

Abstract
Background
Between 40% and 70% of people with treatment‐resistant schizophrenia do not respond to clozapine, despite adequate blood levels. For these people, a number of treatment strategies have emerged, including the prescription of a second anti‐psychotic drug in combination with clozapine.
Objectives
To determine the clinical effects of various clozapine combination strategies with antipsychotic drugs in people with treatment‐resistant schizophrenia both in terms of efficacy and tolerability.
Search methods
We searched the Cochrane Schizophrenia Group's Study‐Based Register of Trials (to 28 August 2015) and MEDLINE (November 2008). We checked the reference lists of all identified randomised controlled trials (RCT). For the first version of the review, we also contacted pharmaceutical companies to identify further trials.
Selection criteria
We included only RCTs recruiting people of both sexes, aged 18 years or more, with a diagnosis of treatment‐resistant schizophrenia (or related disorders) and comparing clozapine plus another antipsychotic drug with clozapine plus a different antipsychotic drug.
Data collection and analysis
We extracted data independently. For dichotomous data, we calculated risk ratios (RRs) and 95% confidence intervals (CI) on an intention‐to‐treat basis using a random‐effects meta‐analysis. For continuous data, we calculated mean differences (MD) and 95% CIs. We used GRADE to create 'Summary of findings' tables and assessed risk of bias for included studies.
Main results
We identified two further studies with 169 participants that met our inclusion criteria. This review now includes five studies with 309 participants. The quality of evidence was low, and, due to the high degree of heterogeneity between studies, we were unable to undertake a formal meta‐analysis to increase the statistical power.
For this update, we specified seven main outcomes of interest: clinical response in mental state (clinically significant response, mean score/change in mental state), clinical response in global state (mean score/change in global state), weight gain, leaving the study early (acceptability of treatment), service utilisation outcomes (hospital days or admissions to hospital) and quality of life.
We found some significant differences between clozapine combination strategies for global and mental state (clinically significant response and change), and there were data for leaving the study early and weight gain. We found no data for service utilisation and quality of life.
Clozapine plus aripiprazole versus clozapine plus haloperidol
There was no long‐term significant difference between aripiprazole and haloperidol combination strategies in change of mental state (1 RCT, n = 105, MD 0.90, 95% CI ‐4.38 to 6.18, low quality evidence). There were no adverse effect data for weight gain but there was a benefit of aripiprazole for adverse effects measured by the LUNSERS at 12 weeks (1 RCT, n = 105, MD ‐4.90, 95% CI ‐8.48 to ‐1.32) and 24 weeks (1 RCT, n = 105, MD ‐4.90, 95% CI ‐8.25 to ‐1.55), but not 52 weeks (1 RCT, n = 105, MD ‐4.80, 95% CI ‐9.79 to 0.19). Similar numbers of participants from each group left the study early (1 RCT, n = 106, RR 1.27, 95% CI 0.72 to 2.22, very low quality evidence).
Clozapine plus amisulpride versus clozapine plus quetiapine
One study showed a significant benefit of amisulpride over quetiapine in the short term, for both change in global state (Clinical Global Impression (CGI): 1 RCT, n = 50, MD ‐0.90, 95% CI ‐1.38 to ‐0.42, very low quality evidence) and mental state (Brief Psychiatric Rating Scale (BPRS): 1 RCT, n = 50, MD ‐4.00, 95% CI ‐5.86 to ‐2.14, low quality evidence). Similar numbers of participants from each group left the study early (1 RCT, n = 56, RR 0.20, 95% CI 0.02 to 1.60, very low quality evidence)
Clozapine plus risperidone versus clozapine plus sulpiride
There was no difference between risperidone and sulpiride for clinically significant response, defined by the study as 20% to 50% reduction in Positive and Negative Syndrome Scale (PANSS) (1 RCT, n = 60, RR 0.82, 95% CI 0.40 to 1.68, very low quality evidence). There were similar equivocal results for weight gain (1 RCT, n = 60, RR 0.40, 95% CI 0.08 to 1.90, very low quality evidence) and mental state (PANSS total: 1 RCT, n = 60, MD ‐2.28, 95% CI ‐7.41 to 2.85, very low quality evidence). No‐one left the study early.
Clozapine plus risperidone versus clozapine plus ziprasidone
There was no difference between risperidone and ziprasidone for clinically significant response (1 RCT, n = 24, RR 0.80, 95% CI 0.28 to 2.27, very low quality evidence), change in global state CGI‐II score (1 RCT, n = 22, MD ‐0.30, 95% CI ‐0.82 to 0.22, very low quality evidence), change in PANSS total score (1 RCT, n = 16, MD 1.00, 95% CI ‐7.91 to 9.91, very low quality evidence) or leaving the study early (1 RCT, n = 24, RR 1.60, 95% CI 0.73 to 3.49, very low quality evidence).
Clozapine plus ziprasidone versus clozapine plus quetiapine
One study found, in the medium term, a superior effect for ziprasidone combination compared with quetiapine combination for clinically significant response in mental state (> 50% reduction PANSS: 1 RCT, n = 63, RR 0.54, 95% CI 0.35 to 0.81, low quality evidence), global state (CGI ‐ Severity score: 1 RCT, n = 60, MD ‐0.70, 95% CI ‐1.18 to ‐0.22, low quality evidence) and mental state (PANSS total score: 1 RCT, n = 60, MD ‐12.30, 95% CI ‐22.43 to ‐2.17, low quality evidence). There was no effect for leaving the study early (1 RCT, n = 63, RR 0.52, CI 0.05 to 5.41, very low quality evidence).
Authors' conclusions
The reliability of results from this review is limited, evidence is of low or very low quality. Furthermore, due to the limited number of included studies, we were unable to undertake formal meta‐analyses. As a consequence, any conclusions drawn from these findings are based on single, small‐sized RCTs with high risk of type II error. Properly conducted and adequately powered RCTs are required. Future trialists should seek to measure patient‐important outcomes such as quality of life, as well as clinical response and adverse effects.
Keywords: Adult; Female; Humans; Male; Amisulpride; Antipsychotic Agents; Antipsychotic Agents/adverse effects; Antipsychotic Agents/therapeutic use; Aripiprazole; Aripiprazole/adverse effects; Aripiprazole/therapeutic use; Clozapine; Clozapine/adverse effects; Clozapine/therapeutic use; Dibenzothiazepines; Dibenzothiazepines/therapeutic use; Drug Resistance; Drug Therapy, Combination; Haloperidol; Haloperidol/adverse effects; Haloperidol/therapeutic use; Piperazines; Piperazines/therapeutic use; Quetiapine Fumarate; Randomized Controlled Trials as Topic; Risperidone; Risperidone/therapeutic use; Schizophrenia; Schizophrenia/drug therapy; Sulpiride; Sulpiride/adverse effects; Sulpiride/analogs & derivatives; Sulpiride/therapeutic use; Thiazoles; Thiazoles/therapeutic use; Weight Gain
Plain language summary
Clozapine combined with different antipsychotic drugs for treatment‐resistant schizophrenia
Background
Schizophrenia is a severe mental illness that includes symptoms of hallucinations (sensations that appear real but are created by a person's mind), delusions (unrealistic beliefs) and apathy (lack of interest) which can significantly impact on people's lives. The main treatment is with antipsychotic medicines; however, some people with schizophrenia do not respond to antipsychotic medicines (called treatment resistance), which is a major challenge in the management of schizophrenia. The antipsychotic medicine, clozapine, is an effective medicine to use if treatment resistance occurs; however, it can cause unwanted side effects that include drowsiness, dizziness, headache, tremor (shaking), and excessive salivation (mouth watering). A more serious side effect is the reduction in the number of white blood cells, which can lead to an increased risk of infection. Clozapine is often used in combination with other antipsychotic medicines for treatment‐resistant schizophrenia, and this review investigated the clinical effects and safety of various clozapine combinations.
Study characteristics
We searched the Cochrane Schizophrenia Group Trial's Register in August 2015 and January 2016 and found five clinical studies involving 309 adults diagnosed with schizophrenia or related illnesses who were resistant to treatment but had shown some response to clozapine. The studies compared clozapine combined with the antipsychotic medicines (haloperidol, aripiprazole, amisulpride, quetiapine, sulpiride, ziprasidone and risperidone).
Key results
It was not possible to perform an overall analysis because the five studies were too different. Therefore, all results were based on data from one study per comparison.
Aripiprazole versus haloperidol combination: there was no overall difference in the effectiveness of the two treatment combinations; however, the aripiprazole combination caused fewer side effects.
Amisulpride versus quetiapine combination: the amisulpride combination was more effective in treating schizophrenia in comparison with the quetiapine combination.
Risperidone versus sulpiride combination: there were no overall differences in clinical effectiveness between these combinations.
Risperidone versus ziprasidone combination: neither combination showed superiority over the other in improving the symptoms of schizophrenia.
Ziprasidone versus quetiapine combination: the ziprasidone combination was more effective in improving both mental and global state than the quetiapine combination.
Quality of the evidence
The reliability of the evidence is questionable and was noted to be low or very low quality. Only a small number of studies, with limited data were available. No data were available for important measures such as quality of life and service use and no firm conclusions could be made. Further good‐quality evidence is needed.
Summary of findings
Summary of findings for the main comparison. CLOZAPINE + ARIPIPRAZOLE versus CLOZAPINE + HALOPERIDOL (Cipriani 2013).
| Clozapine + aripi prazole versus clozapine + haloperidol for treatment‐resistant schizophrenia | ||||||
|
Patient or population: people with treatment‐resistant schizophrenia Setting: inpatients and outpatients Intervention: aripiprazole (+ CLO) Comparison: haloperidol (+ CLO) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with haloperidol (+ CLO) | Risk with aripiprazole (+ CLO) | |||||
| Clinical response: no clinically significant response in mental state | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| Adverse effects: weight gain | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| Clinical response: mean score/change in global state | See comment | See comment | ‐ | ‐ | ‐ | No data reported. |
|
Clinical response: mean score/change in mental state: change in BPRS score from baseline (high = good), Long term (12 months) |
The mean score/change in mental state (change in BPRS from baseline) ‐ long term (12 months) was 0 | The mean score/change in mental state ‐ defined by change in BPRS from baseline ‐ long term (12 months) in the intervention group was 0.9 more (4.38 fewer to 6.18 more) | ‐ | 105 (1 RCT) | ⊕⊕⊝⊝ Low 1,2,3,4 | ‐ |
|
Leaving the study early: acceptability of treatment ‐ as measured by completion of trial Long term (12 months) |
Study population | RR 1.27 (0.72 to 2.22) | 106 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,5,6 | ‐ | |
| 283 per 1000 | 359 per 1000 (204 to 628) | |||||
| Moderate | ||||||
| 283 per 1000 | 359 per 1000 (204 to 628) | |||||
| Service utilisation outcomes: hospital admission or days in hospital | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| Quality of life/satisfaction with care for either recipients of care or carers: significant change in quality of life/satisfaction | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BPRS: Brief Psychiatric Rating Scale; CI: confidence interval; CLO: clozapine; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: downgraded by 1 level because high risk for performance bias (open label), but low risk for other biases (selection, detection, attrition, reporting).
2 Inconsistency and publication bias: not applicable (no meta‐analysis).
3 Indirectness: not downgraded because good applicability in terms of participants and interventions and rating scale measures participant‐important outcome (mental state).
4 Imprecision: downgraded by 1 level because underpowered to detect difference. Not downgraded by 2 levels because CI around mean difference did not include appreciable benefit and appreciable harm (total score on BPRS = 126).
5 Indirectness: downgraded by 1 level because leaving the study early a surrogate measure of acceptability of treatment.
6 Imprecision: downgraded by 2 level because underpowered to detect difference and CI around relative effect included appreciable benefit and harm (from less likely to leave study early to over two times more likely to leave study early).
Summary of findings 2. CLOZAPINE + AMISULPIRIDE versus CLOZAPINE + QUETIAPINE (Genc 2007).
| Clozapine + amisulpride versus clozapine + quetiapine for treatment‐resistant schizophrenia | ||||||
|
Patient or population: people with treatment‐resistant schizophrenia Setting: inpatients and outpatients Intervention: amisulpride (+ CLO) Comparison: quetiapine (+ CLO) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with quetiapine (+ CLO) | Risk with amisulpride (+ CLO) | |||||
| Clinical response: no clinically significant response in mental state | See comment | See comment | Not estimable | (1 RCT) | ‐ | No data reported. |
| Adverse effects: weight gain | See comment | See comment | Not estimable | (1 RCT) | ‐ | No data reported. |
|
Clinical response: mean score/change in global state: mean CGI score (high = poor) Short term (8 weeks) |
The mean score/change in global state (CGI) ‐ short term (8 weeks) was 0 | The mean score/change in global state (CGI) ‐ short term (8 weeks) in the intervention group was 0.9 fewer (1.38 fewer to 0.42 fewer) | ‐ | 50 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,3,4 | ‐ |
|
Clinical response: mean score/change in mental state: mean BPRS score (high = poor) Short term (8 weeks) |
The mean score/change in mental state (BPRS) ‐ short term (8 weeks) was 0 | The mean score/change in mental state (BPRS) ‐ short term (8 weeks) in the intervention group was 4 fewer (5.86 fewer to 2.14 fewer) | ‐ | 50 (1 RCT) | ⊕⊕⊝⊝ Low 1,2,4,5,6 | ‐ |
| Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | Study population | RR 0.20 (0.02 to 1.60) | 56 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,4,7 | ‐ | |
| 179 per 1000 | 36 per 1000 (4 to 286) | |||||
| Moderate | ||||||
| 179 per 1000 | 36 per 1000 (4 to 286) | |||||
| Service utilisation outcomes: hospital admission or days in hospital | See comment | See comment | Not estimable | (1 RCT) | ‐ | No data reported. |
| Quality of life/satisfaction with care for either recipients of care or carers: significant change in quality of life/satisfaction | See comment | See comment | Not estimable | (1 RCT) | ‐ | No data reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression; CI: confidence interval; CLO: clozapine; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: downgraded by 2 levels because high risk of reporting bias and unclear (so potentially high) risk of other biases (selection, performance, attrition).
2 Inconsistency and publication bias: not applicable (no meta‐analysis).
3 Indirectness: not downgraded because good applicability in terms of participants and interventions and rating score measures a participant‐important outcome (global state).
4 Imprecision: downgraded by 1 level because underpowered to detect difference. Not downgraded by 2 levels because CI around mean difference did not include appreciable benefit and appreciable harm (total score on CGI = 7).
5 Indirectness: not downgraded because good applicability in terms of participants and interventions and rating score measures a participant‐important outcome (mental state).
6 Imprecision: not downgraded because powered to detect difference and narrow CI.
7 Indirectness: downgraded by 1 level because leaving study early surrogate measure of participant‐important outcome (acceptability of treatment).
Summary of findings 3. CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE (Kong 2001).
| Clozapine + risperidone versus clozapine + sulpiride for treatment‐resistant schizophrenia | ||||||
|
Patient or population: people with treatment‐resistant schizophrenia Setting: inpatients Intervention: risperidone (+ CLO) Comparison: sulpiride (+ CLO) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with Sulpiride (+ CLO) | Risk with Risperidone (+ CLO) | |||||
| Clinical response: no clinically significant response in mental state: 20% to 50% reduction in PANSS total score | Study population | RR 0.82 (0.40 to 1.68) | 60 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,3,4 | ‐ | |
| 367 per 1000 | 301 per 1000 (147 to 616) | |||||
| Moderate | ||||||
| 367 per 1000 | 301 per 1000 (147 to 616) | |||||
| Adverse effects: weight gain | Study population | RR 0.40 (0.08 to 1.90) | 60 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,4,5 | ‐ | |
| 167 per 1000 | 67 per 1000 (13 to 317) | |||||
| Moderate | ||||||
| 167 per 1000 | 67 per 1000 (13 to 317) | |||||
| Clinical response: mean score/change in global state | See comment | See comment | ‐ | (1 RCT) | ‐ | No data reported. |
| Clinical response: mean score/change in mental state: mean PANSS total score (high = poor) | The mean score/change in mental state (PANSS total) was 0 | The mean score/change in mental state (PANSS total) in the intervention group was 2.28 undefined fewer (7.41 fewer to 2.85 more) | ‐ | 60 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,6,7 | ‐ |
| Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | Study population | Not estimable | 60 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,8,9 | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Service utilisation outcomes: hospital admission or days in hospital | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| Quality of life/satisfaction with care for either recipients of care or carers: significant change in quality of life/satisfaction | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CLO: clozapine; PANSS: Positive and Negative Syndrome Scale; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: downgraded by 2 levels because unclear (so potentially high) risk of biases (selection, performance, detection, reporting).
2 Inconsistency and publication bias: not applicable (no meta‐analysis).
3 Indirectness: downgraded by 1 level because unclear population applicability (inclusion criteria not clearly specified). Not downgraded by 2 levels because rating scale measures participant‐important outcome (mental state).
4 Imprecision: downgraded by 2 levels because underpowered to detect difference and CI around relative effect includes appreciable benefit and harm.
5 Indirectness: downgraded by 1 level because unclear population applicability (inclusion criteria not clearly specified). Not downgraded by 2 levels because weight gain a direct measure of a participant‐important outcome.
6 Indirectness: downgraded by 1 level because unclear population applicability (inclusion criteria not clearly specified). Not downgraded by 2 levels because rating scale measures participant‐important outcome (mental state).
7 Imprecision: downgraded by 1 level because underpowered to detect difference. Not downgraded by 2 levels because CI around mean difference did not include appreciable benefit and appreciable harm (total score on PANSS = 120).
8 Indirectness: downgraded by 2 levels because unclear population applicability (inclusion criteria not clearly specified) and leaving the study early a surrogate measure of acceptability of treatment.
9 Imprecision: downgraded by 1 level because underpowered to detect difference. Not downgraded by 2 levels because no CI.
Summary of findings 4. CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE (Kuwilsky 2010).
| Clozapine + risperidone versus clozapine + ziprasidone for treatment‐resistant schizophrenia | ||||||
|
Patient or population: people with treatment‐resistant schizophrenia Setting: inpatients and outpatients Intervention: risperidone (+ CLO) Comparison: ziprasidone (+ CLO) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with ziprasidone (+ CLO) | Risk with risperidone (+ CLO) | |||||
| Clinical response: no clinically significant response in mental state: 20% reduction in PANSS total score Medium term (26 weeks) | Study population | RR 0.80 (0.28 to 2.27) | 24 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,3,4 | ‐ | |
| 417 per 1000 | 333 per 1000 (117 to 946) | |||||
| Moderate | ||||||
| 417 per 1000 | 333 per 1000 (117 to 946) | |||||
| Adverse effects: weight gain | See comment | See comment | Not estimable | ‐ | ‐ | No SDs reported. |
|
Clinical response: mean score/change in global state: mean CGI‐II Global improvement score (high = poor) Short term (6 weeks) |
The mean score/change in global state (CGI‐II Global improvement) ‐ short term (6 weeks) was 0 | The mean score/change in global state (CGI‐II global improvement) ‐ short term (6 weeks) in the intervention group was 0.3 fewer (0.82 fewer to 0.22 more) | ‐ | 22 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,5,6 | ‐ |
|
Clinical response: mean score/change in mental state: mean PANSS total score (high = poor) Medium term (26 weeks) |
The mean score/change in mental state (PANSS total) ‐ medium term (26 weeks) was 0 | The mean score/change in mental state (PANSS total) ‐ medium term (26 weeks) in the intervention group was 1 more (7.91 fewer to 9.91 more) | ‐ | 16 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,3,7 | ‐ |
|
Leaving the study early: acceptability of treatment ‐ as measured by completion of trial Long term (52 weeks) |
Study population | RR 1.60 (0.73 to 3.49) | 24 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,8,9 | ‐ | |
| 417 per 1000 | 667 per 1000 (304 to 1000) | |||||
| Moderate | ||||||
| 417 per 1000 | 667 per 1000 (304 to 1000) | |||||
| Service utilisation outcomes: hospital admission or days in hospital | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| Quality of life/satisfaction with care for either recipients of care or carers: significant change in quality of life/satisfaction | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; CLO: clozapine; PANSS: Positive and Negative Syndrome Scale; RCT: randomised controlled trial; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: downgraded by 2 levels because high risk of performance bias, detection bias, attrition bias, and reporting bias.
2 Inconsistency and publication bias: not applicable (no meta‐analysis).
3 Indirectness: not downgraded because good applicability in terms of participants and interventions and rating score measures a participant‐important outcome (mental state).
4 Imprecision: downgraded by 2 levels because underpowered to detect difference and CI around relative effect includes appreciable benefit and harm (from less likely to over two times more likely to have no clinical response in mental state defined by PANSS 20% reduction).
5 Indirectness: not downgraded because good applicability (participants and interventions), and rating score measures a participant‐important outcome (global state).
6 Imprecision: downgraded by 1 level because underpowered to detect difference. Not downgraded by 2 levels because CI around mean difference does not include appreciable benefit and appreciable harm (total score on CGI = 7).
7 Imprecision: downgraded by 1 level because underpowered to detect difference. Not downgraded by 2 levels because CI around mean difference does not include appreciable benefit and appreciable harm (total score on PANSS = 120).
8 Indirectness: downgraded by 1 level because leaving the study early a surrogate for participant‐important outcome (acceptability of treatment).
9 Indirectness: downgraded by 2 levels because underpowered to detect difference and CI around relative effect includes appreciable benefit and harm (from less likely to over three times more likely to leave the study early).
Summary of findings 5. CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE (Wen 2015).
| Clozapine + ziprasidone versus clozapine + quetiapine for treatment‐resistant schizophrenia | ||||||
|
Patient or population: people with treatment‐resistant schizophrenia Setting: inpatients and outpatients Intervention: ziprasidone (+ CLO) Comparison: quetiapine (+ CLO) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with quetiapine (+ CLO) | Risk with ziprasidone (+ CLO) | |||||
|
Clinical response: no clinically significant response in mental state: ≥ 50% reduction in PANSS total score Medium term (12 weeks) |
Study population | RR 0.54 (0.35 to 0.81) | 63 (1 RCT) | ⊕⊕⊝⊝ Low 1,2,3,4 | ‐ | |
| 844 per 1000 | 456 per 1000 (295 to 683) | |||||
| Moderate | ||||||
| 844 per 1000 | 456 per 1000 (295 to 683) | |||||
| Adverse effects: weight gain | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
|
Clinical response: mean score/change in global state: mean CGI‐S score (high = poor) Medium term (12 weeks) |
The mean score/change in global state (CGI‐S) ‐ medium term (12 weeks) was 0 | The mean score/change in global state (CGI‐S) ‐ medium term (12 weeks) in the intervention group was 0.7 fewer (1.18 fewer to 0.22 fewer) | ‐ | 60 (1 RCT) | ⊕⊕⊝⊝ Low 1,2,4,5 | ‐ |
|
Clinical response: mean score/change in mental state: mean PANSS total score (high = poor) Medium term (12 weeks) |
The mean score/change in mental state (PANSS total) ‐ medium term (12 weeks) was 0 | The mean score/change in mental state (PANSS total) ‐ medium term (12 weeks) in the intervention group was 12.3 fewer (22.43 fewer to 2.17 fewer) | ‐ | 60 (1 RCT) | ⊕⊕⊝⊝ Low 1 2 3 4 | ‐ |
| Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | Study population | RR 0.52 (0.05 to 5.41) | 63 (1 RCT) | ⊕⊝⊝⊝ Very low 1,2,6,7 | ‐ | |
| 63 per 1000 | 33 per 1000 (3 to 338) | |||||
| Moderate | ||||||
| 63 per 1000 | 33 per 1000 (3 to 338) | |||||
| Service utilisation outcomes: hospital admission or days in hospital | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| Quality of life/satisfaction with care for either recipients of care or carers: significant change in quality of life/satisfaction | See comment | See comment | Not estimable | ‐ | ‐ | No data reported. |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CGI ‐S: Clinical Global Impression – Severity; CI: confidence interval; CLO: clozapine; PANSS: Positive and Negative Syndrome Scale; RCT: randomised controlled trial; RR: risk ratio; SD: standard deviation. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1 Risk of bias: downgraded by 2 levels because unclear (so potentially high) risk of biases (selection, performance, reporting).
2 Inconsistency and publication bias: not applicable (no meta‐analysis).
3 Indirectness: not downgraded because good applicability in terms of participants and interventions and rating scale measures a participant‐important outcome (mental state).
4 Imprecision: not downgraded because powered to detect difference and narrow CI.
5 Indirectness: not downgraded because good applicability (participants and interventions) and rating scale measures a participant‐important outcome (global state).
6 Indirectness: downgraded by 1 level because leaving the study early surrogate measure for participant‐important outcome (acceptability of treatment).
7 Imprecision: downgraded by 2 levels because underpowered to detect difference and CI around relative effect includes appreciable benefit and harm (from less likely to leave study early to five times more likely to leave study early).
Background
Description of the condition
Treatment resistance is one of the most important clinical challenges in the management of schizophrenia (Dold 2014). There is no uniform definition of treatment resistance, however a review by Suzuki 2011 found that the majority of trials stipulated non‐response to at least two previous antipsychotic drugs for at least six weeks. For people with treatment‐resistant schizophrenia, clozapine is considered first‐line (NICE 2014). A large number of randomised trials have demonstrated the superior antipsychotic efficacy of clozapine in both treatment non‐resistant (Leucht 2013) and resistant participants (Samara 2016). However, due the risk of agranulocytosis, clozapine is only recommended for treatment‐resistant people.
Description of the intervention
Between 40% and 70% of people with treatment‐resistant schizophrenia do not respond to clozapine (Taylor 2000). As a result, a number of approaches to clozapine‐resistant schizophrenia have emerged. These include pharmacological and non‐pharmacological methods. For pharmacological methods, Dold 2014 distinguish combination strategies, the simultaneous administration of two different antipsychotic drugs, from augmentation strategies, the addition of a drug of a different class, such as an antidepressant or mood stabiliser. Unfortunately, the terms combination and augmentation are often interchanged.
How the intervention might work
Clozapine is a polyvalent drug that lacks high potency dopamine receptor blockade. It is thought that adding on an antipsychotic drug with strong anti‐dopaminergic activity produces an additive effect and improve clinical response. A number of meta‐analyses have been carried out to determine the efficacy of clozapine combination treatment, with inconsistent results. Barbui 2009 identified 21 studies comparing clozapine combination treatment to clozapine monotherapy or placebo. They found a significant benefit of combination treatment when all studies were included, but no significant effect when the data from the six double‐blind studies were extracted and analysed separately. In comparison, Taylor 2012 conducted a meta‐analysis on 14 double‐blind, randomised, placebo‐controlled trials including 734 participants and found a small but significant benefit of combination treatment over placebo.
Why it is important to do this review
The original version of this review highlighted the paucity of studies comparing different clozapine combination treatment strategies, and the methodological shortcomings of the included trials. In 2015, treatment‐resistant schizophrenia remained a big challenge in clinical practice. One review on the pharmacotherapy of treatment‐resistant schizophrenia concluded that there is no sufficient convincing evidence to recommend combination strategies generally (Dold 2014). However, on the basis of scientific reasoning, the authors suggested choosing two antipsychotic drugs with a different receptor binding profile, for example a multi‐receptor antagonist such as clozapine and a potent D2 antagonist. This pragmatic view is incorporated into treatment guidelines (NICE 2014). This review is needed to provide an evidence base for recommendations on combination treatment in people with schizophrenia who are clozapine resistant.
Objectives
To determine the clinical effects of various clozapine combination strategies with antipsychotic drugs in people with treatment‐resistant schizophrenia both in terms of efficacy and tolerability.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials (RCT). We planned to include 'double‐blind' trials if it was implied that the study was randomised, but none were described as such. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
We included people of both sexes, aged 18 years or more, with a diagnosis of treatment‐resistant schizophrenia or related disorders (e.g. schizoaffective disorder, schizophreniform disorder), however diagnosed. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994).
Types of interventions
Clozapine plus another antipsychotic drug versus
clozapine plus a different other antipsychotic drug.
Any dose and means of administration was acceptable.
Types of outcome measures
We divided outcomes into short term (less than 12 weeks), medium term (12 weeks up to but not including 52 weeks), and long term (52 weeks and longer).
Primary outcomes
1. Clinical response.
1.1. No clinically significant response in global state ‐ as defined by each of the studies.
1.2. No clinically significant response in mental state ‐ as defined by each of the studies.
2. Adverse effect.
2.1. Weight gain.
Secondary outcomes
1. Clinical response.
1.1. Mean score/change in global state ‐ as defined by each of the studies.
1.2. Mean score/change in mental state ‐ as defined by each of the studies.
1.3. No clinically significant response in mental state (positive symptoms) ‐ as defined by each of the studies.
1.4. Mean score/change in mental state (positive symptoms) ‐ as defined by each of the studies.
1.5. No clinically significant response in mental state (negative symptoms) ‐ as defined by each of the studies.
1.6. Mean score/change in mental state (negative symptoms).
1.7. Use of additional medication (other than anticholinergic drugs) for psychiatric symptoms.
2. Adverse effects.
2.1. General adverse events.
2.1.1. Death: suicide or any causes.
2.2. Specific adverse events.
2.2.1. Clinically significant extrapyramidal adverse effects ‐ as defined by each of the studies.
2.2.2. Mean score/change in extrapyramidal adverse effects.
2.2.3. Use of antiparkinsonian drugs (i.e. anticholinergic drugs).
2.2.4. Blood dyscrasias such as agranulocytosis.
2.2.5. Hypersalivation.
2.3. Other adverse effects (general or specific).
3. Leaving the study early.
3.1. Acceptability of treatment ‐ as measured by completion of trial.
4. Service utilisation outcomes.
4.1. Hospital admission.
4.2. Days in hospital.
5. Economic outcomes.
6. Quality of life/satisfaction with care for either recipients of care or carers.
6.1. Clinically important change in quality of life/satisfaction ‐ as defined by each of the studies.
6.2. Mean score/change in quality of life/satisfaction scale.
'Summary of findings' table
We used the GRADE approach to interpret findings (Schünemann 2011) and used GRADE profiler (GRADEpro) to import data from Review Manager 5 (RevMan 2011) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined and the sum of available data on all outcomes we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the 'Summary of findings' table.
Clinical response ‐ no clinically significant response in mental state ‐ as defined by each of the studies.
Adverse effect ‐ weight gain.
Clinical response ‐ mean score/change in global state ‐ as defined by each of the studies.
Clinical response ‐ mean score/change in mental state ‐ as defined by each of the studies.
Leaving the study early ‐ acceptability of treatment ‐ as measured by completion of trial.
Service utilisation outcomes ‐ hospital admission or days in hospital.
Quality of life/satisfaction with care for either recipients of care or carers ‐ significant change in quality of life/satisfaction ‐ as defined by each of the studies ‐ or mean score/change in quality of life/satisfaction.
Search methods for identification of studies
We have updated the methods section of this review in line with latest Cochrane Schizophrenia recommendations. The methods section of the previous versions of this review can be found in Appendix 1.
Electronic searches
Cochrane Schizophrenia Group's Study‐Based Register of Trials
On 28 August 2015, the Information Specialist searched the Register using the following search strategy:
(*Clozapine*) in Title, Abstract, OR Index Terms of REFERENCE OR in Intervention of STUDY
In such a study‐based register, searching the major concept retrieves all the synonyms and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics.
This register is compiled by systematic searches of major resources (including MEDLINE, Embase, AMED, BIOSIS, CINAHL, PsycINFO, PubMed, and registries of clinical trials) and their monthly updates, handsearches, grey literature, and conference proceedings (see Group's Module). There is no language, date, document type, or publication status limitations for inclusion of records into the register.
For previous searches, see Appendix 3.
Searching other resources
1. Reference searching
We checked reference lists of all identified studies for further relevant studies.
2. Personal contact
For the original search, we contacted the first author of each included study for information regarding unpublished trials and additional information. However, this was not done in this update.
Data collection and analysis
Selection of studies
The review authors (SB, OU, and MC) independently inspected all English language citations from the searches to identify relevant abstracts. The Chinese translators, Jun Xia and Juan Juan Ren did the same for the Chinese language citations. We obtained the full reports of the papers for more detailed inspection, before deciding whether the paper met the review criteria. We resolved any disagreement by consensus. There was no blinding to the names of authors, institutions, and journal of publication.
Data extraction and management
1. Extraction
Two review authors (SB and MC) independently extracted data from the newly included studies in English and resolved any disagreement by discussion with another review author (AC or UO). For the new Chinese language paper, translators Jun Xia and Juan Juan Ren extracted the data. Thus, double extraction was possible for all studies.
Where data were presented only in figures, we contacted the authors requesting the raw data. When there was no reply, two review authors (SB and MC) independently made estimations from the figures. Where estimations were within 0.2, they were averaged and rounded to one decimal point. Where there was greater than 0.2 discrepancy, we re‐examined the figure and obtained a third estimate. We felt imprecision was preferable to not including data in the analyses.
2. Management
2.1. Forms
We extracted data onto standard, simple forms.
2.2. Scale‐derived data
We included continuous data from rating scales only if:
the psychometric properties of the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000);
the measuring instrument had not been written or modified by one of the trialists for that particular trial.
Ideally, the measuring instrument should have been either a self‐report or completed by an independent rater or relative (not the therapist).
2.3. Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. However, calculation of change needs two assessments (baseline and endpoint) which can be difficult in unstable and difficult‐to‐measure conditions such as schizophrenia. We decided to primarily use endpoint data and only use change data if endpoint data were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences (Higgins 2011, Chapter 9.4.5.2)
2.4. Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion:
standard deviations (SD) and means are reported in the paper or obtainable from the authors; but see Dealing with missing data;
when a scale starts from the finite number zero, the SD, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996);
if a scale started from a positive value (such as Positive and Negative Syndrome Scale (PANSS) which can have values from 30 to 210) the calculation described above was modified to take the scale starting point into account. In these cases, skew is present if 2SD > (S ‐ Smin), where S is the mean score and Smin is the minimum score.
Endpoint scores on scales often have a finite start and end point and these rules can be applied. When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not.
We planned to enter skewed data from studies of less than 200 participants into additional tables rather than an analysis. This was not required as no meta‐analysis was performed.
2.5. Common measure
To facilitate comparison between trials, we intended to convert variables that could be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (e.g. mean days per month). However, this was not required.
2.6. Conversion of continuous to binary
Where possible, we planned to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It was generally assumed that if there had been a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS, Overall 1962) or the PANSS (Kay 1986), this could be considered as a clinically significant response (Leucht 2005a; Leucht 2005b). Where data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
Assessment of risk of bias in included studies
Two review authors (SB and MC) independently assessed the risk of bias for the new studies using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions to assess trial quality (Higgins 2011). This set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, such as sequence generation, allocation concealment, blinding, incomplete outcome data, and selective reporting. It was decided that where there were no or inadequate details, the risk of bias would be labelled as "unclear". We acknowledge that the risk of bias could alternatively have been labelled "high" in these cases. Indeed, when rating risk of bias as serious or very serious in the 'Summary of Findings' tables, it was decided where the risk of bias was predominantly unclear, this would correspond to a very serious rating, because of the high potential for bias. One review author (SB) updated the assessments made for the original studies to comply with the new format. AC supervised SB and MC in this process. We noted the level of risk of bias in both the text of the review (Risk of bias in included studies) and in the Characteristics of included studies table.
Measures of treatment effect
1. Binary data
For binary outcomes, we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000).
2. Continuous data
For continuous outcomes, we estimated mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference SMD). However, had scales of very considerable similarity been used, we would have presumed there was a small difference in measurement, and we would have calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
We identified no cluster trials. However, had clustering not been accounted for in primary studies, we would have presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intraclass correlation coefficients (ICC) for their clustered data and to adjust for this by using accepted methods (Gulliford 1999). If clustering has been incorporated into the analysis of primary studies, we would present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the ICC (design effect = 1 + (m ‐ 1) × ICC) (Donner 2002). If the ICC was not reported, we assumed it to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed taking into account ICCs and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we intended to only use data of the first phase of cross‐over studies. However, we identified no cross‐over trials.
3. Studies with multiple treatment groups
We identified no studies with more than two treatment arms. However, had this been the case, the additional treatment arms would be presented in comparisons if relevant. The binary data would be simply added and combined within the two‐by‐two table, and the continuous data combined following the formula in Section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systematic Reviews of Interventions. Where the additional treatment arms were not relevant, these data would not have been reproduced.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss of follow‐up, the findings of a trial must lose credibility (Xia 2009). We decided that, for any particular outcome, should more than 50% of data be unaccounted for, we would not reproduce these data or use them within the analyses. However, if more than 50% of those in one arm of a study were lost, but the total loss was less than 50%, we marked such data with (*) to indicate that such a result may well be prone to bias.
2. Binary
If attrition for a binary outcome was between 0% and 50% and where these data were not clearly described, we presented data on an intention‐to‐treat basis. Those leaving the study early are all assumed to have the same rates of negative outcome as those who completed, with the exception of the outcome of death and adverse effects. If any data were identified for these outcomes, we used the rate of those who stayed in the study ‐ in that particular arm of the trial ‐ for those who did not. We planned to undertake a sensitivity analysis testing how prone the primary outcomes were to change when 'completer' data only were compared to the intention‐to‐treat analysis using the above assumptions.
If attrition for a binary outcome was between 0% and 50% and outcomes of these people were described, we included these data as reported. Where these data were not clearly described, for the primary outcome we assumed the worst for each person who was lost, and for adverse effects we assumed rates similar to those among participants who did continue to have their data recorded.
3. Continuous data
3.1. Attrition
In the case where attrition for a continuous outcome was between 0% and 50% and completer‐only data were reported, we have reproduced these.
3.2. Standard deviations
SDs were not reported in one study, and presented only in figures in another. We planned to obtain the missing values from the authors. When this failed, and there were missing measures of variance for continuous data, but an exact standard error (SE) and CIs available for group means, and either P value or 't' value available for differences in mean, we calculated SDs according to the rules described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011): when only the SE is reported, SDs are calculated by the formula SD = SE × square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systematic Reviews of Interventions present detailed formula for estimating SDs from P values, t or F values, CIs, ranges or other statistics (Higgins 2011). If these formulae did not apply, we planned to calculate the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study's outcome and thus to lose information. We planned to examine the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3. Last observation carried forward
We anticipated that some studies would use the method of last observation carried forward (LOCF) within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). Therefore, where LOCF data were used in the trial, if less than 50% of the data were assumed, we reproduced these data and indicated that they were the product of LOCF assumptions.
Assessment of heterogeneity
No formal meta‐analysis was possible. As such, assessment of heterogeneity between trials was not required. The following outlines the methods we would have taken.
1. Clinical heterogeneity
We would have considered all included studies initially, without seeing comparison data, to judge clinical heterogeneity. We would have inspected all studies for clearly outlying situations or people which we had not predicted would arise. Should such outliers have arisen, we would have discussed them.
2. Methodological heterogeneity
We would have considered all included studies initially, without seeing comparison data, to judge methodological heterogeneity. We would have inspected all studies for clearly outlying methods which we had not predicted would arise. Should such outliers have arisen, we would have discussed them.
3. Statistical heterogeneity
3.1. Visual inspection
We would have visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2. Employing the I2 statistic
Heterogeneity between studies could have been investigated by considering the I2 method alongside the Chi2 P value. The I2 statistic provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed I2 statistic depends on magnitude and direction of effects and strength of evidence for heterogeneity (e.g. P value from the Chi2 test, or a CI for the I2 statistic). We interpreted an I2 statistic estimate of 50% of greater accompanied by a statistically significant Chi2 statistic as evidence of substantial levels of heterogeneity (Section 9.5.2 of the Cochrane Handbook for Systematic Reviews of Interventions; Higgins 2011). If we had found substantial levels of heterogeneity in the primary outcome, we would have explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
1. Protocol versus full study
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in Section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We tried to locate protocols of the included RCTs and compared outcomes in the protocol and the published report. This was possible for two out of five studies.
2. Funnel plot
We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We intended not to used funnel plots for outcomes where there were 10 or fewer studies, hence we have not included any.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. The random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. However, there is a disadvantage to the random‐effects model as it puts added weight onto small studies which often are the most biased ones. Depending on the direction of effect these studies can either inflate or deflate the effect size. We chose the random‐effects model for all analyses.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
No subgroup analysis was planned.
2. Investigation of heterogeneity
We planned to report if inconsistency between studies was high. First, we planned to check data had been entered correctly. Second, we planned to visually inspect the graph and successively remove outlying studies to see if heterogeneity was restored. Should this have occurred with no more than 10% of the data being excluded, we planned to present data. If not, we would not have pooled data and would have discussed issues.
Should unanticipated clinical or methodological heterogeneity have been obvious, we would have simply stated hypotheses regarding these for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
No sensitivity analyses were performed. The following describes the procedures we planned to follow.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way as to imply randomisation. For the primary outcomes, we planned to include these studies and if there was no substantive difference when the implied randomised studies were added to those with better description of randomisation, then we planned to use all data from these studies.
2. Assumptions for lost binary data
Where assumptions have to be made regarding people lost to follow‐up, we planned to compare the findings of the primary outcomes when we used our assumption compared with completer data only. If there was a substantial difference, we planned to report results and discuss them but continue to employ our assumption.
Where assumptions had to be made regarding missing SD data, we planned to compare the findings on primary outcomes when we used our assumption compared with completer data only. We planned to undertake a sensitivity analysis testing how prone results were to change when 'completer' data only were compared to the imputed data using the above assumption. If there was a substantial difference, we planned to report results and discuss them but continue to employ our assumption.
3. Risk of bias
We planned to analyse the effects of excluding trials that were at high risk of bias across one or more of the domains of randomisation (implied as randomised with no further details available) allocation concealment, blinding and outcome reporting for the meta‐analysis of the primary outcomes. If the exclusion of trials at high risk of bias did not substantially alter the direction of effect or the precision of the effect estimates, then we planned to include data from these trials in the analysis.
4. Imputed values
If required, we planned to undertake a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster‐randomised trials. If there were substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we would not have pooled data from the excluded trials with the other trials contributing to the outcome, but presented them separately.
5. Fixed and random effects
We used a random‐effects model to synthesis all data; however, we planned to also synthesise data for the primary outcomes using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates altered the significance of the results compared to the more evenly distributed weights in the random‐effects model.
Results
Description of studies
For substantive descriptions of studies see the Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The original search for this review (March and November 2008) yielded 1331 references of potentially eligible studies, of which we obtained 68 full‐text papers for a second assessment after checking titles and abstracts. After exclusion of papers not meeting the inclusion criteria (four studies not randomised, 14 did not include participants with treatment‐resistant schizophrenia, 50 did not meet the intervention criteria e.g. no combination treatment arm comparison), the original review included three RCTs (Genç 2007; Kong 2001; Zink 2009).
The search update (August 2015) yielded 358 additional references. We obtained nine full‐text articles for a second assessment of seven individual new studies, four of which were English language and reviewed by SB, MC and OU, and three of which were Chinese language and reviewed by translators Jun Xia and Juan Juan Ren. After exclusion of papers not meeting the inclusion criteria (two did not include people with treatment‐resistant schizophrenia, three did not meet the intervention criteria, e.g. no combination treatment arm), we added two additional RCTs to the review (Cipriani 2013a; Wen 2015). Cipriani 2013a reported the long‐term data from a study with an earlier reference (Barbui 2011). In addition, an update to Zink 2009 was identified with medium‐term and long‐term data (Kuwilsky 2010).
See Figure 1 which presents the PRISMA flow diagram of the updated version of review.
1.
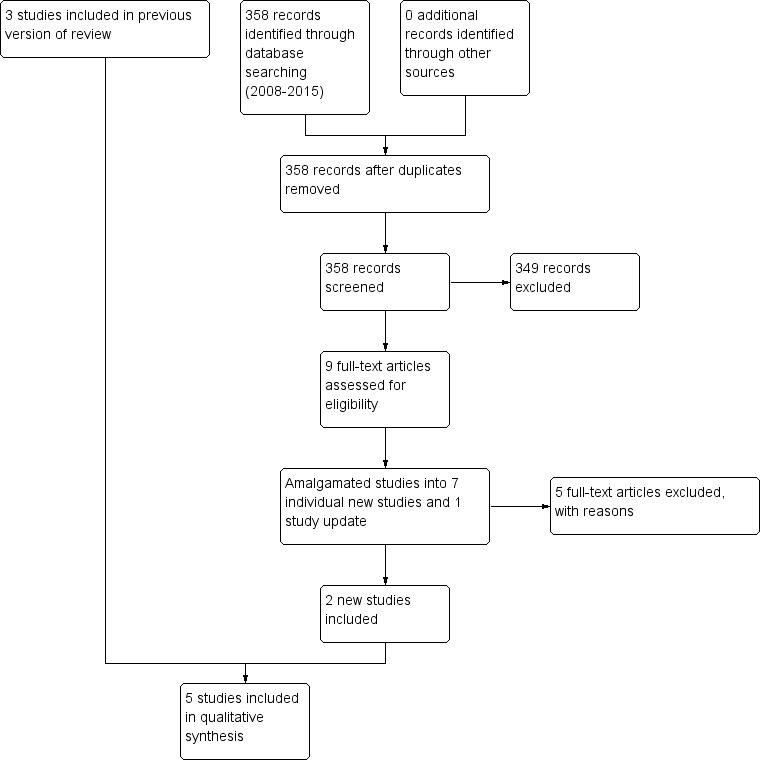
Study flow diagram (2015 update).
Included studies
The current version of the review includes five studies.
1. Study design
All studies used a parallel group design.
2. Length of trials
Genç 2007 and Kong 2001 were short‐term studies with a duration of eight weeks. Wen 2015 was a medium‐term study with a duration of 12 weeks. Both Cipriani 2013a and Kuwilsky 2010 were long‐term studies with a duration of 52 weeks.
3. Participants
All the participants had a diagnosis of schizophrenia or related disorders. Cipriani 2013a, Genç 2007, Kuwilsky 2010, and Wen 2015 used Diagnostic and Statistical Manual of Mental Disorders Fourth Edition (DSM‐IV) to provide diagnostic criteria. Kong 2001 used Chinese criteria (Chinese Classification of Mental Disorders, Second Edition, Revised; CCMD‐2‐R). In addition, the participants were all described as treatment‐resistant with partial response to clozapine.
The definition of partial response varied. Cipriani 2013a used persistent positive symptoms despite at least six months of treatment with clozapine 400 mg/day or greater. Genç 2007 used a score of greater than 45 on the BPRS or a rating of greater than 4 on at least two of the four BPRS positive symptom items, despite at least 12 weeks of clozapine 400 mg/day to 600 mg/day. Kuwilsky 2010 defined partial response as a PANSS total score of 65 or greater despite at least 12 weeks of clozapine 300 mg/day, and Wen 2015 used a PANSS score of 80 or greater and Clinical Global Impression ‐ Severity (CGI‐S) score of 4 or greater after at least 12 weeks of clozapine 400 mg/day or greater. Kong 2001 did not provide details of their definition.
Only Cipriani 2013a and Kuwilsky 2010 reported mean number of hospital admissions prior to randomisation by group, which ranged between three and seven. Most studies included both inpatients and outpatients. Only Kong 2001 included only inpatients. Three studies clearly reported inclusion and exclusion criteria (Genç 2007; Kuwilsky 2010; Wen 2015). All three excluded people with substance abuse. Cipriani 2013a reported only inclusion criteria and Kong 2001 reported neither inclusion or exclusion criteria.
4. Study size
All studies were small. The number of participants in each study were 106 (Cipriani 2013a), 56 (Genç 2007), 60 (Kong 2001), 24 (Kuwilsky 2010), and 63 (Wen 2015). In total, 309 participants participated in the five trials.
5. Interventions
No two studies compared the same two combination treatment strategies. Cipriani 2013a compared clozapine plus haloperidol to clozapine plus aripiprazole. Genç 2007 compared clozapine plus amisulpride to clozapine plus quetiapine. Kong 2001 compared clozapine plus risperidone to clozapine plus sulpiride. Kuwilsky 2010 compared clozapine plus ziprasidone to clozapine plus risperidone. Wen 2015 compared clozapine plus ziprasidone to clozapine plus quetiapine.
6. Dosing
In Cipriani 2013a, clinicians were allowed to prescribe the allocated pharmacological treatments (starting dose and dose changes) according to clinical status and circumstances. The mean baseline dose of clozapine was 413 mg/day (SD 157) for the haloperidol group and 418 mg/day (SD 141) for the aripiprazole group. The mean baseline dose of haloperidol was 2.1 mg/day (SD 1.3) and of aripiprazole was 8.7 mg/day (SD 3.9). Twelve week but not endpoint (52 week) mean doses were reported.
In Genç 2007, the mean baseline dose of clozapine was 550 mg/day (SD 127.09) in the amisulpride group and 536.95 mg/day (SD 125.42) in the quetiapine group. These doses remained stable throughout the study. The mean dose of amisulpride added was 437.03 mg/day (SD 104.32), and the maximum was 600 mg/day. The mean dose of quetiapine added was 595.65 mg/day (SD 125.21), and the maximum was 900 mg/day. Participants judged to be unable to tolerate the dose escalation schedule because of adverse effects were maintained at their maximum tolerated dose for the remainder of the study. No endpoint mean doses were reported.
Kong 2001 did not report the baseline clozapine dose, only the maximum, which was 400 mg/day in the risperidone group and 500 mg/day in the sulpiride group. Risperidone was started at 4 mg/day and the final dose was 6 mg/day; sulpiride was started at 800 mg/day and the final dose was 1200 mg/day. No endpoint mean doses were reported.
In Kuwilsky 2010, the mean baseline dose of clozapine was 437.5 mg/day (SD 140.4) in the risperidone group and 370.8 mg/day (SD 150.0) in the ziprasidone group. Risperidone and ziprasidone were titrated starting with doses of 1 mg and 20 mg respectively. The final doses followed clinical requirements, with the mean dose of risperidone 3.82 mg/day (SD 1.8) and ziprasidone 134 mg/day (SD 34.4). During the trial, reductions of clozapine by 50 mg per week were allowed, and the mean dose of clozapine at the end point (52 weeks) was 325 mg/day (SD 185.4) in the ziprasidone group and 450 mg/day (SD 168.3) in the risperidone group.
In Wen 2015, the baseline mean dose of clozapine was 479 mg/day (SD 56.5) in the ziprasidone group, and 481.3 mg/day (SD 51.7) in the quetiapine group. Ziprasidone and quetiapine were added during the first week. The dose of ziprasidone was titrated from 80 mg/day finishing at 120 mg/day to 160 mg/day, and the dose of quetiapine from 200 mg/day finishing at 400 mg/day to 750 mg/day. One week after ziprasidone or quetiapine was added, the dose of clozapine was reduced accordingly. No end point mean doses were reported.
7. Leaving the study early
In Cipriani 2013a, 19 participants in the aripiprazole group and 15 participants in the haloperidol group left early during the 12‐month follow‐up period. Reasons for leaving by 12 weeks were given and included lack of efficacy, acceptability problems, and lack of adherence. All randomised participants who received at least one dose of the investigational drugs were included in the intention‐to‐treat analysis (except one participant for whom rating scores were not completed at three months and who was subsequently excluded from analysis of these variables).
In Genç 2007, five participants in the quetiapine group discontinued the study within the first two weeks for reasons of exacerbation of psychotic symptoms (four participants) and lack of efficacy (one participant). One participant in the amisulpride group was missed in follow‐up after the second week. These six participants were excluded both from the analysis and from reporting of baseline characteristics.
In Kong 2001 all participants completed the study (no‐one left early).
In Kuwilsky 2010, more than 50% of participants had left by 52 weeks. Seven out of 12 remained in the ziprasidone group and four out of 12 remained in the risperidone group. Reasons for leaving early included akathisia, feelings of agitation and insufficient treatment response. Four participants withdrew their consent with no further explanation given. Participants who left the study early were excluded from further assessment.
In Wen 2015, one participant in the ziprasidone group and two participants in the quetiapine group left early due to adverse effects. These participants were excluded from the analyses of global and mental state outcomes, but included in the analyses of adverse events.
8. Outcomes scales
A variety of scales were used to assess clinical response and adverse events. We present details of the scales that provided useable data below.
8.1. Global state
8.1.1. Clinical Global Impression Scale (CGI)
The CGI is a collection of rating scales commonly used in studies of schizophrenia that enable clinicians to quantify severity of illness, global improvement, therapeutic effect, and adverse effects during therapy (Guy 1976). These mostly 7‐point scales, from 'normal' (1 point) to 'extremely ill' (7 points), require the clinician to compare the person to typical people in their clinical experience.
8.2. Mental state
8.2.1. Brief Psychiatric Rating Scale (BPRS)
The BPRS is used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms (Overall 1962). The scale has 18 items, and each item can be defined on a 7‐point scale varying from 'not present' (1 point) to 'extremely severe' (7 points). Scoring is from 18 to 126.
8.2.2. Positive and Negative Syndrome Scale (PANSS)
The PANSS scale was developed to evaluate the positive, negative, and general symptoms in schizophrenia (Kay 1986). The scale has 30 items and each item can be defined on a 7‐point scoring system varying from 'absent' (1 point) to 'extreme' (7 points). This scale can be divided into subscales for measuring the severity of general psychopathology, positive symptoms, negative symptoms, mania (excited component), and aggression (Supplemental Aggression Risk Profile). Total PANSS score is from 30 to 210. Higher scores indicate more pronounced symptomatology.
8.2.3. Hamilton Rating Scale for Depression (HAMD)
The HAMD instrument is designed to be used only on people already diagnosed as having an affective disorder of depressive type (Hamilton 1960). It is used for quantifying the results of an interview, and its value depends entirely on the skill of the interviewer in eliciting the necessary information. The scale contains 17 variables measured on either a 5‐point, from 'absent' (0 points) to 'very severe' (4 points), or a 3‐point rating scale, the latter being used where quantification of the variable is either difficult or impossible. Among the variables are depressed mood, suicide, work and loss of interest, retardation, agitation, gastrointestinal symptoms, general somatic symptoms, hypochondriasis, loss of insight, and loss of weight. It is useful to have two raters independently scoring a person at the same interview. The scores of the person are obtained by summing the scores of the two raters.
8.2.4. Global Assessment of Functioning (GAF)
GAF is a rating scale for a person's overall capacity of psychosocial functioning scoring from 1 to 100 (APA 2004). Higher scores indicate a higher level of functioning.
8.2.5. Scale for the Assessment of Positive Symptoms (SAPS)
The SAPS measures positive symptoms in schizophrenia (Andreasen 1984a). It has 35 items split into four domains (hallucinations, delusions, bizarre behaviour, and positive formal thought disorder), each rated from 'absent' (0 points) to severe (5 points).
8.2.6. Scale for the Assessment of Negative Symptoms (SANS)
The SANS measures negative symptoms in schizophrenia (Andreasen 1984b). It has 26 items split into five domains (affective flattening or blunting, alogia, avolition, anhedonia, and attention), each rated from 'absent' (0 points) to 'severe' (5 points).
8.3. Adverse effects scales
8.3.1. Simpson Angus Scale (SAS)
The SAS is a 10‐item scale, with a scoring system of 0 points to 4 points for each item, measures drug‐induced parkinsonism, a short‐term drug‐induced movement disorder (Simpson 1970). A low score indicates low levels of parkinsonism.
8.3.2. Extrapyramidal Symptom Rating Scale (EPS)
The EPS consists of a questionnaire relating to parkinsonism, akathisia, dystonia, and dyskinesia, with seven items scored on a 4‐point scale from 'absent' (0 points) to 'severe' (3 points) symptoms, and a physician's examination for parkinsonism and akathisia (seven items), dystonia (one item), and dyskinetic movement (seven items), all scored on a 7‐point scale from 0 to 6 depending on severity and frequency (Chouinard 1980). The clinician also completes four clinical global impression scores for the severity of dyskinesia, parkinsonism, dystonia, and akathisia. High scores indicate severe levels of movement disorder.
8.3.3. Hillside Akathisia Scale (HAS)
The HAS consists of two subjective and three objective items which the assessor rates on a 5‐point scale from 'absent' (0 points) to 'present and not controllable' (4 points) (Fleischhacker 1989). There is also a rating from 0 to 7 of severity of akathisia according to clinical experience, and improvement in condition compared to admission.
8.3.4. Liverpool University Neuroleptic Side Effect Rating Scale (LUNSERS)
The LUNSERS is a self‐assessment tool for measuring the adverse effects of antipsychotic medications (Day 1995). There are 41 questions covering extrapyramidal, psychic, anticholinergic, other autonomic, hormonal, allergic, and miscellaneous adverse effects. In addition, there are 10 'red‐herrings' (questions which are intended to be misleading or distracting) designed to test for over‐rating. It is a check‐box format with a 5‐point scale from 'not at all' (0 points) to 'very much' (4 points). A high score indicates a high adverse‐effect rating.
8.3.5. Udvalg for Kliniske Undersgelser (UKU) Side Effects Rating Scale
The UKU is a clinician‐rated score based on 48 items covering psychic, neurological, autonomic, and miscellaneous adverse effects, scored from 0 points to 3 points in severity over the last three days (Lingjærde 1987). In addition, there is a 4‐point scale for effect on daily performance from 'no side effects' (0 points) to 'side effects that interfere markedly with the participant's performance' (3 points).
9. Missing outcomes
No studies reported data on service utilisation outcomes, economic outcomes, or quality of life/satisfaction with care for either recipients of care or carers.
Excluded studies
In the original review, we obtained 24 full‐text papers for a second assessment, of which 21 studies did not meet inclusion/exclusion criteria. In the update, we obtained nine full‐text papers for a second assessment, two of which were publications of the same study. We excluded five studies (see Characteristics of excluded studies table for details).
Awaiting classification
There are no trials awaiting classification.
Ongoing studies
There are no ongoing studies we are aware of.
Risk of bias in included studies
For graphical representations of our judgements of risk of bias, refer to Figure 2 and Figure 3. For full details of judgements see 'Risk of bias' tables.
2.
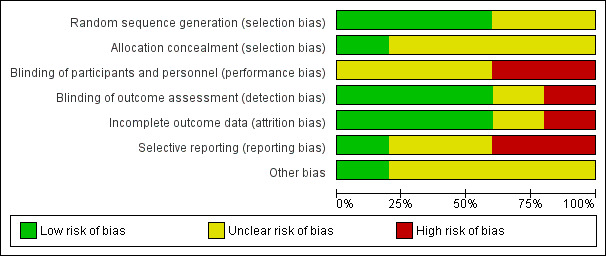
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
3.

Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
Allocation
Four out of five studies were described as randomised, but only Cipriani 2013a and Kuwilsky 2010 provided adequate details to be rated low risk for random sequence generation. In both of these studies, a trial biostatistician was responsible for randomisation using a computer‐based method. Kong 2001 provided insufficient information to comment on allocation.
The remainder were rated as unclear. It was noted that in Kong 2001, the baseline characteristics of participants in the two groups (duration of illness, mean score on PANSS) were very similar. Considering that this study recruited only 30 participants per arm, it is difficult to explain this scenario by means of a proper randomisation or by chance alone.
Only Cipriani 2013a provided details of allocation concealment. In this study, recruiting physicians were asked to contact an administrator at the co‐ordinating site by telephone, who accessed a computerised system that provided the participant's allocated treatment. The administrator had no access to the randomisation lists, and the site investigators did not know the randomisation block size. This way, the treatment allocation was fully concealed, and this study was rated low risk. All other studies were rated as unclear.
Blinding
Both Cipriani 2013a and Kuwilsky 2010 had a naturalistic, open‐label design. The limitation of this study design is a high risk of performance bias, although this may be less problematic in head‐to‐head trials as compared to placebo controlled. Even though not clearly reported in the paper, it seems that Genç 2007 was an open study (participants and providers were probably aware of the allocated treatment) and thus also at high risk of performance bias. Wen 2015 was described as single blind, so rated as unclear risk for performance bias.
Evaluation of outcomes in Cipriani 2013a, Genç 2007, and Wen 2015 was carried out by blinded assessors. As a result, these studies had a low risk of detection bias. This was not the case for Kuwilsky 2010, where assessors were aware of the allocated treatment.
Kong 2001 did not report on blinding, so the risk of both performance and detection bias was rated unclear.
Incomplete outcome data
In Cipriani 2013a, 19 participants in the aripiprazole group and 15 participants in the haloperidol group dropped out during the 12‐month follow‐up period. Reasons for leaving early by 12 weeks were given and were balanced between groups. All randomised participants who received at least one dose of investigation drugs were included in the intention‐to‐treat analysis of their primary outcome (leaving the study early). Only one participant was not included in the analysis of the BPRS and LUNSERS continuous outcomes due to failure to complete these rating scales at three months. Therefore, this study was rated as low risk of attrition bias.
In Genç 2007, five participants in the quetiapine group dropped out and one participant in the amisulpride group was missed at follow‐up at two weeks. These six participants were excluded from the analysis and from the reporting of baseline characteristics. There was no significant difference between groups, but this did not confirm the absence of bias, especially because this is a small study (n = 56). Moreover, reasons for incomplete data are not balanced between groups. Therefore, this study was rated as unclear risk for attrition bias.
In Kong 2001, all participants randomised completed the trial, so was rated low risk for attrition bias.
In Kuwilsky 2010, more than 50% of participants had dropped out by 52 weeks. There was no intention‐to‐treat analysis. As a result, this study is rated high risk for attrition bias. Moreover, the 52‐week data has not been included in our analyses, as per our protocol.
In Wen 2015, the three participants who dropped out were excluded from the analyses of global and mental state outcomes, but included in the analyses of adverse events. Since the attrition rate was less than 5%, and reasons were balanced between groups, the study was rated as low risk.
Selective reporting
All primary outcomes in Cipriani 2013a were prespecified in the trial protocol (Nosè 2009), and so it was rated low risk for reporting bias. However, it was noted that some secondary outcomes (mean dose of clozapine, prolactin, QTc interval) were only reported at 12 weeks.
No protocol was available for Genç 2007. Alone this would lead to an unclear risk. However, because no SDs were given for various scales (BPRS, SAPS, SANS, CGI), this study was rated high risk for reporting bias.
The protocol for Kuwilsky 2010 (NCT00224315) detailed six primary outcome measures (PANSS, SANS, HAMD, GAF, CGI, and QTc) and six secondary outcome measures (bodyweight, extrapyramidal motor symptom, akathisia, prolactin, blood pressure, and heart rate). It is not clear from the protocol that the authors intended to report PANSS total, PANSS positive, PANSS negative, and PANSS global psychopathology separately. In addition, there were no SDs reported for some outcomes, such as CGI and GAF at 26 and 52 weeks, and weight gain. As a result, Kuwilsky 2010 was rated as high risk for reporting bias.
For Kong 2001 and Wen 2015, no protocol was available, so these studies were rated as unclear risk for reporting bias.
Other potential sources of bias
We did not identify any other potential sources of bias in one out of five of the included studies (Cipriani 2013a). We judged the remaining four studies as unclear in this respect (for various reasons).
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
The results of the analyses are presented below. Where there were no data available for the primary outcomes, we have highlighted this. However, we have chosen not to list the missing secondary outcomes, and the reader is referred to the Secondary outcomes section to see the full list. No studies reported data on service utilisation outcomes, economic outcomes, or quality of life/satisfaction with care for either recipients of care or carers.
Two studies reported data on clozapine dose during the follow‐up period. This was not an outcome specified in our protocol, but it has clinical relevance as combination treatment might have the benefit of reducing clozapine dose. In Cipriani 2013a, the mean dose of clozapine in the haloperidol group was 413 mg/day (SD 157) at baseline and 395 mg/day (SD 161) at 12 weeks. In the aripiprazole group, the mean dose was 418 mg/day (SD 141) at baseline and 421 mg/day (SD 142) at 12 weeks. There was no significant difference between groups at 12 weeks. In Kuwilsky 2010, the dose of clozapine was reported at baseline, six weeks, 26 weeks, and 52 weeks. In the risperidone group, the doses were 437.5 mg/day (SD 140.4), 406.8 mg/day (no SD reported), 422.2 mg/day (SD 128.4), and 450 mg/day (SD 168.3), respectively. In the ziprasidone group, the doses were 370.8 mg/day (SD 150.0), 361.4 mg/day (no SD reported), 307.1 mg/day (SD 171.8), and 325 mg/day (SD 185.4), respectively. The dose reduction of clozapine was significant in the ziprasidone group.
1. Comparison 1: CLOZAPINE + ARIPIRAZOLE versus CLOZAPINE + HALOPERIDOL
One study provided data (n = 106) (Cipriani 2013a).
1.1. Clinical response: mean score/change in mental state ‐ mean change in BPRS score from baseline (high = good)
The study reported change data for BPRS score. There was no difference in change in BPRS score between groups at 12 weeks (1 RCT, n = 105, MD ‐1.40, 95% CI ‐5.59 to 2.79), or 52 weeks (1 RCT, n = 105, MD 0.90, 95% CI ‐4.38 to 6.18) (Analysis 1.1).
1.1. Analysis.
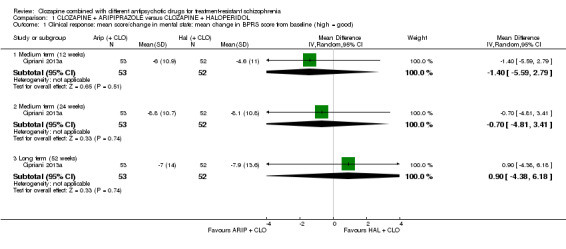
Comparison 1 CLOZAPINE + ARIPIPRAZOLE versus CLOZAPINE + HALOPERIDOL, Outcome 1 Clinical response: mean score/change in mental state: mean change in BPRS score from baseline (high = good).
1.2. Adverse effects: other adverse effects (general or specific) ‐ mean change in LUNSERS score from baseline (high = poor)
The study presented LUNSERS total score as a measure of subjective tolerability and reported mean change in score and SDs. There was a significant difference between groups in change of LUNSERS total score at 12 weeks favouring aripiprazole (1 RCT, n = 105, MD ‐4.90, 95% CI ‐8.48 to ‐1.32). However, at 52 weeks there was no significant difference between groups (1 RCT, n = 105, MD ‐4.80, 95% CI ‐9.79 to 0.19) (Analysis 1.2).
1.2. Analysis.
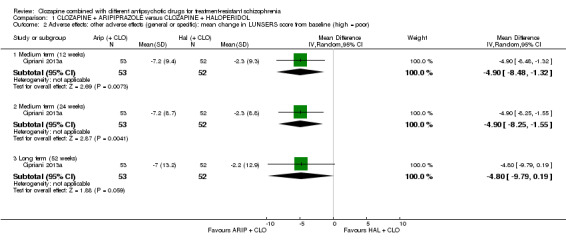
Comparison 1 CLOZAPINE + ARIPIPRAZOLE versus CLOZAPINE + HALOPERIDOL, Outcome 2 Adverse effects: other adverse effects (general or specific): mean change in LUNSERS score from baseline (high = poor).
1.3. Leaving the study early: acceptability of treatment ‐ as measured by completion of trial
There was no significant difference between groups in number of participants withdrawing from allocated treatment at 12 weeks (1 RCT, n = 106, RR 0.88, 95% CI 0.34 to 2.24), or 52 weeks (1 RCT, n = 106, RR 1.27, 95% CI 0.72 to 2.22) (Analysis 1.3).
1.3. Analysis.
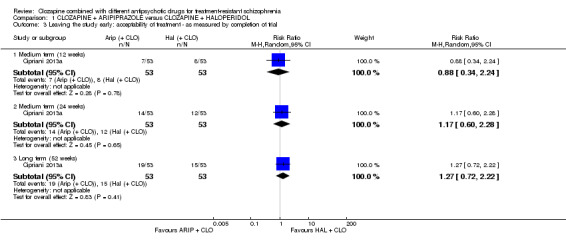
Comparison 1 CLOZAPINE + ARIPIPRAZOLE versus CLOZAPINE + HALOPERIDOL, Outcome 3 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial.
1.4. Missing outcomes
There were no usable data available for any other prespecified outcomes.
2. Comparison 2: CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE
One study provided data (n = 56) (Genç 2007).
2.1. Clinical response: 1. mean score/change in global state ‐ mean CGI score (high = poor)
Mean CGI scores were estimated from the figures, and SDs calculated from MDs and t values. There was a significant difference between groups at eight weeks favouring amisulpride (1 RCT, n = 50, MD ‐0.90, 95% CI ‐1.38 to ‐0.42) (Analysis 2.1).
2.1. Analysis.
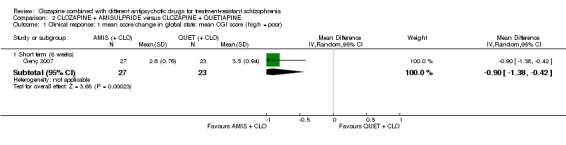
Comparison 2 CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE, Outcome 1 Clinical response: 1 mean score/change in global state: mean CGI score (high = poor).
2.2. Clinical response: 2a. mean score/change in mental state ‐ mean BPRS score (high = poor)
Mean scores were estimated from the figures, and SDs calculated from MDs and t value. There was a significant difference between groups for BPRS at eight weeks favouring amisulpride (1 RCT, n = 50, MD ‐4.00, 95% CI ‐5.86 to ‐2.14) (Analysis 2.2).
2.2. Analysis.
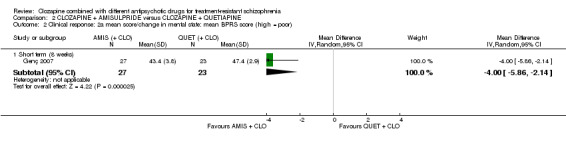
Comparison 2 CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE, Outcome 2 Clinical response: 2a mean score/change in mental state: mean BPRS score (high = poor).
2.3. Clinical response: 2b. mean score/change in mental state ‐ mean SAPS score (high = poor)
Mean scores were estimated from the figures, and SDs calculated from MDs and t value. There was a significant difference between groups for SAPS at eight weeks favouring amisulpride (1 RCT, n = 50, MD ‐6.90, 95% CI ‐12.82 to ‐0.98) (Analysis 2.3).
2.3. Analysis.
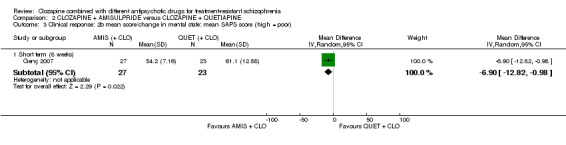
Comparison 2 CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE, Outcome 3 Clinical response: 2b mean score/change in mental state: mean SAPS score (high = poor).
2.4. Clinical response: 2c. mean score/change in mental state ‐ means SANS score (high = poor)
Mean scores were estimated from the figures, and SDs calculated from MDs and t values. There was a significant difference between groups for SANS at eight weeks favouring amisulpride (1 RCT, n = 50, MD ‐5.20, 95% CI ‐7.14 to ‐3.26) (Analysis 2.4).
2.4. Analysis.
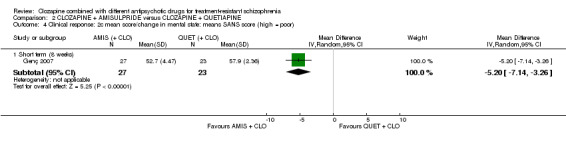
Comparison 2 CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE, Outcome 4 Clinical response: 2c mean score/change in mental state: means SANS score (high = poor).
2.5. Leaving the study early: acceptability of treatment ‐ as measured by completion of trial
There was no significant difference between groups in number of participants leaving the study early (1 RCT, n = 56, RR 0.20, 95% CI 0.02 to 1.60) (Analysis 2.5).
2.5. Analysis.
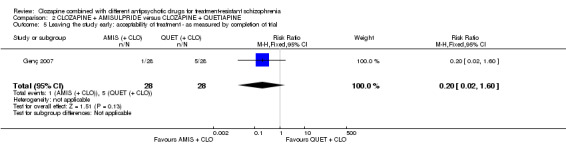
Comparison 2 CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE, Outcome 5 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial.
2.6. Missing outcomes
There were no usable data available for any other prespecified outcomes.
3. Comparison 3: CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE
3.1. Clinical response: no clinically significant response in mental state ‐ reduction in PANSS total score of 20% to 50%
Kong 2001 defined clinical improvement as a 20% to 50% reduction on PANSS total score. There was no significant difference between groups (1 RCT, n = 60, RR 0.82, 95% CI 0.40 to 1.68) (Analysis 3.1).
3.1. Analysis.
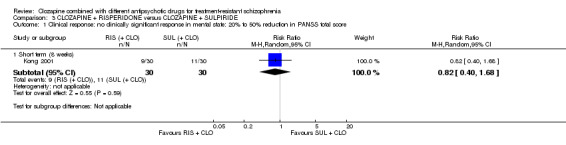
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 1 Clinical response: no clinically significant response in mental state: 20% to 50% reduction in PANSS total score.
3.2. Adverse effect: weight gain
There was no significant difference between groups in weight gain (1 RCT, n = 60, RR 0.40, 95% CI 0.08 to 1.90) (Analysis 3.2).
3.2. Analysis.
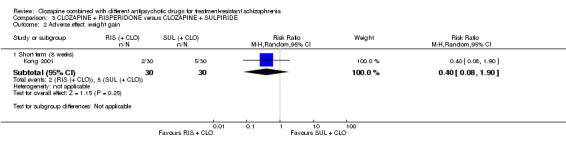
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 2 Adverse effect: weight gain.
3.3. Clinical response: 2a. mean score/change in mental state ‐ mean PANSS total score at endpoint (high = poor)
The mean PANSS total score was reported at the endpoint. There was no significant difference between groups (1 RCT, n = 60, MD ‐2.28, 95% CI ‐7.41 to 2.85) (Analysis 3.3).
3.3. Analysis.
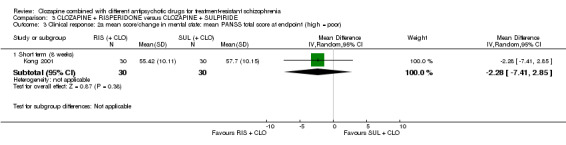
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 3 Clinical response: 2a mean score/change in mental state: mean PANSS total score at endpoint (high = poor).
3.4. Clinical response: 2b. mean score/change in mental state (positive symptoms) ‐ mean PANSS positive score at endpoint (high = poor)
The mean PANSS positive score was reported at the endpoint. This difference was significant, favouring risperidone (1 RCT, n = 60, MD ‐2.55, 95% CI ‐4.64 to ‐0.46) (Analysis 3.4).
3.4. Analysis.
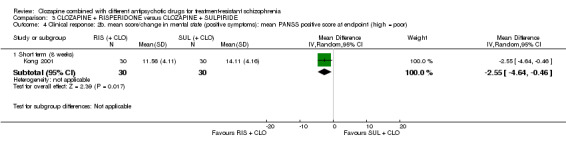
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 4 Clinical response: 2b. mean score/change in mental state (positive symptoms): mean PANSS positive score at endpoint (high = poor).
3.5. Clinical response: 2c. mean score/change in mental state (negative symptoms) ‐ mean PANSS negative score at endpoint (high = poor)
The mean PANSS negative score was reported at the endpoint. There was no significant difference between groups (1 RCT, n = 60, MD ‐0.54, 95% CI ‐3.19 to 2.11) (Analysis 3.5).
3.5. Analysis.
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 5 Clinical response: 2c. mean score/change in mental state (negative symptoms): mean PANSS negative score at endpoint (high = poor).
3.6. Adverse effects: specific adverse effects: hypersalivation
There was no significant difference between groups in hypersalivation (1 RCT, n = 60, RR 0.33, 95% CI 0.04 to 3.03) (Analysis 3.6).
3.6. Analysis.
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 6 Adverse effects: specific adverse effects: hypersalivation.
3.7. Leaving the study early: acceptability of treatment ‐ as measured by completion of trial
No‐one left early in either group.
3.8. Missing outcomes
There were no usable data available for any other prespecified outcomes.
4. Comparison 4: CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE
4.1. Clinical response: no clinically significant response in mental state ‐ reduction in PANSS total score of 20%
Kuwilsky 2010 defined a treatment response as a reduction of the PANSS total score by 20%. There was no significant difference between groups at six weeks (1 RCT, n = 24, RR 0.67, 95% CI 0.13 to 3.30) or 26 weeks (1 RCT, n = 24, RR 0.80, 95% CI 0.28 to 2.27) (Analysis 4.1). Since at 52 weeks more than 50% of the total numbers randomised were not accounted for, we did not include these data.
4.1. Analysis.
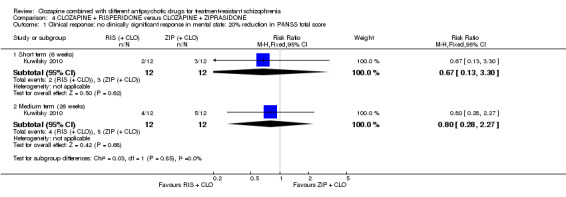
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 1 Clinical response: no clinically significant response in mental state: 20% reduction in PANSS total score.
4.2. Clinical response: no clinically significant response in mental state (positive symptoms) ‐ reduction in PANSS positive subscore of 20%
Kuwilsky 2010 defined a significant response as a 20% decrease in PANSS positive subscore. Only six‐week data were available. There was no significant difference between groups at six weeks (1 RCT, n = 24, RR 3.00, 95% CI 0.36 to 24.92) (Analysis 4.2).
4.2. Analysis.
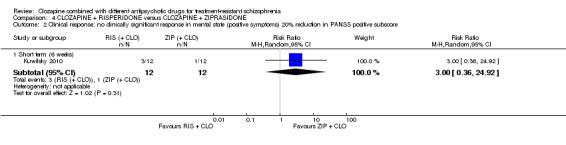
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 2 Clinical response: no clinically significant response in mental state (positive symptoms) 20% reduction in PANSS positive subscore.
4.3. Clinical response: 1a. mean score/change global state ‐ mean CGI subscale score (high = poor)
Kuwilsky 2010 reported mean CGI subscale scores (severity of illness, global improvement, therapeutic efficacy) at six, 26, and 52 weeks. However, SDs were only reported for the six‐week data. There was no significant difference between groups at six weeks (severity of illness: 1 RCT, n = 22, MD 0.20, 95% CI ‐0.32 to 0.72; global improvement: 1 RCT, n = 22, MD ‐0.30, 95% CI ‐0.82 to 0.22; therapeutic efficacy: 1 RCT, n = 22, MD ‐0.30, 95% CI ‐0.79 to 0.19) (Analysis 4.3).
4.3. Analysis.
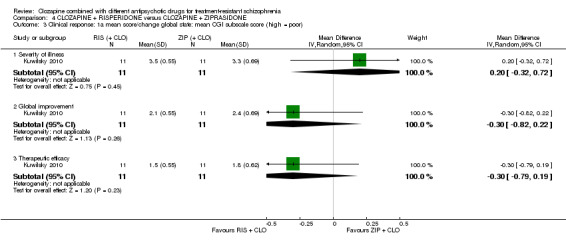
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 3 Clinical response: 1a mean score/change global state: mean CGI subscale score (high = poor).
4.4. Clinical response: 1b. mean score/change global state ‐ mean GAF score (high = good)
Kuwilsky 2010 reported mean GAF scores at six, 26 and 52 weeks. However, SDs were only reported for the six‐week data. There was no significant difference between groups at six weeks (1 RCT, n = 22, MD 0.00, 95% CI ‐7.84 to 7.84) (Analysis 4.4).
4.4. Analysis.
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 4 Clinical response: 1b mean score/change global state: mean GAF score (high = good).
4.5. Clinical response: 2a. mean score/change mental state ‐ mean HAMD score (high = poor)
For the HAMD scale, means and SDs were estimated from figures. There was a significant difference between groups at six weeks favouring risperidone (1 RCT, n = 22, MD ‐3.40, 95% CI ‐6.71 to ‐0.09). There was no significant difference at 26 weeks (1 RCT, n = 16, MD ‐0.70, 95% CI ‐5.35 to 3.95) (Analysis 4.5). Due to attrition of more than 50%, 52‐week data were not included.
4.5. Analysis.
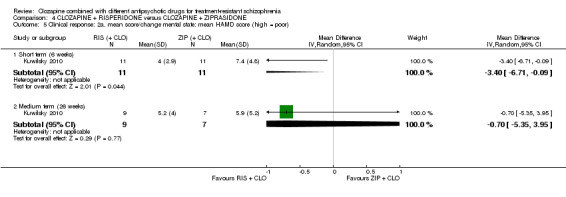
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 5 Clinical response: 2a. mean score/change mental state: mean HAMD score (high = poor).
4.6. Clinical response: 2b. mean score/change mental state ‐ mean PANSS total score (high = poor)
For PANSS total, means and SDs were estimated from the figures. There was no significant difference in PANSS total score between groups at six weeks (1 RCT, n = 22, MD ‐3.10, 95% CI ‐11.38 to 5.18) and 26 weeks (1 RCT, n = 16, MD 1.00, 95% CI ‐7.91 to 9.91) (Analysis 4.6). Due to attrition of more than 50%, 52‐week data were not included.
4.6. Analysis.
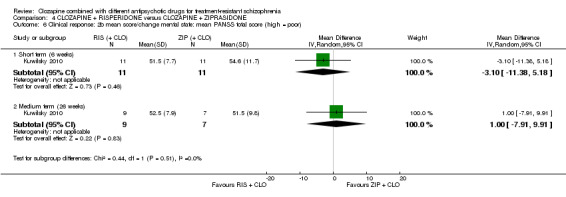
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 6 Clinical response: 2b mean score/change mental state: mean PANSS total score (high = poor).
4.7. Clinical response: 2c. mean score/change in mental state (positive symptoms) ‐ mean PANSS positive score (high = poor)
For PANSS positive scores, means and SDs were estimated from the figures. There was no significant difference in PANSS positive score between groups at six weeks (1 RCT, n = 22, MD ‐0.20, 95% CI ‐1.84 to 1.44) or 26 weeks (1 RCT, n = 16, MD ‐0.20, 95% CI ‐2.58 to 2.18) (Analysis 4.7). Due to attrition of more than 50%, 52‐week data were not included.
4.7. Analysis.
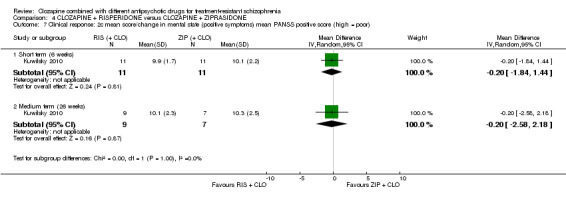
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 7 Clinical response: 2c mean score/change in mental state (positive symptoms) mean PANSS positive score (high = poor).
4.8. Clinical response: 2d. mean score/change in mental state (negative symptoms) ‐ mean PANSS negative score (high = poor)
For PANSS negative scores, means and SDs were estimated from the figures. There was no significant difference in PANSS negative score between groups at six weeks (1 RCT, n = 22, MD ‐1.20, 95% CI ‐4.63 to 2.23) or 26 weeks (1 RCT, n = 16, MD 1.50, 95% CI ‐2.66 to 5.66) (Analysis 4.8). Due to attrition of more than 50%, 52‐week data were not included.
4.8. Analysis.
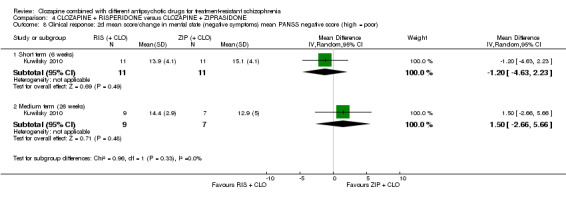
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 8 Clinical response: 2d mean score/change in mental state (negative symptoms) mean PANSS negative score (high = poor).
4.9. Clinical response: 2e. mean score/change in mental state (negative symptoms) ‐ mean SANS score (high = poor)
For SANS, there was no significant difference between groups at six weeks (1 RCT, n = 22, MD ‐4.00, 95% CI ‐17.55 to 9.55) or 26 weeks (1 RCT, n = 16, MD 1.80, 95% CI ‐14.31 to 17.91) (Analysis 4.9). Due to attrition of more than 50%, 52‐week data were not included.
4.9. Analysis.
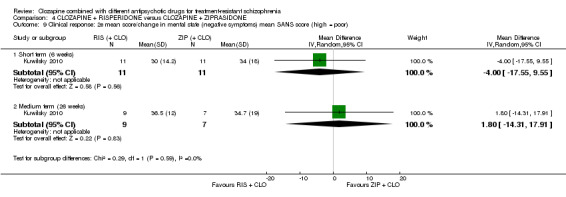
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 9 Clinical response: 2e mean score/change in mental state (negative symptoms) mean SANS score (high = poor).
4.10. Clinical response: 2f. mean score/change in global state ‐ mean PANSS global psychopathology score (high = poor)
For PANSS global psychopathology, there was no significant difference between groups at six weeks (1 RCT, n = 22, MD ‐1.60, 95% CI ‐6.60 to 3.40) or 26 weeks (1 RCT, n = 16, MD 0.00, 95% CI ‐5.83 to 5.83) (Analysis 4.10). Due to attrition of more than 50%, 52‐week data were not analysed.
4.10. Analysis.
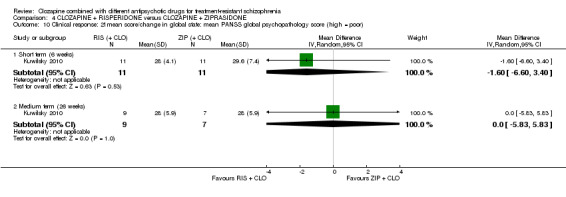
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 10 Clinical response: 2f mean score/change in global state: mean PANSS global psychopathology score (high = poor).
4.11. Adverse effects: specific adverse effects: mean score/change in extrapyramidal adverse effects ‐ mean EPS score (high = poor)
Mean EPS scores and SDs were estimated from the figures. There was no significant difference between groups at six weeks (1 RCT, n = 22, MD 0.60, 95% CI ‐0.67 to 1.87) or 26 weeks (1 RCT, n = 16, MD 0.30, 95% CI ‐0.63 to 1.23) (Analysis 4.11). Due to attrition of more than 50%, 52‐week data were not analysed.
4.11. Analysis.
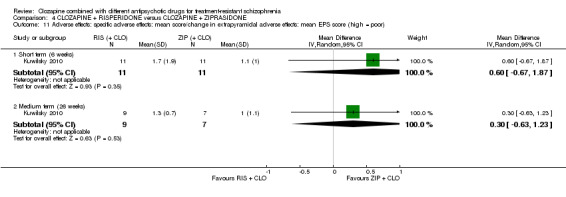
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 11 Adverse effects: specific adverse effects: mean score/change in extrapyramidal adverse effects: mean EPS score (high = poor).
We were not able to estimate the means and SDs of the HAS from the figures.
4.12. Adverse effects: other adverse effects (general or specific) ‐ mean CGI adverse effect scores (high = poor)
Kuwilsky 2010 reported mean CGI adverse effect scores at six, 26, and 52 weeks. However, SDs are only available for the six‐week data. There was no difference between groups for this adverse‐effect rating at six weeks (1 RCT, n = 22, MD ‐0.10, 95% CI ‐0.53 to 0.33) (Analysis 4.12).
4.12. Analysis.
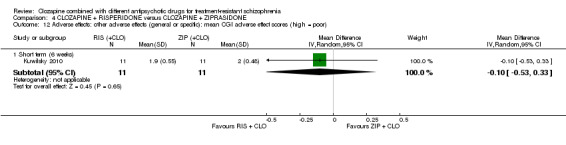
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 12 Adverse effects: other adverse effects (general or specific): mean CGI adverse effect scores (high = poor).
The authors also reported that clozapine adverse effects, such as hypersalivation, sedation, and weight gain, were evaluated on an observer‐based visual analogue scale between 1 (no adverse effects) and 10 (severe and intolerable adverse effects). There were no data available for this scale.
4.13. Leaving the study early: acceptability of treatment ‐ as measured by completion of trial
The sample study changed significantly during the trial. There was no significant difference between groups at six weeks (1 RCT, n = 24, RR 1.00, 95% CI 0.07 to 14.21), 26 weeks (1 RCT, n = 24, RR 0.60, 95% CI 0.18 to 1.97), or 52 weeks (1 RCT, n = 24, RR 1.60, 95% CI 0.73 to 3.49) (Analysis 4.13).
4.13. Analysis.
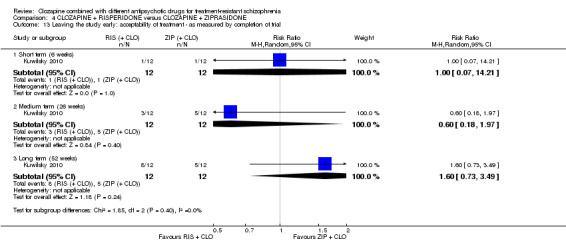
Comparison 4 CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE, Outcome 13 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial.
4.14. Missing outcomes
There were no usable data available for any other prespecified outcomes.
5. Comparison 5: CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE
5.1. Clinical response: 1a. no clinically significant response in mental state ‐ PANSS reduction 50% or greater
There was a significant difference between groups in the number of participants not achieving a 50% reduction or greater in PANSS total by 12 weeks favouring ziprasidone (1 RCT, n = 63, RR 0.54, 95% CI 0.35 to 0.81) (Analysis 5.1).
5.1. Analysis.
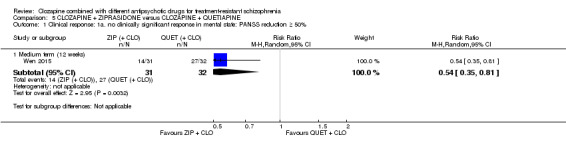
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 1 Clinical response: 1a. no clinically significant response in mental state: PANSS reduction ≥ 50%.
5.2. Clinical response: 1b. no clinically significant response in mental state ‐ PANSS reduction 25% or greater
There was no difference between groups for number not achieving a 25% reduction or greater in PANSS (1 RCT, n = 63, RR 0.65, 95% CI 0.38 to 1.10) (Analysis 5.2).
5.2. Analysis.
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 2 Clinical response: 1b. no clinically significant response in mental state: PANSS reduction ≥ 25%.
5.3. Clinical response: 2a. mean score/change global state ‐ mean CGI‐S score (high = poor)
Wen 2015 reported mean CGI severity of illness scores and SDs for all participants who completed the 12‐week follow‐up period. There was a significant difference between groups on CGI‐S favouring ziprasidone (1 RCT, n = 60, MD ‐0.70, 95% CI ‐1.18 to ‐0.22) (Analysis 5.3).
5.3. Analysis.
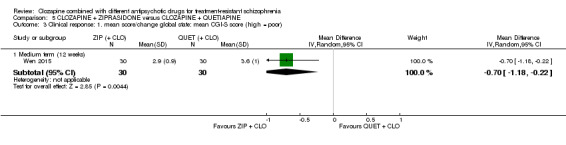
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 3 Clinical response: 1. mean score/change global state: mean CGI‐S score (high = poor).
5.4. Clinical response: 2b. mean score/change mental state ‐ mean PANSS total score (high = poor)
PANSS total means and SDs were reported at 12 weeks. There was a significant difference between groups favouring ziprasidone (1 RCT, n = 60, MD ‐12.30, 95% CI ‐22.43 to ‐2.17) (Analysis 5.4).
5.4. Analysis.
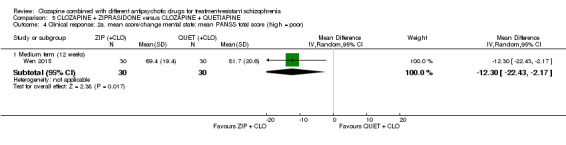
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 4 Clinical response: 2a. mean score/change mental state: mean PANSS total score (high = poor).
5.5. Clinical response: 2c. mean score/change in mental state (positive symptoms) ‐ mean PANSS positive score (high = poor)
There was a significant difference between groups on PANSS positive subscores at 12 weeks favouring ziprasidone (1 RCT, n = 60, MD ‐3.10, 95% CI ‐5.52 to ‐0.68) (Analysis 5.5).
5.5. Analysis.
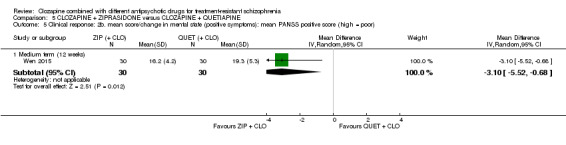
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 5 Clinical response: 2b. mean score/change in mental state (positive symptoms): mean PANSS positive score (high = poor).
5.6. Clinical response: 2d. mean score/change in mental state (negative symptoms) ‐ mean PANSS negative score (high = poor)
There was no significant difference between groups on PANSS negative subscores at 12 weeks (1 RCT, n = 60, MD 0.80, 95% CI ‐1.99 to 3.59) (Analysis 5.6).
5.6. Analysis.
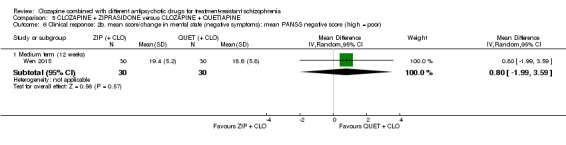
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 6 Clinical response: 2b. mean score/change in mental state (negative symptoms): mean PANSS negative score (high = poor).
5.7. Adverse effects: specific adverse effects ‐ mean score/change in extrapyramidal adverse effects ‐ reported extrapyramidal adverse effects
There was no significant difference between groups for reported extrapyramidal adverse effects (1 RCT, n = 63, RR 2.06, 95% CI 0.41 to 10.47) (Analysis 5.7).
5.7. Analysis.
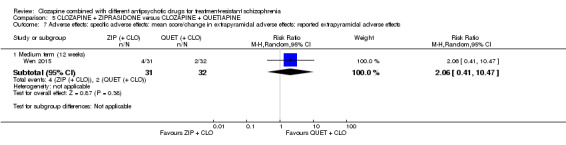
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 7 Adverse effects: specific adverse effects: mean score/change in extrapyramidal adverse effects: reported extrapyramidal adverse effects.
5.8. Adverse effects: other adverse effects (general or specific) ‐ overall adverse effect rate
There was no significant difference between groups for overall adverse effect rate (1 RCT, n = 63, RR 0.75, 95% CI 0.50 to 1.13) (Analysis 5.8).
5.8. Analysis.
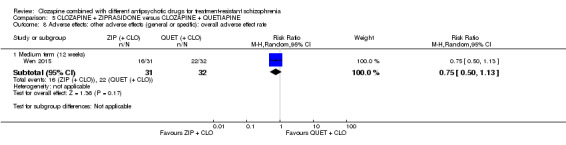
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 8 Adverse effects: other adverse effects (general or specific): overall adverse effect rate.
5.9. to 5.17. Adverse effects: other adverse effects (general or specific) ‐ various
Wen 2015 reported binary data for a number of other adverse effects, and found no significant effects.
Agitation (1 RCT, n = 63, RR 1.03, 95% CI 0.15 to 6.88) (Analysis 5.9), constipation (1 RCT, n = 63, RR 0.15, 95% CI 0.01 to 2.74) (Analysis 5.10), drowsiness (1 RCT, n = 63, RR 0.47, 95% CI 0.18 to 1.19) (Analysis 5.11), dry mouth (1 RCT, n = 63, RR 0.17, 95% CI 0.02 to 1.35) (Analysis 5.12), headache (1 RCT, n = 63, RR 1.03, 95% CI 0.28 to 3.77) (Analysis 5.13), insomnia (1 RCT, n = 63, RR 0.69, 95% CI 0.12 to 3.84) (Analysis 5.14), orthostatic hypotension (1 RCT, n = 63, RR 0.21, 95% CI 0.01 to 4.13) (Analysis 5.15), tachycardia (1 RCT, n = 63, RR 0.69, 95% CI 0.12 to 3.84) (Analysis 5.16), vertigo (1 RCT, n = 63, RR 0.21, 95% CI 0.03 to 1.67) (Analysis 5.17).
5.9. Analysis.
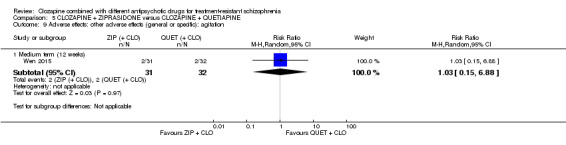
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 9 Adverse effects: other adverse effects (general or specific): agitation.
5.10. Analysis.
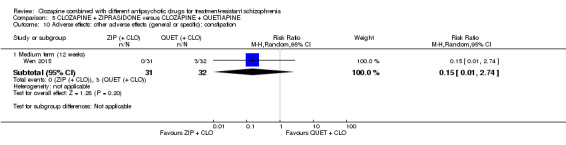
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 10 Adverse effects: other adverse effects (general or specific): constipation.
5.11. Analysis.
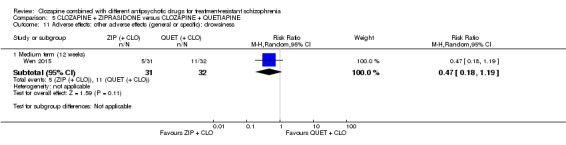
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 11 Adverse effects: other adverse effects (general or specific): drowsiness.
5.12. Analysis.
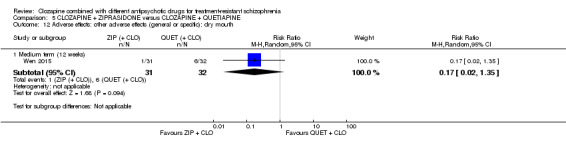
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 12 Adverse effects: other adverse effects (general or specific): dry mouth.
5.13. Analysis.
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 13 Adverse effects: other adverse effects (general or specific): headache.
5.14. Analysis.
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 14 Adverse effects: other adverse effects (general or specific): insomnia.
5.15. Analysis.
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 15 Adverse effects: other adverse effects (general or specific): orthostatic hypotension.
5.16. Analysis.
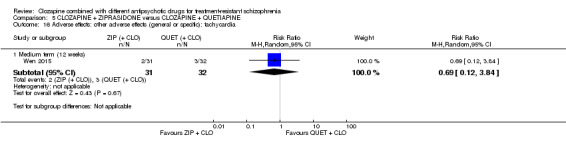
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 16 Adverse effects: other adverse effects (general or specific): tachycardia.
5.17. Analysis.
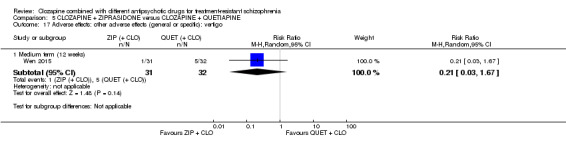
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 17 Adverse effects: other adverse effects (general or specific): vertigo.
5.18. Leaving the study early: acceptability of treatment ‐ as measured by completion of trial
There was no significant difference between groups for leaving the study early at 12 weeks (1 RCT, n = 63, RR 0.52, 95% CI 0.05 to 5.41) (Analysis 5.18).
5.18. Analysis.
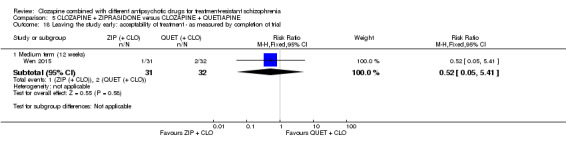
Comparison 5 CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE, Outcome 18 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial.
5.19. Missing outcomes
There were no usable data available for any other prespecified outcomes.
Discussion
Summary of main results
Combination treatment strategies are commonly used for people with an incomplete response to clozapine. However, only a small number of studies have compared one combination treatment to another. The original review identified three RCTs, but the methodological rigour of these studies was felt to be too poor for analysis. In this update, we have analysed the data from these studies and two new RCTs, but we have made clear our judgements on the quality of the data. No formal meta‐analysis was possible.
Comparison 1: CLOZAPINE + ARIPIRAZOLE versus CLOZAPINE + HALOPERIDOL (Cipriani 2013)
There was a significant difference in adverse effects in terms of the LUNSERS total score at three months and six months, favouring aripiprazole. However, at 12 months there was no significant difference between combination treatments. For all other outcomes reported, there was no significant difference (Cipriani 2013a).
Comparison 2: CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE (Genc 2007)
In all measures of clinical response reported (CGI, BPRS, SAPS, and SANS), there was a significant benefit of amisulpride over quetiapine. However, there was no significant difference in acceptability of treatment as measured by completion of the trial. Adverse effect data were not presented in a useable format (Genç 2007).
Comparison 3: CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE (Kong 2001)
There was a significant difference in PANSS positive score at the end point, favouring risperidone. For all other outcomes reported, there was no significant difference (Kong 2001).
Comparison 4: CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE (Kuwilsky 2010)
There was a significant difference between combination treatments in HAMD score at six weeks favouring risperidone, but not at 26 weeks. There was no significant difference between groups in any other outcome (Kuwilsky 2010).
Comparison 5: CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE (Wen 2015)
There was a significant benefit of ziprasidone in four outcomes measured: number of participants with 50% or greater PANSS total reduction, CGI severity of illness score, PANSS total, and PANSS positive score at 12 weeks (Wen 2015).
Overall completeness and applicability of evidence
The aim of this review was to investigate the comparative clinical effects of various clozapine combination strategies in people with treatment‐resistant schizophrenia and partial response to clozapine. We identified five RCTs which included seven different combination treatment strategies (clozapine plus aripiprazole, haloperidol, amisulpiride, quetiapine, sulpiride, risperidone or ziprasidone). Of course, possible combination strategies exist that are not included in this review. Moreover, there is no unique definition of partial responsiveness, with each study stipulating their own criteria (with the exception of one study which provided no detailed information). The outcomes investigated varied across studies. All included measures of mental state, using a variety of rating scales including BPRS, PANSS, SAPS, and SANS, and three out of five studies reported change in global state, using the CGI scale. Few outcomes using rating scales were reported with dichotomous data. All studies investigated adverse effects, either with event rates or rating scales, but results were poorly reported and in some cases unusable. Only one study, Kong 2001, reported data on weight gain that we could analyse. The primary outcome in Cipriani 2013a was leaving the study early. Although not a named outcome in the other studies, all reported on number of withdrawals, which we analysed as a surrogate marker for acceptability of treatment, as in Cipriani 2013a. There were a number of outcomes that were not reported in any study. These included service utilisation outcomes (such as hospital admissions or number of days in hospital), and quality of life measured (for both recipients of care and carers), which have been included in the 'Summary of findings' tables to highlight the need for data on these patient‐important outcomes in future trials. An outcome that was not included in our protocol but reported in two studies was clozapine dose (Cipriani 2013a; Kuwilsky 2010). A third study describes clozapine dose being reduced "accordingly" after introducing a second antipsychotic, but provided no data (Wen 2015). Clozapine dose is certainly an outcome of interest as reduction in dose may decrease the burden of clozapine adverse effects. This in itself may be a reason to initiate combination treatment.
Overall, the completeness and acceptability of the evidence relating to clozapine combination treatments versus other clozapine combination treatments was poor. There were insufficient data to make any recommendations for combination treatment strategy.
Quality of the evidence
We systematically assessed the quality of evidence using the GRADE approach for each outcome presented in the 'Summary of findings' tables. The GRADE approach takes into account study design, study limitation, inconsistency of results, indirectness of evidence, imprecision, and publication bias. As it was not possible to carry out a meta‐analysis, inconsistency and publication bias do not apply to our comparisons. Overall, the quality of evidence assessed in this study was low or very low. All included studies purported to be RCTs, although only two provided detailed information on the means of randomisation. There was generally good applicability in terms of populations and interventions, with all studies including only participants with treatment‐resistant schizophrenia or related disorders, and comparing two different clozapine combination strategies. However, some indirectness was introduced where attrition was used as a surrogate marker for acceptability of treatment. The degree of imprecision is assessed on an outcome‐by‐outcome basis, but it should be noted that for two studies rating scale data were estimated from figures in the table.
Comparison 1: CLOZAPINE + ARIPIPRAZOLE versus CLOZAPINE + HALOPERIDOL (Cipriani 2013)
This study was methodologically rigorous, clearly reported, and contained the largest study sample (Cipriani 2013a). Moreover, this was the only study with useable long‐term data. The authors undertook an intention‐to‐treat analysis resulting in a low risk of attrition bias. However, the quality of evidence was limited by the naturalistic, open‐label study design. As a result, the quality of evidence was rated low on all outcomes assessed using the GRADE approach.
Comparison 2: CLOZAPINE + AMISULPIRIDE versus CLOZAPINE + QUETIAPINE (Genc 2007)
In this study, participants who withdrew from allocated treatment were excluded from reporting on baseline characteristics and the analyses, preventing a true comparison of those randomised to each group (Genç 2007). The quality of evidence was rated very low on the outcomes assessed using the GRADE approach, with the exception of BPRS because it was powered to detect a difference. Moreover, the rating scale data were presented in figures without SDs, and so means were estimated and SDs calculated using the t scores provided in the text. Therefore, the data in the analyses will be imprecise.
Comparison 3: CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE (Kong 2001)
There was insufficient information to assess the risk of selection, performance, and detection bias (Kong 2001). In addition, the authors did not provide a working definition of partial response to clozapine, so the applicability in terms of population was unclear. As a result, the quality of evidence was rated very low on all outcomes assessed using the GRADE approach.
Comparison 4: CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE (Kuwilsky 2010)
The study was limited by the naturalistic, open‐label design (Kuwilsky 2010). In addition, this was the only study rated as high risk for detection bias, as the rating scale assessors were not blinded to the treatment allocation. This study had the smallest sample size and the highest attrition rate. There was no intention‐to‐treat analysis resulting in a high risk of attrition bias. Indeed, the 52‐week data were not analysable as per our protocol. The quality of evidence was rated very low on all outcomes assessed using the GRADE approach. It should also be noted that means and SDs were estimated from the figures for many continuous outcomes, and will therefore be imprecise.
Comparison 5: CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE (Wen 2015)
The study was described as randomised, but no details about the method or allocation concealment were provided (Wen 2015). Since there was low risk of detection bias and the study was powered to detect a difference for a number of outcomes, this quality of evidence was rated low, rather than very low, for the majority of outcomes assessed using the GRADE approach.
Potential biases in the review process
Some relevant data were presented in the text without SDs, and was therefore unusable. All our attempts to obtain this data from the relevant authors were unsuccessful. It should be noted that one of the review authors is the author of an included study (Cipriani 2013a). This author did not extract data from their trial.
Agreements and disagreements with other studies or reviews
We are aware of other meta‐analyses of comparisons of clozapine combination with other anti‐psychotic drugs versus placebo, but we know of no other reviews comparing two different combination strategies.
Unlike in the previous version of this review, we have extracted the data from the trials were possible, and presented the analyses. The previous version called for new, properly conducted RCTs comparing different combination treatments. Two new studies have been conducted since, and additional long‐term data from another have been published. However, sample sizes remain small. Larger studies are still required to detect small effect sizes.
Recently, two important contributions in the field of schizophrenia and clozapine treatment have been published (Samara 2016; Stroup 2016). In a network meta‐analysis including 40 blinded RCTs with 5172 unique participants a pattern of superiority for olanzapine, clozapine, and risperidone was seen in other efficacy outcomes, but results were not consistent and effect sizes were usually small (primary outcome was efficacy as measured by overall change in symptoms of schizophrenia; secondary outcomes included change in positive and negative symptoms of schizophrenia, categorical response to treatment, withdrawals for any reason and for inefficacy of treatment, and important adverse events) (Samara 2016). Therefore, the authors concluded that insufficient evidence exists on which antipsychotic drug is more efficacious for people with treatment‐resistant schizophrenia, and blinded RCTs ‐ in contrast to unblinded, randomised effectiveness studies ‐ provide little evidence of the superiority of clozapine compared with other second‐generation antipsychotic drugs. Network meta‐analysis is very helpful in comparing the relative effectiveness and acceptability of competing treatments (Cipriani 2013b), also in treatment guidelines (Leucht 2016; Rouse 2016). However, several issues still need to be addressed when conducting a network meta‐analysis for the results to be valid and correctly interpreted, especially in mental health (Mavridis 2015).
Slightly different results were found in the second paper, where the authors compared the effectiveness of initiating treatment with either clozapine or a standard antipsychotic drug among adults with evidence of treatment‐resistant schizophrenia in routine clinical practice (Stroup 2016). US national Medicaid data from 2001 to 2009 were used to examine treatment outcomes in a cohort of people with schizophrenia and evidence of treatment resistance that initiated clozapine (n = 3123) or a standard antipsychotic drug (n = 3123). The primary outcome was hospital admission for a mental disorder, while secondary outcomes included discontinuation of the index antipsychotic drug, use of an additional antipsychotic drug, incidence of serious medical conditions, and mortality. Initiation of clozapine was associated with a significantly decreased rate of psychiatric hospital admission (hazard ratio (HR) 0.78, 95% CI 0.69 to 0.88), index antipsychotic discontinuation (HR 0.60, 95% CI 0.55 to 0.65), and use of an additional antipsychotic drug (HR 0.76, 95% CI 0.70 to 0.82). By contrast, clozapine was associated with a non statistically significant increase of incidence of diabetes mellitus (HR 1.63, 95% CI 0.98 to 2.70), hyperlipidaemia (HR 1.40, 95% CI 1.09 to 1.78), and intestinal obstruction (HR 2.50, 95% CI 0.97 to 6.44). According to this study, in adults with schizophrenia and evidence of treatment resistance, initiating clozapine compared with initiating a standard antipsychotic drug was associated with greater effectiveness on several important outcomes, so increasing the judicious use of clozapine should be warranted (together with vigilance to prevent and detect serious medical adverse effects).
Authors' conclusions
Implications for practice.
1. For people with schizophrenia
There is some low quality evidence that certain combination strategies are superior to others for particular outcomes. Aripiprazole may produce fewer adverse effects than haloperidol as an add‐on treatment. Amisulpride and ziprasidone may produce a better short‐term clinical response in terms of global and mental state scores than quetiapine. Risperidone may be superior to sulpiride in reducing delusions and hallucinations, and superior to ziprasidone in ameliorating low mood, but all this evidence is inconclusive. Therefore, it is not possible to show one combination strategy as superior to all the others. In addition, no study measured quality of life, an outcome of utmost importance to people with treatment‐resistant schizophrenia.
2. For clinicians
There is no high quality evidence for the superiority of one combination strategy. What exists is low quality evidence from single randomised controlled trials for the benefit of one add‐on therapy over another for certain outcomes. Clinicians will continue to need to use their judgement when choosing add‐on therapy, interpreting the results of the analyses in this review in light of the individual person and with caution.
3. For policy makers/managers
The evidence is too weak to allow any recommendations for policy makers. In addition, no study measured service utilisation or economic outcomes.
Implications for research.
General: properly conducted and adequately powered randomised controlled trials are required to determine the efficacy and tolerability of combination treatment in people without a partial response to clozapine. Trialists should seek to measure patient‐important outcomes such as quality of life, as well as measures of clinical response and adverse effects.
We have included a table with the details of a suggested study design that, if implemented, would have a low risk of bias and provide data on outcomes of interest to the patient, the clinician, and the policy makers (Table 6).
1. Suggested design of study.
| Methods | Allocation: proper randomisation (e.g. by computer‐generated number sequence) and adequate allocation concealment (e.g. by central randomisation by a third party). Blinding: ideally double blind, but pragmatically blinding the participant and the outcome assessor is adequate. Setting: inpatients and outpatients. Duration: short‐term primary outcome (at 12 weeks), and then medium‐ to long‐term follow‐up (up to 52 week). |
| Participants | Diagnosis: treatment‐resistant schizophrenia, defined by persistent positive symptoms despite at least 6 months of treatment with clozapine ≥ 400 mg/day. N = 200. Sex: men and women. Age: > 18 years. |
| Interventions | 1. Clozapine plus risperidone (or paliperidone). 2. Clozapine plus aripiprazole (or amisulpride). |
| Outcomes | Measure of clinical response to include both dichotomous measures of global (e.g. CGI score) and mental state (e.g. BPRS score). Adverse effects to include weight gain, extrapyramidal symptoms, haematological problems, and hypersalivation. Acceptability assessed by leaving the study early. Service utilisation (e.g. hospital admission). Quality of life/satisfaction measure. |
| Notes | The study should be funded by an independent funding body, such as the National Institute for Health Research or Wellcome Trust. |
BPRS: Brief Psychiatric Rating Scale; CGI: Clinical Global Impression; n: number of participants.
What's new
| Date | Event | Description |
|---|---|---|
| 8 March 2017 | New citation required but conclusions have not changed | Results from update search incorporated into review. Additional data does not change overall conclusions. |
| 30 January 2016 | New search has been performed | Search update, inclusion of two further studies and additional long‐term data from one study, additional data extraction from previous studies, analyses and text update. |
History
Protocol first published: Issue 1, 2007 Review first published: Issue 3, 2009
| Date | Event | Description |
|---|---|---|
| 11 November 2009 | Amended | Contact details updated. |
Acknowledgements
We would like to thank the editorial base of the Cochrane Schizophrenia Group for their help. We thank Jun Xia and Juan Juan Ren for extracting data from the Chinese literature, Marianna Boso and Corrado Barbui (University of Verona) for their help in carrying out the original review, and Clive Adams and Stefan Leucht for their invaluable editorial support and encouragement. Andrea Cipriani is supported by the NIHR Oxford Cognitive Health Clinical Research Facility and was expert witness for a patent issue about quetiapine extended release.
Appendices
Appendix 1. Methods section of original version (2010)
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials. We included trials described as 'double‐blind' if it was implied that the study was randomised. For example, if the demographic details of the participants in each group were similar. We excluded quasi‐randomised studies, such as those allocating by using alternate days of the week.
Types of participants
We included people of both sexes, aged 18 years or more, with a diagnosis of treatment‐resistant schizophrenia or related disorders (e.g. schizoaffective disorder, schizophreniform disorder), however diagnosed. There is no clear evidence that the schizophrenia‐like psychoses are caused by fundamentally different disease processes or require different treatment approaches (Carpenter 1994).
Types of interventions
Clozapine plus another antipsychotic drug.
Clozapine plus a different other antipsychotic drug.
Any dose and means of administration was acceptable.
Types of outcome measures
Primary outcomes
We divided outcomes into short term (less than three months) medium term (three to 12 months), and long term (over one year).
The primary measure of efficacy was clinical improvement on psychotic symptoms, measured either as a dichotomous outcome (proportions of participants with treatment response as defined by each of the studies), or as a continuous outcome (reported either as endpoint score or change from baseline to endpoint).
1. Clinical response.
1.1. No clinically significant response in global state (dichotomous outcome) ‐ as defined by each of the studies.
1.2. Mean score/change in global state (continuous outcome).
1.3. No clinically significant response on positive symptoms (dichotomous outcome) ‐ as defined by each of the studies.
1.4. Mean score/change in positive symptoms (continuous outcome).
1.5. No clinically significant response on negative symptoms (dichotomous outcome) ‐ as defined by each of the studies.
1.6. Mean score/change in negative symptoms (continuous outcome).
1.7. Use of additional medication (other than anticholinergic drugs) for psychiatric symptoms.
Secondary outcomes
1. Death: suicide or any causes.
2. Leaving the study early (acceptability of treatment), as measured by completion of trial.
3. Extrapyramidal adverse effects.
3.1. Incidence of use of antiparkinson drugs (i.e. anticholinergic drugs).
3.2. Clinically significant extrapyramidal adverse effects ‐ as defined by each of the studies.
3.3. Mean score/change in extrapyramidal adverse effects.
4. Blood adverse affects.
4.1. Blood dyscrasias such as agranulocytosis.
5. Other adverse effects, general and specific.
5.1. Hypersalivation.
5.2. Weight gain.
5.3. Other adverse effects.
6. Service utilisation outcomes.
6.1. Hospital admission.
6.2. Days in hospital.
7. Economic outcomes.
8. Quality of life/satisfaction with care for either recipients of care or carers.
8.1. Significant change in quality of life/satisfaction ‐ as defined by each of the studies.
8.2. Mean score/change in quality of life/satisfaction.
Data collection and analysis
Selection of studies
Material downloaded from electronic sources included details of author, institution, or journal of publication. Two review authors (MB and AC) independently inspected all reports of identified studies. We resolved any disagreement by consensus; however, where doubt remained, we acquired the full article. Two review authors (MB and AC) independently decided whether these then met the review criteria. There was no blinding to the names of authors, institutions, and journal of publication. We resolved any further disagreements by consensus with a third review author (CB) and if disagreement could not be resolved by discussion, we sought further information and added these trials to the list of those awaiting assessment.
Data extraction and management
1. Data extraction
Two review authors (MB and AC) independently extracted data and resolved disagreement by discussion with a third review author (CB). When this was not possible, we sought further information from trial authors.
To facilitate comparison between trials, we converted variables (such as days in hospital) that could be reported in different metrics (mean days per year, per week or per month) to a common metric (e.g. mean days per month).
When insufficient data were provided to identify the original group size (prior to dropouts), we contacted the authors. Where possible, we converted continuous scores into dichotomous data.
2. Management
We extracted the data onto standard, simple forms. Where possible, data were entered into Review Manager 5 in such a way that the area to the left of the 'line of no effect' indicated a 'favourable' outcome for clozapine.
3. Scale‐derived data
Many rating scales are available to measure outcomes in mental health trials (Marshall 2000). These scales vary in quality and many are poorly validated. It is generally accepted that measuring instruments should have the properties of reliability (the extent to which a test effectively measures anything at all) and validity (the extent to which a test measures that which it is supposed to measure) (Rust 1989). Before publication of an instrument, most scientific journals insist that its reliability and validity be demonstrated to the satisfaction of referees. As a minimum standard, data were excluded from unpublished rating scales. In addition, the rating scale should be either: a self report; or completed by an independent rater or relative. We presented rating scale data that were provided by the treating physician but marked them with an (*) to indicate potential bias. More stringent standards for instruments may be set in future editions of this review.
Assessment of risk of bias in included studies
We used the latest version of the Cochrane 'Risk of bias' tool to assess the risk of bias in the included studies. This instrument consists of six items. Two of the items assess the strength of the randomisation process in preventing selection bias in the assignment of participants to interventions: adequacy of sequence generation and allocation concealment. The third item (blinding) assesses the influence of performance bias on the study results. The fourth item assesses the likelihood of incomplete outcome data, which raises the possibility of bias in effect estimates. The fifth item assesses selective reporting, the tendency to preferentially report statistically significant outcomes. It requires a comparison of published data with trial protocols, when such are available. The sixth item refers to other sources of bias that are relevant in certain circumstances, for example, in relation to trial design (methodological issues such as those related to cross‐over designs and early trial termination) or setting. Two review authors independently assessed trial quality in accordance with the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008). Where inadequate details of allocation concealment and other characteristics of trials were provided, we contacted the trial authors in order to obtain further information. If the raters disagreed, we made the final rating by consensus with the involvement, if necessary, of another review author.
Measures of treatment effect
1. Binary data
When summation was appropriate with binary outcomes such as improved/not improved, we calculated the risk ratio (RR) statistic with a 95% confidence interval (CI) using a random‐effects model. In addition, as a measure of efficiency, we estimated the number needed to treat for an additional beneficial outcome (NNTB) or harmful outcome (NNTH) from the pooled totals. We calculated the NNTB/NNTH as the inverse of the risk difference.
2. Continuous data
2.1. Summary statistic
For continuous outcomes, we estimated a mean difference (MD) with 95% CI. This analysis was based on the random‐effects model as this takes into account any differences between studies even if there was no statistically significant heterogeneity. If standard deviations were not recorded, we asked authors to supply the data. In the absence of data from the authors, we used the mean standard deviation from other studies (Furukawa 2006). Continuous data may be presented from different scales, rating the same outcome. In this event, we presented all data without summation and inspected the general direction of effect.
2.2. Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we applied the following standards to all data before inclusion:
standard deviations and means reported in the paper or obtainable from the authors;
when a scale starts from the finite number zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996);
if a scale starts from a positive value (such as Positive and Negative Syndrome Scale (PANSS) which can have values from 30 to 210) the calculation described above was modified to take the scale starting point into account. In these cases skew was presented if 2SD > (S ‐ Smin), where S is the mean score and Smin is the minimum score.
Endpoint scores on scales often have a finite start and end point and these rules can be applied to them. When continuous data are presented on a scale which includes a possibility of negative values (such as change on a scale), it is difficult to tell whether data are non‐normally distributed (skewed) or not. We presented skewed data in the 'Other data' tables rather than included in the analysis.
2.3. Endpoint versus change data
For change data (endpoint minus baseline), the situation is even more problematic. In the absence of individual participant data it is impossible to know if data are skewed, though this is likely. According to a previous published review of the Cochrane Schizophrenia Group (Duggan 2005), we presented change data in order to summarise available information. In doing this, it was assumed either that data were not skewed or that the analyses could cope with the unknown degree of skew. Again, without individual participant data it is impossible to test this assumption. Where both change and endpoint data were available for the same outcome category, we presented only endpoint data. We acknowledge that by doing this, much of the published change data could have been excluded, but argue that endpoint data is more clinically relevant and that if change data were to be presented along with endpoint data, it would be given undeserved equal prominence. We contacted authors of studies that only reported change for endpoint figures.
Unit of analysis issues
1. Cluster trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992), whereby P values are spuriously low, CIs unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review, we will seek to contact first authors of studies to obtain intraclass correlation coefficients of their clustered data and to adjust for this by using accepted methods (Gulliford 1999). When clustering was incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological, or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia, we intended to use data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, the additional treatment arms were presented in comparisons. Where the additional treatment arms were not relevant, these data were not reproduced.
Dealing with missing data
1. Overall loss of credibility
At some degree of loss to follow‐up data must lose credibility (Xia 2009). Since there is no evidence as to the degree of attrition which makes a reasonable analysis of the data possible, we included all trials in the main analysis. If, for a given outcome, more than 50% of the total numbers randomised were not accounted for, we did not present results as such data will be impossible to interpret with authority. However, if more than 50% of those in one arm of a study were lost but the total loss was less than 50%, data were marked with a (*) to indicate the result may be prone to bias.
2. Missing data
When data were missing and the method of 'last observation carried forward' (LOCF) had been used to do an intention‐to‐treat analysis, then we used the LOCF data with due consideration of the potential bias and uncertainty introduced. For studies that did not specify the reasons for people leaving the study early (dropouts), we assumed that these people had no change in clinical outcome variables.
Assessment of heterogeneity
1. Clinical heterogeneity
We considered all the included studies within any comparison to judge clinical heterogeneity.
2. Statistical heterogeneity
2.1. Visual inspection
We visually inspected the graphs to investigate the possibility of statistical heterogeneity.
2.2. Employing the I2 statistic
We investigated heterogeneity between studies by the I2 statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I2 estimate was 50% or greater, we interpreted this as indicating the presence of significant heterogeneity (Higgins 2008). If inconsistency was high, data were not summated, but presented separately.
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results. We entered data from all identified and selected trials into a funnel graph (trial effect against trial size) in an attempt to investigate the likelihood of overt publication bias (Egger 1997).
Data synthesis
We employed a random‐effects model for analyses throughout. We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us and as a result significant between trial heterogeneity is implemented in the pooled estimate the random‐effects model is usually more conservative in terms of statistical significance. The disadvantage of the random‐effects model is that it puts added weight onto the smaller of the studies ‐ those trials that are most vulnerable to bias.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analysis
No subgroup analysis was planned.
2. Investigation of heterogeneity
If data were clearly heterogeneous we checked that data were correctly extracted and entered and that we had made no unit of analysis errors. If the high levels of heterogeneity remained, we did not undertake a meta‐analysis at this point for if there is considerable variation in results, and particularly if there is inconsistency in the direction of effect, it may be misleading to quote a mean value for the intervention effect. We would have wanted to explore heterogeneity. We prespecified no characteristics of studies that may have been associated with heterogeneity except quality of trial method. If no clear association could be shown by sorting studies by quality of methods, we performed a random‐effects meta‐analysis. Should another characteristic of the studies be highlighted by the investigation of heterogeneity, perhaps some clinical heterogeneity not hitherto predicted but plausible causes of heterogeneity, we discussed these post‐hoc reasons and analysed and presented the data. However, should the heterogeneity be substantially unaffected by use of random‐effects meta‐analysis and no other reasons for the heterogeneity be clear, we presented the final data without a meta‐analysis.
Sensitivity analysis
No sensitivity analysis was planned.
Appendix 2. Previous versions
Plain language summary
Schizophrenia is a severe mental illness affecting 1% of the population throughout the world. The symptoms of schizophrenia are perceptions without cause (hallucinations), fixed false beliefs (delusions) with or without apathy, and slowing of movement or thought. In most Western countries, people who do not respond to the majority of common antipsychotic medicines (called treatment‐resistant people) are tried on the atypical antipsychotic clozapine. If they do not respond to clozapine alone, then another antipsychotic medicine is usually recommended. This review looks at clinical trials which compare the response to a second antipsychotic medicine in people who are treatment resistant, and on clozapine.
In the present review, we looked at 48 studies, only four fulfilled the criteria to be included, the total number of people randomised was 246. Two studies lasted 52 weeks and the other two studies lasted eight weeks, and all compared different second antipsychotic medicines with clozapine (aripiprazole versus haloperidol, risperidone versus sulpiride, risperidone versus ziprasidone, and amisulpride versus quetiapine).
When specific symptoms of schizophrenia were studied, there was change for the better in all groups but no second antipsychotic was significantly better than the one it was compared to. When looking at side effects, people taking sulpiride were slightly more likely to have excessive salivation and weight gain than those taking risperidone. The people in the aripiprazole group showed an advantage in the perception of side effects, when people on clozapine plus aripiprazole were compared to those on clozapine plus haloperidol.
These four trials contained small numbers of people and the results were often not well recorded. Although there is a suggestion that adding a second antipsychotic medicine may improve general functioning and decrease the symptoms of schizophrenia, it is still not possible to say which antipsychotic medicine would help the most. A large, longer and independent trial should be done on people who have not responded completely to clozapine to find the most effective treatment.
(Plain language summary prepared for this review by Janey Antoniou of RETHINK, UK; www.rethink.org)
Appendix 3. Previous searches
Search in 2008
Electronic searches
Cochrane Schizophrenia Group Trials Register
Search methods for identification of studies Electronic searches We searched the Cochrane Schizophrenia Group Trials Register (March 2008) using the phrase: [((clozapin* or clozaril* or leponex* or denzapin* or zaponex*) in title, abstract and index fields in REFERENCE) OR ((clozapin* or clozaril* or leponex* or denzapin* or zaponex*) in interventions field in STUDY]
This register is compiled by systematic searches of major databases, handsearches, and conference proceedings (see Cochrane Schizophrenia Group module).
MEDLINE
MEDLINE search carried out independently by review authors in November 2008. The MEDLINE (OvidSP) search strategy is below.
| # | Searches | Results | Search type |
| 1 | clozapine.mp. and Clozapine/ | 5550 | Advanced |
| 2 | Schizophrenia/ and schizophrenia.mp | 65799 | Advanced |
| 3 | 1 and 2 | 2388 | Advanced |
| 4 | limit 3 to clinical trial, all | 462 | Advanced |
Searching other resources
1. Reference checking
We checked reference lists of all identified randomised controlled trials.
2. Handsearching
If we found any appropriate journals and conference proceedings relating to clozapine combination strategies for treatment‐resistant schizophrenia, we manually searched these periodicals.
3. Personal communication
We attempted to contact the corresponding author of each included study for information regarding supplemental data and unpublished trials. We contacted a defined list of experts in the field and asked of their knowledge of other studies, published or unpublished, relevant to the review article.
4. Industry
We requested that pharmaceutical companies marketing investigational products provided relevant published and unpublished data.
Search in 2011 and 2012
Electronic searches
1. Cochrane Schizophrenia Group Trials Register
We searched the Cochrane Schizophrenia Group Trials Register (January 2011 and 19 July 2012) using the phrase:
[((*clozapin* or *clozaril* or *leponex* or *denzapin* or *zaponex*) in title, abstract and index fields in REFERENCE) OR ((*clozapin* or *clozaril* or *leponex* or *denzapin* or *zaponex*) in interventions field in STUDY]
This register is compiled by systematic searches of major databases, handsearches, and conference proceedings (see group module).
Searching other resources
1. Reference searching
We checked reference lists of all identified studies for further relevant studies.
2. Personal contact
When appropriate, the first author of each included papers was contacted and additional published and unpublished trials were requested.
Data and analyses
Comparison 1. CLOZAPINE + ARIPIPRAZOLE versus CLOZAPINE + HALOPERIDOL.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: mean score/change in mental state: mean change in BPRS score from baseline (high = good) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Medium term (12 weeks) | 1 | 105 | Mean Difference (IV, Random, 95% CI) | ‐1.40 [‐5.59, 2.79] |
| 1.2 Medium term (24 weeks) | 1 | 105 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐4.81, 3.41] |
| 1.3 Long term (52 weeks) | 1 | 105 | Mean Difference (IV, Random, 95% CI) | 0.90 [‐4.38, 6.18] |
| 2 Adverse effects: other adverse effects (general or specific): mean change in LUNSERS score from baseline (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Medium term (12 weeks) | 1 | 105 | Mean Difference (IV, Random, 95% CI) | ‐4.9 [‐8.48, ‐1.32] |
| 2.2 Medium term (24 weeks) | 1 | 105 | Mean Difference (IV, Random, 95% CI) | ‐4.9 [‐8.25, ‐1.55] |
| 2.3 Long term (52 weeks) | 1 | 105 | Mean Difference (IV, Random, 95% CI) | ‐4.8 [‐9.79, 0.19] |
| 3 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Medium term (12 weeks) | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.34, 2.24] |
| 3.2 Medium term (24 weeks) | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.60, 2.28] |
| 3.3 Long term (52 weeks) | 1 | 106 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [0.72, 2.22] |
Comparison 2. CLOZAPINE + AMISULPRIDE versus CLOZAPINE + QUETIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: 1 mean score/change in global state: mean CGI score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Short term (8 weeks) | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.38, ‐0.42] |
| 2 Clinical response: 2a mean score/change in mental state: mean BPRS score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Short term (8 weeks) | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐5.86, ‐2.14] |
| 3 Clinical response: 2b mean score/change in mental state: mean SAPS score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Short term (8 weeks) | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐6.90 [‐12.82, ‐0.98] |
| 4 Clinical response: 2c mean score/change in mental state: means SANS score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Short term (8 weeks) | 1 | 50 | Mean Difference (IV, Random, 95% CI) | ‐5.20 [‐7.14, ‐3.26] |
| 5 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.02, 1.60] |
Comparison 3. CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: no clinically significant response in mental state: 20% to 50% reduction in PANSS total score | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Short term (8 weeks) | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.40, 1.68] |
| 2 Adverse effect: weight gain | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Short term (8 weeks) | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.4 [0.08, 1.90] |
| 3 Clinical response: 2a mean score/change in mental state: mean PANSS total score at endpoint (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Short term (8 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐2.28 [‐7.41, 2.85] |
| 4 Clinical response: 2b. mean score/change in mental state (positive symptoms): mean PANSS positive score at endpoint (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Short term (8 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐2.55 [‐4.64, ‐0.46] |
| 5 Clinical response: 2c. mean score/change in mental state (negative symptoms): mean PANSS negative score at endpoint (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Short term (8 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐0.54 [‐3.19, 2.11] |
| 6 Adverse effects: specific adverse effects: hypersalivation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Short term (8 weeks) | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.04, 3.03] |
| 7 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Short term (8 weeks) | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
3.7. Analysis.
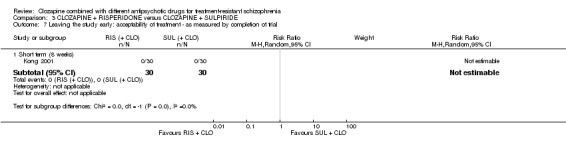
Comparison 3 CLOZAPINE + RISPERIDONE versus CLOZAPINE + SULPIRIDE, Outcome 7 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial.
Comparison 4. CLOZAPINE + RISPERIDONE versus CLOZAPINE + ZIPRASIDONE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: no clinically significant response in mental state: 20% reduction in PANSS total score | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Short term (6 weeks) | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.13, 3.30] |
| 1.2 Medium term (26 weeks) | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.8 [0.28, 2.27] |
| 2 Clinical response: no clinically significant response in mental state (positive symptoms) 20% reduction in PANSS positive subscore | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Short term (6 weeks) | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.36, 24.92] |
| 3 Clinical response: 1a mean score/change global state: mean CGI subscale score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Severity of illness | 1 | 22 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.32, 0.72] |
| 3.2 Global improvement | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.82, 0.22] |
| 3.3 Therapeutic efficacy | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.79, 0.19] |
| 4 Clinical response: 1b mean score/change global state: mean GAF score (high = good) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐7.84, 7.84] |
| 5 Clinical response: 2a. mean score/change mental state: mean HAMD score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐3.40 [‐6.71, ‐0.09] |
| 5.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐5.35, 3.95] |
| 6 Clinical response: 2b mean score/change mental state: mean PANSS total score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐3.10 [‐11.38, 5.18] |
| 6.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 1.0 [‐7.91, 9.91] |
| 7 Clinical response: 2c mean score/change in mental state (positive symptoms) mean PANSS positive score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐1.84, 1.44] |
| 7.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐2.58, 2.18] |
| 8 Clinical response: 2d mean score/change in mental state (negative symptoms) mean PANSS negative score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐1.20 [‐4.63, 2.23] |
| 8.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 1.5 [‐2.66, 5.66] |
| 9 Clinical response: 2e mean score/change in mental state (negative symptoms) mean SANS score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐4.0 [‐17.55, 9.55] |
| 9.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 1.80 [‐14.31, 17.91] |
| 10 Clinical response: 2f mean score/change in global state: mean PANSS global psychopathology score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 10.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐1.60 [‐6.60, 3.40] |
| 10.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐5.83, 5.83] |
| 11 Adverse effects: specific adverse effects: mean score/change in extrapyramidal adverse effects: mean EPS score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | 0.60 [‐0.67, 1.87] |
| 11.2 Medium term (26 weeks) | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.63, 1.23] |
| 12 Adverse effects: other adverse effects (general or specific): mean CGI adverse effect scores (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 12.1 Short term (6 weeks) | 1 | 22 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.53, 0.33] |
| 13 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 Short term (6 weeks) | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.07, 14.21] |
| 13.2 Medium term (26 weeks) | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.18, 1.97] |
| 13.3 Long term (52 weeks) | 1 | 24 | Risk Ratio (M‐H, Random, 95% CI) | 1.6 [0.73, 3.49] |
Comparison 5. CLOZAPINE + ZIPRASIDONE versus CLOZAPINE + QUETIAPINE.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical response: 1a. no clinically significant response in mental state: PANSS reduction ≥ 50% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.35, 0.81] |
| 2 Clinical response: 1b. no clinically significant response in mental state: PANSS reduction ≥ 25% | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.38, 1.10] |
| 3 Clinical response: 1. mean score/change global state: mean CGI‐S score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Medium term (12 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐0.70 [‐1.18, ‐0.22] |
| 4 Clinical response: 2a. mean score/change mental state: mean PANSS total score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Medium term (12 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐12.30 [‐22.43, ‐2.17] |
| 5 Clinical response: 2b. mean score/change in mental state (positive symptoms): mean PANSS positive score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Medium term (12 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐3.10 [‐5.52, ‐0.68] |
| 6 Clinical response: 2b. mean score/change in mental state (negative symptoms): mean PANSS negative score (high = poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Medium term (12 weeks) | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐1.99, 3.59] |
| 7 Adverse effects: specific adverse effects: mean score/change in extrapyramidal adverse effects: reported extrapyramidal adverse effects | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 2.06 [0.41, 10.47] |
| 8 Adverse effects: other adverse effects (general or specific): overall adverse effect rate | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.50, 1.13] |
| 9 Adverse effects: other adverse effects (general or specific): agitation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.15, 6.88] |
| 10 Adverse effects: other adverse effects (general or specific): constipation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 2.74] |
| 11 Adverse effects: other adverse effects (general or specific): drowsiness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.18, 1.19] |
| 12 Adverse effects: other adverse effects (general or specific): dry mouth | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.35] |
| 13 Adverse effects: other adverse effects (general or specific): headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.28, 3.77] |
| 14 Adverse effects: other adverse effects (general or specific): insomnia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.12, 3.84] |
| 15 Adverse effects: other adverse effects (general or specific): orthostatic hypotension | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.01, 4.13] |
| 16 Adverse effects: other adverse effects (general or specific): tachycardia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.69 [0.12, 3.84] |
| 17 Adverse effects: other adverse effects (general or specific): vertigo | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 17.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.03, 1.67] |
| 18 Leaving the study early: acceptability of treatment ‐ as measured by completion of trial | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 18.1 Medium term (12 weeks) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.05, 5.41] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Cipriani 2013a.
| Methods | Allocation: randomised (using centralised randomisation procedure), allocation concealed. Blinding: open‐label (participants and clinicians not blind to treatment, assessors for rating scales blind). Duration: 52 weeks. Design: multicentre, naturalistic, parallel. Setting: inpatients and outpatients. Country: Italy. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 106. Age: mean 41.5 years in haloperidol group, 40.3 years in aripiprazole group. Sex: 32% female in haloperidol group, 37% female in aripiprazole group. History: partial response to clozapine after at least 6 months of treatment stable dose, mean disease duration 18 years in haloperidol group, 14 years in aripiprazole group. |
|
| Interventions | 1. Clozapine + haloperidol: clozapine mean baseline dose = 413 mg/day (SD 157) and haloperidol mean baseline dose = 2.1 mg/day (SD 1.3). N = 53. 2. Clozapine + aripiprazole: clozapine mean baseline dose = 418 mg/day (SD 141) and aripiprazole mean baseline dose = 8.7 mg/day (SD 3.9). N = 53. |
|
| Outcomes | Mental state: change in BPRS score from baseline. Leaving the study early. Adverse effects: change in LUNSERS score from baseline. |
|
| Notes | Underpowered: target sample size 216, total recruited 106. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants randomised (quote) "using a computer generated random number program." |
| Allocation concealment (selection bias) | Low risk | Quote: "A randomisation procedure by telephone was used to keep treatment allocation concealed." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "...the patients and clinicians were not blind to pharmacological treatments." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...all outcome assessments based on rating scales were performed by trained assessors masked to the allocated treatment." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All randomised participants who received at least 1 dose of investigation drugs were included in the intention‐to‐treat analysis of the primary outcome (leaving the study early). Only 1 patient was not included in the analysis of the BPRS and LUNSERS continuous outcomes due to missing data. |
| Selective reporting (reporting bias) | Low risk | All outcomes expected and specified in the protocol were reported. |
| Other bias | Low risk | We found no other bias. |
Genç 2007.
| Methods | Allocation: unclear. Blindness: unclear. Duration: 8 weeks. Design: multicentre, parallel. Setting: inpatients and outpatients. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 56. Age: mean 37.29 years in amisulpride group, mean "7.30" in quetiapine group, likely misreported as no significant difference between groups. Sex: 55.6% female in amisulpride group, 60.9% female in quetiapine group. History: partial response to clozapine after 12 weeks' treatment at stable dose, demonstrated by BPRS total > 45. |
|
| Interventions | 1. Clozapine plus amisulpride: clozapine mean baseline dose 550 mg/day (SD 127.09) and amisulpride mean baseline dose 437.03 mg/day (SD 104.32). N = 27. 2. Clozapine plus quetiapine: clozapine mean baseline dose 536.95 mg/day (SD 125.42) and quetiapine mean baseline dose 595.65 mg/day (SD 125.21). N = 23. |
|
| Outcomes | Clinical response: global state (CGI), mental state (BPRS, SAPS, SANS). Leaving the study early. Unable to use: Extrapyramidal adverse effect: UKU, SAS (no mean endpoint scores). |
|
| Notes | Baseline characteristics reported after participants left early. 8‐week rating scale scores estimated from graph by two review authors (SB and SD) calculated from t score of difference in means. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned...". Probably done. |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment was not mentioned. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information on blindness of participants given. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor blinded. Quote: "The first author, who was the rater remained blind throughout the study." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 5 participants in the quetiapine group dropped out and 1 participant in the amisulpride group was missed at follow‐up at 2 weeks. These 6 participants were excluded from the analysis and from the reporting of baseline characteristics. There was no significant difference between groups, but this does not confirm the absence of bias, especially because this was a small study (N = 56). Moreover, reasons for incomplete data were not balanced between groups. |
| Selective reporting (reporting bias) | High risk | No protocol available, no SDs given for various scales (BPRS, SAPS, SANS, CGI). |
| Other bias | Unclear risk | We could not rule out the potential for other bias. |
Kong 2001.
| Methods | Allocation: unclear. Blindness: unclear. Duration: 8 weeks. Design: multicentre. Setting: inpatients. |
|
| Participants | Diagnosis: schizophrenia (CCMD‐2‐R)*. N = 60. Age: < 42 years. Sex: 38 male and 22 female. History: partial response to clozapine, but criteria not clearly specified. |
|
| Interventions | 1. Clozapine plus risperidone: clozapine mean dose 400 mg/day and risperidone mean dose 4 mg/day to 6 mg/day. SDs not provided. N = 30. 2. Clozapine plus sulpiride: clozapine mean dose 500 mg/day and sulpiride mean dose 800 mg/day to 1200 mg/day. SDs not provided. N = 30. |
|
| Outcomes | Clinically significant response: 20% to 50% reduction PANSS total. Clinical response: mental state (PANSS total, PANSS positive, PANSS negative). Adverse effects: weight gain, hypersalivation. Leaving the study early. Unable to use: Adverse effects: TESS score. |
|
| Notes | *It is unclear whether patients with schizoaffective disorder were enrolled. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information. |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information. Allocation done by hospital number so possibly not concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No one left early. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | We could not rule out the potential for other bias. |
Kuwilsky 2010.
| Methods | Allocation: randomised (by random number generator). Blindness: open label. Duration: 52 weeks. Design: multicentre, naturalistic, parallel. Setting: inpatients and outpatients. Country: Germany. |
|
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐IV). N = 24. Age: mean 31.8 years in risperidone group, 37.25 years in ziprasidone group. Sex: 41.7% female in risperidone group, 41.7% female in ziprasidone group. History: partial response to clozapine after at least 3 months with stable dose as demonstrated by PANSS total score > 65. |
|
| Interventions | 1. Clozapine plus risperidone: clozapine mean dose 437.5 mg/day (SD 140.4) and risperidone mean dose 3.82 mg/day (SD 1.8). N = 12. 2. Clozapine plus ziprasidone: clozapine mean dose 370.8 mg/day (SD 150.0) and ziprasidone mean dose 134 mg/day (SD 34.4). N = 12. |
|
| Outcomes | Clinically significant response: 20% reduction PANSS. Clinical response global state (CGI subscales, GAF), mental state (PANSS total, PANSS positive, PANSS negative, PANS global psychopathology, SANS, HAMD). Leaving the study early. Adverse effects: EPS, CGI adverse effects. Unable to use: Global state: CGI and GAF (26 and 52 weeks) (no SDs reported). Adverse effects: HAS (data from figure not extractable). |
|
| Notes | Dichotomous outcomes available from text. Baseline rating scores provided in table with SDs. 6‐week, 26‐week, and 52‐week scores and SDs estimated from graphs by 1 review author (SB). Baseline and 6‐week CGI and GAF score provided in table with SDs. No SDs for 26‐week or 52‐week scores. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "...the patients were randomized...using a random number generator." |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not mentioned. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open label"; "antipsychotics were applied in an open manner." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No mention of blinded assessors. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | > 50% of participants had dropped out by 52 weeks and there was no intention‐to‐treat analysis. |
| Selective reporting (reporting bias) | High risk | The study protocol was available but not all of the study's prespecified (primary and secondary) outcomes that were of interest in the review were reported in the prespecified way. There were no SDs reported for some outcomes. |
| Other bias | Unclear risk | We could not rule out the potential for other bias. |
Wen 2015.
| Methods | Allocation: randomised (no detailed information). Blindness: single blind. Duration: 12 weeks. Design: multicentre, parallel. Setting: inpatients and outpatients. Country: China. |
|
| Participants | Diagnosis: schizophrenia (DSM‐IV). N = 63. Age: mean age 37.1 years. Sex: 43% female. History: treatment with clozapine for > 12 weeks at a dose > 400 mg with no improvement observed, PANSS score ≥ 80 and CGI‐S ≥ 4. |
|
| Interventions | 1. Clozapine plus ziprasidone: clozapine mean baseline dose = 479 mg/day (SD 56.5), ziprasidone was titrated from 80 mg/day up to 120 mg/day to 160 mg/day, 1 week after ziprasidone was added, the dose of clozapine was reduced accordingly. N = 31. 2. Clozapine plus quetiapine: clozapine mean baseline dose = 481.3 mg/day (SD 51.7), quetiapine was titrated from 200 mg/day up to 400 mg/day to 750 mg/day, 1 week after quetiapine was added, the dose of clozapine was reduced accordingly. N = 32. |
|
| Outcomes | Clinically significant response: > 50% reduction PANSS. Clinical response: global state (CGI‐S), mental state (PANSS total, PANSS positive, PANSS negative). Adverse effects: rate, agitation, constipation, drowsiness, dry mouth, extrapyramidal adverse effects, headache, insomnia, orthostatic hypotension, tachycardia, vertigo. Leaving the study early. |
|
| Notes | Chinese language. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was performed, no detailed information provided. |
| Allocation concealment (selection bias) | Unclear risk | No data. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as single blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The participants were evaluated by 3 doctors who did not have knowledge about the treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3 participants who dropped out were excluded from the analyses of global and mental state outcomes, but included in the analyses of adverse events. Since the attrition rate was < 5%, and reasons were balanced between groups, the study was rated as low risk. |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. |
| Other bias | Unclear risk | We could not rule out the potential for other bias. |
General
n: number of participants
SD: standard deviation
Diagnostic tools
CCDM‐2‐R: Chinese Classification of Mental disorders
DSM‐IV: Diagnostic and Statistical Manual of mental disorders, fourth edition
Global effects scales
CGI: Clinical Global Impression
CGI‐S: Clinical Global Impression ‐ Severity
GAF: Global assessment of functioning
Mental state scales
BPRS: Brief Psychiatric Rating Scale
HAMD: Hamilton Depression Scale
PANSS: Positive and Negative Syndrome Scale
SANS: Scale for the Assessment of Negative Symptoms
SAPS: Scale for the Assessment of Positive Symptoms
Adverse effect scales
EPS: Extrapyramidal Symptoms Scale
HAS: Hillside Akathisia Scale
LUNSERS: Liverpool University Neuroleptic Side Effect Rating Scale
SAS: Simpson‐Angus Extrapyramidal Symptoms Rating Scale
TESS: Treatment Emergent Symptom Scale
UKU: Udvalg for Kliniske Undersgelser Side Effect Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Angst 1971 | Allocation: randomised. Participants: people with schizophrenia and manic psychoses. Intervention: no combination treatment ‐ levomepromazine vs clozapine. |
| Anil 2009 | Allocation: randomised, double‐blind. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ risperidone + clozapine vs placebo + clozapine. |
| Anonymous 2009 | Allocation: non‐randomised (handbook written for the CUTLASS (Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study) trial). |
| Assion 2008 | Allocation: randomised, double‐blind. Participants: people with treatment‐resistant schizophrenia partially responsive or unresponsive to clozapine. Intervention: no active comparison with combination treatment ‐ amisulpride + clozapine vs clozapine + placebo. |
| Bao 1988 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment resistant. |
| Barnes 2013 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia enrolled in 2 separate trials. Intervention: no combination treatment ‐ oral first‐generation antipsychotic drug vs non‐clozapine second‐generation antipsychotic drug or non‐clozapine second‐generation antipsychotic drug vs clozapine. |
| Bender 1997 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ trimipramine vs perazine. |
| Bilder 2001 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ clozapine vs olanzapine vs risperidone vs haloperidol. |
| Bustillo 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ lamotrigine vs placebo. |
| Cao 2003 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ risperidone vs clozapine. |
| Chang 2008 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + aripiprazole vs clozapine + placebo. |
| Cooper 2005 | Allocation: non‐randomised (population‐based study). |
| Dai 2014 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no comparison with other clozapine combination treatment ‐ clozapine + ziprasidone vs ziprasidone + risperidone. |
| Daniel 1994 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ risperidone vs clozapine. |
| Dong 2007 | Allocation: randomised. Participants: people with first‐episode of schizophrenia, not treatment‐resistant. |
| Feifel 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison to combination treatment ‐ antipsychotic + oxytocin vs antipsychotic + placebo (cross‐over design). |
| Fleischhacker 2008a | Allocation: randomised. Participants: people with schizophrenia and suboptimal efficacy/safety on clozapine. Intervention: no comparison with combination treatment ‐ clozapine + aripiprazole vs clozapine. |
| Fleischhacker 2008b | Allocation: randomised. Participants: people with schizophrenia and suboptimal efficacy/safety on clozapine. Intervention: no comparison with combination treatment ‐ clozapine + aripiprazole vs clozapine. |
| Freudenreich 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: placebo controlled trial ‐ clozapine + risperidone vs clozapine + placebo. |
| Gerlach 1978 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no clozapine combination treatment ‐ haloperidol vs haloperidol + biperiden vs thioridazine vs clozapine. |
| Glick 2004 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no randomisation to combination treatments ‐ clozapine vs olanzapine with uncontrolled use of concomitant psychotropic medications in both groups. |
| Goff 1996 | Allocation: randomised. Participants: people with schizophrenia receiving clozapine, not defined as treatment‐resistant. |
| Goff 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: placebo controlled trial ‐ CX516 + clozapine vs placebo. |
| Gunduz‐Bruce 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + pimozide vs clozapine + placebo. |
| Haro 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no comparison with clozapine combination treatment ‐ amisulpride + quetiapine vs clozapine. |
| Hebrani 2008 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + topiramate vs clozapine + placebo. |
| Henderson 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + rosiglitazone vs clozapine + placebo. |
| Honer 2006 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + risperidone vs clozapine + placebo. |
| Honer 2007 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + risperidone vs clozapine + placebo. |
| Honigfeld 1989 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ non‐responders to haloperidol assigned to clozapine vs chlorpromazine. |
| Ji 2005 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ chlorpromazine vs clozapine. |
| Josiassen 2003 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + risperidone vs clozapine + placebo. |
| Klieser 1993 | Allocation: non‐randomised (quasi‐experimental). |
| Kluge 2007 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment resistant. |
| Li 2004 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ loxapine vs clozapine. |
| Liu 2008 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no comparison with combination treatment ‐ clozapine + fluphenazine decanoate vs clozapine. |
| Ma 2007 | Allocation: randomised, double‐blind. Participants: people with treatment‐resistant schizophrenia. Intervention: no comparison with combination treatment ‐ aripiprazole + clozapine vs clozapine. |
| Marder 1998 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ haloperidol vs clozapine. |
| Meltzer 1999 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ risperidone vs clozapine. |
| Millar 2008 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no comparison with active combination treatment ‐ clozapine + aripiprazole vs clozapine + placebo. |
| Nair 1998 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ clozapine 100 mg vs clozapine 300 mg vs clozapine 600 mg. |
| NCT00628420 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ N‐desmethylclozapine vs placebo. |
| NCT00649844 | Allocation: randomised, double‐blind. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ ziprasidone vs clozapine. |
| NCT00654576 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no drug combination treatment ‐ antipsychotic vs antipsychotic + psychosocial intervention. |
| NCT00753051 | Allocation: randomised, double‐blind. Participants: people with treatment‐resistant schizophrenia partially responsive or unresponsive to clozapine. Intervention: no comparison with drug combination treatment ‐ haloperidol + clozapine vs electroconvulsive therapy + clozapine. |
| Petit 1996 | Allocation: randomised. Participants: people with acute exacerbation of schizophrenia. |
| Pickar 1994 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Potkin 1999 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + glycine vs clozapine + placebo. |
| Potter 1989 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ chlorpromazine vs clozapine. |
| Qi 1990 | Allocation: non‐randomised (review). |
| Remington 2009 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + tetrabenazine vs clozapine + placebo. |
| Riera 2004a | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ aripiprazole vs another antipsychotic medication. |
| Riera 2004b | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ aripiprazole vs another antipsychotic medication. |
| Ruan 2008 | Allocation: randomised. Participants: people with schizophrenia and depression. |
| Shen 2004 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Shun 2000 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Sihloh 1997 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + sulpiride vs clozapine + placebo. |
| Small 2003 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Stryjer 2004 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Uzun 2006 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Volavka 2005 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ clozapine vs olanzapine vs risperidone vs haloperidol. |
| Wan 2007 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ olanzapine vs clozapine. |
| Wang 2002 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ risperidone vs clozapine. |
| Welbel 1980 | Allocation: non‐randomised (review). |
| Xu 2008 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no comparison with combination treatment ‐ clozapine + risperidone vs clozapine. |
| Xue 2014 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Yagcioglu 2005 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + risperidone vs clozapine + placebo. |
| Yang 1994 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Zhang 2008a | Allocation: randomised. Participants: people with negative symptoms of schizophrenia. Intervention: no combination treatment ‐ clozapine + paroxetine vs clozapine. |
| Zhang 2008b | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + paroxetine vs clozapine + placebo. |
| Zhang 2013 | Allocation: randomised. Participants: people with no use of antipsychotic medication 2 weeks prior to hospitalisation ‐ not taking clozapine. |
| Zheng 2007 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no combination treatment ‐ quetiapine vs clozapine. |
| Zhu 1999 | Allocation: randomised. Participants: people with schizophrenia but not defined as treatment‐resistant. |
| Zhu 2002 | Allocation: randomised. Participants: people with treatment‐resistant schizophrenia. Intervention: no active comparison with combination treatment ‐ clozapine + pipotiazine palmitate vs clozapine + placebo. |
Differences between protocol and review
The structure of the review has changed since initial publication ‐ in line with methodology changes for all Cochrane Reviews ‐ for example, inclusion of 'Summary of findings' tables. We have re‐ordered and re‐worded our outcomes but not changed the type of outcomes originally listed as outcomes of interest.
Contributions of authors
SB*: screened search results, retrieved papers against eligibility criteria, appraised quality of papers, extracted data from papers, wrote to authors of papers for additional information, entered data into Review Manager 5, analysed data, interpreted data, and drafted the review.
UO*: collected data, designed search strategies, screened search results, screened and retrieved papers against eligibility criteria, appraised quality of papers, wrote to authors of papers for additional information, entered data into Review Manager 5, analysed data, interpreted data, and revised the manuscript.
MC: screened search results, retrieved papers against eligibility criteria, appraised quality of papers, extracted data from papers, interpreted data, and revised the manuscript.
AC: co‐ordinated the update of the review, helped in designing search strategies and collecting supplemental data, analysed data, interpreted review findings and provided a methodological and clinical perspective, and wrote the review.
* Equal contribution as authors for the 2015 search update.
Sources of support
Internal sources
Department of Medicine and Public Health, Section of Psychiatry and Clinical Psychology, University of Verona, Italy.
Department of Applied Health and Behavioral Sciences, Section of Psychiatry, University of Pavia, Italy.
External sources
No sources of support supplied
Declarations of interest
SB: none known.
UO: none known.
MC: none known.
AC: none known. AC was the main author of one of the studies included in this review (Cipriani 2013a), but he was not involved in the data extraction process for this trial.
Co‐lead author with Uwaila Olotu
Co‐lead author with Sarah Barber
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Cipriani 2013a {published data only}
- Barbui C, Accordini S, Nose M, Stroup S, Purgato M, Girlanda F, et al. Aripiprazole versus haloperidol in combination with clozapine for treatment‐resistant schizophrenia in routine clinical care a randomized, controlled trial. Journal of Clinical Psychopharmacology 2011;31(3):266‐73. [DOI] [PubMed] [Google Scholar]
- Cipriani A, Accordini S, Nosè M, Purgato M, Girlanda F, Tansella M, et al. Aripiprazole versus haloperidol in combination with clozapine for treatment‐resistant schizophrenia: a 12‐month, randomized, naturalistic trial. Journal of Clinical Psychopharmacology 2013;33(4):533‐7. [DOI] [PubMed] [Google Scholar]
Genç 2007 {published data only}
- Genç Y, Taner E, Candansayar S. Comparison of clozapine‐amisulpride and clozapine‐quetiapine combinations for patients with schizophrenia who are partially responsive to clozapine: a single‐blind randomized study. Advances in Therapy 2007;24(1):1‐13. [DOI] [PubMed] [Google Scholar]
Kong 2001 {published data only}
- Kong QR. Clozapine combined with risperidone and sulpiride in treatment resistant schizophrenia. Shandong Archives of Psychiatry 2001;14:119‐20. [Google Scholar]
Kuwilsky 2010 {published data only}
- Kuwilsky A, Krumm B, Englisch S, Dressing H, Zink M. Long‐term efficacy and tolerability of clozapine combined with ziprasidone or risperidone. Pharmacopsychiatry 2010;43(6):216‐20. [DOI] [PubMed] [Google Scholar]
- Zink M, Kuwilsky A, Krumm B, Dressing H. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomised controlled clinical trial. Journal of Psychopharmacology 2009;23:305‐14. [DOI] [PubMed] [Google Scholar]
Wen 2015 {published data only}
- Wen RH, Liu S. A comparative study of clozapine combined with ziprasidone or quetiapine for treatment resistant schizophrenia. Medical Journal of Chinese People's Health 2015;27(2):4‐6. [Google Scholar]
References to studies excluded from this review
Angst 1971 {published data only}
- Angst J, Bente D, Berner P, Heimann H, Helmchen H, Hippius H. The clinical effect of clozapine [Das klinische wirkungsbild von clozapin]. Pharmakopsychiatrie und Neuropsychopharmakologie 1971;4:201‐11. [Google Scholar]
Anil 2009 {published data only}
- Anil E. Risperidone augmented with clozapine for schizophrenia. Stanley Foundation Research Programs 2009.
Anonymous 2009 {published data only}
- Anonymous. Clinician's desktop reference for clinical & cost utility of latest antipsychotics cutlass. www.iop.kcl.ac.uk/iop/extras/cutlass/Downloads/cutlass.pdf (date accessed 3 April 2009).
Assion 2008 {published data only}
- Assion HJ, Reinbold H, Lemanski S, Basilowski M, Juckel G. Amisulpride augmentation in patients with schizophrenia partially responsive or unresponsive to clozapine. A randomized, double‐blind, placebo‐controlled trial. Pharmacopsychiatry 2008;41(1):24‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Bao 1988 {published data only}
- Bao XQ. A double‐blind study on the effect of clozapine, penfluridol and chlorpromazine in the treatment of schizophrenia. Chung Hua Shen Ching Ching Shen Ko Tsa Chih [Chinese Journal of Neurology and Psychiatry] 1988;21(5):274‐6. [PubMed] [Google Scholar]
Barnes 2013 {published data only}
- Barnes TR, Drake RJ, Dunn G, Hayhurst KP, Jones PB, Lewis SW. Effect of prior treatment with antipsychotic long‐acting injection on randomised clinical trial treatment outcomes. British Journal of Psychiatry 2013;203(3):215‐20. [DOI] [PubMed] [Google Scholar]
Bender 1997 {published data only}
- Bender S, Olbrich H, Hornstein C, Schöne W, Falkai P. Antipsychotic efficacy of trimipramine: double‐blind comparison with a classical neuroleptic. Pharmacopsychiatry 1997;30(5):151. [DOI] [PubMed] [Google Scholar]
Bilder 2001 {published data only}
- Bilder RM, Goldman RS, Volavka J, Czobor P, Hoptman M, Sheitman B, et al. Neurocognitive effects of clozapine, olanzapine, risperidone, and haloperidol on treatment‐resistant patients with schizophrenia and schizoaffective disorder. International Congress on Schizophrenia Research; 2001 Apr 28 ‐ May 2; Whistler. British Columbia, Canada, 2001.
Bustillo 2009 {published data only}
- Bustillo J. Lamotrigine for schizophrenia. Stanley Foundation Research Programs 2009.
Cao 2003 {published data only}
- Cao HJ, You HF, Fan FL, Zhang J. The control study of risperidone and clozapine for the treatment‐resistant schizophrenia. Chinese Journal of Medicine Research 2003;3(4):316‐9. [Google Scholar]
Chang 2008 {published data only}
Cooper 2005 {published data only}
- Cooper D, Moisan J, Gaudet M, Abdous B, Gregoire JP. Ambulatory use of olanzapine and risperidone: a population‐based study on persistence and the use of concomitant therapy in the treatment of schizophrenia. Canadian Journal of Psychiatry 2005;50(14):901‐8. [DOI] [PubMed] [Google Scholar]
Dai 2014 {published data only}
- Dai X. Ziprasidone combined with small dose of clozapine for 44 cases of treatment resistant schizophrenia [齐拉西酮联合小剂量氯氮平治疗44例难治性精神分裂症效果评价]. For All Health 2014;8(9):176‐7. [Google Scholar]
Daniel 1994 {published data only}
- Daniel DG. Comparison of risperidone and clozapine on clinical and cognitive functions in psychotic disorders. Biological Psychiatry 1994;35:667. [Google Scholar]
Dong 2007 {published data only}
- 董太新, 阮江红, 李永芝, 李昱. Ziprasidone treatment of the first clinical diagnosis of schizophrenia [齐拉西酮治疗首诊精神分裂症临床分析]. Medical Journal of Chinese People's Health 2007;19(12):1061. [Google Scholar]
Feifel 2009 {published data only}
- Feifel D. Oxytocin for schizophrenia. Stanley Foundation Research Programs 2009.
Fleischhacker 2008a {published data only}
- Fleischhacker W, Heikkinen ME, Olie JP, Dewaele P, McQuade R, Hennicken D, et al. Long‐term effect on weight of aripiprazole‐clozapine in schizophrenia patients with suboptimal response on clozapine. European Neuropsychopharmacology 2008;18:S447. [Google Scholar]
Fleischhacker 2008b {published data only}
- Fleischhacker WW, Heikkinen ME, Olie JP, Landsberg W, Dewaele P, McQuade R, et al. Weight change on aripiprazole‐clozapine combination in schizophrenic patients with weight gain and suboptimal response on clozapine: 16‐week double‐blind study. European Psychiatry 2008;23:S114‐5. [Google Scholar]
Freudenreich 2009 {published data only}
- Freudenreich O. Risperidone added to clozapine for schizophrenia. Stanley Foundation Research Programs 2009.
Gerlach 1978 {published data only}
- Gerlach J, Simmelsgaard H. Tardive dyskinesia during and following treatment with haloperidol, haloperidol + biperiden, thioridazine, and clozapine. Psychopharmacology 1978;59(2):105‐12. [DOI] [PubMed] [Google Scholar]
Glick 2004 {published data only}
- Glick ID, Zaninelli R, Hsu C, Young FK, Weiss L, Gunay I, et al. Patterns of concomitant psychotropic medication use during a 2‐year study comparing clozapine and olanzapine for the prevention of suicidal behavior. Journal of Clinical Psychiatry 2004;65(5):679‐85. [DOI] [PubMed] [Google Scholar]
Goff 1996 {published data only}
- Goff DC, Tsai G, Manoach DS, Flood J, Darby DG, Coyle JT. D‐cycloserine added to clozapine for patients with schizophrenia. American Journal of Psychiatry 1996;153(12):1628‐30. [DOI] [PubMed] [Google Scholar]
Goff 2009 {published data only}
- Goff D. CX516 for schizophrenia. Stanley Foundation Research Programs 2009.
Gunduz‐Bruce 2009 {published data only}
- Gunduz‐Bruce H. Pimozide for schizophrenia. Stanley Foundation Research Programs 2009.
Haro 2009 {published data only}
- Haro J. Amisulpride and quetiapine for schizophrenia. Stanley Foundation Research Programs 2009.
Hebrani 2008 {published data only}
- Hebrani P, Behdani F, Rafiee A. Adjunctive topiramate in hospitalized patients with chronic schizophrenia disorder. European Neuropsychopharmacology 2008;18:S466. [Google Scholar]
Henderson 2009 {published data only}
- Henderson D. Rosiglitazone for schizophrenia. Stanley Foundation Research Programs 2009.
Honer 2006 {published data only}
- Honer WG, Thornton AE, Chen EY, Chan RC, Wong JO, Bergmann A, et al. Clozapine alone versus clozapine and risperidone with refractory schizophrenia. New England Journal of Medicine 2006;354(5):472‐82. [DOI] [PubMed] [Google Scholar]
Honer 2007 {published data only}
- Honer W. A randomized controlled trial of antipsychotic polypharmacy: clozapine plus risperidone. European Neuropsychopharmacology 2007;17(Suppl 4):S200. [Google Scholar]
Honigfeld 1989 {published data only}
- Honigfeld G, Patin J. Predictors of response to clozapine therapy. Psychopharmacology 1989;99:64‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Ji 2005 {published data only}
- Ji XW. Serum Level of Homocysteine in Patients with Schizophrenia [Masters thesis]. Shandong University (China), 2005. [Google Scholar]
Josiassen 2003 {published data only}
- Josiassen R, Joseph A, Kohegyi E, Paing W. Clozapine augmentation with risperidone in refractory schizophrenia. Schizophrenia Research 2003;60:288. [Google Scholar]
Klieser 1993 {published data only}
- Klieser E, Strauss WH. Quasi‐experimental comparison of the efficacy of classical and atypical neuroleptics in acute schizophrenia with respect to their cognitive and emotional side effects. Pharmacopsychiatry 1993;26:168. [Google Scholar]
Kluge 2007 {published data only}
- Kluge M, Schuld A, Himmerich H, Dalal M, Schacht A, Wehmeier PM, et al. Clozapine and olanzapine are associated with food craving and binge eating: results from a randomized double‐blind study. Journal of Clinical Psychopharmacology 2007;27(6):662‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Li 2004 {published data only}
- Li YC, Chang HR, Li CS. Control study of loxapine succinate and clozapine on curative effects in treatment of schizophrenics. Health Psychology Journal 2004;3:202. [Google Scholar]
Liu 2008 {published data only}
- 刘友夺, 黄文芳, 黄时金. Fluphenazine decanoate combined clozapine treatment of refractory schizophrenia [氯氮平合癸氟奋乃静治疗难治性精神分裂症对照研究]. Medical Journal of Chinese People's Health 2008;20(12):1274. [Google Scholar]
Ma 2007 {published data only}
- Ma Q, Lian HT, Li LX. A study of aripiprazole combined with clozapine in refractory schizophrenia. Medical Journal of Chinese People's Health 2007;19(12):1046‐8. [Google Scholar]
Marder 1998 {published data only}
- Marder SR. Antipsychotics in treatment‐refractory schizophrenia. In: Marder SR, chairperson. Atypical antipsychotic agents in the treatment of schizophrenia and other psychotic disorders. I. Unique patient populations. Journal of Clinical Psychiatry 1998;59:259‐65. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Meltzer 1999 {published data only}
- Meltzer HY. Risperidone and clozapine for treatment‐resistant schizophrenia. American Journal of Psychiatry 1999;156(7):1126‐7. [DOI] [PubMed] [Google Scholar]
Millar 2008 {published data only}
- Millar H, Felter C, Landsberg W. The effects of aripiprazole in combination with clozapine: patient functioning results from a double‐blind, 16‐week study in patients with schizophrenia (CN138‐170). Journal of Psychopharmacology 2008;22(5):A17. [Google Scholar]
Nair 1998 {published data only}
- Nair CJ, Abraham G, Stanilla JK, Tracy JI, Leon J, Simpson GM, et al. Therapeutic effects of clozapine on tardive dyskinesia. Cognitive and Behavioral Practice 1998;1:123‐31. [Google Scholar]
NCT00628420 {published data only}
- NCT00628420. Safety study of ACP‐104: to demonstrate the safety, tolerability, and pharmacokinetics. www.clinicaltrials.gov/show/NCT00628420 Date first received: 24 February 2008.
NCT00649844 {published data only}
- NCT00649844. A study comparing the efficacy and tolerability of ziprasidone vs clozapine for the treatment of schizophrenia in patients who continue to have symptoms on or cannot tolerate other antipsychotic drugs. www.clinicaltrials.gov/show/NCT00649844 Date first received: 28 March 2008.
NCT00654576 {published data only}
- NCT00654576. Effectiveness of antipsychotic combination with psychosocial intervention on outcome of patients with schizophrenia. www.clinicaltrials.gov/show/NCT00654576 Date first received: 3 April 2008.
NCT00753051 {published data only}
- NCT00753051. Treat clozapine‐resistant schizophrenia comparing between clozapine + haloperidol versus clozapine + electroconvulsive therapy (ECT). www.clinicaltrials.gov/show/NCT00753051 Date first received: 15 September 2008.
Petit 1996 {published data only}
- Petit M, Raniwalla J, Tweed J, Leutenegger E, Dollfus S, Kelly F. A comparison of an atypical and typical antipsychotic, zotepine versus haloperidol in patients with acute exacerbation of schizophrenia: a parallel‐group double‐blind trial. Psychopharmacology Bulletin 1996;32(1):81‐7. [PubMed] [Google Scholar]
Pickar 1994 {published data only}
- Pickar D, Litman RE, Hong WW, Su TP, Weissman EM, Hsiao JK, et al. Clinical response to clozapine in patients with schizophrenia. Archives of General Psychiatry 1994;51(2):159‐60. [DOI] [PubMed] [Google Scholar]
Potkin 1999 {published data only}
- Potkin SG, Jin Y, Bunney BG, Costa J, Gulasekaram B. Effect of clozapine and adjunctive high‐dose glycine in treatment‐resistant schizophrenia. American Journal of Psychiatry 1999;156(1):145‐7. [DOI] [PubMed] [Google Scholar]
Potter 1989 {published data only}
- Potter WZ, Ko GN, Zhang LD, Yan WW. Clozapine in China: a review and preview of US/PRC collaboration. Psychopharmacology 1989;99 Suppl:S87‐91. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Qi 1990 {published data only}
- Qi SX. Clozapine therapy in schizophrenia. Chinese Journal of Nervous and Mental Disorders 1990;16(2):90‐2. [Google Scholar]
Remington 2009 {published data only}
- Remington G. Tetrabenazine for schizophrenia. Stanley Foundation Research Programs 2009.
Riera 2004a {published data only}
- Riera L, Tandon R, Stock EG, Kujawa MJ, Torbeyns AF, Borian FE, et al. Broad effectiveness trial with aripiprazole. 12th Biennial Winter Workshop on Schizophrenia; 2004 Feb 7‐13; Davos, Switzerland. 2004.
Riera 2004b {published data only}
- Riera L, Hu R, Stock E, Nyilas M, Torbeyns A, Borian F, et al. Broad effectiveness trial with aripiprazole. 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, USA. 2004.
Ruan 2008 {published data only}
- Ruan S, Zheng W, Du L. A control study of clozapine plus fluoxetine in schizophrenia with depressive symptoms. Journal of Clinical Psychosomatic Diseases 2008;14(3):211‐3. [Google Scholar]
Shen 2004 {published data only}
- Shen J, Feng Z, Guo J, Wang W, Wang Z. Comparative study on therapy of schizophrenia with quetiapine and clozapine. Tianjin Pharmacy 2004;16(1):38‐9. [Google Scholar]
Shun 2000 {published data only}
- Shun ZJ, Liu BW, Yu FP, Li ZL, Jiang DF, Zhang DQ. A comparison trial of clozapine combined with perphenazine and single clozapine or perphenazine in treatment of schizophrenia. Journal of Hebei Psychological Health 2000;3:142‐4. [Google Scholar]
Sihloh 1997 {published data only}
- Shiloh R, Zemishlany Z, Aizenberg D, Radwan M, Schwartz B, Dorfman‐Etrog P, et al. Sulpiride augmentation in people with schizophrenia partially responsive to clozapine. A double‐blind, placebo‐controlled study. British Journal of Psychiatry 1997;171:569‐73. [DOI] [PubMed] [Google Scholar]
Small 2003 {published data only}
- Small JG, Klapper MH, Malloy FW, Steadman TM. Tolerability and efficacy of clozapine combined with lithium in schizophrenia and schizoaffective disorder. Journal of Clinical Psychopharmacology 2003;23(3):223‐8. [DOI] [PubMed] [Google Scholar]
Stryjer 2004 {published data only}
- Stryjer R, Strous R, Bar F, Shaked G, Shiloh R, Rozencwaig S, et al. Donepezil augmentation of clozapine monotherapy in schizophrenia patients: a double blind cross‐over study. Human Psychopharmacology 2004;19(5):343‐6. [DOI] [PubMed] [Google Scholar]
Uzun 2006 {published data only}
- Uzun S, Kozumplik O, Mimica N, Folnegovic‐Smalc V. Treatment with six different antipsychotics: evaluation by scale for the assessment of negative symptoms (SANS). 13th Biennial Winter Workshop on Schizophrenia Research; 2006 Feb 4‐10; Davos, Switzerland. 2006.
Volavka 2005 {published data only}
- Volavka J, Nolan KA, Kline L, Czobor P, Citrome L, Sheitman B, et al. Efficacy of clozapine, olanzapine, risperidone, and haloperidol in schizophrenia and schizoaffective disorder assessed with nurses observation scale for inpatient evaluation. Schizophrenia Research 2005;76(1):127‐9. [DOI] [PubMed] [Google Scholar]
Wan 2007 {published data only}
- 万好, 李玉芳, 李军, 崔谊. Olanzapine on efficacy in patients with late‐onset schizophrenia and quality of life [奥氮平对晚发精神分裂症患者的疗效及生活质量影响]. 中国医疗前沿 2007;2(23):81‐2. [Google Scholar]
Wang 2002 {published data only}
- Wang R, Geng Y, Pan D, Zhang S. A comparative trial of efficacy of risperidone vs clozapine in treatment of refractory schizophrenia. Chinese Journal of Pharmacoepidemiology 2002;11(5):230‐1. [Google Scholar]
Welbel 1980 {published data only}
- Welbel L. Differences in the clinical effect of various neuroleptics [Roznice w dziallaniu klinicznym niektorych neuroleptykow]. Psychiatria Polska 1980;14(2):113‐8. [PubMed] [Google Scholar]
Xu 2008 {published data only}
- 徐儒瑾, 万学东, 杜春秀, 吴乐平. Joint clozapine risperidone treatment of refractory schizophrenia [利培酮联合氯氮平治疗难治性精神分裂症]. 中国现代药物应用 2008;2(16):78. [Google Scholar]
Xue 2014 {published data only}
- Xue H, Yanbo Y, Fude Y, Zheng L, Chuanyue W, Hong D, et al. The Chinese first‐episode schizophrenia trial: design and methodology of the trial. East Asian Archives Psychiatry 2014;24(4):169‐73. [PubMed] [Google Scholar]
Yagcioglu 2005 {published data only}
- Yagcioglu AE, Kivircik Akdede BB, Turgut TI, Tumuklu M, Yazici MK, Alptekin K, et al. A double‐blind controlled study of adjunctive treatment with risperidone in schizophrenic patients partially responsive to clozapine: efficacy and safety. Journal of Clinical Psychiatry 2005;66(1):63‐72. [DOI] [PubMed] [Google Scholar]
Yang 1994 {published data only}
- Yang X, Hang YL, Jia WLQ, Wei LM. Clozapine combined with chlorpromazine in the treatment of positive symptoms in schizophrenia. Shanghai Psychological Medicine 1994;6(2):71‐2. [Google Scholar]
Zhang 2008a {published data only}
- Zhang YK, Zhao LQ, Huang Q. The influencing of olanzapine to the blood drug level of clozapine and the treatment effect of schizophrenia. Modern Preventive Medicine 2008;35(12):2356‐7. [Google Scholar]
Zhang 2008b {published data only}
- 张仁凯, 王建利, 付学凯, 张春鹏. 60 patients with schizophrenia negative symptoms combined application of clozapine and clozapine compared the efficacy of paroxetine [60例精神分裂症的阴性症状应用氯氮平和氯氮平合并帕罗西汀的疗效比较]. Journal of Qiqihar Medical College [Qiqihar Yixueyuan xuebao] 2008;29(14):1709. [Google Scholar]
Zhang 2013 {published data only}
- Zhang T, Lu M, Ma X. A controlled clinical study of the effect on ECG QT when using clozapine alone and in combination [单用与联合使用氯氮平对心电图QT 间期影响的对照研究]. Journal of Sichuan Mental Health 2013;26(2):120‐1. [Google Scholar]
Zheng 2007 {published data only}
- 郑景莉, 王保国, 付爱玲, 李蓓. Quetiapine combined clozapine in the treatment of negative symptoms of schizophrenia [奎硫平联合氯氮平治疗精神分裂症阴性症状对照研究]. Medical Recapitulate 2007;13(23):1878‐9. [Google Scholar]
Zhu 1999 {published data only}
- Zhu Y, Zhang S, Zhang D. A controlled trial comparing chlorimipramine and sulpiride as adjunct to clozapine in the treatment of negative symptoms of schizophrenia. Journal of Clinical Psychological Medicine 1999;9(4):204‐5. [Google Scholar]
Zhu 2002 {published data only}
- Zhu H, Yu G, Deng D. A study of clozapine combined with or without pipotiazine palmitate in refractory schizophrenia. Journal of Clinical Psychological Medicine 2002;12(1):15‐7. [Google Scholar]
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Andreasen 1984a
- Andreasen NC. Scale for the Assessment of Positive Symptoms (SAPS). Iowa City: University of Iowa, 1984. [Google Scholar]
Andreasen 1984b
- Andreasen NC. Scale for the Assessment of Negative Symptoms (SANS). Iowa City: University of Iowa, 1984. [Google Scholar]
APA 2004
- American Psychiatric Association. Practice Guidelines for the Treatment of Patients with Schizophrenia. 2nd Edition. Arlington, VA: American Journal of Psychiatry, 2004. [Google Scholar]
Barbui 2009
- Barbui C, Signoretti A, Mule S, Boso M, Cipriani A. Does the addition of a second antipsychotic drug improve clozapine treatment?. Schizophrenia Bulletin 2009;35(2):458‐68. [DOI: 10.1093/schbul/sbn030] [DOI] [PMC free article] [PubMed] [Google Scholar]
Barbui 2011
- Barbui C, Accordini S, Nose M, Stroup S, Purgato M, Girlanda F, et al. Aripiprazole versus haloperidol in combination with clozapine for treatment‐resistant schizophrenia in routine clinical care a randomized, controlled trial. Journal of Clinical Psychopharmacology 2011;31(3):266‐73. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'etude des indices d'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT Jr, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Chouinard 1980
- Chouinard G, Ross‐Chouinard A, Annable L. Extrapyramidal Symptom Rating Scale. Canadian Journal of Neurological Science 1980;7:233. [Google Scholar]
Cipriani 2013b
- Cipriani A, Higgins JP, Geddes JR, Salanti G. Conceptual and technical challenges in network meta‐analysis. Annals of Internal Medicine 2013;159(2):130‐7. [DOI] [PubMed] [Google Scholar]
Day 1995
- Day JC, Wood G, Dewey M, Bentall RP. A self‐rating scale for measuring neuroleptic side‐effects. Validation in a group of schizophrenic patients. British Journal of Psychiatry 1995;166(5):650‐3. [DOI] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Dold 2014
- Dold M, Leucht S. Pharmacotherapy of treatment‐resistant schizophrenia: a clinical perspective. Evidence Based Mental Health 2014;17(2):33‐7. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Duggan 2005
- Duggan L, Fenton M, Rathbone J, Dardennes R, El‐Dosoky A, Indran S. Olanzapine for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD001359.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder CSO. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;13:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Fleischhacker 1989
- Fleischhacker WW, Bergmann KJ, Perovich R, Pestreich LK, Borenstein M, Lieberman JA, et al. The Hillside Akathisia Scale: a new rating instrument for neuroleptic‐induced akathisia. Psychopharmacology Bulletin 1989;25(2):222‐6. [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;149:876‐83. [DOI] [PubMed] [Google Scholar]
GRADEpro [Computer program]
- GRADE Working Group, McMaster University. GRADEpro. Version 3.2. Hamilton (ON): GRADE Working Group, McMaster University, 2004.
Gulliford 1999
- Gulliford MC. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy U. ECDEU Assessment Manual for Psychopharmacology. Bethesda (MD): National Institute of Mental Health, 1976. [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale of depression. Journal of Neurology, Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Altman DG. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 (updated September 2008). The Cochrane Collaboration, 2008. Available from handbook.cochrane.org.
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda (NY): Multi‐Health Systems, 1986. [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of Brief Psychiatric Rating Scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2005b
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Leucht 2013
- Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple‐treatments meta‐analysis. Lancet 2013;382(9896):951‐62. [DOI] [PubMed] [Google Scholar]
Leucht 2016
- Leucht S, Chaimani A, Cipriani AS, Davis JM, Furukawa TA, Salanti G. Network meta‐analyses should be the highest level of evidence in treatment guidelines. European Archive of Psychiatry Clinical Neuroscience 2016;266(6):477‐80. [DOI] [PubMed] [Google Scholar]
Lingjærde 1987
- Lingjærde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross‐sectional study of side effects in neuroleptic‐treated patients. Acta Psychatrica Scandinavica 1987;76(Suppl):334. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Mavridis 2015
- Mavridis D, Giannatsi M, Cipriani A, Salanti G. A primer on network meta‐analysis with emphasis on mental health. Evidence Based Mental Health 2015;18(2):40‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00224315
- NCT00224315. Clozapine‐augmentation with ziprasidone or risperidone, a randomized, prospective trial. clinicaltrials.gov/show/NCT00224315 Date first received: 15 September 2005.
NICE 2014
- National Institute for Health and Care Excellence. Psychosis and schizophrenia in adults: prevention and management. www.nice.org.uk/guidance/cg178 (accessed 5 October 2016).
Nosè 2009
- Nosè M, Accordini S, Artioli P, Barale F, Barbui C, Beneduce R, et al. Rationale and design of an independent randomised controlled trial evaluating the effectiveness of aripiprazole or haloperidol in combination with clozapine for treatment‐resistant schizophrenia. Trials 2009;15(10):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Rouse 2016
- Rouse B, Cipriani A, Shi Q, Coleman AL, Dickersin K, Li T. Network meta‐analysis for clinical practice guidelines: a case study on first‐line medical therapies for primary open‐angle glaucoma. Annals of Internal Medicine 2016;164(10):674‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rust 1989
- Rust J, Golombok S. Modern Psychometrics. London: Routledge, 1989. [Google Scholar]
Samara 2016
- Samara MT, Dold M, Gianatsi M, Nikolakopoulou A, Helfer B, Salanti G, et al. Efficacy, acceptability, and tolerability of antipsychotics in treatment‐resistant schizophrenia: a network meta‐analysis. JAMA Psychiatry 2016;73(3):199‐210. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Simpson 1970
- Simpson EN, Angus JWF. A rating scale for extrapyramidal side‐effects. Acta Psychiatrica Scandinavica Supplementum 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Stroup 2016
- Stroup TS, Gerhard T, Crystal S, Huang C, Olfson M. Comparative effectiveness of clozapine and standard antipsychotic treatment in adults with schizophrenia. American Journal of Psychiatry 2016;173(2):166‐73. [DOI] [PubMed] [Google Scholar]
Suzuki 2011
- Suzuki T, Remington G, Mulsant BH, Rajji TK, Uchida H, Graff‐Guerrero A, et al. Treatment resistant schizophrenia and response to antipsychotics: a review. Schizophrenia Research 2011;133:54‐62. [DOI] [PubMed] [Google Scholar]
Taylor 2000
- Taylor DM, Duncan‐McConnell D. Refractory schizophrenia and atypical antipsychotics. Journal of Psychopharmacology 2000;14(4):409‐18. [DOI] [PubMed] [Google Scholar]
Taylor 2012
- Taylor DM, Smith L, Gee SH, Nielsen J. Augmentation of clozapine with a second antipsychotic ‐ a meta‐analysis. Acta Psychiatrica Scandinavica 2012;125(1):15‐24. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]
Zink 2009
- Zink M, Kuwilsky A, Krumm B, Dressing H. Efficacy and tolerability of ziprasidone versus risperidone as augmentation in patients partially responsive to clozapine: a randomised controlled clinical trial. Journal of Psychopharmacology 2009;23:305‐14. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Cipriani 2009
- Cipriani A, Boso M, Barbui C. Clozapine combined with different antipsychotic drugs for treatment resistant schizophrenia. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD006324.pub2; PUBMED: 19588385] [DOI] [PMC free article] [PubMed] [Google Scholar]