

Abstract
Background
Premenstrual syndrome (PMS) is a psychological and somatic disorder of unknown aetiology, with symptoms typically including irritability, depression, mood swings, bloating, breast tenderness and sleep disturbances. About 3% to 10% of women who experience these symptoms may also meet criteria for premenstrual dysphoric disorder (PMDD). PMS symptoms recur during the luteal phase of the menstrual cycle and reduce by the end of menstruation. PMS results from ovulation and may be due to ovarian steroid interactions relating to neurotransmitter dysfunction. Premenstrual disorders have a devastating effect on women, their families and their work.
Several treatment options have been suggested for PMS, including pharmacological and surgical interventions. The treatments thought to be most effective tend to fall into one of two categories: suppressing ovulation or correcting a speculated neuroendocrine anomaly.
Transdermal oestradiol by patch, gel or implant effectively stops ovulation and the cyclical hormonal changes which produce the cyclical symptoms. These preparations are normally used for hormone therapy and contain lower doses of oestrogen than found in oral contraceptive pills. A shortened seven‐day course of a progestogen is required each month for endometrial protection but can reproduce premenstrual syndrome‐type symptoms in these women.
Objectives
To determine the effectiveness and safety of non‐contraceptive oestrogen‐containing preparations in the management of PMS.
Search methods
On 14 March 2016, we searched the following databases: the Cochrane Gynaecology and Fertility Group (CGF) Specialised Register; Cochrane Central Register of Studies (CRSO); MEDLINE; Embase; PsycINFO; CINAHL; ClinicalTrials.gov; metaRegister of Controlled trials (mRCT); and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) Search Portal. In addition, we checked the reference lists of articles retrieved.
Selection criteria
We included published and unpublished randomized placebo or active controlled trials on the efficacy of the use of non‐contraceptive oestrogen‐containing preparations in the management of premenstrual syndrome in women of reproductive age with PMS diagnosed by at least two prospective cycles without current psychiatric disorder.
Data collection and analysis
Two review authors independently selected studies, assessed risk of bias, extracted data on premenstrual symptoms and adverse effects and entered data into Review Manager 5 software. Where possible, intention‐to‐treat or modified intention‐to‐treat analysis was used. Studies were pooled using a fixed‐effect model, analysing cross‐over trials as parallel trials. Standardised mean differences (SMDs) with 95% confidence intervals (CIs) were calculated for premenstrual symptom scores. Risk ratios (RRs) with 95% confidence intervals (CIs) were calculated for dichotomous outcomes. The overall quality of the evidence was assessed using the GRADE working group methods.
Main results
The search resulted in 524 potentially relevant articles. Five eligible randomized controlled trials (RCTs) were identified (305 women). Trials using oral tablets, transdermal patches and implants were identified. No trial used gels.
One small cross‐over trial (11 women, effective sample size 22 women considering cross‐over trials) compared oral luteal‐phase oestrogen versus placebo. Data were very low quality and unsuitable for analysis, but study authors reported that the intervention was ineffective and might aggravate the symptoms of PMS. They also reported that there were no adverse events.
Three studies compared continuous oestrogen with progestogen versus placebo (with or without progestogen). These trials were of reasonable quality, although with a high risk of attrition bias and an unclear risk of bias due to potential carry‐over effects in two cross‐over trials. Continuous oestrogen had a small to moderate positive effect on global symptom scores (SMD −0.34, 95% CI −0.59 to −0.10, P = 0.005, 3 RCTs, 158 women, effective sample size 267 women, I² = 63%, very low quality evidence). The evidence was too imprecise to determine if the groups differed in withdrawal rates due to adverse effects (RR 0.64, 95% CI 0.26 to 1.58, P = 0.33, 3 RCTs, 196 women, effective sample size 284 women, I² = 0%, very low quality evidence). Similarly, the evidence was very imprecise in measures of specific adverse events, with large uncertainties around the true value of the relative risk. None of the studies reported on long‐term risks such as endometrial cancer or breast cancer.
One study compared patch dosage (100 vs 200 µg oestrogen, with progestogen in both arms) and had a high risk of performance bias, detection bias and attrition bias. The study did not find evidence that dosage affects global symptoms but there was much uncertainty around the effect estimate (SMD −1.55, 95% CI −8.88 to 5.78, P = 0.68, 1 RCT, 98 women, very low quality evidence). The evidence on rates of withdrawal for adverse events was too imprecise to draw any conclusions (RR 0.70, 95% CI 0.34 to 1.46, P = 0.34, 1 RCT, 107 women, low‐quality evidence). However, it appeared that the 100 µg dose might be associated with a lower overall risk of adverse events attributed to oestrogen (RR 0.51, 95% Cl 0.26 to 0.99, P = 0.05, 1 RCT, 107 women, very low quality evidence) with a large uncertainty around the effect estimate.
The overall quality of the evidence for all comparisons was very low, mainly due to risk of bias (specifically attrition), imprecision, and statistical and clinical heterogeneity.
Authors' conclusions
We found very low quality evidence to support the effectiveness of continuous oestrogen (transdermal patches or subcutaneous implants) plus progestogen, with a small to moderate effect size. We found very low quality evidence from a study based on 11 women to suggest that luteal‐phase oral unopposed oestrogen is probably ineffective and possibly detrimental for controlling the symptoms of PMS. A comparison between 200 µg and 100 µg doses of continuous oestrogen was inconclusive with regard to effectiveness, but suggested that the lower dose was less likely to cause side effects. Uncertainty remains regarding safety, as the identified studies were too small to provide definite answers. Moreover, no included trial addressed adverse effects that might occur beyond the typical trial duration of 2‐8 months. This suggests the choice of oestrogen dose and mode of administration could be based on an individual woman’s preference and modified according to the effectiveness and tolerability of the chosen regimen.
Plain language summary
Oestrogen for premenstrual syndrome
Review question
Oestrogen is widely used to suppress ovulation, mainly as a contraceptive. This is the first systematic review aiming to evaluate the effectiveness and safety of non‐contraceptive oestrogen‐containing preparations (oral, patch, implant and gel) in controlling symptoms of premenstrual syndrome (PMS).
Background
PMS is characterised by a range of physical, psychological and behavioural symptoms that are not due to any organic disease, and that occur during the luteal phase (second half) of the menstrual cycle and disappear at the onset of menstruation. It is now thought that the condition is due to complex interactions between ovarian steroids and neurotransmitters. A clinical diagnosis requires that the symptoms are confirmed by prospective recording for at least two menstrual cycles and that they cause substantial distress or impairment to daily life (e.g. work, school, social activities, hobbies, interpersonal relationships).
Study characteristics
The review identified 524 potentially relevant articles. Only five randomized controlled trials met our inclusion criteria, and these compared oestrogen with placebo in a total of 305 women who were clinically diagnosed with PMS.
Key results
We found very low quality evidence to suggest that oral unopposed oestrogen given in the luteal phase of the menstrual cycle is probably ineffective for controlling the symptoms of PMS and may even make them worse. There was very low quality evidence to support the effectiveness of continuous oestrogen (in the form of transdermal patches or subcutaneous implants) plus progestogen, with a small to moderate effect size. A comparison between 200 microgram and 100 microgram doses of continuous oestrogen was inconclusive with regard to effectiveness but suggested that the lower dose was less likely to cause side effects. Uncertainty remains regarding safety, as the identified studies were too small to provide definite answers. Moreover, none of the included trials addressed adverse effects that might occur beyond the typical trial duration of 2 to 8 months. This suggests the choice of oestrogen dose and mode of administration could be based on an individual woman’s preference and modified according to the effectiveness and tolerability of the chosen regimen.
Quality of the evidence
The overall quality of the evidence for all comparisons was graded as very low, mainly due to risk of bias in the included studies, imprecision (due to small sample sizes) and differences between the studies.
Summary of findings
Summary of findings for the main comparison. Oestrogen in combination with progestogen (sequential or continuous) compared to placebo for controlling symptoms of premenstrual syndrome.
| Oestrogen in combination with progestogen (sequential or continuous) compared to placebo. for controlling symptoms of premenstrual syndrome | ||||||
| Population: women diagnosed with symptoms of premenstrual syndrome (PMS) Setting: community Intervention: continuous oestrogen (implant or patch) plus progestogen Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with control | Risk with oestrogen in combination with progestogen | |||||
| Symptom scores over 2 to 4 menstrual cycles1 |
The difference in the mean change from baseline in symptom scores suggested a small to moderate benefit in the oestrogen group (SMD 0.27 lower, 95% CI 0.47 lower to 0.07 lower) | ‐ | 158 women, effective sample size 267 women considering cross‐over trials (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 2,3,4 | ||
| Withdrawal due to adverse events over 2 to 4 menstrual cycles1 |
77 per 1000 | 49 per 1000 (20 to 122) | RR 0.64 (0.26 to 1.58) | 196 women, effective sample size 284 women considering cross‐over trials (3 RCTs) | ⊕⊝⊝⊝ VERY LOW 2,4,5 | |
| Specific adverse events over 2 to 4 menstrual cycles1 |
Two RCTs (total 206 women) assessed one or more of nine specific adverse events and all findings were inconclusive. Events assessed were bleeding, breast tenderness, headache, nausea, weight gain, dysmenorrhoea, skin irritation, skin reaction and skin pigmentation. | ⊕⊝⊝⊝ VERY LOW 1,6 | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the mean risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Includes cross‐over trial(s), with cross‐over at 3 or 4 months
2Downgraded one level for serious risk of bias: poor reporting of methods in one study and/or high attrition, includes one or more cross‐over studies with unclear risk of carry‐over effects
3Downgraded one level for serious inconsistency: I² = 63%
4Downgraded one level for questionable applicability: one of the studies administered progestogen in both arms
5 Downgraded one level for serious imprecision: very few events and/or wide confidence intervals compatible with benefit in one or both arms or with no clinically meaningful effect
6Downgraded two levels for very serious imprecision: very few events and/or very wide confidence intervals compatible with benefit in one or both arms or with no clinically meaningful effect
Summary of findings 2. Oestrogen compared to placebo for controlling symptoms of premenstrual syndrome.
| Oestrogen compared to placebo for controlling symptoms of premenstrual syndrome | ||||||
| Population: women diagnosed with symptoms of premenstrual syndrome (PMS) Setting: community Intervention: luteal‐phase oestrogen Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with oestrogen | |||||
| Global symptoms of PMS over 3 menstrual cycles1 |
The study authors reported that luteal‐phase oestrogen had a large negative effect on overall symptoms compared to placebo (P < 0.01) | ‐ | 11 women, effective sample size 22 women considering this was a cross‐over trial (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2,3 | ||
| Adverse events: withdrawal due to adverse events over 3 menstrual cycles1 |
No withdrawals occurred in either arm | ‐ | 11 women, effective sample size 22 women considering this was a cross‐over trial (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2, 3 | ||
| Specific adverse events over 3 menstrual cycles1 |
The study authors reported that "no significant adverse events were encountered". | ‐ | 11 women,effective sample size 22 women considering this was a cross‐over trial (1 RCT) | ⊕⊝⊝⊝ VERY LOW 2, 3 | ||
| *The risk in the intervention group (and its 95% confidence interval) is based on the mean risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Cross‐over study with 11 women, but analyzed as a parallel trial (hence 22 participants). Women had 3 cycles in each arm.
2 Downgraded two levels for very serious risk of bias: poor reporting of methods, no data suitable for analysis as used non‐validated scales
3 Downgraded two levels for very serious imprecision: tiny sample size (but no further downgrading possible)
Summary of findings 3. Dose comparison: oestrogen 100 µg patch compared to 200 µg patch plus progestogen in both groups for controlling symptoms of premenstrual syndrome.
| Dose comparison: oestrogen 100 µg patch compared to 200 µg patch plus progestogen in both groups for controlling symptoms of premenstrual syndrome | ||||||
| Population: women diagnosed with symptoms of premenstrual syndrome (PMS) Setting: community Intervention: oestrogen 100 µg patch plus progestogen Comparison: oestrogen 200 µg patch plus progestogen | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with 200 µg patch plus progestogen | Risk with oestrogen 100 µg patch plus progestogen | |||||
| Symptoms score: Premenstrual Daily Questionnaire (PDQ) Scale from 0 to 30, with a higher score indicating a worse outcome over 8 months follow up |
The difference in the change from baseline in the PDQ score was too imprecise to determine whether dosage had an effect on global symptoms (MD 1.55 lower, 95% CI 8.88 lower to 5.78 higher) | ‐ | 98 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | ||
| Adverse events ‐ withdrawal rates over 8 months' follow‐up |
255 per 1000 | 178 per 1000 (87 to 372) | RR 0.70 (0.34 to 1.46) | 107 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| Adverse events ‐ specific side effects attributed to oestrogen over 8 months' follow‐up |
353 per 1000 | 180 per 1000 (92 to 349) | RR 0.51 (0.26 to 0.99) | 107 (1 RCT) | ⊕⊝⊝⊝ VERY LOW 1 2 | |
| *The risk in the intervention group (and its 95% confidence interval) is based on the mean risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded two levels for very serious risk of bias: unblinded, high attrition
2 Downgraded two levels for very serious imprecision: wide confidence intervals consistent with benefit in one or both arms or with no clinically meaningful effect from the intervention (however no further downgrading possible)
Background
Description of the condition
Premenstrual syndrome (PMS) is a disorder affecting large numbers of women, characterised by a set of symptoms that occur during the second half (luteal phase) of the menstrual cycle. Symptoms disappear by the end of menstruation and do not recur before ovulation, giving a symptom‐free interval of at least one week in the first half (follicular phase) of the menstrual cycle. PMS is cyclical and occurs in most menstrual cycles (O'Brien 2011). PMS is marked by a variety of emotional, physical, and behavioural symptoms. The symptoms typically include irritability, depression, mood swings, bloating, breast tenderness and sleep disturbances (Gianetto‐Berruti 2002; Johnson 2004; O'Brien 2003; Panay 2005). A severe form of PMS is known as premenstrual dysphoria or premenstrual dysphoric disorder (PMDD) and was previously known as late luteal phase dysphoric disorder (O'Brien 2011); it causes significant distress and interferes with normal functioning. The symptoms of PMS and PMDD are similar but the level of impairment is greater in PMDD. Studies have reported that as many as 85% of menstruating women have experienced at least one symptom of PMS (Gianetto‐Berruti 2002; Johnson 2004). In about 3% to 10% of women, the symptoms have adverse effects on activities of daily living and may affect their interpersonal relationships (Gianetto‐Berruti 2002; Halbreich 2003; Johnson 2004; O'Brien 2003; Panay 2005; Smith 1995). About 1.5 million women in the United Kingdom experience such severe PMS that it greatly reduces their quality of life. Studies have demonstrated deterioration in work effectiveness and cognitive function in women with PMS compared to women without PMS (Johnson 2004; Panay 2005).
The precise causation or aetiology of PMS remains unclear. No significant racial or ethnic differences have been shown to exist (Gianetto‐Berruti 2002). PMS tends to be more severe among women aged 25 to 35 years old (Johnson 2004). There appears to be a genetic link with PMS risk; monozygotic twins are twice as likely to suffer from PMS as dizygotic twins (Gianetto‐Berruti 2002). Similarly, daughters of affected mothers have a 70% greater chance of experiencing PMS than those with unaffected mothers (Gianetto‐Berruti 2002). Past history of depressive illness is another significant reported risk factor for PMS (Johnson 2004; Kaur 2004). Previous publications have variably hypothesised that PMS may be associated with excess oestrogen, a deficiency of progesterone or changes in the ratio of oestrogen and progesterone (Gianetto‐Berruti 2002). Attempts to treat the symptoms of PMS by addressing the ratio of oestrogen and progesterone through administering progestogen have not demonstrated benefits (Ford 2012). It is now thought that the condition is due to complex interactions between ovarian steroids and neurotransmitters (Rapkin 2007). Treatment of PMS symptoms by using selective serotonin reuptake inhibitors (SSRIs), which alter the balance of serotonin (a neurotransmitter), has been found effective but comes with dose‐dependent adverse effects (Marjoribanks 2013). Although much has been written on diagnostic criteria over the past 30 years, there is no true consensus on which technique is acceptable. A clinical diagnosis requires symptoms to be confirmed by prospective recording for at least two menstrual cycles and that symptoms cause substantial distress or impairment to daily life (e.g. work, school, social activities, hobbies, interpersonal relationships) (ACOG 2001; O'Brien 2011). Individual studies have used idiosyncratic techniques, this being true particularly for older studies. Typically these methods have included visual analogue scales, categorical scales such as Moos Menstrual Distress Questionnaire, Calendar of Premenstrual Experiences and the Daily Record of Severity of Problems. There remains no established consensus but the Daily Record of Severity of Problems has been used in most recent publications.
Description of the intervention
Oestrogen therapy has been proposed as a method for management of PMS through its ability to suppress ovulation and the subsequent endocrine changes of the cycle. Oestrogen can be delivered as oral tablets, transdermal patches, implants, vaginal pessaries, gels and creams. Various doses and regimens of continuous oestrogen have been used. Because the aim is to suppress ovulation, the rationale for intermittent use is not clear. The oestrogens which have been used and studied in the management of PMS include mainly oestradiol in the form of transdermal patches or subcutaneous implants to suppress ovulation in controlled studies (Green 2017). Both routes of administering oestradiol appear to show positive effects for treating mental and physical symptoms (Green 2017). To prevent endometrial hyperplasia, cyclical progestogens are given to ensure a regular withdrawal bleed. The potential oestrogens which may suppress ovulation include natural and synthetic oestrogens.
How the intervention might work
Though many theories have been proposed, the exact pathway and definitive aetiology of PMS is unknown (O'Brien 2003; Green 2017). PMS is probably related to ovulation as symptoms do not occur before the onset of puberty, during pregnancy or after menopause. PMS can occur in the absence of menstruation and it therefore can occur after hysterectomy (with ovarian conservation) and after endometrial ablation where ovarian function is unaffected (ISPMD 2011). This knowledge has led to the use of several ovulation‐suppressing drugs. The nature of some of these drugs limits their long‐term use because of associated safety issues, thereby also limiting their success. For example, gonadotrophin‐releasing hormone analogues (GnRH agonists) and danazol have both demonstrated efficacy in alleviating several premenstrual symptoms (Green 2017). Though effective, the value of danazol is limited because of its androgenic side effects and so it is rarely used on a long term basis. GnRH is very effective but long‐term use is limited by consequences of the resulting oestrogen‐deficient state.
Controlled trials have demonstrated that 17‐β‐oestradiol combined with cyclical progestogen (for regular withdrawal periods and to prevent endometrial hyperplasia) administered as an implant or patch may be effective in controlling PMS symptoms for long‐term use (Green 2017). Though oestrogen appears to be an effective agent for treating PMS, its use may be limited in individual women by the need for progestogen in women with a uterus and because of the subsequent progestogen PMS‐like side effects. As oestrogen‐only treatment has been found to be associated with endometrial hyperplasia/cancer, progestogen therapy is recommended for endometrial protection. The use of local progestogen (levonorgestrel‐releasing intrauterine system) may reduce the occurrence of PMS‐like side effects but there are no fully published randomized controlled trials (RCTs). We specifically exclude studies on oral contraceptives (OCs), especially those containing drospirenone, as this has been considered in an earlier Cochrane Review (Lopez 2012).
Why it is important to do this review
Whilst there are several published trials of oestrogen treatment of PMS and it is widely used, there is no existing systematic review evaluating the therapeutic effectiveness of non‐contraceptive oestrogen‐containing preparations in the management of PMS.
Objectives
To determine the effectiveness and safety of non‐contraceptive oestrogen‐containing preparations in the management of PMS.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials, either published or unpublished. We excluded non‐randomized studies (for example studies with evidence of inadequate sequence generation such as alternate days, patient numbers) as they are associated with a high risk of bias. Cross‐over trials were eligible if feasible data from both phases could be included in meta‐analyses.
Types of participants
Inclusion
Women in the studies had to be of reproductive age. Diagnosis of PMS had to be confirmed by prospective recording of symptoms for at least two menstrual cycles. Diagnosis through established criteria as set out in the Diagnostic and Statistical Manual of Mental Disorders (DSM‐III, DSM‐IV or DSM‐V) was not specified so as not to exclude older studies.
Exclusion
We excluded studies if participants had only a self‐diagnosis of PMS, were on other medication which resulted in ovulation suppression, had a primary psychiatric diagnosis other than premenstrual dysphoric disorder, or were taking any other hormone therapy (i.e. other than oestrogen and progestogens). Studies with participants already receiving psychotropic medication (e.g. selective serotonin re‐uptake inhibitors) were not excluded.
Types of interventions
We included studies which compared the effects of non‐contraceptive oestrogen‐containing preparations (with or without progestogen) versus placebo or alternative treatment; and studies which compared different dosages of oestrogen.
We included all routes of administration (e.g. oral, vaginal, transdermal patch, cream, gels, intrauterine route) or dosing regimen, provided the duration was longer than one menstrual cycle.
All combined oral contraceptive preparations (oestrogen/progestogen) were excluded, as were all studies of progestogen alone.
Types of outcome measures
The following outcomes were considered in this review.
Primary outcomes
Effectiveness (global symptom scores assessed using a validated prospective screening tool or by pre‐defined medical diagnostic criteria).
Adverse events (specific adverse effects including withdrawals for adverse effects and abnormal uterine bleeding).
Secondary outcomes
Specific symptoms of PMS: psychological, physical and functional symptoms.
Quality of life measures.
Participant satisfaction.
Search methods for identification of studies
We looked up all published and unpublished randomized controlled trials (RCTs) of non‐contraceptive oestrogens versus placebo, non‐contraceptive oestrogens versus alternative treatments and non‐contraceptive oestrogens at different dosages. All searches were conducted without language restriction and in consultation with the Cochrane Gynaecology and Fertility Group Information Specialist. We attempted to identify all relevant trials regardless of language or publication status (published, unpublished, in press and in progress).
Electronic searches
The Information Specialist searched the Cochrane Gynaecology and Fertility (CGF) Specialised Register of Controlled Trials, Cochrane Central Register of Studies (CRSO), MEDLINE, Embase, PsycINFO and CINAHL on 14 March 2016. The complete search strategies for the database searches are provided in Appendix 1, Appendix 2, Appendix 3, Appendix 4, Appendix 5 and Appendix 6. There was no restriction on language.
We searched for ongoing and unpublished studies in trial registers, such as ClinicalTrials.gov (ClinicalTrials.gov/), metaRegister of Controlled trials (mRCT) (www.controlled‐trials.com/mrct/) and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) Search Portal (http://apps.who.int/trialsearch/).
We searched for relevant conference abstracts on the ISI Web of Knowledge.
Searching other resources
Handsearching of conference proceedings
Proceedings from the following main conferences were handsearched.
International Federation of Fertility Societies.
American Society for Reproductive Medicine.
British Fertility Society.
European Society for Human Reproduction and Embryology.
Researchers and organizations
We contacted individual researchers working in the field to identify unpublished and ongoing trials.
We also contacted the UK‐based National Association for Premenstrual Syndrome (NAPS) for relevant articles.
We contacted the following drug and pharmaceutical companies manufacturing oestrogen to request other published or unpublished trials: Organon, Bayer Health Care, Janssen, Abbott, Pfizer, ReSource Medical, Meda, Norvartis, Orion, Teva UK and Marlborough Pharmaceuticals.
Reference lists
We also checked the reference lists of all studies identified by the above methods and examined any systematic reviews or meta‐analyses found.
Data collection and analysis
Data collection and analysis were conducted in accordance with guidelines set out in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Selection of studies
Two authors (BN and OU) independently applied inclusion criteria to all identified trials. We used the titles and abstracts of the identified citations to exclude trials that clearly did not meet the inclusion criteria (Figure 1). If either author judged that the trial might be eligible for inclusion, we obtained the full paper. We independently screened the full articles of selected trials and resolved any disagreements with a third author (PMSO). We gave reasons for excluding potentially relevant trials in Characteristics of excluded studies. We attempted to contact the authors for clarification.
1.
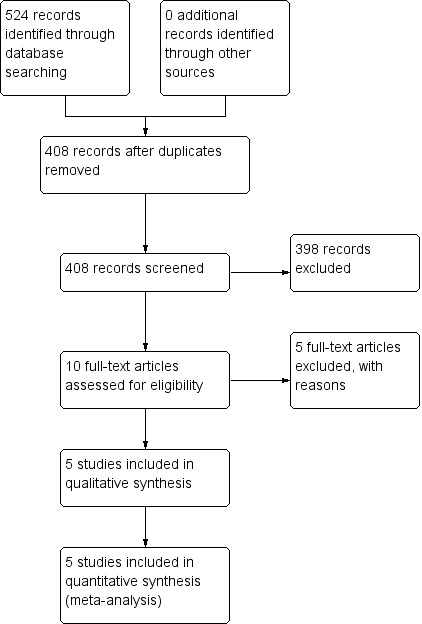
Study flow diagram.
The selection process was illustrated in a flow diagram according to the PRISMA statement (Figure 1) (Moher 2009).
Data extraction and management
Two authors (BN and JHK) independently extracted the data using a pre‐designed data collection form. For each of the studies, we extracted the following data: citation, study design, methodological criteria, inclusion and exclusion criteria, comparison group intervention, participant characteristics, trial setting, elements of intervention, all relevant outcomes measures, and results. Where reports were uncertain or included only summary measures, authors were contacted for clarification.
We also took note of any data that were consistently underreported, and highlighted this deficit along with future research needs. We checked whether authors had conducted an intention‐to‐treat (ITT) analysis (all randomized participants should be analyzed in the groups to which they were originally randomized) or one of its modifications (modified intention‐to‐treat or mITT; Abraha 2010). We calculated the percentage lost to follow‐up and reported this information. For dichotomous outcome measures, we recorded the number of participants experiencing the event and the number analyzed in each group. For continuous outcome measures, we extracted the mean change from baseline, the standard deviation of the mean change, and the number of women for each treatment group at each assessment.
Where changes from baseline were not reported, the mean, standard deviation, and the number of participants for each intervention group at each point in time were extracted. If the data had been reported using geometric means, we recorded this information and extracted a standard deviation on a log scale.
In studies with a cross‐over design, data were taken from both periods. We contacted authors for clarification and missing or insufficient data.
Assessment of risk of bias in included studies
Two review authors (BN and JHK) independently assessed the included studies for risk of bias using the Cochrane 'Risk of bias' assessment tool (Higgins 2011).
This assesses (Appendix 7):
sequence generation;
allocation concealment;
blinding of participants, providers and outcome assessors;
completeness of outcome data;
selective outcome reporting; and
other potential sources of bias, such as inappropriate administration of the intervention or the risk of carry‐over effects in cross‐over trials.
We presented results in both a risk of bias graph and a risk of bias summary. We interpreted the results of meta‐analyses in the light of the findings with respect to risk of bias. We conducted a sensitivity analysis based on risk of bias. We resolved any disagreements by consensus or by discussion with a third author.
Measures of treatment effect
For dichotomous data (e.g. adverse events), we used the numbers of events in the control and intervention groups of each study to calculate Mantel‐Haenszel risk ratios (RRs). For continuous data (e.g. mood score), if all studies reported exactly the same outcomes on the same scale we calculated mean differences (MDs) between treatment groups. If similar outcomes were reported on different scales (e.g. Moos Menstrual Distress Questionnaire (MDQ) and Premenstrual Distress Questionnaire (PDQ)) for premenstrual symptoms we calculated the standardized mean difference (SMD). We reversed the direction of effect of individual studies, if required, to ensure consistency across trials. We presented 95% confidence intervals (CIs) for all outcomes. Where data to calculate RRs or MDs were not available, we utilized the most detailed numerical data available that facilitated similar analyses of included studies (e.g. test statistics, P values). If a separate mean for each treatment arm could not be retrieved but the difference and within‐group standard deviation were available, we used the generic inverse‐variance method in the analyses using Hedges' g to calculate the standardized mean difference (SMD) (Deeks 2010). We compared the magnitude and direction of effect reported by studies with how they are presented in the review, taking account of legitimate differences.
SMDs were interpreted using the following rule of thumb: 0.2 represents a small effect, 0.5 a moderate effect, and 0.8 a large effect (Higgins 2011). A moderate effect (i.e. SMD = 0.5) can be interpreted as the smallest change that an individual person can perceive (Norman 2003).
Unit of analysis issues
The unit of analysis was per woman. We planned that if the woman was not the unit of randomization, such as is the case in cluster randomized trial (general practitioners, for example, might be the unit of randomization), adjustments for clustering would be made following the guidelines in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
In trials with a cross‐over design data were taken from both periods and analyzed as if they were from a parallel trial (Higgins 2011 section 16.4.5). This means that each trial participant contributed to both trial arms unless they dropped out during the second part of the trial, and therefore each cross‐over trial participant was effectively counted as two women. This approach gives rise to a unit of analysis error, but is conservative in that studies are under‐weighted rather than over‐weighted.
Dealing with missing data
The data were analyzed on an intention‐to‐treat basis as far as possible and attempts were made to obtain missing data from the original trialists. Where these were unobtainable, we analyzed only the available data.
Assessment of heterogeneity
The authors considered whether the clinical and methodological characteristics of the included studies were sufficiently similar for meta‐analysis to provide a meaningful summary. Statistical heterogeneity was assessed by measurement of the I² statistic. An I² greater than 50% was taken to indicate substantial heterogeneity (Higgins 2003). If substantial heterogeneity was detected, possible explanations were explored in sensitivity analyses
A rough guide to interpretation of I² values is as follows (Higgins 2011).
0% to 40%: might not be important.
30% to 60%: may represent moderate heterogeneity.
50% to 90%: may represent substantial heterogeneity.
75% to 100%: considerable heterogeneity.
Assessment of reporting biases
In view of the difficulty of detecting and correcting for publication bias and other reporting biases, the authors aimed to minimise their potential impact by ensuring a comprehensive search for eligible studies and by being alert for duplication of data. If there were 10 or more studies in an analysis, we used a funnel plot to explore the possibility of small‐study effects (a tendency for estimates of the intervention effect to be more beneficial in smaller studies).
Data synthesis
All eligible studies were analyzed in Cochrane's statistical software, RevMan 2014. Of the two authors who extracted the data, the first author entered all data into Review Manager 5 in collaboration with a third author, and the second rechecked all entries. Authors resolved disagreements by discussion. They provided a narrative synthesis for all results, along with a statistical meta‐analysis if possible. An increase in the probability of a particular outcome, which may be beneficial (e.g. improvement in global symptoms' scores) or detrimental (e.g. adverse effects), was displayed graphically in the meta‐analyses to the right of the centre‐line and a decrease in the odds of an outcome to the left of the centre‐line.
If the studies were sufficiently similar, we combined the data using a fixed‐effect model in the following comparisons.
Oestrogen versus placebo.
Oestrogen in combination with progestogen (sequential or continuous) versus placebo.
Dose comparisons of oestrogen, with or without progestogen.
Subgroup analysis and investigation of heterogeneity
Where data were available, we conducted a subgroup analysis to determine the separate evidence within the following subgroups.
Subgroups by route of administration of oestrogens.
Subgroups by mode of administration (continuous versus phasic).
Subgroups by route of administration of co‐administered progestogens required for protection of endometrium (not for therapeutic effects as indeed they may re‐introduce PMS‐like symptoms).
If we detected substantial heterogeneity, we planned to explore possible explanations in sensitivity analyses. We planned to take any statistical heterogeneity into account when interpreting the results, especially if there is any variation in the direction of effect.
Sensitivity analysis
We conducted sensitivity analyses for the primary outcomes to determine whether the conclusions are robust to arbitrary decisions made regarding the eligibility and analysis. These analyses included consideration of whether the review conclusions would have differed if:
eligibility were restricted to studies without high risk of bias;
a random‐effects model had been adopted;
the summary effect measure was odds ratio (OR) rather than RR.
Summary of findings table
A 'Summary of findings' table was generated using GRADEPRO software (GRADEpro GDT 2014) and Cochrane methods (Higgins 2011) to evaluate the overall quality of the body of evidence for our main comparisons (oestrogen vs placebo and oestrogen plus progestogen versus placebo) for our primary review outcomes (effectiveness and adverse events). Two review authors independently assessed the studies, using GRADE working group criteria (i.e. study limitations (risk of bias), consistency of effect, imprecision, indirectness and publication bias) (Atkins 2004). Judgements about the quality of the evidence (high, moderate, low or very low) were justified, documented, and incorporated into reporting of results for each outcome.
Results
Description of studies
Results of the search
We prepared a PRISMA flowchart to describe how we processed the references identified through the search results (see Figure 1) (Moher 2009). The literature searches yielded 524 titles of potentially relevant articles. After scanning titles and abstracts, we identified a total of 10 potentially relevant articles and two authors independently assessed full‐text copies against the inclusion criteria. We scanned the reference lists of these studies for further studies, but none were found. Eventually, we excluded five studies with reasons and five studies met the inclusion criteria.
See Characteristics of included studies and Characteristics of excluded studies.
Included studies
Study design and setting
Five trials were eligible for inclusion. All trials had a randomized controlled design (see Characteristics of included studies for detailed information about the individual trials). Three trials used a cross‐over design (Dhar 1990; Panay 2001; Watson 1989); and two a parallel design (Magos 1986; Smith 1995). Four of the trials came from a single unit at King's College Hospital under the direction of Prof J Studd, Consultant Obstetrician and Gynaecologist (Magos 1986; Panay 2001; Smith 1995; Watson 1989).
All but one study reviewed were conducted in the UK. The exception was conducted in Canada (Dhar 1990).
Participants
This review was based on data from 258 participants. The mean number of participants per trial was 65 (range 11 to 112); (see Characteristics of included studies for breakdown per study).
Exclusion criteria varied but most of the studies excluded women with the following characteristics.
Women with only a self‐diagnosis of PMS.
Women who were on other medication which resulted in ovulation suppression.
Women with a primary psychiatric diagnosis (other than premenstrual dysphoric disorder).
Women who were taking any other hormone therapy (i.e. other than oestrogen and progestogens) though women already receiving psychotropic medication (e.g. selective serotonin re‐uptake inhibitors) were not excluded.
Interventions
Magos 1986 investigated a subcutaneous oestrogen implant with 5 mg oral norethisterone for 7 days per cycle. versus placebo with placebo implant for 7 days per cycle.
Watson 1989 investigated oestrogen patches with 5 mg oral norethisterone, day 19 to 26, versus placebo patches plus 5 mg oral norethisterone, day 19 to 26.
Panay 2001 investigated 100 µg oestradiol patches with 1 mg oral norethisterone at 17 to 28 days versus identical placebo patches and tablets.
Smith 1995 compared different doses of oestrogen patches (100 µg versus 200 µg). In each group half of the women were allocated to take dydrogesterone 10 mg daily from day 17 to 26 of the cycle and half to take medroxyprogesterone acetate 5 mg daily from day 17 to 26.
Dhar 1990 investigated oral oestrogen versus placebo.
Outcomes
All studies reported global symptom scores.
Four studies reported adverse events (specific adverse effects including withdrawals as a result of adverse effects and abnormal uterine bleeding).
Primary outcome measures
The primary outcome measures of effectiveness used in the studies were the Moos Menstrual Distress Questionnaire (MDQ) (Magos 1986; Watson 1989), the Premenstrual Distress Questionnaire (PDQ; Panay 2001; Smith 1995; Watson 1989), and a 28‐question mental and physical symptoms daily ratings scale (Dhar 1990). The MDQ and PDQ are validated scales, in contrast to the 28‐question daily ratings scale. For all three scales, a higher score indicated more — or more severe — symptoms, in other words a worse outcome. The studies using MDQ and PDQ could be pooled by using standardized mean differences. One study did not provide an SD but did include the 10th and 90th centile of changes in score, which were therefore used to calculate an SD (Magos 1986). Another study (Watson 1989) did not provide enough information to determine the SD, and we therefore used the pooled SD from another included study (Panay 2001). Although Watson 1989 was a cross‐over trial, the statistical analysis in the paper was performed as for a parallel trial. Too few data were presented in the paper to perform a re‐analysis, and therefore we reported the data in its original parallel trial form. Likewise, the modified ITT analysis reported in the statistical report accompanying Panay 2001, which was based on all women who provided a follow‐up score, performed the analysis as for a parallel trial. The primary outcome measure of adverse effect was the frequency of adverse effects and abnormal laboratory tests mentioned in the publications. All studies had data in a form that allowed a pooled analysis.
Secondary outcome measures
Secondary outcome measures of effectiveness used in the study were psychological and physical symptoms as provided by the MDQ, PDQ and the 28‐question ratings scale subscores. For all these scales, a higher score indicated more or more severe symptoms (or both), in other words a worse outcome. The outcomes for these two symptoms were pooled by using standardized mean differences, again using the pooled SD from one included study — Magos 1986 — to substitute for the missing information in Watson 1989. Further secondary outcomes were a Visual Analog Scale (VAS) of general unwellness or well‐being (Magos 1986; Smith 1995), the GHQ‐60 general health questionnaire (Magos 1986), and participant satisfaction (Smith 1995). For the VAS of feeling unwell and the GHQ‐60, a higher score indicated a worse outcome, whereas for the VAS of feeling well a higher score indicated a better outcome.
Sources of support
Panay 2001 was supported by industry; Magos 1986 by a research charity; and three studies provided no information on support (Dhar 1990; Smith 1995; Watson 1989).
Excluded studies
Five studies were excluded from this review. One randomized trial was excluded because it did not compare oestrogen treatment to either placebo or comparator control (Domoney 2003). Three randomized trials were excluded because they were restricted to specific aspects of PMS instead of global symptoms (de Lignières 1986; Dennerstein 1988; Ensom 2003). One randomized trial was excluded because it made use of oestrogen as part of a contraceptive (Halbreich 2012).
Risk of bias in included studies
See Figure 2 and Figure 3, and Characteristics of included studies.
2.
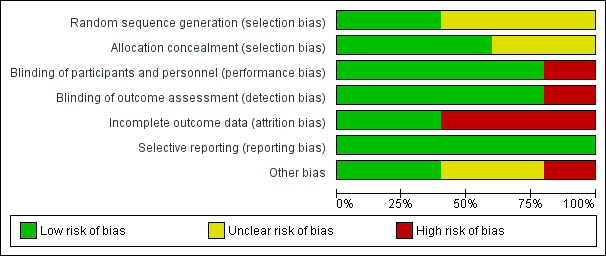
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
Magos 1986 and Smith 1995 were judged to have a low risk and Dhar 1990, Panay 2001 and Watson 1989 to have an unclear risk of bias, the latter because details of the randomisation method were not supplied.
Allocation concealment
Magos 1986, Panay 2001 and Smith 1995 were judged to have a low risk and Dhar 1990 and Watson 1989 to have an unclear risk of this bias.
Blinding
Performance bias
Dhar 1990; Magos 1986; Panay 2001 and Watson 1989 were judged to have a low risk and Smith 1995 to have a high risk of this bias.
Detection bias
Dhar 1990; Magos 1986; Panay 2001 and Watson 1989 were judged to have a low risk and Smith 1995 to have a high risk of this bias.
Incomplete outcome data
Three of the five trials had high rates of dropout or loss to follow up, ranging from 20% to 29% (Magos 1986; Panay 2001; Smith 1995); these were rated at high risk of attrition bias. Dhar 1990 and Watson 1989 were rated at low risk.
A potential risk of bias in cross‐over trials is dropout after the first phase, whereby the participant does not start the second treatment. In Watson 1989, four of the five withdrawals occurred in the first phase and therefore did not bias the results in the two arms differentially. One woman in the active‐to‐placebo group withdrew at three months because she did not want to change treatment because of dramatic improvement. In Panay 2001, all withdrawals but one occurred within the first phase of the trial, Therefore no reasons were found to suspect a systematic difference between the two trial phases due to dropout after the first phase but before the second phase.
Selective reporting
Dhar 1990; Magos 1986; Panay 2001; Smith 1995 and Watson 1989 (all five studies) were judged to have a low risk of bias.
Other potential sources of bias
Three of the studies were cross‐over trials (Watson 1989; Dhar 1990; Panay 2001). The International Society for Premenstrual Disorders (ISPMD) Montreal Consensus agreed that randomized trials of premenstrual disorders should ideally have a parallel design (ISPMD 2011), suggesting these trials were not ideal. The main risks of cross‐over trials are the potential for carry‐over effects from one period to the next, and an increased risk of attrition. The issue of carry‐over effects was not addressed in the three trial reports. The month‐by‐month graphical data of the active‐to‐placebo group in Watson 1989 strongly suggest that the effect of the active treatment has disappeared in the month after the transition from active to placebo, but we rated the risk of bias from carry‐over effects unclear given that only graphical data from a single trial supports it.
Effects of interventions
See: Table 1; Table 2; Table 3
1. Oestrogen versus placebo
One study reported this comparison (Dhar 1990). Luteal‐phase oestrogen was compared with placebo. As the study used non‐validated scales to measure effectiveness and specific (psychological and physical) symptoms of PMS, findings are reported in narrative rather than in forest plots. The quality of the evidence for this comparison was rated as very low (Table 2).
Primary outcomes
1.1 Effectiveness
The study authors reported that luteal‐phase oestrogen had a large negative effect on global (mental and physical) symptoms compared to placebo (P < 0.01).
1.2 Adverse events
None of the 11 women withdrew from the study. The study authors reported that "no significant adverse events were encountered" but did not specify what the nature of a "significant adverse event" would be.
Secondary outcomes
1.3 Specific symptoms of PMS: psychological, physical and functional symptoms
1.3.1 Psychological symptoms
The study authors reported that luteal‐phase oestrogen was significantly less effective than placebo in relieving the severity of mental symptoms of PMS (P < 0.02).
1.3.2 Physical symptoms
The study authors reported that luteal‐phase oestrogen was significantly less effective than placebo in relieving the severity of physical symptoms of PMS (P < 0.02).
1.4 Quality of life
This outcome was not reported.
1.5 Patient satisfaction
This outcome was not reported.
2. Oestrogen in combination with progestogen (sequential or continuous) versus placebo.
Three studies reported this comparison. They compared continuous oestrogen (as an implant or patch) with sequential placebo versus placebo. One of the studies administered progestogen in both arms (Watson 1989). We have included this study in this comparison and conducted a post hoc sensitivity analysis to see whether inclusion of this study influenced the results. The quality of the evidence for this comparison was rated as very low (Table 1)
Primary outcomes
2.1 Effectiveness
Continuous oestrogen (implants and patches) had a moderately positive effect on global symptom scores compared to placebo (SMD −0.27, 95% CI −0.47 to −0.07, P = 0.008, 3 RCTs, 158 women, effective sample size 267 women considering cross‐over trials, I² = 63%, very low quality evidence; Analysis 1.1; Figure 4). The statistical heterogeneity between the three trials appeared to be mainly attributable to Panay 2001, in which a low‐dose patch (100 µg) was used. When Watson 1989 was excluded from the analysis, findings were no longer statistically significant (SMD −0.19, 95% CI −0.40 to 0.03).
1.1. Analysis.
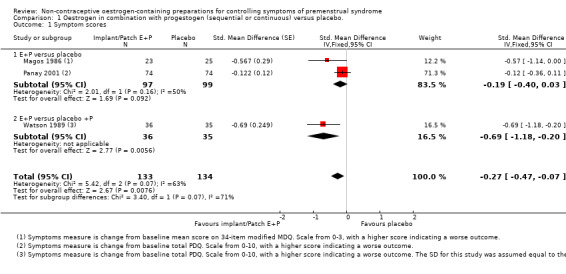
Comparison 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., Outcome 1 Symptom scores.
4.
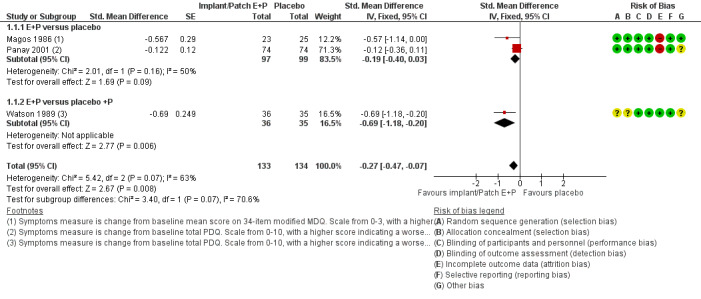
Forest plot of comparison: 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., outcome: 1.1 Symptom scores.
2.2 Adverse events
All three studies reported withdrawals due to adverse events. Confidence intervals were very wide and there were too few events (none in one of the studies), to establish whether there was a difference between the groups (RR 0.64, 95% CI 0.26 to 1.58, P = 0.33, 3 RCTs, 196 women, effective sample size 284 women considering cross‐over trials, I² = 0%, low‐quality evidence; Analysis 1.2; Figure 5). Excluding Watson 1989 from the analysis did not influence the findings.
1.2. Analysis.
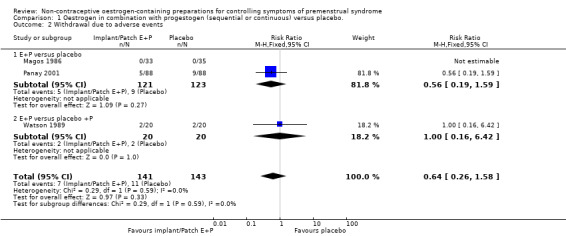
Comparison 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., Outcome 2 Withdrawal due to adverse events.
5.
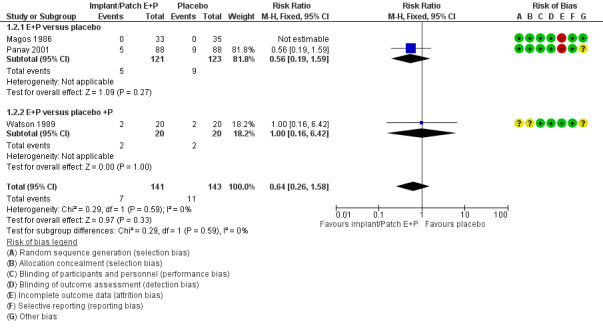
Forest plot of comparison: 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., outcome: 1.2 Withdrawal due to adverse events.
The two studies comparing oestrogen versus placebo also reported specific adverse events (Magos 1986; Panay 2001; Analysis 1.3). There were too few events reported to determine whether there was a difference between the groups.
1.3. Analysis.
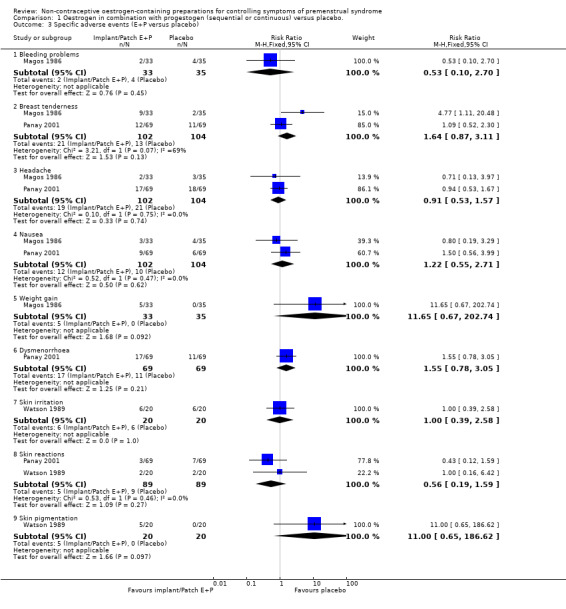
Comparison 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., Outcome 3 Specific adverse events (E+P versus placebo).
Findings for specific events were as follows.
Bleeding problems (RR 0.53, 95% CI 0.10 to 2.70, P = 0.45, 1 RCT, 68 women, very low quality evidence).
Breast tenderness (RR 1.64, 95% CI 0.87 to 3.11, P = 0.13, 2 RCTs, 206 women, I² = 69%, very low quality evidence).
Headache (RR 0.91, 95% CI 0.53 to 1.57, P = 0.74, 2 RCTs, 206 women, I² = 0%, very low quality evidence).
Nausea (RR 1.22, 95% CI 0.55 to 2.71, P = 0.62, 2 RCTs, 206 women, I² = 0%, very low quality evidence).
Weight gain (RR 11.65, 95% CI 0.67 to 202.74, P = 0.09, 1 RCT, 68 women, very low quality evidence).
Dysmenorrhoea (RR 1.55, 95% CI 0.78 to 3.05, P = 0.21, 1 RCT, 138 women, very low quality evidence).
Skin irritation (RR 1.00, 95% CI 0.39 to 2.58, P = 1.0, 1 RCT, 40 women, very low quality evidence).
Skin reactions (RR 0.56, 95% CI 0.19 to 1.59, P = 0.27, 2 RCTs, 178 women, I² = 0%, very low quality evidence).
Skin pigmentation (RR 11.00, 95% CI 0.65 to 186.62, P = 0.10, 1 RCT, 40 women, very low quality evidence).
Secondary outcomes
2.3 Specific symptoms of PMS: psychological, physical and functional symptoms
2.3.1 Psychological symptoms
Two placebo‐controlled RCTs of continuous oestrogen plus progestogen (implants or patches) provided data to determine effects on psychological symptoms (Magos 1986; Watson 1989). Treatment with implants or patches was associated with a reduction in psychological symptoms compared to treatment with placebo (SMD −0.79, 95% Cl −1.17 to −0.40, P < 0.00007, 2 RCTs, 88 women, effective sample size 119 women considering the cross‐over trial, I² = 78%, very low quality evidence). Findings were imprecise and compatible with an effect size ranging from small to large (Analysis 1.4).
1.4. Analysis.
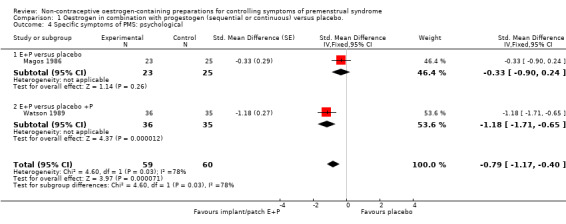
Comparison 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., Outcome 4 Specific symptoms of PMS: psychological.
However, when Watson 1989 was excluded from the analysis, findings were no longer statistically significant (SMD −0.33, 95% CI −0.90 to 0.24).
2.3.2 Physical symptoms
Two placebo‐controlled trials of continuous oestrogen plus progestogen (implants or patches) provided data to determine effects on physical symptoms (Magos 1986; Watson 1989). Physical symptoms were lower in the oestrogen group (SMD −0.60, 95% Cl −0.98 to −0.22, P = 0.0019, 2 RCTs, 88 women, effective sample size 119 women considering the crossover trial, I² = 52%, very low quality evidence). Findings were imprecise and compatible with an effect size ranging from small to large (Analysis 1.5).
1.5. Analysis.
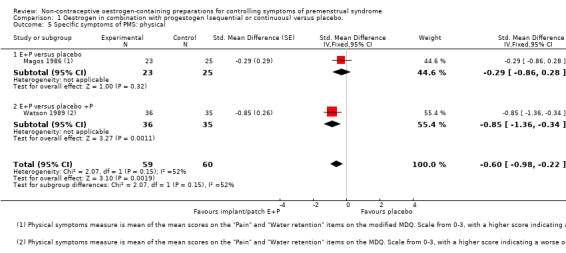
Comparison 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., Outcome 5 Specific symptoms of PMS: physical.
However, when Watson 1989 was excluded from the analysis, findings were no longer statistically significant (SMD −0.29, 95% CI −0.86 to 0.28).
2.4 Quality of life
One placebo‐controlled trial of continuous oestrogen (implants) reported on general quality of life (Magos 1986). There was no conclusive evidence of difference in a VAS for feeling unwell (MD −10.90, 95% CI −22.60 to 0.80, P = 0.07, 1 RCT, 40 women, effective sample size 68 women considering this was a cross‐over trial, low‐quality evidence) and a general health questionnaire (MD −3.30, 95% CI −9.98 to 3.38, P = 0.33, 1 RCT, 40/68 women, very low quality evidence) (Analysis 1.6).
1.6. Analysis.
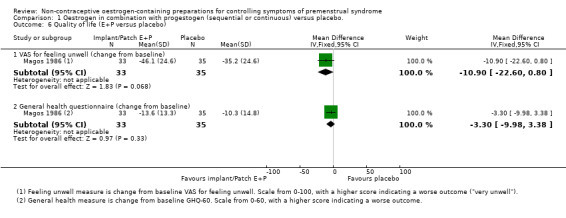
Comparison 1 Oestrogen in combination with progestogen (sequential or continuous) versus placebo., Outcome 6 Quality of life (E+P versus placebo).
2.5 Patient satisfaction
This outcome was not reported in the included studies.
3 Dose comparisons of oestrogen, 100 µg versus 200 µg, with or without progestogen
One study compared different doses of oestrogen patches (100 µg versus 200 µg) (Smith 1995). In each group half of the women were allocated to take dydrogesterone 10 mg daily from day 17 to 26 of the cycle and half to take medroxyprogesterone acetate 5 mg daily from day 17. The quality of the evidence for this comparison was rated as very low (Table 3).
Primary outcomes
3.1 Effectiveness
The evidence was too imprecise to determine whether dosage had an effect on global symptoms (MD −1.55 on a scale of 0 to 30, 95% CI −8.88 to 5.78, P = 0.68, 1 RCT, 98 women, very low quality evidence) (Analysis 2.1).
2.1. Analysis.
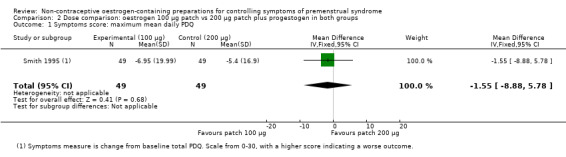
Comparison 2 Dose comparison: oestrogen 100 µg patch vs 200 µg patch plus progestogen in both groups, Outcome 1 Symptoms score: maximum mean daily PDQ.
3.2 Adverse events
The evidence was too imprecise to determine whether there was a difference between the two patch dosages in withdrawal rates for adverse events (RR 0.70, 95% CI 0.34 to 1.46, P = 0.34, 1 RCT, 107 women, very low quality evidence) (Analysis 2.2).
2.2. Analysis.
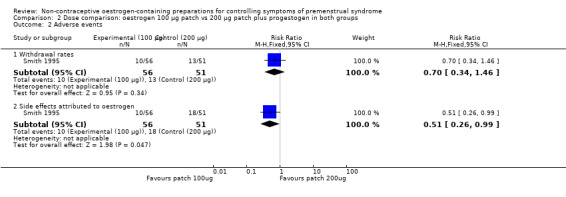
Comparison 2 Dose comparison: oestrogen 100 µg patch vs 200 µg patch plus progestogen in both groups, Outcome 2 Adverse events.
The evidence suggested that there might be a lower overall risk of adverse events in the 100 µg group (RR 0.51, 95% Cl 0.26 to 0.99, P = 0.05, 1 RCT, 107 women, very low quality evidence) (Analysis 2.2). The data did not allow extraction of individual adverse event risks by dosage.
Secondary outcomes
3.3 Specific symptoms of PMS: psychological, physical and functional symptoms
3.3.1 Psychological symptoms
The evidence was too imprecise to determine whether there a difference between the two dosage groups (MD −0.28, 95% CI −1.17 to 0.61, P = 0.54, 1 RCT, 98 women, very low quality evidence) (Analysis 2.3).
2.3. Analysis.
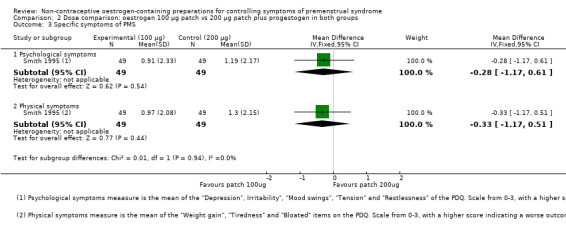
Comparison 2 Dose comparison: oestrogen 100 µg patch vs 200 µg patch plus progestogen in both groups, Outcome 3 Specific symptoms of PMS.
3.3.2 Physical symptoms
The evidence was too imprecise to determine whether there a difference between the two dosage groups (MD −0.33, 95% CI −1.17 to 0.51, P = 0.44, 1 RCT, 98 women, very low quality evidence) (Analysis 2.3).
3.4 Quality of life
This study used a VAS (scale 0 to 100) for feeling well to report on general quality of life. The evidence was too imprecise to determine whether there a difference between the two dosage groups (MD 1.70, 95% CI −19.43 to 22.83, P = 0.87, 1 RCT, 98 women, very low quality evidence) (Analysis 2.4).
2.4. Analysis.
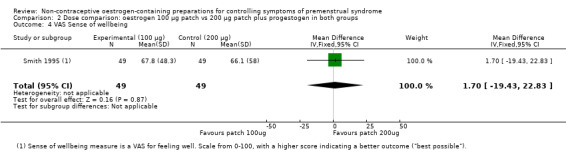
Comparison 2 Dose comparison: oestrogen 100 µg patch vs 200 µg patch plus progestogen in both groups, Outcome 4 VAS Sense of wellbeing.
3.5 Patient satisfaction
The evidence was too imprecise to determine whether there a difference in satisfaction rates between the two dosage groups (RR 1.19, 95% CI 0.85 to 1.68, P = 0.32, 1 RCT, 107 women, very low quality evidence) (Analysis 2.5).
2.5. Analysis.
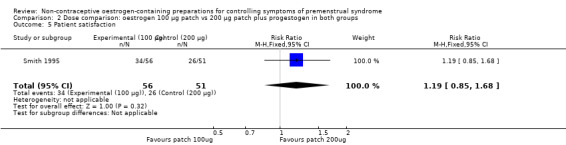
Comparison 2 Dose comparison: oestrogen 100 µg patch vs 200 µg patch plus progestogen in both groups, Outcome 5 Patient satisfaction.
Other analyses
Subgroup analyses
Our planned subgroup analyses were not possible due to lack of data.
Sensitivity analyses
Restricting eligibility to studies without high risk of bias
All five included studies had at least one item with a high risk of bias or multiple items with an unknown risk. Restricting eligibility to studies without an item with high risk of bias was therefore impossible.
The three RCTs comprising the analysis of implants or patches had substantial heterogeneity (I² = 54%), mainly due to one RCT (Panay 2001). Restricting the analyses to Magos 1986 and Watson 1989, the two RCTs that did not show substantial heterogeneity, increased the overall effect size of oestrogen implants or patches, but otherwise did not appreciably change the main findings for the primary outcome.
Adopting a random‐effects model
Pooling of studies was only used for evaluating implants or patches. Use of a random‐effects model rather than a fixed‐effect model to pool the three RCTs did not appreciably change the main findings.
Using OR rather than RR as a summary measure
Use of odds ratios (ORs) instead of risk ratios (RRs) did not affect the results other than the obvious difference in value between the OR and RR.
Funnel plot
There were too few studies to produce a funnel plot.
Discussion
Summary of main results
Effectiveness of oestrogen
This review found one small cross‐over study (n = 11) that compared oral luteal‐phase oestrogen versus placebo. Data were very low quality and unsuitable for analysis, but study authors reported that the intervention was ineffective and might aggravate the symptoms of PMS.
This review found some very low quality evidence that continuous oestrogen combined with progestogen improves global symptoms compared to placebo. The pooled SMD for the effect of continuous oestrogen (patch or implant) on the symptoms of PMS favoured oestrogen over placebo. This applies both to global symptoms and specific symptoms. The overall effect size was small to medium (SMD = 0.34). No evidence was found that quality of life differed between women using continuous oestrogen or placebo.
Finally, one study was found investigating the effect of oestrogen dosage (100 µg vs 200 µg). Data were of very low quality, and there was insufficient evidence to determine whether dosage affected global or specific PMS symptoms, quality of life or patient satisfaction.
Adverse Events
The one trial on oral luteal‐phase oestrogen found no adverse effects, but the quality of evidence was very low due to the very small number of women studied (n = 11).
The four trials on continuous oestrogen combined with progestogen reported a wide range of side effects. There was insufficient evidence to determine a difference in adverse event rates between oestrogen (implant or patch) plus progestogen and placebo. However, the quality of evidence was very low, mainly due to the small sample size and a lack of long‐term follow‐up.
The evidence suggesting that oestrogen patch dose may affect adverse effect rates was also of very low quality. The trial comparing 100 µg versus 200 µg oestrogen patches found a lower overall risk of adverse effects in the lower‐dose group.
Overall completeness and applicability of evidence
There have been no further published studies since 2001, perhaps because clinicians who prescribe oestrogen think that sufficient evidence for the effectiveness of oestrogen is already available. However, a more important reason may be that the pharmaceutical industry sees insufficient commercial potential, particularly outside the UK. Ideally, a large multicentre study should be conducted based on up‐to‐date diagnostic criteria using validated quantification methods.
Very low quality evidence suggested that oral luteal‐phase oestrogen was ineffective (or even harmful) whereas continuous oestrogen plus progestogen was effective. A regimen of oestrogen without progestogen can only be sustained by administering oestrogen periodically, during the luteal phase. Luteal phase delivery of oestrogen will however not suppress ovulation, which may explain why women using oral luteal‐phase oestrogen did not report a beneficial effect on PMS symptoms. On the other hand, if oestrogen is to be administered continuously, progestogen must be added to protect the endometrium.
All four trials of oestrogen plus progestogen alluded to the possible negative effects of oral progestogen, which was used in the trials to protect the endometrium but can cause PMS‐like side effects, reducing treatment efficacy. Indeed, we noted withdrawals from trials due to the side effects of oral progestogen. Unfortunately none of the studies systematically evaluated these side effects, even though they are commonly encountered in clinical practice. In an attempt to overcome the PMS‐like side effects, Panay 2001 used a lower dose (1 mg instead of the usual 5 mg) of oral norethisterone than the dose used in the other two trials of continuous oestrogen (Magos 1986; Watson 1989). However, the low heterogeneity with respect to adverse effects in these three RCTs suggests that this strategy may not influence the rate of adverse effects and therefore no evidence of its success was found.
Interestingly, when oestrogen is used in hormone therapy (HT), progestogens are normally prescribed for 10 to 14 days each month to protect the endometrium. However, a Cochrane Review concluded that smaller doses of progestogens during HT may provide adequate protection of the endometrium (Roberts 2014). One trial of continuous oestrogen in this review (Panay 2001) prescribed the normal duration of 10 days' progestogen but Magos 1986 and Watson 1989 prescribed only 7 to 8 days of progestogen. Whether or not such differences in duration of progestogens will affect the risk of endometrial hyperplasia or the risk of other adverse side effects is unknown. Clearly, further research should be aimed at investigating the optimal dose of progestogens in order to minimize the occurrence of PMS‐like symptoms while still protecting the endometrium.
Using an intrauterine releasing system for progesterone/progestogens instead of oral progestogens for endometrial protection may overcome the problem of PMS‐like side effects and is commonly used in the clinical arena. We found one conference abstract comparing the two methods (Domoney 2003). In this RCT, 37 women who all received transdermal oestradiol patches were randomized to intrauterine progesterone (n = 18) or oral progestogen (n = 19). None of the women in the intrauterine group dropped out due to PMS‐like side effects whereas six women in the oral group did. This provides some evidence to suggest that use of intrauterine progesterone reduces progestagenic PMS‐like side effects, but it is not conclusive.
We searched trial databases and contacted other researchers and relevant drug manufacturers concerning ongoing or past unreported clinical trials of non‐contraceptive oestrogen management for ovulation suppression, and there appeared to be no unreported or ongoing studies. We have received information from one manufacturer (Bayer AG, Leverkussen, Germany), who trialled the use of a levonorgestrel‐releasing intrauterine system (LNG‐IUS) for endometrial protection during oestrogen replacement therapy in postmenopausal women, which led to its licensing for use as the progestogen component of hormone therapy. However, there are no published or ongoing efficacy and safety studies concerning the use of LNG‐IUS for endometrial protection under the higher doses required for ovulation suppression in PMS.
Future clinical trials of the efficacy or effectiveness of continuous oestrogen should therefore include an arm comparing methods to administer progestogen.
Quality of the evidence
The three trials comparing oestrogen plus progestogen versus placebo were at low risk of bias in most domains, though two had a high level of attrition. The trial comparing oestrogen versus placebo was very small (n = 11), reporting of methods was poor and it did not use a validated measure, which meant we could only provide a narrative review. The trial comparing two oestrogen dosages, an open label study, was at high risk of bias in four areas (selection, performance, detection and attrition). All trials reported the number of participants lost to follow‐up; and for all an ITT or a modified ITT analysis was available. We had no evidence of selective outcome reporting by any of the studies. Finally, all four trials in this review reporting on continuous oestrogen (Magos 1986; Panay 2001; Smith 1995; Watson 1989), contributing 247 of the 258 women, were from a single centre (King's College Hospital in London). This potentially limits the external validity of the evidence.
The overall quality of the evidence for the main findings was graded as very low using the GRADE criteria, due to serious risk of bias (related mostly to high attrition rates), imprecision due to the small total sample size and statistical and clinical heterogeneity (I² = 54%) in the comparison of oestrogen plus progestogen versus placebo.
Potential biases in the review process
Efforts were made to retrieve all eligible studies. The studies we found were at least 15 years old, including one only reported in the public literature as a conference abstract. To reduce the risk that we might miss an unpublished study, we contacted all pharmaceutical companies that produce oestrogen tablets, patches or implants for information about unreported past or ongoing trials, They all said there were no such trials, but we cannot exclude that unreported past trials exist because trial databases did not exist or were in their infancy more than 15 years ago.
A specific issue was the use of a cross‐over trial design in three studies: Panay 2001 and Watson 1989, comparing oestrogen plus progestogen versus placebo; and the single trial comparing oestrogen alone versus placebo (Dhar 1990). The ISPMD Montreal Consensus agreed that randomized trials of premenstrual disorders should ideally have a parallel design (O'Brien 2011), suggesting these trials were not ideal. Their main risks would be the potential for carry‐over effects from one period to the next; and an increased risk of attrition, specifically attrition after phase 1 of the trial, preventing women entering phase 2 (Watson 1989). One trial provided month‐by‐month graphical data, which strongly suggested that the effect of the active treatment had disappeared in the month after the transition from active to placebo (Watson 1989). We did therefore consider 3‐month results as valid endpoints for a cross‐over trial in this review, but note that the support for it is small. Overall, we therefore considered all cross‐over trials as having an unclear risk of other bias. Dropouts and loss to follow‐up occurred in two of the three cross‐over trials but almost all occurred within phase 1 of the trials and therefore no reasons were found to suspect they would cause a systematic difference between the two trial phases (Panay 2001; Watson 1989). Overall, we considered this study design suitable for the current review and included data from both periods, which we analyzed as if they were from a parallel trial (Higgins 2011, section 16.4.5). Our main reason for doing so was that two of the three trials did not provide sufficient data to allow a paired analysis, which would have been the preferred way to incorporate the data (Higgins 2011, section 16.4.4). Our chosen method results in confidence intervals that are too wide, with the net effect that the cross‐over trial will be under‐weighted in the final analysis and clinically important heterogeneity may be disguised (Higgins 2011 section 16.4.5). However, we believe a correct analysis would not have affected the reasons for grading the overall quality of the evidence as very low.
Agreements and disagreements with other studies or reviews
All oestrogen studies have been evaluated in this review and we identified no other systematic review of this topic.
Authors' conclusions
Implications for practice.
We found very low quality evidence to support the effectiveness of continuous oestrogen (in the form of transdermal patches or subcutaneous implants) plus progestogen, with a small to moderate effect size. We also found very low quality evidence from a single study of 11 women to suggest that luteal‐phase unopposed oral oestrogen is probably ineffective and possibly detrimental for controlling the symptoms of PMS. A comparison between 200 µg and 100 µg doses of continuous oestrogen was inconclusive with regard to effectiveness, but suggested that the lower dose was less likely to cause side effects. Uncertainty remains regarding safety, as the identified studies were too small to provide definite answers. Moreover, none of the included trials addressed adverse effects that might occur beyond the typical trial duration of 2 to 8 months. This suggests the choice of oestrogen dose and mode of administration could be based on an individual woman’s preference and modified according to the effectiveness and tolerability of the chosen regimen.
Implications for research.
This review found very low quality evidence that continuous oestrogen may be an effective treatment strategy for PMS. However, all trials comparing continuous oestrogen to placebo were published at least 15 years before this review and may have shown a lesser effectiveness due to unwanted progestagenic side effects. This can perhaps be overcome by using local progestogen in the form of an LNG‐IUS or using a different type of progestogen such as drospirenone. Further research comparing various methods of administering progestogens may enable continuous oestrogen to become one of the most effective treatment options for the common condition of PMS. Our review found low‐quality evidence that continuous oestrogen may be considered effective though they are not widely used outside the UK. New and larger randomized clinical trials are justified, in particular to investigate the efficacy and safety of oestrogen in combination with the currently used forms of endometrial protection such as LNG‐IUS or different forms of progestogen. New trials should be based on established diagnostic criteria of the International Society for the Study of Premenstrual Disorders (ISPMD), include a power analysis, have a parallel design and report full statistical information with standard deviations for all continuous outcomes to facilitate their inclusion in meta‐analyses.
Future research should focus on:
direct comparisons between different oestrogen administration regimens, including their safety, tolerability and effectiveness for overall symptoms and for specific symptom types over long‐term follow‐up;
recruiting an adequate number of participants and providing treatment for a sufficient duration and follow up;
developing an agent that provides endometrial protection without regenerating premenstrual symptoms, for example selective progesterone receptor modulators or intrauterine progestogens; and
studies designed according to the established diagnostic criteria of International Society for the Study of Premenstrual Disorders.
Acknowledgements
We are grateful for the support given by the staff of the Cochrane Gynaecology and Fertility Group, especially the Information Specialist.
Appendices
Appendix 1. Gynaecology and Fertility Group search strategy
From inception until 14 March 2016
PROCITE platform
Keywords CONTAINS "premenstrual dysphoric disorder"or"premenstrual aggravation" or"premenstrual pain" or "Premenstrual Syndrome‐Symptoms" or "premenstrual" or "PMDD" or "PMS" or "luteal phase defect" or "luteal phase disorders" or "Mastalgia"or "mastalgia‐outcome"or "dysphoria" or Title CONTAINS "premenstrual dysphoric disorder"or"premenstrual aggravation" or"premenstrual pain" or "Premenstrual Syndrome‐Symptoms" or "premenstrual" or "PMDD" or "PMS" or "luteal phase defect" or "luteal phase disorders" or "Mastalgia"or "mastalgia‐outcome"or "dysphoria"
AND
Keywords CONTAINS "estraderm" or "estradiol" or "estradiol‐cyproterone acetate" or"Estriol‐" or"estrofen"or "estrogen" or"estrogen‐progestogen" or"*Estrogens" or"Estrone" or"estroprogestin" or"esterified conjugated estrogens" or"esterified estrogens"or "esterified conjugated estrogens" or"oestrodiol" or"oestrogen"or "premarin" or"climara" or"17‐beta estradiol"or "conjugated equine estrogens + progesterone" or"conjugated estrogen"or"conjugated estrogens"or "conjugated equine estrogen"or "conjugated equine estrogens"or"CEE"or"CEE + MPA"or "Vivelle+"or"ethinyl‐estradiol"or"ethinyl estradiol‐cyproterone acetate"or"ethinyl estradiol + drospirenone"or Title CONTAINS "estraderm"or"estradiol"or"estradiol‐cyproterone acetate" or"Estriol‐"or"estrofen"or "estrogen"or"estrogen‐progestogen" or"*Estrogens" or"Estrone" or"estroprogestin" or"oestrodiol"or"oestrogen"or "premarin" or"climara" or"17‐beta estradiol"or "CEE" or"CEE + MPA"or "Vivelle+" or"ethinyl‐estradiol" (115 hits)
Appendix 2. Central Register of Studies Online (CRSO) search strategy
From inception until 14 March 2016
CRS ONLINE platform
#1MESH DESCRIPTOR Premenstrual Syndrome EXPLODE ALL TREES (373) #2(Premenstrual Syndrome):TI,AB,KY (573) #3PMS:TI,AB,KY (385) #4PMT:TI,AB,KY (69) #5PMD*:TI,AB,KY (675) #6LLPD*:TI,AB,KY (9) #7(Premenstrual Tension):TI,AB,KY (72) #8(luteal adj2 dysphori*):TI,AB,KY (22) #9(dysphori* adj2 disorder*):TI,AB,KY (187) #10mastalgi*:TI,AB,KY (298) #11(Premenstrual adj2 depressi*):TI,AB,KY (21) #12(Premenstrual adj2 dysphor*):TI,AB,KY (180) #13(luteal adj2 symptom*):TI,AB,KY (6) #14#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 (1816) #15MESH DESCRIPTOR Estradiol Congeners EXPLODE ALL TREES (5175) #16((estrogen* or estradiol)):TI,AB,KY (12460) #17(oestrogen* or oestradiol*):TI,AB,KY (1991) #18vivelle:TI,AB,KY (4) #19Oestrone:TI,AB,KY (96) #20Oestridae:TI,AB,KY (0) #21Estraderm:TI,AB,KY (85) #22#15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 (13378) #23#14 AND #22 (245)
Appendix 3. MEDLINE search strategy
From inception until 14 March 2016
OVID platform
1 Premenstrual Syndrome/ (3713) 2 Premenstrual Syndrome.tw. (2005) 3 PMS.tw. (3980) 4 PMD.tw. (1596) 5 PMDD.tw. (469) 6 LLPDD.tw. (30) 7 Premenstrual Tension.tw. (472) 8 (Dysphor$ adj2 disorder$).tw. (865) 9 (luteal adj2 dysphori$).tw. (82) 10 mastalgi$.tw. (384) 11 (Premenstrual adj2 depressi$).tw. (129) 12 PMT.tw. (1355) 13 (Premenstrual adj2 dysphor$).tw. (799) 14 (luteal adj2 symptom$).tw. (41) 15 or/1‐14 (10754) 16 exp estrogens/ or exp estradiol/ or exp "estrogens, conjugated (usp)"/ or exp ethinyl estradiol/ (150112) 17 (estrogen$ or estradiol).tw. (155913) 18 (oestrogen$ or oestradiol).tw. (27734) 19 vivelle.tw. (6) 20 Oestrone$.tw. (1663) 21 Oestridae.tw. (238) 22 Estraderm.tw. (101) 23 or/16‐22 (232264) 24 15 and 23 (950) 25 randomized controlled trial.pt. (409874) 26 controlled clinical trial.pt. (90289) 27 randomized.ab. (339223) 28 placebo.tw. (172096) 29 clinical trials as topic.sh. (175364) 30 randomly.ab. (244461) 31 trial.ti. (147347) 32 (crossover or cross‐over or cross over).tw. (66472) 33 or/25‐32 (1024402) 34 exp animals/ not humans.sh. (4203554) 35 33 not 34 (942118) 36 24 and 35 (185)
Appendix 4. Embase search strategy
From inception until 14 March 2016
OVID platform 1 exp premenstrual syndrome/ (5230) 2 Premenstrual Syndrome.tw. (2538) 3 PMS.tw. (5273) 4 PMD.tw. (2080) 5 PMDD.tw. (634) 6 LLPDD.tw. (37) 7 Premenstrual Tension.tw. (460) 8 (Dysphor$ adj2 disorder$).tw. (1145) 9 (luteal adj2 dysphori$).tw. (99) 10 mastalgi$.tw. (485) 11 (Premenstrual adj2 depressi$).tw. (147) 12 PMT.tw. (1677) 13 (Premenstrual adj2 dysphor$).tw. (1031) 14 (luteal adj2 symptom$).tw. (57) 15 or/1‐14 (14327) 16 conjugated estrogen/ or estrogen/ or conjugated estrogen plus medroxyprogesterone acetate/ or estrogen therapy/ (111303) 17 (estrogen$ or estradiol).tw. (178860) 18 (oestrogen$ or oestradiol).tw. (28965) 19 vivelle.tw. (196) 20 Oestrone$.tw. (1392) 21 Oestridae.tw. (227) 22 Estraderm.tw. (1043) 23 or/16‐22 (250508) 24 Clinical Trial/ (854056) 25 Randomized Controlled Trial/ (394472) 26 exp randomization/ (69652) 27 Single Blind Procedure/ (21649) 28 Double Blind Procedure/ (126672) 29 Crossover Procedure/ (46270) 30 Placebo/ (271479) 31 Randomi?ed controlled trial$.tw. (130858) 32 Rct.tw. (19565) 33 random allocation.tw. (1491) 34 randomly allocated.tw. (24106) 35 allocated randomly.tw. (2091) 36 (allocated adj2 random).tw. (750) 37 Single blind$.tw. (16992) 38 Double blind$.tw. (159443) 39 ((treble or triple) adj blind$).tw. (528) 40 placebo$.tw. (228518) 41 prospective study/ (324369) 42 or/24‐41 (1545825) 43 case study/ (36684) 44 case report.tw. (300841) 45 abstract report/ or letter/ (951787) 46 or/43‐45 (1282484) 47 42 not 46 (1505241) 48 15 and 23 and 47 (435)
Appendix 5. PsycINFO search strategy
From inception until 14 March 2016
OVID platform
1 exp Premenstrual Syndrome/ (1435) 2 Premenstrual Syndrome.tw. (1049) 3 PMS.tw. (1212) 4 PMD.tw. (442) 5 PMDD.tw. (392) 6 LLPDD.tw. (45) 7 Premenstrual Tension.tw. (168) 8 (Dysphor$ adj2 disorder$).tw. (803) 9 (luteal adj2 dysphori$).tw. (118) 10 mastalgi$.tw. (15) 11 (Premenstrual adj2 depressi$).tw. (131) 12 PMT.tw. (360) 13 (Premenstrual adj2 dysphor$).tw. (688) 14 (luteal adj2 symptom$).tw. (26) 15 or/1‐14 (3384) 16 exp Estrogens/ (5803) 17 (estrogen$ or estradiol).tw. (9757) 18 (oestrogen$ or oestradiol).tw. (903) 19 vivelle.tw. (0) 20 Oestrone$.tw. (10) 21 Oestridae.tw. (1) 22 Estraderm.tw. (1) 23 or/16‐22 (10531) 24 15 and 23 (152) 25 random.tw. (46235) 26 control.tw. (357993) 27 double‐blind.tw. (19679) 28 clinical trials/ (9458) 29 placebo/ (4475) 30 exp Treatment/ (646069) 31 or/25‐30 (993292) 32 24 and 31 (63)
Appendix 6. CINAHL search strategy
From inception until 14 March 2016
EBSCO platform
| # | Query | Results |
| S37 | S24 AND S36 | 41 |
| S36 | S25 OR S26 OR S27 OR S28 OR S29 OR S30 OR S31 OR S32 OR S33 OR S34 OR S35 | 1,032,949 |
| S35 | TX allocat* random* | 4,840 |
| S34 | (MH "Quantitative Studies") | 14,288 |
| S33 | (MH "Placebos") | 9,653 |
| S32 | TX placebo* | 37,534 |
| S31 | TX random* allocat* | 4,840 |
| S30 | (MH "Random Assignment") | 40,672 |
| S29 | TX randomi* control* trial* | 101,812 |
| S28 | TX ( (singl* n1 blind*) or (singl* n1 mask*) ) or TX ( (doubl* n1 blind*) or (doubl* n1 mask*) ) or TX ( (tripl* n1 blind*) or (tripl* n1 mask*) ) or TX ( (trebl* n1 blind*) or (trebl* n1 mask*) ) | 820,358 |
| S27 | TX clinic* n1 trial* | 183,294 |
| S26 | PT Clinical trial | 79,261 |
| S25 | (MH "Clinical Trials+") | 196,591 |
| S24 | S15 AND S23 | 151 |
| S23 | S16 OR S17 OR S18 OR S19 OR S20 OR S21 OR S22 | 16,567 |
| S22 | TX Estraderm | 5 |
| S21 | TX Oestridae | 1 |
| S20 | TX Oestrone | 13 |
| S19 | TX vivelle | 2 |
| S18 | TX oestrogen* or TX oestradiol | 1,139 |
| S17 | TX estrogen* or TX estradiol | 15,502 |
| S16 | (MM "Estrogens, Conjugated") OR (MM "Estrogens+") | 4,943 |
| S15 | S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13 OR S14 | 2,631 |
| S14 | TX(luteal N2 symptom*) | 18 |
| S13 | TX (Premenstrual N2 dysphor*) | 316 |
| S12 | TX PMT | 166 |
| S11 | TX(Premenstrual N2 depressi*) | 27 |
| S10 | TX mastalgi* | 77 |
| S9 | TX(luteal N2 dysphori*) | 12 |
| S8 | TX (Dysphor* N2 disorder*) | 331 |
| S7 | TX PMT | 166 |
| S6 | TX Premenstrual Tension | 44 |
| S5 | TX LLPDD | 7 |
| S4 | TX PMD* | 460 |
| S3 | TX PMS | 974 |
| S2 | TX Premenstrual Syndrome | 1,347 |
| S1 | (MM "Premenstrual Syndrome+") OR (MM "Premenstrual Dysphoric Disorder") | 1,013 |
Appendix 7. The Cochrane's tool for assessing risk of bias
| Domain | Support for judgement | Review authors' judgement |
| Selection bias | ||
| Random sequence generation | Describe the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups. | Selection bias (biased allocation to interventions) due to inadequate generation of a randomized sequence. |
| Allocation concealment | Describe the method used to conceal the allocation sequence in sufficient detail to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. | Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment. |
| Performance bias | ||
| Blinding of participants and personnel Assessments should be made for each main outcome (or class of outcomes) | Describe all measures used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. Provide any information relating to whether the intended blinding was effective. | Performance bias due to knowledge of the allocated interventions by participants and personnel during the study. |
| Detection bias | ||
| Blinding of outcome assessmentAssessments should be made for each main outcome (or class of outcomes) | Describe all measures used, if any, to blind outcome assessors from knowledge of which intervention a participant received. Provide any information relating to whether the intended blinding was effective. | Detection bias due to knowledge of the allocated interventions by outcome assessors. |
| Attrition bias | ||
| Incomplete outcome dataAssessments should be made for each main outcome (or class of outcomes) | Describe the completeness of outcome data for each main outcome, including attrition and exclusions from the analysis. State whether attrition and exclusions were reported, the numbers in each intervention group (compared with total randomized participants), reasons for attrition/exclusions where reported, and any re‐inclusions in analyses performed by the review authors. | Attrition bias due to amount, nature or handling of incomplete outcome data. |
| Reporting bias | ||
| Selective reporting | State how the possibility of selective outcome reporting was examined by the review authors, and what was found. | Reporting bias due to selective outcome reporting. |
| Other bias | ||
| Other sources of bias | State any important concerns about bias not addressed in the other domains in the tool. If particular questions/entries were pre‐specified in the review’s protocol, responses should be provided for each question/entry. |
Bias due to problems not covered elsewhere in the table. |
Data and analyses
Comparison 1. Oestrogen in combination with progestogen (sequential or continuous) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom scores | 3 | 267 | Std. Mean Difference (Fixed, 95% CI) | ‐0.27 [‐0.47, ‐0.07] |
| 1.1 E+P versus placebo | 2 | 196 | Std. Mean Difference (Fixed, 95% CI) | ‐0.19 [‐0.40, 0.03] |
| 1.2 E+P versus placebo +P | 1 | 71 | Std. Mean Difference (Fixed, 95% CI) | ‐0.69 [‐1.18, ‐0.20] |
| 2 Withdrawal due to adverse events | 3 | 284 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.26, 1.58] |
| 2.1 E+P versus placebo | 2 | 244 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.19, 1.59] |
| 2.2 E+P versus placebo +P | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.16, 6.42] |
| 3 Specific adverse events (E+P versus placebo) | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Bleeding problems | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.53 [0.10, 2.70] |
| 3.2 Breast tenderness | 2 | 206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.64 [0.87, 3.11] |
| 3.3 Headache | 2 | 206 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.53, 1.57] |
| 3.4 Nausea | 2 | 206 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.55, 2.71] |
| 3.5 Weight gain | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.65 [0.67, 202.74] |
| 3.6 Dysmenorrhoea | 1 | 138 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.78, 3.05] |
| 3.7 Skin irritation | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.39, 2.58] |
| 3.8 Skin reactions | 2 | 178 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.19, 1.59] |
| 3.9 Skin pigmentation | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.0 [0.65, 186.62] |
| 4 Specific symptoms of PMS: psychological | 2 | 119 | Std. Mean Difference (Fixed, 95% CI) | ‐0.79 [‐1.17, ‐0.40] |
| 4.1 E+P versus placebo | 1 | 48 | Std. Mean Difference (Fixed, 95% CI) | ‐0.33 [‐0.90, 0.24] |
| 4.2 E+P versus placebo +P | 1 | 71 | Std. Mean Difference (Fixed, 95% CI) | ‐1.18 [‐1.71, ‐0.65] |
| 5 Specific symptoms of PMS: physical | 2 | 119 | Std. Mean Difference (Fixed, 95% CI) | ‐0.60 [‐0.98, ‐0.22] |
| 5.1 E+P versus placebo | 1 | 48 | Std. Mean Difference (Fixed, 95% CI) | ‐0.29 [‐0.86, 0.28] |
| 5.2 E+P versus placebo +P | 1 | 71 | Std. Mean Difference (Fixed, 95% CI) | ‐0.85 [‐1.36, ‐0.34] |
| 6 Quality of life (E+P versus placebo) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6.1 VAS for feeling unwell (change from baseline) | 1 | 68 | Mean Difference (IV, Fixed, 95% CI) | ‐10.90 [‐22.60, 0.80] |
| 6.2 General health questionnaire (change from baseline) | 1 | 68 | Mean Difference (IV, Fixed, 95% CI) | ‐3.30 [‐9.98, 3.38] |
Comparison 2. Dose comparison: oestrogen 100 µg patch vs 200 µg patch plus progestogen in both groups.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptoms score: maximum mean daily PDQ | 1 | 98 | Mean Difference (IV, Fixed, 95% CI) | ‐1.55 [‐8.88, 5.78] |
| 2 Adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Withdrawal rates | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.34, 1.46] |
| 2.2 Side effects attributed to oestrogen | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.51 [0.26, 0.99] |
| 3 Specific symptoms of PMS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Psychological symptoms | 1 | 98 | Mean Difference (IV, Fixed, 95% CI) | ‐0.28 [‐1.17, 0.61] |
| 3.2 Physical symptoms | 1 | 98 | Mean Difference (IV, Fixed, 95% CI) | ‐0.33 [‐1.17, 0.51] |
| 4 VAS Sense of wellbeing | 1 | 98 | Mean Difference (IV, Fixed, 95% CI) | 1.70 [‐19.43, 22.83] |
| 5 Patient satisfaction | 1 | 107 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.85, 1.68] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Dhar 1990.
| Methods | Randomized double blind placebo‐controlled cross‐over trial, with cross‐over at 3 months | |
| Participants | 11 women with moderate to severe PMS (a cumulative rating of 50 points over the last 10 days of the luteal phase on a 69 points daily symptoms rating scale, and a cumulative score at least twice that during days 5 to 14 of the menstrual cycle) | |
| Interventions | Luteal‐phase oestrogens versus placebo 6 women given 0.625 mg premarin for 15 days before expected date of menstruation, i.e. luteal phase. 5 women given placebo pills in same manner. Cross‐over after 3 cycles. Duration: 6 menstrual cycles divided into 3 cycles of active treatment and 3 cycles of placebo for each participant. |
|
| Outcomes |
|
|
| Notes | Study was conducted in Canada and supported by the pharmaceutical company St.Laurent, Montreal, Canada | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No detail provided other than that pills were supplied "in random order" |
| Allocation concealment (selection bias) | Unclear risk | Nothing mentioned in the study about methods of allocation concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical looking active and placebo supplied by independent laboratory |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessment by patients |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All patients accounted for |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported, although adverse events poorly defined |
| Other bias | High risk | Unclear risk of carry‐over effects, data presented as percentage of average score at follow‐up and therefore difficult to interpret, main outcome measure not validated |
Magos 1986.
| Methods | Randomized double blind placebo‐controlled parallel trial | |
| Participants | Women aged 25 to 45 years, with regular periods and confirmed PMS on at least 1 of the 6 clusters in the MDQ during prospective daily symptom ratings. | |
| Interventions | Subcutaneous oestradiol implants versus placebo, 33 women given 100 mg subcutaneous oestradiol implant with 5 mg oral norethisterone for 7 days per cycle. 35 women given placebo implant with 5 mg placebo for 7 days per cycle. Duration: 2 consecutive cycles. |
|
| Outcomes |
|
|
| Notes | Trial was conducted in the UK and sponsored by a King's College Voluntary Research Trust. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random number table |
| Allocation concealment (selection bias) | Low risk | Independent physician concealed the allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical looking implants used, dispensed by independent physician |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Patient‐reported scores |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 20% of patients lost to follow‐up due to incomplete data |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | All data reported, no specific reasons to suspect other bias |
Panay 2001.
| Methods | Randomized double blind placebo‐controlled cross‐over trial, with cross‐over at 4 months | |
| Participants | Women with a history of PMS longer than 12 months’ duration, and confirmed from prospective symptom diaries (PDQ). | |
| Interventions | Oestradiol patches versus placebo 100 µg oestradiol patches with 1 mg oral norethisterone 17 to 28 days with identical placebo patches and tablets Duration: 8 months with cross‐over at 4 months. 47 women randomized to active‐first, and 45 women randomized to placebo‐first |
|
| Outcomes |
|
|
| Notes | Results of this trial, based on a per protocol analysis, have been published in abstract form (Panay 2001). However, Dr Panay supplied the full statistical analysis of the trial to us for the purpose of this review. This full analysis includes a modified intention‐to‐treat analysis. This mITT analysis was based on the 74 women (39 in the active‐first and 35 in the placebo‐first arm) for whom at least one assessment was available and was used to determine the effectiveness statistics reported in this review. The analysis of specific adverse effects was based on the 69 women (36 in the active‐first and 33 in the placebo‐first arm) who provided this information. The trial was conducted in the UK, sponsored by a pharmaceutical company. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomization performed outside study centre |
| Allocation concealment (selection bias) | Low risk | Allocation performed remotely and size of randomization block withheld from investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical looking patches and tablets were used. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Patient‐reported scores |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Published abstract reported 41% dropout and 10% lost to follow‐up. However, the intention‐to‐treat analysis provided to the review authors was based on 10% dropout and 10% loss to follow‐up. |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Unclear risk | Risk of carry‐over effects |
Smith 1995.
| Methods | Randomized active controlled parallel trial | |
| Participants | Women diagnosed with PMS and who showed a statistically significant PMS trend in 3 or more symptoms on a daily symptoms rating scale (PDQ) kept prospectively for a 4 to 8 week period of time | |
| Interventions | 100 µg versus 200 µg transdermal oestradiol patches 56 women were allocated to treatment with estraderm TTS 100 µg twice weekly continuously (El00 group), 56 women to treatment with Estraderm TTS 200 µg twice weekly continuously (E200 group). In both the El00 and E200 groups half of the women were allocated to take dydrogesterone 10 mg daily from day 17 to 26 of the cycle and half to take medroxyprogesterone acetate 5 mg daily from day 17 to 26 Duration: follow‐up visits were scheduled at the end of the 4th and 8th months of treatment. |
|
| Outcomes |
|
|
| Notes | Trial conducted in the UK, funding source unknown. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided on implementation of envelope method |
| Allocation concealment (selection bias) | Low risk | Opaque sealed envelope |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Non‐blinded trial |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Non‐blinded trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 21% dropout and 8% lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Low risk | No specific reasons to suspect other bias |
Watson 1989.
| Methods | Randomized double blind placebo‐controlled cross‐over trial, cross‐over at 3 months | |
| Participants | Women with severe PMS characterised by a significant positive and negative trend on at least 3 of the symptoms on a prospectively kept daily rating scale (PDQ). | |
| Interventions | Oestradiol patches versus placebo, 20 women given 100 µg Oestradiol patch with 5 mg oral norethisterone day 19 to 26. 20 women given placebo patch with 5 mg oral norethisterone day 19 to 26. Cross‐over of groups at 3 months Duration: 6 months divided into 3 months' active treatment and 3 months' placebo for each participant |
|
| Outcomes |
|
|
| Notes | Trial conducted in the UK, funding source unknown | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details on randomization method provided |
| Allocation concealment (selection bias) | Unclear risk | No details on concealment method provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo and active treatment looked identical |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Patient‐reported outcomes |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 12.5% of patients withdrew |
| Selective reporting (reporting bias) | Low risk | All expected outcomes were reported |
| Other bias | Unclear risk | Risk of carry‐over effects |
MDQ = Menstrual distress questionnaire
PDQ = Premenstrual distress questionnaire
PMS = Premenstrual syndrome
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| de Lignières 1986 | Study addresses effect on migraine, not on global symptoms |
| Dennerstein 1988 | Study addresses effect on migraine, not on global symptoms |
| Domoney 2003 | Study does not include placebo or oestrogen comparator |
| Ensom 2003 | Study addresses effect on asthma not on global symptoms |
| Halbreich 2012 | Study uses oestrogen as part of oral contraceptive |
Differences between protocol and review
As a result of the time frame between publication of the original protocol and completion of the review, we have updated the Background section to reflect the current state of the evidence in the field and the rationale for the intervention under review.
We stated in the protocol that diagnosis of premenstrual dysphoric disorder must meet established psychiatric diagnostic criteria (DSM‐IV‐TR). None of the studies is recent and therefore all predate such criteria (DSM III, IV or V). We therefore decided to drop this requirement, so that we could include these older studies.
We have included studies comparing different doses of oestrogen. This was not stated in the original search methods, but we felt the question of oestrogen dosage would be clinically relevant.
The protocol did not specify a time point at which to compare study outcomes. In the review, we used the 3‐month time point because it is the minimum recommended treatment duration for randomized controlled trials (ISPMD 2011).
One of the studies made the comparison oestrogen plus progestogen versus placebo plus progestogen. We have included this study in the analysis of oestrogen plus progestogen versus placebo and conducted a post hoc sensitivity analysis to see whether inclusion of this study influenced the results.
We planned to add a subgroup by mode of administration. However there were too few data to conduct this analysis.
The protocol did not specify methods to assess aspects of risk of bias specific to cross‐over trials. In the review, we addressed these aspects under the heading Other potential sources of bias, in particular carry‐over effects and differential dropout rates between the two periods of the cross‐over trial (Higgins 2011, section 16.4.3).
In studies with a cross‐over design, we planned to use only data from the first intervention phase after randomization. However, not all cross‐over trials we found allowed us to extract data from only the first phase. We therefore decided to analyze the data as if it was from a parallel group trial (Higgins 2011, section 16.4.5). This is explained under Unit of analysis issues.
In the case of dichotomous outcomes such as withdrawal rates or risks of adverse events we analyzed the treatment effect as risk ratio (RR) instead of odds ratio (OR), mainly because very few clinicians are adept with ORs (Sackett 1996).
We planned to use the standardized mean difference if outcomes were measured on different scales but did not specify the method to be used if separate baseline and post‐intervention scores could not be extracted. We now specify this methodology in the section Measures of treatment effect because we were not able to extract these scores separately for all studies.
Contributions of authors
BN developed the protocol and review; PMSO initiated the review; BN and JHK managed, extracted and analyzed the data; and JHK, PMSO, OAU and FO reviewed and provided comments on the protocol and the full review.
Sources of support
Internal sources
None, Other.
External sources
None, Other.
Declarations of interest
BN, JHK, OAU and FO have no interests to declare. PMSO has received some travel expenses, lecture fees and consultancy fees from Bayer Women’s Health, Abbvie Pharma, Umecrine Mood and Asarina Pharma for his work related to premenstrual disorders but on topics unrelated to the use of non‐contraceptive oestrogens in PMS.
New
References
References to studies included in this review
Dhar 1990 {published data only}
- Dhar V, Murphy BE. Double‐blind randomized crossover trial of luteal phase estrogens (Premarin) in the premenstrual syndrome (PMS). Psychoneuroendocrinology 1990;15(5‐6):489‐93. [DOI] [PubMed] [Google Scholar]
Magos 1986 {published data only}
- Magos AL, Brincat M, Studd JW. Treatment of the premenstrual syndrome by subcutaneous estradiol implants and cyclical oral norethisterone: placebo controlled study. British Medical Journal 1986;292(6536):1629‐33. [PUBMED: 3087550] [DOI] [PMC free article] [PubMed] [Google Scholar]
Panay 2001 {unpublished data only}
- Panay N, Rees M, Domoney C, Zakaria F, Guilford S, Studd JW. A multicentre double‐blind crossover study comparing 100μg transdermal oestradiol with placebo in the treatment of severe premenstrual syndrome. British Menopause Society Journal 2001;7(Suppl 3):19‐20. [Google Scholar]
Smith 1995 {published data only}
- Smith RN, Studd JW, Zamblera D, Holland EF. A randomised comparison over 8 months of 100 micrograms and 200 micrograms twice weekly doses of transdermal oestradiol in the treatment of severe premenstrual syndrome. British Journal of Obstetrics and Gynaecology 1995;102(6):475‐84. [PUBMED: 7632640] [DOI] [PubMed] [Google Scholar]
Watson 1989 {published data only}
- Watson NR, Studd JWW. Treatment of severe premenstrual syndrome with oestradiol patches and cyclical oral norethisterone. Lancet 1989;334(8665):730‐2. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
de Lignières 1986 {published data only}
- Lignières B, Vincens M, Mauvais‐Jarvis P, Mas JL, Touboul PJ, Bousser MG. Prevention of menstrual migraine by percutaneous oestradiol. British Medical Journal (Research Edition) 1986;293(6561):1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dennerstein 1988 {published data only}
- Dennerstein L, Morse C, Burrows G, Oats J, Brown J, Smith M. Menstrual migraine: a double‐blind trial of percutaneous estradiol. Gynecological Endocrinology 1988;2(2):113‐20. [DOI] [PubMed] [Google Scholar]
Domoney 2003 {published data only}
- Domoney C, Panay N, Hawkins A, Studd J. Treatment of premenstrual syndrome with transdermal oestrogen. International Journal of Gynaecology & Obstetrics 2003;83(suppl 3):37‐8. [Google Scholar]
Ensom 2003 {published data only}
- Ensom MH, Chong G, Zhou D, Beaudin B, Shalansky S, Bai TR. Estradiol in premenstrual asthma: a double‐blind, randomized, placebo‐controlled, crossover study. Pharmacotherapy 2003;23(5):561‐71. [DOI] [PubMed] [Google Scholar]
Halbreich 2012 {published data only}
- Halbreich U, Freeman EW, Rapkin AJ, Cohen LS, Grubb GS, Bergeron R, et al. Continuous oral levonorgestrel/ethinyl estradiol for treating premenstrual dysphoric disorder. Contraception 2012;85(1):19‐27. [DOI] [PubMed] [Google Scholar]
Additional references
Abraha 2010
- Abraha I, Montedori A. Modified intention to treat reporting in randomised controlled trials: systematic review. BMJ 2010;340:C2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
ACOG 2001
- ACOG. ACOG practice bulletin: premenstrual syndrome. International Journal of Gynaecology and Obstetrics 2001;73:183‐91. [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck‐Ytter Y, Flottorp S, et al. GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328:7454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2010
- Deeks JJ, Higgins JPT on behalf of the Statistical Methods Group of The Cochrane Collaboration. Statistical algorithms in Review Manager; August 2010. tech.cochrane.org/revman/documentation/statistical‐methods‐in‐revman‐5.pdf (accessed 11 January 2017).
DSM‐IV‐TR
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (4th ed.). 4th Edition. Arlington, VA: American Psychiatric Press, Inc., 2000. [Google Scholar]
Ford 2012
- Ford O, Lethaby A, Roberts H, Mol BW. Progesterone for premenstrual syndrome. Cochrane Database of Systematic Reviews 2012, Issue 3. [DOI: 10.1002/14651858.CD003415.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gianetto‐Berruti 2002
- Gianetto‐Berruti A, Feyles V. Premenstrual syndrome. Minerva Ginecologica 2002;54(2):85‐95. [PubMed] [Google Scholar]
GRADEpro GDT 2014 [Computer program]
- GRADE Working Group, McMaster University. GRADEpro GDT. Version accessed prior to 4 November 2016. Hamilton (ON): GRADE Working Group, McMaster University, 2014.
Green 2017
- Green LJ, O’Brien PMS, Panay N, Craig M on behalf of the Royal College of Obstetricians and Gynaecologists. Management of premenstrual syndrome. Br J Obst Gynaecol 2017;124:e73‐e105. [Google Scholar]
Halbreich 2003
- Halbreich U, Borenstein J, Pearlstein T, Kahn LS. The prevalence, impairment, impact, and burden of premenstrual dysphoric disorder (PMS/PMDD). Psychoneuroendocrinology 2003;28(Suppl 3):1‐23. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
ISPMD 2011
- O'Brien PMS, Bäckström T, Brown C, Dennerstein L, Endicott J, Epperson CN, et al. Towards a consensus on diagnostic criteria, measurement and trial design of the premenstrual disorders: the ISPMD Montreal consensus. Archives of Women's Mental Health 2011;14(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Johnson 2004
- Johnson S. The epidemiology of premenstrual syndrome. Primary Psychiatry 2004;11(2):27‐32. [Google Scholar]
Kaur 2004
- Kaur G, Gonsalves L, Thacker HL. Premenstrual dysphoric disorder: a review for the treating practitioner. Cleveland Clinic Journal of Medicine 2004;71(4):303‐5, 312‐3, 317‐8 passim. [DOI] [PubMed] [Google Scholar]
Lopez 2012
- Lopez LL, Kaptein AA, Helmerhorst FM. Oral contraceptives containing drospirenone for premenstrual syndrome. Cochrane Database of Systematic Reviews 2012, Issue 2. [DOI: 10.1002/14651858.CD006586.pub4] [DOI] [PubMed] [Google Scholar]
Marjoribanks 2013
- Marjoribanks J, Brown J, O’Brien PMS, Wyatt K. Selective serotonin reuptake inhibitors for premenstrual syndrome. Cochrane Database of Systematic Reviews 2013, Issue 6. [DOI: 10.1002/14651858.CD001396.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: The PRISMA Statement. BMJ 2009;339:2535. [PMC free article] [PubMed] [Google Scholar]
Norman 2003
- Norman GR, Sloan JA, Wyrwich KW. Interpretation of changes in health‐related quality of life: the remarkable universality of half a standard deviation. Medical Care 2003;41(3):582‐92. [DOI] [PubMed] [Google Scholar]
O'Brien 2003
- O'Brien P, Ismail K, Dimmock P. Premenstrual syndrome. In: Shaw R, Soutter W, Stanton S editor(s). Gynaecology. 3rd Edition. London: Churchill Livingston, 2003:401‐11. [Google Scholar]
O'Brien 2011
- O'Brien S, Rapkin A, Dennerstein L, Nevatte T. Diagnosis and Management of Premenstrual Syndrome. BMJ 2011;342:d2994. [DOI] [PubMed] [Google Scholar]
Panay 2005
- Panay N. Premenstrual syndrome: Making sense of the options. Pulse 2004;65(8):50‐4. [Google Scholar]
Rapkin 2007
- Rapkin AJ, Kuo J. The Premenstrual Syndromes. In: O’Brien PMS, Rapkin A, Schmidt P editor(s). PMS & PMDD. London: Informa Healthcare, 2007:69‐83. [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Roberts 2014
- Roberts HE, Hickey M, Lethaby A. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia: A Cochrane review summary. Maturitas 2014;77(1):4‐6. [A Cochrane review summary. Maturitas,] [DOI] [PubMed] [Google Scholar]
Sackett 1996
- Sackett DL, Deeks JJ, Altman DG. Down with odds ratios!. Evidence Based Medicine 1996;1(6):164‐6. [Google Scholar]