

Abstract
Background
Hidradenitis suppurativa (HS) is a chronic inflammatory skin condition characterised by recurrent painful boils in flexural sites, such as the axillae and groin, that affects about 1% of the population, with onset in early adulthood.
Objectives
To assess the effects of interventions for HS in people of all ages.
Search methods
We searched the following databases up to 13 August 2015: the Cochrane Skin Group Specialised Register, CENTRAL in the Cochrane Library (Issue 7, 2015), MEDLINE (from 1946), EMBASE (from 1974), and LILACS (from 1982). We also searched five trials registers and handsearched the conference proceedings of eight dermatology meetings. We checked the reference lists of included and excluded studies for further references to relevant trials.
Selection criteria
Randomised controlled trials (RCTs) of all interventions for hidradenitis suppurativa.
Data collection and analysis
Two review authors independently assessed study eligibility and methodological quality and performed data extraction. Our primary outcomes were quality of life, measured by a validated dermatology‐specific scale, and adverse effects of the interventions.
Main results
Twelve trials, with 615 participants, met our inclusion criteria. The median number of participants in each trial was 27, and median trial duration was 16 weeks. The included studies were conducted over a 32‐year time period, from 1983 to 2015. A single RCT that was underpowered to detect clinically meaningful differences investigated most interventions.
There were four trials of anti‐TNF‐α (tumour necrosis factor‐alpha) therapies, which included etanercept, infliximab, and adalimumab. Adalimumab 40 mg weekly improved the Dermatology Life Quality Index (DLQI) score in participants with moderate to severe HS by 4.0 points relative to placebo (95% confidence interval (CI) ‐6.5 to ‐1.5 points), an effect size approximately equal to the DLQI minimal clinically important difference. We reduced the evidence quality to 'moderate' because the effect size was based on the results of only one study. In a meta‐analysis of two studies with 124 participants, standard dose adalimumab 40 mg every other week was ineffective compared with placebo (moderate quality evidence). In a smaller study of 38 participants, of whom only 33 provided efficacy data, infliximab 5 mg/kg treatment improved DLQI by 8.4 DLQI points after eight weeks. Etanercept 50 mg twice weekly was well tolerated but ineffective.
In a RCT of 200 participants, no difference was found in surgical complications (week one: risk ratio (RR) 0.78, 95% CI 0.58 to 1.05, moderate quality evidence) or risk of recurrence (after three months: RR 0.96, 95% CI 0.68 to 1.34, moderate quality evidence) in those randomised to receive a gentamicin‐collagen sponge prior to primary closure compared with primary closure alone.
RCTs of other interventions, including topical clindamycin 1% solution; oral tetracycline; oral ethinylestradiol 50 mcg with either cyproterone acetate 50 mg or norgestrel 500 mcg; intense pulsed light; neodymium‐doped yttrium aluminium garnet (Nd:YAG) laser; methylene blue gel photodynamic therapy; and staphage lysate, were relatively small studies, preventing firm conclusions due to imprecision.
Authors' conclusions
Many knowledge gaps exist in RCT evidence for HS. Moderate quality evidence exists for adalimumab, which improves DLQI score when 40 mg is given weekly, twice the standard psoriasis dose. However, the 95% confidence interval includes an effect size of only 1.5 DLQI points, which may not be clinically relevant, and the safety profile of weekly dosing has not been fully established. Infliximab also improves quality of life, based on moderate quality evidence.
More RCTs are needed in most areas of HS care, particularly oral treatments and the type and timing of surgical procedures. Outcomes should be validated, ideally, including a minimal clinically important difference for HS.
Plain language summary
Treatments for hidradenitis suppurativa
Background
Hidradenitis suppurativa (HS) is a long‐term, distressing skin condition involving multiple painful boils in skin creases, such as the armpits, groin, and genital region, estimated to affect about 1 in 100 people. It typically begins in early adulthood and has a large impact on quality of life because of pain, scarring, and low self‐esteem. Doctors and the general public have largely ignored the condition, in part because people with HS do not wish to draw attention to their condition, so there is a relative lack of evidence to guide treatment.
Review question
What are the beneficial and harmful effects of treatments for hidradenitis suppurativa in terms of changes in quality of life and side effects?
Study characteristics
Our review included only randomised controlled trials (RCTs); we included 12 trials, containing a total of 615 people. In most cases, only a single trial that was too small to provide meaningful results investigated the treatments. There was no RCT evidence to support several quite commonly used treatments. The average duration of the trials was four months, long enough to check whether a treatment works initially but not long enough to show the duration of disease control or to detect delayed side effects.
Key results
The evidence from two trials for clindamycin lotion applied to the skin and oral tetracyclines was relatively weak, despite these antibiotics being standard treatments for mild to moderate HS. There were four pharmaceutical industry‐sponsored trials of anti‐TNF‐α (tumour necrosis factor‐alpha) therapies, which included etanercept, infliximab, and adalimumab. Of these, a trial of etanercept did not find benefit, whereas a small trial of infliximab reported an improvement in quality of life after eight weeks. A larger trial, including 154 participants, investigated adalimumab. There was no benefit for moderate to severe HS at standard psoriasis doses of 40 mg every other week, but 40 mg weekly did improve quality of life. The estimate of quality of life improvement ranged from a level that probably would help people with HS to a level that might not be enough to justify use of adalimumab. The trial found no increase in serious side effects, including infections, but it was not large enough to detect rare effects. There were no trials investigating when to perform surgery or what surgical procedure to consider. One trial looked at inserting an antibiotic sponge into wounds after removal of HS lesions, but found no benefit compared with surgery without the antibiotic sponge. There were three trials of laser‐type treatments, but the trial quality was too low to recommend these therapies.
Quality of the evidence
Our review has highlighted a need for more clinical trials to give better evidence to guide treatment choices in HS. More trials of oral treatments are required as well as surgical studies. Future trials should include patient‐reported outcomes, such as quality of life and pain.
Summary of findings
Summary of findings for the main comparison. Adalimumab weekly compared with placebo for hidradenitis suppurativa.
| Adalimumab weekly compared with placebo for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: adalimumab weekly Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Adalimumab weekly | |||||
| Change in DLQI score (imputation) Follow‐up: 16 weeks | ‐ | The mean change in DLQI score (imputation) in the intervention groups was 4 lower (6.49 to 1.51 lower) | ‐ | 102 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ |
| Change in DLQI score (LOCF) Follow‐up: 16 weeks | ‐ | The mean change in DLQI score (LOCF) in the intervention groups was 4.1 lower (6.59 to 1.61 lower) | ‐ | 102 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ |
| Frequency of serious adverse effects Follow‐up: 16 weeks | Study population | RR 2 (0.38 to 10.44) | 102 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 39 per 1000 | 78 per 1000 (15 to 409) | |||||
| Moderate | ||||||
| 39 per 1000 | 78 per 1000 (15 to 407) | |||||
| Frequency of treatment discontinuation Follow‐up: 16 weeks | Study population | RR 5 (0.25 to 101.63) | 102 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 0 per 1000 | 39 per 1000² (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 39 per 1000² (0 to 0) | |||||
| Proportion of participants with infectious adverse effects Follow‐up: 16 weeks | Study population | RR 0.94 (0.55 to 1.62) | 102 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 353 per 1000 | 332 per 1000 (194 to 572) | |||||
| Moderate | ||||||
| 353 per 1000 | 332 per 1000 (194 to 572) | |||||
| Proportion with improvement in pain VAS Follow‐up: 16 weeks | Study population | RR 1.77 (1.02 to 3.07) | 96 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 271 per 1000 | 479 per 1000 (276 to 831) | |||||
| Moderate | ||||||
| 271 per 1000 | 480 per 1000 (276 to 832) | |||||
| Change in modified Sartorius scale score (imputation) Follow‐up: 16 weeks | ‐ | The mean change in modified Sartorius scale score (imputation) in the intervention groups was 23 lower (50.16 lower to 4.16 higher) | ‐ | 102 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DLQI: Dermatology Life Quality Index; LOCF: last observation carried forward; RR: risk ratio; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for imprecision because the evidence is based on the results of a single study and subsequent studies are likely to have an important impact on our confidence in the estimate of effect and may change the estimate (Ioannidis 2005). ²Due to the low frequency of events (0) in the control group, the corresponding risk reflects the observed events in the intervention group.
Summary of findings 2. Adalimumab every other week compared with placebo for hidradenitis suppurativa.
| Adalimumab every other week compared with placebo for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: adalimumab every other week Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Adalimumab every other week | |||||
| Change in DLQI score (LOCF) Follow‐up: 16 weeks¹ | ‐ | The mean change in DLQI score (LOCF) in the intervention groups was 1.61 lower (3.86 lower to 0.64 higher) | ‐ | 124 (2 studies) | ⊕⊕⊕⊕ high | ‐ |
| Frequency of serious adverse effects Follow‐up: 16 weeks¹ | Study population | RR 1.47 (0.26 to 8.44) | 124 (2 studies) | ⊕⊕⊕⊕ high | ‐ | |
| 35 per 1000 | 52 per 1000 (9 to 296) | |||||
| Moderate | ||||||
| 20 per 1000 | 29 per 1000 (5 to 169) | |||||
| Frequency of treatment discontinuation Follow‐up: 16 weeks¹ | Study population | RR 4.91 (0.24 to 99.74) | 124 (2 studies) | ⊕⊕⊕⊕ high | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Proportion of participants with infectious adverse effects Follow‐up: 16 weeks¹ | Study population | RR 1.60 (0.57 to 4.53) | 124 (2 studies) | ⊕⊕⊕⊕ high | ‐ | |
| 333 per 1000 | 533 per 1000 (190 to 1000) | |||||
| Moderate | ||||||
| 260 per 1000 | 416 per 1000 (148 to 1000) | |||||
| Change in Pain VAS Follow‐up: 12 weeks | ‐ | The mean change in pain vas in the intervention groups was 16.57 lower (55.28 lower to 22.14 higher) | ‐ | 21 (1 study) | ⊕⊕⊝⊝ low²,³ | ‐ |
| Proportion with improvement in pain Follow‐up: 16 weeks | Study population | RR 1.34 (0.73 to 2.43) | 95 (1 study) | ⊕⊕⊕⊝ moderate³ | ‐ | |
| 271 per 1000 | 363 per 1000 (198 to 658) | |||||
| Moderate | ||||||
| 729 per 1000 | 363 per 1000 (198 to 659) | |||||
| Change in Sartorius scale score (LOCF) Follow‐up: 16 weeks¹ | ‐ | The mean change in Sartorius scale score (LOCF) in the intervention groups was 0.42 standard deviations lower (1.22 lower to 0.37 higher) | ‐ | 124 (2 studies) | ⊕⊕⊕⊝ moderate⁴ | SMD ‐0.42 (‐1.22 to 0.37) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; DLQI: Dermatology Life Quality Index; LOCF: last observation carried forward; RR: risk ratio; SMD: standardised mean difference; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Follow up 12 weeks for 21 participants (Miller 2011). ²Imbalance in baseline disease severity between the 2 groups ‐ downgraded due to indirectness as the results may not be of relevance to the wider population. ³Downgraded one level for imprecision because the evidence is based on the results of a single study (for each of these outcomes) and subsequent studies are likely to have an important impact on our confidence in the estimate of effect and may change the estimate (Ioannidis 2005). ⁴Downgraded one level for inconsistency as the I² statistic of 59% demonstrates substantial study heterogeneity for this outcome.
Summary of findings 3. Infliximab compared with placebo for hidradenitis suppurativa.
| Infliximab compared with placebo for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: infliximab Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Infliximab | |||||
| At least 50% decrease in HS Severity Index Follow‐up: 8 weeks | Study population | RR 4.80 (0.6 to 38.48) | 33 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 56 per 1000 | 267 per 1000 (33 to 1000) | |||||
| Moderate | ||||||
| 56 per 1000 | 269 per 1000 (34 to 1000) | |||||
| Physician global assessment Follow‐up: 8 weeks | Study population | RR 4.80 (1.66 to 13.9) | 33 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 167 per 1000 | 800 per 1000 (277 to 1000) | |||||
| Moderate | ||||||
| 167 per 1000 | 802 per 1000 (277 to 1000) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HS: hidradenitis suppurativa; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for imprecision due to a small number of events in only a single study.
Summary of findings 4. Etanercept compared with placebo for hidradenitis suppurativa.
| Etanercept compared with placebo for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: etanercept Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Etanercept | |||||
| Dermatology Life Quality Index Follow‐up: 12 weeks | ‐ | No significant difference between the 2 groups (P = 0.12, Mantel‐Haenszel test) | ‐ | 17 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for imprecision due to a small number of participants in only a single study.
Summary of findings 5. Topical clindamycin compared with placebo for hidradenitis suppurativa.
| Topical clindamycin compared with placebo for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: topical clindamycin Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Topical clindamycin | |||||
| Adverse effects (non‐serious) Follow‐up: 12 weeks | Study population | RR 0.72 (0.14 to 3.64) | 27 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 214 per 1000 | 154 per 1000 (30 to 780) | |||||
| Moderate | ||||||
| 214 per 1000 | 154 per 1000 (30 to 779) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for imprecision due to a small number of events (five) in only a single study.
Summary of findings 6. Oral tetracycline compared with topical clindamycin for hidradenitis suppurativa.
| Oral tetracycline compared with topical clindamycin for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: oral tetracycline Comparison: topical clindamycin | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Topical clindamycin | Oral tetracycline | |||||
| Participant global assessment VAS Visual analogue scale (VAS) (0 to 100 mm) Follow‐up: 16 weeks | ‐ | The mean participant global assessment VAS in the intervention groups was 28 lower (46.64 to 9.36 lower) | ‐ | 34 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ |
| Pain VAS VAS (0 to 100 mm) Follow‐up: 16 weeks | ‐ | The mean pain VAS in the intervention groups was 3 higher (47.46 lower to 53.46 higher) | ‐ | 34 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ |
| Nodules score Nodule count Follow‐up: 16 weeks | ‐ | The mean nodules score in the intervention groups was 0.3 higher (2.6 lower to 3.2 higher) | ‐ | 34 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ |
| Abscesses score Abscess count Follow‐up: 16 weeks | ‐ | The mean abscesses score in the intervention groups was 0.8 higher (0.83 lower to 2.43 higher) | ‐ | 34 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ |
| Physician global assessment VAS VAS (0 to 100 mm) Follow‐up: 16 weeks | ‐ | The mean physician global assessment VAS in the intervention groups was 9 higher (12.61 lower to 30.61 higher) | ‐ | 34 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for risk of attrition bias due to absence of an intention‐to‐treat analysis, in the context that 12 of 46 participants (26%) dropped out of the study. ²Downgraded one level for imprecision due to a small number of participants (34) in only a single study.
Summary of findings 7. Ethinyloestradiol and cyproterone acetate compared with ethinyloestradiol and norgestrel for hidradenitis suppurativa.
| Ethinyloestradiol and cyproterone acetate compared with ethinyloestradiol and norgestrel for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: ethinyloestradiol and cyproterone acetate Comparison: ethinyloestradiol and norgestrel | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Ethinyloestradiol & norgestrel | Ethinyloestradiol & cyproterone acetate | |||||
| Number of participants reporting non‐serious adverse effects Follow‐up: 6 months | Study population | RR 0.53 (0.29 to 0.98) | 18 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 1000 per 1000 | 530 per 1000 (290 to 980) | |||||
| Moderate | ||||||
| 1000 per 1000 | 530 per 1000 (290 to 980) | |||||
| Participant global assessment VAS Scale from: 0 to 100 Follow‐up: 6 months | ‐ | The mean participant global assessment VAS in the intervention groups was 6 higher (15.98 lower to 27.98 higher) | ‐ | 17 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for imprecision due to a small number of participants in only a single study.
Summary of findings 8. Gentamicin sponge compared with primary closure alone for hidradenitis suppurativa.
| Gentamicin sponge compared with primary closure alone for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: gentamicin sponge Comparison: primary closure alone | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Primary closure alone | Gentamicin sponge | |||||
| Adverse effects ‐ complication rate at 1 week after surgery Follow‐up: 1 weeks | Study population | RR 0.78 (0.58 to 1.05) | 200 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 526 per 1000 | 411 per 1000 (305 to 553) | |||||
| Moderate | ||||||
| 526 per 1000 | 410 per 1000 (305 to 552) | |||||
| Adverse effects ‐ complication rate at 3 months after surgery Follow‐up: 3 months | Study population | RR 0.9 (0.5 to 1.62) | 200 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 197 per 1000 | 178 per 1000 (99 to 320) | |||||
| Moderate | ||||||
| 197 per 1000 | 177 per 1000 (99 to 319) | |||||
| Recurrence rate at 3 months after surgery Follow‐up: 3 months | Study population | RR 0.96 (0.68 to 1.34) | 200 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 421 per 1000 | 404 per 1000 (286 to 564) | |||||
| Moderate | ||||||
| 421 per 1000 | 404 per 1000 (286 to 564) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level due to unclear risk of bias for most domains. In particular, the study report states that there was an imbalance in randomisation due to early cessation of the study, but no further details are provided. Also, no description is provided of any special measures to ensure blinding of personnel, who would otherwise have been aware of treatment allocation from the operative notes.
Summary of findings 9. Intense pulsed light compared with no treatment for hidradenitis suppurativa.
| Intense pulsed light compared with no treatment for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: intense pulsed light Comparison: no treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| No treatment | Intense pulsed light | |||||
| Participant global assessment: satisfaction with treatment Questionnaire Follow‐up: uncertain | Study population | RR 9.67 (2.01 to 46.43) | 34 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Participant global assessment: satisfaction with treatment ‐ axilla Questionnaire Follow‐up: uncertain | Study population | RR 21.00 (1.37 to 322.28) | 24 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Participant global assessment: satisfaction with treatment ‐ groin Questionnaire Follow‐up: uncertain | Study population | RR 5.00 (0.31 to 79.94) | 8 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Participant global assessment: satisfaction with treatment ‐ inframammary Questionnaire Follow‐up: uncertain | Study population | RR 3.00 (0.24 to 37.67) | 2 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level due to performance bias resulting from participants being unblinded, in the absence of a sham treatment for the control side. ²Downgraded one level for imprecision due to a small number of participants in only a single study.
Summary of findings 10. Nd:YAG laser compared with topical control for hidradenitis suppurativa.
| Nd:YAG laser compared with topical control for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: Nd:YAG laser Comparison: topical control | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Topical control | Nd:YAG laser | |||||
| Modified HS‐LASI score after 3 months Follow‐up: 3 months | ‐ | The mean modified HS‐LASI score after 3 months in the intervention groups was 14.03 lower (18.84 to 9.22 lower) | ‐ | 50 (1 study) | ⊕⊝⊝⊝ very low¹,²,³ | ‐ |
| Modified HS‐LASI score after 3 months ‐ axilla Follow‐up: 3 months | ‐ | The mean modified HS‐LASI score after 3 months ‐ axilla ‐ in the intervention groups was 18.7 lower (26.82 to 10.58 lower) | ‐ | 20 (1 study) | ⊕⊝⊝⊝ very low¹,²,³ | ‐ |
| Modified HS‐LASI score after 3 months ‐ groin Follow‐up: 3 months | ‐ | The mean modified HS‐LASI score after 3 months ‐ groin ‐ in the intervention groups was 12.6 lower (20.28 to 4.92 lower) | ‐ | 22 (1 study) | ⊕⊝⊝⊝ very low¹,²,³,⁴ | ‐ |
| Modified HS‐LASI score after 3 months ‐ inframammary Follow‐up: 3 months | ‐ | The mean modified HS‐LASI score after 3 months ‐ inframammary ‐ in the intervention groups was 9.8 lower (19.31 to 0.29 lower) | ‐ | 8 (1 study) | ⊕⊝⊝⊝ very low¹,²,³,⁴ | ‐ |
| Percentage change in modified HS‐LASI score after 5 months compared with baseline Follow‐up: 5 months | ‐ | The mean percentage change in modified HS‐LASI score after 5 months compared with baseline in the intervention groups was 51.4 lower (66.36 to 36.43 lower) | ‐ | 50 (1 study) | ⊕⊝⊝⊝ very low¹,²,³ | ‐ |
| Percentage change in modified HS‐LASI score after 5 months compared with baseline ‐ axilla Follow‐up: 5 months | ‐ | The mean percentage change in modified HS‐LASI score after 5 months compared with baseline ‐ axilla ‐ in the intervention groups was 58.9 lower (78.82 to 38.98 lower) | ‐ | 20 (1 study) | ⊕⊝⊝⊝ very low¹,²,³ | ‐ |
| Percentage change in modified HS‐LASI score after 5 months compared with baseline ‐ groin Follow‐up: 5 months | ‐ | The mean percentage change in modified HS‐LASI score after 5 months compared with baseline ‐ groin ‐ in the intervention groups was 38.7 lower (63.43 to 13.97 lower) | ‐ | 22 (1 study) | ⊕⊝⊝⊝ very low¹,²,³ | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HS‐LASI: Hidradenitis Suppurativa Lesion, Area and Severity Index; Nd:YAG: neodymium‐doped yttrium aluminium garnet. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for risk of attrition bias due to absence of an intention‐to‐treat analysis, in the context that five participants (eight anatomical sites) dropped out and were not included in results. ²Downgraded one level because treating physicians were unblinded, producing a risk of performance bias. ³Downgraded one level for imprecision due to a small number of participants in only a single study.
Summary of findings 11. Niosomal methylene blue gel PDT compared with free methylene blue gel PDT for hidradenitis suppurativa.
| Niosomal methylene blue gel PDT compared with free methylene blue gel PDT for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: niosomal methylene blue gel PDT Comparison: free methylene blue gel PDT | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Free methylene blue gel PDT | Niosomal methylene blue gel PDT | |||||
| HS‐LASI score Follow‐up: mean 6 months | The mean HS‐LASI score in the control groups was 7.9 points | The mean HS‐LASI score in the intervention groups was 4.30 lower (8.36 to 0.24 lower) | ‐ | 20 (1 study) | ⊕⊕⊝⊝ low¹,² | ‐ |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HS‐LASI: Hidradenitis Suppurativa Lesion, Area and Severity Index;PDT: photodynamic therapy. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level because participants and treating physicians were unblinded, producing a risk of performance bias. ²Downgraded one level for imprecision due to a small number of participants in only a single study.
Summary of findings 12. Staphage lysate compared with placebo broth for hidradenitis suppurativa.
| Staphage lysate compared with placebo broth for hidradenitis suppurativa | ||||||
| Patient or population: participants with hidradenitis suppurativa Settings: hospital‐based Intervention: staphage lysate Comparison: placebo broth | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo broth | Staphage lysate | |||||
| Physician global assessment Follow‐up: mean 24 weeks | Study population | RR 6.25 (1.68 to 23.27) | 27 (1 study) | ⊕⊕⊕⊝ moderate¹ | ‐ | |
| 133 per 1000 | 833 per 1000 (224 to 1000) | |||||
| Moderate | ||||||
| 133 per 1000 | 831 per 1000 (223 to 1000) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
¹Downgraded one level for imprecision due to a small number of participants in only a single study.
Background
Please see the glossary in Table 13 for an explanation of the terms we have used.
1. Glossary.
| Term | Description |
| Abscess | Collection of pus within a cavity |
| Apocrine gland | A specialised sweat gland of the skin that produces a viscous secretion |
| Axillae | Arm pits |
| Dehiscence | Breakdown and re‐opening of a wound along the line of stitches |
| Dichotomous data | Binary data with only 2 categories |
| Heterogeneity | The degree of diversity between individual parts that have been grouped together |
| Hurley staging | A static measure of hidradenitis suppurativa (HS) disease severity from isolated lesions (stage I) to involvement of a whole skin region (stage III) |
| Inframammary | Region of skin under the breast |
| Inguinal region | Groin |
| Keratolytics | Topical treatments designed to remove excess keratin from the epidermis |
| Notch cell signalling pathways | Signalling receptors on cell membranes involved in cell differentiation and proliferation |
| Ordinal data | Data containing limited categories that can be ranked from lowest to highest |
| Perineal | The region between the thighs, bounded in the male by the scrotum and anus, and in the female, by the vulva and anus |
| Pilonidal sinus | An abnormal elongated channel in the skin of the buttock region, most often occurring at the top of the cleft of the buttocks |
| Placebo | A dummy treatment designed to mimic an active treatment in appearance |
| Purulent fluid | Pus |
| Sartorius staging | A hidradenitis suppurativa disease severity measure, which involves counting the number of skin lesions in each affected site |
| Seroma | A collection of sterile fluid under the skin following surgery |
| Sinus tract | An abnormal, elongated channel in the skin that permits the escape of fluid |
Description of the condition
Hidradenitis suppurativa (HS) is a chronic, painful inflammatory skin disease involving recurrent deep‐seated lesions; subsequent sinus tract formation; and scarring of apocrine gland‐bearing sites, in particular, the axillary, inguinal, and anogenital regions (Jemec 2012; Revuz 2009). It is also known as 'acne inversa' or 'Verneuil's disease' (Revuz 2009). The skin lesions consist of recurrent tender nodules or subcutaneous abscesses, which can lead to sinus tracts that discharge purulent fluid (Jemec 2012). Diagnosis is based on the clinical features of the skin lesions and their chronicity (Revuz 2009). A consensus disease definition has been proposed, involving a history of at least five discharging or painful skin lesions at typical sites (von der Werth 2000 a).
Epidemiology
The prevalence of HS is about 1% of the adult European population (Revuz 2008), with estimates ranging from 0.33% (Naldi 2006) to 4% (Jemec 1996). A 3:1 female:male ratio has been reported (Revuz 2009), and onset is usually in the second or third decades of life (Jemec 2012). The natural history of HS remains uncertain, but disease severity may be reduced in women after the menopause (von der Werth 2000 b). There is a recognised association with obesity and smoking (Sartorius 2009), although the condition may also be present in non‐smokers with a normal body mass index (Kromann 2014). Associations have also been reported with other inflammatory conditions, such as inflammatory bowel disease (van der Zee 2010), pyoderma gangrenosum (Hsiao 2010), and polycystic ovary syndrome (Kraft 2007).
Pathogenesis
The cause of HS is unknown (Jemec 2012). Potential causes can be grouped into genetic, environmental, endocrine, and microbiological factors.
A genetic cause is implicated in some individuals with a strong family history of HS, in which HS inheritance follows an autosomal dominant pattern (Fitzsimmons 1985). In some Chinese and European families with HS, loss‐of‐function mutations of the gamma‐secretase genes involved in Notch cell signalling pathways have been reported (Pink 2011; Wang 2010).
Environmental factors involve the well‐established associations with smoking and obesity (Sartorius 2009).
An endocrine cause has been suggested because disease onset typically occurs at the time of puberty, and HS severity may be reduced after the menopause in women (von der Werth 2000 b).
In terms of a possible microbiological cause, a number of bacteria may be isolated from affected skin sites, including relatively deep tissue levels, but it is uncertain whether this represents colonisation of sinuses or is pathogenic (Sartorius 2011). Histopathological examination of HS biopsy specimens suggests that follicular occlusion, in which the openings of hair follicles become blocked, is an early pathological event, leading to rupture of the follicle and subsequent inflammation (von Laffert 2011). This potential disease mechanism is supported by an association between HS and three conditions that exhibit this histopathological event, namely, pilonidal sinus; dissecting cellulitis of the scalp; and acne conglobata, a severe form of acne (Scheinfeld 2003).
Impact
Hidradenitis suppurativa has a large impact on peoples' lives because of chronic pain, which may prevent those affected from working during disease flares (Kimball 2012). Purulent discharge can produce odour and stain clothing, resulting in social stigma (Jemec 2012). The condition affects young adults, particularly women of child‐bearing age, and has an impact on sexual functioning, due to perineal involvement and embarrassment (Kurek 2012). Scarring from severe disease can produce considerable disability (Revuz 2009). The overall impact on quality of life is high, with a mean Dermatology Life Quality Index (DLQI) score of 11.3 in those with HS in secondary care (Sartorius 2010), which is equivalent to severe psoriasis (Finlay 2005). Higher rates of depression and anxiety are found in those with HS compared with controls (Shavit 2014). Support from healthcare practitioners, family, and friends is often lacking because of under‐recognition of HS by doctors and society in general (Ingram 2014).
Description of the intervention
More than 40 interventions have been described in the literature for the treatment of HS (Rambhatla 2011), with the evidence in many cases being limited to single case reports or small case series. The large number of interventions reflects a relative lack of effective therapy. Current management typically follows a stepwise approach depending on disease severity, commencing with topical treatment for mild disease, prolonged courses of oral antibiotics for moderate disease, and systemic immunosuppressants or surgery for more severe disease (Jemec 2012).
Systemic pharmacological agents for HS can be divided into a number of groups, namely, antibiotic monotherapy; combination antibiotic therapy; hormonal therapy; oral retinoids; oral immunosuppressants; biologic interventions, such as tumour necrosis factor‐alpha (TNF‐α) antagonists; and a group of other treatments (Jemec 2012).
Surgical interventions involve either the limited excision or radical wide excision of an involved region (Rambhatla 2011). Radical wide excision can be effective (Rambhatla 2011). However, the disease may recur at the edge of the excision margin, and this approach may not be practical if many regions are involved (Harrison 1987). Wound healing and postoperative scarring are further issues (Harrison 1987). Several wound healing methods have been reported, including direct closure, skin grafting, and secondary intention healing with a number of wound‐healing adjuncts (Rambhatla 2011). For the purposes of this review, we considered carbon dioxide laser excision or ablation therapy within the surgical treatment group, as the mode of action is by removal of skin and subcutaneous tissue (Madan 2008).
A group of 'other' interventions includes the neodymium‐doped yttrium aluminium garnet (Nd:YAG) laser, which selectively targets hair follicles; intense pulsed light; phototherapy; intralesional triamcinolone; botulinum toxin (Rambhatla 2011); and staphage lysate derived from lysis of Staphylococcus aureus (S. aureus) (Angel 1987).
Why it is important to do this review
Hidradenitis suppurativa is a relatively common, painful, and disabling skin condition affecting young adults, and it has a large impact on a person's quality of life (Sartorius 2010). Its flexural location means that it is hidden from view and has been largely neglected by society and the research community (Ingram 2014). Treatment is currently unsatisfactory, which has led clinicians to try many different interventions. The evidence base for many of these interventions is relatively weak, and there is little published guidance to aid decision‐making in the treatment of HS. Some randomised controlled trials (RCTs) have been performed, and the aim of this review was to summarise the evidence currently available and highlight knowledge gaps to promote further HS clinical trials.
The plans for this review were published as a protocol 'Interventions for hidradenitis suppurativa' (Ingram 2012).
Objectives
To assess the effects of interventions for hidradenitis suppurativa in people of all ages.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) of interventions for hidradenitis suppurativa (HS). We included the first phase of cross‐over trials, but excluded the second phase. This is because of the relatively long duration of carry‐over effects of HS interventions, such as immunomodulators. The review included within‐participant trials of topical therapies provided that comparison was made between the left and right sides of the same anatomical site.
Types of participants
All individuals of either sex and any age and ethnicity with a clinical diagnosis of HS made by a medical practitioner. Ideally, the clinical diagnosis conformed to the consensus disease definition (von der Werth 2000 a).
Types of interventions
The broad scope of this review meant that we included all interventions provided that they were assessed by at least one RCT. Preliminary literature searches indicated that over 40 interventions have been used for HS, although many lack RCT evidence. In order to structure the review, we grouped interventions into three categories, namely, pharmacological, surgical, and other interventions.
Pharmacological interventions
We subdivided these into topical and systemic therapies.
Topical treatments included antibiotics, keratolytics, and anti‐inflammatory agents.
Systemic treatments included single‐agent antibiotics, such as tetracyclines; combination antibiotic therapy, such as clindamycin and rifampicin; non‐steroidal anti‐inflammatories; the oral contraceptive pill; cyproterone acetate; finasteride; metformin; spironolactone; zinc gluconate; acitretin; isotretinoin; dapsone; prednisolone; methotrexate; azathioprine; ciclosporin; efalizumab; etanercept; adalimumab; infliximab; ustekinumab; and anakinra.
Surgical interventions
These included limited excision with primary closure, limited excision with primary closure including gentamicin implant, de‐roofing of sinus tracts, wide excision closed by a musculocutaneous flap, wide excision closed by a split skin graft, wide excision closed by a biosynthetic skin substitute, wide excision healed by secondary intention, wide excision healed by secondary intention using negative pressure dressing, wide excision healed by secondary intention using silastic foam dressing, carbon dioxide laser excision, and ablation.
Other interventions
These included neodymium‐doped yttrium aluminium garnet (Nd:YAG laser), 1450 nm diode laser, intense pulsed light, intralesional triamcinolone acetate, intralesional botulinum toxin, staphage lysate, bath psoralen‐UVA (ultraviolet A) phototherapy, photodynamic therapy, cryotherapy, radiotherapy, non‐ablative radiofrequency device, and chemical peels.
Comparisons
We compared the outcomes of an intervention with those of placebo or no intervention. Where head‐to‐head RCT data existed, we compared the efficacy of two interventions and permitted one of these interventions to include combination treatment with two therapies.
Types of outcome measures
Primary outcomes
Quality of life, measured by a validated dermatology‐specific scale.
Adverse effects (AEs) of interventions.
Secondary outcomes
Participant global self‐assessment.
Pain score.
Hidradenitis Severity Score (Sartorius 2009 or any alternative physician‐scoring system).
Physician Global Assessment.
Duration of remission, measured by the number of days until first new lesion or disease flare.
Timing of outcome assessments
We considered both the short‐term and longer‐term impact of the interventions. We defined the timing of the short‐term impact as 12 weeks after commencement of pharmacological interventions or 12 weeks after surgery or ablative laser treatment. If a 12‐week outcome measurement was not available, we selected the closest measurement greater than 12 weeks after onset of the intervention. We defined the timing of the longer‐term impact as nine months after onset of the intervention or the closest measurement greater than nine months after the intervention commenced.
Adverse outcomes
We divided adverse effects of interventions into serious ‐ if they resulted in death, hospital admission, or increased duration of hospital stay ‐ or non‐serious. For surgical complications, we subdivided adverse effects into immediate (less than two weeks after surgery) and late (greater than two weeks after surgery).
Economic data
There is a large variation in the costs of interventions for HS. We incorporated health resource usage data in the review if provided by the included studies.
'Summary of findings' table
We summarised the review results in 'Summary of findings' tables, which detail the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the main outcomes (Higgins 2011).
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
We searched the following databases up to 13 August 2015:
the Cochrane Skin Group Specialised Register using the following terms: (acne and invers*) or (hidradeniti* and suppurativ*) or velpeau* or verneuil*;
the Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library (Issue 7, 2015) using the search strategy in Appendix 1;
MEDLINE via Ovid (from 1946) using the strategy in Appendix 2;
EMBASE via Ovid (from 1974) using the strategy in Appendix 3; and
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy in Appendix 4.
Trials registers
We searched the following trials registers, using the terms hidradenitis, acne inversa, inverse acne, velpeau and verneuil, up to 18 August 2015:
The metaRegister of Controlled Trials (www.controlled‐trials.com).
The US National Institutes of Health Ongoing Trials Register (www.clinicaltrials.gov).
The Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
The World Health Organization International Clinical Trials Registry platform (www.who.int/trialsearch).
The EU Clinical Trials Register (https://www.clinicaltrialsregister.eu/).
Searching other resources
In order to identify other potential RCTs for inclusion, we (JRI, PNW, SLC, and ACK) handsearched the abstracts of proceedings from the following major dermatology conferences, which the Cochrane Skin Group Specialised Register does not already handsearch:
American Academy of Dermatology (AAD) (2008/2009);
British Association of Dermatologists (BAD) (2008/2009/2010);
European Academy of Dermatology and Venereology (EADV) (from 2006);
European Academy of Dermatology and Venereology Spring Symposium (from 2006);
European Society for Dermatological Research (ESDR) (2005/2006/2007/2008/2009);
International Investigative Dermatology (IID) (from 2003);
Society for Investigative Dermatology (SID) (2007/2008/2009); and
World Congress of Dermatology (from 2002).
Reference lists
We checked the reference lists of included and excluded studies for further references to relevant trials. We corresponded with authors where necessary to determine if a study met the criteria for inclusion.
Adverse effects
We did not perform a separate search for adverse effects of the target interventions. We examined data on adverse effects described in the included studies only.
Data collection and analysis
Selection of studies
Two authors (JRI and ACK) independently compared the titles and abstracts of the studies retrieved by the searches with the inclusion criteria. The two authors examined full texts for studies that potentially met the criteria or for studies whose abstracts did not provide sufficient information. We resolved any disagreements in terms of final study selection by referral to a third author (FK). We recorded the reasons for exclusion of studies in the 'Characteristics of excluded studies' tables.
Data extraction and management
Two pairs of authors (JRI and either PNW, SLC, or ADO) independently extracted data using a data extraction form based on the 'Checklist of items to consider in data collection or data extraction' found in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). There were no disagreements that required input from a third author. Two authors (JRI and PNW) piloted the data collection form prior to use. We entered the information collected into the 'Characteristics of included studies' tables.
We used the Grading of Recommendations Assessment, Development and Evaluation (GRADE) profiler (GRADEpro) to assess the quality of evidence for each review outcome. We downgraded evidence from the included RCTs from 'high quality' by one level for each serious study limitation found in the domains of risk of bias, inconsistency, indirectness, imprecision, and publication bias.
Assessment of risk of bias in included studies
Two authors (JRI and either PNW, SLC, or ADO) independently assessed the methodological quality of included studies using Cochrane's 'Risk of bias' tool (Higgins 2011). We graded the risk of bias as 'low', 'high', or 'unclear' for each of the following potential sources of bias:
(a) random sequence generation; (b) allocation concealment; (c) blinding of participants, personnel, and outcome assessment; (d) intention‐to‐treat analysis and incomplete outcome data; (e) selective outcome reporting (we checked trial databases to ensure that reported outcomes matched those prospectively listed); and (f) other sources of bias.
Measures of treatment effect
We expressed dichotomous outcome measures as risk ratios (RR) with 95% confidence intervals (CIs). We expressed continuous outcome measures as mean differences (MD) with 95% CIs. We analysed ordinal data from short outcome scales using the methods for dichotomous data, by combining adjacent categories. We treated longer outcome scales as continuous data.
We aimed to analyse time‐to‐event data, namely, the duration of remission, using survival analysis methods to express these as hazard ratios (HR), but did not find these data in our included studies.
Unit of analysis issues
We permitted the first phase of cross‐over trials and pooled the results with those from equivalent parallel group RCTs. We excluded the second phase of cross‐over trials in the context that adequate washout periods are relatively long and difficult to define for many of the HS interventions. We also excluded cluster‐randomised trials.
We permitted within‐participant trials of topical therapies, provided that a systemic effect of the intervention(s) was considered unlikely. For within‐participant trials, we considered as the unit of analysis one side of a particular anatomical location, such as the axillae or inguinal regions. We intended to perform a paired analysis, but paired data were unavailable, so we used parallel group analytical methods. We stipulated that within‐participant trials must randomise the left and right sides of the same anatomic site because different sites may respond differently to a particular treatment, and HS clinical scoring systems may result in different disease severity values depending on the site.
For trials with multiple intervention groups, we performed several pair‐wise comparisons if it was not appropriate to combine the intervention groups.
Dealing with missing data
Whenever possible, we contacted the original trial investigators to request missing data. We intended to attempt the imputation of missing data and explore the impact of missing data through sensitivity analyses, but did not attempt this because of the relatively small number of studies included in our review.
Assessment of heterogeneity
We assessed statistical heterogeneity using the I² statistic. If the value of the I² statistic exceeded 75%, we intended to avoid a meta‐analysis because of considerable heterogeneity and take a narrative approach instead (O'Rourke 1989). However, we found no I² statistic values in this range. An I² statistic of between 40% and 75% may represent substantial heterogeneity (Higgins 2011). For these outcomes, there were too few studies to allow adequate exploration of causes with subgroup analyses, so we used a random‐effects model and interpreted the results with caution.
Assessment of reporting biases
We intended to perform funnel plots and the Egger's test for publication bias (Egger 1997) and present funnel plots for an outcome measure if 10 or more studies contributed data. However, there were insufficient included studies to permit this assessment.
Data synthesis
We used a fixed‐effect model for an I² statistic value less than 40%. We used a random‐effects model for an I² statistic of between 40% and 75%. For dichotomous outcomes, we pooled risk ratios. For continuous outcomes, we combined either the weighted mean difference or standardised mean difference, depending on whether different scales had been used.
Subgroup analysis and investigation of heterogeneity
If the I² statistic suggested substantial heterogeneity, we intended to perform the following subgroup analyses of participant factors:
use of consensus HS disease definition versus no requirement for this definition;
disease duration less than five years versus disease duration greater than five years; and
disease severity of mild to moderate versus severe.
However, there were insufficient studies to permit these analyses.
Sensitivity analysis
We intended to perform an analysis in which we excluded studies at higher risk of bias and compared the results with the overall findings, the risk of bias being determined by allocation concealment quality (high, low, or unclear) and blinding of outcome assessment (high, low, or unclear). Again, there were too few included studies to allow this type of analysis.
Results
Description of studies
Results of the search
Please see the 'Characteristics of included studies' tables, the 'Characteristics of excluded studies' tables, and the 'Characteristics of ongoing studies' tables.
Electronic database searches retrieved 125 references, and we identified a further 15 references from trial registers and by handsearching conference abstracts. No duplicate publications were found, so we screened 140 titles and abstracts. Of these, three are studies awaiting classification (see the 'Characteristics of studies awaiting classification' tables), and a further 15 records relate to eight ongoing studies (see the 'Characteristics of ongoing studies' tables). We excluded 91 references based on the titles and abstracts. We obtained the full text for the 31 remaining references and excluded a further five studies: three were not randomised, one used another body site as the control, and one study was terminated due to lack of recruitment (NCT00722800) (see the 'Characteristics of excluded studies' tables). The remaining 26 records reported 12 studies, which we included. We summarise in Figure 1 the process of screening and selecting studies.
1.
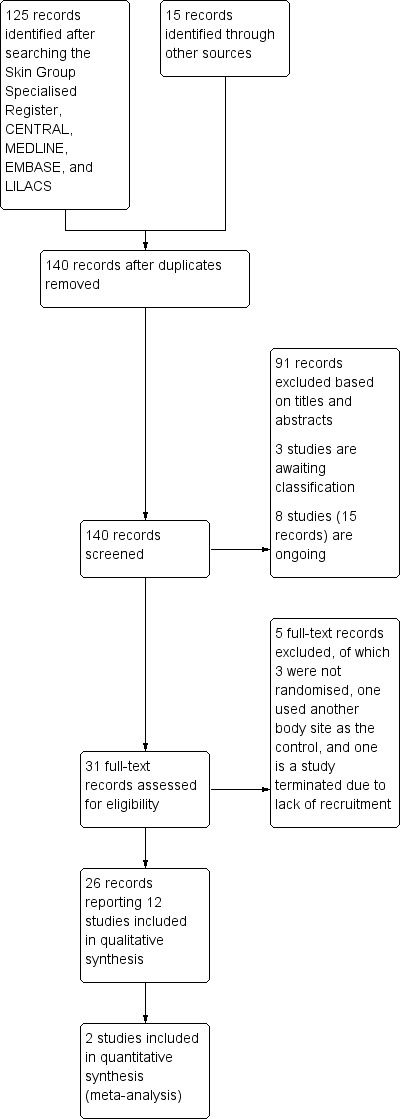
Study flow diagram.
Included studies
The 26 records selected report a total of 12 studies (see the 'Characteristics of included studies' tables), which included 615 participants. Two published articles (listed under Tierney 2009) reported initial and final results of the same trial, and Tierney 2009 has been used as the primary reference. Results from Kimball 2012 have been presented in abstract form at eight different conferences, accounting for most of the difference between the number of studies and the number of records available.
Designs
There were eight parallel group studies, seven with two arms, Adams 2010; Angel 1987; Buimer 2008; Clemmensen 1983; Grant 2010; Jemec 1998; Miller 2011, and one with three arms (Kimball 2012). Three studies investigating topical photodynamic therapy, intense pulsed light, and the neodymium‐doped yttrium aluminium garnet (Nd:YAG) laser, Fadel 2015; Highton 2011; Tierney 2009, were within‐participant studies comparing the left and right sides of the same anatomical site. Highton 2011 reported pooled results for three different anatomical sites, but following e‐mail contact, the authors were able to provide their results subdivided by anatomical location, permitting inclusion in our review. One study, Mortimer 1986, was a cross‐over study of systemic endocrine interventions with no washout period, so we included the results of the first phase only, because of the potential for carry‐over effects to alter the results of the second phase.
Of the 12 trials, three had an active comparator (Fadel 2015; Jemec 1998; Mortimer 1986), with the rest being controlled by no treatment (Highton 2011), by placebo (Adams 2010; Angel 1987; Clemmensen 1983; Grant 2010; Kimball 2012; Miller 2011), by a topical therapy that was also received by those in the intervention arm (Tierney 2009), or by surgery without the postoperative adjunct under investigation (Buimer 2008). The three arms of Kimball 2012 compared two different dosing schedules for adalimumab with a placebo arm.
Sample sizes
The number of participants in the included studies varied considerably. Most randomised less than 50 participants, but there were two larger trials of 154 participants, Kimball 2012, and 200 participants (Buimer 2008), respectively. The median number of participants for all of the included studies was 27.
Participants
The included studies all involved adults aged 18 years and over with a clinical diagnosis of hidradenitis suppurativa (HS). Most studies included men and women, with the exception of Mortimer 1986, which permitted women only because of the nature of the endocrine interventions. None of the studies made specific use of the consensus hidradenitis suppurativa (HS) definition available (von der Werth 2000 a). Six studies required baseline HS severity to be moderate to severe (Grant 2010; Highton 2011; Kimball 2012; Miller 2011; Mortimer 1986; Tierney 2009); one study required baseline HS severity to be mild to moderate (Hurley 1989 stage I to II) (Jemec 1998). Of the remaining five trials, three required 'active' disease (Adams 2010; Angel 1987; Buimer 2008), one permitted a range from mild to severe HS (Fadel 2015), and one did not stipulate a specific disease severity but required a HS disease duration of at least six months (Clemmensen 1983).
Interventions
We present the study results in the three intervention categories specified in the review methods: pharmacological, subdivided into topical and systemic; surgical; and other. There was one placebo‐controlled trial of topical therapy, clindamycin 1% solution (Clemmensen 1983). Another trial used clindamycin 1% solution as the active comparator in a head‐to‐head comparison with oral tetracycline (Jemec 1998). The systemic pharmacological category also includes a comparison of oral ethinylestradiol 50 mcg and norgestrel 500 mcg daily with oral ethinylestradiol 50 mcg and cyproterone acetate 50 mg (Mortimer 1986). Four studies investigated the anti‐TNF‐α (tumour necrosis factor‐alpha) therapies etanercept (Adams 2010), infliximab (Grant 2010), and adalimumab (Kimball 2012; Miller 2011), and we present these as a subgroup of systemic pharmacological therapies. Adalimumab was investigated at a dosing frequency of weekly after initial doses of 160 mg at week zero and 80 mg at week two (one arm of Kimball 2012) and every other week (EOW) after an initial dose of 80 mg at week zero (Miller 2011 and one arm of Kimball 2012). We have presented the two dosing frequencies as two distinct interventions because of the two‐fold difference in cumulative dose received. Efficacy and safety are likely to differ as a result, and because adalimumab is an expensive drug, economic considerations are also pertinent.
There was one surgical trial, which randomised participants who had undergone local excision of active HS lesions to insertion of a gentamicin‐collagen sponge prior to primary closure of the wound or primary closure alone (Buimer 2008). We placed four trials in the 'other' category. Highton 2011 investigated intense pulsed light twice per week for four weeks (420 nm; fluence: 7 to 10 J/cm²; pulse width: 30 to 50 msec) using a Harmony Laser. Tierney 2009 compared Nd:YAG laser treatment and topical clindamycin 1% with topical clindamycin alone. The Nd:YAG laser settings were fluence of 40 to 50 J/cm2, pulse duration of 20 ms, spot size of 10 mm for skin types I to III, and fluence of 25 to 35 J/cm2, pulse duration of 35 ms, spot size of 10 mm for skin types IV to VI. Fadel 2015 investigated 0.01% methylene blue gel photodynamic therapy (PDT), activated using intense pulsed light (630 nm; fluence: 25 J/cm²; pulse width: 20 msec) once every two weeks for a maximum of six months. The study compared free and niosomal methylene blue gel, the latter formulation being a surfactant‐based liposome intended to increase topical delivery of the photosensitiser. Angel 1987 compared staphage lysate given once weekly for 20 weeks, 0.3 ml delivered subcutaneously and 0.6 ml as an aerosol, with the same volumes of vehicle placebo. Staphage lysate was obtained by lysis of broth cultures of two strains of S. aureus using the Gratia bacteriophage, followed by ultrafiltration, and the vehicle placebo was the broth without the bacterial component. The study authors proposed that the therapeutic mechanism of the active intervention is induction of delayed type hypersensitivity.
Outcomes
In terms of our prespecified primary efficacy outcome, quality of life (QoL) measured by a validated dermatology‐specific scale, all four of the studies that investigated anti‐TNF‐α therapies, Adams 2010; Grant 2010; Kimball 2012; Miller 2011, included this outcome, but none of the other studies did. The Dermatology Life Quality Index (DLQI) (Finlay 1994), in which a lower score indicates better quality of life, was the scale used in each case.
All 12 trial reports included data ‐ with varying degrees of detail ‐ regarding our primary outcome to assess harm: the adverse effects of interventions. Buimer 2008 reported complications of surgery divided into immediate, i.e., one week after surgery, and delayed, i.e., at three months. Highton 2011 and Jemec 1998 reported only those adverse effects that led to treatment discontinuation. Kimball 2012 gave a detailed breakdown of all adverse effects including type and severity.
Considering our secondary prespecified outcomes, six studies included a participant global self‐assessment (Adams 2010; Clemmensen 1983; Highton 2011; Jemec 1998; Mortimer 1986; Tierney 2009). There was some variation in the method of assessment, including a visual analogue scale (VAS) (Jemec 1998) and an unvalidated participant questionnaire (Tierney 2009). Four studies measured pain using a VAS (Grant 2010; Jemec 1998; Kimball 2012 in a posthoc analysis; Miller 2011), and a further study also assessed pain with an ordinal scale from zero to five (Adams 2010). Five studies measured a Hidradenitis Severity Score based on the Sartorius 2003 scale (Fadel 2015; Highton 2011; Kimball 2012; Miller 2011; Tierney 2009). Four studies included a Physician Global Assessment as an ordinal scale, Adams 2010; Angel 1987; Grant 2010; Kimball 2012, and one study included a Physician Global Assessment as a VAS (Jemec 1998). Buimer 2008 formally assessed duration of remission in terms of the risk of HS recurrence three months after surgery, but the other studies did not systematically measure this.
The duration of the randomised controlled trial (RCT) phase of most of the included studies fitted with our prespecified short‐term impact timing definition of 12 weeks or more after onset of the intervention. One trial of intense pulsed light, Highton 2011, provided follow‐up data after 12 months, conforming to our longer‐term impact definition. The longest initial RCT phase for a pharmacological trial was six months (Fadel 2015; Mortimer 1986), and the median duration of all of the included studies was 16 weeks. Several pharmacological studies were longer in duration overall but incorporated a subsequent phase of open active treatment for all participants, without a control group, preventing inclusion of the efficacy data. The primary end point for one study, Grant 2010, was only eight weeks after onset of the infliximab intervention. We decided to still include the results of the study in our review because infliximab is known to have a rapid onset of action in other inflammatory dermatoses, such as psoriasis.
One study, Kimball 2012, provided economic outcome data, which measured the Total Work Productivity Impairment (TWPI) score from the Work Productivity and Activity Impairment‐Specific Health Problem (WPAI‐SHP) questionnaire.
Excluded studies
The 'Characteristics of excluded studies' tables contain the details of five studies that we excluded at the full text stage and one that was terminated early. We excluded three studies, Morgan 1983; Puri 2011; Soldin 2000, after the full text demonstrated that the trials were not randomised. We excluded Xu 2011 because in most participants, the Nd:YAG laser intervention was given to both sides of a body site, such as the axillae, and another site, such as the groin, acted as the untreated control. This failed to meet our unit of analysis inclusion criterion. We excluded a further study, NCT00722800, after reading the clinical trials database entry, which stated that the RCT recruited only four participants, of whom only two (both on placebo) completed the trial, so the study was too small to provide meaningful results.
Studies awaiting classification
Servant 2002 is a conference proceeding for which only the abstract title was available, and attempts to contact the authors were unsuccessful, preventing us from obtaining any further information. EUCTR2006‐005405‐67 and EUCTR2007‐000534‐39, trials of oral zinc and botulinum toxin injections, respectively, were both registered in a clinical trials database in 2007, and it was unclear whether the trials have been completed or are ongoing.
Ongoing studies
We found eight ongoing studies from our search of trial registries, summarised in the 'Characteristics of ongoing studies' tables, including the PIONEER I placebo‐controlled adalimumab phase three study, which has been reported in two conference proceedings but has not yet been reported in full in a peer‐reviewed journal.
Risk of bias in included studies
We made a judgement about the risk of bias for each study, which we presented in the 'Characteristics of included studies' tables, alongside the summary of each trial. Figure 2 reports our judgements about each 'Risk of bias' item, namely, random sequence generation, allocation concealment, performance bias, detection bias, attrition bias, reporting bias, and other factors, presented as percentages across all included studies. Figure 3 presents the 'Risk of bias' data for each individual study.
2.
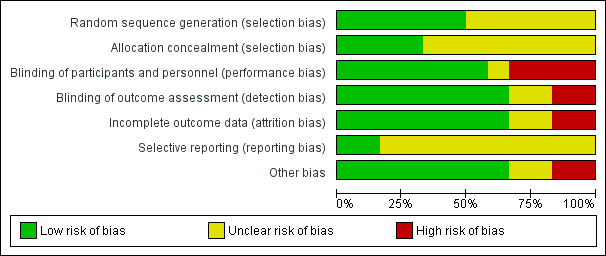
'Risk of bias' graph: review authors' judgements about each 'risk of bias' item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each 'risk of bias' item for each included study.
Allocation
Random sequence generation
We subdivided selection bias into the two distinct elements of random sequence generation and allocation concealment. Six of the included studies employed a reliable method of random sequence generation. Of these, four studies utilised computer randomisation (Grant 2010; Jemec 1998; Kimball 2012; Miller 2011), and two studies employed a coin toss for each participant (Fadel 2015; Tierney 2009). For the remaining six studies, no details of random sequence generation were available. Buimer 2008 reported an imbalance in baseline randomisation but did not explicitly state the intended randomisation ratio. If the ratio was intended to be 1:1, the probability of 124 of the 200 randomised individuals being allocated to the active intervention and only 76 to the control intervention is less than 0.001. The report stated that the imbalance occurred because of early cessation of the study, and it may be that the imbalance was due to lack of block randomisation, but the magnitude of the imbalance is surprising.
Allocation concealment
Eight of the 12 included study reports omitted sufficient details of allocation concealment. There was a low risk of allocation concealment bias in two of the studies, which used a pharmacy assignment code, Grant 2010, or a web/voice‐response system (Kimball 2012), and in two that used coin tossing (Fadel 2015; Tierney 2009), which we judged would not be a problem if participants had already been enrolled into the trial at that point in time. Miller 2011 used sequentially numbered containers, but we classified the risk of bias as 'unclear' because it was not specified whether the containers were opaque.
Blinding
Performance bias
We considered blinding of participants and personnel as effective in seven of the included studies (Adams 2010; Angel 1987; Clemmensen 1983; Grant 2010; Jemec 1998; Kimball 2012; Miller 2011), with comparators that were identical in appearance and had a similar adverse effect profile. In particular, Angel 1987 and Jemec 1998 employed double‐blind double‐dummy designs with subcutaneous injection and aerosol placebos, or oral and topical placebos, respectively. In addition, Kimball 2012 ensured that participants in each study arm received the same frequency of injections. Blinding was probably compromised in Buimer 2008 because a placebo was not used, and postoperatively, participants in the active intervention arm may have been able to detect the gentamicin‐collagen sponge inserted into the wound. In addition, no measures were described to ensure blinding of personnel. The study report for Mortimer 1986 did not provide sufficient details to assess blinding. There was no attempt to ensure either participant or study personnel blinding for the studies of Nd:YAG laser, intense pulsed light, and topical photodynamic therapy (Tierney 2009; Highton 2011; and Fadel 2015, respectively), which did not employ a sham treatment for the control side of the within‐participant comparison.
Detection bias
The same seven included studies that achieved a low risk of performance bias also achieved effective blinding of the outcome assessments (Adams 2010; Angel 1987; Clemmensen 1983; Grant 2010; Jemec 1998; Kimball 2012; Miller 2011), as a result of their identical comparators and the similar adverse effect profiles of the interventions compared. Fadel 2015; Highton 2011; and Tierney 2009 achieved a low risk of detection bias for investigator‐reported outcomes by ensuring that blinded outcome assessors performed the scoring evaluations. However, for the purposes of our review, we still graded Highton 2011 and Tierney 2009 as high risk for detection bias because only participant‐reported outcome results were in a suitable format for inclusion in our review, and they did not blind the participants. We rated detection bias as 'unclear' for Buimer 2008 and Mortimer 1986 because insufficient information was available.
Incomplete outcome data
Of the included studies that provided attrition bias data (all except Buimer 2008), 53/415 participants dropped out prior to measurement of their primary efficacy outcome, representing 13% of the total number of participants in these studies. Buimer 2008 randomised 200 participants but did not give the number of participants evaluated at each follow‐up point, resulting in an unclear risk of bias.
Mortimer 1986 provided the total number of participants who dropped out of the study but did not give details of their treatment allocation. Incomplete outcome data with no intention‐to‐treat analysis and a greater than 20% attrition rate resulted in a high risk of attrition bias in two of the included studies (Jemec 1998 and Tierney 2009). Kimball 2012 and Miller 2011 performed an explicit intention‐to‐treat analysis.
Selective reporting
We judged two studies, Adams 2010; Kimball 2012, to be at low risk of reporting bias as the type and timing of their outcomes were prospectively registered in a clinical trials database and were consistent with the final trial publication. Three other studies, Grant 2010; Miller 2011; Tierney 2009, were also prospectively registered, but insufficient details were provided to assess reporting bias, which we graded as 'unclear'. We could not find prospective trial registration for Angel 1987; Buimer 2008; Clemmensen 1983; Fadel 2015; Highton 2011; Jemec 1998; and Mortimer 1986, in the context that four out of seven of these studies were performed prior to widespread use of clinical trial databases. We also graded these seven studies as 'unclear' for risk of reporting bias.
Other potential sources of bias
In Miller 2011, baseline disease severity was higher in the adalimumab group than the control group, with a mean baseline DLQI of 16.1 for the adalimumab group compared with 8.3 for the placebo group, so the 'regression to the mean' phenomenon is likely to have affected the results. In Tierney 2009, three participants experienced episodes of cellulitis at non‐treatment sites requiring antibiotic therapy that may have affected study results. The report of Buimer 2008 was unclear regarding whether the unit of randomisation was at the level of participants or surgical procedures, so there was potential for bias if some participants underwent more than one procedure. Angel 1987 did not provide a funding source declaration, so we could not assess this potential source of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; Table 12
We summarised the review results in 12 'Summary of findings tables', which detail the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the main outcomes (Higgins 2011). We employed Grading of Recommendations Assessment, Development and Evaluation (GRADE) methodology to provide an assessment of the quality of the evidence for each of the primary and secondary outcomes listed below. In each case, we have given our assessment of the importance of the outcome ranging from nine (critical) to one (unimportant) in parentheses:
For the primary outcomes
Quality of life, measured by a validated dermatology‐specific scale (nine)
Adverse effects (AEs) of interventions (nine for serious AEs, seven for AEs leading to treatment discontinuation, four for non‐serious AEs)
For the secondary outcomes
Pain measured on a visual analogue scale (seven)
Hidradenitis Severity Score (Sartorius 2009 or any alternative physician‐scoring system) (six)
Participant global self‐assessment (five)
Physician Global Assessment (five)
Duration of remission, measured by the number of days until first new lesion or disease flare (five)
In this section, we present the results of studies that incorporated our prespecified primary and secondary outcomes (Types of outcome measures), subdivided into topical and systemic therapies, surgical interventions, and other interventions. We considered Jemec 1998, which compared oral tetracycline with topical clindamycin, in the systemic therapy group of studies because oral tetracycline was the intervention principally under investigation, with topical clindamycin as the active control intervention. Within systemic treatments, we dealt with anti‐TNF‐α therapies as a separate group because four of our included studies were of anti‐TNF‐α therapies. Meta‐analysis of results was possible only for the adalimumab every other week (EOW) intervention, combining the results of Kimball 2012 and Miller 2011. Data for all other comparisons were restricted to only single studies. We have provided forest plots for each intervention where outcome data were available in the required format, and we discuss other outcome data in the narrative. It was not possible to present funnel plots for any outcome measure because less than 10 studies contributed data in each case, and sensitivity and subgroup analyses were also impossible to conduct because of the paucity of included studies.
Topical therapies
(1) Topical clindamycin versus placebo
Clemmensen 1983 randomised 30 participants to receive clindamycin 1% solution or vehicle solution for 12 weeks, the frequency of application being unstated. The study report gave only P values for efficacy results without providing standard deviations, so only a forest plot for adverse effects was possible.
Clemmensen 1983 did not assess the primary outcome 'Quality of life' or the secondary outcomes 'Pain', 'Physician Global Assessment', and 'Duration of remission'.
Primary outcomes
2. Adverse effects
The study reported non‐serious adverse effects: "Local slight burning pain after application on a few occasions" for three participants treated with vehicle solution and two participants who received clindamycin 1% solution (risk ratio (RR) 0.72, 95% confidence interval (CI) 0.14 to 3.64; Analysis 1.1).
1.1. Analysis.
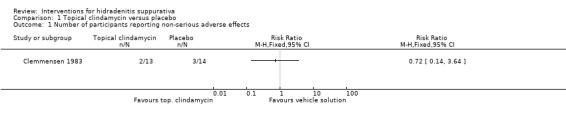
Comparison 1 Topical clindamycin versus placebo, Outcome 1 Number of participants reporting non‐serious adverse effects.
Secondary outcomes
1. Participant global self‐assessment
"Based on a diary, where the intensity and the number of elements and the frequency and the duration of recurrences were recorded," the following scoring system was used: much improved = + 2, improved = + 1, no change = 0, worse = ‐ 1, much worse = ‐ 2. There was no significant difference between the two groups, with the "cumulated score" at 12 weeks being + 8 and + 4 for 13 participants given clindamycin 1% solution and 14 vehicle‐treated participants, respectively.
3. Hidradenitis Severity Score
This was a composite scale composed of participant global assessment and the number of inflammatory nodules, abscesses, and pustules. Pustules had a lower weighting of one point per lesion compared with five points for the other lesion types and five points for a change in one level of the participant global self‐assessment ordinal scale. The authors reported a change in the overall score of all participants in each group from baseline and gave positive scores for a reduction in each lesion type and an improvement in the participant global assessment rating. A significant difference between the two groups was reported in favour of topical clindamycin (P < 0.01, statistical test not specifically indicated), with the "cumulated score" at 12 weeks being + 311 and ‐ 91 for 13 participants given clindamycin 1% solution and 14 vehicle‐treated participants, respectively.
Systemic therapies
(2) Oral tetracycline versus topical clindamycin
Jemec 1998 randomised 46 participants with mild to moderate HS (Hurley stage one or two) to receive oral tetracycline 500 mg twice daily and vehicle solution or the comparator of oral placebo and clindamycin 1% solution for 16 weeks. Thirty‐four participants completed the study and were included in the efficacy analyses.
Jemec 1998 did not assess the primary outcome 'Quality of life' and the secondary outcome 'Duration of remission'.
Primary outcomes
2. Adverse effects
Gastrointestinal upset resulted in treatment discontinuation by two participants, and another withdrew because of a suspected allergic reaction to a topical medication; however, the paper did not provide the treatment allocation in each case. Overall, there were three adverse events in the oral tetracycline group and five events in the topical clindamycin group, but the type, causality, and severity of the events were not specified.
Secondary outcomes
1. Participant global self‐assessment
This was measured on a visual analogue scale (VAS) from 0 to 100 mm, where 100 mm represented maximum disease severity. There was a statistically significant difference in favour of oral tetracycline with an effect size of ‐ 28 mm (mean difference (MD) ‐28, 95% CI ‐46.64 to ‐9.36; Analysis 2.1).
2.1. Analysis.
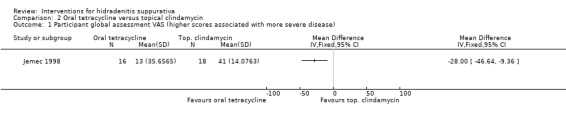
Comparison 2 Oral tetracycline versus topical clindamycin, Outcome 1 Participant global assessment VAS (higher scores associated with more severe disease).
2. Pain
This was measured on a VAS from 0 to 100 mm, where 100 mm represented maximum pain. No statistically significant difference was reported, with an effect size of 3 mm (MD 3.00, 95% CI ‐47.46 to 53.46; Analysis 2.2).
2.2. Analysis.
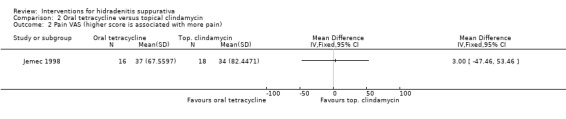
Comparison 2 Oral tetracycline versus topical clindamycin, Outcome 2 Pain VAS (higher score is associated with more pain).
3. Hidradenitis Severity Score
The number of nodules and abscesses was measured separately, with no statistically significant difference found for either lesion type between the two groups (MD 0.30, 95% CI ‐2.60 to 3.20; Analysis 2.3, and MD 0.80, 95% CI ‐0.83 to 2.43; Analysis 2.4, respectively).
2.3. Analysis.
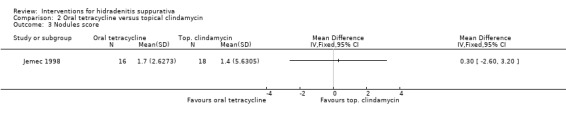
Comparison 2 Oral tetracycline versus topical clindamycin, Outcome 3 Nodules score.
2.4. Analysis.
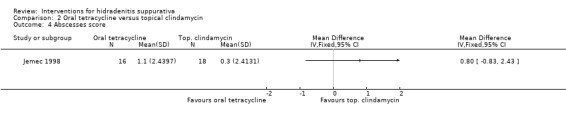
Comparison 2 Oral tetracycline versus topical clindamycin, Outcome 4 Abscesses score.
4. Physician Global Assessment
This was measured on a VAS from 0 to 100 mm, where 100 mm represented maximum disease severity. No statistically significant difference was reported, with an effect size of 9 mm (MD 9.00, 95% CI ‐12.61 to 30.61; Analysis 2.5).
2.5. Analysis.
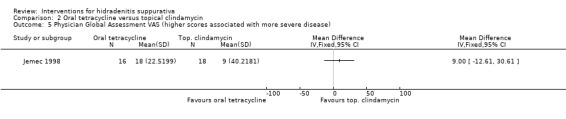
Comparison 2 Oral tetracycline versus topical clindamycin, Outcome 5 Physician Global Assessment VAS (higher scores associated with more severe disease).
(3) Ethinyloestradiol and cyproterone acetate versus ethinylestradiol and norgestrel
Mortimer 1986 was a 12‐month cross‐over study involving 24 female participants with moderate to severe HS comparing ethinylestradiol 50 mcg and norgestrel 500 mcg daily on days five to 25 of each menstrual cycle (E50 group) with ethinylestradiol 50 mcg and cyproterone acetate 50 mg on days five to 14 of each menstrual cycle (cyproterone acetate (CPA) group). In accordance with our review protocol, we present only the six‐months' results, at the end of the first phase of the study immediately prior to treatment cross‐over. At the six‐months' time point, efficacy results were available for 17 of the 24 participants.
Mortimer 1986 did not assess the primary outcome 'Quality of life' and the secondary outcomes 'Pain', 'Physician Global Assessment', and 'Duration of remission'.
Primary outcomes
2. Adverse effects
Two participants in the E50 group and two participants in the CPA group discontinued treatment because of "drug intolerance", but no details were given. Of the 18 participants completing the 12‐month trial treatment period, mild unspecified adverse effects (AEs) were reported for eight participants whilst they were taking E50, and five had AEs whilst taking CPA, which was statistically significant in favour of CPA (RR 0.53, 95% CI 0.29 to 0.98; Analysis 3.1). E50 caused "a variety of non‐specific side effects", with no further details available, while CPA caused weight gain, headaches, and breast soreness.
3.1. Analysis.
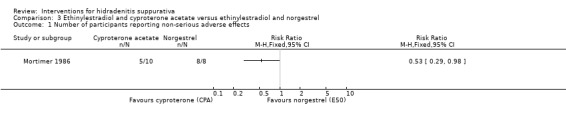
Comparison 3 Ethinylestradiol and cyproterone acetate versus ethinylestradiol and norgestrel, Outcome 1 Number of participants reporting non‐serious adverse effects.
Secondary outcomes
1. Participant global self‐assessment
This was measured on a VAS from 0 to 100 mm, where 50 mm represented baseline disease severity, and 100 mm represented being completely better. Results were read from figures published in the study report, which did not include the values in the body of the text. No significant difference was found, with an effect size of 6.00 mm (MD 6.00, 95% CI ‐15.98 to 27.98; Analysis 3.2).
3.2. Analysis.
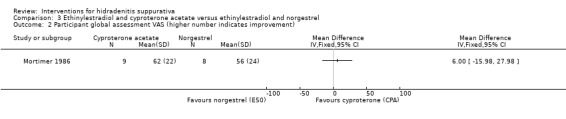
Comparison 3 Ethinylestradiol and cyproterone acetate versus ethinylestradiol and norgestrel, Outcome 2 Participant global assessment VAS (higher number indicates improvement).
3. Hidradenitis Severity Score
Mortimer 1986 did not report the six‐months' results prior to cross‐over.
Systemic therapies ‐ anti‐TNF‐α therapies
(4) Etanercept versus placebo
Adams 2010 randomised 20 HS participants with "active disease" to receive etanercept 50 mg twice weekly by subcutaneous injection or placebo injections for 12 weeks, followed by a second 12‐week phase during which all participants received open label etanercept at the 50 mg twice weekly dose. Seventeen participants completed the randomised phase of the trial, of whom 14 completed the subsequent open label phase. The study report gave only P values for results without providing original data and standard deviations, so we could not produce forest plots. We present efficacy results at the end of the randomised trial phase, at 12 weeks, below.
Adams 2010 did not assess the secondary outcomes 'Hidradenitis Severity Score' and 'Duration of remission'.
Primary outcomes
1. Quality of life
The Dermatology Life Quality Index (DLQI) score was measured and not found to differ significantly between the two groups (P = 0.12, Mantel‐Haenszel test).
2. Adverse effects
There were no serious AEs, and no participants withdrew from treatment because of AEs. The trial report states: "Mild injection site reactions only" associated with treatment.
Secondary outcomes
1. Participant global self‐assessment
An ordinal participant global assessment scale was used from zero (good) to five (severe). No significant difference was found between the two groups (P = 0.41, Mantel‐Haenszel test).
2. Pain
An ordinal pain scale was used from zero (good) to five (severe). No significant difference was found between the two groups (P = 0.77, Mantel‐Haenszel test).
4. Physician Global Assessment
A global assessment was made based on discharge from lesions, erythema, and tenderness on palpation. Clinical response was defined as the proportion of participants with clear or mild HS at the 12‐week assessment, and there was no discernible difference between the two groups (P > 0.99, Fisher's exact test).
(5) Infliximab versus placebo
Grant 2010 randomised 38 participants with moderate to severe HS to receive intravenous injections of infliximab 5 mg/kg at weeks zero, two, and six or placebo injections, reporting their primary outcomes at week eight. Five patients in the placebo arm withdrew prior to week eight, mainly due to lack of efficacy, and were not included in the results, in the absence of an intention‐to‐treat analysis. The total trial duration was 52 weeks, with a subsequent open label treatment phase during which all participants received infliximab, followed by an observation phase.
Grant 2010 did not assess the secondary outcome 'Participant global self‐assessment'.
Primary outcomes
1. Quality of life
There was a decrease in mean DLQI score from baseline to week eight in the infliximab group compared with those on placebo, with an effect size of 8.4 points (P = 0.003, Wilcoxon rank sum test).
2. Adverse effects
There were two serious adverse events in the infliximab group prior to week eight, a pregnancy (outcome not reported) and hypertension requiring hospitalisation, compared with none in the placebo group. Other adverse effects reported in the first eight weeks were only mild in both groups. In the subsequent phase of open infliximab treatment, four participants previously given placebo experienced infusion reactions, of whom three withdrew from treatment as a result. No tuberculosis reactivation or opportunistic infections were reported during the total 12‐month trial period.
Secondary outcomes
2. Pain
This was measured on a VAS from 0 to 100 mm, where 100 mm represented maximum pain. At week eight, there was a significant decrease in pain in the infliximab group compared with those given placebo, with an effect size of 39.2 mm (P < 0.001, Wilcoxon rank sum test).
3. Hidradenitis Severity Score
An unvalidated "Hidradenitis Suppurativa Severity Index" (HSSI) was used, incorporating the number of body sites and body surface area involved; total number of HS lesions; number of dressing changes; and pain VAS score, in which a lower overall score represented improvement in disease severity. The prespecified primary outcome of Grant 2010 was the proportion of participants with ≥ 50% improvement in HSSI score from week zero to week eight. In the absence of the raw trial data, we used this result as our Hidradenitis Severity Score secondary outcome and presented the data as follows: 4/15 participants on infliximab and 1/18 participants on placebo had at least a 50% decrease in the HS Severity Index. No significant difference was found between the two groups (RR 4.80, 95% CI 0.60 to 38.48; Analysis 4.1).
4.1. Analysis.
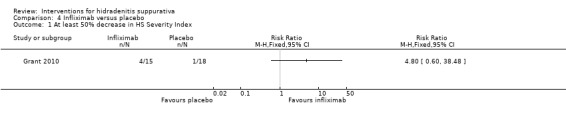
Comparison 4 Infliximab versus placebo, Outcome 1 At least 50% decrease in HS Severity Index.
4. Physician Global Assessment
A six‐point scale was used to assess disease severity relative to baseline: clear (100% improvement), excellent (75% to 99% improvement), good (50% to 74% improvement), fair (25% to 49% improvement), slight (1% to 24% improvement), or worse. We defined clinical response as a clear, excellent, or good response to treatment at week eight, regardless of baseline severity, and found a statistically significant difference in favour of infliximab at week eight (RR 4.80, 95% CI 1.66 to 13.90; Analysis 4.2).
4.2. Analysis.
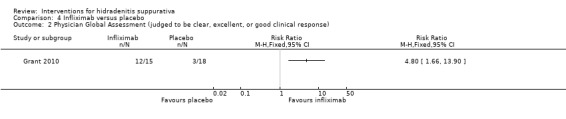
Comparison 4 Infliximab versus placebo, Outcome 2 Physician Global Assessment (judged to be clear, excellent, or good clinical response).
5. Duration of remission
This was defined by the study authors as the time period during the observation phase until an increase of at least 40% in the HSSI score obtained after eight weeks of infliximab. Only five participants entered the observation phase, and of these, three participants relapsed (two originally on infliximab, one originally on placebo).
(6) Adalimumab every other week versus placebo
Two studies, Kimball 2012 and Miller 2011, assessed this comparison, permitting a meta‐analysis of results for several outcome measures. One arm of Kimball 2012 compared adalimumab 40 mg given subcutaneously every other week (EOW), after loading doses of 80 mg at week zero and 40 mg at week one, with placebo injections, reporting primary outcomes at week 16. There were 52 participants in the adalimumab EOW arm and 51 participants in the control arm. Miller 2011 differed slightly in that adalimumab 40 mg EOW was given to those in the active treatment study arm after a single loading dose of 80 mg at week zero, with no extra loading dose at week one, and outcomes were assessed earlier, at week 12. Miller 2011 was a smaller study that originally intended to recruit 30 participants in a 2:1 active:placebo ratio but was halted earlier than planned due to reaching the expiration date of study medication, recruiting 15 participants to the adalimumab EOW arm and six participants for the placebo arm. Kimball 2012 provided data analysis using two methods for handling missing data: last observation carried forward (LOCF) and imputation. However, Miller 2011 used only LOCF, so for this meta‐analysis, we used the LOCF Kimball 2012 data.
These studies did not assess the secondary outcomes 'Participant global self‐assessment' and 'Duration of remission'.
Primary outcomes
1. Quality of life
Data for change in DLQI score were available for 124 participants, and there was no statistically significant difference between adalimumab EOW and placebo upon pooling results of the two trials: effect size ‐1.61 in favour of active treatment (MD ‐1.61, 95% CI ‐3.86 to 0.64; Analysis 5.1).
5.1. Analysis.
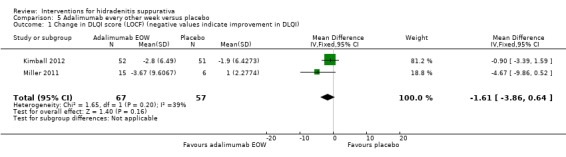
Comparison 5 Adalimumab every other week versus placebo, Outcome 1 Change in DLQI score (LOCF) (negative values indicate improvement in DLQI).
2. Adverse effects
Meta‐analysis of the frequency of serious adverse effects found no statistically significant difference between adalimumab EOW and placebo (RR 1.47, 95% CI 0.26 to 8.44; Analysis 5.2). There was also no statistically significant difference in the frequency of treatment discontinuation due to AEs (RR 4.91, 95% CI 0.24 to 99.74; Analysis 5.3). We meta‐analysed the proportion of participants with infectious adverse effects using a random‐effects model, because of an I² statistic of 40%, and we again found no statistically significant difference between the two groups (RR 1.60, 95% CI 0.57 to 4.53; Analysis 5.4).
5.2. Analysis.
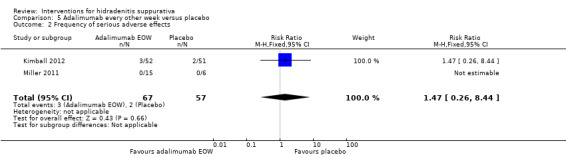
Comparison 5 Adalimumab every other week versus placebo, Outcome 2 Frequency of serious adverse effects.
5.3. Analysis.
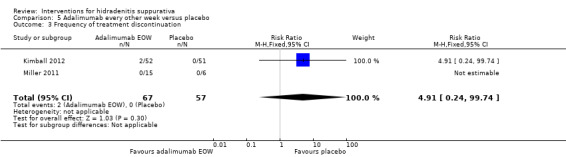
Comparison 5 Adalimumab every other week versus placebo, Outcome 3 Frequency of treatment discontinuation.
5.4. Analysis.
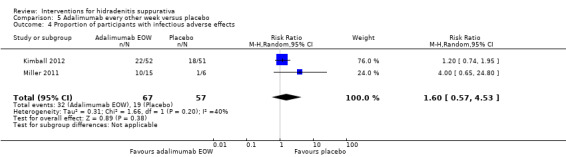
Comparison 5 Adalimumab every other week versus placebo, Outcome 4 Proportion of participants with infectious adverse effects.
Secondary outcomes
2. Pain
Both Kimball 2012 and Miller 2011 measured participant pain on a VAS; however, the results were not suitable for combination in a meta‐analysis. Miller 2011 provided mean pain scores whereas Kimball 2012 performed a posthoc analysis of the proportion of participants achieving a 30% or greater reduction and a 10 mm or greater absolute reduction in VAS score among participants with at least a 10 mm VAS score at baseline. In Miller 2011, there was no statistically significant difference in the change in VAS score between adalimumab EOW and placebo groups at week 12: effect size ‐16.57 mm, (MD ‐16.57, 95% CI ‐55.28 to 22.14; Analysis 5.5). Of the 95 participants in Kimball 2012 with a baseline pain score greater than 10 mm, there was no statistically significant difference in the proportion of participants achieving at least a 30% reduction at week 16 (RR 1.34, 95% CI 0.73 to 2.43; Analysis 5.6).
5.5. Analysis.
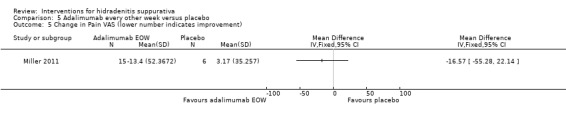
Comparison 5 Adalimumab every other week versus placebo, Outcome 5 Change in Pain VAS (lower number indicates improvement).
5.6. Analysis.
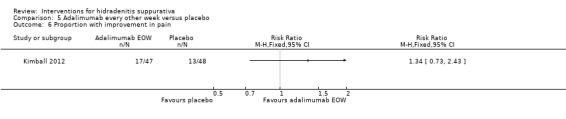
Comparison 5 Adalimumab every other week versus placebo, Outcome 6 Proportion with improvement in pain.
3. Hidradenitis Severity Score
Standardised mean difference was used to pool the results of the two trials because Miller 2011 used the unmodified Sartorius 2003 scale whereas Kimball 2012 used a modified version of the scale. We employed a random‐effects model for the meta‐analysis because of an I² statistic of 59% and downgraded the outcome in evidence quality for this reason. There was no statistically significant difference between adalimumab EOW and placebo for the change in Sartorius scale score: effect size ‐0.42 (MD‐0.42, 95% CI ‐1.22 to 0.37; Analysis 5.7).
5.7. Analysis.
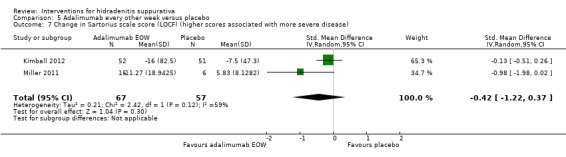
Comparison 5 Adalimumab every other week versus placebo, Outcome 7 Change in Sartorius scale score (LOCF) (higher scores associated with more severe disease).
4. Physician Global Assessment
Only Kimball 2012 provided Physician Global Assessment (PGA) data for 103 participants. The PGA was classified as clear, minimal, mild, moderate, severe, or very severe disease, and clinical response was defined as clear, minimal, or mild disease with at least a two‐grade improvement from baseline. There was no statistically significant difference between the adalimumab EOW group and placebo group (RR 2.45, 95% CI 0.50 to 12.07; Analysis 5.8).
5.8. Analysis.
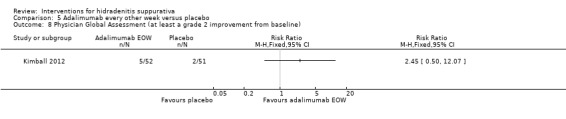
Comparison 5 Adalimumab every other week versus placebo, Outcome 8 Physician Global Assessment (at least a grade 2 improvement from baseline).
Economic outcomes
Kimball 2012 measured the Total Work Productivity Impairment (TWPI) score from the Work Productivity and Activity Impairment‐Specific Health Problem (WPAI‐SHP) questionnaire, with scores ranging from zero (no impairment) to 100. There was no significant difference between adalimumab EOW and placebo: effect size using imputation ‐5.40 (MD ‐5.40, 95% CI ‐14.69 to 3.89; Analysis 5.9); effect size using LOCF ‐3.80 (MD ‐3.80, 95% CI ‐15.17 to 7.57; Analysis 5.10).
5.9. Analysis.
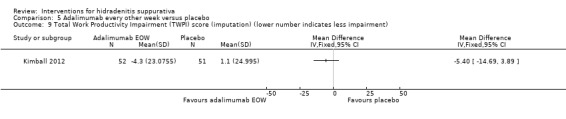
Comparison 5 Adalimumab every other week versus placebo, Outcome 9 Total Work Productivity Impairment (TWPI) score (imputation) (lower number indicates less impairment).
5.10. Analysis.

Comparison 5 Adalimumab every other week versus placebo, Outcome 10 Total Work Productivity Impairment (TWPI) score (LOCF) (lower number indicates less impairment).
(7) Adalimumab weekly versus placebo
The third arm of Kimball 2012 compared subcutaneous adalimumab 40 mg weekly, from weeks four to 15 after loading doses of 160 mg at week zero and 80 mg at week two, with placebo injections, reporting primary end points after 16 weeks. There were 51 participants in the adalimumab weekly arm and 51 participants in the control arm. For missing data, the authors had stated in their protocol that they would use the LOCF method; however, they preferred the imputation method at the time of results analysis. Both analyses were included in the trial publication, and we included results using both methods in our review, particularly because it has an effect on the modified Sartorius scale score results (see below).
Kimball 2012 did not assess the secondary outcomes 'Participant global self‐assessment' and 'Duration of remission'.
Primary outcomes
1. Quality of life
At 16 weeks, adalimumab weekly produced a statistically significantly greater reduction in DLQI score compared with placebo, the effect size being approximately four points: imputation method ‐4.00 points, (MD ‐4.00, 95% CI ‐6.49 to ‐1.51; Analysis 6.1), LOCF method ‐4.10 points (MD ‐4.10, 95% CI ‐6.59 to ‐1.61; Analysis 6.2).
6.1. Analysis.
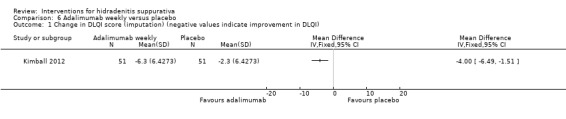
Comparison 6 Adalimumab weekly versus placebo, Outcome 1 Change in DLQI score (imputation) (negative values indicate improvement in DLQI).
6.2. Analysis.
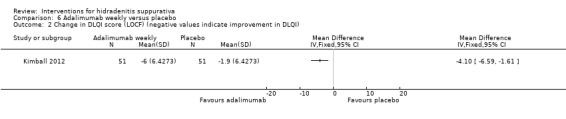
Comparison 6 Adalimumab weekly versus placebo, Outcome 2 Change in DLQI score (LOCF) (negative values indicate improvement in DLQI).
2. Adverse effects
There was no significant difference in the frequency of serious adverse events between the adalimumab weekly and placebo groups (RR 2.00, 95% CI 0.38 to 10.44; Analysis 6.3). There was also no difference in the frequency of treatment discontinuation between the two groups (RR 5.00, 95% CI 0.25 to 101.63; Analysis 6.4). The proportion of participants with infectious adverse effects did not differ between the two groups (RR 0.94, 95% CI 0.55 to 1.62; Analysis 6.5).
6.3. Analysis.
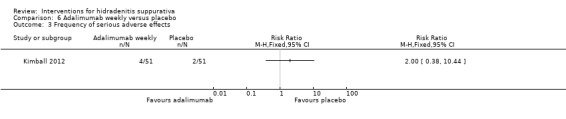
Comparison 6 Adalimumab weekly versus placebo, Outcome 3 Frequency of serious adverse effects.
6.4. Analysis.
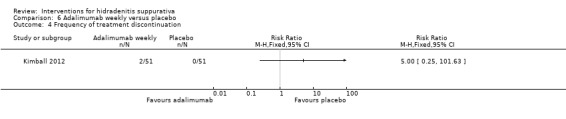
Comparison 6 Adalimumab weekly versus placebo, Outcome 4 Frequency of treatment discontinuation.
6.5. Analysis.
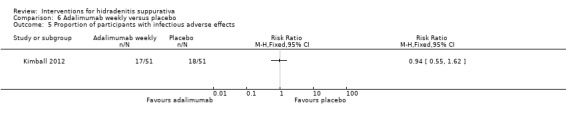
Comparison 6 Adalimumab weekly versus placebo, Outcome 5 Proportion of participants with infectious adverse effects.
Secondary outcomes
2. Pain
As explained previously, Kimball 2012 defined treatment success for this domain as a 30% or greater reduction and a 10 mm or greater absolute reduction in pain VAS score compared with baseline. For the 96 participants with at least a 10 mm VAS score at baseline, there was a statistically significant improvement in the adalimumab weekly group compared with placebo, (RR 1.77, 95% CI 1.02 to 3.07; Analysis 6.6).
6.6. Analysis.

Comparison 6 Adalimumab weekly versus placebo, Outcome 6 Proportion of participants with improvement in pain.
3. Hidradenitis Severity Score
Using the imputation method to handle missing data, there was an effect size of ‐23.00 points for the change in modified Sartorius scale score between the two groups in favour of adalimumab weekly, but the result failed to reach significance at the 95% confidence level (MD ‐23.00, 95% CI ‐50.16 to 4.16; Analysis 6.7). Using the LOCF method, there was a significant difference in favour of adalimumab weekly with an effect size of ‐22.50 points (MD ‐22.50, 95% CI ‐41.93 to ‐3.07; Analysis 6.8).
6.7. Analysis.
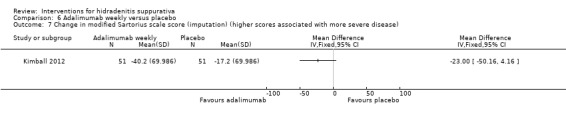
Comparison 6 Adalimumab weekly versus placebo, Outcome 7 Change in modified Sartorius scale score (imputation) (higher scores associated with more severe disease).
6.8. Analysis.
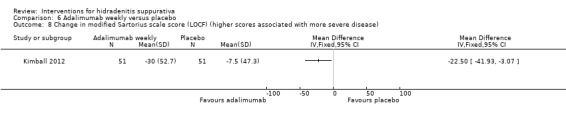
Comparison 6 Adalimumab weekly versus placebo, Outcome 8 Change in modified Sartorius scale score (LOCF) (higher scores associated with more severe disease).
4. Physician Global Assessment
As described above, clinical response was defined as clear, minimal, or mild disease with at least a two‐grade improvement from baseline. Adalimumab weekly was statistical significantly superior to placebo (RR 4.50, 95% CI 1.02 to 19.81; Analysis 6.9).
6.9. Analysis.
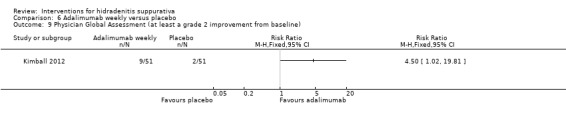
Comparison 6 Adalimumab weekly versus placebo, Outcome 9 Physician Global Assessment (at least a grade 2 improvement from baseline).
Economic outcomes
Both the imputation method and LOCF method found a statistically significant difference in TWPI scores between the two groups in favour of adalimumab weekly. The imputation method effect size was ‐19.50 points (MD ‐19.50, 95% CI ‐30.07 to ‐8.93; Analysis 6.10), and the LOCF method effect size was ‐20.30 (MD ‐20.30, 95% CI ‐32.51 to ‐8.09; Analysis 6.11).
6.10. Analysis.

Comparison 6 Adalimumab weekly versus placebo, Outcome 10 Total Work Productivity Impairment (TWPI) score (imputation) (lower number indicates less impairment).
6.11. Analysis.

Comparison 6 Adalimumab weekly versus placebo, Outcome 11 Total Work Productivity Impairment (TWPI) score (LOCF) (lower number indicates less impairment).
Surgical interventions
(8) Gentamicin sponge versus primary closure alone
Buimer 2008 performed excision and primary closure in 200 HS participants of "Symptomatic lesion(s), i.e. those with discharge, inflammation, infiltration, or suspected abscesses", randomising 124 to insertion of a gentamicin‐collagen sponge prior to closure and 76 to primary closure alone. Study participants were reviewed one week and then three months after surgery.
Buimer 2008 did not assess the primary outcome 'Quality of life' and the secondary outcomes 'Participant global self‐assessment', 'Pain', 'Hidradenitis Severity Score', and 'Physician Global Assessment'.
Primary outcomes
2. Adverse effects
Wound complications were classified as dehiscence, infection, dehiscence and infection, or seroma. At one week, there was no difference in the overall rate of surgical complications (RR 0.78, 95% CI 0.58 to 1.05; Analysis 7.1). There was also no difference in complication rates between the two groups after three months (RR 0.90, 95% CI 0.50 to 1.62; Analysis 7.2).
7.1. Analysis.
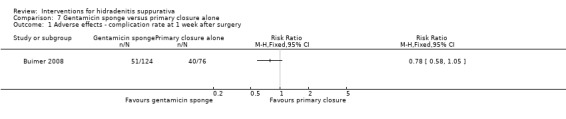
Comparison 7 Gentamicin sponge versus primary closure alone, Outcome 1 Adverse effects ‐ complication rate at 1 week after surgery.
7.2. Analysis.
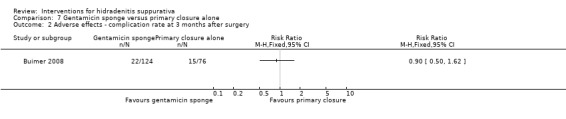
Comparison 7 Gentamicin sponge versus primary closure alone, Outcome 2 Adverse effects ‐ complication rate at 3 months after surgery.
Secondary outcome
5. Duration of remission
Recurrence rate was assessed three months after surgery, and there was no difference between the gentamicin sponge and primary closure alone groups (RR 0.96, 95% CI 0.68 to 1.34; Analysis 7.3).
7.3. Analysis.
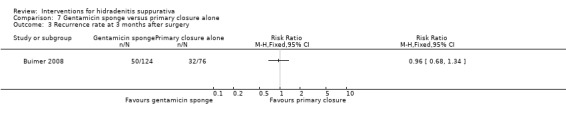
Comparison 7 Gentamicin sponge versus primary closure alone, Outcome 3 Recurrence rate at 3 months after surgery.
Other interventions
(9) Intense pulsed light versus no treatment
Highton 2011 performed a within‐participant comparison, randomising one side of an anatomical region bilaterally affected by HS to receive intense pulsed light (IPL) twice per week for four weeks and the other side to receive no treatment. Seventeen participants underwent treatment: 12 with axillary HS, four with groin involvement, and one with inframammary disease. Outcomes were measured immediately post‐treatment and at three, six, and 12 months later, with no primary outcome time point stated. Following e‐mail communication, the authors provided a breakdown of results for each anatomical region. Ideally, paired data should be analysed for a within‐participant study (Higgins 2011); however, these data were not available, so we employed parallel group methods in the context that the interventions were unlikely to exert an effect beyond the borders of the treated area.
Highton 2011 did not assess the primary outcome 'Quality of life' and the secondary outcomes 'Pain', 'Physician Global Assessment', and 'Duration of remission'.
Primary outcomes
2. Adverse effects
One participant withdrew due to treatment‐related pain (site of treatment unknown). No other details were provided regarding AEs.
Secondary outcomes
1. Participant global self‐assessment
Participant satisfaction with IPL treatment was measured using an unvalidated Likert scale that asked participants to give a rating of worse, unchanged, fair, good, excellent, or clear compared with baseline for each side of the anatomical region of interest. Data were entered as a dichotomous variable, defining treatment success as good, excellent, or cleared. Overall, IPL produced significantly higher participant satisfaction than no treatment (RR 9.67, 95% CI 2.10 to 46.43; Analysis 8.1). Results from subgroup analysis of the skin region treated were statistically significant for the axillary site alone (RR 21.00, 95% CI 1.37 to 322.28) but not for groin treatment, in part due to a smaller effect size and also because of the small number of participants (RR 5.00, 95% CI 0.31 to 79.94).
8.1. Analysis.
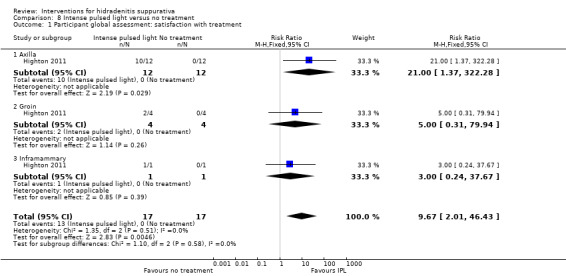
Comparison 8 Intense pulsed light versus no treatment, Outcome 1 Participant global assessment: satisfaction with treatment.
3. Hidradenitis Severity Score
The "fitted mean" percentage change from baseline in Sartorius 2003 score was measured, and we report the data after three months and 12 months of follow up, in keeping with the prespecified short‐term and longer‐term outcome definitions of our review. Standard deviations were not available, so we report the raw data for each anatomical location. At three months, there was a mean effect size difference of ‐46% for axillae and ‐58% for the groin, comparing IPL‐treated sides with no treatment. At 12 months, there was a mean effect size difference of ‐37% for axillary sites and ‐38% for groin sites. Data for the single participant with inframammary disease were not available.
(10) Nd:YAG laser versus topical control
Tierney 2009 performed a within‐participant RCT in which one side of an anatomical site received four treatments with the Nd:YAG laser at monthly intervals and topical antimicrobials, and the other side received only topical antimicrobial therapy. Benzoyl peroxide wash 10% and clindamycin 1% gel or 1% lotion were given as the topical antimicrobial control, with an unstated frequency of application. Thirty‐four anatomical sites were randomised in 22 participants, and results were available for 25 anatomical sites in 17 participants at the initial assessment point, three months after the first treatment (10 axillary sites, 11 groin sites, and four inframammary). Assessments were repeated after a subsequent two‐month observation phase.
Tierney 2009 did not assess the primary outcome 'Quality of life' and the secondary outcomes 'Pain', 'Physician Global Assessment', and 'Duration of remission'. Paired data were unavailable for this within‐participant study, so we undertook a parallel group analysis in its absence.
Primary outcomes
2. Adverse effects
One serious adverse event was reported, a pregnancy of unknown outcome. No participants withdrew because of treatment‐related AEs. Forty per cent of participants experienced pain related to laser treatment; however, "this pain did not interfere with their daily activities and was transient in nature." There were three episodes of cellulitis at non‐treatment sites, which required antibiotic therapy.
Secondary outcomes
1. Participant global self‐assessment
In answer to an unvalidated questionnaire, 92% of the unblinded participants indicated that the laser treatment was more effective than other medical treatment (significance not stated).
3. Hidradenitis Severity Score
The Sartorius 2003 scale was used, modified by addition of erythema, oedema, pain, and purulent discharge domains for each anatomic site ("modified HS‐LASI"). At three months, for all the treated sites combined, there was a significant difference in modified HS Lesion, Area and Severity Index (HS‐LASI) score between the two sides in favour of Nd:YAG laser therapy, with an effect size of ‐14.03 points (MD ‐14.03, 95% CI ‐18.84 to ‐9.22 points; Analysis 9.1). Subgroup analysis showed that the benefit remained significant for each of the three regions (axilla ‐18.70, 95% CI ‐26.82 to ‐10.58; groin ‐12.60, 95% CI ‐20.28 to ‐4.92; inframammary ‐9.80, 95% CI ‐19.31 to ‐0.29).
9.1. Analysis.
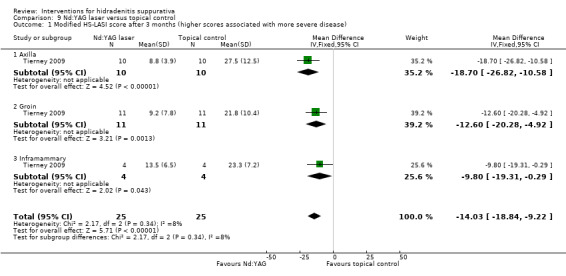
Comparison 9 Nd:YAG laser versus topical control, Outcome 1 Modified HS‐LASI score after 3 months (higher scores associated with more severe disease).
Following the subsequent two‐month observation period, results were given as the percentage change in modified HS‐LASI score compared with baseline. At this five‐month time point, for all the treated sites combined, there was a significant difference between the two sides in favour of Nd:YAG laser therapy, with an effect size of ‐51.40% (MD ‐51.40, 95% CI ‐66.36 to ‐36.43%; Analysis 9.2). Breakdown of results demonstrated that the benefit remained significant for each anatomical site (axilla ‐58.90%, 95% CI ‐78.82 to ‐38.98%; groin ‐38.70%, 95% CI ‐63.43 to ‐13.97%; inframammary ‐57.30%, 95% CI ‐113.86 to ‐0.74%).
9.2. Analysis.
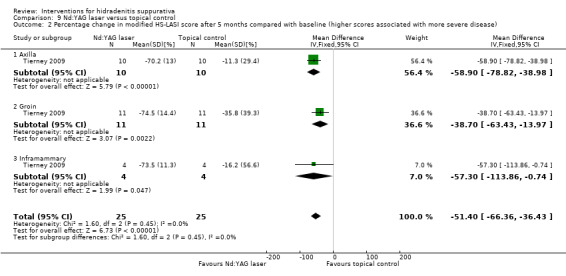
Comparison 9 Nd:YAG laser versus topical control, Outcome 2 Percentage change in modified HS‐LASI score after 5 months compared with baseline (higher scores associated with more severe disease).
(11) Niosomal methylene blue gel PDT versus free methylene blue gel PDT
Fadel 2015 was a within‐participant trial in which one side of an anatomical site was randomised to receive niosomal methylene blue (NMB) gel PDT or free methylene blue (FMB) gel PDT once every two weeks for up to six months. Treatment was discontinued early if there was no improvement after two consecutive sessions. One of the 11 randomised participants was withdrawn prior to receiving any study treatments because of a change in diagnosis to Crohn's disease. Of the remaining 10 participants, four had disease predominantly located in the axilla, two in the groin, three in the buttock region, and one in the inframammary region. In two participants, disease was localised to either one groin or one buttock, and direct contact with the authors confirmed that in these cases, one intervention was randomised to the upper part of the lesion, and the lower part received the other intervention.
Fadel 2015 did not assess the primary outcome 'Quality of life' and the secondary outcomes 'Participant global self‐assessment', 'Pain', and 'Physician Global Assessment'. Paired data were unavailable for this within‐participant study, so we undertook a parallel group analysis in its absence.
Primary outcomes
2. Adverse effects
No serious adverse events were reported, and no participants withdrew because of adverse effects of the interventions. The report did not provide a full breakdown of adverse events, but included a statement that there were no reports of pain, erythema, or hyperpigmentation associated with treatment.
Secondary outcomes
3. Hidradenitis Severity Score
The HS‐LASI score (Sartorius 2009) was significantly improved in lesions treated with NMB gel at the end of the study compared with lesions treated with FMB gel (MD ‐4.30, 95% CI ‐8.36 to ‐0.24; Analysis 10.1).
10.1. Analysis.
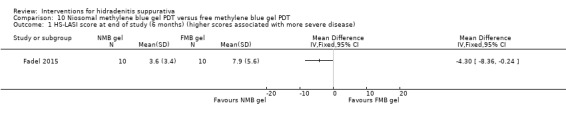
Comparison 10 Niosomal methylene blue gel PDT versus free methylene blue gel PDT, Outcome 1 HS‐LASI score at end of study (6 months) (higher scores associated with more severe disease).
5. Duration of remission
The article narrative reports that there were three recurrences in lesions successfully treated with FMB gel, two in the buttock region after three months, and one in the axilla after six months. There was no mention of any recurrences in lesions treated with NMB gel.
(12) Staphage lysate versus placebo broth
Angel 1987 conducted a two‐arm parallel group RCT lasting 24 weeks in 31 HS participants who had not responded to antibiotics and narrow margin local surgery. Participants were randomised to receive staphage lysate in both a subcutaneous injection and an inhaled aerosol once weekly for 20 weeks or the same volume of vehicle broth placebo via both the subcutaneous and aerosol routes. Outcomes were measured at 24 weeks in the 12 participants who reached the end of the study in the staphage lysate arm and the 15 participants in the placebo arm.
Angel 1987 did not assess the primary outcome 'Quality of life' and the secondary outcomes 'Participant global self‐assessment', 'Pain', 'Hidradenitis Severity Score', and 'Duration of remission'.
Primary outcomes
2. Adverse effects
There were no serious adverse events in either treatment group, but no details were provided regarding treatment discontinuation due to adverse effects or the overall breakdown of adverse events.
4. Physician Global Assessment
Participant response to treatment was graded as improved, the same, or worse by a trial physician, and for our analysis, 'improved' was taken as treatment success. Staphage lysate was more effective than placebo broth, producing improvement in 10 of the 12 participants on active treatment (RR 6.25, 95% CI 1.68 to 23.27; Analysis 11.1).
11.1. Analysis.
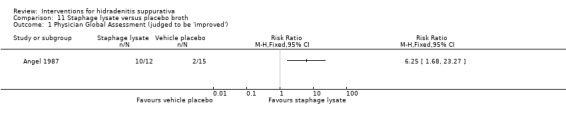
Comparison 11 Staphage lysate versus placebo broth, Outcome 1 Physician Global Assessment (judged to be 'improved').
Discussion
Summary of main results
Our searches identified 101 references for potential inclusion, of which 12 trials met our inclusion criteria, with a total of 615 participants. In most cases, the effects of an intervention were assessed by a single randomised controlled trial (RCT), and meta‐analysis was only possible for the results of two studies that investigated adalimumab administered every other week (EOW). We included trials from each of our intervention groups: one trial of topical therapy; two studies of oral systemic agents; four trials of anti‐TNF‐α (tumour necrosis factor‐alpha) therapies; and four trials of other interventions, including laser and light therapies. The 12 trials were conducted over a 32‐year time period, from 1983 to 2015, which may in part explain the wide variation noted in quality of trial evidence and the outcome measures employed. Only four trials included our primary efficacy outcome measure, quality of life measured on a validated dermatology‐specific scale, and all were recent industry‐sponsored trials of biologic therapies. All 12 trials included some information about our primary outcome for potential harm, namely, the adverse effects of an intervention. However, there was wide variation in the level of detail provided, from a brief narrative discussion to providing figures for rates of serious adverse events, likelihood of causality, rates of treatment discontinuation due to adverse effects, and a breakdown by type of adverse effect.
One trial, Clemmensen 1983, was primarily designed to investigate topical therapy for hidradenitis suppurativa (HS). Thirty participants were randomised to receive clindamycin 1% solution or vehicle solution for 12 weeks. Active treatment was well tolerated, but we found it difficult to comment on efficacy because both outcome measures, a participant diary and a physician‐reported severity score, were unvalidated, and whereas the diary showed no significant difference between groups, the composite scale used for the severity score reported in favour of clindamycin.
Jemec 1998 investigated oral tetracycline in an RCT of 46 participants with mild to moderate hidradenitis suppurativa, using clindamycin 1% solution as an active control. After 16 weeks, there was a difference in the participant global self‐assessment outcome of 28 mm on a visual analogue scale (VAS) from 0 to 100 mm in favour of oral tetracycline, but no significant difference was found in pain, HS Severity Score, or Physician Global Assessment. We downgraded evidence quality to 'low' because of a 26% dropout rate in the absence of an intention‐to‐treat analysis and imprecision arising from the relatively small number of trial participants. Mortimer 1986 compared two endocrine interventions, ethinylestradiol and cyproterone acetate (CPA group), versus ethinylestradiol and norgestrel (E50 group) in a 12‐month cross‐over study. We only included efficacy results for the first six months of the trial due to the risk of carry‐over effects without a washout period. No difference in efficacy was found between the two treatment regimens, although we downgraded the quality of evidence to 'moderate', again because of imprecision arising from the small study size. There was a slight difference in mild adverse effects (AEs) in favour of the CPA group.
Within the anti‐TNF‐α therapy studies, one trial, Adams 2010, compared etanercept with placebo injections. The dose of etanercept, 50 mg twice weekly, was double the dose licensed for psoriasis. Treatment was well tolerated, but no significant difference was found in efficacy relative to placebo. Grant 2010 did find a significant difference in our primary efficacy outcome measure 'Quality of life' comparing standard dose infliximab 5 mg/kg treatment with placebo. There was an effect size of 8.4 Dermatology Life Quality Index (DLQI) points after eight weeks in this relatively small trial of 38 participants with moderate to severe HS. We downgraded the quality of the evidence to 'moderate' because of the small number of events leading to imprecision. Two studies investigated adalimumab EOW compared with placebo (Kimball 2012 and Miller 2011) where meta‐analyses of the combined 124 participants found no difference in both quality of life and the secondary outcome measures of our review. A third arm of Kimball 2012 investigated adalimumab dosed weekly, which is double the dose licensed for psoriasis. There was a significant reduction in DLQI score in favour of adalimumab relative to placebo after 16 weeks, with an effect size of 4.0 points (95% confidence interval (CI) 6.49 to 1.51 points lower), which is approximately equal to the DLQI minimal clinically important difference (Basra 2015). We downgraded the quality of evidence to moderate in the context that the effect size was based on the results of a single study, and subsequent studies are likely to have an important impact on our confidence in the estimate of effect and may change the estimate (Ioannidis 2005).
Buimer 2008 randomised 124 HS lesion excisions to insertion of a gentamicin‐collagen sponge prior to closure and 76 to primary closure alone. There was no difference in complication rates or recurrence rates at one week and three months after surgery. However, there was high risk of performance bias and unclear risk for the other bias domains, downgrading the quality of the evidence. We downgraded the evidence quality of all three within‐participant trials of laser/light interventions to 'low' because of imprecision and a lack of adequate blinding, with no attempt to provide a sham intervention. Highton 2011 reported significant benefit of intense pulsed light (IPL) compared with no treatment in 18 participants, measuring their Sartorius score and satisfaction with treatment on an unvalidated Likert scale. Tierney 2009 gave four neodymium‐doped yttrium aluminium garnet (Nd:YAG) laser treatments at monthly intervals to one side of 25 anatomical sites in 17 participants and found a statistically significant difference in the modified Sartorius score, particularly for axillary sites. Fadel 2015 found that NMB gel was superior to FMB gel when used as a topical photodynamic therapy (PDT) photosensitiser to treat HS lesions. An early small study of staphage lysate, involving subcutaneous injection of the products of S. aureus lysis, showed potential benefit relative to vehicle placebo; however, we downgraded evidence quality to 'moderate' because of imprecision.
Overall completeness and applicability of evidence
Our review has demonstrated that, with the exception perhaps of adalimumab treatment, there remains incomplete RCT evidence for most interventions used to treat HS. Current standard practice for mild to moderate HS is to consider topical antimicrobials and oral tetracyclines; however, this is supported by only two single, moderate quality trials each of less than 50 participants. No RCTs meeting our inclusion criteria were identified for many of the oral systemic agents currently in use to treat HS, including other antibiotics; immunomodulators, such as dapsone, methotrexate, and ciclosporin; retinoids; metformin; and spironolactone. In the anti‐TNF‐α therapy group, etanercept has not been investigated at the 'standard' psoriasis dose of 50 mg weekly, and the evidence for infliximab therapy is limited to one relatively small trial. Although our review included one RCT considering postoperative care in HS, it included no RCTs investigating the timing of surgery and what type of procedure should be performed. The three RCTs of laser or light treatment for HS were small and judged to be of low quality, so it is difficult make treatment recommendations as a result. The trial of staphage lysate injections was small, and there have been no further trials of this intervention since 1987. In the 'other' group of HS therapies, there were no RCTs for phototherapy, intralesional injections of triamcinolone acetate, or botulinum toxin.
We noted considerable outcome measure heterogeneity in the included studies, in part due to evolution of outcome measures during the 32‐year period in which the trials were conducted. This did not affect opportunities for meta‐analysis in our review because only two studies investigated the same intervention and both did include our primary outcome, quality of life. However, updates of our review may be affected by outcome measure variation in the absence of agreement about the key outcomes that should be included. For example, the recent trials of laser, topical PDT, and intense pulsed light (IPL) treatment omitted a validated quality of life measure. A related issue is the validation and clinical meaningfulness of HS outcome measures. The modified Sartorius scale has undergone some validation (Sartorius 2009), but data for a minimal clinically important difference is lacking for this measure and for most of the other outcomes used to grade HS severity. As a result, it is currently difficult to assess treatment success or failure in HS clinical trials.
Despite recognition that HS is a relatively common and disabling condition, economic data regarding impact on society and health resource utilisation is sparse. Kimball 2012 was the only included study to record an economic outcome, namely, the Total Work Productivity Impairment (TWPI) score from the Work Productivity and Activity Impairment‐Specific Health Problem (WPAI‐SHP) questionnaire. Adalimumab every other week did not produce a change in the score, in keeping with its lack of clinical efficacy, whereas adalimumab weekly decreased the score by 20 points. (No details were provided regarding the absolute economic gains.)
Current ongoing studies (Characteristics of ongoing studies) identified from trial registries may address some of the identified knowledge gaps. Of the eight ongoing studies identified, five are investigating biologic therapies, including two further trials of adalimumab, NCT01468207; NCT01468233, and a trial of anakinra (anti‐IL‐1) therapy, NCT01558375, as well as trials of the novel biologics CJM112, NCT02421172, and MEDI8968, NCT01838499. Another trial is assessing Nd:YAG laser treatment after initial clindamycin and rifampicin combination treatment compared with the clindamycin and rifampicin combination as an active control, in a small trial with a recruitment target of 18 participants (NCT01063270). There is also an ongoing trial comparing povidone‐iodine cream with 10% benzoyl peroxide wash for mild to moderate HS (NCT01818167), as well as a comparison of carbon dioxide laser therapy versus surgical deroofing for axillary HS (NCT02163746).
Quality of the evidence
We present 'Summary of findings' tables to summarise the quality of the body of evidence using the five Grading of Recommendations Assessment, Development and Evaluation (GRADE) considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias). See Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8; Table 9; Table 10; Table 11; and Table 12.
There was wide variation in the quality of evidence. We downgraded several early studies and those investigating laser and light treatments to 'low' quality evidence. Imprecision was the most frequently encountered reason to downgrade evidence quality because most HS trials have been relatively small, which is an issue for 10 of the 12 included studies. Although effect size estimates for the comparison of adalimumab weekly versus placebo were based on 102 participants from Kimball 2012, we downgraded the quality of evidence to moderate (Table 1) as the estimates rely on a single study, and subsequent studies may change the estimates (Ioannidis 2005).
In terms of risk of bias, lack of blinding was an issue for the laser and IPL trials, which did not use a sham control treatment for these within‐participant trials. Fadel 2015 and Tierney 2009 achieved a low risk of detection bias for investigator‐reported outcomes by ensuring that blinded outcome assessors performed the scoring evaluations; however, the participant‐reported outcome measures were still at high risk of detection bias. Risk of bias was difficult to assess in nearly all domains for Buimer 2008 because of a lack of information. In particular, the study reported an imbalance in baseline randomisation due to early cessation of the study but did not explicitly state the intended randomisation ratio or the random sequence generation method. Miller 2011 may have been affected by bias due to regression to the mean, resulting from an imbalance in baseline disease severity in which the DLQI score of those allocated to active treatment was twice that of those given placebo.
Unexplained heterogeneity in the meta‐analysis of adalimumab EOW for the change in Sartorius score outcome, with an I² statistic of 59%, led to downgrading of the quality of evidence for this outcome because of inconsistency (Table 2). No occurrences of indirect evidence were noted because most of the included studies were placebo‐controlled trials. We found it difficult to assess the potential for publication bias because of the paucity of RCTs reported in HS, so we downgraded no outcomes for evidence quality as a result.
Potential biases in the review process
During the process of selecting studies, there were some relatively marginal decisions that should be highlighted because of their potential to introduce bias into our review process. We excluded Morgan 1983, a surgical study involving 10 HS participants, from our review because it was a 'quasi‐randomised' trial in which the two interventions were assigned on an alternate basis. We also excluded Xu 2011, a within‐participant study of Nd:YAG laser treatment in 20 participants, because in the unit of analysis section of our review protocol, we had decided to accept only those within‐participant trials that randomised the left and right sides of the same anatomical site. For most participants in Xu 2011, both sides of a body site were treated, and another body site acted as the control. We included Grant 2010 in our review despite the trial being relatively short in duration, lasting eight weeks. We decided to include this study as it was felt that the onset of infliximab effects is rapid, and the majority of its efficacy effects should have occurred by this time point, but the short duration of the trial has potential impact on the detection of adverse effects. However, this issue affected nearly all of the trials in our review, which were relatively short in duration, with a median length of 16 weeks.
On review of the outcome measures employed by our included studies, we did broaden the definition of two outcomes, in order to include as much data as possible, which may have introduced bias. In our published protocol, we stated that pain data would only be accepted in the form of VAS scores; however, Adams 2010 used an ordinal scale from zero to five, which we accepted. We had also specified that physician‐reported HS disease severity should be in the form of modified Sartorius scale scores (Sartorius 2009). However, several studies used the original Sartorius scale (Sartorius 2003), and some older studies used different scales. In the absence of general consensus about HS outcome measures, we decided to include all of these results in our review.
One area of difficulty encountered during our review process was attempts to contact the authors of studies to obtain further information. In several cases, there was no response to our enquiries, which may relate to some of the studies being performed several decades ago.
We attempted to conduct a comprehensive search for studies, but the fact that three studies are awaiting classification and have not yet been incorporated may be a source of potential bias.
Agreements and disagreements with other studies or reviews
Our review has identified wide knowledge gaps in the RCT evidence base for the management of HS, in agreement with the results of a recent HS Priority Setting Partnership (PSP) (Ingram 2014). The PSP was conducted using James Lind Alliance methodology and undertook two online surveys followed by a face to face workshop of HS patients, carers, and their clinicians, in order to identify a top 10 list of HS research priorities. In keeping with the findings of our review, the highest priority of the PSP was "What is the most effective and safe group of oral treatments in treating HS? (e.g. antibiotics, hormonal treatments, retinoids, immunosuppressants, metformin, steroids)". The relative lack of evidence for surgical treatment was reflected by two surgical priorities featuring in the top 10: "What is the best surgical procedure to perform in treating HS e.g. incision & drainage, local excision, wide excision?" (6th priority) and "What is the best method of wound care after surgery or for active disease? (e.g. skin grafts, secondary intention, dressings)" (8th priority).
There have been a number of other systematic reviews of HS treatments. van Rappard 2013 conducted a systematic review of anti‐TNF‐α therapies for severe HS. The authors identified 65 studies, with a total of 459 participants. The four RCTs contained in our review were included, as well as all relevant case series and case reports. 'Moderate to good' responses were found in 82%, 76%, and 68% of participants treated with infliximab, adalimumab, and etanercept, respectively. The relatively high response rate with etanercept may reflect publication bias from inclusion of case reports and case series.
Blok 2013 undertook a HS systematic review that examined all publications of oral retinoid therapy, including acitretin and isotretinoin, and immunosuppressive agents, including biologic therapies, colchicine, ciclosporin, dapsone, and methotrexate. A total of 87 articles were included, containing results for 518 treated participants. The primary efficacy outcome of the review was the proportion of 'responders', divided into those with a 'moderate' response or those with a 'significant' response. Secondary outcomes were relapse rate and adverse events. The authors commented that, overall, the quality of evidence was low, but infliximab and adalimumab were probably the most effective agents. Results for etretinate and its metabolite acitretin were combined, and 16 of the 22 participants reported in a total of six case reports and small case series experienced 'significant' improvement. No response was observed in 112 of 174 participants given isotretinoin, and the authors concluded that the therapeutic effect of this agent in HS is questionable. Our review is unable to comment on retinoid therapy for HS as there are no RCTs available, so no studies met our inclusion criteria.
Alhusayen 2012 performed a systematic review of all pharmacological treatments for HS. Outcomes measures included in their review were as follows: clinical remission, participant global assessment, Physician Global Assessment, number of skin lesions, change in Hurley's stage, and Sartorius score. The authors did not include quality of life and pain scores in contrast to our review. From their review results, the authors concluded that there was moderate quality evidence to support antibacterial treatment and infliximab infusions for mild to moderate and severe HS, respectively. In addition, they concluded that antiandrogen therapy could be considered for women with mild to moderate HS who have failed antibacterial therapy or have an abnormal hormone profile. This latter conclusion is in disagreement with our review, which failed to find sufficient RCT evidence to support endocrine therapy for HS.
Another systematic review of all interventions for HS, including medical, surgical, and miscellaneous interventions, has also been published (Rambhatla 2011). The authors included all HS articles reporting treatment outcomes for at least four individuals and found 62 studies, commenting on a relative lack of RCTs. Treatment recommendations were based on "Morbidity, mortality, symptom improvement, cost reduction, and quality of life". For Hurley stage I (mild) disease, they recommended topical clindamycin 1% solution and consideration of Nd:YAG or carbon dioxide laser therapy. Oral zinc was suggested as a treatment adjunct, and there was a recommendation to avoid isotretinoin because of evidence of a lack of efficacy from case series. Combination treatment with clindamycin 300 mg twice daily and rifampicin 300 mg twice daily was recommended for Hurley stage II (moderate) HS, again based on case series evidence. For refractory stage II disease, biologic therapy was recommended, with infliximab favoured, in part because the review was published before RCT evidence for adalimumab was available. Surgery was recommended for stage III (severe) disease after failure of medical therapy. Our review is more circumspect with regard to treatment recommendations (see below) because it is restricted to the limited number of RCTs available, and in several cases, we have downgraded the quality of the RCT evidence using GRADE methodology.
Authors' conclusions
Implications for practice.
Overall, our review found that knowledge gaps predominate over robust evidence for the treatment of hidradenitis suppurativa (HS). Only 12 RCTs met our inclusion criteria, with a total of 615 participants. Imprecision due to small numbers of participants led us to downgrade the quality of evidence for several of our comparisons. In the context that most interventions were investigated by a single randomised controlled trial (RCT), for which a median of 27 participants were included for 16 weeks, it is difficult to draw meaningful conclusions about adverse effects, particularly rare or delayed effects.
Jemec 2012 and the systematic reviews discussed above provide a framework for HS treatment based on disease severity and lack of response to previous treatments. However, it is difficult to provide strong recommendations from our review because the RCT data are often limited in terms of study size and quality. For example, we have not found sufficient evidence to determine the effects of topical clindamycin or oral tetracycline, despite these being standard treatments for mild to moderate HS. Only one small trial of 30 participants has investigated topical clindamycin 1% solution compared with vehicle placebo (Clemmensen 1983), and the results of the two outcome measures, an unvalidated participant diary and a physician‐reported severity score, were not consistent. We found no placebo‐controlled RCTs of oral tetracyclines for inclusion in our review. Jemec 1998 provided low quality evidence regarding the effect of oral tetracycline compared with topical clindamycin in 46 participants. The study reported no difference in efficacy between the two groups in terms of pain, HS Severity Score, and Physician Global Assessment, but there was a difference in the participant global self‐assessment outcome of 28 mm on a visual analogue scale (VAS) from 0 to 100 mm in favour of oral tetracycline. Overall, the evidence is too limited to provide a recommendation. Similarly, we cannot use the data from Mortimer 1986 to provide a recommendation regarding the endocrine interventions under investigation because of its small size.
Moderate quality evidence does exist for adalimumab therapy. Kimball 2012 demonstrates that adalimumab weekly improves quality of life, although the 95% confidence interval includes an effect size of only 1.5 Dermatology Life Quality Index (DLQI) points, which may not be clinically relevant (Basra 2008; Basra 2015). Meta‐analysis of Kimball 2012 and Miller 2011 shows that adalimumab every other week (EOW) is ineffective. This evidence is likely to affect the treatment of only a relatively small subset of HS patients because anti‐TNF‐α therapy is usually reserved for severe HS refractory to other treatments because of high cost and the potential for serious adverse effects. In particular, the safety profile of weekly treatment has not been established because Kimball 2012 was not powered to detect rare or delayed adverse effects (AEs), and ongoing psoriasis biologic safety registers do not include recipients of adalimumab weekly therapy. In addition, adalimumab weekly therapy is likely to cost twice as much as the standard dosing used for psoriasis, which may further restrict its availability. Results from the PIONEER studies of adalimumab therapy for HS, which should be reported in full in the near future, may help to improve confidence in the estimates of effect size for adalimumab weekly therapy. The evidence for infliximab is less robust, being based on a smaller study; however, the effect size of 8.4 DLQI points is likely to be clinically relevant. The evidence regarding etanercept is only moderate quality in a study of 20 participants with results suggesting that, even at a dose double that licensed for psoriasis, etanercept may not provide benefit in HS.
We downgraded the within‐participant studies of neodymium‐doped yttrium aluminium garnet (Nd:YAG) laser, intense pulsed light (IPL), and topical photodynamic therapy (PDT) to low quality evidence because of imprecision and a lack of blinding in the absence of sham treatments. Implications for practice arising from Buimer 2008 are limited by a high risk of performance bias and unclear risk of bias in most of the other domains due to incomplete study reporting. Furthermore, there was no difference found in the rate of surgical complications or risk of recurrence in the group randomised to receive a gentamicin‐collagen sponge prior to primary closure compared with primary closure alone, so a change in practice in this case cannot be recommended. The trial of staphage lysate was a small study, and there is insufficient evidence to warrant a change of practice based on the results of Angel 1987 alone.
The three studies in 'Studies awaiting classification' may alter the conclusions of the review once assessed.
Implications for research.
Our review has highlighted a need for further RCTs to improve the evidence base for most interventions in HS. One exception perhaps is in the field of biologic therapies, where the evidence is already of higher quality than for other interventions, and there are ongoing studies of adalimumab and anakinra therapy. The HS Priority Setting Partnership (PSP) (Ingram 2014) recently gave highest priority to the question "What is the most effective and safe group of oral treatments in treating HS? (e.g. antibiotics, hormonal treatments, retinoids, immunosuppressants, metformin, steroids)", and our review has highlighted important gaps in the evidence base for these commonly used treatment options.
In terms of RCT design, trials should include a power calculation and recruit sufficient participants to avoid problems with imprecision due to being underpowered. We also found that outcome measure heterogeneity is likely to be an issue in the absence of agreement about the key outcomes that should be included. Selected outcomes should be validated, and ideally, the minimal clinically important difference for the primary outcome should be determined in HS, to ensure that treatment success or failure is clearly defined. The outcomes of a trial should be prospectively declared in a clinical trial database, including the nature and timing of the primary outcome. An intention‐to‐treat analysis, with a predetermined method for dealing with missing data, should be incorporated to minimise the potential for attrition bias.
As well as trials of medical therapy, our review has demonstrated a need for more surgical RCTs. In particular, the RCT evidence base remains weak for timing of surgery, type of surgical procedure, and optimal postoperative wound care in HS. The HS PSP rated both "What is the best surgical procedure to perform in treating HS e.g. incision & drainage, local excision, wide excision?" and "What is the best method of wound care after surgery or for active disease? (e.g. skin grafts, secondary intention, dressings)" in the top 10 priorities for HS research. Although we included three laser or light RCTs in our review, we were not able to make treatment recommendations due to low quality evidence. Future trials in this area should incorporate a sham treatment control to minimise performance and detection bias, and if a within‐participant design is chosen, we recommend that the unit of randomisation should be the left and right sides of the same anatomical site, selecting participants with bilateral HS.
Comparison with a skin disease, such as vitiligo, with a similar prevalence demonstrates the need for more RCTs in HS to guide treatment. The updated Cochrane review for vitiligo (Whitton 2015) included 96 trials containing 4512 participants, which represents eight times as many RCTs compared with our HS review.
What's new
| Date | Event | Description |
|---|---|---|
| 27 February 2017 | Amended | A search of MEDLINE and Embase in February 2017 has identified three new trial reports and the lead author is aware of ongoing studies which have not yet published. The conclusions of this Cochrane Review are therefore still considered up to date, but this decision will be reassessed in February 2018 as potentially relevant trials are not yet reported. |
Notes
A search of MEDLINE and Embase in February 2017 has identified three new trial reports and the lead author is aware of ongoing studies which have not yet published. The conclusions of this Cochrane Review are therefore still considered up to date, but this decision will be reassessed in February 2018 as potentially relevant trials are not yet reported.
Acknowledgements
The authors would like to thank the Cochrane Skin Group editorial base for their methodological support.
The Cochrane Skin Group editorial base wishes to thank Michael Bigby who was the Dermatology Editor for this review; Matthew Grainge and Thomas Chu who were the Statistical Editors; Ching‐Chi Chi who was the Methods Editor; the clinical referees, Noah Sheinfeld and Areti Makrgeorgou; and the consumer referee, who wishes to remain anonymous.
Appendices
Appendix 1. CENTRAL (the Cochrane Library) search strategy
#1 MeSH descriptor: [Hidradenitis Suppurativa] explode all trees #2 acne invers* #3 invers* acne #4 hidradeniti* suppurativ* #5 suppurativ* hidradeniti* #6 velpeau* disease #7 verneuil* disease #8 #1 or #2 or #3 or #4 or #5 or #6 or #7
Appendix 2. MEDLINE (Ovid) search strategy
1. exp Hidradenitis Suppurativa/ 2. acne invers$1.ti,ab. 3. invers$ acne.ti,ab. 4. hidradeniti$ suppurativ$.ti,ab. 5. suppurativ$ hidradeniti$.ti,ab. 6. velpeau$ disease.ti,ab. 7. verneuil$ disease.ti,ab. 8. or/1‐7 9. randomized controlled trial.pt. 10. controlled clinical trial.pt. 11. randomized.ab. 12. placebo.ab. 13. clinical trials as topic.sh. 14. randomly.ab. 15. trial.ti. 16. 9 or 10 or 11 or 12 or 13 or 14 or 15 17. exp animals/ not humans.sh. 18. 16 not 17 19. 8 and 18
[Lines 9‐18: Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision)]
Appendix 3. EMBASE (Ovid) search strategy
1. acne invers$1.ti,ab. 2. invers$ acne.ti,ab. 3. hidradeniti$ suppurativ$.ti,ab. 4. suppurativ$ hidradeniti$.ti,ab. 5. velpeau$ disease.ti,ab. 6. verneuil$ disease.ti,ab. 7. exp suppurative hidradenitis/ 8. or/1‐7 9. crossover procedure.sh. 10. double‐blind procedure.sh. 11. single‐blind procedure.sh. 12. (crossover$ or cross over$).tw. 13. placebo$.tw. 14. (doubl$ adj blind$).tw. 15. allocat$.tw. 16. trial.ti. 17. randomized controlled trial.sh. 18. random$.tw. 19. or/9‐18 20. (ANIMAL/ or NONHUMAN/ or ANIMAL EXPERIMENT/) and HUMAN/ 21. ANIMAL/ or NONHUMAN/ or ANIMAL EXPERIMENT/ 22. 21 not 20 23. 19 not 22 24. 8 and 23
Appendix 4. LILACS search strategy
(acne and invers$) or (hidradeniti$ and suppurativ$) or velpeau$ or verneuil$ or (hidrosadenitis supurativa)
Combined with the LILACS Controlled clinical trials topic‐specific query filter.
Data and analyses
Comparison 1. Topical clindamycin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants reporting non‐serious adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 2. Oral tetracycline versus topical clindamycin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant global assessment VAS (higher scores associated with more severe disease) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2 Pain VAS (higher score is associated with more pain) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3 Nodules score | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4 Abscesses score | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 5 Physician Global Assessment VAS (higher scores associated with more severe disease) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 3. Ethinylestradiol and cyproterone acetate versus ethinylestradiol and norgestrel.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of participants reporting non‐serious adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Participant global assessment VAS (higher number indicates improvement) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 4. Infliximab versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 At least 50% decrease in HS Severity Index | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Physician Global Assessment (judged to be clear, excellent, or good clinical response) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 5. Adalimumab every other week versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in DLQI score (LOCF) (negative values indicate improvement in DLQI) | 2 | 124 | Mean Difference (IV, Fixed, 95% CI) | ‐1.61 [‐3.86, 0.64] |
| 2 Frequency of serious adverse effects | 2 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.26, 8.44] |
| 3 Frequency of treatment discontinuation | 2 | 124 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.91 [0.24, 99.74] |
| 4 Proportion of participants with infectious adverse effects | 2 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.57, 4.53] |
| 5 Change in Pain VAS (lower number indicates improvement) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6 Proportion with improvement in pain | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7 Change in Sartorius scale score (LOCF) (higher scores associated with more severe disease) | 2 | 124 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.42 [‐1.22, 0.37] |
| 8 Physician Global Assessment (at least a grade 2 improvement from baseline) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9 Total Work Productivity Impairment (TWPI) score (imputation) (lower number indicates less impairment) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 10 Total Work Productivity Impairment (TWPI) score (LOCF) (lower number indicates less impairment) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 6. Adalimumab weekly versus placebo.
Comparison 7. Gentamicin sponge versus primary closure alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse effects ‐ complication rate at 1 week after surgery | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 Adverse effects ‐ complication rate at 3 months after surgery | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Recurrence rate at 3 months after surgery | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 8. Intense pulsed light versus no treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant global assessment: satisfaction with treatment | 1 | 34 | Risk Ratio (M‐H, Fixed, 95% CI) | 9.67 [2.01, 46.43] |
| 1.1 Axilla | 1 | 24 | Risk Ratio (M‐H, Fixed, 95% CI) | 21.0 [1.37, 322.28] |
| 1.2 Groin | 1 | 8 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.31, 79.94] |
| 1.3 Inframammary | 1 | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.24, 37.67] |
Comparison 9. Nd:YAG laser versus topical control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Modified HS‐LASI score after 3 months (higher scores associated with more severe disease) | 1 | 50 | Mean Difference (IV, Fixed, 95% CI) | ‐14.03 [‐18.84, ‐9.22] |
| 1.1 Axilla | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | ‐18.7 [‐26.82, ‐10.58] |
| 1.2 Groin | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐12.60 [‐20.28, ‐4.92] |
| 1.3 Inframammary | 1 | 8 | Mean Difference (IV, Fixed, 95% CI) | ‐9.8 [‐19.31, ‐0.29] |
| 2 Percentage change in modified HS‐LASI score after 5 months compared with baseline (higher scores associated with more severe disease) | 1 | 50 | Mean Difference (IV, Fixed, 95% CI) | ‐51.40 [‐66.36, ‐36.43] |
| 2.1 Axilla | 1 | 20 | Mean Difference (IV, Fixed, 95% CI) | ‐58.90 [‐78.82, ‐38.98] |
| 2.2 Groin | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐38.7 [‐63.43, ‐13.97] |
| 2.3 Inframammary | 1 | 8 | Mean Difference (IV, Fixed, 95% CI) | ‐57.30 [‐113.86, ‐0.74] |
Comparison 10. Niosomal methylene blue gel PDT versus free methylene blue gel PDT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 HS‐LASI score at end of study (6 months) (higher scores associated with more severe disease) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
Comparison 11. Staphage lysate versus placebo broth.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Physician Global Assessment (judged to be 'improved') | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Adams 2010.
| Methods | This was a 2‐arm, parallel group RCT The RCT phase lasted 12 weeks, followed by a 12‐week open label phase |
|
| Participants | The trial included 20 participants with a clinical diagnosis of HS This was a single centre outpatient study in the USA Disease severity was described as "active" but not quantified The mean BMI was 32.8 |
|
| Interventions | 2 groups, randomised in a 1:1 ratio:
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Amgen/Wyeth, manufacturer of etanercept, provided study medication, but had no other role in the study | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper provided no details |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebo injections used the same dosing schedule; mild injection site reactions were the only adverse drug reactions reported |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | A single investigator assessed the primary outcome but was blinded to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | An intention‐to‐treat analysis was not performed, but only 2 participants in the active group and 1 participant in the placebo group did not complete the 12‐week study period |
| Selective reporting (reporting bias) | Low risk | The primary outcome was prospectively declared (ClinicalTrials.gov: NCT00949546) |
| Other bias | Low risk | We found no other significant bias |
Angel 1987.
| Methods | This was a 2‐arm, parallel group RCT lasting 24 weeks | |
| Participants | The trial included 31 HS participants who had not responded to antibiotics and local surgery. The paper did not state the number of participants randomised to each intervention | |
| Interventions | 2 groups, randomised in a 1:1 ratio:
|
|
| Outcomes |
|
|
| Notes | Staphage lysate was obtained by lysis of broth cultures of 2 strains of S. aureus using the Gratia bacteriophage, followed by ultrafiltration. The method of action was thought to be the induction of delayed type hypersensitivity. There was no declaration regarding trial sponsorship or funding source. There were several gaps in the reporting of the study; we were unable to contact the authors for clarification | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper did not state a method of random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Placebos were used for both the aerosol and s/c injections consisting of the broth in which the intervention was carried, and no significant adverse effects were reported |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The placebos were effective, and the paper reported no significant adverse effects |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The paper gave no reasons for study withdrawals, but only 4/31 participants withdrew (13%) |
| Selective reporting (reporting bias) | Unclear risk | There was no reference to prospective trial registration. (The study was performed before this was common) |
| Other bias | Unclear risk | The paper did not declare a funding source |
Buimer 2008.
| Methods | This was a 2‐arm, parallel group RCT of wound healing method after local excision and primary closure of active HS lesions | |
| Participants | The trial included 200 participants with a clinical diagnosis of HS and symptomatic lesion(s), "i.e. those with discharge, inflammation, infiltration, or suspected abscesses" | |
| Interventions | 2 groups, randomised to the following:
|
|
| Outcomes |
|
|
| Notes | The authors stated in the paper that there was no significant commercial sponsor involvement We were unable to contact the authors to clarify risks of bias and other study details |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper gave no details on the random sequence generation; however, the study report stated that there was an imbalance in randomisation due to early cessation of the study. The report did not mention whether the randomisation was intended to be in a 1:1 ratio, so we could not quantify the degree of imbalance in randomisation (76 primary closures, 124 gentamicin sponges inserted) |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants were blinded under general anaesthetic but postoperatively may have been able to detect the sponge inserted in the wound. There was no description of any special measures to ensure blinding of personnel, who would otherwise have been aware of treatment allocation from the operation notes |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The paper provided no details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The paper provided no details, including no participant flow diagram |
| Selective reporting (reporting bias) | Unclear risk | There was no mention of prospective registration |
| Other bias | Unclear risk | It was unclear whether participants or surgical procedures were the unit of randomisation, i.e., was more than 1 procedure performed in any individuals? |
Clemmensen 1983.
| Methods | This was a 2‐arm, parallel group RCT lasting 12 weeks. The number of participants randomised to each intervention was not stated | |
| Participants | The trial included 30 participants with a clinical diagnosis of HS and disease onset more than 6 months prior to study entry | |
| Interventions | 2 groups, randomised in a 1:1 ratio:
|
|
| Outcomes |
|
|
| Notes | We were unable to contact the study authors to clarify the frequency of application of interventions. There was no declaration regarding study sponsorship | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper provided no details except that stratification was by baseline disease severity |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | A vehicle placebo was used, which was identical in physical characteristics to the active intervention, and there were similar reports of local irritancy in both groups |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The paper provided no specific details, but the vehicle placebo should have been sufficient |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no intention‐to‐treat analysis, but only a 10% dropout rate due to loss to follow up |
| Selective reporting (reporting bias) | Unclear risk | There was no mention of prospective registration. (The trial was conducted prior to databases being available) |
| Other bias | Low risk | We found no other significant bias |
Fadel 2015.
| Methods | This was a within‐participant trial in which 1 side of the body was randomised to receive 1 intervention and the other side received the other intervention. In 2 participants, disease was localised to either 1 groin or 1 buttock, and in this case, 1 intervention was randomised to the upper part of the lesion, and the other lower part received the other intervention | |
| Participants | The trial included 11 participants with a clinical diagnosis of HS; 1 participant was withdrawn prior to receiving any study treatment due to a change in the diagnosis to Crohn's disease. Of the 10 remaining participants, 4 had disease predominantly located in the axilla, 2 in the groin, 3 in the buttock region, and 1 inframammary. 4 had mild disease, 4 had moderate disease, and 2 had severe disease, classified via the Hurley system | |
| Interventions |
Treatment was continued for up to 6 months or discontinued early if there was no improvement after 2 consecutive sessions |
|
| Outcomes |
|
|
| Notes | The trial had no commercial sponsor. The niosomal delivery system is a surfactant‐based liposome designed to increase topical delivery of the photosensitiser. Dr Tawfik provided additional information via e‐mail on 19 April 2015: all participants were randomised, including the 2 participants who had only a single area of involvement, which was split into the upper and lower half. 10 participants were included in the final efficacy assessment | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A coin toss was used for all participants |
| Allocation concealment (selection bias) | Low risk | A coin toss was used |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and treating clinicians were not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The trial authors clarified that 10 of the 11 randomised participants provided data at the final outcomes assessment |
| Selective reporting (reporting bias) | Unclear risk | There was no reference to prospective trial registration |
| Other bias | Low risk | We identified no other biases |
Grant 2010.
| Methods | This was a 2‐arm, parallel group RCT The RCT phase lasted 8 weeks, followed by an open label treatment phase lasting 22 weeks for those originally given placebo and 14 weeks for those originally given infliximab. There was a subsequent observation phase lasting 22 weeks for those originally given placebo and 30 weeks for those originally given infliximab |
|
| Participants | The trial randomised 38 participants with a clinical diagnosis of HS. However, 5 participants in the placebo group dropped out prior to week 8 for a number of reasons and were omitted from efficacy analyses This was a single centre outpatient study in the USA Participants had moderate to severe HS as defined by a HS Severity Index (HSSI) score greater than 8 |
|
| Interventions | 2 groups, randomised in a 1:1 ratio:
Subsequent open label phase:
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | Only 5 participants took part in the observation phase of the study, so the durations of the remission data were not reliable. Centocor, Inc., which markets infliximab in the USA, supported the trial. Disclosure: Dr Kerdel, Ms Grant, and Ms Cardenas received research support from Centocor Inc.; Drs Gonzalez and Montgomery were employees of Centocor, Inc. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random sequence generation was computer generated |
| Allocation concealment (selection bias) | Low risk | The assignment code was forwarded to the study pharmacist only |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Only the study pharmacist was aware of treatment allocation, and infusions were probably identical |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Detection bias was at low risk for all outcomes ‐ the treating clinician assessed the outcomes but was likely to be blinded; in particular, there were no infusion reactions during the placebo‐controlled phase |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was no intention‐to‐treat analysis, and 5 participants on placebo dropped out during the 8‐week RCT period compared with zero on infliximab. However, on contact with the study authors, placebo participants dropped out because of lack of efficacy or worsening of disease severity, which is likely to under‐ rather than overestimate the efficacy of infliximab |
| Selective reporting (reporting bias) | Unclear risk | The trial was registered on ClinicalTrials.gov as NCT00795574, but no prospective details regarding outcomes were provided |
| Other bias | Low risk | We found no other significant bias |
Highton 2011.
| Methods | This was a within‐patient RCT in which 1 side of an anatomical site was treated, and the other received no treatment There was a 4‐week treatment period followed by a 12‐month observation phase |
|
| Participants | The trial randomised 18 participants with a clinical diagnosis of HS with moderate to severe disease, defined as Hurley stage II or III Participants were required to have bilateral disease in an affected region, and overall, there was no significant difference in disease severity at the control and intervention sites at baseline (P value = 0.31, paired t test). However, information was not provided regarding whether disease severity was comparable on both sides of an affected region for individual participants Disease locations were axillary (12 participants), groin (4 participants), and inframammary (2 participants). 1 participant with inframammary disease dropped out after a single treatment |
|
| Interventions | Left and right sides of a single anatomical location were randomised in a 1:1 ratio:
|
|
| Outcomes |
Primary outcome
Secondary outcome
|
|
| Notes | The publication gave only pooled results for all anatomical locations, but following correspondence, the authors provided a breakdown based on the site of involvement. The trial authors declared no financial conflicts of interest | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper provided no details ‐ "patients were randomised" |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Detection bias was low risk for the Sartorius score – the outcome assessor was not the treating clinician and was blinded to treatment allocation. Scoring was repeated from photographs by 2 additional blinded assessors. The trial was high risk for participant satisfaction; participants were unblinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 participant dropped out of the study, due to treatment‐related pain |
| Selective reporting (reporting bias) | Unclear risk | The study was not registered prospectively |
| Other bias | Low risk | We found no other significant bias |
Jemec 1998.
| Methods | This was a 2‐arm, parallel group RCT lasting 16 weeks | |
| Participants | The trial randomised 46 participants with a clinical diagnosis of HS. However, 12 participants dropped out for a number of reasons and were omitted from efficacy analyses This was a 2‐centre outpatient study in Denmark Participants had mild to moderate HS as defined by Hurley stage 1 or 2 |
|
| Interventions | 2 groups, randomised in a 1:1 ratio:
|
|
| Outcomes |
|
|
| Notes | The primary outcome was not stated, and there was no declaration regarding study sponsorship | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random sequence generation was computer generated |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The trial used uniform containers, placebo tablets, and placebo lotion in a double‐dummy study design |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators and participants assessed outcomes but were blinded to treatment allocation, and adverse effects were unlikely to compromise this |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There was no intention‐to‐treat analysis, and 12 participants (26%) dropped out of the study |
| Selective reporting (reporting bias) | Unclear risk | The study was performed prior to widespread prospective trial registration |
| Other bias | Low risk | We found no other significant bias |
Kimball 2012.
| Methods | This was a 3‐arm, parallel group RCT The RCT phase lasted 16 weeks, followed by a 36‐week open label phase |
|
| Participants | The trial included 154 participants with a clinical diagnosis of HS Participants were recruited from 26 centres in Denmark, Germany, the Netherlands, and the USA Disease severity was moderate in one third of participants and severe in two thirds of participants The mean weight of the participants was 97.2 kg, and just over half were current smokers |
|
| Interventions | 3 groups, randomised in a 1:1:1 ratio:
|
|
| Outcomes |
Primary outcome
Secondary outcomes
|
|
| Notes | During the RCT phase, 2 rescue treatments with either an injection of intralesional triamcinolone or incision and drainage were permitted During the 36‐week open label phase, all participants received adalimumab 40 mg EOW. At weeks 28 or 31, any participant with a HS‐PGA score of moderate or worse was eligible to escalate to weekly dosing for the remainder of the study Abbott Laboratories, manufacturers of adalimumab, sponsored the trial |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random sequence generation was computer generated |
| Allocation concealment (selection bias) | Low risk | An interactive voice‐response/web‐response system was used |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical syringes were used, and all participants received an equal number of injections for each dosing |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators assessed the PGA primary outcome but were blinded to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat analysis was performed |
| Selective reporting (reporting bias) | Low risk | Primary and secondary outcomes were prospectively declared (NCT00918255) |
| Other bias | Low risk | We found no other significant bias |
Miller 2011.
| Methods | This was a 2‐arm, parallel group RCT The RCT phase lasted 12 weeks, followed by a 12‐week observational follow‐up phase with no treatment |
|
| Participants | The trial included 21 participants with moderate to severe HS, defined as Hurley stage II or III, for at least 6 months | |
| Interventions | 2 groups randomised in a 1:2 ratio (placebo:active):
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | The study did not reach its recruitment target of 30 participants because the study medication exceeded its expiry date Abbott, manufacturers of adalimumab, provided the active drug, placebo, and computer randomisation. No salary was paid to the investigators or to the department performing the study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The random sequence generation was computer generated |
| Allocation concealment (selection bias) | Unclear risk | Sequentially numbered containers were used, but the paper did not specify if the containers were opaque |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical syringes were used as well as the same dosing schedule. The adverse effects were unlikely to have caused unblinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Treating clinicians assessed the outcomes but were unlikely to be unblinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The trial undertook ITT analysis with LOCF and first observation carried backwards. The authors stated: "The combination of last observation carried forward and first observation carried backward ensures that for a given patient both missing values after the last observation as well as missing values prior to the first observation are imputed" |
| Selective reporting (reporting bias) | Unclear risk | EudraCT: 2006‐005297‐48, but a search for the study was unsuccessful |
| Other bias | High risk | Baseline disease severity was higher in the adalimumab group |
Mortimer 1986.
| Methods | This was a 2‐arm, cross‐over study The first phase lasted 6 months, followed by a treatment cross‐over phase also lasting 6 months |
|
| Participants | The trial included 24 outpatients with a clinical diagnosis of HS Disease severity was described as moderate to severe, but no definition was provided |
|
| Interventions | 2 groups, randomised in a 1:1 ratio to receive the following for the first 6 months:
The treatment groups crossed over for the subsequent 6 months |
|
| Outcomes |
Primary outcomes (at the end of the initial phase prior to cross‐over at 6 months)
|
|
| Notes | HS severity scale results were not provided at 6‐month time points. Schering Chemicals Ltd., manufacturer of Eugynon 50 (ethinyloestradiol + norgestrel), gave financial support and supplied and packaged the tablets | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The paper provided no details – described as a "double‐blind" study |
| Allocation concealment (selection bias) | Unclear risk | The paper provided no details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The trial was described as a "double‐blind" study, and a commercial company was involved in packaging study medications |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | 1 of the study investigators performed the objective assessments |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was no intention‐to‐treat analysis, and 2 participants dropped out due to treatment failure, their intervention group being unreported |
| Selective reporting (reporting bias) | Unclear risk | The study was performed prior to widespread prospective trial registration |
| Other bias | Low risk | We found no other significant bias |
Tierney 2009.
| Methods | This was a within‐participant RCT in which 1 side of an anatomical site received the intervention, and the other side received a control topical therapy. 4 laser treatments were given at monthly intervals, followed by a 2‐month observation period | |
| Participants | The trial recruited 22 HS participants with moderate to severe disease, as defined by a Hurley grade of II or III. 34 bilaterally affected anatomical sites (axilla/groin/inframammary) were randomised | |
| Interventions | Left and right sides of the same anatomical site received either:
Laser treatment settings:
|
|
| Outcomes |
Primary outcomes
Tierney 2009 reported results 3 months after the first laser treatment, and Mahmoud 2010 (a secondary publication) reported results after a further 2‐month observation period Secondary outcomes
|
|
| Notes | Upon discussion with the study authors, we were told that during the first month of the trial, participants received the topical therapy only. At the end of the first month, they received their first laser treatment, and this continued monthly for the next 3 months, administering a total of 4 treatments. Following this, there was a 2‐month observation period with no laser treatment The authors indicated that there was no support from commercial sponsors. The study was funded in part by an American Society for Dermatologic Surgery Cutting Edge Research Grant and the Shahani Fund. The authors acknowledged support from the Hidradenitis Suppurativa Foundation |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A coin toss was used |
| Allocation concealment (selection bias) | Low risk | A coin toss was used |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and personnel were unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Detection bias was low risk for investigator‐reported outcomes – "blinded nontreating physician investigators performed scoring evaluations" ‐ but high risk for participant‐reported outcomes |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There was no intention‐to‐treat analysis, and 5 participants (23% of total), including 8 anatomical sites (24% of total), dropped out and their results were not included |
| Selective reporting (reporting bias) | Unclear risk | The study was registered prospectively on ClinicalTrials.gov, NCT00494351. The primary outcome was stated to be Hidradenitis Severity Score, but both the original Sartorius 2003 score and a version modified by the authors were measured. The timing of primary end points was not stated |
| Other bias | High risk | 3 episodes of cellulitis at non‐treatment sites required antibiotic therapy, which may have affected the results |
BD: twice daily. BMI: body mass index. CRP: C‐reactive protein. DLQI: Dermatology Life Quality Index. EOW: every other week. ESR: erythrocyte sedimentation rate. GC: gentamicin‐collagen. HS: hidradenitis suppurativa. HS‐LASI: HS Lesion, Area and Severity Index. HS‐PGA: Hidradenitis Suppurativa Physician's Global Assessment. HSSI: Hidradenitis Suppurativa Severity Index. IPL: intermittent pulsed light. ITT: intention‐to‐treat. LOCF: last observation carried forward. Nd:YAG: neodymium‐doped yttrium aluminium garnet. PC: primary closure PDT: photodynamic therapy. PGA: Physician Global Assessment. RCT: randomised controlled trial. S. aureus: Staphylococcus aureus. S/c: subcutaneous. TWPI: Total Work Productivity Impairment. VAS: visual analogue scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Morgan 1983 | This study was not randomised: alternate allocation was used |
| NCT00722800 | This study was terminated early because of poor recruitment ‐ only 4 participants were recruited |
| Puri 2011 | This study was not randomised |
| Soldin 2000 | This was a non‐randomised, retrospective case series |
| Xu 2011 | In most cases, both sides of a body site were treated, and another body site acted as the control |
Characteristics of studies awaiting assessment [ordered by study ID]
EUCTR2006‐005405‐67.
| Methods | This is a parallel group RCT |
| Participants | The trial included adults aged 18 to 64 with Hurley stage I to II HS |
| Interventions |
|
| Outcomes |
|
| Notes | The trial was registered in 2007; we are uncertain if it has been completed |
EUCTR2007‐000534‐39.
| Methods | This is a parallel group RCT |
| Participants | The trial included adults aged 18 to 70 with Hurley stage I to II HS |
| Interventions |
|
| Outcomes | ‐ |
| Notes | The trial was registered in 2007; we are uncertain if it has been completed |
Servant 2002.
| Methods | Unknown |
| Participants | Unknown |
| Interventions |
|
| Outcomes | Unknown |
| Notes | We were unable to contact the authors to obtain any further details other than the title of the conference proceedings |
HS: hidradenitis suppurativa. RCT: randomised controlled trial.
Characteristics of ongoing studies [ordered by study ID]
NCT01063270.
| Trial name or title | Randomized Control Trial Comparing Efficacy of Antibiotic Therapy Alone Versus Antibiotic Therapy in Conjunction With Quadruple Pulse Therapy Using NdYag Laser in Treatment of Hidradenitis Suppurativa |
| Methods | This is a parallel RCT |
| Participants | The trial is including adults with Hurley stage II HS |
| Interventions |
|
| Outcomes |
|
| Starting date | February 2010 |
| Contact information | Iltefat Hamzavi MD Henry Ford Health System |
| Notes | Recruitment target = 18 participants |
NCT01468207.
| Trial name or title | A Phase 3 Multicenter Study of the Safety and Efficacy of Adalimumab in Subjects With Moderate to Severe Hidradenitis Suppurativa ‐ PIONEER I |
| Methods | This is a phase 3 parallel RCT |
| Participants | The trial is including adults with moderate to severe HS |
| Interventions |
|
| Outcomes |
|
| Starting date | November 2011 |
| Contact information | David Williams MD Study Chair, Abbott Andrea L Byars (andrea.byars@abbott.com) |
| Notes | AbbVie sponsor the trial (prior sponsor: Abbott). Results are currently available in 2 conference abstracts, which have not yet been published in full in a peer‐reviewed journal |
NCT01468233.
| Trial name or title | A Phase 3 Multicenter Study of the Safety and Efficacy of Adalimumab in Subjects With Moderate to Severe Hidradenitis Suppurativa ‐ PIONEER II |
| Methods | This is a phase 3 parallel RCT |
| Participants | The trial is including adults with moderate to severe HS |
| Interventions |
|
| Outcomes |
|
| Starting date | November 2011 (likely finish May 2014) |
| Contact information | Martin Okun MD Study Chair, Abbott Andrea L Byars (andrea.byars@abbott.com) |
| Notes | AbbVie sponsor the trial (prior sponsor: Abbott) |
NCT01558375.
| Trial name or title | A Double‐blind, Randomized, Placebo‐controlled Clinical Trial of the Safety and Efficacy of Anakinra in Patients With Hidradenitis Suppurativa |
| Methods | This is a phase 2 placebo‐controlled trial |
| Participants | The trial is including adults with moderate to severe HS (Hurley stage II to III) |
| Interventions |
|
| Outcomes |
|
| Starting date | March 2012 (likely finish: March 2014) |
| Contact information | Evangelos J Giamarellos‐Bourboulis MD, PhD (egiamarel@med.uoa.gr) |
| Notes | The University of Athens sponsors the trial |
NCT01818167.
| Trial name or title | A Prospective Multi‐Center Blinded, Randomized, Controlled Clinical Trial Comparing the Efficacy of Provodine Topical Body Wash Versus 10% Benzoyl Peroxide Topical Body Wash for the Treatment of Hidradenitis Suppurativa |
| Methods | This is a RCT with a cross‐over design |
| Participants | Participants must be aged 13 years and over (Hurley stage I to II) |
| Interventions |
|
| Outcomes |
|
| Starting date | March 2013 |
| Contact information | Dr Virginia Reeder (vreeder1@hfhs.org) |
| Notes | ‐ |
NCT01838499.
| Trial name or title | A Phase IIa Randomized, Double‐Blind, Placebo‐controlled, Multicenter Study to Assess the Safety, Tolerability and Preliminary Efficacy of MEDI8968 in Subjects With Moderate to Severe Hidradenitis Suppurativa |
| Methods | This is a phase 2 parallel RCT |
| Participants | Participants must be aged 18 to 65 years with moderate to severe HS |
| Interventions |
|
| Outcomes |
|
| Starting date | May 2013 |
| Contact information | Dr Robert AK Lee |
| Notes | AstraZeneca sponsors the trial |
NCT02163746.
| Trial name or title | A Prospective, Randomized, Controlled Clinical Trial Comparing the Efficacy Carbon Dioxide (CO2) Laser Excision Versus Surgical Deroofing in the Treatment of Hidradenitis Suppurativa |
| Methods | This is a non‐blinded parallel RCT |
| Participants | Participants must be aged 13 and over (Hurley Stage II HS affecting the axilla) |
| Interventions |
|
| Outcomes |
The timing of primary outcomes is not stated |
| Starting date | March 2014 |
| Contact information | Dr Samreen Choudhry (schoudh5@hfhs.org) |
| Notes | ‐ |
NCT02421172.
| Trial name or title | Efficacy, Safety, and Pharmacokinetics Study of CJM112 in Hidradenitis Suppurativa Patients |
| Methods | This is a phase 2, double‐blind, multicentre RCT with 3 parallel arms |
| Participants | Participants are men and women 18 to 65 years of age (Hurley stages II to III) |
| Interventions |
CJM112 is a fully human IgG1 monoclonal antibody |
| Outcomes |
|
| Starting date | April 2015 |
| Contact information | ‐ |
| Notes | ‐ |
BD: twice daily. DLQI: Dermatology Life Quality Index. HISERG: Hidradenitis Suppurativa European Research Group. HS: hidradenitis suppurativa. HS‐PGA: Hidradenitis Suppurativa Physician's Global Assessment. IgG1: immunoglobulin G1. Nd:YAG: neodymium‐doped yttrium aluminium garnet. PGA: Physician Global Assessment. OD: once daily. RCT: randomised controlled trial.
Differences between protocol and review
In our published protocol, we stated that we would only accept pain data in the form of visual analogue scale (VAS) scores; however, Adams 2010 used an ordinal scale from zero to five, which we accepted. We had also specified that physician‐reported HS disease severity should be in the form of modified Sartorius scale scores (Sartorius 2009). However, several studies used the original Sartorius scale (Sartorius 2003), and some older studies used different scales. In the absence of general consensus about hidradenitis suppurativa (HS) outcome measures, we decided to include all of these results in our review.
We added a stipulation that within‐participant trials must randomise the left and right sides of the same anatomic site because different sites may respond differently to a particular treatment, and HS clinical scoring systems may result in different disease severity values depending on the site.
After publication of our protocol, Cochrane adopted Grading of Recommendations Assessment, Development and Evaluation (GRADE) methodology, and this has been incorporated into our review methodology, results, and discussion.
Compared with the published protocol, there were some alterations in the tasks completed by review authors: PNW handsearched conference proceedings rather than ND; ACK contributed to selection of studies rather than ND; PNW, SLC, and ADO performed independent data extraction along with JRI rather than ACK; and PNW, SLC, and ADO performed independent 'Risk of bias' assessments along with JRI rather than ND.
Several data analyses stated in the protocol were not possible to perform due to a lack of data in the included studies, as follows: duration of remission; paired analysis of data for within‐participant trials (so we used parallel group analytical methods instead); imputation of missing data; subgroup analyses (so we used a random‐effects model and interpreted the results with caution); funnel plots and Egger's test for publication bias; and sensitivity analyses.
We did not encounter any analyses for which the I² statistic exceeded 75% so did not need to take the narrative approach intended in the protocol. In one analysis where we found substantial heterogeneity, there were too few studies to permit a subgroup analyses of participant factors, as originally intended.
In our review, we did not perform a separate search for any adverse effects, rather than only rare or delayed adverse effects as stated in the protocol, because we decided that we would identify common, quickly‐occurring adverse effects from our included studies.
Contributions of authors
JRI was the contact person with the editorial base. JRI co‐ordinated contributions from the co‐authors and wrote the final draft of the review, with advice from VP. JRI and ACK screened papers against eligibility criteria. JRI obtained data on ongoing and unpublished studies. JRI, PNW, SLC, and ADO appraised the quality of papers. JRI, PNW, SLC, and ADO extracted data for the review. JRI and FK sought additional information about papers. JRI entered data into RevMan. JRI and KH analysed and interpreted data. JRI, ND, SLC, and PNW worked on the methods sections. JRI, ND, and PNW drafted the clinical sections of the background and responded to the clinical comments of the referees. KH and JRI responded to the methodology and statistics comments of the referees. TB was the consumer co‐author and checked the review for readability and clarity, as well as ensuring outcomes are relevant to consumers. All authors reviewed the final draft of the manuscript. JRI is the guarantor of the review update.
Disclaimer
The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, NHS or the Department of Health, UK.
Sources of support
Internal sources
No sources of support supplied
External sources
-
The National Institute for Health Research (NIHR), UK.
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
John R Ingram: John R Ingram is a local PI (principal investigator) for an observational study sponsored by AbbVie, manufacturers of adalimumab. He has not acted as a consultant for the manufacturer or taken part in paid Advisory Boards. Pick‐Ngor Woo: nothing to declare. Ser Ling Chua: nothing to declare. Anthony D Ormerod: nothing to declare. Nemesha Desai: "I have received consulting fees for advisory board work for AbbVie, which produces adalimumab." Anneke C Kai: nothing to declare. Kerry Hood: nothing to declare. Tara Burton: "I have received honorarium for speaking at AbbVie meetings in July 2014 and January 2015 about my personal patient journey. In June 2015, the HS Trust received a £5000 core funding grant from AbbVie, which produces adalimumab." Francisco Kerdel: "I have received honorariums for research or speaking agreements from Janssen Biotech, Inc., which produces Remicade® (infliximab); AbbVie, formally Abbott, which produces Humira® (adalimumab); and Amgen/Pfizer, which produces Enbrel® (etanercept)." Sarah E Garner: nothing to declare. Vincent Piguet: "I undertake personal advisory work with Pfizer, AbbVie, Janssen, Novartis, and Almirall. I have received support to my Department from AbbVie, Almirall, Alliance, Beiersdorf UK Ltd, Biotest, Celgene, Galderma, Genus Pharma, Janssen, LEO Pharma, Meda, MSD, Novartis, Pfizer, Sinclair Pharma, Spirit Pharmaceuticals, Stiefel, Sumed, and TyPham. The Department also received financial support from Dermatology Life Quality Index (DLQI) copyright. Please note: adalimumab is produced by AbbVie; ustekinumab is produced by Janssen; infliximab is produced by MSD; and etanercept is produced by Pfizer."
Edited (no change to conclusions)
References
References to studies included in this review
Adams 2010 {published data only}
- Adams DR, Yankura JA, Fogelberg AC, Anderson BE. Treatment of hidradenitis suppurativa with etanercept injection. Archives of Dermatology 2010;146(5):501‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Angel 1987 {published data only}
- Angel MF, Ramasastry SS, Manders EK, Ganfield D, Futrell JW. Beneficial effects of staphage lysate in the treatment of chronic recurrent hidradenitis suppurativa. Surgical Forum 1987;38:111‐2. [EMBASE: 1988094243] [Google Scholar]
Buimer 2008 {published data only}
- Buimer MG, Ankersmit MF, Wobbes T, Klinkenbijl JH. Surgical treatment of hidradenitis suppurativa with gentamicin sulfate: a prospective randomized study. Dermatologic Surgery 2008;34(2):224‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Clemmensen 1983 {published data only}
- Clemmensen OJ. Topical treatment of hidradenitis suppurativa with clindamycin. International Journal of Dermatology 1983;22(5):325‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Fadel 2015 {published and unpublished data}
- Fadel MA, Tawfik AA. New topical photodynamic therapy for treatment of hidradenitis suppurativa using methylene blue niosomal gel: a single‐blind,randomized, comparative study. Clinical & Experimental Dermatology 2015;40(2):116‐22. [EMBASE: 2015763458] [DOI] [PubMed] [Google Scholar]
Grant 2010 {published data only}
- Grant A, Gonzalez T, Montgomery MO, Cardenas V, Kerdel FA. Infliximab therapy for patients with moderate to severe hidradenitis suppurativa: a randomized, double‐blind, placebo‐controlled crossover trial. Journal of the American Academy of Dermatology 2010;62(2):205‐17. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Grant A, Kerdel FA, Gonzalez T. Long‐term efficacy and safety results (60 weeks) of a double‐blind, placebo‐controlled, crossover trial of infliximab for patients with moderate to severe hidradenitis suppurativa. 67th Annual Meeting of the American Academy of Dermatology. AAD San Francisco, CA United States. Conference Start: 20090306 Conference End: 20090310. Journal of the American Academy of Dermatology 2009;60(3 Suppl 1):AB106. [EMBASE: 70142143] [Google Scholar]
Highton 2011 {published data only}
- Highton L, Chan WY, Khwaja N, Laitung JK. Treatment of hidradenitis suppurativa with intense pulsed light: a prospective study. Plastic & Reconstructive Surgery 2011;128(2):459‐65. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Highton L, Laitung G, Khwaja N, Chan W‐Y, Chapel K, Bates K. Treatment of hidradenitis suppurativa using intense pulsed light. 29th Annual Conference of the American Society for Laser Medicine and Surgery National Harbor, MD United States. Conference Start: 20090401 Conference End: 20090405. Lasers in Surgery and Medicine 2009;41:101. [EMBASE: 70332914] [Google Scholar]
Jemec 1998 {published data only}
- Jemec GB, Wendelboe P. Topical clindamycin versus systemic tetracycline in the treatment of hidradenitis suppurativa. Journal of the American Academy of Dermatology 1998;39(6):971‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kimball 2012 {published data only}
- Gottlieb A, Menter A, Armstrong A, Cui N, Yamaguchi Y. Adalimumab treatment in women with moderate to severe hidradenitis suppurativa using a novel endpoint, HISCR (hidradenitis suppurativa clinical response): Analysis from the placebo‐controlled portion of a phase ii, randomized, double‐blind study. 61st Annual Scientific Meeting of the Society for Gynecologic Investigation, SGI 2014 Florence Italy. Conference Start: 20140326 Conference End: 20140329. Reproductive Sciences 2014;21(3 Suppl 1):220A‐221A. [EMBASE: 71380256] [Google Scholar]
- Gottlieb AB, Menter A, Belknap K, Chen K, Teixeira HD. Efficacy and safety of adalimumab treatment in women with moderate to severe hidradenitis suppurativa: Analysis from the placebo‐controlled portion of a phase ii, randomized, double‐blind study. 20th FIGO World Congress of Gynecology and Obstetrics Rome Italy. Conference Start: 20121007 Conference End: 20121012. International Journal of Gynaecology and Obstetrics 2012;119:S360. [EMBASE: 70905615] [Google Scholar]
- Jemec G, Sobell J, Scheinfeld N, Sood S, Gu Y. Efficacy results using a novel hidradenitis suppurativa endpoint, HiSCR (hidradenitis suppurativa clinical response), from the placebo‐controlled phase of a phase 2 adalimumab study. Conference: 72nd Annual Meeting of the American Academy of Dermatology Denver, CO United States. Conference Start: 20140321 Conference End: 20140325. Journal of the American Academy of Dermatology 2014;70(5 Suppl 1):AB42. [EMBASE: 71390261] [Google Scholar]
- Kimball A, Jemec G, Okun M, Gu Y. Efficacy and safety of adalimumab in treatment of moderate to severe hidradenitis suppurativa: results from the placebo‐controlled portion of a phase ii, randomized, double‐blind study. Conference: 69th Annual Meeting of the American Academy of Dermatology New Orleans, LA United States. Conference Start: 20110204 Conference End: 20110208. Journal of the American Academy of Dermatology 2011;64(2 Suppl 1):AB155. [EMBASE: 70331898] [Google Scholar]
- Kimball A, Williams D, Jemec G, Gu Y. Efficacy and safety of adalimumab for moderate to severe hidradenitis suppurativa: Results from the open‐label phase of a 52‐week phase II, randomized, study. 70th Annual Meeting of the American Academy of Dermatology San Diego, CA United States. Conference Start: 20120316 Conference End: 20120320. Journal of the American Academy of Dermatology 2012;66(4 Suppl 1):AB50. [EMBASE: 70704053] [Google Scholar]
- Kimball AB, Chen K, Okun M, Sundaram M. Adalimumab reduces pain in patients with hidradenitis suppurativa: Results from a placebo‐controlled phase II trial. 70th Annual Meeting of the American Academy of Dermatology San Diego, CA United States. Conference Start: 20120316 Conference End: 20120320. Journal of the American Academy of Dermatology 2012;66(4 Suppl 1):AB42. [EMBASE: 70704020] [Google Scholar]
- Kimball AB, Jemec G, Gu Y, Okun M. Efficacy and safety of adalimumab in the treatment of moderate to severe hidradenitis suppurativa: results from the placebo‐controlled portion of phase II, randomized, double‐blind study. 91st Annual Meeting of the British Association of Dermatologists London United Kingdom. Conference Start: 20110705 Conference End: 20110707. British Journal of Dermatology 2011;165(Suppl 1):6. [EMBASE: 70479499] [Google Scholar]
- Kimball AB, Kerdel F, Adams D, Mrowietz U, Gelfand JM, Gniadecki R, et al. Adalimumab for the treatment of moderate to severe Hidradenitis suppurativa: a parallel randomized trial. Annals of Internal Medicine 2012;157(12):846‐55. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Scheinfeld N, Teixeira H, Okun M, Sundaram M, Cui N. Adalimumab treatment is associated with pain reduction in patients with hidradenitis suppurativa, regardless of the presence of depression: Results from a phase II, randomized, placebo‐controlled trial. 72nd Annual Meeting of the American Academy of Dermatology Denver, CO United States. Conference Start: 20140321 Conference End: 20140325. Journal of the American Academy of Dermatology 2014;70(5 Suppl 1):AB35. [EMBASE: 71390235] [Google Scholar]
- Zouboulis CD, Adams DA, Gu Y, Goldblum OM. High‐sensitivity C‐reactive protein response to adalimumab in hidradenitis suppurativa patients. 42nd Annual Meeting of the European Society for Dermatological Research Venice Italy. Conference Start: 20120919 Conference End: 20120922. Journal of Investigative Dermatology 2012;132:S67. [EMBASE: 71015109] [Google Scholar]
Miller 2011 {published data only}
- Miller I, Lynggaard CD, Lophaven S, Zachariae C, Dufour DN, Jemec GB. A double‐blind placebo‐controlled randomized trial of adalimumab in the treatment of hidradenitis suppurativa. British Journal of Dermatology 2011;165(2):391‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mortimer 1986 {published data only}
- Mortimer PS, Dawber RP, Gales MA, Moore RA. A double‐blind controlled cross‐over trial of cyproterone acetate in females with hidradenitis suppurativa. British Journal of Dermatology 1986;115(3):263‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Tierney 2009 {published data only}
- Hamzavi I, Mahmoud B, Tierney E, Hexsel C, Pui J, Ozog O. Prospective clinical and histopathologic study of hidradenitis suppurativa treated with laser. 29th Annual Conference of the American Society for Laser Medicine and Surgery National Harbor, MD United States. Conference Start: 20090401 Conference End: 20090405. Lasers in Surgery and Medicine 2009;41:100. [EMBASE: 70332912] [Google Scholar]
- Mahmoud B, Hexsel C, Ozog D, Tierney E, Hamzavi I. Clinical and histopathologic evaluation of hidradenitis suppurativa treated with long‐pulsed Nd:YAG laser. 67th Annual Meeting of the American Academy of Dermatology, AAD San Francisco, CA United States. Conference Start: 20090306 Conference End: 20090310. Journal of the American Academy of Dermatology 2009;60(3 Suppl 1):AB192. [EMBASE: 70142480] [Google Scholar]
- Mahmoud BH, Tierney E, Hexsel CL, Pui J, Ozog DM, Hamzavi IH. Prospective controlled clinical and histopathologic study of hidradenitis suppurativa treated with the long‐pulsed neodymium:yttrium‐aluminium‐garnet laser. Journal of the American Academy of Dermatology 2010;62(4):637‐45. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Tierney E, Mahmoud BH, Hexsel C, Ozog D, Hamzavi I. Randomized control trial for the treatment of hidradenitis suppurativa with a neodymium‐doped yttrium aluminium garnet laser. Dermatologic Surgery 2009;35(8):1188‐98. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Morgan 1983 {published data only}
- Morgan WP, Harding KG, Hughes LE. A comparison of skin grafting and healing by granulation, following axillary excision for hidradenitis suppurativa. Annals of the Royal College of Surgeons of England 1983;65(4):235‐6. [MEDLINE: ] [PMC free article] [PubMed] [Google Scholar]
NCT00722800 {unpublished data only}
- NCT00722800. A Study to Examine the Safety and Efficacy of Drospirenone and Ethinyl Estradiol (YAZ) Versus Placebo In HS. clinicaltrials.gov/ct2/show/NCT00722800 (accessed 2 April 2015).
Puri 2011 {published data only}
- Puri N, Talwar A. A study on the management of hidradenitis suppurativa with retinoids and surgical excision. Indian Journal of Dermatology 2011;56(6):650‐1. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Soldin 2000 {published data only}
- Soldin MG, Tulley P, Kaplan H, Hudson DA, Grobbelaar AO. Chronic axillary hidradenitis‐‐the efficacy of wide excision and flap coverage. British Journal of Plastic Surgery 2000;53(5):434‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Xu 2011 {published data only}
- Xu LY, Wright DR, Mahmoud BH, Ozog DM, Mehregan DA, Hamzavi IH. Histopathologic study of hidradenitis suppurativa following long‐pulsed 1064‐nm Nd:YAG laser treatment. Archives of Dermatology 2011;147(1):21‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
EUCTR2006‐005405‐67 {published data only}
- EUCTR2006‐005405‐67. Study of the effectiveness and tolerance of L35 versus placebo in the treatment of hidradenitis suppurativa (Verneuil 's Disease) [Etude de l'efficacité et de la tolérance du L35 versus placebo dans le traitement de l'hidradénite suppurée (Maladie de Verneuil)]. www.clinicaltrialsregister.eu/ctr‐search/search?query=2006‐005405‐67 (accessed 2 April 2015).
EUCTR2007‐000534‐39 {published data only}
- EUCTR2007‐000534‐39. Comparative randomised study on the effectiveness of the treatment of hidradenitis suppurative disease or verneuil by sub cutaneous injections of botulinum toxin versus placebo [Etude comparative randomisee intra‐individuelle de l'efficacite du traitement d'hidradenite suppuree ou maladie de verneuil par injections sous cutanee de toxine botulinique versus placebo]. www.clinicaltrialsregister.eu/ctr‐search/trial/2007‐000534‐39/FR (accessed 2 April 2015).
Servant 2002 {published data only (unpublished sought but not used)}
- Servant JM, Couturaud B, Briand E. Algosteril range (calcium alginate rope/dressing/powder) versus tulle gras lumiere/vaseline (vaseline gauze) in the treatment of lesions due to Verneuil's disease. 4th Australian Wound Management Association Conference, Adelaide, South Australia. Adelaide, 2002.
References to ongoing studies
NCT01063270 {unpublished data only}
- NCT01063270. Trial Comparing Efficacy of Treatments for Hidradenitis Suppurativa. clinicaltrials.gov/show/NCT01063270 (accessed 17 September 2014).
NCT01468207 {published and unpublished data}
- Jemec GB, Sundaram M, Pinsky B, Gu Y, Williams D, Bao Y. Adalimumab improves treatment satisfaction with medication (TS‐M) in patients with moderate to severe Hidradenitis Suppurativa (HS) in a 12‐week randomised controlled trial (PIONEER I). 44th Annual Meeting of the European Society for Dermatological Research Copenhagen Denmark. Conference Start: 20140910 Conference End: 20140913. Journal of Investigative Dermatology 2014;134:S31. [EMBASE: 71606497] [Google Scholar]
- Kimball A, Zouboulis C, Armstrong A, Korman N, Crowley J, Lynde C, et al. Safety and efficacy of adalimumab in patients with moderate to severe hidradenitis suppurativa: Results from first 12 weeks of PIONEER I, a phase 3, randomized, placebo‐controlled trial. 73rd Annual Meeting of the American Academy of Dermatology San Francisco, CA United States. Journal of the American Academy of Dermatology 2015;72(5 Suppl 1):AB60. [EMBASE: 71895003] [Google Scholar]
- Kimball A, Zouboulis CC, Armstrong AW, Korman N, Crowley JJ, Lynde C, et al. Safety and efficacy of adalimumab in patients with moderate to severe hidradenitis suppurativa: results from first 12 weeks of PIONEER I, a phase 3, randomized, placebo‐controlled trial. 44th Annual Meeting of the European Society for Dermatological Research Copenhagen Denmark. Conference Start: 20140910 Conference End: 20140913. Journal of Investigative Dermatology 2014;134:S36. [EMBASE: 71606530] [Google Scholar]
- NCT01468207. Efficacy and Safety Study of Adalimumab in Treatment of Hidradenitis Suppurativa (PIONEER I). clinicaltrials.gov/show/NCT01468207 (accessed 17 September 2014).
NCT01468233 {unpublished data only}
- Armstrong A, Pinsky B, Sundaram M, Shu L, Okun M, Bao Y. Adalimumab improves health‐related quality of life (HRQoL) in patients with moderate to severe hidradenitis suppurativa (HS): results from the first 12 weeks of PIONEER II. 73rd Annual Meeting of the American Academy of Dermatology San Francisco, CA United States. Conference Start: 20150320 Conference End: 20150324. Journal of the American Academy of Dermatology 2015;72(5 Suppl 1):AB38. [EMBASE: 71894918] [Google Scholar]
- Jemec G, Gottlieb A, Forman S, Giamarellos‐Bourboulis E, Reguiai Z, Gu Y, et al. Efficacy and safety of adalimumab in patients with moderate to severe hidradenitis suppurativa: Results from PIONEER II, a phase 3, randomized, placebo‐controlled trial. 73rd Annual Meeting of the American Academy of Dermatology San Francisco, CA United States. Conference Start: 20150320 Conference End: 20150324. Journal of the American Academy of Dermatology 2015;72(5 Suppl 1):AB45. [EMBASE: 71894944] [Google Scholar]
- Jemec GBE, Sundaram M, Pinsky B, Shu L, Okun, M, Bao Y. Adalimumab improves treatment satisfaction with medication (TS‐M) in patients with moderate to severe hidradenitis suppurativa (HS) in a 12‐week randomized controlled trial (PIONEER II). Journal of the American Academy of Dermatology 2015;72(5 Suppl 1):AB39. [EMBASE: 71894919] [Google Scholar]
- Kimball AB, Pinsky B, Sundaram M, Geng Z, Okun M, Bao Y. Adalimumab is associated with reduced skin pain in patients with moderate to severe hidradenitis suppurativa (HS): Results from the first 12 weeks of PIONEER II. 73rd Annual Meeting of the American Academy of Dermatology San Francisco, CA United States. Journal of the American Academy of Dermatology 2015;72(5 Suppl 1):AB39. [EMBASE: 71894920] [Google Scholar]
- NCT01468233. Efficacy and Safety Study of Adalimumab in the Treatment of Hidradenitis Suppurativa (PIONEER II). clinicaltrials.gov/show/NCT01468233 (accessed 17 September 2014).
NCT01558375 {unpublished data only}
- NCT01558375. Anakinra in Hidradenitis Suppurativa. clinicaltrials.gov/show/NCT01558375 (accessed 17 September 2014).
NCT01818167 {published data only}
- NCT01818167. An Investigation Into the Efficacy of Provodine Topical Cream as Compared to 10% Benzoyl Peroxide Wash for the Treatment of Hidradenitis Suppurativa. clinicaltrials.gov/ct2/show/NCT01818167 (accessed 2 April 2015).
NCT01838499 {published data only}
- NCT01838499. Assessment of the Safety, Tolerability and Efficacy of MEDI8968 in Subjects With Moderate to Severe Hidradenitis Suppurativa. clinicaltrials.gov/ct2/show/NCT01838499 (accessed 2 April 2015).
NCT02163746 {published data only}
- NCT02163746. Study to Compare Two Treatments for Axillary Hidradenitis Suppurativa: Carbon Dioxide Laser Versus Surgical Deroofing. clinicaltrials.gov/ct2/show/NCT02163746 (accessed 2 April 2015).
NCT02421172 {published data only}
- NCT02421172. Efficacy, Safety, and Pharmacokinetics Study of CJM112 in Hidradenitis Suppurativa Patients. clinicaltrials.gov/ct2/show/NCT02421172 (accessed 2 April 2015).
Additional references
Alhusayen 2012
- Alhusayen R, Shear NH. Pharmacologic interventions for hidradenitis suppurativa: what does the evidence say?. American Journal of Clinical Dermatology 2012;13(5):283‐91. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Basra 2008
- Basra MK, Fenech R, Gatt RM, Salek MS, Finlay AY. The Dermatology Life Quality Index 1994‐2007: a comprehensive review of validation data and clinical results. British Journal of Dermatology 2008;159(5):997‐1035. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Basra 2015
- Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology 2015;230(1):27‐33. [EMBASE: 2015703605] [DOI] [PubMed] [Google Scholar]
Blok 2013
- Blok JL, Hattem S, Jonkman MF, Horváth B. Systemic therapy with immunosuppressive agents and retinoids in hidradenitis suppurativa: a systematic review. British Journal of Dermatology 2013;168(2):243‐52. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [MEDLINE: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Finlay 1994
- Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI) ‐ a simple practical measure for routine clinical use. Clinical & Experimental Dermatology 1994;19(3):210‐6. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Finlay 2005
- Finlay AY. Current severe psoriasis and the Rule of Tens. British Journal of Dermatology 2005;152(5):861‐7. [EMBASE: 2005228623] [DOI] [PubMed] [Google Scholar]
Fitzsimmons 1985
- Fitzsimmons JS, Guilbert PR, Fitzsimmons EM. Evidence of genetic factors in hidradenitis suppurativa. British Journal of Dermatology 1985;113(1):1‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Harrison 1987
- Harrison BJ, Mudge M, Hughes LE. Recurrence after surgical treatment of hidradenitis suppurativa. British Medical Journal 1987;294(6570):487‐9. [EMBASE: 1987087966] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. The Cochrane Collaboration.
Hsiao 2010
- Hsiao JL, Antaya RJ, Berger T, Maurer T, Shinkai K, Leslie KS. Hidradenitis suppurativa and concomitant pyoderma gangrenosum: a case series and literature review. Archives of Dermatology 2010;146(11):1265‐70. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Hurley 1989
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH editor(s). Dermatologic Surgery. New York: Marcel Dekker, 1989:729‐39. [Google Scholar]
Ingram 2012
- Ingram JR, Desai N, Kai AC, Chua SL, Woo PN, Kerdel F, Burton T, Ormerod AD, Garner SE, Hood K, Piguet V. Interventions for hidradenitis suppurativa. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD010081] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ingram 2014
Ioannidis 2005
- Ioannidis JP. Contradicted and initially stronger effects in highly cited clinical research. Journal of the American Medical Association 2005;294(2):218‐228. [EMBASE: 2005336655] [DOI] [PubMed] [Google Scholar]
Jemec 1996
- Jemec GB, Heidenheim M, Nielsen NH. The prevalence of hidradenitis suppurativa and its potential precursor lesions. Journal of the American Academy of Dermatology 1996;35(2 Pt 1):191‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Jemec 2012
- Jemec GB. Clinical practice. Hidradenitis suppurativa. New England Journal of Medicine 2012;366(2):158‐64. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kraft 2007
- Kraft JN, Searles GE. Hidradenitis suppurativa in 64 female patients: retrospective study comparing oral antibiotics and antiandrogen therapy. Journal of Cutaneous Medicine & Surgery 2007;11(4):125‐31. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kromann 2014
- Kromann CB, Deckers IE, Esmann S, Boer J, Prens EP, Jemec GB. Risk factors, clinical course and long‐term prognosis in hidradenitis suppurativa: a cross‐sectional study. British Journal of Dermatology 2014;171(4):819‐24. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Kurek 2012
- Kurek A, Peters EM, Chanwangpong A, Sabat R, Sterry W, Schneider‐Burrus S. Profound disturbances of sexual health in patients with acne inversa. Journal of the American Academy of Dermatology 2012;67(3):422‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Madan 2008
- Madan V, Hindle E, Hussain W, August PJ. Outcomes of treatment of nine cases of recalcitrant severe hidradenitis suppurativa with carbon dioxide laser. British Journal of Dermatology 2008;159(6):1309‐14. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Mahmoud 2010
- Mahmoud BH, Tierney E, Hexsel CL, Pui J, Ozog DM, Hamzavi IH. Prospective controlled clinical and histopathologic study of hidradenitis suppurativa treated with the long‐pulsed neodymium:yttrium‐aluminium‐garnet laser. Journal of the American Academy of Dermatology 2010;62:637‐45. [DOI] [PubMed] [Google Scholar]
Naldi 2006
- Naldi L. Epidemiology. In: Jemec GB, Revuz J, Leyden JJ editor(s). Hidradenitis Suppurativa. 1. Berlin: Springer, 2006:58‐64. [Google Scholar]
O'Rourke 1989
- O'Rourke K, Detsky AS. Meta‐analysis in medical research: strong encouragement for higher quality in individual research efforts. Journal of Clinical Epidemiology 1989;42(10):1021‐4. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pink 2011
- Pink AE, Simpson MA, Brice GW, Smith CH, Desai N, Mortimer PS, et al. PSENEN and NCSTN mutations in familial hidradenitis suppurativa (acne inversa). Journal of Investigative Dermatology 2011;131(7):1568‐70. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Rambhatla 2011
- Rambhatla PV, Lim HW, Hamzavi I. A Systematic Review of Treatments for Hidradenitis Suppurativa. Archives of Dermatology 2012;148(4):439‐46. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Revuz 2008
- Revuz JE, Canoui‐Poitrine F, Wolkenstein P, Viallette C, Gabison G, Pouget F, et al. Prevalence and factors associated with hidradenitis suppurativa: results from two case‐control studies. Journal of the American Academy of Dermatology 2008;59(4):596‐601. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Revuz 2009
- Revuz J. Hidradenitis suppurativa. Journal of the European Academy of Dermatology & Venereology 2009;23(9):985‐98. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sartorius 2003
- Sartorius K, Lapins J, Emtestam L, Jemec GB. Suggestions for uniform outcome variables when reporting treatment effects in hidradenitis suppurativa. British Journal of Dermatology 2003;149(1):211‐3. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sartorius 2009
- Sartorius K, Emtestam L, Jemec GB, Lapins J. Objective scoring of hidradenitis suppurativa reflecting the role of tobacco smoking and obesity. British Journal of Dermatology 2009;161(4):831‐9. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sartorius 2010
- Sartorius K, Killasli H, Heilborn J, Jemec GB, Lapins J, Emtestam L. Interobserver variability of clinical scores in hidradenitis suppurativa is low. British Journal of Dermatology 2010;162(6):1261‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Sartorius 2011
- Sartorius K, Killasli H, Oprica C, Sullivan A, Lapins J. Bacteriology of hidradenitis suppurativa exacerbations and deep tissue cultures obtained during carbon dioxide laser treatment. British Journal of Dermatology 2012;166(4):879‐83. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Scheinfeld 2003
- Scheinfeld NS. A case of dissecting cellulitis and a review of the literature. Dermatology Online Journal 2003;9(1):8. [MEDLINE: ] [PubMed] [Google Scholar]
Shavit 2014
van der Zee 2010
- Zee HH, Woude CJ, Florencia EF, Prens EP. Hidradenitis suppurativa and inflammatory bowel disease: are they associated? Results of a pilot study. British Journal of Dermatology 2010;162(1):195‐7. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
van Rappard 2013
- Rappard DC, Limpens J, Mekkes JR. The off‐label treatment of severe hidradenitis suppurativa with TNF‐α inhibitors: a systematic review. Journal of Dermatological Treatment 2013;24(5):392‐404. [PUBMED: 22397574] [DOI] [PubMed] [Google Scholar]
von der Werth 2000 a
- Werth JM, Williams HC, Raeburn JA. The clinical genetics of hidradenitis suppurativa revisited. British Journal of Dermatology 2000;142(5):947‐53. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
von der Werth 2000 b
- Werth JM, Williams HC. The natural history of hidradenitis suppurativa. Journal of the European Academy of Dermatology & Venereology 2000;14(5):389‐92. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
von Laffert 2011
- Laffert M, Stadie V, Wohlrab J, Marsch WC. Hidradenitis suppurativa/acne inversa: bilocated epithelial hyperplasia with very different sequelae. British Journal of Dermatology 2011;164(2):367‐71. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Wang 2010
- Wang B, Yang W, Wen W, Sun J, Su B, Liu B, et al. Gamma‐secretase gene mutations in familial acne inversa. Science 2010;330(6007):1065. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Whitton 2015
- Whitton ME, Pinart M, Batchelor J, Leonardi‐Bee J, González U, Jiyad Z, et al. Interventions for vitiligo. Cochrane Database of Systematic Reviews 2015, Issue 2. [DOI: 10.1002/14651858.CD003263.pub5] [DOI] [PMC free article] [PubMed] [Google Scholar]