

Abstract
Background
Primary sclerosing cholangitis is a chronic cholestatic liver disease that is associated with both hepatobiliary and colorectal malignancies, which can result in liver cirrhosis and its complications. The optimal pharmacological treatment for patients with primary sclerosing cholangitis remains controversial.
Objectives
To assess the comparative benefits and harms of different pharmacological interventions in people with primary sclerosing cholangitis by performing a network meta‐analysis, and to generate rankings of available pharmacological interventions according to their safety and efficacy. Given that it was not possible to assess whether potential effect modifiers were similar across comparisons, we did not perform the network meta‐analysis but instead used standard Cochrane methods.
When trials begin to provide an adequate description of potential effect modifiers, we will attempt to conduct network meta‐analysis.
Search methods
We searched CENTRAL, MEDLINE, Embase, Science Citation Index ‐ Expanded, the WHO International Clinical Trials Registry Platform, and randomised controlled trials registers until February 2017 to identify randomised clinical trials (RCT) on pharmacological interventions for primary sclerosing cholangitis.
Selection criteria
We included only RCTs, irrespective of language, blinding, or publication status, in which participants were given a diagnosis of primary sclerosing cholangitis. We excluded trials that included previously liver‐transplanted participants. We considered any of various pharmacological interventions compared with one other or with placebo. We excluded trials that compared different doses of various pharmacological interventions or that reported different treatment durations, except for ursodeoxycholic acid (UDCA). As UDCA is the drug most commonly investigated for primary sclerosing cholangitis, we performed a second analysis in which we stratified the dose of UDCA.
Data collection and analysis
We calculated the odds ratio and the rate ratio with 95% confidence intervals (CIs) using both fixed‐effect and random‐effects models based on available‐participant analysis with Review Manager. We assessed risk of bias according to Cochrane, controlled risk of random errors with Trial Sequential Analysis, and assessed the quality of the evidence using GRADE.
Main results
We identified 22 RCTs in which 1211 participants were randomised to 13 different interventions. Most were placebo‐controlled trials. Trials had few restrictions apart from an established diagnosis of primary sclerosing cholangitis, evidence of cholestasis, absence of decompensated liver disease, and absence of malignancy. However, some trials included symptomatic participants only, and others included both symptomatic and asymptomatic participants. A total of 11 RCTs (706 participants) provided data for one or more outcomes. The period of follow‐up ranged from three months to three years in most trials. Only three trials reported follow‐up longer than three years. Investigators found no evidence of differences in important clinical benefits such as reduction in mortality at maximal follow‐up and improvement in health‐related quality of life.
Primary outcomes Mortality:Effect estimates: colchicine versus placebo: odds ratio 0.44, 95% CI 0.04 to 5.07, participants = 84, one trial; penicillamine versus placebo: odds ratio 1.18, 95% CI 0.39 to 3.58, participants = 70, one trial; steroids versus placebo: odds ratio 3.00, 95% CI 0.10 to 90.96, participants = 11, one trial; ursodeoxycholic acid versus placebo: odds ratio 1.51, 95% CI 0.63 to 3.63, participants = 348, two trials, I2 = 0%; vancomycin versus placebo: not estimable because no events in either group, participants = 29, one trial.
Serious adverse events (proportion):Effect estimates: infliximab versus placebo: odds ratio not estimable (because of zero events in both arms), participants = 7, one trial; steroids versus placebo: odds ratio 20.00, 95% CI 0.93 to 429.90, participants = 11, one trial; vancomycin versus placebo: not estimable because no events in either group, participants = 29, one trial.
Serious adverse events (number):Effect estimates: infliximab versus placebo: rate ratio 0.80, 95% CI 0.02 to 40.44, participants = 7, one trial; penicillamine versus placebo: rate ratio 13.60, 95% CI 0.78 to 237.83, participants = 70, one trial; steroids versus placebo: rate ratio 3.32, 95% CI 0.71 to 15.62, participants = 11, one trial.
Adverse events (proportion):Effect estimates: steroids versus placebo: odds ratio 20.00, 95% CI 0.93 to 429.90, participants = 11, one trial; ursodeoxycholic acid versus placebo: odds ratio 1.22, 95% CI 0.68 to 2.17, participants = 198, one trial; vancomycin versus placebo: not estimable because no events in either group, participants = 29, one trial.
Adverse events (number):Effect estimates: cyclosporin versus placebo: rate ratio 2.64, 95% CI 0.99 to 7.03, participants = 26, one trial; steroids versus placebo: rate ratio 3.32, 95% CI 0.71 to 15.62, participants = 11, one trial; ursodeoxycholic acid plus metronidazole versus ursodeoxycholic acid: rate ratio 2.36, 95% CI 0.98 to 5.71, participants = 71, one trial.
Health‐related quality of life: ursodeoxycholic acid versus placebo: mean difference 1.30, 95% CI ‐5.61 to 8.21, participants = 198, one trial (Short Form (SF)‐36 General Health Scale).
Secondary outcomes Studies provided no evidence of differences in clinical benefits such as a reduction in the requirement for liver transplantation or a reduction in the incidence proportion of cholangiocarcinoma. One small trial (29 participants) comparing vancomycin versus placebo reported no malignancies, no liver decompensation, and no liver transplantation in either group after a very short follow‐up period of 12 weeks after treatment. None of the remaining trials clearly reported other clinical benefits such as decreased development of all malignancies, colorectal cancer, liver decompensation, time to liver decompensation, time to liver transplantation, or requirement for cholecystectomy to allow comparisons between different interventions.
Source of funding: Fifteen trials reported the source of funding; three were funded by parties without vested interest in results of the trial, and 12 were funded in part or in full by drug companies.
Authors' conclusions
Evidence is currently insufficient to show differences in effectiveness measures such as mortality, health‐related quality of life, cirrhosis, or liver transplantation between any active pharmacological intervention and no intervention. However, trials were at high risk of bias and included small numbers of participants, had short follow‐up periods, and reported few clinical outcomes. An urgent need exists to identify an effective medical treatment for primary sclerosing cholangitis through well‐designed RCTs with adequate follow‐up that aim to identify differences in outcomes important to people with primary sclerosing cholangitis.
Plain language summary
Medical treatment for people with primary sclerosing cholangitis
Background
Primary sclerosing cholangitis is a disease that affects the bile ducts. Bile ducts are tubes that transport the bile produced by liver cells. Primary sclerosing cholangitis is a relatively uncommon disease, with 1 in 10,000 people affected. It is more common among men, and most people receive the diagnosis at between 30 and 50 years of age. Primary sclerosing cholangitis can lead to liver damage, liver failure, and bile duct cancer, and it decreases a person's longevity. Various medical treatments for primary sclerosing cholangitis have been tested. The best way to treat patients with primary sclerosing cholangitis remains unclear. We sought to resolve this issue by searching for studies conducted to explore this topic. We included all randomised clinical trials whose results were reported until February 2017. We included only trials in which participants had not undergone liver transplantation before participating in the trial. Apart from using standard Cochrane methods, which allow comparison of only two treatments at a time (direct comparison), we planned to use an advanced method (network meta‐analysis) that would allow comparison of many different individual treatments as reported by research trials. However, because of the nature of the available information, we could not determine whether results of the network meta‐analysis were reliable. So, we used standard Cochrane methods instead.
Study characteristics
We identified 22 randomised clinical trials with a total of 2211 participants that met our inclusion criteria. Participants in these trials were randomised to 13 different treatments. In most trials, placebo (dummy treatment) was provided as one of the treatments. Trials applied few restrictions apart from confirmation of primary sclerosing cholangitis, evidence of bile stagnation, which is an early marker of primary sclerosing cholangitis, absence of liver failure, and absence of cancer. However, only 11 trials (706 participants) provided the information that we sought. The remaining trials, which were conducted in people with primary sclerosing cholangitis, compared different treatments but did not report important information on deaths, complications, health‐related quality of life, liver failure, liver transplantation, or cancer. Participants in most of these trials were followed‐up only for three months to three years. Only three trials followed‐up trial participants for longer than three years.
Source of funding: Fifteen trials reported their source of funding; three of these were funded by parties without vested interest in results of the trial, and 12 were funded in part or in full by drug companies.
Key results
Differences in important clinical benefits such as reduction in mortality (deaths) at maximal follow‐up, improvement in health‐related quality of life, reduction in the requirement for liver transplantation, or reduction in development of cholangiocarcinoma were imprecise in all comparisons. Other important clinical benefits such as incidence proportion of all malignancies, colorectal cancer, liver decompensation, time to liver decompensation, and time to liver transplantation and requirement for cholecystectomy were not reported in any trial in a format that could be analysed to allow comparison between different treatments. No evidence currently suggests that any medical treatment for primary sclerosing cholangitis is effective. An urgent need exists to identify an effective medical treatment for patients with primary sclerosing cholangitis by performing additional well‐designed randomised clinical trials.
Quality of evidence
The overall quality of evidence was very low, and all trials were judged to be at high risk of bias, which means that wrong conclusions may overestimate benefits or underestimate harms of one treatment or another because of the way the trials were conducted.
Summary of findings
Summary of findings for the main comparison. Ursodeoxycholic acid versus placebo for primary sclerosing cholangitis.
| Ursodeoxycholic acid versus placebo for primary sclerosing cholangitis | |||||
| Patient or population: people with primary sclerosing cholangitis Settings: secondary or tertiary care Intervention: ursodeoxycholic acid Comparison: placebo | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | Number of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Placebo | Ursodeoxycholic acid | ||||
|
Mortality Follow‐up: 60 months |
72 per 1000 | 105 per 1000 (47 to 220) | OR 1.51 (0.63 to 3.63) | 348 (2 trials) | ⊕⊝⊝⊝ very low1,2,3 |
| Serious adverse events | No trials reported the number of participants with serious adverse events or numbers of serious adverse events. | ||||
|
Proportion of people with adverse events Follow‐up: 60 months |
337 per 1000 | 358 per 1000 (237 to 498) | OR 1.22 (0.68 to 2.17) | 198 (1 trial) | ⊕⊝⊝⊝ very low1,2,3 |
| Number of adverse events | No trials reported the number of adverse events. | ||||
|
Health‐related quality of life Follow‐up: 5 years Scale: SF‐36 General Health Scale (Limits: 0 to 100; higher = better) |
Mean in the placebo group was 61.10. | Mean in the ursodeoxycholic acid group was 1.30 higher (5.61 lower or 8.21 higher). | ‐ |
198 (1 trial) |
⊕⊝⊝⊝ very low1,2,3 |
|
Liver transplantation Follow‐up: 60 months |
123 per 1000 | 120 per 1000 (68 to 202) | OR 0.97 (0.52 to 1.81) | 348 (2 trials) | ⊕⊝⊝⊝ very low1,2,3,4 |
| Any malignancy | No trials reported this outcome. | ||||
|
Cholangiocarcinoma Follow‐up: 60 months |
43 per 1000 | 57 per 1000 (21 to 142) | OR 1.34 (0.48 to 3.68) | 348 (2 trials) | ⊕⊝⊝⊝ very low1,2,3 |
| Colorectal cancer | No trials reported this outcome. | ||||
| Cholecystectomy | No trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion. The corresponding risk (and its 95% confidence interval) is based on assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Downgraded one level for risk of bias: the trial(s) were at high risk of bias. 2 Downgraded one level for imprecision: the sample size was small. 3 Downgraded one level for imprecision: the confidence intervals were wide and overlapped a clinically significant reduction or increase (25% reduction or increase) and no effect. 4 Downgraded two levels for inconsistency: I2 was high and overlap of confidence intervals was poor.
Background
Description of the condition
Primary sclerosing cholangitis is a chronic inflammatory disease of the liver involving intrahepatic or extrahepatic bile ducts, or both, that is characterised by fibrosis with bile duct strictures, stasis of bile (cholestasis), liver fibrosis, and liver cirrhosis (NCBI 2014). Global variation has been noted in the incidence and prevalence of primary sclerosing cholangitis, with annual incidence varying from 0.07 to 1.3 per 100,000 people, prevalence ranging from 0.2 to 13.6 per 100,000 people, and a trend showing increasing incidence (Boonstra 2012). Primary sclerosing cholangitis is more common in men (Boonstra 2012). Most people with this disease receive the diagnosis when they are between 30 and 50 years of age (Talwalkar 2001). A significant association has been observed between inflammatory bowel disease (ulcerative colitis and Crohn's disease that predominantly affects the colon) and primary sclerosing cholangitis, with about 20% to 75% of people with primary sclerosing cholangitis having inflammatory bowel disease (O'Mahony 2006; Chapman 2008; Boonstra 2012). The cause of primary sclerosing cholangitis is unclear. Although genetic and environmental factors are recognised, the main hypotheses regarding cause are that primary sclerosing cholangitis is an autoimmune disorder (i.e. immune system incorrectly recognises bile ducts as foreign material and attacks them), and that it is an immune‐mediated inflammatory disease (i.e. some triggering factor incites activation of the immune mechanism, leading to damage to the bile ducts) (O'Mahony 2006). People with certain human leukocyte antigen (HLA) typing are more likely to develop primary sclerosing cholangitis, although genes outside the HLA also play a role in its development (O'Mahony 2006; Chapman 2008; Liu 2013). The role of bacteria and viruses in the development of primary sclerosing cholangitis in susceptible people remains unclear (O'Mahony 2006; Chapman 2008).
Diagnosis of primary sclerosing cholangitis is based on the presence of biochemical features of cholestasis (i.e. elevated alkaline phosphatase or serum bilirubin) or elevation of both alkaline phosphatase and serum bilirubin with characteristic bile duct changes (i.e. multiple short segment strictures and segmental dilatation) on cholangiography, when other causes of biliary stricture and cholestasis have been excluded (EASL 2009; Chapman 2010). Causes of biliary stricture that need to be excluded include malignancy, iatrogenic causes (i.e. bile duct injury during cholecystectomy (Stewart 2014)), ischaemia (i.e. injury during cholecystectomy (Stewart 2014) or following liver transplantation (Jay 2011)), immunoglobulin (Ig)G4‐related cholangitis, recurrent pyogenic cholangitis, and recurrent pancreatitis (Chapman 2010). Biliary strictures not caused by primary sclerosing cholangitis are called secondary sclerosing cholangitis. The distinction between primary and secondary sclerosing cholangitis may be difficult, particularly for people who have undergone previous surgery on the liver and biliary tract. Inflammatory bowel disease in people with bile duct stricture favours a diagnosis of primary sclerosing cholangitis (Chapman 2010). It should be noted that bilirubin or alkaline phosphatase may be normal in people with primary sclerosing cholangitis (Chapman 2010). Currently, antibody tests including perinuclear antineutrophil cytoplasmic antibody (pANCA) have no role in the diagnosis of primary sclerosing cholangitis because these antibodies are non‐specific (Chapman 2010). Although various prognostic models have been developed for people with primary sclerosing cholangitis, use of these models has not been recommended because experts have not reached consensus on their usefulness (Chapman 2010). Approximately 50% of people die or require liver transplantation after about 20 years (Boonstra 2013).
Variant forms of primary sclerosing cholangitis include small duct primary sclerosing cholangitis (wherein bile duct dilatation is not noted on endoscopic retrograde cholangiopancreatography but liver biopsy reveals the diagnosis of primary sclerosing cholangitis) and autoimmune hepatitis (a primary sclerosing cholangitis variant syndrome in which cholangiographic features suggest primary sclerosing cholangitis but biochemical and histological features suggest autoimmune hepatitis) (Yimam 2014).
Description of the intervention
Various pharmacological interventions have been tried to treat people with primary sclerosing cholangitis. These include the bile acids ursodeoxycholic acid (UDCA) and tauro‐ursodeoxycholic acid (TUDCA) (Perez 2009; Poropat 2011), immunosuppressants or immunomodulators such as glucocorticosteroids (Giljaca 2010), methotrexate (Novak 2008), mycophenolate mofetil (Talwalkar 2005), etanercept (Epstein 2004), probiotics (Vleggaar 2008), and copper chelating agents (agents that remove copper) such as D‐penicillamine (Klingenberg 2006). Endoscopic interventions such as balloon dilatation of localised strictures, endoscopic stenting to relieve cholestasis (Koro 2013), surgical interventions such as extrahepatic biliary resection for relief of symptoms in people with primary sclerosing cholangitis without advanced cirrhosis (Pawlik 2008), liver resection for hilar cholangiocarcinoma associated with primary sclerosing cholangitis (Valero 2012), and liver transplantation in cases of cirrhosis (Klose 2014) or cholangiocarcinoma (Gores 2013) are the other interventions used to treat patients with primary sclerosing cholangitis.
How the intervention might work
Given the presumed mechanism of the disease, which is centred around the bile ducts, and knowledge of when immune mechanisms are implicated in development and/or progression of the disease, many interventions evaluated for primary sclerosing cholangitis have been immunomodulatory; others have been known to modify the enterohepatic circulation of bile acids.
Certain bile acids are protective of, and others are harmful to, hepatocytes (liver cells), cholangiocytes (cells that line the bile duct), and other gastrointestinal cells lining the oesophagus and stomach (Perez 2009). Bile acids such as UDCA and TUDCA may protect cholangiocytes from damage caused by hydrophobic bile acids by decreasing oxidative stress (through a direct antioxidant effect or an increase in antioxidant defences) (Paumgartner 2002; Perez 2009). Bile acids also stimulate choleresis (secretion of bile acids from liver cells (hepatocytes)), thereby decreasing cholestasis and resulting damage to cells and inhibiting apoptosis (programmed cell death) (Paumgartner 2002; Perez 2009). Primary sclerosing cholangitis is considered an autoimmune disorder or an immune‐mediated inflammatory disease (O'Mahony 2006); therefore, altering immunity and the inflammatory response with glucocorticoids and other immunosuppressants may decrease damage resulting from the inflammatory response.
Alternative treatment strategies that have been explored in primary sclerosing cholangitis include modifying the metabolism of copper. It is recognised that people with sclerosing cholangitis experience an accumulation of copper in the liver (Gross 1985). D‐pencillamine might remove the excess copper, thereby protecting hepatocytes from damage caused by copper accumulation. Endoscopic interventions and extrahepatic biliary resections work by relieving cholestasis when a dominant bile duct stricture is present. In addition, extrahepatic biliary resections result in excision of diseased tissue. Liver resection for hilar cholangiocarcinoma results in excision of cancers that develop in people with primary sclerosing cholangitis. Liver transplantation is aimed at replacing the liver of the person with advanced liver cirrhosis with a functioning liver. We have included only pharmacological interventions in this Cochrane review (i.e. we have excluded endoscopic and surgical interventions).
Why it is important to do this review
The optimal pharmacological intervention for primary sclerosing cholangitis is not known. Currently, no pharmacological intervention is recommended for the treatment of individuals with primary sclerosing cholangitis, except for the variant form, namely, autoimmune hepatitis‐primary sclerosing cholangitis variant syndrome, for which glucocorticoid treatment is recommended (Chapman 2010). Through this systematic review and attempted network meta‐analysis, we intend to provide evidence of highest quality showing the role of different interventions used to treat people with primary sclerosing cholangitis.
Objectives
To assess the comparative benefits and harms of different pharmacological interventions in people with primary sclerosing cholangitis by performing a network meta‐analysis, and to generate rankings of available pharmacological interventions according to their safety and efficacy. Given that it was not possible to assess whether potential effect modifiers were similar across comparisons, we did not perform the network meta‐analysis but instead used standard Cochrane methods to assess the benefits and harms of different interventions.
When trials begin to provide an adequate description of potential effect modifiers, we will attempt to conduct network meta‐analysis to generate rankings of available pharmacological interventions according to their safety and efficacy. For this reason, we have retained (in Appendix 1) the plan to perform network meta‐analysis. Once sufficient data are available for network meta‐analysis, we will move Appendix 1 back into the Methods section of this review.
Methods
Criteria for considering studies for this review
Types of studies
We considered only randomised clinical trials for this systematic review, irrespective of language, publication status, or date of publication. We excluded studies of other design because of the risk of bias associated with such studies. We are aware that such exclusions make us focus much more on potential benefits while not fully assessing risks of serious adverse events and risks of adverse events.
Types of participants
We included randomised clinical trials with participants with primary sclerosing cholangitis, irrespective of method of diagnosis, presence of symptoms, or whether primary sclerosing cholangitis is associated with inflammatory bowel disease. We excluded randomised clinical trials in which participants had previously undergone liver transplantation.
Types of interventions
We included studies comparing any of the following pharmacological interventions used alone or in combination for treatment of primary sclerosing cholangitis versus each other or versus placebo or no intervention.
We considered the following interventions.
Ursodeoxycholic acid (UDCA).
Tauro‐ursodeoxycholic acid (TUDCA).
Glucocorticosteroids.
Methotrexate.
Mycophenolate mofetil.
Etanercept.
Probiotics.
D‐penicillamine.
Colchicine.
Infliximab.
Vancomycin.
Metronidazole.
The above list is not exhaustive. If we identified pharmacological interventions of which we were not aware, we considered them as eligible and included them in the review if they are used primarily for treatment of individuals with primary sclerosing cholangitis.
Types of outcome measures
We assessed the benefits and harms of available pharmacological interventions used to treat people with primary sclerosing cholangitis in terms of the following outcomes.
Primary outcomes
-
Mortality.
Short‐term mortality (up to one year).
Medium‐term mortality (one to five years).
Mortality at maximum follow‐up.
-
Adverse events. We defined an adverse event as any untoward medical occurrence not necessarily having a causal relationship with treatment but resulting in a dose reduction or discontinuation of treatment (ICH‐GCP 1997) (at any time after commencement of treatment). We defined a serious adverse event as any event that would increase mortality; is life‐threatening; required inpatient hospitalisation; resulted in persistent or significant disability; or was a congenital anomaly/birth defect; or any important medical event that might have jeopardised the person or required intervention to prevent it. We used the definitions used by trial authors for adverse events and serious adverse events.
Proportion of participants with serious adverse events.
Number of serious adverse events.
Proportion of participants with any type of adverse event.
Numbers of adverse events of any type.
-
Quality of life as defined by the authors of included trials using a validated scale such as the EuroQol Group Quality of Life Questionnaire (EQ‐5D) or the Short Form (SF)‐36 General Health Scale (EuroQol 2014; Ware 2014).
Short‐term (up to one year).
Medium‐term (one to five years).
Long‐term (beyond five years).
Secondary outcomes
-
Liver transplantation.
Proportion of participants with liver transplantation.
Time to liver transplantation.
-
Decompensated liver disease (long‐term).
Proportion of participants with decompensated liver disease.
Time to liver decompensation.
Any malignancy (long‐term), in particular, cholangiocarcinoma and colorectal cancer.
Cholecystectomy (long‐term).
Search methods for identification of studies
Electronic searches
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, and Science Citation Index ‐ Expanded (Royle 2003) from inception to 22 February 2017 for randomised clinical trials comparing two or more of the above interventions. We searched for all possible comparisons including the interventions of interest. To identify additional ongoing or completed trials, we searched the World Health Organization International Clinical Trials Registry Platform search portal, which includes trials from various trial registers, including International Standard Randomized Controlled Trials Number (ISRCTN) and ClinicalTrials.gov. Search strategies are available in Appendix 2.
Searching other resources
We searched the references of identified trials and existing Cochrane reviews on primary sclerosing cholangitis to identify additional trials for inclusion.
Data collection and analysis
Selection of studies
Three review authors (FS, KG, and CT) independently identified trials for inclusion by screening titles and abstracts yielded by the search. We sought full‐text articles for all references that at least one of the review authors had identified for potential inclusion. We selected trials for inclusion on the basis of review of full‐text articles. We listed excluded full‐text references along with reasons for their exclusion in the Characteristics of excluded studies table. We planned to list for further follow‐up any ongoing trials identified primarily via search of clinical trial registers.We resolved discrepancies through discussion.
Data extraction and management
Three review authors (FS, KG, and CT) independently extracted the following data.
-
Outcome data (for each outcome and for each treatment arm when applicable).
Number of participants randomised.
Number of participants included for analysis.
Number of participants with events for binary outcomes, mean and standard deviation for continuous outcomes, number of events for count outcomes, and number of participants with events and average follow‐up period for time‐to‐event outcomes.
Definition of outcomes or scale used, if appropriate.
-
Data on potential effect modifiers.
Participant characteristics such as age, sex, comorbidity, presence of symptoms, and number and proportion of participants with inflammatory bowel disease.
Details of intervention and control (including dose, frequency, and duration) such as treatment for inflammatory bowel disease.
Risk of bias (assessment of risk of bias in included studies).
-
Other data.
Year and language of publication,
Country in which participants were recruited.
Year(s) in which trial was conducted.
Inclusion and exclusion criteria.
Follow‐up time points of the outcome.
We planned to obtain data separately for symptomatic participants and asymptomatic participants, if available from the report. We also planned to obtain data separately for participants with inflammatory bowel disease and those without inflammatory bowel disease, if available. We sought unclear or missing information by contacting trial authors. If we had any doubt whether trials shared the same participants ‐ completely or partially (by identifying common trial authors and centres) ‐ we made attempts to contact trial authors to clarify whether the trial report was duplicated. We resolved differences in opinion through discussion.
Assessment of risk of bias in included studies
We followed guidance as provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and described in the Cochrane Hepato‐Biliary Group Module (Gluud 2013) to assess risk of bias in included studies. Specifically, we assessed risk of bias in included trials for the following domains using the methods below (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Lundh 2017).
Allocation sequence generation
Low risk of bias: trial authors performed sequence generation using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if an independent person not otherwise involved in the study performed them.
Unclear risk of bias: trial authors did not specify the method of sequence generation.
High risk of bias: the sequence generation method was not random. We planned to include such studies only for assessment of harms.
Allocation concealment
Low risk of bias: the participant allocations could not have been foreseen in advance of, or during, enrolment. A central and independent randomisation unit controlled allocation. The investigators were unaware of the allocation sequence (e.g. if the allocation sequence was hidden in sequentially numbered, opaque, and sealed envelopes).
Unclear risk of bias: the trial authors did not describe the method used to conceal the allocation so the intervention allocations may have been foreseen before, or during, enrolment.
High risk of bias: it is likely that the investigators who assigned the participants knew the allocation sequence. We will include such studies only for assessment of harms.
Blinding of participants and personnel
Low risk of bias: any of the following: no blinding or incomplete blinding, but review authors judge that the outcome is not likely to be influenced by lack of blinding; or blinding of participants and key study personnel ensured, and it is unlikely that blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of ‘low risk’ or ‘high risk’; or the trial did not address this outcome.
High risk of bias: any of the following: no blinding or incomplete blinding, and the outcome is likely to be influenced by lack of blinding; or blinding of key study participants and personnel attempted, but it is likely that blinding could have been broken, and the outcome is likely to be influenced by lack of blinding.
Blinded outcome assessment
Low risk of bias: any of the following: no blinding of outcome assessment, but review authors judge that outcome measurement is not likely to be influenced by lack of blinding; or blinding of outcome assessment ensured, and it is unlikely that blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of ‘low risk’ or ‘high risk’; or the trial did not address this outcome.
High risk of bias: any of the following: no blinding of outcome assessment, and outcome measurement is likely to be influenced by lack of blinding; or blinding of outcome assessment, but it is likely that blinding could have been broken, and the outcome measurement is likely to be influenced by lack of blinding.
Incomplete outcome data
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. The study used sufficient methods, such as multiple imputation, to handle missing data.
Unclear risk of bias: there was insufficient to assess whether missing data in combination with the method used to handle missing data was likely to induce bias on results.
High risk of bias: results were likely to be biased owing to missing data.
Selective outcome reporting
Low risk of bias: the trial reported the following predefined outcomes: mortality, or decompensated liver disease, or requirement for transplantation along with treatment‐related adverse events. If the original trial protocol was available, outcomes should be those called for in that protocol. If the trial protocol was obtained from a trial registry (e.g. www.clinicaltrials.gov), outcomes sought should be those enumerated in the original protocol if the trial protocol was registered before or at the time the trial was begun. If the trial protocol was registered after the trial was begun, we will not consider those outcomes to be reliable.
Unclear risk of bias: not all predefined, or clinically relevant and reasonably expected, outcomes were reported fully, or it was unclear whether data on these outcomes were recorded.
High risk of bias: one or more predefined or clinically relevant and reasonably expected outcomes were not reported, although data on these outcomes should have been available and even recorded.
For‐profit bias
Low risk of bias: the trial appeared to be free of industry sponsorship or another type of for‐profit support that could manipulate trial design, conductance, or results.
Unclear risk of bias: the trial may or may not be free of for‐profit bias, as no information on clinical trial support or sponsorship was provided.
High risk of bias: the trial was sponsored by industry or received another type of for‐profit support.
Other bias
Low risk of bias: the trial appeared to be free of other components (e.g. inappropriate control or dose or administration of control, baseline differences, early stopping) that could put it at risk of bias.
Unclear risk of bias: the trial may or may not be free of other components that could put it at risk of bias.
High risk of bias: other factors in the trial could put it at risk of bias (e.g. inappropriate control or dose or administration of control, baseline differences, early stopping).
We considered a trial to be at low risk of bias if we assessed it to be at low risk of bias across all domains. Otherwise, we considered a trial to be at high risk of bias regarding one or more domains.
Measures of treatment effect
For dichotomous variables (e.g. short‐term and medium‐term mortality or liver transplantation, proportion of participants with adverse events, decompensated liver disease, cirrhosis, hepatocellular carcinoma), we calculated odds ratios with 95% confidence intervals (CIs). For continuous variables (e.g. quality of life reported on the same scale), we planned to calculate mean differences with 95% CIs. We planned to use standardised mean difference values with 95% CIs for quality of life, if included trials used different scales. For count outcomes (e.g. numbers of adverse events), we calculated rate ratios with 95% CIs. For time‐to‐event data (e.g. mortality at maximal follow‐up or requirement for liver transplantation, time to liver decompensation, time to cirrhosis), we planned to use hazard ratios with 95% CIs. We also calculated Trial Sequential Analysis‐adjusted CIs to control random errors (Thorlund 2011).
Unit of analysis issues
The unit of analysis was people with primary sclerosing cholangitis according to the intervention group to which they were randomly assigned.
Cluster‐randomised clinical trials
As expected, we did not find cluster‐randomised clinical trials. However, if we had found them, we planned to include them, provided that the effect estimate adjusted for cluster correlation was available.
Cross‐over randomised clinical trials
We found one cross‐over randomised clinical trial (Rasmussen 1998). We planned to include outcomes after the period of first treatment because primary sclerosing cholangitis is a chronic disease and treatments could potentially have a residual effect.
Trials with multiple treatment groups
We collected data for all trial intervention groups that met the inclusion criteria.
Dealing with missing data
We performed an intention‐to‐treat analysis (Newell 1992) when possible. Otherwise, we used data that were available to us (e.g. a trial may have reported only per‐protocol analysis results). Given that such 'per‐protocol' analyses may be biased, we planned to conduct best/worst‐case scenario (good outcome in intervention group and bad outcome in control group) and worst/best‐case scenario (bad outcome in intervention group and good outcome in control group) analyses as sensitivity analyses when possible.
For continuous outcomes, we used analysis of available cases. We planned to impute the standard deviation from P values according to guidance given in Higgins 2011. If data were likely to be normally distributed, we planned to use the median for meta‐analysis when the mean was not available. When it was impossible to calculate the standard deviation from the P value or the confidence intervals, we planned to impute the standard deviation using the largest standard deviation in other trials for that outcome. This form of imputation may decrease the weight of the study for calculation of mean differences and may bias the effect estimate to no effect for calculation of standardised mean differences (Higgins 2011).
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity by carefully examining the characteristics and design of included trials. We planned to assess the presence of clinical heterogeneity by comparing effect estimates in the presence or absence of symptoms, the presence or absence of inflammatory bowel disease along with primary sclerosing cholangitis, and doses of pharmacological interventions. Different study designs and risk of bias may contribute to methodological heterogeneity. We used the I2 test and the Chi2 test and overlapping of CIs to assess for heterogeneity.
Assessment of reporting biases
We planned to use visual asymmetry on a funnel plot to explore reporting bias when we could include at least 10 trials for direct comparison (Egger 1997; Macaskill 2001). In the presence of heterogeneity that could be explained by subgroup analysis, we planned to prepare the funnel plot for each subgroup with an adequate number of trials. We planned to use the linear regression approach described by Egger 1997 to determine funnel plot asymmetry. None of the comparisons involved 10 or more trials, so we did not explore reporting biases.
We considered selective reporting as evidence of reporting bias.
Data synthesis
We performed meta‐analyses according to Cochrane recommendations (Higgins 2011), using the software package Review Manager 5 (RevMan 2014). We used a random‐effects model (DerSimonian 1986) and a fixed‐effect model (Demets 1987). When we found discrepancy between the two models, we reported both results; otherwise, we reported only results from the fixed‐effect model.
Calculation of required information size and Trial Sequential Analysis
For calculation of required information size, see Appendix 3. We performed Trial Sequential Analysis to control risks of random error (Wetterslev 2008; Thorlund 2011; TSA 2011) when we included at least two trials in the meta‐analysis. We used an alpha error as per guidance provided by Jakobsen 2014, power of 90% (beta error of 10%), relative risk reduction of 20%, control group proportions observed in trials, and diversity as observed in the meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We planned to assess differences in effect estimates between the following subgroups.
Trials with low risk of bias compared to trials with high risk of bias.
Participants with symptomatic compared to participants with asymptomatic primary sclerosing cholangitis.
Participants with present inflammatory bowel disease compared to participants with absent inflammatory bowel disease.
Different doses of pharmacological interventions. For example, doses of ursodeoxycholic acid used in randomised clinical trials include 13 mg to 15 mg/kg/d for low‐dose (Lindor 1997), 17 mg to 23 mg/kg/d for moderate‐dose (Olsson 2005), and 28 mg to 30 mg/kg/d for high‐dose ursodeoxycholic acid (Lindor 2009).
We planned to use the Chi2 test for subgroup differences to identify subgroup differences.
Sensitivity analysis
If a trial reported only per‐protocol analysis results, we planned to re‐analyse these results using best/worst‐case scenario and worst/best‐case scenario analyses as sensitivity analyses when possible. We did not do this because we found insufficient information.
Presentation of results and GRADE assessments
We reported all outcomes in a 'Summary of findings' table format, downgrading the quality of evidence for risk of bias, inconsistency, indirectness, imprecision, and publication bias using GRADE (Guyatt 2011) for comparisons with at least two trials.
Results
Description of studies
Results of the search
We identified 3320 references through electronic searches of CENTRAL (N = 277), MEDLINE (N = 1612), Embase (N = 458), Science Citation Index ‐ Expanded (N = 908), the World Health Organization International Clinical Trials Registry Platform (N = 37) and randomised controlled trials registers (N = 28). After we removed 689 duplicates, 2631 references remained. We then excluded 2528 clearly irrelevant references by screening titles and reading abstracts. We retrieved 103 references for further assessment. We identified no references by scanning the reference lists of identified randomised trials. We excluded 48 references (37 studies) for the reasons listed in the Characteristics of excluded studies table. Three trials are awaiting classification (Anonymous 2006; ISRCTN16531030; NCT00059202). Ten are ongoing trials without interim data (EUCTR2012‐004170‐26‐IT; EUCTR2015‐003310‐24‐SE; EUCTR2015‐003392‐30‐GB; NCT01672853; NCT01688024; NCT01755507; NCT02177136; NCT02704364; NCT02943460; NCT03035058). In total, 22 trials (42 references) met the inclusion criteria of this review (Allison 1986; LaRusso 1988; Stiehl 1989; Beuers 1992; Lo 1992; Sandborn 1993; Knox 1994; Olsson 1995; Bansi 1996; De Maria 1996; Lindor 1997; Rasmussen 1998; Mitchell 2001; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Tabibian 2013; Rahimpour 2016; Trauner 2016). The reference flow is summarised in the study flow diagram (Figure 1).
1.
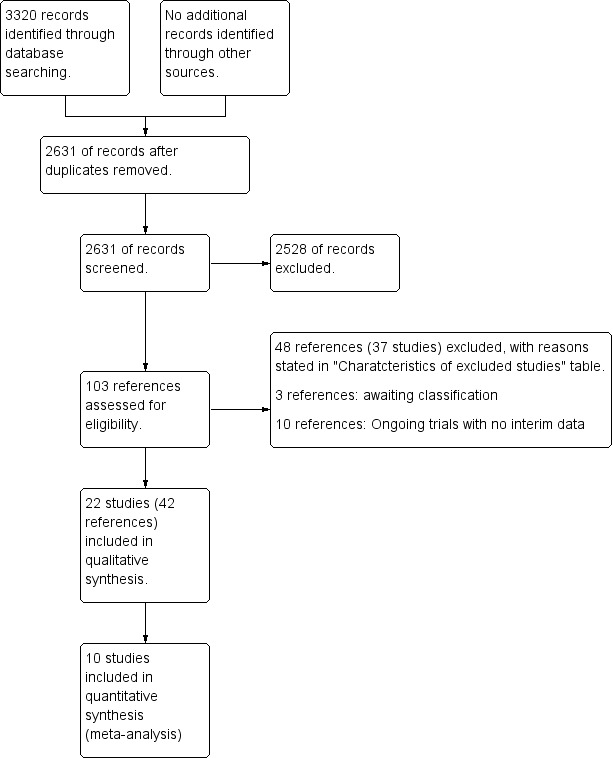
Study flow diagram.
Included studies
We have summarised the interventions used in the 22 randomised clinical trials in the Characteristics of included studies tables. All trials assessed potential pharmacological interventions, given alone or in combination, for primary sclerosing cholangitis. Twenty‐one trials were parallel randomised clinical trials (Allison 1986; LaRusso 1988; Stiehl 1989; Beuers 1992; Lo 1992; Sandborn 1993; Knox 1994; Olsson 1995; Bansi 1996; De Maria 1996; Lindor 1997; Mitchell 2001; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Tabibian 2013; Rahimpour 2016; Trauner 2016), and one was a cross‐over randomised clinical trial (Rasmussen 1998). Of the 21 parallel randomised clinical trials, 17 were two‐arm trials (Allison 1986; LaRusso 1988; Stiehl 1989; Beuers 1992; Lo 1992; Sandborn 1993; Knox 1994; Olsson 1995; Bansi 1996; Lindor 1997; Mitchell 2001; Farkkila 2004; Sterling 2004; Olsson 2005; Hommes 2008; Lindor 2009; Rahimpour 2016), two were three‐arm trials (De Maria 1996; Cullen 2008), and two were four‐arm trials (Tabibian 2013; Trauner 2016). The cross‐over randomised clinical trial was a two‐armed trial (Rasmussen 1998).
A total of 1211 participants were randomised to 13 different interventions in the 22 trials. Comparisons included the following.
Colchicine versus placebo
Olsson 1995: colchicine (44 participants) versus placebo (44 participants); follow‐up 36 months.
Cyclosporin versus placebo
Sandborn 1993: cyclosporin (16 participants) versus placebo (16 participants); follow‐up 35 months.
Infliximab versus placebo
Hommes 2008: infliximab (4 participants) versus placebo (4 participants); follow‐up 13 months.
Methotrexate versus placebo
Knox 1994: methotrexate (11 participants) versus placebo (11 participants); follow‐up 48 months.
Rasmussen 1998: methotrexate (five participants) versus placebo (eight participants); follow‐up 24 months (Note: This was a cross‐over randomised clinical trial, and participants crossed over to the opposite arm at one year).
NorUrsodeoxycholic acid versus placebo
Trauner 2016: NorUrsodeoxycholic acid (randomised to 500 mg/d or 1000 mg/d or 1500 mg/d) (participants: not stated) versus placebo (participants: not stated); follow‐up one month.
Penicillamine versus placebo
LaRusso 1988: penicillamine (39 participants) versus placebo (39 participants); follow‐up 36 months.
Steroids versus placebo
Allison 1986: steroids (six participants) versus placebo (six participants); follow‐up three months.
UDCA (high) versus placebo
Lindor 2009: UDCA (high) (76 participants) versus placebo (76 participants); follow‐up 60 months (in some participants).
UDCA (moderate) versus placebo
Bansi 1996: UDCA (moderate) (11 participants) versus placebo (11 participants); follow‐up 12 months.
Mitchell 2001: UDCA (moderate) (13 participants) versus placebo (13 participants); follow‐up 24 months.
Olsson 2005: UDCA (moderate) (97 participants) versus placebo (97 participants); follow‐up 60 months.
UDCA (low) versus placebo
Beuers 1992: UDCA (low) (six participants) versus placebo (six participants); follow‐up 12 months.
Lindor 1997: UDCA (low) (51 participants) versus placebo (51 participants); follow‐up 27 months.
Lo 1992: UDCA (low) (seven participants) versus placebo (seven participants); follow‐up 24 months.
Stiehl 1989: UDCA (low) (six participants) versus placebo (six participants); follow‐up not stated clearly.
UDCA (low) versus UDCA (moderate) versus UDCA (high)
Cullen 2008: UDCA (low) (11 participants) versus UDCA (moderate) (11 participants) versus UDCA (high) (nine participants); follow‐up 24 months.
UDCA (low) versus colchicine versus placebo
De Maria 1996: UDCA (low) (20 participants) versus colchicine (20 participants) versus placebo (20 participants); follow‐up 24 months.
UDCA (low) plus metronidazole versus UDCA (low)
Farkkila 2004: UDCA (low) plus metronidazole (37 participants) versus UDCA (low) (37 participants); follow‐up 36 months.
UDCA (low) plus mycophenolate versus UDCA (low)
Sterling 2004: UDCA (low) plus mycophenolate (six participants) versus UDCA (low) (six participants); follow‐up 24 months.
Vancomycin versus metronidazole
Tabibian 2013: vancomycin (randomised to 125 mg or 250 mg thrice daily) (16 participants) versus metronidazole (randomised to 250 mg or 500 mg thrice daily) (16 participants); follow‐up 24 months.
Vancomycin versus placebo
Rahimpour 2016: vancomycin (18 participants) versus placebo (11 participants); follow‐up three months.
The mean or median age of participants ranged from 31 years to 53 years in the 19 trials that reported this information (Allison 1986; LaRusso 1988; Beuers 1992; Lo 1992; Sandborn 1993; Knox 1994; Olsson 1995; Bansi 1996; De Maria 1996; Lindor 1997; Mitchell 2001; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Tabibian 2013; Rahimpour 2016). The proportion of females ranged from 21.4% to 62.5% in the 19 trials that reported this information (Allison 1986; LaRusso 1988; Beuers 1992; Lo 1992; Sandborn 1993; Knox 1994; Olsson 1995; Bansi 1996; De Maria 1996; Lindor 1997; Mitchell 2001; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Tabibian 2013; Rahimpour 2016). The follow‐up period in most trials ranged from one month to five years, and only three trials had a follow‐up period longer than three years (Knox 1994; Olsson 2005; Lindor 2009). Of these, one trial reported follow‐up of five years in selected participants only, and the period of follow‐up in remaining participants was not clear (Lindor 2009). A total of 11 trials (706 participants) provided data for one or more outcomes (Allison 1986; LaRusso 1988; Sandborn 1993; Olsson 1995; Farkkila 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Tabibian 2013; Rahimpour 2016). Trials did not provide information on whether participants were symptomatic. Similarly, trials did not report whether people with inflammatory bowel disease were included, although one trial excluded participants with severe inflammatory bowel disease or required specific treatment other than mesalazine (Hommes 2008). Information on potential effect modifiers such as presence of symptoms and proportion of participants with inflammatory bowel disease was missing from many trials.
Table 2 presents the intervention and control used in these trials and risk of bias arranged according to each pair‐wise comparison.
1. Characteristics table (according to comparisons).
| Study name | Number of people in intervention group | Number of people in control group | Risk of bias | Overall risk of bias | ||||||
| Random sequence generation | Allocation concealment | Blinding of participants and personnel | Blinding of outcome assessment | Incomplete outcome data | Selective reporting | Vested interest bias | ||||
| Colchicine vs placebo | ||||||||||
| Olsson 1995 | 44 | 40 | Unclear | Unclear | Low | Low | Unclear | High | Unclear | High |
| Cyclosporin vs placebo | ||||||||||
| Sandborn 1993 | 16 | 10 | Unclear | Unclear | Low | Low | High | High | High | High |
| Infliximab vs placebo | ||||||||||
| Hommes 2008 | 4 | 3 | Unclear | Unclear | Low | Low | High | High | High | High |
| Methotrexate vs placebo | ||||||||||
| Knox 1994 | 11 | 10 | Unclear | Unclear | Low | Low | High | High | High | High |
| Rasmussen 1998 | 5 (crossed over after 1 year) | 8 (crossed over after 1 year) |
Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | High |
| NorUrsodeoxycholic acid vs placebo | ||||||||||
| Trauner 2016 | Not stated | Not stated | Unclear | Unclear | Unclear | Unclear | Unclear | High | High | High |
| Penicillamine vs placebo | ||||||||||
| LaRusso 1988 | 39 | 31 | Unclear | Unclear | Low | Low | Unclear | Low | High | High |
| Steroids vs placebo | ||||||||||
| Allison 1986 | 6 | 5 | Unclear | Low | Low | Low | High | High | Low | High |
| UDCA (high) vs placebo | ||||||||||
| Lindor 2009 | 76 | 74 | Low | Low | Low | Low | Low | High | High | High |
| UDCA (moderate) vs placebo | ||||||||||
| Bansi 1996 | 11 | 11 | Unclear | Unclear | Unclear | Unclear | High | High | Unclear | High |
| Mitchell 2001 | 13 | 13 | Unclear | Unclear | Low | Low | Low | High | Unclear | High |
| Olsson 2005 | 97 | 101 | Unclear | Low | Low | Low | High | Low | High | High |
| UDCA (low) vs placebo | ||||||||||
| Beuers 1992 | 6 | 8 | Low | Unclear | Low | Low | Unclear | High | High | High |
| Lindor 1997 | 51 | 51 | Low | Unclear | Low | Low | High | High | High | High |
| Lo 1992 | 7 | 7 | Unclear | Unclear | Unclear | Unclear | High | High | Unclear | High |
| Stiehl 1989 | 6 | 6 | Unclear | Unclear | Unclear | Unclear | High | High | Unclear | High |
| UDCA (low) vs UDCA (moderate) vs UDCA (high) | ||||||||||
| Cullen 2008 | 11 | 11 (UDCA (moderate)) and 9 (UDCA (high)) | Low | Low | Low | Low | High | High | High | High |
| UDCA (low) vs colchicine vs placebo | ||||||||||
| De Maria 1996 | 20 | 19 (colchicine) and 20 (placebo) | Unclear | Unclear | High | Unclear | Unclear | High | Unclear | High |
| UDCA (low) plus metronidazole vs UDCA (low) | ||||||||||
| Farkkila 2004 | 37 | 34 | Low | Low | Low | Low | High | High | High | High |
| UDCA (low) plus mycophenolate vs UDCA (low) | ||||||||||
| Sterling 2004 | 6 | 10 | Unclear | Unclear | High | High | Unclear | High | High | High |
| Vancomycin vs metronidazole | ||||||||||
| Tabibian 2013 | 16 | 13 | Unclear | Unclear | Low | Low | High | High | Low | High |
| Vancomycin vs placebo | ||||||||||
| Rahimpour 2016 | 18 | 11 | Low | Low | Low | Low | Low | High | Low | High |
Source of funding: Fifteen trials reported the source of funding; three were funded by parties without vested interest in results of the trial (Allison 1986; Tabibian 2013; Rahimpour 2016), and 12 were funded in part or in full by the pharmaceutical industry (LaRusso 1988; Beuers 1992; Sandborn 1993; Knox 1994; Lindor 1997; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Trauner 2016).
Excluded studies
Of 37 excluded studies, we excluded 16 because they were not randomised clinical trials (Wagner 1971; Stiehl 1989a; Tabibian 1989; Stiehl 1994; Lindor 1995; Stiehl 1996; Eisenburg 1997; Harnois 2001; Kurihara 2003; Lankarani 2003; Chapman 2005; Lindor 2005; Tada 2006; Silveira 2008; Lindor 2009a; Imam 2011); seven because they provided comments on randomised clinical trials and other published experiences (Fromm 1992; Goldberg 1992; Gross 1993; Spengler 1993; Beuers 1998; Lankarani 2005; Triantos 2012); and one because it was an editorial (Chapman 2009). We excluded one study because it was conducted to examine non‐pharmacological agents (Vleggaar 2008). One study investigated an intervention that was not targeted at control of primary sclerosing cholangitis (Vleggaar 2001). In three trials, participants in different arms received the same pharmacological agent in different doses (Stiehl 1994a; van Hoogstraten 1998; van Hoogstraten 2000), and one study did not provide separate data for trial participants in the control group who received colchicine or no intervention (Van Thiel 1992). Therefore, we excluded these studies. We excluded one study because people with liver transplantation were included (Hay 2001) and we excluded six trials on cholestatic liver disease because investigators did not provide separate data for trial participants with primary sclerosing cholangitis (van de Meeberg 1996; Vleggaar 2001; Ter Borg 2004; Villamil 2005; Mayo 2007; Kuiper 2010).
Risk of bias in included studies
We have summarised the risk of bias in included trials in Figure 2 and Figure 3. Except for one small trial including 29 participants who were followed‐up for 12 weeks, at the end of which none had died or developed treatment‐related or disease‐related adverse events (Rahimpour 2016), all trials were at high risk of bias.
2.
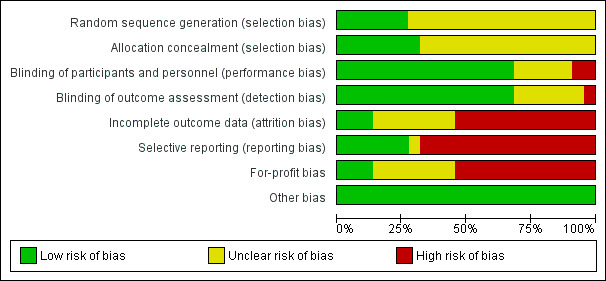
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Six trials (27.3%) had adequate sequence generation (Beuers 1992; Lindor 1997; Farkkila 2004; Cullen 2008; Lindor 2009; Rahimpour 2016). The remaining 16 trials did not report the sequence generation and were considered to be at unclear risk of sequence generation bias.
Seven trials (31.8%) had adequate allocation concealment (Allison 1986; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Lindor 2009; Rahimpour 2016). The remaining 15 trials did not report the allocation concealment and were considered to be at unclear risk of allocation concealment bias.
Thus, four trials (18.2%) had low risk of selection bias (Farkkila 2004; Cullen 2008; Lindor 2009; Rahimpour 2016). The remaining 21 trials were at unclear risk of bias.
Blinding
Fifteen trials (68.2%) reported adequate blinding of participants, personnel, and outcome assessors and were at low risk of performance and detection biases (Allison 1986; LaRusso 1988; Beuers 1992; Sandborn 1993; Knox 1994; Olsson 1995; Lindor 1997; Mitchell 2001; Farkkila 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Tabibian 2013; Rahimpour 2016). Two trials were at high risk of performance bias (De Maria 1996; Sterling 2004), as one group of participants in one trial did not receive any intervention (De Maria 1996), and participants or investigators in the other trial were not blinded to the intervention (Sterling 2004). The remaining five trials were at unclear risk of performance bias. One trial was at high risk of detection bias, as investigators were not blinded to the intervention in another trial (Sterling 2004). The remaining trials were at unclear risk of performance bias and detection bias.
Incomplete outcome data
Only three of the 22 trials (13.6%) were free from bias owing to incomplete outcome data (Mitchell 2001; Lindor 2009; Rahimpour 2016). Post‐randomisation drop‐outs may be related to the intervention in 12 trials; therefore, we considered these 12 trials to be at high risk of bias (Allison 1986; Stiehl 1989; Lo 1992; Sandborn 1993; Knox 1994; Bansi 1996; Lindor 1997; Farkkila 2004; Olsson 2005; Cullen 2008; Hommes 2008; Tabibian 2013). Participant flow was not available for the remaining seven trials; therefore, we considered these seven trials to be at unclear risk of bias (LaRusso 1988; Beuers 1992; Olsson 1995; De Maria 1996; Rasmussen 1998; Sterling 2004; Trauner 2016).
Selective reporting
A pre‐published protocol was not available for any trial. Only six trials (27.3%) reported mortality and liver transplantation; hence we considered these trials to be free from reporting bias (Allison 1986; LaRusso 1988; Olsson 1995; Olsson 2005; Lindor 2009; Rahimpour 2016). We considered the remaining trials to be at high risk of bias, as they reported neither mortality nor liver transplantation.
Other potential sources of bias
Thirteen trials (68.4%) reported the source of funding, and we rated for‐profit bias as low in only three of these (13.6%) (Allison 1986; Tabibian 2013; Rahimpour 2016). Twelve trials were at high risk of for‐profit bias because they were funded in part or in full by pharmaceutical industries with vested interest in study results (LaRusso 1988; Beuers 1992; Sandborn 1993; Knox 1994; Lindor 1997; Farkkila 2004; Sterling 2004; Olsson 2005; Cullen 2008; Hommes 2008; Lindor 2009; Trauner 2016).
Effects of interventions
See: Table 1
Mortality
Six trials (542 participants) provided data on mortality (Allison 1986, LaRusso 1988; Olsson 1995; Olsson 2005; Lindor 2009; Rahimpour 2016). Mortality was reported at different time points; therefore, we have analysed mortality at maximal follow‐up. Median follow‐up times in these six trials were:
0.25 year (Allison 1986);
4 years (LaRusso 1988);
3 years (Olsson 1995);
14 years (Olsson 2005);
3 years (Lindor 2009); and
0.25 year (Rahimpour 2016).
As shown in Analysis 1.1, studies provided no evidence of differences in any comparisons.
1.1. Analysis.
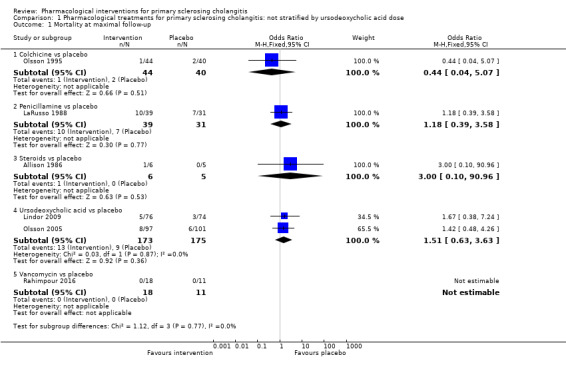
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 1 Mortality at maximal follow‐up.
Colchicine versus placebo: odds ratio 0.44, 95% CI 0.04 to 5.07, participants = 84, one trial.
Penicillamine versus placebo: odds ratio 1.18, 95% CI 0.39 to 3.58, participants = 70, one trial.
Steroids versus placebo: odds ratio 3.00, 95% CI 0.10 to 90.96, participants = 11, one trial.
Vancomycin versus placebo: odds ratio not estimable, participants = 29, one trial.
Ursodeoxycholic acid versus placebo: odds ratio 1.51, 95% CI 0.63 to 3.63, participants = 348, two trials, I2 = 0%.
Studies found no evidence of heterogeneity for ursodeoxycholic acid versus placebo (I2 = 0; Chi2 test for heterogeneity P = 0.87). Analysis revealed no differences in interpretation of results when the fixed‐effect versus the random‐effects model was used for comparison.
Proportion of people with serious adverse events
Three trials (47 participants) provided data on proportions of participants with serious adverse events (Allison 1986; Hommes 2008; Rahimpour 2016). Analysis 1.2shows no differences in any of these comparisons.
1.2. Analysis.
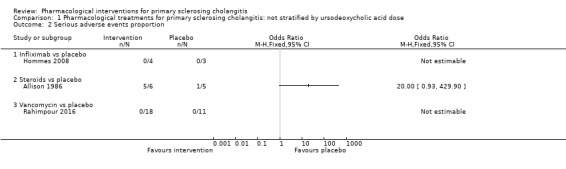
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 2 Serious adverse events proportion.
Infliximab versus placebo: odds ratio not estimable (because of zero events in both arms), participants = 7, one trial.
Steroids versus placebo: odds ratio 20.00, 95% CI 0.93 to 429.90, participants = 11, one trial.
Vancomycin versus placebo: odds ratio not estimable, participants = 29, one trial.
Lindor 2009 did not report the proportion of participants with serious adverse events; however, trial authors stated that "serious adverse events were more with UDCA group".
Number of serious adverse events
Three trials (88 participants) provided data on numbers of serious adverse events (Allison 1986; LaRusso 1988; Hommes 2008). Analysis 1.3 shows no differences in any of these comparisons.
1.3. Analysis.
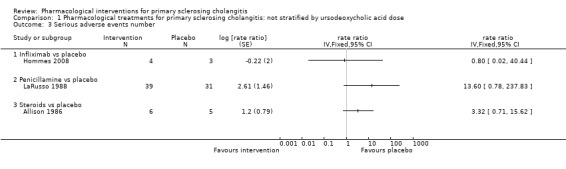
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 3 Serious adverse events number.
Infliximab versus placebo: rate ratio 0.80, 95% CI 0.02 to 40.44, participants = 7, one trial.
Penicillamine versus placebo: rate ratio 13.60, 95% CI 0.78 to 237.83, participants = 70, one trial.
Steriods versus placebo: rate ratio 3.32, 95% CI 0.71 to 15.62, participants = 11, one trial.
Vancomycin versus placebo: odds ratio not estimable, participants = 29, one trial.
Proportion of people with adverse events
Three trials (238 participants) provided data on proportions of participants with adverse events (Allison 1986; Olsson 2005; Rahimpour 2016). Analysis 1.4 shows no differences in any of these comparisons.
1.4. Analysis.
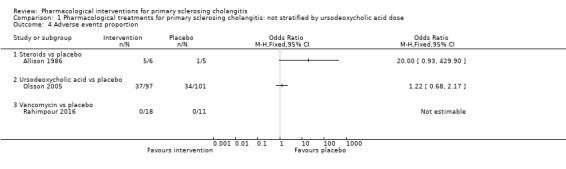
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 4 Adverse events proportion.
Steroids versus placebo: odds ratio 20.00, 95% CI 0.93 to 429.90, participants = 11, one trial.
Ursodeoxycholic acid versus placebo: odds ratio 1.22, 95% CI 0.68 to 2.17, participants = 198, one trial.
Vancomycin versus placebo: odds ratio not estimable, participants = 29, one trial.
Number of total adverse events
Five trials (207 participants) reported the number of adverse events (Allison 1986; LaRusso 1988; Sandborn 1993; Farkkila 2004; Tabibian 2013). As shown in Analysis 1.5, the number of adverse events was higher with penicillamine versus placebo (rate ratio 2.48, 95% CI 1.18 to 5.23, participants = 70, one trial) and with vancomycin versus metronidazole (rate ratio 0.41, 95% CI 0.19 to 0.87, 29 participants, one trial). Analysis revealed no differences in any of the remaining comparisons.
1.5. Analysis.
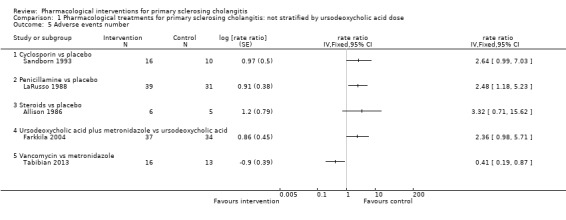
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 5 Adverse events number.
Cyclosporin versus placebo: rate ratio 2.64, 95% CI 0.99 to 7.03, participants = 26, one trial.
Steroids versus placebo: rate ratio 3.32, 95% CI 0.71 to 15.62, participants = 11, one trial.
Ursodeoxycholic acid plus metronidazole versus ursodeoxycholic acid: rate ratio 2.36, 95% CI 0.98 to 5.71, participants = 71, one trial.
Quality of life
Only one trial estimated quality of life using a validated scale (Olsson 2005). Investigators found no evidence of differences between the mean value of the SF‐36 General Health Scale in ursodeoxycholic acid versus placebo groups (mean difference 1.30, 95% CI ‐5.61 to 8.21, participants = 198, one trial) after a median follow‐up of five years (Analysis 1.6).
1.6. Analysis.
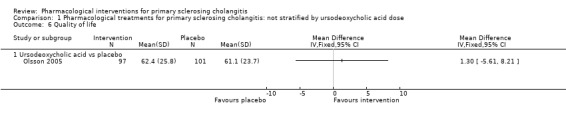
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 6 Quality of life.
Liver transplantation
Seven trials (613 participants) reported liver transplantation (Allison 1986; LaRusso 1988; Olsson 1995; Farkkila 2004; Olsson 2005; Lindor 2009; Rahimpour 2016). Liver transplantation was reported at different time points; therefore, we analysed liver transplantation at maximal follow‐up. Median follow‐up times in these six trials were:
0.25 year (Allison 1986);
4 years (LaRusso 1988);
3 years (Olsson 1995);
3 years (Farkkila 2004);
14 years (Olsson 2005);
3 years (Lindor 2009); and
0.25 year (Rahimpour 2016).
Analysis 1.7 shows no differences in any of these comparisons.
1.7. Analysis.
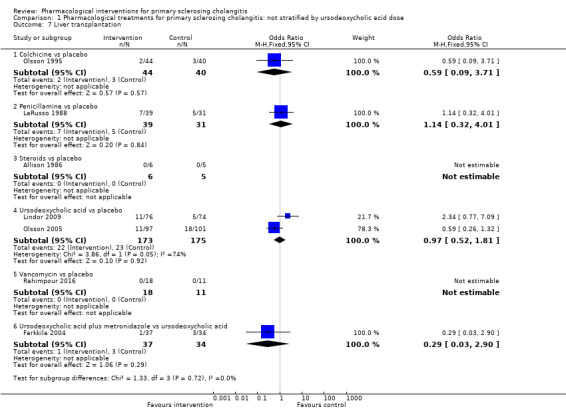
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 7 Liver transplantation.
Colchicine versus placebo: odds ratio 0.59, 95% CI 0.09 to 3.71, participants = 84, one trial.
Penicillamine versus placebo: odds ratio 1.18, 95% CI 0.39 to 3.58, participants = 70, one trial.
Steroids versus placebo: odds ratio not estimable (zero events in both groups), participants = 11, one trial.
Ursodeoxycholic acid versus placebo: odds ratio 0.97, 95% CI 0.52 to 1.81, participants = 348, two trials, I2 = 74%.
Vancomycin versus placebo: odds ratio not estimable, participants = 29, one trial.
Ursodeoxycholic acid plus metronidazole versus ursodeoxycholic acid: odds ratio 0.29, 95% CI 0.03 to 2.90, participants = 71, one trial.
Evidence shows heterogeneity in the ursodeoxycholic acid versus placebo group (I2 = 74%, Chi2 test for heterogeneity P = 0.05) and no difference in interpretation of results for fixed‐effect versus random‐effects models for this comparison.
Decompensated liver disease
One trial (29 participants) reported no decompensated liver disease in the vancomycin group nor in the placebo group after three months of follow‐up (Rahimpour 2016). None of the remaining trials reported this information adequately for analysis of data.
Any malignancy
One trial (29 participants) reported no malignancy in the vancomycin group nor in the placebo group after three months of follow‐up (Rahimpour 2016). None of the remaining trials reported this information adequately for analysis of data.
Cholangiocarcinoma
Four trials (403 participants) reported the proportion of cholangiocarcinoma (Sandborn 1993; Olsson 2005; Lindor 2009; Rahimpour 2016). Analysis 1.8 shows no differences in any of these comparisons.
1.8. Analysis.
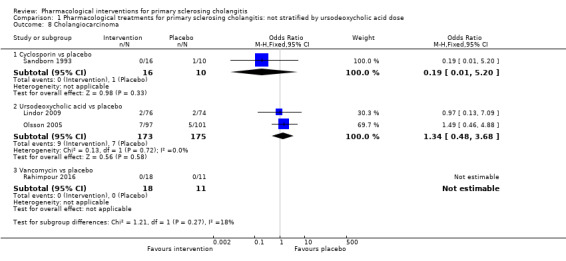
Comparison 1 Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose, Outcome 8 Cholangiocarcinoma.
Cyclosporin versus placebo: odds ratio 0.19, 95% CI 0.01 to 5.20, participants = 26, one trial.
Ursodeoxycholic acid versus placebo: odds ratio 1.34, 95% CI 0.48 to 3.68, participants = 348, two trials, I2 = 0%.
Vancomycin versus placebo: odds ratio not estimable, participants = 29, one trial.
Evidence shows no heterogeneity in the ursodeoxycholic acid versus placebo group (I2 = 0, Chi2 test for heterogeneity P = 0.13) and no difference in interpretation of for fixed‐effect versus random‐effects models for this comparison.
Colorectal cancer
One trial (29 participants) reported no colorectal cancer in the vancomycin group nor in the placebo group after three months of follow‐up (Rahimpour 2016). None of the remaining trials reported this information adequately for analysis of data.
Cholecystectomy
None of the included trials reported this information.
Subgroup analysis
We were unable to perform any subgroup analysis because of the paucity of data. However, we were able to perform an analysis stratified by doses of UDCA (low, moderate, or high) (Analysis 2.1; Analysis 2.2; Analysis 2.3; Analysis 2.4; Analysis 2.5; Analysis 2.6; Analysis 2.7; Analysis 2.8). We included all studies with the exception of Cullen 2008 in both the main analysis and the stratified analysis. Cullen 2008 compared three different doses of UDCA without including any other control. So we included this trial only in the stratified analysis and stratified the UDCA dose. The stratified analysis did not change our interpretation of results.
2.1. Analysis.
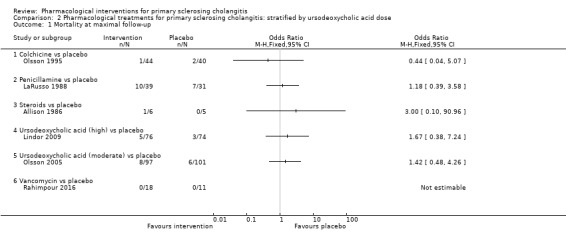
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 1 Mortality at maximal follow‐up.
2.2. Analysis.
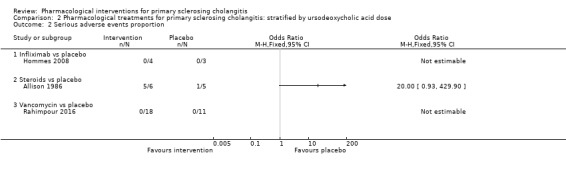
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 2 Serious adverse events proportion.
2.3. Analysis.
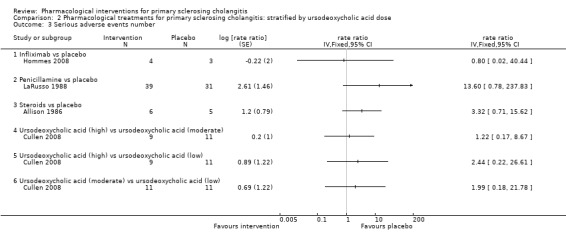
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 3 Serious adverse events number.
2.4. Analysis.
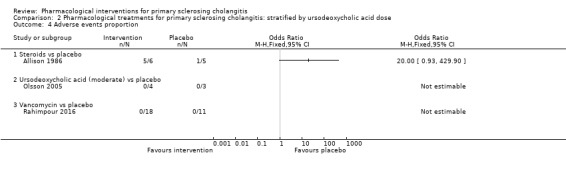
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 4 Adverse events proportion.
2.5. Analysis.
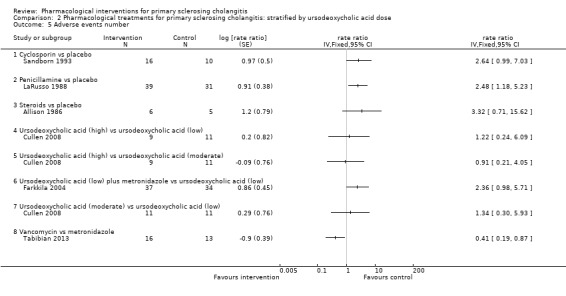
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 5 Adverse events number.
2.6. Analysis.
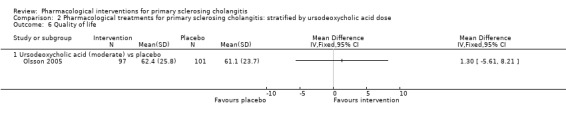
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 6 Quality of life.
2.7. Analysis.
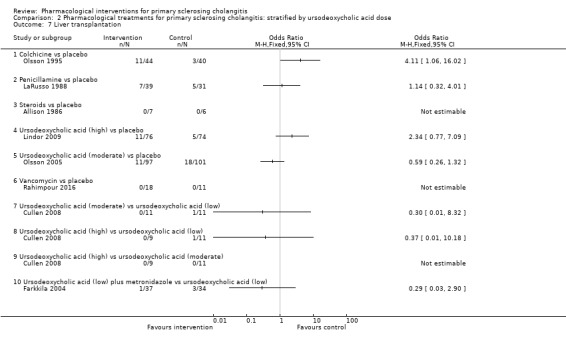
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 7 Liver transplantation.
2.8. Analysis.
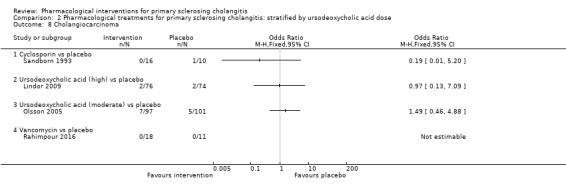
Comparison 2 Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose, Outcome 8 Cholangiocarcinoma.
Sensitivity analysis
We did not perform the planned sensitivity analysis because data were sparse, and because we did not impute the mean or the standard deviation for continuous outcomes.
Trial Sequential Analysis
Only three comparisons included more than one trial under the outcome.
Mortality at maximal follow‐up: UDCA versus placebo.
Liver transplantation: UDCA versus placebo.
Cholangiocarcinoma: UDCA versus placebo.
On the basis of an alpha error of 2.5%, power of 90% (beta error of 10%), relative risk reduction of 20%, control group proportion observed in trials (mortality at maximal follow‐up: 7.2%; liver transplantation: 12.3%; and cholangiocarcinoma 4.3%), and heterogeneity observed in analyses, required information sizes were 14,509; 34,179; and 24,972. As shown in Figure 4, only a small fraction of the required information size was reached and trial sequential monitoring boundaries were not drawn. The Z‐curve did not cross any boundaries. This indicates that risk of random error is high for all outcomes included in this review. We could not calculate trial sequential adjusted boundaries because sample sizes in these trials were small.
4.
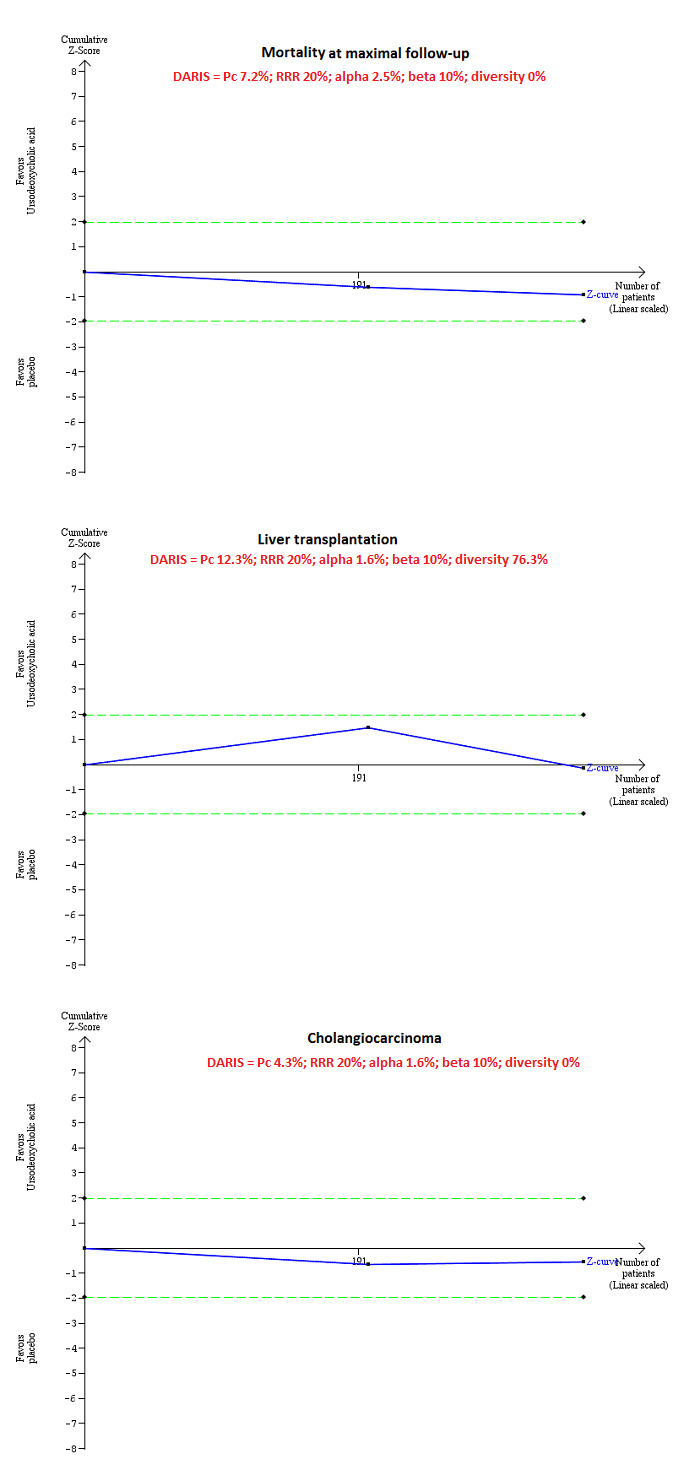
Based on an alpha error of 2.5%, power of 90% (beta error of 10%), relative risk reduction (RRR) of 20%, control group proportion observed in the trials (Pc), and heterogeneity observed in the analyses, only a small fraction of the diversity‐adjusted required information size (DARIS) has been reached (required information size = 348; DARIS = 14,509 for mortality at maximal follow‐up; required information size = 348; DARIS = 35,846 for liver transplantation; required information size = 348; DARIS = 29,191 for cholangiocarcinoma), and trial sequential monitoring boundaries were not drawn. The Z‐curves (blue lines) do not cross conventional boundaries (dotted green lines). This indicates high risk of random errors for all outcomes included in this review.
Quality of evidence
The overall quality of evidence was low or very low for all outcomes unless otherwise indicated. We downgraded the quality of evidence because of risk of bias (downgraded by two levels) for most comparisons, imprecision (small sample size: downgraded by one level), imprecision (wide confidence intervals: downgraded by one level), and inconsistency (downgraded by two levels) (Table 1). Ursodeoxycholic acid was the only comparison performed by at least two trials; we have presented this in Table 1. The remaining comparisons are presented in the text.
Discussion
Summary of main results
In this systematic review of pharmacological interventions for people with primary sclerosing cholangitis, we included 22 randomised clinical trials, 10 of which provided information on one or more outcomes of interest for this review. We found no evidence of differences between any of the interventions and placebo for important clinical benefits such as reduction in mortality at maximal follow‐up, improvement in health‐related quality of life, reduction in the requirement for liver transplantation, or reduction in the incidence proportion of cholangiocarcinoma. Other important clinical benefits such as incidence proportion of all malignancies, colorectal cancer, liver decompensation, time to liver decompensation, time to liver transplantation, and requirement for cholecystectomy were not reported clearly enough in any of the included trials to allow comparison of different interventions. However, it should be pointed out that primary sclerosing cholangitis is a slowly progressive disease, and that follow‐up in these trials was short. Future trials should provide a follow‐up period of 10 years or longer and should include important clinical outcomes.
Overall completeness and applicability of evidence
This review included randomised clinical trials in people with primary sclerosing cholangitis. Trials applied few restrictions apart from an established diagnosis of primary sclerosing cholangitis, evidence of cholestasis, absence of decompensated liver disease, and absence of malignancy. Therefore, the findings of this review are applicable to most people with primary sclerosing cholangitis without decompensated liver disease.
Quality of the evidence
The overall quality of evidence was very low. Risk of bias was unclear or high in all included trials. Selection bias was related mainly to unclear description of random sequence generation and of allocation concealment. Appropriate methods of randomisation and adequate reporting of the method of randomisation used will decrease selection bias. Most of the performance bias detected was due to missing, incomplete, or unclear information. A more detailed description of blinding will improve the quality of evidence. Drop‐outs were due mainly to participants' lack of compliance (missing follow‐up, treatment discontinuation, or withdrawal of consent). This reflects the real‐life situation in which lack of compliance with treatment is prevalent, and because the primary aim of researchers is to recommend or not recommend a specific treatment (or combination of treatments), an intention‐to‐treat analysis should be performed to avoid a biased estimate of treatment effect. Currently, no validated surrogate outcomes have been used to evaluate interventions for people with primary sclerosing cholangitis (Ponsioen 2016). Reporting all important clinical outcomes with appropriate follow‐up can decrease selective reporting bias. Funding from parties without vested interest in the results or at least publication of a full protocol before recruitment along with adherence to Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) guidelines (Chan 2013) and the Consolidated Standards of Reporting Trials (CONSORT) statement (Schulz 2010) in the final report will decrease risk of bias in these trials.
Results showed imprecision of treatment effects for all outcomes because of small sample size, along with wide confidence intervals that overlap no effect and clinically significant improvement or deterioration or both. Overall, the quality of evidence was very low (Table 1).
Potential biases in the review process
We followed guidance provided in the Cochrane Handbook for Systematic Reviews of Interventions. Two review authors independently selected studies and extracted data. We performed a thorough search of the literature. However, the search period included the pre‐mandatory trial registration era, and some trials on interventions that were not effective or were harmful may not have been reported at all.
A major limitation of this review was the high risk of bias in included trials, resulting in low or very low quality of evidence. Another major limitation of this review was the paucity of available data. We included few trials under each comparison. Many comparisons included only one trial, making it difficult for review authors to assess whether effect estimates are reproducible, and making assessment of inconsistency underpowered in comparisons involving more than one trial. Lack of evidence of inconsistency should not be considered synonymous with lack of inconsistency. This paucity of data decreases confidence in the results.
We excluded studies that compared variations among different interventions. Hence, this review has not provided information on whether one variation is better than another.
We included only randomised clinical trials known to focus on benefits without collecting and reporting harms in a detailed manner. Our choice of studies for inclusion (i.e. only randomised clinical trials) might have caused us to miss a large number of studies that addressed reporting of harms. Accordingly, this review is biased towards focusing on benefits and ignoring harms. We did not search for interventions and trials registered at regulatory authorities (e.g. FDA (US Food and Drug Administration); EMA (European Medicines Agency)). This may have led us to overlook trials; as such trials usually are unpublished, lack of their inclusion may make our comparisons appear more advantageous than they really are. However, this topic is of academic interest only because study results show no evidence of benefit of any intervention for people with primary biliary cholangitis (i.e. there is no reason to suggest that any interventions should be used in routine clinical practice regardless of their adverse event profile).
We planned to perform a network meta‐analysis. However, it was not possible to assess whether potential effect modifiers such as presence of symptoms and presence of inflammatory bowel disease were similar across different comparisons, and performing a network meta‐analysis in this scenario can be misleading. Therefore, we did not perform the network meta‐analysis but instead assessed comparative benefits and harms of different interventions using standard Cochrane methods.
Agreements and disagreements with other studies or reviews
A few systematic reviews have examined pharmacological interventions for primary sclerosing cholangitis; all have evaluated single classes of drugs. Owing to the cholestatic nature of the disease, bile acids have always been given particular attention in primary sclerosing cholangitis and their use has been evaluated in another Cochrane systematic review (Poropat 2011). This review concluded that evidence was insufficient to support or refute clinical effects of ursodeoxycholic (UDCA)/bile acids in patients with primary sclerosing cholangitis. Another meta‐analysis of randomised clinical trials comparing standard or high doses of UDCA (> 15 mg/kg body weight per day) versus placebo or no intervention in primary sclerosing cholangitis found that neither standard nor high doses of UDCA favourably influence progression of primary sclerosing cholangitis (Triantos 2011). In fact, review authors found no significant differences in outcomes (mortality, cholangiocarcinoma, histology stage progression) nor in symptoms (pruritus and fatigue) between the group treated with UDCA and the placebo/untreated group (Triantos 2011). However, a paper (Tabibian 2014) published after analysis of the available literature including an uncontrolled trial reporting negative effects of UDCA withdrawal on the biochemical and symptomatic picture of patients with primary sclerosing cholangitis (Wunsch 2014) argued for potential reconsideration of the use of UDCA in primary sclerosing cholangitis.
One Cochrane systematic review has explored the use of glucocorticosteroids for primary sclerosing cholangitis (Giljaca 2010). This review concluded that no evidence was available to support or refute oral glucocorticosteroids for patients with primary sclerosing cholangitis, and that intrabiliary application of corticosteroids via a nasobiliary tube seemed to induce severe adverse effects (Giljaca 2010).
This Cochrane systematic review identified only one randomised trial examining D‐penicillamine for primary sclerosing cholangitis (Klingenberg 2006). Results showed that D‐penicillamine had no significant effect on mortality nor on liver transplantation.
The present systematic review is the first to include all pharmacological interventions for primary sclerosing cholangitis. Our conclusions reflect and summarise those reported in the up‐to‐date evidence‐based literature: Effective medical treatment for primary sclerosing cholangitis is not available.
Authors' conclusions
Implications for practice.
Evidence is currently insufficient to show differences in effectiveness measures such as mortality, health‐related quality of life, cirrhosis, or liver transplantation between any active pharmacological intervention versus no intervention. However, confidence intervals were wide and follow‐up was short; therefore, important clinical benefits or harms could not be ruled out.
Implications for research.
The timing of this report is important, as we are entering a period when clinical trials are evaluating several new potential treatments for primary sclerosing cholangitis. An understanding of the limitations of previous studies will guide researchers as they design current and future studies. We have identified an urgent need for effective medical intervention for patients with primary sclerosing cholangitis. Currently, the three compartments providing treatment targets for clinical trials are bile acid manipulation, biological modulators of immune cell activation, and recruitment and antifibrotic therapies. High‐quality randomised clinical trials designed to measure clinically important differences in accordance with SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials; Chan 2013) and CONSORT (Consolidated Standards of Reporting Trials) guidelines (Schulz 2010) are necessary. However, researchers must take into account specific considerations regarding primary sclerosing cholangitis as they design future trials, including the rarity and phenotypical heterogeneity of the disease, its prolonged natural history, our limited understanding of risk stratification, and the lack of validated surrogate endpoints and quality of life/patient‐reported outcome measures for this disorder. Aspects of trial design that need to be addressed in future studies include:
ensuring that patients recruited into trials are phenotypically similar across randomised groups, and that biological plausibility can be found for the treatment under evaluation in the cohort of patients studied;
stratifying risk of trial participants to ensure balance in trial groups while reducing the risk of type 1 and 2 errors;
embedding several exploratory endpoints into the design to assess whether these are good surrogate outcomes; and
appropriately powering studies with adequate follow‐up with potential to conduct record linkage studies to identify long‐term effects of interventions for primary sclerosing cholangitis.
What's new
| Date | Event | Description |
|---|---|---|
| 12 April 2017 | Amended | The Cochrane Central Editorial Unit requested removal of the 'attempted network meta‐analysis' phrase from the end of the review title, as this further description of the review might create confusion in the reader. Although we followed the planned methodology for network meta‐analysis, it was not possible to assess whether the potential effect modifiers were similar across different comparisons. Therefore, we did not perform the network meta‐analysis and instead assessed the comparative benefits and harms of different interventions using standard Cochrane methodology. |
Notes
We have noted considerable overlap between the Methods of this review and those of several other protocols and reviews written by the same group of review authors.
Acknowledgements
We thank the Cochrane Comparing of Multiple Interventions Methods Group and the Cochrane Hepato‐Biliary Group for their support and advice. We thank the copy editor for improving this review.
Cochrane Review Group funding acknowledgement: The Danish State is the largest single funder of the Cochrane Hepato‐Biliary Group through its investment in the Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Copenhagen University Hospital, Denmark. Disclaimer: The views and opinions expressed in this review are those of the review authors and do not necessarily reflect those of the Danish State or the Copenhagen Trial Unit.
Peer Reviewers: Georgia Salanti,Switzerland; Ming Hua Zheng, P. R. China; Adriani Nikolakopoulou, Switzerland. Contact Editor: Christian Gluud, Denmark. Sign‐off Editor: Christian Gluud, Denmark.
Appendices
Appendix 1. Methods for network meta‐analysis if we find this is possible in the future
Measures of treatment effect
Relative treatment effects
For dichotomous variables (e.g. proportion of participants with serious adverse events or any adverse events), we will calculate the odds ratio with 95% credible interval (or Bayesian confidence interval) (Severini 1993). For continuous variables (e.g. quality of life reported on the same scale), we will calculate the mean difference with 95% credible interval. We will use standardised mean difference values with 95% credible interval for quality of life if included trials use different scales. For count outcomes (e.g. numbers of adverse events and serious adverse events), we will calculate the rate ratio with 95% credible interval. For time‐to‐event data (e.g. mortality at maximal follow‐up), we will calculate hazard ratio with 95% credible interval.
Relative ranking
We will estimate ranking probabilities for all treatments of being at each possible rank for each intervention. Then, we will obtain the surface under the cumulative ranking curve (SUCRA) (cumulative probability) and rankogram (Salanti 2011; Chaimani 2013).
Unit of analysis issues
We will collect data for all trial treatment groups that meet the inclusion criteria. The codes for analysis that we will use account for the correlation between effect sizes from trials with more than two groups.
Assessment of heterogeneity
We will assess clinical and methodological heterogeneity by carefully examining the characteristics and design of included trials. We will assess the presence of clinical heterogeneity by comparing effect estimates under different categories of potential effect modifiers. Different study designs and risk of bias may contribute to methodological heterogeneity.
We will assess the statistical heterogeneity by comparing results of the fixed‐effect model meta‐analysis and the random‐effects model meta‐analysis, and between‐study standard deviation (tau2 and comparing this with values reported in the study of the distribution of between‐study heterogeneity (Turner 2012)), and by calculating I2 (using Stata/SE 14.2). If we identify substantial heterogeneity ‐ clinical, methodological, or statistical ‐ we will explore and address heterogeneity in a subgroup analysis (see ‘Subgroup analysis and investigation of heterogeneity for network meta‐analysis’ section).
Assessment of transitivity across treatment comparisons
We will evaluate the plausibility of the transitivity assumption (the assumption that participants included in different studies with different immunosuppressive regimens can be considered part of a multi‐arm randomised clinical trial and could potentially have been randomised to any treatment) (Salanti 2012). In other words, any participant who meets the inclusion criteria is, in principle, equally likely to be randomised to any of the above eligible interventions. If we have any concern that clinical safety and effectiveness are dependent upon effect modifiers, we will continue to do traditional Cochrane pair‐wise comparisons and will not perform a network meta‐analysis on all participant subgroups.
Assessment of reporting biases
For the network meta‐analysis, we will judge reporting bias by completeness of the search (i.e. searching various databases and including conference abstracts), as we do not currently find any meaningful order to performing a comparison‐adjusted funnel plot, as suggested by Chaimani 2012. However, if we find any meaningful order, for example, the control group used depended upon the year of conduct of the trial, we will use a comparison‐adjusted funnel plot, as suggested by Chaimani 2012.
Data synthesis
Methods for indirect and mixed comparisons
We will conduct network meta‐analyses to compare multiple interventions simultaneously for each of the primary and secondary outcomes. Network meta‐analysis combines direct evidence within trials and indirect evidence across trials (Mills 2012). We will obtain a network plot to ensure that trials were connected by treatments using Stata/SE 14.2 (Chaimani 2013). The network plot for mortality at maximal follow‐up for this review is presented in Figure 21. We will exclude any trials that were not connected to the network. We will conduct a Bayesian network meta‐analysis using the Markov chain Monte Carlo method in OpenBUGS 3.2.3, as per guidance from the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) documents (Dias 2014a). We will model treatment contrast (i.e. log odds ratio for binary outcomes, mean difference or standardised mean difference for continuous outcomes, log rate ratio for count outcomes, and log hazard ratio for time‐to‐event outcomes) for any two interventions ('functional parameters') as a function of comparisons between each individual intervention and an arbitrarily selected reference group ('basic parameters') (Lu 2006) using appropriate likelihood functions and links. We will use binomial likelihood and logit link for binary outcomes, Poisson likelihood and log link for count outcomes, binomial likelihood and complementary log‐log link for time‐to‐event outcomes, and normal likelihood and identity link for continuous outcomes. We will apply a fixed‐effect model and a random‐effects model for the network meta‐analysis. We will report both models for comparison with the reference group in a forest plot. For pair‐wise comparison, we will report the fixed‐effect model if the two models reported similar results; otherwise, we will report the more conservative model.
5.
Network plot for mortality at maximal follow‐up. The size of the node (circle) provides a measure of the number of trials in which the particular treatment was included in one of the arms. The thickness of the line provides a measure of the number of direct comparisons between two nodes (treatments).
We will use a hierarchical Bayesian model using three different initial values and codes provided by NICE DSU (Dias 2014a). We will use a normal distribution with large variance (10,000) for treatment effect priors (vague or flat priors). For the random‐effects model, we will use a prior distributed uniformly (limits: 0 to 5) for between‐trial standard deviation but assumed similar between‐trial standard deviation across treatment comparisons (Dias 2014a). We will use a 'burn‐in' of 5000 simulations, check for convergence visually, and run models for another 10,000 simulations to obtain effect estimates. If we did not obtain convergence, we will increase the number of simulations for 'burn‐in'. If we do not obtain convergence still, we will use alternate initial values and priors according to methods suggested by van Valkenhoef 2012. We will also estimate the probability that each intervention ranks at one of the possible positions using the NICE DSU codes (Dias 2014a).
Assessment of inconsistency
We will assess inconsistency (statistical evidence of violation of the transitivity assumption) by fitting both an inconsistency model and a consistency model. We will use inconsistency models described in the NICE DSU manual, as we plan to use a common between‐study deviation for comparisons (Dias 2014b). In addition, we will use the design‐by‐treatment full interaction model (Higgins 2012) and IF (inconsistency factor) plots (Chaimani 2013) to assess inconsistency. In the presence of inconsistency, we will assess whether it is due to clinical or methodological heterogeneity by performing separate analyses for each of the different subgroups mentioned in the ‘Subgroup analysis and investigation of heterogeneity for network meta‐analysis’ section below.
If we find evidence of inconsistency, we will identify areas in the network where substantial inconsistency might be present in terms of clinical and methodological diversities between trials and, when appropriate, will limit network meta‐analysis to a more compatible subset of trials.
Direct comparison
We will perform direct comparisons using the same codes and the same technical details.
Sample size calculations
To control for risk of random errors, we will interpret information with caution when the accrued sample size in the network meta‐analysis (i.e. across all treatment comparisons) was less than the required sample size (required information size). For calculation of the required information size, see Appendix 3.
Subgroup analysis and investigation of heterogeneity for network meta‐analysis
We will assess differences in effect estimates between subgroups listed in subgroup analysis and investigation of heterogeneity using meta‐regression with the help of the OpenBUGS code (Dias 2012a) if we include a sufficient number of trials. We will use potential modifiers as study level co‐variates for meta‐regression. We will calculate a single common interaction term (Dias 2012a). If 95% credible intervals of the interaction term do not overlap zero, we will consider this as evidence of difference in subgroups.
Presentation of results
We will present effect estimates with 95% CrI for each pair‐wise comparison calculated from direct comparisons and network meta‐analysis. We will present the cumulative probability of treatment ranks (i.e. the probability that the treatment is within the top two, the probability that the treatment is within the top three, etc.) in graphs (surface under the cumulative ranking curve, or SUCRA) (Salanti 2011). We will plot the probability that each treatment is best, second best, third best, etc., for each of the different outcomes (rankograms), which generally are considered more informative (Salanti 2011; Dias 2012b).
We will present 'Summary of findings' tables for mortality. In Table 1, we will follow the approach suggested by Puhan et al. (Puhan 2014). First, we will calculate direct and indirect effect estimates and 95% credible intervals using the node‐splitting approach (Dias 2010) (i.e. calculate the direct estimate for each comparison by including only trials that performed direct comparisons of treatments, and the indirect estimate for each comparison by excluding trials that performed direct comparisons of treatments). Then we will rate the quality of direct and indirect effect estimates using GRADE, which takes into account risk of bias, inconsistency, directness of evidence, imprecision, and publication bias (Guyatt 2011). We will present estimates of the network meta‐analysis and will rate the quality of network meta‐analysis effect estimates as the best quality of evidence between direct and indirect estimates (Puhan 2014). In addition, in the same table, we will present illustrations and information on numbers of trials and participants, as per the standard 'Summary of findings' table.
Appendix 2. Search strategies
| Database | Time span | Search strategy |
| Central Register of Controlled Trials (CENTRAL) (Wiley). | Issue 2, 2017. | #1 MeSH descriptor: [Cholangitis, Sclerosing] explode all trees #2 primary sclerosing cholangitis or PSC #3 #1 or #2 |
| MEDLINE (OvidSP). | January 1947 to February 2017. | 1. exp Cholangitis, Sclerosing/ 2. (primary sclerosing cholangitis or PSC).mp. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier] 3. 1 or 2 4. randomised controlled trial.pt. 5. controlled clinical trial.pt. 6. randomised.ab. 7. placebo.ab. 8. drug therapy.fs. 9. randomly.ab. 10. trial.ab. 11. groups.ab. 12. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 13. exp animals/ not humans.sh. 14. 12 not 13 15. 3 and 14 |
| Embase (OvidSP). | January 1974 to February 2017. | 1. exp primary sclerosing cholangitis/ 2. (primary sclerosing cholangitis or PSC).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] 3. 1 or 2 4. exp crossover‐procedure/ or exp double‐blind procedure/ or exp randomised controlled trial/ or single‐blind procedure/ 5. (((((random* or factorial* or crossover* or cross over* or cross‐over* or placebo* or double*) adj blind*) or single*) adj blind*) or assign* or allocat* or volunteer*).af. 6. 4 or 5 7. 3 and 6 |
| Science Citation Index ‐ Expanded (Web of Knowledge) | January 1945 to February 2017. | #1 TS=(primary sclerosing cholangitis or PSC) #2 TS=(random* OR rct* OR crossover OR masked OR blind* OR placebo* OR meta‐analysis OR systematic review* OR meta‐analys*) #3 #1 AND #2 |
| World Health Organization International Clinical Trials Registry Platform Search Portal (apps.who.int/trialsearch/Default.aspx) | February 2017. | Condition: primary sclerosing cholangitis |
| ClinicalTrials.gov | February 2017. | Interventional Studies | primary sclerosing cholangitis | Phase 2, 3, 4 |
Appendix 3. Sample size calculation
Five‐year mortality in patients with primary sclerosing cholangitis is 18% (Talwalkar 2001). The required information size is based on a control group proportion of 18%, a relative risk reduction of 20% in the experimental group, type I error of 5%, and type II error of 20% in 3396 participants. Network analyses may be more prone to risk of random error than direct comparisons (Del Re 2013). Accordingly, a larger sample size is required in indirect comparisons than in direct comparisons (Thorlund 2012). Power and precision in indirect comparisons depend upon various factors, such as the number of participants included under each comparison and heterogeneity between the trials (Thorlund 2012). If no heterogeneity is evident across trials, the sample size in indirect comparisons would be equivalent to the sample size in direct comparisons. The effective indirect sample size can be calculated using the number of participants included in each direct comparison (Thorlund 2012). For example, a sample size of 2500 participants in the direct comparison A versus C (nAC) and a sample size of 7500 participants in the direct comparison B versus C (nBC) result in an effective indirect sample size of 1876 participants. However, in the presence of heterogeneity within comparisons, the sample size required is greater. In the above scenario, for an I2 statistic for each of the comparisons A versus C (IAC2) and B versus C (IBC2) of 25%, the effective indirect sample size is 1407 participants. For an I2 statistic for each of the comparisons A versus C and B versus C of 50%, the effective indirect sample size is 938 participants (Thorlund 2012). If the study includes only three groups and sample size is greater than required information size, we will calculate the effective indirect sample size using the following generic formula (Thorlund 2012):
((nAC × (1 ‐ IAC2)) × (nBC × (1 ‐ IBC2))/((nAC × (1 ‐ IAC2)) + (nBC × (1 ‐ IBC2)).
No method is currently known to calculate the effective indirect sample size for a network analysis involving more than three intervention groups.
Data and analyses
Comparison 1. Pharmacological treatments for primary sclerosing cholangitis: not stratified by ursodeoxycholic acid dose.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality at maximal follow‐up | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Colchicine vs placebo | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.44 [0.04, 5.07] |
| 1.2 Penicillamine vs placebo | 1 | 70 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.39, 3.58] |
| 1.3 Steroids vs placebo | 1 | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.10, 90.96] |
| 1.4 Ursodeoxycholic acid vs placebo | 2 | 348 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.51 [0.63, 3.63] |
| 1.5 Vancomycin vs placebo | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Serious adverse events proportion | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Infliximab vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Steroids vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events number | 3 | rate ratio (Fixed, 95% CI) | Totals not selected | |
| 3.1 Infliximab vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Penicillamine vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Steroids vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Adverse events proportion | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Steroids vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Ursodeoxycholic acid vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events number | 5 | rate ratio (Fixed, 95% CI) | Totals not selected | |
| 5.1 Cyclosporin vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Penicillamine vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Steroids vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Ursodeoxycholic acid plus metronidazole vs ursodeoxycholic acid | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.5 Vancomycin vs metronidazole | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Quality of life | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.1 Ursodeoxycholic acid vs placebo | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Liver transplantation | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 Colchicine vs placebo | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.59 [0.09, 3.71] |
| 7.2 Penicillamine vs placebo | 1 | 70 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.32, 4.01] |
| 7.3 Steroids vs placebo | 1 | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.4 Ursodeoxycholic acid vs placebo | 2 | 348 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.52, 1.81] |
| 7.5 Vancomycin vs placebo | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.6 Ursodeoxycholic acid plus metronidazole vs ursodeoxycholic acid | 1 | 71 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.03, 2.90] |
| 8 Cholangiocarcinoma | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Cyclosporin vs placebo | 1 | 26 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.01, 5.20] |
| 8.2 Ursodeoxycholic acid vs placebo | 2 | 348 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.48, 3.68] |
| 8.3 Vancomycin vs placebo | 1 | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 2. Pharmacological treatments for primary sclerosing cholangitis: stratified by ursodeoxycholic acid dose.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality at maximal follow‐up | 6 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Colchicine vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Penicillamine vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Steroids vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 Ursodeoxycholic acid (high) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 Ursodeoxycholic acid (moderate) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Serious adverse events proportion | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Infliximab vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Steroids vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Serious adverse events number | 4 | rate ratio (Fixed, 95% CI) | Totals not selected | |
| 3.1 Infliximab vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Penicillamine vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Steroids vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Ursodeoxycholic acid (high) vs ursodeoxycholic acid (moderate) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 Ursodeoxycholic acid (high) vs ursodeoxycholic acid (low) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.6 Ursodeoxycholic acid (moderate) vs ursodeoxycholic acid (low) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Adverse events proportion | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1 Steroids vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Ursodeoxycholic acid (moderate) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Adverse events number | 6 | rate ratio (Fixed, 95% CI) | Totals not selected | |
| 5.1 Cyclosporin vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Penicillamine vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Steroids vs placebo | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.4 Ursodeoxycholic acid (high) vs ursodeoxycholic acid (low) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.5 Ursodeoxycholic acid (high) vs ursodeoxycholic acid (moderate) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.6 Ursodeoxycholic acid (low) plus metronidazole vs ursodeoxycholic acid (low) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.7 Ursodeoxycholic acid (moderate) vs ursodeoxycholic acid (low) | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.8 Vancomycin vs metronidazole | 1 | rate ratio (Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Quality of life | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 6.1 Ursodeoxycholic acid (moderate) vs placebo | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Liver transplantation | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1 Colchicine vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Penicillamine vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Steroids vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Ursodeoxycholic acid (high) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Ursodeoxycholic acid (moderate) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.6 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.7 Ursodeoxycholic acid (moderate) vs ursodeoxycholic acid (low) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.8 Ursodeoxycholic acid (high) vs ursodeoxycholic acid (low) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.9 Ursodeoxycholic acid (high) vs ursodeoxycholic acid (moderate) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.10 Ursodeoxycholic acid (low) plus metronidazole vs ursodeoxycholic acid (low) | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Cholangiocarcinoma | 4 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1 Cyclosporin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Ursodeoxycholic acid (high) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 Ursodeoxycholic acid (moderate) vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 Vancomycin vs placebo | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Allison 1986.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK.
Number randomised: 17.
Post‐randomisation drop‐outs: 6 (35.3%).
Revised sample size: 11.
Mean age: 52 years.
Females: 3 (27.3%). Ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 3 months after completion of 2‐week treatment. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: continuous nasobiliary irrigation with normal saline plus hydrocortisone (100 mg/d) for 2 weeks (n = 6). Group 2: continuous nasobiliary irrigation with saline alone (1 L/d) for 2 weeks (n = 5). | |
| Outcomes | 1. Mortality. 2. Proportion of participants with any type of adverse events. 3. Proportion of participants with severe adverse events. 4. Number of any type of adverse events. 5. Number of severe adverse events. 6. Liver transplantation. | |
| Notes | Reasons for post‐randomisation drop‐out: "technical failures":
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised by sealed envelope to receive continuous nasobiliary irrigation with either normal saline alone or normal saline plus hydrocortisone. [.] The randomisation code was blocked to ensure an approximately equal number of patients in each group at any stage of the trial". |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomised by sealed envelope". Comment: "Opaque sealed envelopes manually shuffled" (trial author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients and interpreters blinded to allocation" (trial author's reply). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients and interpreters blinded to allocation" (trial author's reply). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | Low risk | Comment: No published protocol was available; mortality and liver transplantation were reported. |
| For‐profit bias | Low risk | Comment: "Patients were cared for and followed within normal NHS founded hospital stay. No additional grants were sought" (trial author's reply). |
| Other bias | Low risk | Comment: no other bias. |
Bansi 1996.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK.
Number randomised: 23.
Post‐randomisation drop‐outs: 1 (4.3%).
Revised sample size: 22.
Mean age: 53 years.
Females: 7 (31.8%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria: not stated. Follow‐up: 12 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: moderate‐dose UDCA (20 mg/kg/d) over the period of follow‐up of the study (n = 11). Group 2: placebo over the period of follow‐up of the study (n = 11). | |
| Outcomes | No outcomes of interest were reported. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind placebo‐controlled trial". Comment: Further details were not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Beuers 1992.
| Methods | Randomised clinical trial. | |
| Participants | Country: Germany.
Number randomised: 14.
Post‐randomisation drop‐outs: unclear.
Revised sample size: 14.
Mean age: 39 years.
Females: 3 (21.4%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 12 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: low‐dose UDCA (13‐15 mg/kg/d) over the period of follow‐up of the study (n = 6). Group 2: identical‐appearing placebo over the period of follow‐up of the study (n = 8). | |
| Outcomes | No outcomes of interest were reported. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were assigned with a computer generated block randomisation to receive UDCA or identical‐appearing placebo". |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was a double‐blind, randomized trial comparing the efficacy and safety of UDCA with that of placebo treatment…… Patients were assigned with a computer generated block randomization to receive UDCA or identical appearing placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The study was a double‐blind, randomized trial comparing the efficacy and safety of UDCA with that of placebo treatment…… Patients were assigned with a computer generated block randomization to receive UDCA or identical appearing placebo". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: Two patients (1 for each group) were excluded from the analysis (withdrawal), but adverse events were reported. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | High risk | Quote: "Patients were assigned with a computer generated block randomization to receive UDCA or identical appearing placebo in 250‐mg capsules (13 to 15 mg/kg body wt/day; provided by Dr. Falk GmbH, Frei‐burg, Germany)". Comment: The trial was funded by a party with a vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
Cullen 2008.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK/Germany.
Number randomised: 33.
Post‐randomisation drop‐outs: 2 (6%).
Revised sample size: 31.
Mean age: 47 years.
Females: 8. Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 24 months. |
|
| Interventions | Participants were randomly assigned to 1 of 3 groups.
Group 1: low‐dose UDCA (10 mg/kg/d) plus placebo over the period of follow‐up of the study (n = 11).
Group 2: moderate‐dose UDCA (20 mg/kg/d) plus placebo over the period of follow‐up of the study (n = 11). Group 3: high‐dose UDCA (30 mg/kg/d) over the period of follow‐up of the study (n = 9). |
|
| Outcomes | 1. Number of any type of adverse events. 2. Number of severe adverse events. 3. Liver transplantation. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "This randomisation was carried out by an independent blinded trial pharmacist in each centre using a predetermined randomisation scheme. Patient numbers were issued sequentially within a centre". |
| Allocation concealment (selection bias) | Low risk | Quote: "This randomisation was carried out by an independent blinded trial pharmacist in each centre using a predetermined randomisation scheme. Patient numbers were issued sequentially within a centre". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "A proportion of the capsules taken by patients in the low and standard dose arms of the trials were placebos. The trial was a randomised, double blinded, dose‐finding study". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "A proportion of the capsules taken by patients in the low and standard dose arms of the trials were placebos. The trial was a randomised, double blinded, dose‐finding study". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | High risk | Quote: "Dr. Falk Pharma (Freiburg, Germany) provided drugs and placebos for this trial as well as financial support for the statistical calculations performed at ClinResearch (Koln, Germany), an independent institute for biostatistics of clinical trials". Comment: The trial was funded by a party with vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
De Maria 1996.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 59.
Post‐randomisation drop‐outs: 0 (0%).
Revised sample size: 59.
Mean age: 31 years.
Females: 17 (28.8%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria: not stated. Follow‐up: 24 months. |
|
| Interventions | Participants were randomly assigned to 1 of 3 groups.
Group 1: low‐dose UDCA (300 mg twice a day) over the period of follow‐up of the study (n = 20).
Group 2: colchicine (60 mg twice a day) over the period of follow‐up of the study (n = 19). Group 3: no active intervention (n = 20). |
|
| Outcomes | No outcomes of interest were reported. | |
| Notes | "No statistical differences in the various outcome measures for the colchicine and the untreated group were evident after 2 years of follow‐up. As a result, these data were collapsed as a single controlled group (n = 39) and were compared against the UDCA group (n = 20)". | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: A group of participants received no treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Farkkila 2004.
| Methods | Randomised clinical trial. | |
| Participants | Country: Finland.
Number randomised: 80.
Post‐randomisation drop‐outs: 9 (11.3%).
Revised sample size: 71.
Mean age: 39 years.
Females: 38 (53.6%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 36 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups.
Group 1: low‐dose UDCA (15 mg/kg/d) and placebo over the period of follow‐up of the study (n = 37). Group 2: low‐dose UDCA (15 mg/kg/d) and metronidazole 600 to 800 mg/d over the period of follow‐up of the study (n = 34). |
|
| Outcomes | 1. Number of any type of adverse events. 2. Liver transplantation. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was done centrally with computer generated blocks". |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomisation was done centrally with computer generated blocks". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "In this multicenter, randomized, double‐blind, placebo‐controlled trial, the patients were randomized either to UDCA and placebo (n = 41) or UDCA and MTZ (n = 39)". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "In this multicenter, randomized, double‐blind, placebo‐controlled trial, the patients were randomized either to UDCA and placebo (n = 41) or UDCA and MTZ (n = 39). Endoscopic retrograde cholangiopancreatography findings were analysed by two radiologists independently, specialised in hepatobiliary disease, and blinded to clinical data and the order of examinations". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | High risk | Quote: "Mary and Georg C. Ehnrooth Foundation.
Medications were supplied, free of charge, by Orion Pharma and Leiras, Finland". Comment: The trial was funded by a party with vested interest in the results: Orion Pharma produces metronidazole, and Leiras produces UDCA. |
| Other bias | Low risk | Comment: no other bias. |
Hommes 2008.
| Methods | Randomised clinical trial. | |
| Participants | Country: The Netherlands.
Number randomised: 10.
Post‐randomisation drop‐outs: 3 (30%).
Revised sample size: 7.
Mean age: 45 years.
Females: 4 (57.1%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 13 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups.
Group 1: infliximab (5 mg/kg) at weeks 0, 2, 6, 12, 18, and 24 (n = 4). Group 2: placebo at weeks 0, 2, 6, 12, 18, and 24 (n = 3). |
|
| Outcomes | 1. Proportion of participants with severe adverse events 2. Number of severe adverse events. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised in a 2:1 ratio to receive infliximab or placebo at weeks 0, 2, 6,12, 18, and 24". Comment: Additional details were not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Infliximab was supplied in 20‐mL vials containing 100mg of the lyophilized concentrate; placebo was identically formulated. The infusion solution was administered by blinded investigators using an infusion set". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Infliximab was supplied in 20‐mL vials containing 100mg of the lyophilized concentrate; placebo was identically formulated. The infusion solution was administered by blinded investigators using an infusion set". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | High risk | Quote: "Daan Hommes has served as consultant and speaker for both Centocor and Schering Plough. Supported by a Research Grant from Centocor, Inc (Malvern, USA)". Comment: The trial was funded by a party with vested interest in the results (this company produces infliximab). |
| Other bias | Low risk | Comment: no other bias. |
Knox 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 24.
Post‐randomisation drop‐outs: 3 (12.5%).
Revised sample size: 21.
Mean age: 37 years.
Females: 7 (33.3%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 48 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups.
Group 1: methotrexate 5 mg every 12 hours (15 mg/wk) for 24 months (n = 11). Group 2: identical placebo for 24 months (n = 10). |
|
| Outcomes | No outcomes of interest were reported. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The code was broken on patients who were judged to be treatment failures". Comment: Additional details were not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "A double‐blind controlled trial of oral‐pulse methotrexate therapy in the treatment of primary sclerosing cholangitis.…Methotrexate (or placebo) was administered orally each week in three divided doses of 5 mg every 12 hours (15 mg/wk) for 2 years in a double‐blind manner. Identical methotrexate and placebo tablets were kindly provided by Lederle laboratories". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "A double‐blind controlled trial of oral‐pulse methotrexate therapy in the treatment of primary sclerosing cholangitis.Methotrexate (or placebo) was administered orally each week in three divided doses of 5 mg every 12 hours (15 mg/wk) for 2 years in a double‐blind manner. Identical methotrexate and placebo tablets were kindly provided by Lederle laboratories". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | High risk | Quote: "Identical methotrexate and placebo tablets were kindly provided by Lederle laboratories..Supported by General Research Center grant MOlRR00054 from the National Institutes of Health and Lederle Laboratories, Pearl River, New York". Comment: The trial was funded by a party with vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
LaRusso 1988.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 70.
Post‐randomisation drop‐outs: unclear.
Revised sample size: 70.
Mean age: 42 years.
Females: 26 (37.1%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 36 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups.
Group 1: penicillamine 750 mg/d over the period of follow‐up of the study (n = 39). Group 2: placebo over the period of follow‐up of the study (n = 31). |
|
| Outcomes | 1. Mortality. 2. Number of any type of adverse events. 3. Number of severe adverse events. 4. Liver transplant. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "We initiated in 1980 a randomized double‐blind trial of penicillamine versus placebo. Patients were randomly assigned to drug or placebo groups. Randomization was weighted in favour of the drug group in anticipation of possible drug toxicity requiring severance from the study. Penicillamine and placebo (furnished to us through the courtesy of Merck Sharp & Dohme, West Point, Pa.) were dispensed in identical yellow capsules by one pharmacist". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "We initiated in 1980 a randomized double‐blind trial of penicillamine versus placebo. Patients were randomly assigned to drug or placebo groups. Randomization was weighted in favour of the drug group in anticipation of possible drug toxicity requiring severance from the study. Penicillamine and placebo (furnished to us through the courtesy of Merck Sharp & Dohme, West Point, Pa.) were dispensed in identical yellow capsules by one pharmacist". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Selective reporting (reporting bias) | Low risk | Comment: No published protocol was available; mortality and liver transplantation were reported. |
| For‐profit bias | High risk | Quote: "This work was supported by the Mayo Foundation, by a grant‐in‐aid from Merck Sharp & Dohme Research Laboratories and in part by a grant from the National Institutes of Health (RR585)". Comment: The trial was funded by a party with vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
Lindor 1997.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 105.
Post‐randomisation drop‐outs: 3 (2.9%).
Revised sample size: 102.
Mean age: 43 years.
Females: 44 (43.1%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: mean follow‐up 27 months (minimum 3 months). |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups.
Group 1: low‐dose UDCA (13‐15 mg/kg/d) over the period of follow‐up of the study (n = 51). Group 2: identical‐appearing placebo over the period of follow‐up of the study (n = 51). |
|
| Outcomes | Time to liver transplantation. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was carried out separately for each of the eight strata (combination of variables) with a computer generated, blocked, randomised drug/assignment schedule. Patient groups were stratified according to histologic stage, serum bilirubin and the presence or absence of oesophageal varices". |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients, physicians, nurses and study coordinators were blinded as to whether active drug or placebo was being administrated". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The patients, physicians, nurses and study coordinators were blinded as to whether active drug or placebo was being administrated". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | High risk | Quote: "Supported in part by Axcan Pharma (produces UDCA)". Comment: The trial was funded by a party with vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
Lindor 2009.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 150.
Post‐randomisation drop‐outs: 0 (0%).
Revised sample size: 150.
Mean age: 47 years.
Females: 64 (42.7%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: planned 60 months, but study stopped earlier owing to futility. Only 50 participants had a cholangiography at 60 months. Biochemical follow‐up. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: high‐dose UDCA (28‐30 mg/kg/d) continued even after primary endpoint was reached, except for liver transplantation or death (n = 76). Group 2: identical placebo continued even after primary endpoint was reached, except for liver transplantation or death (n = 74). | |
| Outcomes | 1. Mortality. 2. Cholangiocarcinoma. 3. Liver transplant. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Computer‐based dynamic allocation used to assign patients to study groups via the coordinating centre in Rochester, MN". |
| Allocation concealment (selection bias) | Low risk | Quote: "Computer‐based dynamic allocation used to assign patients to study groups via the coordinating centre in Rochester, MN". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The physician, study coordinator, and patient were blinded as to whether active drug or placebo was being administered". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The physician, study coordinator, and patient were blinded as to whether active drug or placebo was being administered". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All randomised participants were included in the group to which they were allocated (i.e. intention‐to‐treat analysis was performed). |
| Selective reporting (reporting bias) | Low risk | Comment: No published protocol was available; mortality and liver transplantation were reported. |
| For‐profit bias | High risk | Quote: "Supported by National Institute of Diabetes and Digestive and Kidney diseases Grant 56924 and Axcan Pharma (produces UDCA) as well as well as Grant M01RR00065 from the National Center for Research resources." Comment: The trial was funded by a party with vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
Lo 1992.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK.
Number randomised: 18.
Post‐randomisation drop‐outs: 4 (22.2%).
Revised sample size: 14.
Mean age: 47 years.
Females: 7 (38.9%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria: not stated. Follow‐up: 24 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: low‐dose UDCA (10 mg/kg/d) over the period of follow‐up of the study (n = 7). Group 2: placebo over the period of follow‐up of the study (n = 7). | |
| Outcomes | No outcomes of interest were reported. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Mitchell 2001.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK/Germany.
Number randomised: 26.
Post‐randomisation drop‐outs: 0 (0%).
Revised sample size: 26.
Mean age: 52 years.
Females: 7 (26.9%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 24 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: moderate‐dose (20 mg/kg/d) UDCA over the period of follow‐up of the study (n = 13). Group 2: identical‐appearing placebo over the period of follow‐up of the study (n = 13). | |
| Outcomes | No outcomes of interest were reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "This preliminary study was designed as a double blind, randomized trial comparing the efficacy and safety of UDCA with that of placebo treatment….The placebo was an identical‐appearing capsule administered in the same quantity and manner". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "This preliminary study was designed as a double blind, randomized trial comparing the efficacy and safety of UDCA with that of placebo treatment. . .The placebo was an identical‐appearing capsule administered in the same quantity and manner". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote:"Patients who were lost to follow‐up or died during the study period were included in the final analysis, provided that at least one set of follow‐up data was available". Comment: No post‐randomisation drop‐outs were reported. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; nooutcomes of interest were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Olsson 1995.
| Methods | Randomised clinical trial. | |
| Participants | Country: Sweden.
Number randomised: 84.
Post‐randomisation drop‐outs: unclear.
Revised sample size: 84.
Mean age: 42 years.
Females: 28 (37.8%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria: not stated. Follow‐up: 36 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: colchicine 1 mg/d over the period of follow‐up of the study (n = 44). Group 2: placebo identical in appearance over the period of follow‐up of the study (n = 40). | |
| Outcomes | 1. Mortality. 2. Liver transplant. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The randomization procedure was performed for each center using the sealed envelope technique". Comment: Further information on sealed envelope technique is not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "The randomization procedure was performed for each center using the sealed envelope technique". Comment: Further information on sealed envelope technique is not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The results of a double‐blind, randomized, controlled study comparing colchicine with placebo for 36 months in 84 patients with PSC are reported. After giving informed consent, the patients in each center were randomized to receive 1 mg colchicine daily or a placebo identical in appearance". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The results of a double‐blind, randomized, controlled study comparing colchicine with placebo for 36 months in 84 patients with PSC are reported. After giving informed consent, the patients in each center were randomized to receive 1 mg colchicine daily or a placebo identical in appearance". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Selective reporting (reporting bias) | Low risk | Comment: No published protocol was available; mortality and liver transplant were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Olsson 2005.
| Methods | Randomised clinical trial. | |
| Participants | Country: Sweden/Norway.
Number randomised: 219.
Post‐randomisation drop‐outs: 21 (9.6%).
Revised sample size: 198.
Mean age: 43 years.
Females: 58 (29.3%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 60 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: moderate‐dose UDCA (17‐23 mg/kg/d) over the period of follow‐up of the study (n = 97). Group 2: placebo (250 mg gelatin capsules containing microcrystalline cellulose, cornstarch, and magnesium stearate) over the period of follow‐up of the study (n = 101). | |
| Outcomes | 1. Mortality. 2. Proportion of participants with any type of adverse events. 3. Cholangiocarcinoma. 4. Liver transplant. 5. Quality of life. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "The trial code was kept at the pharmacies in the hospitals. The code was not broken until data from all patients had been collected". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "We conducted a randomized, double‐blind, placebo controlled, multicenter….At that time we had recruited 219 patients (121 from Sweden, 77 from Norway, and 21 from Denmark) who were randomized to either UDCA (in a daily dose of 17–23 mg/kg of body weight divided in 2 doses) or placebo in identical 250‐mg gelatin capsules containing microcrystalline cellulose". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "We conducted a randomized, double‐blind, placebo controlled, multicenter….At that time we had recruited 219 patients (121 from Sweden, 77 from Norway, and 21 from Denmark) who were randomized to either UDCA (in a daily dose of 17–23 mg/kg of body weight divided in 2 doses) or placebo in identical 250‐mg gelatin capsules containing microcrystalline cellulose". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | Low risk | Comment: No published protocol was available; mortality and liver transplant were reported. |
| For‐profit bias | High risk | Quote: "Supported by Dr Falk Pharma GmbH". Comment: The trial was funded by a party with vested interest in the results (this company produces UDCA). |
| Other bias | Low risk | Comment: no other bias. |
Rahimpour 2016.
| Methods | Randomised clinical trial. | |
| Participants | Country: Iran.
Number randomised: 29.
Post‐randomisation drop‐outs: 0 (0%).
Revised sample size: 29.
Average age: 36 years.
Females: 12 (41.4%).
Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 12 weeks after 12 weeks of treatment. |
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: vancomycin 125 mg QDS (n = 18). Group 2: placebo (n = 11). | |
| Outcomes |
|
|
| Notes | Trial authors provided additional information on outcomes in February 2017. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "An independent investigator who was blinded to the treatment group made random allocation cards by using computer‐generated random numbers". |
| Allocation concealment (selection bias) | Low risk | Quote: "Another investigator who was also blinded was responsible for the patients’ enrolments and data collection". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "We used the triple blinding method which meant that patients, investigators who were responsible for the patients’ enrolment and the analyzer of the data at the end of the study were unaware of identities to reduce the chance of bias occurrence in the study". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "We used the triple blinding method which meant that patients, investigators who were responsible for the patients’ enrolment and the analyzer of the data at the end of the study were unaware of identities to reduce the chance of bias occurrence in the study". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: No post‐randomisation drop‐outs were reported. |
| Selective reporting (reporting bias) | Low risk | Comment: No published protocol was available; mortality and morbidity were reported. |
| For‐profit bias | Low risk | Quote: "This study was supported by a grant from the Tehran University of Medical Sciences". |
| Other bias | Low risk | Comment: no other bias. |
Rasmussen 1998.
| Methods | Cross‐over randomised clinical trial. | |
| Participants | Country: Denmark.
Number randomised: 13.
Post‐randomisation drop‐outs: not stated.
Revised sample size: 13.
Mean age: not stated.
Females: not stated. Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria: not stated. Follow‐up: 24 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: methotrexate (10 mg/m2 body area/wk) for the first year followed by placebo (n = 5). Group 2: placebo followed by methotrexate (10 mg/m2 body area/wk) (n = 8). | |
| Outcomes | No outcomes of interest were reported before cross‐over. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Trial authors stated double‐blind and have used placebo. However, the groups blinded were not reported. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: Trial authors stated double‐blind and have used placebo. However, the groups blinded were not reported. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Sandborn 1993.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 35.
Post‐randomisation drop‐outs: 9 (25.7%).
Revised sample size: 26.
Mean age: 39 years.
Females: 10 (38.5%). Separate data for the subgroup with ulcerative colitis: yes. Inclusion criteria:
Exclusion criteria:
Follow‐up: final analysis performed after mean follow‐up of 34 months in the placebo group and 36 months in the cyclosporin group. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: low‐dose cyclosporin (initial dose 5 mg/kg/d) for at least 1 year (mean 2.8 years) (n = 16). Group 2: placebo for at least 1 year (mean 3 years) (n = 10). | |
| Outcomes | 1. Numbers of any types of adverse events. 2. Cholangiocarcinoma. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "From 27 June 1985 to 13 July 1988, 35 patients with precirrhotic primary sclerosing cholangitis were randomly allocated to receive low dose cyclosporin (initial dose 5 mg/kg/day) or placebo in a double blind trial". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "From 27 June 1985 to 13 July 1988, 35 patients with precirrhotic primary sclerosing cholangitis were randomly allocated to receive low dose cyclosporin (initial dose 5 mg/kg/day) or placebo in a double blind trial". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs were reported. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | High risk | Quote: "Supported by grants from the Sandoz Corporation and the Mayo Foundation". Comment: The trial was funded by parties with vested interest in the results. |
| Other bias | Low risk | Comment: no other bias. |
Sterling 2004.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 25.
Post‐randomisation drop‐outs: 9 (36%).
Revised sample size: 16.
Mean age: 44 years.
Females: 10 (62.5%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 24 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: mycophenolate mofetil 1000 mg twice/d and low‐dose UDCA (13–15 mg/kg/d) combined treatment over the period of follow‐up of the study (n = 6). Group 2: low‐dose UDCA (13–15 mg/kg/d) over the period of follow‐up of the study (n = 10). | |
| Outcomes | No outcomes of interest were reported. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "Concealed randomisation via investigational pharmacy or by concealed envelopes" (study author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Neither patient nor investigator was blinded to study medication". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Neither patient nor investigator was blinded to study medication". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "All data were analysed by the intention‐to‐treat method". Comment: Post‐randomisation drop‐outs were reported. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | High risk | Quote: "Supported in part by a NIH grant to the General Clinical Research Center of Virginia Commonwealth University Medical Center, M01‐RR‐00065‐35 and by the generous support of Roche Laboratory, Nutley, NJ and Axcan Scandipharm, Birmingham, AL, USA". Comment: The trial was funded by a party with vested interest in the results (Roche produces mycophenolate mofetil). |
| Other bias | Low risk | Comment: no other bias. |
Stiehl 1989.
| Methods | Randomised clinical trial. | |
| Participants | Country: Germany.
Number randomised: 16.
Post‐randomisation drop‐outs: 4 (25%).
Revised sample size: 12.
Mean age: data not available.
Females: data not available. Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria: not stated. Exclusion criteria: not stated. Follow‐up: unclear: definitive analysis planned for 12 months and interim analysis at 3 months. |
|
| Interventions | Participants were randomly assigned to 1 of 2 groups. Group 1: low‐dose UDCA (8‐10 mg/kg/d) over the period of follow‐up of the study (n = 6). Group 2: placebo over the period of follow‐up of the study (n = 6). | |
| Outcomes | No outcomes of interest were reported. | |
| Notes | Reasons for post randomisation drop‐out not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; no outcomes of interest were reported. |
| For‐profit bias | Unclear risk | Comment: This information was not available. |
| Other bias | Low risk | Comment: no other bias. |
Tabibian 2013.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA.
Number randomised: 35.
Post‐randomisation drop‐outs: 7 (20%).
Revised sample size: 28.
Mean age: 40 years. Females: 14 (40%). Separate data for the subgroup with ulcerative colitis: no. Inclusion criteria:
Exclusion criteria:
Follow‐up: 3 months. |
|
| Interventions | Participants were randomly assigned to 1 of 4 groups. Group 1: vancomycin 125 or 250 mg orally 4 times a day for 12 weeks (n = 15). Group 2: metronidazole 250 or 500 mg orally 3 times a day for 12 weeks (n = 13). |
|
| Outcomes | Numbers of any types of adverse events. | |
| Notes | Reasons for post‐randomisation drop‐out:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Drugs were packaged in identical gelatin capsules, and patients and investigators were blinded to the type and dose of the drug". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Drugs were packaged in identical gelatin capsules, and patients and investigators were blinded to the type and dose of the drug". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Post‐randomisation drop‐outs may be related to the treatment that participants received. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | Low risk | Quote: "Funded by the PSC Partners Seeking a Cure 2009–2010 Research Grant". |
| Other bias | Low risk | Comment: no other bias. |
Trauner 2016.
| Methods | Randomised clinical trial. | |
| Participants | Country: international, multi‐centric.
Number randomised: 159.
Post‐randomisation drop‐outs: not stated.
Revised sample size: 159.
Average age: not stated
Females: not stated Inclusion criteria:
Follow‐up: 4 weeks after 12 weeks of treatment. |
|
| Interventions | Participants were randomly assigned to 4 groups. Group 1: 3 randomised doses of norursodeoxycholic acid (500 mg/d, 1000 mg/d, and 1500 mg/d) (n = not stated). Group 2: placebo (n = not stated). | |
| Outcomes | 1. Serious adverse events. | |
| Notes | Given that the number of participants in each group was not reported, it was not possible to include this trial in the analysis. The proportion of serious adverse events was not reported so that we could report this information in a narrative manner. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: This information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: This information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: Placebo was used, but blinding was not mentioned. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: Placebo was used, but blinding was not mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: This information was not available. |
| Selective reporting (reporting bias) | High risk | Comment: No published protocol was available; mortality was not reported. |
| For‐profit bias | High risk | Quote: "Employment: Dr. Falk Pharma GmbH". Comment: Two of the co‐authors were employed by the company that manufactures the drug. |
| Other bias | Low risk | Comment: no other bias. |
AMA = antimitochondrial antibody; PSC = primary sclerosing cholangitis; UDCA = ursodeoxycholic acid
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Beuers 1998 | Not a RCT (comments on Lindor 1997). |
| Chapman 2005 | Not a RCT. |
| Chapman 2009 | Editorial on Lindor 2009. |
| Eisenburg 1997 | Not an RCT. |
| Fromm 1992 | Comment on a non‐RCT. |
| Goldberg 1992 | Comment on a non‐RCT. |
| Gross 1993 | Comment on a non‐RCT. |
| Harnois 2001 | Not a RCT. |
| Hay 2001 | The study includes transplanted patients. |
| Imam 2011 | Not an RCT. |
| Kuiper 2010 | No separate data for participants with primary sclerosing cholangitis. |
| Kurihara 2003 | Not a RCT. |
| Lankarani 2003 | Not a RCT. |
| Lankarani 2005 | Comment on an included trial (Sterling 2004). |
| Lindor 1995 | Not an RCT. |
| Lindor 2005 | Not a RCT. |
| Lindor 2009a | Review, not a RCT. |
| Mayo 2007 | No separate data for participants with primary sclerosing cholangitis. |
| Silveira 2008 | Not a RCT. |
| Spengler 1993 | Comments on Beuers 1992 and other published experiences. |
| Stiehl 1989a | Not a RCT. |
| Stiehl 1989b | Not a RCT. |
| Stiehl 1994 | Not a RCT. |
| Stiehl 1994a | All participants received the same treatment (UDCA) for 1 year before the randomised period (UDCA and placebo groups). |
| Stiehl 1996 | Review, not a RCT. |
| Tabibian 1989 | Not a RCT. |
| Tada 2006 | Not a RCT. |
| Ter Borg 2004 | No separate data for participants with primary sclerosing cholangitis. |
| Triantos 2012 | Comment on an excluded study (Imam 2011). |
| van de Meeberg 1996 | No separate data for participants with primary sclerosing cholangitis. |
| van Hoogstraten 1998 | No comparison between different treatments: Participants in both arms received the same dose of UDCA once a day or in divided doses. |
| van Hoogstraten 2000 | In this RCT, participants received different types and doses of steroids in combination with UDCA. |
| Van Thiel 1992 | Control group received colchicine or no treatment, and no separate data were available for participants who received no treatment. |
| Villamil 2005 | No separate data for participants with primary sclerosing cholangitis. |
| Vleggaar 2001 | Treatment was not targeted at improving outcomes related to primary sclerosing cholangitis. |
| Vleggaar 2008 | No pharmacological agents were studied. |
| Wagner 1971 | Not a RCT. |
RCT = randomised clinical trial; UDCA = ursodeoxycholic acid
Characteristics of studies awaiting assessment [ordered by study ID]
Anonymous 2006.
| Methods | Awaiting full text. |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes |
ISRCTN16531030.
| Methods | Randomised, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | Trial of low‐dose, medium‐dose, and high‐dose ursodeoxycholic acid with placebo in primary sclerosing cholangitis. |
| Outcomes | Not available. |
| Notes | Recruitment status: completed. |
NCT00059202.
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | High‐dose UDCA (28‐30 mg/kg/d) vs placebo. |
| Outcomes | Cirrhosis, decompensated cirrhosis, cholangiocarcinoma, liver transplantation, quality of life, and mortality. |
| Notes | Recruitment status: completed. |
UDCA = ursodeoxycholic acid
Characteristics of ongoing studies [ordered by study ID]
EUCTR2012‐004170‐26‐IT.
| Trial name or title | EUCTR2012‐004170‐26‐IT. |
| Methods | Randomised double‐blinded placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | N‐acetylcysteine 600 mg vs placebo. |
| Outcomes | Quality of life. |
| Starting date | Not stated. |
| Contact information | agasbarrini@RM.UNICATT.IT |
| Notes | Not recruiting. |
EUCTR2015‐003310‐24‐SE.
| Trial name or title | UDCAPSCSURV. |
| Methods | Phase 3, open‐label, randomised, prospective clinical trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | 17‐23 mg/kg/d UDCA vs placebo. |
| Outcomes | Decompensated liver cirrhosis and liver transplantation. |
| Starting date | Not stated. |
| Contact information | hanns‐ulrich.marschall@gu.se |
| Notes |
EUCTR2015‐003392‐30‐GB.
| Trial name or title | EUCTR2015‐003392‐30‐GB. |
| Methods | Phase 2, randomised, double‐blind, placebo‐controlled, parallel‐group, multiple‐centre study. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | NGM282 vs placebo. |
| Outcomes | No outcomes of interest for this review. |
| Starting date | Not stated. |
| Contact information | clinical@ngmbio.com |
| Notes |
NCT01672853.
| Trial name or title | NCT01672853. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | GS‐6624, a monoclonal antibody against Lysyl Oxidase Like 2 (LOXL2), vs placebo. |
| Outcomes | Adverse events. |
| Starting date | February 2013. |
| Contact information | Rob Myers, M.D. Gilead Sciences. |
| Notes |
NCT01688024.
| Trial name or title | NCT01688024. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | Mitomycin C vs placebo. |
| Outcomes | Adverse events. |
| Starting date | September 2012. |
| Contact information | chen37@jhmi.edu |
| Notes |
NCT01755507.
| Trial name or title | NCT01755507. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | Norursodeoxycholic acid vs placebo. |
| Outcomes | Adverse events. |
| Starting date | December 2012. |
| Contact information | roels@drfalkpharma.de |
| Notes |
NCT02177136.
| Trial name or title | NCT02177136. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | Obeticholic acid vs placebo. |
| Outcomes | Adverse events. |
| Starting date | December 2014. |
| Contact information | kate.mckeown@interceptpharma.com |
| Notes |
NCT02704364.
| Trial name or title | NCT02704364. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | NGM282 vs placebo. |
| Outcomes | No outcomes of interest for this review. |
| Starting date | February 2016. |
| Contact information | kkim@ngmbio.com |
| Notes |
NCT02943460.
| Trial name or title | NCT02943460. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | GS‐9674 vs placebo. |
| Outcomes | Adverse events. |
| Starting date | November 2016. |
| Contact information | GS‐US‐428‐4025@Gilead.com |
| Notes |
NCT03035058.
| Trial name or title | NCT03035058. |
| Methods | Randomised, double‐blind, placebo‐controlled trial. |
| Participants | Patients with primary sclerosing cholangitis. |
| Interventions | Vedolizumab vs placebo. |
| Outcomes | No outcomes of interest for this review. |
| Starting date | February 2017. |
| Contact information | medicalinformation@tpna.com |
| Notes |
vs = versus
Differences between protocol and review
It was not possible to assess whether potential effect modifiers were similar across different comparisons. Therefore, we did not perform the network meta‐analysis but instead assessed comparative benefits and harms of different interventions using standard Cochrane methods. The method that we plan to use if we conduct a network meta‐analysis in the future is available in Appendix 1.
We performed Trial Sequential Analysis in addition to conventional methods of assessing risk of random errors using the P value.
Contributions of authors
Francesca Saffioti and Kurinchi Gurusamy selected the studies, extracted data, and wrote the review. Clare Toon selected some studies and extracted some data. Neil Hawkins, Emmanuel Tsochatzis, Brian Davidson, and Douglas Thorburn critically commented on the review. All review authors approved this version before publication.
Sources of support
Internal sources
University College London, UK.
External sources
-
National Institute for Health Research, UK.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure, Cochrane Programme Grant or Cochrane Incentive funding to the Cochrane Hepato‐Biliary and Upper Gastrointestinal and Pancreatic Diseases Groups. The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS, or the Department of Health. The funding covered salary, equipment, and other resources needed to complete this review. The review was supported by the Complex Reviews Support Unit, also funded by the National Institute for Health Research (project number 14/178/29).
Declarations of interest
This report is independent research funded by the National Institute for Health Research (NIHR Cochrane Programme Grants, 13/89/03 ‐ Evidence‐based diagnosis and management of upper digestive, hepato‐biliary, and pancreatic disorders). The views expressed in this publication are those of the review authors and are not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
Francesca Saffioti, Kurinchi Gurusamy, Clare Toon, Neil Hawkins, Emmanuel Tsochatzis, and Brian Davidson have no financial disclosures. Astellas funded Douglas Thorburn for his attendance at the International Liver Transplantation Society meeting in 2014. Douglas Thorburn has also received £25,000 from Boston Scientific to fund a clinical research fellow in 2013. We have no other financial disclosures to report.
Edited (no change to conclusions)
References
References to studies included in this review
Allison 1986 {published data only}
- Allison MC, Burroughs AK, Noone P, Summerfield JA. Biliary lavage with corticosteroids in primary sclerosing cholangitis. A clinical, cholangiographic and bacteriological study. Journal of Hepatology 1986;3(1):118‐22. [DOI] [PubMed] [Google Scholar]
Bansi 1996 {published data only}
- Bansi D, Christie J, Fleming K, Chapman R. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a randomised double‐blind placebo‐controlled trial. Gastroenterology 1996;110(4):A1146. [Google Scholar]
- Bansi D, Christie J, Fleming K, Chapman R. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a randomised double‐blind, placebo‐controlled trial. Gut 1996;38(Suppl 1):A54. [Google Scholar]
Beuers 1992 {published data only}
- Beuers U, Spengler U, Kruis W, Aydemir U, Wiebecke B, Heldwein W, et al. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: a placebo‐controlled trial. Hepatology 1992;16(3):707‐14. [DOI] [PubMed] [Google Scholar]
- Beuers U, Spengler U, Kruis W, Aydemir Û, Heldwein W, Weinzierl M, et al. Effect of ursodeoxycholic acid in primary sclerosing cholangitis: a controlled trial [abstract]. Hepatology 1991;14(4 Pt 2):64a. [DOI] [PubMed] [Google Scholar]
- Spengler U, Beuers U, Kruis W, et al. Ursodeoxycholic acid in primary sclerosing cholangitis. Bile Acids and the Hepatobiliary System From Basic Science to Clinical Practice Proceedings of Falk Symposium 68th Held in Basel, Switzerland, October 12‐14, 1992.. 1993:316‐22.
Cullen 2008 {published data only}
- Beuers U, Cullen SN, Fleming K, Rust C, Chapman RW. Tolerance and efficacy of a high‐dose treatment with urosodeoxycholic acid in primary sclerosing cholangitis. A double‐blind randomised, placebo‐controlled dose‐finding study. Zeitschrift fur Gastroenterologie 2006;44(8):760. [Google Scholar]
- Cullen SN, Rust C, Fleming K, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. Journal of Hepatology 2006;44(2 Suppl):S235‐6. [DOI] [PubMed] [Google Scholar]
- Cullen SN, Rust C, Fleming K, Edwards C, Beuers U, Chapman RW. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis is safe and effective. Journal of Hepatology 2008;48(5):792‐800. [DOI] [PubMed] [Google Scholar]
De Maria 1996 {published data only}
- Maria N, Colantoni A, Rosenbloom E, Thiel DH. Ursodeoxycholic acid does not improve the clinical course of primary sclerosing cholangitis over a 2‐year period. Hepatogastroenterology 1996;43(12):1472‐9. [PubMed] [Google Scholar]
Farkkila 2004 {published data only}
- Farkkila M, Karvonen AL, Nurmi H, Nuutinen H, Taavitsainen M, Pikkarainen P, et al. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo‐controlled trial. Hepatology 2004;40(6):1379‐86. [DOI] [PubMed] [Google Scholar]
Hommes 2008 {published data only}
- Hommes DW, Erkelens W, Ponsioen C, Stokkers P, Rauws E, Spek M, et al. A double‐blind, placebo‐controlled, randomized study of infliximab in primary sclerosing cholangitis. Journal of Clinical Gastroenterology 2008;42(5):522‐6. [DOI] [PubMed] [Google Scholar]
Knox 1994 {published data only}
- Knox TA, Kaplan MM. A double‐blind controlled trial of oral‐pulse methotrexate therapy in the treatment of primary sclerosing cholangitis. Gastroenterology 1994;106(2):494‐9. [DOI] [PubMed] [Google Scholar]
LaRusso 1988 {published data only}
- LaRusso N, Wiesner R, Ludwig J, MacCarty R, Beaver S, Zinsmeister A, et al. Randomized trial of penicillamine in primary sclerosing cholangitis. Hepatology 1986;6(5):1205. [DOI] [PubMed] [Google Scholar]
- LaRusso NF, Wiesner RH, Ludwig J, MacCarty RL, Beaver SJ, Zinsmeister AR. Prospective trial of penicillamine in primary sclerosing cholangitis. Gastroenterology 1988;95(4):1036‐42. [DOI] [PubMed] [Google Scholar]
Lindor 1997 {published data only}
- Hilscher M, Tabibian JH, Enders FB, Carey EJ, Lindor KD. Long‐term follow‐up of a multicenter, randomized controlled trial of ursodeoxycholic acid in primary sclerosing cholangitis. Hepatology 2015;62:509A. [Google Scholar]
- Lindor KD. A randomized trial of ursodeoxycholic acid (UDCA) in the treatment of primary sclerosing cholangitis (PSC). Gastroenterology 1996;110(4):A1252. [Google Scholar]
- Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis‐Ursodeoxycholic Acid Study Group. The New England Journal of Medicine 1997;336(10):691‐5. [DOI] [PubMed] [Google Scholar]
- Pardi DS, Loftus EV, Kremers WK, Keach J, Lindor KD. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003;124(4):889‐93. [DOI] [PubMed] [Google Scholar]
Lindor 2009 {published data only}
- Eaton JE, Silveira MG, Pardi DS, Sinakos E, Kowdley KV, Luketic VA, et al. High‐dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. The American Journal of Gastroenterology 2011;106(9):1638‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor KD, Enders FB, Schmoll JA, Hoskin TL, Jorgensen RL, Petz JL. Randomized, double‐blind controlled trial of high‐dose ursodeoxycholic acid (UCDA) for primary sclerosing cholangitis (PSC). Hepatology 2008;48(4Suppl):378a. [Google Scholar]
- Lindor KD, Kowdley KV, Luketic VAC, Harrison ME, McCashland T, Befeler AS, et al. High‐dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009;50(3):808‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinakos E, Marshall HU, Kowdley KV, Befler A, Keach J, Lindor K. Bile acid changes after high‐dose ursodeoxycholic acid treatment in primary sclerosing cholangitis: relation to disease progression. Hepatology 2010;52:197‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lo 1992 {published data only}
- Lo SK, Herrmann R, Chapman RW, Fleming KA, Shearman J, Cusick P, et al. Ursodeoxycholic acid in primary sclerosing cholangitis: a double‐blind placebo controlled trial. Hepatology 1992;16(2 Pt 2):92a. [Google Scholar]
Mitchell 2001 {published data only}
- Mitchell S, Bansi D, Hunt N, Christie J, Fleming K, Chapman R. High dose ursodeoxycholic acid (UDCA) in primary sclerosing cholangitis (PSC): results after two years of a randomised double‐blind, placebo‐controlled trial. Gastroenterology 1997;112(4):A1335. [Google Scholar]
- Mitchell SA, Bansi D, Hunt N, Christie J, Fleming K, Chapman R. High dose ursodeoxycholic acid (UDCA) in primary sclerosing cholangitis (PSC): results after two years of a randomised double‐blind, placebo‐controlled trial. Gut 1997;40(Suppl 1):A29. [Google Scholar]
- Mitchell SA, Bansi DS, Hunt N, Bergmann K, Fleming KA, Chapman RW. A preliminary trial of high‐dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology 2001;121(4):900‐7. [DOI] [PubMed] [Google Scholar]
Olsson 1995 {published data only}
- Olsson R, Broome U, Danielsson A, Hagerstrand I, Jarnerot G, Loof L, et al. Colchicine treatment of primary sclerosing cholangitis. Gastroenterology 1995;108(4):1199‐203. [DOI] [PubMed] [Google Scholar]
Olsson 2005 {published data only}
- Lindstrom L, Boberg KM, Friis‐Liby I, Hultcrantz RW, Bergquist A. A reduction in alkaline phosphatase levels is associated to improved prognosis in primary sclerosing cholangitis: a 14 year follow up of the Scandinavian ursodeoxycholic acid trial. Hepatology 2012;56:247a. [Google Scholar]
- Lindstrom L, Boberg KM, Wikman O, Friis‐Liby I, Hultcrantz R, Prytz H, et al. High dose ursodeoxycholic acid in primary sclerosing cholangitis does not prevent colorectal neoplasia. Alimentary Pharmacology and Therapeutics 2012;35(4):451‐7. [DOI] [PubMed] [Google Scholar]
- Olsson R, Boberg KM, Muckadell OS, Lindgren S, Hultcrantz R, Folvik G, et al. High‐dose ursodeoxycholic acid in primary sclerosing cholangitis: a 5‐year multicenter, randomized, controlled study. Gastroenterology 2005;129(5):1464‐72. [DOI] [PubMed] [Google Scholar]
- Olsson RG, Boberg KM, Schaffalitzky de Muckadel O, Lindgren S, Hultcrantz R, Folvik G, et al. Five‐year treatment with high‐dose UDCA in PSC. Journal of Hepatology 2004;40(Suppl 1):161. [Google Scholar]
Rahimpour 2016 {published data only}
- Rahimpour S, Nasiri‐Toosi M, Khalili H, Daryani NE, Taromlou MKN, Azizi Z. A triple blinded, randomized, placebo‐controlled clinical trial to evaluate the efficacy and safety of oral vancomycin in primary sclerosing cholangitis: a pilot study. Journal of Gastrointestinal and Liver Diseases 2016;25(4):457‐64. [DOI] [PubMed] [Google Scholar]
Rasmussen 1998 {published data only}
- Rasmussen HH, Tage‐Jensen U, Vyberg M, Schlichting P, Schaffalitzky de Mucadell O, Bonnevie O. Methotrexate for treatment of primary sclerosing cholangitis. Journal of Hepatology 1998;28(1 Suppl 1):128. [Google Scholar]
Sandborn 1993 {published data only}
- Sandborn WJ, Wiesner RH, Tremaine WJ, Larusso NF. Ulcerative colitis disease activity following treatment of associated primary sclerosing cholangitis with cyclosporin. Gut 1993;34(2):242‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner RH, Steiner B, LaRusso NF, Lindor KD, Baldus WP. A controlled clinical trial evaluating cyclosporine in the treatment of primary sclerosing cholangitis. Hepatology 1991; Vol. 14, issue 4 Pt 2:63a.
Sterling 2004 {published data only}
- Sterlin RK, Salvatori JJ, Luketic VA, Sanyal AJ, Stravitz RT, Fulcher AS, et al. A prospective, randomized controlled pilot study of ursodeoxycholic acid (UDCA) combined with mycophenolate mofetil (MMF) in the treatment of primary sclerosing cholangitis. Hepatology 2003;38(4 Suppl 1):204a. [DOI] [PubMed] [Google Scholar]
- Sterling RK, Salvatori JJ, Luketic VA, Sanyal AJ, Fulcher AS, Stravitz RT, et al. A prospective, randomized‐controlled pilot study of ursodeoxycholic acid combined with mycophenolate mofetil in the treatment of primary sclerosing cholangitis. Alimentary Pharmacology and Therapeutics 2004;20(9):943‐9. [DOI] [PubMed] [Google Scholar]
Stiehl 1989 {published data only}
- Stiehl A, Raedsch R, Rudolph G, Theilmann L. Treatment of primary sclerosing cholangitis with ursodeoxycholic acid: first results of a controlled study. Hepatology 1989;10(4):602. [Google Scholar]
Tabibian 2013 {published data only}
- Tabibian JH, Lindor KD. Long‐term outcomes following a randomized study of oral antibiotics in primary sclerosing cholangitis. Gastroenterology 2016;150(4 Suppl 1):S1069‐S70. [Google Scholar]
- Tabibian JH, Weeding E, Jorgensen RA, Petz JL, Keach JC, Talwalkar JA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis ‐ a pilot study. Alimentary Pharmacology and Therapeutics 2013;37(6):604‐12. [DOI] [PubMed] [Google Scholar]
Trauner 2016 {published data only}
- Trauner MH, Fickert P, Hirschfield G, Denk G, Altorjay I, Marschall HU, et al. Norursodeoxycholic acid (NORUDCA) improves cholestasis in primary sclerosing cholangitis (PSC) independent of ursodeoxycholic acid (UDCA) pre‐treatment and response. Hepatology 2016;64(Suppl 1):111A. [Google Scholar]
References to studies excluded from this review
Beuers 1998 {published data only}
- Beuers U, Sackmann FM. Treatment of primary sclerosing cholangitis: drug, endoscopic, combined or no treatment at all?. Zeitschrift für Gastroenterologie 1998;36(2):189‐91. [PubMed] [Google Scholar]
Chapman 2005 {published data only}
- Chapman RW. High‐dose ursodeoxycholic acid in the management of primary sclerosing cholangitis. Bile Acid Biology and Its Therapeutic Implications 2005;141:230‐41. [Google Scholar]
Chapman 2009 {published data only}
- Chapman RW. High‐dose ursodeoxycholic acid in the treatment of primary sclerosing cholangitis: throwing the urso out with the bathwater?. Hepatology 2009;50(3):671‐3. [DOI] [PubMed] [Google Scholar]
Eisenburg 1997 {published data only}
- Eisenburg J. Primary sclerosing cholangitis. Ursodeoxycholic acid is without clinical effect. Fortschritte der Medizin 1997;115(18):7‐8. [PubMed] [Google Scholar]
Fromm 1992 {published data only}
- Fromm H. New treatment for primary sclerosing cholangitis: promising results with ursodiol. Gastroenterology 1992;103(1):343‐4. [DOI] [PubMed] [Google Scholar]
Goldberg 1992 {published data only}
- Goldberg E, Gerdes H. Primary sclerosing cholangitis: is medical therapy on the way?. Gastroenterology 1992;102(2):729‐31. [DOI] [PubMed] [Google Scholar]
Gross 1993 {published data only}
- Gross JB. Promises, promises: ursodeoxycholic acid for primary sclerosing cholangitis. Gastroenterology 1993;104(3):941‐3. [DOI] [PubMed] [Google Scholar]
Harnois 2001 {published data only}
- Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High‐dose ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. The American Journal of Gastroenterology 2001;96(5):1558‐62. [DOI] [PubMed] [Google Scholar]
Hay 2001 {published data only}
- Hay JE, Malinchoc M, Dickson ER. A controlled trial of calcitonin therapy for the prevention of post‐liver transplantation atraumatic fractures in patients with primary biliary cirrhosis and primary sclerosing cholangitis. Journal of Hepatology 2001;34(2):292‐8. [DOI] [PubMed] [Google Scholar]
Imam 2011 {published data only}
- Imam MH, Sinakos E, Gossard AA, Kowdley KV, Luketic VA, Edwyn Harrison M, et al. High‐dose ursodeoxycholic acid increases risk of adverse outcomes in patients with early stage primary sclerosing cholangitis. Alimentary Pharmacology and Therapeutics 2011;34(10):1185‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kuiper 2010 {published data only}
- Kuiper EMM, Erpedum KJ, Beuers U, Hansen BE, Thio HB, Man RA, et al. The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: results of a double‐blind, randomized, placebo‐controlled trial. Hepatology 2010;52(4):1334‐40. [DOI] [PubMed] [Google Scholar]
Kurihara 2003 {published data only}
- Kurihara T, Maeda A, Shigemoto M, Yamashita K, Kamatani N. Efficacy of bezafibrate in a patient with primary sclerosing cholangitis. Journal of Gastroenterology 2003;38(3):300‐1. [DOI] [PubMed] [Google Scholar]
Lankarani 2003 {published data only}
- Lankarani KB. Use of mycophenolate mofetil in the treatment of primary sclerosing cholangitis. Journal of Clinical Gastroenterology 2003;36(1):86. [DOI] [PubMed] [Google Scholar]
Lankarani 2005 {published data only}
- Lankarani KB. Mycophenolate mofetil for the treatment of primary sclerosing cholangitis. Alimentary Pharmacology & Therapeutics 2005;21(10):1279‐80. [DOI] [PubMed] [Google Scholar]
Lindor 1995 {published data only}
- Lindor K. Long‐term experience with ursodeoxycholic acid for patients with primary biliary cirrhosis and primary sclerosing cholangitis. International Falk Workshop Bile Acids in Liver Diseases 1995:141‐5. [Google Scholar]
Lindor 2005 {published data only}
- Lindor KD. Dose effect of ursodeoxycholic acid used in the treatment of primary biliary cirrhosis and primary sclerosing cholangitis. Bile Acid Biology and Its Therapeutic Implications 2005;141:225‐9. [Google Scholar]
Lindor 2009a {published data only}
- Lindor KD. Ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Bile Acid Biology and Therapeutic Actions. Vol. 165, Dordrecht: Springer, 2009:255‐8. [Google Scholar]
Mayo 2007 {published data only}
- Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first‐line treatment for cholestatic pruritus. Hepatology 2007;45(3):666‐74. [DOI] [PubMed] [Google Scholar]
Silveira 2008 {published data only}
- Silveira MG, Lindor KD. High dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Journal of Hepatology 2008;48(5):692‐4. [DOI] [PubMed] [Google Scholar]
Spengler 1993 {published data only}
- Spengler U, Beuers U, Kruis W, Aydemir Û, Wiebecke B, Heldwein W, et al. Ursodeoxycholic acid in primary sclerosing cholangitis. Bile Acids and the Hepatobiliary System. From Basic Science to Clinical Practice. Proceedings of Falk Symposium 68 1993:316‐22. [Google Scholar]
Stiehl 1989a {published data only}
- Stiehl A, Raedsch R. Treatment of primary sclerosing cholangitis with ursodeoxycholic acid. Zeitschrift fur Gastroenterologie ‐ Verhandlungsband 1989;24:136. [PubMed] [Google Scholar]
Stiehl 1989b {published data only}
- Stiehl A, Raedsch R. [Treatment of primary sclerosing cholangitis with ursodeoxycholic acid]. Z Gastroenterol Verh 1989;24:136. [PubMed] [Google Scholar]
Stiehl 1994 {published data only}
- Stiehl A. Ursodeoxycholic acid therapy in treatment of primary sclerosing cholangitis. Scandinavian Journal of Gastroenterology. Supplement 1994;204:59‐61. [DOI] [PubMed] [Google Scholar]
Stiehl 1994a {published data only}
- Stiehl A, Walker S, Stiehl L, Rudolph G, Hofmann WJ, Theilmann L. Effect of ursodeoxycholic acid on liver and bile duct disease in primary sclerosing cholangitis. A 3‐year pilot study with a placebo‐controlled study period. Journal of Hepatology 1994;20(1):57‐64. [DOI] [PubMed] [Google Scholar]
- Stiehl L, Rudolph G, Raedsch R, Stiehl A. Effects of ursodeoxycholic acid in patients with primary sclerosing cholangitis. Bile Acids as Therapeutic Agents. From Basic Science to Clinical Practice. Proceedings of Falk Symposium 58 1991, (37):305‐7. [Google Scholar]
Stiehl 1996 {published data only}
- Stiehl A. Ursodeoxycholic acid in the treatment of primary sclerosing cholangitis. Italian Journal of Gastroenterology 1996;28(3):178‐80. [PubMed] [Google Scholar]
Tabibian 1989 {published data only}
- Tabibian N. Rifampin as antipruritic agent in primary sclerosing cholangitis. American Journal of Gastroenterology 1989;84(3):340. [PubMed] [Google Scholar]
Tada 2006 {published data only}
- Tada S, Ebinuma H, Saito H, Hibi T. Therapeutic benefit of sulfasalazine for patients with primary sclerosing cholangitis. Journal of Gastroenterology 2006;41(4):388‐9. [DOI] [PubMed] [Google Scholar]
Ter Borg 2004 {published data only}
- ter Borg PC, Os E, Broek WW, Hansen BE, Buuren HR. Fluvoxamine for fatigue in primary biliary cirrhosis and primary sclerosing cholangitis: a randomised controlled trial [ISRCTN88246634]. BMC Gastroenterology 2004;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Triantos 2012 {published data only}
- Triantos CK, Koukias N, Nikolopoulou V, Burroughs AK. Ursodeoxycholic acid in primary sclerosing cholangitis. Alimentary Pharmacology & Therapeutics 2012;35(5):622‐3. [DOI] [PubMed] [Google Scholar]
van de Meeberg 1996 {published data only}
- Meeberg PC, Wolfhagen FH, Berge‐Henegouwen GP, Salemans JM, Tangerman A, Buuren HR, et al. Single or multiple dose ursodeoxycholic acid for cholestatic liver disease: biliary enrichment and biochemical response. Journal of Hepatology 1996;25(6):887‐94. [DOI] [PubMed] [Google Scholar]
van Hoogstraten 1998 {published data only}
- Hoogstraten HJ, Wolfhagen FH, Meeberg PC, Kuiper H, Nix GA, Becx MC, et al. Ursodeoxycholic acid therapy for primary sclerosing cholangitis: results of a 2‐year randomized controlled trial to evaluate single versus multiple daily doses. Journal of Hepatology 1998;29(3):417‐23. [DOI] [PubMed] [Google Scholar]
- Hoogstraten HJF, Meeberg PC, Wolfhagen FHJ, Kuiper H, Hop WCJ, Berge‐Henegouwen GP, et al. Ursodeoxycholic acid (UDCA) for primary sclerosing cholangitis (PSC): results of a 2‐year randomized controlled trial evaluating single versus multiple daily doses. Hepatology 1997;26(4 Pt 2):401a. [DOI] [PubMed] [Google Scholar]
- Hoogstraten HJF, Wolfhagen FJH, Meeberg PC, Buuren HR, Berge Henegouwen GP, Schalm SW. Single versus three times daily ursodeoxycholic acid (UDCA) for primary sclerosing cholangitis (PSC): results of a randomized controlled trial. European Journal of Gastroenterology and Hepatology 1996;8(Suppl 12):A 7. [Google Scholar]
- Vleggaar FP, Hoogstraten HJF, Boland GJ, Steenbergen W, Hattum J, Henegouwen GPV, et al. Treatment with budesonide or prednisone in combination with ursodeoxycholic acid (UDCA) in primary sclerosing cholangitis (PSC): a randomized controlled pilot study. Hepatology 1998;28(4):647A. [Google Scholar]
- Hoogstraten HJ, Wolfhagen FH, Meeberg PC, Kuiper H, Nix GA, Becx MC, et al. Ursodeoxycholic acid therapy for primary sclerosing cholangitis: results of a 2‐year randomized controlled trial to evaluate single versus multiple daily doses. Journal of Hepatology 1998;29(3):417‐23. [DOI] [PubMed] [Google Scholar]
- Hoogstraten HJF, Wolfhagen FJH, Meeberg PC, Buuren HR, Henegouwen GPB, Schalm SW. Single versus three times daily ursodeoxycholic acid (UDCA) for primary sclerosing cholangitis (PSC): results of a randomized controlled trial. Gastroenterology 1996;110(4):A1352. [Google Scholar]
- Hoogstraten HJF, Meeberg PC, Wolfhagen FHJ, Kuiper H, Hop WCJ, Berge Henegouwen GP, et al. Ursodeoxycholic acid (UDCA) for primary sclerosing cholangitis (PSC): results of a 2‐year randomized controlled trial evaluating single versus multiple daily doses. Hepatology 1997;26(4):1089. [Google Scholar]
van Hoogstraten 2000 {published data only}
- Hoogstraten HJ, Vleggaar FP, Boland GJ, Steenbergen W, Griffioen P, Hop WC, et al. Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double‐blind pilot study. Belgian‐Dutch PSC Study Group. The American Journal of Gastroenterology 2000;95(8):2015‐22. [DOI] [PubMed] [Google Scholar]
- Hoogstraten HJ, Vleggaar FP, Boland GJ, Steenbergen W, Griffioen P, Hop WC, et al. Budesonide or prednisone in combination with ursodeoxycholic acid in primary sclerosing cholangitis: a randomized double‐blind pilot study. Belgian‐Dutch PSC Study Group. American Journal of Gastroenterology 2000;95(8):2015‐22. [DOI] [PubMed] [Google Scholar]
Van Thiel 1992 {published data only}
- Vanthiel DH, Wright HI, Gavaler JS. Ursodeoxycholic acid (UDCA) therapy for primary sclerosing cholangitis (PSC) ‐ preliminary report of a randomized controlled trial. Hepatology 1992;16(4):A62. [Google Scholar]
Villamil 2005 {published data only}
- Villamil AG, Bandi JC, Galdame OA, Gerona S, Gadano AC. Efficacy of lidocaine in the treatment of pruritus in patients with chronic cholestatic liver diseases. The American Journal of Medicine 2005;118(10):1160‐3. [DOI] [PubMed] [Google Scholar]
Vleggaar 2001 {published data only}
- Vleggaar FP, Buuren HR, Berge Henegouwen GP, Hop WC, Erpecum KJ. No beneficial effects of transdermal nicotine in patients with primary sclerosing cholangitis: results of a randomized double‐blind placebo‐controlled cross‐over study. European Journal of Gastroenterology and Hepatology 2001;13(2):171‐5. [DOI] [PubMed] [Google Scholar]
- Vleggaar FP, Buuren HR, Henegouwen GPV, Erpecum KJ. No beneficial effects of transdermal nicotine in primary sclerosing cholangitis (PSC): results of a randomized double‐blind cross‐over study. Hepatology 1999;30(4):563A. [DOI] [PubMed] [Google Scholar]
Vleggaar 2008 {published data only}
- Vleggaar F, Monkelbaan J, Erpecum K. No beneficial effects of probiotics in primary sclerosing cholarigitis (PSC): a randomized placebo‐controlled cross‐over study. Gastroenterology 2007;132(4):A767. [Google Scholar]
- Vleggaar FP, Monkelbaan JF, Erpecum KJ. Probiotics in primary sclerosing cholangitis: a randomized placebo‐controlled crossover pilot study. European Journal of Gastroenterology and Hepatology 2008;20(7):688‐92. [DOI] [PubMed] [Google Scholar]
- Vleggaar FP, Monkelbaan JF, Erpecum KJ. No beneficial effects of probiotics in primary sclerosing cholangitis: a randomized placebo‐controlled crossover study. European Journal of Gastroenterology and Hepatology 2008;20(7):A67. [DOI] [PubMed] [Google Scholar]
Wagner 1971 {published data only}
- Wagner A. Azathioprine treatment in primary sclerosing cholangitis. Lancet 1971;2(7725):663‐4. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Anonymous 2006 {published data only}
- Anonymous. Primary sclerosing cholangitis. To slow progression and reduce carcinoma risk. MMW Fortschritte der Medizin 2006;148(24):42‐3. [PubMed] [Google Scholar]
ISRCTN16531030 {published data only}
- Randomised trial of low dose, medium dose and high dose ursodeoxycholic acid with placebo in primary sclerosing cholangitis. http://isrctn.com/ISRCTN16531030 (accessed 5 July 2015).
NCT00059202 {published data only}
- Trial of high‐dose urso in primary sclerosing cholangitis. http://clinicaltrials.gov/show/NCT00059202 (accessed 5 July 2015).
References to ongoing studies
EUCTR2012‐004170‐26‐IT {published data only}
- Randomized double‐blind placebo‐controlled trial to evaluate the efficacy of N‐acetylcystein in patients with chronic pancreatitis and primary sclerosing cholangitis.. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2012‐004170‐26 (accessed 5 July 2015).
EUCTR2015‐003310‐24‐SE {published data only}
- A phase 3, open‐label, randomized, prospective clinical trial evaluating the efficacy of stratified treatment with ursodeoxycholic acid (UDCA) in preventing hepatobiliary and colorectal malignancy in surveillance patients with primary sclerosing cholangitis (PSC). https://www.clinicaltrialsregister.eu/ctr‐search/trial/2015‐003310‐24/SE (accessed 22 February 2017).
EUCTR2015‐003392‐30‐GB {published data only}
- A phase 2, randomized, double blind, placebo controlled, parallel group, multiple center study to evaluate the safety, tolerability, and efficacy of NGM282 administered for 12 weeks in patients with primary sclerosing cholangitis (PSC). https://www.clinicaltrialsregister.eu/ctr‐search/trial/2015‐003392‐30/GB/ (accessed 22 February 2017).
NCT01672853 {published data only}
- Simtuzumab (GS‐6624) in the prevention of progression of liver fibrosis in subjects with primary sclerosing cholangitis (PSC). http://clinicaltrials.gov/show/NCT01672853 (accessed 5 July 2015).
NCT01688024 {published data only}
- Mitomycin C therapy for patients with primary sclerosing cholangitis. http://clinicaltrials.gov/show/NCT01688024 (accessed 5 July 2015).
NCT01755507 {published data only}
- Norursodeoxycholic acid in the treatment of primary sclerosing cholangitis NUC‐3. http://clinicaltrials.gov/show/NCT01755507 (accessed 5 July 2015).
NCT02177136 {published data only}
- Obeticholic acid (OCA) in primary sclerosing cholangitis (PSC). https://clinicaltrials.gov/show/NCT02177136 2015 (accessed 30 June 2015).
NCT02704364 {published data only}
- Phase 2 study of NGM282 in patients with primary sclerosing cholangitis. https://clinicaltrials.gov/ct2/show/NCT02704364 (accessed 22 February 2017).
NCT02943460 {published data only}
- Safety, tolerability, and efficacy of GS9674 in adults with primary sclerosing cholangitis without cirrhosis (PSCphase 2). https://clinicaltrials.gov/ct2/show/NCT02943460 (accessed 22 February 2017).
NCT03035058 {published data only}
- Efficacy and safety of vedolizumab intravenous (IV) in the treatment of primary sclerosing cholangitis in subjects with underlying inflammatory bowel disease. https://clinicaltrials.gov/ct2/show/NCT03035058 (accessed 22 February 2017).
Additional references
Boonstra 2012
- Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. Journal of Hepatology 2012;56(5):1181‐8. [DOI] [PubMed] [Google Scholar]
Boonstra 2013
- Boonstra K, Weersma RK, Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population‐based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013;58(6):2045‐55. [DOI] [PubMed] [Google Scholar]
Chaimani 2012
- Chaimani A, Salanti G. Using network meta‐analysis to evaluate the existence of small‐study effects in a network of interventions. Research Synthesis Methods 2012;3(2):161‐76. [DOI] [PubMed] [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta‐analysis in STATA. PloS One 2013;8(10):e76654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chan 2013
- Chan A‐W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža‐Jerić K, et al. Spirit 2013 statement: defining standard protocol items for clinical trials. Annals of Internal Medicine 2013;158(3):200‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chapman 2008
- Chapman R, Cullen S. Etiopathogenesis of primary sclerosing cholangitis. World Journal of Gastroenterology 2008;14(21):3350‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chapman 2010
- Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010;51(2):660‐78. [DOI] [PubMed] [Google Scholar]
Del Re 2013
- Re AC, Spielmans GI, Flückiger C, Wampold BE. Efficacy of new generation antidepressants: differences seem illusory. PLoS One 2013;8(6):e63509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Demets 1987
- DeMets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Dias 2012a
- Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 3: Heterogeneity: subgroups, meta‐regression, bias and bias‐adjustment, September 2011 (last updated April 2012). www.nicedsu.org.uk/TSD3%20Heterogeneity.final%20report.08.05.12.pdf (accessed 27 March 2014).
Dias 2012b
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 1: Introduction to evidence synthesis for decision making, April 2011 (last updated April 2012). www.nicedsu.org.uk/TSD1%20Introduction.final.08.05.12.pdf (accessed 27 March 2014).
Dias 2014a
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta‐analysis of randomised controlled trials, August 2011 (last updated April 2014). http://www.nicedsu.org.uk/TSD2%20General%20meta%20analysis%20corrected%2015April2014.pdf (accessed 8 October 2014). [PubMed]
Dias 2014b
- Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: Inconsistency in networks of evidence based on randomised controlled trials, May 2011 (last updated April 2014). http://www.nicedsu.org.uk/TSD4%20Inconsistency.final.15April2014.pdf (accessed 8 October 2014). [PubMed]
EASL 2009
- European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases. Journal of Hepatology 2009;51(2):237‐67. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Epstein 2004
- Epstein MP, Kaplan MM. A pilot study of etanercept in the treatment of primary sclerosing cholangitis. Digestive Diseases and Sciences 2004;49(1):1‐4. [DOI] [PubMed] [Google Scholar]
EuroQol 2014
- EuroQol. About EQ‐5D. http://www.euroqol.org/about‐eq‐5d.html (accessed 8 October 2014).
Giljaca 2010
- Giljaca V, Poropat G, Stimac D, Gluud C. Glucocorticosteroids for primary sclerosing cholangitis. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD004036.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gluud 2013
- Gluud C, Nikolova D, Klingenberg SL, Alexakis N, Als‐Nielsen B, Colli A, et al. Cochrane Hepato‐Biliary Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)). 2013, Issue 7. Art. No.: LIVER.
Gores 2013
- Gores GJ, Darwish Murad S, Heimbach JK, Rosen CB. Liver transplantation for perihilar cholangiocarcinoma. Digestive Diseases 2013;31(1):126‐9. [DOI] [PubMed] [Google Scholar]
Gross 1985
- Gross JB Jr, Ludwig J, Wiesner RH, McCall JT, LaRusso NF. Abnormalities in tests of copper metabolism in primary sclerosing cholangitis. Gastroenterology 1985;89(2):272‐8. [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction ‐ GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
Higgins 2012
- Higgins JPT, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta‐analysis: concepts and models for multi‐arm studies. Research Synthesis Methods 2012;3(2):98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice CFR & ICH Guidelines. Vol. 1, Pennsylvania, USA: Barnett International/PAREXEL, 1997. [Google Scholar]
Jakobsen 2014
- Jakobsen JC, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta‐analytic methods. BMC Medical Research Methodology 2014;14(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jay 2011
- Jay CL, Lyuksemburg V, Ladner DP, Wang E, Caicedo JC, Holl JL, et al. Ischemic cholangiopathy after controlled donation after cardiac death liver transplantation: a meta‐analysis. Annals of Surgery 2011;253(2):259‐64. [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
Klingenberg 2006
- Klingenberg SL, Chen W. D‐penicillamine for primary sclerosing cholangitis. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD004182.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Klose 2014
- Klose J, Klose MA, Metz C, Lehner F, Manns MP, Klempnauer J, et al. Outcome stagnation of liver transplantation for primary sclerosing cholangitis in the Model for End‐Stage Liver Disease era. Langenbecks Archives of Surgery 2014;399(8):1021‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Koro 2013
- Koro NS, Alkaade S. Role of endoscopy in primary sclerosing cholangitis. Current Gastroenterology Reports 2013;15(12):361. [DOI] [PubMed] [Google Scholar]
Liu 2013
- Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, et al. Dense genotyping of immune‐related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nature Genetics 2013;45(6):670‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lu 2006
- Lu G, Ades AE. Assessing evidence inconsistency in mixed treatment comparisons. Journal of the American Statistical Association 2006;101(474):447‐59. [Google Scholar]
Lundh 2017
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Macaskill 2001
- Macaskill P, Walter SD, Irwig L. A comparison of methods to detect publication bias in meta‐analysis. Statistics in Medicine 2001;20(4):641‐54. [DOI] [PubMed] [Google Scholar]
Mills 2012
- Mills EJ, Ioannidis JP, Thorlund K, Schünemann HJ, Puhan MA, Guyatt GH. How to use an article reporting a multiple treatment comparison meta‐analysis. JAMA 2012;308(12):1246‐53. [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
NCBI 2014
- NCBI. Cholangitis, Sclerosing. http://www.ncbi.nlm.nih.gov/mesh/68015209 (accessed 10 June 2014).
Newell 1992
- Newell DJ. Intention‐to‐treat analysis: implications for quantitative and qualitative research. International Journal of Epidemiology 1992;21(5):837‐41. [DOI] [PubMed] [Google Scholar]
Novak 2008
- Novak K, Swain MG. Role of methotrexate in the treatment of chronic cholestatic disorders. Clinics in Liver Disease 2008;12(1):81‐96. [DOI] [PubMed] [Google Scholar]
O'Mahony 2006
- O'Mahony CA, Vierling JM. Etiopathogenesis of primary sclerosing cholangitis. Seminars in Liver Disease 2006;26(1):3‐21. [DOI] [PubMed] [Google Scholar]
OpenBUGS 3.2.3 [Computer program]
- Members of OpenBUGS Project Management Group. OpenBUGS. Version 3.2.3. Members of OpenBUGS Project Management Group, 2014.
Paumgartner 2002
- Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36(3):525‐31. [DOI] [PubMed] [Google Scholar]
Pawlik 2008
- Pawlik TM, Olbrecht VA, Pitt HA, Gleisner AL, Choti MA, Schulick RD, et al. Primary sclerosing cholangitis: role of extrahepatic biliary resection. Journal of the American College of Surgeons 2008;206(5):822‐30. [DOI] [PubMed] [Google Scholar]
Perez 2009
- Perez MJ, Briz O. Bile‐acid‐induced cell injury and protection. World Journal of Gastroenterology 2009;15(14):1677‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ponsioen 2016
- Ponsioen CY, Chapman RW, Chazouilleres O, Hirschfield GM, Karlsen TH, Lohse AW, et al. Surrogate endpoints for clinical trials in primary sclerosing cholangitis: review and results from an international PSC study group consensus process. Hepatology 2016;63(4):1357‐67. [DOI] [PubMed] [Google Scholar]
Poropat 2011
- Poropat G, Giljaca V, Stimac D, Gluud C. Bile acids for primary sclerosing cholangitis. Cochrane Database of Systematic Reviews 2011, Issue 1. [DOI: 10.1002/14651858.CD003626.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Puhan 2014
- Puhan MA, Schünemann HJ, Murad MH, Li T, Brignardello‐Petersen R, Singh JA, et al. A GRADE Working Group approach for rating the quality of treatment effect estimates from network meta‐analysis. BMJ 2014;349:g5630. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Salanti 2011
- Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple‐treatment meta‐analysis: an overview and tutorial. Journal of Clinical Epidemiology 2011;64(2):163‐71. [DOI] [PubMed] [Google Scholar]
Salanti 2012
- Salanti G. Indirect and mixed‐treatment comparison, network, or multiple‐treatments meta‐analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Research Synthesis Methods 2012;3(2):80‐97. [DOI] [PubMed] [Google Scholar]
Savović 2012a
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials: combined analysis of meta‐epidemiological studies. Health Technology Assessment 2012;16(35):1‐82. [DOI] [PubMed] [Google Scholar]
Savović 2012b
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. PLoS Medicine 2010;7(3):e1000251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stata/SE 14.2 [Computer program]
- StataCorp LP. Stata/SE 14.2 for Windows[64‐bit x86‐64]. Version 14. College Station: StataCorp LP, 2017.
Stewart 2014
- Stewart L. Iatrogenic biliary injuries: identification, classification, and management. Surgical Clinics of North America 2014;94(2):297‐310. [DOI] [PubMed] [Google Scholar]
Tabibian 2014
- Tabibian JH, Lindor KD. Ursodeoxycholic acid in primary sclerosing cholangitis: if withdrawal is bad, then administration is good (right?). Hepatology 2014;60(3):785‐8. [DOI] [PubMed] [Google Scholar]
Talwalkar 2001
- Talwalkar JA, Lindor KD. Natural history and prognostic models in primary sclerosing cholangitis. Best Practice & Research: Clinical Gastroenterology 2001;15(4):563‐75. [DOI] [PubMed] [Google Scholar]
Talwalkar 2005
- Talwalkar JA, Angulo P, Keach JC, Petz JL, Jorgensen RA, Lindor KD. Mycophenolate mofetil for the treatment of primary sclerosing cholangitis. American Journal of Gastroenterology 2005;100(2):308‐12. [DOI] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User Manual for Trial Sequential Analysis (TSA). Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, Denmark. www.ctu.dk/tsa 2011:1‐115.
Thorlund 2012
- Thorlund K, Mills EJ. Sample size and power considerations in network meta‐analysis. Systematic Reviews 2012;1:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Triantos 2011
- Triantos CK, Koukias NM, Nikolopoulou VN, Burroughs AK. Meta‐analysis: ursodeoxycholic acid for primary sclerosing cholangitis. Alimentary Pharmacology & Therapeutics 2011;34(8):901‐10. [DOI] [PubMed] [Google Scholar]
TSA 2011 [Computer program]
- Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen. TSA version 0.9. Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, 2011.
Turner 2012
- Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JP. Predicting the extent of heterogeneity in meta‐analysis, using empirical data from the Cochrane Database of Systematic Reviews. International Journal of Epidemiology 2012;41(3):818‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Valero 2012
- Valero V 3rd, Cosgrove D, Herman JM, Pawlik TM. Management of perihilar cholangiocarcinoma in the era of multimodal therapy. Expert Review of Gastroenterology & Hepatology 2012;6(4):481‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Valkenhoef 2012
- Valkenhoef G, Lu G, Brock B, Hillege H, Ades AE, Welton NJ. Automating network meta‐analysis. Research Synthesis Methods 2012;3(4):285‐99. [DOI] [PubMed] [Google Scholar]
Ware 2014
- Ware JE. SF‐36® Health Survey Update. http://www.sf‐36.org/tools/sf36.shtml 2014 (accessed 8 October 2014).
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wunsch 2014
- Wunsch E, Trottier J, Milkiewicz M, Raszeja‐Wyszomirska J, Hirschfield GM, Barbier O, et al. Prospective evaluation of ursodeoxycholic acid withdrawal in patients with primary sclerosing cholangitis. Hepatology 2014;60(9):31‐940. [DOI] [PubMed] [Google Scholar]
Yimam 2014
- Yimam KK, Bowlus CL. Diagnosis and classification of primary sclerosing cholangitis. Autoimmunity Reviews 2014;13(4‐5):445‐50. [DOI] [PubMed] [Google Scholar]