

Abstract
The proposed mechanism of fibril formation of transthyretin (TTR) involves self-assembly of partially unfolded monomers. However, the mechanism(s) of disassembly to monomer and potential intermediates involved in this process are not fully understood. In this study, native mass spectrometry and surface-induced dissociation (SID) are used to investigate the TTR disassembly mechanism(s) and the effects of temperature and ionic strength on the kinetics of TTR complex formation. Results from the SID of hybrid tetramers formed during subunit exchange provide strong evidence for a two-step mechanism whereby the tetramer dissociates to dimers that then dissociate to monomers. Also, the SID results uncovered a hidden pathway in which a specific topology of the hybrid tetramer is directly produced by assembly of dimers in the early steps of TTR disassembly. Implementation of SID to dissect protein topology during subunit exchange provides unique opportunities to gain unparalleled insight into disassembly pathways.
Graphical Abstract

Protein interactions are involved in many cellular processes, and their function(s) are modulated by the overall structure dynamics and topology of the complex. Both structure(s) and function(s) of these complexes can be modified by mutation and environmental factors: i.e., temperature, pH, hydration, and crowding.1–3 During the course of evolution, assembly of native polypeptides has resulted in protein complexes with increased biological functionality and diversity; however, self-assembly of non-native/partially unfolded proteins give rise to aberrant behavior, including some of the most challenging diseases to treat, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases.4–6 Knowledge regarding the driving forces and molecular details involved in such interactions along with a better understanding of conformational diversity of the individual subunits of the complex may potentially guide the development of novel drugs, vaccines, and biological nanostructures.7,8 Conventional biophysical approaches, such as X-ray crystallography, nuclear magnetic resonance, and cryo-EM that provide exquisite molecular level structural information, are often ensemble measurements and struggle with analyzing dynamic systems.
Transthyretin (TTR), a homotetrameric protein complex (MW ≈ 56 kDa), is mainly involved in retinol binding protein (RBP) transport.9 TTR is also implicated in both hereditary and nonhereditary amyloidosis.10,11 Although TTR has been extensively studied for decades with some potent drugs developed, such as tafamidis,12–14 its mechanisms of subunit exchange and overall stability are not fully understood. Previous studies have suggested that both monomers and dimers serve as intermediates during fibril formation; however, which is the main constituent of fibrils remains contentious.15,16 Regardless, aggregation of TTR has shown a direct correlation with the self-assembly of unfolded monomers.
Two models for TTR disassembly have been proposed by Kelly et al. (model A)17 and Robinson et al. (model B)18 (Figure S1) using subunit exchange (SUE) experiments wherein mixing untagged (U4) and tagged (T4) TTR (or isotopically labeled in the case of model B) and the kinetics of forming mixed TTR complexes (Figure 1A) were monitored under native conditions. U2T2 can possess three different topologies, UU/TT, UT/TU, and UT/UT, where “/” denotes the weaker of the two dimeric interfaces,19 but none of the aforementioned models were able to distinguish and quantify the different topologies. These models also differ in that there is an additional tetramer to monomer dissociation in model B and dimer to monomer dissociation is included only in model A. It is important to note that subunit reassembly is fast and experimental observation of monomers and/or dimers is not possible. In addition, resolution of conflicting mechanisms is further complicated owing to the differences in tags, which further complicates comparisons of the reported kinetics.
Figure 1.
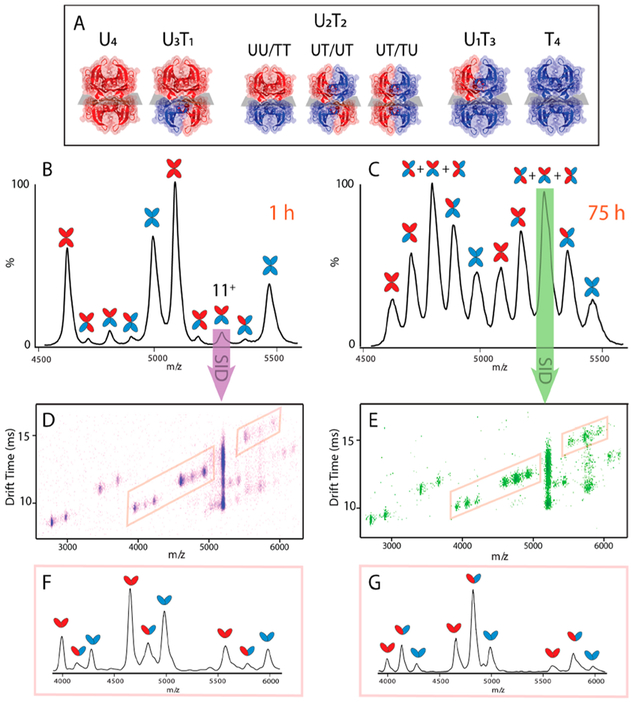
(A) Tetramers involved in TTR subunit exchange experiments and corresponding abbreviations. Untagged and tagged subunits are shown in red and blue, respectively. Mass spectra of an equimolar solution of WT-TTR and CT-TTR incubated at 4 °C after 1 h (B) and after 75 h (C). SID-IM-MS of U2T2 (11+) and mass spectra of corresponding dimer peaks extracted from highlighted trend line after 1 h (D, F) and after 75 h (E, G).
Here, we employ native mass spectrometry (MS),20–22 ion mobility (IM),23,24 and surface-induced dissociation (SID)25 to monitor TTR SUE at the topological level. SID is an invaluable approach for dissecting the topology of protein complexes owing to direct correlation between SID energy and interface cleavage area.19,26 A lower energy SID of TTR has yielded dimers, its building block, wherein the dimer–dimer interface has the smallest surface area. Such a gas-phase product cannot be obtained using collision-induced dissociation, a commonly used activation method in MS.25,27
EXPERIMENTAL SECTION
Materials and Protein Preparation.
Constructs containing either the C-terminal GFP-6xHis (pET15b) or the N-terminal 6xHis-MBP/dual Flag-tagged (pET28b) fusion proteins on TTR that are TEV protease cleavable were transformed into BL21 (DE3) RIPL E. coli cells (Agilent). Colonies were grown in LB at 37 °C until an OD 600 nm value of 0.6–0.8. The cells were induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubated at 37 °C for 3 h. Cells were harvested by centrifugation and lysed on ice in the presence of complete protease inhibitor tablet (Roche) and 5 mM β-mercaptoethanol (β-ME) using a M-110P microfluidizer (Microfluidics) running at 20000 psi. Cellular debris was cleared by centrifugation at 25000g for 25 min. The supernatant was filtered with a 0.45 μm syringe filter before loading onto a HisTrap HP column (GE Healthcare). The column was equilibrated at 4 °C with 50 mM potassium phosphate buffer (pH 8.0), 500 mM NaCl, and 30 mM imidazole (buffer A), and the bound protein was eluted in the same buffer with 250 mM imidazole (buffer B). The protein was immediately buffer exchanged back into buffer A using a HiPrep 26/10 desalting column (GE Healthcare). The fusion tags were cleaved from TTR by incubation with TEV protease 5 mM β-ME overnight at 4 °C. The His-labeled tags were separated from the nonlabeled TTR by reverse immobilized-metal affinity chromatography on a HisTrap HP column using buffer A. The C-terminal tag on TTR consists of the following amino acid sequence: …ASGENLFYQ. The flow-through was collected and concentrated using a 50 kDa MWCO centrifuge concentrator (Millipore) in the presence of 5 mM β-ME and filtered using a 0.45 μm syringe filter. Aggregates and small-molecular-weight contaminants were separated at 4 °C on a Superdex HiLoad 16/600 75 pg size-exclusion column (GE Healthcare) equilibrated with 50 mM Tris and 10% glycerol, pH 7.5 (at room temperature), and 150 mM NaCl. Peaks corresponding to the tetrameric state were confirmed by mass spectrometry prior to pooling. Aliquots of TTR (50 uL) were flash-frozen with liquid nitrogen and kept at −80 °C.
Subunit Exchange of Wild Type (WT) TTR and C-Terminal Tagged (CT) TTR (or Dual Flag-Tagged (FT2) TTR) Using Native MS.
Fresh TTR samples (untagged and tagged) were buffer-exchanged into ammonium acetate (EMD, Millipore Corp) using Micro Bio-Spin 6 Columns (Bio-Rad). Subunit exchange of WT-TTR+CT-TTR and WT-TTR+FT2-TTR was investigated using three different ratios (1:1, 1:2, and 5:2), and each sample was incubated at 4, 24, 30, and 37 °C. At various time points, aliquots (2 uL) were taken and triethylammonium acetate (TEAA) or ethylendiammonium diacetate (EDDA, Sigma; 50–100 mM) was added for charge reduction. TEAA was found to be more effective than EDDA as a charge reducer. Charge reducers used in this study offer two advantages: (1) better resolved m/z peaks for quantification and (2) satisfaction of the charge reduction requirement for native SID experiments, as the numbers of charges of protein have a strong effect on the population of subunits released via SID, and charge-reduction protein complexes better resemble the solution structure.28
Samples were introduced via static-spray nano-ESI into a Synapt G2 (Waters, U.K.) modified with a SID cell.29 Capillary-voltage (~1.2–1.5 kV) was applied to a Pt wire inserted into borosilicate tips pulled in house (Sutter 1000). A moderately high sampling voltage (100–180 V) was used to strip off adducts without unfolding ions in the gas phase. Source pressure (5–6 mbar), trap flow (4 L/min fly through and 2 L/min in SID mode), and He flow (120 mL/min) were also adjusted for maximum transmission.
Along with MS data, at each data point, SID of U2T2 (11+) was collected using a collision voltage of 40 V (Elab = 440 eV). The peaks corresponding to dimers in the IM-MS were extracted using DriftScope (Waters, U.K.), and UniDec was used for mass deconvolution to determine the relative abundance of tetramers (MS spectrum) and dimers (SID spectrum).30 These results were then plotted over time and used for kinetics fitting.
To determine whether results for subunit exchange between WT-TTR and FT2-TTR can be reproduced using native MS, the exchange rate (kex) was calculated (0.0427 ± 0.0130 h−1) and gave a result relatively similar to the previously reported value (0.0400 ± 0.0025 h−1) using anion exchange chromatography at 24 °C and in 130 mM salt.31
Kinetic Fitting.
Kintek Explorer (v 7.6) was used for global fitting with and without SID data.32 The relative abundance of UU/TT and (UT/UT + UT/TU) was calculated using the eqs 1 and 2 and used as constraints in kinetic fitting as an additional criteria for fitting parameters.
| (1) |
| (2) |
When only MS data were implemented, both models could successfully fit the experimental data: R2 = 91.1 and 92.6 for models A and B, respectively. Also, due to multiple steps involved in each model, without inclusion of SID data, a large number of good fits were achieved with different rate constants. Conversely, global kinetic fitting with the SID data imposed additional constraints on individual rate constants and improved the fit with R2 = 93.7 and 94.1 for models A and B, respectively, at 4 °C. Kinetic models were analyzed using identical rate values, i.e. tetramer to dimer and dimer to monomer, unless independent rate constants provided a better fit. A diffusion-limited rate (1.0 × 105 h−1 M−1) was chosen for reassembly of subunits.
RESULTS AND DISCUSSION
Native mass spectrometry has been extensively employed to study protein–protein interactions22,33 because it offers both identification and quantification of individual species while preserving noncovalent interactions.34–36 Such information can be utilized to study kinetics of interactions and further identify involved intermediates. Here, we also used native IM-MS and surface-induced dissociation (SID) to study the products formed from TTR complexes during subunit exchange.18,37–39
Incubation of WT-TTR and CT-TTR at 4 °C resulted in a mass spectrum with homotetramers (U4 and T4) as the most abundant peaks (Figure 1B). As the SUE reaction proceeds, the peak intensities decrease and approach a minimum at equilibrium (75 h). Meanwhile, heterotetramers evolve and U2T2 becomes the predominant peak (Figure 1C). The newly developed CT tag offered better gas- and solution-phase similarity to WT-TTR as opposed to the previously used dual-Flag tag (FT2-TTR) (Figure S2). Thus, we used the CT tag for further study.
Topology Information Obtained from SID Data.
To assess the topology of U2T2 assemblies (Figure 1A), we performed SID on U2T2 and related resulting dimers to the corresponding topology. For instance, SID-MS of U2T2 at 4 °C mostly consisted of homodimers (UU and TT) at the starting point, t0 (Figure 1F). Due to the low-energy regime used in our SID experiments, only the UU/TT tetramer would produce a spectrum that contains UU and TT dimers in a 1:1 ratio (Figure 2A). However, the abundances of these products decrease over time and at equilibrium the UT dimer becomes the main peak, resulting in a ratio of 1:4:1 (UU:UT:TT) (Figure 1G). UT dimer can only be obtained from SID of two topologies (UT/UT and UT/TU), which means an equimolar solution containing all three topologies is required to yield 1:4:1 abundances of UU, UT, and TT dimers, respectively (Figure 2B).
Figure 2.
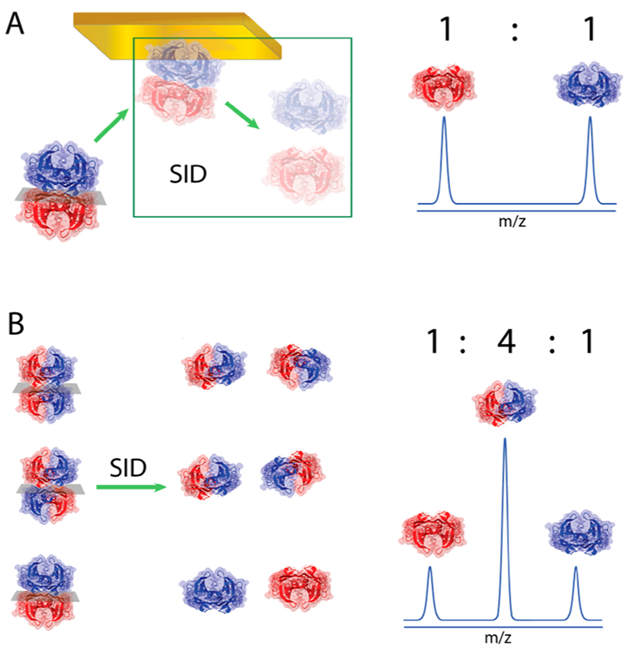
(A) SID of a TTR solution with just UU/TT tetramer yielding an MS spectrum with equal signal intensity for UU and TT dimers. (B) SID-MS spectrum of an equimolar solution of UU/TT, UT/UT, and UT/TU.
This direct correlation between dimers resulted from SID of U2T2 and its topologies was used to calculate the relative abundance of UU/TT and (UT/UT + UT/TU). The topological data provide an additional constraint in kinetic fitting (see the Experimental Section for details). Also, we noticed that an increase in temperature altered the ratio of dimers. While an increase in temperature did not change the equilibrium ratio, the relative abundance of UU/TT tetramer at t0 decreased from ~82% at 4 °C to ~65% at 24 °C and ~50% at 37 °C (Figure 3B,D,F).
Figure 3.

Fitting curves obtained from model A for SUE between WT-TTR (5 μM) and CT-TTR (5 μM) at 4 °C (A), 24 °C in 50 mM A (C), and 37 °C in 25 mM AA (E). Relative abundance of UU/TT and UT/UT+UT/TU obtained from SID at 4 °C (B), 24 °C (D), and 37 °C (F). A stepwise increase in tetramer dissociation rates is observed upon addition of CT-TTR monomer at 37 °C (G). Blue and red ovals denote the tagged and untagged monomers, respectively.
Kinetic modeling was used to globally fit and compare model A (Figure 3) and model B (Figure S3). To improve the reliability of kinetic parameters, two other ratios (1:2 and 5:2 (WT:CT)) at each temperature along with SID information (Figure 3B,D,F) were included in global fitting (Figures S4 and S5 for models A and B, respectively). Interestingly, both models provide good fits for MS data but upon close inspection of SID data reveal some discrepancies (vide infra).
Although similar fits were obtained for MS data, each model provided different fits for SID data at t0 (Figure 3B,D,F for model A and Figure S3B,D,F for model B). Interestingly, the best fit was obtained using model A, which can precisely predict dimer ratios at t0 at various temperatures. Individual rate constants measured using model A are summarized in Table 1, which will be used to justify the variation of dimer ratios.
Table 1.
Rate Constants Measured for Subunit Exchange between WT-TTR and CT-TTR in 50 mM AA Using Model A
| temp (°C) | ktet-dim (h−1) | ktet-dim (h−1) (UT/UT+UT/TU) | kdim-mon (h−1) (UU and TT) | kdim-mon (h−1) (UT) |
|---|---|---|---|---|
| 4 | 0.20 ± 0.029 | 0.81 ± 0.15 | 68 ± 15 | 37 ± 12 |
| 24 | 0.075 ± 0.007 | 0.44 ± 0.049 | 107 ± 17 | 48 ± 10 |
Temperature Effect on TTR Subunit Exchange.
The temperature dependence of SUE was also investigated (at 4, 24, and 37 °C) to explore the effects of TTR dynamics and to address the origin of variation of dimer ratios at different temperatures. Few studies (<3) have reported SUE at physiological temperature (37 °C), quite possibly because elevated temperatures reduce the rate of SUE and destabilize the protein. To increase the rate of exchange, a lower concentration (25 mM) of ammonium acetate (AA) was used for these studies, since there is an inverse relationship between ionic strength and exchange rate (Figure S6). Fitting parameters in 25 mM AA are provided in Table 2.
Table 2.
Fitted Rate Constants Using Model A for Subunit Exchange between WT-TTR and CT-TTR in 25 mM AA
| temp (°C) | ktet-dim (h−1) | ktet-dim (h−1) (UT/UT+UT/TU) | kdim-mon (h−1) (UU and TT) | kdim-mon (h−1) (UT) |
|---|---|---|---|---|
| 4 | 0.52 ± 0.11 | 2.4 ± 0.74 | 130 ± 33 | 65 ± 29 |
| 24 | 0.17 ± 0.10 (0.34 ± 0.18)a | 1.53 ± 0.81 | 156 ± 64 | 80 ± 61 |
| 30 | 0.09 ± 0.06 (0.20 ± 0.12) | 1.24 ± 2.4 | 241 ± 53 | 85 ± 106 |
| 37 | 0.04 ± 0.03 (0.16 ± 0.18) | 0.59 ± 0.24 | 249 ± 80 | 124 ± 60 |
Numbers in parentheses correspond to CT-TTR.
Temperature can affect protein stability, as shown by the faster disappearance rate for the tagged protein in comparison to untagged TTR at 37 °C (Figure 3E, cyan vs red curve). On the other hand, they have similar kinetic stabilities at lower temperatures (4 and 24 °C), as indicated by the similar relative abundances of homotetramers (and also U3T1 and U1T3). This tag-induced destabilizing effect at 37 °C was also found to be an accumulative effect (Figure 3G) wherein the rate of tetramer dissociation (ktet-dim) is increased with the number of tagged subunits. A similar trend has been previously described for subunit exchange of WT and L55P-TTR, an amyloidogenic mutant, in which the rate of exchange was directly proportional to the number of L55P monomers in the tetramer.18
Temperature can also destabilize the secondary structure of proteins, as non-native monomer formation has been reported under native conditions at 37 °C.40,41 This additional pathway can delay monomer association and alter the exchange rate. To quantify the contribution of unfolding in TTR dynamics at higher temperatures, ideally a tag with no effect on the stability of TTR is required. However, such a requirement can be challenging, as even deuteration has been shown to alter TTR stability.42
Information derived from the data contained in both Tables 1 and 2 clearly indicates that increased temperatures favor dissociation of dimers. This observation is consistent with destabilizing hydrogen bonding of the monomer–monomer interactions in the individual dimers with increases in temperature.43 The increase in monomer abundances favors heterodimer production (UT), as shown in Figure S7, in which the relative abundance of UT in comparison to UU (or TT) is increased with an increase in temperature (4 and 24 °C). As more UT is produced, it eventually yields more UT/UT and UT/TU heterotetramers. Therefore, while SUE at higher temperatures is slowed by hydrophobic interactions that facilitate dimer–dimer formation, the relative abundance of UT/UT and UT/TU at t0 increases in comparison to UU/TT (Figure 3).
Interestingly, the kdim-mon value of the UT dimer is found to be lower in comparison to those for the UU and TT dimers. This measured higher stability is consistent with our SID data, as high-energy SID yields a relatively higher abundance of heterodimer, UT (Figure S8). This suggests that UU/TT can dissociate to monomers more extensively due to the lower stability of the constituent dimers (UU and TT). Yet, more experimental evidence is required to explicitly confirm or reject the UT stability. Molecular modeling simulations can further validate this observation.
SID Reveals a Hidden Pathway in TTR Disassembly Mechanism.
A detailed topological analysis of both models also offered insight into the mechanism of TTR disassembly. In the original model A,31 equal amounts of three topologies of U2T2 are produced during the entire SUE reaction (Figure S1) that should yield an SID spectrum with 1:4:1 ratio of UU, UT, and TT dimers. These ratios are observed only at equilibrium in our SID experiments. To justify the 1:1 ratio of homotetramers (UU and TT) observed at 4 °C, an additional reaction pathway is required, in which UU/TT can be formed from assembly of UU and TT dimers resulting from dissociation of homotetramers (Figure 4, purple box). UU/TT can also be produced along with UT/UT and UT/TU from monomer assembly, but this occurs later in the SUE process (Figure 4, green box). This hidden pathway is only revealed by the SID results. To our knowledge, there is no other biophysical method to offer such detailed information on dynamics and mechanism of protein assembly.
Figure 4.
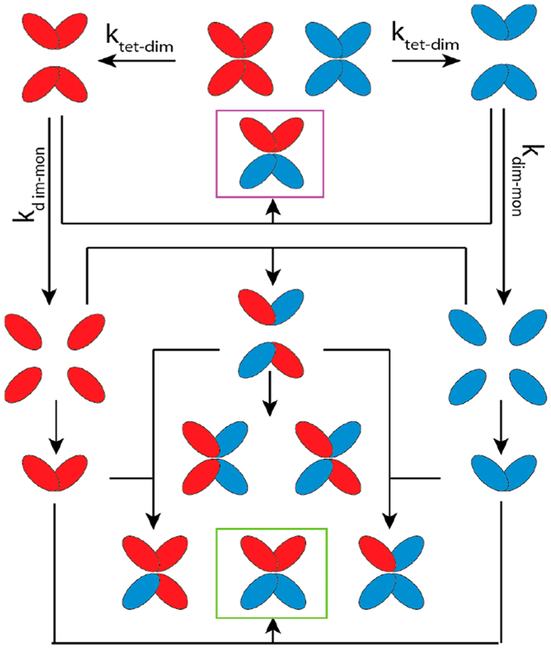
Revised model for TTR disassembly. Purple and green boxes indicate UU/TT tetramers formed from two different routes.
Unlike model A, which perfectly predicts topologies of U2T2 at t0 for different temperatures, model B is unable to provide such correlation (increase in temperature and decrease in UU/TT) (Figure S3); at all temperatures investigated, similar fit curves are produced at t0 (with 100% of UU/TT). Thus, the results obtained from SID provide convincing evidence that the mechanism proposed by Kelly et al. (model A) with revision can accurately predict experimental observations.
CONCLUSION
In conclusion, our SID data validates the model proposed by Kelly et al., in which TTR monomers are produced from dimer dissociation44 rather than direct tetramer dissociation. Furthermore, the model was revised with the addition of parallel and early formation of the heterotetramer with two tags from the dimer assembly. This pathway is disfavored at higher temperatures and was experimentally hidden in previous studies. Topological information provided by SID results was necessary to extract such details regarding TTR dynamics. Taken together, our results are in accord with the mechanism of tafamidis inhibition, where it binds and stabilizes the weaker dimer–dimer interface,13 an interface that we find to dissociate first.
We speculate that the utility of SID serves to dissect the topology of subunits/ligands in complexes formed by protein–protein and protein–ligand interactions. We also foresee this approach to be instrumental in further elucidating reaction mechanisms and intermediates, including those that may be hidden using other techniques. Finally, further studies with higher resolution MS coupled with IM is poised to provide insight into conformational changes that are potentially involved in protein assembly as well as how small molecule/ligand binding and mutation affect the formation of protein complexes.45
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the National Institutes of Health (grants R01GM121751-01A1 and P41GM128577-01).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b05066.
Schematic of two previously proposed models, IM-MS plot of SUE between WT-TTR and FT2-TTR, plots obtained for different ratios fitted with both models, and plot for monomer and dimer changes during SU (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Stefani M; Dobson CM J. Mol. Med 2003, 81 (11), 678–699. [DOI] [PubMed] [Google Scholar]
- (2).Ellis RJ Curr. Opin. Struct. Biol 2001, 11 (1), 114–119. [DOI] [PubMed] [Google Scholar]
- (3).White DA; Buell AK; Knowles TPJ; Welland ME; Dobson CM J. Am. Chem. Soc 2010, 132 (14), 5170–5175. [DOI] [PubMed] [Google Scholar]
- (4).Knowles TPJ; Vendruscolo M; Dobson CM Nat. Rev. Mol. Cell Biol 2014, 15, 384. [DOI] [PubMed] [Google Scholar]
- (5).Jucker M; Walker LC Nature 2013, 501, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Iadanza MG; Jackson MP; Hewitt EW; Ranson NA; Radford SE Nat. Rev. Mol. Cell Biol 2018, 19, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang S Nat. Biotechnol 2003, 21, 1171. [DOI] [PubMed] [Google Scholar]
- (8).King NP; Sheffler W; Sawaya MR; Vollmar BS; Sumida JP; André I; Gonen T; Yeates TO; Baker D Science 2012, 336 (6085), 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Monaco H; Rizzi M; Coda A Science 1995, 268 (5213), 1039–1041. [DOI] [PubMed] [Google Scholar]
- (10).Westermark P; Sletten K; Johansson B; Cornwell GG Proc. Natl. Acad. Sci. U. S. A 1990, 87 (7), 2843–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Connors LH; Lim A; Prokaeva T; Roskens VA; Costello CE Amyloid 2003, 10 (3), 160–184. [DOI] [PubMed] [Google Scholar]
- (12).Petrassi HM; Klabunde T; Sacchettini J; Kelly JW J. Am. Chem. Soc 2000, 122 (10), 2178–2192. [Google Scholar]
- (13).Bulawa CE; Connelly S; DeVit M; Wang L; Weigel C; Fleming JA; Packman J; Powers ET; Wiseman RL; Foss TR; Wilson IA; Kelly JW; Labaudinière R Proc. Natl. Acad. Sci. U. S. A 2012, 109 (24), 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grimster NP; Connelly S; Baranczak A; Dong J; Krasnova LB; Sharpless KB; Powers ET; Wilson IA; Kelly JW J. Am. Chem. Soc 2013, 135 (15), 5656–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jiang X; Smith CS; Petrassi HM; Hammarstrom P; White JT; Sacchettini JC; Kelly JW Biochemistry 2001, 40 (38), 11442–11452. [DOI] [PubMed] [Google Scholar]
- (16).Serag AA; Altenbach C; Gingery M; Hubbell WL; Yeates TO Nat. Struct. Biol 2002, 9, 734. [DOI] [PubMed] [Google Scholar]
- (17).Wiseman RL; Green NS; Kelly JW Biochemistry 2005, 44 (25), 9265–9274. [DOI] [PubMed] [Google Scholar]
- (18).Keetch CA; Bromley EHC; McCammon MG; Wang N; Christodoulou J; Robinson CV J. Biol. Chem 2005, 280 (50), 41667–41674. [DOI] [PubMed] [Google Scholar]
- (19).Quintyn RS; Yan J; Wysocki VH Chem. Biol 2015, 22 (5), 583–592. [DOI] [PubMed] [Google Scholar]
- (20).Ruotolo BT; Benesch JLP; Sandercock AM; Hyung S-J; Robinson CV Nat. Protoc 2008, 3, 1139. [DOI] [PubMed] [Google Scholar]
- (21).Benesch JLP; Ruotolo BT; Simmons DA; Robinson CV Chem. Rev 2007, 107 (8), 3544–3567. [DOI] [PubMed] [Google Scholar]
- (22).Heck AJR Nat. Methods 2008, 5, 927. [DOI] [PubMed] [Google Scholar]
- (23).Bleiholder C; Dupuis NF; Wyttenbach T; Bowers MT Nat. Chem 2011, 3 (2), 172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).May JC; McLean JA Anal. Chem 2015, 87 (3), 1422–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jones CM; Beardsley RL; Galhena AS; Dagan S; Cheng G; Wysocki VH J. Am. Chem. Soc 2006, 128 (47), 15044–15045. [DOI] [PubMed] [Google Scholar]
- (26).Sahasrabuddhe A; Hsia Y; Busch F; Sheffler W; King NP; Baker D; Wysocki VH Proc. Natl. Acad. Sci. U. S. A 2018, 115, 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Jurchen JC; Williams ER J. Am. Chem. Soc 2003, 125 (9), 2817–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhou MW; Dagan S; Wysocki VH Analyst 2013, 138 (5), 1353–1362. [DOI] [PubMed] [Google Scholar]
- (29).Zhou MW; Huang CS; Wysocki VH Anal. Chem 2012, 84 (14), 6016–6023. [DOI] [PubMed] [Google Scholar]
- (30).Marty MT; Baldwin AJ; Marklund EG; Hochberg GKA; Benesch JLP; Robinson CV Anal. Chem 2015, 87 (8), 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Rappley I; Monteiro C; Novais M; Baranczak A; Solis G; Wiseman RL; Helmke S; Maurer MS; Coelho T; Powers ET; Kelly JW Biochemistry 2014, 53 (12), 1993–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Johnson KA Fitting Enzyme Kinetic Data with KinTek Global Kinetic Explorer In Methods in Enzymology; Academic Press: 2009; Vol. 467, Chapter 23, pp 601–626. [DOI] [PubMed] [Google Scholar]
- (33).Loo JA Mass Spectrom. Rev 1997, 16 (1), 1–23. [DOI] [PubMed] [Google Scholar]
- (34).Cong X; Liu Y; Liu W; Liang X; Russell DH; Laganowsky AJ Am. Chem. Soc 2016, 138 (13), 4346–9. [DOI] [PubMed] [Google Scholar]
- (35).Zhang S; Van Pelt CK; Wilson DB Anal. Chem 2003, 75 (13), 3010–3018. [DOI] [PubMed] [Google Scholar]
- (36).Cubrilovic D; Haap W; Barylyuk K; Ruf A; Badertscher M; Gubler M; Tetaz T; Joseph C; Benz J; Zenobi R ACS Chem. Biol 2014, 9 (1), 218–226. [DOI] [PubMed] [Google Scholar]
- (37).Uetrecht C; Watts NR; Stahl SJ; Wingfield PT; Steven AC; Heck AJR Phys. Chem. Chem. Phys 2010, 12 (41), 13368–13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sobott F; Benesch JL; Vierling E; Robinson CV J. Biol. Chem 2002, 277 (41), 38921–9. [DOI] [PubMed] [Google Scholar]
- (39).Schneider F; Hammarström P; Kelly JW Protein Sci. 2001, 10 (8), 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Groenning M; Campos RI; Hirschberg D; Hammarstrom P; Vestergaard B Sci. Rep 2015, 5, 11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Quintas A; Saraiva MJM; Brito RMM J. Biol. Chem 1999, 274 (46), 32943–32949. [DOI] [PubMed] [Google Scholar]
- (42).Yee AW; Moulin M; Breteau N; Haertlein M; Mitchell EP; Cooper JB; Boeri Erba E; Forsyth VT Angew. Chem 2016, 128 (32), 9438–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ross PD; Rekharsky MV Biophys. J 1996, 71 (4), 2144–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Foss TR; Wiseman RL; Kelly JW Biochemistry 2005, 44 (47), 15525–15533. [DOI] [PubMed] [Google Scholar]
- (45).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Clowers BH; Russell DH Anal. Chem 2018, 90 (17), 10472–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.