

Abstract
Background:
Platelet-neutrophil interactions contribute to vascular occlusion and tissue damage in thromboinflammatory disease. Platelet glycoprotein Ibα (GPIbα), a key receptor for cell-cell interactions, is believed to be constitutively active for ligand-binding. Here we established the role of platelet-derived protein disulfide isomerase (PDI) in reducing the allosteric disulfide bonds in glycoprotein Ibα (GPIbα) and enhancing the ligand-binding activity under thromboinflammatory conditions.
Methods:
Bioinformatic analysis identified two potential allosteric disulfide bonds in GPIbα. Agglutination assays, flow cytometry, surface plasmon resonance analysis, a protein-protein docking model, proximity ligation assays, and mass spectrometry were utilized to demonstrate a direct interaction between PDI and GPIbα and to determine a role for PDI in regulating GPIbα function and platelet-neutrophil interactions. Real-time microscopy and animal disease models were employed to study the pathophysiological role of PDI-GPIbα signaling under thromboinflammatory conditions.
Results:
Deletion or inhibition of platelet PDI significantly reduced GPIbα-mediated platelet agglutination. Studies using PDI-null platelets and recombinant PDI or Anfibatide, a clinical-stage GPIbα inhibitor, revealed that the oxidoreductase activity of platelet surface-bound PDI was required for the ligand-binding function of GPIbα. PDI directly bound to the extracellular domain of GPIbα on the platelet surface and reduced the Cys4-Cys17 and Cys209-Cys248 disulfide bonds. Real-time microscopy using platelet-specific PDI conditional knockout and sickle cell disease mice demonstrated that PDI-regulated GPIbα function was essential for platelet-neutrophil interactions and vascular occlusion under thromboinflammatory conditions. Studies using a mouse model of ischemia/reperfusion-induced stroke indicated that PDI-GPIbα signaling played a crucial role in tissue damage.
Conclusions:
Our results demonstrate that PDI-facilitated cleavage of the allosteric disulfide bonds tightly regulates GPIbα function, promoting platelet-neutrophil interactions, vascular occlusion, and tissue damage under thromboinflammatory conditions.
Keywords: protein disulfide isomerase, glycoprotein Ibα, platelet-neutrophil interactions, thromboinflammation
Introduction
Protein disulfide isomerase (PDI) catalyzes modification of thiol-disulfide bonds during protein folding in the endoplasmic reticulum (ER).1 The oxidoreductase activity of PDI requires two CysGlyHisCys active sites at the N- and C-termini. Despite having the ER-retention sequence, PDI is present in the circulation of healthy humans and bear the reductase activity,2 implicating constitutive secretion of PDI under resting conditions. PDI is also secreted from activated platelets in a manner dependent on actin polymerization.3 Genomic and histologic analyses indicate that cellular PDI is up-regulated at the myocardial infarct region,4 suggesting the pathological role in cardiovascular disease. Using intravital microscopy with PDI conditional knockout (CKO) mice, blocking antibodies, and a small-molecule inhibitor, we and others have demonstrated the importance of platelet surface-bound PDI in αIIbβ3 integrin activation and thrombus formation following arteriolar injury.5–7 However, the molecular basis for the role of extracellular PDI has remained speculative.
Platelets primarily contribute to hemostasis and thrombosis. When endothelial cells are damaged, platelets adhere to von Willebrand factor (vWF) and collagen through glycoprotein Ibα (GPIbα), the major ligand-binding subunit of the GPIb-IX-V complex and GPVI, respectively,8 followed by platelet-platelet aggregation via the fibrinogen-αIIbβ3 interaction. Platelets also interact with leukocytes at sites of vascular injury and release pro-thrombotic and pro-inflammatory molecules, exacerbating thromboinflammatory conditions in which inflammation is coupled to thrombosis.9 Platelet-leukocyte adhesion is mainly mediated by the interactions of platelet P-selectin and GPIbα with neutrophil P-selectin glycoprotein ligand-1 and αMβ2 integrin, respectively. Despite advances in our knowledge of the major receptors and counterreceptors for platelet-neutrophil interactions,9 the molecular mechanisms regulating the receptor-counterreceptor association remain poorly understood.
GPIbα binds to numerous molecules, including vWF,10 thrombin,11 P-selectin,12 and αMβ2 integrin,13 enabling platelets to participate in a wide range of disease processes. In particular, the GPIbα-αMβ2 interaction stabilizes platelet-leukocyte association, resulting in the initiation and progression of thromboinflammatory diseases.14, 15 However, it was reported that the extracellular domain of GPIbα is dispensable for platelet-leukocyte interactions during development of atherosclerosis under high-fat diets.16 These results imply that the contribution of GPIbα to platelet-leukocyte interactions may depend on the disease condition.
Function of mature proteins can be controlled by oxidation or reduction of allosteric disulfide bonds.17 There are approximately 3,700 human proteins with a known 3D structure, and they collectively contain >15,000 disulfide bonds.18 Notably, 45% of all disulfide bonds are found in cell surface proteins. Disulfide bonds are classified based on the geometry of the five dihedral angles that define the cystine residue.19 Twenty disulfide bond configurations are possible using this classification scheme. The allosteric disulfides are being associated with one of three configurations; –RHStaple, –/+RHHook, and –LHHook.17 Interestingly, the Cys4-Cys17 disulfide bond in GPIbα has an allosteric –RHStaple configuration.19 A study showed that PDI and GPIbα are sufficiently close on the platelet surface to allow fluorescence resonance energy transfer and treatment of platelets with polyclonal anti-PDI antibodies reduces fluorescently-labeled vWF binding.20 A single amino acid substitution (Gly233Val, Asp235Val, Lys237Val, or Met239Val) in the Cys209-Cys248 disulfide loop in GPIbα results in a gain-of-function mutation.21 We reported that reactive oxygen species generated by platelet NADPH oxidase 2 influence GPIbα function.15 These results suggest that the structure and function of GPIbα are modulated under oxidative stress and PDI is involved in this regulation.
In the present study, we demonstrate for the first time that PDI-facilitated cleavage of two allosteric disulfide bonds in GPIbα tightly regulates the ligand-binding activity and the PDI-promoted function of GPIbα is critical for platelet-neutrophil interactions, vascular occlusion, and tissue damage under thromboinflammatory conditions.
Methods
Extended methods are given in the online-only Data Supplement.
The data, analytic methods, and study materials will be/have been made available to other researchers for purposes of reproducing the results or replicating the procedure (available at the authors’ laboratories).
Mice
Megakaryocyte (platelet)-specific PDI CKO mice (PDIflox/flox;platelet factor 4 (PF4)-cre) were generated by crossing PF4-Cre mice with PDIflox/flox mice as we reported.5 PDIflox/flox mice were backcrossed 10 generations to C57BL/6 mice prior to generation of CKO mice. Mice in which the extracellular domain of GPIbα is replaced with the α subunit of human interleukin-4 receptor (hIL4R/GPIbα mice) were generously provided by Dr. Jerry Ware.22 WT (C57BL/6), hemizygous control (Tg(Hu-miniLCRα1GγAγδβS)Hba−/−Hbb+/−), and Berkeley sickle (Tg(Hu-miniLCRα1GγAγδβS)Hba−/−Hbb−/−) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Chimeric control and Berkeley mice were generated by transplantation of Berkeley bone marrow into lethally irradiated C57BL/6 mice (6 weeks old), and the chimerism was confirmed by PCR and electrophoresis analyses 3 months after transplantation as described previously.23 These chimeric Berkeley mice were hereafter referred to as sickle cell disease (SCD) mice and used at the age of 18–22 weeks old in this study. Age-matched mice of both genders were used in all studies. The University of Illinois Institutional Animal Care and Use Committee approved all animal care and experimental procedures (Protocols 18–015 and 16–172).
Intravital microscopy
Intravital microscopy was performed in a mouse model of TNF-α-induced cremaster venular inflammation as we described previously.24 Control and PDI CKO male mice (6–8 weeks old) were anesthetized with intraperitoneal injection of ketamine and xylazine. The cremaster muscle was exteriorized and superfused with 37ºC bicarbonate-buffered saline throughout the experiment. Platelets and neutrophils were visualized by infusion of DyLight 488-conjugated anti-CD42c (0.2 µg/g body weight (BW)) and Alexa Fluor 647-conjugated anti-Ly-6G (0.1 μg/g BW) antibodies, respectively. In some experiments, wtPDI or dmPDI (4 μg/g BW) was infused into platelet-specific PDI CKO mice 3 hours after intrascrotal injection of TNF-α. In other experiments, bovine serum albumin (BSA) or Anfibatide (5 – 50 ng/g BW) was infused into WT mice 3 hours after intrascrotal injection of TNF-α. The experiments were performed in a single-blind fashion in which the investigators did not know the identity of the sample or mouse. Images were captured in 6–8 different cremaster venules with a diameter of 25–40 μm in each mouse and recorded using an Olympus BX61W microscope with a water immersion objective (60 × 1.0 NA) and a high-speed camera (ORCA-Flash4.0 V2, C11440, Hamamatsu). The numbers of rolling and adherent neutrophils were determined in an area of 0.02 mm2 over 5 minutes, followed by normalization to vessel length. The kinetics of platelet accumulation was monitored in the integrated median fluorescence intensities of the anti-CD42c antibody. The fluorescence signal was normalized to the number of adherent neutrophils and the vessel length and plotted as a function of time. Time “0” was set to when image capture began on each vessel. Blood flow rates were calculated by centerline velocity of Alexa Fluor 488-conjugated microspheres (diameter: 200 nm). Data were analyzed using Slidebook (version 6.0, Intelligent Imaging Innovations).
In adoptive transfer experiments, WT platelets were labeled with calcein-AM (1 μM) for 15 minutes at 37°C. Platelets were treated with control IgG or anti-PDI antibodies (10 μg/ml), BSA or Anfibatide (0.2 μg/ml), or both inhibitors, followed by incubation with 0.025 U/ml thrombin for 5 minutes at 37°C and with 50 μM D-Phe-Pro-Arg-chloromethylketone. After washing, the labeled platelets (2 × 108 cells/0.1 ml) were infused into TNF-α-challenged WT mice which were pretreated without or with eptifibatide (10 μg/g BW) to block ligand-αIIbβ3 interactions. The drug was reinfused every 20 minutes for the duration of the experiment. Endogenous neutrophils were visualized by infusion of an Alexa Fluor 647-conjugated anti-Ly-6G antibody. The number of infused platelets adhered to neutrophils was counted and normalized to the number of adherent neutrophils.
For SCD mice, severe inflammation was induced by intraperitoneal injection of TNF-α (500 ng) as we described previously.25 Control IgG or a blocking anti-PDI antibody (BD34, 1.5 μg/g BW), BSA or Anfibatide (25 ng/g BW), or both inhibitors were infused 3 hours after TNF-α injection, followed by intravital microscopy as described above. During or after intravital microscopic studies, survival times for each mouse were recorded. Each time point began at TNF-α injection and ended when the mouse died or up to 6 hours after TNF-α injection. The experiments were performed in a single-blind manner.
Statistics
Data were analyzed using the GraphPad Prism 7 software. Statistical significance was assessed by Student’s t-test, ANOVA and Dunnett’s or Tukey’s test, Kruskal-Wallis test with post hoc Dunn correction, and Mantle-Cox log-rank test. A P value less than 0.05 was considered significant.
Results
Bioinformatic analysis identifies potential allosteric disulfide bonds in platelet surface receptors
Using a database on disulfide bonds (http://149.171.101.136/python/disulfideanalysis/index.html)26 and Protein Data Bank identifiers (PDB IDs) of platelet surface receptors, bioinformatic analysis was performed to search potential allosteric bonds in each molecule. Our results showed that many receptor proteins, including integrins, GPIbα, and CD40, contain at least one potential allosteric disulfide bond that has not been reported (Table S1). Among them, GPIbα attracted our interest because it is an essential platelet receptor involved in numerous vascular disease27 and has been known to be constitutively active for ligand-binding. In addition to the Cys4-Cys17 disulfide bond (–RHStaple) in GPIbα,19 we identified the Cys209-Cys248 bond as a putative allosteric one with a –LHHook configuration (PDB ID: 1M0Z).10
The oxidoreductase activity of platelet surface-bound PDI is required for GPIbα-mediated agglutination and its ligand-binding function
Since PDI facilitates thiol-disulfide bond shuffling and is released from platelets,3, 5 we tested whether platelet PDI regulates GPIbα function. The assay of vWF-induced platelet agglutination and aggregation was performed in the presence of botrocetin or ristocetin which induces vWF binding to GPIbα. Deletion of platelet PDI exhibited a significant reduction in agglutination and aggregation (Figure 1A). Treatment of PDI-null platelets with wtPDI but not dmPDI restored decreased platelet interactions to the WT level. In control experiments, PDI deletion did not affect the surface level of GPIbα and P-selectin (Figure S1A-B). Inhibition of extracellular PDI activity with a blocking anti-PDI antibody (BD34, 10 μg/ml) that specifically inhibits the activity of PDI but not other oxidoreductases5 impaired agglutination and aggregation of mouse and human platelets (Figure 1B and1E).
Figure 1. The oxidoreductase activity of platelet surface-bound PDI is crucial for vWF-mediated agglutination/aggregation and its ligand-binding.
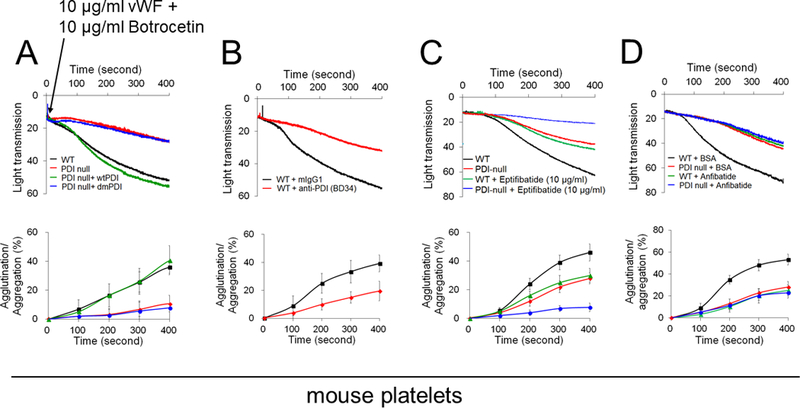
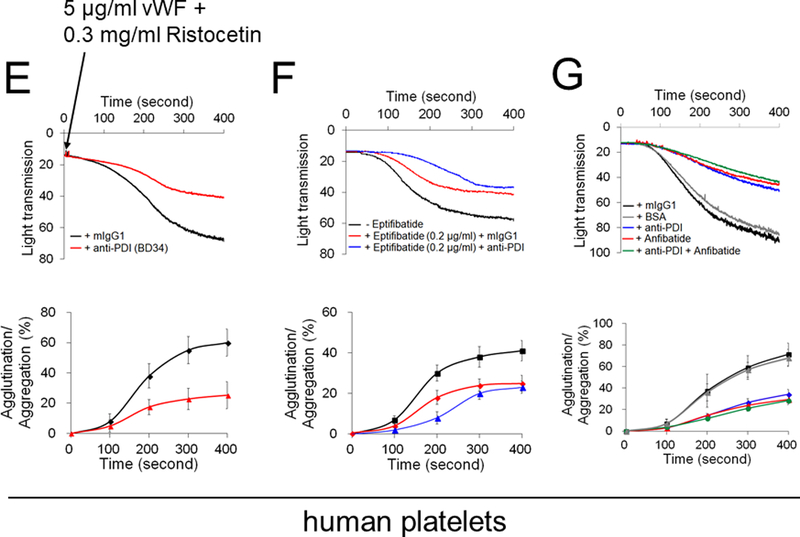
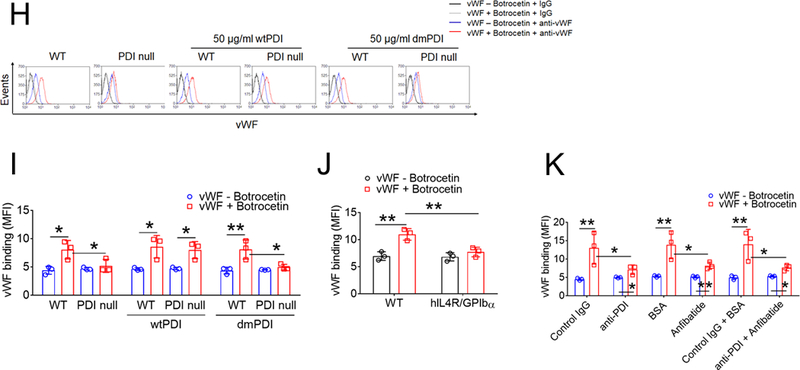
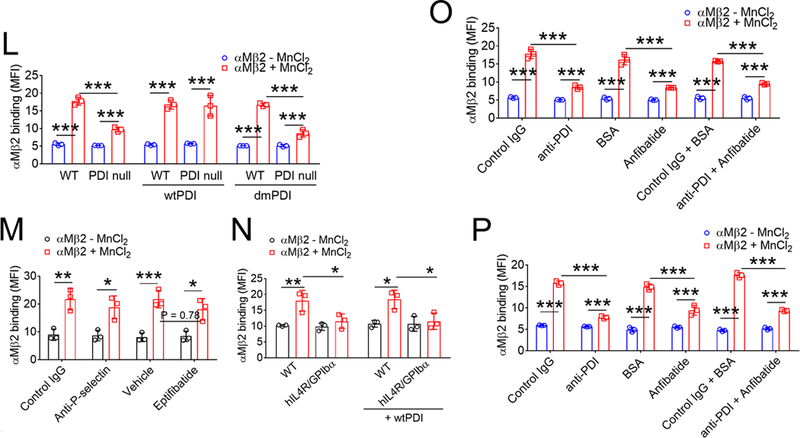
(A-D) Mouse and (E-G) human platelets were incubated with vWF and either botrocetin (A-D) or ristocetin (E-G), followed by measurement of agglutination and aggregation. (A) PDI-null platelets were pretreated with or without wtPDI or dmPDI, 50 µg/ml. (B-G) Platelets were pretreated with (B, E, and F) 10 µg/ml control IgG or a blocking anti-PDI antibody (BD34), (C and F) vehicle or 10 (C) or 0.2 (F) µg/ml eptifibatide, (D and G) 0.2 µg/ml BSA or Anfibatide, or both an anti-PDI antibody and Anfibatide. The representative agglutination/aggregation traces (upper panel) and quantitative graphs (bottom panel) were obtained from 3 independent experiments. (H-O) Mouse and (P) human platelets were pretreated with or without (H, I, and L) recombinant PDI or (K, M, O, and P) control IgG, an anti-PDI antibody, an anti-P-selectin, eptifibatide, BSA, Anfibatide, or both an anti-PDI antibody and Anfibatide. (J and N) WT and hIL4R/GPIbα platelets were used. After washing, platelets were incubated with (H-K) 10 μg/ml vWF and 10 mM EDTA in the presence or absence of 10 μg/ml botrocetin or with (L-P) 10 μg/ml αMβ2 in the presence or absence of 0.5 mM MnCl2 and (N) 50 μg/ml wtPDI. Flow cytometry was performed using fluorescently-labeled antibodies against vWF or β2 integrin. The data are shown as the geometric mean fluorescence intensity (MFI) value. All data represent the mean ± SD (n = 3). *P < 0.05, **P < 0.01, or ***P < 0.001 after ANOVA and Tukey’s test.
Since platelet PDI is crucial for αIIbβ3 integrin activation,5 we further determined the role of PDI in GPIbα-mediated agglutination by blocking platelet aggregation with eptifibatide, an αIIbβ3 antagonist. We confirmed that thrombin-induced aggregation of mouse and human platelets was abolished by eptifibatide at a concentration of 10 and 0.2 μg/ml, respectively (Figure S2). Compared to WT platelets, when PDI-null platelets were pretreated with 10 μg/ml eptifibatide, platelet-platelet interactions were further impaired (Figure 1C). Similarly, vWF-induced initial agglutination of eptifibatide-treated human platelets was inhibited by co-treatment with a blocking anti-PDI antibody (Figure 1F). Moreover, we tested the effect of Anfibatide, a clinical-stage GPIbα inhibitor28 in mouse and human platelets. Pretreatment with this drug displayed a dose-dependent inhibition in platelet agglutination, with around 50% inhibition at 0.2 μg/ml (Figure S3). When used in PDI-null platelets or human platelets treated with anti-PDI antibodies, Anfibatide showed no further effect (Figure 1D and1G). Together, these results suggest that in addition to the role in αIIbβ3-mediated platelet aggregation,5, 6 the oxidoreductase activity of platelet surface-bound PDI promotes GPIbα-mediated agglutination.
A previous study using Alexa Fluor 488-conjugated vWF suggested the regulation of vWF binding to platelet GPIbα by PDI.20 However, due to the importance of several Lys residues in vWF for the binding to GPIbα,29 labeling primary amines might alter vWF activity. Thus, we assessed binding of unlabeled vWF to platelets by flow cytometry. In addition to GPIbα, vWF binds to platelet αIIbβ3 integrin and P-selectin, and both interactions are Ca2+-dependent.30, 31 To eliminate this possibility, EDTA was used in this assay. We found that soluble vWF binding to WT platelets increased in the presence of botrocetin and the binding was markedly reduced by PDI deletion (Figure 1H-I). WTPDI but not dmPDI rescued the defect in PDI-null platelets, and both PDIs had no effect in WT platelets. We confirmed that vWF binding was abolished by loss of the GPIbα extracellular domain (Figure 1J) and treatment of WT platelets with a blocking anti-P-selectin antibody did not affect vWF binding (Figure S4). Further, we used an anti-PDI antibody and Anfibatide in this study. To eliminate the possibility that platelet PDI alters vWF conformation, platelets were pretreated with each inhibitor or both and washed out before the addition of vWF and botrocetin. Compared to the control, treatment with the anti-PDI antibody or Anfibatide alone showed a significant reduction in vWF binding to WT platelets (Figure 1K). The combination of both inhibitors had no additional effect. In control experiments, PDI was detected on the surface of unstimulated platelets, but the surface amount significantly increased upon thrombin stimulation (Figure S5).
Activated αMβ2 integrin interacts with GPIbα, mediating platelet-leukocyte interactions in vascular disease.14, 24 PDI deletion significantly diminished binding of MnCl2-activated αMβ2 to platelets, which was rescued by the addition of wtPDI but not dmPDI (Figure 1L). We found that treatment of platelets with an anti-P-selectin antibody and eptifibatide minimally inhibited αMβ2 binding, whereas deletion of the extracellular domain of GPIbα abrogated αMβ2 binding to platelets despite the addition of wtPDI (Figure 1M-N), indicating the specific interaction between αMβ2 and GPIbα. Treatment with the anti-PDI antibody or Anfibatide alone reduced αMβ2 binding to mouse and human platelets in comparison with each control, and no additional effect was observed by treatment with both inhibitors (Figure 1O-P). These results suggest that PDI on the surface of unstimulated platelets plays a role in the initial binding of vWF or activated αMβ2 to GPIbα.
PDI directly binds to the extracellular domain of GPIbα on the platelet surface and the binding increases during cell activation
As determined by surface plasmon resonance (SPR) analysis, wtPDI and dmPDI bound to immobilized GPIbα with a Kd of 0.9 ± 0.3 and 1.2 ± 0.3 µM, respectively (Figure 2A-B), indicating that PDI directly binds to GPIbα in a manner independent of the isomerase activity. Co-immunoprecipitation assays using lysates of platelets labeled with sulfo-N-hydroxysulfosuccinimide-biotin (SSB), a cell-impermeable, biotin-containing probe reacting with primary amines showed that GPIbα interacted with PDI in unstimulated platelets and the binding increased after stimulation with 0.025 U/ml thrombin (Figure 2C-D) at which GPIbα was not shed (Figure S1A). When the blot was re-probed with avidin, the 140-kDa band (GPIbα) was detected even in unstimulated platelets, and the band density increased upon thrombin stimulation. These results suggest that the surface interaction between PDI and GPIbα is enhanced during platelet activation.
Figure 2. PDI directly binds to GPIbα in an oxidoreductase activity-independent manner.
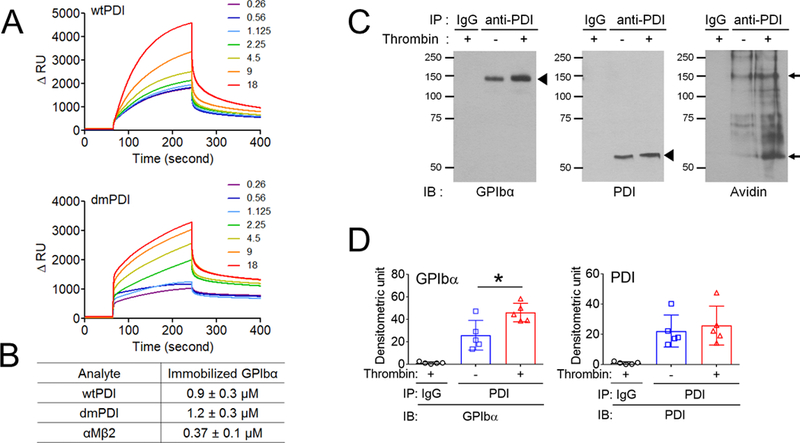
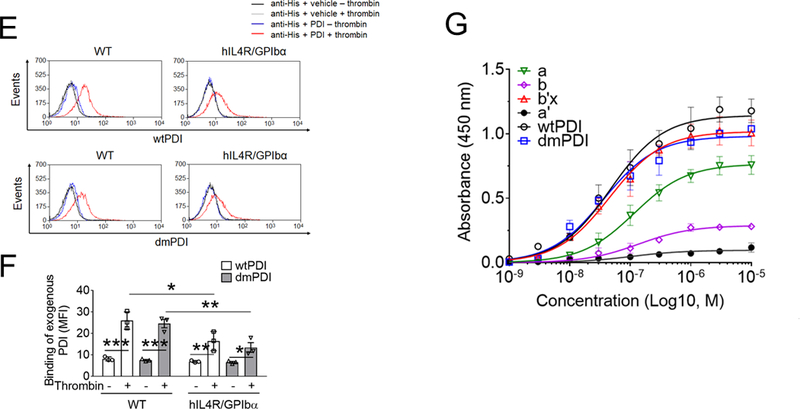
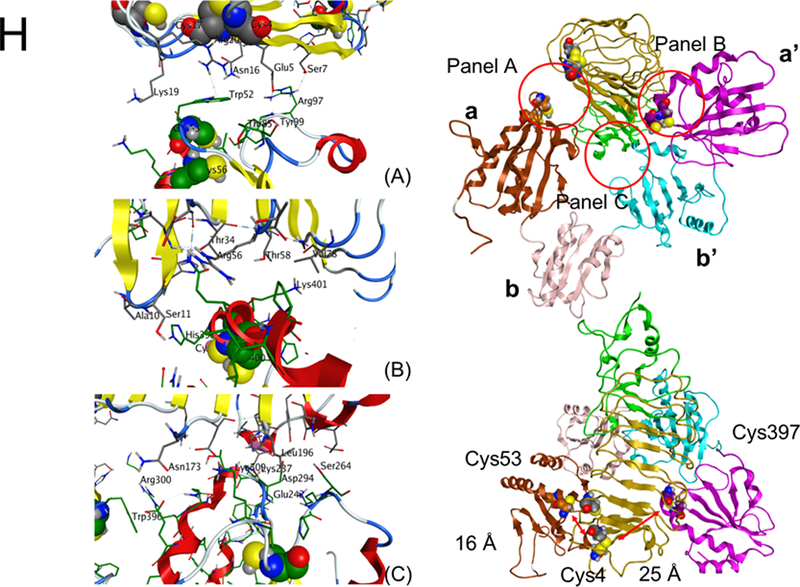
(A-B) Recombinant wtPDI or dmPDI (0.26 to 18 µM), or αMβ2 (0.03 to 21 µM, data not shown) was infused over the reference and surfaces immobilized with the extracellular domain of GPIbα for 180 seconds, followed by dissociation phase of 120 seconds. The dissociation constant, Kd, was calculated based on the Kon and Koff value. Data represent the mean ± SD (n = 3). (C-D) Mouse platelets were stimulated with or without 0.025 U/ml of thrombin and labeled with SSB. Lysates were immunoprecipitated with control IgG or anti-PDI antibodies and immunoblotted with indicated antibodies, followed by densitometry (mean ± SD, n = 5). The blots were re-probed with peroxidase-conjugated avidin. The arrowheads show the band of GPIbα and PDI, and arrows show the 140- (GPIbα) and 60-kD (PDI) bands. (E-F) Platelets isolated from WT and hIL4R/GPIbα Tg mice were treated with or without 0.025 U/ml thrombin, followed by incubation with His-tagged wtPDI or dmPDI (50 μg/ml). The bound PDI was determined by flow cytometry using Alexa Fluor 488-conjugated anti-polyHis antibodies. The data are shown as the MFI value. (G) ELISA was conducted using 1 nM to 10 μM of His-tagged PDI and its domain fragments and immobilized GST-tagged full-length GPIbα. Bound PDI or its domain was measured by anti-polyHis antibodies. Values obtained from BSA-coated surfaces were subtracted as a negative control. Data represent the mean ± SD (n = 3–4). *P < 0.05, **P < 0.01, or ***P < 0.001 after Student’s t-test (for D) and ANOVA and Tukey’s test (for F). (H) A docking model of PDI-GPIbα complex. A set of representative residues at the interface between GPIbα and reduced PDI. Panel A: the a-domain of PDI and the β-finger of GPIbα, Panel B: the a’-domain of PDI and the β-finger of GPIbα, and Panel C: the b’-domain of PDI and the β-switch of GPIbα.
We found that wtPDI and dmPDI equally bound to resting WT platelets and the binding increased to 3-fold following thrombin stimulation (Figure 2E-F). The binding of both PDIs was significantly decreased to thrombin-stimulated hIL4R/GPIbα platelets, with a slight reduction (85% of control) in unstimulated hIL4/GPIbα platelets. There were no differences between WT and hIL4R/GPIbα platelets in thrombin-induced aggregation, αIIbβ3 activation, and P-selectin exposure (Figure S6). These results support our finding of the direct interaction between PDI and the extracellular domain of GPIbα. To further identify the binding site of PDI for GPIbα, an enzyme-linked immunosorbent assay (ELISA) was carried out using His-tagged PDI and its four domains (a, b, b’x, and b’), and GST-tagged full-length GPIbα. As detected by anti-polyHis antibodies, PDI and its b’x domain had similar binding affinities, and the a-domain in part contributed to PDI-GPIbα binding (Figure 2G). In contrast, the b- and a’-domains were minimally involved in binding to GPIbα.
To gain additional insights into the binding conformation between GPIbα and PDI, protein-protein docking of the X-ray models (PDB ID: 3P72 (GPIbα),32 4EKZ (reduced PDI), and 4EL1 (oxidized PDI)33) was conducted. In the top-scored pose between GPIbα and reduced PDI, the β-finger portion of GPIbα was positioned between the a- and a’-domains of PDI, whereas the β-switch region of GPIbα formed an extended contact with the b’-domain of PDI (Figure 2H). At all the interfaces, the binding was driven by a mixture of hydrophobic, electrostatic, and hydrogen bonding interactions. The Cys53 residue in the active site of the PDI a-domain was positioned within 16 Å of the Cys4 residue in GPIbα. The Cys397 residue in the PDI a’-domain was further, at approximately 25 Å. The possible flexibility of the β-finger region of GPIbα, an ability to undergo conformational changes associated with reduction or oxidation of Cys residues by PDI as well as the shorter distance to the Cys 53 and Cys56 than to the Cys397 and Cys400 catalytic pair, suggests that the former pair is likely to participate in reduction of the Cys4-Cys17 disulfide bond in GPIbα. Because of the wider gap between a and a’ domains in oxidized PDI, GPIbα was positioned deeper between the two domains and was oriented in the opposite direction compared to that in the complex with reduced PDI. The distance between the Cys4-Cys17 bond in GPIbα and the PDI active sites (Cys53-Cys56 and Cys397-Cys400 pairs) was 28 Å and 33 Å, respectively (Figure S7), suggesting that much larger conformational changes are needed for oxidized PDI to modify the disulfide bond.
PDI reduces allosteric disulfide bonds in GPIbα and enhances the ligand-binding activity
Biochemical studies using Nα-(3-maleimidylpropionyl) biocytin (MPB), a biotin-containing probe reacting with free Cys residues34 showed that only wtPDI increased MPB labeling on GPIbα under different redox conditions, with a greater extent in a reducing environment (Figure 3A-C). No MPB labeling on GPIbα was observed in the absence of PDI. These results suggest that the oxidoreductase activity of PDI facilitates thiol exposure in GPIbα. The GPIbα disulfide bonds cleaved by PDI was further identified using mass spectrometry (Figure 3D-F). Eleven Cys-containing peptides (Table 1) reporting on the redox state of the three GPIbα disulfides were resolved and quantified. GPIbα was treated with 2- or 10-fold molar excess of wtPDI or dmPDI and the redox state of the disulfides was determined. In accordance with crystal structures of GPIbα,10 the three disulfide bonds in untreated GPIbα were > 98% oxidized (Figure 3G). The Cys4-Cys17 and Cys209-Cys248, but not Cys211-Cys264, disulfide bonds are dose-dependently cleaved by wtPDI. Redox-inactive dmPDI did not cleave the bonds.
Figure 3. PDI reduces the allosteric disulfide bonds in GPIbα, enhancing the ligand-binding function.
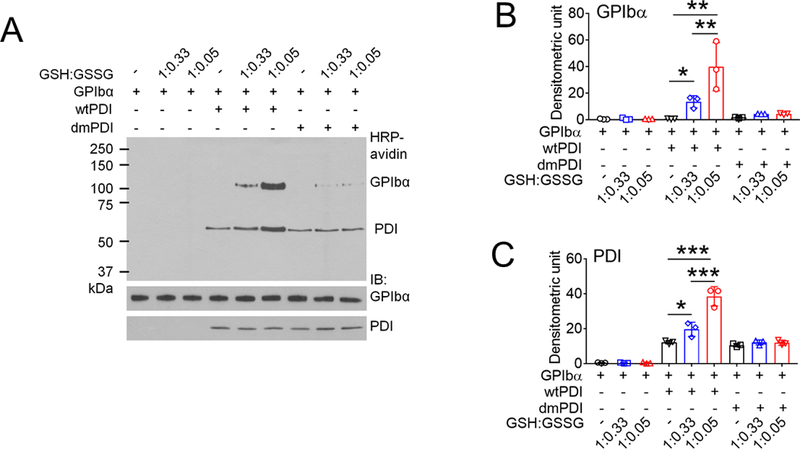
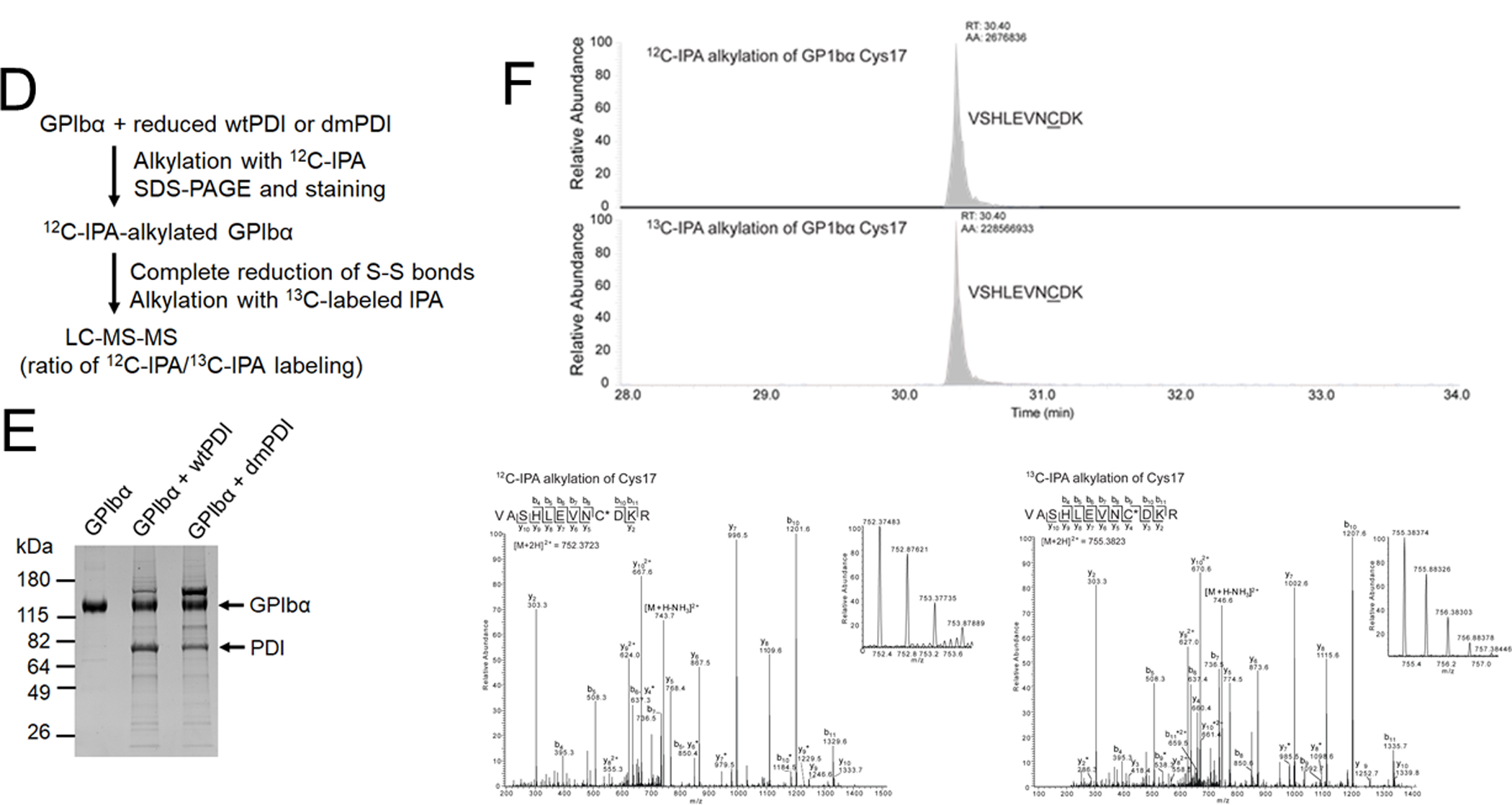
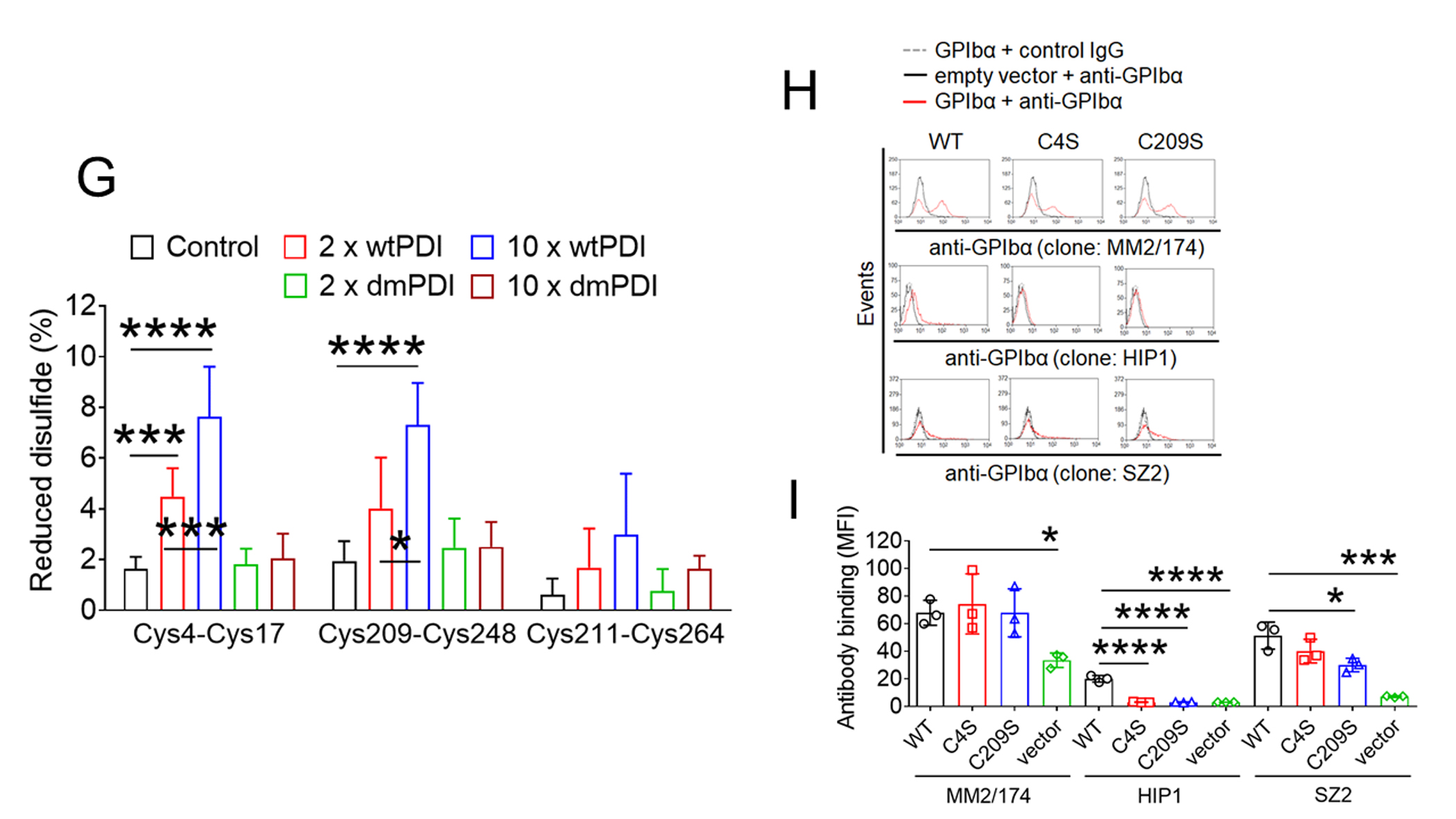
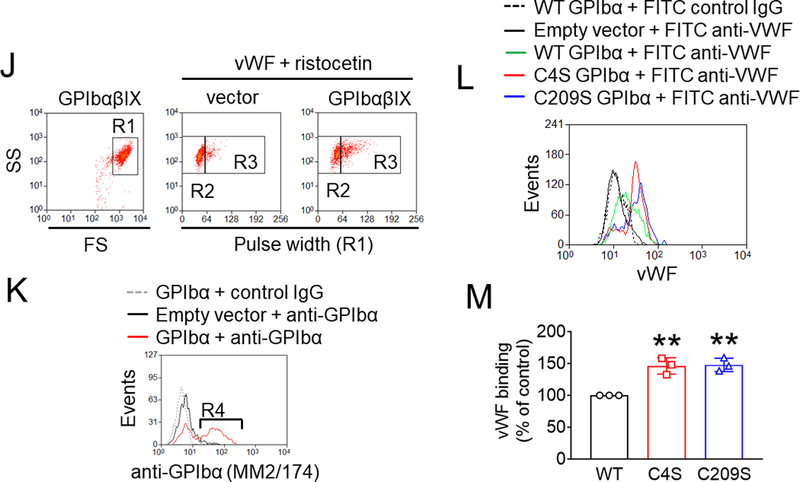
(A-C) GPIbα (50 nM) and either wtPDI or dmPDI (100 nM) were incubated with MPB under different redox conditions (GSH:GSSG = 0:0, 1:0.33, or 1:0.05 mM). Proteins were electrophoresed under reduced conditions and blotted with peroxidase-conjugated avidin (upper panel) or antibodies against GPIbα or PDI (lower panel). Representative blot and quantitated graphs of densitometry (mean ± SD, n = 3). (D) A schematic of mass spectrometric analysis (see Methods). (E-H) Recombinant GPIbα extracellular domain (1.8 µM) was incubated with or without reduced wtPDI or dmPDI (3.6 or 18 μM), followed by incubation with 12C-IPA. (E) Proteins were separated on SDS-PAGE and stained with Sypro Ruby. The band of GPIbα was excised, reduced with 100 mM dithiothreitol, and alkylated with 13C-IPA. (F) HPLC resolution of the GPIbα VASHLEVNCDKR peptide containing Cys17 (underlined) labelled with 12C-IPA or 13C-IPA (top panel). Representative tandem mass spectra of VASHLEVNCDKR peptide with Cys17 (indicated with an asterisk) labelled with 12C-IPA or 13C-IPA. The accurate mass spectrum of the peptide is shown in the inset (observed [M+2H]2+ = 752.3748 m/z and expected [M+2H]2+ = 752.3723 m/z; observed [M+2H]2+ = 755.3837m/z and expected [M+2H]2+ = 755.3823 m/z). Ions with a loss of water are labeled with an asterisk. (G) Redox state of the GPIbα Cys4-Cys17, Cys209-Cys248 and Cys211-Cys264 disulfide bonds in the absence or presence of 2- or 10-fold molar excess of wtPDI or dmPDI. Data represent the mean ± SD of the analysis of 3–9 Cys-containing peptides from 2 different GPIbα preparations. (H-I) CHO cells expressing WT or mutant GPIbαβ/IX or GPIbβ/IX (with an empty vector for GPIbα) were incubated with anti-GPIbα antibodies (MM2/174, HIP1, or SZ2), followed by flow cytometry. (J-M) The CHO cells were incubated with 35 μg/ml vWF and 2 mg/ml ristocetin under a stirring condition (1,000 rpm) for 10 minutes. After washing, cells were treated with isotype control IgG or anti-GPIbα antibodies (MM2/174) and then with Alexa Fluor 647-conjugated anti-mouse IgG antibodies. Cells were incubated with FITC-conjugated control IgG or anti-vWF antibodies, followed by flow cytometry. R1, CHO cells; R2, single cell population; and R3, cell aggregates. (K) The surface level of GPIbα in CHO cells. (L-M) Histograms of cell surface-bound vWF and the quantitative graph. Data represent the mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 or ****P < 0.0001 after ANOVA and Tukey’s test (B-C, G, and M) or Dunnett’s test (I).
Table 1.
List of Cys-containing peptides generated from tryptic digest of recombinant GPIbα that are detected by mass spectrometry.
| Disulfide bond | Cys position | Peptide sequence* |
|---|---|---|
| 4–17 | 4 17 |
HPICEVSK VASHLEVNCDK VASHLEVNCDKR |
| 209–248 | 248 | AMTSNVASVQCDNSDK AM(oxidation)TSNVASVQCDNSDK AMTSNVASVQCDNSDKFPVYK AM(oxidation)TSNVASVQCDNSDKFPVYK |
| 211–264 | 264 | GCPTLGDEGDTDLYDYYPEEDTEGDK GCPTLGDEGDTDLYDYYPEEDTEGDKVR |
| N/A | 65 | LTQLNLDRCELTK CELTK |
Cys was labelled with 12C-IPA (133.05276) or 13C-IPA (139.07289).
Previous findings that treatment of platelets with polyclonal anti-PDI antibodies alters thiol exposure in GPIbα upon agonist stimulation and influences binding of monoclonal antibodies against GPIbα imply that PDI-mediated modification of disulfide bonds induces a conformational change in GPIbα.20 To study the role of each disulfide bond in GPIbα function, Chinese hamster ovary (CHO) cells35 were transfected with a vector containing WT GPIbα or the mutant in which the Cys4 or Cys209 residue was mutated to Ser, along with vectors containing GPIbβ or GPIX. As analyzed by flow cytometry with different monoclonal anti-GPIbα antibodies, MM2/174 recognizing the elastase-resistant part of GPIbα36 equally detected WT and mutant GPIbα on CHO cells (Figure 3H-I). Both WT and mutant GPIbαβ/IX were equally expressed on the surface of CHO cells. Interestingly, HIP1 binding to the second leucine-rich repeat of GPIbα37 detected WT GPIbα but not two mutants (Figure 3H-I), whereas SZ2 recognizing the anionic sulfated tyrosine region13, 37 equally bound to WT GPIbα and the Cys4Ser mutant but showed a significantly reduced binding to the Cys209Ser mutant (Figure 3H-I). These results indicate that GPIbα undergoes a conformational change by reduction of the Cys4-Cys17 or Cys209-Cys248 disulfide bond. As determined in single cells expressing a high level of GPIbα (both R2/R4 gates) (Figure 3J-K), vWF binding significantly increased in cells expressing either mutant, compared to WT GPIbα under stirring conditions (Figure 3L-M). These results suggest that reduction of the Cys4-Cys17 or Cys209-Cys248 disulfide bond in GPIbα enhances the ligand binding activity.
Platelet PDI-regulated GPIbα is crucial for platelet-neutrophil aggregation under inflammatory conditions
Since the interaction between GPIbα and activated αMβ2 integrin stabilizes platelet-neutrophil adhesion,15 the role of platelet PDI in platelet-neutrophil aggregation was determined under shear-mimicking conditions. Consistent with previous reports,15, 24 a new cell population of platelet-neutrophil aggregates appeared in the R1 gate (above the neutrophil population, R2 gate), and most cells (>95%) in the R1 gate were positive for both CD42c and Ly-6G (Figure 4A). Deletion of platelet PDI significantly inhibited the platelet-neutrophil aggregation as assessed by the number of cell-cell aggregates (R3 gate) and the fluorescence intensity of anti-CD42c antibodies in the R1 gate (Figure 4A-C). Immunofluorescence microscopy also showed that the size of platelet-neutrophil aggregates was reduced by platelet PDI deletion (Figure 4D). Treatment of WT platelets with either a blocking anti-PDI antibody or Anfibatide impaired the formation of platelet-neutrophil aggregates as measured by the fluorescence intensity of the anti-CD42c antibody in platelet-neutrophil aggregates (Figure 4E). When platelets were treated with both inhibitors, the inhibitory effect was comparable to that of a single inhibitor. Similar results were obtained with human cells (Figure 4F). These data suggest that platelet-derived PDI promotes platelet-neutrophil aggregation by regulating GPIbα function.
Figure 4. Platelet PDI-regulated GPIbα function is important for platelet-neutrophil aggregation under inflammatory conditions.
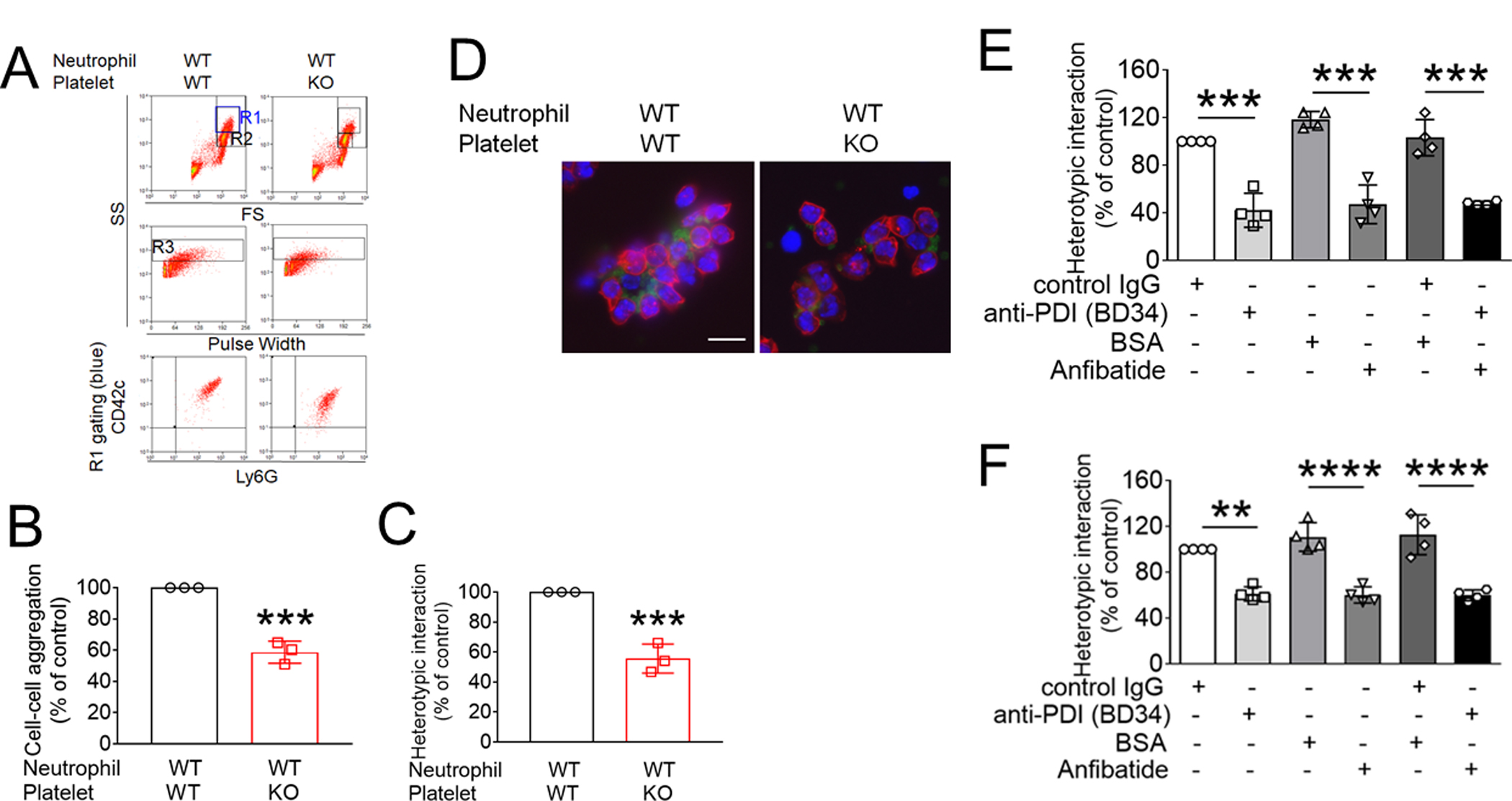
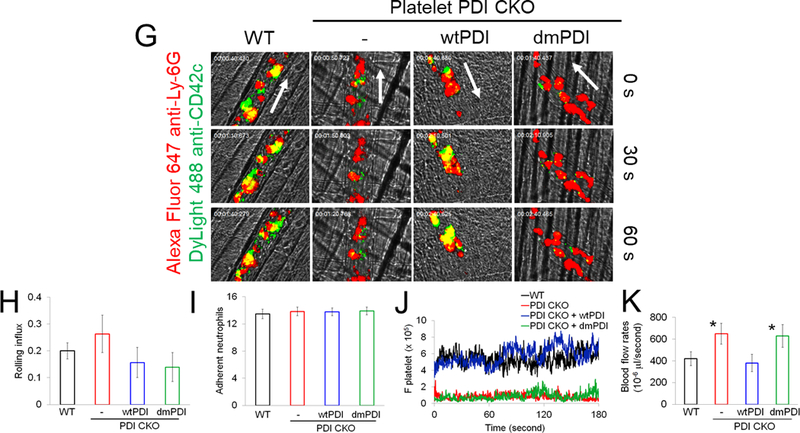
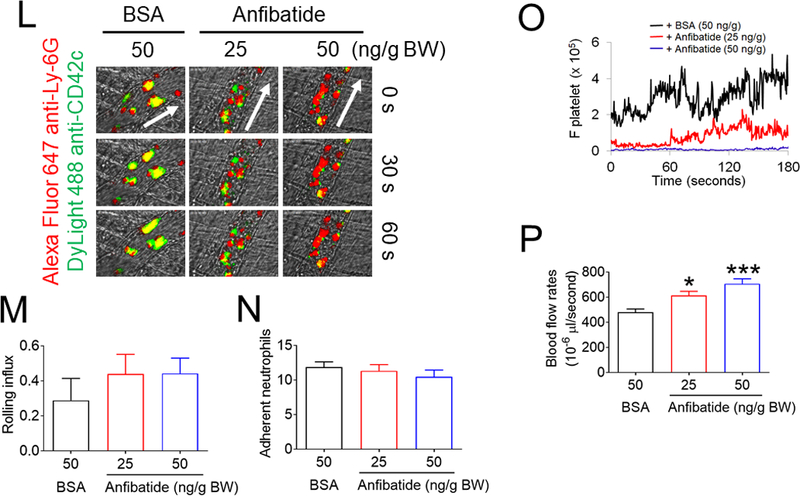
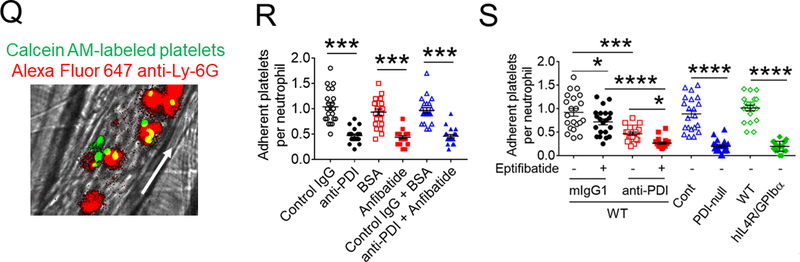
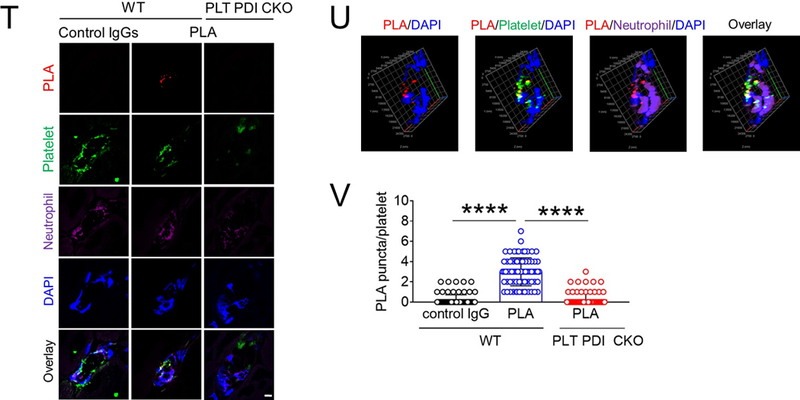
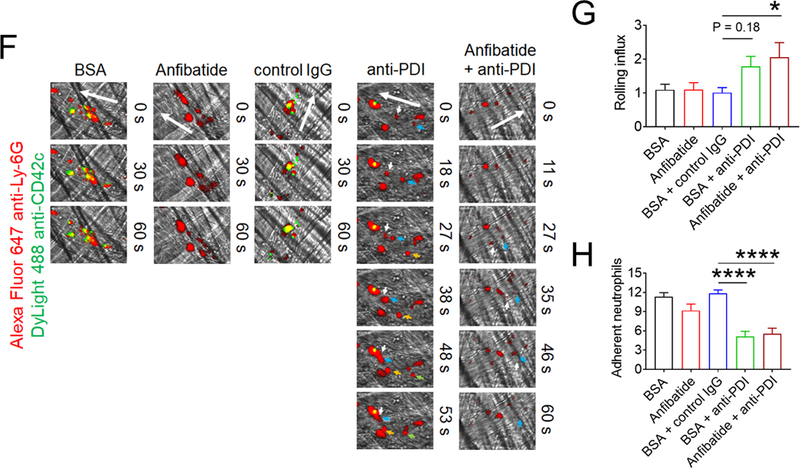
(A-F) In vitro platelet-neutrophil aggregation assay was performed using (A-E) mouse and (F) human cells under shear-mimicking conditions as described in Methods. (A) R1, platelet-neutrophil aggregates; R2, neutrophils; and R3, the number of cell aggregates in the R1 gate. Cell-cell aggregation was measured by (B) the number of cell-cell aggregates and (C, E, and F) the fluorescence intensities of anti-CD42c antibodies in the R1 gate (Heterotypic interaction). The number of cell-cell aggregates and the fluorescence intensity of an anti-CD42c antibody in (B-C) WT platelets and neutrophils or (E-F) control IgG-treated cells were shown as 100%. (D) Antibody-labeled neutrophils and platelets were mixed under a stirring condition, followed by cytospin and fluorescence microscopy. Neutrophils: red, platelets: green, and DAPI: blue. Bar = 10 μm. (G-P) Vascular inflammation was induced by intrascrotal injection of TNF-α into (G-K) WT littermate control and platelet-specific PDI CKO mice or (L-P) WT (C57BL/6) mice, followed by real-time intravital microscopy as described in Methods. (G-K) In some experiments, wtPDI or dmPDI (4 μg/g BW) were infused into PDI CKO mice. (L-P) BSA (50 ng/g BW) or Anfibatide (25–50 ng/g BW) was infused into WT mice 3 hours after intrascrotal injection of TNF-α. (G and L) Representative images at various time points. Arrows show the direction of blood flow. (H, I, M, and N) The numbers of rolling and adherent neutrophils. (J and O) The integrated median fluorescence intensities of anti-CD42c antibodies (F platelets) were normalized to the number of adherent neutrophils and plotted as a function of time. (K and P) Blood flow rates were calculated by centerline velocity of Alexa Fluor 488-conjugated microspheres. (Q-R) WT platelets were labeled with calcein AM and incubated without or with 10 µg/ml of control IgG or anti-PDI antibodies, 0.2 µg/ml of BSA or Anfibatide, or both inhibitors. The labeled platelets (2 × 108 in 100 μl per mouse) were infused into WT (C57BL/6) mice treated with intrascrotal injection of TNF-α. Endogenous neutrophils were visualized by infusion of Alexa Fluor 647-conjugated anti-Ly-6G antibodies. A representative image (Q) was obtained in mice infused with control IgG-treated platelets. (S) The same adoptive transfer experiment was performed with WT, hIL4R/GPIbα, littermate WT control (cont), or PDI-null platelets. The recipient TNF-α-challenged WT mice were treated without or with eptifibatide (10 μg/g BW), followed by infusion of platelets. The number of infused platelets that attach to adherent neutrophils was counted and normalized to the number of neutrophils. (T-V) The proximity ligation assay (PLA) was performed as described in Methods. Representative immunohistochemical stainings of the cremaster muscle sections and PLA signals were obtained from 6 different sections (20 different vessels) in 3 different mice per group. Platelets: green, PLA signal: red, and neutrophils: purple (pseudocolor). (U) Z-stacks of PLA signal were obtained from WT mice. Bar = 5 μm. Data represent the mean ± SD (B-F and V, n = 4) or SEM (H-S, n = 30–36 venules in 6 mice per group (H-P) or n = 20–22 venules in 4–5 mice per group (Q-S)). *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001 after student’s t-test (for B-C) or ANOVA and Tukey’s test (for E-V).
To study the physiological role of platelet PDI-regulated GPIbα in platelet-neutrophil interactions, intravital microscopy was performed in platelet-specific PDI CKO mice. Deletion of platelet PDI did not alter rolling and adhesion of neutrophils on TNF-α-activated endothelium but markedly reduced platelet adhesion to adherent neutrophils (Figure 4G-J, Videos 1-2). Infusion of wtPDI but not dmPDI into the CKO mice rapidly rescued the defect in platelet-neutrophil association (Videos 3-4). Moreover, blood flow rates were significantly enhanced by loss of platelet PDI, which was restored to the WT level by infusion of wtPDI (Figure 4K). These results suggest the contribution of platelet-derived PDI to platelet-neutrophil interactions and microvascular occlusion during inflammation.
We further tested the effect of Anfibatide on platelet-neutrophil interactions in TNF-α-challenged WT mice. In a control experiment, GPIbα was not shed from circulating platelets isolated from WT and platelet-specific PDI CKO mice after TNF-α treatment (Figure S8). When infused 3-hour after TNF-α injection, Anfibatide, 25–50 ng/g BW, had minimal effect on neutrophil rolling and adhesion on the vessel wall but dose-dependently reduced platelet adhesion to adherent neutrophils and even induced detachment of adherent platelets from neutrophils (Figure 4L-O). Compared to BSA control, Anfibatide significantly enhanced blood flow rates (Figure 4P). The drug (50 ng/g BW) had no effect on blood counts in mice (Table 2). These results suggest GPIbα-mediated platelet-neutrophil aggregation slows down blood flow during vascular inflammation.
Table 2.
The number of circulating blood cells in mice treated with Anfibatide.
| WBC (x103/μl) | NE (x103/μl) | LY (x103/μl) | MO (x103/μl) | RBC (x106/μl) | PLT (x103/μl) | MPV (fl) | |
|---|---|---|---|---|---|---|---|
| BSA | 9.1 ± 2.7 | 1.0 ± 0.3 | 7.0 ± 1.2 | 0.2 ± 0.1 | 7.4 ± 0.7 | 902 ± 197 | 4.3 ± 0.2 |
| Anfibatide | 9.3 ± 2.0 | 0.7 ± 0.3 | 7.1 ± 1.5 | 0.2 ± 0.1 | 7.1 ± 1.2 | 838 ± 160 | 4.4 ± 0.3 |
Male WT (C57BL/6, 6–7 weeks old) mice were treated by iv infusion of BSA or Anfibatide, 50 ng/g BW. One hour later, blood was drawn and counted using an automated hematology analyzer (Hemavet 950, Drew Scientific). Data represent the mean ± SD (n = 6 mice per group).
WBC, white blood cells; NE, neutrophils; LY, lymphocytes; MO, monocytes; RBC, red blood cells; PLT, platelets; and MPV, mean platelet volume.
To further examine the contribution of PDI-regulated GPIbα to platelet-neutrophil interactions, we employed adoptive transfer experiments. Fluorescently-labeled WT platelets were treated with or without 0.025 U/ml thrombin in the presence of a blocking anti-PDI antibody, Anfibatide, or both, followed by infusion into TNF-α-challenged WT mice. We found that most infused platelets directly attached to adherent neutrophils without forming thrombi (Figure 4Q). Treatment with either inhibitor inhibited 50–55% of platelet adhesion to neutrophils (Figure 4R), and co-treatment with both inhibitors had no further effect. Since platelet PDI is important for αIIbβ3-mediated platelet accumulation in arteriolar thrombosis,5 we attempted to eliminate the possibility that platelet PDI contributes to platelet-neutrophil interactions through αIIbβ3 integrin. Using the same adoptive transfer experiment, we found that when infused into TNF-α-challenged WT mice treated with 10 μg/g BW of eptifibatide at which αIIbβ3-mediated platelet-platelet interactions are abolished, adhesion of infused platelets to neutrophils was moderately but significantly reduced (Figure 4S), implicating the role of αIIbβ3 in platelet-neutrophil interactions as reported previously.38 When isolated platelets were treated with a blocking anti-PDI antibody, platelet-neutrophil interactions were further inhibited in eptifibatide-treated mice, compared to vehicle-treated mice (Figure 4S). Deletion of platelet PDI or the extracellular GPIbα domain markedly reduced platelet adhesion to neutrophils.
Finally, we examined whether extracellular PDI directly interacts with GPIbα under in vivo conditions using a proximity ligation assay in which oligonucleotides conjugated to antibodies form circular DNA strands when bound in close proximity.39 The circular DNA serves as a template for localized rolling-circle amplification, allowing us to visualize PDI-GPIbα interactions at single molecule resolution. The PDI-GPIbα complex was detected at the interface of adherent platelets and neutrophils in cremaster vessels of WT control mice (Figure 4T-V). In contrast, the PLA signal was minimal when the section of WT mice was stained with control IgGs or in the section of platelet-specific PDI CKO mice. Overall, these data suggest that extracellular PDI enhances the ligand-binding function of GPIbα by direct interaction, inducing platelet-neutrophil interactions under thromboinflammatory conditions.
Platelet PDI contributes to the pathogenesis of ischemia/reperfusion-induced thromboinflammation by regulating GPIbα function
Since previous studies have demonstrated a critical role for GPIbα in the pathogenesis of ischemia/reperfusion (I/R)-induced stroke,40 we further tested whether PDI-regulated GPIbα is crucial for stroke. Consistent with a previous report,41 when infused into mice challenged with middle cerebral artery occlusion (MCAO) and subsequent reperfusion (Figure 5A), Anfibatide, 5 to 25 ng/g BW, dose-dependently reduced the infarct volume and ameliorated neurological deficits as assessed by the Bederson score and the grip test, as compared to BSA control (Figure S9). WT and platelet-specific PDI CKO mice were treated with post-ischemic infusion of BSA or Anfibatide (5 ng/g BW). Compared to BSA-treated WT mice, BSA-treated PDI CKO mice exhibited a significant decrease in the infarct volume and improvement in neurological deficits (Figure 5B-E). Anfibatide treatment did not have additional effects in PDI CKO mice, supporting a role for platelet-derived PDI-regulated GPIbα in tissue damage during stroke.
Figure 5. Extracellular PDI and GPIbα contribute to tissue damage in I/R-induced stroke and cell-cell aggregation and vaso-occlusion in sickle cell disease.
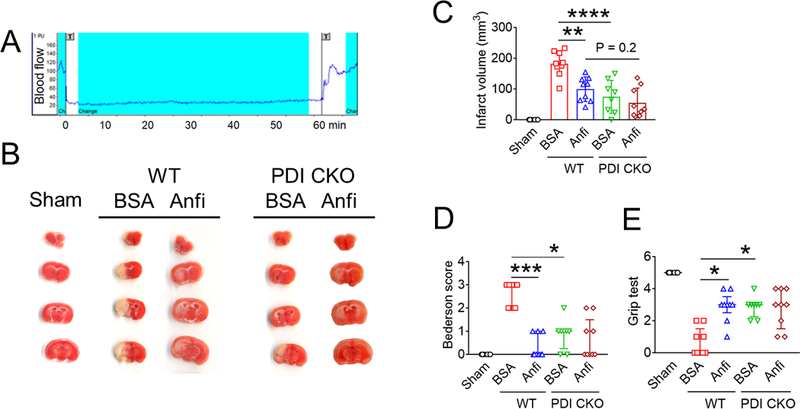
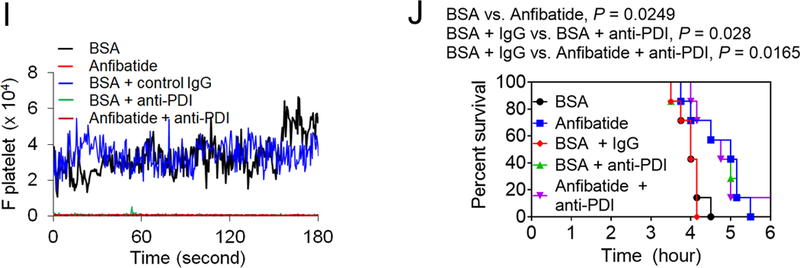
(A-E) Stroke was induced in WT and platelet-specific PDI CKO mice by transient MCAO and reperfusion as described in Methods. BSA or Anfibatide (5 ng/g BW) was intravenously infused into mice right after 1-hour MCAO. (A) MCAO and reperfusion were confirmed by a laser Doppler flowmeter. (B) Staining of coronal brain sections with 2,3,5-triphenyltetrazolium chloride. (C) The infarct volume was measured. (D-E) The Bederson score and grip test were used to analyze neurologic deficits in the surviving animals 24 hours after MCAO. Data represent the mean ± SD except for the Bederson score and the grip test score, which are shown as scatter plots including median with interquartile range (n = 8–9 mice per group). (F-J) SCD mice were treated with intraperitoneal injection of TNF-α. BSA or Anfibatide (25 ng/g BW), control IgG or blocking anti-PDI antibodies (1.5 μg/g BW), or both inhibitors were infused into mice 3 hours after TNF-α injection. Platelets and neutrophils were visualized as described in Figure 4. (F) Representative images. Large and small arrows show the direction of blood flow and rolling neutrophils, respectively. (G-H) The numbers of rolling and adherent neutrophils were counted. (I) The integrated median fluorescence intensities of anti-CD42c antibodies (F platelets) were normalized to the number of adherent neutrophils and plotted as a function of time. (J) Survival curves. Survival times were significantly prolonged in the groups treated with an anti-PDI antibody, Anfibatide, or both inhibitors, compared to each control. Data represent the mean ± SEM (n = 7–8 mice per group), *:P<0.05, **:P<0.01, ***:P<0.001, or ****:P<0.0001 after ANOVA and Tukey’s test (C, G and H), Kruskal-Wallis test with post hoc Dunn correction (D and E), or Mantel-Cox log-rank test (J).
SCD, an inherited blood disorder, causes red blood cell hemolysis, intravascular cell activation, and chronic inflammation.42 The hallmark of SCD is recurrent vaso-occlusion that results from cell-cell aggregation and induces severe pain crisis in the patients. Since platelet-neutrophil aggregation contributes to vaso-occlusion in SCD mice,23, 25 the effect of Anfibatide, anti-PDI antibodies, or both were tested in SCD mice challenged with TNF-α that induces severe inflammation and vaso-occlusion.25 In control experiments, the surface level of PDI was elevated on platelets isolated from SCD mice following agonist stimulation (Figure S10A-B). Further, platelet GPIbα in SCD mice was expressed and shed upon stimulation with 0.05 U/ml thrombin (Figure S10A and S10C) as shown in WT or PDI-null platelets (Figure S1A).
When platelets of SCD mice were treated with an anti-PDI antibody or Anfibatide, platelet-neutrophil aggregation was reduced under shear-mimicking conditions and treatment of platelets with both inhibitors did not potentiate the inhibitory effects (Figure S11). Compared to BSA, Anfibatide did not affect neutrophil-endothelial cell interactions but abrogated platelet adhesion to neutrophils on the vessel wall of TNF-α-challenged SCD mice (Figure 5F-I). Inhibition of PDI activity with the anti-PDI antibody significantly reduced neutrophil adhesion to inflamed ECs and abolished platelet-neutrophil interactions, with a moderate increase in the rolling influx probably due to the reduced neutrophil adhesion.34 Compared to infusion of each inhibitor alone, co-infusion of Anfibatide and an anti-PDI antibody did not enhance the inhibitory effects on neutrophil adhesion and platelet-neutrophil interactions (Figure 5F-I). The TNF-α challenge with surgical trauma for intravital microscopy causes death in most SCD mice within several hours as a result of acute vaso-occlusion.23, 25 Compared to each control, treatment with Anfibatide, an anti-PDI antibody, or both significantly prolonged survival times in TNF-α-challenged SCD mice (Figure 5J, P = 0.0249 between BSA and Anfibatide, P = 0.028 between BSA + IgG and BSA + anti-PDI, and P = 0.0165 between BSA + IgG and Anfibatide + anti-PDI). Fifty percent of mice died at 4.0, 5.0, 4.2, 5.0, and 4.8 hours after TNF-α injection in SCD mice treated with BSA, Anfibatide, BSA + control IgG, BSA + an anti-PDI antibody, and both inhibitors, respectively. Overall, these results indicate that platelet GPIbα and extracellular PDI play a critical role in inducing vaso-occlusion in SCD and imply that specific inhibitors of GPIbα or PDI could potentially provide significant benefit for acute vaso-occlusive events in SCD patients.
Discussion
Growing evidence shows that the initiation and progression of vascular disease are controlled by reduction or oxidation of allosteric disulfide bonds in plasma proteins and cell surface receptors.17 In the present study, we demonstrate for the first time that platelet PDI directly binds to the extracellular domain of GPIbα and reduces the allosteric disulfide bonds, enhancing the ligand-binding function of GPIbα and inducing platelet-neutrophil aggregation under thromboinflammatory disease. Importantly, our studies using a specific anti-PDI antibody and a clinical-stage GPIbα inhibitor clearly show that the selective inhibition of these molecules could be an effective therapeutic approach to attenuate the pathogenesis of thromboinflammatory disease. Since GPIbα has been considered constitutively active, our findings represent a paradigm shift since GPIbα function is tightly regulated by a conformational change induced by PDI.
PDI modulates the function of platelet surface receptors such as integrins.43, 44 We reported that the initial binding of PDI to β3 integrin is independent of the CysGlyHisCys active sites but the oxidoreductase activity of PDI is necessary for the subsequent regulation of integrin function.5 Using multiple orthogonal approaches including SPR, flow cytometry, ELISA, and a computer-aided protein-protein docking model, we have demonstrated that platelet-derived PDI directly binds to the extracellular domain of GPIbα mainly through the interaction between the b’-domain of PDI and the β-switch region of GPIbα. In addition, the PDI a-domain is likely to contribute to binding and modifying the Cys4-Cys17 disulfide bond in GPIbα. Our results suggest that the b’-domain of PDI is the essential core for the initial binding to GPIbα in the extracellular region and the CysGlyHisCys active site probably in the a-domain is indispensable for the subsequent binding and regulatory function.
It is reported that PDI is present in circulating platelet-derived microparticles as a catalytically active form in healthy people and the level of PDI-containing microparticles is elevated in patients with diabetes.2 We have found that activated αMβ2 or vWF binds to unstimulated platelets and the binding is significantly impaired by loss or inhibition of platelet PDI (Figure 1F-K). Since the small amount of PDI is detected on unstimulated platelets (Figure S5 and Figure 2C), these results suggest that PDI on the surface of resting platelets could play a role in the initial binding of vWF and αMβ2 to GPIbα. Ligand binding-induced GPIbα signaling is likely to further release PDI from platelets, enhancing PDI-regulated GPIbα function. In addition to this finding, our results show that platelet PDI does not regulate P-selectin exposure (Figure S1) and inhibition of P-selectin has no effect on vWF binding to the platelet surface (Figure S4). Thus, we rule out the possibility of vWF binding to P-selectin in our assays. Furthermore, our findings that inhibition or deletion of PDI impairs binding of vWF and αMβ2 to platelet GPIbα (Figure 1H-P) and the interaction between PDI and GPIbα (Figure 2C-F) under static conditions suggest that shear is not required for the regulation of GPIbα function by PDI. It should be noted that PDI binding to GPIbα is significantly enhanced upon thrombin stimulation (Figure 2C-F). Since reactive oxygen species produced from platelet NADPH oxidase 2 regulate GPIbα function,15 the enhanced PDI-GPIbα binding may result from the altered redox environment following platelet activation. Further studies are required to determine whether different redox environments influence the binding of PDI to GPIbα and other binding partners.
Oxidoreductases modify allosteric disulfide bonds in target proteins.17 The Cys4-Cys17 and Cys209-Cys248 disulfide bonds in GPIbα have –RHStaple and –LHHook allosteric configurations, respectively. The conformational constraints imposed on –RHStaple bonds by topological features stress the bond.17 The stress is a result of direct stretching of the disulfide bond and neighboring α angles and likely underpins the susceptibility of this bond type to cleavage. In contrast, the –LHHook configuration is no more stressed than the other configurations, and it remains to be determined the reason for its allosteric association. Notably, our studies using CHO cells expressing GPIb-IX with WT or mutant GPIbα demonstrate that cleavage of either Cys4-Cys17 or Cys209-Cys248 disulfide bond enhances vWF binding and both mutants undergo a conformational change as evaluated by different monoclonal antibodies. Therefore, binding of PDI to GPIbα and subsequent reduction in the disulfide bonds induce a conformational change in GPIbα, increasing the ligand-binding activity. Nevertheless, we cannot eliminate the possibility that these two GPIbα disulfides may also be targeted by other thiol isomerases. Furthermore, it is noticeable that although Cys209Ser mutation in the GPIbα gene has been associated with Bernard-Soulier syndrome45 and Cys209Ser mutant GPIbα was not transiently expressed in CHO cells already expressing GPIbβ and GPIX,46 under our conditions the mutant protein along with GPIbβ and GPIX was expressed on the surface of CHO cells as detectable by anti-GPIbα antibodies or vWF binding. The reason for these apparent differences between the studies is not known.
Many efforts have been made to develop specific inhibitors of PDI and GPIbα for the treatment of thrombotic diseases. Although several PDI inhibitors have been reported,47 there are challenges including off-target effects, selectivity, and impairment of the hemostatic function.5–7 For example, rutin, an anti-oxidant PDI inhibitor,48 is being tested in clinical trials as an anti-thrombotic agent. We have found that treatment with 50 μM rutin inhibits GPIbα-mediated agglutination of PDI-null platelets (Figure S12), indicating the off-target effect. GPIbα has been considered an attractive target for treating thromboinflammatory diseases. However, only a limited number of GPIbα inhibitors are currently being developed.28 Anfibatide, a novel snake venom-derived GPIbα antagonist, is in Phase II clinical trial to evaluate the efficacy in patients with myocardial infarction.28 It specifically binds to GPIbα49 and blocks platelet adhesion, agglutination, and aggregation induced by vWF-GPIbα binding.41, 50, 51 Interestingly, Anfibatide treatment further impaired platelet thrombus formation in vWF KO mice,50 suggesting that this drug inhibits other thrombotic pathways induced by binding of other ligands to GPIbα. Our studies show that Anfibatide blocks binding of both vWF and activated αMβ2 to GPIbα and abolishes platelet-neutrophil interactions (Figures 1 and 4). Furthermore, our findings that extracellular PDI interacts with GPIbα at the interface of adherent platelets and neutrophils on the vessel wall (Figure 4T-V), post-ischemic infusion of Anfibatide into mice effectively mitigates brain tissue damage during reperfusion injury, and no additional effect of this drug is observed in platelet-specific PDI CKO mice (Figure 5) support the role of PDI-regulated GPIbα in the pathogenesis of thromboinflammatory diseases.
Platelet-neutrophil aggregation contributes to vaso-occlusion and tissue damage in SCD.23, 24 A recent study shows that CCP-224, a specific GPIbα inhibitor, attenuates platelet-neutrophil interactions in SCD patient blood.52 Our in vivo studies using SCD mice demonstrate that Anfibatide treatment abrogates platelet-neutrophil interactions without affecting neutrophil recruitment to the inflamed vessel wall and prolongs survival times in TNF-α-challenged SCD mice (Figure 5). These results indicate that platelet-neutrophil aggregation is a crucial inducer for vaso-occlusion in SCD and Anfibatide might be a novel therapeutic agent to treat acute vaso-occlusion in the patients. Notably, there is no potentiated beneficial effect of Anfibatide and an anti-PDI antibody in the survival of SCD mice. Using myeloid-specific PDI CKO mice, we reported that neutrophil surface-bound PDI promotes the ligand binding activity of αMβ2 integrin and neutrophil recruitment to sites of inflammation.34 The fact that platelet GPIbα is the counter-receptor of neutrophil αMβ2 for the cell-cell interaction may account for no additional inhibition by co-treatment of SCD mice with inhibitors of GPIbα and PDI. However, there are some limitations of our in vivo studies. Since inhibition of extracellular PDI decreases both platelet-neutrophil and neutrophil-endothelial cell interactions, extracellular PDI possibly regulates the function of other surface molecules that may participate in cell-cell interactions. Also, we could not unequivocally prove that PDI-regulated GPIbα signaling mediates platelet-neutrophil interactions under in vivo conditions. The development of inhibitors specifically blocking PDI-GPIbα binding will be of great value to further confirm this intriguing concept.
Overall, our study demonstrates the molecular mechanism controlling the structure and function of GPIbα via cleavage of two allosteric disulfide bonds by PDI, enhancing the ligand-binding activity. Our results provide compelling evidence that platelet PDI is required for not only thrombosis5 but also other diseases, including stroke and SCD, in which platelet GPIbα contributes to disease initiation and propagation.
Supplementary Material
Clinical Perspective.
What is new?
Prior studies have linked PDI to thrombotic disease, but the molecular basis for extracellular PDI in regulating platelet function has remained speculative.
Platelet PDI directly binds to the extracellular domain of GPIbα that has been considered constitutively active for ligand-binding, regulating the structure and function of GPIbα by reducing two allosteric Cys4-Cys17 and Cys209-Cys248 disulfide bonds.
PDI-GPIbα signaling plays a critical role in platelet-neutrophil aggregation and vascular occlusion under thromboinflammatory conditions.
What are the clinical implications?
This work identifies platelet PDI as a switch that enhances the ligand-binding activity of GPIbα under thromboinflammatory conditions, such as vasculitis, stroke, and sickle cell disease.
Selective inhibition of PDI and GPIbα could be an effective therapeutic approach to attenuate the pathogenesis of thromboinflammatory disease.
Acknowledgments
J.L., K.K., S.J., J.C., P.A.P., and P.J.H. designed and performed research, collected and analyzed data, and wrote a draft of the manuscript; B.X.: performed research; X.D., X.L., R.K.A., and X.D. provided important reagents; J.C. initiated, designed, and performed research, collected and analyzed data, and wrote the manuscript. The authors thank Dr. Deane Mosher for providing critical suggestions on PDI-GPIbα binding assays, Drs. Jerry Ware and Robert Flaumenhaft for providing hIL4R/GPIbα mice and constructs of PDI domains, respectively, and Drs. Seockwon Youn and Alan Tseng for their technical assistance.
Sources of Funding
This work was supported by grants from the National Institutes of Health, American Society of Hematology Bridge Award, and American Heart Association Grant-in-Aid. Drs. Jing Li and Kyungho Kim are recipients of American Heart Association Career Development Award. Dr. Joyce Chiu is supported by the Helen and Robert Ellis Postdoctoral Fellowship and the Professor Tony Basten Postdoctoral Fellowship from the Sydney Medical School Foundation.
Footnotes
Disclosures
X.D and X.L are employees of Lee’s Pharmaceutical Holdings Limited. Other authors declare no competing financial interests.
References
- 1.Cho J Protein disulfide isomerase in thrombosis and vascular inflammation. J Thromb Haemost 2013;11:2084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raturi A, Miersch S, Hudson JW and Mutus B. Platelet microparticle-associated protein disulfide isomerase promotes platelet aggregation and inactivates insulin. Biochim Biophys Acta 2008;1778:2790–6. [DOI] [PubMed] [Google Scholar]
- 3.Crescente M, Pluthero FG, Li L, Lo RW, Walsh TG, Schenk MP, Holbrook LM, Louriero S, Ali MS, Vaiyapuri S, Falet H, Jones IM, Poole AW, Kahr WH and Gibbins JM. Intracellular Trafficking, Localization, and Mobilization of Platelet-Borne Thiol Isomerases. Arterioscler Thromb Vasc Biol 2016;36:1164–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Severino A, Campioni M, Straino S, Salloum FN, Schmidt N, Herbrand U, Frede S, Toietta G, Di Rocco G, Bussani R, Silvestri F, Piro M, Liuzzo G, Biasucci LM, Mellone P, Feroce F, Capogrossi M, Baldi F, Fandrey J, Ehrmann M, Crea F, Abbate A and Baldi A. Identification of protein disulfide isomerase as a cardiomyocyte survival factor in ischemic cardiomyopathy. J Am Coll Cardiol 2007;50:1029–37. [DOI] [PubMed] [Google Scholar]
- 5.Kim K, Hahm E, Li J, Holbrook LM, Sasikumar P, Stanley RG, Ushio-Fukai M, Gibbins JM and Cho J. Platelet protein disulfide isomerase is required for thrombus formation but not essential for hemostasis in mice. Blood 2013;122:1052–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou J, Wu Y, Wang L, Rauova L, Hayes VM, Poncz M and Essex DW. The C-terminal CGHC motif of protein disulfide isomerase supports thrombosis. J Clin Invest 2015;125:4391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho J, Furie BC, Coughlin SR and Furie B. A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J Clin Invest 2008;118:1123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Delaney MK, O’Brien KA and Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 2010;30:2341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Kim K, Barazia A, Tseng A and Cho J. Platelet-neutrophil interactions under thromboinflammatory conditions. Cell Mol Life Sci 2015;72:2627–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ and Gros P. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain. Science 2002;297:1176–9. [DOI] [PubMed] [Google Scholar]
- 11.Lechtenberg BC, Freund SM and Huntington JA. GpIbalpha interacts exclusively with exosite II of thrombin. J Mol Biol 2014;426:881–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, McIntire LV, Berndt MC and Lopez JA. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med 1999;190:803–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC and Lopez JA. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med 2000;192:193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, D AS, Bledzka K, Ustinov V, Zhu L, Qin J, Munday AD, Lopez J, Plow E and Simon DI. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun 2017;8:15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim K, Li J, Tseng A, Andrews RK and Cho J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood 2015;126:1952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koltsova EK, Sundd P, Zarpellon A, Ouyang H, Mikulski Z, Zampolli A, Ruggeri ZM and Ley K. Genetic deletion of platelet glycoprotein Ib alpha but not its extracellular domain protects from atherosclerosis. Thromb Haemost 2014;112:1252–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butera D, Cook KM, Chiu J, Wong JW and Hogg PJ. Control of blood proteins by functional disulfide bonds. Blood 2014;123:2000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogg PJ. Targeting allosteric disulphide bonds in cancer. Nat Rev Cancer 2013;13:425–31. [DOI] [PubMed] [Google Scholar]
- 19.Chen VM and Hogg PJ. Allosteric disulfide bonds in thrombosis and thrombolysis. J Thromb Haemost 2006;4:2533–41. [DOI] [PubMed] [Google Scholar]
- 20.Burgess JK, Hotchkiss KA, Suter C, Dudman NP, Szollosi J, Chesterman CN, Chong BH and Hogg PJ. Physical proximity and functional association of glycoprotein 1balpha and protein-disulfide isomerase on the platelet plasma membrane. J Biol Chem 2000;275:9758–66. [DOI] [PubMed] [Google Scholar]
- 21.Dong J, Schade AJ, Romo GM, Andrews RK, Gao S, McIntire LV and Lopez JA. Novel gain-of-function mutations of platelet glycoprotein IBalpha by valine mutagenesis in the Cys209-Cys248 disulfide loop. Functional analysis under statis and dynamic conditions. J Biol Chem 2000;275:27663–70. [DOI] [PubMed] [Google Scholar]
- 22.Kanaji T, Russell S and Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood 2002;100:2102–7. [DOI] [PubMed] [Google Scholar]
- 23.Kim K, Li J, Barazia A, Tseng A, Youn SW, Abbadessa G, Yu Y, Schwartz B, Andrews RK, Gordeuk VR and Cho J. ARQ 092, an orally-available, selective AKT inhibitor, attenuates neutrophil-platelet interactions in sickle cell disease. Haematologica 2017;102:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li J, Kim K, Hahm E, Molokie R, Hay N, Gordeuk VR, Du X and Cho J. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. J Clin Invest 2014;124:1483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barazia A, Li J, Kim K, Shabrani N and Cho J. Hydroxyurea with AKT2 inhibition decreases vaso-occlusive events in sickle cell disease mice. Blood 2015;126:2511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong JW and Hogg PJ. Analysis of disulfide bonds in protein structures. J Thromb Haemost 2010;8:2345. [DOI] [PubMed] [Google Scholar]
- 27.Franco AT, Corken A and Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood 2015;126:582–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metharom P, Berndt MC, Baker RI and Andrews RK. Current state and novel approaches of antiplatelet therapy. Arterioscler Thromb Vasc Biol 2015;35:1327–38. [DOI] [PubMed] [Google Scholar]
- 29.Matsushita T, Meyer D and Sadler JE. Localization of von willebrand factor-binding sites for platelet glycoprotein Ib and botrocetin by charged-to-alanine scanning mutagenesis. The Journal of biological chemistry 2000;275:11044–9. [DOI] [PubMed] [Google Scholar]
- 30.Weisel JW, Nagaswami C, Vilaire G and Bennett JS. Examination of the platelet membrane glycoprotein IIb-IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. The Journal of biological chemistry 1992;267:16637–43. [PubMed] [Google Scholar]
- 31.Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, Lopez JA and Dong JF. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood 2004;103:2150–6. [DOI] [PubMed] [Google Scholar]
- 32.McEwan PA, Andrews RK and Emsley J. Glycoprotein Ibalpha inhibitor complex structure reveals a combined steric and allosteric mechanism of von Willebrand factor antagonism. Blood 2009;114:4883–5. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Li W, Ren J, Fang J, Ke H, Gong W, Feng W and Wang CC. Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid Redox Signal 2013;19:36–45. [DOI] [PubMed] [Google Scholar]
- 34.Hahm E, Li J, Kim K, Huh S, Rogelj S and Cho J. Extracellular protein disulfide isomerase regulates ligand-binding activity of alphaMbeta2 integrin and neutrophil recruitment during vascular inflammation. Blood 2013;121:3789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Estevez B, Kim K, Delaney MK, Stojanovic-Terpo A, Shen B, Ruan C, Cho J, Ruggeri ZM and Du X. Signaling-mediated cooperativity between glycoprotein Ib-IX and protease-activated receptors in thrombin-induced platelet activation. Blood 2016;127:626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuijpers RW, Ouwehand WH, Bleeker PM, Christie D and von dem Borne AE. Localization of the platelet-specific HPA-2 (Ko) alloantigens on the N-terminal globular fragment of platelet glycoprotein Ib alpha. Blood 1992;79:283–8. [PubMed] [Google Scholar]
- 37.Shen Y, Romo GM, Dong JF, Schade A, McIntire LV, Kenny D, Whisstock JC, Berndt MC, Lopez JA and Andrews RK. Requirement of leucine-rich repeats of glycoprotein (GP) Ibalpha for shear-dependent and static binding of von Willebrand factor to the platelet membrane GP Ib-IX-V complex. Blood 2000;95:903–10. [PubMed] [Google Scholar]
- 38.Weber C and Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest 1997;100:2085–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG and Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 2006;3:995–1000. [DOI] [PubMed] [Google Scholar]
- 40.De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F and Kleinschnitz C. Thromboinflammation in Stroke Brain Damage. Stroke 2016;47:1165–72. [DOI] [PubMed] [Google Scholar]
- 41.Li TT, Fan ML, Hou SX, Li XY, Barry DM, Jin H, Luo SY, Kong F, Lau LF, Dai XR, Zhang GH and Zhou LL. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br J Pharmacol 2015;172:3904–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang D, Xu C, Manwani D and Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016;127:801–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho J, Kennedy DR, Lin L, Huang M, Merrill-Skoloff G, Furie BC and Furie B. Protein disulfide isomerase capture during thrombus formation in vivo depends on the presence of beta3 integrins. Blood 2012;120:647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lahav J, Wijnen EM, Hess O, Hamaia SW, Griffiths D, Makris M, Knight CG, Essex DW and Farndale RW. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin alpha2beta1. Blood 2003;102:2085–92. [DOI] [PubMed] [Google Scholar]
- 45.Simsek S, Noris P, Lozano M, Pico M, von dem Borne AE, Ribera A and Gallardo D. Cys209 Ser mutation in the platelet membrane glycoprotein Ib alpha gene is associated with Bernard-Soulier syndrome. Br J Haematol 1994;88:839–44. [DOI] [PubMed] [Google Scholar]
- 46.Gonzalez-Manchon C, Larrucea S, Pastor AL, Butta N, Arias-Salgado EG, Ayuso MS and Parrilla R. Compound heterozygosity of the GPIbalpha gene associated with Bernard-Soulier syndrome. Thromb Haemost 2001;86:1385–91. [PubMed] [Google Scholar]
- 47.Flaumenhaft R, Furie B and Zwicker JI. Therapeutic implications of protein disulfide isomerase inhibition in thrombotic disease. Arterioscler Thromb Vasc Biol 2015;35:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC and Flaumenhaft R. Protein disulfide isomerase inhibitors constitute a new class of antithrombotic agents. J Clin Invest 2012;122:2104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao Y, Ge H, Chen H, Li H, Liu Y, Chen L, Li X, Liu J, Niu L and Teng M. Crystal structure of agkisacucetin, a Gpib-binding snake C-type lectin that inhibits platelet adhesion and aggregation. Proteins 2012;80:1707–11. [DOI] [PubMed] [Google Scholar]
- 50.Lei X, Reheman A, Hou Y, Zhou H, Wang Y, Marshall AH, Liang C, Dai X, Li BX, Vanhoorelbeke K and Ni H. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb Haemost 2014;111:279–89. [DOI] [PubMed] [Google Scholar]
- 51.Zheng L, Mao Y, Abdelgawwad MS, Kocher NK, Li M, Dai X, Li B and Zheng XL. Therapeutic efficacy of the platelet glycoprotein Ib antagonist anfibatide in murine models of thrombotic thrombocytopenic purpura. Blood Adv 2016;1:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jimenez MA, Novelli E, Shaw GD and Sundd P. Glycoprotein Ibalpha inhibitor (CCP-224) prevents neutrophil-platelet aggregation in Sickle Cell Disease. Blood Adv 2017;1:1712–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.