

Summary
B cell responses are dynamic processes that depend on multiple types of interactions. Rare antigen-specific B cells must encounter antigen and specialized systems are needed – unique to each lymphoid tissue type – to ensure this happens efficiently. Lymphoid tissue barrier cells act to ensure that pathogens, while being permitted entry for B cell recognition, are blocked from replication or dissemination. T follicular helper (Tfh) cells often need to be primed by dendritic cells (DCs) before supporting B cell responses. For most responses, antigen-specific helper T cells and B cells need to interact, first to initiate clonal expansion and the plasmablast response, and later to support the germinal center (GC) response. Newly formed plasma cells need to travel to supportive niches. GC B cells must become confined to the follicle center, organize into dark and light zones and interact with Tfh cells. Memory B cells need to be positioned for rapid responses following reinfection. Each of these events requires the actions of multiple G-protein coupled receptors (GPCRs) and their ligands, including chemokines and lipid mediators. This review will focus on the guidance cue code underlying B cell immunity, with an emphasis on findings from our laboratory and on newer advances in related areas. We will discuss our recent identification of geranylgeranyl-glutathione as a ligand for P2RY8. Our goal is to provide the reader with a focused knowledge about the GPCRs guiding B cell responses and how they might be therapeutic targets, while also providing examples of how multiple types of GPCRs can cooperate or act iteratively to control cell behavior.
Keywords: chemoattractant, migration-inhibition, CXCR5, CXCR4, EBI2, S1PR2, P2RY8
Introduction
Humoral immune responses in mice and man are typically initiated and supported within secondary lymphoid organs – spleen, lymph nodes (LNs) or mucosal lymphoid tissues such as Peyer’s patches (PPs). Lymphocytes continually recirculate from blood to lymphoid tissue and back into blood, spending several hours to a day in each lymphoid tissue they enter, surveying for antigen. The spleen, the largest secondary lymphoid organ, has an open circulation system and filters antigens from the blood. LNs, of which there are several hundred in humans, filter lymph fluid while mucosal lymphoid tissues such as intestinal PPs sample antigens from the mucosa. The lymphoid regions of spleen, known as the white pulp cords, share a similar organization with LNs and PPs that includes B cell rich follicles surrounding the T cell zone. The rodent spleen also has a specialized compartment known as the marginal zone (MZ) that sits between the white pulp and the blood-rich red pulp, where some of the blood entering the spleen is released before percolating through the red pulp to return to circulation. In spleen and skin-draining LNs of mice in barrier facilities, the follicles contain quiescent B cells that are undergoing a random walk-like migration, moving at ~6μm/min, surveying for antigen. Following immunization with a complex antigen, small numbers of antigen-specific B cells become activated and, after receiving T cell help, some differentiate into plasma cells while others move to the follicle interior to form a germinal center (GC). GCs are the sites of antibody somatic hypermutation and affinity maturation and they give rise to diversified and higher affinity plasma cells and memory B cells specific for the inducing antigen. In PPs the follicles are large and typically contain GCs due to their chronic exposure to mucosal antigens. The gut-draining mesenteric LNs also harbor chronic GC responses. Plasma cells have specialized trafficking properties that cause them to lodge in niches where they can receive trophic factors and secrete antibody into circulation.
Underlying the lymphocytes are the stromal cells that guide and support the movements and cell-cell interactions needed for humoral immunity (1, 2). This includes specialized vascular endothelial cells in the high endothelial venules (HEVs) of LNs and PPs that support lymphocyte entry, and lymphatic endothelial cells (LECs) that bring lymph to LNs and support the exit of cells from LNs and PPs. The parenchyma contains a network of fibroblastic reticular cells (FRCs) and these take on specialized features in each lymphoid compartment. The T zone reticular cells (TRCs) produce CCL21 and CCL19, the chemokine ligands for CCR7. Single cell RNA sequencing (scRNAseq) analysis of LN stroma has shown heterogeneity in the TRC population (3). FRCs associated with the follicle include the centrally located follicular dendritic cells (FDCs) that are specialized in capturing opsonized antigens for display to B cells, and a population of marginal reticular cells (MRCs) that are situated in LNs between the follicle and the lymph-filled subcapsular sinus (SCS); both FDCs and MRCs produce the CXCR5 ligand CXCL13. The medullary region of LNs, subepithelial dome (SED) of PPs, and MZ and red pulp of spleen contain additional fibroblastic stromal cell types. Each lymphoid tissue contains multiple types of myeloid cells including many dendritic cells (DCs) in the T zone and interfollicular regions, and macrophages in the medulla and SCS region of LNs, red pulp and MZ of spleen, and SED of PPs. As well as FDCs, DCs and macrophages contribute to the antigen capture and display properties of lymphoid tissues.
Chemokines form a large family of secreted chemoattractant proteins that engage 7-transmembrane G-protein-coupled receptors (GPCRs) (4, 5). All chemokine receptors couple to Gαi-containing heterotrimeric G-proteins and transmit signals via Rac that allow cell polarization and directed movement up the ligand gradient. Some chemokine receptors also couple to other G-proteins or downstream signaling molecules that influence additional properties of the cell such as gene expression. As well as chemokines, a large number of lipid mediators can engage specific GPCRs to trigger Gαi-signaling and promote chemotaxis. In the context of B cell responses, important lipid chemoattractants include sphingosine-1-phosphate (S1P) and oxysterols. Additionally, chemotaxis can be regulated by molecules that tell cells when to stop moving or to turn. Molecules in this class include ligands for receptors that couple to Gα13-containing heterotrimeric G-proteins that activate Rho and antagonize Rac-mediated migration.
This review will focus on the GPCRs and their ligands at the core of the guidance cue code needed for B cell immunity. Many additional classes of molecules are important in supporting cell movements during immune responses, including integrins and their ligands, selectins, intercellular adhesion molecules of the Ig superfamily, and neuronal-type migration regulatory molecules such as plexins and ephrins – for updates about these molecules the reader is referred to other recent reviews and studies (6–10).
Naïve B cell trafficking and antigen encounter
CXCL13 as a follicle-defining factor.
After entering lymphoid tissues from the blood, naïve B cells travel rapidly into lymphoid follicles. This process depends on the chemokine CXCL13 and its receptor CXCR5 (11). CXCL13 has long been known to be expressed by follicular stromal cells (Fig. 1) and scRNAseq analysis has shown that FDCs and MRCs express the highest amounts of this chemokine (3). CXCL13 has multiple positive charges and binds strongly to heparin in vitro, and tissue staining suggests the protein is concentrated near the cells that produce it (1), but the molecules involved in binding and displaying CXCL13 in vivo are not defined. CXCR5 is upregulated along with other maturation markers such as IgD and CD21 as B cells mature in the BM (Immgen.org). As well as attracting B cells into follicles, CXCL13 signaling in B cells promotes their expression of LTα1β2, a cytokine that helps maintain follicular stromal cells in a mature CXCL13-expressing state, thus forming a positive feedback loop (12). CXCL13 expression is not entirely LTα1β2 dependent and expression can be promoted in some contexts by retinoic acid (13). CXCR5 function is not desensitized by CXCL13, allowing this receptor to promote motility of B cells in an ongoing manner (14). Evidence that this occurs in vivo is difficult to collect because of the critical role of the receptor for B cell access to follicles. However, two-photon laser scanning microscopy (TPLSM) of CXCL13-deficient LNs showed a reduced motility of B cells in GCs (15). In accord with CXCR5 signaling via Gαi-proteins to promote motility, B cells deficient in Gαi show reduced motility within LNs (16). The extent to which CXCL13 is soluble versus substrate-bound while being engaged by B cell CXCR5, and thus the extent to which the B cell response is haptotactic versus chemotactic (17, 18) needs investigation. A single molecule tracking study of fluorescently labeled CXCL13 in LN tissue sections showed its behavior in the follicle was multimodal, with an immobile and a mobile fraction (19). A recent solution structure of CXCL13 revealed an extended 19 amino acid C-terminus that included a heparan sulfate (HS)-binding site. Testing of 25 forms of HS tetrasaccharide identified only one that bound with high consistency, suggesting possible selectivity at the HS binding level that might contribute to chemokine compartmentalization. The study also showed that CXCL13 could engage CXCR5 while in the HS-bound form (20). Thus, CXCL13 may support both chemotactic and haptotactic movement of follicular B cells.
Figure 1. In situ hybridization for CXCL13 defines B cell follicles in the spleen.
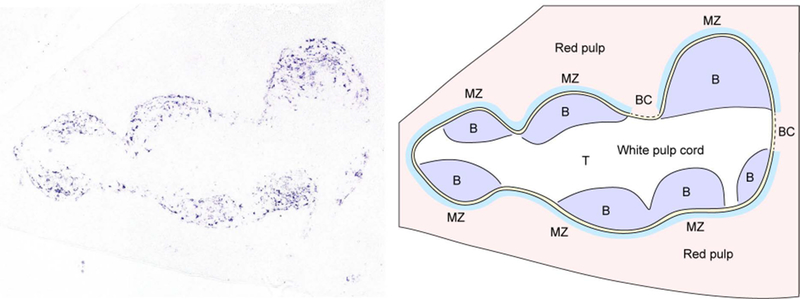
CXCL13 mRNA expression (left panel, purple signal) was identified in spleen sections using in situ hybridization. Shown is a single white pulp cord. CXCL13 is abundantly expressed by follicular stromal cells. The right panel shows a diagram corresponding to the splenic architecture of the section shown on the left. B cell follicles (B, purple) can be distinguished from the T cell zone (T, white) at the center of the white pulp cord. The marginal zone (MZ, light blue) and marginal sinus (yellow) surrounds the outer areas of the white pulp cord. Bridging channels (BC) are areas in which the T cell zone directly contacts the red pulp.
An oxysterol refines follicular organization.
While CXCL13 is the dominant organizer of B cell follicles, it does not work alone. This was evident in the initial characterization of CXCR5 and CXCL13-deficient mice, where B cells could still be observed segregating from other cell types (12, 21). A second ligand receptor system that organizes lymphoid follicles is EBI2 (GPR183) and its oxysterol ligand, 7α,25-dihydroxycholesterol (7α,25-HC) (22, 23). When mice lack both CXCR5 and EBI2, B cell clustering is almost completely disrupted; full disruption occurs when CCR7 is also removed (24). EBI2 becomes expressed on B cells at a similar stage of maturation as CXCR5 (25). 7α,25-HC is made from cholesterol by the actions of two enzymes, Ch25h and Cyp7b1. Both of these enzymes are required in stromal cells for the generation of 7α,25-HC in lymphoid tissues (26). Laser capture microdissection provided evidence that Ch25h expression was enriched in the outer regions of the B cell follicle and was low in the center (26). The pattern of Ch25h mRNA expression has since been more precisely mapped through RNAscope-based in situ hybridization (3, 27) (Fig. 2). scRNAseq analysis of LN stromal cells showed that Ch25h was expressed by two main subsets, MRCs and a subset of CCL19lo TRCs (3). The latter likely include the Ch25h+ cells present at the follicle – T zone interface. Cyp7b1 is also expressed in this region but is less tightly demarcated (27) (Fig. 2). It is inferred from this enzyme distribution that 7α,25-HC is made in greater amounts at the follicle perimeter and is low in the follicle center. This distribution matches with the finding that EBI2 over-expression in B cells promotes their movement to the outer follicle. While it appears that EBI2 is redundant in organizing primary follicles since the size and distribution of follicles appears unaffected in EBI2 knockout (KO) mice, this chemoattractant does influence the efficiency with which naïve B cells access the outer region of the follicles since in mixed BM chimeras, EBI2 KO B cells are under-represented in the outer follicle and over-represented in the center (25, 28).
Figure 2. Model of EBI2-mediated B and T cell co-localization.
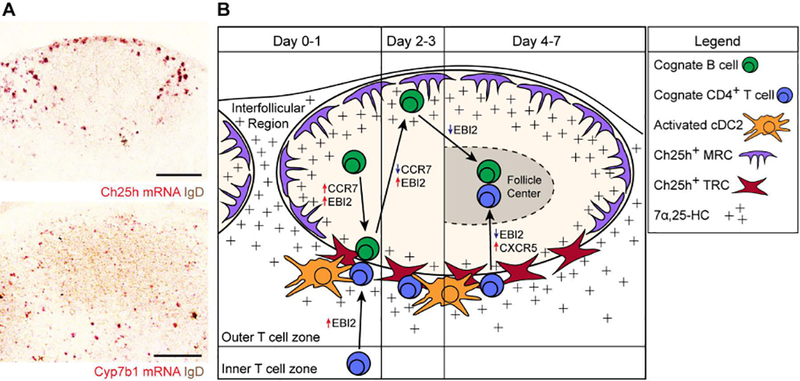
(A) RNAscope in situ hybridization for Ch25h and Cyp7b1 mRNA expression (red signal) relative to endogenous B cells (IgD, brown) in lymph node sections. Ch25h signal is enriched in outer- and inter-follicular regions and at the follicle-T zone interface. Cyp7b1 signal is also detected in these regions. (B) Model for cognate B cell and CD4+ T cell movement after immunization. Early after immunization (Day 0–1), B cells upregulate EBI2 and CCR7 to promote positioning along the follicle-T zone interface. Concurrently, cognate CD4+ T cells also upregulate EBI2 to position at this interface, where they interact with activated cDC2s as well as the activated B cells. A subset of T zone fibroblastic reticular cells (TRCs) expresses Ch25h, supporting the 7α,25-HC gradient that guides cells to this location. During days 2–3, B cells downregulate CCR7 and maintain upregulated EBI2 expression, promoting their positioning at outer-follicular and inter-follicular locations. In these locations, Ch25h-expressing marginal reticular cells (MRCs) support the 7α,25-HC gradient. The cognate CD4+ T cells continue to receive signals from activated cDC2s to promote their differentiation into T follicular helper (Tfh) cells. During days 4–7, the cognate B cells downregulate EBI2 to promote their positioning at the center of the follicle to form GCs. The cognate CD4+ T cells that have developed into Tfh cells utilize CXCR5 to move into the follicle, where they support the GC response. These cells have been shown to express lower levels of EBI2, supporting their positioning in GCs (144).
Multiple chemokines promote HEV crossing.
B cell entry into LNs involves CCR7-CCL21 and CXCR4-CXCL12; in PPs and to a lesser extent in mesenteric LNs, CXCR5-CXCL13 also functions at this step (29). Each of these chemokines can be displayed on the luminal surface of HEVs. B cell movement out of blood into lymphoid tissues also depends on G-protein coupled receptor kinase-2 (GRK2)-mediated desensitization of S1PR1 (30). Lysophosphatidic acid (LPA), generated by HEV-expressed autotaxin (Enpp2), promotes T cell movement from blood into LNs but it has not been determined whether LPA and LPARs also function during B cell transmigration across HEVs (31, 32). Following lymphoid tissue entry, B cells move rapidly into B cell follicles in what can be a multistep process, sometimes involving guidance cues in addition to CXCL13 (33).
LPA acting via LPAR2 and Rho promotes T cell motility in the LN T zone (33, 34). Autotaxin is abundantly expressed by MRCs around follicles as well as by HEVs and stromal cells in the T zone (3, 34). Since LPAR2 is similarly expressed by B cells and T cells (Immgen.org), it is possible that LPA contributes to B cell motility within follicles or adjacent locations but this remains to be determined.
S1P promotes lymphoid tissue egress.
Egress of naïve B cells out of lymphoid tissues and back into circulation requires S1PR1, a Gαi-coupled receptor, and S1P (35). S1PR1 mRNA is upregulated in B cells as they reach the mature follicular state. B cell egress from the BM is only mildly dependent on S1PR1 (36, 37). Instead, mobilization from the BM is thought to more strongly depend on reduced retention by CXCR4 (38). Real-time imaging studies of lymphocyte egress from LNs support a multistep model where lymphocytes approach cortical and medullary lymphatic sinuses in a largely S1PR1-independent manner, probe the sinus and commit to entering in an S1PR1-dependent manner (16, 39). S1P is produced by lymphatic endothelium and is abundant in lymph and blood (35). Once within the sinus, the cells become rounded and are carried with lymph to the efferent lymphatic and then back to circulation. Exit from Peyer’s patches has similar requirements though CXCR4 is involved in guiding B cells into proximity with lymphatic sinuses (40). Egress from the spleen occurs into blood rather than lymphatics (41). Real time imaging of the mouse spleen showed that S1PR1 was important for B cell movement out of the white pulp into the red pulp and hence for access to venous sinuses that return the cells to circulation (42). These broad requirements for S1PR1 ensure that functional antagonism of S1PR1 with the small molecule FTY720 (Fingolimod) causes a strong block in B cell egress from lymphoid organs and is associated with marked B lymphopenia (35).
LN egress of naïve B cells can be regulated by β2-adrenergic receptor (AR) signaling. B and T cells responding to engagement of this GPCR showed increased retention-promoting signals through CCR7 and CXCR4 (43). How β2-AR signaling modifies lymphocyte movement is not yet clear but may involve chemokine receptor-heterodimerization (43). GPCR heterodimerization has been described for a range of GPCRs though the understanding of its physiological significance and of how the heterodimer transmits modified signals has only been established in a small number of cases (44, 45). Adrenergic nerves directly innervate lymphoid organs and release noradrenaline in a circadian manner (46, 47). In accord with these observations, the increased adrenergic signaling at night in rodents augmented B and T lymphocyte numbers in LNs and this was correlated with enhanced antibody responses following immunization at night; a similar cycle is thought to occur in humans but with peak LN accumulation occurring during the day (48). These cycles additionally involve intrinsic actions of circadian clock genes causing small oscillations in S1pr1 mRNA and protein expression that are thought to enhance mouse LN egress during the day (49). This study also provided evidence for small circadian fluctuations in LN CCL21 mRNA, with expression of this LN homing and retention factor being higher during the night. How the β2-AR inputs and clock gene oscillations are integrated to control the circadian trafficking of lymphocytes remains to be established. However, these observations do suggest that attention should be given to circadian patterns when performing vaccinations.
Unique shuttling behavior of naïve MZ B cells.
MZ B cells are specialized cells that occupy the blood-exposed MZ niche in the rodent spleen and respond rapidly and often T cell-independently to blood borne antigens (50). Although MZ B cells are resident in the spleen, they are highly motile and continually shuttle between MZ and follicle. This behavior allows them to mediate efficient delivery of opsonized antigens into follicles for encounter by the larger population of recirculating B cells (41). The mechanism of shuttling depends on the cooperative behavior of at least two GPCRs (42, 51). The MZ is rich in S1P and MZ B cells are attracted into this compartment in an S1PR1 (and partially S1PR3) dependent manner. S1PR1 is highly prone to ligand-mediated desensitization and within minutes of entering the compartment, S1PR1 is downregulated in a GRK2-dependent manner, causing the cells to lose much of their attraction for this location. Instead, CXCL13-mediated attraction becomes dominant and the B cells travel into the follicle. Within the S1P-low follicle, MZ B cells recover their S1PR1 expression and this allows their return to the MZ, thereby establishing the shuttling behavior (41). A further Gαi-coupled receptor, CB2, contributes to MZ B cell retention in the MZ since its antagonism leads to appearance of MZ B cells in the blood. CB2 responds to a number of endocannabinoids and 2-arachidonylglycerol (2-AG) is a candidate ligand acting in the spleen (52), though the impact of disrupting 2-AG production on MZ B cell compartmentalization has not been determined. MZ B cells depend on Notch2 for their differentiation. A recent study found that MZ B cells are dependent on Gαi signaling for ADAM10 expression and activation of Notch2, helping explain the strong MZ B cell developmental defect in mice treated to block Gαi signaling (53). The Gαi-coupled receptors necessary for the ADAM10 activation remain to be determined.
Follicle-associated innate-like lymphocytes provide a LN protective barrier
LN follicles are situated immediately below the SCS. Particles, including pathogens, that arrive in afferent lymph first encounter SCS-associated cells. While much of the molecular material arriving in afferent lymph travels to the macrophage-rich medullary sinuses where it may be phagocytosed and destroyed, some is captured by SCS macrophages. One purpose for this capture is to allow antigen to be delivered into follicles for encounter by cognate B cells (54). However, this also means that SCS macrophages and LECs are exposed to potentially high doses of pathogens. It is therefore not surprising that this region is constantly surveyed by a population of innate-like lymphocytes. In peripheral LNs, many of these cells are CCR6hi, CXCR6+, IL7Rαhi, RORγt+ IL17-committed cells (55). This population was found to be quite heterogeneous, including γδT cells, CD4 and CD8 double negative αβT cells, and non-T cells. Positioning of the cells adjacent to the SCS required CCR6 and the ligand CCL20 was found to be made by SCS-associated LECs (56). Subcutaneous injection of CCL20 was sufficient to increase the association of the cells with the SCS. Given that CCL20 can be expressed by cells in the skin and its levels increase in inflamed skin (57), it is possible that CCL20 arriving in the afferent lymph can amplify CCR6hi innate-like lymphocyte association with the SCS. Intralymphatic labeling procedures showed that a fraction of the CXCR6+ innate-like lymphocyte population is situated within the SCS lumen and intravital TPLSM showed that some cells exchange between the parenchyma and the lumen. However, the innate-like cells are largely LN resident, indicating that this exchange behavior is likely at equilibrium and constitutes a type of shuttling akin to that described for MZ B cells in the spleen. Indeed, as found for MZ B cells, movement of the innate-like lymphocytes from the parenchyma into the sinus showed an intrinsic dependence on S1PR1 (56). We speculate that as observed for MZ B cells, once in the sinus, exposure to the high amounts of S1P in the lymph causes downregulation of S1PR1 and alleviates the attraction of the cells into this compartment, allowing their return to the parenchyma. When innate-like lymphocytes were unable to closely associate with the SCS due to CCR6-deficiency, mice showed a diminished LN IL17 response to a range of subcutaneously administered pathogens (56).
NK cells are also located in the periphery of LNs, predominantly in the medulla. Their positioning requires S1PR5 and CXCR4 (58). S1PR5 is more resistant to S1P-mediated desensitization than S1PR1 and this may allow it to function more effectively in guiding cells chemotactically through S1P gradients. S1PR5 is also resistant to CD69-mediated repression (59), allowing activated CD69+ NK cells to continue sensing S1P. When NK positioning in the outer cortex was defective due to S1PR5-deficiency, NK cell activation and IFNγ production 2 hrs after Salmonella typhimurium infection was compromised (58). These combined defense systems are likely to help ensure that intact and potentially viable pathogens can arrive to LNs for stimulation of B cells but are prevented from overrunning the LN. Rapid cytokine production by innate-like lymphocytes can induce anti-bacterial peptides (60) and promote neutrophil recruitment (61), and NK cells can directly kill infected SCS macrophages (62).
Acute positional changes after B cell activation and T cell encounter
Upon antigen encounter and BCR signaling, B cells move within a few hours to the follicle-T zone interface. This occurs through directed migration up a CCL21 gradient and depends on a 2–3 fold increase in CCR7 (63). Given that CCR7 ligands are distributed throughout the T zone, it had been unclear what caused the B cells to align at the interface. More recent work has established that EBI2 and 7α,25-HC cooperate with CCR7 (and likely CXCR5) to distribute activated B cells along the B-T zone interface (24, 64). Although the precise distribution of the oxysterol is not known, the expression of Ch25h by stromal cells along this interface but not deeper within the T zone or follicle is thought to ensure that EBI2 ligand is enriched in this region (Fig. 2).
Interestingly, EBI2 is upregulated more promptly than CCR7 following BCR engagement (24, 64). When examined in the first 2–3 hours after antigen exposure, activated B cells in LNs show a transient accumulation just beneath the SCS (64). Ch25hhi MRCs are present in this region, making it likely that 7α,25-HC is made locally. Imaging studies have shown that B cells may capture antigens from the surface of SCS macrophages (54). Given that some amount of antigen encounter needs to occur before EBI2 is upregulated, it remains unclear whether the transient attraction to this potentially antigen-laden region is to facilitate capture of more (newly arriving) antigen, perhaps to better sample associated innate stimuli, or whether interactions with SCS macrophages allows the transfer of other types of signals (perhaps signals that influence the subsequent differentiation of the B cell).
Activation also causes the retention of B cells in the responding lymphoid organ. Exposure to inflammatory stimuli such as TLR ligands or type I IFN causes prompt expression of the lymphocyte activation antigen CD69, and this type II transmembrane protein physically interacts with and inhibits the function of S1PR1 (35, 65, 66). Activation by BCR engagement will induce CD69 and, at a slower pace, cause downregulation of S1PR1 transcription (51). Thus, egress is often regulated as a two-tiered process, with initial global retention of any lymphocytes exposed to inflammatory stimuli – enhancing the chance of rare responders being present to encounter antigen – followed by more prolonged retention of cells that have received a cognate BCR signal. B cell retention in the responding LN can last for extended periods or even be terminal as S1PR1 remains downregulated in GC B cells and in many plasma cells.
cDC2 positioning and priming of Tfh cell responses
Positioning of Tfh-inducing cDC2s.
In most T cell-dependent antibody responses, CD4 T cells must first be activated by encounter with antigen-presenting DCs. Conventional DCs (cDC) are divided into two main classes, cDC1 and cDC2, that can be distinguished based on a number of surface markers and by their dependence on different transcription factors (67). Several recent studies have shown that cDC2s are more effective than cDC1s in promoting CD4 T cell activation and the development of Tfh cells (68). Within the spleen, sentinel cDC2s are enriched in MZ bridging channels, gaps in the MZ that connect the T zone directly to the red pulp (Fig. 1). Splenic cDC2 homeostasis and positioning in MZ bridging channels depends on intrinsic EBI2 expression, and cDC2s migrate in response to 7α,25-HC (69, 70). In the course of studies to determine the enzyme requirements for EBI2-dependent cDC2 positioning and homeostasis, we found that Ch25h-deficient mice had a less severe cDC2 deficiency than EBI2-deficient mice. This led us to discover that a second EBI2 ligand, 7α,27-HC, is involved in promoting splenic cDC2 positioning and homeostasis (27). 7α,27-HC synthesis requires Cyp27a1 and this enzyme is expressed by stromal cells in and around the splenic white pulp, being most concentrated in the bridging channels (27). Although Cyp27a1 deficiency alone did not affect cDC2 distribution or numbers, when mice lacked both Ch25h and Cyp27a1 they suffered a cDC2 deficiency that matched that observed in EBI2-deficient mice. 7α,27-HC shares with 7α,25-HC a dependence on Cyp7b1 for its production and on Hsd3b7 for its degradation (27). Intriguingly, cDCs abundantly express Cyp27a1 but hematopoietic deficiency in the enzyme does not affect their properties, leaving it unclear what role the enzyme plays in these cells. Since it is a mitochondrially-associated enzyme it will be of interest to determine if Cyp27a1 deficiency in DCs leads to alterations in their metabolic properties.
Within LNs many resident cDC2s, identified by CD11b staining, are situated at the follicle-T zone interface (Fig. 2). EBI2 deficiency disrupted the distribution of cDC2s (identified by DCIR2 expression) in mesenteric LNs but had little effect on their distribution in peripheral LNs ((27) and unpublished data), indicating that additional factors are involved in guiding DC distribution in peripheral LNs. Migratory cDC2s in peripheral LNs, including lung draining LNs, also localize preferentially to the follicle-T zone interface. DC migration from the periphery to the T zone depends strongly on CCR7 and CCL21 and also on the scavenger receptor ACKR4 that helps prevent CCL21 accumulation in LN SCS regions (71). Work on the response to an inhaled antigen showed the importance of Irf4- and Dock8-dependent cDC2s in Tfh cell priming in mediastinal LNs (72). Recent work revealed that when cDC2s are mobilized under type-2 immunizing conditions, CCR8 cooperates with CCR7 to promote cDC2 movement from the LN SCS into the parenchyma (73). The CCR8 ligand, CCL8, is made by CD169+ SCS macrophages under these conditions.
Following exposure to inflammatory mediators, splenic cDC2s and cDC1s move rapidly into the T zone. Under some conditions, such as following activation by LPS or by missing self-CD47 cells, DCIR2+ cDC2s become enriched at the follicle-T zone interface while cDC1s are in a mutually excluded location at the T zone center (27, 74). This patterning depends on EBI2 upregulation in the cDC2 and its cooperation with CCR7, such that the cDC2 are thought to navigate to the location where the combined concentration of EBI2 and CCR7 ligands is highest. In addition to the compartmentalized expression of Ch25h at the follicle-T zone interface, the expression of the 7α,25-HC metabolizing enzyme Hsd3b7 was enriched in the T zone center. Remarkably, an important fraction of the Hsd3b7 expression was by the cDC1s and removal of the enzyme from these cells disrupted the organized positioning of cDC2s (27). When DCs lacked EBI2 and activated cDC2s could not position appropriately, the magnitude of the Tfh cell and B cell response was diminished.
Following immunization with the type I IFN inducing agent, poly I:C, activated cDC2s became uniformly dispersed through the splenic T zone rather than positioning at the follicle—T zone interface. This positioning was shown to reflect type I IFN-mediated upregulation of Ch25h within the T zone and increased production of 7α,25-HC, likely disrupting the organizing gradient of this EBI2 ligand (27). Interestingly, although EBI2 was important for effective Tfh cell induction in response to missing self-CD47 cells, strong induction of Tfh cells occurred following poly I:C and SRBC co-injection (unpublished data). Under these conditions, both the antigen specific T cells and cDC2s were dispersed throughout the T cell zone. These data suggest that the microenvironments that most effectively support Tfh cell development can differ depending on the inducing stimulus.
Localization of activated CD4 T cells to the follicle – T zone interface.
EBI2 is rapidly upregulated in activated CD4 T cells and plays a role in their repositioning early after activation to the follicle-T zone interface (75) (Fig. 2). Indeed, even at baseline, EBI2 causes CD4 T cells to favor the outer T zone compared to CD8 T cells (74). When CD4 T cells lack EBI2, the magnitude of the early Tfh cell response to a number of antigens is reduced (75). Although CXCR5 can be expressed quite early during CD4 T cell activation, it is not essential for CD4 T cell positioning at the follicle-T zone interface (76), though it may contribute to the efficiency of this process under some conditions (77). As CD4 T cells engage with cDC2s they receive poorly defined signals that promote Tfh cell differentiation. The follicle-T zone interface is also the location where activated T cells begin interacting with antigen-engaged B cells. In most models this is thought to follow the initial activation of CD4 T cells by cDC2s (or possibly other DCs), with the B cell encounters helping stabilize the Tfh state while simultaneously promoting proliferation and differentiation of the B cells. In some cases, it is possible that naïve T cells first encounter antigen presented by cognate B cells, bypassing the involvement of DCs (78).
B cell relocation after T cell help
B cell – T cell interactions lead to relocalization of activated B cells (Fig. 2). In some responses the cells relocate to interfollicular regions and possibly even to the ‘back’ of the follicle (away from the T zone) (79–81). These behaviors depend on maintained or possibly slightly elevated EBI2 expression together with CCR7 downregulation (64). It is likely that CXCR5 also helps distribute the cells during this step, especially the cells that travel to the outer follicle. CD40 signaling is sufficient to promote movement of antigen-engaged B cells to inter- and outer- follicular regions (64). The signals and transcription factors involved in downregulating CCR7 and upregulating EBI2 in B cells have not been defined. Despite the striking nature of this relocalization event, its physiological significance remains unclear. B cells in the outer follicle have been shown to undergo proliferation (79). In interfollicular regions they may continue to interact with T cells that also localize to this region. It seems likely that additional cell-cell interactions occur within these niches that help guide or support the ongoing proliferation and differentiation of the activated B cells. For example, type-3 innate lymphoid cells (ILC3) were recently found to enrich in interfollicular regions of mesenteric LNs in an EBI2-dependent manner (82). After a short period (perhaps 1 day) in this region, activated B cells differentiate into plasmablasts or GC B cells.
Plasma cell homing
Plasma cells occupy specialized niches in primary and secondary lymphoid tissues. In the spleen most plasma cells are situated near large vessels and reticular fibers in the red pulp; in LNs they are situated near lymphatic sinuses in medullary cords; in the BM they locate near sinusoids. These locations ensure rapid access of secreted antibodies to circulation. As B cells differentiate in to plasma cells they downregulate CXCR5 and CCR7 and highly express CXCR4 (83). The CXCR4 ligand, CXCL12, also known as stromal cell derived factor-1 (SDF1) is strongly expressed by stromal cells in the splenic red pulp, LN medullary cords, and BM; CXCR4 is critical for plasma cell positioning in these compartments (84) (Fig. 3). The transcription factor cMyb is needed for CXCR4 function in plasma cells though the cMyb target genes involved are not yet known (85).
Figure 3. Plasma cell subsets, egress and tissue tropism.
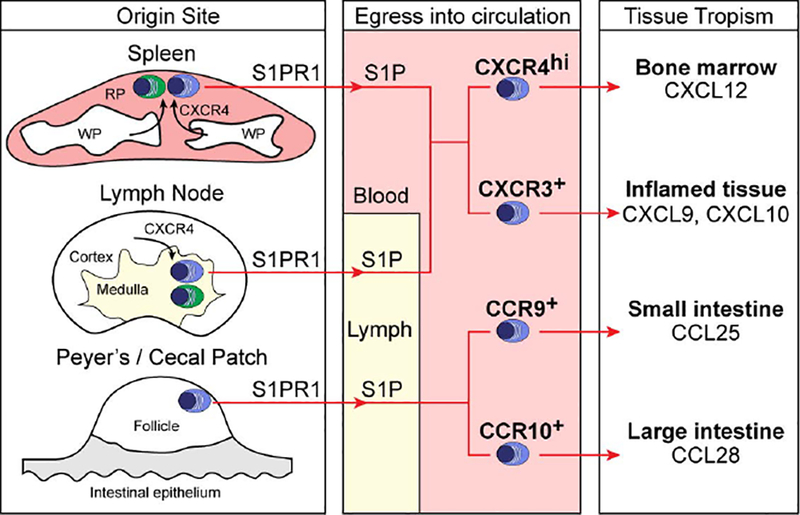
Plasma cells (PCs) in the spleen utilize CXCR4 to position in the red pulp, and those in the LN use CXCR4 to position in the medulla. PCs egress into circulation using S1PR1 (blue cells), while a subset remains resident in spleen and LNs (green cells). Depending on the type of immune response, different subsets of PCs develop. These subsets express distinct chemokine receptors that govern their tissue tropism, with CXCR4hi PCs predominantly homing to BM sinusoids, CXCR3+ PCs homing to inflamed tissue, and CCR9 / CCR10+ PCs from Peyer’s or cecal patches homing to gut tissues.
Early in situ hybridization (83) and protein staining (86) data showed CXCL12 expression was high in splenic red pulp and LN medullary cords. Using CXCL12-DsRed mice and CXCL12-GFP mice, CXCL12 expression was detected in perivascular stromal cells around sinusoids and possibly other stromal cells in the red pulp; lower expression was detected around white pulp central arterioles (87, 88). Analysis of LNs in CXCL12-GFP reporter mice showed CXCL12 mRNA is expressed by most stromal cells within LNs (88, 89) and this has been supported by scRNAseq data, though the medullary stromal cell cluster showed the strongest expression (3). A recent publication reported isolation of LN medullary stromal cells (medRC) as CD45−CD31−MAdCAM1−BP3lo cells and found by bulk RNAseq that these cells abundantly expressed CXCL12 while expressing little CCL21 or CXCL13 (90). Although not all studies have shown that CXCL12 protein is most abundant in the medulla, we found different CXCL12 staining patterns depending on the antibody reagents used, and mouse K15C antibody staining on rat tissue gave the most sensitive detection of protein distribution (86). If we assume sensitivity correlates with the most accurate representation of protein distribution, and assuming rat and mouse are similar, then we can infer that CXCR4 guides plasmablasts and plasma cells to the medulla because of its higher expression of CXCL12. It remains important to accurately map the distribution of CXCL12 in human lymphoid tissues.
It is likely that factors beyond CXCL12 mRNA expression determine its high protein abundance in the medulla since CXCL12 binds strongly to heparin and HS-containing proteoglycans, though the role of such retention factors in the medulla is not known. Mice harboring a GAG binding mutant of CXCL12 in place of the wild-type gene have disrupted GC organization, but the distribution of PCs was not described (91). Moreover, there are three splice variants of CXCL12, and the CXCL12γ isoform has a 30 amino acid extension at the C-terminus that mediates very strong GAG binding and relies on such binding to be active (92). However, the expression pattern of the different splice variants in lymphoid tissues is unknown. Reciprocally, CXCL12 may be kept low in other parts of the tissue by proteolytic turnover. Dipeptidylpeptidase 4 (DPP4), a transmembrane protein also known as CD26, is widely expressed by lymphocytes and DCs (Immgen.org) and is able to proteolytically process multiple chemokines including CXCL12 and CXCL10 (93). DPP4-mediated cleavage of CXCL12 reduces its activity on CXCR4 and such cleavage is thought to contribute to G-CSF-mediated mobilization of HSCs from the BM (93). However, whether DPP4 acts to locally promote the turnover of CXCL12 in a way that controls the distribution of PCs (or B cells at other stages of development) has not been established, though the widespread distribution of DPP4 makes this seem likely.
Atypical chemokine receptors (ACKRs) have important roles in scavenging or transcytosing chemokines and thereby help control their distribution (94). ACKR3 (CXCR7/RDC1) can bind CXCL12 and more weakly CXCL11, and is expressed by various cells in the splenic red pulp and MZ (likely including endothelium), as well as by MZ B cells (95) (unpublished data). The abundant ACKR3 expression by cells in the splenic MZ is likely to help maintain a low CXCL12 concentration in this compartment, which may be important for ensuring CXCL12 gradients direct PCs to appropriate regions of the red pulp (95). ACKR3 antagonism causes alterations in the MZ architecture and is associated with elevated serum CXCL12 indicating a role for this receptor in scavenging circulatory CXCL12 (95). Flow cytometry of human B cells revealed ACKR3 expression by tonsil plasmablasts (96) and mRNA for ACKR3 is present in mouse splenic plasma cells ((97) and Immgen.org). Given that plasma cells move to areas of high CXCL12 expression in a CXCR4-dependent manner (84), the role of ACKR3 in these cells is unclear. The early phase of plasmablast movement out of GCs was suggested to be chemokine receptor independent (98) and it is possible that ACKR3 helps antagonize CXCR4 function during this phase.
A challenge in treating systemic autoimmune diseases has been developing methods to deplete plasma cells. A study in the lupus-prone NZB/W mouse strain found that treatment with the CXCR4 antagonist, AMD3100, displaces PCs into blood and causes a reduction in numbers within BM and spleen. Combining the treatment with bortezomib, a proteasome inhibitor used to treat multiple myeloma, caused a more marked reduction in BM and spleen PCs and ameliorated disease in the mice (99). Further work is needed to understand the therapeutic potential of CXCR4 antagonism for the depletion of long-lived plasma cells.
Plasma cell tropism for the intestine is promoted by CCR9 and CCR10 responding to CCL25 and CCL28, respectively (Fig. 3). CCR9 and CCR10 appear to cooperate in promoting PC homing to the small intestine. CCL25 is highly expressed in the small intestine and is minimally expressed in the large intestine, while CCL28 is more strongly expressed in the large intestine (100). For the colon, CCR10-CCL28 is the dominant chemokine requirement and CCR9 has not been shown to have a role. A recent study found that mice lacking CCL28 have markedly reduced colonic IgA PCs and fecal IgA (101). Interestingly, the PCs that were present produced less IgA suggesting that CCR10 signaling in gut PCs may augment their secretory function. In accord with the site of antigen drainage being mapped on to the subsequent tropism of the plasma cell, IgA+ cells from PPs showed tropism for the small intestine while those from colon-draining cecal patches homed to both the small intestine and colon. IgA+ cells arising in cecal patches had higher CCR10 than those in PPs (102). A recent study has obtained evidence that IgA plasma cells may become mobilized from the gut following immunizations that affect epithelial integrity, and these plasma cells can then travel to the central nervous system (103). This study adds to other evidence that plasma cell retention in tissues can be modulated by external factors, something that may be important in allowing seeding of previously occupied niches by newly generated plasma cells (104).
Plasma cells generated under IFNγ-positive conditions express CXCR3 (and T-bet) and this can contribute to their migration to inflamed tissues such as the kidneys in lupus nephritis models (104) (Fig. 3). However, this may not be a universal requirement as CXCR3-deficiency on the autoantibody prone NZB/W background did not alter cellular infiltrates and glomerulonephritis, suggesting that other factors can promote plasma cell accumulation at inflamed sites (105). Beyond control of plasma cell homing, a recent study found that over-expression of CXCR3 in GC B cells (occurring due to cMyb-deficiency) favored early development of plasma cells (106). It will be interesting to see if this is a consequence of CXCR3 positioning GC B cells in a niche that favors the plasma cell fate or whether CXCR3 signaling directly augments plasma cell differentiation.
Plasma cell egress from secondary lymphoid tissues and homing to BM or gut mucosa depends on S1PR1 (107, 108) (Fig. 3). In the case of spleen and LN responses, the factors controlling whether a PC upregulates S1PR1 may be central to determining whether they home to the BM or stay in the peripheral lymphoid tissue. Since BM-tropic PCs are enriched for long-lived PCs compared to those that stay in the secondary lymphoid organ, it remains of interest to understand whether S1PR1 expression is coupled to becoming a long-lived cell.
GC organization and confinement
GCs form in the central region of lymphoid follicles, overlapping with the pre-existing FDC network. Movement of activated B cells to this location is promoted by coordinated changes in GPCR function. EBI2 becomes transcriptionally downregulated, reducing B cell attraction to the outer regions of the follicle (Fig. 2). The cells continue to express CXCR5 and this supports their migration within the follicle, and they maintain a low CCR7 expression, reducing their attraction to the follicle-T zone interface. At the same time, the early GC cells upregulate S1PR2, a receptor that couples to Gα13 and supports migration-inhibition in response to S1P (109). GC B cells also have higher Gα13 expression than naïve follicular B cells suggesting an augmented utilization of the Gα13 signaling pathway. S1P is made by almost all cell types as part of their sphingolipid metabolism program but it is only secreted by some cell types. Blood and lymphatic endothelium are known to generate extracellular S1P due to their expression of spinster-2 (Spns2), a S1P transporter (110). It is not known whether other lymphoid stromal cells express levels of Spns2 needed for S1P export though some stromal cell subsets, most prominently MRCs, showed detectable Spns2 expression in a scRNAseq analysis (3). S1P has a very short half-life (likely a few minutes) in the interstitial space due to the presence of several widely expressed lipid-phosphatases and an S1P lyase (110). In accord with endothelium and MRCs being sources of S1P, S1PR1 staining in tissue and an S1PR1-fluorescent protein bio-reporter provided evidence that S1P was present in highest amounts in the outer follicle and was lower in the follicle interior (33, 111). Over-expression experiments have shown that S1PR2 is sufficient to promote B cell clustering at the follicle center (112). Reciprocally, if S1PR2 is deleted, GC B cells show less complete confinement to the GC than wild-type B cells.
Mice lacking Gα13 were found to have greater deconfinement of GC B cells than S1PR2-deficient mice (113). This led to a search for another Gα13 coupled receptor in GC B cells. The efforts to identify such a receptor were facilitated by the finding that S1PR2 and Gα13 are often mutated in human GC B cell type DLBCL (113). Analysis of DLBCL sequencing datasets revealed the presence of another GPCR named P2RY8, an orphan, which was frequently mutated in GCB-DLBCL and BL. Although this receptor was named for its similarity to purinergic receptors, it was not found to respond to nucleotides (114). Given that we had searched for a second receptor because of the stronger GC B cell deconfinement phenotype in Gα13- compared to S1PR2-deficient mice, we were surprised to find that mice (and other rodents) lacked a P2RY8 ortholog. Remarkably, however, when human P2RY8 was over-expressed in mouse B cells it promoted a clustering behavior similar to S1PR2 over-expression. An interesting difference between the two receptors was that the follicle center-confining actions of P2RY8 were dependent on FDCs (115) whereas those of S1PR2 were not (112). P2RY8 also shared with S1PR2 an ability to restrain the growth of GC B cells in vivo in mice (113). This finding was consistent with the idea that P2RY8 mutations in human GC B cells were relieving a growth-restraining activity, permitting GC B cell overgrowth and contributing to malignancy.
In recent work we have deorphanized P2RY8. Using biochemical fractionation methods we identified a novel molecule, S-geranylgeranyl-L-glutathione (Ggg) as a nM potent P2RY8 ligand (116). Ggg was present in mouse and human lymphoid tissues as well as other tissue types, being particularly abundant in bile (116). Using sensitive bioassay and mass spectrometry assays, it appeared that many cell types could produce Ggg. One possibility is that Ggg is made in most cell types during processes associated with geranylgeranyl metabolism, but it is only secreted by some cell types. Identification of the enzyme(s) involved in Ggg biosynthesis and the transporter(s) involved in its export will be necessary to test this idea and to understand the cell types generating extracellular Ggg, as well as to define the range of conditions under which it is produced. Glutathione conjugates can be metabolized by γ-glutamyltransferase class enzymes and we found that Ggt5, also known as γ-glutamyl leukotrienase (GGL) due to its ability to metabolize LTC4 to LTD4, could metabolize Ggg to a form inactive on P2RY8 (116). Strikingly, Ggt5 is highly expressed by FDCs in primary follicles and in GCs (116). These findings have led to our current model (Fig. 4) where follicles contain, remarkably, gradients of three lipid mediators: 7α,25-HC, S1P and Ggg. All three gradients are likely to cooperate to ensure GCs form at the follicle center. By having the center of the follicle as a low point in the 7α,25-HC gradient, EBI2 downregulation favors B cell positioning in this region. S1P and Ggg both exert migration-inhibitory actions on GC B cells and thereby cooperate to restrain their movement into the outer follicle where the concentrations of these lipids are highest. Why two cues acting through the same signaling pathway are needed is not yet clear but it seems likely that the shapes of each of these lipid gradients will be different. S1P gradients appear not to depend on FDCs and may therefore allow organization of follicles to occur even without FDCs; Ggg gradients depend on FDCs and this may provide a different shape to the gradient. Perhaps by responding to both migration inhibitory factors and their distinctly determined gradients, a sharper boundary can be achieved. It is also possible that the two gradients allow a higher level of organization within the GC. In this regard it is notable that tonsil GCs organize into additional compartments beyond the dark and light zones that are discussed below (117). Perhaps S1P and Ggg work together with local chemoattractants to achieve this added level of compartmentalization (Fig. 4).
Figure 4. Model of P2RY8-mediated B cell confinement and proposed distribution of B cell guidance cues.
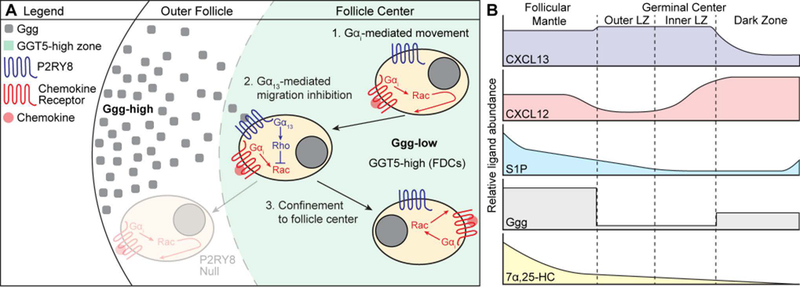
(A) A gradient of Ggg is formed through GGT5-mediated degradation of Ggg at the center of the follicle, resulting in higher levels of Ggg in the outer follicle and lower levels at the center. When P2RY8-expressing B cells experience movement (either random or chemokine-directed) towards outer areas of the follicle, Ggg engages P2RY8 to induce Gα13-mediated activation of Rho, which inhibits Rac-based movement. This confines a P2RY8-expressing cell to the center of the follicle. Movement towards the center of the follicle is not inhibited by Ggg, promoting central clustering. The distribution of chemokine (CXCL13, CXCL12 – red dot) is not shown. (B) Proposed model of B cell guidance cue distribution within GC-containing follicles. We speculate that the high density of GGT5 expression within GCs (being higher on LZ than DZ FDCs) results in a sharp, stepwise drop in Ggg levels. In contrast, degradation of S1P does not depend on FDCs, but may be mediated by many cell types (including B cells), suggesting that the S1P gradient may decay in a more linear fashion. The interplay between these two migration-inhibitory ligands may help support the formation of inner and outer zones in the GC LZ that have been reported in tonsil GCs (117, 145). CXCL13 is present throughout the follicular mantle (region around the GC) and is abundant within the LZ, while CXCL12 is enriched in the DZ. As suggested by Figure 2, the 7α,25-HC gradient is high in the follicular mantle and low at the follicle center. These cues together support GC confinement and zonal organization.
The only previously known glutathione-conjugated lipid that functions as an intercellular signaling molecule is LTC4, a ligand for CysLTR1 and CystLTR2. Interestingly, LTC4 was active on P2RY8 though with 100-fold lower potency than Ggg (116). In the course of measuring Ggg levels in lymphoid tissues, we noted the presence of comparable amounts of LTC4 (unpublished data). Since Ggt5 can metabolize LTC4, it is possible that this lipid mediator also exists in gradients within the follicle. However, the LTC4 receptors CysLTR1 and CysLTR2 are not highly expressed by naïve or GC B cells or Tfh cells making it unclear whether such a gradient would have any consequential effects on B cell responses.
GCs are organized into a LZ and a DZ in a manner that depends on CXCR4 (86) and the importance of this organization has been extensively reviewed (118, 119). CXCL12 is expressed by specialized stromal cells present in the GC DZ that have been termed CXCL12-reticular cells (CRCs). The GAG binding property of CXCL12 is critical for correct GC organization, likely reflecting local retention of CXCL12 by CRCs (88, 89). We chose not to call these CXCL12+ DZ cells FDCs at the time of describing them because early studies had defined FDCs by their antigen-capturing activity (120) and CRCs showed minimal complement receptor-2 (CR2) expression or evidence of antigen capture. However, CRCs were morphologically more similar to FDCs than to other stromal cell types in terms of having extensive fine dendritic processes (88). In our recent scRNAseq analysis of LN stromal cells we identified a subset that was enriched for FDC markers that we initially considered to be purely FDCs, though we were surprised by the amount of CXCL12 transcript contained within this subset (3). One of the novel markers in this population was Sox9. When tissue sections were stained for Sox9 it was found to be present not only in the nuclei of CR2+ FDCs but also in the nearby CXCL12+ CRCs (3). While this analysis is not definitive due to the low numbers of cells within this subset in the scRNAseq dataset, these findings provide further evidence that FDCs and CRCs are closely related. Given this information, for simplicity, GC CRCs could also be referred to as DZ FDCs.
Like FDCs, CRCs are present within the primary follicle (88). Whether they influence the migration of naïve B cells or other cell types within the primary follicle has not been determined. However, it seems likely that their pre-existence helps establish the initial orientation of the GC with the DZ near the T zone and the LZ facing the outer follicle and thus sites of antigen entry.
ACKR4 (CCRL1) is a scavenger receptor that can bind CCL19, CCL21 and CCL25. ACKR4 is notable for its high expression in GC B cells leading us to speculate that it might help ensure the CCL21 gradient that extends into lymphoid follicles (63) is prevented from becoming established in a potentially disruptive way within GCs. However, analysis of ACKR4 KO mice did not reveal increased CCL21 within LN GCs and the GC response in these mice was unaltered (121). Unexpectedly, however, ACKR4-deficient mice showed exaggerated early B cell proliferative responses and this was suggested to reflect exaggerated movement of the cells to interfollicular regions as a consequence of heightened CCL21 responsiveness (121). What remains puzzling is that high ACKR4 mRNA expression was restricted to the GC state; whether the lower amounts that might be present on early activated B cells are sufficient to intrinsically alter CCR7 function needs further investigation.
Memory B cells – remembering where things are
Memory B cells are generated most efficiently in T cell-dependent B cell responses and they arise predominantly, although not uniquely, from GCs (122). Despite the long appreciation of humoral immune memory, the understanding of the properties of memory B cells, including their trafficking behavior is incomplete. This reflects in part the difficulty of tracking memory B cells by surface markers alone; while small B cells that are isotype switched are considered memory B cells, not all memory B cells are isotype switched.
An early step in memory B cell maturation from a GC precursor is likely the need to exit the GC microenvironment. Recent work in mice examining the earliest-forming memory B cells found that they downregulate S1PR2 and upregulate EBI2 and CCR6 while continuing to express CXCR4, CXCR5 and CCR7 (123, 124). The downregulation of S1PR2 (and in humans P2RY8) presumably helps the cells leave the GC and upregulation of EBI2 is likely to cooperate with this change in promoting their movement away from GCs. Although CCR6 has been highlighted as a marker of memory B cells (125), it is also expressed by pre-GC B cells (126, 127), requiring careful assessment of additional markers or properties of the CCR6hi cells to discern their differentiation state. Pre-plasma cells arising in GCs were also found to upregulate EBI2 and downregulate S1PR2 (128), but their early movement out of the follicle has been suggested to occur in a Gαi-independent manner (98).
A function for CCR6 expression by pre-GC B cells has been established in PPs, where it guides early-activated B cells to the SED to foster interactions with αvβ8+ DCs that promote TGFβ activation and IgA class switching (129). Whether CCR6 upregulation on B cells has a function in other lymphoid tissues has been obscure. The finding that CCL20 helps position innate-like lymphocytes near the LN SCS (discussed above) (56) suggests that CCR6 may help guide activated or memory B cells to this region. Experiments with CCR6-deficient memory B cells are needed to determine the function of this receptor during recall responses.
An early study of memory B cells in rats provided evidence that they can be enriched in the splenic MZ (122). For memory cells induced in the spleen, this is a satisfying observation as the MZ is a site of early exposure to blood borne antigens. However, this behavior may be rather specific to certain types of immune response or may only represent a transient state, as other studies have not observed an enrichment of memory B cells in the MZ of mice but instead observe cells in both the MZ and follicle (122). The possibility that some of these cells are undergoing a MZ B cell-like shuttling behavior needs to be considered. In one study, IgG1+ memory B cells were found concentrated within the follicle around contracted GCs (130); further experiments under different immunization or infection conditions are needed to discern if this is a common behavior.
A recent study of B cells responding to subcutaneously injected antigen plus adjuvant found that memory B cells had a longer dwell time in the draining LN than naïve B cells (131). The higher expression of CD69 and associated antagonism of S1PR1 was considered a likely factor contributing to this increased dwell time. Within the LN, the memory cells showed a strong preference to migrate in the B cell follicle in close association with the SCS and the cells often contacted CD169+ SCS macrophages, likely surveying for antigen. Tfh cells were also found in these regions and memory B cell – Tfh cell conjugates formed quickly following secondary immunization and gave rise to subcapsular proliferative foci that rapidly produced plasma cells (131). The memory B cells in this study expressed CXCR3 in addition to CCR6 and EBI2 and it was suggested that all three of these receptors may contribute to the SCS distribution of the memory cells. The CXCR3 ligands CXCL9 and CXCL10 are expressed by various LN stromal cells, including cells in interfollicular regions (3, 132). Following inflammation or infection CXCL9 has been associated with positioning of memory CD8 T cells in subcapsular regions making it likely that CXCR3 expression also influences memory B cell positioning during such responses (133).
In other studies, memory B cells are found to be part of the recirculating B cell compartment or to be present in the peripheral tissue (122, 134, 135). Recent parabiosis experiments have established that many memory B cells in the lung following influenza infection are resident and their induction requires local antigen encounter (136). While CXCR5 and EBI2 are presumably involved in the trafficking of recirculating memory B cells, the receptors involved in occupancy of non-lymphoid niches need further study. The B resident memory cells in the lung are CD69+ and CXCR3+, hinting at receptors likely involved in their localization (134, 136). Another report has shown that CCR10 is required for mice to maintain a lamina propria memory B cell population. The unique expression of CCR10 by memory-like B cells in the gut suggested that these were tissue resident cells (137). Residency at surfaces associated with their sites of initial induction should place memory B cells in locations where re-encounter with the invader is most efficient, contributing to more rapid and robust immune responses.
Conclusions
Through work over the last several decades, a rich understanding has emerged of the molecular cues choreographing immune cell migration during the humoral immune response. A general theme is that each cell positioning event involves the concerted action of multiple cues. Often both protein (chemokine) and lipid mediators work together, their different biophysical properties possibly encoding distinct cell-scale spatial information. The extent to which chemokines function as immobilized haptotactic versus soluble chemotactic cues is still being worked out. Much needs to be done to understand how the distribution of chemokines is controlled, including defining the contributions of glycosaminoglycans and other scaffolding factors, of scavenger receptors, and of proteases. Moreover, the understanding of the transcriptional control of lymphoid tissue chemokine and chemokine receptor expression is in its infancy. A challenge with studying lipid guidance factors is determining their distribution as antibodies are not available to detect them in situ within tissues. Bioreporters have been used to study S1P distribution and S1PR1 activity (58, 138, 139) and more such approaches are needed. The emerging examples of intrinsic cross-talk between chemoattractant-type GPCRs (43, 73) suggests there will be further mechanisms acting to provide fine control of immune cell migration and localization. Studies in a number of model systems are advancing our understanding of how cells integrate information from multiple attractive inputs, with differences between receptors in their sensitivity to desensitization often having an important influence (18, 140). Mutations that disrupt chemokine receptor desensitization can cause human disease (141–143). How cells integrate inputs from pro-migratory and migration-inhibitory receptors to achieve tissue level organization needs more detailed cell biological study. Through deeper investigations of these topics, we can anticipate emergence of an ability to precisely describe every organizing event needed for humoral immune responses to occur, how we can control these events to therapeutically steer such responses, and how we can accurately engineer them in tissue reconstruction systems to allow ex vivo study of human antibody responses.
Acknowledgements
We thank past and present members of the Cyster lab for helpful discussions. E.L. was supported by the UCSF BMS Graduate Program and NSF grant 1144247. J.G.C is an investigator of the Howard Hughes Medical Institute. This work was supported in part by NIH grants AI040098 and AI045073. The authors have no financial or personal relationships that could be viewed as a conflict of interest.
References
- 1.Cyster JG, Ansel KM, Reif K, et al. Follicular stromal cells and lymphocyte homing to follicles. Immunol Rev.2000;176:181–193. [DOI] [PubMed] [Google Scholar]
- 2.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol.2009;9:618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodda LB, Lu E, Bennett ML, et al. Single-Cell RNA Sequencing of Lymph Node Stromal Cells Reveals Niche-Associated Heterogeneity. Immunity.2018;48:1014–1028 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol.2014;32:659–702. [DOI] [PubMed] [Google Scholar]
- 5.Schulz O, Hammerschmidt SI, Moschovakis GL, Forster R. Chemokines and Chemokine Receptors in Lymphoid Tissue Dynamics. Annu Rev Immunol.2016;34:203–242. [DOI] [PubMed] [Google Scholar]
- 6.Rose DM, Alon R, Ginsberg MH. Integrin modulation and signaling in leukocyte adhesion and migration. Immunol Rev.2007;218:126–134. [DOI] [PubMed] [Google Scholar]
- 7.Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol.2012;24:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res.2015;107:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan H, Wu L, Shih C, et al. Plexin B2 and Semaphorin 4C Guide T Cell Recruitment and Function in the Germinal Center. Cell Rep.2017;19:995–1007. [DOI] [PubMed] [Google Scholar]
- 10.Lu P, Shih C, Qi H. Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science.2017;356 eaai9264. [DOI] [PubMed] [Google Scholar]
- 11.Cyster JG. B cell follicles and antigen encounters of the third kind. Nat Immunol.2010;11:989–996. [DOI] [PubMed] [Google Scholar]
- 12.Ansel KM, Ngo VN, Hyman PL, et al. A chemokine driven positive feedback loop organizes lymphoid follicles. Nature.2000;406:309–314. [DOI] [PubMed] [Google Scholar]
- 13.van de Pavert SA, Olivier BJ, Goverse G, et al. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol.2009;10:1193–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stachowiak AN, Wang Y, Huang YC, Irvine DJ. Homeostatic lymphoid chemokines synergize with adhesion ligands to trigger T and B lymphocyte chemokinesis. J Immunol.2006;177:2340–2348. [DOI] [PubMed] [Google Scholar]
- 15.Allen CD, Okada T, Tang HL, Cyster JG. Imaging of germinal center selection events during affinity maturation. Science.2007;315:528–531. [DOI] [PubMed] [Google Scholar]
- 16.Sinha RK, Park C, Hwang IY, Davis MD, Kehrl JH. B lymphocytes exit lymph nodes through cortical lymphatic sinusoids by a mechanism independent of sphingosine-1-phosphate-mediated chemotaxis. Immunity.2009;30:434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber M, Hauschild R, Schwarz J, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science.2013;339:328–332. [DOI] [PubMed] [Google Scholar]
- 18.Schwarz J, Bierbaum V, Vaahtomeri K, et al. Dendritic Cells Interpret Haptotactic Chemokine Gradients in a Manner Governed by Signal-to-Noise Ratio and Dependent on GRK6. Curr Biol.2017;27:1314–1325. [DOI] [PubMed] [Google Scholar]
- 19.Miller H, Cosgrove J, Wollman AJM, et al. High-Speed Single-Molecule Tracking of CXCL13 in the B-Follicle. Front Immunol.2018;9:1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monneau YR, Luo L, Sankaranarayanan NV, et al. Solution structure of CXCL13 and heparan sulfate binding show that GAG binding site and cellular signalling rely on distinct domains. Open Biol.2017;7 170133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forster R, Mattis AE, Kremmer E, et al. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell.1996;87:1037–1047. [DOI] [PubMed] [Google Scholar]
- 22.Gatto D, Brink R. B cell localization: regulation by EBI2 and its oxysterol ligand. Trends Immunol.2013;14:446–453. [DOI] [PubMed] [Google Scholar]
- 23.Cyster JG, Dang EV, Reboldi A, Yi T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol.2014;14:731–743. [DOI] [PubMed] [Google Scholar]
- 24.Gatto D, Wood K, Brink R. EBI2 operates independently of but in cooperation with CXCR5 and CCR7 to direct B cell migration and organization in follicles and the germinal center. J Immunol.2011;187:4621–4628. [DOI] [PubMed] [Google Scholar]
- 25.Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature.2009;460:1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi T, Wang X, Kelly LM, et al. Oxysterol gradient generation by lymphoid stromal cells guides activated B cell movement during humoral responses. Immunity.2012;37:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu E, Dang EV, Cyster JG. Distinct oxysterol requirements for positioning naive and activated dendritic cells in the spleen. Science Immunology.2017;2:eaal5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity.2009;31:259–269. [DOI] [PubMed] [Google Scholar]
- 29.Cyster JG, von Andrian U. Dynamics of B Cell Migration to and within Secondary Lymphoid Organs In: Honjo T, Alt FW, Neuberger MS, eds. Molecular Biology of B cells. London: Elsevier, 2004:203–246. [Google Scholar]
- 30.Arnon TI, Xu Y, Lo C, et al. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science.2011;333:1898–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanda H, Newton R, Klein R, et al. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nat Immunol.2008;9:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bai Z, Cai L, Umemoto E, et al. Constitutive lymphocyte transmigration across the basal lamina of high endothelial venules is regulated by the autotaxin/lysophosphatidic acid axis. J Immunol.2013;190:2036–2048. [DOI] [PubMed] [Google Scholar]
- 33.Park C, Hwang IY, Sinha RK, et al. Lymph node B lymphocyte trafficking is constrained by anatomy and highly dependent upon chemoattractant desensitization. Blood.2012;119:978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katakai T, Kondo N, Ueda Y, Kinashi T. Autotaxin produced by stromal cells promotes LFA-1-independent and Rho-dependent interstitial T cell motility in the lymph node paracortex. J Immunol.2014;193:617–626. [DOI] [PubMed] [Google Scholar]
- 35.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol.2007;8:1295–1301. [DOI] [PubMed] [Google Scholar]
- 36.Pereira JP, Xu Y, Cyster JG. A Role for S1P and S1P1 in Immature-B Cell Egress from Mouse Bone Marrow. PLoS One.2010;5:e9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allende ML, Tuymetova G, Lee BG, et al. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J Exp Med.2010;207:1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beck TC, Gomes AC, Cyster JG, Pereira JP. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J Exp Med.2014;211:2567–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grigorova IL, Schwab SR, Phan TG, et al. Cortical sinus probing, S1P1-dependent entry and flow-based capture of egressing T cells. Nat Immunol.2009;10:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt TH, Bannard O, Gray EE, Cyster JG. CXCR4 promotes B cell egress from Peyer’s patches. J Exp Med.2013;210:1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnon TI, Cyster JG. Blood, sphingosine-1-phosphate and lymphocyte migration dynamics in the spleen. Curr Top Microbiol Immunol.2014;378:107–128. [DOI] [PubMed] [Google Scholar]
- 42.Arnon TI, Horton RM, Grigorova IL, Cyster JG. Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature.2013;493:684–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through beta2-adrenergic receptors. J Exp Med.2014;211:2583–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fribourg M, Moreno JL, Holloway T, et al. Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell.2011;147:1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gomes I, Ayoub MA, Fujita W, et al. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol.2016;56:403–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Felten DL, Felten SY, Carlson SL, Olschowka JA, Livnat S. Noradrenergic and peptidergic innervation of lymphoid tissue. J Immunol.1985;135:755s–765s. [PubMed] [Google Scholar]
- 47.Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev.2000;52:595–638. [PubMed] [Google Scholar]
- 48.Suzuki K, Hayano Y, Nakai A, Furuta F, Noda M. Adrenergic control of the adaptive immune response by diurnal lymphocyte recirculation through lymph nodes. J Exp Med.2016;213:2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Druzd D, Matveeva O, Ince L, et al. Lymphocyte Circadian Clocks Control Lymph Node Trafficking and Adaptive Immune Responses. Immunity.2017;46:120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kraal G, Mebius R. New insights into the cell biology of the marginal zone of the spleen. Int Rev Cytol.2006;250:175–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cinamon G, Matloubian M, Lesneski MJ, et al. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat Immunol.2004;5:713–720. [DOI] [PubMed] [Google Scholar]
- 52.Muppidi JR, Arnon TI, Bronevetsky Y, et al. Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J Exp Med.2011;208:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang IY, Boularan C, Harrison K, Kehrl JH. Galphai Signaling Promotes Marginal Zone B Cell Development by Enabling Transitional B Cell ADAM10 Expression. Front Immunol.2018;9:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phan TG, Gray EE, Cyster JG. The microanatomy of B cell activation. Curr Opin Immunol.2009;21:258–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gray EE, Friend S, Suzuki K, Phan TG, Cyster JG. Subcapsular sinus macrophage fragmentation and CD169+ bleb acquisition by closely associated IL-17-committed innate-like lymphocytes. PLoS One.2012;7:e38258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Roth TL, Gray EE, et al. Migratory and adhesive cues controlling innate-like lymphocyte surveillance of the pathogen-exposed surface of the lymph node. Elife.2016;5 e18156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Huang Z, Yan R, et al. Vgamma4 gammadelta T Cells Provide an Early Source of IL-17A and Accelerate Skin Graft Rejection. J Invest Dermatol.2017;137:2513–2522. [DOI] [PubMed] [Google Scholar]
- 58.Fang V, Chaluvadi VS, Ramos-Perez WD, et al. Gradients of the signaling lipid S1P in lymph nodes position natural killer cells and regulate their interferon-gamma response. Nat Immunol.2017;18:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bankovich AJ, Shiow LR, Cyster JG. CD69 suppresses sphingosine-1-phosophate receptor-1 function through interaction with membrane helix 4. J Biol Chem.2010;285:22328–22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell.2010;140:845–858. [DOI] [PubMed] [Google Scholar]
- 61.Bogoslowski A, Butcher EC, Kubes P. Neutrophils recruited through high endothelial venules of the lymph nodes via PNAd intercept disseminating Staphylococcus aureus. Proc Natl Acad Sci U S A.2018;115:2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chtanova T, Schaeffer M, Han SJ, et al. Dynamics of Neutrophil Migration in Lymph Nodes during Infection. Immunity.2008;29:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Okada T, Miller MJ, Parker I, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol.2005;3:e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelly LM, Pereira JP, Yi T, Xu Y, Cyster JG. EBI2 guides serial movements of activated B cells and ligand activity is detectable in lymphoid and nonlymphoid tissues. J Immunol.2011;187:3026–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shiow LR, Rosen DB, Brdickova N, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature.2006;440:540–544. [DOI] [PubMed] [Google Scholar]
- 66.Grigorova IL, Panteleev M, Cyster JG. Lymph node cortical sinus organization and relationship to lymphocyte egress dynamics and antigen exposure. Proc Natl Acad Sci U S A.2010;107:20447–20452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murphy KM. Transcriptional control of dendritic cell development. Adv Immunol.2013;120:239–267. [DOI] [PubMed] [Google Scholar]
- 68.Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol.2019; 19:89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gatto D, Wood K, Caminschi I, et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nat Immunol.2013. 14:446–453. [DOI] [PubMed] [Google Scholar]
- 70.Yi T, Cyster JG. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. Elife.2013;2:e00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulvmar MH, Werth K, Braun A, et al. The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol.2014;15:623–630. [DOI] [PubMed] [Google Scholar]
- 72.Krishnaswamy JK, Gowthaman U, Zhang B, et al. Migratory CD11b(+) conventional dendritic cells induce T follicular helper cell-dependent antibody responses. Sci Immunol.2017;2 eaam9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sokol CL, Camire RB, Jones MC, Luster AD. The Chemokine Receptor CCR8 Promotes the Migration of Dendritic Cells into the Lymph Node Parenchyma to Initiate the Allergic Immune Response. Immunity.2018;49:449–463 e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Calabro S, Liu D, Gallman A, et al. Differential Intrasplenic Migration of Dendritic Cell Subsets Tailors Adaptive Immunity. Cell Rep.2016;16:2472–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature.2016;533:110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haynes NM, Allen CD, Lesley R, et al. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol.2007;179:5099–5108. [DOI] [PubMed] [Google Scholar]
- 77.Chen X, Ma W, Zhang T, Wu L, Qi H. Phenotypic Tfh development promoted by CXCR5-controlled re-localization and IL-6 from radiation-resistant cells. Protein Cell.2015;6:825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hong S, Zhang Z, Liu H, et al. B Cells Are the Dominant Antigen-Presenting Cells that Activate Naive CD4(+) T Cells upon Immunization with a Virus-Derived Nanoparticle Antigen. Immunity.2018. 49:695–708. [DOI] [PubMed] [Google Scholar]
- 79.Coffey F, Alabyev B, Manser T. Initial clonal expansion of germinal center B cells takes place at the perimeter of follicles. Immunity.2009;30:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kerfoot SM, Yaari G, Patel JR, et al. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity.2011;34:947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kitano M, Moriyama S, Ando Y, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity.2011;34:961–972. [DOI] [PubMed] [Google Scholar]
- 82.Chu C, Moriyama S, Li Z, et al. Anti-microbial Functions of Group 3 Innate Lymphoid Cells in Gut-Associated Lymphoid Tissues Are Regulated by G-Protein-Coupled Receptor 183. Cell Rep.2018;23:3750–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hargreaves DC, Hyman PL, Lu TT, et al. A coordinated change in chemokine responsiveness guides plasma cell movements. J Exp Med.2001;194:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cyster JG. Homing of antibody secreting cells. Immunol Rev.2003;194:48–60. [DOI] [PubMed] [Google Scholar]
- 85.Good-Jacobson KL, O’Donnell K, Belz GT, Nutt SL, Tarlinton DM. c-Myb is required for plasma cell migration to bone marrow after immunization or infection. J Exp Med.2015;212:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Allen CD, Ansel KM, Low C, et al. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol.2004;5:943–952. [DOI] [PubMed] [Google Scholar]
- 87.Inra CN, Zhou BO, Acar M, et al. A perisinusoidal niche for extramedullary haematopoiesis in the spleen. Nature.2015;527:466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rodda LB, Bannard O, Ludewig B, Nagasawa T, Cyster JG. Phenotypic and Morphological Properties of Germinal Center Dark Zone Cxcl12-Expressing Reticular Cells. J Immunol.2015. 195:4781–4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bannard O, Horton RM, Allen CD, et al. Germinal center centroblasts transition to a centrocyte phenotype according to a timed program and depend on the dark zone for effective selection. Immunity.2013;39:912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Huang HY, Rivas-Caicedo A, Renevey F, et al. Identification of a new subset of lymph node stromal cells involved in regulating plasma cell homeostasis. Proc Natl Acad Sci U S A.2018;115:E6826–E6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barinov A, Luo L, Gasse P, et al. Essential role of immobilized chemokine CXCL12 in the regulation of the humoral immune response. Proc Natl Acad Sci U S A.2017;114:2319–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Connell BJ, Sadir R, Baleux F, et al. Heparan sulfate differentially controls CXCL12alpha- and CXCL12gamma-mediated cell migration through differential presentation to their receptor CXCR4. Sci Signal.2016;9:ra107. [DOI] [PubMed] [Google Scholar]
- 93.Mortier A, Gouwy M, Van Damme J, Proost P, Struyf S. CD26/dipeptidylpeptidase IV-chemokine interactions: double-edged regulation of inflammation and tumor biology. J Leukoc Biol.2016;99:955–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nibbs RJ, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol.2013;13:815–829. [DOI] [PubMed] [Google Scholar]
- 95.Wang H, Beaty N, Chen S, et al. The CXCR7 chemokine receptor promotes B-cell retention in the splenic marginal zone and serves as a sink for CXCL12. Blood.2012;119:465–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Humpert ML, Pinto D, Jarrossay D, Thelen M. CXCR7 influences the migration of B cells during maturation. Eur J Immunol.2014;44:694–705. [DOI] [PubMed] [Google Scholar]
- 97.Shi W, Liao Y, Willis SN, et al. Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol.2015;16:663–673. [DOI] [PubMed] [Google Scholar]
- 98.Fooksman DR, Schwickert TA, Victora GD, et al. Development and migration of plasma cells in the mouse lymph node. Immunity.2010;33:118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng Q, Khodadadi L, Taddeo A, et al. CXCR4-CXCL12 interaction is important for plasma cell homing and survival in NZB/W mice. Eur J Immunol.2018;48:1020–1029. [DOI] [PubMed] [Google Scholar]
- 100.Brandtzaeg P, Johansen FE. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev.2005;206:32–63. [DOI] [PubMed] [Google Scholar]
- 101.Matsuo K, Nagakubo D, Yamamoto S, et al. CCL28-Deficient Mice Have Reduced IgA Antibody-Secreting Cells and an Altered Microbiota in the Colon. J Immunol.2018;200:800–809. [DOI] [PubMed] [Google Scholar]
- 102.Masahata K, Umemoto E, Kayama H, et al. Generation of colonic IgA-secreting cells in the caecal patch. Nature communications.2014;5:3704. [DOI] [PubMed] [Google Scholar]
- 103.Rojas OL, Probstel AK, Porfilio EA, et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell.2018. 176:610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hiepe F, Dorner T, Hauser AE, et al. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol.2011;7:170–178. [DOI] [PubMed] [Google Scholar]
- 105.Moser K, Kalies K, Szyska M, et al. CXCR3 promotes the production of IgG1 autoantibodies but is not essential for the development of lupus nephritis in NZB/NZW mice. Arthritis Rheum.2012;64:1237–1246. [DOI] [PubMed] [Google Scholar]
- 106.Piovesan D, Tempany J, Di Pietro A, et al. c-Myb Regulates the T-Bet-Dependent Differentiation Program in B Cells to Coordinate Antibody Responses. Cell Rep.2017;19:461–470. [DOI] [PubMed] [Google Scholar]
- 107.Kabashima K, Haynes NM, Xu Y, et al. Plasma cell S1P1 expression determines secondary lymphoid organ retention versus bone marrow tropism. J Exp Med.2006;203:2683–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gohda M, Kunisawa J, Miura F, et al. Sphingosine 1-phosphate regulates the egress of IgA plasmablasts from Peyer’s patches for intestinal IgA responses. J Immunol.2008;180:5335–5343. [DOI] [PubMed] [Google Scholar]
- 109.Green JA, Cyster JG. S1PR2 links germinal center confinement and growth regulation. Immunol Rev.2012;247:36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baeyens A, Fang V, Chen C, Schwab SR. Exit Strategies: S1P Signaling and T Cell Migration. Trends Immunol.2015;36:778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramos-Perez WD, Fang V, Escalante-Alcalde D, Cammer M, Schwab SR. A map of the distribution of sphingosine 1-phosphate in the spleen. Nat Immunol.2015;16:1245–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Green JA, Suzuki K, Cho B, et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol.2011;12:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muppidi JR, Schmitz R, Green JA, et al. Loss of signalling via Galpha13 in germinal centre B-cell-derived lymphoma. Nature.2014;516:254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Inoue A, Ishiguro J, Kitamura H, et al. TGFalpha shedding assay: an accurate and versatile method for detecting GPCR activation. Nat Methods.2012;9:1021–1029. [DOI] [PubMed] [Google Scholar]
- 115.Muppidi JR, Lu E, Cyster JG. The G protein-coupled receptor P2RY8 and follicular dendritic cells promote germinal center confinement of B cells, whereas S1PR3 can contribute to their dissemination. J Exp Med.2015;212:2213–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lu E, Woflreys F, Muppidi JR, Xu Y, Cyster JG. S-geranylgeranyl-L-glutathione is a ligand for human B-cell confinement receptor P2RY8. Nature.2019; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hardie DL, Johnson GD, Khan M, MacLennan IC. Quantitative analysis of molecules which distinguish functional compartments within germinal centers. Eur J Immunol.1993;23:997–1004. [DOI] [PubMed] [Google Scholar]
- 118.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol.2012;30:429–457. [DOI] [PubMed] [Google Scholar]
- 119.Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Curr Opin Immunol.2017;45:21–30. [DOI] [PubMed] [Google Scholar]
- 120.Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol.2008;20:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kara EE, Bastow CR, McKenzie DR, et al. Atypical chemokine receptor 4 shapes activated B cell fate. J Exp Med.2018;215:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weisel F, Shlomchik M. Memory B Cells of Mice and Humans. Annu Rev Immunol.2017;35:255–284. [DOI] [PubMed] [Google Scholar]
- 123.Laidlaw BJ, Schmidt TH, Green JA, et al. The Eph-related tyrosine kinase ligand Ephrin-B1 marks germinal center and memory precursor B cells. J Exp Med.2017;214:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Y, Shi J, Yan J, et al. Germinal-center development of memory B cells driven by IL-9 from follicular helper T cells. Nat Immunol.2017;18:921–930. [DOI] [PubMed] [Google Scholar]
- 125.Suan D, Krautler NJ, Maag JLV, et al. CCR6 Defines Memory B Cell Precursors in Mouse and Human Germinal Centers, Revealing Light-Zone Location and Predominant Low Antigen Affinity. Immunity.2017;47:1142–1153 e1144. [DOI] [PubMed] [Google Scholar]
- 126.Taylor JJ, Pape KA, Jenkins MK. A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med.2012;209:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schwickert TA, Alabyev B, Manser T, Nussenzweig MC. Germinal center reutilization by newly activated B cells. J Exp Med.2009;206:2907–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ise W, Fujii K, Shiroguchi K, et al. T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity.2018;48:702–715 e704. [DOI] [PubMed] [Google Scholar]
- 129.Reboldi A, Arnon TI, Rodda LB, et al. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science.2016;352:aaf4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aiba Y, Kometani K, Hamadate M, et al. Preferential localization of IgG memory B cells adjacent to contracted germinal centers. Proc Natl Acad Sci U S A.2010;107:12192–12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moran I, Nguyen A, Khoo WH, et al. Memory B cells are reactivated in subcapsular proliferative foci of lymph nodes. Nature communications.2018;9:3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Groom JR, Richmond J, Murooka TT, et al. CXCR3 Chemokine Receptor-Ligand Interactions in the Lymph Node Optimize CD4(+) T Helper 1 Cell Differentiation. Immunity.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell.2012;150:1235–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Onodera T, Takahashi Y, Yokoi Y, et al. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc Natl Acad Sci U S A.2012;109:2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bagnara D, Squillario M, Kipling D, et al. A Reassessment of IgM Memory Subsets in Humans. J Immunol.2015;195:3716–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Allie SR, Bradley JE, Mudunuru U, et al. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol.2018. 20:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hu S, Yang K, Yang J, Li M, Xiong N. Critical roles of chemokine receptor CCR10 in regulating memory IgA responses in intestines. Proc Natl Acad Sci U S A.2011;108:E1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kono M, Tucker AE, Tran J, et al. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J Clin Invest.2014;124:2076–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kono M, Conlon EG, Lux SY, et al. Bioluminescence imaging of G protein-coupled receptor activation in living mice. Nature communications.2017;8:1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sarris M, Sixt M. Navigating in tissue mazes: chemoattractant interpretation in complex environments. Curr Opin Cell Biol.2015;36:93–102. [DOI] [PubMed] [Google Scholar]
- 141.Murphy PM, Heusinkveld L. Multisystem multitasking by CXCL12 and its receptors CXCR4 and ACKR3. Cytokine.2018;109:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakagawa M, Schmitz R, Xiao W, et al. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J Exp Med.2014;211:2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kataoka K, Nagata Y, Kitanaka A, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet.2015;47:1304–1315. [DOI] [PubMed] [Google Scholar]
- 144.Suan D, Nguyen A, Moran I, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity.2015;42:704–718. [DOI] [PubMed] [Google Scholar]
- 145.Brachtel EF, Washiyama M, Johnson GD, et al. Differences in the germinal centres of palatine tonsils and lymph nodes. Scand J Immunol.1996;43:239–247. [PubMed] [Google Scholar]