

ABSTRACT
Senescence is a tumor suppressor program characterized by a stable growth arrest while maintaining cell viability. Senescence-associated ribogenesis defects (SARD) have been shown to regulate senescence through the ability of the ribosomal protein S14 (RPS14 or uS11) to bind and inhibit the cyclin-dependent kinase 4 (CDK4). Here we report another ribosomal protein that binds and inhibits CDK4 in senescent cells: L22 (RPL22 or eL22). Enforcing the expression of RPL22/eL22 is sufficient to induce an RB and p53-dependent cellular senescent phenotype in human fibroblasts. Mechanistically, RPL22/eL22 can interact with and inhibit CDK4-Cyclin D1 to decrease RB phosphorylation both in vitro and in cells. Briefly, we show that ribosome-free RPL22/eL22 causes a cell cycle arrest which could be relevant during situations of nucleolar stress such as cellular senescence or the response to cancer chemotherapy.
KEYWORDS: Ribosome biogenesis, senescence, RPL22/eL22, CDK4, cyclin D1, RPS14/uS11
Graphical Abstract
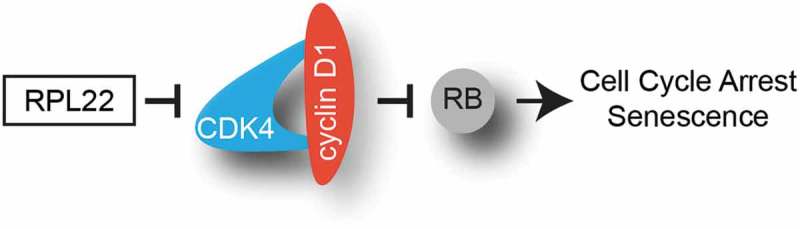
Introduction
Ribosome biogenesis is a complex multistep process implicating numerous cofactors and requiring proper coordination of the three major RNA polymerases [1]. The process is regulated by several oncogenes and/or tumor suppressors according to the cellular state whether proliferative, apoptotic, arrested or senescent [2,3]. In the nucleolus, polymerase I is responsible for 47S rRNA synthesis which is maturated into 18S, 5.8S and 28S rRNA while polymerase III synthetizes the 5S rRNA in the nucleoplasm [4,5]. Moreover, polymerase II transcribes the mRNAs of other protein components or cofactors implicated in processing and assembly of functional ribosomes. Several ribosomal proteins, which include RPL5/uL18, RPL11/uL5, RPS14/uS11 and RPL22/eL22, link nucleolar stress to p53 activation [6–9]. One physiological process leading to nucleolar stress is cellular senescence, a process that plays a role in embryonic development [10,11], wound healing [12] and tumor suppression [13].
During senescence, aberrant signaling triggers the degradation of multiple proteins by a process dubbed senescence-associated proteins degradation (SAPD). Targets of SAPD include several ribosome biogenesis factors and their down-regulation explains in part the senescence-associated ribogenesis defects (SARD) [14–16]. While SARD characterizes senescence in response to a variety of stresses, triggering the process by depleting individual ribogenesis factors is sufficient to induce senescence [16]. Analysis of the interactome of CDK4 in senescent cells induced by depleting one particular ribosome biogenesis factor, namely ribosomal L1 domain-containing protein 1 (RSL1D1) identified RPS14/uS11 and other ribosomal proteins as potential partners [16]. Ribosomal proteins are frequently mutated or their expression pattern is altered in various types of cancers [17,18]. RPL22/eL22 is often mutated in T-cell acute lymphoblastic leukemia (T-ALL) and in endometrial, colorectal and gastric cancers [19–23]. A large-scale analysis demonstrated that RPL22/eL22 hemizygous gene deletions are also frequent in numerous types of cancers and cell lines [24]. Moreover, RPL22/eL22 down-regulation is observed in non-small cell lung carcinoma and its haploinsufficiency promotes transformation and thymic lymphoma development in a mouse model [19,25]. All these facts suggest a tumor suppressor role for RPL22/eL22 which has previously been linked to p53 stabilization and activation [9]. In this report, we demonstrate that following ribosome biogenesis defects in senescence, RPL22/eL22 can control cell cycle progression through the inhibition of CDK4-cyclin D1 complex in a manner similar to RPS14/uS11 [16]. This demonstration provides an additional explanation for the tumor suppressor activity of RPL22/eL22.
Materials and methods
Plasmids
Retroviral vector pBABE was previously described [26]. pLXSN, pLXSN-E6, pLXSN-E7 were provided by Dr. Denise A. Galloway (Fred Hutchinson Cancer Center). pBABE-CDK4(WT) was a gift from Dr. Scott W. Lowe (Memorial Sloan-Kettering Cancer Center, New-York, NY). pcDNA3.1-3xFLAG, pcDNA3-Myc-Cyclin D1(WT), pMSCV-shNTC and pMSCV-shRB were previously described in [16]. Shp53 under H1 promoter regulation in pRS-shp53 was described in [27] and subcloned in EcoRI/XhoI restriction sites to create retroviral vector MSCV-shp53. pCMV6-RPS14(WT)-Myc-FLAG (RC223055) and pCMV6-RPL22(WT)-Myc-FLAG (RC208910) were purchased from Origene, Atlanta, GA. RPL22(WT)-Myc was PCR amplified from the vector pCMV6-RPL22(WT)-Myc-FLAG and subcloned in BamHI/SalI restriction sites to create retroviral vector pBABE-RPL22(WT)-Myc. For PCR primers used for cloning see Supplementary Table S1.
Lentiviruses pLKO expressing shRPL22 (shRPL22-A, shRPL22-B) and shCTR were purchased from Sigma-Aldrich (#75015, #75017 and SHC002). Finally, pCMV-VSV-G (Addgene #8454) and pCMV-dR.91 (Delta8.9) were obtained from Dr. R. Weinberg’s laboratory (Whitehead Institute, Cambridge, MA). ShRNA target sequences are presented in Supplementary Table S1.
Cells, reagents and viral gene transfer
Palbociclib (PD0332991) was purchased from Sigma-Aldrich (Oakville, ON). Phoenix Ampho packaging cells were a gift from Dr. Scott W. Lowe. Human embryonic kidney HEK-293T cells were obtained from Invitrogen. Prostate cancer PC3 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and human diploid fibroblasts IMR90 were obtained from Coriell Institute for Medical Research (Camden, NJ). PC3 cells were cultured in RPMI medium (Wisent, Montreal, QC) supplemented with 10% fetal bovine serum (FBS; Wisent), 1% penicillin/streptomycin sulfate (Wisent) and 2 mM L-glutamine (Wisent). All other cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM; Wisent) supplemented with 10% FBS (Wisent) and 1% penicillin/streptomycin sulfate (Wisent) and 2 mM L-glutamine (Wisent). Retroviral and lentiviral-mediated gene transfer were done as described before [16,26].
Immunoblotting
Immunoblotting was performed as previously described in [15]. The following primary antibodies were used: anti-phospho-H3S10 rabbit polyclonal (1:1000, #06–570, lot: 2,517,793, Millipore, Billerica, MA), anti-MCM6 rabbit polyclonal (1:1000, A300-194A, lot: A300-194A-2, Bethyl Laboratories), anti-p53 mouse monoclonal (1:1000, clone DO-1, Sc-126, lot: C1413, Santa Cruz Biotechnology), anti-phospho-p53S15 rabbit polyclonal (1:500, #9284, lot: 9, Cell Signaling), anti-FLAG mouse monoclonal (1:1000, F1804, M2, lot: SLBK1346V, Sigma-Aldrich), anti-c-Myc rabbit polyclonal (1:1000, clone A-14, sc-789, lot: K0215, Santa Cruz Biotechnology), anti-Myc tag mouse monoclonal (1:2000, 9E10, lot: 114K4821, Sigma, Saint Louis, Missouri), anti-phospho-RBS795 rabbit polyclonal (1:500, #9301, lot: 13, Cell Signaling), anti-RB mouse monoclonal (1:1000, clone 4H1, #9309, lot: 9, Cell Signaling), anti-CDK4 rabbit polyclonal (1:2000, A304-225A, lot: A304-225A-1, Bethyl Laboratories), anti-α-Tubulin mouse monoclonal (1:20000, clone B-5-1-2, T6074, lot: 023M4813, Sigma-Aldrich), anti-GST mouse monoclonal (1:2000, clone GST-2, G1160, lot: 012M4814, Sigma-Aldrich), anti-RPL22 mouse monoclonal (1:500, clone D-7, sc-373993, lot: D2216, Santa Cruz Biotechnology), anti-RPL22 rabbit polyclonal (1:1000, cat#: 25002–1-AP, Immunogen cat#: Ag21851, Proteintech) and anti-RPS3 mouse monoclonal (1:1000, clone C-7, sc-376008, lot: D2116, Santa Cruz Biotechnology).
The following secondary antibodies were used: goat anti-rabbit IgG conjugated to HRP (1:3000, #170–6515, lot: 64,126,042, Bio-Rad, Mississauga, ON), goat anti-mouse IgG conjugated to HRP (1:3000, #170–6516, lot: 64,132,955, Bio-Rad, Mississauga, ON).
Cell proliferation and senescence analysis
Proliferation was assessed from estimations of cell number according to a crystal violet retention assay [26]. Senescence-associated-β-galactosidase (SA-β-gal) activity was assayed as described before [26]. Separation of ribosomal RPL22-Myc from non-ribosomal RPL22-Myc was done as previously described [16]. Source data for growth curves and SA-β-gal staining are presented in Supplementary Table S2.
Immunofluorescence
Immunofluorescence images were performed as described in [16,26]. Primary antibodies used were: anti-PML rabbit polyclonal (1:200, A301-167A, lot: A301-167A-2, Bethyl Laboratories), anti-53BP1 rabbit polyclonal (1:200, Ab-1, Cat# PC712, lot: D00137736, Calbiochem, EMD Bios-ciences, San Diego, CA), anti-phospho-γH2A.XS139 mouse monoclonal (1:100, JBW-301, lot: 2552645, Millipore, Billerica, MA) and anti-Myc tag mouse monoclonal (1:100, 9E10, lot: 114K4821, Sigma, Saint Louis, Missouri).
The following secondary antibodies were used: goat anti-rabbit IgG (H + L) conjugated to Alexa Fluor 488 (1:1000, #A11008, lot: 1166843, Molecular Probes, Invitrogen, Eugene, OR), goat anti-mouse IgG (H + L) conjugated to Alexa Fluor 488 (1:1000, #A11029, lot: 1423008, Life Technologies, Eugene, OR), goat anti-rabbit IgG (H + L) conjugated to Alexa Fluor 568 (1:1000, #A11036, lot: 1504529, Life Technologies, Eugene, OR), goat anti-mouse IgG (H + L) conjugated to Alexa Fluor 568 (1:1000, #A11031, lot: 1398018, Life Technologies, Eugene, OR).
Images were acquired with a FV300 Olympus confocal microscope with a PMT 1st generation and Fluoview V4.2 or a Zeiss Axio Imager Z2 upright microscope with a CoolSNAP FX camera (Photometrics) and/or Axiocam camera and ZEN 2 Imager (2.0.14283.302). Images were processed with ImageJ (2.0.0-rc-49/1.51g). Source data for immunofluorescence are presented in Supplementary Table S2.
Immunoprecipitation (IP)
HEK-293T cells (5 × 106) were seeded in 10 cm-cell culture dishes and grown for 24 hours. Cells were transiently transfected using the calcium phosphate method with 15 µg of pcDNA3.1(C1)-3xFLAG, pCMV6-RPL22(WT)-Myc-FLAG or pCMV6-RPS14(WT)-Myc-FLAG in combination with either pBABE-CDK4(WT) or pcDNA3-Myc-cyclin D1(WT). Cells were harvested 24 hours post-transfection in IP buffer (50 mM Tris-HCl, pH 7.9, 1 mM EDTA, 0.1 mM EGTA, 12.5 mM MgCl2, 400 mM NaCl, 20% glycerol, 1% Triton X-100 [BioShop], 0.1% SDS and 1X Complete-EDTA free-protease inhibitor cocktail [Roche Applied Science]). Cell lysates were kept on ice for 15 min and then sonicated for 40 seconds at the lowest intensity. Cell lysates were cleared by centrifugation at 13,000 rpm for 1 min and immunoprecipitations were performed with anti-FLAG M2 Affinity Gel (#A2220-5ML, lot: SLBT8835, Sigma-Aldrich, St. Louis, MO) for 30 min at 4°C. Of note, anti-FLAG M2 Affinity Gel was previously blocked for 1 hour at 4°C in IP buffer containing 2.5% BSA, 0.16 µg/µl salmon sperm DNA (Sigma-Aldrich) and 0.16 µg/µl E. coli tRNA (Sigma-Aldrich) and then washed twice with IP buffer before being used for immunoprecipitation. Immunoprecipitates were recovered after 30 min of incubation at 4°C and washed three times for 30 min in IP buffer. Proteins of immunoprecipitates and total cell lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes and analyzed by Western Blotting.
In vitro protein phosphorylation
Human active CDK4-cyclin D1 (Cat#: C0620, lot: SLBK7657V, Sigma-Aldrich, St. Louis, MO) was incubated alone or with human recombinant GST-RB (773–928) (Cat#: R05-55G, lot: M166-3, SignalChem) in the presence or absence of palbociclib (1 µM) or with a gradient of human recombinant His-RPL22 (Cat#: NBP2-23419, lot: 12221401, Novus Biologicals, Oakville, ON) in kinase assay buffer I (Cat#: K01-09, Signal-Chem): 25 mM MOPS, pH 7.2, 12.5 mM ß-glycerol-phosphate, 25 mM MgCl2, 5 mM EGTA, 2 mM EDTA, 0.25 mM dithiothreitol (DTT), 40 µM ATP; at 30°C for 30 min according to SignalChem protocol. Then, Laemmli buffer was added to stop the reaction. Samples were boiled at 98°C for 5 min and the reaction products were separated by SDS-PAGE, transferred to nitrocellulose membranes and analyzed by Western Blotting.
In vitro protein interaction (GST pull-down assay)
GST (50 ng) (Cat#: SRP5348, lot: F664-2, Sigma-Aldrich, St. Louis, MO) or GST tagged human active CDK4-cyclin D1 (50 ng) (Cat#: C0620, lot: SLBK7657V, Sigma-Aldrich) or GST tagged human CDK4 (50 ng) (Cat#: C31-14G, lot: K127-1, Signal Chem, Richmond, BC) or GST tagged human cyclin D1 (50 ng) (Cat#: 009-001-153S, lot: 37136, Rockland antibodies & assays, Limerick, PA) were incubated with human recombinant His-RPL22 (375 ng) (Cat#: NBP2-23419, lot: 12221401, Novus Biologicals, Oakville, ON) in 500 μl of PB buffer (20 mM Hepes, pH 7.5, 130 mM KCl, 5 mM MgCl2, 1 mM DTT, 0.5 mM EDTA, 0.05% NP40) and mixed using a rotating machine at 30°C for 2 hours. Proper amounts of glutathione-Sepharose beads (Cat#: 17–0756-01, GE Healthcare, Sweden) were washed three times with PB buffer. Then, 10 μl of glutathione beads and 5 μl of BSA (25% stock solution) were added to the mix of proteins and incubation continued at room temperature for 30 min with rotation. The beads were then washed three times for 30 min with PB buffer at room temperature with rotation. Then, the appropriate quantity of 6X loading buffer (0.5 M Tris-HCl pH6.8, 30% glycerol, 10% SDS, 1% bromophenol blue and 15% β-mercaptoethanol) was added. The samples were boiled for 5 min and separated by SDS-PAGE for Western Blotting.
Real-time PCR
Total RNA extracts were prepared in TransZol (Civic Bioscience) according to the manufacturer’s instructions. Total RNA was reverse transcribed and gene expression level was determined with a LightCycler 96 (software version 1.1) Real-Time PCR (Roche Applied Science) was performed using SYBR Green technologies as described before [26]. QPCR primers are presented in Supplementary Table S1. Source data for qPCR are presented in Supplementary Table S2.
Statistics and reproducibility
Statistical analysis (two-tailed Student’s t-test) was performed using Excel. A value of p < 0.05 was considered statistically significant. Experiments were repeated at least three times.
Data availability
Source data for Figures 1(e), 2(a,b,d,e), 3(a,b), S1(b–d) and S2(a,c) can be found in Supplementary Table S2. All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Figure 1.
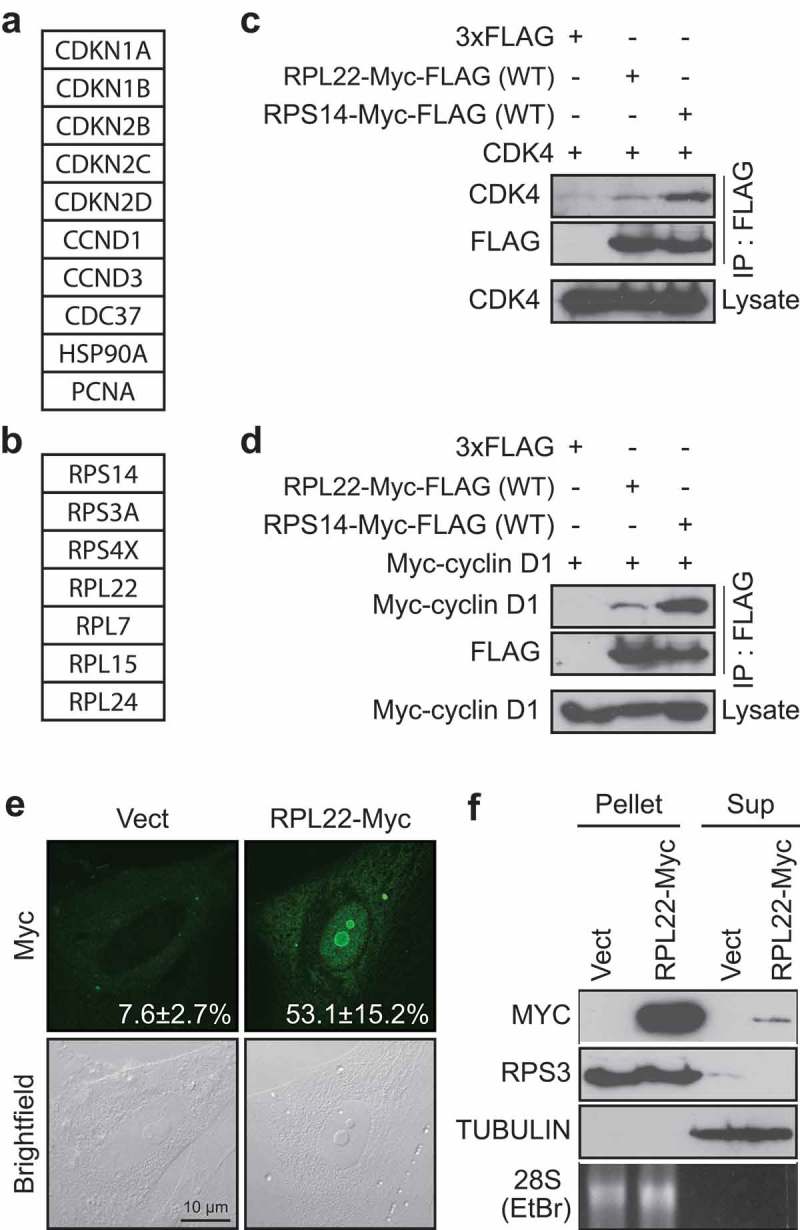
RPL22/eL22 can interact with CDK4 and/or cyclin D1 (a, b) List of selected proteins found to immunoprecipitate with a FLAG-tagged version of CDK4(K35M) from RSL1D1 knockdown-induced senescent cells classified as known interactors of CDK4 (a) and ribosomal proteins interactors (b). (c) HEK-293T cells were transfected with vectors expressing CDK4 and 3xFLAG control, Myc-FLAG tagged RPL22 wild type (RPL22-Myc-FLAG(WT)) or Myc-FLAG tagged RPS14 wild type (RPS14-Myc-FLAG(WT)) and immunoprecipitated with an anti-FLAG antibody. Lysates and immunoprecipitates were immunoblotted for the indicated proteins. (d) HEK-293T cells were transfected with vectors expressing Myc-cyclin D1 and 3xFLAG control, RPL22-Myc-FLAG(WT) or RPS14-Myc-FLAG(WT) and immunoprecipitated with an anti-FLAG antibody. Lysates and immunoprecipitates were immunoblotted for the indicated proteins. (e) Indirect immunofluorescence (IF) with a specific anti-Myc tag antibody showing RPL22-Myc in IMR90 cells expressing an empty control vector (Vect) or RPL22-Myc at day 12 post-infection (representative images). Data were quantified from 3-independent cell counts up to a total of at least 100 cells in triplicate and are presented as the mean percentage of positive cells for nucleolar localization of RPL22-Myc ± SD. Brightfield images are shown alongside. Scale bar, 10 µm. (f) Immunoblots for the indicated proteins and ethidium bromide detection of 28S rRNA in ribosome purification by sedimentation (Pellet) or its supernatant (Sup) obtained from extracts of IMR90 cells expressing RPL22-Myc or an empty control vector (Vect) at day 7 post-infection. Blots in C, D and F are representative of 3 independent experiments with similar results.
Figure 2.
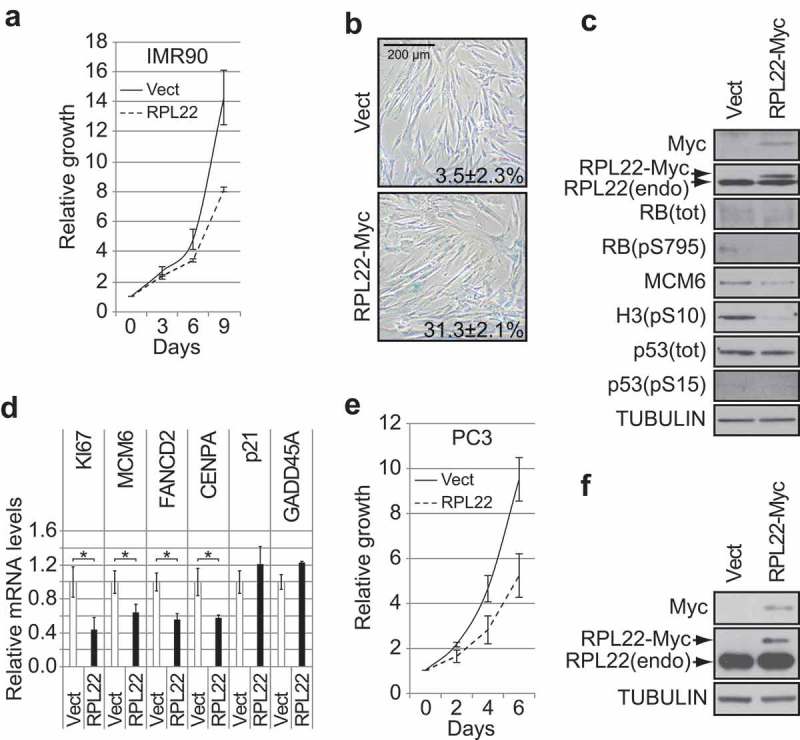
RPL22/eL22 regulates senescence in normal human diploid fibroblasts and decreases proliferation of p53-null PC3 cells. (a) Growth curves of normal human diploid fibroblasts IMR90, expressing RPL22-Myc or an empty control vector (Vect). Data are presented as means normalized to day 0 of each condition and error bars indicate SEM, n = 3 independent experiments. (b) SA-β-gal for cells as in (a) at day 12 post-infection. Data were quantified from 5-independent cell counts up to a total of at least 150 cells in triplicate and are presented as the mean percentage of positive cells ± SD. Scale bar, 200 µm. (c) Immunoblots of the indicated proteins performed on cells as in (a) at day 12 post-infection. RB(pS795): RB phosphorylated on serine 795; RB(tot): total RB; H3(pS10): histone H3 phosphorylated on serine 10; p53(pS15): p53 phosphorylated on serine 15; p53(tot): total p53; RPL22(endo): endogenous RPL22. (d) QPCR for the indicated genes performed on reverse transcribed total RNA extracted from cells as in (a) at day 12 post-infection. Data are normalized over TBP and HMBS, and presented as means relative to vector infected cells. Error bars indicate SD of triplicates. * = p < 0.05, using two-tailed Student’s t-test. (e) Growth curves of PC3 cells expressing RPL22-Myc or an empty control vector (Vect). Data are presented as means normalized to day 0 of each condition and error bars indicate SEM, n = 3 independent experiments. (f) Immunoblots of the indicated proteins performed on cells as in (e) at day 7 post-infection. RPL22(endo): endogenous RPL22. Blots in C and F are representative of 3 independent experiments with similar results.
Figure 3.
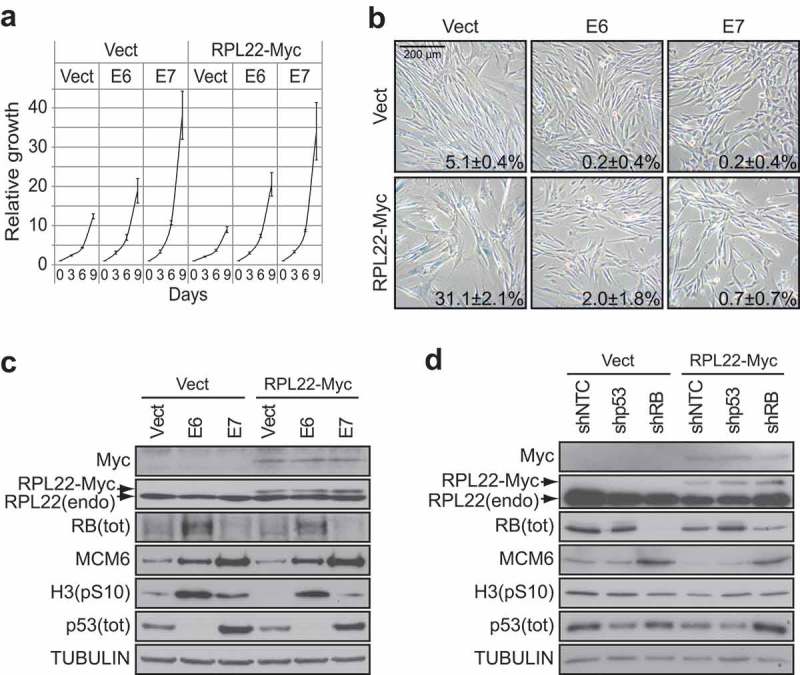
E6 or E7 can bypass RPL22/eL22-induced senescence. (a) Growth curves of IMR90 cells expressing RPL22-Myc or an empty control vector (Vect) in combination with the expression of the viral oncoproteins E6, E7 or an empty control vector (Vect). Data are presented as means normalized to day 0 of each condition and error bars indicate SEM, n = 3 independent experiments. (b) SA-β-gal for cells as in (a) at day 12 post-infection. Data were quantified from 5-independent cell counts up to a total of at least 150 cells in triplicate and are presented as the mean percentage of positive cells ± SD. Scale bar, 200 µm. (c) Immunoblots of indicated proteins performed on cells as in (a) at day 12 post-infection. RB(tot): total RB; H3(pS10): histone H3 phosphorylated on serine 10; p53(tot): total p53; RPL22(endo): endogenous RPL22. (d) Immunoblots of indicated proteins performed on IMR90 cells expressing RPL22-Myc or an empty control vector (Vect) in combination with the expression of a control shRNA (shNTC), a shRNA against p53 (shp53) or a shRNA against RB (shRB) at day 12 post-infection. RB(tot): total RB; H3(pS10): histone H3 phosphorylated on serine 10; p53(tot): total p53; RPL22(endo): endogenous RPL22. Blots in C and D are representative of 3 independent experiments with similar results.
Results
RPL22/eL22 as a possible regulator of CDK4-cyclin D1 complex
We recently published the interactome of the CDK4(K35M) mutant in RSL1D1 knockdown-induced senescent cells [16]. By using a less stringent cut-off, we found other known partners of CDK4 such as the cyclin-dependent kinase inhibitor 2D (CDKN2D or INK4D), the cell division cycle 37 (CDC37), the heat shock protein 90A (HSP90A) and the proliferating cell nuclear antigen (PCNA) (Figure 1(a) and Supplementary Table S3). In addition to the discovery of RPS14/uS11 as a CDK4 partner [16], we also found other ribosomal proteins namely S3A (RPS3A or eS1), S4X (RPS4X), L7 (RPL7 or uL30), L15 (RPL15 or eL15), L24 (RPL24 or eL24) and L22 (RPL22 or eL22) (Figure 1(b) and Supplementary Table S3). Considering that RPL22/eL22 is an haploinsufficient tumor suppressor gene [19], we decided to study its ability to regulate the cell cycle by interacting and regulating the CDK4-cyclin D1 complex. In order to confirm and determine the strength of the interaction of RPL22/eL22 with CDK4 and/or cyclin D1, we overexpressed and immunoprecipitated flag-tagged versions of RPL22/eL22 or RPS14/uS11 in the presence of overexpressed CDK4 or cyclin D1 (Figure 1(c,d) respectively). Much as with RPS14/uS11, RPL22/eL22 can interact with CDK4 (Figure 1(c)) and with cyclin D1 (Figure 1(d)) in HEK-293T cells. However, in both cases, RPL22/eL22 interacted less strongly than RPS14/uS11 (Figure 1(c,d)). Immunofluorescence in normal human fibroblasts (IMR90 cells) after retroviral gene transfer of a myc-tagged version of RPL22/eL22 showed its strong nucleolar accumulation and limited nucleoplasmic accumulation (Figure 1(e)) while RPS14/uS11 accumulated in the nucleoplasm in the same experimental setting [16]. Following ribosome purification of IMR90 cells expressing RPL22-Myc, we detected the myc-tagged version of RPL22/eL22 in the ribosomal fraction showing its incorporation into ribosomes but we also detected it in the supernatant (Figure 1(f)) pointing to ribosome-free RPL22/eL22. Taken together, these results suggest that RPL22/eL22 could regulate CDK4-cyclin D1 complex.
RPL22/eL22 induces senescence and biomarkers of RB pathway activation
The ability of RPL22/eL22 to interact with CDK4 and/or cyclin D1 suggested that RPL22/eL22 could induce senescence in an RB-dependent manner similarly to RPS14/uS11 [16]. To investigate this possibility, we used retroviral gene transfer to express RPL22/eL22 in IMR90 cells. We found that RPL22/eL22 induced proliferation defects (Figure 2(a)) and many senescence biomarkers, including: an accumulation of cells positive for senescence-associated beta-galactosidase (SA-β-gal) (Figure 2(b)), a reduction in RB phosphorylation, a decrease in the levels of the E2F target genes MCM6, FANCD2 and CENPA, a reduction in the levels of the mitotic marker H3-pS10 and a downregulation of KI67 expression (Figure 2(c,d) and Supplementary Figure S1(a)). Despite the detection of many markers of senescence, RPL22/eL22 expression did not lead to p53 accumulation or to its phosphorylation on serine 15 (Figures 2Cc and Supplementary Figure S1(a)) and we did not detect transcriptional induction of its target genes p21 (also known as CDKN1A) or GADD45A (Figure 2(d)). Moreover, we did not observe strong evidence of PML bodies or DNA damage foci accumulation (Supplementary Figure S1(b,c)) nor did we detect any strong increase in the expression of SASP components (Supplementary Figure S1(d)). In addition, low and stable expression of RPL22/eL22 reduced proliferation in p53-null PC3 cells (Figure 2(e,f)). The tumor suppressor role of RPL22/eL22 suggests that its depletion could lead to better proliferation capability but in fact its depletion decreased proliferation of IMR90 or PC3 cells (Supplementary Figure S2) reflecting its essential role in protein biosynthesis and cell growth. Together, these results suggest that RPL22/eL22, as RPS14/uS11 [16], induces cellular senescence through CDK4 inhibition and RB hypophosphorylation.
RPL22/eL22-induced senescence engages both the RB and the p53 tumor suppressor pathways
RPL22/eL22 has been shown to form a complex with the E3 ligase MDM2, the ribosomal proteins RPL5/uL18 and RPL11/uL5 and to synergize with RPL11/uL5 in the activation of p53 [9]. Considering that p53 can be activated without significant stabilization [28], it is impossible to completely exclude a role for p53 in RPL22/eL22-induced senescence from our initial results (Figure 2). In order to investigate this possibility and the preponderance of the RB or the p53 tumor suppressor pathways in the establishment of RPL22/eL22 senescence, we generated IMR90 cells stably expressing RPL22/eL22 in combination with papillomavirus oncoproteins E6 or E7. The E6 oncoprotein is known to disables p53 functions, whereas E7 inactivates the RB pathway [29]. In this model, we found that both E6 or E7 efficiently prevented reduced proliferation (Figure 3(a)) and accumulation of flat cells positive for SA-β-gal (Figure 3(b)) caused by enforced expression of RPL22/eL22. These oncoproteins also rescued the levels of the E2F target gene MCM6 and of the mitotic marker H3-pS10 (Figure 3(c)). Then, we generated IMR90 cells stably expressing RPL22/eL22 in combination with shRNAs targeting p53 or RB. Once again, we found that depletion of p53 or RB rescued the levels of MCM6 and H3-pS10 (Figure 3(d)) consolidating the result obtained with E6 and E7 (Figure 3(a–c)). Taken together, these results suggest that RPL22/eL22-induced senescence relies in part on activation of both p53 and RB pathways.
RPL22/eL22 is a CDK4 inhibitor
Because RPL22/eL22 and RPS14/uS11 were both recovered from the same CDK4 immunoprecipitate in RSL1D1 knockdown-induced senescence, were both found to interact with CDK4 and/or cyclin D1 (Figure 1) and were each able to induce a senescence phenotype in normal cells (Figure 2 and [16]), we hypothesized that RPL22/eL22 could be a direct CDK inhibitor much like RPS14/uS11 [16]. To demonstrate this, we established an in vitro kinase assay using purified recombinant proteins. We observed that RPL22/eL22 can effectively inhibit the CDK4-cyclin D1 phosphorylation of an RB fragment in a dose-dependent manner although less effectively than palbociclib (Figure 4(a)). Finally, to investigate a direct interaction of RPL22/eL22 with CDK4, cyclin D1 or the complex CDK4-cyclin D1, we performed GST-pull down assays with purified recombinant proteins. As reported for RPS14/uS11 [16], RPL22/eL22 can bind to both CDK4 or cyclin D1 alone but also with the complex CDK4-cyclin D1 (Figure 4(b)). These results suggest that RPL22/eL22 can act like a cyclin-dependent kinase inhibitor (CKI) of the INK family that binds CDK4 and also of the CIP/KIP family which binds to CDK-cyclin complexes (Figure 4(c)). The fact that RPL22/eL22 and RPS14/uS11 bind to cyclin D1 alone suggest another family we named PRICC (Protein Ribosomal Inhibitor of CDK-Cyclin) (Figure 4(b,c)) [16]. Taken together, these results show that RPL22/eL22 can directly bind and inhibit the CDK4-cyclin D1 kinase activity.
Figure 4.
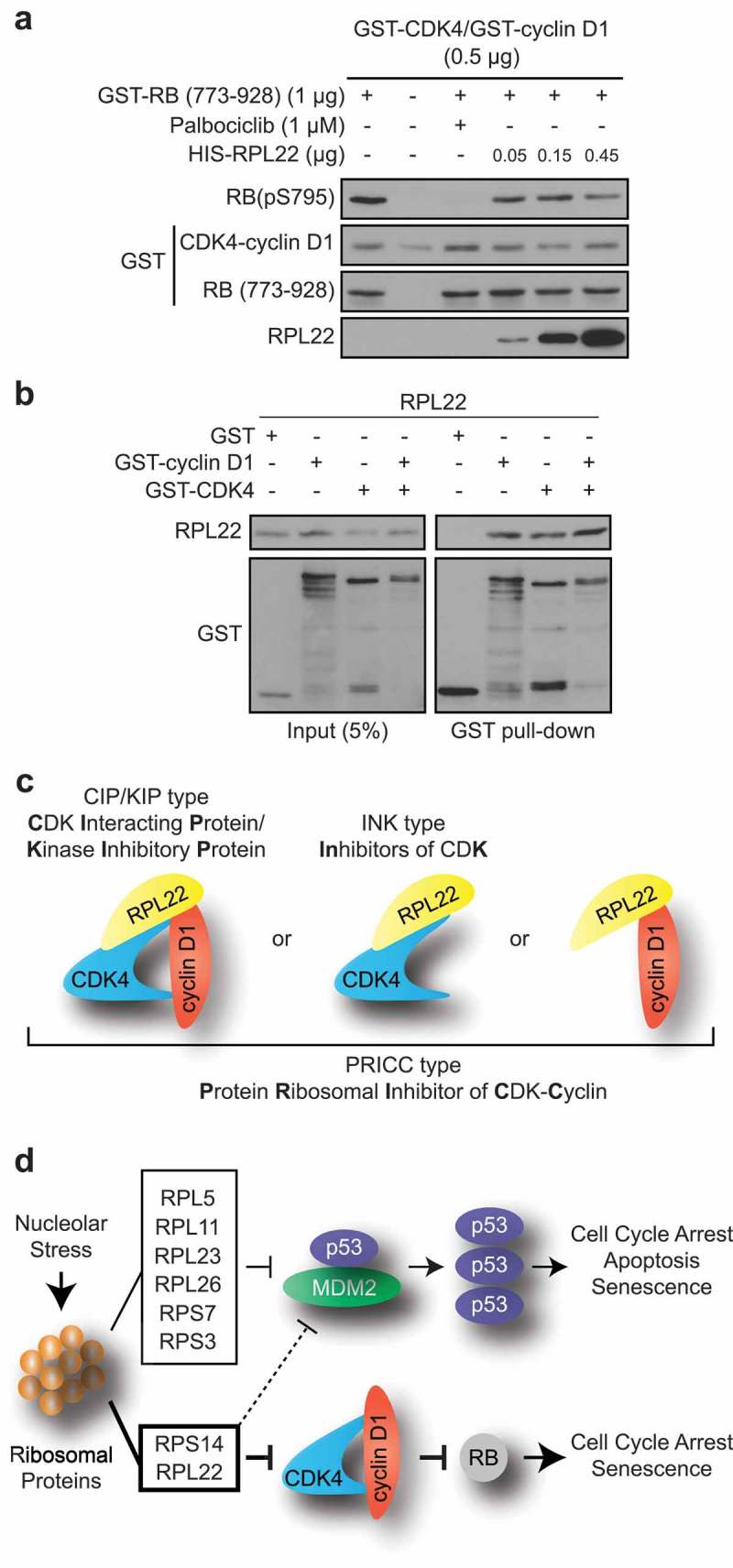
RPL22/eL22 directly binds and inhibits CDK4-cyclin D1 complex. (a) Immunoblots of the indicated proteins after in vitro kinase assay containing ATP and GST-CDK4 and GST-cyclin D1, with or without GST-RB (773–928), palbociclib and HIS-RPL22. RB(pS795): RB phosphorylated on serine 795. (b) In vitro GST pull-down of recombinant GST, GST-cyclin D1 and/or GST-CDK4 and recombinant HIS-RPL22 using glutathione beads. Lysate and pull-down were immunoblotted for the indicated proteins. (c) Model showing interactions between RPL22 and cyclin D1, CDK4 or CDK4-cyclin D1. (d) Model showing how nucleolar stress can lead to activation of p53 and RB tumor suppressor pathways. Blots in A and B are representative of 3 independent experiments with similar results.
Discussion
Senescent cells are characterized as non-proliferative with a high rate of proteins synthesis and defects in ribosome biogenesis [16,30,31]. The SARD phenotype led to the discovery that seven ribosomal proteins can interact with CDK4 in senescent cells. Among them, we previously characterized RPS14/uS11, which can accumulate in the nucleoplasm of senescent cells and contribute to the cell cycle arrest through inhibition of CDK4-cyclin D1 complex and RB phosphorylation (Figure 4(d)) [16]. In parallel, RPL22/eL22 was identified as a partner of CDK4, suggesting an additional mechanism that explains the tumor suppressor role of this protein (Figure 1(b–d)) [9]. Effectively, RPL22/eL22 can directly inhibit CDK4-cyclin D1 kinase activity and its expression induces senescence in IMR90 cells through hypophosphorylation of RB without significant activation or stabilization of p53 (Figures 2(a–d) and 4). Moreover, RPL22/eL22 can decrease proliferation of p53-null PC3 prostate cancer cells, suggesting a p53-independent control of cell cycle progression (Figure 2(e,f)).
It is reported that RPL22/eL22, like RPS14/uS11 and many other ribosomal proteins, can bind MDM2 to inhibit its E3 ligase activity, leading to p53 stabilization after nucleolar stress (Figure 4(d)) [8,9,32–35]. We show that RPL22/eL22-induced senescence can be overcome by the papillomavirus oncoproteins E6 or E7 which, respectively, inactivate the p53 or RB tumor suppressor pathways (Figure 3(a–c)). These results imply that both pathways are implicated in the establishment of RPL22/eL22 senescence and that RPL22/eL22 expression could have subtle effects on p53 activation or p53/MDM2 interaction, which were not detected in our experimental setting (Figure 4(d)). Short treatment with Nutlin-3 can cause p53 activation without significant stabilization [28] and we propose that RPL22/eL22 could act in a similar manner. An alternative explanation is based on the report that RPL22/eL22 binds p53 mRNA and inhibits its translation [36]. In this scenario, any stabilization of p53 via MDM2 inhibition by RPL22/eL22 is cancelled by inhibition of p53 mRNA translation.
The discovery of a second ribosomal protein linking the SARD to cell cycle regulation is significant and could lead to the development of small molecules or peptides mimicking their CDK inhibitory effects. Unlike RPS14/uS11 which accumulates in the nucleoplasm, RPL22/eL22 accumulates in the nucleolus when overexpressed. It is plausible that RPL22/eL22 control a nucleolar pool of CDK4-cyclin D, which is known to phosphorylate the nucleolar transcription factor UBF [37]. We anticipate that other ribosomal proteins might target different CDKs to strengthen the anti-proliferative effects induced by nucleolar stresses or ribosome biogenesis defects. Our work provides a rationale for identification of cell cycle regulators and development of anticancer therapy.
Funding Statement
This work was supported by the Canadian Cancer Society [704223]; Canadian Institute of Health and Research: CIHR [MOP11151]; Cancer Research Society of Canada [CRS].
Acknowledgments
We thank Scott Lowe (MSK), Denise A. Galloway (Fred Hutchinson Cancer Center), Andrew Koff (MSK), Bob Weinberg (MIT) and Tom Moss (U. Laval) for comments and/or reagents. We thank Éric Bonneil, Francis McManus and the IRIC Proteomics Core Facility for proteomic analysis. This work was funded by grants from the CIHR (Canadian Institute of Health and Research: CIHR MOP11151) to G.F. and the CCSRI (Canadian Cancer Society Research Institute: 704223) to G.F. F.L. is supported by FRQS (Fonds de Recherche du Québec-Santé) and CRS (Cancer Research Society). G.F. is supported by the CIBC chair for breast cancer research at the CR-CHUM.
Author Contributions
F.L. performed most of the cell culture experiments, immunoprecipitation, kinase assays, pull-down, western blots, SA-β-gal, growth curves and data analysis. N.D.T performed all qPCR, ribosome purification and reproduced RPL22/eL22 senescence and Figure 3(d). A.F.R confirmed RPL22/eL22 senescence and bypass with E6 and E7 and cell culture experiment leading to Supplementary Figure S1(a). N.D.T., A.F.R., M.-C.R., and/or F.L. performed parallel cell culture experiments for Figures 1(e,f) and 3(d) and Supplementary Figure S1(a). L.M constructed pBabe-RPL22-Myc, confirmed the PC3 experiment and did Supplementary Figure S2. P.K. performed the immunofluorescences in Supplementary Figure S1(b,c). E.S.-G. confirmed the immunoprecipitation. M.B. helped in quantification of SA-β-gal. S.L.-P. contributed by technical input with immunofluorescences. S.I performed the immunofluorescence in Figure 1(e) and bioinformatics analysis. F.L., L.B.-G., V.B. and G.F. participated in manuscript corrections and experimental design. F.L. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Tafforeau L, Zorbas C, Langhendries JL, et al. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of pre-rRNA processing factors. Mol Cell. 2013;51(4):539–551. Epub 2013/08/27 PubMed PMID: 23973377. [DOI] [PubMed] [Google Scholar]
- [2].Ruggero D, Pandolfi PP.. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3(3):179–192. Epub 2003/ 03/04 PubMed PMID: 12612653. [DOI] [PubMed] [Google Scholar]
- [3].White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6(1):69–78. Epub 2005/02/03 PubMed PMID: 15688068. [DOI] [PubMed] [Google Scholar]
- [4].Mullineux ST, Lafontaine DL. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie. 2012;94(7):1521–1532. Epub 2012/ 02/22 PubMed PMID: 22342225. [DOI] [PubMed] [Google Scholar]
- [5].White RJ. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008;24(12):622–629. Epub 2008/11/05 PubMed PMID: 18980784. [DOI] [PubMed] [Google Scholar]
- [6].Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279(43):44475–44482. Epub 2004/08/17 PubMed PMID: 15308643. [DOI] [PubMed] [Google Scholar]
- [7].Lohrum MAE, Ludwig RL, Kubbutat MHG, et al. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell. 2003;3(6):577–587. [DOI] [PubMed] [Google Scholar]
- [8].Zhou X, Hao Q, Liao J, et al. Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene. 2013;32(3):388–396. Epub 2012/ 03/07 PubMed PMID: 22391559; PubMed Central PMCID: PMCPMC3736832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cao B, Fang Z, Liao P, et al. Cancer-mutated ribosome protein L22 (RPL22/eL22) suppresses cancer cell survival by blocking p53-MDM2 circuit. Oncotarget. 2017;8(53):90651–90661. Epub 2017/ 12/07 PubMed PMID: 29207594; PubMed Central PMCID: PMCPMC5710875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Munoz-Espin D, Canamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155(5):1104–1118. Epub 2013/11/19 PubMed PMID: 24238962. [DOI] [PubMed] [Google Scholar]
- [11].Storer M, Mas A, Robert-Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155(5):1119–1130. Epub 2013/11/19 PubMed PMID: 24238961. [DOI] [PubMed] [Google Scholar]
- [12].Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31(6):722–733. Epub 2014/12/17 PubMed PMID: 25499914; PubMed Central PMCID: PMCPMC4349629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436(7051):720–724. Epub 2005/ 08/05 PubMed PMID: 16079850. [DOI] [PubMed] [Google Scholar]
- [14].Deschenes-Simard X, Lessard F, Gaumont-Leclerc MF, et al. Cellular senescence and protein degradation: breaking down cancer. Cell Cycle. 2014;13(12):1840–1858. Epub 2014/ 05/29 PubMed PMID: 24866342; PubMed Central PMCID: PMCPMC4111748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Deschenes-Simard X, Gaumont-Leclerc MF, Bourdeau V, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27(8):900–915. Epub 2013/04/20. PubMed PMID: 23599344; PubMed Central PMCID: PMCPMC3650227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lessard F, Igelmann S, Trahan C, et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol. 2018;20(7):789–799. Epub 2018/06/27 10.1038/s41556-018-0127-y. PubMed PMID: 29941930. . [DOI] [PubMed] [Google Scholar]
- [17].Goudarzi KM, Lindstrom MS. Role of ribosomal protein mutations in tumor development (Review). Int J Oncol. 2016;48(4):1313–1324. Epub 2016/ 02/20 PubMed PMID: 26892688; PubMed Central PMCID: PMCPMC4777597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lessard F, Brakier-Gingras L, Ferbeyre G. Ribosomal proteins control tumor suppressor pathways in response to nucleolar stress. Bioessays. 2019;e1800183 Epub 2019/02/02 PubMed PMID: 30706966 DOI: 10.1002/bies.201800183 [DOI] [PubMed] [Google Scholar]
- [19].Rao S, Lee S-Y, Gutierrez A, et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood. 2012;120(18):3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Novetsky AP, Zighelboim I, Thompson DM Jr., et al. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol Oncol. 2013;128(3):470–474. Epub 2012/ 11/07 PubMed PMID: 23127973; PubMed Central PMCID: PMCPMC3845021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. Epub 2013/ 10/18 PubMed PMID: 24132290; PubMed Central PMCID: PMCPMC3927368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ferreira AM, Tuominen I, van Dijk-Bos K, et al. High frequency of RPL22 mutations in microsatellite-unstable colorectal and endometrial tumors. Hum Mutat. 2014;35(12):1442–1445. Epub 2014/ 09/10 PubMed PMID: 25196364. [DOI] [PubMed] [Google Scholar]
- [23].Nagarajan N, Bertrand D, Hillmer AM, et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012;13(12):R115 Epub 2012/ 12/15 PubMed PMID: 23237666; PubMed Central PMCID: PMCPMC4056366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ajore R, Raiser D, McConkey M, et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol Med. 2017;9(4):498–507. Epub 2017/03/08 PubMed PMID: 28264936; PubMed Central PMCID: PMCPMC5376749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yang M, Sun H, Wang H, et al. Down-regulation of ribosomal protein L22 in non-small cell lung cancer. Med Oncol. 2013;30(3):646 Epub 2013/06/26 PubMed PMID: 23797773. [DOI] [PubMed] [Google Scholar]
- [26].Vernier M, Bourdeau V, Gaumont-Leclerc MF, et al. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev. 2011;25(1):41–50. Epub 2011/01/06 PubMed PMID: 21205865; PubMed Central PMCID: PMCPMC3012935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Moiseeva O, Bourdeau V, Roux A, et al. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29(16):4495–4507. Epub 2009/06/17 PubMed PMID: 19528227; PubMed Central PMCID: PMCPMC2725737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Allen MA, Andrysik Z, Dengler VL, et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife. 2014;3:e02200 Epub 2014/05/29 PubMed PMID: 24867637; PubMed Central PMCID: PMCPMC4033189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mallette FA, Goumard S, Gaumont-Leclerc MF, et al. Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene. 2004;23(1):91–99. Epub 2004/01/09 PubMed PMID: 14712214. [DOI] [PubMed] [Google Scholar]
- [30].Dorr JR, Yu Y, Milanovic M, et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature. 2013;501(7467):421–425. Epub 2013/ 08/16 PubMed PMID: 23945590. [DOI] [PubMed] [Google Scholar]
- [31].Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. Epub 2008/12/05 PubMed PMID: 19053174; PubMed Central PMCID: PMCPMC2592359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Donati G, Peddigari S, Mercer CA, et al. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013;4(1):87–98. Epub 2013/07/09 PubMed PMID: 23831031; PubMed Central PMCID: PMCPMC3928573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013;5(1):237–247. Epub 2013/10/15 PubMed PMID: 24120868; PubMed Central PMCID: PMCPMC3808153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Horn HF, Vousden KH. Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene. 2008;27(44):5774–5784. Epub 2008/06/19 PubMed PMID: 18560357. [DOI] [PubMed] [Google Scholar]
- [35].Marechal V, Elenbaas B, Piette J, et al. The ribosomal L5 protein is associated with MDM-2 and MDM-2-P53 complexes. Mol Cell Biol. 1994;1411:7414–7420. PubMed PMID: WOS:A1994PM68400040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rashkovan M, Vadnais C, Ross J, et al. Miz-1 regulates translation of Trp53 via ribosomal protein L22 in cells undergoing V(D)J recombination. Proc Natl Acad Sci U S A. 2014;111(50):E5411–9. Epub 2014/ 12/04 PubMed PMID: 25468973; PubMed Central PMCID: PMCPMC4273400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Voit R, Hoffmann M, Grummt I. Phosphorylation by G1-specific cdk-cyclin complexes activates the nucleolar transcription factor UBF. Embo J. 1999;18(7):1891–1899. Epub 1999/04/15 PubMed PMID: 10202152; PubMed Central PMCID: PMCPMC1171274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figures 1(e), 2(a,b,d,e), 3(a,b), S1(b–d) and S2(a,c) can be found in Supplementary Table S2. All data that support the findings of this study are available from the corresponding authors upon reasonable request.
Figure 1.
RPL22/eL22 can interact with CDK4 and/or cyclin D1 (a, b) List of selected proteins found to immunoprecipitate with a FLAG-tagged version of CDK4(K35M) from RSL1D1 knockdown-induced senescent cells classified as known interactors of CDK4 (a) and ribosomal proteins interactors (b). (c) HEK-293T cells were transfected with vectors expressing CDK4 and 3xFLAG control, Myc-FLAG tagged RPL22 wild type (RPL22-Myc-FLAG(WT)) or Myc-FLAG tagged RPS14 wild type (RPS14-Myc-FLAG(WT)) and immunoprecipitated with an anti-FLAG antibody. Lysates and immunoprecipitates were immunoblotted for the indicated proteins. (d) HEK-293T cells were transfected with vectors expressing Myc-cyclin D1 and 3xFLAG control, RPL22-Myc-FLAG(WT) or RPS14-Myc-FLAG(WT) and immunoprecipitated with an anti-FLAG antibody. Lysates and immunoprecipitates were immunoblotted for the indicated proteins. (e) Indirect immunofluorescence (IF) with a specific anti-Myc tag antibody showing RPL22-Myc in IMR90 cells expressing an empty control vector (Vect) or RPL22-Myc at day 12 post-infection (representative images). Data were quantified from 3-independent cell counts up to a total of at least 100 cells in triplicate and are presented as the mean percentage of positive cells for nucleolar localization of RPL22-Myc ± SD. Brightfield images are shown alongside. Scale bar, 10 µm. (f) Immunoblots for the indicated proteins and ethidium bromide detection of 28S rRNA in ribosome purification by sedimentation (Pellet) or its supernatant (Sup) obtained from extracts of IMR90 cells expressing RPL22-Myc or an empty control vector (Vect) at day 7 post-infection. Blots in C, D and F are representative of 3 independent experiments with similar results.
Figure 2.
RPL22/eL22 regulates senescence in normal human diploid fibroblasts and decreases proliferation of p53-null PC3 cells. (a) Growth curves of normal human diploid fibroblasts IMR90, expressing RPL22-Myc or an empty control vector (Vect). Data are presented as means normalized to day 0 of each condition and error bars indicate SEM, n = 3 independent experiments. (b) SA-β-gal for cells as in (a) at day 12 post-infection. Data were quantified from 5-independent cell counts up to a total of at least 150 cells in triplicate and are presented as the mean percentage of positive cells ± SD. Scale bar, 200 µm. (c) Immunoblots of the indicated proteins performed on cells as in (a) at day 12 post-infection. RB(pS795): RB phosphorylated on serine 795; RB(tot): total RB; H3(pS10): histone H3 phosphorylated on serine 10; p53(pS15): p53 phosphorylated on serine 15; p53(tot): total p53; RPL22(endo): endogenous RPL22. (d) QPCR for the indicated genes performed on reverse transcribed total RNA extracted from cells as in (a) at day 12 post-infection. Data are normalized over TBP and HMBS, and presented as means relative to vector infected cells. Error bars indicate SD of triplicates. * = p < 0.05, using two-tailed Student’s t-test. (e) Growth curves of PC3 cells expressing RPL22-Myc or an empty control vector (Vect). Data are presented as means normalized to day 0 of each condition and error bars indicate SEM, n = 3 independent experiments. (f) Immunoblots of the indicated proteins performed on cells as in (e) at day 7 post-infection. RPL22(endo): endogenous RPL22. Blots in C and F are representative of 3 independent experiments with similar results.
Figure 3.
E6 or E7 can bypass RPL22/eL22-induced senescence. (a) Growth curves of IMR90 cells expressing RPL22-Myc or an empty control vector (Vect) in combination with the expression of the viral oncoproteins E6, E7 or an empty control vector (Vect). Data are presented as means normalized to day 0 of each condition and error bars indicate SEM, n = 3 independent experiments. (b) SA-β-gal for cells as in (a) at day 12 post-infection. Data were quantified from 5-independent cell counts up to a total of at least 150 cells in triplicate and are presented as the mean percentage of positive cells ± SD. Scale bar, 200 µm. (c) Immunoblots of indicated proteins performed on cells as in (a) at day 12 post-infection. RB(tot): total RB; H3(pS10): histone H3 phosphorylated on serine 10; p53(tot): total p53; RPL22(endo): endogenous RPL22. (d) Immunoblots of indicated proteins performed on IMR90 cells expressing RPL22-Myc or an empty control vector (Vect) in combination with the expression of a control shRNA (shNTC), a shRNA against p53 (shp53) or a shRNA against RB (shRB) at day 12 post-infection. RB(tot): total RB; H3(pS10): histone H3 phosphorylated on serine 10; p53(tot): total p53; RPL22(endo): endogenous RPL22. Blots in C and D are representative of 3 independent experiments with similar results.