

Abstract
Background
Hereditary haemochromatosis is a genetic disorder related to proteins involved in iron transport, resulting in iron load and deposition of iron in various tissues of the body. This iron overload leads to complications including liver cirrhosis (and related complications such as liver failure and hepatocellular carcinoma), cardiac failure, cardiac arrhythmias, impotence, diabetes, arthritis, and skin pigmentation. Phlebotomy (venesection or 'blood letting') is the currently recommended treatment for hereditary haemochromatosis. The optimal treatment of hereditary haemochromatosis remains controversial.
Objectives
To assess the comparative benefits and harms of different interventions in the treatment of hereditary haemochromatosis through a network meta‐analysis and to generate rankings of the available treatments according to their safety and efficacy. However, we found only one comparison. Therefore, we did not perform the network meta‐analysis and we assessed the comparative benefits and harms of different interventions using standard Cochrane methodology.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, Science Citation Index Expanded, World Health Organization International Clinical Trials Registry Platform, and randomised clinical trials registers to March 2016 to identify randomised clinical trials on treatments for hereditary haemochromatosis.
Selection criteria
We included only randomised clinical trials (irrespective of language, blinding, or publication status) in participants with hereditary haemochromatosis. We excluded trials which included participants who had previously undergone liver transplantation. We considered any of the various interventions compared with each other or with inactive treatment.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. We calculated the odds ratio (OR) and rate ratio with 95% confidence intervals (CI) using both fixed‐effect and random‐effects models with RevMan 5 based on available‐participant analysis. We assessed risk of bias according to Cochrane, controlled risk of random errors with Trial Sequential Analysis, and assessed the quality of the evidence using GRADE.
Main results
Three trials with 146 participants met the inclusion criteria of this review. Two parallel group trials with 100 participants provided information on one or more outcomes. The remaining trial was a cross‐over trial, with no usable data for analysis. All the trials were at high risk of bias. Overall, all the evidence was of very low quality. All three trials compared erythrocytapheresis (removal of red cells only, instead of whole blood) versus phlebotomy. Two of the trials shared the same first author. The mean or median age in the three trials ranged from 42 to 55 years. None of the trials reported whether the included participants were symptomatic or asymptomatic or a mixture of both. Two trials were conducted in people who were haemochromatosis treatment‐naive. The trial that provided most data for this review excluded people with malignancy, heart failure, and serious cardiac arrhythmias. We found no trials assessing iron‐chelating agents.
Only one of the trials with 38 participants reported no short‐term mortality and no serious adverse events at the end of the short‐term follow‐up (eight months). Two trials reported the proportion of people with adverse events: 10/49 (20.4%) in the erythrocytapheresis group versus 11/51 (21.6%) in the phlebotomy group. One of these two trials provided data on adverse event rates (42.1 events per 100 participants with erythrocytapheresis versus 52.6 events per 100 participants with phlebotomy). There was no evidence of differences in the proportion of people with adverse events and the number of adverse events (serious and non‐serious) between the groups (proportion of people with adverse events: OR 0.93, 95% CI 0.36 to 2.43; participants = 100; trials = 2; number of adverse events: rate ratio 0.80, 95% CI 0.32 to 2.03; participants = 38; trial = 1). There was no difference between the groups regarding short‐term health‐related quality of life (mean difference (MD) 1.00, 95% CI ‐10.80 to 12.80; participants = 38; trials = 1). This outcome was measured using EQ‐VAS (range: 0 to 100 where a higher score indicates better health‐related quality of life). None of the trials reported mortality beyond one year, health‐related quality of life beyond one year, liver transplantation, decompensated liver disease, cirrhosis, hepatocellular carcinoma, diabetes, or cardiovascular complications during the long‐term follow‐up.
The two trials that provided data for this review were funded by parties with no vested interest in the results; the source of funding of the third trial was not reported.
Authors' conclusions
There is currently insufficient evidence to determine whether erythrocytapheresis is beneficial or harmful compared with phlebotomy. Phlebotomy has less equipment requirements and remains the treatment of choice in people with hereditary haemochromatosis who require blood letting in some form. However, it should be noted that there is no evidence from randomised clinical trials that blood letting in any form is beneficial in people with hereditary haemochromatosis. Having said this, a trial including no treatment is unlikely to be conducted. Future trials should compare different frequencies of phlebotomy and erythrocytapheresis versus phlebotomy with and without different iron‐chelating agents compared with each other, and with placebo. Such trials should include long‐term follow‐up of participants (e.g. using national record linkage databases) to determine whether treatments are beneficial or harmful in terms of clinical outcomes such as deaths, health‐related quality of life, liver damage and its consequences, heart damage and its consequences, and other outcomes that are of importance to people with hereditary haemochromatosis.
Plain language summary
Treatments for hereditary haemochromatosis
Background
Hereditary haemochromatosis is an inherited genetic disorder (derived from one's parents) resulting in excessive iron accumulation in the body. Some people develop liver damage leading to liver failure, heart damage leading to heart failure, impotence (inability for a man to have an erection or orgasm), diabetes, arthritis (joint pain and swelling), and skin pigmentation (colouring) because of excessive iron accumulation. Several treatments are used to treat hereditary haemochromatosis but the best way is not clear. We searched for randomised clinical trials (well‐design clinical studies where people are randomly put into one of two or more treatment groups) reported to March 2016. We included trials in which participants had not had a liver transplant. Apart from using standard Cochrane methods which allow comparison of only two treatments at a time (direct comparison), we planned to use an advanced method which allows comparison of the many different treatments which are individually compared in the trials (network meta‐analysis). However, because there was only one comparison, we used standard Cochrane methodology.
Study characteristics
We identified three trials. Two trials with 100 participants provided information on one or more outcomes (measures of how well the treatments worked). The trials compared phlebotomy (removal of blood or 'blood letting') versus erythrocytapheresis (removal of blood, separation of red cells (which carry oxygen in the blood), and return of the remaining parts of the blood). Two trials were conducted in people who had not undergone previous treatment for haemochromatosis. The trial that provided most data for this review excluded people with cancer, heart failure, and serious irregular heartbeats.
Source of funding: the two trials that provided data for this review were funded by parties with no vested interest in the results; the source of funding of the third trial was not reported.
Key results
There were no deaths or serious complications in the short term in either group in the only trial that reported this information. There was no evidence of any difference in the percentage of people with any complications, the number of complications per person, and short‐term health‐related quality of life (a measure of a person's satisfaction with their life and health) between the treatments. None of the trials reported deaths beyond one year, health‐related quality of life beyond one year, liver transplantation, severe liver damage, liver failure, liver cancer, diabetes, heart failure, or stroke during the long term. There is currently insufficient evidence to determine whether erythrocytapheresis is beneficial or harmful compared with phlebotomy. Erythrocytapheresis requires special equipment, while phlebotomy does not. So, phlebotomy remains the treatment of choice in people with hereditary haemochromatosis even though there is no evidence from randomised clinical trials that blood letting is beneficial. Having said this, a randomised clinical trial including no treatment is unlikely to be conducted.
Quality of evidence
The overall quality of evidence was very low as the trials were at high risk of bias, which means that there is possibility of making wrong conclusions overestimating benefits or underestimating harms of treatments because of the way that the studies were conducted. Further high‐quality randomised clinical trials to identify how often blood letting should be performed and those comparing erythrocytapheresis versus blood letting are required. Such trials should include long‐term monitoring of participants (perhaps by linking health records in some countries).
Summary of findings
Summary of findings for the main comparison. Erythrocytapheresis versus phlebotomy for hereditary haemochromatosis.
| Erythrocytapheresis versus phlebotomy for hereditary haemochromatosis | |||||
| Patient or population: people with hereditary haemochromatosis Settings: secondary or tertiary Intervention: erythrocytapheresis Comparison: phlebotomy | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| Phlebotomy | Therapeutic erythrocytapheresis | ||||
| Long‐term mortality | None of the included trials reported mortality beyond 1 year. | ||||
|
Mortality Follow‐up period: 8 months |
There was no mortality in either group in the short‐term in the 1 trial that reported this information. | 38 (1 study) | ⊕⊝⊝⊝ Very low1,2 | ||
|
Serious adverse events Follow‐up period: 8 months |
There were no serious adverse events in either group in the 1 trial that reported this information. | 38 (1 study) | ⊕⊝⊝⊝ Very low1,2 | ||
|
Health‐related quality of life
EQ‐VAS. Scale from: 0 to 100. Follow‐up period: 8 months |
The mean health‐related quality of life in the control groups was 68 | The mean health‐related quality of life in the intervention groups was 1 higher (10.8 lower to 12.8 higher) | ‐ | 38 (1 study) | ⊕⊝⊝⊝ Very low1,2 |
| Health‐related quality of life beyond one year | None of the included trials reported health‐related quality of life beyond one year | ||||
| *The basis for the assumed risk is the mean control group proportion or control event rate. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval. | |||||
| GRADE Working Group grades of evidence High quality: further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: we are very uncertain about the estimate. | |||||
1 Downgraded one level for risk of bias. 2 Downgraded two levels for imprecision (one level for small sample size and one level for wide confidence intervals).
Background
Description of the condition
Hereditary haemochromatosis is a genetic disorder related to proteins involved in iron transport, resulting in iron load and deposition of iron in various tissues of the body (Adams 2007). The most common mutation causing hereditary haemochromatosis is substitution of cysteine with tyrosine at position 282 (C282Y) of the HFE gene (Feder 1996; Pietrangelo 2004; Adams 2007; Bardou‐Jacquet 2014). This is an autosomal‐recessive genetic disorder (i.e. it can manifest itself only when both alleles (copies of the gene in both chromosomes) carry the mutation) (Feder 1996; Pietrangelo 2004; Adams 2007; Bardou‐Jacquet 2014). However, compound heterozygosity with another allele H63D (substitution of histamine with aspartic acid at position 63) (i.e. one copy of the C282Y mutated human haemochromatosis protein (HFE) gene and one copy of the H63D mutated HFE gene) can also result in manifestation of haemochromatosis (Feder 1996; van Bokhoven 2011). Several other mutations related to the HFE protein and other proteins involved in iron transport, namely ferroportin, hepcidin, transferrin receptor‐2, haemojuvelin, and ceruloplasmin, can lead to hereditary haemochromatosis (Pietrangelo 2004; Adams 2007; van Bokhoven 2011). Carriers of the autosomal‐recessive mutated HFE gene (either the C282Y or H63D allele in one of the chromosomes) varies globally and depends on ethnic origin. In the US, the prevalence of the carrier state is about 5.4% for C282Y and 13.5% for H63D alleles (Steinberg 2001). Approximately 0.3% of the general population in the US are homozygous for C282Y, 1.9% are homozygous for H63D, and 2% have C282Y/H63D compound heterozygosity (Steinberg 2001). The frequencies of C282Y and H63D alleles are more common in non‐Hispanic white people compared to non‐Hispanic black people and Mexican‐American people (Steinberg 2001). In Europe, there is significant variation in different countries with the frequency of C282Y more common in countries such as Ireland and the UK (Lucotte 2003). Overall, 0% to 28% of people carry at least one C282Y allele in different countries in Europe (Mercier 1998; Cassanelli 2001; Lucotte 2003; Ropero 2006; Voicu 2009); and 23% to 30% carry at least one H63D allele (Cassanelli 2001; Ropero 2006; Voicu 2009). However, on average, only 0.3% of people are homozygous for the C282Y allele. In Australia, screening of people of Northern Europe ancestry revealed that 0.7% of people were homozygous for the C282Y mutation and an additional 2.4% had C282Y/H63D compound heterozygosity (Allen 2008).
Diagnosis of hereditary haemochromatosis is suspected by abnormal serum iron studies such as serum ferritin and transferrin saturation, and established by the presence of C282Y homozygous gene products and the presence of other known rarer mutations (van Bokhoven 2011). Liver iron stores measured by magnetic resonance imaging or liver biopsy may be helpful in identifying elevated iron stores in the liver (Bacon 2011; van Bokhoven 2011). The proportion of people with haemochromatosis‐predisposing mutations who develop clinical symptoms of iron overload is very controversial. Asymptomatic elevation of serum ferritin and transferrin saturation (i.e. screen‐detected hereditary haemochromatosis) is a common mode of clinical presentation (Bardou‐Jacquet 2014). In symptomatic people, common symptoms related to hereditary haemochromatosis at the time of diagnosis are poor general health, fatigue, malaise, diabetes, and arthralgia (Pietrangelo 2004; Allen 2008; van Bokhoven 2011). Complications related to hereditary haemochromatosis include liver cirrhosis (and related complications such as liver failure and hepatocellular carcinoma), cardiac failure, cardiac arrhythmias, impotence, diabetes, arthritis, and skin pigmentation (Pietrangelo 2004; Schmitt 2005; van Bokhoven 2011; Bardou‐Jacquet 2014). While some researchers state that 28% to 50% of men and 1.4% to 44% of women homozygous for haemochromatosis‐predisposing mutations develop symptoms (Bradley 1996; Allen 2008), other researchers point out that the frequency of symptoms commonly attributed to haemochromatosis such as poor general health, fatigue, malaise, diabetes, and arthralgia were similar between people homozygous for haemochromatosis‐predisposing mutations and the general population (Beutler 2002). Therefore, it is not clear whether these symptoms are related to haemochromatosis at all. However, it should be pointed out that people homozygous for haemochromatosis‐predisposing mutations had more frequent liver disorders compared to the general population (Beutler 2002). Overall, the odds of developing hepatocellular carcinoma and porphyria cutanea tarda (skin blisters in areas of the body exposed to sunlight) were higher in C282Y homozygotes and C282Y/H63D compound heterozygotes compared to people in control groups (Ellervik 2007). Approximately one‐third of symptomatic people with C282Y homozygosity and a mean age of 50 years referred to a tertiary care centre die over 20 years (Wojcik 2002; Schmitt 2005). Although the symptoms related to hereditary haemochromatosis are thought to be due to iron overload and some studies have indicated a relationship between symptoms and a serum ferritin level of 1000 µg/L (Allen 2008), there is currently no firm evidence for a relationship between symptoms and the degree of iron overload (Beutler 2002; van Bokhoven 2011). While screening of family members and the general population have been advocated by some researchers (Pietrangelo 2004; Bacon 2011; de Graaff 2015), other researchers found no evidence of any tangible benefit of screening based on systematic reviews of clinical effectiveness (Schmitt 2005; Whitlock 2006). However, asymptomatic elevation of serum ferritin and transferrin saturation (i.e. screen‐detected hereditary haemochromatosis) is a common mode of clinical presentation (Bardou‐Jacquet 2014).
Description of the intervention
The main treatments for hereditary haemochromatosis include phlebotomy (venesection or blood letting), erythrocytapheresis (removal of red cells only instead of removal of whole blood), and administration of iron‐chelating agents such as desferrioxamine (van Bokhoven 2011). Removal of 500 mL of blood per week guided by serum transferrin levels and haemoglobin levels is recommended (van Bokhoven 2011). The major problems with regular phlebotomy are venous access and the requirement to visit a healthcare facility for treatment (van Bokhoven 2011). While there are no absolute contraindications for phlebotomy, the relative contraindications include severe heart disease and anaemia (Assi 2014), and possibly hypoproteinaemia.
Erythrocytapheresis involves removal of red cells only instead of whole blood and requires specialist equipment. However, the number of treatments can be reduced compared to regular phlebotomy as more iron can be removed per session (van Bokhoven 2011; Rombout‐Sestrienkova 2012). Desferrioxamine is usually administered subcutaneously, intramuscularly, or intravenously (Martindale 2011). A starting dose of 500 mg is recommended and the drug may be administered three to seven times a week (Martindale 2011). Adverse reactions of desferrioxamine include severe allergy, arthralgia, pain at injection site, gastrointestinal symptoms, tachycardia, and thrombocytopenia (Martindale 2011; van Bokhoven 2011). Newer iron‐chelating agents such as deferasirox have also been used for the treatment of primary hereditary haemochromatosis (Cancado 2015). Deferasirox is an oral chelating agent and appears to have equivalent efficacy and safety profiles as desferrioxamine (Vichinsky 2007; Pennell 2014). This review will not cover lifestyle modifications such as reduced alcohol consumption and dietary changes.
How the intervention might work
Since red blood cells contain iron as a component of haemoglobin, their removal (by erythrocytapheresis or phlebotomy) reduces body iron content, which could potentially diminish iron deposition in tissues and the subsequent complications. Desferrioxamine is an iron‐chelating agent that might work by removing iron deposition from the tissues (Martindale 2011).
Why it is important to do this review
The optimal treatment of hereditary haemochromatosis is not known. Currently, both the European Association for the Study of the Liver (EASL) and American Association for the Study of Liver Diseases (AASLD) recommend phlebotomy as the treatment of choice (EASL 2010; Bacon 2011). One randomised clinical trial reported that erythrocytapheresis required fewer sessions than venesection to decrease iron overload (Rombout‐Sestrienkova 2012). It is also not clear whether any of these measures decrease the development of complications. So, there is clearly discordance between the evidence and recommendations. Network meta‐analysis allows combination of the direct evidence and indirect evidence, and allows ranking of different treatments in terms of the different outcomes (Salanti 2011; Salanti 2012). There has been no network meta‐analysis on the comparative effectiveness of different interventions in the treatment of hereditary haemochromatosis. This systematic review and attempted network meta‐analysis provides evidence from randomised clinical trials on the role of different medical interventions in the treatment of people with hereditary haemochromatosis.
Objectives
To assess the comparative benefits and harms of different interventions in the treatment of hereditary haemochromatosis through meta‐analysis and to generate rankings of the available treatments according to their safety and efficacy. However, we found only one comparison. When more trials become available, we will attempt to conduct network meta‐analysis in order to generate rankings of the available treatments according to their safety and efficacy. This is why we retain the planned methodology for network meta‐analysis in our Appendix 1. Once data appear allowing for the conduct of network meta‐analysis, this Appendix 1 will be moved back into the Methods section.
Methods
Criteria for considering studies for this review
Types of studies
We considered only randomised clinical trials for this network meta‐analysis irrespective of language, publication status, or date of publication. We excluded studies of other design because of the risk of bias in such studies. We are all aware that such exclusions make us focus much more on potential benefits and not fully assess the risks of serious adverse events as well as risks of adverse events.
Types of participants
We included participants with hereditary haemochromatosis irrespective of the method of diagnosis of the disease or the presence of symptoms. We exclude randomised clinical trials in which participants had undergone liver transplantation previously.
Types of interventions
We planned to include the following interventions that are possible treatments for hereditary haemochromatosis and can be compared with each other or with no active treatment.
The interventions that we considered were:
phlebotomy;
desferrioxamine;
erythrocytapheresis.
The above list was not exhaustive. If we identified any other interventions that we were not aware of, we planned to consider them as eligible and include them in the review if they were used primarily for the treatment of hereditary haemochromatosis. We excluded trials that did not include at least two or more of the included interventions.
Types of outcome measures
We planned to assess the comparative benefits and harms of available pharmacological interventions aimed at treating people with hereditary haemochromatosis for the following outcomes.
Primary outcomes
Long‐term mortality (time to death; maximal follow‐up).
-
Mortality:
short‐term mortality (up to one year);
medium‐term mortality (one to five years).
-
Adverse events (within three months after cessation of treatment). Depending on the availability of data, we attempted to classify adverse effects as serious or non‐serious. We defined a non‐serious adverse event as any untoward medical occurrence not necessarily having a causal relationship with the treatment but resulting in a dose reduction or discontinuation of treatment (any time after commencement of treatment) (ICH‐GCP 1997). We defined a serious adverse event as any event that would increase mortality; was life threatening; required hospitalisation; resulted in persistent or significant disability; was a congenital anomaly/birth defect; or any important medical event that might have jeopardised the person or required intervention to prevent it. We used the definition used by trial authors for non‐serious and serious adverse events:
proportion of participants with serious adverse events;
number of serious adverse events;
proportion of participants with any type of adverse event;
number of any type of adverse event.
-
Health‐related quality of life as defined in the included trials using a validated scale such as EQ‐5D or 36‐item Short Form (SF‐36) (EuroQol 2014; Ware 2014):
short‐term (up to one year);
medium‐term (one to five years);
long‐term (beyond five years).
We planned to consider long‐term health‐related quality of life more important than short‐term or medium‐term health‐related quality of life, although short‐term and medium‐term health‐related quality of life are also important primary outcomes.
Secondary outcomes
-
Liver transplantation (maximal follow‐up):
proportion of participants with liver transplantation;
time to liver transplantation.
-
Decompensated liver disease (maximal follow‐up):
proportion of participants with decompensated liver disease;
time to liver decompensation.
-
Cirrhosis (any cirrhosis with or without clinical symptoms and with or without decompensation) (maximal follow‐up):
proportion of participants with cirrhosis;
time to cirrhosis.
Hepatocellular carcinoma (maximal follow‐up).
Diabetes (maximal follow‐up).
Cardiovascular complications such as cardiac failure, myocardial infarction, and stroke (maximal follow‐up).
Search methods for identification of studies
Electronic searches
We searched the Cochrane Central Register of Controlled Trials (CENTRAL; 2016, Issue 3), MEDLINE (OvidSP), Embase (OvidSP), and Science Citation Index Expanded (Web of Knowledge) (Royle 2003) from inception to 29 March 2016 for randomised clinical trials comparing two or more of the above interventions. We searched for all possible comparisons formed by the interventions of interest. To identify further ongoing or completed trials, we also searched the World Health Organization International Clinical Trials Registry Platform search portal (www.who.int/ictrp/en), which searches various trial registers, including ISRCTN and ClinicalTrials.gov on 29 March 2016. Appendix 2 shows the search strategies used.
Searching other resources
We searched the references of the identified trials and the existing Cochrane Reviews on hereditary haemochromatosis to identify additional trials for inclusion.
Data collection and analysis
Selection of studies
Two review authors (EB and MK) independently identified the trials for inclusion by screening the titles and abstracts. We sought full‐text articles for any references that at least one of the review authors identified for potential inclusion. We selected the trials for inclusion based on the full‐text articles. We planned to list the excluded full‐text references with reasons for their exclusion in the 'Characteristics of excluded studies' table. We listed any ongoing trials identified primarily through the search of the clinical trial registers for further follow‐up. We resolved discrepancies through discussion and by arbitration with KG, DT, and ET.
Data extraction and management
Two review authors (EB and MK) independently extracted the following data.
-
Outcome data (for each outcome and for each treatment arm whenever applicable):
number of participants randomised;
number of participants included for the analysis;
number of participants with events for binary outcomes, mean and standard deviation for continuous outcomes, number of events for count outcomes, and the number of participants with events and the mean follow‐up period for time‐to‐event outcomes;
definition of outcomes or scale used if appropriate.
-
Data on potential effect modifiers:
participant characteristics such as age, sex, comorbidities, proportion of symptomatic participants, method of diagnosis, proportion of people with C282Y homozygosity, previous use of treatments;
details of the intervention and control (including dose (in the case of desferrioxamine) or target reduction (in the case of phlebotomy and erythrocytapheresis, frequency, and duration);
details of any cointerventions;
risk of bias (assessment of risk of bias in included studies).
-
Other data:
year and language of publication;
country in which the participants were recruited;
year(s) in which the trial was conducted;
inclusion and exclusion criteria;
follow‐up time points of the outcome.
If available, we planned to obtain the data separately for symptomatic participants and asymptomatic participants from the report. We attempted to contact trial authors when there was unclear or missing information, or when there was doubt whether trials shared the same participants, completely or partially (by identifying common authors and centres), or if we needed clarification whether the trial report was duplicated. We resolved any differences in opinion through discussion.
Assessment of risk of bias in included studies
We followed the guidance given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and in the Cochrane Hepato‐Biliary Module (Gluud 2015) to assess the risk of bias in included studies. Specifically, we assessed the risk of bias in included trials for the following domains (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Lundh 2017).
Allocation sequence generation
Low risk of bias: the study authors performed sequence generation using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if an independent person not otherwise involved in the study performed them.
Unclear risk of bias: the study authors did not specify the method of sequence generation.
High risk of bias: the sequence generation method was not random. We planned to only include such studies for assessment of harms.
Allocation concealment
Low risk of bias: the participant allocations could not have been foreseen in advance of, or during, enrolment. A central and independent randomisation unit controlled allocation. The investigators were unaware of the allocation sequence (e.g. if the allocation sequence was hidden in sequentially numbered, opaque, and sealed envelopes).
Unclear risk of bias: the study authors did not describe the method used to conceal the allocation so the intervention allocations may have been foreseen before, or during, enrolment.
High risk of bias: it is likely that the investigators who assigned the participants knew the allocation sequence. We planned to only include such studies for assessment of harms.
Blinding of participants and personnel
Low risk of bias: any of the following: no blinding or incomplete blinding, but the review authors judged that the outcome was not likely to be influenced by lack of blinding; or blinding of participants and key study personnel ensured, and it was unlikely that the blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk'; or the trial did not address this outcome.
High risk of bias: any of the following: no blinding or incomplete blinding, and the outcome was likely to be influenced by lack of blinding; or blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome was likely to be influenced by lack of blinding.
Blinded outcome assessment
Low risk of bias: any of the following: no blinding of outcome assessment, but the review authors judged that the outcome measurement was not likely to be influenced by lack of blinding; or blinding of outcome assessment ensured, and unlikely that the blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk'; or the trial did not address this outcome.
High risk of bias: any of the following: no blinding of outcome assessment, and the outcome measurement was likely to be influenced by lack of blinding; or blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement was likely to be influenced by lack of blinding.
Incomplete outcome data
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. The study used sufficient methods, such as multiple imputation, to handle missing data.
Unclear risk of bias: there was insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias on the results.
High risk of bias: the results were likely to be biased due to missing data.
Selective outcome reporting
Low risk of bias: the trial reported the following predefined outcomes: mortality, decompensated liver disease, requirement for transplantation, and treatment‐related adverse events. If the original trial protocol was available, the outcomes should have been those called for in that protocol. If the trial protocol was obtained from a trial registry (e.g. www.clinicaltrials.gov), the outcomes sought should have been those enumerated in the original protocol if the trial protocol was registered before or at the time that the trial was begun. If the trial protocol was registered after the trial was begun, those outcomes were not considered to be reliable.
Unclear risk of bias: not all predefined, or clinically relevant and reasonably expected, outcomes were reported fully, or it was unclear whether data on these outcomes were recorded or not.
High risk of bias: one or more predefined or clinically relevant and reasonably expected outcomes were not reported, despite the fact that data on these outcomes should have been available and even recorded.
For‐profit bias
Low risk of bias: the trial appeared to be free of industry sponsorship or other type of for‐profit support that could manipulate the trial design, conductance, or results of the trial.
Uncertain risk of bias: the trial may or may not have been free of for‐profit bias as no information on clinical trial support or sponsorship was provided.
High risk of bias: the trial was sponsored by industry or received other type of for‐profit support.
Other bias
Low risk of bias: the trial appeared to be free of other components (e.g. inappropriate control or dose or administration of control, baseline differences, early stopping) that could put it at risk of bias.
Uncertain risk of bias: the trial may or may not have been free of other components that could put it at risk of bias.
High risk of bias: there were other factors in the trial that could put it at risk of bias (e.g. inappropriate control or dose or administration of control, baseline differences, early stopping).
We considered a trial at low risk of bias if we assessed the trial as at low risk of bias across all domains. We considered a trial at low risk of bias for an outcome if we assessed the trial as at low risk of bias across all study level domains. Otherwise, we considered the trials at uncertain risk of bias or at high risk of bias regarding one or more domains as at high risk of bias.
Measures of treatment effect
For dichotomous variables (e.g. short‐term and medium‐term mortality or liver transplantation, proportion of participants with adverse events, decompensated liver disease, cirrhosis, hepatocellular carcinoma, or diabetes), we calculated the odds ratio (OR) with 95% confidence intervals (CI). For continuous variables (e.g. quality of life reported on the same scale), we calculated the mean difference (MD) with 95% CI. We planned to use standardised mean differences with 95% CI for quality of life if included trials used different scales. For count outcomes (e.g. number of adverse events), we calculated the rate ratio with 95% CI. For time‐to‐event data (e.g. long‐term mortality or requirement for liver transplantation, time to liver decompensation, and time to cirrhosis), we planned to use the hazard ratio with 95% CIs. We also calculated Trial Sequential Analysis‐adjusted CI to control random errors (Thorlund 2011).
Unit of analysis issues
The unit of analysis was the person with hereditary haemochromatosis according to the intervention group to which they were randomly assigned.
Cluster randomised clinical trials
We found no cluster randomised clinical trials. If we found them, we planned to include them provided that the effect estimate adjusted for cluster correlation was available.
Cross‐over randomised clinical trials
We found one cross‐over randomised clinical trial. We planned to only include the outcomes after the period of first treatment since hereditary haemochromatosis is a chronic disease and the treatments could potentially have a residual effect. However, the cross‐over trial did not report any outcomes prior to the cross‐over.
Trials with multiple treatment groups
We collected data for all trial treatment groups that met the inclusion criteria.
Dealing with missing data
We performed an intention‐to‐treat analysis whenever possible (Newell 1992). Otherwise, we planned to use the data that were available to us (e.g. a trial might have reported only per‐protocol analysis results). As such 'per‐protocol' analyses may be biased, we planned to conduct best‐worst case scenario analyses (good outcome in intervention group and bad outcome in control group) and worst‐best case scenario analyses (bad outcome in intervention group and good outcome in control group) as sensitivity analyses whenever possible but did not perform this because there were no post‐randomisation dropouts in either trial that provided data.
For continuous outcomes, we planned to impute the standard deviation from P values according to guidance given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). If the data were likely to be normally distributed, we planned to use the median for meta‐analysis when the mean was not available. If it was not possible to calculate the standard deviation from the P value or the CIs, we planned to impute the standard deviation using the largest standard deviation in other trials for that outcome. This form of imputation may decrease the weight of the study for calculation of MDs and may bias the effect estimate to no effect for calculation of standardised mean differences (Higgins 2011).
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity by carefully examining the characteristics and design of included trials. We planned to assess the presence of clinical heterogeneity by comparing effect estimates in the presence or absence of symptoms, different targets of iron reduction, and different doses of desferrioxamine or different methods of erythrocytapheresis or phlebotomy, and the doses of the pharmacological treatments. Different study designs and risk of bias may contribute to methodological heterogeneity. If we identified substantial heterogeneity, clinical, methodological, or statistical, we explored and address heterogeneity in a subgroup analysis (see Subgroup analysis and investigation of heterogeneity). We used the I2 test and Chi2 test for heterogeneity, and overlapping of CIs to assess heterogeneity.
Assessment of reporting biases
We planned to use visual asymmetry on a funnel plot to explore reporting bias in the presence of at least 10 trials that could be included for a direct comparison (Egger 1997; Macaskill 2001). In the presence of heterogeneity that could be explained by subgroup analysis, we planned to produce a funnel plot for each subgroup in the presence of the adequate number of trials. We planned to use the linear regression approach described by Egger 1997 to determine funnel plot asymmetry.
We also considered selective reporting and non‐reporting of trials (identified from searching the trial registers) as evidence of reporting bias.
Data synthesis
We performed the meta‐analyses according to the recommendations of The Cochrane Collaboration (Higgins 2011), using the software package Review Manager 5 (RevMan 2014). We used a random‐effects model (DerSimonian 1986) and a fixed‐effect model (DeMets 1987). In the case of a discrepancy between the two models, we have reported both results; otherwise, we have reported only the results from the fixed‐effect model.
Calculation of required information size and Trial Sequential Analysis
For calculation of the required information size, see Appendix 3. We performed Trial Sequential Analysis to control the risks of random errors (Wetterslev 2008; Thorlund 2011; TSA 2011) when there were at least two trials included in the meta‐analysis. We used an alpha error of 2.5% (Jakobsen 2014), power of 90% (beta error of 10%), a relative risk reduction of 20%, a control group proportion observed in the trials, and the diversity observed in the meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We planned to assess the differences in the effect estimates between the following subgroups.
Trials with low risk of bias compared to trials with high risk of bias.
Participants with symptomatic compared to participants with asymptomatic hereditary haemochromatosis.
Different targets of iron reduction.
Different doses of desferrioxamine or different methods of erythrocytapheresis or phlebotomy.
We planned to use the chi2 test for subgroup differences to identify subgroup differences.
Sensitivity analysis
If a trial reported only per‐protocol analysis results, we planned to re‐analyse the results using the best‐worst case scenario and worst‐best case scenario analyses as sensitivity analyses whenever possible. However, we did not perform this because both trials that provided data for this review had no post‐randomisation dropouts.
Presentation of results and GRADE assessments
We reported mortality, serious adverse events, and health‐related quality of life in a 'Summary of findings' table format, downgrading the quality of evidence for risk of bias, inconsistency, indirectness, imprecision, and publication bias using GRADE (Guyatt 2011).
Results
Description of studies
Results of the search
We identified 3852 references through electronic searches of CENTRAL (N = 312), MEDLINE (N = 2245), Embase (N = 489), Science Citation Index Expanded (N = 690), World Health Organization International Clinical Trials Registry Platform (N = 30), and randomised controlled trials registers (N = 86). After the removal of 814 duplicates, we obtained 3038 references. We then excluded 3033 clearly irrelevant references through screening titles and reading abstracts. We retrieved five references for further assessment. No references were identified through scanning reference lists of the identified randomised trials. Four of the references were reports of three trials which fulfilled the inclusion criteria of our review (Rombout‐Sestrienkova 2012; Sundic 2014; Rombout‐Sestrienkova 2016; Characteristics of included studies table). One reference is an ongoing trial without any interim data, comparing erythrocytapheresis versus plasmapheresis (Ong 2015; Characteristics of ongoing studies table). The reference flow is summarised in the study flow diagram (Figure 1).
1.
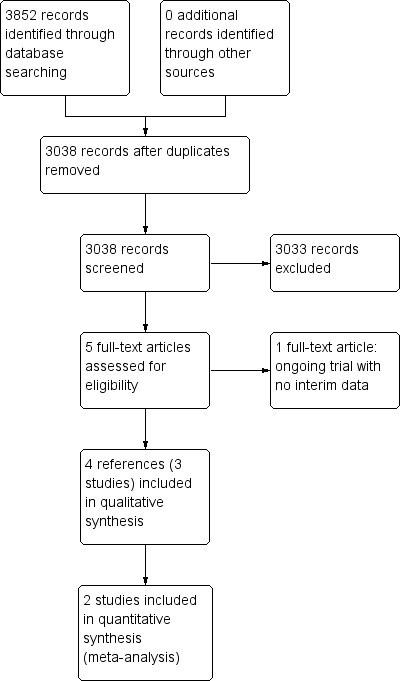
Study flow diagram.
Included studies
Three trials included 146 participants (Rombout‐Sestrienkova 2012; Sundic 2014; Rombout‐Sestrienkova 2016). All the three trials were two‐armed and compared erythrocytapheresis versus phlebotomy. Two trials were simple parallel randomised clinical trials (Rombout‐Sestrienkova 2012; Sundic 2014). The remaining trial was a cross‐over randomised clinical trial in which participants were randomised to receive therapeutic erythrocytapheresis or phlebotomy (Rombout‐Sestrienkova 2016). After one year, the participants were crossed‐over to receive the opposite treatment (Rombout‐Sestrienkova 2016). Two trials with 100 participants provided data for analyses (Rombout‐Sestrienkova 2012; Sundic 2014).
None of the trials reported whether they included symptomatic or asymptomatic participants, or a mixture of both. Two trials which provided data for this review were conducted in people who had not undergone previous treatment for haemochromatosis (Rombout‐Sestrienkova 2012; Sundic 2014). The trial which did not provide data for this review included only people on maintenance therapy for haemochromatosis (Rombout‐Sestrienkova 2016). The trial that provided most data for this review excluded people with malignancy, heart failure, and serious cardiac arrhythmias (Rombout‐Sestrienkova 2012).
One trial carried out erythrocytapheresis bi‐weekly (not clear whether the authors meant this to be once every two weeks or twice weekly) (Sundic 2014), one trial once every two weeks (Rombout‐Sestrienkova 2012), and on trial variably depending upon serum ferritin level (Rombout‐Sestrienkova 2016). Two trials carried out phlebotomy once a week (Rombout‐Sestrienkova 2012; Sundic 2014), and one trial variably depending upon serum ferritin level (Rombout‐Sestrienkova 2016). The amount of red blood cells withdrawn during each treatment of erythrocytapheresis was 350 mL to 800 mL (Rombout‐Sestrienkova 2012; Sundic 2014; Rombout‐Sestrienkova 2016). The amount of blood withdrawn during each treatment of phlebotomy was 450 mL to 500 mL (Rombout‐Sestrienkova 2012; Sundic 2014; Rombout‐Sestrienkova 2016). The treatment duration in one trial was 12 weeks (Sundic 2014), was one year (after which the people crossed‐over) in one trial (Rombout‐Sestrienkova 2016), and was variable depending upon the amount of iron to be removed in one trial (Rombout‐Sestrienkova 2012). All the trials used serum ferritin level of 50 μg/L or less as the target for treatment.
The mean or median age in the trials ranged from 42 to 55 years. The proportion of females was 9.7% in Sundic 2014 and 26.3% in Rombout‐Sestrienkova 2012. Rombout‐Sestrienkova 2016 did not report this information.
Two trials that provided data for this review were funded by parties with no vested interest in the results (Rombout‐Sestrienkova 2012; Sundic 2014); the other trial did not report the source of funding (Rombout‐Sestrienkova 2016).
Excluded studies
We excluded no studies that we sought full text for.
Risk of bias in included studies
The risk of bias is summarised in Figure 2 and Figure 3. As shown in Figure 3, all the trials were at overall high risk of bias.
2.
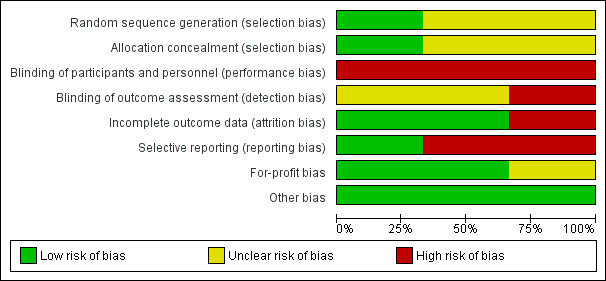
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
One trial was at low risk of selection bias due to random sequence generation (Sundic 2014). The remaining trials were at unclear risk of random sequence generation (Rombout‐Sestrienkova 2012; Rombout‐Sestrienkova 2016). One trial was at low risk of selection bias due to allocation concealment (Rombout‐Sestrienkova 2012). The remaining trials were at unclear risk of allocation concealment (Sundic 2014; Rombout‐Sestrienkova 2016).
Blinding
None of the trials were at low risk of performance or detection bias due to blinding of participants, personnel, and outcome assessors. All the trials were at high risk of performance bias (Rombout‐Sestrienkova 2012; Sundic 2014; Rombout‐Sestrienkova 2016). One trial was at high risk of detection bias (Rombout‐Sestrienkova 2016); the remaining two trials were at unclear risk of detection bias (Rombout‐Sestrienkova 2012; Sundic 2014).
Incomplete outcome data
Two trials were at low risk of attrition bias due to incomplete outcome data (Rombout‐Sestrienkova 2012; Sundic 2014). The remaining trial was at high risk of attrition bias due to incomplete outcome data (Rombout‐Sestrienkova 2016).
Selective reporting
One trial was at low risk of reporting bias due to selective outcome reporting (Rombout‐Sestrienkova 2012). The remaining two trials were at high risk of reporting bias due to selective outcome reporting (Sundic 2014; Rombout‐Sestrienkova 2016).
Other potential sources of bias
Two trials were at low risk of for‐profit bias (Rombout‐Sestrienkova 2012; Sundic 2014). One trial was at unclear risk of for‐profit bias since the source of funding was not reported (Rombout‐Sestrienkova 2016). All the trials were at low risk of 'other' bias.
Effects of interventions
See: Table 1
Long‐term mortality
None of the trials reported long‐term mortality.
Mortality
One trial (38 participants) reported short‐term mortality at eight months (Rombout‐Sestrienkova 2012). There was no mortality in either group. None of the trials reported mortality beyond one year.
Serious adverse events
One trial (38 participants) reported serious adverse events (Rombout‐Sestrienkova 2012). There were no serious adverse events in either group.
All adverse events
Two trials with 100 participants reported proportion of participants with any adverse events (Rombout‐Sestrienkova 2012; Sundic 2014). The proportion of people with any adverse events in the erythrocytapheresis group was 10/49 (20.4%) versus 11/51 (21.6%) in the phlebotomy group. There was no evidence of difference in all adverse events between the groups (OR 0.93, 95% CI 0.36 to 2.43; participants = 100; trials = 2; I2 = 0%). There was no alteration in the results by using the random‐effects model.
One of these two trials with 38 participants also reported the number of adverse events (Rombout‐Sestrienkova 2012). The adverse event rate was 42.1 events per 100 participants in the erythrocytapheresis group and 52.6 events per 100 participants in the phlebotomy groups. There was no evidence of difference in all adverse events between the groups (rate ratio 0.80, 95% CI 0.32 to 2.03; participants = 38; trials = 1).
Health‐related quality of life
One trial with 38 participants reported short‐term health‐related quality of life (up to one year) (Rombout‐Sestrienkova 2012) using EQ‐VAS (EuroQol 2014) on a scale of 0 to 100 with higher numbers indicating better health‐related quality of life. There was no significant difference in health‐related quality of life between the groups (MD 1.00, 95% CI ‐10.80 to 12.80; participants = 38; trials = 1). None of the trials reported health‐related quality of life beyond one year.
Liver transplantation
None of the trials reported liver transplantation.
Decompensated liver disease
None of the trials reported decompensated liver disease.
Cirrhosis
None of the trials reported cirrhosis.
Hepatocellular carcinoma
None of the trials reported hepatocellular carcinoma.
Diabetes
None of the trials reported diabetes.
Cardiovascular complications
None of the trials reported cardiovascular complications such as cardiac failure, myocardial infarction, and stroke at maximal follow‐up.
Subgroup analysis
We did not perform any of the subgroup analyses because none of the trials were at low risk of bias, the trials did not report whether the participants were symptomatic or asymptomatic, all the trials used serum ferritin level of 50 μg/L or less as the target, and the trials used similar methods of erythrocytapheresis and phlebotomy.
Trial Sequential Analysis
Only one comparison had more than one trial and was eligible for Trial Sequential Analysis. As shown in Figure 4, the accrued sample size was only a small fraction of the diversity‐adjusted required information size (DARIS); therefore, the boundaries could not be drawn. There was a high risk of random errors. The TSA‐adjusted CI could not be calculated as there was too little information to be used.
4.
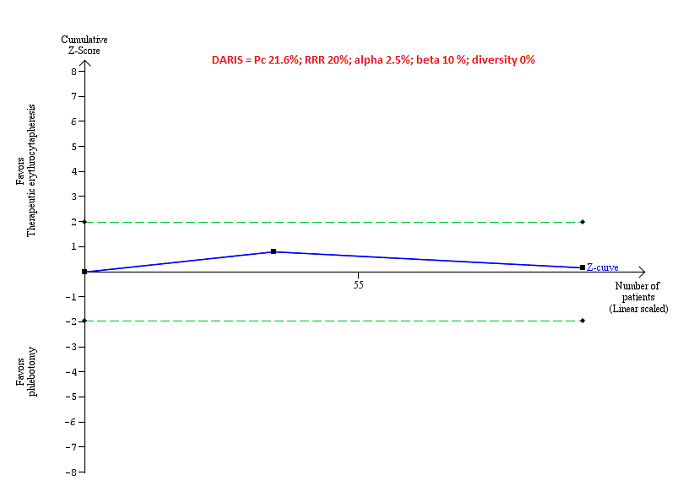
Trial Sequential Analysis of adverse events (proportion) performed using an alpha error of 2.5%, power of 90% (beta error of 10%), relative risk reduction of 20%, control group proportion (Pc) observed in trials (21.6% for proportion of people with adverse events), and observed diversity (0%) shows that the accrued sample size was only a small fraction of the diversity‐adjusted required information size (DARIS) that the boundaries could not be drawn. The Z‐curve (blue line) does not cross the conventional boundaries (dotted green line). There was a high risk of random errors.
Reporting bias
We did not explore reporting bias using a funnel plot because of few trials included in the review.
Quality of evidence
All the evidence available was downgraded to very low quality of evidence because of the risk of bias in the trials (downgraded one level for risk of bias) and imprecision (downgraded one level for small sample size and one more level for wide CIs). There was no evidence of indirectness, heterogeneity, or publication bias. So, we did not downgrade for these domains (Table 1).
Discussion
Summary of main results
In this review, we included three trials (146 participants) (Rombout‐Sestrienkova 2012; Sundic 2014; Rombout‐Sestrienkova 2016). However, one of these trials provide no information for this review (Rombout‐Sestrienkova 2016). Only one trial was included in most of the outcomes (Rombout‐Sestrienkova 2012). The only outcome of interest for this review and reported in more than one trial was proportion of people with adverse events (Rombout‐Sestrienkova 2012; Sundic 2014). There was no short‐term mortality or serious adverse events in either group in the one trial that reported these outcomes (Rombout‐Sestrienkova 2012). There were no statistically significant differences between erythrocytapheresis and phlebotomy in the proportion of people with adverse events, number of adverse events, and short‐term health‐related quality of life. None of the trials reported mortality beyond one year, health‐related quality of life beyond one year, liver transplantation, decompensated liver disease, cirrhosis, hepatocellular carcinoma, diabetes, or cardiovascular complications in the long‐term. In summary, there was no evidence of a difference between erythrocytapheresis and phlebotomy in people with hereditary haemochromatosis.
Overall completeness and applicability of evidence
We planned to include all treatments used for hereditary haemochromatosis but found that the only comparison reported was erythrocytapheresis versus phlebotomy. The current recommended treatment for hereditary haemochromatosis is phlebotomy and the trials were conducted relatively recently. Therefore, the findings of this review are applicable in the current clinical setting. The trials did not report whether the participants were symptomatic or asymptomatic. They probably included participants who required blood letting of some form regardless of symptoms based on the ferritin and transferrin levels. Therefore, the findings of the review are likely to be applicable in symptomatic and asymptomatic people. Both the trials that contributed data for this review included only treatment‐naive people, that is, people who had not received previous treatments for hereditary haemochromatosis. Therefore, the findings of the review are applicable only in people with hereditary haemochromatosis who had not received treatment previously. Finally, the trial that provided most of the information for this review excluded people with malignancy, serious cardiac arrhythmias, heart failure, and epilepsy. Therefore, the findings of this review are not applicable in such people.
Phlebotomy requires minimal equipment while erythrocytapheresis requires special equipment to perform the procedure. Since there is no evidence to suggest that erythrocytapheresis is beneficial versus phlebotomy, there is no need for hospitals to buy special equipment, based on currently available evidence.
Quality of the evidence
The overall quality of evidence was very low for all the outcomes. All the trials were at high risk of bias, mainly because blinding of participants and healthcare providers was not performed in any of the trials. The sample size was small for all the comparisons. The only outcome in which Trial Sequential Analysis was attempted showed that the sample size was less than 5% of the required information size to identify a relative risk reduction of 20%. There were also wide CIs for all the comparisons.
Potential biases in the review process
We followed the guidance of the Cochrane Handbook for Systematic Reviews of Interventions with two review authors independently selecting studies and extracting data. We performed a thorough search of literature. However, the search period included the pre‐mandatory trial registration era and it is possible that we missed some trials on treatments that were not effective or were harmful or were not reported at all.
We only included randomised clinical trials which are known to focus mostly on benefits and do not collect and report harms in a detailed manner. According to our choice of studies (i.e. only randomised clinical trials), we might have missed a large number of studies that address reporting of harms. Accordingly, this review is biased towards benefits ignoring harms. We did not search for interventions and trials registered at regulatory authorities (e.g., FDA (US Food and Drug Administration); EMA (European Medicines Agency), etc). This may have overlooked trials and as such trials usually are unpublished, the lack of inclusion of such trials may make our comparisons look more advantageous than they really are.
Agreements and disagreements with other studies or reviews
There have been no previous systematic reviews on this topic. The authors of the trial that provided most information for this review concluded that erythrocytapheresis is a highly effective treatment to reduce iron overload, and that from a societal perspective, it might potentially also be a cost‐saving therapy (Rombout‐Sestrienkova 2012). Our findings show that it is too early to know whether erythrocytapheresis is beneficial or harmful compared with phlebotomy. While we did not collect the cost information for this review, we noted that the costs of purchasing the equipment and maintenance of the equipment was not included in the cost calculations. This is likely to alter the conclusions about the difference between treatment costs. It is also not clear whether blinding of participants was performed. Lack of blinding of participants to treatment may cause biased estimate of the costs related to productivity loss. Therefore, the existing evidence did not allow us to support the trial authors' conclusions.
Authors' conclusions
Implications for practice.
There is currently insufficient evidence to determine whether erythrocytapheresis is beneficial or harmful compared with phlebotomy. Phlebotomy has less equipment requirements and remains the treatment of choice in people with hereditary haemochromatosis who require blood letting in some form. However, it should be noted that there is no evidence from randomised clinical trials that blood letting in any form is beneficial in people with hereditary haemochromatosis. Having said this, a trial including no treatment is unlikely to be conducted in future.
Implications for research.
Future trials should compare different frequencies of phlebotomy and erythrocytapheresis versus phlebotomy with or without iron‐chelating agents compared with each other and with placebo. Such trials should include long‐term follow‐up of participants (e.g. using national record linkage databases) to determine whether treatments are beneficial or harmful in terms of clinical outcomes such as deaths, health‐related quality of life, liver damage and its consequences, heart damage and its consequences, and other outcomes that are of importance to people with hereditary haemochromatosis. The trials should be designed using guidance from SPIRIT statement (Standard Protocol Items: Recommendations for Interventional Trials; Chan 2013) and reported according to the CONSORT statement (Schulz 2010).
What's new
| Date | Event | Description |
|---|---|---|
| 12 April 2017 | Amended | The Cochrane Central Editorial Unit requested removal of the 'attempted network meta‐analysis' phrase from the end of the review title, as this further description of the review might create confusion in the reader. Although we followed the planned methodology for network meta‐analysis, we found only one comparison. Therefore, we did not perform the network meta‐analysis and assessed the comparative benefits and harms of different interventions using standard Cochrane methodology. |
Notes
There was considerable overlap between the 'Methods' of this review and those of several other reviews written by the same group of authors.
Acknowledgements
We thank the Cochrane Comparing of Multiple Interventions Methods Group and the Cochrane Hepato‐Biliary Group for their support and advice. We also thank the copy editors who improved the clarity of the review.
Peer reviewers of the review: Barbara de Graaff, Australia; Dario Conte, Italy; Ronald Koretz, USA; Theis Lange, Denmark.
Contact editor: Janus Christian Jakobsen, Denmark. Sign‐off editor: Christian Gluud, Denmark.
Cochrane Review Group funding acknowledgement: the Danish State is the largest single funder of The Cochrane Hepato‐Biliary Group through its investment in The Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Copenhagen University Hospital, Denmark.
Disclaimer: the views and opinions expressed in this review are those of the review authors and do not necessarily reflect those of the Danish State or The Copenhagen Trial Unit.
Appendices
Appendix 1. Methods for network meta‐analysis if we find this is possible in the future
Measures of treatment effect
Relative treatment effects
For dichotomous variables (e.g. proportion of participants with serious adverse events or any adverse events), we will calculate the odds ratio with 95% credible interval (or Bayesian confidence interval) (Severini 1993). For continuous variables (e.g. quality of life reported on the same scale), we will calculate the mean difference with 95% credible interval. We will use standardised mean difference values with 95% credible interval for quality of life if included trials use different scales. For count outcomes (e.g. number of adverse events and serious adverse events), we will calculate the rate ratio with 95% credible interval. For time‐to‐event data (e.g. mortality at maximal follow‐up), we will calculate hazard ratio with 95% credible interval.
Relative ranking
We will estimate the ranking probabilities for all treatments of being at each possible rank for each intervention. Then, we will obtain the surface under the cumulative ranking curve (SUCRA) (cumulative probability) and rankogram (Salanti 2011; Chaimani 2013).
Unit of analysis issues
We will collect data for all trial treatment groups that meet the inclusion criteria. The codes for analysis, that we will use, accounts for the correlation between the effect sizes from trials with more than two groups.
Assessment of heterogeneity
We will assess clinical and methodological heterogeneity by carefully examining the characteristics and design of included trials. We will assess the presence of clinical heterogeneity by comparing effect estimates under different categories of potential effect modifiers. Different study designs and risk of bias may contribute to methodological heterogeneity.
We will assess the statistical heterogeneity by comparing the results of the fixed‐effect model meta‐analysis and the random‐effects model meta‐analysis, between‐study standard deviation (tau2 and comparing this with values reported in the study of the distribution of between‐study heterogeneity (Turner 2012)), and by calculating I2 (using Stata/SE 14.2). If we identify substantial heterogeneity, clinical, methodological, or statistical, we will explore and address heterogeneity in a subgroup analysis (see ‘Subgroup analysis and investigation of heterogeneity for network meta‐analysis’ section).
Assessment of transitivity across treatment comparisons
We will evaluate the plausibility of transitivity assumption (the assumption that the participants included in the different studies with different immunosuppressive regimens can be considered to be a part of a multi‐arm randomised clinical trial and could potentially have been randomised to any of the treatments) (Salanti 2012). In other words, any participant that meets the inclusion criteria is, in principle, equally likely to be randomised to any of the above eligible interventions. If there is any concern that the clinical safety and effectiveness are dependent upon the effect modifiers, we will continue to do traditional Cochrane pairwise comparisons and we will not perform a network meta‐analysis on all participant subgroups.
Assessment of reporting biases
For the network meta‐analysis, we will judge the reporting bias by the completeness of the search (i.e. searching various databases and including conference abstracts), as we do not currently find any meaningful order to perform a comparison‐adjusted funnel plot as suggested by Chaimani 2012. However, if we find any meaningful order, for example, the control group used depended upon the year of conduct of the trial, we will use comparison‐adjusted funnel plot as suggested by Chaimani 2012.
Data synthesis
Methods for indirect and mixed comparisons
We will conduct network meta‐analyses to compare multiple interventions simultaneously for each of the primary and secondary outcomes. Network meta‐analysis combines direct evidence within trials and indirect evidence across trials (Mills 2012). We will obtain a network plot to ensure that the trials were connected by treatments using Stata/SE 14.2 (Chaimani 2013). We will exclude any trials that were not connected to the network. We will conduct a Bayesian network meta‐analysis using the Markov chain Monte Carlo method in OpenBUGS 3.2.3 as per the guidance from the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) documents (Dias 2014a). We will model the treatment contrast (i.e. log odds ratio for binary outcomes, mean difference or standardised mean difference for continuous outcomes, log rate ratio for count outcomes, and log hazard ratio for time‐to‐event outcomes) for any two interventions ('functional parameters') as a function of comparisons between each individual intervention and an arbitrarily selected reference group ('basic parameters') (Lu 2006) using appropriate likelihood functions and links. We will use binomial likelihood and logit link for binary outcomes, Poisson likelihood and log link for count outcomes, binomial likelihood and complementary log‐log link for time‐to‐event outcomes, and normal likelihood and identity link for continuous outcomes. We will perform a fixed‐effect model and random‐effects model for the network meta‐analysis. We will report both models for comparison with the reference group in a forest plot. For pairwise comparison, we will report the fixed‐effect model if the two models reported similar results; otherwise, we will report the more conservative model.
We will use a hierarchical Bayesian model using three different initial values using codes provided by NICE DSU (Dias 2014a). We will use a normal distribution with large variance (10,000) for treatment effect priors (vague or flat priors). For the random‐effects model, we will use a prior distributed uniformly (limits: 0 to 5) for between‐trial standard deviation but assumed similar between‐trial standard deviation across treatment comparisons (Dias 2014a). We will use a 'burn‐in' of 5000 simulations, check for convergence visually, and run the models for another 10,000 simulations to obtain effect estimates. If we did not obtain convergence, we will increase the number of simulations for 'burn‐in'. If we do not obtain convergence still, we will use alternate initial values and priors using methods suggested by van Valkenhoef 2012. We will also estimate the probability that each intervention ranks at one of the possible positions using the NICE DSU codes (Dias 2014a).
Assessment of inconsistency
We will assess inconsistency (statistical evidence of the violation of transitivity assumption) by fitting both an inconsistency model and a consistency model. We will use the inconsistency models used in the NICE DSU manual, as we plan to use a common between‐study deviation for the comparisons (Dias 2014b). In addition, we will use the design‐by‐treatment full interaction model (Higgins 2012) and IF (inconsistency factor) plots (Chaimani 2013) to assess inconsistency. In the presence of inconsistency, we will assess whether the inconsistency is because of clinical or methodological heterogeneity by performing separate analyses for each of the different subgroups mentioned in the ‘Subgroup analysis and investigation of heterogeneity for network meta‐analysis’ section below.
If there is evidence of inconsistency, we will identify areas in the network where substantial inconsistency might be present in terms of clinical and methodological diversities between trials and, when appropriate, limit network meta‐analysis to a more compatible subset of trials.
Direct comparison
We will perform the direct comparisons using the same codes and the same technical details.
Sample size calculations
To control for the risk of random errors, we will interpret the information with caution when the accrued sample size in the network meta‐analysis (i.e. across all treatment comparisons) was less than the required sample size (required information size). For calculation of the required information size, see Appendix 3.
Subgroup analysis and investigation of heterogeneity for network meta‐analysis
We will assess the differences in the effect estimates between the subgroups listed in Subgroup analysis and investigation of heterogeneity using meta‐regression with the help of the OpenBUGS code (Dias 2012a) if we include a sufficient number of trials. We will use the potential modifiers as study level co‐variates for meta‐regression. We will calculate a single common interaction term (Dias 2012a). If the 95% credible intervals of the interaction term do not overlap zero, we will consider this as evidence of difference in subgroups.
Presentation of results
We will present the effect estimates with 95% CrI for each pairwise comparisons calculated from the direct comparisons and network meta‐analysis. We will also present the cumulative probability of the treatment ranks (i.e. the probability that the treatment is within the top two, the probability that the treatment is within the top three, etc.) in graphs (surface under the cumulative ranking curve or SUCRA) (Salanti 2011). We will also plot the probability that each treatment is best, second best, third best etc for each of the different outcomes (rankograms), which are generally considered more informative (Salanti 2011; Dias 2012b).
We will present the 'Summary of findings' tables for mortality. In the 'Table 1', we will follow the approach suggested by Puhan et al. (Puhan 2014). First, we will calculate the direct and indirect effect estimates and 95% credible intervals using the node‐splitting approach (Dias 2010), i.e. calculate the direct estimate for each comparison by including only trials in which there was direct comparison of treatments and the indirect estimate for each comparison by excluding the trials in which there was direct comparison of treatments. Then we will rate the quality of direct and indirect effect estimates using GRADE which takes into account the risk of bias, inconsistency, directness of evidence, imprecision, and publication bias (Guyatt 2011). Then, we will present the estimates of the network meta‐analysis and rate the quality of network meta‐analysis effect estimates as the best quality of evidence between the direct and indirect estimates (Puhan 2014). In addition, in the same table, we will present illustrations and information on the number of trials and participants as per the standard 'Summary of Findings' Table.
Appendix 2. Search strategies
| Database | Time span | Search strategy |
| The Central Register of Controlled Trials (CENTRAL) (Wiley) | 2016, Issue 3 | #1 MeSH descriptor: [Hemochromatosis] explode all trees #2 (hemochromatos* or hemochromatos* or iron overload or ironoverload) #3 #1 or #2 |
| MEDLINE (OvidSP) | January 1947 to March 2016 | 1. exp Hemochromatosis/ 2. (hemochromatos* or hemochromatos* or iron overload or ironoverload).ti,ab. 3. 1 or 2 4. randomized controlled trial.pt. 5. controlled clinical trial.pt. 6. randomized.ab. 7. placebo.ab. 8. drug therapy.fs. 9. randomly.ab. 10. trial.ab. 11. groups.ab. 12. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 13. exp animals/ not humans.sh. 14. 12 not 13 15. 3 and 14 |
| Embase (OvidSP) | January 1974 to March 2016 | 1. exp hemochromatosis/ 2. (hemochromatos* or hemochromatos* or iron overload or ironoverload).ti,ab. 3. 1 or 2 4. exp crossover‐procedure/ or exp double‐blind procedure/ or exp randomized controlled trial/ or single‐blind procedure/ 5. (((((random* or factorial* or crossover* or cross over* or cross‐over* or placebo* or double*) adj blind*) or single*) adj blind*) or assign* or allocat* or volunteer*).af. 6. 4 or 5 7. 3 and 6 |
| Science Citation Index Expanded (Web of Knowledge) | January 1945 to March 2016 | #1 TS=(hemochromatos* or hemochromatos* or iron overload or ironoverload) #2 TS=(random* OR rct* OR crossover OR masked OR blind* OR placebo* OR meta‐analysis OR systematic review* OR meta‐analys*) #3 #1 AND #2 |
| World Health Organization International Clinical Trials Registry Platform Search Portal (apps.who.int/trialsearch/Default.aspx) | March 2016 | Condition: hemochromatos* or hemochromatos* or iron overload or ironoverload |
| ClinicalTrials.gov | March 2016 | Interventional Studies | (hemochromatos* OR hemochromatos* OR iron overload OR ironoverload) | Phase 2, 3, 4 |
Appendix 3. Sample size calculation
The five‐year mortality in people with hereditary haemochromatosis is 5% (Wojcik 2002). The required information size based on a control group proportion of 20%, a relative risk reduction of 20% in the intervention group, type I error of 5%, and type II error of 20% is 13,492 participants. Network analyses are more prone to the risk of random errors than direct comparisons (Del Re 2013). Accordingly, a greater sample size is required in indirect comparisons than direct comparisons (Thorlund 2012). The power and precision in indirect comparisons depends upon various factors, such as the number of participants included under each comparison and the heterogeneity between the trials (Thorlund 2012). If there is no heterogeneity across the trials, the sample size in indirect comparisons would be equivalent to the sample size in direct comparisons. The effective indirect sample size can be calculated using the number of participants included in each direct comparison (Thorlund 2012). For example, a sample size of 2500 participants in the direct comparison A versus C (nAC) and a sample size of 7500 participants in the direct comparison B versus C (nBC) results in an effective indirect sample size of 1876 participants. However, in the presence of heterogeneity within the comparisons, the sample size required is higher. In the above scenario, for an I2 statistic for each of the comparisons A versus C (IAC2) and B versus C (IBC2) of 25%, the effective indirect sample size is 1407 participants. For an I2 statistic for each of the comparisons A versus C and B versus C of 50%, the effective indirect sample size is 938 participants (Thorlund 2012). If there were only three groups and the sample size in the trials is more than the required information size, we will calculate the effective indirect sample size using the following generic formula (Thorlund 2012):
((nAC x (1 ‐ IAC2)) x (nBC x (1 ‐ IBC2))/((nAC x (1 ‐ IAC2)) + (nBC x (1 ‐ IBC2)).
There is currently no method to calculate the effective indirect sample size for a network analysis involving more than three intervention groups.
Data and analyses
Comparison 1. Therapeutic erythrocytapheresis versus phlebotomy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Adverse events (proportion) | 2 | 100 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.36, 2.43] |
| 2 Adverse events (number) | 1 | Rate Ratio (Fixed, 95% CI) | Totals not selected | |
| 3 Health‐related quality of life (EQ‐VAS) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected |
1.1. Analysis.
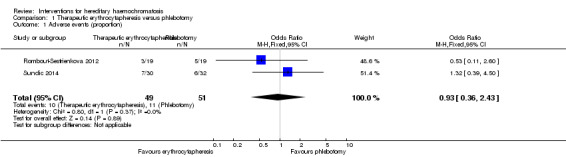
Comparison 1 Therapeutic erythrocytapheresis versus phlebotomy, Outcome 1 Adverse events (proportion).
1.2. Analysis.
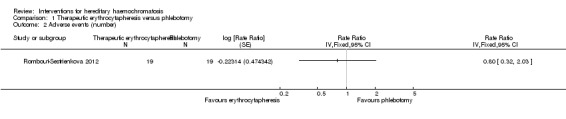
Comparison 1 Therapeutic erythrocytapheresis versus phlebotomy, Outcome 2 Adverse events (number).
1.3. Analysis.
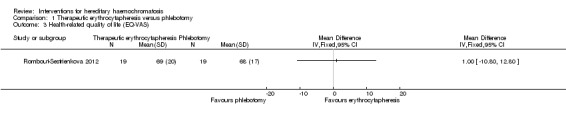
Comparison 1 Therapeutic erythrocytapheresis versus phlebotomy, Outcome 3 Health‐related quality of life (EQ‐VAS).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Rombout‐Sestrienkova 2012.
| Methods | Randomised clinical trial. | |
| Participants | Country: Netherlands. Number randomised: 38. Post‐randomisation dropouts: 0 (0%). Revised sample size: 38. Mean age: 52 years. Number of women: 10 (26.3%). Symptomatic: not stated. Asymptomatic: not stated. Mean follow‐up period: 8 months. Target used for iron reduction: serum ferritin ≤ 50 μg/L. Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: therapeutic erythrocytapheresis (n = 19). Further details: 350 mL to 800 mL of red blood cells once every 2 weeks. Group 2: phlebotomy (n = 19). Further details: 500 mL of whole blood once weekly. Treatment duration: variable depending upon the iron to be removed. |
|
| Outcomes | Mortality, adverse events, and health‐related quality of life. | |
| Notes | We attempted to contact the corresponding author to obtain additional information on risk of bias and outcomes in June 2015. We did not receive any reply. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned in a 1:1 ratio by an independent person working as quality assurance manager". |
| Allocation concealment (selection bias) | Low risk | Quote: "patients were randomly assigned in a 1:1 ratio by an independent person working as quality assurance manager". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "single‐blind". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote: "single‐blind". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: there were no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: all important outcomes were reported. |
| For‐profit bias | Low risk | Quote: "This work was performed with the support of the Sanquin Blood Bank grants 03‐006". |
| Other bias | Low risk | Comment: no other risk of bias. |
Rombout‐Sestrienkova 2016.
| Methods | Randomised clinical trial. | |
| Participants | Country: Netherlands. Number randomised: 53. Post‐randomisation dropouts: 7 (13.2%). Revised sample size: 46. Mean age: 55 years. Number of women: not stated. Symptomatic: not stated. Asymptomatic: not stated. Mean follow‐up period: 1 year (after this there was cross‐over). Target used for iron reduction: serum ferritin ≤ 50 μg/L. Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: therapeutic erythrocytapheresis (n = 20). Further details: 350 mL to 800 mL red blood cells; variable frequency depending on serum ferritin level. Group 2: phlebotomy (n = 26). Further details: 500 mL per single treatment; variable frequency depending on serum ferritin level. Treatment duration: 1 year. |
|
| Outcomes | None of our outcomes of interest were reported at the end of the first treatment. | |
| Notes | Reasons for post‐randomisation dropouts: not stated clearly. We attempted to contact the corresponding author in June 2015 to obtain additional information on risk of bias and outcomes. We did not receive any reply. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: this information was not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: this information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Comment: probably not blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Comment: probably not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: important outcomes such as mortality and complications were not reported. |
| For‐profit bias | Unclear risk | Comment: this information was not available. |
| Other bias | Low risk | Comment: no other risk of bias. |
Sundic 2014.
| Methods | Randomised clinical trial. | |
| Participants | Country: Norway. Number randomised: 62. Post‐randomisation dropouts: 0 (0%). Revised sample size: 62. Mean age: 42 years. Number of women: 6 (9.7%). Symptomatic: not stated. Asymptomatic: not stated. Mean follow‐up period (for all groups): 3 months. Target used for iron reduction: serum ferritin ≤ 50 μg/L. Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: therapeutic erythrocytapheresis (n = 30). Further details: 400 mL per single treatment bi‐weekly*. Group 2: phlebotomy (n = 32). Further details: 450 mL per single treatment weekly. Treatment duration: 12 weeks. |
|
| Outcomes | Adverse events. | |
| Notes | We attempted to contact the corresponding author in June 2015 to obtain additional information on risk of bias and outcomes. We did not receive any reply. * It was unclear whether the authors meant 'bi‐weekly' to be once every two weeks or twice weekly. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation procedure was centralised to one of the participating centres, using a randomly generated sequence". |
| Allocation concealment (selection bias) | Unclear risk | Comment: this information was not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "different treatment". |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: this information was not available. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: there were 6 dropouts/withdrawal but data regarding tolerance reported. |
| Selective reporting (reporting bias) | High risk | Comment: no data about mortality but just tolerability. |
| For‐profit bias | Low risk | Quote: "Grants from Helse Vest RHF and Helse Fonna HF (public hospital trusts)". |
| Other bias | Low risk | Comment: no other risk of bias. |
Characteristics of ongoing studies [ordered by study ID]
Ong 2015.
| Trial name or title | Mi‐iron |
| Methods | Randomised clinical trial |
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions | Erythrocytapheresis versus plasmapheresis |
| Outcomes | Modified Fatigue Impact Scale Medical Outcomes Study 36‐item short form V.2 Hospital Anxiety and Depression Scale Arthritis Impact Measurement Scales 2 short form |
| Starting date | June 2012 |
| Contact information | Martin Delatycki (martin.delatycki@ghsv.org.au) |
| Notes |
HFE = human haemochromatosis protein.
Differences between protocol and review
There was only one comparison. So we did not perform the network meta‐analysis, and assessed the comparative benefits and harms of different interventions using standard Cochrane methodology. The methodology that we plan to use if we conduct a network meta‐analysis in future is available in Appendix 1.
We performed Trial Sequential Analysis in addition to conventional method of assessing the risk of random errors using P‐value.
Contributions of authors
EB and MK extracted data and provided them in a format that could be analysed. KG wrote the review. ET, DT, and BD critically commented on the review. All review authors approved this version before publication.
Sources of support
Internal sources
University College London, UK.
External sources
-
National Institute for Health Research, UK.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure, Cochrane Programme Grant, or Cochrane Incentive funding to the Cochrane Hepato‐Biliary and Upper Gastrointestinal and Pancreatic Diseases Groups. The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, the National Health Service, or the Department of Health. The funding covered salary, equipment, and other resources to complete this review.
-
Elena Buzzetti, Italy.
Dr Elena Buzzetti was supported by an educational grant from AIGO (Associazione Italiana Gastroenterologi Ospedalieri) and M.I.M.I (Montecatini Interactive Medicine International).
Declarations of interest
This report is independent research funded by the National Institute for Health Research (NIHR Cochrane Programme Grants, 13/89/03 ‐ Evidence‐based diagnosis and management of upper digestive, hepato‐biliary, and pancreatic disorders). The views expressed in this publication are those of the review authors and not necessarily those of the National Health Service (NHS), the NIHR, or the Department of Health.
EB: has no financial disclosures. MK: has no financial disclosures. DT: funded by Astellas for his attendance at the International Liver Transplantation Society meeting in 2014; received GBP 25,000 from Boston Scientific to fund a clinical research fellow in 2013. BD: has no financial disclosures. ET: has no financial disclosures. KG: has no financial disclosures.
Edited (no change to conclusions)
References
References to studies included in this review
Rombout‐Sestrienkova 2012 {published data only}
- Rombout‐Sestrienkova E, Nieman FH, Essers BA, Noord PA, Janssen MC, Deursen CT, et al. Erythrocytapheresis versus phlebotomy in the initial treatment of HFE hemochromatosis patients: results from a randomized trial. Transfusion 2012;52(3):470‐7. [DOI] [PubMed] [Google Scholar]
- Rombout‐Sestrienkova E, Noord P, Reuser E, Heeremans J, Deursen C, Janssen M, et al. Therapeutic erythrocytapheresis (TE) versus phlebotomy (P) in the treatment of hereditary hemochromatosis (HH) patients: preliminary results from an ongoing randomized clinical trial (NCT 00202436). Transfusion and Apheresis Science 2009;40(2):135‐6. [Google Scholar]
Rombout‐Sestrienkova 2016 {published data only}
- Rombout‐Sestrienkova E, Winkens B, Essers BA, Nieman FH, Noord PA, Janssen MC, et al. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE hemochromatosis patients: results from a randomized crossover trial. Transfusion 2016;56(1):261‐70. [DOI] [PubMed] [Google Scholar]
Sundic 2014 {published data only}
- Sundic T, Hervig T, Hannisdal S, Assmus J, Ulvik RJ, Olaussen RW, et al. Erythrocytapheresis compared with whole blood phlebotomy for the treatment of hereditary haemochromatosis. Blood Transfusion 2014;12(Suppl 1):S84‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
Ong 2015 {published data only}
- Ong SY, Dolling L, Dixon JL, Nicoll AJ, Gurrin LC, Wolthuizen M, et al. Should HFE p.C282y homozygotes with moderately elevated serum ferritin be treated? A randomised controlled trial comparing iron reduction with sham treatment (Mi‐iron). BMJ Open 2015;5(8):e008938. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Adams 2007
- Adams PC, Barton JC. Haemochromatosis. Lancet 2007;370(9602):1855‐60. [DOI] [PubMed] [Google Scholar]
Allen 2008
- Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, et al. Iron‐overload‐related disease in HFE hereditary hemochromatosis. New England Journal of Medicine 2008;358(3):221‐30. [DOI] [PubMed] [Google Scholar]
Assi 2014
- Assi TB, Baz E. Current applications of therapeutic phlebotomy. Blood Transfusion 2014;12(Suppl 1):s75‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bacon 2011
- Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS, American Association for the Study of Liver Diseases. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology (Baltimore, Md.) 2011;54(1):328‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bardou‐Jacquet 2014
- Bardou‐Jacquet E, Brissot P. Diagnostic evaluation of hereditary hemochromatosis (HFE and non‐HFE). Hematology/Oncology Clinics of North America 2014;28(4):625‐35. [DOI] [PubMed] [Google Scholar]
Beutler 2002
- Beutler E, Felitti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G → A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002;359(9302):211‐8. [DOI] [PubMed] [Google Scholar]
Bradley 1996
- Bradley LA, Haddow JE, Palomaki GE. Population screening for haemochromatosis: a unifying analysis of published intervention trials. Journal of Medical Screening 1996;3(4):178‐84. [DOI] [PubMed] [Google Scholar]
Cancado 2015
- Cancado R, Melo MR, Moraes Bastos R, Santos PC, Guerra‐Shinohara EM, Ballas SK, et al. Deferasirox in patients with iron overload secondary to hereditary hemochromatosis: results of a 1‐year Phase 2 study. European Journal of Haematology 2015;95(6):545‐50. [DOI] [PubMed] [Google Scholar]
Cassanelli 2001
- Cassanelli S, Pignatti E, Montosi G, Garuti C, Mariano M, Campioli D, et al. Frequency and biochemical expression of C282Y/H63D hemochromatosis (HFE) gene mutations in the healthy adult population in Italy. Journal of Hepatology 2001;34(4):523‐8. [DOI] [PubMed] [Google Scholar]
Chaimani 2012
- Chaimani A, Salanti G. Using network meta‐analysis to evaluate the existence of small‐study effects in a network of interventions. Research Synthesis Methods 2012;3(2):161‐76. [DOI] [PubMed] [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta‐analysis in STATA. PloS one 2013;8(10):e76654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chan 2013
- Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza‐Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Annals of Internal Medicine 2013;158(3):200‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
de Graaff 2015
- Graaff B, Neil A, Sanderson K, Si L, Yee KC, Palmer AJ. A systematic review and narrative synthesis of health economic studies conducted for hereditary haemochromatosis. Applied Health Economics and Health Policy 2015;13(5):469‐83. [DOI] [PubMed] [Google Scholar]
Del Re 2013
- Re AC, Spielmans GI, Flückiger C, Wampold BE. Efficacy of new generation antidepressants: differences seem illusory. PLoS One 2013;8(6):e63509. [DOI] [PMC free article] [PubMed] [Google Scholar]
DeMets 1987
- DeMets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Dias 2010
- Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta‐analysis. Statistics in Medicine 2010;29(7‐8):932‐44. [DOI] [PubMed] [Google Scholar]
Dias 2012a
- Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 3: heterogeneity: subgroups, meta‐regression, bias and bias‐adjustment, September 2011 (last updated April 2012). www.nicedsu.org.uk/TSD3%20Heterogeneity.final%20report.08.05.12.pdf (accessed 27 March 2014).
Dias 2012b
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 1: introduction to evidence synthesis for decision making, April 2011 (last updated April 2012). www.nicedsu.org.uk/TSD1%20Introduction.final.08.05.12.pdf (accessed 27 March 2014).
Dias 2014a
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: a generalised linear modelling framework for pairwise and network meta‐analysis of randomised controlled trials, August 2011 (last updated April 2014). www.nicedsu.org.uk/TSD2%20General%20meta%20analysis%20corrected%2015April2014.pdf (accessed 8 October 2014). [PubMed]
Dias 2014b
- Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: inconsistency in networks of evidence based on randomised controlled trials, May 2011 (last updated April 2014). www.nicedsu.org.uk/TSD4%20Inconsistency.final.15April2014.pdf (accessed 8 October 2014). [PubMed]
EASL 2010
- European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. Journal of Hepatology 2010;53(1):3‐22. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ (Clinical Research Ed.) 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ellervik 2007
- Ellervik C, Birgens H, Tybjaerg‐Hansen A, Nordestgaard BG. Hemochromatosis genotypes and risk of 31 disease endpoints: meta‐analyses including 66,000 cases and 226,000 controls. Hepatology 2007;46(4):1071‐80. [DOI] [PubMed] [Google Scholar]
EuroQol 2014
- EuroQol. About EQ‐5D, 2014. www.euroqol.org/about‐eq‐5d.html (accessed 8 October 2014).
Feder 1996
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I‐like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics 1996;13(4):399‐408. [DOI] [PubMed] [Google Scholar]
Gluud 2015
- Gluud C, Nikolova D, Klingenberg SL. Cochrane Hepato‐Biliary Group. About Cochrane (Cochrane Review Groups (CRGs)). 2015, Issue 11. Art. No.: LIVER.
Guyatt 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction ‐ GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.handbook.cochrane.org.
Higgins 2012
- Higgins JPT, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta‐analysis: Concepts and models for multi‐arm studies. Research Synthesis Methods 2012;3(2):98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice CFR & ICH Guidelines. Vol. 1, Pennsylvania: Barnett International/PAREXEL, 1997. [Google Scholar]
Jakobsen 2014
- Jakobsen JC, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta‐analytic methods. BMC Medical Research Methodology 2014;14(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
Lu 2006
- Lu G, Ades AE. Assessing evidence inconsistency in mixed treatment comparisons. Journal of the American Statistical Association 2006;101(474):447‐59. [Google Scholar]
Lucotte 2003
- Lucotte G, Dieterlen F. A European allele map of the C282Y mutation of hemochromatosis: Celtic versus Viking origin of the mutation?. Blood, Cells, Molecules & Diseases 2003;31(2):262‐7. [DOI] [PubMed] [Google Scholar]
Lundh 2017
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Macaskill 2001
- Macaskill P, Walter SD, Irwig L. A comparison of methods to detect publication bias in meta‐analysis. Statistics in Medicine 2001;20(4):641‐54. [DOI] [PubMed] [Google Scholar]
Martindale 2011
- Sweetman S, editor. Martindale: the complete drug reference (online version), 37th edition, 2011. www.pharmpress.com/product/MC_MART/martindale‐the‐complete‐drug‐reference (accessed 23 September 2014).
Mercier 1998
- Mercier G, Bathelier C, Lucotte G. Frequency of the C282Y mutation of hemochromatosis in five French populations. Blood, Cells, Molecules & Diseases 1998;24(2):165‐6. [DOI] [PubMed] [Google Scholar]
Mills 2012
- Mills EJ, Ioannidis JP, Thorlund K, Schünemann HJ, Puhan MA, Guyatt GH. How to use an article reporting a multiple treatment comparison meta‐analysis. JAMA 2012;308(12):1246‐53. [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
Newell 1992
- Newell DJ. Intention‐to‐treat analysis: implications for quantitative and qualitative research. International Journal of Epidemiology 1992;21(5):837‐41. [DOI] [PubMed] [Google Scholar]
OpenBUGS 3.2.3 [Computer program]
- Members of OpenBUGS Project Management Group. OpenBUGS. Version 3.2.3. Members of OpenBUGS Project Management Group, 2014.
Pennell 2014
- Pennell DJ, Porter JB, Piga A, Lai Y, El‐Beshlawy A, Belhoul KM, et al. A 1‐year randomized controlled trial of deferasirox vs deferoxamine for myocardial iron removal in beta‐thalassemia major (CORDELIA). Blood 2014;123(10):1447‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pietrangelo 2004
- Pietrangelo A. Hereditary hemochromatosis ‐ a new look at an old disease. New England Journal of Medicine 2004;350(23):2383‐97. [DOI] [PubMed] [Google Scholar]
Puhan 2014
- Puhan MA, Schünemann HJ, Murad MH, Li T, Brignardello‐Petersen R, Singh JA, et al. A GRADE Working Group approach for rating the quality of treatment effect estimates from network meta‐analysis. BMJ (Clinical Research Ed.) 2014;349:g5630. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Ropero 2006
- Ropero P, Briceno O, Mateo M, Polo M, Mora A, Gonzalez FA, et al. Frequency of the C282Y and H63D mutations of the hemochromatosis gene (HFE) in a cohort of 1,000 neonates in Madrid (Spain). Annals of Hematology 2006;85(5):323‐6. [DOI] [PubMed] [Google Scholar]
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Salanti 2011
- Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple‐treatment meta‐analysis: an overview and tutorial. Journal of Clinical Epidemiology 2011;64(2):163‐71. [DOI] [PubMed] [Google Scholar]
Salanti 2012
- Salanti G. Indirect and mixed‐treatment comparison, network, or multiple‐treatments meta‐analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Research Synthesis Methods 2012;3(2):80‐97. [DOI] [PubMed] [Google Scholar]
Savović 2012a
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials: combined analysis of meta‐epidemiological studies. Health Technology Assessment 2012;16(35):1‐82. [DOI] [PubMed] [Google Scholar]
Savović 2012b
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [DOI] [PubMed] [Google Scholar]
Schmitt 2005
- Schmitt B, Golub RM, Green R. Screening primary care patients for hereditary hemochromatosis with transferrin saturation and serum ferritin level: systematic review for the American College of Physicians. Annals of Internal Medicine 2005;143(7):522‐36. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ (Clinical Research Ed.) 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Severini 1993
- Severini TA. Bayesian interval estimates which are also confidence intervals. Journal of the Royal Statistical Society. Series B (Methodological) 1993;55(2):533‐40. [Google Scholar]
Stata/SE 14.2 [Computer program]
- StataCorp LP. Stata/SE 14.2 for Windows[64‐bit x86‐64]. Version 14. College Station: StataCorp LP, 2017.
Steinberg 2001
- Steinberg KK, Cogswell ME, Chang JC, Caudill SP, McQuillan GM, Bowman BA, et al. Prevalence of C282Y and H63D mutations in the hemochromatosis (HFE) gene in the United States. JAMA 2001;285(17):2216‐22. [DOI] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for trial sequential analysis (TSA). Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, Denmark. 2011. p. 1‐115. Available from www.ctu.dk/tsa.
Thorlund 2012
- Thorlund K, Mills EJ. Sample size and power considerations in network meta‐analysis. Systematic Reviews 2012;1:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
TSA 2011 [Computer program]
- Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen. TSA version 0.9. Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, 2011.
Turner 2012
- Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JP. Predicting the extent of heterogeneity in meta‐analysis, using empirical data from the Cochrane Database of Systematic Reviews. International Journal of Epidemiology 2012;41(3):818‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
van Bokhoven 2011
- Bokhoven MA, Deursen CT, Swinkels DW. Diagnosis and management of hereditary haemochromatosis. BMJ (Clinical Research Ed.) 2011;342:c7251. [DOI] [PubMed] [Google Scholar]
van Valkenhoef 2012
- Valkenhoef G, Lu G, Brock B, Hillege H, Ades AE, Welton NJ. Automating network meta‐analysis. Research Synthesis Methods 2012;3(4):285‐99. [DOI] [PubMed] [Google Scholar]
Vichinsky 2007
- Vichinsky E, Onyekwere O, Porter J, Swerdlow P, Eckman J, Lane P, et al. A randomised comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. British Journal of Haematology 2007;136(3):501‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Voicu 2009
- Voicu PM, Cojocariu C, Petrescu‐Danila E, Covic M, Stanciu C, Rusu M. Prevalence of HFE (hemochromatosis) gene mutations C282Y and H63D in a Romanian population. Blood, Cells, Molecules & Diseases 2009;42(1):14‐5. [DOI] [PubMed] [Google Scholar]
Ware 2014
- Ware JE. SF‐36® health survey update, 2014. www.sf‐36.org/tools/sf36.shtml (accessed on 8 October 2014).
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [DOI] [PubMed] [Google Scholar]
Whitlock 2006
- Whitlock EP, Garlitz BA, Harris EL, Beil TL, Smith PR. Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Annals of Internal Medicine 2006;145(3):209‐23. [DOI] [PubMed] [Google Scholar]
Wojcik 2002
- Wojcik JP, Speechley MR, Kertesz AE, Chakrabarti S, Adams PC. Natural history of C282Y homozygotes for hemochromatosis. Canadian Journal of Gastroenterology 2002;16(5):297‐302. [DOI] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]