhospitines A−D from K. hospita.6
Table 1.
1H NMR Data of Compounds 1−7 (400 MHz, CDCl3)
| Position |
δH (J in Hz) |
||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| 1 | 6.79 d (10.1) | 6.37 d (10.1) | 6.37 d (10.1) | 6.80 d (10.1) | 6.79 d (10.1) | 6.35 d (10.1) | 6.73 d (10.1) |
| 2 | 5.97 d (10.1) | 5.89 d (10.1) | 5.89 d (10.1) | 5.97 d (10.1) | 5.96 d (10.1) | 5.99 d (10.1) | 5.93 d (10.1) |
| 5 | 2.11 dd (4.1, 12.8) | 2.24 m | 2.25 dd (12.8, 7.8) | 2.10 m | 2.15 dd (3.7, 12.8) | 2.25 t (8.9) | 2.13 m |
| 6a | 1.11 m | 1.25 m | 1.24 overlap | 1.11 m | 1.11 m | 1.24 m | 1.01 m |
| 6b | 1.56 m | 1.37 overlap | 1.58 m | 1.59 m | 1.58 m | 1.54 overlap | |
| 7a | 1.58 overlap | 1.30 overlap | 1.25 m overlap | 1.21 m | 1.22 m | 1.38 m | 1.24 m |
| 7b | 1.74 overlap | 1.74 m | 1.58 m | 1.56 m | 1.69 m | 1.57 m | |
| 8 | 2.09 t (8.0) | 1.99 m | 2.06 dd (15.8, 7.8) | 2.05 m | 1.97 m | 2.03 t (7.8) | 2.05 t (8.5) |
| 11a | 1.61 m | 1.23 overlap | 1.69 m | 1.67 m | 1.64 m | 1.68 m | 1.64 m |
| 11b | 1.86 m | 2.10 m | 2.14 m | 1.85 m | 1.99 m | 2.12 m | 1.84 m |
| 12a | 1.25 m | 1.42 m | 1.22 m | 1.21 m | 1.48 m | 1.19 m | 1.22 m |
| 12b | 1.84 m | 1.61 m | 1.68 m overlap | 1.69 m | 1.76 m | 1.65 m | 1.54 m |
| 15a | 1.42 m | 1.21 m | 1.26 m | 1.36 m | 1.32 m | 1.28 m | 1.31 m |
| 15b | 1.54 m | 1.37 m | 1.39 m | 1.43 m | |||
| 16 | 1.83 m | 1.31 overlap | 1.84 m | 1.36 m | 1.62 m | 1.41 m | 1.62 m |
| 1.98 m | 1.88 overlap | 1.67 m | 1.92 m | 1.89 m | 1.94 m | ||
| 17 | 3.17 t (9.4) | 1.58 m | 1.87 m | 2.15 m | 2.26 dd (8.7, 20.2) | 1.81 m | 2.11 m |
| 18 | 0.83 s | 0.95 s | 0.99 s | 1.02 s | 1.03 s | 0.94 s | 1.13 s |
| 19a | 0.68 d (5.0) | 0.79 d (5.0) | 0.79 d (5.0) | 0.71 d (5.0) | 0.77 d (5.0) | 0.78 d (5.0) | 0.71 d (4.9) |
| 19b | 1.32 d (5.0) | 1.38 d (5.0) | 1.39 d (5.0) | 1.35 d (5.0) | 1.31 d (5.0) | 1.38 d (5.0) | 1.31 d (4.9) |
| 20 | 2.02 m | 2.67 m | 2.48−2.57 brd | 2.38 m | 2.74 m | 2.79 m | |
| 21 | 0.90 d (5.9) | 4.88 d (5.0) | 5.03 d (4.6) | 5.08 d (2.3) | a. 3.59 t (8.9) | ||
| b. 4.32 t (8.0) | |||||||
| 22 | 6.64 s | a. 2.09 m | a. 2.03 m | a. 2.03 m | a. 1.97 m | a. 1.82 m | a. 2.67 d (14.2) |
| b. 2.51 d (12.4) | b. 2.25 dd (6.4, 12.8) | b. 2.11 m | b. 2.47 dd (9.2, 13.7) | b. 2.18 m | b. 2.78 t (10.5) | ||
| 24 | 5.65 s | 6.06 s | 6.66 d (1.4) | 6.69 d (1.4) | 6.72 d (1.4) | 6.69 s | 6.05 s |
| 26 | 2.28 s | 1.88 s | 1.94 d (1.4) | 1.94 d (1.4) | 1.93 d (1.4) | 1.93 s | 1.89 s |
| 27 | 0.96 s | 2.15 s | 2.14 s | ||||
| 28 | 1.09 s | 1.06 s | 1.06 s | 1.09 s | 1.11 s | 1.06 s | 1.09 s |
| 29 | 1.01 s | 1.07 s | 1.07 s | 0.97 s | 0.963 s | 1.07 s | 0.95 s |
| 30 | 0.88 s | 0.89 s | 0.89 s | 0.955 s | 0.89 s | 0.88 s | |
| NH: 9.58 s | MeO: 3.38 s | MeO: 3.34 s | MeO: 3.40 s | ||||
Figure 1.
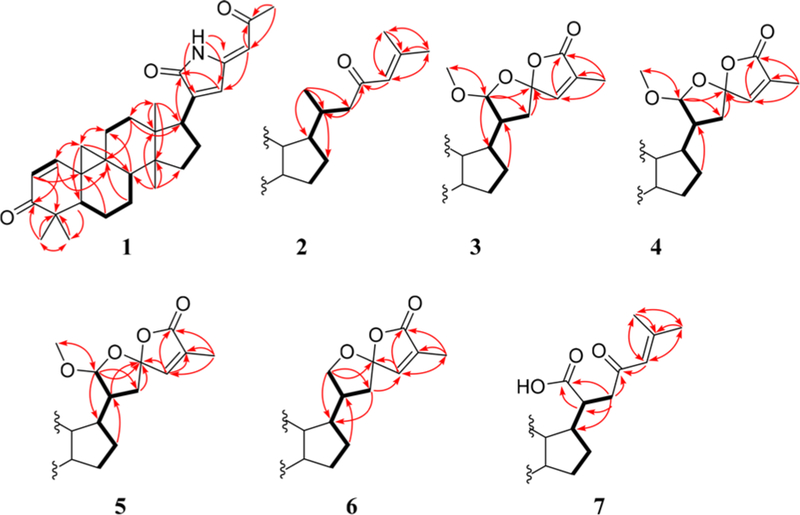
1H−H COSY (bold lines) and key HMBC (arrows) correlations of compounds 1−7.
Figure 2.

Key NOESY correlations (dotted arrows) of compounds 1−7.
Figure 3.
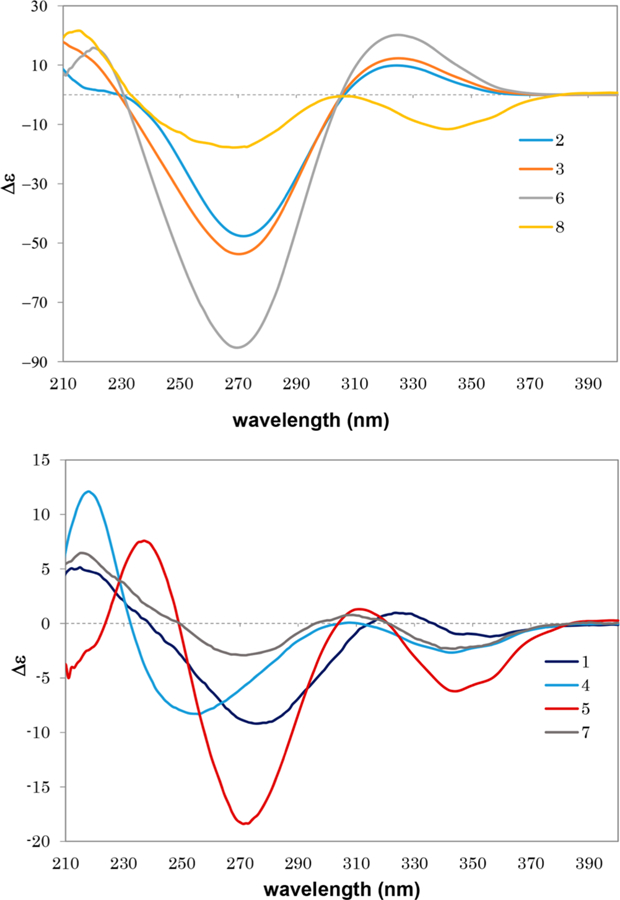
Experimental ECD spectra of compounds 1−8 in MeOH.
Table 2.
13C NMR Data of Compounds 1−7 (100 MHz, CDCl3)
| position | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 153.5 | 156.7 | 156.5 | 153.5 | 153.5 | 156.3 | 153.6 |
| 2 | 126.9 | 124.4 | 124.5 | 126.8 | 126.9 | 124.5 | 126.8 |
| 3 | 205.2 | 205.1 | 205.0 | 205.2 | 205.2 | 204.9 | 205.2 |
| 4 | 45.8 | 45.0 | 45.7 | 45.9 | 45.9 | 45.0 | 45.9 |
| 5 | 43.7 | 40.3 | 40.3 | 43.8 | 44.2 | 40.5 | 43.9 |
| 6 | 18.9 | 20.2 | 20.4 | 19.05 | 19.4 | 20.3 | 19.2 |
| 7 | 22.7 | 23.2 | 23.2 | 22.7 | 23.3 | 23.1 | 22.9 |
| 8 | 42.7 | 40.3 | 40.3 | 42.4 | 43.7 | 40.3 | 42.8 |
| 9 | 24.9 | 33.2 | 33.1 | 25.1 | 24.5 | 32.9 | 24.8 |
| 10 | 30.2 | 27.1 | 27.1 | 30.1 | 29.9 | 27.0 | 29.9 |
| 11 | 27.7 | 29.5 | 29.3 | 27.89 | 27.6 | 29.2 | 27.6 |
| 12 | 30.0 | 32.0 | 30.0 | 30.9 | 31.6 | 31.0 | 29.2 |
| 13 | 47.9 | 45.6 | 45.0 | 45.4 | 45.5 | 45.6 | 45.2 |
| 14 | 49.6 | 49.3 | 48.7 | 48.9 | 49.2 | 48.5 | 49.2 |
| 15 | 34.4 | 32.9 | 32.7 | 34.4 | 34.2 | 33.2 | 33.7 |
| 16 | 25.8 | 28.1 | 26.9 | 26.9 | 27.4 | 27.1 | 27.2 |
| 17 | 43.1 | 51.1 | 48.8 | 44.1 | 46.8 | 49.9 | 48.2 |
| 18 | 18.6 | 14.5 | 15.4 | 18.2 | 17.8 | 15.9 | 16.1 |
| 19 | 27.7 | 29.7 | 29.6 | 27.96 | 29.2 | 29.5 | 28.4 |
| 20 | 144.3 | 33.3 | 45.6 | 44.6 | 46.7 | 40.2 | 42.5 |
| 21 | 172.5 | 19.7 | 111.9 | 106.2 | 110.2 | 74.3 | 179.9 |
| 22 | 133.1 | 51.7 | 42.0 | 38.9 | 37.9 | 42.4 | 46.6 |
| 23 | 147.9 | 201.4 | 110.6 | 110.9 | 111.7 | 111.8 | 198.9 |
| 24 | 103.0 | 124.3 | 144.0 | 146.2 | 146.1 | 144.4 | 123.1 |
| 25 | 199.2 | 154.9 | 133.8 | 132.4 | 132.0 | 133.5 | 156.9 |
| 26 | 31.1 | 27.7 | 10.6 | 10.4 | 10.4 | 10.6 | 27.8 |
| 27 | 21.4 | 20.7 | 171.2 | 171.2 | 171.2 | 171.4 | 20.9 |
| 28 | 19.1 | 21.4 | 21.3 | 21.4 | 21.4 | 21.3 | 21.4 |
| 29 | 18.4 | 20.4 | 20.4 | 19.13 | 19.1 | 20.4 | 19.1 |
| 30 | 17.1 | 17.2 | 18.4 | 18.6 | 17.1 | 18.4 | |
| MeO | 56.2 | 55.0 | 56.1 |
Compound 2 was isolated as a white, amorphous powder. Its molecular formula, C30H45O2, was deduced from HRMS analysis that showed a protonated molecular ion peak at m/z 437.3397 [M + H]+ (calcd 437.3420). The 1H NMR spectrum (Table 1) displayed seven methyl signals at δH 2.15 (3H, s), 1.88 (3H, s), 1.07 (3H, s), 1.06 (3H, s), 0.95 (3H, s), 0.90 (3H, d, J = 5.9 Hz), and 0.88 (3H, s). Two AB doublets at δH 0.79 and 1.38 (each 1H, both J = 5.0 Hz) suggested the presence of cyclopropyl protons at H-19α and H-19β. The 13C NMR spectrum (Table 2) displayed signals for 30 carbons, including seven methyls, eight methylenes, seven methines, and six quaternary carbons as well as two carbonyls as established by DEPT analysis. These data suggested that 2 has a cycloartane skeleton. Two α,β-unsaturated ketones were indicated by three olefinic protons at δH 6.37 (1H, d, J = 10.1 Hz), 5.89 (1H, d, J = 10.1 Hz), and 6.06 (1H, s), two carbonyl carbons at δC 205.1 and 201.4, and four olefinic carbons at δC 156.7, 154.9, 124.4, and 124.3. The presence of a linear side chain with oxygen at C-23 was assigned from the HMBC correlations of H-20 with C-16, H3-21 with C-17/C-20/C-22, H2-22 with C-20/C-21/C-23, H-24 with C-23/C-26/C-27, and H3-26/H3-27 with C-24/C-25 (Figure 1). While the spectroscopic data of 2 and cycloartan-1,24-diene-3,23-dione (8)5 were similar, the differences in the 13C NMR signals of C-1, C-5, C-8, C-9, and C-10 suggested that compound 2 is an isomer of 8. Furthermore, analysis of the NOESY spectrum of 2 demonstrated several key correlations: H2-19 with H-5/H3-30, H3-28 with H-5, H3-18 with H-8, and H-17 with H3-30/H3-21. These data suggested strongly that H2-19, H3-28, H-5, H-17, H-21, and H3-30 have α-orientations, while H-8, H3-29, and H3-18 have β-configurations (Figure 2). The different optical rotations, (MeOH, c 0.01) and −22.6 (MeOH, c 0.35), for 2 and 8, respectively, also supported the conclusion that compounds 2 and 8 are stereoisomers. This was also supported by the ECD spectrum of 2. According to the above-mentioned theory,6,7 an opposite Cotton effect of 2 with that of 8 in the 310−400 nm region indicated a difference of absolute configuration for the cyclopropane ring. The similar negative Cotton effects at around 270 nm, attributed to the side chain with an α,β-unsaturated ketone, suggested the configuration at C-20 could be the same for both compounds 2 and 8. In addition, the positive Cotton effect of 2 at around 325 nm was identical to those of kleinhospitines C and D in having an α-cyclopropyl ring. Based on the above observations, compound 2 was determined as (9R,10R)-cycloartan-1,24-diene-3,23-dione.
Compound 3 was isolated as a white, amorphous powder, and its HRMS showed a protonated molecular ion at m/z 495.3101 [M + H]+ (calcd 495.3110), corresponding to the molecular formula C31H43O5. The 1H and 13C NMR spectra (Tables 1 and 2) for positions 1 to 19 of 3 were very close to those of 2, which suggested strongly a cycloartane skeleton with an α,β-unsaturated ketone in ring A. The HMQC, COSY, and HMBC spectra of 2 and 3 also agreed well; thus, the two compounds could be proposed as having the same skeleton and differing only in their side-chain structures. With signals for an olefin at δH 6.66 (1H, d, J = 1.4 Hz) and δC 144.0, a carbonyl at δC 171.2, two acetal carbons at δC 110.6 and 111.9, a methoxy at δH 3.38, and a methyl at δH 1.94 (3H, d, J = 1.4 Hz), compound 3 was assigned with a spiro α,β-unsaturated γ-lactone containing methyl and methoxy groups at C-25 and C-21, respectively; this conclusion was supported by appropriate HMBC correlations (Figure 1). The same side chain is also found in the known cycloartane 9,5 and all spectroscopic data for the spiro lactone moieties in 3 and 9 were found to be almost identical (Table 2). The differences in the C-1, C-5, C-8, C-9, and C-10 carbon signals suggested that 3 and 9 have different orientations of the 9,10-cyclopropyl ring. In 3, the NOESY correlations of H2-19 with H-5/H3-30, H3-28 with H-5, and H-17 with H3-30/H-21 implied that these protons are α-oriented, while the correlations of H3-18 with H-8/H-20 indicated β-orientations (Figure 2). The different specific rotations, +158.6 (CHCl3, c 0.16) for 3 and −4.1 (CHCl3, c 0.16) for 9, also indicated that the two compounds are diastereomers. The absolute configuration of 3 was confirmed by the ECD spectrum, which showed a positive first Cotton effect at around 325 nm as typical of an α-cyclopropyl ring.6,7 Based on the total evidence obtained, the structure of 3 was proposed as (9R,10R,21S,23R)-2½3,23/ 27-diepoxy-21-methoxycycloartan-1,24-diene-3,27-dione.
Compounds 4 and 5 were isolated as white, amorphous powders, and both gave the same molecular formula, C31H43O5, based on positive HRMS protonated molecular ion peaks at m/z 495.3101 and 495.3114 [M + H]+ (calcd 495.3110), respectively. The 1H NMR and 13C NMR spectra of the two compounds indicated a cycloartane skeleton (Tables 1 and 2). The 13C NMR data for C-1, C-5, C-8, and C-9, as well as C-10 in 4 and 5 were different from those of 3, most likely due to a different configuration at the cyclopropyl ring. In addition, differences were also observed at C-17, C-22, and C-24. In the 13C NMR spectrum of 4, a slightly high-field shifted C-21 signal suggested an α-orientation for the attached methoxy group. Moreover, an essential correlation between H-21 and H-20 appeared only in the NOESY spectrum of 4, confirming a β-orientation for both protons and an α-orientation for the methoxy group on C-21 (Figure 2). Furthermore, the key correlations of H-22β with H-24 and H-20 indicated that the oxygen at C-23 in the lactone ring is oriented to the α-position in both 4 and 5, compared with its β-orientation in 3. Compounds 4 and 5 had opposite specific rotations, +77.4 (CHCl3, c 0.16) and −26.6 (CHCl3, c 0.16), respectively. The ECD spectrum of both 4 and 5 showed first negative Cotton effects around 340 nm, confirming the ring A and β-cyclopropyl ring. Subsequently, in the 200−250 nm range, positive Cotton effects around 220 and 235 nm for 4 and 5, respectively, suggested a 23S configuration for both compounds.6,7 Based on the available evidence, the structures of 4 and 5 were elucidated as (9S,10S,21R,23S)-2½3,23/27-diepoxy-21-methoxycyloartan-1,24-diene-3,27-dione and (9S,10S,21S,23S)-2½3,23/27-diepoxy-21-methoxycyloartan-1,24-diene-3,27-dione, respectively.
Compound 6 was isolated as a white, amorphous powder and exhibited a positive HRMS protonated molecular ion peak at m/z 465.2996 [M + H]+ (calcd 465.3005), consistent with the molecular formula C30H41O4. Its 1H and 13C NMR spectra were close to those of the known cycloartane triterpenoid (9S,10S,23R)-21,23:23,27-diepoxycycloarta-1,24-diene-3,27-dione (10),7 except for the signals around the cyclopropyl ring Tables 1 and 2). These discrepancies indicated different orientations of the C-9,10-cyclopropane ring. The NOESY correlations of H2-19 with H-5/H3-30, H-5 with H3-28, H3-30 with H-17, and H3-18 with H-8/H-20 for 6 suggested an α-oriented cyclopropane unit, while compound 10 contains a β-oriented cyclopropane (Figure 2). The two stereoisomers showed different optical rotations, −205.9 (CHCl3, c 0.13) for 6 and −92.4 (CHCl3, c 0.16) for 10, respectively. Therefore, the structure of 6 was assigned as (9R,10R, 23R)-21,23:23,27-diepoxycycloarta-1,24-diene-3,27-dione.
Compound 7 was isolated as a white, amorphous powder. Its molecular formula was determined as C30H43O4 based on the HRMS protonated molecular ion peak at m/z 467.3170 [M + H]+ (calcd 467.3161). An analysis of all NMR spectroscopic data obtained, including the 1H, 13C, COSY, HMBC, and NOESY spectra, indicated that this compound has a cycloartane skeleton with a β-oriented cyclopropane ring. The presence of signals for two methyl groups (δH 1.89/δC 27.8 and δH 2.14/δC 20.9), an olefin (δH 6.05/δC 123.1 and δC 156.9), and a carbonyl (δC 198.9) indicated a 3,3-dimethylacryl unit in the side chain at C-17, as also found in 2 and 8 and confirmed by consistent HMBC correlations (Figure 1). However, a low-field carbon signal at δC 179.9 suggested the presence of a carboxylic acid group attached to C-20 in the side chain of 7 rather than the methyl group present in 2 and 8. From all the data obtained, including COSY, HMBC, and NOESY spectra (Figures 1 and 2), the structure of the side chain was fully determined as 6-methyl-4-oxohept-5-enoic acid, attached at its C-20 position to the cycloartane skeleton. The 1H and 13C NMR signal patterns of C-20 to C-27 were very close to those of tirucallane triterpenes with the same side chain.32–34 Although the configuration at C-20 was not clear, the structure of 7 was concluded to be cycloarta-1,24-diene-3,23-dion-21-oic acid.
Compounds 1−11 were evaluated for antiproliferative activity against five human tumor cell lines, A549, MDA-MB-231, MCF-7, KB, and a KB-subline, KB-VIN, showing the MDR phenotype with overexpression of P-glycoprotein (P-gp) (Table 3). Most compounds exhibited IC50 values greater than 10 μM against all tested cell lines. However, all compounds evaluated were either equipotent or more potent against the MDR versus the non-MDR tumor cell line. The orientation of the cyclopropyl ring affected the resultant activity since compound 9, with a β-cyclopropyl unit, demonstrated more potent antiproliferative activity against the MDR subline than 3, with an α-cyclopropyl ring. When the configuration of the spiro carbon at C-23 was reversed (cf. 9 versus 5), the KB-VIN potency dropped significantly. However, when the orientation of the methoxy group on C-21 changed from β in 5 to α in 4, the KB-VIN potency increased. Compounds 6 and 10 without a methoxy group on C-21 were among the less potent compounds. In turn, compound 1, with a γ-lactam ring in the side chain, was among the most potent compounds, with antiproliferative IC50 values of 6.9 to 10 μM, but it was essentially equipotent against the KB and KB-VIN cancer cell lines. Based on these results, the configuration of the cyclopropyl ring and the type of side chain substitution, including the orientation of the methoxy group and spiro carbon, could be seen to influence the antiproliferative activities of the cycloartane triterpenoids isolated.
Table 3.
Antiproliferative Data of Isolated Compounds
| compound | cell linea (IC50
μM)b |
||||
|---|---|---|---|---|---|
| A549 | MDA-MB-231 | MCF-7 | KB | KB-VIN | |
| 1 | 9.4 ± 0.4 | 10 | >10 | 8.0 ± 0.5 | 6.9 ± 0.3 |
| 2 | >10 | >10 | >10 | >10 | 8.4 ± 0.1 |
| 3 | >10 | >10 | >10 | >10 | >10 |
| 4 | 9.1 ± 0.5 | >10 | >10 | >10 | 5.7 ± 0.1 |
| 5 | >10 | >10 | >10 | >10 | >10 |
| 6 | >10 | >10 | >10 | >10 | >10 |
| 7 | >10 | >10 | >10 | >10 | >10 |
| 8 | >10 | >10 | >10 | >10 | >10 |
| 9 | 8.9 ± 0.2 | >10 | 10 | >10 | 6.1 ± 0.5 |
| 10 | 8.2 ± 0.2 | >10 | >10 | >10 | >10 |
| 11 | >10 | >10 | >10 | >10 | >10 |
| PXL (nM)c | 6.3 ± 0.3 | 8.1 ± 0.2 | 11.2 ± 0.3 | 6.8 ± 0.3 | 2786 ± 6.2 |
A549 (lung carcinoma), MDA-MB-231 (triple-negative breast cancer), MCF-7 (estrogen receptor-positive and HER2-negative breast cancer), KB, KB-VIN (P-gp-overexpressing MDR subline of KB).
Antiproliferative activity expressed as IC50 values for each cell line, the concentration of compound that caused 50% reduction relative to untreated cells determined by the SRB assay.
The IC50 data of paclitaxel (PXL) are expressed in nM.
All new compounds except 7 were also tested for anti-HIV activity in MT4 cells, with azidothymidine (AZT) as a control. Compound 3 demonstrated some activity, with an IC50 value of 2.4 μM (SI = 5.3), while 6 showed submicromolar inhibitory activity, with an IC50 value of 0.8 μM (SI = 5.8) (Table 4). Although based on the limited assay results available at present, an α-oriented cyclopropane and spiro-furan ring system are likely important for such antiviral activity.
Table 4.
Anti-HIV Data of Compounds 1−6
| compound | IC50a (μM) (MT4 cells) | CC50b (μM) (MT4 cells) | selectivity index (SI)c | statusd |
|---|---|---|---|---|
| 1 | − | − | − | I |
| 2 | − | − | − | I |
| 3 | 2.4 ± 0.1 | 12.6 ± 0.4 | 5.3 | A |
| 4 | − | − | − | I |
| 5 | − | − | − | I |
| 6 | 0.8 ± 0.1 | 4.6 ± 0.1 | 5.8 | A |
| AZTe | 0.014 ± 0.01 | >3.74 | >267.1 | A |
IC50 = 50% HIV inhibition concentration.
CC50 = 50% antiproliferative concentration.
Selectivity index (SI) = CC50/IC50; − = no selective anti-HIV activity (SI < 5).
A = active; I = inactive.
AZT: azidothymidine.
EXPERIMENTAL SECTION
General Experimental Procedures.
A JASCO P-2200 digital polarimeter was used for the determination of optical rotations. ECD spectra were detected on a JASCO J-820 spectrometer. NMR spectra were taken on JEOL JMN-ECA600 and JMN-ECS400 NMR spectrometers. Tetramethylsilane was used as an internal standard, and chemical shifts are stated as δ values. HRMS data were obtained on a JMS-SX102A (FAB) or JMS-T100TD (DART) mass spectrometer. Analytical and preparative TLC were conducted on RP-18F254 plates and precoated silica gel 60F254 (0.50 or 0.25 mm thickness; Merck). Column chromatography was performed with silica gel 60 N (63–210 μm, spherical, neutral, Kanto Chemical). Silica gel 60 RP-18 F254S (0.25 mm, Merck) was applied for reversed-phase and 60 F254 (1 mm) for normal-phase preparative TLC.
Plant Material.
Kleinhovia hospita leaves were collected at Banyuanyara Village, Sanrobone Subdistrict, Takalar District, South Sulawesi Province, Indonesia, in May 2015, and identified by Joko Santoso, Department of Biology Pharmacy, Faculty of Pharmacy, Gadjah Mada University. A voucher specimen has been deposited in the Pharmacognosy-Phytochemistry Laboratory, Hasanuddin University (AR-FFUH-03).
Extraction and Isolation.
Air-dried and powdered K. hospital leaves (300 g) were extracted three times with MeOH (2.5 L) at room temperature for 48 h. The extract was concentrated in vacuo to yield 30.3 g of a semisolid, from which 26 g was further partitioned between n-hexane and MeOH−H2O (9:1) to afford an n-hexane extract (6.97 g) and a MeOH−H2O layer. The MeOH−H2O layer was evaporated and partitioned with EtOAc and H2O to produce EtOAc (5.57 g) and water-soluble (11.59 g) extracts. The n-hexane extract (6.5 g) was subjected to medium-pressure liquid chromatography (MPLC) on silica gel eluted with n-hexane, n-hexane−EtOAc (90:10, 70:30, 50:50, v/v), EtOAc, and MeOH to afford 195 fractions of 50 mL each. Those fractions with similar TLC profiles were combined to give 10 further fractions (FA−FJ). Fraction FB (750 mg) was subjected to column chromatography (CC) on silica gel eluted with n-hexane−EtOAc (20:1, v/v) to provide 22 subfractions of 50 mL each, which were combined into five subfractions (FB-1− FB-5) based on their similar TLC profiles. Subfraction FB-3 was separated further by CC on silica gel with n-hexane−EtOAc (90:1, v/ v) to furnish 22 additional subfractions of 20 mL each. Subfractions with similar TLC profiles were combined to give three subfractions, FB-3A−FB-3C. Compound 11 (10.6 mg) was isolated from subfraction FB-3A by recrystallization from n-hexane. Fraction D (FD, 670 mg) was further separated by CC on silica gel using n-hexane−acetone (15:1 and 10:1, v/v) for elution, to yield 29 subfractions of 20 mL each, which were combined into four subfractions (FD-1−FD-4) based on the similarity of their TLC profiles. Subfraction FD-2 (14.5 mg) was further separated by preparative reversed-phase TLC with acetone−MeOH (1:1, v/v) to yield 8 (3.9 mg). Repeated chromatography of subfraction FD-3 (169.4 mg) with CC on silica gel eluted with n-hexane−acetone (15:1, v/v) afforded 23 subfractions of 20 mL each. Subfractions with similar TLC profiles were combined to give four additional subfractions (FD-3A−FD-3D). Compounds 8 (2.4 mg) and 2 (2.4 mg) were isolated from subfraction FD-3C (55.7) by preparative normal-phase TLC with n-hexane−acetone (15:1). Fraction FF (437.7 mg) was fractionated by MPLC with MeOH−H2O (4:1, v/v), MeOH, and acetone, to afford 30 subfractions of 20 mL each. Combination based on their similar TLC profiles resulted in seven subfractions (FF-1−FF-7). Subfraction FF-3 was further separated by preparative normal-phase TLC with n-hexane−EtOAc (3:1, v/v) to yield five subfractions, FF-3A−FF-3E. Repeated chromatography with preparative normal-phase TLC of subfraction FF-3B (21.6 mg) using n-hexane−CHCl3 (1:4, v/v) afforded 4 (5.7 mg) and 5 (4.5 mg). Subfraction G (320 mg) was separated using silica gel reversed-phase MPLC eluted with MeOH−H2O (4:1, v/v), MeOH, and acetone to give 39 subfractions. Eight combined subfractions (FG-1−FG-8) were produced based on the similarity of their TLC profiles. Subfraction FG-2 (15.8 mg) was purified by preparative normal-phase TLC with the mobile phase n-hexane−EtOAc (3:1, v/v) to yield 3 (1.3 mg). Subfraction FG-3 (81.5 mg) was added to MeOH to yield soluble and insoluble portions of 19.2 mg and 60.9 mg, respectively. The insoluble MeOH portion was purified by preparative normal-phase TLC with the solvent system n-hexane−CHCl3 (1:5, v/v) to yield 9 (11.4) and 10 (27.4 mg).
Compounds 1, 6, and 7 were isolated from an EtOAc extract of K. hospita. The EtOAc extract (4.60 g) was fractionated by MPLC on normal-phase silica gel eluting with n-hexane−EtOAc (5:1, 1:1, 1:5, v/v), EtOAc, and MeOH, to afford 52 fractions of 20 mL each. They were combined into nine fractions (FE-1−FE-9) based on the similarity of their TLC profiles. Repeated chromatography of subfraction FE-4 (890.7 mg) with silica gel reversed-phase MPLC eluted with MeOH−H2O (4:1, v/v), MeOH, and acetone yielded 48 subfractions, which were combined into eight subfractions (FE-4A− FE-4H) based on the similarity of their TLC profiles. Subfraction FE-4B (448.5 mg) was subjected to CC on normal-phase silica gel with the mobile phases n-hexane−EtOAc (2:1, 1:1, v/v), EtOAc, and MeOH to afford 62 subfractions, which were combined into eight subfractions (FE-4B-1−FE-4B-8) based on their TLC profiles. Subfraction FE-4B-2 (13.1 mg) was purified by preparative normalphase TLC with the solvent system n-hexane−CHCl3 (1:10, v/v) to yield 10 (1.5 mg) and 6 (3.9 mg). Subfraction FE-4B-3 (37.7 mg) was further separated by semipreparative HPLC RP-18 chromatography, eluting with MeOH−H2O (4:1, v/v), to afford five subfractions (FE-4B-3A−FE-4B-3E). Subfraction FE-4B-3B (4.7 mg) was subjected to preparative normal-phase TLC eluting with n-hexane−CHCl3 (1:20, v/v) to afford 1 (4.1 mg). The same solvent system and repeated preparative normal-phase TLC of subfractions FE-4B-3C and FE-4B-3D afforded 3 (2.2 mg) and 6 (3.0 mg), respectively. Subfraction FE-4B-5 (62.6 mg) was separated further by column chromatography on normal-phase silica gel with n-hexane−EtOAc (2;1, 1:2 v/v), EtOAc, and MeOH, to afford six subfractions (FE-4B-5A−FE-4B-5F). Compound 7 (5.1 mg) was isolated from subfraction FE-4B-5C by semipreparative HPLC RP-18 with CH3CN−MeOH−H2O (1:6:0.5).
Compound 1: pale yellow, amorphous powder; +88.6 (CHCl3, c 0.10); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 448.2 [M + H]+; HRMS m/z 448.2854 [M + H]+ (calcd for C29H38NO3, 448.2852).
Compound 2: white, amorphous powder; −116.2 (MeOH, c 0.01); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 437.4 [M + H]+; HRMS m/z 437.3397 [M + H]+ (calcd for C30H45O2, 437.3420).
Compound 3: white, amorphous powder; +158.6 (CHCl3, c 0.16); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 495.3 [M + H]+; HRMS m/z 495.3101 [M + H]+ (calcd for C31H43O5, 495.3110).
Compound 4: white, amorphous powder; +77.4 (CHCl3, c 0.16); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 495.2 [M + H]+; HRMS m/z 495.3101 [M + H]+ (calcd for C31H43O5, 495.3110).
Compound 5: white, amorphous powder; −26.6 (CHCl3, c 0.16); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 495.2 [M + H]+; HRMS m/z 495.3114 [M + H]+ (calcd for C31H43O5, 495.3110).
Compound 6: white, amorphous powder; −205.9 (CHCl3, c 0.13); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 465.3 [M + H]+; HRMS m/z 465.2996 [M + H]+ (calcd for C30H41O4, 465.3005).
Compound 7: white, amorphous powder; −13.9 (CHCl3, c 0.25); 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz), see Tables 1 and 2; MS m/z 467.3 [M + H]+; HRMS m/z 467.3170 [M + H]+ (calcd for C30H43O4, 467.3161).
Antiproliferative Activity Assay.
The antiproliferative activity assay using five human tumor cell lines, namely, A549 (lung carcinoma), MDA-MB-231 (estrogen receptor-negative, progesterone receptor-negative, HER2-negative breast cancer), MCF-7 (estrogen receptor-positive, HER2-negative breast cancer), KB (originally isolated from epidermoid carcinoma of the nasopharynx), and KB-VIN (vincristine (VIN)-resistant KB subline showing MDR phenotype by overexpressing P-gp), was performed as described previously.27 We confirmed that the KB and KB-VIN cell lines were identical to AV-3 (ATCC number, CCL-21) as a HeLa (cervical carcinoma) contaminant by short tandem repeat profiling. All cell lines were obtained from the Lineberger Comprehensive Cancer Center (UNC-CH) or from ATCC, except KB-VIN, which was obtained from Professor Y.-C. Cheng (Yale University). Briefly, 4000−12 000 freshly trypsinized cells were seeded in 96-well microtiter plates with each test compound prepared in DMSO. After 72 h of culture, the treated cells were fixed in 10% trichloroacetic acid followed by staining with 0.04% sulforhodamine B. The highest concentration of vehicle (DMSO) used in our assay was 0.1% v/v, which showed no effect on cell growth. Paclitaxel (Sigma-Aldrich, purity >95%) was used as a reference compound. The mean IC50 is the concentration of compound that inhibited cell growth by 50% compared with a vehicle control under the experimental conditions used and is the average from at least three independent experiments with duplicate samples (n = 6). All values listed in Table 3 were calculated by Excel (Microsoft).
Multicycle Viral Replication in a MT4 Cellular Assay.34
MT4 cells (1 × 105 cells/mL) in 96-well plates were infected by HIV-1 NL4–3 Nanoluc-sec at a dose of 50 TCID50/well in the presence of each test compound at various concentrations. On day 3 postinfection, supernatants were collected and assayed for luciferase activity using the Nano-Glo Luciferase Assay System (Promega). The antiviral potency is defined as the compound concentration that reduces the luciferase activity by 50% (EC50).
Cytotoxicity Assay (MT4 Cells).35
The cytotoxicity of the compound against MT4 cells was assessed by a CytoTox-Glo cytotoxicity assay (Promega). MT4 cells were cultured for 3 days in the presence of various concentrations of each test compound, and the 50% cytotoxic concentration (CC50) causing a 50% reduction of cell viability was determined by following the manufacturer’s protocol.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by JSPS KAKENHI Grant Number 16H05811 awarded to K.N.G. and Indonesia Endowment Fund for Education (LPDP) and Ministry of Research, Technology and Higher Education of the Republic of Indonesia (BUDI-LN Scholarship) awarded to A.R. Partial support from NIH grants CA177584 and AI033066 awarded to K.H.L., as well as the Eshelman Institute for Innovation (Chapel Hill, North Carolina) awarded to M.G., is also acknowledged.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.8b00211.
1D- and 2D-NMR spectra for 1–7 and 1D-NMR spectra for 8–11 (PDF)
REFERENCES
- (1).Paramita S Jurnal Tumbuhan Obat Indonesia 2016, 9, 29–36. [Google Scholar]
- (2).Latiff A Plant Resources of South-East Asia; Faridah HI, van der Maesen LJG, Eds.; Prosea Foundation: Bogor, Indonesia, 1997; Vol. 11, pp 166–167. [Google Scholar]
- (3).Soekamto NH; Noor A; Dini I; Garson RA Proceedings of the International Seminar on Chemistry; Jakarta, Indonesia, October 30−31, 2008; pp 231–234. [Google Scholar]
- (4).Arung ET; Kusuma IW; Kim YU; Shimizu K; Kondo R J. Wood Sci 2012, 58, 77–80. [Google Scholar]
- (5).Gan LS; Ren G; Mo JX; Zhang XY; Yao W; Zhou CX J. Nat. Prod 2009, 72, 1102–1105. [DOI] [PubMed] [Google Scholar]
- (6).Zhou CX; Zou L; Gan LS; Cao YL Org. Lett 2013, 15, 2734–2737. [DOI] [PubMed] [Google Scholar]
- (7).Mo JX; Baib Y; Liu B; Zhou CX; Zou L; Gan LS Helv. Chim. Acta 2014, 97, 887–894. [Google Scholar]
- (8).Boar RB; Romer CR Phytochemistry 1975, 14, 1143–1146. [Google Scholar]
- (9).Chen JY; Li PL; Tang XL; Wang SJ; Jiang YT; Shen L; Xu BM; Shao YL; Li GQ J. Nat. Prod 2014, 77, 1997–2005. [DOI] [PubMed] [Google Scholar]
- (10).Gao J; Huang F; Zhang J; Zhu G; Yang M; Xiao P J. Nat. Prod 2006, 69, 1500–1502. [DOI] [PubMed] [Google Scholar]
- (11).Grougnet R; Magiatis P; Mitaku S; Loizou S; Moutsatsou P; Terzis A; Cabalion P; Tillequin F; Michel S J. Nat. Prod 2006, 69, 1711–1714. [DOI] [PubMed] [Google Scholar]
- (12).Shen T; Yuan HQ; Wan WZ; Wang XL; Wang XN; Ji M; Lou HX J. Nat. Prod 2008, 71, 81–86. [DOI] [PubMed] [Google Scholar]
- (13).Lim SH; Mahmood K; Komiyama K; Kam TS J. Nat. Prod 2008, 71, 1104–1106. [DOI] [PubMed] [Google Scholar]
- (14).Choudhary MI; Jan S; Abbaskhan A; Musharraf SG; Samreen SSA; Atta-ur-Rahman. J. Nat. Prod 2008, 71, 1557–1560. [DOI] [PubMed] [Google Scholar]
- (15).Banskota AH; Tezuka Y; Tran KQ; Tanaka K; Saiki I; Kadota S J. Nat. Prod 2000, 63, 57–64. [DOI] [PubMed] [Google Scholar]
- (16).Kongkum N; Tuchinda P; Pohmakotr M; Reutrakul V; Piyachaturawat P; Jariyawat S; Suksen K; Akkarawongsapat R; Kasisit J; Napaswad C J. Nat. Prod 2013, 76, 530–537. [DOI] [PubMed] [Google Scholar]
- (17).Nian Y; Zhang YL; Chen JC; Lu L; Qiu MH; Qing C J. Nat. Prod 2010, 73, 93–98. [DOI] [PubMed] [Google Scholar]
- (18).Prawat U; Chairerk O; Lenthas R; Salae AW; Tuntiwachwuttikul P Phytochem. Lett 2013, 6, 286–290. [Google Scholar]
- (19).Reddy SD; Siva B; Phani Babu VS; Vijaya M; Nayak VL; Mandal R; Tiwari AK; Shashikala P; Babu KS Eur. J. Med. Chem 2017, 136, 74–84. [DOI] [PubMed] [Google Scholar]
- (20).Toume K; Nakazawa T; Ohtsuki T; Arai MA; Koyano T; Kowithayakorn T; Ishibashi M J. Nat. Prod 2011, 74, 249–255. [DOI] [PubMed] [Google Scholar]
- (21).Kikuchi T; Akihisa T; Tokuda H; Ukiya M; Watanabe K; Nishino H J. Nat. Prod 2007, 70, 918–922. [DOI] [PubMed] [Google Scholar]
- (22).Long C; Beck J; Cantagrel F; Marcourt L; Vendier L; David B; Plisson F; Derguini F; Vandenberghe I; Aussagues Y; Ausseil F; Lavaud C; Sautel F; Massiot G J. Nat. Prod 2012, 75, 34–47. [DOI] [PubMed] [Google Scholar]
- (23).Gutierrez-Lugo MT; Singh MP; Maiese WM; Timmermann BN J. Nat. Prod 2002, 65, 872–875. [DOI] [PubMed] [Google Scholar]
- (24).Gromova AS; Lutsky VI; Li D; Wood SG; Owen NL; Semenov AA; Grant DM J. Nat. Prod 2000, 63, 911–914. [DOI] [PubMed] [Google Scholar]
- (25).Zhang HJ; Tan GT; Hoang VD; Hung NV; Cuong NM; Soejarto DD; Pezzuto JM; Fong HH J. Nat. Prod 2003, 66, 263–268. [DOI] [PubMed] [Google Scholar]
- (26).Truong NB; Pham CV; Doan HT; Nguyen HV; Nguyen CM; Nguyen HT; Zhang HJ; Fong HH; Franzblau SG; Soejarto DD; Chau MV J. Nat. Prod 2011, 74, 1318–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Suzuki A; Saito Y; Fukuyoshi S; Goto M; Miyake K; Newman DJ; O’Keefe BR; Lee KH; Nakagawa-Goto K J. Nat. Prod 2017, 80, 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Suzuki A; Miyake K; Saito Y; Rasyid FA; Tokuda H; Takeuchi M; Suzuki N; Ichiishi E; Fujie T; Goto M; Sasaki S; Nakagawa-Goto K Planta Med 2017, 83, 300–305. [DOI] [PubMed] [Google Scholar]
- (29).Suzuki Y; Saito Y; Goto M; Newman DJ; O’Keefe BR; Lee KH; Nakagawa-Goto K J. Nat. Prod 2017, 80, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rasyid FA; Fukuyoshi S; Ando H; Miyake K; Atsumi T; Fujie T; Saito Y; Goto M; Shinya T; Mikage M; Sasaki Y; Nakagawa-Goto K Chem. Pharm. Bull 2017, 65, 116–120. [DOI] [PubMed] [Google Scholar]
- (31).Miyake K; Suzuki A; Morita C; Goto M; Newman DJ; O’Keefe BR; Morris-Natschke SL; Lee KH; Nakagawa-Goto K J. Nat. Prod 2016, 79, 2883–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Happi GM; Kouam SF; Talontsi FM; Zühlke S; Lamshöft M; Spiteller M Fitoterapia 2015, 102, 35–40. [DOI] [PubMed] [Google Scholar]
- (33).Orisadipe AT; Adesomoju AA; D’Ambrosio M; Guerriero A; Okogun JI Phytochemistry 2005, 66, 2324–2328. [DOI] [PubMed] [Google Scholar]
- (34).Mireku EA; Kusari S; Eckelmann D; Mensah AY; Talontsi FM; Spiteller M Fitoterapia 2015, 106, 84–91. [DOI] [PubMed] [Google Scholar]
- (35).Dang Z; Zhu L; Lai W; Bogerd H; Lee KH; Huang L; Chen CH ACS Med. Chem. Lett 2016, 7, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.