

Abstract
Background
Burning mouth syndrome (BMS) is a term used for oral mucosal pain (burning pain or discomfort in the tongue, lips or entire oral cavity) without identifiable cause. General population prevalence varies from 0.1% to 3.9%. Many BMS patients indicate anxiety, depression, personality disorders and impaired quality of life (QoL). This review updates the previous versions published in 2000 and 2005.
Objectives
To determine the effectiveness and safety of any intervention versus placebo for symptom relief and changes in QoL, taste, and feeling of dryness in people with BMS.
Search methods
Cochrane Oral Health's Information Specialist searched the following databases: Cochrane Oral Health's Trials Register (to 31 December 2015), the Cochrane Central Register of Controlled Trials (CENTRAL; 2015, Issue 11) in the Cochrane Library (searched 31 December 2015), MEDLINE Ovid (1946 to 31 December 2015), and Embase Ovid (1980 to 31 December 2015). We searched ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform for ongoing trials. We placed no restrictions on the language or date of publication when searching the electronic databases
Selection criteria
Randomised controlled trials (RCTs) comparing any treatment against placebo in people with BMS. The primary outcomes were symptom relief (pain/burning) and change in QoL. Secondary outcomes included change in taste, feeling of dryness, and adverse effects.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. Outcome data were analysed as short‐term (up to three months) or long‐term (three to six months).
Main results
We included 23 RCTs (1121 analysed participants; 83% female). Interventions were categorised as: antidepressants and antipsychotics, anticonvulsants, benzodiazepines, cholinergics, dietary supplements, electromagnetic radiation, physical barriers, psychological therapies, and topical treatments.
Only one RCT was assessed at low risk of bias overall, four RCTs' risk of bias was unclear, and 18 studies were at high risk of bias. Overall quality of the evidence for effectiveness was very low for all interventions and all outcomes.
Twenty‐one RCTs assessed short‐term symptom relief. There is very low‐quality evidence of benefit from electromagnetic radiation (one RCT, 58 participants), topical benzodiazepines (two RCTs, 111 participants), physical barriers (one RCT, 50 participants), and anticonvulsants (one RCT, 100 participants). We found insufficient/contradictory evidence regarding the effectiveness of antidepressants, cholinergics, systemic benzodiazepines, dietary supplements or topical treatments. No RCT assessing psychological therapies evaluated short‐term symptom relief.
Four studies assessed long‐term symptom relief. There is very low‐quality evidence of a benefit from psychological therapies (one RCT, 30 participants), capsaicin oral rinse (topical treatment) (one RCT, 18 participants), and topical benzodiazepines (one RCT, 66 participants). We found no evidence of a difference for dietary supplements or lactoperoxidase oral rinse. No studies assessing antidepressants, anticonvulsants, cholinergics, electromagnetic radiation or physical barriers evaluated long‐term symptom relief.
Short‐term change in QoL was assessed by seven studies (none long‐term).The quality of evidence was very low. A benefit was found for electromagnetic radiation (one RCT, 58 participants), however findings were inconclusive for antidepressants, benzodiazepines, dietary supplements and physical barriers.
Secondary outcomes (change in taste and feeling of dryness) were only assessed short‐term, and the findings for both were also inconclusive.
With regard to adverse effects, there is very low‐quality evidence that antidepressants increase dizziness and drowsiness (one RCT, 37 participants), and that alpha lipoic acid increased headache (two RCTs, 118 participants) and gastrointestinal complaints (3 RCTs, 138 participants). We found insufficient/contradictory evidence regarding adverse events for anticonvulsants or benzodiazepines. Adverse events were poorly reported or unreported for cholinergics, electromagnetic radiation, and psychological therapies. No adverse events occurred from physical barriers or topical therapy use.
Authors' conclusions
Given BMS' potentially disabling nature, the need to identify effective modes of treatment for sufferers is vital. Due to the limited number of clinical trials at low risk of bias, there is insufficient evidence to support or refute the use of any interventions in managing BMS. Further clinical trials, with improved methodology and standardised outcome sets are required in order to establish which treatments are effective. Future studies are encouraged to assess the role of treatments used in other neuropathic pain conditions and psychological therapies in the treatment of BMS.
Plain language summary
Interventions for treating burning mouth syndrome
Review question
Which treatments help to relieve symptoms for people with burning mouth syndrome (BMS)?
Background
BMS is a common painful condition. Symptoms include burning, dryness or uncomfortable sensations in the mouth and changes to taste, with no obvious underlying medical or dental cause. BMS is usually persistent and suffered long term, and can lead to a reduced quality of life (QoL). Currently, scientific research suggests that BMS is caused by underlying damage to the nerves. There are many treatments available including drugs for anxiety, other psychological conditions and increasing saliva production, protective barriers and treatments applied to the mouth surface amongst others.
Study characteristics
This review of studies was carried out through Cochrane Oral Health, and the evidence is current up to 31 December 2015.
We found 23 studies (assessing 1121 people; 83% were women), published between 1995 and 2015 to include in this review. Twenty‐one studies assessed short‐term (up to three months) symptom relief, and four studies assessed long‐term (from three to six months) symptom relief. Seventeen studies provided information about side effect occurrence, seven studies assessed a measure of QoL, and two studies assessed changes in taste and feeling of dryness.
All of the 23 treatments included in this review were compared to a placebo (fake treatment): antidepressants and antipsychotics (two studies), antiseizure drugs (one study), types of tranquillisers (four studies), saliva stimulants (one study), dietary supplements (12 studies), directed energy waves (one study), physical barriers (one study), psychological therapies (one study), and treatments applied to the mouth surface (five studies).
Key results
Short‐term symptom relief
We found evidence of short‐term symptom relief for directed energy waves (one study, 58 participants), a type of tranquilliser used topically (that is held in the mouth before being removed, and which also acts as an antiseizure drug) called clonazepam (two studies, 111 participants), thin plastic tongue covers (one study, 50 participants), and an antiseizure drug called gabapentin (one study, 100 participants).
There was no difference in short‐term symptom relief found for antidepressants, saliva stimulants, and another type of tranquilliser used systemically (one that is swallowed) also called clonazepam. We were unable to show whether dietary supplements or treatments applied to the mouth surface provide symptom relief in the short term or not.
Short‐term relief was not reported for the single study that assessed a psychological therapy.
Long‐term symptom relief
We found evidence of long‐term symptom relief for psychological therapy (one study, 30 participants), chili pepper mouthrinse (one study, 18 participants) and the topical tranquilliser called clonazepam (one study, 66 participants).
We found there was no difference in long‐term symptom relief for dietary supplements or treatments applied to the mouth surface.
Studies which assessed antidepressants, directed energy waves, saliva stimulants, antiseizure drugs, or physical barriers did not evaluate long‐term symptom relief.
Change in QoL
There was evidence of short‐term improvement in QoL for directed energy waves (one study, 58 patients), although no difference was found for antidepressants, tranquillisers, dietary supplements and physical barriers. No study assessed long‐term QoL changes.
Change in taste or feeling of dryness
A few studies assessed short‐term change in taste or feeling of dryness (none evaluated these outcomes long‐term), but there was not enough evidence to judge the effects of treatment on these outcomes.
Side effects
Side effects were more likely to be experienced with antidepressants (dizziness and drowsiness more likely: one study, 37 people), and with a dietary supplement called alpha lipoic acid (also known as ALA) with or without other ingredients (headaches more likely: two studies, 118 people; and upset stomachs more likely: three studies, 138 people).
Quality of the evidence Overall, we found very low‐quality evidence for each short‐ and long‐term outcome we investigated (symptom relief; changes in QoL, taste and feeling of dryness; and side effects) in all types of assessed treatment: antidepressants and antipsychotics, antiseizure drugs, types of tranquillisers, saliva stimulants, dietary supplements, directed energy waves, physical barriers (except side effects, which was assessed as low quality), psychological therapies, and treatments applied to the mouth surface. As we found so few studies at low risk of bias, we are currently unable to prove or disprove the effectiveness of any treatments for managing BMS.
Summary of findings
Background
Description of the condition
Burning mouth syndrome (BMS) is defined as burning or painful sensations from an oral mucosa with no clinical signs of pathology or identifiable medical or dental causes (IHS 2013). In addition to pain, many BMS patients also report subjective xerostomia (dryness), oral paraesthesia and/or altered taste (Bergdahl 1999; Woda 1999).
There is confusion in the literature as a wide variety of different terms have been used to describe the sensation of a burning mouth (Buchanan 2010; Fortuna 2013). These include glossodynia, glossopyrosis, stomatodynia, stomatopyrosis, sore tongue, burning mouth and oral dysaesthesia. A sensation of oral burning can be associated with systemic or local causes such as hyposalivation, oral candidiasis, oral parafunction, some deficiency states or side effects of drug treatments (Buchanan 2010; Scala 2003). In these instances the treatment of the underlying cause results in resolution of the burning mouth symptom, and a diagnosis of BMS cannot be made. The diagnosis of primary BMS is thus a diagnosis of exclusion.
The International Association for the Study of Pain (IASP) classification of chronic pain defines BMS as a "distinctive nosological entity characterized by unremitting oral burning or similar pain in the absence of detectable oral mucosal changes" (Merksey 1994); however, it does not draw the distinction between burning as a symptom and primary BMS. The International Headache Society (IHS) describes BMS as "an intraoral burning or dysaesthetic sensation, recurring daily for more than 2 hours per day over more than 3 months, without clinically evident causative lesions" (IHS 2013).
The epidemiological data on BMS are generally poor, due in part to lack of strict adherence to diagnostic criteria (Zakrzewska 1999). Reported prevalence rates of burning mouth symptoms in general populations vary from 0.1% to 3.9% (Bergdahl 1999; Kohorst 2015). A recent epidemiological study using strict diagnostic criteria (albeit reporting from a predominantly Caucasian population) estimated a BMS incidence rate of 11.4 cases per 100,000 person‐years (18.8 cases per 100,000 person‐years for women, and 3.7 cases per 100,000 person‐years for men) (Kohorst 2014). The incidence of BMS sharply increases after the age of 50 in both women and men, with the highest incidence in women (70.3 cases per 100,000 person‐years) aged 70 to 79 years (Kohorst 2014). The natural history of BMS has not been clearly defined and there are no reports of longitudinal cohort studies (Zakrzewska 1999). There is anecdotal evidence of at least partial spontaneous remission in approximately half of these patients within six to seven years (Grushka 1987a). In another study, only two out of 53 patients reported complete spontaneous remission of their symptoms within five years after onset (Sardella 2006).
The prominent feature of BMS is the symptom of burning pain or discomfort, which can be localised just to the tongue or lips or both but can be more widespread and involve the whole of the oral cavity (Grushka 1987b; Scala 2003). In most patients the symptoms are bilateral. Often words such as 'pricking', 'tingling', 'numbness' or 'itching' instead of 'burning' are used to describe the pain (Braud 2013). In most cases the symptoms have continued for many months and the intensity of pain tends to increase towards the end of the day (Forssell 2012). Altered taste sensation and a symptom of oral dryness (in patients with no alteration of the salivary flux) are frequently reported (Bergdahl 1999; Grushka 1987b; Scala 2003), and recent studies indicate that 67% to 80% of BMS patients suffer from sleep disturbance (Almoznino 2016; López‐Jornet 2015).
Recent neurophysiologic, psychophysical, neuropathological functional imaging studies have elucidated that several neuropathic, mainly subclinical mechanisms, act at different levels of the nervous system and contribute to the pathophysiology of primary BMS (Jääskeläinen 2012). Thermal quantitative sensory threshold (QST) studies have demonstrated signs of small‐fibre mediated neuropathy (Forssell 2002; Granot 2005; Svensson 1993), sometimes together with extrasegmental sensory alterations; suggesting possibly more generalised somatosensory dysfunction in BMS (Grémeau‐Richard 2010; Puhakka 2016; Svensson 1993). Furthermore, blink reflex studies have demonstrated subclinical trigeminal nerve lesions in approximately 20% of BMS patients (Forssell 2002; Jääskeläinen 1997). The frequent report of taste dysfunction in patients with BMS has prompted the hypothesis that there could be hyperactivity of the somatosensory fibres of the trigeminal nerve, following loss of central inhibition due to taste fibre damage (Kolkka‐Palomaa 2016). Supporting this, three electrogustatometric studies have reported evidence for chorda tympani hypofunction in BMS (Eliav 2007; Grémeau‐Richard 2010; Just 2010). In line with the thermal QST and electrogustatometric evidence for focal small fibre hypofunction in BMS, several studies have demonstrated loss of epithelial nerve fibres in tongue mucosal biopsies from BMS patients (Lauria 2005; Penza 2010; Puhakka 2016; Yilmaz 2007).
Central nervous system pathology seems also to be involved in the generation of BMS pain symptoms. Giving further evidence for the neuropathic nature of BMS, the characteristics in functional magnetic resonance imaging (fMRI) activation patterns to painful stimuli have been shown to be similar in BMS patients and patients with other neuropathic pain conditions (Albuquerque 2006). A recent study on cerebral reorganisation demonstrated altered grey and white matter volumes in the hippocampus and medial prefrontal cortex in BMS patients, as well as altered functional connectivity patterns of these regions (Khan 2014). Two positron emission tomography (PET) studies have demonstrated a decline in endogenous dopamine levels in BMS, suggesting deficiencies in central pain modulation (Hagelberg 2003; Jääskeläinen 2001).
The question concerning the relative involvement of peripheral versus central mechanisms in BMS pain has important implications, especially concerning different treatment approaches. A study investigating the effects of peripheral lingual nerve block on spontaneous burning pain in BMS showed that in half of the patients the lingual nerve block relieved the pain, suggesting predominantly peripheral mechanisms acting in this subgroup. In some cases, lingual nerve anaesthesia had no effect or even increased the pain intensity, indicating that central mechanisms may be more important in the pathophysiology of pain in some BMS patients (Grémeau‐Richard 2010).
Most studies on the pathophysiology of BMS have explored the neuropathic background of these pains, but many other factors may be involved. A hypothesis linking BMS with dysregulation of adrenal, gonadal and neuroactive steroids has been presented (Woda 2009). Autonomic nervous system impairment (Heckmann 2001; Koszewicz 2012) or immune function suppression (Koike 2014) have also been suggested to have a role in BMS pathophysiology. It has also been speculated that salivary dysfunction plays a role in BMS because more than half of BMS patients complain of dry mouth (i.e. xerostomia) (Bergdahl 1999). However, while some investigators have demonstrated decreased salivary gland output in BMS (Lee 2015), some studies have indicated that the salivary flow rate in BMS patients is the same as in controls (De Moura 2009). Methodological and patient population differences may explain the contradictory results; according to the current definition, patients with hyposalivation should not receive the primary BMS diagnosis. Moreover, studies concerning the composition of saliva in BMS have yielded conflicting results (Scala 2003).
BMS is well studied from the psychological perspective, showing convincing evidence for close relationship between psychological factors and the pain experience (Galli 2016). Many BMS patients show evidence of anxiety, depression or personality disorders. One study demonstrated that when compared with a control group, BMS patients had a significantly lower scores in socialisation, significantly higher scores in somatic anxiety and more negative thoughts (Bergdahl 1995b). It has been demonstrated that patients with BMS show an increased tendency for somatisation, as well as several other psychiatric features when measured on the SCL‐90 (Symptom Checklist‐90) questionnaire (Eli 1994). Psychological disorders could theoretically be associated with BMS by several mechanisms. While one such mechanism, dopaminergic hypofunction has been shown to be related to BMS pain, it has been suggested that the high psychological or psychiatric comorbidity in BMS can be understood in terms of shared vulnerability to both chronic pain and psychiatric disorders, mediated by dysfunctional brain dopamine activity (Taiminen 2011).
Description of the intervention
These recent findings from BMS research suggest both central and peripheral neuropathological changes are present in the condition. Consequently, it could be proposed that BMS may respond to those treatments offered for other neuropathic conditions, such as antidepressants, anticonvulsants, dietary/nutritional supplements and topical anaesthetic or analgesic agents (Finnerup 2015; Foster 2007; NICE 2013). Moreover, the increasing evidence associating BMS with psychological comorbidities such as anxiety and depression, would suggest anxiolytics, antidepressants, and psychological therapies may be helpful in the management of BMS. The application of a physical barrier may also work to reduce the impact of parafunctional habits which may induce or sustain BMS (López‐Jornet 2009a).
How the intervention might work
Topical anaesthetic treatments would reduce BMS pain by blocking peripheral pain pathways, while topical capsaicin therapy aims to desensitise peripheral nerves. Antidepressant drugs produce blockade of various central nervous system (CNS) receptors, such as serotonin and norepinephrine, thereby increasing the activity of the descending inhibitory pain pathways. Some older antidepressants used commonly in pain management, such as the tricyclic antidepressants (TCAs), have complex pharmacodynamics and act by inhibiting multiple CNS receptors. This lack of specificity may in part be why the TCAs are clinically effective in managing pain, but also why they tend to produce adverse effects. More modern antidepressants such as the selective serotonin reuptake inhibitors (SSRIs), are more selective in their effects on CNS receptors, hence tend to produce less side effects than the TCAs. The roles of SSRIs in pain management have yet to be fully explored.
Anticonvulsant medications achieve their analgesic effects through a variety of mechanisms which include blockade of voltage‐dependent sodium and calcium channels in peripheral neurones and actions on neuropeptides such as glutamate and substance P. The benzodiazepine class of drugs possesses anxiolytic and anticonvulsant properties, achieved through enhancing the effect of the neurotransmitter gamma‐Aminobutyric acid (GABA) (Lopez‐D'alessandro 2011).
Clinical psychology is known to help patients with chronic pain conditions improve their quality of life, despite having a background of persistent pain (Bergdahl 1995a). Recently, electromagnetic radiation (by low‐level laser and transcranial magnetic stimulation) has been used as a non‐invasive analgesic intervention for chronic pain, and its application in treating drug‐resistant BMS is now being explored (Spanemberg 2015; Umezaki 2016).
Several topical agents are used in the management of BMS, with varying degrees of biological plausibility in how they exert their effects: benzydamine hydrochloride is known to have topical analgesic properties (Sardella 1999); lactoperoxidase oral rinse is considered because of its previous role in the management of xerostomia symptoms (Marino 2010); topical urea is thought to exert a hydrating effect on the oral mucosa in a similar way to its potential effects on the skin (Alvarenga da Silva 2014); and, capsaicin has been found to act as a topical desensitising agent in other neuropathic pain conditions (Marino 2010).
The mechanisms of many treatments used in BMS are unclear and not well described: bethanecol is a parasympathomimetic which is reported to alleviate dry mouth symptoms; alpha lipoic acid is reported to exert "a neuro‐regenerative action" (Cavalcanti 2009; Palacios‐Sánchez 2015); hypericum perforatum extract is used due to its previous role in the management of depression (Sardella 2008); 'Catuama' is reported to have analgesic and antidepressant properties (Spanemberg 2012); lycopene and olive oil compound are used due to their antioxidant properties (Cano‐Carrillo 2014); and lastly, 'tongue protectors' have been studied alone and also in conjunction with topical aloe vera ‐ they are reported to control 'parafunctional habits' that may cause mucosal trauma, coupled with the potential mucosal healing benefits of aloe vera (López‐Jornet 2013).
It is likely that there is substantial diversity amongst neuropathic pain patients with respect to clinical presentation, sensory examination features and possibly the underlying pain mechanisms (Chaparro 2012). This diversity in pain mechanisms may be one reason for the limited analgesic efficacy of monotherapy pharmacological agents. Moreover, dose‐related drug side effects (e.g. drowsiness, dizziness) may limit the tolerability of higher and more effective dosages. Therefore, combining medications with different pharmacological mechanisms may result in greater effectiveness and relatively less side effects (Chaparro 2012).
Why it is important to do this review
Burning mouth syndrome is a common, often chronic, condition that appears to have a negative impact on quality of life. Several investigators have found reduced quality of life in BMS patients compared to controls when using SF‐36 (36‐Item Short Form Health Survey) and OHIP‐49 (Oral Health Impact Profile‐49) outcome measures (López‐Jornet 2008; Souza 2011), although we highlight that neither measure's use is specific to BMS.
This is an update of the Cochrane review first published in 2000 and previously updated in 2005 (Zakrzewska 2000; Zakrzewska 2005).
Objectives
To determine the effectiveness and safety of any intervention versus placebo for symptom relief and changes in quality of life, taste, and feeling of dryness in people with burning mouth syndrome.
Methods
Criteria for considering studies for this review
Types of studies
All randomised controlled trials (RCTs) comparing any treatment against placebo.
Types of participants
People with primary burning mouth syndrome (BMS), that is, oral mucosal pain with no dental or medical cause for such symptoms. Trials recruiting participants with other types of pain were only to be included if data on BMS participants could be separated out.
Types of interventions
All treatments that were evaluated in placebo‐controlled RCTs.
Types of outcome measures
Primary outcomes
Relief of burning or discomfort (symptom relief).
Change in quality of life (e.g. depression, anxiety).
Secondary outcomes
Change in taste.
Change in feeling of dryness.
Adverse effects. We assessed treatment safety from reported adverse events.
Search methods for identification of studies
Electronic searches
Cochrane Oral Health's Information Specialist conducted systematic searches in the following databases for randomised controlled trials and controlled clinical trials. There were no language, publication year or publication status restrictions:
Cochrane Oral Health's Trials Register (searched 31 December 2015) (Appendix 2);
Cochrane Central Register of Controlled Trials (CENTRAL; 2015, Issue 11) in the Cochrane Library (searched 31 December 2015) (Appendix 1);
MEDLINE Ovid (1946 to 31 December 2015) (Appendix 3);
Embase Ovid (1980 to 31 December 2015) (Appendix 4).
Subject strategies were modelled on the search strategy designed for MEDLINE Ovid. Where appropriate, they were combined with subject strategy adaptations of the highly sensitive search strategy designed by Cochrane for identifying randomised controlled trials and controlled clinical trials as described in the Cochrane Handbook for Systematic Reviews of Interventions, Chapter 6 (Lefebvre 2011).
Searching other resources
We searched the following trial registries for ongoing studies:
US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (clinicaltrials.gov; searched 31 December 2015) (Appendix 5);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch; searched 31 December 2015) (Appendix 6).
The review authors handsearched the following conference proceedings from January 2005 to December 2015:
British Society for Oral Medicine (BSOM);
British Society for Dental Research (BSDR);
International Association for Dental Research (IADR).
We also scrutinised bibliographies of identified publications and reviews for potentially relevant references, and attempted to contact authors of relevant studies to identify missing data from unreported trials.
We did not perform a separate search for adverse effects of interventions used, we considered adverse effects described in included studies only.
Data collection and analysis
Selection of studies
Teams of two review authors independently screened the titles and abstracts retrieved from the initial electronic searches. Reports from the studies that fulfilled the inclusion criteria were obtained. When there was insufficient information available to determine whether a study fulfilled the inclusion criteria, the full report was obtained and assessed independently by the same review authors. Disagreements were resolved by discussion. The review authors were not blinded to the studies' authorship.
Data extraction and management
At least two review authors independently extracted data from each study included using a tool developed for the review. All studies meeting the inclusion criteria underwent data extraction and an assessment of risk of bias using a standardised data extraction form. Studies rejected at this and subsequent stages were recorded in the Characteristics of excluded studies table. Differences were resolved by discussion. For each study with more than one control or comparison group for the intervention, the results were extracted for each intervention arm. The review authors were not blinded to the studies' authorship.
We extracted the following data.
Year of publication, country of origin, number of centres, source of study funding and any conflicts of interest.
Details of the participants including demographic characteristics and criteria for inclusion/exclusion.
Details on the type of intervention and comparisons.
Details on the study design.
Details on the outcomes reported which included method and timings of assessments and adverse outcomes.
We contacted authors of the relevant studies, to supply missing information or data where necessary. We contacted trial authors for missing data if the study was published from the year 2000 onwards. We considered it unfeasible to obtain data for trials published prior to this cut‐off date.
Assessment of risk of bias in included studies
Teams of two review authors independently assessed the risk of bias for each included study. The 'Risk of bias' assessment was conducted using Cochrane's tool for assessing risk of bias (Higgins 2011a) and in accordance with guidance included in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Higgins 2011b). For each included study we assessed the following seven key domains.
Sequence generation (selection bias).
Allocation concealment (selection bias).
Blinding of participants and personnel (performance bias).
Blinding of outcome assessment (detection bias).
Completeness of outcome data (attrition bias).
Selective outcome reporting (reporting bias).
Risk of other potential sources of bias (other bias).
For each study, we determined the overall risk of bias according to the following criteria:
low risk of bias ‐ when there was a low risk of bias across all seven key domains;
unclear risk of bias ‐ when there was an unclear risk of bias in one or more of the seven key domains (no domains judged to be at high risk of bias);
high risk of bias ‐ when there was a high risk of bias in one or more of the seven key domains.
For consistent rating application, one author (Anne‐Marie Glenny) arbitrated all assessments. We completed a 'Risk of bias' table for each included study, a 'Risk of bias' summary and 'Risk of bias' graph.
Measures of treatment effect
We analysed outcome data as short‐term (≤ 3 months from baseline) or long‐term (> 3 to ≤ 6 months from baseline) as a manageable cut‐off threshold. For continuous outcomes (e.g. pain/burning on a visual analogue scale (VAS)), we used mean differences (MDs) and 95% confidence intervals (CIs) to summarise the data; in the event that different studies measured outcomes using different scales, we would have expressed the estimate of effect of an intervention as standardised mean differences (SMDs) and 95% CIs. Dichotomous outcomes (e.g. greater or less than 50% reduction in pain intensity as measured by a VAS, or improvement from baseline versus no change/worsened score), we expressed the estimate of effect of an intervention as risk ratios (RRs) together with 95% CIs.
Unit of analysis issues
If cluster‐randomised trials had been included, we would have undertaken data analysis, whenever feasible, at the same level as the randomisation, or at the individual level accounting for the clustering. Analysis of cross‐over studies took into account the two‐period nature of the data using for example, a paired t‐test (Elbourne 2002). We entered MDs and standard errors into Review Manager (RevMan) software (Review Manager 2014), using the generic inverse variance method (Higgins 2011b).
Dealing with missing data
When required we contacted trial authors for missing data if the study had been published in the year 2000 or after. The review authors considered it unfeasible to request data for trials published prior to this date. We used methods outlined in the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) to estimate missing standard deviations (Higgins 2011b).
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity. We further assessed the significance of any discrepancies in the estimates of the treatment effects from the different trials, by means of Cochran's test for heterogeneity ‐ heterogeneity would have been considered significant if P value < 0.1 (Higgins 2011b). We also utilised the I2 statistic, which describes the percentage total variation across studies due to heterogeneity rather than chance, to quantify heterogeneity with I2 over 50% being considered substantial heterogeneity (Higgins 2011b).
Assessment of reporting biases
If there had been a sufficient number of trials (more than 10) included in any meta‐analysis, we would have assessed publication bias in accordance with the recommendations on testing for funnel plot asymmetry, described by the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Higgins 2011b).
Data synthesis
Where data allowed, we performed meta‐analysis of studies which assessed the same comparisons and outcomes. We combined RRs for dichotomous outcomes, and MDs (we would have produced SMDs if different scales had been used) for continuous outcomes, using a random‐effects model where there were four or more studies, or a fixed‐effect model for less than four studies.
We included data from cross‐over studies (provided they incorporated a washout period) in meta‐analyses using the generic inverse variance method described in the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Higgins 2011b), combining them with parallel studies using the methods described in Elbourne 2002. For cross‐over studies not incorporating a washout period, we utilised the first period data only, in accordance with Section 16.4.5 of the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Higgins 2011b).
Where single studies compared more than one active intervention with a placebo within the same analysis, the number of participants in the study's control group was halved before combining study data to avoid double‐counting control participants within a single meta‐analysis.
Subgroup analysis and investigation of heterogeneity
If there were sufficient studies, we would have used sensitivity analyses and meta‐analysis regression (using Stata software (Stata 2015)) to explore, quantify, and control for sources of heterogeneity between studies for type of therapy.
Sensitivity analysis
If the number (and quality) of studies had allowed, we would have undertaken a sensitivity analysis for each intervention and outcome limiting the analysis to studies at low overall risk of bias.
Presentation of main results
We produced a 'Summary of findings' table for the main outcomes. We assessed the quality of the body of evidence, taking into account the overall risk of bias of the included studies, the directness of the evidence, the inconsistency of the results, the precision of the estimates, the risk of publication bias and the magnitude of the effect. We categorised the quality of the body of evidence of each of the main outcomes as high, moderate, low or very low.
Results
Description of studies
Details of the trial participants, interventions and outcomes measured can be seen in the Characteristics of included studies table.
Results of the search
The literature search for this review identified 377 records after the duplicates were removed. These 377 records were screened independently and in duplicate. After screening, we retained 64 records for further assessment and categorised 313 records as not relevant.
We obtained published papers for 60 records. Following our assessment of the 60 full‐text articles, we excluded a total of 37 studies (37 articles) with reasons provided (Characteristics of excluded studies table).
We included 23 studies (a total of 25 articles, including eight already included studies from the previous version of the review), of which 21 studies (all except Bogetto 1999 and Silvestre 2012) provided useable data. Figure 1 shows the study selection process.
1.
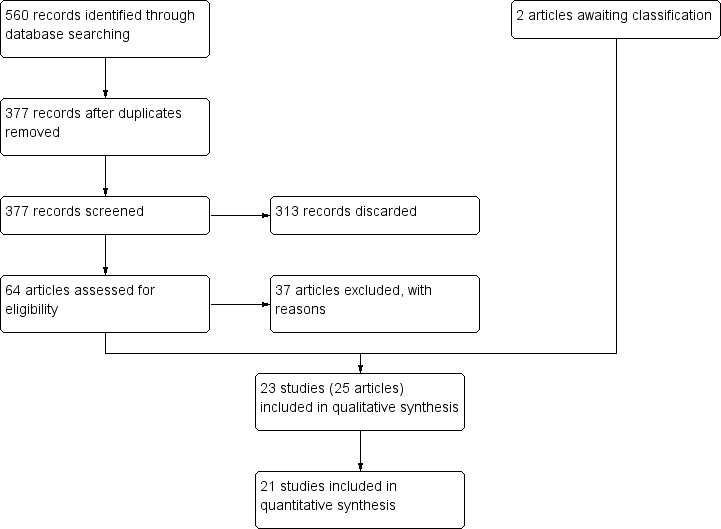
Study flow diagram.
Included studies
Previously, this review included nine trials (Bergdahl 1995a; Bogetto 1999; Femiano 2000; Femiano 2002a; Femiano 2002b; Grémeau‐Richard 2004; Pisanty 1975; Sardella 1999; Tammiala‐Salonen 1999). This update excludes a previously included trial (Pisanty 1975), due to insufficiently indicating whether its patients had burning mouth syndrome and also not being a randomised controlled trial (RCT), and includes an additional 15 RCTs (Alvarenga da Silva 2014; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Heckmann 2012; Lopez‐D'alessandro 2011; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Sardella 2008; Silvestre 2012; Spanemberg 2012; Spanemberg 2015).
A total of 23 RCTs (1285 patients included; 1121 patients assessed) were included in this latest update of the review (Alvarenga da Silva 2014; Bergdahl 1995a; Bogetto 1999; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Grémeau‐Richard 2004; Heckmann 2012; Lopez‐D'alessandro 2011; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Sardella 2008; Silvestre 2012; Spanemberg 2012; Spanemberg 2015; Tammiala‐Salonen 1999).
Characteristics of studies
Included studies were undertaken in several countries.
Eight trials (35%) were conducted in Italy (Bogetto 1999; Carbone 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Marino 2010; Sardella 1999; Sardella 2008).
Six trials (26%) in Spain (Cano‐Carrillo 2014; López‐Jornet 2009b; López‐Jornet 2011; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Silvestre 2012).
Four trials (17%) in Brazil (Alvarenga da Silva 2014; Cavalcanti 2009; Spanemberg 2012; Spanemberg 2015).
The remaining five single studies (4% each, totalling 22% of included studies) were conducted in Argentina (Lopez‐D'alessandro 2011), Finland (Tammiala‐Salonen 1999), France (Grémeau‐Richard 2004), Germany (Heckmann 2012), and Sweden (Bergdahl 1995a).
All trials were published in English, except one Italian study (Bogetto 1999).
All studies were placebo‐controlled parallel RCTs, except three (13%) cross‐over studies (Cavalcanti 2009; Femiano 2000; Silvestre 2012). Of these three studies, only two incorporated a washout period into the cross‐over study design (Cavalcanti 2009: 20 days; Silvestre 2012: 1 week).
Grémeau‐Richard 2004 was a multicentre trial, and all other studies operated from a single centre. Sixteen trials (70%) contained two arms (intervention versus placebo), one three‐armed trial (4%) contained a 'no treatment' arm in addition to comparing intervention with placebo (Sardella 1999), and the remaining six trials (26%) were multi‐armed to investigate several interventions (five arms: Bogetto 1999; four arms: Femiano 2002b; Lopez‐D'alessandro 2011; Marino 2010; Spanemberg 2015; three arms: Carbone 2009).
Follow‐up varied greatly between included studies, and ranged between one week (Silvestre 2012) to six months (Bergdahl 1995a; Grémeau‐Richard 2004; Rodríguez de Rivera‐Campillo 2010). Four trials (17%) followed up their patients for longer than three months (Bergdahl 1995a; Carbone 2009; Marino 2010; Rodríguez de Rivera‐Campillo 2010).
Nine studies (39%) provided information on source of funding:
São Paulo State Research Foundation (Alvarenga da Silva 2014; Cavalcanti 2009);
funded by universities (Grémeau‐Richard 2004; Sardella 2008);
national dental professional body (Tammiala‐Salonen 1999);
mixed funding from a university and national government bodies (Spanemberg 2015);
mixed funding from a university and a national dental professional body (Bergdahl 1995a);
unfunded (Cano‐Carrillo 2014; Heckmann 2012).
The remaining 14 studies (61%) did not report on source of funding.
Five trials (22%) reported no conflict of interests (Alvarenga da Silva 2014; Cano‐Carrillo 2014; Heckmann 2012; Marino 2010; Palacios‐Sánchez 2015), while 18 trials (78%) omitted reporting their authors' conflicts of interests.
Characteristics of participants
All 23 included RCTs appropriately defined their participants as having burning mouth syndrome (BMS) ‐ that is, persistent oral mucosal pain with no dental or medical cause for such symptoms (IHS 2013). Excerpted details of diagnosis and duration are presented in Additional Table 10.
1. Diagnosis of burning mouth syndrome (BMS) and duration of disease in included studies.
| Study ID | Diagnosis description | Duration of disease |
| Alvarenga da Silva 2014 | "..patients with BMS diagnosed according to the International Association for the Study of Pain (IASP) guidelines. They underwent laboratory tests and a careful examination to exclude other causes of burning mouth. The exclusion criteria were other facial pain syndromes, other causes of abnormal salivation, other neuropathies or primary diseases associated with burning mouth" | Mean duration of BMS (+/‐ SD): topical urea 10% (n = 19) 6.97 years (+/‐ 4.93); placebo (n = 19) 2.78 years (+/‐ 2.61) |
| Bergdahl 1995a | "The patients were odontologically and medically examined and treated according to the protocol for the management of patients with BMS proposed by Bergdahl et al, including complete anamnesis, general medical and odontological examination, laboratory investigation and an epicutaneous patch test. [...] All the odontologically and medically diagnosed diseases were treated, but the treatment had no influence on the burning sensations and therefore these patients were labelled as suffering from resistant BMS" | Not reported |
| Bogetto 1999 | (Translated from Italian) "Inclusion criteria: diagnosis of BMS, according to the criteria provided by the literature [six references]" | (Translated from Italian) "The total average duration of the disorder was 2.7 ( +/‐ 3.2 ) years" |
| Cano‐Carrillo 2014 | "Inclusion criteria for participating in the study were as follows: a clinical history of continuous symptoms of oral burning or pain on a daily or almost daily basis, during all or part of the day for more than 6 months, without paroxysms, and independent of the nervous pathway; an absence of clinical abnormalities that might account for the symptoms; and normal blood test findings (complete blood count, blood glucose, serum, iron and transferrin levels, serum vitamin B12, and folate. Patients with pain attributable to other conditions (angiotensin‐converting enzyme inhibitor use, candidiasis, lichenoid reactions, sores, tongue atrophy, etc.) were excluded" | "The majority of patients had severe burning sensation and had suffered from BMS over a long period" (Inclusion criteria required continuous symptoms for longer than 6 months) |
| Carbone 2009 | "The study was prospectively performed on patients with previously untreated BMS referred to the Oral Medicine Section of the University of Turin [...], approximately 90 patients reporting oral symptoms suggestive of BMS were screened for participation [...]. Consistent with Gremeau‐Richard et al (2004), the inclusion criteria were the presence of an isolated complaint of chronic pain in the oral mucosa with a normal clinical examination, and pain present for more than 4 months, which was continuous throughout all or part of the day, with no paroxysms and not following a nerve trajectory. Candida infection was ruled out and any organic conditions that could be considered as causative factors for similar oral symptoms were ruled out in all subjects by laboratory examinations (e.g. full blood cell count, and serum levels of iron, ferritine, folate, vitamin B12, and glucose)" |
Not reported (Inclusion criteria required continuous symptoms for longer than 4 months) |
| Cavalcanti 2009 | "Subjects [...] who reported a history of oral burning pain for more than 6 months and absence of oral findings were assessed for eligibility.[...] The patients underwent detailed clinical evaluation and laboratory tests [complete blood cell count; blood glucose level; serum iron and ferritin levels; serum vitamin B12; folic acid levels; salivary flow rate measurement; exfoliative cytology; detection of local abnormalities] to exclude possible local and ⁄ or systemic causes for oral burning" | "The mean duration of BMS was 37.43 months (range 6‐132 months)" |
| Femiano 2000 | "Only BMS patients with objective evidence of a normal‐looking oral mucosa, with absence of identifiable oral mucosal pathological lesions, with normal salivary secretion (.15 ml/15 min unstimulated and .1 ml min‐1 after 5% citric acid stimulation), and with normal laboratory results [refers to earlier quote: "full blood count, serum ferritin, vitamin B12, SGOT, SGPT, serum total IgE (PRIST) and IgE specific for methacrylate, corrected whole blood folate and random blood sugars"] were included. [...] The final patient group thus consisted of persons with BMS and neither clinical nor laboratory evidence of organic disease" | Not reported |
| Femiano 2002a | "..patients, diagnosed with BMS from a history of constant burning discomfort in the anterior tongue, lower lip or hard palate, for more than two months, with no relevant drug or medical history, were examined for evidence of clinical oral mucosal lesions and alterations in laboratory parameters (whole blood folate, serum vitamin B12, serum ferritin, serum glucose, thyroid hormone levels) that could be responsible for the BMS. A final study subgroup of 60 subjects with BMS [...] was identified with no clinical or laboratory evidence of disease" | Not reported (Inclusion criteria required continuous symptoms for longer than 2 months) |
| Femiano 2002b | "The study population consisted of persons with BMS as defined elsewhere [reference indicates van der Waal 1990] and with neither clinical nor laboratory evidence of organic disease. [...] the final test subjects were BMS patients only with objective evidence of a normal‐looking oral mucosa, with absence of identifiable oral mucosal pathological lesions, with normal salivary secretion and with normal laboratory results" | Not reported |
| Grémeau‐Richard 2004 | "..patients with stomatodynia were screened for participation [...]. The inclusion criteria were the presence of an isolated complaint of chronic pain in the oral mucosa with a normal clinical examination. Pain was present for more than 4 months, was continuous throughout all or part of the day, with no paroxysms and did not follow a nerve trajectory. Patients presenting with an organic condition that could be considered as a causative factor such as diabetes or anaemia were not included. Such local or systemic conditions were sought with laboratory examinations only when suspected from the clinical approach (e.g. blood cell count, serum iron folate level or detection of Candida). Also, patients with abnormal neurological conditions and those regularly treated on a daily basis by anti‐depressants, anti‐convulsants, other psychotropic drugs or psychological therapy were also excluded from this study. [...] Reliability for diagnosis of stomatodynia had been assessed in a previous study involving the same experimentors" | Not reported (Inclusion criteria required continuous symptoms for longer than 4 months) |
| Heckmann 2012 | "Twenty‐three patients suffering from BMS were referred to the oral pain clinic of Erlangen University Dental School. [...] Inclusion was restricted to idiopathic cases. [...] the patients received a physical examination of their oral cavity including a test for possible pathological infections with candida" | Mean duration of disease: clonazepam (n = 10) 2.8 years (SD 1.9); placebo (n = 10) 3.6 years (SD 2.4) |
| Lopez‐D'alessandro 2011 | "..patients with idiopathic BMS of more than three months duration. [...] Patients with deficiencies of folic acid, vitamin B, carriers of anemias of any kind and patients with Sjögren syndrome were also excluded" | Not reported (Inclusion criteria required continuous symptoms for longer than 3 months) |
| López‐Jornet 2009b | "..patients attending our service with symptomatology compatible with BMS were invited to participate [...]. Inclusion criteria to participate in the study were presentation of a clinical history of continuous symptomatology of oral burning or pain, daily or almost daily, during all or part of the day for more than 6 months evolution, without paroxysms, and independent of the nervous pathway; likewise, no clinical abnormality that would justify the symptomatology. Furthermore, the patients had to present a normal blood analysis (completed blood cell counts, blood glucose levels, serum iron and transferrin levels, serum Vit B12 and folate) [...]. Patients with pain attributable to other entities (candidiasis, lichenoid reactions, sores, etc.) were excluded" | "The average time suffering BMS was 3 years, with a minimum of 6 months and a maximum of 5 years" |
| López‐Jornet 2011 | "Inclusion criteria for participating in the study were a clinical history of continuous symptoms of oral burning or pain on a daily or almost daily basis, during all or part of the day for more than 6 months, without paroxysms, and independent of the nervous pathway. Likewise, the included patients presented no clinical abnormalities that could account for the symptoms. Furthermore, the patients had to present normal blood test findings (complete blood count, blood glucose, serum iron and transferrin levels, serum vitamin B12, and folate) [...]. Patients with pain attributable to other conditions (angiotensin‐converting enzyme inhibitor use, candidiasis, lichenoid reactions, sores, tongue atrophy, etc.) were excluded" | Not reported (Inclusion criteria required continuous symptoms for longer than 6 months) |
| Marino 2010 | "..patients who referred [...] for otherwise idiopathic BMS [...].They all complained of a burning, stinging or painful sensation in the mouth in the absence of alterations in the appearance of the oral mucosa or any local or systemic diseases. [...] exclusion criteria were: (i) evidence of any local disorders that may be responsible for the burning mouth sensation, such as infection by Candida species, parafunctional habits, temporomandibular joint disorders, allergic contact stomatitis, benign migratory glossitis and lichen planus" "Inclusion criteria: Symptoms of diffuse burning pain of the tongue and ⁄ or oral mucosa associated or unassociated with subjective oral dryness or loss or alteration of taste or sensation; Burning pain almost every day; Normal‐looking mucosa in the region of burning; Absence of systemic disorders or laboratory alterations known to be associated with orofacial pain; Daily bilateral oral burning (or pain‐like sensation); Pain is unremitting for at least 4–6 months [...] Exclusion criteria: Presence of specific local etiologic evidence for the burning (e.g. disease of the oral mucosa, hyposalivation); Presence of specific systemic etiologic evidence for the burning (e.g. diabetes, anemia); Use of medications known to be associated with oral burning and ⁄ or alteration of taste or sensation" |
"The mean time from symptom onset to enrolment was 18 months" (Inclusion criteria required continuous symptoms for longer than 4 months) |
| Palacios‐Sánchez 2015 | "Diagnosis was made during the first screening phase. [...] patients over 18 years of age clinically diagnosed with BMS who reported a history of continuous oral burning pain for more than 4 months with no clinical signs that could justify the syndrome (Scala 2003) [...] Exclusion criteria included: patients whose burning sensation could be related to local alterations [...] All patients were assessed for salivary flow rates, at rest and stimulated, complete blood count and biochemistry values, including ferritin, vitamin B12 and folic acid levels. [...] According to Lamey and Lewis's BMS classification, 38 patients (63.3%) belonged to type I, 17 patients (28.3%) to type II, and only 5 patients (8.3%) to type III" | "The evolution time of symptomatology varied between 4 months and 20 years" |
| Rodríguez de Rivera‐Campillo 2010 | "..adults with BMS [...]. Some patients attended the clinic to receive dental or medical treatment, while others were referred by colleagues after unsuccessful treatments. All subjects reported oral burning in the absence of apparent oral lesions. [...] We excluded patients with disorders in the oral mucosa that could explain the symptoms, those who were receiving treatment for BMS [...]" | Duration of disease (all patients (n = 66)): < 6 months n = 4 (6%); 6‐12 months n = 12 (18%); > 12 months n = 50 (76%) |
| Sardella 1999 | "The criterion for admission was the diagnosis of "idiopathic" or "essential" burning mouth syndrome. We use this term to refer to all forms of burning sensation in the mouth, including complaints described as stinging sensation or pain, in association with an oral mucosa that appears clinically normal in the absence of local or systemic diseases or alterations; these include nutritional and hematologic deficiencies, diabetes mellitus, the presence of Candida albicans or candidiasis infection, xerostomia, denture design faults, parafunctional habits, contact allergy to dental materials, oral lichen planus, and geographic tongue. To identify the "essential" BMS cases, the patients' medical and dental histories were carefully taken, particular attention being paid to the characteristics of the complaint (type, localization, duration), the clinical oral inspection performed, and the laboratory evaluations requested. In particular, the laboratory data included complete blood cell counts, blood glucose levels, serum iron and transferrin levels, and serum vitamin B12 and folate levels. Furthermore, patch testing for allergy to dental material was performed, a tongue and palate smear for the detection of Candida was taken, and salivary gland flow rates, resting and stimulated, were determined. When altered parameters were detected, an appropriate therapy was proposed. [...] Patients experiencing symptomatic improvement after correction of their deficiencies were excluded from the investigations, the assumption being made that they did not have "essential" BMS. This clinical design led to the identification of 30 patients with "essential" BMS. [...] With reference to the classification suggested by Lamey and Lewis,13 BMS type II was present in 16 patients (53%), BMS type III in 10 patients (33%), and BMS type I in the remaining 4 patients (14%)" |
"The duration of the syndrome was a matter of months or even years, with a mean duration before the beginning of the clinical trial of 18 months" |
| Sardella 2008 | "Subjects referred [...] who reported a history of oral burning pain for at least 6 months and who lacked oral findings were considered for this study. [...] Demographic and medical questionnaires asking for information related to the presence of current systemic diseases and on‐going medications were administered. To confirm the diagnosis of essential BMS, the patients underwent a standard set of evaluations to exclude local or systemic conditions that could be considered causative factors for an oral burning sensation (salivary flow rates; laboratory tests [complete blood cell counts, blood glucose levels, serum iron and transferrin levels, serum vitamin B12 folate levels]; isolation of Candida species; detection of parafunctional activities)" | Mean duration of BMS (+/‐ SD): hypericum perforatum (n = 19) 28.8 months (+/‐ 8.9); placebo (n = 19) 32.4 months (+/‐ 9.9) |
| Silvestre 2012 | "BMS was diagnosed according to the current criteria, and the discomfort had been present on a daily basis for at least 6 months (Scala 2003). [...] excluded from the study [...] were those [...] patients with oral mucosal lesions that might explain the burning sensation" | "The mean duration of the disease was 5.43 ± 3.23 years (range 1‐14 years). Patients with a BMS duration of 4 and 5 years represented 39.1% of the total (n = 9)" |
| Spanemberg 2012 | "The sample comprised 72 patients of both sexes with a diagnosis of BMS [...]. The study included patients [...] who reported symptoms of burning or pain in the oral mucosa of at least 6 months' duration and who presented with a clinically normal mucosa. [...] Patients who showed hyposalivation (salivary flow rate at rest of 0.1 mL/min), as well as alterations in their hemogram, serum levels of glucose, iron, folic acid, and vitamin B12, were also excluded" | "The time of development of BMS ranged from 6 months to 20 years, with a median of 24 months" |
| Spanemberg 2015 | "The study included patients [...] who reported having had symptoms of burning or pain in the oral mucosa for at least six months and who presented a clinically normal mucosa" | "The duration of the symptoms ranged from 6 months to 30 years; 33.3% [n = 26] of the patients had been presenting the disorder for one to three years" |
| Tammiala‐Salonen 1999 | "..patients who were referred [...] because of oral mucosal burning pain. [...] The patients underwent a thorough clinical examination, including measurement of whole salivary flow, blood samples (blood count and levels of glucose, B12 vitamins, and folate), and diagnosis of candidiasis. The investigators asked patients about pain intensity and duration, overall health, and medications. [...] Criteria for inclusion were daily, or almost daily, oral burning pain that had lasted 6 months or longer and had a moderate to severe intensity" | "The mean duration of pain in the trazodone group was 3.0 years (6 months to 17 years) and in the placebo group it was 2.8 years (6 months to 20 years)" |
SD = standard deviation.
In total, 1061 women (83%) and 221 men (17%) are reported to have participated in the included studies.
The mean age of participants ranged from 45 years (Femiano 2002a) to 73 years old (Silvestre 2012); however, the average age of participants was older than 60 years in 19 trials (83%: Alvarenga da Silva 2014; Bogetto 1999; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2000; Femiano 2002b; Grémeau‐Richard 2004; Heckmann 2012; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Sardella 2008; Silvestre 2012; Spanemberg 2012; Spanemberg 2015).
Twenty‐one studies (91%) did not report any data relating to socioeconomic status. Of the two studies which did, Bogetto 1999 reported participants' number of schooling years, and Alvarenga da Silva 2014 reported data on race, occupation, and marital status.
Characteristics of interventions
A broad range of interventions were investigated by the included studies, categorised into nine groups.
-
Antidepressants and antipsychotics
Paroxetine (Bogetto 1999)
Amitriptyline (Bogetto 1999)
Amisulpride (Bogetto 1999)
Trazodone (Tammiala‐Salonen 1999).
-
Anticonvulsants
Gabapentin (Lopez‐D'alessandro 2011)
Gabapentin + alpha lipoic acid (ALA) (Lopez‐D'alessandro 2011).
-
Benzodiazepines
Systemic clordemetildiazepam (Bogetto 1999)
Topical clonazepam (Grémeau‐Richard 2004; Rodríguez de Rivera‐Campillo 2010)
Systemic clonazepam (Heckmann 2012).
-
Cholinergics (parasympathomimetics)
Bethanechol (Femiano 2002b).
-
Dietary supplements
ALA without adjunctive active ingredients (Carbone 2009; Cavalcanti 2009; Lopez‐D'alessandro 2011)
ALA + vitamins (Tiobec) (Carbone 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Marino 2010)
ALA + adjunctive lycopene + green tea extract (Thioderm) (López‐Jornet 2009b; Palacios‐Sánchez 2015)
Hypericum perforatum (St John's Wort) (Sardella 2008)
'Catuama' herbal compound (Spanemberg 2012)
Lycopene (Cano‐Carrillo 2014).
-
Electromagnetic radiation
Low‐level laser therapy (Spanemberg 2015).
-
Physical barriers
Tongue protector + adjunctive reinforced self‐control instruction (RS‐CI)) (López‐Jornet 2011).
-
Psychological therapies
Cognitive therapy (CT) (Bergdahl 1995a).
Topical treatments
Benzydamine hydrochloride oral rinse (Sardella 1999)
Lactoperoxidase oral rinse (Biotene) (Femiano 2002b; Marino 2010)
Topical urea (Alvarenga da Silva 2014)
Capsaicin oral rinse (Marino 2010; Silvestre 2012).
There was heterogeneity in the administration of some treatments. For example, alpha lipoic acid was issued in total daily dosages ranging from 600 to 800 mg, either as single or as split doses; and topical clonazepam was given as 1 mg three times daily or 0.5 mg up to four times daily as required (Characteristics of included studies table).
Characteristics of outcome assessment
The outcomes used by the included studies are described in the Characteristics of included studies table. A wide variety of outcome measures were employed by the various studies (Additional Table 11).
2. Outcome measure scales.
| Study ID | Primary: Symptom relief (symptom intensity/pain), scales used | Primary: Change in quality of life (QoL) (anxiety, depression), scales used | Secondary: Change in taste, scales used | Secondary: Change in feeling of dryness; scales used | Additional assessment scales not relevant to this review |
| Alvarenga da Silva 2014 | EDOF‐HC protocol (Orofacial Pain Clinic ‐ Hospital das Clinicas) | Outcome not assessed | Quantitative Sensory Testing (QST): gustative threshold | Xerostomia questionnaire1 | Quantitative Sensory Testing (QST): olfactory threshold (1); thermal detection thresholds for cold (2) and warm (3) sensations; mechanical detection thresholds for touch (4), vibration (5), and electrical perception (6); mechanical pain sensitivity ‐ superficial (7) and deep pain thresholds (8); electrical pain threshold at the teeth (9), corneal reflex (10), salivary flow (11) |
| Bergdahl 1995a | VAS2 (ranked 1‐7) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Bogetto 1999 | Clinical Global Impression I (CGI I) |
Anxiety: Hamilton Anxiety Rating Scale (HARS) Depression: Montgomery–Åsberg Depression Rating Scale (MADRS) |
Outcome not assessed | Outcome not assessed | n/a |
| Cano‐Carrillo 2014 | VAS (0‐10) |
General health assessment: 36‐Item Short Form Health Survey (SF‐36) Oral health impact on QoL: Oral Health Impact Profile‐14 (OHIP‐14) Anxiety/depression: Hospital Anxiety and Depression (HAD) scale |
Outcome not assessed | Outcome not assessed | n/a |
| Carbone 2009 | 1. VAS (0‐10) 2. McGill Pain Questionnaire (MPQ) |
Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Cavalcanti 2009 | 1. VAS (0‐100 mm) 2. Global Perceived Effect (GPE) |
Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Femiano 2000 | Bespoke burning mouth syndrome (BMS) symptomology change scale | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Femiano 2002a | Bespoke burning mouth syndrome (BMS) symptomology change scale | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Femiano 2002b | Bespoke burning mouth syndrome (BMS) symptomology change scale | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Grémeau‐Richard 2004 | Numerical Pain Scale (0‐10) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Heckmann 2012 | Numerical pain ratings scale (0‐10) | Depression: Beck Depression Inventory (BDI) | Taste test score (0‐16)3 | Outcome not assessed | 1. Smell test score 2. Zerssen Mood Scale 3. Dementia: Mini‐Mental State Examination 4. Salivary flow score (swab method: weight g/min)4 |
| Lopez‐D'alessandro 2011 | Bespoke geographical burning distribution numerical scale (0‐4) |
Anxiety: Hamilton Anxiety Rating Scale (HARS) ‐ baseline only Anxiety/depression: Hospital Anxiety and Depression (HAD) scale ‐ baseline only |
Outcome not assessed | Outcome not assessed | n/a |
| López‐Jornet 2009b | VAS (0‐10) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| López‐Jornet 2011 | VAS (0‐10) |
General health assessment: 36‐Item Short Form Health Survey (SF‐36) Oral health impact on QoL: Oral Health Impact Profile‐49 (OHIP‐49) Anxiety/depression: Hospital Anxiety and Depression (HAD) scale |
Outcome not assessed | Outcome not assessed | n/a |
| Marino 2010 | VAS (0‐10) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Palacios‐Sánchez 2015 | VAS (0‐10) | Depression: Beck Depression Inventory (BDI) ‐ baseline only, as covariate | Outcome not assessed | Outcome not assessed | n/a |
| Rodríguez de Rivera‐Campillo 2010 | VAS (0‐10) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Sardella 1999 | VAS (0‐8) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Sardella 2008 | VAS (0‐10) | Assessed by posing simple questions. No standard QoL questionnaire used | Outcome not assessed | Outcome not assessed | n/a |
| Silvestre 2012 | VAS (0‐10) | Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Spanemberg 2012 | 1. Visual numeric scale (VNS) (0‐10) 2. Faces scale (FS) (0‐5) |
Outcome not assessed | Outcome not assessed | Outcome not assessed | n/a |
| Spanemberg 2015 | 1. VAS (0‐100 mm) 2. Visual numeric scale (VNS) (0‐10) |
Oral Health Impact Profile‐14 (OHIP‐14) | Outcome not assessed | Outcome not assessed | n/a |
| Tammiala‐Salonen 1999 | 1. VAS (0‐100 mm) 2. McGill Pain Questionnaire (MPQ) |
Depression: Beck Depression Inventory (BDI) | Outcome not assessed | Outcome not assessed | n/a |
1 No defined title. See original reference: Korn 2002. 2 VAS: visual analogue scale. 3 No defined title. See original reference: Mueller 2003. 4 No defined title. See original reference: Navazesh 1982.
Due to heterogeneity in follow‐up duration between included studies (ranging from one week (Silvestre 2012) to six months (Bergdahl 1995a; Marino 2010; Rodríguez de Rivera‐Campillo 2010)), it was agreed it would be more clinically useful to separate outcome assessment by short term (≤ 3 months) and long term (> 3 to ≤ 6 months).
Twenty‐one studies (91%) reported short‐term outcome assessment (Alvarenga da Silva 2014; Bogetto 1999; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Grémeau‐Richard 2004; Heckmann 2012; Lopez‐D'alessandro 2011; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Palacios‐Sánchez 2015; Sardella 1999; Sardella 2008; Silvestre 2012; Spanemberg 2012; Spanemberg 2015; Tammiala‐Salonen 1999). Three studies (Carbone 2009; Marino 2010; Rodríguez de Rivera‐Campillo 2010) which reported short‐term outcome assessment also reported long‐term outcomes. The remaining study (Bergdahl 1995a) only reported long‐term outcome assessment.
We were unable to include data from two studies, due to excessive attrition in the placebo arm (79%) in one study (Bogetto 1999), and in the other study there was not only substantial attrition (23%) during the cross‐over trial's first phase, but all patients that developed side effects were also withdrawn (Silvestre 2012). All other trials were included in the quantitative analysis.
Primary outcomes
Symptom relief
A broad range of scales were used to assess symptom relief (characterised by included studies as change in burning/symptom intensity/pain).
Visual analogue scale (VAS), or an alternatively named variation, of varying widths (n = 17) (Bergdahl 1995a; Cano‐Carrillo 2014 (reported separately for pain and burning); Carbone 2009; Cavalcanti 2009; Grémeau‐Richard 2004; Heckmann 2012; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Sardella 2008; Silvestre 2012; Spanemberg 2012; Spanemberg 2015; Tammiala‐Salonen 1999).
Bespoke BMS symptomology change scale (n = 3) (Femiano 2000; Femiano 2002a; Femiano 2002b).
Clinical Global Impression I (CGI I) scale (n = 1) (Bogetto 1999).
Bespoke geographical burning distribution numerical scale (0 to 4) (n = 1) (Lopez‐D'alessandro 2011).
EDOF‐HC (Orofacial Pain Clinic ‐ Hospital das Clinicas) protocol (n = 1) (Alvarenga da Silva 2014).
Furthermore, four studies supplemented their VAS assessment of symptom relief with:
McGill Pain Questionnaire (MPQ) (Carbone 2009; Tammiala‐Salonen 1999);
Faces scale (FS) (0 to 5) (Spanemberg 2012);
visual numeric scale (VNS) (Spanemberg 2015);
Global Perceived Effect (GPE) scale (Cavalcanti 2009).
Change in quality of life (QoL)
Seven studies either directly assessed change in QoL or used surrogate markers for its assessment.
Four studies directly assessed change in QoL.
Two studies used a combination of assessing both general QoL (by using the 36‐Item Short Form Health Survey (SF‐36)) and the impact of oral health on patients' QoL (by using one of two versions of the Oral Health Impact Profile (OHIP‐14 or OHIP‐49)) (Cano‐Carrillo 2014 (SF‐36 and OHIP‐14); López‐Jornet 2011 (SF‐36 and OHIP‐49)).
One study singularly used OHIP‐14 to assess change in QoL (Spanemberg 2015).
The fourth study assessed QoL by posing simple questions to patients, rather than using a standardised or validated QoL questionnaire (Sardella 2008).
The remaining three studies used surrogate markers to assess change in QoL.
Two studies assessed change in depression using the Beck Depression Inventory (BDI) (Heckmann 2012; Tammiala‐Salonen 1999).
The third study assessed change in anxiety using the HARS scale, and change in depression using the Montgomery Asberg Depression Rating Scale (MADRS) (Bogetto 1999).
Furthermore, two studies supplemented their direct assessment of change in QoL with adjunctive use of the HAD (Hospital Anxiety and Depression) scale to also assess anxiety and depression as surrogate markers of QoL (Cano‐Carrillo 2014; López‐Jornet 2011).
Secondary outcomes
Change in taste
Only two studies assessed change in taste. One study used a taste test score (Heckmann 2012), and the other assessed change in taste by use of the gustative threshold from the Quantitative Sensory Testing (QST) protocol's combination battery of 12 tests (Alvarenga da Silva 2014). One study contributed data to quantitative synthesis (Heckmann 2012).
Change in feeling of dryness
A single study (Alvarenga da Silva 2014) assessed change in feeling of dryness, using a xerostomia questionnaire; however no data contributed to quantitative synthesis due to it being narratively reported only.
Adverse effects
Side effects were included as an outcome measure in this review update, despite not formally being included as an outcome in the original protocol.
Seven studies clearly reported the occurrence of adverse effects experienced in their trials (Cavalcanti 2009; Grémeau‐Richard 2004; Marino 2010; Rodríguez de Rivera‐Campillo 2010; Silvestre 2012; Spanemberg 2012; Tammiala‐Salonen 1999).
Six studies did not report adverse effects (Alvarenga da Silva 2014; Bergdahl 1995a; Bogetto 1999; Heckmann 2012; Palacios‐Sánchez 2015; Spanemberg 2015).
Five studies reported that no adverse effects occurred (Cano‐Carrillo 2014; Carbone 2009; Femiano 2000; López‐Jornet 2011; Sardella 1999).
The remaining five studies provided a partial narrative report of the occurrence of adverse effects.
Three studies indicated potential missing adverse effects data ("without notable adverse effects" (Femiano 2002a); "very mild" (although data were provided by author upon request) (Lopez‐D'alessandro 2011); "minimal" (López‐Jornet 2009b)).
One multi‐armed trial omitted reporting adverse effects data for one of the arms (lactoperoxidase data missing (Femiano 2002b)).
One trial reported the occurrence of an adverse event as rationale for a patient withdrawal but made no further mention of adverse effects experienced (Sardella 2008).
Nine included studies contributed adverse events data towards quantitative synthesis (Cavalcanti 2009; Femiano 2002b; Grémeau‐Richard 2004; Lopez‐D'alessandro 2011; López‐Jornet 2009b; Rodríguez de Rivera‐Campillo 2010; Silvestre 2012; Spanemberg 2012; Tammiala‐Salonen 1999).
Excluded studies
We excluded a total of 37 studies (Characteristics of excluded studies table). The main reasons for exclusion were that the trial did not have a placebo arm (13 studies: Bai 2010; Bessho 1998; Campisi 1997; Grechko 1996; Huang 2006; Huang 2009; López‐Jornet 2013; Lu 2002; Maina 2002; Peng 2001; Pisanty 1975; Qui 2010; Yong 2003); was not an RCT (10 studies: Ferguson 1981; Forabosco 1992; Grushka 1998; Hirsch 2011; Hugoson 1991; Ito 2010; Kho 2010; Petruzzi 2004; Romeo 2010; Woda 1998); there were insufficient details to permit inclusion as the articles were conference abstracts or letters to the editor (three studies: Palacios‐Sanchez 2010; Pellegrini 2010; Vukoja 2011); we were unable to locate the articles (three studies: Li 2002; Ma 2006; Mo 2003); inappropriate design (two studies: placebo group outcome assessed after one month and intervention group assessed after three months (Miziara 2009) and intervention/placebo immediately assessed, no clinical application (Grémeau‐Richard 2010)); the diagnosis of BMS was uncertain (two studies: Bogetto 1997; Toida 2009); or data for BMS participants were combined with other non‐BMS diagnoses (four studies: Hansen 1990; Lamey 1986; Lindholm 2015; Loldrup 1989).
Risk of bias in included studies
A summary of the risk of bias for each individual study is presented in Figure 2, and overall across studies for each risk of bias domain in Figure 3. One study was deemed to have a low overall risk of bias (Sardella 2008), four studies were deemed to have an unclear overall risk of bias (Carbone 2009; Grémeau‐Richard 2004; Sardella 1999; Tammiala‐Salonen 1999), and the remainder were deemed to have a high overall risk of bias.
2.
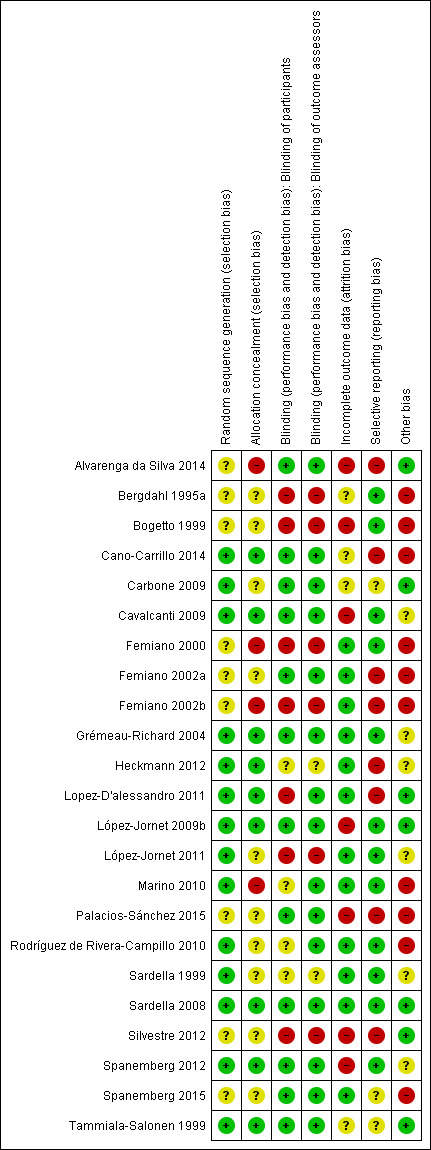
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
3.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Nine studies were deemed to have a low risk of selection bias, after taking into account both random sequence generation and allocation concealment (Cano‐Carrillo 2014; Cavalcanti 2009; Grémeau‐Richard 2004; Heckmann 2012; Lopez‐D'alessandro 2011; López‐Jornet 2009b; Sardella 2008; Spanemberg 2012; Tammiala‐Salonen 1999). Ten studies were found to have an unclear risk of selection bias, after taking into account both random sequence generation and allocation concealment (Bergdahl 1995a; Bogetto 1999; Carbone 2009; Femiano 2002a; López‐Jornet 2011; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Silvestre 2012; Spanemberg 2015). The remaining four studies were deemed to be at high risk of selection bias (Alvarenga da Silva 2014; Femiano 2000; Femiano 2002b; Marino 2010).
Blinding
Twelve studies were assessed as being at low risk of performance bias (Alvarenga da Silva 2014; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2002a; Grémeau‐Richard 2004; López‐Jornet 2009b; Palacios‐Sánchez 2015; Sardella 2008; Spanemberg 2012; Spanemberg 2015; Tammiala‐Salonen 1999); seven were assessed as being at high risk of performance bias (Bergdahl 1995a; Bogetto 1999; Femiano 2000; Femiano 2002b; Lopez‐D'alessandro 2011; López‐Jornet 2011; Silvestre 2012). All other studies were assessed as at unclear risk of performance bias.
With regard to detection bias, 15 studies were assessed as being at low risk of bias (Alvarenga da Silva 2014; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2002a; Grémeau‐Richard 2004; Lopez‐D'alessandro 2011; López‐Jornet 2009b; Marino 2010; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Sardella 2008; Spanemberg 2012; Spanemberg 2015; Tammiala‐Salonen 1999); six studies were assessed as being at high risk of bias (Bergdahl 1995a; Bogetto 1999; Femiano 2000; Femiano 2002b; López‐Jornet 2009b; Silvestre 2012). All other studies were assessed as unclear risk of detection bias.
Twelve studies were assessed as being at low risk of bias for both performance and detection bias (Alvarenga da Silva 2014; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2002a; Grémeau‐Richard 2004; López‐Jornet 2009b; Palacios‐Sánchez 2015; Sardella 2008; Spanemberg 2012; Spanemberg 2015; Tammiala‐Salonen 1999).
Reasons for assessing studies at unclear or high risk of bias for performance and detection bias included vast differences in treatment arm modalities, authors of studies describing them as 'open label', and adverse events likely to have precluded blinding of participants.
Incomplete outcome data
Twelve studies were deemed to have a low risk of attrition bias, after accounting for incomplete outcome data (Femiano 2000; Femiano 2002a; Femiano 2002b; Grémeau‐Richard 2004; Heckmann 2012; Lopez‐D'alessandro 2011; López‐Jornet 2011; Marino 2010; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Sardella 2008; Spanemberg 2015).
Seven studies were deemed to have a high risk of attrition bias, after accounting for incomplete outcome data (Alvarenga da Silva 2014; Bogetto 1999; Cavalcanti 2009; López‐Jornet 2009b; Palacios‐Sánchez 2015; Silvestre 2012; Spanemberg 2012). In five studies, there was inadequate explanation as to why dropouts occurred (Alvarenga da Silva 2014; Bogetto 1999; López‐Jornet 2009b; Palacios‐Sánchez 2015; Spanemberg 2012); one study presented data in a flow diagram (which stated two participants in each arm were excluded from the analysis), which did not match details provided in the text (which stated seven patients had their data excluded from analysis) ‐ hence the study was assessed as at high risk of bias (Cavalcanti 2009); one study removed participants who had experienced adverse events from the analysis (Silvestre 2012).
Four studies were deemed to have an unclear risk of attrition bias, after accounting for incomplete outcome data (Bergdahl 1995a; Cano‐Carrillo 2014; Carbone 2009; Tammiala‐Salonen 1999).
Selective reporting
Twelve studies were deemed to have a low risk of reporting bias, after accounting for selective outcome reporting (Bergdahl 1995a; Bogetto 1999; Cavalcanti 2009; Femiano 2000; Grémeau‐Richard 2004; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Sardella 2008; Spanemberg 2012).
Eight studies were deemed to have a high risk of reporting bias, after accounting for selective outcome reporting (Alvarenga da Silva 2014; Cano‐Carrillo 2014; Femiano 2002a; Femiano 2002b; Heckmann 2012; Lopez‐D'alessandro 2011; Palacios‐Sánchez 2015; Silvestre 2012).
Five studies failed to report on prespecified or expected outcomes (Cano‐Carrillo 2014; Femiano 2002a; Femiano 2002b; Lopez‐D'alessandro 2011; Palacios‐Sánchez 2015).
Three studies did not report their prespecified outcomes appropriately or fully (Alvarenga da Silva 2014; Heckmann 2012; Silvestre 2012).
Three studies were deemed to have an unclear risk of reporting bias, after accounting for selective outcome reporting (Carbone 2009; Spanemberg 2015; Tammiala‐Salonen 1999).
Other potential sources of bias
Seven studies were determined to have a low risk of other potential sources of bias (Alvarenga da Silva 2014; Carbone 2009; Lopez‐D'alessandro 2011; López‐Jornet 2009b; Sardella 2008; Silvestre 2012; Tammiala‐Salonen 1999)
Ten studies were considered to have a high risk of other potential sources of bias (Bergdahl 1995a; Bogetto 1999; Cano‐Carrillo 2014; Femiano 2000; Femiano 2002a; Femiano 2002b; Marino 2010; Palacios‐Sánchez 2015; Rodríguez de Rivera‐Campillo 2010; Spanemberg 2015).
Eight studies indicated cause for concern over baseline comparability in addition to their study's unclearly described randomisation process (Bergdahl 1995a; Bogetto 1999; Femiano 2000; Femiano 2002a; Femiano 2002b; Marino 2010; Palacios‐Sánchez 2015; Spanemberg 2015).
We had lesser concerns over baseline comparability in one study (Cano‐Carrillo 2014) due to reporting appropriate randomisation; however the distribution of anxiolytic use amongst patients was unclear.
One study (Rodríguez de Rivera‐Campillo 2010) indicated wide use of psychoactive medication within sample and adjunctive non‐standardised psychotherapy delivered at the discretion of three uncalibrated clinicians; however, the distribution of patients in receipt of psychotherapy was not reported.
Six were judged to have an unclear risk of other potential sources of bias (Cavalcanti 2009; Grémeau‐Richard 2004; Heckmann 2012; López‐Jornet 2011; Sardella 1999; Spanemberg 2012).
Three studies (Cavalcanti 2009; Grémeau‐Richard 2004; Sardella 1999) indicated concerns over baseline comparability, although an appropriate randomisation process was undertaken.
One study's intervention was a systemic anxiolytic drug (Heckmann 2012), correspondingly baseline anxiety should have been assessed to account for confounding.
One study (López‐Jornet 2011) allowed patient use of anxiolytics within sample although the distribution between groups was unclear.
The remaining study (Spanemberg 2012) presented contradictory text within paper, raising concerns of reporting accuracy.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9
Summary of findings for the main comparison. Antidepressants versus placebo for treating people with burning mouth syndrome.
| Antidepressants compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: antidepressants (trazodone) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Antidepressants | |||||
|
Symptom relief: short‐term (≤ 3 months) ‐ Mean VAS pain score (Scale 0‐10: lower better) |
4.66 | 1.26 higher (0.24 lower to 2.76 higher) | ‐ | 37 (1 RCT) | ⊕⊝⊝⊝ very low1 | Only trazodone was assessed by a single study No data were available to estimate long‐term symptom relief No data were available to estimate the effect of other antidepressants |
| Change in quality of life (QoL): short‐term (≤ 3 months) ‐ Mean Beck Depression Inventory score(Scale 0‐63: lower better) | This single study narratively reported that both intervention and placebo participants were less depressed at trial completion, but there was no evidence of a difference between groups | 37 (1 RCT) |
⊕⊝⊝⊝ very low2 | Single study assessing trazodone No data were available to estimate long‐term change in QoL |
||
| Change in taste | No included studies reported change in taste | |||||
| Change in feeling of dryness | No included studies reported change in feeling of dryness | |||||
| Adverse effects | There was evidence of an increase in dizziness (RR 11.61, 95% CI 1.66 to 81.04) and drowsiness (RR 4.75, 95% CI 1.18 to 19.07) in people treated with antidepressants | 37 (1 RCT) | ⊕⊝⊝⊝ very low3 | Only trazodone was assessed by a single study No data were available to estimate the harms of other antidepressants |
||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: confidence interval; OIS: optimal information size; RCT: randomised controlled trial; RR: risk ratio; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Assumed placebo risk based on mean placebo group pain score at short‐term (≤ 3 months) follow‐up; downgraded once due to risk of bias: unclear risk of attrition and reporting biases; downgraded once due to indirectness: concerns relating to applicability, only 1 type of antidepressant assessed, effects of other antidepressants may differ; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect. 2Downgraded once due to unclear risk of attrition and reporting biases; downgraded twice due to indirectness: use of surrogate measure, and also concerns relating to applicability (only 1 type of antidepressant assessed, effects of other antidepressants may differ); downgraded once due to imprecision: OIS not met. 3Downgraded once due to risk of bias: unclear risk of attrition and reporting biases; downgraded once due to indirectness: concerns relating to applicability, only 1 type of antidepressant assessed, effects of other antidepressants may differ; downgraded twice due to imprecision: OIS not met, and narrative report did not permit estimation of effect or 95% CI.
Summary of findings 2. Anticonvulsants versus placebo for treating people with burning mouth syndrome.
| Anticonvulsants compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: anticonvulsants (gabapentin: +/‐ alpha lipoic acid (ALA)) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Anticonvulsants | |||||
|
Symptom relief: short‐term (≤ 3 months) ‐ Bespoke geographical burning distribution numerical scale (0‐4) (Dichotomised) |
150 per 1000 | 600 per 1000 (313 to 1000) | RR 4.00 (2.09 to 7.67) | 100 (1 RCT) | ⊕⊝⊝⊝ very low1 | No data were available to estimate long‐term symptom relief Only gabapentin (+/‐ adjunctive ALA) was assessed by a single study No data were available to estimate the effect of other anticonvulsants |
| Change in quality of life (QoL) | No included studies reported change in QoL | |||||
| Change in taste | No included studies reported change in taste | |||||
| Change in feeling of dryness | No included studies reported change in feeling of dryness | |||||
| Adverse effects | There was evidence of an increase in drowsiness (RR 31.95, 95% CI 1.84 to 553.64) in people treated with gabapentin alone (1 RCT, 80 participants). This effect was not found for people treated with gabapentin + adjunctive ALA (1 RCT, 80 participants), nor was there evidence of an increase in mild headaches | 100 (1 RCT) |
⊕⊝⊝⊝ very low2 | Only gabapentin (+/‐ adjunctive ALA) was assessed by a single study No data were available to estimate the harms of other anticonvulsants | ||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: confidence interval; OIS: optimal information size; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Assumed placebo risk based on control estimate at short‐term (≤ 3 months) follow‐up: total of control participants experiencing symptom relief, divided by total number of participants in control group multiplied by 100; downgraded twice due to high risk of bias across multiple domains; downgraded once due to indirectness: concerns relating to applicability, only 1 type of anticonvulsant assessed, effects of other anticonvulsants may differ; downgraded once due to imprecision: OIS not met. 2Single 4‐armed study (totalling 120 people), with 3 of 4 groups included in this comparison (placebo group: 60 people; gabapentin group: 20 people; gabapentin + ALA group: 20 people), the remaining group (ALA: 20 people) and placebo group (60 people) are included within Table 5; downgraded twice due to high risk of bias across multiple domains; downgraded once due to indirectness: concerns relating to applicability, only 1 type of anticonvulsant assessed, effects of other anticonvulsants may differ; downgraded once due to imprecision: OIS not met.
Summary of findings 3. Benzodiazepines versus placebo for treating people with burning mouth syndrome.
| Benzodiazepines compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: benzodiazepines (topical/systemic clonazepam) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Benzodiazepines | |||||
|
Symptom relief: short‐term (≤ 3 months) ‐ Mean VAS pain score (Scale 0‐10: lower better) |
4.76 | 1.89 lower (2.19 to 1.59 lower) | ‐ | 111 (2 RCTs) | ⊕⊝⊝⊝ very low1 | There was evidence of a difference between subgroups between the 2 subgroups: systemic and topical clonazepam Short‐term relief is estimated from 2 topical clonazepam studies. Long‐term symptom relief (> 3 to ≤ 6 months) was 1.39 lower (1.96 to 0.83 lower) than the placebo group (1 RCT, 66 participants) No evidence of short‐term symptom relief from a single study assessing systemic clonazepam (≤ 3 months): mean VAS score in both placebo and intervention groups was 4.5 (MD 0.00 lower, 95% CI 1.86 lower to 1.86 higher) (1 RCT, 20 participants) No data were available to estimate the effect of other benzodiazepines |
| Change in quality of life (QoL): short‐term (≤ 3 months) ‐ Mean depression score(Scale 0‐4: lower better) | 0.8 | 0.20 lower (0.95 lower to 0.55 higher) | ‐ | 20 (1 RCT) | ⊕⊝⊝⊝ very low2 | Single study assessing systemic clonazepam No data were available to estimate long‐term change in QoL |
|
Change in taste: short‐term (≤ 3 months) ‐ Mean taste test score (Scale 0‐16: higher better) |
12.3 | 1.00 lower (3.11 lower to 1.11 higher) | ‐ | 20 (1 RCT) | ⊕⊝⊝⊝ very low3 | Single study assessing systemic clonazepam No data were available to estimate long‐term change in taste |
| Change in feeling of dryness | No included studies reported change in feeling of dryness | |||||
| Adverse effects | There was no difference in the adverse events reported (drowsiness: 2 RCTs, 114 participants; dry mouth, spasmophilia, or euphoric behaviour: 1 RCT, 48 participants) for people treated with benzodiazepines | ⊕⊝⊝⊝ very low4 | Both studies used topical clonazepam We were unable to estimate the harms of systemic clonazepam as adverse events were not reported by its study No data were available to estimate the harms of other benzodiazepines |
|||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: confidence interval; MD: mean difference; OIS: optimal information size; RCT: randomised controlled trial; RR: risk ratio; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Assumed placebo risk based on mean placebo group pain score at short‐term (≤ 3 months) follow‐up; downgraded once due to studies being assessed at unclear risk of bias across multiple domains; downgraded once due to inconsistency: substantial heterogeneity (I2 = 56%); downgraded once due to indirectness: concerns relating to applicability, only 1 type of benzodiazepine (clonazepam ‐ irrespective of mode of administration) assessed, effects of other benzodiazepines may differ; downgraded once due to imprecision: OIS not met. 2Assumed placebo risk based on mean placebo group depression score at short‐term (≤ 3 months) follow‐up; downgraded once due to high risk of selective reporting bias; downgraded twice due to indirectness: use of surrogate measure, and also concerns relating to applicability, only 1 type of benzodiazepine (clonazepam ‐ irrespective of mode of administration) assessed, effects of other benzodiazepines may differ; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect. 3Assumed placebo risk based on mean placebo group taste score at short‐term (≤ 3 months) follow‐up; downgraded once due to indirectness: concerns relating to applicability, only 1 type of benzodiazepine (clonazepam ‐ irrespective of mode of administration) assessed, effects of other benzodiazepines may differ; downgraded once due to high risk of selective reporting bias; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect. 4Downgraded once due to studies being assessed at unclear risk of bias across multiple domains; downgraded once due to indirectness: concerns relating to applicability, only 1 type of benzodiazepine (clonazepam ‐ irrespective of mode of administration) assessed, effects of other benzodiazepines may differ; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect.
Summary of findings 4. Cholinergics versus placebo for treating people with burning mouth syndrome.
| Cholinergics compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: cholinergics (bethanechol) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Cholinergics | |||||
|
Symptom relief: short‐term (≤ 3 months) ‐ Bespoke BMS symptomology change scale (Dichotomised) |
150 per 1000 | 750 per 1000 (39 to 1000) | RR 5.00 (0.26 to 98.00) | 40 (1 RCT) | ⊕⊝⊝⊝ very low1 | No data were available to estimate long‐term symptom relief Only bethanechol was assessed by a single study No data were available to estimate the effect of other cholinergics |
| Change in quality of life (QoL) | No included studies reported change in QoL | |||||
| Change in taste | No included studies reported change in taste | |||||
| Change in feeling of dryness | No included studies reported change in feeling of dryness | |||||
| Adverse effects | This single study narratively reported adverse effect data. 4 participants (20%) in the bethanechol arm reported adverse events (nausea, dizziness, blood pressure fall, cold perspiration or sporadic abdominal pain). Reported data did not present the distribution of these outcomes by participant | 40 (1 RCT) | ⊕⊝⊝⊝ very low2 | Only bethanechol was assessed by a single study No data were available to estimate the harms of other cholinergics | ||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) BMS: burning mouth syndrome; CI: confidence interval; OIS: optimal information size; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1No placebo group participants experienced pain relief in the single study assessing cholinergics. Consequently, in order to calculate the corresponding risk, the assumed placebo risk for cholinergics is based on the assumed placebo risk for anticonvulsants, as detailed in Table 2; downgraded twice: risk of bias concerns across multiple domains (allocation, blinding, selective reporting and absence of baseline data); downgraded once due to indirectness: concerns relating to applicability, only 1 type of anticholinergic assessed, effects of other cholinergics may differ; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect. 2Downgraded twice: risk of bias concerns across multiple domains (allocation, blinding, selective reporting and absence of baseline data); downgraded once due to indirectness: concerns relating to applicability, only 1 type of anticholinergic assessed, effects of other cholinergics may differ; downgraded twice due to imprecision: OIS not met, and narrative report did not permit estimation of effect or 95% CI.
Summary of findings 5. Dietary supplements versus placebo for treating people with burning mouth syndrome.
| Dietary supplements compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: dietary supplements (ALA with (+ vitamins, + lycopene + green tea extract) or without adjunctive active ingredients; hypericum perforatum (St John's Wort); 'Catuama' herbal compound; lycopene) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Dietary supplements | |||||
| Symptom relief: short‐term (≤ 3 months) | There is insufficient or contradictory evidence regarding the benefit of any of the evaluated dietary supplements over placebo to evaluate short‐term symptom relief | 628 (12 RCTs) | ⊕⊝⊝⊝ very low1 | Interventions evaluated for short‐term symptom relief included:
The mean VAS pain score (scale 0‐10: lower better) for long‐term symptom relief (> 3 to ≤ 6 months) was 0.89 lower (2.37 lower to 0.59 higher) for people treated with ALA (with or without adjunctive vitamins) than the placebo group (2 RCTs, 94 participants) |
||
|
Change in quality of life (QoL): short‐term (≤ 3 months) ‐ Mean OHIP‐14 score (Scale 0‐70: lower better) |
17.38 | 0.93 higher (3.14 lower to 5.00 higher) | ‐ | 50 (1 RCT) | ⊕⊝⊝⊝ very low2 | Single study assessing short‐term QoL, anxiety and depression compared lycopene with placebo No data were available to estimate long‐term change in QoL, or its surrogate markers: anxiety or depression No data were available to estimate the effect of other dietary supplements; however, 1 study (43 patients), comparing hypericum perforatum extract with placebo, evaluated QoL from patient self‐reports and narratively indicated that both intervention and placebo participants were better able to cope at trial completion |
|
Change in QoL ‐ anxiety: short‐term (≤ 3 months) ‐ Mean HAD anxiety score (Scale 0‐21: lower better) |
11.5 | 2.85 lower (5.28 to 0.42 lower) | ⊕⊝⊝⊝ very low3 | |||
|
Change in QoL ‐ depression: short‐term (≤ 3 months) ‐ Mean HAD depression score (Scale 0‐21: lower better) |
6.25 | 1.87 lower (4.23 lower to 0.49 higher) | ⊕⊝⊝⊝ very low4 | |||
| Change in taste | No included studies reported change in taste | |||||
| Change in feeling of dryness | No included studies reported change in feeling of dryness | |||||
| Adverse effects |
|
⊕⊝⊝⊝ very low5 | ||||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) ALA: alpha lipoic acid; CI: confidence interval; HAD: Hospital Anxiety and Depression scale; OHIP‐14: Oral Health Impact Profile‐14; OIS: optimal information size; RCT: randomised controlled trial; RR: risk ratio; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Downgraded twice due to high risk of bias across multiple domains; downgraded twice due to inconsistency between, and within, subgroups; 1 RCT (Carbone 2009) contributes to 2 subgroups: ALA with vitamins and ALA without adjunctive active ingredients. 2Assumed placebo risk based on mean placebo group QoL score at short‐term (≤ 3 months) follow‐up; downgraded once due to high risk of bias relating to selective reporting and lack of baseline data; downgraded once due to indirectness: concerns relating to applicability, only 1 type of dietary supplement assessed, effects of other dietary supplements may differ; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect. 3Assumed placebo risk based on mean placebo group anxiety score at short‐term (≤ 3 months) follow‐up; downgraded once due to high risk of bias relating to selective reporting and lack of baseline data; downgraded twice due to indirectness: use of surrogate measure, and also concerns relating to applicability, only 1 type of dietary supplement assessed, effects of other dietary supplements may differ; downgraded once due to imprecision: OIS not met. 4Assumed placebo risk based on mean placebo group depression score at short‐term (≤ 3 months) follow‐up; downgraded once due to high risk of bias relating to selective reporting and lack of baseline data; downgraded twice due to indirectness: use of surrogate measure, and also concerns relating to applicability, only 1 type of dietary supplement assessed, effects of other dietary supplements may differ; downgraded once due to imprecision: OIS not met. 5Downgraded twice due to high risk of bias across multiple domains; downgraded twice due to inconsistency between, and within, subgroups; downgraded once for imprecision: wide CIs estimated around effect sizes.
Summary of findings 6. Electromagnetic radiation versus placebo for treating people with burning mouth syndrome.
| Electromagnetic radiation compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: electromagnetic radiation (low‐level laser therapy) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Electromagnetic radiation | |||||
|
Symptom relief: short‐term (≤ 3 months) ‐ Mean VAS score (Scale 0‐100: lower better) |
62.84 | 30.36 lower (44.22 to 16.50 lower) | ‐ | 58 (1 RCT) | ⊕⊝⊝⊝ very low1 | No data were available to estimate long‐term symptom relief Only low‐level laser therapy (infrared laser and red laser) was assessed by a single study No data were available to estimate the effect of other types of electromagnetic radiation (e.g. transcranial magnetic stimulation) |
|
Change in quality of life (QoL): short‐term (≤ 3 months) ‐ Mean OHIP‐14 score (Scale 0‐56: lower better) |
13.39 | 5.24 lower (7.38 to 3.09 lower) | ⊕⊝⊝⊝ very low2 | |||
| Change in taste | No included study reported change in taste | |||||
| Change in feeling of dryness | No included study reported change in feeling of dryness | |||||
| Adverse effects | No included study reported the occurrence of adverse events | |||||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OHIP‐14: Oral Health Impact Profile‐14; OIS: optimal information size; RCT: randomised controlled trial; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Assumed placebo risk based on mean placebo group pain score at short‐term (≤ 3 months) follow‐up; downgraded once due to the single study being assessed at unclear risk of bias across multiple domains and high risk of bias relating to other bias; downgraded once due to indirectness: concerns relating to applicability, only 1 type of electromagnetic radiation assessed, effects of other types of electromagnetic radiation may differ; downgraded once due to imprecision: OIS not met. 2Assumed placebo risk based on mean placebo group QoL score at short‐term (≤ 3 months) follow‐up; downgraded once due to the single study being assessed at unclear risk of bias across multiple domains and high risk of bias relating to other bias; downgraded once due to indirectness: concerns relating to applicability, only 1 type of electromagnetic radiation assessed, effects of other types of electromagnetic radiation may differ; downgraded once due to imprecision: OIS not met.
Summary of findings 7. Physical barriers versus placebo for treating people with burning mouth syndrome.
| Physical barriers compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: physical barriers (tongue protector) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Physical barriers | |||||
| Symptom relief: short‐term (≤ 3 months) ‐ Mean VAS pain score (Scale 0‐10: lower better) | 5.6 | 1.10 lower (2.14 to 0.06 lower) | ‐ | 50 (1 RCT) | ⊕⊝⊝⊝ very low1 | Single study assessing tongue protectors No data were available to estimate long‐term symptom relief |
| Change in quality of life (QoL): short‐term (≤ 3 months) ‐ Mean OHIP‐49 score (Scale 0‐196: lower better) | 53.72 | 9.20 lower (26.90 lower to 8.50 higher) | ‐ | 50 (1 RCT) | ⊕⊝⊝⊝ very low2 | Single study assessing tongue protectors No data were available to estimate long‐term change in QoL, or its surrogate markers: anxiety or depression |
|
Change in QoL ‐ anxiety: short‐term (≤ 3 months) ‐ Mean HAD anxiety score (Scale 0‐21: lower better) |
11.04 | 0.16 higher (3.19 lower to 3.51 higher) | ‐ | ⊕⊝⊝⊝ very low3 | ||
|
Change in QoL ‐ depression: short‐term (≤ 3 months) ‐ Mean HAD depression score (Scale 0‐21: lower better) |
8.92 | 0.64 lower (3.98 lower to 2.70 higher) | ‐ | ⊕⊝⊝⊝ very low4 | ||
| Change in taste | No included studies reported change in taste | |||||
| Change in feeling of dryness | No included studies reported change in feeling of dryness | |||||
| Adverse effects | The single study narratively reported that no adverse events occurred from the use of tongue protectors | 50 (1 RCT) | ⊕⊕⊝⊝ low5 | |||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: confidence interval; HAD: Hospital Anxiety and Depression scale; OHIP‐49: Oral Health Impact Profile‐49; OIS: optimal information size; RCT: randomised controlled trial; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Assumed placebo risk based on mean placebo group pain score at short‐term (≤ 3 months) follow‐up; downgraded twice due to serious concerns of risk of bias, relating to blinding and potential confounding influence of participants in receipt of anxiolytics; downgraded once due to imprecision: OIS not met. 2Assumed placebo risk based on mean placebo group quality of life score at short‐term (≤ 3 months) follow‐up; downgraded twice due to serious concerns of risk of bias, relating to blinding and potential confounding influence of participants in receipt of anxiolytics; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect. 3Assumed placebo risk based on mean placebo group anxiety score at short‐term (≤ 3 months) follow‐up; downgraded twice due to serious concerns of risk of bias, relating to blinding and potential confounding influence of participants in receipt of anxiolytics; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect; downgraded once due to indirectness: use of surrogate measure. 4Assumed placebo risk based on mean placebo group depression score at short‐term (≤ 3 months) follow‐up; downgraded twice due to serious concerns of risk of bias, relating to blinding and potential confounding influence of participants in receipt of anxiolytics; downgraded twice due to imprecision: OIS not met, and 95% CI includes no effect; downgraded once due to indirectness: use of surrogate measure. 5Downgraded twice due to serious concerns of risk of bias, relating to blinding and potential confounding influence of participants in receipt of anxiolytics; downgraded once due to imprecision: OIS not met.
Summary of findings 8. Psychological therapies versus placebo for treating people with burning mouth syndrome.
| Psychological therapies compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: psychological therapies (cognitive therapy) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Psychological therapies | |||||
|
Symptom relief: long‐term (> 3 to ≤ 6 months) ‐ Mean VAS pain score (Scale 1‐7: lower better) |
4.6 | 3.20 lower (4.22 to 2.18 lower) | ‐ | 30 (1 RCT) | ⊕⊝⊝⊝ very low1 | Single study assessing cognitive therapy No data was available to estimate short‐term symptom relief No data was available to estimate the effect of other psychological therapies |
| Change in quality of life (QoL) | No included study reported change in QoL | |||||
| Change in taste | No included study reported change in taste | |||||
| Change in feeling of dryness | No included study reported change in feeling of dryness | |||||
| Adverse effects | No included study reported the occurrence of adverse events | |||||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: confidence interval; OIS: optimal information size; RCT: randomised controlled trial; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Assumed placebo risk based on mean placebo group pain score at long‐term (> 3 to ≤ 6 months) follow‐up; downgraded twice due to serious concerns of risk of bias, relating to blinding and lack of baseline data; downgraded once due to indirectness: concerns relating to applicability, only 1 type of psychological therapy assessed, effects of other psychological therapies may differ; downgraded once due to imprecision: OIS not met.
Summary of findings 9. Topical treatments versus placebo for treating people with burning mouth syndrome.
| Topical treatments compared with placebo for treating burning mouth syndrome | ||||||
|
Patient or population: people diagnosed with burning mouth syndrome Settings: secondary care Intervention: topical treatments (benzydamine hydrochloride oral rinse, lactoperoxidase oral rinse (Biotene), topical urea (10%), capsaicin oral rinse) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Topical treatments | |||||
| Symptom relief: short‐term (≤ 3 months) | There is insufficient or contradictory evidence regarding the benefit of any of the evaluated topical treatments over placebo to evaluate short‐term symptom relief | 150 (4 RCTs) |
⊕⊝⊝⊝ very low1 | Interventions evaluated for short‐term symptom relief included:
The mean VAS pain score (scale 0‐10: lower better) for long‐term symptom relief (> 3 to ≤ 6 months) was 2.60 lower (5.11 to 0.09 lower) for people treated with capsaicin oral rinse than the placebo group (1 RCT, 18 participants) No evidence of long‐term symptom relief from a single study assessing lactoperoxidase oral rinse (Biotene): mean VAS score in intervention group was 1.50 lower than people treated with placebo (3.91 lower to 0.91 higher) (1 RCT, 18 participants) |
||
| Change in quality of life (QoL) | No included studies reported change in QoL | |||||
|
Change in taste: short‐term (≤ 3 months) ‐ Mean Quantitative Sensory Testing (QST) score: gustative threshold (Scale: unknown) |
A single study narratively reported that there was no difference in short‐term gustation thresholds | 38 (1 RCT) |
⊕⊝⊝⊝ very low2 | Single study assessing topical urea (10%) No data were available to estimate long‐term change in taste No data were available to estimate the effect of other topical treatments | ||
|
Change in feeling of dryness: short‐term (≤ 3 months) ‐ Xerostomia questionnaire (Scale 1‐5: lower better) |
A single study narratively reported that there was no difference in short‐term xerostomia questionnaire assessment before or after treatment | 38 (1 RCT) |
⊕⊝⊝⊝ very low3 | Single study assessing topical urea (10%) No data were available to estimate long‐term change in feeling of dryness No data were available to estimate the effect of other topical treatments | ||
| Adverse effects | A single study narratively reported that no adverse events occurred from the use of benzydamine hydrochloride oral rinse | 20 (1 RCT) | ⊕⊝⊝⊝ very low4 | No data were available to estimate the effect of other topical treatments | ||
| *The basis for the assumed risk is provided in footnotes. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: confidence interval; OIS: optimal information size; RCT: randomised controlled trial; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate Very low quality: we are very uncertain about the estimate | ||||||
1Downgraded twice due to high risk of bias across multiple domains; downgraded twice due to inconsistency between, and within, subgroups; downgraded once due to imprecision: OIS not met; 1 RCT (Marino 2010) contributes to 2 subgroups: capsaicin oral rinse and lactoperoxidase oral rinse (Biotene). 2Downgraded twice due to high risk of bias relating to attrition and selective reporting; downgraded once due to indirectness: concerns relating to applicability, only 1 type of topical therapy assessed, effects of other topical therapies may differ; downgraded once due to imprecision: OIS not met. 3Downgraded twice due to high risk of bias relating to attrition and selective reporting; downgraded once for indirectness: concerns relating to applicability, only 1 type of topical therapy assessed, effects of other topical therapies may differ; downgraded once due to imprecision: OIS not met. 4Downgraded once due to unclear risk of bias across multiple domains; downgraded once due to indirectness: concerns relating to applicability, only 1 type of topical therapy assessed, effects of other topical therapies may differ; downgraded once due to imprecision: OIS not met.
Interventions were allocated between nine groups to assess their efficacy: antidepressants and antipsychotics, anticonvulsants, benzodiazepines, cholinergics, dietary supplements, electromagnetic radiation, physical barriers, psychological therapies, and topical treatments. The quality of the evidence was assessed as very low for all outcomes (both short (up to three months from baseline) and long term (three to six months from baseline)) in all intervention categories (with the exception of adverse effects for physical barriers, which were assessed as low‐quality evidence). See Additional Table 12 for full details of reported adverse effects, Additional Table 13 for adverse effect outcome data, and Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9.
3. Reported adverse effects.
| Study ID | Comparison | Adverse effects reported |
| Alvarenga da Silva 2014 | Topical treatments | Comment: no adverse event data reported, nor is there any indication that it was collected by investigators |
| Bergdahl 1995a | Psychological therapies | Not reported |
| Bogetto 1999 | Antidepressants and antipsychotics | Not reported |
| Cano‐Carrillo 2014 | Dietary supplements | Quote: "No patients experienced any adverse effects resulting from treatment at any of the evaluation times" |
| Carbone 2009 | Dietary supplements | Quote: "No adverse events were reported in any of the three groups" (refers to ALA; ALA + vitamins; placebo) |
| Cavalcanti 2009 | Dietary supplements | Quote: "Seven patients had their data excluded from analysis, four of them did not complete the proposed treatment because of the symptoms that they judged as connected to treatment and these symptoms are presented with side‐effects data" Comment: adverse events fully reported in Table 3 in the paper. Of the 4 patients withdrawing due to severe adverse events, 2 were from each group (ALA and placebo) |
| Femiano 2000 | Dietary supplements | Quote: "No adverse events were recorded in any of the groups" |
| Femiano 2002a | Dietary supplements | Quote: "..symptomatic improvements in BMS treated with alpha‐lipoic acid were seen without notable adverse effects" Comment: Narrative report, indicating adverse events occurred, potentially missing data. No other adverse event data reported |
| Femiano 2002b | Cholinergics Dietary supplements Systemic treatments Topical treatments |
Quotes:
Bethanechol: "Four of the 20 patients complained of adverse effects (nausea, dizziness, blood pressure fall, cold perspiration or sporadic abdominal pain) but not severe enough to demand the suspension of treatment"
ALA: "Four patients reported heartburn but this was corrected by ranitidine 150 mg" Comment: bethanechol data not usable in analysis as distribution of 5 adverse events between 4 complainants was unclear. Adverse events for lactoperoxidase not reported |
| Grémeau‐Richard 2004 | Benzodiazepines | Quotes: "Side effects (Table 2) were not significantly more frequent in the active treatment group: they were reported by nine out of 24 subjects in the clonazepam group and six out of 24 in the placebo group (P > 0.05, X2‐test). Two subjects in the active treatment group and one in the control group dropped out from the trial because of these side effects" Comment: adverse events fully reported in 'Table 2' in the paper |
| Heckmann 2012 | Benzodiazepines | Not reported |
| Lopez‐D'alessandro 2011 | Anticonvulsants Dietary supplements |
Quote: "the adverse effects that appeared were very mild" Comment: narrative report, indicating adverse events occurred; data obtained from contact author Gabapentin arm (n = 20): drowsiness (n = 5) Gabapentin + ALA arm (n = 20): drowsiness (n = 2); mild headache (n = 1) ALA arm (n = 20): mild headache (n = 2); intermittent facial skin rash (n = 1) Placebo arm: no adverse effects reported |
| López‐Jornet 2009b | Dietary supplements | Quotes: "All patients responded to a questionnaire on each of the visits (0, 1, and 2 months) in which they were asked about the possible adverse effects of the medication"; "adverse effects were minimal; only one patient [ALA group] abandoned treatment because of having gastrointestinal upset as the side effect of the medication" Comment: narrative report, indicating adverse events occurred, potentially missing data. No other adverse event data reported, despite investigators repeatedly recording adverse events, and the statement relates only to the patient who withdrew from the trial due to severe side effects |
| López‐Jornet 2011 | Physical barriers | Quote: "No adverse effects were observed" |
| Marino 2010 | Dietary supplements Topical treatments |
Quote: "All the patients successfully finished study I [the treatment phase, distinct from follow‐up phase which 64% (36/56) completed] and no untoward effect occurred in any of the groups" Re: Capsaicin: "We did not observe any adverse effects in our patients and the treatment was well tolerated by the patients despite the need for repeated applications because of its short duration of action" Comment: no other adverse event data reported |
| Palacios‐Sánchez 2015 | Dietary supplements | Quote: "All patients were assessed every 15 days [...] for the occurrence of side effects" Comment: despite specifying assessing occurrence of adverse effects in the Methods section, there is no detail reporting their occurrence in the results |
| Rodríguez de Rivera‐Campillo 2010 | Benzodiazepines | Quotes: "All the patients were scheduled for a visit after 1 week for the sole purpose of detecting undesirable side effects. They were again scheduled for visits after 1 months and 6 months , which allowed the clinicians to monitor their evolution"; "Some patients reported a sensation of effervescence and numbness of the tongue when the tablet was dissolved. [...] The only side effect registered was some degree of sleepiness in 5 patients in the clonazepam group, which did not require the clinicians to suspend the treatment" |
| Sardella 1999 | Topical treatments | Quote: "No adverse effects were noted in the groups using the [Benzydamine] and placebo solutions" |
| Sardella 2008 | Dietary supplements | Quotes: "During the follow‐up visits [1 month, 2 months and 3 months], the patients were explicitly asked about the occurrence of adverse events"; "One subject in the test group failed to finish the trial because a severe headache that developed during the fifth week was considered a side effect of the therapy" Comment: despite recording adverse effects at 3 time points, no other adverse event data are reported except for explaining Group A dropout patient ‐ not sufficient to use for estimation of risk ratio, as other data likely to be omitted from paper |
| Silvestre 2012 | Topical treatments | Quotes: "Patients were also questioned about possible adverse effects or discomfort caused by the treatments"; "Those subjects who developed adverse effects were removed from the study"; "The study initially involved 30 patients, of which 7 abandoned the trial in the first week of treatment [...] two complained of greatly increased burning sensation when using the [capsaicin] rinse"; "intense burning sensation was described by one‐third of the subjects during and for a few minutes (maximum 20 minutes) after application of the capsaicin rinse" Comment: reports patients developing adverse effects were removed from the study; however, numbers for analysis estimated from text to be 8 patients ("one‐third" of group n = 23 to estimate whole patient integer) from intervention arm, in addition to the further 2 intervention arm patients who withdrew from the trial due to the increased burning sensation. Data combined for analysis |
| Spanemberg 2012 | Dietary supplements | Quotes: "12 withdrew from the study. Exacerbation of symptoms was reported by 6 individuals: 3 in the control group and 3 in the test group. 6 withdrew from the study for reasons unrelated to the treatment"; "One patient complained of somnolence and weight gain and another of insomnia. Two patients who took the test substance reported exacerbation of the symptoms in the first week of treatment, but this was also observed in 4 patients in the control group" Comment: adverse effects experienced by dropout patients referred to separately under rationale for withdrawals. Data combined for analysis |
| Spanemberg 2015 | Electromagnetic radiation | Not reported |
| Tammiala‐Salonen 1999 | Antidepressants and antipsychotics | Quote: "Patients in the trazodone group reported significantly more dizziness (P < 0.0001) and drowsiness (P < 0.05) than patients in the placebo group (Table 1). Two patients in the trazodone group and 8 in the placebo group reported no side effects" Comment: adverse events fully reported in Table 1 in the paper; however, effect P values estimated from reported data (tabulated in Additional Table 13) are more conservative than the effect estimates narratively reported in the paper |
ALA = alpha lipoic acid.
4. Adverse event outcomes.
| Antidepressants/antipsychotics versus placebo | ||||||
| Intervention | Outcome | No of studies | No of patients | Risk ratio (M‐H, Fixed, 95% CI) | Overall effect (P value) | Heterogeneity (P value; I2) |
| Antidepressants (trazodone) |
Dizziness | 1 | 37 | 11.61 (1.66 to 81.04) | P = 0.01 | Not applicable |
| Drowsiness | 1 | 37 | 4.75 (1.18 to 19.07) | P = 0.03 | Not applicable | |
| Abdominal pains | 1 | 37 | 1.32 (0.42 to 4.15) | P = 0.64 | Not applicable | |
| Headache | 1 | 37 | 1.58 (0.30 to 8.40) | P = 0.59 | Not applicable | |
| Palpitations | 1 | 37 | 1.06 (0.17 to 6.72) | P = 0.95 | Not applicable | |
| Tremor | 1 | 37 | 2.11 (0.21 to 21.32) | P = 0.53 | Not applicable | |
| Dry mouth | 1 | 37 | 3.17 (0.36 to 27.72) | P = 0.30 | Not applicable | |
| Urinary incontinence | 1 | 37 | 3.16 (0.14 to 72.84) | P = 0.47 | Not applicable | |
| Antipsychotics | Adverse effect data were not available for analysis from the single included study comparing antipsychotics versus placebo (Bogetto 1999) | |||||
| Anticonvulsants versus placebo | ||||||
| Intervention | Outcome | No of studies | No of patients | Risk ratio (M‐H, Fixed, 95% CI) | Overall effect (P value) | Heterogeneity (P value; I2) |
| Gabapentin | Drowsiness | 1 | 80 | 31.95 (1.84 to 553.64) | P = 0.02 | Not applicable |
| Gabapentin + ALA | Drowsiness | 1 | 80 | 14.52 (0.73 to 290.44) | P = 0.08 | Not applicable |
| Mild headache | 1 | 80 | 8.71 (0.37 to 205.80) | P = 0.18 | Not applicable | |
| Benzodiazepines versus placebo | ||||||
| Intervention | Outcome | No of studies | No of patients | Risk ratio (M‐H, Fixed, 95% CI) | Overall effect (P value) | Heterogeneity (P value; I2) |
| Topical clonazepam | Drowsiness | 2 | 114 | 2.71 (0.84 to 8.74) | P = 0.09 | P = 0.16; I2 = 48% |
| Dry mouth | 1 | 48 | 3.00 (0.13 to 70.16) | P = 0.49 | Not applicable | |
| Spasmophilia | 1 | 48 | 3.00 (0.13 to 70.16) | P = 0.49 | Not applicable | |
| Euphoric behaviour | 1 | 48 | 3.00 (0.13 to 70.16) | P = 0.49 | Not applicable | |
| Cholinergics versus placebo | ||||||
| Bethanechol | Adverse effect data presented collectively and not usable for analysis from the single included study comparing cholinergics versus placebo (Femiano 2002b) | |||||
| Dietary supplements versus placebo | ||||||
| Intervention | Outcome | No of studies | No of patients | Risk ratio (M‐H, Fixed, 95% CI) | Overall effect (P value) | Heterogeneity (P value; I2) |
| ALA (+/‐ adjunctive ingredients) | Gastrointestinal complaints | 3 | 138 | 4.00 (1.21 to 13.27) | P = 0.02 | P = 0.78; I2 = 0% |
| Headache | 2 | 118 | 10.87 (1.36 to 87.03) | P = 0.02 | P = 0.82; I2 = 0% | |
| Drowsiness | 1 | 38 | 1.00 (0.07 to 14.85) | P = 1.00 | Not applicable | |
| Increase in blood pressure | 1 | 38 | 1.00 (0.07 to 14.85) | P = 1.00 | Not applicable | |
| Intermittent facial skin rash | 1 | 80 | 8.71 (0.37 to 205.80) | P = 0.18 | Not applicable | |
| 'Catuama' herbal compound | Drowsiness | 1 | 72 | 2.69 (0.11 to 63.96) | P = 0.54 | Not applicable |
| Weight gain | 1 | 72 | 2.69 (0.11 to 63.96) | P = 0.54 | Not applicable | |
| Insomnia | 1 | 72 | 2.69 (0.11 to 63.96) | P = 0.54 | Not applicable | |
| Exacerbation of symptoms | 1 | 72 | 1.12 (0.33 to 3.83) | P = 0.86 | Not applicable | |
| Electromagnetic radiation versus placebo | ||||||
| Low‐level laser therapy | Adverse effect data were not available for analysis from the single included study comparing electromagnetic radiation versus placebo (Spanemberg 2015) | |||||
| Physical barriers versus placebo | ||||||
| Tongue protector | Adverse effect data were not available for analysis from the single included study comparing physical barriers versus placebo (López‐Jornet 2011) | |||||
| Psychological therapies versus placebo | ||||||
| Cognitive therapy | Adverse effect data were not available for analysis from the single included study comparing psychological therapies versus placebo (Bergdahl 1995a) | |||||
| Topical treatments versus placebo | ||||||
| Benzydamine hydrochloride oral rinse | Adverse data only presented for benzydamine hydrochloride oral rinse against placebo; narratively reported that no adverse effects occurred (Sardella 1999) | |||||
ALA = alpha lipoic acid; CI = confidence interval; M‐H = Mantel‐Haenszel.
Antidepressants and antipsychotics
Two studies (104 participants), at either high or unclear risk of bias, provided very low‐quality evidence comparing antidepressants and/or antipsychotics against placebo (Bogetto 1999 (paroxetine 20 mg daily; amitriptyline 25 mg daily; amisulpride 50 mg daily); Tammiala‐Salonen 1999 (trazodone 200 mg daily)).
Data from Bogetto 1999 are not presented due to excessive attrition (19/24; 79%) in the study's placebo arm.
Primary outcomes
Symptom relief
Short term (≤ 3 months)
One study (37 participants: Tammiala‐Salonen 1999) compared an antidepressant (trazodone 200 mg daily) against placebo. There was no evidence to demonstrate a difference in short‐term symptom relief (mean visual analogue scale (VAS) pain score, scale 0 to 10: lower better; mean difference (MD) 1.26, 95% confidence interval (CI) ‐0.24 to 2.76; effect P = 0.10) (Analysis 1.1).
1.1. Analysis.
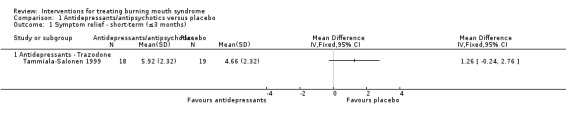
Comparison 1 Antidepressants/antipsychotics versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
Long term (> 3 to ≤ 6 months)
No included studies, comparing antidepressants and antipsychotics versus placebo, provided data on long‐term symptom relief.
Change in quality of life (QoL)
A single study (37 participants: Tammiala‐Salonen 1999) narratively reported assessment of short‐term change in depression as a surrogate measure for QoL, and indicated that both intervention and placebo participants were less depressed at trial completion, but there was no evidence of a difference between groups. No included study assessed long‐term change in QoL.
Secondary outcomes
No included studies, comparing antidepressants or antipsychotics against placebo, provided data on short‐ or long‐term change in taste or feeling of dryness.
Adverse effects
One study (37 participants: Tammiala‐Salonen 1999) reported adverse effect data, comparing an antidepressant (trazodone) against placebo. There was evidence to suggest trazodone resulted in an increase in dizziness (risk ratio (RR) 11.61, 95% CI 1.66 to 81.04; effect P = 0.01) and drowsiness (RR 4.75, 95% CI 1.18 to 19.07; effect P = 0.03); however, there was no evidence of a difference between trazodone and placebo in terms of abdominal pains (RR 1.32, 95% CI 0.42 to 4.15; effect P = 0.64), headache (RR 1.58, 95% CI 0.30 to 8.40; effect P = 0.59), palpitations (RR 1.06, 95% CI 0.17 to 6.72; effect P = 0.95), tremor (RR 2.11, 95% CI 0.21 to 21.32; effect P = 0.53), dry mouth (RR 3.17, 95% CI 0.36 to 27.72; effect P = 0.30) or urinary incontinence (RR 3.16, 95% CI 0.14 to 72.84; effect P = 0.47).
Anticonvulsants
One multi‐armed study (100 participants: Lopez‐D'alessandro 2011 ), at high risk of bias, provided very low‐quality evidence comparing an anticonvulsant (gabapentin 300 mg daily ± alpha lipoic acid (ALA) 600 mg daily) against placebo. This study did not assess long‐term outcome data.
Primary outcomes
Symptom relief
Short term (≤ 3 months)
Overall, there was evidence of short‐term symptom relief in favour of the anticonvulsant (bespoke geographical burning distribution numerical scale 0 to 4, dichotomised; RR 4.00, 95% CI 2.09 to 7.67; effect P < 0.0001). Two subgroups were formed: gabapentin only versus placebo and gabapentin with adjunctive ALA versus placebo. No between‐subgroup difference was shown (Analysis 2.1).
2.1. Analysis.
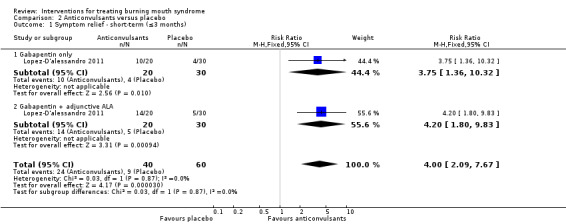
Comparison 2 Anticonvulsants versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
Change in QoL
Neither short‐ nor long‐term change in QoL data were available for the comparison of anticonvulsants (with or without ALA) against placebo.
Secondary outcomes
No short‐ or long‐term data on change in taste or feeling of dryness were available for the comparison of anticonvulsants (with or without ALA) against placebo.
Adverse effects
The single study (Lopez‐D'alessandro 2011) comparing an anticonvulsant (gabapentin, without and with adjunctive ALA) against placebo, narratively reported adverse effects as being "very mild". Data obtained from the study author indicated that drowsiness was more likely to be experienced when using gabapentin (without adjunctive ALA) (80 participants; RR 31.95, 95% CI 1.84 to 553.64; effect P = 0.02). There was no evidence of a difference in drowsiness (80 participants; RR 14.52, 95% CI 0.73 to 290.44; effect P = 0.08) or mild headache (80 participants; RR 8.71, 95% CI 0.37 to 205.80; effect P = 0.18) when using gabapentin with ALA.
Benzodiazepines
Primary outcomes
Four studies (166 participants), at either high or unclear risk of bias, provided very low‐quality evidence comparing benzodiazepines against placebo (Bogetto 1999 (systemic clordemetildiazepam 1 mg daily); Grémeau‐Richard 2004 (topical clonazepam 3 mg daily); Heckmann 2012 (systemic clonazepam 0.5 mg daily); Rodríguez de Rivera‐Campillo 2010 (topical clonazepam 0.5 to 2 mg daily)).
Data from Bogetto 1999 are not presented due to excessive attrition (19/24; 79%) in the study's placebo arm.
Symptom relief
Short term (≤ 3 months)
Three studies (131 participants: Grémeau‐Richard 2004; Heckmann 2012; Rodríguez de Rivera‐Campillo 2010) provided short‐term data on symptom relief for the comparison of benzodiazepines versus placebo. Two studies (111 participants) evaluated topical benzodiazepine use and showed an improvement in symptom relief between clonazepam and placebo, in favour of clonazepam (mean VAS pain score, scale 0 to 10: lower better; MD ‐1.89, 95% CI ‐2.19 to ‐1.59; effect P < 0.00001) (Grémeau‐Richard 2004; Rodríguez de Rivera‐Campillo 2010). No difference in symptom relief was shown between systemic clonazepam and placebo (mean VAS pain score, scale 0 to 10: lower better; 20 participants; MD 0.00, 95% CI ‐1.86 to 1.86; effect P = 1.00) (Heckmann 2012).
The pooled results for the topical and systemic administration of clonazepam showed evidence of short‐term symptom relief however, there was substantial statistical heterogeneity between subgroups.
Long term (> 3 to ≤ 6 months)
Only one study (66 participants: Rodríguez de Rivera‐Campillo 2010) assessed benzodiazepines (topical clonazepam) against placebo for long‐term symptom relief. There was evidence of symptom improvement in favour of clonazepam (mean VAS pain score, scale 0 to 10: lower better; MD ‐1.39, 95% CI ‐1.96 to ‐0.83; effect P < 0.00001) (Analysis 3.2).
3.2. Analysis.
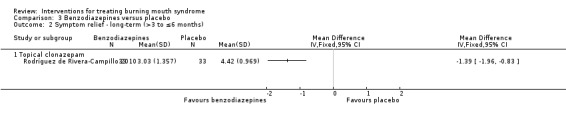
Comparison 3 Benzodiazepines versus placebo, Outcome 2 Symptom relief ‐ long‐term (>3 to ≤6 months).
Change in QoL
Heckmann 2012 (20 participants) assessed depression, as a surrogate marker of QoL. No difference was shown of short‐term change in depression (mean depression score, scale 0 to 4: lower better; MD ‐0.20, 95% CI ‐0.95 to 0.55; effect P = 0.60) (Analysis 3.3). No included studies assessed long‐term change in QoL, or its surrogate markers.
3.3. Analysis.
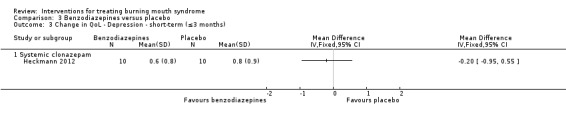
Comparison 3 Benzodiazepines versus placebo, Outcome 3 Change in QoL ‐ Depression ‐ short‐term (≤3 months).
Secondary outcomes
Change in taste
One study (20 participants: Heckmann 2012) compared benzodiazepines (systemic clonazepam) against placebo. No difference was shown of short‐term change in taste (mean taste test score, scale 0 to 16: higher better; MD ‐1.00, 95% CI ‐3.11 to 1.11; effect P = 0.35) (Analysis 3.4). No included studies assessed long‐term change in taste.
3.4. Analysis.
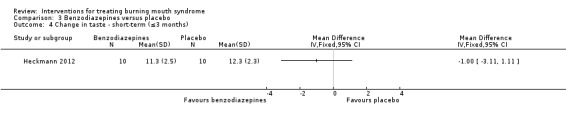
Comparison 3 Benzodiazepines versus placebo, Outcome 4 Change in taste ‐ short‐term (≤3 months).
Change in feeling of dryness
No included studies assessed short‐ or long‐term change in feeling of dryness.
Adverse effects
Two studies (114 participants: Grémeau‐Richard 2004; Rodríguez de Rivera‐Campillo 2010), comparing benzodiazepines (both topical clonazepam) against placebo, demonstrated no difference in drowsiness (RR 2.71, 95% CI 0.84 to 8.74; effect P = 0.09).
One of the two studies (48 participants: Grémeau‐Richard 2004) also showed no difference in dry mouth, spasmophilia, or euphoric behaviour (all three side effects independently: RR 3.00, 95% CI 0.13 to 70.16; effect P = 0.49).
Cholinergics
One study (40 participants: Femiano 2002b), at high risk of bias, provided very low‐quality evidence comparing a cholinergic (bethanechol 15 mg daily) against placebo. This study did not assess long‐term outcome data.
Primary outcomes
Symptom relief
Short term (≤ 3 months)
The single study evaluating a cholinergic showed no difference in short‐term symptom relief between bethanechol and placebo (bespoke burning mouth syndrome (BMS) symptomology change scale, dichotomised; RR 5.00, 95% CI 0.26 to 98.00; effect P = 0.29) (Analysis 4.1).
4.1. Analysis.
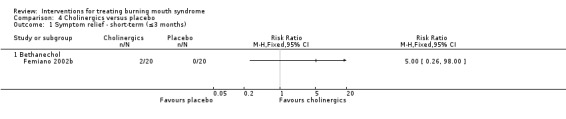
Comparison 4 Cholinergics versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
Change in QoL
Neither short‐ nor long‐term change in QoL data were available to compare cholinergics against placebo.
Secondary outcomes
No short‐ or long‐term data on change in taste or feeling of dryness were available for the comparison of cholinergics versus placebo.
Adverse effects
Femiano 2002b narratively reported adverse effect data, comparing a cholinergic (bethanechol) against placebo. Four participants (20%) in the bethanechol arm reported adverse events (nausea, dizziness, blood pressure fall, cold perspiration or sporadic abdominal pain). Reported data did not present the distribution of these outcomes by participant.
Dietary supplements
Twelve studies (628 participants) provided very low‐quality evidence comparing dietary supplements against placebo (ALA without adjunctive active ingredients (Cavalcanti 2009; Carbone 2009; Lopez‐D'alessandro 2011); ALA + vitamins (Tiobec) (Carbone 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Marino 2010); ALA + adjunctive lycopene + green tea extract (Thioderm) (López‐Jornet 2009b; Palacios‐Sánchez 2015); hypericum perforatum (St John's Wort) (Sardella 2008); 'Catuama' herbal compound (Spanemberg 2012); lycopene (Cano‐Carrillo 2014)). All studies were at either high or unclear risk of bias, apart from one assessed at low risk of bias (Sardella 2008).
Primary outcomes
Symptom relief
Short term (≤ 3 months)
Twelve studies (628 participants) compared dietary supplements against placebo (Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Lopez‐D'alessandro 2011; López‐Jornet 2009b; Marino 2010; Palacios‐Sánchez 2015; Sardella 2008; Spanemberg 2012). There was insufficient or contradictory evidence regarding the benefit of any of the evaluated dietary supplements over placebo (Analysis 5.1; Analysis 5.2; Analysis 5.3).
5.1. Analysis.
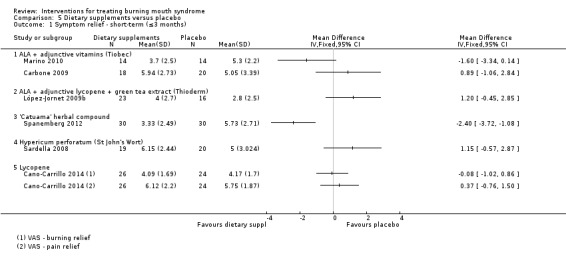
Comparison 5 Dietary supplements versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
5.2. Analysis.
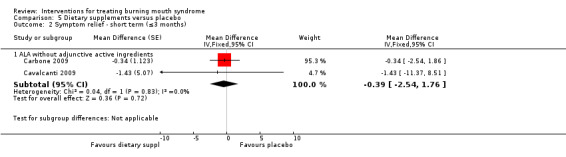
Comparison 5 Dietary supplements versus placebo, Outcome 2 Symptom relief ‐ short term (≤3 months).
5.3. Analysis.
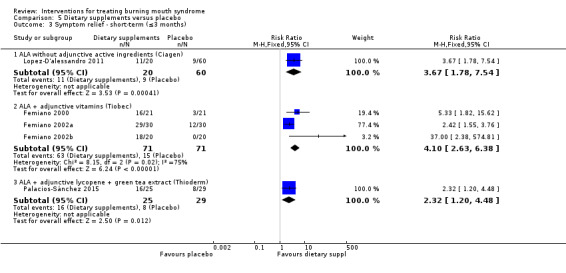
Comparison 5 Dietary supplements versus placebo, Outcome 3 Symptom relief ‐ short‐term (≤3 months).
No difference in symptom relief was found between ALA + adjunctive vitamins and placebo when assessed using continuous outcome data (two studies, 66 participants: Carbone 2009; Marino 2010; mean VAS pain score, scale 0 to 10: lower better; MD ‐0.49, 95% CI ‐1.79 to 0.81; effect P = 0.46) (not shown in Analysis 5.1). Three studies (142 participants: Femiano 2000; Femiano 2002a; Femiano 2002b) reported data for relief/no relief. There was a short‐term benefit for ALA with adjunctive vitamins (bespoke BMS symptomology change scale, dichotomised; RR 4.10, 95% CI 2.63 to 6.38; effect P < 0.00001). There was a substantial amount of heterogeneity (P = 0.02; I2 = 75%) between the results of these three studies (Analysis 5.3).
Long term (> 3 to ≤ 6 months)
Two studies (94 participants: Carbone 2009; Marino 2010) provided long‐term data comparing dietary supplements against placebo. Both studies compared ALA (either with or without adjunctive vitamins). Overall, there was no difference in long‐term symptom relief (mean VAS pain score, scale 0 to 10: lower better; MD ‐0.89, 95% CI ‐2.37 to 0.59; P = 0.24). There was no evidence of statistical heterogeneity (P = 0.83; I2 = 0%) (Analysis 5.4).
5.4. Analysis.
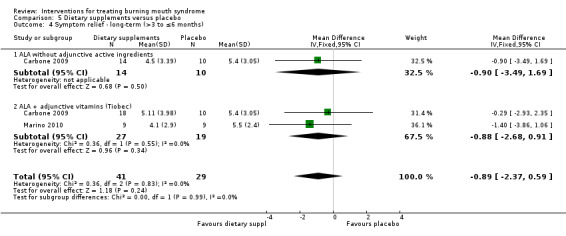
Comparison 5 Dietary supplements versus placebo, Outcome 4 Symptom relief ‐ long‐term (>3 to ≤6 months).
Change in QoL
One included study (50 participants: Cano‐Carrillo 2014), comparing lycopene against placebo, provided data assessing short‐term change in QoL. Overall QoL was assessed by OHIP‐14 (Oral Health Impact Profile 14), and anxiety and depression were also assessed adjunctively, as surrogate markers of QoL (Analysis 5.5).
5.5. Analysis.
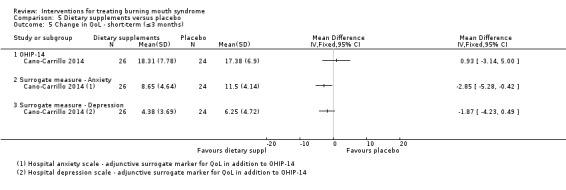
Comparison 5 Dietary supplements versus placebo, Outcome 5 Change in QoL ‐ short‐term (≤3 months).
There was no difference in short‐term change in QoL (mean OHIP‐14 score, scale 0 to 70: lower better; MD 0.93, 95% CI ‐3.14 to 5.00; effect P = 0.65), or in depression (as a surrogate marker of QoL: mean HAD (Hospital Anxiety and Depression) depression score, scale 0 to 21: lower better; MD ‐1.87, 95% CI ‐4.23 to 0.49; effect P = 0.12); however, there was short‐term benefit for lycopene with a mean score reduction in anxiety of ‐2.85 (mean HAD anxiety score, scale 0 to 21: lower better; 95% CI ‐5.28 to ‐0.42; effect P = 0.02).
One study (43 participants: Sardella 2008), comparing hypericum perforatum extract with placebo, evaluated QoL from patient self‐reports and narratively indicated that both intervention and placebo participants were better able to cope at trial completion.
No included studies assessed long‐term change in QoL, or its surrogate markers.
Secondary outcomes
No included studies assessed short‐ or long‐term change in taste or feeling of dryness, when comparing dietary supplements against placebo.
Adverse effects
Three studies (138 participants: Cavalcanti 2009; Femiano 2002b; López‐Jornet 2009b), comparing dietary supplements (all three studies used ALA, with or without adjunctive active ingredients) against placebo, provided evidence that gastrointestinal complaints were four times more likely to be experienced (RR 4.00, 95% CI 1.21 to 13.27; effect P = 0.02). There was no evidence of heterogeneity (P = 0.78; I2 = 0%). The gastrointestinal effect was sufficiently severe to cause an intervention‐allocated participant to abandon treatment in one trial (López‐Jornet 2009b).
Two studies (118 participants: Cavalcanti 2009; Lopez‐D'alessandro 2011), comparing dietary supplements (ALA, with or without adjunctive active ingredients) against placebo, indicated evidence of an 11‐fold increase in headache occurrence (RR 10.87, 95% CI 1.36 to 87.03; effect P = 0.02). There was no evidence of heterogeneity (P = 0.82; I2 = 0%). One study (43 participants: Sardella 2008), comparing hypericum perforatum with placebo, failed to report adverse effect occurrence; however, the study authors indicated a severe headache was sufficient cause for an intervention‐allocated participant to abandon treatment.
Two studies (110 participants: Cavalcanti 2009; Spanemberg 2012), comparing dietary supplements (ALA (Cavalcanti 2009); 'Catuama' herbal compound (Spanemberg 2012)) against placebo, showed no difference in drowsiness (RR 1.58, 95% CI 0.21 to 11.71; effect P = 0.65). There was no evidence of heterogeneity (P = 0.64; I2 = 0%).
One study (38 participants: Cavalcanti 2009), comparing dietary supplements (ALA without adjunctive active ingredients) against placebo, demonstrated there was no difference in blood pressure (RR 1.00, 95% CI 0.07 to 14.85; effect P = 1.00); however the study authors reported increase in blood pressure was sufficient to cause an intervention‐ and a placebo‐allocated participant to abandon treatment.
One study (80 participants: Lopez‐D'alessandro 2011), comparing dietary supplements (ALA) against placebo, indicated there was no difference in intermittent facial skin rash (RR 8.71, 95% CI 0.37 to 205.80; effect P = 0.18).
One study (72 participants: Spanemberg 2012), comparing dietary supplements ('Catuama' herbal compound) against placebo, showed no difference in exacerbation of symptoms (RR 1.12, 95% CI 0.33 to 3.83; effect P = 0.86), insomnia or weight gain (both side effects independently: RR 2.69, 95% CI 0.11 to 63.96; effect P = 0.54).
Four studies (224 participants) comparing dietary supplements (ALA alone and ALA with adjunctive vitamins (Tiobec) (Carbone 2009; Femiano 2000; Marino 2010) and lycopene (Cano‐Carrillo 2014)) against placebo, narratively reported that no adverse effects occurred.
Femiano 2002a (60 participants, ALA with adjunctive vitamins) reported no "notable adverse effects", and Femiano 2002b (40 participants, ALA with adjunctive vitamins) narratively reported four intervention participants (20%) had experienced heartburn, before being corrected by administration of ranitidine (150 mg). Sardella 2008 (43 participants, hypericum perforatum extract) reported that one intervention participant (5%) developed severe headache and discontinued treatment.
Electromagnetic radiation
One study (58 participants: Spanemberg 2015) at high risk of bias, provided very low‐quality evidence comparing electromagnetic radiation (infrared laser; red laser) against placebo. This study did not assess long‐term outcome data.
Primary outcomes
Symptom relief
Short term (≤ 3 months)
Spanemberg 2015 compared two types of low‐level laser therapy (infrared laser; red laser) against placebo. It demonstrated a short‐term benefit in symptom relief (mean VAS pain score, scale 0 to 100: lower better; MD ‐30.36, 95% CI ‐44.22 to ‐16.50; effect P < 0.0001) (Analysis 6.1).
6.1. Analysis.
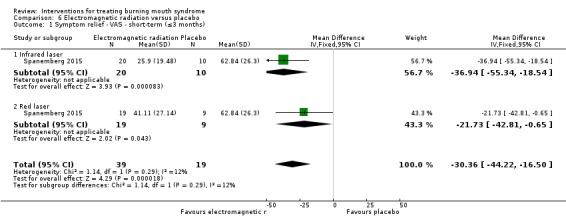
Comparison 6 Electromagnetic radiation versus placebo, Outcome 1 Symptom relief ‐ VAS ‐ short‐term (≤3 months).
Change in QoL
Short term (≤ 3 months)
There was evidence of a short‐term improvement in QoL from low‐level laser therapy (58 participants: Spanemberg 2015) compared with placebo (mean OHIP‐14 score, scale 0 to 56: lower better; MD ‐5.24, 95% CI ‐7.38 to ‐3.09; effect P < 0.00001) (Analysis 6.2).
6.2. Analysis.
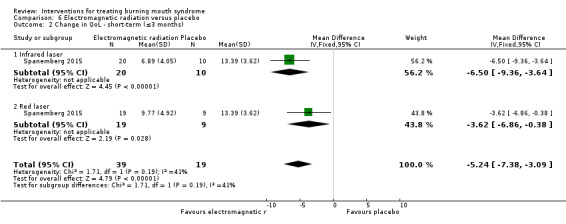
Comparison 6 Electromagnetic radiation versus placebo, Outcome 2 Change in QoL ‐ short‐term (≤3 months).
Secondary outcomes
Spanemberg 2015 did not assess change in taste, feeling of dryness, or report the occurrence of adverse effects.
Physical barriers
One study (50 participants: López‐Jornet 2011) at high risk of bias, provided very low‐quality evidence comparing a physical barrier (tongue protector + adjunctive reinforced self‐control instruction (RS‐CI)) against placebo. This study did not assess long‐term outcome data.
Primary outcomes
Symptom relief
Short term (≤ 3 months)
In this single study (50 participants: López‐Jornet 2011), there was a short‐term benefit for physical barriers with a mean score reduction in symptoms of ‐1.10 (mean VAS pain score, scale 0 to 10: lower better; 95% CI ‐2.14 to ‐0.06; effect P = 0.04) (Analysis 7.1).
7.1. Analysis.
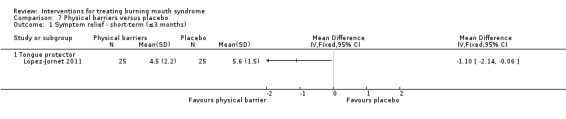
Comparison 7 Physical barriers versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
Change in QoL
Short term (≤ 3 months)
López‐Jornet 2011 assessed short‐term change in overall QoL by OHIP‐49, and adjunctively assessed anxiety and depression as surrogate QoL markers. There was no difference in short‐term change in overall QoL (mean OHIP‐49 score, scale 0 to 196: lower better; MD ‐9.20, 95% CI ‐26.90 to 8.50; effect P = 0.31), or in anxiety (mean HAD anxiety score, scale 0 to 21: lower better; MD 0.16, 95% CI ‐3.19 to 3.51; effect P = 0.93) or depression (mean HAD depression score, scale 0 to 21: lower better; MD ‐0.64, 95% CI ‐3.98 to 2.70; effect P = 0.71) either (Analysis 7.2).
7.2. Analysis.
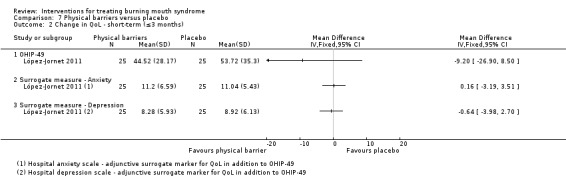
Comparison 7 Physical barriers versus placebo, Outcome 2 Change in QoL ‐ short‐term (≤3 months).
Secondary outcomes
This study did not assess change in taste or feeling of dryness.
Adverse effects
López‐Jornet 2011 narratively reported that no adverse effects occurred.
Psychological therapies
Primary outcomes
One study (30 participants: Bergdahl 1995a) at high risk of bias, provided very low‐quality evidence comparing a psychological therapy (cognitive therapy (CT)) against placebo. The study presented long‐term outcome data only.
Symptom relief
Long term (> 3 to ≤ 6 months)
Bergdahl 1995a demonstrated a long‐term benefit for psychological therapy with a mean score reduction in symptoms of ‐3.20 (mean VAS pain score, scale 1 to 7: lower better; 95% CI ‐4.22 to ‐2.18; effect P < 0.00001) (Analysis 8.1).
8.1. Analysis.

Comparison 8 Psychological therapies versus placebo, Outcome 1 Symptom relief ‐ long‐term (>3 to ≤6 months).
Change in QoL
Neither short‐ nor long‐term change in QoL data were available to compare psychological therapy against placebo.
Secondary outcomes
Bergdahl 1995a did not assess change in taste, feeling of dryness, or report the occurrence of adverse effects.
Topical treatments
Five studies (180 participants), at either high or unclear risk of bias, provided very low‐quality evidence comparing topical treatments (benzydamine hydrochloride oral rinse (Sardella 1999); lactoperoxidase oral rinse (Biotene) (Femiano 2002b; Marino 2010); topical urea (10%) (Alvarenga da Silva 2014); and capsaicin oral rinse (Marino 2010; Silvestre 2012)) against placebo.
Data from Silvestre 2012 (30 participants) are not presented due to attrition (7/30; 23%) in this cross‐over study's first phase and their protocol requirement for participants who develop adverse effects to be withdrawn from the trial.
Primary outcomes
Symptom relief
Short term (≤ 3 months)
Four studies (150 participants: Alvarenga da Silva 2014; Femiano 2002b; Marino 2010; Sardella 1999) compared topical treatments with placebo to estimate their short‐term effect on symptom relief. Pooling of data was not undertaken due to symptom data being presented in both dichotomous and continuous form. There is insufficient or contradictory evidence regarding the benefit of any of the evaluated topical interventions over placebo (Analysis 9.1; Analysis 9.2).
9.1. Analysis.
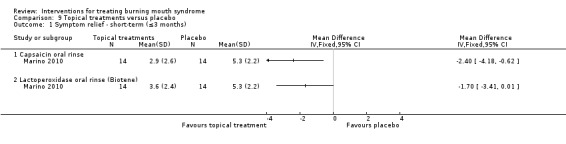
Comparison 9 Topical treatments versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
9.2. Analysis.
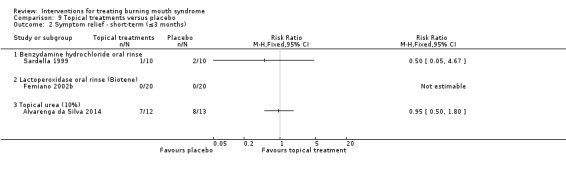
Comparison 9 Topical treatments versus placebo, Outcome 2 Symptom relief ‐ short‐term (≤3 months).
Long term (> 3 to ≤ 6 months)
One multi‐armed study (27 participants: Marino 2010) provided data on long‐term symptom relief of topical treatments (capsaicin oral rinse and lactoperoxidase oral rinse (Biotene)) compared with placebo. This study, assessed at high risk of bias, demonstrated a long‐term benefit for capsaicin oral rinse (mean VAS pain score, scale 0 to 10: lower better; MD ‐2.60, 95% CI ‐5.11 to ‐0.09; effect P = 0.04)). No effect was shown for the lactoperoxidase oral rinse (Biotene) data (MD ‐1.50, 95% CI ‐3.91 to 0.91; effect P = 0.22) (Analysis 9.3).
9.3. Analysis.
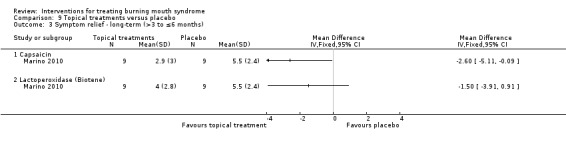
Comparison 9 Topical treatments versus placebo, Outcome 3 Symptom relief ‐ long‐term (>3 to ≤6 months).
Change in QoL
No included studies comparing topical treatments against placebo provided data assessing short‐ or long‐term change in QoL.
Secondary outcomes
Change in taste
One study (38 participants: Alvarenga da Silva 2014) comparing a topical treatment (topical urea (10%)) against placebo reported narratively that there were no differences in short‐term gustation thresholds after treatment (Mann‐Whitney: sweet P = 0.376; salty P = 0.689; sour P = 0.689; bitter P = 0.689). No included studies comparing topical treatments assessed long‐term change in taste.
Change in feeling of dryness
The same single study (Alvarenga da Silva 2014) reported narratively that there were no differences in short‐term xerostomia questionnaire assessment after treatment. No included studies comparing topical treatments assessed long‐term change in feeling of dryness.
Adverse events
One study (20 participants: Sardella 1999) comparing benzydamine hydrochloride oral rinse against placebo, reported narratively that no adverse effects occurred.
Discussion
Summary of main results
A total of 23 placebo‐controlled randomised controlled trials (RCTs) were included in this review, evaluating the effectiveness of 23 different interventions for the treatment of burning mouth syndrome (BMS) symptoms (distributed between nine intervention categories: antidepressants and antipsychotics; anticonvulsants; benzodiazepines; cholinergics; dietary supplements; electromagnetic radiation; physical barriers; psychological therapies; and topical treatments). There was a considerable amount of heterogeneity in the types of interventions studied, and how the interventions were delivered.
There was some evidence of a benefit in short‐term symptom relief for electromagnetic radiation, topical benzodiazepines, physical barriers and anticonvulsants. We found insufficient/contradictory evidence regarding the short‐term effectiveness of antidepressants, systemic benzodiazepines, cholinergics, dietary supplements or topical treatments. No RCT assessing psychological therapies evaluated short‐term symptom relief.
We also found some evidence of long‐term symptom relief for psychological therapies, capsaicin oral rinse (topical treatment) and topical benzodiazepines. We found no evidence of a difference for dietary supplements or lactoperoxidase oral rinse. No studies assessing antidepressants, anticonvulsants, cholinergics, electromagnetic radiation, or physical barriers evaluated long‐term symptom relief.
We found some evidence of a short‐term quality of life (QoL) improvement for electromagnetic radiation, however findings were inconclusive for antidepressants, benzodiazepines, dietary supplements and physical barriers. No studies assessing anticonvulsants, cholinergics, psychological therapies, or topical treatments evaluated short‐term change in QoL, and no RCTs from any category assessed long‐term change in QoL.
Changes in taste and feeling of dryness were assessed by included studies in the short term only, and the findings for each were inconclusive.
With regard to adverse events, there is very low‐quality evidence that antidepressants increase dizziness and drowsiness, and that alpha lipoic acid increased headache and gastrointestinal complaints. We found insufficient/contradictory evidence regarding adverse events for anticonvulsants or benzodiazepines. Adverse events were poorly reported or unreported for cholinergics, electromagnetic radiation, and psychological therapies. No adverse events occurred from physical barriers or topical therapy use.
See Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9.
Overall completeness and applicability of evidence
All of the study participants included in the current review were deemed to adhere to an appropriate diagnostic classification of BMS (Additional Table 10).
We had serious concerns regarding the applicability of the evidence for seven of the nine intervention categories contained in this review (excepting dietary supplements and physical barriers), as studies from these seven categories assessed only one type of each intervention and the effects of other types of these interventions (antidepressants and antipsychotics; anticonvulsants; benzodiazepines; cholinergics; electromagnetic radiation; psychological therapies; and topical treatments) may differ.
While we were able to assess short‐term symptom relief for all intervention categories except psychological therapies, fewer data were available for long‐term assessment. Consequently, we are currently unable to estimate long‐term symptom relief resulting from use of antidepressants, anticonvulsants, cholinergics, electromagnetic radiation, or physical barriers.
Furthermore, we were only able to assess:
short‐term change in QoL for dietary supplements, electromagnetic radiation, and physical barriers (no interventions long term),
short‐term change in depression (as a surrogate measure of QoL) for antidepressants and benzodiazepines,
short‐term change in taste for benzodiazepines and topical treatments (no interventions long term),
short‐term change in feeling of dryness for topical treatments alone (no interventions long term).
Nor do we have any data to assess adverse event occurrence from electromagnetic radiation or psychological therapies.
Most studies provided baseline demographics of their included participants. A single study (Alvarenga da Silva 2014) reported participants' race. In terms of the gender and age distribution presented, the majority of studies were consistent; both in comparison to each other and when compared to previously published epidemiological data (Bergdahl 1999). Several studies, however, were inconsistent with expected male‐to‐female (M/F) ratios: Femiano 2002a (M/F: 18/42), Femiano 2002b (M/F: 32/48), Heckmann 2012 (M/F: 7/13), Tammiala‐Salonen 1999 (M/F: 0/37). There were no obvious reasons as to why these studies had unusual gender spreads.
There is a paucity of RCTs evaluating neuropathic pain medications (Finnerup 2015; NICE 2013) in the management of BMS ‐ with only amitriptyline (Bogetto 1999) and very low‐dosage gabapentin (Lopez‐D'alessandro 2011) being included in this review. This is in spite of the growing body of evidence to support the role of neuropathic mechanisms in BMS (Jääskeläinen 2012). Moreover, only a single included RCT looked at clinical psychology as a way of managing BMS (Bergdahl 1995a); despite a wealth of evidence supporting the use of psychological therapies in chronic pain management (Sturgeon 2014).
Some of the treatments assessed within this review, or other interventions not covered by the included studies, may be effective in the management of BMS symptoms. However, until further high‐quality evidence is forthcoming we will not be able to make any recommendations for the treatment for BMS.
Quality of the evidence
The quality of the evidence was assessed as very low for all outcomes (both short‐ and long‐term) in all intervention categories (with the exception of adverse events for physical barriers, which were assessed as low‐quality evidence).
Of the 23 studies included in this review, only one was deemed to have a low overall risk of bias; four were found to have an unclear risk of bias; and the remaining 18 studies were judged to be at high overall risk of bias. 'Other bias' was the most frequent cause of a high risk of bias assessment of a study (n = 10; 43%), followed by selective outcome reporting (n = 8; 35%) and blinding of participants (n = 7; 30%). Trials with open assessment of the outcome, as described by Femiano and colleagues (Femiano 2000; Femiano 2002b) and Bogetto and colleagues (Bogetto 1999), have been shown to overestimate the treatment effects by 35% (Jüni 1999). Indeed, this may explain why the two open‐label trials of alpha‐lipoic acid (Femiano 2000; Femiano 2002b) provided greater estimates of effect than the double‐blinded trial of the same intervention (Femiano 2002a).The most commonly occurring cause for an 'other bias' assessment was due to failing to present baseline demographics for each study arm (n = 8; 35%).
We were unable to perform sensitivity analyses with regard to risk of bias due to the fact that only one included study was assessed to have a low overall risk of bias.
Inappropriate and misleading use of graphs and tables which did not contain usable data was commonplace, although we sought data directly from study authors where possible to do so. More recently, authors have been able to publish online supplements containing all of the relevant study data; they no longer have to select which restricted set of data to report. In many of the studies a high risk of reporting bias could have been avoided if additional raw data had been made available to the review group.
Potential biases in the review process
The review authors strictly adhered to the prespecified methodology for conducting systematic reviews included in the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Higgins 2011b). Two review authors screened all abstracts independently and in duplicate, and all identified papers were then assessed and had the risk of bias assessment conducted by at least two review authors.
After discussion, the review authors agreed that the placebo comparator arms employed by two studies were suitable for inclusion as they were considered to be non‐active placebos (Bergdahl 1995a (psychological therapies: cognitive therapy versus motivation/oral examination); López‐Jornet 2011 (physical barriers: tongue protector + reinforced self‐control instruction (RS‐CI) versus RS‐CI)).
Data from Bogetto 1999 were not included within the quantitative synthesis due to excessive attrition (19/24; 79%) in the study's placebo arm, or from Silvestre 2012 due to attrition (7/30; 23%) in this cross‐over study's first phase and their protocol requirement for participants who developed adverse effects to be withdrawn from the trial.
We decided it was inappropriate to use reported six‐month data from Grémeau‐Richard 2004 and one‐year data from Femiano 2002a, as both studies only presented data at these time points from participants who had positively responded to treatment at their earlier assessment, and consequently comprised a highly biased subset of data.
Due to one of the included cross‐over RCTs containing no washout period (Femiano 2000), we only incorporated data from the first phase of their study within our analyses.
We identified that two studies reported different length scales from the same outcome assessment measure (OHIP‐14 (Oral Health Impact Profile‐14): Cano‐Carrillo 2014 scale 0 to 70, lower better; Spanemberg 2015 scale 0 to 56, lower better). Despite the inconsistency in scale length, we used the length reported for each study rather than assuming that either one was incorrect.
Lastly, despite all studies initially assessing symptom relief by use of a continuous outcome metric, seven studies (Femiano 2000; Femiano 2002a; Femiano 2002b; Lopez‐D'alessandro 2011; Palacios‐Sánchez 2015; Sardella 1999; Alvarenga da Silva 2014) transformed their data to report symptom relief categorically instead, which only permitted us to estimate their effect estimates dichotomously as risk ratios. Consequently, we had to present results from continuous and dichotomous data separately; as such, we were unable to estimate heterogeneity across the dietary supplements and topical treatments categories (except where permitted within subgroups), and were also unable to estimate subgroup differences.
Agreements and disagreements with other studies or reviews
This review largely concurs with the findings of two other systematic reviews of placebo‐controlled RCTs:
de Moraes 2012 (12 RCTs compared to this review's inclusion of 23 RCTs; includes Carbone 2009; Cavalcanti 2009; Femiano 2002a; Femiano 2002b; Grémeau‐Richard 2004; Lopez‐D'alessandro 2011; López‐Jornet 2009b; Marino 2010; Sardella 1999; Sardella 2008; Tammiala‐Salonen 1999 and our excluded study Petruzzi 2004), and
Kisely 2016 (24 RCTs compared to this review's inclusion of 23 RCTs; includes Bergdahl 1995a; Cano‐Carrillo 2014; Carbone 2009; Cavalcanti 2009; Femiano 2000; Femiano 2002a; Femiano 2002b; Grémeau‐Richard 2004; Heckmann 2012; Lopez‐D'alessandro 2011; López‐Jornet 2009b; López‐Jornet 2011; Marino 2010; Rodríguez de Rivera‐Campillo 2010; Sardella 1999; Sardella 2008; Silvestre 2012; Spanemberg 2012; Tammiala‐Salonen 1999 and five of our excluded studies Grémeau‐Richard 2010; López‐Jornet 2013; Miziara 2009; Petruzzi 2004; Toida 2009).
While neither de Moraes 2012 or Kisely 2016 undertook meta‐analyses of included studies, both reviews concluded there was limited quality evidence to support the efficacy of any one treatment over another in the treatment of primary BMS. We also highlight agreement with their observations that few RCTs provide long‐term follow‐up (longer than three months), which we endorse for the sustained management of this chronic painful condition.
A systematic review (Cui 2016) concentrating specifically on clonazepam (a benzodiazepine) estimated short‐term relief more conservatively than this review did (from the same three RCTs (Grémeau‐Richard 2004; Heckmann 2012; Rodríguez de Rivera‐Campillo 2010) assessed by a visual analogue scale (VAS) (0 to 10, lower better); Cui 2016: mean difference (MD) ‐1.44, 95% confidence interval (CI) ‐2.06 to ‐0.82 versus this review's Analysis 3.1: MD ‐1.84, 95% CI ‐2.14 to ‐1.54). However, we attribute this difference to Cui 2016's selective use of interim data from Heckmann 2012 (instead of the ultimately assessed outcome), standard deviation errors from Grémeau‐Richard 2004's data (up to four times larger, we assume from being incorrectly copied across), and their decision to pool topical and systemic benzodiazepines in a single group. Considering these differences, we maintain confidence in our larger estimate of effectiveness despite originating from very low‐quality evidence.
3.1. Analysis.
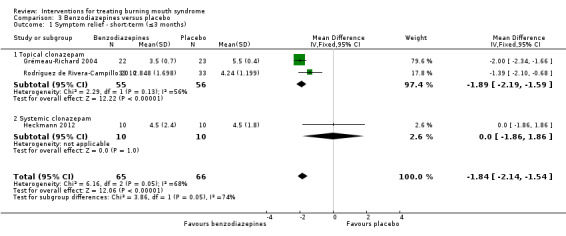
Comparison 3 Benzodiazepines versus placebo, Outcome 1 Symptom relief ‐ short‐term (≤3 months).
Authors' conclusions
Implications for practice.
Currently there is a dearth of high‐quality research evidence to allow the development of clear guidance for those charged with treating burning mouth syndrome (BMS) patients. Clinicians may use the results and detailed data from this systematic review to assist in discussions with their BMS patients, about which treatment options to undertake in the management of their condition.
Implications for research.
Further randomised controlled trials (RCTs), of longer duration (a minimum of three months follow‐up) and high methodological quality, are required in order to establish the effective forms of treatment for patients suffering from BMS. Future high‐quality research, which further assesses the treatments included in this review and incorporates novel BMS therapies, would very likely result in a change in clinical practice. Furthermore, treatments established in the management of other neuropathic pain conditions and psychological therapies should be considered for clinical trials in BMS.
Comparability of groups at baseline is of great importance, particularly with regard to intensity of symptoms, gender and psychological background. True randomisation with concealed allocation of treatment groups should provide comparable groups, although details of baseline characteristics should still be provided and an estimate of comparability undertaken. Given the subjectivity of the symptoms to be assessed, participants, healthcare providers and outcome assessors should be blinded to the intervention.
Visual analogue scales (VAS) or validated patient‐reported outcome measures should be used for the assessment of pain intensity. A decision regarding how large a treatment effect constitutes an adequate outcome requires to be made. Most treatments for chronic pain aim for a 50% reduction in pain scores from baseline; however, it could be that this is too high and 30% would be more pragmatic. Farrar et al (Farrar 2000) argue that use of consistent clinically important cut‐off points for pain outcomes would not only enhance validity and comparability, but would also have more clinical applicability. Other outcome measures looking at improvements in quality of life, anxiety and depression are imperative, as the negative impact of this condition on mood and on daily life is potentially high. The development of a core outcome set as described by COMET (Core Outcome Measures in Effectiveness Trials Initiative; www.comet‐initiative.org), may be a way of facilitating the production of valid and homogeneous outcome data from BMS clinical trials.
All participants included in a trial should be accounted for in the analysis of the results, with the analysis undertaken on an intention‐to‐treat basis. It should be acknowledged that the conduct and adequate funding of high‐quality RCTs in this field will impact the volume of research undertaken due to funding agencies' limited resources. Larger studies are essential and multicentre studies may be one way of ensuring that the study power is great enough to yield statistically significant results.
More detailed reporting of the adverse effects of treatments are required, as tolerability is an important factor for patients when making treatment choices (Ioannidis 2004).
What's new
| Date | Event | Description |
|---|---|---|
| 9 November 2016 | New citation required but conclusions have not changed | 15 new studies added. New authors. Methods updated |
| 31 December 2015 | New search has been performed | Searches updated to 31 December 2015 |
History
Protocol first published: Issue 4, 2000 Review first published: Issue 3, 2001
| Date | Event | Description |
|---|---|---|
| 19 August 2008 | Amended | Converted to new review format |
| 15 November 2004 | New search has been performed | Searches updated to October 2004 |
| 15 November 2004 | New citation required and conclusions have changed | Substantive amendment. 2 studies previously awaiting assessment have been excluded; a further 4 randomised controlled trials have been included and 1 previously included trial excluded |
Acknowledgements
The review authors would like to thank the external referees who provided comments for each version of this review. Moreover, we would like to thank the study authors who responded to requests for further information (Desiree Cavalcanti (Cavalcanti 2009), Felice Femiano (Femiano 2002a), Edgardo López‐D'alessandro (Lopez‐D'alessandro 2011), Pia Lopez‐Jornet (Cano‐Carrillo 2014), Luis Alberto Moreno Lopez (Palacios‐Sánchez 2015) and Alain Woda (Grémeau‐Richard 2004)), and translators who assisted with foreign language studies (Mónica Ballesteros, Ming Jian, Maddalena Manfredi and May Wong). Joanna Zakrzewska undertook this work at University College London (UCL) Hospitals who received a proportion of funding from the UK National Institute for Health Research (NIHR) Biomedical Research Centre funding. The review authors would also like to thank members of Cochrane Oral Health's editorial base for their support, particularly Janet Lear (Group Administrator), Anne Littlewood (Information Specialist), Laura MacDonald (Managing Editor) and Luisa Fernandez‐Mauleffinch (Managing Editor and Copy Editor).
Appendices
Appendix 1. Cochrane Oral Health's Trials Register search strategy
From February 2014, searches of Cochrane Oral Health's Trials Register for this review were undertaken using the Cochrane Register of Studies and the search strategy below:
1 ("burning mouth":ti,ab) AND (INREGISTER) 2 ("burning tongue":totalling,ab) AND (INREGISTER) 3 (glossodynia:ti,ab) AND (INREGISTER) 4 (glossopyrosis:ti,ab) AND (INREGISTER) 5 ((stomatodynia or stomatopyrosis):ti,ab) AND (INREGISTER) 6 (("oral dysaesthesia" or "oral dysesthesia"):ti,ab) AND (INREGISTER) 7 (BMS:ti,ab) AND (INREGISTER) 8 (#1 or #2 or #3 or #4 or #5 or #6 or #7) AND (INREGISTER)
Previous searches of Cochrane Oral Health's Trials Register were undertaken using the Procite software and the search strategy below:
("Burning mouth syndrome" OR "Burning mouth" OR "Burning tongue" OR Glossodynia OR Glossopyrosis OR Stomatodynia OR Stomatopyrosis OR "oral dysaesthesia" OR "oral dysesthesia" OR ( #4 CONTAINS BMS) OR (#43 CONTAINS BMS))
Appendix 2. Cochrane Central Register of Controlled Trials (CENTRAL) search strategy
#1 MeSH descriptor Burning Mouth Syndrome this term only #2 (burning in All Text near/3 mouth in All Text) #3 (burning in All Text near/3 tongue in All Text) #4 MeSH descriptor Glossalgia this term only #5 Glossalgia* in All Text #6 Glossodynia* in All Text #7 Glossopyros* in All Text #8 Stomatodynia* in All Text #9 Stomatopyros* in All Text #10 "oral dysaesthesia" in All Text #11 "oral dysesthesia" in All Text #12 (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11)
Appendix 3. MEDLINE Ovid search strategy
1. Burning Mouth Syndrome/ 2. (burning adj3 mouth).mp. 3. (burning adj3 tongue).mp. 4. Glossalgia/ 5. Glossalgia$.mp. 6. Glossodynia$.mp. 7. Glossopyros$.mp. 8. Stomatodynia$.mp. 9. Stomatopyros$.mp. 10. (oral adj dysaesthesia).mp. 11. (oral adj dysesthesia).mp. 12. or/1‐11
This subject search was linked to the Cochrane Highly Sensitive Search Strategy (CHSSS) for identifying randomised trials (RCTs) in MEDLINE: sensitivity‐maximising version (2008 revision) as referenced in Chapter 6.4.11.1 and detailed in box 6.4.c of theCochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Lefebvre 2011).
1. randomized controlled trial.pt. 2. controlled clinical trial.pt. 3. randomized.ab. 4. placebo.ab. 5. drug therapy.fs. 6. randomly.ab. 7. trial.ab. 8. groups.ab. 9. or/1‐8 10. exp animals/ not humans.sh. 11. 9 not 10
Appendix 4. Embase Ovid search strategy
1. Burning Mouth Syndrome/ 2. (burning adj3 mouth).mp. 3. (burning adj3 tongue).mp. 4. Glossalgia/ 5. Glossalgia$.mp. 6. Glossodynia$.mp. 7. Glossopyros$.mp. 8. Stomatodynia$.mp. 9. Stomatopyros$.mp. 10. (oral adj dysaesthesia).mp. 11. (oral adj dysesthesia).mp. 12. or/1‐11
This subject search was linked to Cochrane Oral Health's filter for identifying RCTs in Embase Ovid:
1. random$.ti,ab. 2. factorial$.ti,ab. 3. (crossover$ or cross over$ or cross‐over$).ti,ab. 4. placebo$.ti,ab. 5. (doubl$ adj blind$).ti,ab. 6. (singl$ adj blind$).ti,ab. 7. assign$.ti,ab. 8. allocat$.ti,ab. 9. volunteer$.ti,ab. 10. CROSSOVER PROCEDURE.sh. 11. DOUBLE‐BLIND PROCEDURE.sh. 12. RANDOMIZED CONTROLLED TRIAL.sh. 13. SINGLE BLIND PROCEDURE.sh. 14. or/1‐13 15. (exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti.) 16. 14 NOT 15
Appendix 5. US National Institutes of Health Ongoing Trials Register (ClinicalTrials.gov) search strategy
"burning mouth" or "burning tongue"
Appendix 6. World Health Organization International Clinical Trials Registry Platform search strategy
"burning mouth"
"burning tongue"
Data and analyses
Comparison 1. Antidepressants/antipsychotics versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Antidepressants ‐ Trazodone | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Anticonvulsants versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.0 [2.09, 7.67] |
| 1.1 Gabapentin only | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.75 [1.36, 10.32] |
| 1.2 Gabapentin + adjunctive ALA | 1 | 50 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.2 [1.80, 9.83] |
Comparison 3. Benzodiazepines versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 3 | 131 | Mean Difference (IV, Fixed, 95% CI) | ‐1.84 [‐2.14, ‐1.54] |
| 1.1 Topical clonazepam | 2 | 111 | Mean Difference (IV, Fixed, 95% CI) | ‐1.89 [‐2.19, ‐1.59] |
| 1.2 Systemic clonazepam | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐1.86, 1.86] |
| 2 Symptom relief ‐ long‐term (>3 to ≤6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 Topical clonazepam | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Change in QoL ‐ Depression ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Systemic clonazepam | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Change in taste ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 4. Cholinergics versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Bethanechol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 5. Dietary supplements versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 6 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 ALA + adjunctive vitamins (Tiobec) | 2 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 ALA + adjunctive lycopene + green tea extract (Thioderm) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 'Catuama' herbal compound | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Hypericum perforatum (St John's Wort) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Lycopene | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Symptom relief ‐ short term (≤3 months) | 2 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 2.1 ALA without adjunctive active ingredients | 2 | Mean Difference (Fixed, 95% CI) | ‐0.39 [‐2.54, 1.76] | |
| 3 Symptom relief ‐ short‐term (≤3 months) | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 ALA without adjunctive active ingredients (Ciagen) | 1 | 80 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.67 [1.78, 7.54] |
| 3.2 ALA + adjunctive vitamins (Tiobec) | 3 | 142 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.10 [2.63, 6.38] |
| 3.3 ALA + adjunctive lycopene + green tea extract (Thioderm) | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.32 [1.20, 4.48] |
| 4 Symptom relief ‐ long‐term (>3 to ≤6 months) | 2 | 70 | Mean Difference (IV, Fixed, 95% CI) | ‐0.89 [‐2.37, 0.59] |
| 4.1 ALA without adjunctive active ingredients | 1 | 24 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐3.49, 1.69] |
| 4.2 ALA + adjunctive vitamins (Tiobec) | 2 | 46 | Mean Difference (IV, Fixed, 95% CI) | ‐0.88 [‐2.68, 0.91] |
| 5 Change in QoL ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5.1 OHIP‐14 | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Surrogate measure ‐ Anxiety | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Surrogate measure ‐ Depression | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 6. Electromagnetic radiation versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ VAS ‐ short‐term (≤3 months) | 1 | 58 | Mean Difference (IV, Fixed, 95% CI) | ‐30.36 [‐44.22, ‐16.50] |
| 1.1 Infrared laser | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐36.94 [‐55.34, ‐18.54] |
| 1.2 Red laser | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐21.73 [‐42.81, ‐0.65] |
| 2 Change in QoL ‐ short‐term (≤3 months) | 1 | 58 | Mean Difference (IV, Fixed, 95% CI) | ‐5.24 [‐7.38, ‐3.09] |
| 2.1 Infrared laser | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐6.50 [‐9.36, ‐3.64] |
| 2.2 Red laser | 1 | 28 | Mean Difference (IV, Fixed, 95% CI) | ‐3.62 [‐6.86, ‐0.38] |
Comparison 7. Physical barriers versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Tongue protector | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Change in QoL ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 OHIP‐49 | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Surrogate measure ‐ Anxiety | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Surrogate measure ‐ Depression | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 8. Psychological therapies versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ long‐term (>3 to ≤6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 9. Topical treatments versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom relief ‐ short‐term (≤3 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 1.1 Capsaicin oral rinse | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Lactoperoxidase oral rinse (Biotene) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Symptom relief ‐ short‐term (≤3 months) | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Benzydamine hydrochloride oral rinse | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Lactoperoxidase oral rinse (Biotene) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Topical urea (10%) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Symptom relief ‐ long‐term (>3 to ≤6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Capsaicin | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Lactoperoxidase (Biotene) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alvarenga da Silva 2014.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 38 BMS patients Group 1 mean age 66.32 SD 12.01, Group 2 mean age 58.42 SD 13.70 years (no overall age data provided) Sex: 35 F:3 M (F 92%:M 8%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: topical treatments Group 1: (n = 19) topical medication comprised of urea 10% to be applied at the oral cavity 3‐4 times per day for 3 months Group 2: (n = 19) placebo (5% sodium carboxymethylcellulose, 0.15% methyl paraben, and 10% glycerol in distilled water quantity sufficient to make 100 g) to be applied at the oral cavity 3‐4 times per day for 3 months |
|
| Outcomes | "EDOF‐HC protocol (Orofacial Pain Clinic ‐ Hospital das Clinicas): a standardized orofacial pain questionnaire to detail the following: 1) chief complaint, 2) general pain characteristics (location, quality, duration, pain relief, pain triggering), 3) headache and/or body pain complaints, and 4) patient's medical history and comorbidities" at baseline and 3 months Xerostomia questionnaire at baseline and 3 months Quantitative sensory testing (QST) at baseline and 3 months
|
|
| Source of funding | São Paulo State Research Foundation (FAPESP) | |
| Notes | All participants had been treated with 25‐50 mg of amitriptyline within the last 3 months The duration of pain was significantly longer in the study group (6.97, SD 4.93 years) compared to 5 placebo group (2.78, SD 2.61 years) Insufficient results data were provided not sure how well validated all the QST are and whether there are age/gender related norms SES: data reported on race, occupation, and marital status: "Color: study group (n = 19) white 16, black 2, mulatto 1; control group (n = 19) white 16, black 1, mulatto 2; occupation: study group (n = 19) housekeeper 7, retired 6, domestic 2, seamstress 2, biomedical 1, unemployed 1; control group (n = 19) housekeeper 9, retired 5, unemployed 2, nanny 1, secretary 1, seller 1; marital status: study group (n = 19) married 8, widowed 6, single 4, divorced 1; control group (n = 19) married 8, widowed 7, single 3, divorced 1" Conflict of interests: authors reported no conflict of interests exist Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "The subjects were randomly divided into two groups" Comment: insufficient information to make a judgement |
| Allocation concealment (selection bias) | High risk | Comment: nothing stated about allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotation: "double‐blind clinical trial" Comment: no further information, although placebo administered as for intervention |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "double‐blind clinical trial" Comment: no further information, although placebo administered as for intervention |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quotation: "Among the 38 patients that were included in this sample, 25 (65.9%) returned for the re‐evaluation (12 from the study and 13 from the control group)" Comment: no further details were provided – inadequate data provided on dropouts |
| Selective reporting (reporting bias) | High risk | Comment: no appropriate outcome data were reported despite being assessed – study authors only commented on whether there was any significant differences or not with P values |
| Other bias | Low risk | Comment: no other sources of bias were identified |
Bergdahl 1995a.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 30 BMS patients
Sex: 24 F:6 M (F 80%:M 20%) 24/30 (80%) were women with a mean age of 56 years (range: 40‐69) and 6/30 (20%) were men with a mean age of 46 years (range: 38‐57) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: psychological therapies Group 1: (n = 15) "therapy group" (TG) ‐ Phase 1: an introductory session consisting of a motivational input and an oral examination. The patients were given time to decide whether or not to participate in the study. Phase 2: evaluation of BMS intensity (pre‐treatment). Phase 3: cognitive therapy (CT) for 12‐15 sessions; 1 hour once a week. Phase 4: evaluation of BMS intensity and oral examination immediately after completed CT (post‐treatment). Phase 5: evaluation of BMS intensity and oral examination 6 months after completed CT (6‐month follow‐up). 2 psychologists, dentist estimated outcome Group 2: (n = 15) "attention/placebo group" (APG) ‐ Phase 1: an introductory session consisting of a motivational input and an oral examination. The patients were given time to decide whether or not to participate in the study. Phase 2: evaluation of BMS intensity (pre‐treatment). Phase 3: return visits 3 times during 12‐15 weeks for evaluation of BMS intensity and oral examination. Phase 4: evaluation of BMS intensity and oral examination (post‐treatment). Phase 5: evaluation of BMS intensity and oral examination 6 months later (6‐month follow‐up). 2 psychologists, dentist estimated outcome Duration: 12 to 15 weeks |
|
| Outcomes |
VAS ‐ graded 1‐7, "graded from endurable to unendurable" pre‐treatment, post‐treatment, 6 months post‐treatment ‐ "All the patients evaluated their burning mouth intensity with the same dentist" Global symptom reduction post‐treatment and 6/12 – not prespecified |
|
| Source of funding | Swedish Dental Society and the Faculty of Odontology, Umeå University, Sweden | |
| Notes | Groups comparable at baseline SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "divided at random into equal groups" Comment: no details provided of exactly how this was conducted |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details provided on allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Quotation: "All the patients evaluated their burning mouth intensity with the same dentist" Comment: the study was unblinded to the participants.There was no mention of blinding ‐ given that both arms employed completely different treatment modalities, it is unlikely that it would be possible to blind the participants or investigators |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | High risk | Quotation: "All the patients evaluated their burning mouth intensity with the same dentist" Comment: the study was unblinded to the participants.There was no mention of blinding ‐ given that both arms employed completely different treatment modalities, it is unlikely that it would be possible to blind the participants or investigators |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: it was not stated if all participants completed the study/no mention of missing data |
| Selective reporting (reporting bias) | Low risk | Comment: the prespecified outcome measure was reported appropriately |
| Other bias | High risk | Commnent: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out |
Bogetto 1999.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 121 BMS patients
Sex: 91 F:30 M (F 75%:M 25%) Mean age 65.4 years (SD 10.6 years) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: antidepressants and antipsychotics + benzodiazepines Group 1: (n = 24) paroxetine 20 mg/day Group 2: (n = 23) amitriptyline 25 mg/day Group 3: (n = 26) clordemetildiazepam 1 mg/day Group 4: (n = 24) amisulpride 50 mg/day Group 5: (n = 24) placebo Duration: 8 weeks |
|
| Outcomes | Hamilton Anxiety Rating Scale (HARS) (clinician‐rated) Montgomery–Åsberg Depression Rating Scale (MADRS) Clinical Global Impression I (CGI I) scale (clinician‐rated) |
|
| Source of funding | Not reported | |
| Notes | All participants were given a 2‐week washout period from prior medication prior to being randomised SES: participants' average number of schooling years reported (5.7 years (SD 2.3) (min 0 ‐ max 13)) Conflict of interests: not reported Data analysis: per‐protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "...patients were subdivided in 5 groups using a random criteria..." Comment: the method used to generate the randomisation is not reported in the text |
| Allocation concealment (selection bias) | Unclear risk | Comment: no details provided on allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Quotation: "open label" Comment: open study hence participants not blinded |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | High risk | Quotation: "open label" Comment: open study hence assessors not blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Group 1 paroxetine 9/24 (37.5%) dropouts Group 2 amitriptyline 14/23 (60.8%) dropouts Group 3 clordemetildiazepam 11/26 (42.3%) dropouts Group 4 amisulpride 1/24 (4.2%) dropouts Group 5 placebo 19/24 (79.2%) dropouts Explanation provided by study authors ‐ possible reasons for dropouts: "patients with BMS tend to change medical doctor frequently, side effects of drug tested apart from amisulpride that is characterized by few side effects" Comment: insufficient details on why participants dropped out ‐ reason for missing outcome data likely to be related to true outcome |
| Selective reporting (reporting bias) | Low risk | Comment: outcomes reported in prespecified way |
| Other bias | High risk | Commnent: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out |
Cano‐Carrillo 2014.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 60 BMS patients Sex: 48 F:12 M (F 80%:M 20%) Mean age 63.3, SD 12.9 years Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 30) extra virgin olive oil (VOO) with lycopene 300 ppm: applied as a spray to the mouth, to be swallowed afterwards: 1.5 mL 3 times a day for 12 weeks Group 2: (n = 30) identical placebo formulation: as above – "consisted of a formulation (water and dye) identical to that of the study product but without the active agents" |
|
| Outcomes |
VAS "pain": "A visual analogue scale (VAS) was used to evaluate symptoms at the start of treatment (Day 0) and after 12 weeks of treatment. In this way, the difference between baseline and endpoint scores numerically expresses any symptomatic improvement (0 = none and 10 = extreme). The subjects were asked to mark a vertical line through a 10 cm horizontal line to indicate their level of symptoms. The scores for pain were classified into: slight (≤ 3.3), moderate (3.4–6.6), and severe (≥ 6.7)" VAS "burning": "A visual analogue scale (VAS) was used to evaluate symptoms at the start of treatment (Day 0) and after 12 weeks of treatment. In this way, the difference between baseline and endpoint scores numerically expresses any symptomatic improvement (0 = none and 10 = extreme). The subjects were asked to mark a vertical line through a 10 cm horizontal line to indicate their level of symptoms. The scores for pain were classified into: slight (≤ 3.3), moderate (3.4–6.6), and severe (≥ 6.7)" General health assessment ‐ 36‐Item Short Form Health Survey (SF‐36) at baseline – 12 weeks Oral Health Impact Profile‐14 (OHIP‐14) at baseline – 12 weeks The Hospital Anxiety and Depression (HAD) scale at baseline – 12 weeks "Lipid profile blood test" at 12 weeks "Patient‐rated benefit and satisfaction" ‐ Not stated how this was measured |
|
| Source of funding | Study authors reported no funding received | |
| Notes | Outcome measures and their use were partly very confusing SF‐36 normally compared to norms for age and gender SES: not reported Conflict of interests: study authors reported no conflict of interests exist Data analysis: per‐protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Randomization was performed using the website http://randomization.com to assign participants to either the intervention group or the placebo group" Comment: appropriate method |
| Allocation concealment (selection bias) | Low risk | Quotation: "A code for randomization was kept in an opaque envelope in a safe environment and was not consulted until the end of the study" Comment: appropriate method |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "A randomized, double‐blind and placebo‐controlled study design was adopted"; "Both patients and researchers were blind to treatment assignment (treatment/placebo)"; "The placebo consisted of a formulation (water and dye) identical to that of the study product but without the active agent"; "The products were coded by an operator external to the study in identical opaque containers (without any brand name)" Comment: probably achieved |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotations: "In all cases, data were collected by a single researcher blind to the group to which each patient belonged"; "Data were analysed by a third party blinded to the allocation results" Comment: probably achieved |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quotations: "4/30 patients in the treatment group left the study; 6/30 patients in the placebo group left the study"; "Patients were lost from the sample due to lack of compliance" (both groups) Comment: dropouts not accounted for in analysis |
| Selective reporting (reporting bias) | High risk | Not all prespecified outcomes reported in the results (e.g. tolerability, compliance with treatment)
All reported outcomes not specified in the methods (e.g. patient‐rated benefit and satisfaction) Definition of outcome "evolution of pain symptoms" unclear; numbers of participants in each of these groups are not given |
| Other bias | High risk | The baseline data provided for the arms are incomplete, however, given appropriate randomisation method balance/imbalance less of a concern Participants occasionally using anxiolytics to induce sleep were accepted ‐ results as to the use of anxiolytics within the cohort were not given, thus it is not known whether groups were comparable at baseline with regards to anxiolytics. There were also errors noted in the presented outcome data (tables 3 and 4) ‐ however the corrected data were obtained from the study authors after contacting them |
Carbone 2009.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 66 BMS patients Sex: 54 F:12 M (F 82%:M 18%) Of the 52 patients who completed the trial, mean age was reported as 67.3 years (SD 11.9) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 22) 400 mg alpha lipoic acid (ALA) and vitamins B (1, 2, 6, 12), C, E, and folic acid (vitamin B9) ‐ 1 pill twice/day by mouth 30 minutes after food for 8 weeks followed by phase II which was a 2‐month follow‐up period without therapy (Tiobec, produced by Laborest) Group 2: (n = 22) 400 mg alpha lipoic acid ‐ 1 pill twice/day by mouth 30 minutes after food for 8 weeks followed by phase II which was a 2‐month follow‐up period without therapy (produced by Laborest as standalone ALA specifically for this trial) Group 3: (n = 22) placebo pill 1 pill twice/day 30 minutes after food (containing dicalcium phosphate, microcrystalline cellulose, hydroxipropylmethyl cellulose, silicon dioxide, vegetal magnesium stearate, shellac and stearic acid) for 8 weeks. Followed by phase II which was a further 2‐month follow‐up period without 'therapy' |
|
| Outcomes | Proportion of participants achieving a 50% improvement in BMS symptoms from baseline to T3 and T4, measured by the VAS score. Each participant was examined by the same examiner (blinded to treatment) at the beginning of therapy (T0), 2 weeks (T1) and 4 weeks (T2) after the start of treatment, at the end of treatment (T3), and after 2 months of follow‐up (T4) Quality of pain experienced by participants was also assessed using the McGill Pain Questionaire. Similarly, each participant was examined by the same examiner (blinded to treatment) at the beginning of therapy (T0), at 2 weeks (T1) and at 4 weeks (T2) after the start of treatment, at the end of treatment (T3), and after 2 months of follow‐up (T4) | |
| Source of funding | Not reported | |
| Notes | The study authors query whether all participants should have been treated with a 7‐day course of antifungal therapy to eradicate subclinical candidosis. Current clinical practice would consider this unnecessary and irrelevant if the diagnosis is BMS. Is 8 weeks treatment sufficient time to provide a definitive answer? SES: not reported Conflict of interests: not reported Data analysis: per‐protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Randomization was performed using computer‐generated random number tables in order to assign patients to receive one of the three 8‐week standardized treatments" Comment: satisfactory |
| Allocation concealment (selection bias) | Unclear risk | Comment: allocation concealment was not discussed |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "Double blind"; "The medication (pills) was distributed in identical containers. During treatment, neither the physician nor the patients knew which of the three medications they were using" Comment: satisfactory |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "Each patient was examined by the same examiner (blind to treatment)" Comment: satisfactory – patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quotation: "Fourteen patients did not complete the study and were excluded from the efficacy sample. No side effects were reported as being a reason for withdrawal from the trial: 10 patients dropped out because of lack of compliance, and four patients because of lack of efficacy. No significant difference in the rate of dropout was observed among the three groups (p = 0.079, v2‐test)" Intention‐to‐treat was also considered Comment: reasons for dropouts were described overall, but not for each arm. 10 participants "dropped out" due to a "lack of compliance" – it is unclear exactly what "lack of compliance" meant and how these 10 were spread across each of the study arms |
| Selective reporting (reporting bias) | Unclear risk | Comment: full McGill Pain Questionnaire data not provided but alluded to and partially described Quotation: "The McGill Pain Questionnaire scores showed some improvements compared to the baseline measurements (Friedman test), but significant differences among the three groups were never observed (Kruskall–Wallis test). In particular, the affective, and the mixed affective/evaluative subscales slightly improved in Group C (Friedman test: p = 0.004 and 0.022, respectively); conversely, the evaluative subscale improved in all three groups (Friedman test: Group A, p = 0.007; Group B, p = 0.003; Group C, p = 0.046)" Comment: no baseline characteristic data were presented (only P values of statistical tests), full McGill Pain Questionnaire data not provided (again only P values) |
| Other bias | Low risk | Comment: no other obvious risks of bias |
Cavalcanti 2009.
| Methods | Single centre, placebo‐controlled cross‐over RCT | |
| Participants | 38 BMS patients Sex: 34 F:4 M (F 89%:M 11%) Mean age 63.1 years (range: 36–78) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 17) 200 mg alpha lipoic acid (ALA) capsules 3 times daily for 30 days. A 20‐day washout period followed before participants received placebo (cellulose starch 100 mg), administered in identical capsules 3 times daily for a further 30 days. Participants who reported any improvement with the proposed treatment were contacted after 60 days to assess maintenance or loss of the results (follow‐up) Group 2: (n = 14) placebo (cellulose starch 100 mg), administered in identical capsules 3 times daily for 30 days. A 20‐day washout period followed before 200 mg of ALA capsules 3 times daily for a further 30 days. Participants who reported any improvement with the proposed treatment were contacted after 60 days to assess maintenance or loss of the results (follow‐up) |
|
| Outcomes | Extent of reduction of symptoms based on VAS: rating of burning was evaluated by measurements of the VAS, ranging from 0 (no burning) to 100 mm (maximum burning), before and after each cycle (i.e. before the beginning of the treatment (baseline T0) and at follow‐up visits: after completing the first cycle of 30 days (T1), at the end of washout period of 20 days (T2) and at the end of the second cycle of 30 days (T3)) Self‐reported description of improvement Global Perceived Effect (GPE). Adapted from Femiano and colleagues and scored by the participant according to a 5‐point scale, ranging from: 1 = worse; 0 = no change; +1 = slight improvement; +2 = decided improvement; +3 = no burning anymore (resolution) after each cycle |
|
| Source of funding | São Paulo State Research Foundation (FAPESP) | |
| Notes | Participants taking antidepressants, possibly anxiolytics, angiotensin‐converting enzyme inhibitors and hormone replacement therapy were included in the study population – although they appeared to be distributed evenly across the arms SES: not reported Conflict of interests: not reported Data analysis: per‐protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "appropriate random‐number generator software (available at www.graphpad.com/quickcalcs/RandMenu.csm)" Comment: appropriate |
| Allocation concealment (selection bias) | Low risk | Quotation: "After determining the eligibility and obtaining the consent, to guarantee the blinding, the researcher sent the patient's study number to the pharmacist, who then allocated patients" Comment: participants and investigators enrolling participants could not foresee assignment because pharmacy‐controlled central allocation was used to conceal allocation |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "placebo‐controlled double‐blind crossover trial"; "Patients started with 200 mg of ALA or placebo (cellulose starch 100 mg), administered in identical capsules three times daily for 30 days" Comment: blinding of participants and key study personnel ensured, and unlikely that the blinding could have been broken. Side effects from ALA may have led to participants being suspicious that they might be taking the active treatment – results suggest no significant difference in reported adverse events between groups, hence unlikely that adverse events introduced significant bias |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotations: "placebo‐controlled double‐blind crossover trial"; "Patients started with 200 mg of ALA or placebo (cellulose starch 100 mg), administered in identical capsules three times daily for 30 days" Comment: blinding of key study personnel ensured, and unlikely that the blinding could have been broken |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 38 BMS patients commenced the trial. 2 appear to have been lost to follow‐up and 5 discontinued the interventions. 4 of them did not complete the proposed treatment because of the symptoms that they felt were due to treatment (these adverse effects were listed). 4 participants were excluded from the analysis. Proportions appear similar across the 2 groups Data from flow diagram (stating 2 participants in each arm were excluded from the analysis) did not match details in text (which stated 7 patients had their data excluded from analysis) |
| Selective reporting (reporting bias) | Low risk | Comment: standard deviations not reported; data by treatment cycle obtained from study authors |
| Other bias | Unclear risk | Comment: errors in reporting within paper (flow diagram, and n values), although clarified by authors in correspondence; data received from study authors revealed a baseline imbalance in relation to the VAS scores between the 2 study arms however randomisation process conducted appropriately |
Femiano 2000.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 42 BMS patients
Median age 63 years (range 43 to 78)
Sex: 32 F:10 M (F 76%:M 14%)
20 had removable prostheses Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 21) alpha lipoic acid and vitamins B (1, 2, 6, 12), C, E, and folic acid (vitamin B9) 600 mg/day for 20 days, followed by 200 mg/day for 10 days (Tiobec, produced by Laborest) Group 2: (n = 21) placebo cellulose starch 100 mg/day for 30 days Duration: 30 days As a second stage study, the original controls were then treated with the active regimen for 30 days |
|
| Outcomes | "All patients were reviewed at 15‐day intervals"; bespoke BMS symptomology change scale: "change in symptomatology scored as Worsening ‐ Unchanged +/‐ Slight improvement + Decided improvement + + Resolution + + +" ‐ unclear as to exactly how these data were recorded and by whom | |
| Source of funding | Not reported | |
| Notes | Unvalidated outcome measure used
Comparability of groups at baseline is unclear SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "randomly divided into two groups (Test and Control) each of 21 subjects, matched for age and sex" Comment: unclear as to how the randomisation was conducted and what was meant by "matched" |
| Allocation concealment (selection bias) | High risk | Comment: allocation concealment was not discussed |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Quotation: "open controlled clinical study" Comment: described as an open‐label study hence non‐blinded |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | High risk | Quotation: "open controlled clinical study" Comment: described as an open‐label study hence non‐blinded ‐ how reported outcome was recorded (i.e. self‐reported by patient or by clinician) is unclear |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no dropouts or missing data reported |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes were reported in the prespecified format |
| Other bias | High risk | Commnent: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out |
Femiano 2002a.
| Methods | Placebo‐controlled parallel RCT (unclear where study was conducted or whether single or multicentre) | |
| Participants | 60 BMS patients
Median age 45 years (range 22 to 68)
Sex: 42 F:18 M (F 70%:M 30%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 30) alpha lipoic acid and vitamins B (1, 2, 6, 12), C, E, and folic acid (vitamin B9) in 200 mg oral pills, 3 times a day for 8 weeks (Tiobec, produced by Laborest) Group 2: (n = 30 ) placebo cellulose starch 100 mg/day, 3 times a day Duration: 2 months (note: those showing improvement in symptoms at 2 months given a further month of treatment and followed for 1 year) |
|
| Outcomes |
Bespoke BMS symptomology change scale ‐ change in symptomatology scored as: Worsening ‐ Unchanged +‐ Slight improvement + Decided improvement ++ Resolution +++ "Results at 1 year follow‐up for changes in burning symptomatology in all subjects who showed an improvement at 2 months, as shown in Table 2, using alpha lipoic acid (test) or placebo control (starch)" – categories = No change, Slight deterioration, Significant deterioration |
|
| Source of funding | Not reported | |
| Notes | Outcome measures were confusing and unvalidated
Comparability of groups at baseline unclear It is unclear whether other ALA studies published by this study author around the same time used the same participants or not SES: not reported Conflict of interests: not reported Data analysis: per‐protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "divided at random for two groups of patients" Comment: no detail provided about how randomisation was conducted |
| Allocation concealment (selection bias) | Unclear risk | Comment: nothing stated about allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotation: "using lipoic acid as test, and cellulose starch as control, where neither the patient nor doctor could distinguish the substance used" Comment: appropriate placebo control |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "The patients were clinically assessed every 15 days, and symptomatology was recorded"..."neither the patient nor doctor could distinguish the substance used" Comment: blinded outcome assessment, although how reported outcomes were recorded (i.e. self‐reported by patient or by clinician) is unclear |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: according to the results presented, no dropouts occurred |
| Selective reporting (reporting bias) | High risk | Quotation: "patients were clinically assessed every 15 days, and symptomatology was recorded" Comment: the above was prespecified but no data were provided in results Quotation: "without notable adverse effects" Comment: no prespecified method of assessing adverse effects was provided and above comment suggests some adverse effects were reported, but no data provided in results Quotation: "The study was concluded with a re‐evaluation of the results 1 year after commencement of the trial" Comment: the study only analysed results for the participants who "showed an improvement at 2 months", despite the methods section suggesting "Patients that reported any amelioration within 4 months (12 of Control group and 29 of Test group) were given further therapy for 1 month, with a protocol identical to that used previously". Therefore the methods and results do not correlate with each other. One should expect that all participants should be followed up at 1 year, not only the responders |
| Other bias | High risk | Commnent: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out |
Femiano 2002b.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 80 BMS patients Median age 63 years (range 30 to 74) Sex: 48 F:32 M (F 60%:M 40%) | |
| Interventions |
Intervention category: dietary supplements + topical treatments + cholinergics Group 1: (n = 20) bethanechol (urecholine) 5 mg oral dose every 8 hours between meals Group 2: (n = 20) lactoperoxidase oral solution (Biotene oral rinse) topically 5‐6 times daily Group 3: (n = 20) alpha lipoic acid and vitamins B (1, 2, 6, 12), C, E, and folic acid (vitamin B9) in 200 mg oral pills, 3 times a day (every 8 hours) for 60 days (Tiobec, produced by Laborest) Group 4: (n = 20)placebo ‐ xylitol 3% in distilled water (no further details provided) Study conducted over 60 days |
|
| Outcomes |
Bespoke BMS symptomology change scale scored as: Worsening, Unchanged, Slight improvement, Decided improvement, Resolution – unclear as to how this was assessed Weekly "assessments" performed – further details not provided. Unclear whether symptomatology or side effects were assessed at every visit |
|
| Source of funding | Not reported | |
| Notes | 16/80 participants reported to have "used anxiolytic drugs to control the BMS" – these were allocated equally amongst the study arms 2 participants were reported in results section to be ongoing with anxiolytics during trial Unusually high number of male participants included within the study cohort It is unclear whether other ALA studies published by the author around the same time used the same participants or not SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotations: "This BMS cohort was then randomly divided into 4 groups"; "matched for age and sex" Comment: no detail provided about how randomisation was conducted |
| Allocation concealment (selection bias) | High risk | Comment: no detail given regarding allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Quotation: "This open controlled study of α‐lipoic acid" Comment: the study is described as open‐label |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | High risk | Quotation: "This open controlled study of α‐lipoic acid" Comment: the study is described as open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quotation: no dropouts reported. No missing data reported |
| Selective reporting (reporting bias) | High risk | Comment: no data provided for BMS symptoms during trial (e.g. at week 4) only after trial completion scores were given. "All patients reported increased salivation" was reported in Group 1 ‐ but this was not a prespecified outcome |
| Other bias | High risk | Commnent: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out |
Grémeau‐Richard 2004.
| Methods | Multicentre (6 centres), placebo‐controlled parallel RCT (open‐label study continued for 6 months following initial 14‐day study – data not included in this analysis) | |
| Participants | 48 patients with "stomatodynia" ("isolated complaint of chronic pain in the oral mucosa with normal clinical examination, with duration of pain greater than 4 months")
Mean age 65 years SD 2.1
Sex: 44 F:4 M (F 92%:M 8%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: benzodiazepines Group 1: (n = 24) clonazepam tablet 1 mg to be sucked without swallowing for 3 minutes, 3 times a day (after each meal) for 2 weeks Group 2: (n = 24) placebo tablet to be sucked without swallowing for 3 minutes, 3 times a day (after each meal) for 2 weeks Duration: 2 weeks intervention, 6‐month open follow‐up |
|
| Outcomes |
Numerical Pain Scale (0‐10): "The primary criterion used to evaluate drug efficacy was the difference in pain intensity score observed for each subject before and after 14 days of treatment (NS 0 – NS 2)" ‐ mean pain intensity (0 "no pain" to 10 "maximal pain imaginable") "As a secondary outcome criterion, the immediate effects of clonazepam were evaluated by comparing, between the active treatment and control groups, the average of the differences (NS 0 – NS 1) in pain intensity score before and 5 min after topical 5 application" Compliance and adverse events were also recorded |
|
| Source of funding | "The authors thank Laboratoire Roche for clonazepam and placebo tablets, C Cadène and the association 'Langue de feu' for their help, and the Teaching Hospital of Clermont‐Ferrand for financial and administrative support" | |
| Notes | Groups comparable at baseline ‐ baseline anxiety or depression status were not known The active treatment was continued for all participants for 6 months after the initial trial finished. It would have been interesting to note if there were any adverse events upon withdrawal of the active treatment SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Randomisation was performed in blocks of eight by the hospital pharmacy" Comment: third party randomisation |
| Allocation concealment (selection bias) | Low risk | Quotations: "Randomisation was performed in blocks of eight by the hospital pharmacy"; "Experimentors were blinded to patient allocation" Comment: appropriate method |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "Experimentors were blinded to patient allocation"; "All the tablets looked identical" Comment: side effects could potentially unblind participants, however, no significant difference in side effects reported between 2 arms. Unblinding mentioned: "After unblinding, five patients identified as clonazepam receivers .... "– however, this was after the initial trial period of 14 days |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotations: "Experimentors were blinded to patient allocation"; "All the tablets looked identical" Comment: patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quotations: "At fourteen days: Two Group A patients discontinued intervention because of side effects. One group B patient discontinued intervention because of side effects". ITT analysis performed – assumption "that the three patients who did not complete the study were not modified by the treatment". The pain intensity outcome at 5 minutes only reported on active arm "n = 22" and placebo arm "n = 23" i.e. suggesting that the 3 dropouts occurred within the first 5 minutes of the study. No ITT analysis was provided for the 5 minute outcome. Unclear if this was a simple error in notation or otherwise Comment: no real imbalance in dropouts and number of them unlikely to influence result |
| Selective reporting (reporting bias) | Low risk | Comment: all outcomes reported in prespecified way |
| Other bias | Unclear risk | Comment: baseline anxiety levels were not assessed ‐hence the spread of baseline anxiety between the study arms was not known. Bearing in mind that clonazepam is an anxiolytic drug which was noted to have a systemic uptake within the participants, the presence of baseline anxiety is a potential confounder and should have been taken into account (potential omitted variable bias) |
Heckmann 2012.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 20 BMS patients Overall ages not presented ‐ Group 1 (clonazepam): from text ‐ mean 67.5 (range 49‐89) / from table ‐ mean 65.0 SD: 12.4. Group 2 (placebo): from text ‐ mean 65.4 (49‐78) / from table ‐ mean 62.9 SD: 8.7 Sex: 13 F:7 M (F 65%:M 35%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: benzodiazepines Group 1: (n = 10) clonazepam 0.5 mg capsules ‐ 1 daily Group 2: (n = 10) placebo capsules ‐ 1 daily Duration: 9 weeks "As the intake of clonazepam can cause dependency, the medication was tapered off at the end of study in those subjects who had received verum. They took drops (0.1 mg clonazepam per drop) for a period of 10 days starting with five drops; this dose was reduced by one drop every 2 days" |
|
| Outcomes |
Beck Depression Inventory (BDI) – 2 weeks before treatment, beginning of treatment, 3 weeks after starting treatment, at end of treatment and 2 weeks after end of treatment Zerssen Mood Scale – 2 weeks before treatment, beginning of treatment, 3 weeks after starting treatment, at end of treatment and 2 weeks after end of treatment Taste test score (0‐16): "Taste test. For quantitative assessment of gustatory function, a standardized validated test based on filter papers impregnated with tastants was used. Strips with the basic tastes sweet, sour, salty, and bitter (in four concentrations each) were applied onto the extended tongue, which was then taken back into the closed mouth. Before application of each taste strip, patients rinsed their mouths with water. Following presentation of the strip, patients were asked to identify the taste from a list of four descriptors (sweet, sour, salty, and bitter). The sum of correct identifications was used for further statistical analysis" ‐ performed 2 weeks before treatment and at end of treatment and 2 weeks after end of treatment (methods fully detailed in Mueller 2003: scale 0‐16 (each taste component 0‐4), higher better) Smell test score: "Smell test. The odour identification part of the Sniffin'‐ Sticks test battery were used to screen for changes in olfactory function. Following presentation of a common odour, subjects were each asked to identify it from a list of descriptors. The sum of correct identifications was used for further analysis" ‐ performed 2 weeks before treatment and at end of treatment and 2 weeks after end of treatment "Salivary flow rate. The salivary flow rate was measured using a cotton swab. It was weighed and placed onto the patient's tongue for 1 minute. After that, the cotton swab was weighed again and the resulting difference was used to calculate salivary flow rate" ‐ performed 2 weeks before treatment and at end of treatment and 2 weeks after end of treatment (methods fully detailed in Navazesh 1982: weight scale, higher better. Swab method is 1 of 4 methods (draining, spitting, suction, swab) compared to calculate salivary flow. (Note Navazesh 1982 authors state swab method is least reliable and most variable of the 4 options) Numerical pain ratings scale (0‐10): "Pain ratings. Patients rated the sensation of burning pain in the mouth on a scale ranging between 0 and 10, with 0 indicating no pain and 10 indicating maximum possible pain" ‐ performed 2 weeks before treatment and at end of treatment and 2 weeks after end of treatment |
|
| Source of funding | Study authors reported no funding was received | |
| Notes | SES: not reported Conflict of interests: authors reported no conflict of interests exist Data analysis: ITT | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotations: "performed by an independent individual using a specialized software program (RANDLIST; DatInf, Tübingen, Germany)"; "enrolment numbers were established, and the subjects to be investigated were randomized in such a way as to form five groups made up of four participants each (i.e., two were assigned clonazepam and two were assigned a placebo)" Comment: appropriate method |
| Allocation concealment (selection bias) | Low risk | Comment: randomisation and blinding was conducted by an independent person |
| Blinding (performance bias and detection bias) Blinding of participants | Unclear risk | Quotations: "The bottles were sealed and labelled with the study code and the enrolment number"; "When the study was complete, unblinding was carried out by an independent individual" Comment: it is unclear to the review authors at what point was the "study complete"; was it at the end of treatment or at the final session 5 visit? According to the data provided, the participants were unblinded for the outcomes taken during the final (session 5) |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Unclear risk | Quotation: "When the study was complete, unblinding was carried out by an independent individual" Comment: according to above comment, the investigators were still blinded at the session 5 assessment point. Again there was uncertainty about when exactly study completion occurred |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all prespecified outcomes were reported in the prespecified way |
| Selective reporting (reporting bias) | High risk | Comment: side effects were not mentioned as a prespecified outcome and were not adequately reported in the outcomes ("The drugs were tolerated very well by all participants"; "does not cause major side effects"). This comment would suggest that some side effects were noted, but not formally reported. One would expect side effects from clonazepam and should be able to expect that side effects would be more thoroughly reported, as side effects may have unblinded participants |
| Other bias | Unclear risk | Comment: discrepancy between baseline ages of participants when comparing text and table data |
Lopez‐D'alessandro 2011.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 120 BMS patients Mean age 14.1 SD 57.5 years, median: 57 Sex: 94 F:26 M (F 78%:M 22%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: anticonvulsants + dietary supplements Group 1: (n = 20) 600 mg daily alpha lipoic acid ‐ for 2 months (Ciagen 600, produced by Craveri). Supplement includes excipients (sodium lauryl sulfate 30.00 mg; 329.50 mg lactose monohydrate; cornstarch 115.50 mg; croscarmellose sodium ( AC‐DI‐SOL) 72.00 mg; colloidal silicon dioxide (Aerosil 200) 5.50 mg; 35.00 mg magnesium stearate; povidone K‐30 12.50 mg; methocel E15 25.08 mg; 10.05 mg lactose monohydrate; 0.39 mg titanium dioxide; polyethylene glycol 6000 0.78 mg; yellow iron oxide 3.68 mg) Group 2: (n = 20) 300 mg daily gabapentin (GABA) Group 3: (n = 20) 600 mg daily alpha lipoic acid (ALA) + 300 mg daily gabapentin (GABA) Group 4: (n = 60) 100 mg daily starch and cellulose placebo |
|
| Outcomes |
Number of sites affected ‐ "evaluated the presence of burning through a numerical scale especially created for this work, describing the burning from 0 to 4, where the 0 value corresponded to the absence of burning, the 1 value to the presence of burning in a single area of the tongue, the 2 value to two distinctive areas (tongue and gums, tongue and lips or tongue and palate), the 3 value to three areas and the 4 value corresponded to burning spread throughout the mouth. This specific designed scale, which considered the geographical distribution of burning in different areas of the mouth, allowed us to distinguish improvements or deteriorations of burning sensation in the various assessments" "Evaluation of the effects… the day before the start of treatment and thirty and sixty days, respectively. To evaluate the changes that occurred with the taking of the different drugs, it was established that the improvements (positive changes) involved the passage of a certain level or numerical category of burning to a lower one, the deteriorations (negative changes) involved an increase of a certain level of burning to a higher one and the total resolution indicated the total absence of burning, that is to say the transition from any higher value to zero value. In this way four categories were obtained for the analysis of the 5 results: Category 1: with negative changes (deterioration), Category 2: no changes; Category 3: with positive changes (improvements) and Category 4: with total recovery" Change in quality of life (QoL) was assessed at baseline only (consequently unable to assess change) by use of 2 surrogate (measuring anxiety and depression) scales: ‐ anxiety: Hamilton Anxiety Rating Scale (HARS) ‐ baseline only; and ‐ anxiety/depression: Hospital Anxiety and Depression (HAD) scale ‐ baseline only |
|
| Source of funding | Not reported | |
| Notes | The dosage of gabapentin given is not therapeutic as an anticonvulsant Outcome evaluation was based on the numerical scale which was based on the number of sites affected. Thus only 1 (not validated) outcome measure was used in the study SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "The 120 patients were randomly divided into 6 groups of 20 members each" Comment: unclear as to how the randomisation was conducted |
| Allocation concealment (selection bias) | Low risk | Quotations: "The support staff of our service made a draw with 6 balls to link the groups with the cycles of treatment"; "allocation that was always masked to both patients and researcher" Comment: assumption that "support staff" were independent to investigators – seems unlikely that allocation concealment would be compromised |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Quotation: "through the use of capsules of similar size and appearance so that just the support staff was the one who recorded the information until the end of the treatment (blind)" Comment: "similar size and appearance" ‐ unlikely that formulations were identical, as weights were different and they may not look the same. Unclear how preparations were provided to participants and in what packaging. Placebo only group involved 1 tablet while group 3 had 2 tablets to take – hence unblinding group 3 that they were on the combined treatment arm |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Comment: patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no dropouts were reported |
| Selective reporting (reporting bias) | High risk | Quotation: "adverse effects that appeared were very mild" Comment: clarification sought from contact author who provided adverse effect data. No baseline characteristics were presented for each arm of the study. Baseline anxiety and depression was presented for all participants but not for each arm. Outcome data were not presented in the prespecified way – graphs were presented which were difficult to interpret as the actual data values were not presented. The study authors combined "positive changes" with "full recovery" data for the active arms and "no change" with "worsened" data with the placebo – these outcomes should have been presented separately as combining them is very misleading. No useable data were presented for month 1, only month 2. Clarification sought from contact author who provided missing data from month 1 |
| Other bias | Low risk | Comment: possible selection bias caused by exclusion criteria – "patients using more than 3 systemically daily drugs, those ones taking psychotropic and antihypertensives drugs as well as patients with serious psychiatric conditions previously diagnosed were excluded" |
López‐Jornet 2009b.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 60 BMS patients Mean age 64.37 SD 11.61 years Sex: 54 F:6 M (F 90%:M 10%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 23) 800 mg in oral pills per day for 8 weeks: each containing alpha lipoic acid (0.05 mg) + adjunctive lycopene (100 mg)+ green tea extract (40%, 50 mg) (Thioderm, produced by Sesderma) Group 2: (n = 16) placebo ‐ cellulose tablets of the same appearance, shape, texture and colour as the treatment for 8 weeks |
|
| Outcomes | Pain intensity as recorded on a 10 cm VAS. The scores for pain were classified into: slight (≤ 3.3), moderate (3.4–6.6) and severe (≥ 6.7). Pain scores were recorded before treatment (day 0) and at 1 and 2 months (only participants completing treatment protocol for the 2 months were included i.e. n = 39) | |
| Source of funding | Not reported | |
| Notes | SES: not reported Conflict of interests: not reported Data analysis: per‐protocol | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Having met the eligibility criteria, participants were randomly divided into two groups according to a list made by simple randomization block design, generated using a randomization table. A simple block randomization list with a block size of four was prepared by a team member not involved in the recruitment and follow‐up of the patients" Comment: satisfactory |
| Allocation concealment (selection bias) | Low risk | Quotation: "Randomization allocation concealment was performed by sending the randomization numbers in sealed envelopes to the investigator responsible for giving the assigned treatment after each eligible patient was enrolled" Comment: satisfactory |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "double blind"; "as placebo, cellulose tablets of the same appearance shape, texture and colour as the treatment" [were used] Comment: satisfactory |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotations: "double blind"; "as placebo, cellulose tablets of the same appearance shape, texture and colour as the treatment" Comment: satisfactory; patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 7/30 missing from intervention group (1 due to gastrointestinal side effects, other 6 reasons not given); 14/30 missing from control group (authors suggest that withdrawal may have been due to lack of efficacy of placebo) Proportions of dropouts different across 2 groups |
| Selective reporting (reporting bias) | Low risk | Comment: all prespecified outcomes were reported appropriately |
| Other bias | Low risk | Comment: baseline inequalities in gender and age are present, however randomisation appropriate |
López‐Jornet 2011.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 50 BMS patients Mean age 61.18, SD 12.27 (range 37‐84) Sex: 46 F:4 M (F 92%:M 8%) Inclusion and diagnostic criteria appropriate ‐ exclusion of all "patients with known neurological disorders and those previously treated, even irregularly, with antidepressants, anticonvulsants, other psychotropic drugs, or psychological therapy" represents exclusion of a significant proportion of the BMS population, hence reducing applicability to all BMS patients (i.e. possible reduced external validity) |
|
| Interventions |
Intervention category: physical barriers Group 1: (n = 25) control ‐ "Patients were informed in detail about their illness, and were instructed not to rub their tongue against their teeth and/or dentures. A self‐control technique was used to this effect, the patients being given 10 printed habit‐modifying reminder points to be placed in visible place" Group 2: (n = 25) tongue protector ‐ same as group 1 with tongue protector: "The protector consisted of a transparent, low‐density polyethylene sheath covering the tongue from 4 the tip to the posterior third. These tongue protectors were single‐use devices measuring 0.1 mm in thickness, with a standard size (67 mm in length and 66 mm wide), and were custom manufactured by our group. Each patient received a kit with the protectors and the reminder points for treatment"; "Instructions were provided on their use – the protector being worn during the daytime for period of 2 months. We recommended use of the protector 15 min/three times a day" Duration of intervention: 2 months |
|
| Outcomes | Oral symptoms VAS (0: no pain, 10: most severe pain experienced): "Patients were asked to indicate the mean pain intensity for the 2 weeks preceding the consultation. The difference between baseline and the endpoint scores numerically expressed symptoms variation" ‐ measured at baseline and 2 months Hospital Anxiety and Depression scale (HAD) ‐ measured at baseline and 2 months Oral Health Impact Profile‐49 (OHIP‐49) ‐ measured at baseline and 2 months 36‐Item Short Form Health Survey (SF‐36) ‐ measured at baseline and 2 months |
|
| Source of funding | Not reported | |
| Notes | The biological plausibility of the active treatment is not clear SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "…were randomly allocated to one of the two arms of the study. The random allocation sequence was generated using software available online at…" Comment: appropriate method |
| Allocation concealment (selection bias) | Unclear risk | Comment: nothing stated about allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Comment: nothing stated about blinding of participants or study personnel. The treatment modalities differed vastly from each other, hence unblinding participants and the investigator |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | High risk | Comment: nothing stated about blinding of participants or study personnel. The treatment modalities differed vastly from each other, hence unblinding participants and the investigator |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Commnent: no missing outcome data |
| Selective reporting (reporting bias) | Low risk | Comment: all prespecified outcomes were reported in the prespecified way |
| Other bias | Unclear risk | Quotation: "Patients occasionally using anxiolytics to induce sleep were accepted" Comment: no data provided on baseline use of anxiolytics between groups – prespecified as inclusion criteria but not reported upon. Important data as could induce baseline inequality or act as confounder |
Marino 2010.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 56 BMS patients Mean age 62 years (SD 9.8) Sex: 46 F:10 M (F 82%:M 18%) Inclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements + topical treatments Group 1: (n = 14) thrice daily oral rinses with capsaicin, 250 mg of red pepper emulsion in 50 ml of water for 8 weeks followed by 8‐week post‐treatment observation Group 2: (n = 14) alpha lipoic acid and vitamins B (1, 2, 6, 12), C, E, and folic acid (vitamin B9) in 400 mg oral pills, twice a day for 60 days (Tiobec, produced by Laborest) Group 3: (n = 14) lysozyme lactoperoxidase oral rinse (Biotene; GlaxoSmithKline Consumer Healthcare; GlaxoSmithKline S.P.A., Verona, Italy), 5 times a day for 8 weeks followed by 8‐week post‐treatment observation Group 4: (n = 14) 0.05 g of boric acid dissolved in 100 ml of distilled water (placebo), thrice a day for mouthwash for 8 weeks followed by 8‐week post‐treatment observation |
|
| Outcomes | Severity of burning mouth sensation was assessed at the beginning and at the end of both the studies by means of a subjective VAS ranging from 0 (no burning mouth sensation) to 10 (severe burning mouth sensation) | |
| Source of funding | Not reported | |
| Notes | Tiobec contains vitamin C, B1, 2, 6, 12, PP, folic acid, E (this is not mentioned in the paper) No side effects reported ‐ unexpected given the nature of capsaicin SES: not reported Conflict of interests: study authors report no conflict of interests exist Data analysis: unclear |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Patients were single‐blindly randomised by means of computer‐generated random number" Comment: probably undertaken |
| Allocation concealment (selection bias) | High risk | Quotation: "Patients were single‐blindly randomised by means of computer‐generated random number tables" Comment: study is single‐blind hence allocation was not concealed to investigators. Participants and investigators enrolling participants appear not to have been able to foresee assignment |
| Blinding (performance bias and detection bias) Blinding of participants | Unclear risk | Comment: no blinding attempted or possible ‐ mouthwashes (capsaicin) and Biotene gel are being compared against a systemic option (ALA) |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Comment: patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: dropouts were fully reported – they only occurred after phase 1 (active treatment) was completed. Although attrition rate was high, missing outcome data balanced in numbers across intervention groups, with similar reasons for missing data across groups |
| Selective reporting (reporting bias) | Low risk | Comment: prespecified outcomes were reported appropriately |
| Other bias | High risk | Commnent: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out |
Palacios‐Sánchez 2015.
| Methods | Single centre, parallel‐arm, placebo‐controlled RCT | |
| Participants | 60 BMS patients Mean age: 62.13 years (range 36‐86) Sex: 55 F:5 M (F 92%:M 8%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 30) 600 mg (3 x 200 mg every 8 hours) in oral pills per day for 8 weeks: each containing alpha lipoic acid (0.05 mg) + adjunctive lycopene (100 mg)+ green tea extract (40%, 50 mg) (Thioderm, produced by Sesderma) Group 2: (n = 30) placebo ‐ similar looking cellulose tablets to group 1 intervention, for 8 weeks (provided by ALA manufacturer) |
|
| Outcomes | Symptom relief, surrogately measured by VAS (scale 0‐10, but expressed dichotomously in reporting) to measure change in symptoms, at baseline, 1 month and 2 months Depression by Beck Depression Inventory (BDI), at baseline (unknown if recorded latterly) |
|
| Source of funding | Not reported, although all treatment (intervention and placebo) provided by Sesderma Laboratories | |
| Notes | Adverse effects not reported despite stating a priori that their occurrence would be recorded Baseline mean symptom intensity presented for all participants, rather than each group separately. May be baseline imbalance Depression reported by 54% of participants (32/59) Use of medication: antidepressants/anxiolytics 53%; antihypertensives 25%; thyroid treatment 12%; other medication 43% SES: not reported Conflict of interests: study authors report no conflict of interests exist Data analysis: per‐protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly allocated to one of the two different sequence groups" Comment: no detail provided about sequence generation method used |
| Allocation concealment (selection bias) | Unclear risk | Comment: not reported |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Comment: described as a "double‐blind placebo‐controlled study", but no further detail provided. Provision of identical placebo treatment by manufacturer would have allowed blinding of participants and outcome assessors, but not confirmed in text |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Comment: described as a "double‐blind placebo‐controlled study", but no further detail provided. Provision of identical placebo treatment by manufacturer would have allowed blinding of participants and outcome assessors, but not confirmed in text |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 54 patients (90%) complete trial (Group 1 n = 25; Group 2 n = 29); however, rationale for losses not reported |
| Selective reporting (reporting bias) | High risk | Comment: secondary outcomes (depression and adverse effects) not reported after receipt of treatment: depression reported at baseline, but unknown if recorded latterly, as appears to only have been used as a covariate within the authors' logistic regression analyses Adverse effects not reported despite stating a priori that their occurrence would be recorded |
| Other bias | High risk | Comment: no baseline data presented; given lack of detail regarding randomisation process substantial inequalities at baseline cannot be ruled out Wide use of medication by participants, and over half reporting depression |
Rodríguez de Rivera‐Campillo 2010.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 66 BMS patients Mean age 64.9 years (range 48–85) Sex: 64 F:2 M (F 97%:M 3%) Inclusion and diagnostic criteria appropriate Baseline characteristics for each group provided in graphs and table were difficult to interpret – no obvious differences between groups at baseline |
|
| Interventions |
Intervention category: benzodiazepines Group 1: (n = 33) topical clonazepam ‐ "Each patient was given a sealed envelope containing 32 tablets of 0.5 mg of clonazepam. They were instructed to take a single tablet at the first sign of discomfort in the morning. The tablet should be dissolved in the mouth for three minutes, and then the remaining saliva should be spat out. The patient should then note his or her sensations and the evolution of the symptoms. If there was improvement, the procedure was to be repeated when the symptoms reappeared. Patients were advised not to exceed four tablets a day (that is, a total dose of 2 mg of clonazepam)" Group 2: (n = 33) placebo ‐ "lactose tablets, of the same shape and size as those given to Group A. Their instructions were the same as those given to Group A" The amount of tablets used varied between participants |
|
| Outcomes | VAS from 0 to 10 (no scale descriptors provided) – baseline, 1 month, 6 months | |
| Source of funding | Not reported | |
| Notes | No email correspondence details provided – therefore, we could not contact authors to determine VAS descriptors SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "table of random numbers was used in order to ensure the randomization of the treatments" Comment: appropriate method |
| Allocation concealment (selection bias) | Unclear risk | Comment: nothing stated about allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | Unclear risk | Quotations: "double blind study"; "Each patient was given a sealed envelope containing 32 tablets of 0.5 mg of clonazepam"; "Group B: 33 patients, placebo group. They were given 32 lactose tablets, of the same shape and size as those given to Group A" Comment: from information provided, blinding method appears appropriate ‐ adverse effects (only reported in active arm – "sleepiness in 5 patients of the clonazepam group, which did not require the clinicians to suspend the treatment") and some participants in active arm reported sensation of effervescent and numbness for up to 3 hours – possible unblinding of participants due to side effects of clonazepam |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotations: "double blind study"; "Each patient was given a sealed envelope containing 32 tablets of 0.5 mg of clonazepam"; "Group B: 33 patients, placebo group. They were given 32 lactose tablets, of the same shape and size as those given to Group A" Comment: blinded outcome assessment; patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no dropouts or missing data reported ‐ over a 6‐month study period one would expect some to drop out |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported in the prespecified way |
| Other bias | High risk | Quotations: "Three clinicians, with extensive experience in oral medicine, examined the patients"; "They were again scheduled for visits after 1 month and 6 months, which allowed the clinicians to monitor their evolution"; "Both groups of patients showed improvement, which was partially due to the psychotherapy. The management of patients with BMS should be focused on two aspects. On one hand, clinicians could treat the symptoms; on the other hand, they could use basic psychotherapy customized to each person, which can be carried out in our dental office. The aforementioned psychotherapy is focused on listening to the patient, emphasizing affectivity, security and tranquility, and transmitting the feeling that we know exactly what the patient is going through as well as the difficulties we face in giving him/her solutions to his/her problems. Our personal experience has shown us that, if we manage to calm the patient with our attitude, the possibility of improvement increases; this is particularly true in patients who are relatively stable from an emotional point of view" Comment: the above statements suggest that there was additional treatment in the form of psychological intervention for each participant, undertaken by 3 different clinicians. No mention of standardisation of consultations or calibration of clinicians, hence possible confounding factor in terms of "psychotherapy" provided in a non‐standardised/uncalibrated way. Unclear if adverse events were recorded by investigators (see above) – possible unblinding of investigating clinicians, as a result of this which could influence the consultation and "psychotherapy" provided to each participant Quotation: "None of the patients was treated in the last month before their inclusion in the study" Comment: despite the above quotation, the authors reported most participants were taking adjuvant medications (e.g. antidepressants, anxiolytics) during the study. Therefore the former statement is misleading and inaccurate, as most would consider antidepressants and anxiolytics to be active treatments for BMS. No data were provided on type of drugs used, any dose changes during the study period – hence unclear whether there were confounder inequalities between groups during the study |
Sardella 1999.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 30 BMS patients
Mean age 69 years (range 54‐85)
Sex: 26 F:4 M (F 87%:M 13%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: topical treatments Group 1: (n = 10) benzydamine hydrochloride 0.15% oral rinse, 15 ml rinse 3 times a day for 4 weeks Group 2: (n = 10) placebo oral rinse 3 times a day for 4 weeks Group 3: (n = 10) no treatment Duration: 4 weeks |
|
| Outcomes | Change in severity of symptoms VAS (0 = no symptoms, 8 = a severe burning sensation) before and after treatment scored as: "ineffective" (same dot value for the symptoms at the 2 visits), "partially effective" (reduction in dot value of the symptoms), "effective" (complete absence of symptoms) | |
| Source of funding | Not reported | |
| Notes | Groups comparable at baseline Unvalidated outcome measure used Study only double‐blind for groups 1 and 2 SES: not reported Conflict of interests: not reported Data analysis: ITT |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Each patient was assigned to one of 3 management modalities (A, B or C) through use of a table of random numbers. To avoid subgroups of different sizes, a block randomization was used" Comment: appropriate method |
| Allocation concealment (selection bias) | Unclear risk | Comment: nothing stated about allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | Unclear risk | Quotation: "This study was a double‐blind, randomized, longitudinal investigation" Comment: nothing further stated about the blinding procedure. Group C was a no treatment control group, thus knew which group they have been allocated, and were not blinded to treatment, however this treatment arm not relevant to this review |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Unclear risk | Quotation: "This study was a double‐blind" Comment: nothing further stated about the blinding procedure; patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quotation: ".. all patients completed the study period" Comment: no dropouts reported |
| Selective reporting (reporting bias) | Low risk | Comment: outcome reported in prespecified way |
| Other bias | Unclear risk | Comment: insufficient data provided on baseline characteristics of participants. Baseline symptom severity VAS data were given for each individual participant. The mean VAS scores differed between treatment arms (Group A = 6.7, Group B = 6.1 and Group C = 5.7 ). It is unclear whether there were any significant differences regarding the symptom severity at the baseline |
Sardella 2008.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 43 BMS patients Mean age 64.9 SD 4.7 years Sex: 35 F:4 M (reported per protocol) (F 90%:M 10%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 21) hypericum perforatum extract 300 mg capsules (hypericin 0.31%, hyperforin 3.0%) 3 times a day for 12 weeks Group 2: (n = 22) placebo capsules "identically appearing" 3 times a day for 12 weeks |
|
| Outcomes | 10 cm VAS consisting of a horizontal line marked from 0 (no pain) to 10 (the worst pain ever experienced) measured at first visit (t0) and at 3 follow‐up visits (after 4 (t28), 8 (t56), and 12 (t84) weeks) Number of oral mucosa sites with reported burning symptoms measured at first visit (t0) and at 3 follow‐up visits (after 4 (t28), 8 (t56), and 12 (t84) weeks) Non‐standardised assessment of quality of life ‐ self‐reported descriptions in response to "simple questions (i.e. did you feel irritable/depressed/worry? or did oral burning sensations interfere with your daily activities? or have you had difficult in concentrating on things as reading or watching a TV movie?)" |
|
| Source of funding | "This study has been supported by a grant of the University of Milan (FIRST, Fondo Interno per la Ricerca Scientifica e Tecnologica, no 12‐1‐5201001‐540). We also thank Body Spring (Ancona, Italy) that kindly supplied both hypericum extract and placebo" | |
| Notes | SES: not reported Conflict of interests: not reported Data analysis: per protocol | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "random allocation sequence was generated using online software available at http://graphpad.com/quickcalcs/randomise1.cfm" Comment: appropriate |
| Allocation concealment (selection bias) | Low risk | Quotation: "To guarantee allocation concealment, the researchers deciding on patient eligibility did not know the sequence, and a researcher who was not involved in patient enrolment assigned the patients to one of the two arms" Comment: concealed allocation |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "double‐blind, placebo‐controlled study"; "Patients were randomized to receive indistinguishable 300‐mg capsules of H. perforatum extract (hypericin 0.31%, hyperforin 3.0%; Test Group) or placebo (Control Group)" Comment: probably achieved |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "double‐blind, placebo‐controlled study" Comment: patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all dropouts and missing data accounted for Proportions similar across groups Reasons similar across groups |
| Selective reporting (reporting bias) | Low risk | Comment: all primary outcomes were presented in the prespecified format |
| Other bias | Low risk | Comment: no other obvious risk of bias |
Silvestre 2012.
| Methods | Single centre, placebo‐controlled cross‐over RCT | |
| Participants | 30 BMS patients Mean age 72.65 SD 12.10 years (range 40‐90 years) Sex: 19 F:4 M (reported per protocol) (F 83%:M 17%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: topical treatments Group 1: (n = 30) capsaicin ‐ 0.02% capsaicin rinse administered 3 times a day – applied for 30 seconds in volumes of 15 ml – 1 week; "After this first week of treatment, the patients completed a one‐week washout period (no rinses, only regular dental hygiene in the form of tooth brushing), after which they were assigned to the opposite group for a further week of treatment"; "During this cross‐over period, treatment and the scoring of discomfort were carried out in the same way as in the first week, but administering the opposite oral rinse". No details of whether this was before or after meals Group 2: (n = 30) placebo ‐ "Placebo" rinse (unspecified preparation) ‐ applied for 30 seconds in volumes of 15 ml – 1 week; "After this first week of treatment, the patients completed a one‐week washout period (no rinses, only regular dental hygiene in the form of tooth brushing), after which they were assigned to the opposite group for a further week of treatment"; "During this cross‐over period, treatment and the scoring of discomfort were carried out in the same way as in the first week, but administering the opposite oral rinse". No details of whether this was before or after meals |
|
| Outcomes | VAS ‐ "discomfort at different times during the study (0 cm = no discomfort, 10 cm = unbearable or maximum discomfort)" – measured "morning before treatment in both the capsaicin group (AM1) and in the placebo group (BM1). The VAS score was then again recorded in the afternoon of the first day of treatment in both groups (AA1 and BA1) and at the end of one week of treatment in both groups, in the morning (AM7 and BM7) and in the afternoon (AA7 and BA7)" | |
| Source of funding | Not reported | |
| Notes | No baseline characteristics provided for each study arm SES: not reported Conflict of interests: not reported Data analysis: Per protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "The patients were randomized to two groups" Comment: not enough information to make a judgement |
| Allocation concealment (selection bias) | Unclear risk | Comment: no mention of allocation concealment |
| Blinding (performance bias and detection bias) Blinding of participants | High risk | Comment: likely unblinding of participants due to burning sensation of capsaicin |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | High risk | Comment: likely unblinding of participants due to burning sensation of capsaicin; patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quotation: "where indicated, those subjects who developed adverse effects were removed from the study" Comment: removal of participants who have had adverse events from the analysis is likely to introduce significant bias |
| Selective reporting (reporting bias) | High risk | Comment: VAS scores data were not presented appropriately – graph only showing capsaicin group with no raw data values (i.e. no means, standard deviations or ranges provided). Paper suggested quantitative data would be presented as median and range – unclear why this was as the distribution of the VAS data was not stated (i.e. no mention that it was non‐normally distributed) |
| Other bias | Low risk | Comment: given that participants had a mean of 5 years duration of BMS, 1 week treatment duration is very short |
Spanemberg 2012.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 72 BMS patients Mean ages ‐ Group 1: 63.6 SD 9.61 (range 41–79); Group 2: 61.5 SD 6.76 (range 46–73) Sex: 53 F:7 M (reported per protocol) (F 88%:M 12%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: dietary supplements Group 1: (n = 38) 'Catuama' herbal compound ‐ 310 mg capsules ‐ Paullinia cupana ('guarana': 125 mg), Trichilia catigua ('catuaba': 87.5 mg), Zingiber officinalis ('ginger root': 10 mg), and Ptychopetalum olacoides ('potency wood': 87.5 mg) ‐ 2 capsules a day, before lunch and dinner, for 8 weeks after the first evaluation Group 2: (n = 34) placebo capsules containing magnesium silicate, with the same colour and shape as those taken by the group test ‐ 2 capsules a day, before lunch and dinner, for 8 weeks after the first evaluation |
|
| Outcomes | Visual numeric scale (VNS) consists of a ruler divided into 11 equal parts, numbered successively from 0 (without symptoms) to 10 (maximum intensity of the symptoms). At baseline, 4, 8, and 12 weeks after the treatment onset. The assessment at 12 weeks was carried out 30 days after the end of treatment Faces scale (FS): "the individual classified the intensity of their symptoms according to the expression shown 4 in each pictured face. The expression of happiness corresponds to 0 (without symptoms) and the expression of maximum unhappiness to 5 (maximum intensity of the symptoms)". At baseline, 4, 8, and 12 weeks after the treatment onset. The assessment at 12 weeks was carried out 30 days after the end of treatment |
|
| Source of funding | Not reported | |
| Notes | No data provided as to how quickly one would expect the active treatment to work and how long it works for ‐ presumably a prolonged benefit was expected as the study looked at outcomes 4 weeks after cessation of treatment SES: not reported Conflict of interests: not reported Data analysis: per protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "The herbal and placebo were stored in identical vials and properly coded to blind both researcher and patients. The researcher chose the vial by lot and thus were patients randomly allocated to treatment groups" Comment: appropriate method |
| Allocation concealment (selection bias) | Low risk | Comment: as above, likely that allocation was concealed |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotation: "All patients were evaluated by the same investigator, who had no information about the medicine codes. The blinding was maintained throughout the trial" Comment: probably achieved |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "All patients were evaluated by the same investigator, who had no information about the medicine codes. The blinding was maintained throughout the trial" Comment: blinded outcome assessment; patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quotation: "6 withdrew from the study for reasons unrelated to the treatment" – 5 in active, 1 in control – no reasons stated. Lost to follow‐up ‐ 8 in active arm, 4 in control. According to flow chart – 13 in active arm and 5 in control arm dropped out Comment: the paper states that 8 in active and 4 in control arms were not evaluated. Hence, there is a mismatch between dropouts and evaluated participants which is not explained. Higher attrition in active arm with inadequate data provided as to why this was |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported in the prespecified manner |
| Other bias | Unclear risk | Quotation: "Two patients who took the test substance reported exacerbation of the symptoms in the first week of treatment, but this was also observed in 4 patients in the control group" Comment: the text above is contradicted by the flow chart which states 3 patients in each arm reported exacerbation of symptoms |
Spanemberg 2015.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 78 BMS patients
Mean age 62.8 years Sex: 67 F:11 M (F 86%:M 14%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: electromagnetic radiation Group 1: (n = 20) infrared weekly laser: 1 session weekly for 10 weeks (10 sessions: Diode laser (Thera Lase, DMC Equipamentos LTDA, São Carlos, Brazil. Spot tip: 0.028 cm²), aluminium gallium arsenide (GaAlAs), 830 nm wavelength, 100 mW output, continuous emissions, 3.57 W/cm², 5 J energy p/point, 176 J/cm² radiant exposure, applied 50 seconds p/point) + OHI (mucosal hydration and irritant avoidance (spicy/citric food, alcohol, tobacco)) Group 2: (n = 20) infrared laser 3 times per week: 3 sessions weekly for 3 weeks (9 sessions: Diode laser (Thera Lase, DMC Equipamentos LTDA, São Carlos, Brazil. Spot tip: 0.028 cm²), GaAlAs, 830 nm wavelength, 100 mW output, continuous emissions, 3.57 W/cm², 5 J energy p/point, 176 J/cm² radiant exposure, applied 50 seconds p/point) + OHI (as above) Group 3: (n = 19) red laser 3 times per week: 3 sessions weekly for 3 weeks (9 sessions: Diode laser (Thera Lase, DMC Equipamentos LTDA, São Carlos, Brazil. Spot tip: 0.028 cm²), aluminium gallium indium phosphide (InGaAlP), 685 nm wavelength, 35 mW output, continuous emissions, 1.25 W/cm², 2 J energy p/point, 72 J/cm² radiant exposure, applied 58 seconds p/point) + OHI (as above) Group 4: (n = 19) placebo laser 3 times per week: 3 sessions weekly for 3 weeks (9 sessions: Diode laser (Thera Lase, DMC Equipamentos LTDA, São Carlos, Brazil. Spot tip: 0.028 cm²), plastic‐tipped with rubber interior to block radiation emission, duration of application per point not reported) + OHI (as above) Duration: Groups 2‐4 ‐ used in this review: 3 weeks; Group 1 ‐ data not used within this review due to no comparable placebo group: 10 weeks |
|
| Outcomes |
VAS and visual numeric scale (VNS): "Both scales were applied to check whether patients would be consistent in their responses" at baseline, 3 weeks (end of treatment) and 11 weeks (2 months after treatment cessation) using 100 mm VAS scale (endpoints were 0 = no pain, 100 = worst pain possible) and VNS scale (endpoints were 0 = no pain, 10 = worst pain possible) Oral Health Impact Profile‐14 (OHIP‐14) (Portuguese language version) at baseline and 3 weeks (end of treatment) Adverse effects |
|
| Source of funding | Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; process number 4906‐13‐6), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS) ‐ all in Brazil | |
| Notes | SES: not reported
Conflict of interests: not reported
Data analysis: ITT QoL not assessed at 11‐week follow‐up, only immediately after cessation of treatment (3 weeks) Note: While VAS (0‐100 mm) and VNS (0‐10) were both assessed to ensure response consistency, VAS was used in this review's analyses due to the greater precision in reported results and reassurance from the study authors: "In both scales, the patients were consistent in their responses, presenting Pearson correlation coefficient > 0.9" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quotation: "randomly allocated into four groups" Comment: not reported |
| Allocation concealment (selection bias) | Unclear risk | Comment: not reported |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotation: "the present randomized, blind, placebo‐controlled study" Comment: although not explicitly reported, it is assumed participants were blinded |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "the present randomized, blind, placebo‐controlled study" Comment: not reported who was blinded, however only patient‐reported outcomes assessed |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quotation: "All the patients in the sample (n = 78) completed the study" |
| Selective reporting (reporting bias) | Unclear risk | Comment: adverse events were not reported despite being indicated to have been assessed in the final session |
| Other bias | High risk | Comment: baseline inequality in OHIP and VNS present between infrared and placebo groups Study authors did not assess OHIP‐14 at 11‐week follow‐up, but did assess VAS and VNS |
Tammiala‐Salonen 1999.
| Methods | Single centre, placebo‐controlled parallel RCT | |
| Participants | 37 BMS patients
Mean age 58.6 years (range 39 to 71) Sex: 37 F (F 100%) Inclusion/exclusion and diagnostic criteria appropriate |
|
| Interventions |
Intervention category: antidepressants and antipsychotics Group 1: (n = 18) trazodone 200 mg daily Group 2: (n = 19) placebo Duration: 8 weeks |
|
| Outcomes |
VAS at 0, 2, 4 and 8 weeks using 100 mm VAS scale (endpoints were 0 = no pain, 100 = worst pain possible) Short McGill Pain Questionnaire (MPQ) at baseline and 8 weeks ‐ "intensity and character of the pain were further defined by the use of the Finnish version of the McGill Pain Questionnaire" Beck Depression Inventory (BDI) Global assessment at 8 weeks |
|
| Source of funding | Finnish Dental Society | |
| Notes | Groups differed at baseline with regard to pain intensity Number of dropouts was higher in those who were depressed SES: not reported Conflict of interests: not reported Data analysis: per protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quotation: "Randomization was performed in blocks of 6 by the manufacturer of the drug (Orion)" Comment: third party randomisation |
| Allocation concealment (selection bias) | Low risk | Quotations: "The randomization code was not opened during the trial"; "The examiner could not guess the treatment of the subjects" Comment: probably achieved |
| Blinding (performance bias and detection bias) Blinding of participants | Low risk | Quotations: "Identical capsules of trazodone and of passive placebo were packed in the same way"; "nothing suggested that the blinding had not remained intact for the patients"; "Seven subjects in the trazodone group and 2 in the placebo group failed to finish the trial because of side effects, mainly because of dizziness" Comment: probably achieved |
| Blinding (performance bias and detection bias) Blinding of outcome assessors | Low risk | Quotation: "The examiner could not guess the treatment of the subjects" Comment: blinded outcome assessment; patient‐reported outcomes only |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: reasons for dropouts were reported appropriately. Unclear as to how these data were handled – no mention of numbers analysed for the outcomes |
| Selective reporting (reporting bias) | Unclear risk | Comment: prespecified outcomes were reported but not completely – although VAS measurement on speaking, eating and suffering – no data provided – only mention that there was no significant differences between the groups. McGill pain score raw data not provided, reported to be no significant difference in standard deviations for VAS and Beck data only shown graphically with no raw data provided |
| Other bias | Low risk | Comment: no other risks of bias were noted |
ALA = alpha lipoic acid; BMS = burning mouth syndrome; F = female; ITT = intention‐to‐treat; M = male; OHI = oral hygiene instruction; ppm = parts per million; QoL = quality of life; RCT = randomised controlled trial; SD = standard deviation; SES = socioeconomic status; VAS = visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bai 2010 | Does not use a placebo group ‐ electric acupuncture therapy versus oral oryzanol |
| Bessho 1998 | Does not use a placebo group ‐ compares Kampo medicine with diazepam |
| Bogetto 1997 | Amisulpride versus paroxetine ‐ conference abstract only ‐ insufficient detail provided to confirm that participants had burning mouth syndrome |
| Campisi 1997 | Does not use a placebo group ‐ compares 2 different forms of sucralfate ‐ 20% suspension versus 1 g chewable tablet |
| Ferguson 1981 | Not an RCT ‐ single‐centre, double‐blind CCT of 145 oophorectomised patients comparing mestranol with placebo |
| Forabosco 1992 | Not an RCT ‐ the diagnosis of BMS was uncertain in this study, all subjects included in the study with BMS symptoms received the same intervention (hormone replacement therapy) |
| Grechko 1996 | Does not use a placebo group ‐ compares electrical stimulation therapy with standard methods of treatment (novocaine blockade, analgesics, etc.) |
| Grushka 1998 | Not an RCT ‐ all 30 subjects received clonazepam (starting dose was 0.25 mg daily, with an increase in dose of 0.25 mg on a weekly basis if symptoms continued) |
| Grémeau‐Richard 2010 | Inappropriate design ‐ only immediate assessment, no clinical application. "The spontaneous burning was measured with a visual analogue scale (VAS) just before and 15 min after injection" |
| Hansen 1990 | Not true cross‐over; not all participants received both interventions. Mixed diagnosis; unable to separate out results for BMS sufferers |
| Hirsch 2011 | Not an RCT ‐ pilot study reporting 3 cases of open‐label application of topical sucralose |
| Huang 2006 | Does not use a placebo group ‐ acupoint injection of vitamin B1 and B12 versus oral oryzanol |
| Huang 2009 | Does not use a placebo group ‐ acupoint injection of vitamin B1 and B12 versus oral oryzanol and vitamin B2 complex |
| Hugoson 1991 | Not an RCT ‐ participants grouped according to presence of BMS symptoms or vitamin deficiency or both. Only those with both symptoms and vitamin deficiency received therapy |
| Ito 2010 | Not an RCT ‐ open‐label case series in 22 patients with BMS who were given varying doses of milnacipram over a 12‐week period |
| Kho 2010 | Not an RCT ‐ not primary BMS, some participants had anaemia, diabetes mellitus and hyposalivation |
| Lamey 1986 | Participants initially divided according to whether they were vitamin deficient or not. The non‐vitamin deficient group were randomly allocated to various vitamin replacement regimens, although results are not broken down according to regimen |
| Li 2002 | Unable to locate a copy of the original article from any location worldwide after repeated search through the British Library's Inter‐Library Loans service |
| Lindholm 2015 | The trial included participants with various neuropathic orofacial pain conditions. Data for BMS sufferers (n = 5; 31%) could not be separated out from other types of pain |
| Loldrup 1989 | Participants randomly allocated to 1 of 3 groups: clomipramine, mianserin or placebo. The trial included patients with pain of no known organic origin. Data for BMS sufferers could not be separated out from other types of pain |
| Lu 2002 | Does not use a placebo group ‐ acupoint injection of vitamin B1 and B12 versus oral oryzanol and vitamin B complex |
| López‐Jornet 2013 | Comparison between tongue protector and tongue protector plus aloe vera gel ‐ therefore study is not placebo‐controlled, as the tongue protector is considered to be an active treatment |
| Ma 2006 | Unable to locate a copy of the original article from any location worldwide after repeated search through the British Library's Inter‐Library Loans service |
| Maina 2002 | Does not use a placebo group ‐ compares SSRIs (paroxetine 20 mg/day or sertraline 50 mg/day) with amisulpride 50 mg/day |
| Miziara 2009 | Inappropriate design ‐ placebo group outcome assessed after 1 month while intervention group outcome assessed after 3 months |
| Mo 2003 | Unable to locate a copy of the original article from any location worldwide after repeated search through the British Library's Inter‐Library Loans service |
| Palacios‐Sanchez 2010 | Insufficient details to permit entry to review as only conference abstract available (no inclusion/exclusion criteria or diagnostic classifications) and possibly not a randomised study ‐ alpha lipoic acid versus placebo |
| Pellegrini 2010 | Insufficient details to permit entry to review as only conference abstract available (no inclusion/exclusion criteria or diagnostic classifications) |
| Peng 2001 | Does not use a placebo group ‐ compares livial (a synthetic hormone) with oryzanol and vitamin E |
| Petruzzi 2004 | Error in MEDLINE reference. Not an RCT, sample were alternately assigned to arms. Clarification received from lead author |
| Pisanty 1975 | Not an RCT. Also insufficient data provided to determine whether participants had BMS or other diagnoses causing such symptoms ‐ estrone 10,000 U and estrone 50,000 U ointments versus placebo |
| Qui 2010 | Does not use a placebo group ‐ compares laser acupuncture versus acupoint injection of vitamin B1 and B12 |
| Romeo 2010 | Not an RCT ‐ no control group, case series ‐ laser therapy |
| Toida 2009 | Not primary BMS, methods state all participants were being treated for gastritis for 4 months and had oral burning for at least a month |
| Vukoja 2011 | Letter to the editor, insufficient details on diagnostic classification, methods or outcomes ‐ laser therapy versus placebo |
| Woda 1998 | Not an RCT ‐ all 25 subjects received clonazepam (0.5 or 1 mg) 2 or 3 times daily |
| Yong 2003 | Does not use a placebo group ‐ acupoint injection of vitamin B1 and B12 combined with oral oryzanolum versus oral oryzanolum |
BMS = burning mouth syndrome; CCT = controlled clinical trial; RCT = randomised controlled trial; SSRIs = selective serotonin reuptake inhibitors.
Characteristics of studies awaiting assessment [ordered by study ID]
NCT02580734.
| Methods | Placebo‐controlled, triple‐blinded cross‐over RCT |
| Participants | 20 BMS patients
Mean age: Unknown until published (eligible age range 18 to 90) Sex: Distribution unknown until published Inclusion/exclusion and diagnostic criteria appropriate |
| Interventions |
Intervention category: Dietary supplements Group 1: (n = 20) Melatonin 12 mg daily (3 mg capsules taken 4 times p/day) Group 2: (n = 20) Placebo (capsules taken 4 times p/day) Duration: 5 months (2 months, followed by 1 month washout, and then final 2 months) |
| Outcomes |
Outcomes not of interest to this review
|
| Source of funding | Unknown until published |
| Notes | Study currently unpublished Contact: Andrea Sardella (andrea.sardella@unimi.it) |
Umezaki 2016.
| Methods | Single‐centre, placebo‐controlled, single‐blinded parallel RCT |
| Participants | 26 BMS patients
Mean age 63.9 years (SD 9.56) Sex: 24F:2M (F92%:M8%) Inclusion/exclusion and diagnostic criteria appropriate |
| Interventions |
Intervention category: Electromagnetic radiation Group 1: (n = 14) High‐frequency transcranial magnetic stimulation (TMS): 10 daily sessions (1 session p/day for 5 days, 2 days untreated and then a further 5 days of 1 session p/day) totalling 30,000 single‐pulse stimulations at 10 Hz (MagVenture MagPro x100 Stimulator ‐ MagVenture Inc., Denmark; Cool‐B65 A/P figure 8 coil, positioned over left dorsolateral prefrontal cortex (DLPFC) at medial frontal 10‐20 system EEG‐electrode location (F3); unconnected ECT electrodes placed under coil ‐ Natus Neurology, Middleton, Wisconsin) Group 2: (n = 12) Placebo: 10 daily sessions (1 session p/day for 5 days, 2 days untreated and then a further 5 days of 1 session p/day) totalling 30,000 single‐pulse stimulations (MagVenture MagPro x100 Stimulator ‐ MagVenture Inc., Denmark; shielded Cool‐B65 A/P figure 8 coil, positioned over left DLPFC at location F3; connected ECT electrodes placed under coil to stimulate when TMS was triggered ‐ Natus Neurology, Middleton, Wisconsin) Duration: 2 months |
| Outcomes |
Outcomes not of interest to this review
|
| Source of funding | Not reported |
| Notes | No numbers (tables 1‐3), means or SDs (text and figures 2‐3) associated with the study's results were available in the published study paper. Until this information is obtained we are unable to incorporate this study within our analyses Quotation: "SSRIs were prescribed for around 40% of the patients, but these did not adequately relieve the BMS pain. Although 30% of the patients had a prior history of depression, none currently met diagnostic criteria for depression" Comment: Unclear if SSRIs were prescribed before the trial commenced, or during the trial as a co‐intervention SES: Not reported Conflict of interests: Not reported Data analysis: Per‐protocol (according to Figure 1). 6 participants did not complete the assigned intervention duration due to competing commitments which allowed them to attend only 2 sessions (of 10 scheduled) each. Group 1: 2 patients abandoned study; Group 2: 4 patients left study |
BMS = burning mouth syndrome; F = female; M = male; QoL = quality of life; RCT = randomised controlled trial; SD = standard deviation; SES = socioeconomic status; SSRIs = selective serotonin reuptake inhibitors; VAS = visual analogue scale.
Differences between protocol and review
The current review has employed Cochrane's tool for assessing risk of bias in randomised trials (Higgins 2011a). Papers identified in previous versions were reformatted using this new template. In keeping with current best practice (Higgins 2011b), previously included controlled clinical trials were excluded in this update (Pisanty 1975), and a GRADE 'Summary of findings' table has been produced for each comparison. Adverse effects were included as an outcome measure in this version of the review, despite not formally being included as an outcome in the original protocol. After feedback, we determined the substantial effect of burning mouth syndrome upon patients' quality of life is of sufficient direct impact to justify upgrading quality of life from a secondary to a primary outcome. Due to heterogeneity in study follow‐up periods and outcome assessment time points, we analysed outcome data as short‐term (≤ 3 months from baseline) or long‐term (> 3 to ≤ 6 months from baseline) for a manageable cut‐off threshold. In accordance with Section 16.4.5 of the Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011) (Higgins 2011b), we utilised first period data only of cross‐over trials which did not incorporate a washout period.
Contributions of authors
Development and co‐ordination of the update: Roddy McMillan (RM). Running electronic searches: Anne Littlewood (Cochrane Oral Health). Communication with authors and organisations: RM and Jo Weldon (JW). Screening titles, abstracts and full‐text papers: RM, Joanna Zakrzewska (JZ), Anne‐Marie Glenny (AMG), JW. Reviewing papers: RM, Heli Forssell (HF), JZ, John Buchanan (JB), AMG, JW. Extracting data: RM, HF, JZ, JB, JW. Appraising quality/risk of bias: RM, HF, JZ, JB, AMG, JW. Inputting data: RM, JW. Data validation: AMG. Analysis of data: JW, AMG. Risk of bias analysis: RM, AMG. Interpretation of data: JW, AMG. Summary of findings tables: AMG, JW. Writing the review: RM, HF, JW, AMG.
Sources of support
Internal sources
St Bartholomew's and the Royal London, UK.
University Dental Hospital of Manchester, UK.
Turku University, Finland.
-
National Institute for Health Research (NIHR) University College London (UCL) Hospitals Biomedical Research Centre, UK.
Research time
School of Dentistry, The University of Manchester, UK.
-
Manchester Academic Health Sciences Centre (MAHSC), UK.
Cochrane Oral Health is supported by MAHSC and the NIHR Manchester Biomedical Research Centre
External sources
-
National Institute for Health Research (NIHR), UK.
This project was supported by the NIHR, via Cochrane Infrastructure funding to Cochrane Oral Health. The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health
-
Cochrane Oral Health Global Alliance, Other.
The production of Cochrane Oral Health reviews has been supported financially by our Global Alliance since 2011 (ohg.cochrane.org/partnerships‐alliances). Contributors over the past year have been the British Association for the Study of Community Dentistry, UK; the British Society of Paediatric Dentistry, UK; the Canadian Dental Hygienists Association, Canada; the Centre for Dental Education and Research at All India Institute of Medical Sciences, India; the National Center for Dental Hygiene Research & Practice, USA; New York University College of Dentistry, USA; and NHS Education for Scotland, UK
Declarations of interest
Roddy McMillan: none known. Heli Forssell: none known. John AG Buchanan: John Buchanan has previously co‐authored an evidence‐based review for BMJ publications Clinical Evidence on burning mouth syndrome with Joanna M Zakrzewska. Anne‐Marie Glenny: none known. Anne‐Marie Glenny is an editor with Cochrane Oral Health. Joanna M Zakrzewska: Joanna M Zakrzewska has previously co‐authored an evidence‐based review for BMJ publications Clinical Evidence on burning mouth syndrome with John Buchanan. Jo C Weldon: none known. Jo Weldon is a salaried member of staff of Cochrane Oral Health.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Alvarenga da Silva 2014 {published data only}
- Alvarenga da Silva L, Siqueira JT, Teixeira MJ, Siqueira SR. The role of xerostomia in burning mouth syndrome: a case‐control study. Arquivos de Neuro‐psiquiatria 2014;72(2):91‐8. [DOI] [PubMed] [Google Scholar]
Bergdahl 1995a {published data only}
- Bergdahl J, Anneroth G, Perris H. Cognitive therapy in the treatment of patients with resistant burning mouth syndrome: a controlled study. Journal of Oral Pathology & Medicine 1995;24(5):213‐5. [DOI] [PubMed] [Google Scholar]
Bogetto 1999 {published data only}
- Bogetto F, Bonatto Revello R, Ferro G, Maina L, Ravizza L. Psychopharmacological treatment of burning mouth syndrome (BMS). A study sample of 121 patients [Trattamento psicofarmacologico della burning mouth syndrome (BMS). Studio su di un campione di 121 pazienti]. Minerva Psichiatrica 1999;40(1):1‐10. [Google Scholar]
Cano‐Carrillo 2014 {published and unpublished data}
- Cano‐Carrillo P, Pons‐Fuster A, López‐Jornet P. Efficacy of lycopene‐enriched virgin olive oil for treating burning mouth syndrome: a double‐blind randomised. Journal of Oral Rehabilitation 2014;41(4):296‐305. [DOI] [PubMed] [Google Scholar]
Carbone 2009 {published data only (unpublished sought but not used)}
- Carbone M, Pentenero M, Carrozzo M, Ippolito A, Gandolfo S. Lack of efficacy of alpha‐lipoic acid in burning mouth syndrome: a double‐blind, randomized, placebo‐controlled study. European Journal of Pain 2009;13(5):492‐6. [DOI] [PubMed] [Google Scholar]
Cavalcanti 2009 {published and unpublished data}
- Cavalcanti DR, Silveira FR. Alpha lipoic acid in burning mouth syndrome ‐ a randomized double‐blind placebo‐controlled trial. Journal of Oral Pathology & Medicine 2009;38(3):254‐61. [DOI] [PubMed] [Google Scholar]
Femiano 2000 {published data only}
- Femiano F, Gombos F, Scully C, Busciolano M, Luca P. Burning mouth syndrome (BMS): controlled open trial of the efficacy of alpha‐lipoic acid (thioctic acid) on symptomatology. Oral Diseases 2000;6(5):274‐7. [DOI] [PubMed] [Google Scholar]
Femiano 2002a {published and unpublished data}
- Femiano F, Scully C. Burning mouth syndrome (BMS): double blind controlled study of alpha‐lipoic acid (thioctic acid) therapy. Journal of Oral Pathology & Medicine 2002;31(5):267‐9. [DOI] [PubMed] [Google Scholar]
Femiano 2002b {published data only}
- Femiano F. Burning mouth syndrome (BMS): an open trial of comparative efficacy of alpha‐lipoic acid (thioctic acid) with other therapies. Minerva Stomatologica 2002;51(9):405‐9. [PubMed] [Google Scholar]
Grémeau‐Richard 2004 {published data only}
- Grémeau‐Richard C, Woda A, Navez ML, Attal N, Bouhassira D, Gagnieu MC, et al. Topical clonazepam in stomatodynia: a controlled study. Journal of Dental Research 2003;82(Spec Iss B):B‐159 (Abstract No 1174). [Google Scholar]
- Grémeau‐Richard C, Woda A, Navez ML, Attal N, Bouhassira D, Gagnieu MC, et al. Topical clonazepam in stomatodynia: a randomised placebo‐controlled study. Pain 2004;108(1‐2):51‐7. [DOI] [PubMed] [Google Scholar]
Heckmann 2012 {published data only}
- Heckmann SM, Kirchner E, Grushka M, Wichmann MG, Hummel T. A double‐blind study on clonazepam in patients with burning mouth syndrome. Laryngoscope 2012;122(4):813‐6. [DOI] [PubMed] [Google Scholar]
Lopez‐D'alessandro 2011 {published and unpublished data}
- López‐D'alessandro E, Escovich L. Combination of alpha lipoic acid and gabapentin, its efficacy in the treatment of Burning Mouth Syndrome: a randomized, double‐blind, placebo controlled trial. Medicina Oral, Patología Oral y Cirugía Bucal 2011;16(5):e635‐40. [DOI] [PubMed] [Google Scholar]
López‐Jornet 2009b {published data only}
- López‐Jornet P, Camacho‐Alonso F, Leon‐Espinosa S. Efficacy of alpha lipoic acid in burning mouth syndrome: a randomized, placebo‐treatment study. Journal of Oral Rehabilitation 2009;36(1):52‐7. [DOI] [PubMed] [Google Scholar]
López‐Jornet 2011 {published data only}
- López‐Jornet P, Camacho‐Alonso F, Andujar‐Mateos P. A prospective, randomized study on the efficacy of tongue protector in patients with burning mouth syndrome. Oral Diseases 2011;17(3):277‐82. [DOI] [PubMed] [Google Scholar]
Marino 2010 {published data only}
- Marino R, Torretta S, Capaccio P, Pignataro L, Spadari F. Different therapeutic strategies for burning mouth syndrome: preliminary data. Journal of Oral Pathology & Medicine 2010;39(8):611‐6. [DOI] [PubMed] [Google Scholar]
Palacios‐Sánchez 2015 {published and unpublished data}
- Palacios‐Sánchez B, Moreno‐López LA, Cerero‐Lapiedra R, Llamas‐Martínez S, Esparza‐Gómez G. Alpha lipoic acid efficacy in burning mouth syndrome. A controlled clinical trial. Medicina Oral, Patología Oral y Cirugía Bucal 2015;20(4):e435‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rodríguez de Rivera‐Campillo 2010 {published data only (unpublished sought but not used)}
- Rodríguez De Rivera‐Campillo ME, López‐López J, Chimenos‐Kustner E. Treatment of burning mouth syndrome with topical clonazepam [Tratamiento del Síndrome de Boca Ardiente con clonacepam tópico]. Piel 2011;26(6):263‐8. [Google Scholar]
- Rodríguez de Rivera‐Campillo E, López‐López J, Chimenos‐Küstner E. Response to topical clonazepam in patients with burning mouth syndrome: a clinical study. Bulletin de Groupement Europeen pour la Recherche Scientifique en Stomatologie et Odontologie 2010;49(1):19‐29. [PubMed] [Google Scholar]
Sardella 1999 {published data only}
- Sardella A, Uglietti D, Demarosi F, Lodi G, Bez C, Carrassi A. Benzydamine hydrochloride oral rinses in management of burning mouth syndrome. A clinical trial. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics 1999;88(6):683‐6. [DOI] [PubMed] [Google Scholar]
Sardella 2008 {published data only}
- Sardella A, Lodi G, Demarosi F, Tarozzi M, Canegallo L, Carrassi A. Hypericum perforatum extract in burning mouth syndrome: a randomized placebo‐controlled study. Journal of Oral Pathology & Medicine 2008;37(7):395‐401. [DOI] [PubMed] [Google Scholar]
Silvestre 2012 {published data only}
- Silvestre FJ, Silvestre‐Rangil J, Tamarit‐Santafé C, Bautista D. Application of a capsaicin rinse in the treatment of burning mouth syndrome. Medicina Oral, Patología Oral y Cirugía Bucal 2012;17(1):e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spanemberg 2012 {published data only}
- Spanemberg JC, Cherubini K, Figueiredo MA, Gomes AP, Campos MM, Salum FG. Effect of an herbal compound for treatment of burning mouth syndrome: randomized, controlled, double‐blind clinical trial. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics 2012;113(3):373‐7. [DOI] [PubMed] [Google Scholar]
Spanemberg 2015 {published data only}
- Spanemberg JC, López López J, Figueiredo MA, Cherubini K, Salum FG. Efficacy of low‐level laser therapy for the treatment of burning mouth syndrome: a randomized, controlled trial. Journal of Biomedical Optics 2015;20(9):098001. [DOI] [PubMed] [Google Scholar]
Tammiala‐Salonen 1999 {published data only}
- Tammiala‐Salonen T, Forssell H. Trazodone in burning mouth pain: a placebo‐controlled, double‐blind study. Journal of Orofacial Pain 1999;13(2):83‐8. [PubMed] [Google Scholar]
References to studies excluded from this review
Bai 2010 {published data only}
- Bai H, Yu P. Observation of effectiveness of electrical acupuncture on burning mouth syndrome. Journal of Clinical Acupuncture and Moxibustion 2010;26(7):28‐9. [Google Scholar]
Bessho 1998 {published data only}
- Bessho K, Okubo Y, Hori S, Murakami K, Iizuka T. Effectiveness of kampo medicine (sai‐boku‐to) in treatment of patients with glossodynia. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics 1998;86(6):682‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bogetto 1997 {published data only}
- Bogetto F, Ferro G, Maina G, Gandolfo S. Amisulpride vs paroxetine in the treatment of burning mouth syndrome (BMS). Biological Psychiatry 1997;42:27S (Abstract No 14‐29). [Google Scholar]
Campisi 1997 {published data only}
- Campisi G, Spadari F, Salvato A. Sucralfate in oral medicine. Our clinical findings [Il sucralfato in odontostomatologia. Nostra esperienza clinica]. Minerva Stomatologica 1997;46(6):297‐305. [PubMed] [Google Scholar]
Ferguson 1981 {published data only}
- Ferguson MM, Carter J, Boyle P, Hart DM, Lindsay R. Oral complaints related to climacteric symptoms in oophrorectomized women. Journal of the Royal Society of Medicine 1981;74(7):492‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Forabosco 1992 {published data only}
- Forabosco A, Criscuolo M, Coukos G, Uccelli E, Weinstein R, Spinato S, et al. Efficacy of hormone replacement therapy in postmenopausal women with oral discomfort. Oral Surgery, Oral Medicine, and Oral Pathology 1992;73(5):570‐4. [DOI] [PubMed] [Google Scholar]
Grechko 1996 {published data only}
- Grechko VE, Borisova EG. Use of transcutaneous electrical nerve stimulation in the complex treatment of glossalgia. Neuroscience & Behavioral Physiology 1996;26(6):584‐6. [DOI] [PubMed] [Google Scholar]
Grémeau‐Richard 2010 {published data only}
- Grémeau‐Richard C, Dubray C, Aublet‐Cuvelier B, Ughetto S, Woda A. Effect of lingual nerve block on burning mouth syndrome (stomatodynia): A randomised crossover trial. Pain 2010;149(1):27‐32. [DOI] [PubMed] [Google Scholar]
Grushka 1998 {published data only}
- Grushka M, Epstein J, Mott A. An open‐label, dose escalation pilot study of the effect of clonazepam in burning mouth syndrome. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, & Endodontics 1998;86(5):557‐61. [DOI] [PubMed] [Google Scholar]
Hansen 1990 {published data only}
- Hansen HJ, Thorøe U. Low power laser biostimulation of chronic oro‐facial pain. A double‐blind placebo controlled cross‐over study in 40 patients. Pain 1990;43(2):169‐79. [DOI] [PubMed] [Google Scholar]
Hirsch 2011 {published data only}
- Hirsch AR, Ziad A, Kim AY, Lail NS, Sharma S. Pilot study: alleviation of pain in burning mouth syndrome with topical sucralose. Headache 2011;51(3):444‐6. [DOI] [PubMed] [Google Scholar]
Huang 2006 {published data only}
- Huang J, Huo HY. Clinical observation of effectiveness of acupoint injection on 23 cases of BMS. Journal of Practical Traditional Chinese Internal Medicine 2006;20:77. [Google Scholar]
Huang 2009 {published data only}
- Huang MZ, Zong JJ. Therapeutic efficacy of acupointinjection of vitamins B1 and B12 in the treatment of burning mouth syndrome. Acta Academiae Medicinae Jiangxi 2009;49:90‐3. [Google Scholar]
Hugoson 1991 {published data only}
- Hugoson A, Thorstensson B. Vitamin B status and response to replacement therapy in patients with burning mouth syndrome. Acta Odontologica Scandinavica 1991;49(6):367‐75. [DOI] [PubMed] [Google Scholar]
Ito 2010 {published data only}
- Ito M, Kimura H, Yoshida K, Kimura Y, Ozaki N, Kurita K. Effectiveness of milnacipran for the treatment of chronic pain in the orofacial region. Clinical Neuropharmacology 2010;33(2):79‐83. [DOI] [PubMed] [Google Scholar]
Kho 2010 {published data only}
- Kho HS, Lee JS, Lee EJ, Lee JY. The effects of parafunctional habit control and topical lubricant on discomforts associated with burning mouth syndrome (BMS). Archives of Gerontology and Geriatrics 2010;51(1):95–9. [DOI] [PubMed] [Google Scholar]
Lamey 1986 {published data only}
- Lamey PJ, Hammond A, Allam BF, McIntosh WB. Vitamin status of patients with burning mouth syndrome and the response to replacement therapy. British Dental Journal 1986;160(3):81‐4. [DOI] [PubMed] [Google Scholar]
Li 2002 {published data only}
- Li YQ, Luo HY. Observation of effectiveness of acupoint injection on burning mouth syndrome. Journal of Mudanjiang Medical College 2002;23(6):34‐5. [Google Scholar]
Lindholm 2015 {published data only}
- Lindholm P, Lamusuo S, Taiminen T, Pesonen U, Lahti A, Virtanen A, et al. Right secondary somatosensory cortex ‐ a promising novel target for the treatment of drug‐resistant neuropathic orofacial pain with repetitive transcranial magnetic stimulation. Pain 2015;156(7):1276‐83. [DOI] [PubMed] [Google Scholar]
Loldrup 1989 {published data only}
- Loldrup D, Langemark M, Hansen HJ, Olesen J, Bech P. Clomipramine and mianserin in chronic idiopathic pain syndrome. A placebo controlled study. Psychopharmacology 1989;99(1):1‐7. [DOI] [PubMed] [Google Scholar]
López‐Jornet 2013 {published data only}
- López‐Jornet P, Camacho‐Alonso F, Molino‐Pagan D. Prospective, randomized, double‐blind, clinical evaluation of aloe vera barbadensis, applied in combination with a tongue protector to treat burning mouth syndrome. Journal of Oral Pathology & Medicine 2013;42(4):295‐301. [DOI] [PubMed] [Google Scholar]
Lu 2002 {published data only}
- Lu HR, Li HW. Therapeutic efficacy of acupointInjection of vitamins B1 and B12 in the treatment of burning mouth syndrome. Acta Academiae Medicinae Jiangxi 2002;42:74‐5. [Google Scholar]
Ma 2006 {published data only}
- Ma GT. Acupoint injection of vitamin B1 and B12 in the treatment of burning mouth syndrome. Journal of Integrative Medicine 2006;14:254. [Google Scholar]
Maina 2002 {published data only}
- Maina G, Vitalucci A, Gandolfo S, Bogetto F. Comparative efficacy of SSRIs and amisulpride in burning mouth syndrome: a single‐blind study. Journal of Clinical Psychiatry 2002;63(1):38‐43. [DOI] [PubMed] [Google Scholar]
Miziara 2009 {published data only}
- Miziara ID, Filho BC, Oliveira R, Rodrigues dos Santos RM. Group psychotherapy: an additional approach to burning mouth syndrome. Journal of Psychosomatic Research 2009;67(5):443‐8. [DOI] [PubMed] [Google Scholar]
Mo 2003 {published data only}
- Mo ZY, Liu J. Acupoint injection of vitamins B1 and B12 in the treatment of burning mouth syndrome. Journal of Stomatology 2003;23:121. [Google Scholar]
Palacios‐Sanchez 2010 {published data only}
- Palacios‐Sanchez B, Moreno‐Lopez LA, Esparza‐Gomez G, Cerero‐Lapiedra R, Llamas‐Martinez S. Efficacy of ALA in the treatment of burning mouth syndrome. Oral Diseases 2010;16(6):549 (Abstract No 148). [Google Scholar]
Pellegrini 2010 {published data only}
- Pellegrini VD, Silva EFP, Kato IT, Prates RA, Sugaya NN. Low level laser therapy in BMS patients: a preliminary report. Oral Diseases 2010;16(6):550 (Abstract No 150). [Google Scholar]
Peng 2001 {published data only}
- Peng JY, Wu Y, Han W. Clinical efficacy of burning mouth syndrome treated by livial. Bulletin of Hunan Medical University 2001;26(2):157‐8. [PubMed] [Google Scholar]
Petruzzi 2004 {published and unpublished data}
- Petruzzi M, Lauritano D, Benedittis M, Baldoni M, Serpico R. Systemic capsaicin for burning mouth syndrome: short‐term results of a pilot study. Journal of Oral Pathology & Medicine 2004;33(2):111‐4. [DOI] [PubMed] [Google Scholar]
Pisanty 1975 {published data only}
- Pisanty S, Rafaely B, Polishuk W. The effect of steroid hormones on buccal mucosa of menopausal women. Oral Surgery, Oral Medicine, and Oral Pathology 1975;40(3):346‐53. [DOI] [PubMed] [Google Scholar]
Qui 2010 {published data only}
- Qiu HL 2010. Therapeutic efficacy of combined acupuncture and laser acupuncture in treating burning mouth syndrome. Chinese Journal of Physical Medicine and Rehabilitation 2010;32:229‐30. [Google Scholar]
Romeo 2010 {published data only}
- Romeo U, Vecchio A, Capocci M, Maggiore C, Ripari M. The low level laser therapy in the management of neurological burning mouth syndrome. A pilot study. Annali di Stomatologia 2010;1(1):14‐8. [PMC free article] [PubMed] [Google Scholar]
Toida 2009 {published data only}
- Toida M, Kato K, Makita H, Long NK, Takeda T, Hatakeyama D, et al. Palliative effect of lafutidine on oral burning sensation. Journal of Oral Pathology & Medicine 2009;38(3):262‐8. [DOI] [PubMed] [Google Scholar]
Vukoja 2011 {published data only}
- Vukoja D, Alajbeg I, Vučićević Boras V, Brailo V, Alajbeg IZ, Andabak Rogulj A. Is effect of low‐level laser therapy in patients with burning mouth syndrome result of a placebo?. Photomedicine and Laser Surgery 2011;29(9):647‐8. [DOI] [PubMed] [Google Scholar]
Woda 1998 {published data only}
- Woda A, Navez ML, Picard P, Gremeau C, Pichard‐Leandri E. A possible therapeutic solution for stomatodynia (burning mouth syndrome). Journal of Orofacial Pain 1998;12(4):272‐8. [PubMed] [Google Scholar]
Yong 2003 {published data only}
- Yong M, Zhu YM, Wu L, Wang YR. Point injection of vitamin B1 and B12 in burning mouth syndrome. Inner Mongolia Medical Journal 2003;35:493‐4. [Google Scholar]
References to studies awaiting assessment
NCT02580734 {published data only (unpublished sought but not used)}
- NCT02580734. Melatonin to treat burning mouth syndrome (BMS): a randomized, cross‐over, placebo‐controlled, triple‐blind clinical trial. clinicaltrials.gov/show/NCT02580734 (first received 20 December 2013).
Umezaki 2016 {published data only (unpublished sought but not used)}
- Umezaki Y, Badran BW, DeVries WH, Moss J, Gonzales T, George MS. The efficacy of daily prefrontal repetitive transcranial magnetic stimulation (rTMS) for burning mouth syndrome (BMS): a randomized controlled single‐blind study. Brain Stimulation 2016;9(2):234‐42. [DOI] [PubMed] [Google Scholar]
Additional references
Albuquerque 2006
- Albuquerque RJC, Leeuw R, Carlson CR, Okeson JP, Miller CS, Andersen AH. Cerebral activation during thermal stimulation of patients who have burning mouth disorder: An fMRI study. Pain 2006;122(3):223‐34. [DOI] [PubMed] [Google Scholar]
Almoznino 2016
Bergdahl 1995b
- Bergdahl J, Anneroth G, Perris H. Personality characteristics of patients with resistant burning mouth syndrome. Acta Odontologica Scandinavica 1995;53(1):7‐11. [DOI] [PubMed] [Google Scholar]
Bergdahl 1999
- Bergdahl M, Bergdahl J. Burning mouth syndrome: prevalence and associated factors. Journal of Oral Pathology & Medicine 1999;28(8):350‐4. [DOI] [PubMed] [Google Scholar]
Braud 2013
- Braud A, Toure B, Agbo‐Godeau S, Descroix V, Boucher Y. Characteristics of pain assessed with visual analog scale and questionnaire in burning mouth syndrome patients: a pilot study. Journal of Orofacial Pain 2013;27(3):235‐42. [DOI] [PubMed] [Google Scholar]
Buchanan 2010
- Buchanan JA, Zakrzewska JM. Burning mouth syndrome. BMJ Clinical Evidence 2010;06:1301. [PMC free article] [PubMed] [Google Scholar]
Chaparro 2012
- Chaparro LE, Wiffen PJ, Moore RA, Gilron I. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 7. [DOI: 10.1002/14651858.CD008943.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cui 2016
- Cui Y, Xu H, Chen F, Liu J, Jiang L, Zhou Y, et al. Efficacy evaluation of clonazepam for symptom remission in burning mouth syndrome: a meta‐analysis. Oral Diseases 2016;22(6):503‐11. [DOI: 10.1111/odi.12422] [DOI] [PubMed] [Google Scholar]
de Moraes 2012
- Moraes M, do Amaral Bezerra BA, Rocha Neto PC, Oliveira Soares AC, Pinto LP, Lisboa Lopes Costa A. Randomized trials for the treatment of burning mouth syndrome: an evidence‐based review of the literature. Journal of Oral Pathology & Medicine 2012;41(4):281‐7. [DOI] [PubMed] [Google Scholar]
De Moura 2009
- Moura SA, Sousa JM, Lima DF, Negreiros AN, Silva Fde V, Costa LJ. Burning mouth syndrome (BMS): sialometric and sialochemical analysis and salivary protein profile. Gerodontology 2007;24(3):173‐6. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Eli 1994
- Eli I, Baht R, Littner MM, Kleinhauz M. Detection of psychopathologic trends in glossodynia patients. Psychosomatic Medicine 1994;56(5):389‐94. [DOI] [PubMed] [Google Scholar]
Eliav 2007
- Eliav E, Kamran B, Schaham R, Czerninski R, Gracely RH, Benoliel R. Evidence of chorda tympani dysfunction in patients with burning mouth syndrome. Journal of the American Dental Association 2007;138(5):628‐33. [DOI] [PubMed] [Google Scholar]
Farrar 2000
- Farrar JT, Portenoy RK, Berlin JA, Kinman JL, Strom BL. Defining the clinically important difference in pain outcome measures. Pain 2000;88(3):287‐94. [DOI] [PubMed] [Google Scholar]
Finnerup 2015
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurology 2015;14(2):162‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Forssell 2002
- Forssell H, Jääskeläinen S, Tenovuo O, Hinkka S. Sensory dysfunction in burning mouth syndrome. Pain 2002;99(1‐2):41‐7. [DOI] [PubMed] [Google Scholar]
Forssell 2012
- Forssell H, Teerijoki‐Oksa T, Kotiranta U, Kantola R, Bäck M, Vuorjoki‐Ranta TR, et al. Pain and pain behavior in burning mouth syndrome: a pain diary study. Journal of Orofacial Pain 2012;26(2):117‐25. [PubMed] [Google Scholar]
Fortuna 2013
- Fortuna G, Lorenzo M, Pollio A. Complex oral sensitivity disorder: a reappraisal of current classification of burning mouth syndrome. Oral Diseases 2013;19(7):730‐2. [DOI] [PubMed] [Google Scholar]
Foster 2007
- Foster TS. Efficacy and safety of alpha‐lipoic acid supplementation in the treatment of symptomatic diabetic neuropathy. Diabetes Educator 2007;33(1):111‐7. [DOI] [PubMed] [Google Scholar]
Galli 2016
Granot 2005
- Granot M, Nagler RM. Association between regional idiopathic neuropathy and salivary involvement as the possible mechanism for oral sensory complaints. Journal of Pain 2005;6(9):581‐7. [DOI] [PubMed] [Google Scholar]
Grushka 1987a
- Grushka M, Katz RL, Sessle BJ. Spontaneous remission in burning mouth syndrome. Journal of Dental Research 1987;66(2):274. [Google Scholar]
Grushka 1987b
- Grushka M. Clinical features of burning mouth syndrome. Oral Surgery, Oral Medicine, and Oral Pathology 1987;63(1):30‐6. [DOI] [PubMed] [Google Scholar]
Hagelberg 2003
- Hagelberg N, Forssell H, Rinne JO, Scheinin H, Taiminen T, Aalto S, et al. Striatal dopamine D1 and D2 receptors in burning mouth syndrome. Pain 2003;101(1‐2):149‐54. [DOI] [PubMed] [Google Scholar]
Heckmann 2001
- Heckmann SM, Heckmann JG, HiIz MJ, Popp M, Marthol H, Neundörfer B, et al. Oral mucosal blood flow in patients with burning mouth syndrome. Pain 2001;90(3):281‐6. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 2011;343:889‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011b
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
IHS 2013
- Headache Classification Subcommittee of the International Headache Society (IHS). The international classification of headache disorders 3rd edition. Cephalalgia 2013;33(9):629‐808. [DOI] [PubMed] [Google Scholar]
Ioannidis 2004
- Ioannidis JP, Evans SJ, Gøtzsche PC, O'Neill RT, Altman DG, Schulz K, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Annals of Internal Medicine 2004;141(10):781‐8. [DOI] [PubMed] [Google Scholar]
Just 2010
- Just T, Steiner S, Pau HW. Oral pain perception and taste in burning mouth syndrome. Journal of Oral Pathology & Medicine 2010;39(1):22‐7. [DOI] [PubMed] [Google Scholar]
Jääskeläinen 1997
- Jääskeläinen SK, Forssell H, Tenovuo O. Abnormalities of the blink reflex in burning mouth syndrome. Pain 1997;73(3):455‐60. [DOI] [PubMed] [Google Scholar]
Jääskeläinen 2001
- Jääskeläinen SK, Rinne JO, Forssell H, Tenovuo O, Kaasinen V, Sonninen P, et al. Role of the dopaminergic system in chronic pain ‐ a fluorodopa‐PET study. Pain 2001;90(3):257‐60. [DOI] [PubMed] [Google Scholar]
Jääskeläinen 2012
- Jääskeläinen SK. Pathophysiology of primary burning mouth syndrome. Clinical Neurophysiology 2012;123(1):71‐7. [DOI] [PubMed] [Google Scholar]
Jüni 1999
- Jüni P, Witschi A, Bloch R, Egger M. The hazards of scoring the quality of clinical trials for meta‐analysis. JAMA 1999;282(11):1054‐60. [DOI] [PubMed] [Google Scholar]
Khan 2014
- Khan SA, Keaser ML, Meiller TF, Seminowicz DA. Altered structure and function in the hippocampus and medial prefrontal cortex in patients with burning mouth syndrome. Pain 2014;155(8):1472‐80. [DOI] [PubMed] [Google Scholar]
Kisely 2016
- Kisely S, Forbes M, Sawyer E, Black E, Lalloo R. A systematic review of randomized trials for the treatment of burning mouth syndrome. Journal of Psychosomatic Research 2016;86:39‐46. [DOI] [PubMed] [Google Scholar]
Kohorst 2014
- Kohorst JJ, Bruce AJ, Torgerson RR, Schenk LA, Davis MDP. A population‐based study of the incidence of burning mouth syndrome. Mayo Clinic Proceedings 2014;89(11):1545‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kohorst 2015
- Kohorst JJ, Bruce AJ, Torgerson RR, Schenck LA, Davis MD. The prevalence of burning mouth syndrome: a population‐based study. British Journal of Dermatology 2015;172(6):1654‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Koike 2014
- Koike K, Shinozaki T, Hara K, Noma N, Okada‐Ogawa A, Asano M, et al. Immune and endocrine function in patients with burning mouth syndrome. Clinical Journal of Pain 2014;30(2):168‐73. [DOI] [PubMed] [Google Scholar]
Kolkka‐Palomaa 2016
- Kolkka‐Palomaa M, Jääskeläinen SK, Laine MA, Teerijoki‐Oksa T, Sandell M, Forssell H. Pathophysiology of primary burning mouth syndrome with special focus on taste dysfunction: a review. Oral Diseases 2015;21(8):937‐48. [DOI] [PubMed] [Google Scholar]
Korn 2002
- Korn GP, Pupo DB, Quedas A, Filho IB. Correlation between xerostomia degree and sialometry in patients with Sjögren's Syndrome [Correlação entre o grau de xerostomia e o resultado da sialometria em pacientes com Síndrome de Sjögren]. Revista Brasileira de Otorinolaringologia 2002;68(5):1‐2. [Google Scholar]
Koszewicz 2012
- Koszewicz M, Mendak M, Konopka T, Koziorowska‐Gawron E, Budrewicz S. The characteristics of autonomic nervous system disorders in burning mouth syndrome and Parkinson disease. Journal of Orofacial Pain 2012;26(4):315‐20. [PubMed] [Google Scholar]
Lauria 2005
- Lauria G, Majorana A, Borgna M, Lombardi R, Penza P, Padovani A, et al. Trigeminal small‐fibre sensory neuropathy causes burning mouth syndrome. Pain 2005;115(3):332‐7. [DOI] [PubMed] [Google Scholar]
Lee 2015
- Lee YC, Hong IK, Sy NA, Eun YG. Evaluation of salivary function in patients with burning mouth syndrome. Oral Diseases 2015;21(3):308‐13. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
López‐Jornet 2008
- López‐Jornet P, Camacho‐Alonso F, Lucero‐Berdugo M. Quality of life in patients with burning mouth syndrome. Journal of Oral Pathology & Medicine 2008;37(7):389‐94. [DOI] [PubMed] [Google Scholar]
López‐Jornet 2009a
- López‐Jornet P, Camacho‐Alonso F, Leon‐Espinosa S. Burning mouth syndrome, oral parafunctions, and psychological profile in a longitudinal case study. Journal of the European Academy of Dermatology and Venereology 2009;23(3):363‐5. [DOI] [PubMed] [Google Scholar]
López‐Jornet 2013
- López‐Jornet P, Camacho‐Alonso F, Molino‐Pagan D. Prospective, randomized, double‐blind, clinical evaluation of aloe vera barbadensis, applied in combination with a tongue protector to treat burning mouth syndrome. Journal of Oral Pathology & Medicine 2013;42(4):295‐301. [DOI] [PubMed] [Google Scholar]
López‐Jornet 2015
- López‐Jornet P, Lucero‐Berdugo M, Castillo‐Felipe C, Zamora Lavella C, Ferrandez‐Pujante A, Pons‐Fuster A. Assessment of self‐reported sleep disturbance and psychological status in patients with burning mouth syndrome. Journal of the European Academy of Dermatology and Venereology 2015;29(7):1285‐90. [DOI] [PubMed] [Google Scholar]
Merksey 1994
- Merksey H, Bogduk N. Classification of Chronic Pain. 2nd Edition. Seattle: International Association for the Study of Pain Press, 1994. [Google Scholar]
Mueller 2003
- Mueller C, Kallert S, Renner B, Stiassny K, Temmel AF, Hummel T, et al. Quantitative assessment of gustatory function in a clinical context using impregnated 'taste strips'. Rhinology 2003;41(1):2‐6. [PubMed] [Google Scholar]
Navazesh 1982
- Navazesh M, Christensen CM. A comparison of whole mouth resting and stimulated salivary measurement procedures. Journal of Dental Research 1982;61(10):1158‐62. [DOI] [PubMed] [Google Scholar]
NICE 2013
- National Institute for Health and Care Excellence (NICE). Neuropathic pain in adults: pharmacological management in non‐specialist settings. Clinical guideline CG173. Available from www.nice.org.uk/guidance/cg173 (accessed prior to 24 May 2016).
Penza 2010
- Penza P, Majorana A, Lombardi R, Camozzi F, Bonadeo S, Sapelli P, et al. 'Burning tongue' and 'burning tip': the diagnostic challenge of the burning mouth syndrome. Clinical Journal of Pain 2010;26(6):528‐32. [DOI] [PubMed] [Google Scholar]
Puhakka 2016
- Puhakka A, Forssell H, Soinila S, Virtanen A, Röyttä M, Laine M, et al. Peripheral nervous system involvement in primary burning mouth syndrome ‐ results of a pilot study. Oral Diseases 2016;21(8):338‐44. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Sardella 2006
- Sardella A, Lodi G, Demarosi F, Bez C, Cassano S, Carrassi A. Burning mouth syndrome: a retrospective study investigating spontaneous remission and response to treatments. Oral Diseases 2006;12(2):152‐5. [DOI] [PubMed] [Google Scholar]
Scala 2003
- Scala A, Checchi L, Montevecchi M, Marini I, Giamberardino MA. Update on burning mouth syndrome: overview and patient management. Critical Reviews in Oral Biology and Medicine 2003;14(4):275‐91. [DOI] [PubMed] [Google Scholar]
Souza 2011
- Souza FT, Santos TP, Bernardes VF, Teixeira AL, Kümmer AM, Silva TA, et al. The impact of burning mouth syndrome on health‐related quality of life. Health and Quality of Life Outcomes 2011;29(9):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stata 2015 [Computer program]
- StataCorp LP. Stata Data Analysis and Statistical Software: release 14. StataCorp LP, 2015.
Sturgeon 2014
- Sturgeon JA. Psychological therapies for the management of chronic pain. Psychology Research and Behavior Management 2014;10(7):115‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Svensson 1993
- Svensson P, Bjerring P, Arendt‐Nielsen L, Kaaber S. Sensory and pain thresholds to orofacial argon laser stimulation in patients with chronic burning mouth syndrome. Clinical Journal of Pain 1993;9(3):207‐15. [DOI] [PubMed] [Google Scholar]
Taiminen 2011
- Taiminen T, Kuusalo L, Lehtinen L, Forssell H, Hagelberg N, Tenovuo O, et al. Psychiatric (axis I) and personality (axis II) disorders in patients with burning mouth syndrome or atypical facial pain. Scandinavian Journal of Pain 2011;2(4):155‐60. [DOI] [PubMed] [Google Scholar]
van der Waal 1990
- Waal I. The Burning Mouth Syndrome. Copenhagen: Munksgaard, 1990. [Google Scholar]
Woda 1999
- Woda A, Pionchon P. A unified concept of idiopathic orofacial pain: Clinical features. Journal of Orofacial Pain 1999;13(3):172‐84. [PubMed] [Google Scholar]
Woda 2009
- Woda A, Dao T, Grémeau‐Richard C. Steroid dysregulation and stomatodynia (burning mouth syndrome). Journal of Orofacial Pain 2009;23(3):202‐10. [PubMed] [Google Scholar]
Yilmaz 2007
- Yilmaz Z, Renton T, Yiangou Y, Zakrzewska J, Chessell IP, Bountra C, et al. Burning mouth syndrome as a trigeminal small fibre neuropathy: increased heat and capsaicin receptor TRPV1 in nerve fibres correlates with pain score. Journal of Clinical Neuroscience 2007;14(9):864‐71. [DOI] [PubMed] [Google Scholar]
Zakrzewska 1999
- Zakrzewska JM, Hamlyn PJ. Facial pain. In: Crombie IK, Croft PR, Linton SJ, Resche L, Korff M editor(s). Epidemiology of Pain. Seattle: International Association for the Study of Pain Press, 1999:177‐202. [Google Scholar]
References to other published versions of this review
Zakrzewska 2000
- Zakrzewska JM, Glenny A‐M, Forssell H. Interventions for the treatment of burning mouth syndrome. Cochrane Database of Systematic Reviews 2000, Issue 4. [DOI: 10.1002/14651858.CD002779] [DOI] [Google Scholar]
Zakrzewska 2005
- Zakrzewska JM, Forssell H, Glenny AM. Interventions for the treatment of burning mouth syndrome. Cochrane Database of Systematic Reviews 2005, Issue 1. [DOI: 10.1002/14651858.CD002779.pub2] [DOI] [PubMed] [Google Scholar]