

Abstract
The multistep process regulating the maturation of membrane proteins in the endoplasmic reticulum (ER) and the secretory pathway is disrupted in many protein misfolding disorders. Mutations in the ion channel CFTR that impair its folding and subsequent localization to the plasma membrane cause cystic fibrosis (CF), an inherited and eventually lethal disease that impairs the function of multiple organs, mostly the lungs. Here, we found that proper maturation of CFTR is dependent on crosstalk between phosphorylation and methylation events in the regulatory insertion (RI) element of the protein. Manipulating these posttranslational modifications (PTMs) prevented the maturation of wild-type CFTR and instead induced its degradation by ER quality control systems. Deletion of Phe508 (ΔF508), the most prevalent mutation in CF, and other mutations in CFTR that impair its trafficking, such as N1303K, also led to quantitative and qualitative PTM changes that prevented the maturation of misfolded CFTR. Further analysis revealed that a wild-type CFTR–like PTM pattern and function was restored in ΔF508 CFTR when cells were cultured at 28°C but only in the presence of the kinase CK2α. Furthermore, the ability to replicate this PTM pattern predicted the efficacy of treatments in restoring ΔF508 CFTR activity. Accordingly, evaluation of patient information revealed that point mutations of several of the modification sites are associated with clinical CF. These findings identify a minimal quantitative and qualitative PTM code for CFTR maturation that distinguishes correctly folded from misfolded CFTR.
Introduction
Cystic fibrosis (CF) is a fatal protein misfolding disease that affects over 80,000 people worldwide and is characterized by progressive lung disease and pancreatic dysfunction (1–3). The most prevalent mutation in CF patients is a deletion of Phe508 (ΔF508) in the cystic fibrosis transmembrane conductance regulator (CFTR), an ion channel (4–6). The deletion impairs proper folding and trafficking of the mutant protein. Misfolded ΔF508 CFTR is energetically unstable and quickly degraded, resulting in nearly complete loss of channel function (7–13). Misfolding of ΔF508 CFTR has previously been suggested to affect posttranslational modification (PTM)-dependent regulation of CFTR channel activity (14, 15) (16). However, PTMs and functional cross-talk between different PTMs do not only regulate protein activity but also influence protein turn-over, conformation and localization, as well as interactions with other proteins (17, 18). For example, PTM changes may play a role during partial restoration of ΔF508 CFTR ion channel function induced by a lower (“permissive”) temperature (28°C) (12), which also markedly remodels ΔF508 CFTR-protein interactions (19). Although CFTR PTMs were among the first identified by mass spectrometry (14, 20–27) and a number of studies show enhanced ubiquitination of ΔF508 CFTR (28–30), it remains unclear how misfolding precisely affects the PTMs of ΔF508 CFTR and other misfolded CFTR mutants in vivo. Consequently, it’s unclear what role PTMs might play during the rescue of misfolded CFTR function at permissive temperature (12) or how PTMs might regulate differential protein interactions that govern ΔF508 CFTR biogenesis and degradation (19). Identification of ΔF508 CFTR PTMs, particularly those that are differentially regulated in comparison to wild-type CFTR, will be key to a better understanding of ΔF508 CFTR channel and cellular processing defects, and may enable the development of new strategies for therapeutic intervention in CF patients.
In this study, we investigated how protein misfolding changes the PTM landscape of ΔF508 CFTR, analyzed the functional relevance of such changes, and evaluated what role those PTMs had during rescue of function at permissive temperature or upon treatment with small molecules. By comprehensively identifying CFTR PTMs in patient-derived bronchial epithelial cell lines, we found that misfolding of ΔF508 CFTR leads to differential site modification and crosstalk between PTMs within previously unknown PTM hotspots. Our data further revealed how such changes affect CFTR maturation and function in primary bronchial epithelial cells and the Fischer rat thyroid (FRT) epithelial cell line and that specific PTMs determine successful maturation and trafficking of not only ΔF508 CFTR but also of other CFTR mutants and can be used as predictors of ΔF508 CFTR function.
Results
Characterization of the wild-type and ΔF508 CFTR PTM landscape
To determine the PTM landscape of wild-type and ΔF508 CFTR in bronchial epithelial cells and identify misfolding-induced differential modifications that might contribute to CF, we performed coimmunoprecipitated CFTR using CoPIT (co-interacting protein identification technology) from 16HBE and CFBE41o- cell lines, derived from CF patients (31). Affinity-purified CFTR and interacting proteins were digested with various endoproteases, and peptides were fragmented by collision- induced dissociation (CID), higher collisional energy dissociation (HCD), or electron transfer dissociation (ETD) to confidently determine localization of PTMs (32). To overcome any inherent search bias in the identification of PTMs and enable a comprehensive identification of the CFTR PTM landscape, the dataset (consisting of 90 experiments with over 60,000 CFTR spectra) was searched using two different approaches. In the first approach, the most common PTMs—ubiquitination, phosphorylation, acetylation, and methylation—were included as differential masses in a standard peptide search (33). In the second approach, all mass spectra that did not yield a peptide identification were searched again with ProLuCID Blind-PTM (34), a wide mass window search strategy that detects and reports systematic mass shifts of peptide fragment ions from the theoretical fragment ion masses of the corresponding unmodified peptide (35) and, therefore, allows the identification of modified peptides without prior knowledge of the modifying event(s). The mass differences reported by Blind-PTM were also used to confirm or correct PTM assignments reported in the initial forward search approach (fig. S1).
Together, the experiments yielded more than 80 % sequence coverage of the CFTR protein and identified 37 different PTM sites in either wild-type or ΔF508 CFTR, which included 20 phosphorylation, 6 methylation and 11 ubiquitination sites. Although many other modifications of CFTR were also detected, most of these were either exceedingly rare (<0.03% of total CFTR spectra) and therefore not further considered here, or they reflect chemical modifications that commonly occur during sample handling, such as carbamidomethylation or loss of ammonia (fig. S1D). Of the 37 identified PTMs, > 54 % had not been reported previously (table S1). In particular, almost none of the methylation and ubiquitination sites have been reported. Most methylation sites were present only in wild-type CFTR, whereas most ubiquitination sites were detectable only in ΔF508 CFTR, likely reflecting increased targeting of ΔF508 CFTR for proteasomal and lysosomal degradation (28). Mapping the PTM sites to the CFTR sequence revealed several “PTM hotspots” of roughly 50 amino acids length, in which multiple PTMs cluster together (Fig. 1A). The first hotspot from Lys420 to Lys464 covered part of the regulatory insertion (RI) element, an approximately 39 amino acid stretch of the nucleotide binding domain 1 (NBD1) that is intrinsically disordered and likely involved in folding of NBD1 (36–39). Several other hotspots (Lys564 to Ser670, Lys698 to Ser737, and Arg751 to Ser839) overlapped either partially or entirely with the intrinsically disordered, regulatory (R) region. Wild-type CFTR hotspots were phosphorylated and contained methylation sites at the N- or C-terminus of each hotspot. However, the same sites in ΔF508 CFTR hotspots were preferentially ubiquitinated instead of methylated and the ΔF508 CFTR hotspots contained altered phosphorylation amounts as well as additional phosphorylation and ubiquitination sites. Furthermore, hotspots overlapped with additional sequence motifs. For example, the hotspot Lys564 to Lys584 overlaps with the previously described di-acidic CFTR ER exit motif (Tyr563 to Asp567, or YKDAD) (40). This di-acidic ER exit motif was differentially modified in wild-type and ΔF508 CFTR. Whereas the wild-type CFTR ER exit motif was methylated at Lys564, the same lysine residue (Lys564) in the ΔF508 CFTR ER exit motif was mainly ubiquitinated, along with an additional, ubiquitinated lysine downstream of the ΔF508 CFTR ER exit motif (Lys584) (Fig. 1B). A similar pattern of replacement of methylation with ubiquitination was observed for the RI element of ΔF508 CFTR. ΔF508 CFTR was ubiquitinated at Lys420 in the RI element and at Lys442, just C-terminal of the RI element. Wild-type CFTR was only methylated at Lys442. Methylation of wild-type CFTR at Lys442 correlated with increased phosphorylation at residues Thr421, Ser422 and Ser427 in the RI element (Fig. 1C). This pattern of mutually exclusive methylation (wild-type) and ubiquitylation (ΔF508) points to a more elaborate PTM code in which phosphorylation sites are preceded or followed by lysines that can be differentially modified to regulate specific functions or steps in CFTR biogenesis.
Figure 1. Identification of CFTR post-translational modification hotspots.

A. PTMs identified for wild-type CFTR (below) or ΔF508 CFTR (above) isolated from human bronchial epithelial cell lines 16HBE41o- and CFBE41o- were mapped onto the CFTR amino acid sequence (center). Phosphorylation is indicated in green, methylation in black, ubiquitination in lilac/urple, and O-glycosylation in orange. PTM hotspots are indicated in yellow, transmembrane domains are shown in dark blue, and the regulatory (R)-region and nucleotide binding domains (NBDs) are labeled. B. PTM spectrum of the tryptic peptide (boxed inset) covering part of the ER exit motif (blue). The abundance of mass shifts detected by Blind-PTM is shown, and major modifications are indicated. The inset further shows N-terminal as well as C-terminal overlapping peptides with their respective modifications shown in red. ΔF508 CFTR PTMs are above the sequence, wild-type PTMs below. C. PTM spectrum of peptide covering the N-terminal NBD1 sequence (RI element) is shown. N- or C-terminal overlapping peptides containing additional modifications are indicated in the inset, whereby ΔF508 CFTR PTMs are above and wild-type PTMs are below the sequence. Abbreviations: Ox, oxidation; Me, mono-methylation; di-Methyl, di-methylation; ac, acetylation; +Lys, mis-cleavage upstream of lysine; Ub, ubiquitination. Data represent n=14 independent biological replicates.
Mis-regulation of phosphorylation and methylation events in ΔF508 CFTR
To quantify differences in abundance of specific PTMs between wild-type and ΔF508 CFTR, we combined CoPIT with stable isotope labeling (SIL) in a novel “balanced SIL approach” (fig. S2). In this approach, the eluate of ΔF508 CFTR IPs from “heavy”-labeled CFBE41o- cells was mixed in a 1:1 ratio with the eluate of wild-type CFTR IPs from non-isotope labeled HBE41o- cells. Relative quantification of modified CFTR peptides was carried out after normalization to the average ratio of all non-modified CFTR peptides identified for the wild-type and ΔF508 CFTR protein (Fig. 2A). If a peptide sequence is quantitatively modified by a PTM, the corresponding non-modified peptide would deviate significantly from a 1:1 ratio in this approach, thus allowing the detection of modification changes regardless of whether the PTM modified peptide was identified or not. For proof of principle, ΔF508 CFTR data were searched against a database containing only the wild-type CFTR sequence. In this case, the ratio for the peptide containing the F508 deletion drastically deviated from 1:1 with log2RΔF508:wt = 5.28 ± 0.11, indicating exclusive presence in ΔF508 CFTR expressing cells.
Figure 2. Mis-regulation of domain-specific phosphorylation in CF.
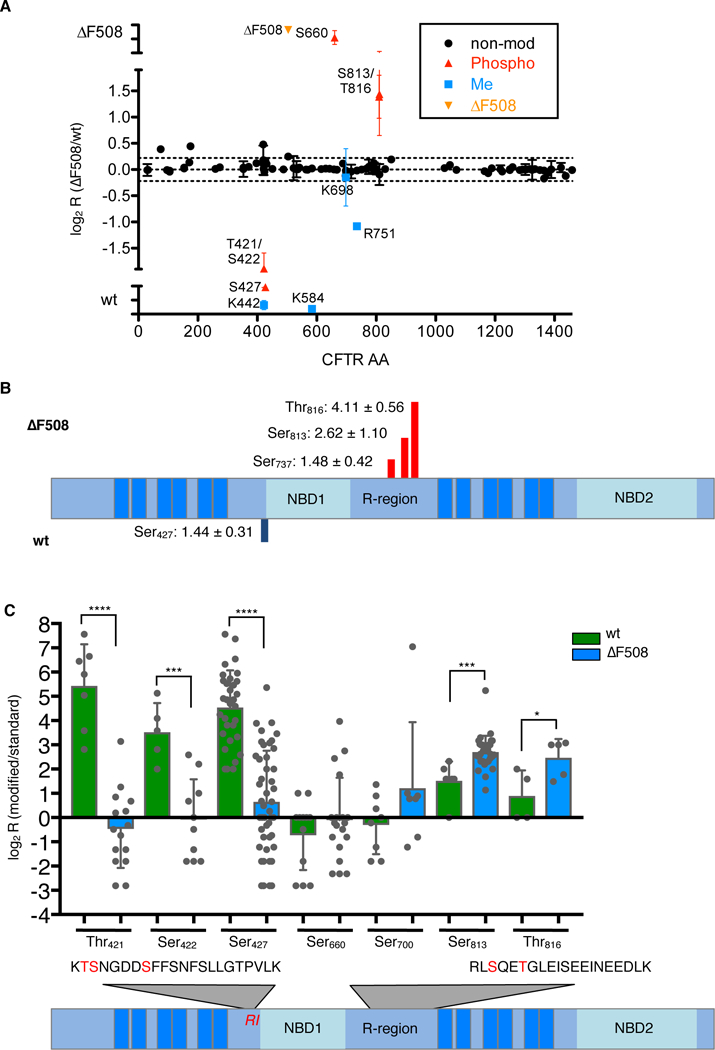
A. SILAC ratios and differential PTMs of peptides across the CFTR sequence from 16HBE41o- and CFBE41o- cells. Data are representative of n=4 experiments. B. TMT-based quantification of phosphorylation of sites in ΔF508 CFTR relative to wild-type (wt) in 16HBE41o- and CFBE41o- cells. Values are log2(ratio). non-mod, not modified; Phospho, phosphorylated; Me, methylated. Data are representative of n=7 experiments. C. Differential phosphorylation of Thr421, Ser422, Ser427 (RI element) and Ser660, Ser700, Ser813 and Thr816 (R-region) was confirmed by simultaneous detection of spiked-in synthetic heavy (13C615N2 Lys; 13C615N4 Arg)-labeled peptides in 16HBE41o- and CFBE41o- cells. Data are mean ± S.D. from n=5 independent biological replicates. *P < 0.0332, **P < 0.0021, ***P < 0.0002, and ****P < 0.0001 by unpaired, two-tailed t-tests, analyzed and corrected by the Bonferroni-Sidak method.
Balanced stable isotope labeling confirmed reduced methylation of ΔF508 CFTR at Lys442 and Lys584 as well as a two-fold decrease in Arg751 methylation. The approach further revealed that phosphorylation of the RI element at Thr421, Ser422 and Ser427 was strongly decreased in ΔF508 CFTR. The change in ratio of the corresponding non-modified peptide indicated that while more than 40% of wild-type CFTR molecules are modified at Thr421 to Ser427, ΔF508 CFTR is preferentially non-modified at this first PTM hotspot. In contrast, the ΔF508 CFTR R-region showed strongly increased phosphorylation at Ser660, Ser813 and Thr816. Subsequent relative quantification experiments using TMT-labeling confirmed decreased phosphorylation of ΔF508 CFTR in the RI element and increased phosphorylation of the ΔF508 R-domain (Fig. 2B). Additionally, we observed increased phosphorylation of ΔF508 CFTR at the inhibitory site Ser737. Notably, it’s been shown that phosphorylation of Ser737 by either AMPK (adenosine monophosphate-activated protein kinase) or LMTK2 (lemur tyrosine kinase 2) increases endocytosis and, thus, removal of CFTR from the plasma membrane, which leads to an overall decreased CFTR chloride conductance at the plasma membrane (22, 23, 41–43).
To precisely determine the amount of phosphorylation at each site in peptides containing multiple phosphorylation sites, we used synthetic, stable isotope-labeled peptides phosphorylated at either one of the sites, which were spiked into the sample prior to mass spectrometric data acquisition. Quantification with these synthetic peptides showed that phosphorylation of the wild-type RI element is at least 10-fold higher at all three phosphorylated sites (Thr421, Ser422 and Ser427) than for ΔF508 CFTR (Fig. 2C). Furthermore, no doubly or triply phosphorylated variants were identified, indicating that the RI element phosphorylation events are separate events that occur either at different points in time or in different cellular compartments. Additionally, we confirmed a 2- to 3-fold increase in ΔF508 CFTR phosphorylation at both Ser813 and Thr816 over wild-type CFTR.
Phosphorylation in the RI element by the kinase CK2α
Exploration of a previously published CFTR interactome (19) provided evidence for ~25 protein kinases and kinase proteoforms that interact with wild-type or ΔF508 CFTR and which may mediate the differential phosphorylations. Many of those previously identified kinases belong to the CAMK and CMGC serine/threonine protein kinase family (fig. S3), including casein kinase 2α (CK2α), a catalytic subunit (encoded by CSNK2A1) of the constitutively active serine/threonine-protein kinase complex CK2. CK2 has been shown to phosphorylate CFTR in vitro and influence channel function of wild-type, but not ΔF508, CFTR; however, the importance of CK2 phosphorylation and the actual phosphorylation sites, as well as which catalytic isoform may phosphorylate CFTR, are somewhat unclear (44, 45). Because the CK2α consensus recognition motif (T/SXXE/D) is present in the differentially phosphorylated RI element peptide TSNGDDSFFSNFSLLGTPVLK, which contains the phosphorylated sites Thr421, Ser422 and Ser427, we further explored the involvement of CK2α in mediating differential phosphorylation of wild-type and ΔF508 CFTR RI elements (46). Treatment of cultured HBE41o- cells with CX-4945 (also known as silmitasertib), a competitive inhibitor of CK2α, markedly reduced the phosphorylation of wild-type CFTR at all three sites (Thr421, Ser422 and Ser427) to an extent similar to that of ΔF508 CFTR (Fig. 3A). Additionally, R-domain phosphorylation of wild-type CFTR was slightly reduced upon treatment of cells with CX-4945, except for phosphorylation at Ser700. This experiment indicated that CK2α mediates phosphorylation of Thr421, Ser422 and Ser427 in the RI element, and that the observed increase in R-domain phosphorylation of ΔF508 CFTR is likely not a direct consequence of its reduced RI element phosphorylation.
Figure 3. CSNK2A1-mediated phosphorylation of Thr421 to Ser427 is essential for CFTR maturation.


A. CX-4945 mediated inhibition of CK2α prevents phosphorylation of wild-type CFTR at Thr421, Ser422 and Ser427 as quantified with spiked-in synthetic, heavy isotope-labeled peptides. Data are means ± S.D. of n=3 experiments; statistical analysis noted below. B. Experimental outline for AHA labeling and enrichment of newly synthesized CFTR by click chemistry. C. Western blot of input lysate for CFTR-IP before click reaction. D. Western blot showing that wild-type CFTR maturation is dependent on CK2α phosphorylation in the RI element. Blots are representative of n=3 experiments. E. Quantification of newly synthesized immature (band B) and mature (band C) CFTR relative to time point 0 (h, hours). F. Western blot of mature CFTR (band C) in FRT cells expressing various CFTR mutants. G. Phosphorylation at Thr421, Ser422 and Ser427 in the various CFTR mutants described in (F) was quantified with spiked-in synthetic heavy isotope-labeled peptides, relative to the amount of mature CFTR present. Data are mean ± S.D. of ≥ n=4 independent measurements per site; statistical analysis noted below. (H to K) AHA pulse-chase assays in primary human bronchial epithelial (NHBE) cells (H) and FRT cells expressing G551D or wild-type CFTR (K) treated with CX-4945 or DMSO. Subsequent quantification of newly synthesized immature CFTR (band B) and mature CFTR (band C) at the time points indicated relative to time 0 (I and K). Top panels show input and loading control. SA-HRP, streptavidin-conjugated horse radish peroxidase. Time of AHA labeling before CFTR immunoprecipitation is indicated. Blots are representative, and the data are means ± S.D. of n=3 independent biological replicates. *P < 0.0332, **P < 0.0021, ***P < 0.0002, and ****P < 0.0001 by unpaired, two-tailed t- tests with Bonferroni-Sidak correction (A) or one-Way ANOVA with Tukey post-test (I and K).
Crucial influence of RI element phosphorylation on wild-type CFTR maturation
To reveal the functional significance of the difference in phosphorylation abundance between wild-type and ΔF508 CFTR at sites Thr421, Ser422 and Ser427, we performed pulse-chase experiments either in the presence of CX-4945 or of control vehicle (DMSO). Treatment of wild-type CFTR expressing HBE41o- cells with CX-4945 and simultaneous pulse-chase labeling with the methionine homolog azido-homoalanine (AHA) (47–50) revealed that phosphorylation by CK2α is essential for full maturation of wild-type CFTR (Fig. 3, B to E). In the absence of CX-4945, newly synthesized, AHA-labeled wild-type CFTR molecules were first detected 30 min after the AHA pulse. These represented the nascent and coreglycosylated CFTR forms (bands A and B, hereafter A/B, in the blots) that are found in the endoplasmic reticulum (ER) (11, 51, 52). Fully mature CFTR (band C in the blots), which has been complex-glycosylated in the Golgi apparatus (53), was observed approximately 120 min after the AHA pulse. CX-4945 treatment substantially delayed appearance of nascent and core-glycosylated CFTR forms and reduced or prevented appearance of mature, fully glycosylated CFTR, indicating that upon loss of RI phosphorylation wild-type CFTR is no longer able to fully mature. Therefore, these phosphorylation events appear to be essential for correct folding and trafficking of wild-type CFTR in the early secretory pathway. In agreement, CK2α is well known to be localized to the cytoplasmic side of the ER and Golgi apparatus (54).
To test whether decreased phosphorylation in the RI element correlates with known impairments in CFTR trafficking, we quantified Thr421, Ser422 and Ser427 phosphorylation in the different CFTR mutants G551D, R117H, N1303K and AF508 (55–57). Whereas G551D and R117H mutations affect CFTR ion channel function but do not affect biogenesis as evidenced by the presence of Band C, which represents fully glycosylated CFTR (Fig. 3F), N1303K and ΔF508 CFTR both result in misfolding and impaired trafficking (57). Quantification of the phosphorylation of synthetic stable-isotope labeled peptides showed that G551D and R117H CFTR mutants expressed in FRT cells were phosphorylated at Thr421, Ser422’ and Ser427 in FRT cells as is seen in wild-type CFTR. In contrast, phosphorylation at Thr421 to Ser427 was either absent or drastically reduced for N1303K and ΔF508 CFTR (Fig. 3G). Thus, CFTR mutants that mature normally and are trafficked correctly to the plasma membrane, also display abundant phosphorylation at Thr421 to Ser427, whereas misfolded CFTR mutants with impaired trafficking are either less or not at all phosphorylated at Thr421 to Ser427. Furthermore, the amount of observed phosphorylation at Thr421 to Ser427 may be correlated to the amount of fully glycosylated CFTR (band C in the blot) that is present.
To validate that phosphorylation by CK2α is required for CFTR maturation in primary human cells, and in tissue other than lung, we performed AHA pulse chase experiments in primary human bronchial epithelial (NHBE) cells isolated from healthy individuals and in thyroid-derived FRT cells that express either wild-type or G551D CFTR. Complex glycosylation of wild-type and G551D CFTR was largely impaired or abrogated in these experiments in the presence of CX-4945 (Fig. 3, H to K), confirming that maturation of CFTR is dependent on CK2α phosphorylation during early biogenesis in a native system as well as in thyroid derived cell lines. Because inhibiting the CK2α–mediated phosphorylation of Thr421-Ser427 prevented full CFTR maturation in the Golgi, and delayed core-glycosylation of CFTR, these results argue for a phosphorylation of the RI element by CK2α in the ER or in the early secretory pathway.
To identify the mechanisms that are influenced by CK2α phosphorylation of the RI element during early biogenesis, we used CoPIT to determine the wild-type CFTR interactome upon CX-4945 treatment (data file S1), and compared it with the previously published interactomes of wild-type and ΔF508 CFTR (19). Results showed that CK2α inhibition induced protein-protein interactions that were characteristic for the ΔF508 CFTR interactome (Fig. 4). In particular, treatment of wild-type CFTR with CX-4945 induced 61 out of 76 previously observed ΔF508 CFTR-specific protein-protein interactions involved in ER quality control, retrograde trafficking, as well as selective degradation of misfolded proteins and degradation by the ubiquitin-proteasome system (19). Furthermore, methylation of wild-type CFTR at Lys442 was no longer detected upon treatment with CX-4945. The experiments showed that wild-type CFTR fails ER quality control upon treatment with CX-4945, indicating that crosstalk between phosphorylation and methylation is used to distinguish between correctly folded and misfolded CFTR proteins.
Figure 4. CK2α inhibition induces wild-type CFTR degradation.
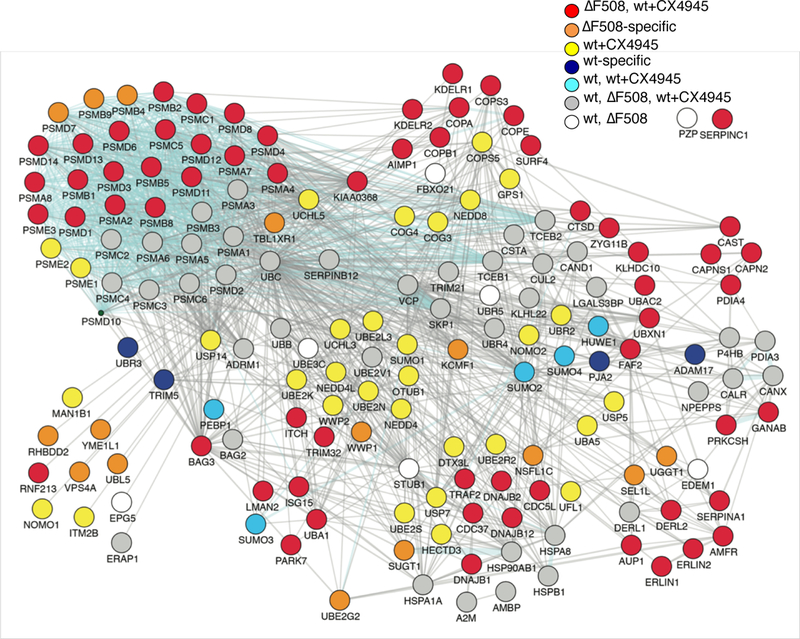
Degradation network identified for wild-type CFTR, wild-type CFTR treated with CX-4945, or ΔF508 CFTR in 16HBE41o- and CFBE41o- cells shows recruitment of ΔF508- specific interactions in wild-type CFTR expressing cells treated with CX-4945 (red nodes). Data represent independent biological replicates (wt n=8, ΔF508 n=8, wt+CX-4945 n=3).
Rescue of function by phosphorylation of the RI element in ΔF508 CFTR
A temperature of 24 – 30°C (a so-called “permissive temperature”) partially restores ion channel function to ΔF508 CFTR in cells (12). We therefore hypothesized this might also change the crosstalk between phosphorylation, methylation and ubiquitination within the RI-element. To test if such PTM changes occur and contribute to the rescue of ΔF508 CFTR function, a balanced SIL experiment was carried out comparing the PTM pattern of ΔF508 CFTR in cells cultured for 24 hours at 30°C with that of ΔF508 CFTR at 37°C. The permissive temperature (30°C) at least partially restored wild-type-like phosphorylation in the ΔF508 CFTR RI element, with relative phosphorylation at Ser422 and Ser427 increasing more than 1.8-fold upon the temperature shift. This increase in phosphorylation was accompanied by replacement of ubiquitination with methylation at Lys442. Furthermore, ubiquitination at Lys420 was no longer detected at the permissive temperature (Fig. 5A).
Figure 5. Rescue of ΔF508 CFTR function at permissive temperature depends on CSNK2A1 phosphorylation of Thr421 to Ser427.

A. Quantification of phosphorylated sites Ser422 and Ser427, methylation site Lys442 and ubiquitination site Lys420 at 37°C (red bars) or at permissive temperature of 30°C (blue bars). B. Quantification of ΔF508 CFTR phosphorylation at Thr421, Ser422 and Ser427 by synthetic heavy-labeled peptides shows reduced phosphorylation upon shRNA- mediated knockdown of CSNK2A1. C. Western blot showing that shRNA-mediated knockdown of CSNK2A1 impairs ΔF508 CFTR maturation at permissive temperature. Detection of b-actin was used as loading control. D. Representative traces of forskolin (F) and genistein (G) stimulated ΔF508 CFTR short circuit currents (/sc) showing that knockdown of CSNK2A1 (right) or treatment with 10 μM CX-4945 (left) prevent temperature shift (28°C) induced rescue of ΔF508 CFTR channel activity. Specificity of the current is established by treatment with CFTR inhibitor 172 (I). E. Quantification of Δ/sc currents as log2 fold change relative to control cells. All data are representative or mean ± S.D. of n=3 independent biological replicates. *P < 0.0332, **P < 0.0021, ***P < 0.0002, and ****P < 0.0001 by one-Way ANOVA with Bonferroni post-test.
We then stably expressed a CK2α (CSNK2A1 )-specific shRNA in CFBEo41- cells (CFBEo41-CSNK2A1shRNA cells), which reduced CK2α protein abundance to < 40 % of that in controls, as quantified from Western blotting (fig. S4). Knockdown of CK2α almost completely suppressed the phosphorylation of ΔF508 CFTR at Thr421, Ser422 and Ser427, suggesting that CK2α mediates the phosphorylation of ΔF508 CFTR at these residues (Fig. 5B). Loss of CK2α further affected ΔF508 CFTR phosphorylation in the R-region, as it increased Ser660 phosphorylation and decreased phosphorylation at Ser700. This differential phosphorylation might be specific to ΔF508 CFTR, because phosphorylation of wild-type CFTR at Ser660 did not increase, but instead decreased upon CX-4945 treatment. These results showed that permissive temperature at least partially restored CK2α mediated phosphorylation of the RI element and méthylation at Lys442, and that phosphorylation and ubiquitination of the RI element are functionally coupled to phosphorylation of the N-terminal part of the R-region. In contrast, the extent of phosphorylation in the C-terminal part of the R-region (Ser813 and Thr816) remained almost unaltered upon CK2α knockdown.
To evaluate the importance of CK2α mediated phosphorylation for functional rescue of ΔF508 CFTR at permissive temperature, CFBE41o- cells either stably expressing the CSKN2A1 shRNA or a scrambled control shRNA were cultured at permissive temperature of 28 – 30°C. Whereas the permissive temperature induced ΔF508 CFTR maturation in control shRNA expressing cells as expected (Fig. 5C, left panel), loss of CK2α prevented the low temperature-induced maturation of ΔF508 CFTR (Fig. 5C, right panel). To further understand the physiological consequences of CK2α mediated phosphorylation of the RI element, we also determined chloride channel activity of ΔF508 CFTR at a slightly lower permissive temperature (28°C) and upon inhibition of CK2α activity. As expected, the permissive temperature promoted an increase in ΔF508 CFTR chloride channel activity in CFBE41o- cells treated with the vehicle (DMSO) alone (Fig. 5D, left panel). In contrast, CX-4945 treatment prevented the increase in ΔF508 CFTR- specific chloride current at a permissive temperature (Fig. 5, D and E). Moreover, shRNA-mediated knockdown of CK2α also prevented an increase of ΔF508 CFTR chloride channel activity at permissive temperature, and no significant difference in ΔF508 CFTR chloride channel activity was observed between CFBEo41- CSNK2A1shRNA cells at 37°C and at 28°C (Fig. 5D, right panel). These results show that CK2α-mediated phosphorylation of the RI element is essential for rescue of ΔF508 CFTR maturation and function at permissive temperature.
Direct correlation between CFTR PTM pattern and function
To test if increased phosphorylation at Thr421, Ser422 and Ser427 in the RI element and methylation of Lys442 in the first PTM hotspot is a universal feature of successful CFTR maturation as indicated by the results of the pulse chase experiments and the G551D and R117H CFTR mutants, and to find out if other PTM changes further contribute to functional rescue of ΔF508 CFTR, we analyzed the CFTR PTM landscape at permissive temperature, upon treatment with different compounds (VX-809, SAHA, TSA, Corr-4a, DMSO, CX-494), or upon knockdown of CK2α (Fig. 6A). The results revealed that the relative PTM abundance or site occupancy correlated with the average relative increase or decrease in ΔF508 CFTR channel activity at permissive temperature or upon treatment with the different compounds (58–62) (Fig. 6A). Permissive temperature or treatment with compounds that promote ΔF508 CFTR function generally increased phosphorylation of RI phosphorylation sites Thr421, Ser422, or Ser427 and decreased ubiquitination or replaced ubiquitination with methylation or acetylation of the two lysines (Lys420 and Lys442) that flank Thr421/Ser422/Ser427. Permissive temperature also replaced ubiquitination of ΔF508 CFTR at the ER exit motif lysine (Lys564) with methylation as observed in wild-type CFTR. Unexpectedly though, ubiquitination of ΔF508 CFTR was not reduced uniformly in all conditions that increased channel activity, and some compounds such as VX-809 and SAHA even led to ubiquitylation of ΔF508 CFTR at additional sites, located mainly in the NBD2. For example, treatment with 5 μM VX-809 as well as knockdown of CK2α both induced ubiquitination of Lys1284, which in wild-type CFTR can be cross-linked with Cys276 and forms an inter-domain contact between NBD2 and cytoplasmic loop 2 (CL2) (63, 64). Ubiquitylation of Lys1284 would most likely abolish this interdomain contact. Such individual differences in the PTM landscape may help to explain the limited success of some compounds to induce a complete rescue ΔF508 CFTR function.
Figure 6. A minimal PTM code for CFTR maturation.
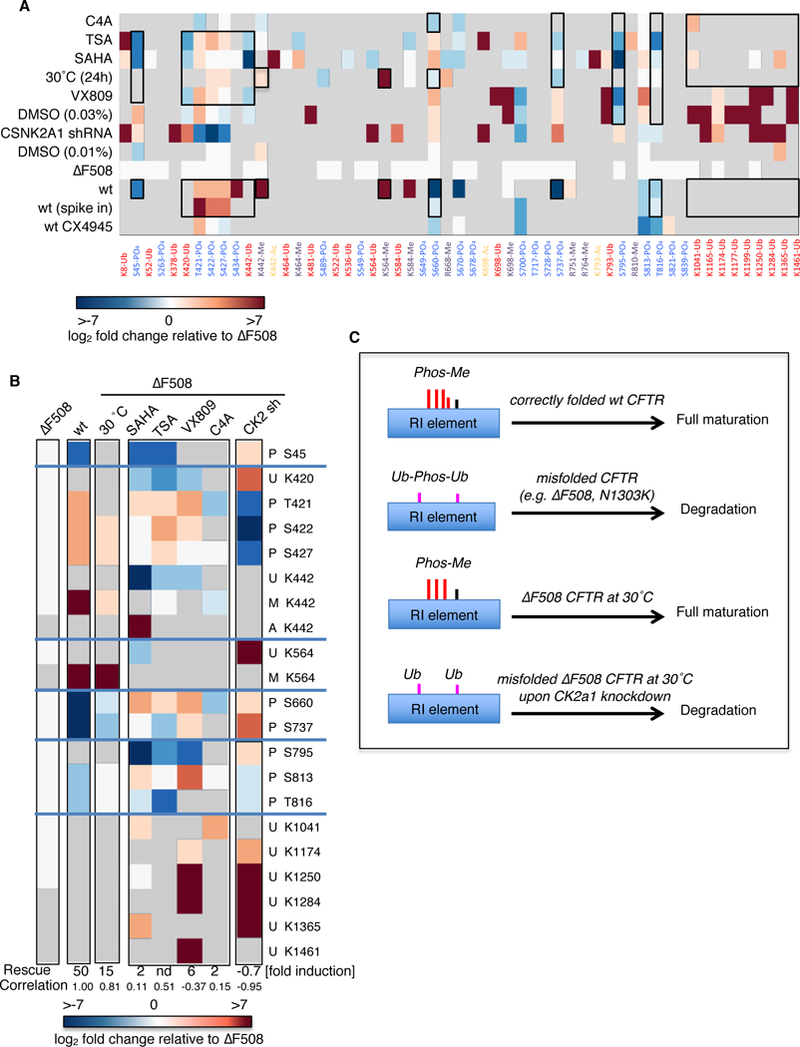
A. Heatmap of CFTR PTMs at permissive temperature or upon treatment with different CFTR modifier drugs. B. Heatmap of signature PTMs organized by CFTR domains. Modified residues are indicated either below (A) or next to the heatmap (B). Abbreviations: P: Phosphorylation; U: Ubiquitination; M; Methylation; A: Acetylation. Grey color in the heatmap represents values not measured (NaN) whereas all additional values are normalized to ΔF508 CFTR whenever possible. The increase in ΔF508 CFTR anion channel activity is indicated for each of the different conditions and the Pearson correlation coefficient between the different PTM signatures and the wild-type CFTR PTM signature is shown. Data represent ≥2 independent biological replicates per condition. nd, not determined. C. Schematic representation of the proposed PTM code for CFTR maturation. Correctly folded CFTR displays a methylation-phosphorylation pattern in the RI element PTM hotspot that allows maturation of the protein and functions as a combinatorial PTM code. Upon misfolding this code is quantitatively and qualitatively altered, which leads to degradation of the CFTR protein. At a permissive temperature, the wild-type code is re-instated and enables maturation of ΔF508 CFTR. However, if phosphorylation in the RI element is inhibited, maturation at permissive temperature is blocked and the code resembles that of ΔF508 CFTR at 37°C.
Closer inspection of the data revealed that rescue efficiency correlated with how well the wild-type CFTR PTM pattern at 17 sites can be mimicked by ΔF508 CFTR, as the change in PTM abundances at these sites recapitulated the fold change observed in channel function increase (Fig. 6B). Indeed, these select PTMs may be used as a molecular signature. Features of this PTM signature include increased phosphorylation of Thr421 to Ser427 in the RI element with concomitant reduced or absent ubiquitination at flanking lysines 420 and 442 and increased methylation at Lys442, reduced R-region phosphorylation at residues Ser795, as well as reduced inhibitory phosphorylation at Ser737, and decreased ubiquitylation of lysines in the NBD2 (Fig. 6B). When correlating the relative change of ΔF508 CFTR PTM abundances to the wild-type CFTR PTM signature, permissive temperature re-established this PTM signature most effectively (correlation coefficient rwt,ts = 0.81). If ΔF508 CFTR channel function rescue was weak, the correlation of PTM signatures was low (rwt,SAHA = 0.11 and rwt,C4A = 0.15) and in the absence of channel function upon CK2α knockdown the correlation was even strongly negative (rwt,CSNK2A1sh = –0.95). Changes in PTM abundance or occupancy at the signature sites upon treatment with VX809, which promotes ΔF508 CFTR channel function to a moderate degree, also did not correlate with the PTM signature of wild-type CFTR (rwt,VX809 = –0.37). Only phosphorylation at the first PTM hotspot (Thr421/Ser422) was re-established upon VX809 treatment and led to concomitant reduced ubiquitination of the flanking lysines Lys420 and Lys442.
These findings emphasize that enhanced phosphorylation of the RI element sites with concomitant reduced ubiquitination is necessary and a minimal prerequisite for promoting ΔF508 trafficking and channel function.
Clinical relevance of PTM-site point mutations
Querying the CFTR mutation database (www.genet.sickkids.on.ca) and CFTR 2.0 database (57) revealed that point mutations that prevent phosphorylation (T421A, S549F, S549I, S549N or S549R) or lysine methylation (K464N) lead to severe clinical phenotypes associated with CF and congenital bilateral absence of the vas deferens (CBAVD) (Table 1). Furthermore, mutation of the methylation site Arg751 (to R751P, R751C or R751L) causes lung disease and is included in the CF carrier testing panel. These mutational analyses argue that the previously unknown phosphorylation and methylation sites are crucial for CFTR function in vivo and that loss of any of these sites is clinically relevant.
Table 1. Clinical significance of naturally occurring point mutants of CFTR PTM sites.
Given are the residue, observed point mutation, and clinical significance of point mutations as reported in the CFTR mutation databases (30).
| Modified peptide sequence | Site | A-score (−10×log(P)) |
Mutation in CF patients |
Clinical significance |
|---|---|---|---|---|
| T*SNGDDSLFFSNFSLLGTPVLK | T421 | 19.21 | T421A | CBAVD |
| GQLLAVAGSTGAGK(me)T | K464 | K464N | CF, severe phenotype at early age with pancreatic insufficiency, chronic cough and bronchial infection, 3659delC mutation on the other chromosome (expected to lead to pancreatic insufficiency) | |
| FAEK(ub)DNIVLGEGGITLSGGQR | K536 | K536E | parent of a child with a positive newborn screening test | |
| DNIVLGEGGITLS*GGQR | S549 | 64.79 | S549N, S549I, S549F, S549R | CF, severe clinical phenotype |
| AVYK(ub)DADLYLLDSPFGYLDVLTEK | K564 | K564E | CBAVD | |
| NS*ILTETLHR | S660 | 56.02 | S660T | asymptomatic |
| NSILTETHR(me) | R668 | R668C | does not cause CF | |
| LS*LVPDSEQGEAILPR | S737 | 94.12 | S737F | elevated sweat chloride |
| LSLVPDSEQGEAILPR(me)I | R751 | R751P, R751C, R751L | lung disease, carrier testing for R751C | |
| VSLAPQANLTELDIYSR(me)R | R810 | R810G | CBAVD (ΔF508 on other allele) | |
| LS*QETGLEISEEINEEDLK | S813 | 69.89 | S813P | very mild CF |
| AYFLQTSQQLK(ub)QLESEGR | K1041 | K1081R | reduction of band C (McClure 2014) | |
| TGSGK(ub)STLLSAFLR | K1250 | K1250A mutation dramatically prolonged burst duration (abolishes ATP hydrolysis) | ||
Discussion
Phosphorylation has previously been thought to mainly regulate CFTR channel activity through a rather complex interplay between channel activating and inhibitory sites. Here, we show that phosphorylation of CFTR in the RI element serves a different role: it is crucial for proper CFTR biogenesis. Three different sites in the RI element were found phosphorylated (Thr421, Ser422 and Ser427) by CK2α, a kinase that is localized to the cytoplasmic side of the ER and Golgi apparatus (54). It is likely that phosphorylation of all three sites is required for successful maturation, because wild-type CFTR and the G551D and R117H mutants that are correctly trafficked to the plasma membrane were phosphorylated at all three sites. Data from the CF mutation database, www.genet.sickkids.on.ca reaffirm the importance of phosphorylation sites in the RI element for full CFTR function in vivo, as a single point mutation of T421 to A421 has been identified in a CF patient with CBAVD.
The amount of phosphorylation at a given site in the RI element further correlated with how much fully mature CFTR (band C) was present, as shown in the different CFTR mutants G551D, R117H, N1303K and ΔF508 CFTR. This suggested, that the amount of phosphorylation directly determines the amount of fully glycosylated CFTR, for example by determining the number of CFTR molecules that mature. As a possible mechanism, we suggest that marginally and non-phosphorylated CFTR is recognized by the cellular degradation machinery, because reduced amounts or absence of the phosphorylation lead to ubiquitination of Lys420 and Lys442, which might mark CFTR for degradation, and because inhibition of wild-type RI phosphorylation by CX-4945 caused wild-type CFTR to interact with proteins that either recognize misfolded proteins, are involved in their retrograde trafficking, or that promote CFTR degradation by the ubiquitin-proteasomal system. Such a mechanism would also explain why deletion of the entire RI element including the ubiquitination site Lys420 leads to increased maturation of ΔF508 and wild-type CFTR in BHK cells (65), as ΔRI CFTR might escape recognition by the degradation machinery during early biogenesis.
Furthermore, we discovered that changes in phosphorylation sites and amounts upon misfolding of ΔF508 CFTR are accompanied by replacement of adjacent methylation with ubiquitination sites. As methylation-phosphorylation hotspots overlapped with protein sequence motifs, such as the ER exit motif, phosphorylation accompanied by methylation could provide the cell with a specific signal to regulate different steps during CFTR biogenesis and may function as a “proceed” or “stop/degrade” signal. For example, ubiquitination instead of methylation of Lys564 and Lys584 surrounding the CFTR ER exit motif (AA 566 to 568) might signal that the protein is misfolded, preventing its association with Sec24 for cargo loading into COPII vesicles and thus its exit from the ER (40). Indeed, a K564E point mutation, which prevents methylation and changes the polarity of the site, has been associated with CBAVD in CF patients (Cystic Fibrosis Mutation Database, www.genet.sickkids.on.ca). Similarly, misfolded CFTR mutants with impaired trafficking showed not only reduced phosphorylation at Thr421-Ser427 in the RI element, but also replacement of methylation with ubiquitination at the flanking lysines Lys420 and Lys442. Thus, replacement of methylation with ubiquitination might further signal that the protein is not correctly folded and should be degraded, whereby the replacement of methylation with ubiquitin indicates that the degradation at this point during the biogenesis is by ubiquitin dependent mechanisms.
Rescue of ΔF508 CFTR function was also tightly correlated with increased phosphorylation of Thr421-Ser427 in the ΔF508 CFTR RI element and was always accompanied by reduced ubiquitination of the flanking lysines Lys420 and Lys442, and in some cases by increased methylation of Lys442. Inhibiting phosphorylation of the RI element either by chemical inhibition or by knockdown of the responsible kinase, prevented temperature rescue of ΔF508 CFTR, showing the importance of the phosphorylation sites not only in wild-type CFTR biogenesis, but also showing that the ΔF508 CFTR phosphorylation defect in fact constitutes the trafficking defect that leads to CF. We thus suggest that crosstalk between methylation, ubiquitination and phosphorylation events in the CFTR RI element represents a quantitative, combinatorial PTM code that regulates CFTR maturation and that the ΔF508 CFTR phosphorylation defect in fact constitutes the trafficking defect that leads to CF (Fig. 6C).
In summary, this study reveals that crosstalk between specific PTMs contributes to protein folding and quality control during protein maturation. It will be interesting to explore if such a PTM code can be generalized and exists in other membrane proteins to regulate biogenesis.
Because misfolding of ΔF508 CFTR alters the CFTR methylation-phosphorylation pattern, and these changes crucially contribute to ΔF508 CFTR degradation, assessing how misfolding mutations affect PTMs and the respective protein networks in other misfolding diseases should not only lead to new insight into how misfolded proteins are distinguished from correctly folded ones and to new insight into membrane protein maturation in general, but may open new avenues for therapy as well.
Materials and Methods
Cell lines and cell culture
CFBE41o- and WT-HBE41o- bronchial epithelial cell lines, which express ΔF508 or wild-type CFTR, respectively, as well as FRT cells expressing either wild-type, ΔF508, N1303K, G551D or R117H CFTR were kindly provided by Dr. J. Clancy (University of Alabama, Birmingham, AL) (66). The CFBE41o- cell line was derived from a CF patient homozygous for ΔF508, whereas the wild-type cell line (HBE) was generated from CFBE41o- cells by re-introduction of F508 using SFHR (67). CFBE41o- and WT-HBE41o- cells were cultured at 37 °C, 5% CO2 in Advanced- MEM (GIBCO, Carlsbad, CA) supplemented with 1% Penicillin/Streptomycin (GIBCO), 10% fetal bovine serum (GIBCO) and 2 mM L-Glutamine (GIBCO) and appropriate selective antibiotics. For SILAC labeling experiments, cells were cultured for > 6 passages in lysine and arginine free SILAC-A-MEM (Gibco, custom media), supplemented with heavy-labeled 13C615N2 Lysine and 13C615N4 Arginine (Cambridge Isotope Laboratories, Tewksbury, MA), 2% FBS, 1% Penicillin/Streptomycin, and 2 mM L-Glutamine. FRT cells were cultured at 37 °C, 5% CO2 in Ham’s F12, Coon’s modification (Sigma-Aldrich) supplemented with 5% FBS and 100 μ/ml hygromycin (Invitrogen). Treatment with 5 – 10 μM VX809 (Selleck, Houston, TX), 10 μM CX-4945 (Santa Cruz Biotechnologies, Dallas, TX) or vehicle (DMSO), was carried out for 20 hours or as indicated.
Lentiviral infection and stable cell lines
CFBE41o-, wt-HBE41o- or primary bronchial epithelial cells were infected with Mission shRNA containing lentiviral particles (Sigma-Aldrich) at MOI 3 for 16 hours before seeding onto 12 mm Snapwell membranes (Corning, Cambridge, MA) coated with rat tail collagen I (BD Biosciences). Lentivirus infection is covered under TSRI approval #01–13-10–07 and all steps were carried out in a BSL2/3 certified laboratory. Stable cell lines were created by puromycin selection (20 μg/ml) and serial dilution after infection. Quantification of CK2 knockdown was carried out with ImageJ (NIH, http://imagej.nih.gov/ij) (68).
Co-Immunoprecipitation
Co-Immunoprecipitation was essentially carried out as described in Pankow, Bamberger et al., 2015 (19). Approximately 1 × 108 cells per IP were lysed on ice for 20 min in lysis buffer (0.5 % Igepal CA-630 [Sigma-Aldrich], 50 mM Tris [pH 7.5], 250 mM NaCl, 1 mM EDTA, 1× Complete EDTA-free Protease Inhibitor mix and 1× PhosSTOP [Roche]). Insoluble material was removed by centrifugation (30 min, 14,000 rpm, 4 °C) subsequent to sonication in a 55 kHz water-bath (Branson) for 3 min. The supernatant was pre-cleared by incubation with CL4B- Sepharose (GE Healthcare) for 1–2 hours at 4 °C. The pre-cleared lysate was then incubated overnight at 4 °C with 50 μl (approx. 250 μg) of 3G11 rat monoclonal, or 13.1 or 24.1 mouse monoclonal antibody recognizing CFTR (both ATCC, Manassas, VA, produced at TSRI) covalently coupled to ProteinG-Sepharose (GE Healthcare). Immunoprecipitates were washed three times with lysis buffer and two times with lysis buffer containing no detergent. Bound proteins were eluted twice with 0.2 M Glycine [pH 2.3] / 0.5 % Igepal CA-630 at 37 °C for 20 min and neutralized with 1 M NH4CO3 (final concentration 100 mM). Subsequently, proteins in the eluate were precipitated by standard Methanol/Chloroform precipitation with 4 volumes Methanol/volume sample and 1 volume Chloroform/volume sample. The precipitate was washed with methanol and re-solubilized in 100 mM Tris, pH 8.5, 0.2 % Rapigest (Waters) for further digestion by endoproteases trypsin or chymotrypsin.
Coupling of antibodies to Sepharose beads
Antibodies recognizing CFTR were coupled to ProteinG Sepharose 4 Fast Flow beads (GE Healthcare, Piscataway, NJ) at 6 mg/ml packed beads and allowed to bind for 2 hours at room temperature. After washing twice with sodium borate, pH 9.0, bound antibodies were covalently crosslinked to the beads with 20 mM Dimethylpimelimidate (DMP, Pierce) for 30 min, RT. The crosslinking was stopped by washing with 200 mM Ethanolamine (Sigma-Aldrich), pH 8.0.
Azidohomoalanine (AHA)-labeling and click chemistry
HBE41o-, FRT, or NHBE cells at 90 – 95 % confluence were methionine starved for 1 h by replacing the growth medium with AHA labeling media (1× HBSS (GibCo), 10 % dialyzed FBS (Pierce), 2mM GlutaMAX (GibCo), 4mM MgCl2, 4mM CaCl2 and 1mM sodium pyruvate (GibCo)). Subsequently, media was replaced by AHA labeling medium containing 4 mM AHA (Life Technologies or Click Chemistry Solutions) or 4mM AHA and 10 μM CX-4945 (Santa Cruz Biotechnology) to label newly synthesized CFTR. At indicated time points (15 min to 4 h) cells were harvested in TNI lysis buffer and lysates stored at −80°C before carrying out CFTR- IPs using 50 μg of mouse monoclonal 24.1 antibody (ATCC, produced at TSRI) or M3A7 (EMD Millipore) as described above. Following CFTR-Co-IP, beads were washed with TNI lysis buffer, followed by washing with TNI containing no detergent and re-buffering into 100 mM HEPES, pH 7.3. The click reaction was then carried out on-beads in de-oxygenized 100 mM HEPES, pH 7.3, with 100 μM Tris(benzyltriazolylmethyl)amine (TBTA, Sigma-Aldrich), 1 mM CuSO4 (Sigma-Aldrich), 100 μM Biotin-Alkyne (Invitrogen, or Setareh Biotech, Eugene, Oregon), and Cu wire (49) and allowed to proceed for 2 hours to 4 hours at 4°C. Excess Biotin-Alkyne was removed by 3 washes with 100 mM HEPES, pH 7.3. Maturing CFTR was detected by western blotting using Streptavidin coupled to horseradish peroxidase (HRP) at 1:40,0000 dilution (Pierce, Cat.No. 21130). Quantification of the AHA labeling experiments was carried out with ImageJ.
Western blotting
Protein lysates were prepared in TNI lysis buffer as described above and denatured in SDS sample buffer (69) for 30 min at 37°C for detecting CFTR or at 95°C for 5 min, separated by SDS-PAGE and transferred onto nitrocellulose (Protran, Schleicher&Schuell, Germany). CFTR was detected with mouse monoclonal antibodies M3A7 (EMD Millipore) or 24.1 (ATCC) or rat monoclonal 3G11 (TSRI). β-actin was detected with mouse monoclonal antibody AC-15 (Sigma), Casein kinase II alpha with mouse monoclonal antibody 8E5 (Thermo Fisher, Waltham, MA) and Na+/K+ATPase with antibody H-300 (sc28800, Santa Cruz). HRP conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were detected with enhanced chemiluminescence reagent (ECL, Pierce, Rockford, IL). Quantification was carried out with ImageJ (NIH,http://imagej.nih.gov/ij) (68) and statistically analyzed in Prism 7 (GraphPad Software, Inc.) using One-Way-ANOVA with Bonferroni post-correction.
Ussing chamber measurements
CFBE41o- cells were plated on 12 mm Snapwell membranes (Corning, Cambridge, MA) coated with rat tail collagen I (BD Biosciences) at a density of 2×105cells/cm2 and cultured in A-MEM supplemented with 10 % FBS, 1% Penicillin/Streptomycin and 2 mM Glutamax (GIBCO, Carlsbad, CA). When cells reached confluence, cells were maintained in B-ALI differentiation medium under air-liquid interface conditions for at least 21 days. Transepithelial resistance (TEER, RT) was measured with a Millicell ERS2 Volt-Ohmmeter (Millipore, Billerica, MA) and was between 200 and 3200 Ω. cm2. Polarized cultures were mounted in EasyMount Ussing chambers (Physiological Instruments, San Diego, CA), bathed bilaterally with Krebs-bicarbonate Ringer solution (in mM, 140 Na+, 119.8 Cl-, 25 HCO3-, 2.8 K+, 2.4 HPO4, 0.4 PO4, 1.2 Mg+, 1.2 Ca+, 5 Glucose) and bubbled continuously with 95% O2 / 5% CO2. The epithelial sodium channel was blocked with 100 μM amiloride (Sigma-Aldrich). To isolate CFTR specific apical Cl- current, CFTR was stimulated by forskolin (10 μM, apical) and genistein (50 μM, apical) followed by addition of CFTR Inhibitor 172 (20 μM, EMD Biosciences, apical). Measurements were carried out at 37°C and short circuit current (/sc) was recorded and analyzed with Acquire and Analyze 2.0 (Physiological Instruments). The Δ/sc was calculated as the difference between the maximum /sc value reached upon Forskolin/Genistein stimulation and the /sc value obtained upon addition of the CFTR Inhibitor 172.
Sample preparation for MudPIT analysis and LC/LC-MS/MS
Samples were reduced with 5 mM TCEP (Pierce) for 30 min at 37°C, alkylated with 10 mM lodoacetamide (Pierce) or Chloroacetamide (Sigma) for 20 min at 25°C and digested overnight with 3 μg of sequencing-grade recombinant trypsin (Promega, Madison, Wl). For double digests with trypsin and chymotrypsin, samples were split in half. One half was digested with 0.5 μg chymotrypsin for 3 hours and the other half was digested with trypsin as described above. To inactivate, Rapigest, 9 % formic acid was added after the digest, insoluble material removed by centrifugation (15 min, 14,000 rpm), and samples reduced to near dryness in vacuum. Purified peptides were resuspended in a small volume of buffer A (95% H2O, 5% MeCN, 0.1% formic acid) and pressure loaded onto the back-end of a preparative MudPIT microcapillary column consisting of fused silica (i.d. 250 μm) packed in-house with 2.5 cm of 5 μm Aqua C18 resin (Phenomenex, Ventura, CA) and 2.5 cm of strong cation exchange resin (Partisphere SCX, Whatman, Clifton, NJ). For spike-in experiments, 3 fmol each of 13C615N2-lysine or 13C615N4-arginine (heavy) labeled peptides (Thermo Fisher, custom synthesis) were mixed into the sample before loading onto the preparative column. The preparative column was connected by a small union body (Upchurch, Oak Harbor, WA) to an analytical reversed-phase column (115 mm fused silica i.d. 100 μm) packed with 3 μm Aqua C18 resin (Phenomenex). Samples were analyzed by nano-ESI-MudPIT on an LTQ-Orbitrap XL, LTQ-Velos-Orbitrap or Orbitrap-Elite (Thermo Fisher) by placing the triphasic MudPIT column in-line with an Agilent 1100 quaternary HPLC pump (Agilent, Palo Alto, CA) and separating the peptides in multiple dimensions with a modified 10-step gradient (0%, 10%, 20%, 30%, 40%, 50%, 70%, 80%, 90%, 100% Buffer C, (500 mM ammonium acetate/5% acetonitrile/0.1% formic acid)) over 20 hours as described previously (70). Each full scan mass spectrum (400–2000 m/z) was followed by 10–20 data- dependent MS/MS scans at 35% normalized collisional energy and an ion count threshold of 1000 or 10 data-dependent MS/MS scans using higher collisional energy (HCD) and an ion count threshold of 5000 using either an LTQ Orbitrap XL or LTQ Orbitrap Velos Pro (Thermo Fisher). Dynamic exclusion was used with an exclusion list of 500, repeat time of 60 s and asymmetric exclusion window of −0.51 m/z and +1.5 m/z. Each sample was loaded onto a freshly prepared MudPIT column.
Mass spectrometry data analysis, PTM site confirmation and quantification
Raw-files were extracted with RawConverter using monoisotopic peak extraction (71). MS/MS spectra were searched with ProLuCID (34) against the human UniProtKB/Swiss-Prot database release date 2012–04, using a target-decoy approach in which each protein sequence is reversed and concatenated to the normal database (72) and in which sequence variations of CFTR were manually annotated. Using the Integrated Proteomics Pipeline version 3 (Integrated Proteomics Applications, Inc., San Diego, CA), search parameters were set to 50 ppm precursor mass tolerance, half-tryptic specificity, with an allowed maximum number of internal missed cleavages of 1 or 2, minimum peptide length of 7 amino acids and carboxyamidomethylation (m = 57.021464 Da) as a static modification. Phosphorylation (m = 79.9663 Da, residues S, T, Y), Ubiquitination (m = 114.042927, residue K), Acetylation (m = 42.0106, residue K), monomethylation (m = 14.015650, residues K, R), dimethylation (m = 28.0313, residues K, R) and oxidation of methionine (m = 15.9949) were set as differential modifications with a maximum of three internal differential modifications allowed per peptide. Search results were filtered with DTASelect version 2.1 (73) to a peptide false discovery rate (FDR) of less than 0.5% (corresponding to a protein FDR of less than 1%), allowing either only fully tryptic or half-tryptic peptides, requiring a precursor mass deviation of less than 8 ppm from the theoretical calculated mass, and a minimum deltaCN score of 0.1. Spectra of modified peptides were manually validated to confirm modification and modification sites. To validate phosphorylation sites automatically, we additionally used Ascore requiring a score > 15 (74). The subset of non-identified spectra was further searched with Blind-PTM, a large window mass-tolerant search algorithm that uses MS/MS information and the high mass accuracy and resolution of precursor scans to determine possible mass shifts and identify unexpected PTMs (Xu, T., unpublished, http://www.proteomicswiki.com/wiki/index.php/BlindPTM). The search window was set to – 200 Da and + 600 Da and results were filtered with DTASelect2.1 Mass shifts were interpreted using Unimod (www.unimod.org) and manual interpretation of spectra.
Quantitative analysis was carried out with Census (75) using the SILAC option in high resolution mode (10 ppm tolerance for peak extraction), proline conversion option (for SILAC samples), and requiring a determinant factor score of ≥ 0.5 and regression value of ≥ 0.8, except for singleton peptides for which we required a singleton profile score of ≥ 0.85.
Statistical analysis
Data are presented as mean with S.D. or S.E.M. of independent biological replicates indicated by n in each legend. Also as indicated in each figure legend, data were analyzed for significance in Prism 7 (GraphPad Software, Inc.) with one-Way-ANOVA with Tukey post-correction or two-tailed unpaired t-tests corrected by the Bonferroni-Sidak method. P values < 0.0332 were considered significant and are indicated in the data as: *P < 0.0332, **P < 0.0021, ***P < 0.0002, and ****P < 0.0001.
Supplementary Material
Acknowledgements:
We thank T. Xu for making Blind-PTM available and for discussing data analysis strategies. We further thank C. Delahunty for editing of the manuscript.
Funding: The study was supported by National Institutes of Health grants 5R01 HL079442, 1R01 HL 131697–01A1 and P41 GM103533 to J.R.Y.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability: Raw files were deposited in the MassIVE repository under ID MSV000082908 and can be accessed through ftp://massive.ucsd.edu/MSV000082908. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.WHO. 2004. The molecular genetic epidemiology of cystic fibrosis. In www.who.int/genomics/publications/en/HGNWB04.02report.pdf.
- 2.Zolin A, M. E., van Rens J et al. 2016. ECFSPR Annual Report 2014.
- 3.CFF. 2016. 2014 Annual Data Report.
- 4.Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, and Tsui LC. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245: 1073–1080. [DOI] [PubMed] [Google Scholar]
- 5.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL, and et al. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245: 1066–1073. [DOI] [PubMed] [Google Scholar]
- 6.Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, and et al. 1989. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 245: 1059–1065. [DOI] [PubMed] [Google Scholar]
- 7.Dalemans W, Barbry P, Champigny G, Jallat S, Dott K, Dreyer D, Crystal RG, Pavirani A, Lecocq JP, and Lazdunski M. 1991. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 354: 526–528. [DOI] [PubMed] [Google Scholar]
- 8.Drumm ML, Wilkinson DJ, Smit LS, Worrell RT, Strong TV, Frizzell RA, Dawson DC, and Collins FS. 1991. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science 254: 1797–1799. [DOI] [PubMed] [Google Scholar]
- 9.Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, and Grinstein S. 1993. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem 268: 21592–21598. [PubMed] [Google Scholar]
- 10.Li C, Ramjeesingh M, Reyes E, Jensen T, Chang X, Rommens JM, and Bear CE. 1993. The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet 3: 311–316. [DOI] [PubMed] [Google Scholar]
- 11.Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, and Grinstein S. 1994. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J 13: 6076–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, and Welsh MJ. 1992. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358: 761–764. [DOI] [PubMed] [Google Scholar]
- 13.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, and Riordan JR. 1995. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83: 129–135. [DOI] [PubMed] [Google Scholar]
- 14.Gadsby DC, and Nairn AC. 1999. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 79: S77–S107. [DOI] [PubMed] [Google Scholar]
- 15.Winter MC, and Welsh MJ. 1997. Stimulation of CFTR activity by its phosphorylated R domain. Nature 389: 294–296. [DOI] [PubMed] [Google Scholar]
- 16.Hwang TC, Wang F, Yang IC, and Reenstra WW. 1997. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am J Physiol 273: C988–998. [DOI] [PubMed] [Google Scholar]
- 17.Mann M, and Jensen ON. 2003. Proteomic analysis of post-translational modifications. Nat Biotechnol 21: 255–261. [DOI] [PubMed] [Google Scholar]
- 18.Beltrao P, Bork P, Krogan NJ, and van Noort V. 2013. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol 9: 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pankow S, Bamberger C, Calzolari D, Martinez-Bartolome S, Lavallee- Adam M, Balch WE, and Yates JR 3rd. 2015. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528: 510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bozoky Z, Krzeminski M, Muhandiram R, Birtley JR, Al-Zahrani A, Thomas PJ, Frizzell RA, Ford RC, and Forman-Kay JD. 2013. Regulatory R region of the CFTR chloride channel is a dynamic integrator of phospho- dependent intra- and intermolecular interactions. Proc Natl Acad Sci U S A 110: E4427–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia Y, Mathews CJ, and Hanrahan JW. 1997. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J Biol Chem 272: 4978–4984. [DOI] [PubMed] [Google Scholar]
- 22.King JD Jr., Fitch AC, Lee JK, McCane JE, Mak DO, Foskett JK, and Hallows KR. 2009. AMP-activated protein kinase phosphorylation of the R domain inhibits PKA stimulation of CFTR. Am J Physiol Cell Physiol 297: C94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkinson DJ, Strong TV, Mansoura MK, Wood DL, Smith SS, Collins FS, and Dawson DC. 1997. CFTR activation: additive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am J Physiol 273: L127–133. [DOI] [PubMed] [Google Scholar]
- 24.Townsend RR, Lipniunas PH, Tulk BM, and Verkman AS. 1996. Identification of protein kinase A phosphorylation sites on NBD1 and R domains of CFTR using electrospray mass spectrometry with selective phosphate ion monitoring. Protein Sci 5: 1865–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neville DC, Rozanas CR, Price EM, Gruis DB, Verkman AS, and Townsend RR. 1997. Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci 6: 2436–2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClure M, DeLucas LJ, Wilson L, Ray M, Rowe SM, Wu X, Dai Q, Hong JS, Sorscher EJ, Kappes JC, and Barnes S. 2012. Purification of CFTR for mass spectrometry analysis: identification of palmitoylation and other post- translational modifications. Protein Eng Des Sel 25: 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Picciotto MR, Cohn JA, Bertuzzi G, Greengard P, and Nairn AC. 1992. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J Biol Chem 267: 12742–12752. [PubMed] [Google Scholar]
- 28.Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, and Lukacs GL. 2010. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329: 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward CL, Omura S, and Kopito RR. 1995. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83: 121–127. [DOI] [PubMed] [Google Scholar]
- 30.Lee S, Henderson MJ, Schiffhauer E, Despanie J, Henry K, Kang PW, Walker D, McClure ML, Wilson L, Sorscher EJ, and Zeitlin PL. 2014. Interference with ubiquitination in CFTR modifies stability of core glycosylated and cell surface pools. Mol Cell Biol 34: 2554–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pankow S, Bamberger C, Calzolari D, Bamberger A, and Yates JR 3rd. 2016. Deep interactome profiling of membrane proteins by co-interacting protein identification technology. Nat Protoc 11: 2515–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI, and Yates JR 3rd. 2002. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A 99: 7900–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yates JR 3rd, Eng JK, McCormack AL, and Schieltz D. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67: 1426–1436. [DOI] [PubMed] [Google Scholar]
- 34.Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, Wong CC, Fonslow B, Delahunty C, Gao Y, Shah H, and Yates JR 3rd. 2015. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chick JM, Kolippakkam D, Nusinow DP, Zhai B, Rad R, Huttlin EL, and Gygi SP. 2015. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat Biotechnol 33: 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanelis V, Hudson RP, Thibodeau PH, Thomas PJ, and Forman-Kay JD. 2010. NMR evidence for differential phosphorylation-dependent interactions in WT and DeltaF508 CFTR. EMBO J 29: 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker JM, Hudson RP, Kanelis V, Choy WY, Thibodeau PH, Thomas PJ, and Forman-Kay JD. 2007. CFTR regulatory region interacts with NBD1 predominantly via multiple transient helices. Nat Struct Mol Biol 14: 738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, and Emtage S. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23: 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mornon JP, Lehn P, and Callebaut I. 2008. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci 65: 2594–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Matteson J, An Y, Moyer B, Yoo JS, Bannykh S, Wilson IA, Riordan JR, and Balch WE. 2004. COPII-dependent export of cystic fibrosis transmembrane conductance regulator from the ER uses a di-acidic exit code. J Cell Biol 167: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baldursson O, Berger HA, and Welsh MJ. 2000. Contribution of R domain phosphoserines to the function of CFTR studied in Fischer rat thyroid epithelia. Am J Physiol Lung Cell Mol Physiol 279: L835–841. [DOI] [PubMed] [Google Scholar]
- 42.Kongsuphol P, Cassidy D, Hieke B, Treharne KJ, Schreiber R, Mehta A, and Kunzelmann K. 2009. Mechanistic insight into control of CFTR by AMPK. J Biol Chem 284: 5645–5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luz S, Cihil KM, Brautigan DL, Amaral MD, Farinha CM, and Swiatecka-Urban A. 2014. LMTK2-mediated phosphorylation regulates CFTR endocytosis in human airway epithelial cells. J Biol Chem 289: 15080–15093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pagano MA, Arrigoni G, Marin O, Sarno S, Meggio F, Treharne KJ, Mehta A, and Pinna LA. 2008. Modulation of protein kinase CK2 activity by fragments of CFTR encompassing F508 may reflect functional links with cystic fibrosis pathogenesis. Biochemistry 47: 7925–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Treharne KJ, Xu Z, Chen JH, Best OG, Cassidy DM, Gruenert DC, Hegyi P, Gray MA, Sheppard DN, Kunzelmann K, and Mehta A. 2009. Inhibition of protein kinase CK2 closes the CFTR Cl channel, but has no effect on the cystic fibrosis mutant deltaF508CFTR. Cell Physiol Biochem 24: 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meggio F, and Pinna LA. 2003. One-thousand-and-one substrates of protein kinase CK2? FASEB J 17: 349–368. [DOI] [PubMed] [Google Scholar]
- 47.Dieterich DC, Link AJ, Graumann J, Tirrell DA, and Schuman EM. 2006. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc Natl AcadSci US A 103: 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, and Schuman EM. 2010. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci 13: 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Presolski SI, Hong VP, and Finn MG. 2011. Copper-Catalyzed Azide- Alkyne Click Chemistry for Bioconjugation. Curr Protoc Chem Biol 3: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McClatchy DB, Ma Y, Liu C, Stein BD, Martinez-Bartolome S, Vasquez D, Hellberg K, Shaw RJ, and Yates JR 3rd. 2015. Pulsed Azidohomoalanine Labeling in Mammals (PALM) Detects Changes in Liver-Specific LKB1 Knockout Mice. J Proteome Res 14: 4815–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellgaard L, McCaul N, Chatsisvili A, and Braakman I. 2016. Co- and Post- Translational Protein Folding in the ER. Traffic 17: 615–638. [DOI] [PubMed] [Google Scholar]
- 52.Aebi M 2013. N-linked protein glycosylation in the ER. Biochim Biophys Acta 1833: 2430–2437. [DOI] [PubMed] [Google Scholar]
- 53.Ward CL, and Kopito RR. 1994. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem 269: 25710–25718. [PubMed] [Google Scholar]
- 54.Faust M, Jung M, Gunther J, Zimmermann R, and Montenarh M. 2001. Localization of individual subunits of protein kinase CK2 to the endoplasmic reticulum and to the Golgi apparatus. Mol Cell Biochem 227: 73–80. [PubMed] [Google Scholar]
- 55.Nitsch L, Tramontano D, Ambesi-Impiombato FS, Quarto N, and Bonatti S. 1985. Morphological and functional polarity of an epithelial thyroid cell line. Eur J Cell Biol 38: 57–66. [PubMed] [Google Scholar]
- 56.Zurzolo C, Lisanti MP, Caras IW, Nitsch L, and Rodriguez-Boulan E. 1993. Glycosylphosphatidylinositol-anchored proteins are preferentially targeted to the basolateral surface in Fischer rat thyroid epithelial cells. J Cell Biol 121: 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, Masica DL, Karchin R, Millen L, Thomas PJ, Patrinos GP, Corey M, Lewis MH, Rommens JM, Castellani C, Penland CM, and Cutting GR. 2013. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 45: 1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, Hoch B, Kellner W, Kelly JW, Schmidt A, Thomas PJ, Matsumura Y, Skach WR, Gentzsch M, Riordan JR, Sorscher EJ, Okiyoneda T, Yates JR 3rd, Lukacs GL, Frizzell RA, Manning G, Gottesfeld JM, and Balch WE. 2010. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol 6: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, Roldan A, Verkman AS, Kurth M, Simon A, Hegedus T, Beekman JM, and Lukacs GL. 2013. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol 9: 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, and Negulescu PA. 2011. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci US A 108: 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang D, Ciciriello F, Anjos SM, Carissimo A, Liao J, Carlile GW, Balghi H, Robert R, Luini A, Hanrahan JW, and Thomas DY. 2012. Ouabain Mimics Low Temperature Rescue of F508del-CFTR in Cystic Fibrosis Epithelial Cells. Front Pharmacol 3: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pilewski JM, and Frizzell RA. 1999. Role of CFTR in airway disease. Physiol Rev 79: S215–255. [DOI] [PubMed] [Google Scholar]
- 63.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, and Riordan JR. 2008. Phenylalanine-508 mediates a cytoplasmic- membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci U S A 105: 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu F, Zhang Z, Csanady L, Gadsby DC, and Chen J. 2017. Molecular Structure of the Human CFTR Ion Channel. Cell 169: 85–95 e88. [DOI] [PubMed] [Google Scholar]
- 65.Aleksandrov AA, Kota P, Aleksandrov LA, He L, Jensen T, Cui L, Gentzsch M, Dokholyan NV, and Riordan JR. 2010. Regulatory insertion removal restores maturation, stability and function of DeltaF508 CFTR. J Mol Biol 401: 194–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bebok Z, Collawn JF, Wakefield J, Parker W, Li Y, Varga K, Sorscher EJ, and Clancy JP. 2005. Failure of cAMP agonists to activate rescued deltaF508 CFTR in CFBE41o- airway epithelial monolayers. J Physiol 569: 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gruenert DC, Bruscia E, Novelli G, Colosimo A, Dallapiccola B, Sangiuolo F, and Goncz KK. 2003. Sequence-specific modification of genomic DNA by small DNA fragments. J Clin Invest 112: 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schneider CA, Rasband WS, and Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laemmli UK 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 70.Washburn MP, Wolters D, and Yates JR 3rd. 2001. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19: 242–247. [DOI] [PubMed] [Google Scholar]
- 71.He L, Diedrich J, Chu YY, and Yates JR 3rd. 2015. Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal Chem 87: 11361–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elias JE, Haas W, Faherty BK, and Gygi SP. 2005. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat Methods 2: 667–675. [DOI] [PubMed] [Google Scholar]
- 73.Cociorva D, L. TD, and Yates JR. 2007. Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics Chapter 13: Unit 13 14. [DOI] [PubMed] [Google Scholar]
- 74.Beausoleil SA, Villen J, Gerber SA, Rush J, and Gygi SP. 2006. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol 24: 1285–1292. [DOI] [PubMed] [Google Scholar]
- 75.Park SK, Venable JD, Xu T, and Yates JR 3rd. 2008. A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods 5: 319–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.