

Abstract
Background
Primary biliary cholangitis (previously primary biliary cirrhosis) is a chronic liver disease caused by the destruction of small intra‐hepatic bile ducts resulting in stasis of bile (cholestasis), liver fibrosis, and liver cirrhosis. The optimal pharmacological treatment of primary biliary cholangitis remains uncertain.
Objectives
To assess the comparative benefits and harms of different pharmacological interventions in the treatment of primary biliary cholangitis through a network meta‐analysis and to generate rankings of the available pharmacological interventions according to their safety and efficacy. However, it was not possible to assess whether the potential effect modifiers were similar across different comparisons. Therefore, we did not perform the network meta‐analysis and instead assessed the comparative benefits and harms of different interventions using standard Cochrane methodology.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL; 2017, Issue 2), MEDLINE, Embase, Science Citation Index Expanded, World Health Organization International Clinical Trials Registry Platform, and randomised controlled trials registers to February 2017 to identify randomised clinical trials on pharmacological interventions for primary biliary cholangitis.
Selection criteria
We included only randomised clinical trials (irrespective of language, blinding, or publication status) in participants with primary biliary cholangitis. We excluded trials which included participants who had previously undergone liver transplantation. We considered any of the various pharmacological interventions compared with each other or with placebo or no intervention.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. We calculated the odds ratio (OR) and rate ratio with 95% confidence intervals (CI) using both fixed‐effect and random‐effects models based on available‐participant analysis with Review Manager 5. We assessed risk of bias according to Cochrane, controlled risk of random errors with Trial Sequential Analysis, and assessed the quality of the evidence using GRADE.
Main results
We identified 74 trials including 5902 participants that met the inclusion criteria of this review. A total of 46 trials (4274 participants) provided information for one or more outcomes. All the trials were at high risk of bias in one or more domains. Overall, all the evidence was low or very low quality. The proportion of participants with symptoms varied from 19.9% to 100% in the trials that reported this information. The proportion of participants who were antimitochondrial antibody (AMA) positive ranged from 80.8% to 100% in the trials that reported this information. It appeared that most trials included participants who had not received previous treatments or included participants regardless of the previous treatments received. The follow‐up in the trials ranged from 1 to 96 months.
The proportion of people with mortality (maximal follow‐up) was higher in the methotrexate group versus the no intervention group (OR 8.83, 95% CI 1.01 to 76.96; 60 participants; 1 trial; low quality evidence). The proportion of people with mortality (maximal follow‐up) was lower in the azathioprine group versus the no intervention group (OR 0.56, 95% CI 0.32 to 0.98; 224 participants; 2 trials; I2 = 0%; low quality evidence). However, it has to be noted that a large proportion of participants (25%) was excluded from the trial that contributed most participants to this analysis and the results were not reliable. There was no evidence of a difference in any of the remaining comparisons. The proportion of people with serious adverse events was higher in the D‐penicillamine versus no intervention group (OR 28.77, 95% CI 1.57 to 526.67; 52 participants; 1 trial; low quality evidence). The proportion of people with serious adverse events was higher in the obeticholic acid plus ursodeoxycholic acid (UDCA) group versus the UDCA group (OR 3.58, 95% CI 1.02 to 12.51; 216 participants; 1 trial; low quality evidence). There was no evidence of a difference in any of the remaining comparisons for serious adverse events (proportion) or serious adverse events (number of events). None of the trials reported health‐related quality of life at any time point.
Funding: nine trials had no special funding or were funded by hospital or charities; 31 trials were funded by pharmaceutical companies; and 34 trials provided no information on source of funding.
Authors' conclusions
Based on very low quality evidence, there is currently no evidence that any intervention is beneficial for primary biliary cholangitis. However, the follow‐up periods in the trials were short and there is significant uncertainty in this issue. Further well‐designed randomised clinical trials are necessary. Future randomised clinical trials ought to be adequately powered; performed in people who are generally seen in the clinic rather than in highly selected participants; employ blinding; avoid post‐randomisation dropouts or planned cross‐overs; should have sufficient follow‐up period (e.g. five or 10 years or more); and use clinically important outcomes such as mortality, health‐related quality of life, cirrhosis, decompensated cirrhosis, and liver transplantation. Alternatively, very large groups of participants should be randomised to facilitate shorter trial duration.
Plain language summary
Medical treatment of primary biliary cholangitis
Background
Primary biliary cholangitis (previously called primary biliary cirrhosis) is a chronic liver disease caused by the destruction of small bile ducts within the liver (tubes that carry the bile produced by the liver) resulting in stagnation of bile (cholestasis) and liver damage and replacement of liver cells with scar tissue (liver cirrhosis). The best way to treat people with primary biliary cholangitis is unclear. We sought to resolve this issue by searching for existing trials on the topic. We included all randomised clinical trials (clinical studies where people are randomly put into one of two or more intervention groups) reported to February 2017. We included only trials in which participants with primary biliary cholangitis had not undergone liver transplantation previously. Apart from using standard Cochrane methods which allow comparison of only two treatments at a time (direct comparison), we planned to use an advanced method which allows comparison of the many different treatments that are individually compared in the trials (network meta‐analysis). However, because of the nature of the information available, we could not determine whether the network meta‐analysis results were reliable. Therefore, we used standard Cochrane methodology.
Study characteristics
We identified 74 randomised clinical trials (5902 participants). Of these, 46 randomised clinical trials (4274 participants) provided information for one or more measures (outcomes). The trials included people with primary biliary cholangitis with and without symptoms; with and without antimitochondrial antibody (AMA) (an indicator of primary biliary cholangitis) regardless of whether they received previous treatments. The average follow‐up period in the trials ranged from one month to eight years in the trials that reported this information.
Funding: nine trials receive no additional funding or were funded by parties with no vested interest in the results. Thirty‐one trials were partially or fully funded by the pharmaceutical companies that would benefit based on the results of the trial. The source of funding was not available from the remaining trials.
Quality of evidence
The overall quality of evidence was very low and all the trials were at high risk of bias, which means that there is possibility of making wrong conclusions overestimating benefits or underestimating harms of one treatment or the other because of the way that the trials were conducted.
Key results
There was no reliable evidence of decrease in the deaths between any of the interventions versus no intervention. There was no evidence of decrease in serious complications or complications of any severity between any of the treatments and no treatment. None of the trials reported health‐related quality of life (a measure of a person's satisfaction with their life and health) at any time point.
Overall, there is currently no evidence of benefit of any intervention in primary biliary cholangitis. There is significant uncertainty in this issue and further high‐quality randomised clinical trials are required.
Summary of findings
Background
Description of the condition
Primary biliary cholangitis (previously named primary biliary cirrhosis) is a chronic liver disease caused by the destruction of small intrahepatic bile ducts resulting in stasis of bile (cholestasis), liver fibrosis, and liver cirrhosis (NCBI 2014). There is global variation in the incidence and prevalence of primary biliary cholangitis with annual incidence varying from 1.6 to 3.2 per 100,000 people and prevalence varying from 5 to 38 per 100,000 people, with a trend of increasing incidence and prevalence in many countries (Metcalf 1997; Boberg 1998; Kim 2000; Sood 2004; Lazaridis 2007; Pla 2007; Rautiainen 2007; Myers 2009; Baldursdottir 2012; Boonstra 2014). It is more common in women, particularly aged 25 to 40 years (Metcalf 1997; Kim 2000; Gershwin 2005; Pla 2007; Myers 2009; Baldursdottir 2012). The mean age at diagnosis is 40 to 60 years (Kim 2000; Parikh‐Patel 2001; Gershwin 2005; Myers 2009; Baldursdottir 2012).
The aetiology of primary biliary cholangitis is unclear. The associations with primary biliary cholangitis include family history of primary biliary cholangitis, Sjögren's syndrome (autoimmune disease characterised by dry mouth and dry eyes), systemic lupus erythematosus (autoimmune connective tissue disorder), autoimmune thyroid disease, multiple sclerosis (autoimmune disorder of the central nervous system), scleroderma (autoimmune disease affecting the skin and internal organs), polymyositis (chronic inflammation of the muscles, possibly an autoimmune disease), history of cigarette smoking, history of hair dye use, and urinary tract infections (Parikh‐Patel 2001; Gershwin 2005; Lazaridis 2007; Prince 2010; Lammert 2013). People with primary biliary cholangitis have other coexisting autoimmune disorders such as rheumatoid arthritis, systemic lupus erythematosus, autoimmune thyroid disease, multiple sclerosis, scleroderma, and polymyositis (Parikh‐Patel 2001; Gershwin 2005; Prince 2010; Lammert 2013). Although the strong association between personal and family history of autoimmune diseases suggests that primary biliary cholangitis may have an autoimmune aetiology, the clustering of primary biliary cholangitis in certain areas and associations between primary biliary cholangitis and hair dye use, past smoking, and history of urinary tract infections have prompted people to consider environmental factors such as toxins and infections as possible aetiologies or triggering factors for primary biliary cholangitis (Leung 2005; Dronamraju 2010; Prince 2010; Selmi 2010).
A significant proportion of people with primary biliary cholangitis are asymptomatic at the time of diagnosis (up to about 60% in some studies (Pla 2007)). Itching and fatigue are the most common symptoms (Pla 2007; Myers 2009). Other ways of clinical presentation include Raynaud's syndrome (bluish discolouration of the fingers and toes due to vasospasm in response to cold or emotional stress); features of portal hypertension; osteoporosis; high cholesterol (particularly high ratio of high‐density lipoprotein cholesterol (which is considered protective for the heart) to low‐density lipoprotein cholesterol); and rarely deficiencies of vitamin A, vitamin D, vitamin E, and vitamin K (Kim 2000; Gershwin 2005; Pla 2007; Myers 2009; Baldursdottir 2012). Approximately 3% to 8% of people require liver transplantation in about five to six years from diagnosis (Kim 2000; Lindor 2009; Myers 2009; Baldursdottir 2012). Approximately 3% to 4% of people with primary biliary cholangitis die every year, usually because of liver‐related causes such as decompensated liver disease or hepatocellular carcinoma (Rautiainen 2007; Myers 2009). Overall, approximately 21% to 50% of people are dead in about 10 to 11 years from diagnosis (Kim 2000; Rautiainen 2007; Myers 2009; Floreani 2011; Baldursdottir 2012).
The diagnosis of primary biliary cholangitis is made in the presence of any two of the following three criteria (Lindor 2009).
Elevation of alkaline phosphatases.
Presence of antimitochondrial antibody (AMA).
Liver biopsy demonstrating non‐suppurative destructive cholangitis and destruction of interlobular bile ducts.
Some variations of primary biliary cholangitis are AMA‐negative primary biliary cholangitis that requires liver biopsy for establishing the diagnosis and the primary biliary cholangitis ‐ autoimmune hepatitis overlap syndrome (Lindor 2009). However, there is currently no strong evidence that the course of the disease is different between the classic primary biliary cholangitis and these variants (Lindor 2009).
Description of the intervention
Various pharmacological interventions have been tried to treat people with primary biliary cholangitis. These include bile acids such as ursodeoxycholic acid (UDCA) (Kaplan 2004; Combes 2005; Rautiainen 2005; Rudic 2012a); fibrates such as bezafibrate (Kurihara 2000; Rudic 2012b); immunosuppressants or immunomodulators such as glucocorticosteroids (Prince 2005; Rautiainen 2005), colchicine (Almasio 2000; Gong 2004a; Kaplan 2004), methotrexate (Kaplan 2004; Combes 2005; Giljaca 2010), azathioprine (Gong 2007a), ciclosporin (Gong 2007b), chlorambucil (Li Wei 2012), mycophenolate mofetil (Jones 1999; Talwalkar 2005), and thalidomide (McCormick 1994); and copper‐chelating agents such as D‐penicillamine (Gong 2004b) and tetrathiomolybdate (Askari 2010). Several other interventions such as bisphosphonates and hormonal replacement to prevent or treat osteoporosis (Ormarsdottir 2004; Rudic 2011a; Rudic 2011b; Guanabens 2013); antidepressants such as fluoxetine and fluvoxamine to overcome fatigue (Ter Borg 2004; Talwalkar 2006); cholesterol‐lowering agents such as simvastatin to decrease the high cholesterol (Cash 2013); and cholestyramine, rifampicin, and S‐adenosyl methionine for pruritus (Bergasa 2000) have been evaluated for control of various symptoms. Liver transplantation is performed in some people with decompensated liver disease due to primary biliary cholangitis (Kim 2000; Lindor 2009; Myers 2009; Baldursdottir 2012).
How the intervention might work
Certain bile acids are protective while other bile acids are harmful to hepatocytes (liver cells), cholangiocytes (cells that line the bile duct), and gastrointestinal cells lining the oesophagus and stomach (Perez 2009). Bile acids such as UDCA may protect the cholangiocytes from the damage caused by hydrophobic bile acids by decreasing the oxidative stress (by direct antioxidant effect or an increase in antioxidant defences) (Paumgartner 2002; Perez 2009). Bile acids also stimulate the secretion of bile acids from hepatocytes, thereby decreasing their stasis and the resulting damage to the cells and inhibit apoptosis (programmed cell death) (Paumgartner 2002; Perez 2009). Fibrates inactivate hydrophobic bile acids and, therefore, decrease the damage to the cells (Kurihara 2000). Since primary biliary cholangitis is considered an autoimmune disorder, altering the immunity and inflammatory response using glucocorticoids and other immunosuppressants may decrease the damage resulting from the inflammatory response. D‐Penicillamine and tetrathiomolybdate might remove the excess copper, thereby protecting the cells from the damage caused by copper accumulation. They also have antifibrotic properties (Song 2008). In this Cochrane Review, we included only pharmacological interventions aimed at controlling the liver disease (i.e. we excluded symptomatic treatments, lifestyle modifications, and liver transplantation).
Why it is important to do this review
The optimal pharmacological treatment of primary biliary cholangitis is unknown. Currently, both the European Association for the Study of the Liver (EASL) and American Association for the Study of Liver Diseases (AASLD) recommend UDCA for the management of primary biliary cholangitis (EASL 2009; Lindor 2009). However, one Cochrane Review that compared UDCA versus placebo or no intervention reported that there was no survival or symptomatic benefit for UDCA (Rudic 2012a). Therefore, there is clearly a discordance between the evidence and guideline recommendation. Network meta‐analysis allows combination of the direct evidence and indirect evidence, and allows ranking of different interventions in terms of the different outcomes (Salanti 2011; Salanti 2012). There has been no Cochrane Review on the different pharmacological interventions for primary biliary cholangitis. This systematic review and attempted network meta‐analysis provides the best level of evidence for the role of different interventions used in the treatment of people with primary biliary cholangitis.
Objectives
To assess the comparative benefits and harms of different pharmacological interventions in the treatment of primary biliary cholangitis through a network meta‐analysis and to generate rankings of the available pharmacological interventions according to their safety and efficacy. However, it was not possible to assess whether the potential effect modifiers were similar across different comparisons. Therefore, we did not perform the network meta‐analysis, and, instead, assessed the comparative benefits and harms of different interventions using standard Cochrane methodology.
When more trials become available with adequate description of potential effect modifiers, we will attempt to conduct network meta‐analysis to generate rankings of the available interventions according to their safety and efficacy. This is why we retained the planned methodology for network meta‐analysis in our Appendix 1. Once data appear allowing for the conduct of network meta‐analysis, this Appendix 1 will be moved back into the Methods section.
Methods
Criteria for considering studies for this review
Types of studies
We considered randomised clinical trials only for this network meta‐analysis, irrespective of the language, publication status, or date of publication. We excluded studies of other design because of the risk of bias in such studies. We are all aware that such exclusions make us focus much more on potential benefits and not fully assess the risks of serious adverse events as well as risks of adverse events.
Types of participants
We included randomised clinical trials with participants with primary biliary cholangitis irrespective of the method of diagnosis of the disease or the presence of symptoms. We excluded randomised clinical trials in which participants had undergone liver transplantation previously.
Types of interventions
Any of the following pharmacological interventions that are possible treatments used either alone or in combination for primary biliary cholangitis and can be compared with each other or with placebo or no intervention.
The interventions that we considered were:
UDCA;
obeticholic acid;
bezafibrate;
glucocorticosteroids;
colchicine;
methotrexate;
azathioprine;
ciclosporin;
chlorambucil;
mycophenolate mofetil;
thalidomide;
D‐penicillamine;
tetrathiomolybdate.
The above list was not exhaustive. If we identified pharmacological interventions that we were not aware of, we considered them as eligible and included them in the review if they were used primarily for the treatment of primary biliary cholangitis.
Types of outcome measures
We assessed the comparative benefits and harms of available pharmacological interventions aimed at treating people with primary biliary cholangitis for the following outcomes.
Primary outcomes
Mortality at maximal follow‐up.
-
Mortality:
short‐term mortality (up to one year);
medium‐term mortality (one to five years).
-
Adverse events (within three months after cessation of treatment). Depending on the availability of data, we attempted to classify adverse events as serious or non‐serious. We defined a non‐serious adverse event as any untoward medical occurrence not necessarily having a causal relationship with the treatment but resulting in a dose reduction or discontinuation of treatment (any time after commencement of treatment) (ICH‐GCP 1997). We defined a serious adverse event as any event that would increase mortality; was life threatening; required hospitalisation; resulted in persistent or significant disability; was a congenital anomaly/birth defect; or any important medical event that might jeopardise the person or require intervention to prevent it. We used the definition used by study authors for non‐serious and serious adverse events:
proportion of participants with serious adverse events;
number of serious adverse events;
proportion of participants with any type of adverse event;
number of any type of adverse event.
-
Health‐related quality of life as defined in the included trials using a validated scale such as EQ‐5D or 36‐item Short Form (SF‐36) (EuroQol 2014; Ware 2014):
short‐term (up to one year);
medium‐term (one to five years);
long‐term (beyond five years).
We considered long‐term quality of life more important than short‐term or medium‐term quality of life, although short‐term and medium‐term quality of life are also important primary outcomes.
Secondary outcomes
-
Liver transplantation (maximal follow‐up):
proportion of participants with liver transplantation;
time to liver transplantation.
-
Decompensated liver disease (maximal follow‐up):
proportion of participants with decompensated liver disease;
time to liver decompensation.
-
Cirrhosis (maximal follow‐up):
proportion of participants with cirrhosis;
time to cirrhosis.
Hepatocellular carcinoma (maximal follow‐up).
Search methods for identification of studies
Electronic searches
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, Embase, and Science Citation Index Expanded (Royle 2003) from inception to 27 February 2017 for randomised clinical trials comparing two or more of the above interventions without applying any language restrictions. We searched for all possible comparisons formed by the interventions of interest. To identify further ongoing or completed trials, we also searched the World Health Organization International Clinical Trials Registry Platform Search Portal (apps.who.int/trialsearch/), which searches various trial registers, including ISRCTN and ClinicalTrials.gov. Appendix 2 shows the search strategies we used.
Searching other resources
We searched the references of the identified trials and existing Cochrane Reviews on primary biliary cholangitis to identify additional trials for inclusion.
Data collection and analysis
Selection of studies
Two review authors (KG and FS) independently identified the trials for inclusion by screening the titles and abstracts. We sought full‐text articles for any references that at least one of the review authors identified for potential inclusion. We selected trials for inclusion based on the full‐text articles. We listed the excluded full‐text references with reasons for their exclusion in the Characteristics of excluded studies table. We have also listed any ongoing trials identified primarily through the search of the clinical trial registers for further follow‐up. We resolved discrepancies through discussion.
Data extraction and management
Two review authors (KG and FS or LHE) independently extracted the following data.
-
Outcome data (for each outcome and for each treatment arm whenever applicable):
number of participants randomised;
number of participants included for the analysis;
number of participants with events for binary outcomes, mean and standard deviation for continuous outcomes, number of events for count outcomes, and the number of participants with events and the mean follow‐up period for time‐to‐event outcomes;
definition of outcomes or scale used if appropriate.
-
Data on potential effect modifiers:
participant characteristics such as age, sex, comorbidities, proportion of symptomatic participants, proportion with AMA‐positive status, proportion of participants with overlap syndrome, and responders;
details of the intervention and control (including dose, frequency, and duration);
risk of bias (assessment of risk of bias in included studies).
-
Other data:
year and language of publication;
country in which the participants were recruited;
year(s) in which the trial was conducted;
inclusion and exclusion criteria;
follow‐up time points of the outcome.
If available, we planned to obtain the data separately for symptomatic participants and asymptomatic participants from the report. If available, we also planned to obtain the data separately for people with AMA‐positive status and people with AMA‐negative status and for responders and non‐responders separately. We sought unclear or missing information by contacting the trial authors. If there was any doubt whether trials shared the same participants, completely or partially (by identifying common authors and centres), we attempted to contact the trial authors to clarify whether the trial report was duplicated. We resolved any differences in opinion through discussion.
Assessment of risk of bias in included studies
We followed the guidance given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and described in the Cochrane Hepato‐Biliary Module (Gluud 2017) to assess the risk of bias in included trials. Specifically, we assessed the risk of bias in included trials for the following domains using the methods below (Schulz 1995; Moher 1998; Kjaergard 2001; Wood 2008; Savović 2012a; Savović 2012b; Lundh 2017).
Allocation sequence generation
Low risk of bias: sequence generation was achieved using computer random number generation or a random number table. Drawing lots, tossing a coin, shuffling cards, and throwing dice were adequate if performed by an independent person not otherwise involved in the trial.
Unclear risk of bias: the method of sequence generation was not specified.
High risk of bias: the sequence generation method was not random.
Allocation concealment
Low risk of bias: the participant allocations could not have been foreseen in advance of, or during, enrolment. Allocation was controlled by a central and independent randomisation unit. The allocation sequence was unknown to the investigators (e.g. if the allocation sequence was hidden in sequentially numbered, opaque, and sealed envelopes).
Unclear risk of bias: the method used to conceal the allocation was not described so that intervention allocations may have been foreseen in advance of, or during, enrolment.
High risk of bias: the allocation sequence was likely to be known to the investigators who assigned the participants.
Blinding of participants and personnel
Low risk of bias: any of the following: no blinding or incomplete blinding, but the review authors judged that the outcome was not likely to be influenced by lack of blinding; or blinding of participants and key study personnel ensured, and it was unlikely that the blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk'; or the trial did not address this outcome.
High risk of bias: any of the following: no blinding or incomplete blinding, and the outcome was likely to be influenced by lack of blinding; or blinding of key study participants and personnel attempted, but likely that the blinding could have been broken, and the outcome was likely to be influenced by lack of blinding.
Blinding of outcome assessors
Low risk of bias: any of the following: no blinding of outcome assessment, but the review authors judged that the outcome measurement was not likely to be influenced by lack of blinding; or blinding of outcome assessment ensured, and unlikely that the blinding could have been broken.
Unclear risk of bias: any of the following: insufficient information to permit judgement of 'low risk' or 'high risk'; or the trial did not address this outcome.
High risk of bias: any of the following: no blinding of outcome assessment, and the outcome measurement was likely to be influenced by lack of blinding; or blinding of outcome assessment, but likely that the blinding could have been broken, and the outcome measurement was likely to be influenced by lack of blinding.
Incomplete outcome data
Low risk of bias: missing data were unlikely to make treatment effects depart from plausible values. Sufficient methods, such as multiple imputation, were employed to handle missing data.
Unclear risk of bias: there was insufficient information to assess whether missing data in combination with the method used to handle missing data were likely to induce bias on the results.
High risk of bias: the results were likely to be biased due to missing data.
Selective outcome reporting
Low risk of bias: the trial reported at least the following predefined outcomes: mortality, decompensated liver disease, requirement for transplantation, or treatment‐related adverse events. If the original trial protocol was available, the outcomes should have been those called for in that protocol. If the trial protocol was obtained from a trial registry (e.g. www.clinicaltrials.gov), the outcomes sought should have been those enumerated in the original protocol if the trial protocol was registered before or at the time that the trial was begun. If the trial protocol was registered after the trial was begun, those outcomes were not considered to be reliable.
Unclear risk: not all predefined, or clinically relevant and reasonably expected, outcomes were reported fully, or it was unclear whether data on these outcomes were recorded or not.
High risk: one or more predefined or clinically relevant and reasonably expected outcomes were not reported, even though data on these outcomes were likely to have been available and even recorded.
For‐profit bias
Low risk of bias: the trial appeared to be free of industry sponsorship or other type of for‐profit support that may manipulate the trial design, conductance, or results of the trial.
Unclear risk of bias: the trial may or may not have been free of for‐profit bias as no information on clinical trial support or sponsorship was provided.
High risk of bias: the trial was sponsored by industry or received other type of for‐profit support.
Other bias
Low risk of bias: the trial appeared to be free of other components (e.g. inappropriate control or dose or administration of control) that could put it at risk of bias.
Unclear risk of bias: the trial may or may not have been free of other components that could put it at risk of bias.
High risk of bias: there are other factors in the trial that could put it at risk of bias (e.g. inappropriate control or dose or administration of control).
We considered a trial at low risk of bias if we assessed the trial to be at low risk of bias across all domains. Otherwise, we considered trials to be at unclear risk of bias or at high risk of bias regarding one or more domains as at high risk of bias.
Measures of treatment effect
For dichotomous variables (e.g. short‐term and medium‐term mortality, liver transplantation, proportion of participants with adverse events, decompensated liver disease, cirrhosis, or hepatocellular carcinoma), we calculated the odds ratio (OR) with 95% confidence intervals (CI). For continuous variables (e.g. quality of life reported on the same scale), we planned to calculate the mean difference with 95% CI. We planned to use standardised mean difference values with 95% CI for quality of life if included trials used different scales. For count outcomes (e.g. number of adverse events), we calculated the rate ratio with 95% CI. For time‐to‐event data (e.g. mortality at maximal follow‐up or requirement for liver transplantation, time to liver decompensation, and time to cirrhosis), we planned to use the hazard ratio (HR) with 95% CIs. We also calculated Trial Sequential Analysis‐adjusted CI to control random errors (Thorlund 2011).
Unit of analysis issues
The unit of analysis was people with primary biliary cholangitis according to the intervention group to which they were randomly assigned.
Cluster randomised clinical trials
We found no cluster randomised clinical trials. However, if we had found them, we would have included them provided that the effect estimate adjusted for cluster correlation was available.
Cross‐over randomised clinical trials
If we found cross‐over randomised clinical trials, we included the outcomes after the period of first intervention only since primary biliary cholangitis is a chronic disease and the interventions could potentially have a residual effect.
Trials with multiple treatment groups
We collected data for all trial intervention groups that met our inclusion criteria.
Dealing with missing data
We performed an intention‐to‐treat analysis whenever possible (Newell 1992). Otherwise, we used the data that were available to us (e.g. a trial may have reported only per‐protocol analysis results). As such per‐protocol analyses may be biased, we planned to conduct best‐worst case scenario analysis (good outcome in intervention group and bad outcome in control group) and worst‐best case scenario analysis (bad outcome in intervention group and good outcome in control group) as sensitivity analyses whenever possible.
For continuous outcomes, we planned to impute the standard deviation from P values according to guidance given in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). If the data were likely to be normally distributed, we planned to use the median for meta‐analysis when the mean was not available. If it was not possible to calculate the standard deviation from the P value or the CIs, we planned to impute the standard deviation using the largest standard deviation in other trials for that outcome. This form of imputation may decrease the weight of the study for calculation of mean differences and may bias the effect estimate to no effect for calculation of standardised mean differences (Higgins 2011).
Assessment of heterogeneity
We assessed clinical and methodological heterogeneity by carefully examining the characteristics and design of included trials. We assessed the presence of clinical heterogeneity by comparing effect estimates in the presence or absence of symptoms, the presence or absence of AMA, responders versus non‐responders, and the doses of the pharmacological interventions. Different study designs and risk of bias may contribute to methodological heterogeneity. We used the I2 test and Chi2 test for heterogeneity, and overlapping of CIs to assess heterogeneity.
Assessment of reporting biases
We planned to use visual asymmetry on a funnel plot to explore reporting bias in the presence of at least 10 trials that could be included for a direct comparison (Egger 1997; Macaskill 2001). In the presence of heterogeneity that could be explained by subgroup analysis, we planned to produce a funnel plot for each subgroup in the presence of an adequate number of trials (at least 10 trials). We planned to use the linear regression approach described by Egger 1997 to determine funnel plot asymmetry.
We also considered selective reporting as evidence of reporting bias.
Data synthesis
We performed the meta‐analyses according to the recommendations of Cochrane (Higgins 2011), using the software package Review Manager 5 (RevMan 2014). We used a random‐effects model (DerSimonian 1986) and a fixed‐effect model (DeMets 1987). In the case of a discrepancy between the two models, we reported both results; otherwise, we reported only the results from the fixed‐effect model.
Calculation of required information size and Trial Sequential Analysis
For calculation of the required information size, see Appendix 3. We performed Trial Sequential Analysis to control the risk of random errors when there were at least two trials included for mortality at maximal follow‐up, serious adverse events (proportion) and health‐related quality of life, the three outcomes that determine whether an intervention should be used (Wetterslev 2008; Thorlund 2011; TSA 2011; Wetterslev 2017). We used an alpha error as per guidance of Jakobsen 2014, power of 90% (beta error of 10%), a relative risk reduction of 20%, a control group proportion observed in the trials, and the diversity observed in the meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We planned to assess the differences in the effect estimates between the following subgroups.
Trials at low risk of bias compared to trials at high risk of bias.
Participants with symptomatic compared to participants with asymptomatic primary biliary cholangitis.
AMA‐positive participants compared to AMA‐negative participants.
Responders compared to non‐responders to bile acids.
Different doses of pharmacological interventions. For example, various doses of UDCA used in randomised clinical trials include 5 mg/kg to 7 mg/kg, 13 mg/kg to 15 mg/kg (moderate dose), and 23 mg/kg to 25 mg/kg (high dose) (Angulo 1999a; Lindor 1997).
We planned to use the Chi2 test for subgroup differences to identify subgroup differences.
Sensitivity analysis
If a trial reported only per‐protocol analysis results, we planned to re‐analyse the results using the best‐worst case scenario and worst‐best case scenario analyses as sensitivity analyses whenever possible.
Presentation of results and GRADE assessments
We reported mortality, serious adverse events, and health‐related quality of life, the three most important outcomes that determine the use of an intervention in a 'Summary of findings' table format, downgrading the quality of evidence for risk of bias, inconsistency, indirectness, imprecision, and publication bias using GRADE (Guyatt 2011). We have presented the 'Summary of findings' tables for all comparisons in which two trials were included for one of mortality at maximal follow‐up, serious adverse events, or health‐related quality of life.
Results
Description of studies
Results of the search
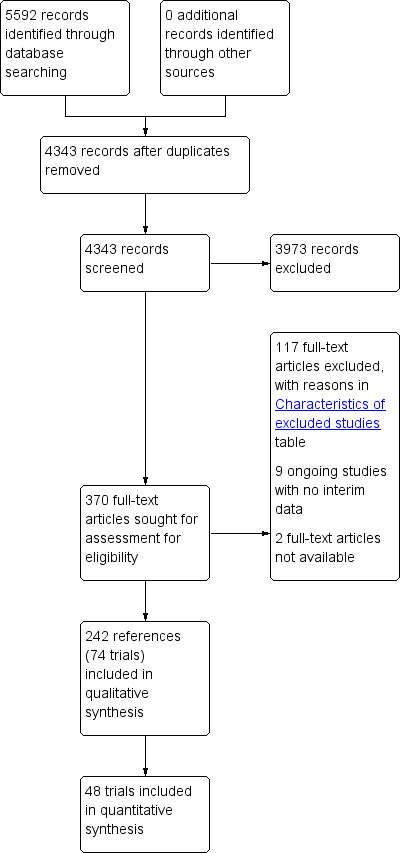
We identified 5592 references through electronic searches of CENTRAL (n = 1104), MEDLINE (n = 2383), Embase (n = 604), Science Citation Index Expanded (n = 1362), World Health Organization International Clinical Trials Registry Platform (n = 88), and ClinicalTrials.gov (n = 51). After the removal of 1249 duplicates we obtained 4343 references. We then excluded 3973 clearly irrelevant references through screening titles and reading abstracts. We retrieved 370 references for further assessment. No references were identified through scanning reference lists of the identified randomised trials. We excluded 117 references for the reasons stated in the Characteristics of excluded studies table. Nine references are an ongoing trial without any interim data (ChiCTR‐IPR‐16008935; EUCTR2015‐002698‐39‐GB; NCT02308111; NCT02701166; NCT02823353; NCT02823366; NCT02937012; NCT02943447; NCT02965911). We were unable to obtain the full texts for two references (O'Brian 1990; Zaman 2006). In total, 242 references (74 trials) met the inclusion criteria. The reference flow is summarised in the study flow diagram (Figure 1).
1.
Study flow diagram.
Included studies
The 74 trials that met the inclusion criteria for this review included 5902 participants. Some 28 trials did not contribute any information for this review leaving 4274 participants included in one or more outcomes in the review (Bodenheimer 1988; Arora 1990; Oka 1990; Smart 1990; Poupon 1991a; Senior 1991; Battezzati 1993; Manzillo 1993a; Manzillo 1993b; Bobadilla 1994; Goddard 1994; Lim 1994; McCormick 1994; Steenbergen 1994; Lindor 1997; Kaplan 1999; Leuschner 1999; Nakai 2000; Mazzarella 2002; Ueno 2005; Iwasaki 2008a; Iwasaki 2008b; Askari 2010; Liberopoulos 2010; Cash 2013; Bowlus 2014; Kowdley 2014a; Mayo 2015). In the main review unstratified by the dose of UDCA or obeticholic acid, 4060 participants were included in one or more outcomes in the review. The mean or median age of the participants ranged from 46 to 64 years in the trials that reported this information. The proportion of females ranged from 77.8% to 100% in the trials that reported this information. The proportion of participants with symptoms varied from 19.9% to 100% in the trials that reported this information. The proportion of participants who were AMA positive ranged from 80.8% to 100% in the trials that reported this information. Ten trials included non‐responders to bile acids only (Van Hoogstraten 1998; Wolfhagen 1998; Kanda 2003; Ueno 2005; Iwasaki 2008b; Mason 2008; Liberopoulos 2010; Hirschfield 2015; Hosonuma 2015; Nevens 2016). The remaining trials did not state whether they included responders or non‐responders, or both. However, it appeared that most trials included participants who had not received previous treatments or regardless of the previous treatments received. The interventions, controls, number of participants included in each trial, and the follow‐up period reported in the different trials are listed in Table 8.
1. Characteristics of included studies arranged by comparison.
| Study name | No participants randomised | Post‐randomisation dropouts | No participants for whom outcome was reported | Intervention(s) | Control | Mean follow‐up period (months) |
| Smart 1990 | 20 | Not stated | 20 | Antioxidants | No intervention | Not stated |
| Christensen 1985 | 248 | 63 | 185 | Azathioprine | No intervention | 63 |
| Heathcote 1976 | 45 | 6 | 39 | Azathioprine | No intervention | Not stated |
| Hoofnagle 1986 | 24 | 0 | 24 | Chlorambucil | No intervention | 52 |
| Bodenheimer 1988 | 57 | 10 | 47 | Colchicine | No intervention | 33 |
| Kaplan 1986 | 60 | 3 | 57 | Colchicine | No intervention | 24 |
| Warnes 1987 | 64 | Not stated | 64* | Colchicine | No intervention | 19 (median) |
| Bobadilla 1994 | 40 | Not stated | 40 | Colchicine + UDCA | No intervention | 12 |
| Lombard 1993 | 349 | 0 | 349 | Ciclosporin | No intervention | 31 (median) |
| Minuk 1988 | 12 | 0 | 12 | Ciclosporin | No intervention | Not stated |
| Wiesner 1990 | 40 | 11 | 29 | Ciclosporin | No intervention | 35 (median) |
| Dickson 1985 | 309 | 82 | 227 | D‐Penicillamine | No intervention | 60 (median) |
| Epstein 1979 | 98 | Not stated | 98 | D‐Penicillamine | No intervention | 66 |
| Macklon 1982 | 60 | 0 | 60 | D‐Penicillamine | No intervention | 37 |
| Matloff 1982 | 52 | 0 | 52 | D‐Penicillamine | No intervention | 24 |
| Neuberger 1985 | 189 | Not stated | 189 | D‐Penicillamine | No intervention | Not stated |
| Taal 1983 | 24 | Not stated | 24 | D‐Penicillamine | No intervention | 18 |
| Triger 1980 | 35 | Not stated | 35 | D‐Penicillamine | No intervention | Not stated |
| Mitchison 1989 | 36 | 0 | 36 | Glucocorticosteroids | No intervention | 36 |
| Ueno 2005 | 20 | Not stated | 20 | Lamivudine | No intervention | Not stated |
| Mitchison 1993 | 104 | 3 | 101 | Malotilate | No intervention | 25 (median) |
| Hendrickse 1999 | 60 | Not stated | 60 | Methotrexate | No intervention | 68 |
| Steenbergen 1994 | 14 | Not stated | 14 | Methotrexate + UDCA | No intervention | 24 |
| Mayo 2015 | 45 | 3 | 42 | NGM282 | No intervention | Not stated |
| Bowlus 2014 | 216 | Not stated | 216 | Obeticholic acid | No intervention | 12 |
| Hirschfield 2015 | 165 | 0 | 165 | Obeticholic acid | No intervention | 3 |
| Kowdley 2014a | 59 | Not stated | 59 | Obeticholic acid | No intervention | Not stated |
| Manzillo 1993a | 32 | Not stated | 32 | S‐Adenosyl methionine | No intervention | 1 |
| Manzillo 1993b | 6 | Not stated | 6 | S‐Adenosyl methionine | No intervention | 2 |
| Cash 2013 | 21 | 8 | 13 | Simvastatin | No intervention | 12 |
| Askari 2010 | 28 | 0 | 28 | Tetrathiomolybdate | No intervention | Not stated |
| McCormick 1994 | 18 | 0 | 18 | Thalidomide | No intervention | Not stated |
| Arora 1990 | 9 | Not stated | 9 | UDCA | No intervention | 5 |
| Battezzati 1993 | 88 | 2 | 86 | UDCA | No intervention | 6 |
| Combes 1995a | 151 | 0 | 151 | UDCA | No intervention | 24 |
| Eriksson 1997 | 116 | 15 | 101 | UDCA | No intervention | 24 |
| Heathcote 1994 | 222 | Not stated | 222 | UDCA | No intervention | 24 |
| Leuschner 1989 | 20 | 0 | 18 | UDCA | No intervention | 12 |
| Lim 1994 | 32 | Not stated | 32 | UDCA | No intervention | Not stated |
| Lindor 1994 | 180 | 10 | 170 | UDCA | No intervention | 24 |
| Oka 1990 | 52 | 7 | 45 | UDCA | No intervention | Not stated |
| Papatheodoridis 2002 | 92 | 6 | 86 | UDCA | No intervention | 89 |
| Pares 2000 | 192 | 0 | 192 | UDCA | No intervention | 41 (median) |
| Poupon 1991a | 149 | 3 | 146 | UDCA | No intervention | Not stated |
| Senior 1991 | 20 | 1 | 19 | UDCA | No intervention | 18 |
| Turner 1994 | 46 | 0 | 46 | UDCA | No intervention | 24 |
| Goddard 1994 | 57 | Not stated | 57 | Intervention 1: UDCA Intervention 2: colchicine Intervention 3: colchicine + UDCA | No intervention | 15 |
| Wolfhagen 1998 | 50 | Not stated | 50 | Azathioprine + glucocorticosteroids + UDCA | UDCA | 12 |
| Iwasaki 2008a | 45 | Not stated | 45 | Bezafibrate | UDCA | 12 |
| Kurihara 2000 | 24 | Not stated | 24 | Bezafibrate | UDCA | Not stated |
| Hosonuma 2015 | 27 | 0 | 27 | Bezafibrate + UDCA | UDCA | 96 |
| Iwasaki 2008b | 22 | Not stated | 22 | Bezafibrate + UDCA | UDCA | 12 |
| Kanda 2003 | 22 | 0 | 22 | Bezafibrate + UDCA | UDCA | 7 |
| Nakai 2000 | 23 | Not stated | 23 | Bezafibrate + UDCA | UDCA | 12 |
| Almasio 2000 | 90 | 6 | 84 | Colchicine + UDCA | UDCA | Not stated |
| Ikeda 1996 | 22 | 0 | 22 | Colchicine + UDCA | UDCA | 24 |
| Poupon 1996 | 74 | Not stated | 74 | Colchicine + UDCA | UDCA | 24 |
| Raedsch 1993 | 28 | 8 | 20 | Colchicine + UDCA | UDCA | 24 |
| Yokomori 2001 | 11 | Not stated | 11 | Colestilan + UDCA | UDCA | Not stated |
| Liberopoulos 2010 | 10 | Not stated | 10 | Fenofibrate + UDCA | UDCA | Not stated |
| Leuschner 1999 | 40 | 0 | 39 | Glucocorticosteroids + UDCA | UDCA | 24 |
| Rautiainen 2005 | 77 | 8 | 69 | Glucocorticosteroids + UDCA | UDCA | 36 |
| Gao 2012 | 79 | Not stated | 79 | Intervention 1: glucocorticosteroids + UDCA Intervention 2: azathioprine + UDCA | UDCA | Not stated |
| Mason 2008 | 59 | 0 | 59 | Lamivudine + zidovudine + UDCA | UDCA | 6 |
| Combes 2005 | 265 | 0 | 265 | Methotrexate + UDCA | UDCA | 91 (median) |
| Gonzalezkoch 1997 | 25 | Not stated | 25 | Methotrexate + UDCA | UDCA | 11 |
| Nevens 2016 | 217 | Not stated | 216 | Obeticholic acid + UDCA | UDCA | 12 |
| Ferri 1993 | 30 | 0 | 30 | TUDCA | UDCA | 6 |
| Ma 2016 | 199 | 8 | 191 | TUDCA | UDCA | 6 |
| Kaplan 1999 | 87 | 2 | 85 | Colchicine | Methotrexate | 24 |
| Comparison of doses | ||||||
| Lindor 1997 | 150 | Not stated | 150 | Intervention 1: UDCA (high) Intervention 2: UDCA (moderate) |
UDCA (low) | 12 |
| Angulo 1999a | 155 | Not stated | 155 | Intervention 1: UDCA (high) Intervention 2: UDCA (moderate) |
UDCA (low) | 12 |
| Van Hoogstraten 1998 | 61 | 2 | 59 | UDCA (moderate) | UDCA (low) | Not stated |
| Mazzarella 2002 | 42 | Not stated | 42 | UDCA (high) | UDCA (moderate) | 72 |
TUDCA: taurodeoxycholic acid; UDCA: ursodeoxycholic acid.
Source of funding: nine trials receive no additional funding or were funded by parties with no vested interest in the results (Heathcote 1976; Hoofnagle 1986; Almasio 2000; Nakai 2000; Iwasaki 2008a; Iwasaki 2008b; Askari 2010; Cash 2013; Hosonuma 2015). Thirty‐one trials were partially or fully funded by the pharmaceutical companies that would benefit based on the results of the trial (Triger 1980; Matloff 1982; Christensen 1985; Dickson 1985; Bodenheimer 1988; Minuk 1988; Oka 1990; Wiesner 1990; Poupon 1991a; Senior 1991; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; McCormick 1994; Combes 1995a; Poupon 1996; Eriksson 1997; Van Hoogstraten 1998; Wolfhagen 1998; Leuschner 1999; Pares 2000; Papatheodoridis 2002; Combes 2005; Rautiainen 2005; Mason 2008; Bowlus 2014; Kowdley 2014a; Mayo 2015; Ma 2016; Nevens 2016). The source of funding was not available from the 34 remaining trials.
Excluded studies
The reasons for exclusion are summarised in the Characteristics of excluded studies table. While the reasons for exclusion for most references were self‐explanatory, the reasons for exclusion of 15 references required some explanation (Poupon 1994; Lindor 1995a; Emond 1996; Lindor 1996; Angulo 1999b; Angulo 1999c; Degott 1999; Corpechot 2000; Jorgensen 2002; Kaplan 2004; Combes 2005b; Leung 2010; Leung 2011; Kowdley 2015; Carbone 2016). These 15 references were long‐term follow‐up reports of included trials, but the randomisation was not maintained and the 'no intervention' group received the intervention. While this is acceptable if some participants crossed over for specific reasons in an intention‐to‐treat analysis, it is not acceptable if the cross‐over from one group to another was done in a systematic manner. Therefore, we excluded these references.
Risk of bias in included studies
The risk of bias is summarised in Figure 2, Figure 3, and Table 9.
2.
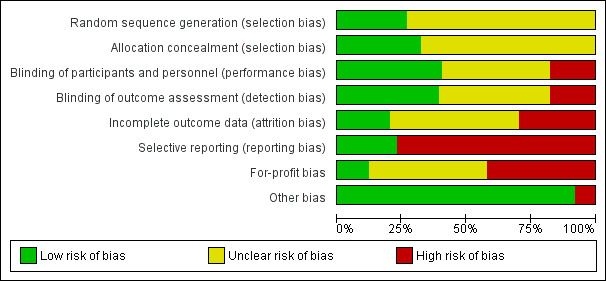
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
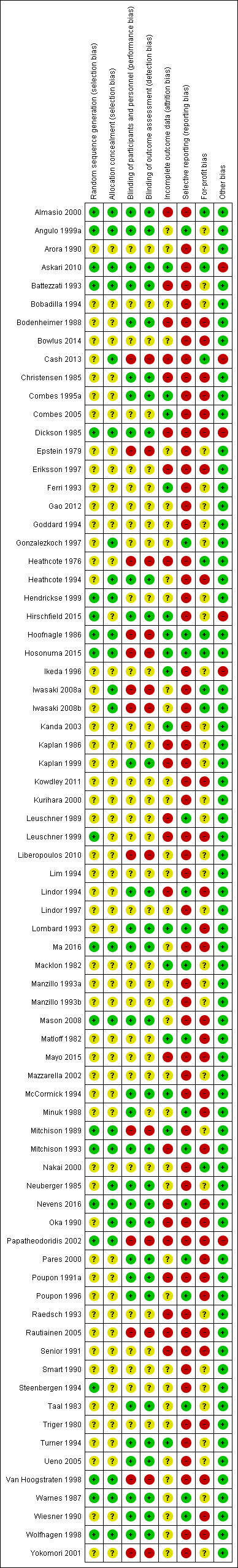
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
2. Risk of bias arranged according to comparisons.
| Name of studies | Intervention(s) | Control | Random sequence generation | Allocation concealment | Blinding of participants and health professionals | Blinding of outcome assessors | Missing outcome bias | Selective outcome reporting | For‐profit bias | Other bias |
| Smart 1990 | Antioxidants | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Christensen 1985 | Azathioprine | No intervention | Unclear | Unclear | Low | Low | High | High | High | Low |
| Heathcote 1976 | Azathioprine | No intervention | Unclear | Unclear | High | High | High | High | Low | Low |
| Hoofnagle 1986 | Chlorambucil | No intervention | Low | Low | High | High | Low | Low | Low | Low |
| Bodenheimer 1988 | Colchicine | No intervention | Unclear | Unclear | Low | Low | High | High | High | Low |
| Kaplan 1986 | Colchicine | No intervention | Unclear | Unclear | Unclear | Unclear | High | High | Unclear | Low |
| Warnes 1987 | Colchicine | No intervention | Low | Low | Low | Low | Unclear | Low | Unclear | Low |
| Bobadilla 1994 | Colchicine + UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Lombard 1993 | Ciclosporin | No intervention | Unclear | Unclear | Low | Low | Low | Low | High | Low |
| Minuk 1988 | Ciclosporin | No intervention | Unclear | Unclear | Low | Unclear | Unclear | Low | High | Low |
| Wiesner 1990 | Ciclosporin | No intervention | Unclear | Unclear | Low | Low | Unclear | Low | High | Low |
| Dickson 1985 | D‐Penicillamine | No intervention | Low | Low | Low | Low | High | High | High | High |
| Epstein 1979 | D‐Penicillamine | No intervention | Unclear | Unclear | High | High | Unclear | High | Unclear | Low |
| Macklon 1982 | D‐Penicillamine | No intervention | Unclear | Unclear | Unclear | Unclear | Low | Low | Unclear | Low |
| Matloff 1982 | D‐Penicillamine | No intervention | Unclear | Unclear | Unclear | Unclear | Low | Low | High | Low |
| Neuberger 1985 | D‐Penicillamine | No intervention | Unclear | Low | Low | Low | Unclear | High | Unclear | Low |
| Taal 1983 | D‐Penicillamine | No intervention | Unclear | Unclear | Low | Low | Unclear | Low | Unclear | Low |
| Triger 1980 | D‐Penicillamine | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | High | Low |
| Mitchison 1989 | Glucocorticosteroids | No intervention | Low | Low | High | High | Low | High | Unclear | Low |
| Ueno 2005 | Lamivudine | No intervention | Unclear | Unclear | Low | Low | Unclear | High | Unclear | Low |
| Mitchison 1993 | Malotilate | No intervention | Low | Low | Low | Low | High | Low | High | Low |
| Hendrickse 1999 | Methotrexate | No intervention | Low | Low | Unclear | Unclear | Unclear | High | Unclear | Low |
| Steenbergen 1994 | Methotrexate + UDCA | No intervention | Low | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Mayo 2015 | NGM282 | No intervention | Unclear | Unclear | Unclear | Unclear | High | High | High | Low |
| Bowlus 2014 | Obeticholic acid | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | High | Low |
| Hirschfield 2015 | Obeticholic acid | No intervention | Low | Unclear | Low | Low | Low | High | Unclear | High |
| Kowdley 2011 | Obeticholic acid | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | High | Low |
| Manzillo 1993a | S‐Adenosyl methionine | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Manzillo 1993b | S‐Adenosyl methionine | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Cash 2013 | Simvastatin | No intervention | Unclear | Low | High | High | High | High | Low | High |
| Askari 2010 | Tetrathiomolybdate | No intervention | Low | Low | Low | Low | Low | High | Low | High |
| McCormick 1994 | Thalidomide | No intervention | Unclear | Unclear | Low | Low | Low | High | High | Low |
| Arora 1990 | UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Battezzati 1993 | UDCA | No intervention | Low | Low | Low | Low | High | High | Unclear | Low |
| Combes 1995a | UDCA | No intervention | Unclear | Unclear | Low | Low | Low | High | High | Low |
| Eriksson 1997 | UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | High | High | High | Low |
| Heathcote 1994 | UDCA | No intervention | Unclear | Low | Low | Low | Unclear | High | High | Low |
| Leuschner 1989 | UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | High | Low | Unclear | Low |
| Lim 1994 | UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Lindor 1994 | UDCA | No intervention | Unclear | Unclear | Low | Low | High | Low | High | Low |
| Oka 1990 | UDCA | No intervention | Unclear | Low | Low | Low | High | High | High | Low |
| Papatheodoridis 2002 | UDCA | No intervention | Low | Low | High | High | High | High | High | High |
| Pares 2000 | UDCA | No intervention | Unclear | Unclear | Low | Low | Unclear | Low | High | Low |
| Poupon 1991a | UDCA | No intervention | Unclear | Unclear | Low | Low | High | High | High | Low |
| Senior 1991 | UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | High | High | High | Low |
| Turner 1994 | UDCA | No intervention | Unclear | Unclear | Low | Low | Low | High | Unclear | Low |
| Goddard 1994 | Intervention 1: UDCA Intervention 2: colchicine Intervention 3: colchicine + UDCA | No intervention | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Wolfhagen 1998 | Azathioprine + glucocorticosteroids + UDCA | UDCA | Low | Low | Low | Low | Unclear | High | High | Low |
| Iwasaki 2008a | Bezafibrate | UDCA | Unclear | Low | High | High | Unclear | High | Low | Low |
| Kurihara 2000 | Bezafibrate | UDCA | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Hosonuma 2015 | Bezafibrate + UDCA | UDCA | Low | Low | High | High | Low | Low | Low | Low |
| Iwasaki 2008b | Bezafibrate + UDCA | UDCA | Unclear | Low | High | High | Unclear | High | Low | Low |
| Kanda 2003 | Bezafibrate + UDCA | UDCA | Unclear | Unclear | Unclear | Unclear | Low | High | Unclear | Low |
| Nakai 2000 | Bezafibrate + UDCA | UDCA | Unclear | Unclear | Unclear | Unclear | Unclear | High | Low | Low |
| Almasio 2000 | Colchicine + UDCA | UDCA | Low | Low | Low | Low | High | High | Low | Low |
| Ikeda 1996 | Colchicine + UDCA | UDCA | Unclear | Unclear | Unclear | Unclear | Low | High | Unclear | High |
| Poupon 1996 | Colchicine + UDCA | UDCA | Unclear | Unclear | Low | Low | Unclear | Low | High | Low |
| Raedsch 1993 | Colchicine + UDCA | UDCA | Unclear | Unclear | Unclear | Unclear | High | High | Unclear | Low |
| Yokomori 2001 | Colestilan + UDCA | UDCA | Unclear | Unclear | High | High | Unclear | High | Unclear | Low |
| Liberopoulos 2010 | Fenofibrate + UDCA | UDCA | Unclear | Unclear | High | High | Unclear | High | Unclear | Low |
| Leuschner 1999 | Glucocorticosteroids + UDCA | UDCA | Low | Unclear | Unclear | Unclear | High | High | High | Low |
| Rautiainen 2005 | Glucocorticosteroids + UDCA | UDCA | Unclear | Unclear | High | High | High | High | High | Low |
| Gao 2012 | Glucocorticosteroids + UDCA Azathioprine + UDCA | UDCA | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Mason 2008 | Lamivudine + zidovudine + UDCA | UDCA | Low | Low | Low | Low | Unclear | High | High | Low |
| Combes 2005 | Methotrexate + UDCA | UDCA | Unclear | Unclear | Unclear | Unclear | Low | High | High | Low |
| Gonzalezkoch 1997 | Methotrexate + UDCA | UDCA | Unclear | Low | Unclear | Unclear | Unclear | Low | Unclear | Low |
| Nevens 2016 | Obeticholic acid + UDCA | UDCA | Low | Low | Low | Low | High | Low | High | Low |
| Ferri 1993 | TUDCA | UDCA | Unclear | Unclear | Unclear | Unclear | Low | High | Unclear | Low |
| Ma 2016 | TUDCA | UDCA | Low | Low | Low | Low | Unclear | High | High | Low |
| Kaplan 1999 | Colchicine | Methotrexate | Unclear | Unclear | Low | Low | High | High | Unclear | Low |
| Comparison of doses | ||||||||||
| Lindor 1997 | Intervention 1: UDCA (high) Intervention 2: UDCA (moderate) |
UDCA (low) | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
| Angulo 1999a | Intervention 1: UDCA (high) Intervention 2: UDCA (moderate) |
UDCA (low) | Low | Low | Low | Low | Unclear | Low | Unclear | Low |
| Van Hoogstraten 1998 | UDCA (moderate) | UDCA (low) | Low | Low | High | High | Unclear | High | High | Low |
| Mazzarella 2002 | UDCA (high) | UDCA (moderate) | Unclear | Unclear | Unclear | Unclear | Unclear | High | Unclear | Low |
TUDCA: taurodeoxycholic acid; UDCA: ursodeoxycholic acid.
Allocation
Twenty trials were at low risk of bias due to random sequence generation (Dickson 1985; Hoofnagle 1986; Warnes 1987; Mitchison 1989; Battezzati 1993; Mitchison 1993; Steenbergen 1994; Van Hoogstraten 1998; Wolfhagen 1998; Angulo 1999a; Hendrickse 1999; Leuschner 1999; Almasio 2000; Papatheodoridis 2002; Mason 2008; Askari 2010; Hirschfield 2015; Hosonuma 2015; Ma 2016; Nevens 2016). The remaining trials were at unclear risk of bias.
Twenty‐four trials were at low risk of bias due allocation concealment (Dickson 1985; Neuberger 1985; Hoofnagle 1986; Warnes 1987; Mitchison 1989; Oka 1990; Battezzati 1993; Mitchison 1993; Heathcote 1994; Gonzalezkoch 1997; Van Hoogstraten 1998; Wolfhagen 1998; Angulo 1999a; Hendrickse 1999; Almasio 2000; Papatheodoridis 2002; Iwasaki 2008a; Iwasaki 2008b; Mason 2008; Askari 2010; Cash 2013; Hosonuma 2015; Ma 2016; Nevens 2016). The remaining trials were at unclear risk of bias.
Sixteen trials were at low risk of both random sequence generation bias and allocation concealment bias (Dickson 1985; Warnes 1987; Mitchison 1989; Battezzati 1993; Mitchison 1993; Van Hoogstraten 1998; Wolfhagen 1998; Angulo 1999a; Hendrickse 1999; Almasio 2000; Papatheodoridis 2002; Mason 2008; Askari 2010; Hosonuma 2015; Ma 2016; Nevens 2016); these trials were considered to be at low risk of selection bias. The remaining trials were at unclear risk of selection bias.
Blinding
Thirty trials were at low risk of performance bias (Taal 1983; Christensen 1985; Dickson 1985; Neuberger 1985; Warnes 1987; Bodenheimer 1988; Minuk 1988; Oka 1990; Wiesner 1990; Poupon 1991a; Battezzati 1993; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; McCormick 1994; Turner 1994; Combes 1995a; Poupon 1996; Wolfhagen 1998; Angulo 1999a; Kaplan 1999; Almasio 2000; Pares 2000; Ueno 2005; Mason 2008; Askari 2010; Hirschfield 2015; Ma 2016; Nevens 2016). Thirteen trials were at high risk of performance bias (Heathcote 1976; Epstein 1979; Hoofnagle 1986; Mitchison 1989; Van Hoogstraten 1998; Yokomori 2001; Papatheodoridis 2002; Rautiainen 2005; Iwasaki 2008a; Iwasaki 2008b; Liberopoulos 2010; Cash 2013; Hosonuma 2015). The remaining trials were at unclear risk of performance bias.
Twenty‐nine trials were at low risk of detection bias (Taal 1983; Christensen 1985; Dickson 1985; Neuberger 1985; Warnes 1987; Bodenheimer 1988; Oka 1990; Wiesner 1990; Poupon 1991a; Battezzati 1993; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; McCormick 1994; Turner 1994; Combes 1995a; Poupon 1996; Wolfhagen 1998; Angulo 1999a; Kaplan 1999; Almasio 2000; Pares 2000; Ueno 2005; Mason 2008; Askari 2010; Hirschfield 2015; Ma 2016; Nevens 2016). Thirteen trials were at high risk of detection bias (Heathcote 1976; Epstein 1979; Hoofnagle 1986; Mitchison 1989; Van Hoogstraten 1998; Yokomori 2001; Papatheodoridis 2002; Rautiainen 2005; Iwasaki 2008a; Iwasaki 2008b; Liberopoulos 2010; Cash 2013; Hosonuma 2015). The remaining trials were at unclear risk of detection bias.
Twenty‐nine trials were at low risk of performance bias and detection bias (Taal 1983; Christensen 1985; Dickson 1985; Neuberger 1985; Warnes 1987; Bodenheimer 1988; Oka 1990; Wiesner 1990; Poupon 1991a; Battezzati 1993; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; McCormick 1994; Turner 1994; Combes 1995a; Poupon 1996; Wolfhagen 1998; Angulo 1999a; Kaplan 1999; Almasio 2000; Pares 2000; Ueno 2005; Mason 2008; Askari 2010; Hirschfield 2015; Ma 2016; Nevens 2016). Thirteen trials were at high risk of performance bias and detection bias (Heathcote 1976; Epstein 1979; Hoofnagle 1986; Mitchison 1989; Van Hoogstraten 1998; Yokomori 2001; Papatheodoridis 2002; Rautiainen 2005; Iwasaki 2008a; Iwasaki 2008b; Liberopoulos 2010; Cash 2013; Hosonuma 2015). The remaining trials were at unclear risk of performance and detection bias.
Incomplete outcome data
Fifteen trials were at low risk of attrition bias (Macklon 1982; Matloff 1982; Hoofnagle 1986; Mitchison 1989; Ferri 1993; Lombard 1993; McCormick 1994; Turner 1994; Combes 1995a; Ikeda 1996; Kanda 2003; Combes 2005; Askari 2010; Hirschfield 2015; Hosonuma 2015). Twenty‐two trials were at high risk of attrition bias due to dropouts which may have been related to the intervention that the participant received (Heathcote 1976; Christensen 1985; Dickson 1985; Kaplan 1986; Bodenheimer 1988; Leuschner 1989; Oka 1990; Poupon 1991a; Senior 1991; Battezzati 1993; Mitchison 1993; Raedsch 1993; Lindor 1994; Eriksson 1997; Kaplan 1999; Leuschner 1999; Almasio 2000; Papatheodoridis 2002; Rautiainen 2005; Cash 2013; Mayo 2015; Nevens 2016). The remaining trials were at unclear risk of attrition bias.
Selective reporting
We were unable to find any protocols published prior to the full study reports. Seventeen trials were at low risk of due to selecting outcome reporting (Macklon 1982; Matloff 1982; Taal 1983; Hoofnagle 1986; Warnes 1987; Minuk 1988; Leuschner 1989; Wiesner 1990; Lombard 1993; Mitchison 1993; Lindor 1994; Poupon 1996; Gonzalezkoch 1997; Angulo 1999a; Pares 2000; Hosonuma 2015; Nevens 2016). The remaining trials were at high risk of bias due to selective reporting (reporting bias).
Other potential sources of bias
For profit bias: nine trials receive no additional funding or were funded by parties with no vested interest in the results and were at low risk of for‐profit bias (Heathcote 1976; Hoofnagle 1986; Almasio 2000; Nakai 2000; Iwasaki 2008a; Iwasaki 2008b; Askari 2010; Cash 2013; Hosonuma 2015). Thirty‐one trials partially or fully funded by the pharmaceutical companies that would benefit based on the results of the trial were at high risk of for‐profit bias (Triger 1980; Matloff 1982; Christensen 1985; Dickson 1985; Bodenheimer 1988; Minuk 1988; Oka 1990; Wiesner 1990; Poupon 1991a; Senior 1991; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; McCormick 1994; Combes 1995a; Poupon 1996; Eriksson 1997; Van Hoogstraten 1998; Wolfhagen 1998; Leuschner 1999; Pares 2000; Papatheodoridis 2002; Combes 2005; Rautiainen 2005; Mason 2008; Bowlus 2014; Kowdley 2014a; Mayo 2015; Ma 2016; Nevens 2016). The remaining trials were at unclear risk of for‐profit bias.
Six trials were at high risk of other bias: authors presented the results of only a subgroup of participants without explaining the reason for this approach (Dickson 1985; Ikeda 1996); a significant proportion of participants crossed over from placebo to UDCA (Papatheodoridis 2002); it was unclear whether the participants continued to take UDCA in both groups (Askari 2010); participants continued to take varying doses of UDCA (Hirschfield 2015); and participants were allowed to continue previous prescriptions for primary biliary cholangitis (it was unclear whether this was balanced across groups) (Cash 2013). The remaining trials were at low risk of other bias.
Overall risk of bias
All trials were at high risk of bias in one or more domains.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7
Summary of findings for the main comparison. Ursodeoxycholic acid (UDCA) versus no intervention for primary biliary cholangitis.
| UDCA versus no intervention for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: UDCA Comparison: no intervention | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| No intervention | UDCA | ||||
|
Mortality at maximal follow‐up Follow‐up: 12 to 89 months |
208 per 1000 | 206 per 1000 (136 to 301) | OR 0.99 (0.60 to 1.64) | 734 (6 trials) | ⊕⊝⊝⊝ Very low1,2 |
|
Serious adverse events (proportion) Follow‐up: 12 to 41 months |
There were no events in either group | 380 (3 trials) | ⊕⊝⊝⊝ Very low1,2,3 | ||
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; UDCA: ursodeoxycholic acid. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
3 There was moderate heterogeneity (downgraded by one level).
Summary of findings 2. Azathioprine versus no intervention for primary biliary cholangitis.
| Azathioprine versus no intervention for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: azathioprine Comparison: no intervention | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| No intervention | Azathioprine | ||||
|
Mortality at maximal follow‐up Follow‐up: 63 months in 1 trial and not stated in 1 trial |
208 per 1000 | 128 per 1000 (78 to 205) | OR 0.56 (0.32 to 0.98) | 224 (2 trials) | ⊕⊝⊝⊝ Very low1,2 |
| Serious adverse events (proportion) | None of the trials reported this outcome. | ||||
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
Summary of findings 3. Colchicine versus no intervention for primary biliary cholangitis.
| Colchicine versus no intervention for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: colchicine Comparison: no intervention | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| No intervention | Colchicine | ||||
|
Mortality at maximal follow‐up Follow‐up: 12 to 24 months |
208 per 1000 | 168 per 1000 (78 to 327) | OR 0.77 (0.32 to 1.85) | 122 (2 trials) | ⊕⊝⊝⊝ Very low1,2 |
|
Serious adverse events (proportion) Follow‐up: 12 months |
There were no events in either group | 64 (1 trial) | ⊕⊝⊝⊝ Very low1,2,3 | ||
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
3 There was moderate heterogeneity (downgraded by one level).
Summary of findings 4. Ciclosporin versus no intervention for primary biliary cholangitis.
| Ciclosporin versus no intervention for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: ciclosporin Comparison: no intervention | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| No intervention | Ciclosporin | ||||
|
Mortality at maximal follow‐up Follow‐up: 31 to 35 months |
208 per 1000 | 188 per 1000 (118 to 283) | OR 0.88 (0.51 to 1.50) | 390 (3 trials) | ⊕⊝⊝⊝ Very low1,2 |
| Serious adverse events (proportion) | None of the trials reported this outcome. | ||||
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
Summary of findings 5. D‐Penicillamine versus no intervention for primary biliary cholangitis.
| D‐Penicillamine versus no intervention for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: D‐penicillamine Comparison: no intervention | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| No intervention | D‐Penicillamine | ||||
|
Mortality at maximal follow‐up (Follow‐up 24 to 66 months) |
208 per 1000 | 191 per 1000 (130 to 274) | OR 0.90 (0.57 to 1.44) | 423 (5 trials) | ⊕⊝⊝⊝ Very low1,2,3 |
|
Serious adverse events (proportion) (Follow‐up 24 months) |
4 per 1000 | 104 per 1000 (6 to 679) | OR 28.77 (1.57 to 526.67) | 52 (1 trial) | ⊕⊝⊝⊝ Very low1,2,3 |
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
3 There was moderate heterogeneity (downgraded by one level).
Summary of findings 6. Colchicine plus ursodeoxycholic acid (UDCA) versus UDCA for primary biliary cholangitis.
| Colchicine plus UDCA versus UDCA for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: colchicine + UDCA Comparison: UDCA | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| UDCA | Colchicine + UDCA | ||||
|
Mortality at maximal follow‐up Follow‐up: 24 months in 1 trial; not reported in 1 trial |
110 per 1000 | 185 per 1000 (45 to 524) | OR 1.84 (0.38 to 8.91) | 158 (2 trials) | ⊕⊝⊝⊝ Very low1,2 |
|
Serious adverse events (proportion) Follow‐up: not stated |
14 per 1000 | 42 per 1000 (2 to 526) | OR 3.08 (0.12 to 78.14) | 74 (1 trial) | ⊕⊝⊝⊝ Very low1,2,3 |
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; UDCA: ursodeoxycholic acid. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
3 There was moderate heterogeneity (downgraded by one level).
Summary of findings 7. Methotrexate plus ursodeoxycholic acid (UDCA) versus UDCA for primary biliary cholangitis.
| Methotrexate plus UDCA versus UDCA for primary biliary cholangitis | |||||
|
Patient or population: people with primary biliary cholangitis Settings: secondary or tertiary care Intervention: methotrexate + UDCA Comparison: UDCA | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (trials) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| UDCA | Methotrexate + UDCA | ||||
|
Mortality at maximal follow‐up Follow‐up: 11 to 91 months |
110 per 1000 | 126 per 1000 (64 to 237) | OR 1.17 (0.55 to 2.51) | 290 (2 trials) | ⊕⊝⊝⊝ Very low1,2 |
| Serious adverse events (proportion) | None of the trials reported this outcome. | ||||
| Serious adverse events (number of events) | None of the trials reported this outcome. | ||||
| Health‐related quality of life | None of the trials reported this outcome. | ||||
| *The basis for the assumed risk is the mean control group proportion across all the trials. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; OR: odds ratio; UDCA: ursodeoxycholic acid. | |||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | |||||
1 Risk of bias in the trial(s) was high (downgraded by two levels). 2 Sample sizes were small and 95% confidence intervals overlapped clinically significant and clinically insignificant or no effect (downgraded by two levels).
3 There was moderate heterogeneity (downgraded by one level).
Mortality at maximal follow‐up
Twenty‐eight trials including 2823 participants reported mortality at maximal follow‐up (Heathcote 1976; Epstein 1979; Macklon 1982; Matloff 1982; Taal 1983; Christensen 1985; Neuberger 1985; Hoofnagle 1986; Kaplan 1986; Warnes 1987; Minuk 1988; Leuschner 1989; Mitchison 1989; Wiesner 1990; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; Turner 1994; Poupon 1996; Gonzalezkoch 1997; Hendrickse 1999; Almasio 2000; Pares 2000; Papatheodoridis 2002; Combes 2005; Hosonuma 2015; Nevens 2016). The period of follow‐up in these trials varied between 11 and 96 months. The proportion of people with mortality (maximal follow‐up) was higher in the methotrexate group (adjusted proportion: 23.3%) than in the no intervention group (1/30 (3.3%)) (OR 8.83, 95% CI 1.01 to 76.96; 60 participants; 1 trial). The proportion of people with mortality (maximal follow‐up) was lower in the azathioprine group (adjusted proportion: 53.5%) than in the no intervention group (72/107 (67.3%)) (OR 0.56, 95% CI 0.32 to 0.98; 224 participants; 2 trials; I2 = 0%). There was no evidence of a difference in any of the remaining comparisons (Analysis 1.1).
1.1. Analysis.
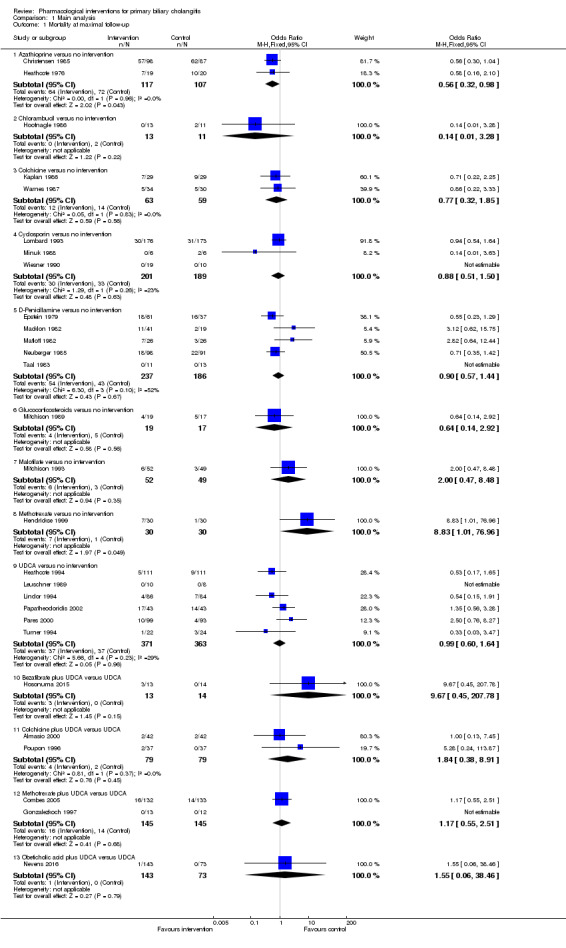
Comparison 1 Main analysis, Outcome 1 Mortality at maximal follow‐up.
Mortality (up to one year)
Eight trials including 655 participants reported mortality (up to year) (Heathcote 1976; Neuberger 1985; Warnes 1987; Minuk 1988; Leuschner 1989; Gonzalezkoch 1997; Almasio 2000; Nevens 2016). There was no evidence of a difference in any of the comparisons (Analysis 1.2).
1.2. Analysis.
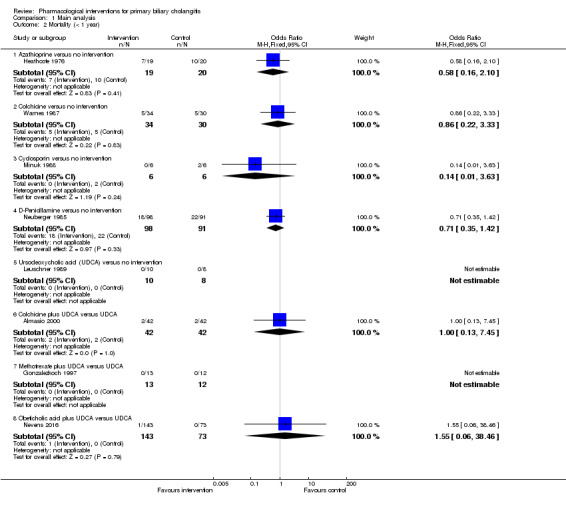
Comparison 1 Main analysis, Outcome 2 Mortality (< 1 year).
Mortality (one to five years)
Twenty trials including 2168 participants reported mortality (one to five years) (Epstein 1979; Macklon 1982; Matloff 1982; Taal 1983; Christensen 1985; Hoofnagle 1986; Kaplan 1986; Mitchison 1989; Wiesner 1990; Lombard 1993; Mitchison 1993; Heathcote 1994; Lindor 1994; Turner 1994; Poupon 1996; Hendrickse 1999; Pares 2000; Papatheodoridis 2002; Combes 2005; Hosonuma 2015). The proportion of people with mortality (one to five years) was higher in the methotrexate group (adjusted proportion: 23.3%) than in the no intervention group (1/30 (3.3%)) (OR 8.83, 95% CI 1.01 to 76.96; 60 participants; 1 trial). There was no evidence of a difference in any of the remaining comparisons (Analysis 1.3).
1.3. Analysis.
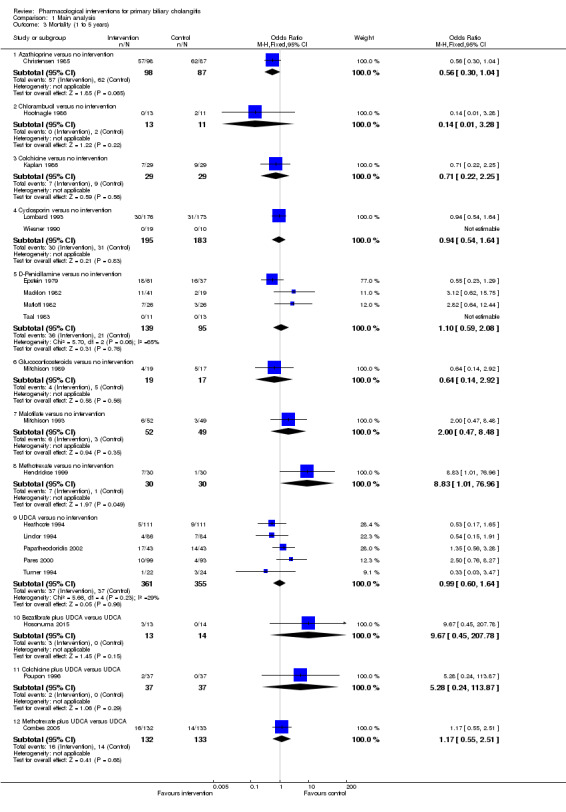
Comparison 1 Main analysis, Outcome 3 Mortality (1 to 5 years).
Serious adverse events (proportion)
Eleven trials including 1076 participants reported serious adverse events (proportion) (Matloff 1982; Warnes 1987; Leuschner 1989; Lindor 1994; Poupon 1996; Kurihara 2000; Pares 2000; Kanda 2003; Mason 2008; Hirschfield 2015; Nevens 2016). The period of follow‐up varied from three to 41 months. The proportion of people with serious adverse events (proportion) was higher in the D‐penicillamine group (adjusted proportion: 28.8%; based on a control group proportion of 1%) versus the no intervention group (0/26 (0.0%)) (OR 28.77, 95% CI 1.57 to 526.67; 52 participants; 1 trial). The proportion of people with serious adverse events (proportion) was higher in the obeticholic acid plus UDCA group (adjusted proportion: 4.1%) versus the UDCA group (19/143 (13.3%)) (OR 3.58, 95% CI 1.02 to 12.51; 216 participants; 1 trial). There was no evidence of a difference in any of the remaining comparisons (Analysis 1.4).
1.4. Analysis.
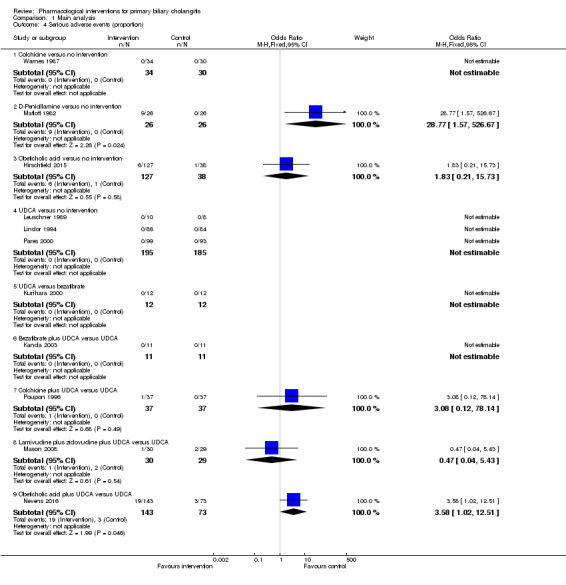
Comparison 1 Main analysis, Outcome 4 Serious adverse events (proportion).
Serious adverse events (number of events)
One trial including 216 participants reported serious adverse events (number of events) (Nevens 2016). The period of follow‐up was 12 months. There was no evidence of a difference between the UDCA plus obeticholic acid versus the UDCA groups (Analysis 1.5).
1.5. Analysis.
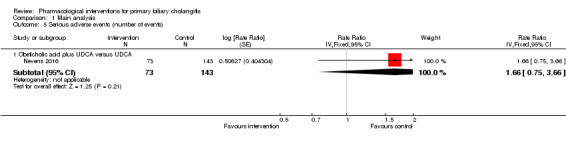
Comparison 1 Main analysis, Outcome 5 Serious adverse events (number of events).
Adverse events (proportion)
Nineteen trials including 1652 participants reported adverse events (proportion) (Macklon 1982; Dickson 1985; Minuk 1988; Leuschner 1989; Wiesner 1990; Ferri 1993; Lombard 1993; Mitchison 1993; Raedsch 1993; Lindor 1994; Ikeda 1996; Gonzalezkoch 1997; Kurihara 2000; Pares 2000; Yokomori 2001; Kanda 2003; Rautiainen 2005; Gao 2012; Hirschfield 2015). The proportion of people with adverse events (proportion) was higher in the ciclosporin group (adjusted proportion: 76.2%) versus the no intervention group (97/189 (51.3%) (OR 3.04, 95% CI 1.98 to 4.68; 390; 3 trials; I2 = 27%), D‐penicillamine group (adjusted proportion: 50.6%) versus the no intervention group (25/135 (18.5%)) (OR 4.51, 95% CI 2.56 to 7.93; 287 participants; 2 trials; I2 = 0%); malotilate group (adjusted proportion: 19.2%) versus the no intervention group (1/49 (2.0%)) (OR 11.43, 95% CI 1.40 to 93.04; 101 participants; 1 trial); and obeticholic acid group (adjusted proportion: 96.1%) versus the no intervention group (32/38 (84.2%)) (OR 4.58, 95% CI 1.31 to 15.95; 165 participants; 1 trial). The proportion of people with adverse events (proportion) was higher in the glucocorticosteroids plus UDCA (adjusted proportion: 15.8%) versus the UDCA group (2/61 (3.3%) (OR 5.54, 95% CI 1.35 to 22.84; 135 participants; 2 trials; I2 = 0%) and methotrexate plus UDCA (adjusted proportion: 100.0%) versus the UDCA group (0/12 (0.0%)) (OR 115.00, 95% CI 4.98 to 2657.48; 25 participants; 1 trial). The proportion of people with adverse events (proportion) was higher in the taurodeoxycholic acid (TUDCA) group (adjusted proportion: 60.0%) versus the UDCA group (1/15 (6.7%)) (OR 21.00, 95% CI 2.16 to 204.61; 30 participants; 1 trial). There was no evidence of a difference in any of the remaining comparisons (Analysis 1.6).
1.6. Analysis.
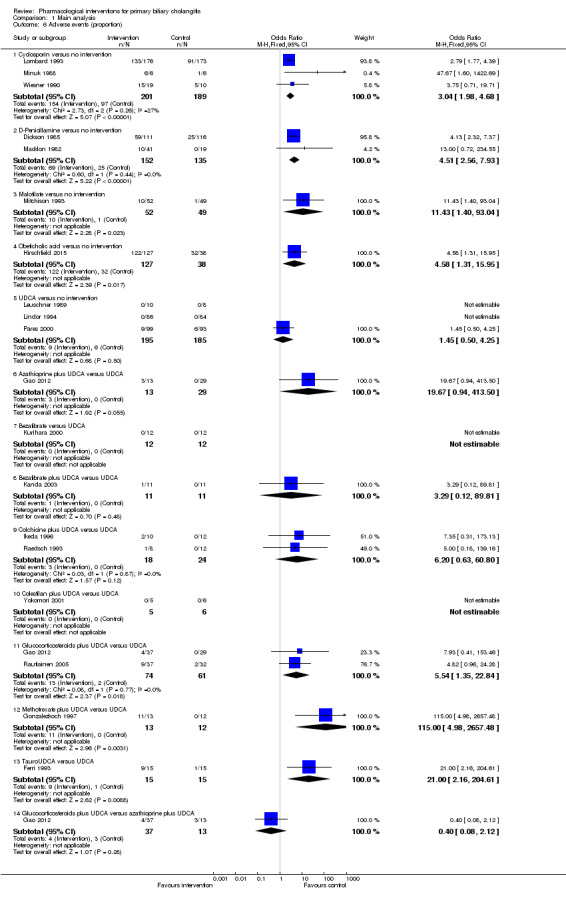
Comparison 1 Main analysis, Outcome 6 Adverse events (proportion).
Adverse events (number)
Fourteen trials including 1304 participants reported adverse events (number) (Matloff 1982; Taal 1983; Dickson 1985; Hoofnagle 1986; Minuk 1988; Wiesner 1990; Lombard 1993; Mitchison 1993; Ikeda 1996; Gonzalezkoch 1997; Wolfhagen 1998; Hirschfield 2015; Hosonuma 2015; Ma 2016). The number of adverse events was higher in the chlorambucil group (adjusted rate: 57.9 events per 100 participants) versus the no intervention group (3/11 (27.3 events per 100 participants)) (rate ratio 3.67, 95% CI 1.04 to 12.87; 24 participants; 1 trial); ciclosporin group (adjusted rate: 84.4 events per 100 participants) versus the no intervention group (128/189 (67.7 events per 100 participants)) (rate ratio 2.58, 95% CI 1.26 to 5.31; 390 participants; 3 trials; I2= 69%); D‐penicillamine group (adjusted rate: 48.4 events per 100 participants) versus the no intervention group (37/155 (23.9 events per 100 participants)) (rate ratio 2.99, 95% CI 1.04 to 8.63; 303 participants; 3 trials; I2 = 75%), malotilate group (adjusted rate: 20.7 events per 100 participants) versus the no intervention group (2/49 (4.1 events per 100 participants)) (rate ratio 6.13, 95% CI 1.38 to 27.14; 101 participants; 1 trial); and obeticholic acid group (adjusted rate: 175.0 events per 100 participants) versus the no intervention group (96/38 (252.6 events per 100 participants)) (rate ratio 1.41, 95% CI 1.13 to 1.75; 76 participants; 1 trial); ; ; ; . The number of adverse events was higher in the methotrexate plus UDCA group (adjusted rate: 30.6 events per 100 participants) versus the UDCA group (0/12 (0.0 events per 100 participants)) (rate ratio 30.64, 95% CI 1.84 to 510.76; 27 participants; 1 trial). There was no evidence of a difference in any of the remaining comparisons (Analysis 1.7).
1.7. Analysis.
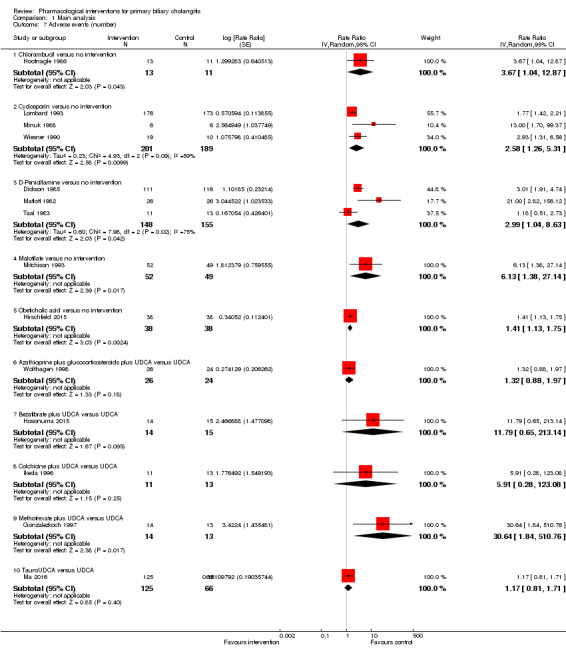
Comparison 1 Main analysis, Outcome 7 Adverse events (number).
Health‐related quality of life
None of the trials reported health‐related quality of life at any time point.
Liver transplantation
Eleven trials including 1561 participants reported liver transplantation (Neuberger 1985; Wiesner 1990; Lombard 1993; Heathcote 1994; Lindor 1994; Turner 1994; Eriksson 1997; Hendrickse 1999; Papatheodoridis 2002; Combes 2005; Hosonuma 2015). There was no evidence of a difference in any of the comparisons (Analysis 1.8).
1.8. Analysis.
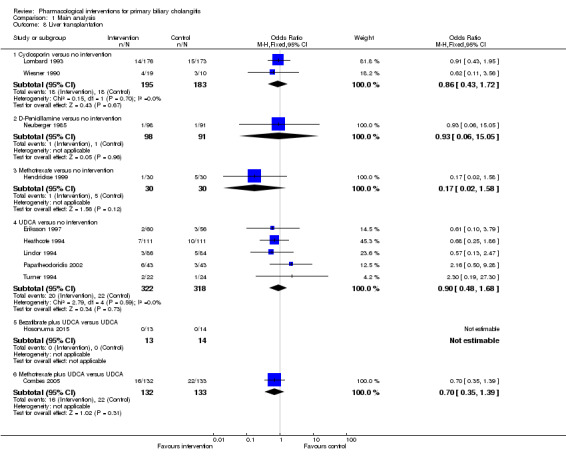
Comparison 1 Main analysis, Outcome 8 Liver transplantation.
Decompensated liver disease
Seven trials including 909 participants reported decompensated liver disease (Taal 1983; Combes 1995a; Almasio 2000; Papatheodoridis 2002; Combes 2005; Gao 2012; Nevens 2016). There was no evidence of a difference in any of the comparisons (Analysis 1.9).
1.9. Analysis.
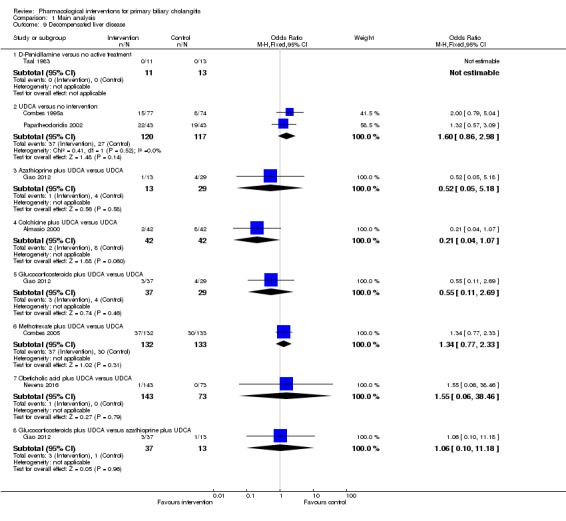
Comparison 1 Main analysis, Outcome 9 Decompensated liver disease.
Cirrhosis
Three trials including 103 participants reported cirrhosis (Heathcote 1976; Turner 1994; Wolfhagen 1998). There was no evidence of a difference in any of the comparisons (Analysis 1.10).
1.10. Analysis.
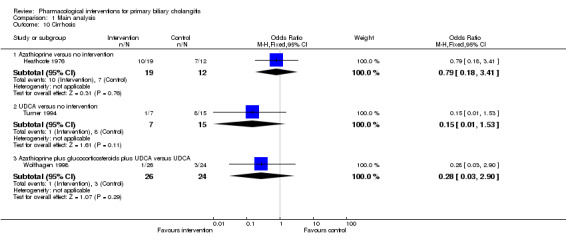
Comparison 1 Main analysis, Outcome 10 Cirrhosis.
Hepatocellular carcinoma
None of the trials reported hepatocellular carcinoma.
Subgroup analysis
All the trials were at high risk of bias for one or more domains. None of the trials reported separate data for symptomatic and asymptomatic participants, AMA‐positive and AMA‐negative participants, or for responders and non‐responders to bile acids. A secondary analysis performed by stratifying for the doses of UDCA and obeticholic acid revealed no differences between the main analysis except for the following.
There was no evidence of differences in the proportion of people with adverse events when stratified by the dose of obeticholic acid (obeticholic acid (high) versus no intervention: OR 16.60, 95% CI 0.90 to 305.59; 79 participants; 1 trial; obeticholic acid (moderate) versus no intervention: OR 8.81, 95% CI 1.01 to 76.73; 86 participants; 1 trial; and obeticholic acid (low) versus no intervention: OR 1.59, 95% CI 0.41 to 6.17; 76 participants; 1 trial). In addition, when stratified by dose, only obeticholic acid (high) had higher number of adverse events than no intervention (rate ratio 1.91, 95% CI 1.50 to 2.44; 79 participants; 1 trial). It also had higher number of adverse events than obeticholic acid (moderate) and obeticholic acid (low) (obeticholic acid (moderate) versus obeticholic acid (high): rate ratio 0.66, 95% CI 0.53 to 0.81; 89 participants; 1 trial; obeticholic acid (low) versus obeticholic acid (high): rate ratio 0.55, 95% CI 0.43 to 0.70; 79 participants; 1 trial).
Sensitivity analysis
We did not perform a sensitivity analysis of imputing information based on different scenarios because of paucity of data to carry out these analyses. We did not impute standard deviation; therefore, we did not perform a sensitivity analysis to assess the impact of imputing the standard deviation.
Reporting bias
We did not assess reporting bias by creating a funnel plot because of the few trials included under each comparison.
Using fixed‐effect model versus random‐effects model
The interpretation of results was not altered based on the model used for analysis.
Quality of evidence
The overall quality of evidence was very low for all the outcomes (Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7). This was because of the high risk of bias in all the trials (downgraded by two levels); small sample sizes for all outcomes and wide CIs (downgraded by two levels for imprecision) and heterogeneity (downgraded by two levels) for some of the outcomes.
Sample size calculations and Trial Sequential Analysis
The required sample size for identifying a 20% relative risk reduction in the different outcomes based on an alpha error of 5%, a beta error of 20%, and the control group proportion observed across trials were as follows.
Mortality (up to one year) (control group proportion: 25.2%): 2166 participants.
Mortality (one to five years) (control group proportion: 20.0%): 2894 participants.
Mortality at maximal follow‐up (control group proportion: 20.8%): 2758 participants.
Serious adverse events (proportion) (control group proportion: 0.4%): 175,996 participants.
Adverse events (proportion) (control group proportion: 27%): 1978 participants.
Liver transplantation (control group proportion: 7.4%): 8910 participants.
Decompensated liver disease (control group proportion: 20.8%): 2758 participants.
Cirrhosis (control group proportion: 55.6%): 632 participants.
The above mentioned are sample sizes uncorrected for heterogeneity. In the presence of heterogeneity, for example, in the presence of a heterogeneity of 27%, the required information size for adverse events (proportion) is 1978/(1 ‐ 0.27) = 2710 participants.
As shown in Figure 4, Figure 5, and Figure 6, the accrued sample sizes were only small fractions of the diversity‐adjusted required information size (DARIS) and therefore, the boundaries could not be drawn. There was a high risk of random errors. The TSA‐adjusted CI could not be calculated as there was too little information for the calculation (i.e. the CIs were wide).
4.
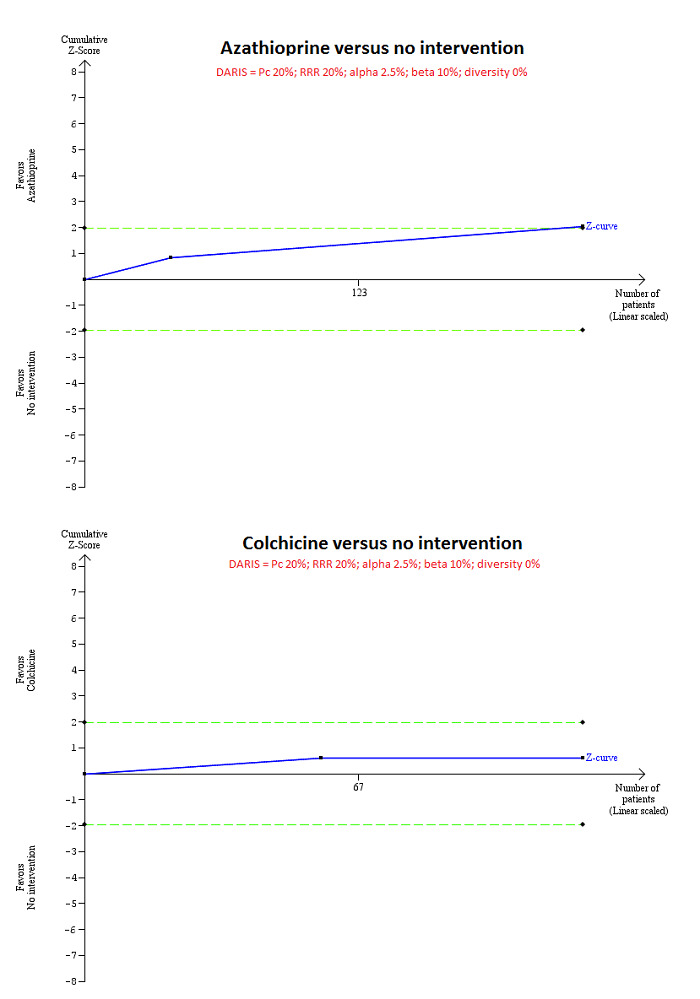
Trial Sequential Analysis of mortality at maximal follow‐up: azathioprine versus no intervention and colchicine versus no intervention.
Based on an alpha error of 2.5%, power of 90% (beta error of 10%), a relative risk reduction (RRR) of 20%, a control group proportion observed in the trials (Pc = 20%), and diversity observed in the analyses (0%), the accrued sample size (224 for azathioprine versus intervention and 122 for colchicine versus no intervention) was only a small fraction of the diversity adjusted required information size (DARIS) (4580 for both comparisons); therefore, the trial sequential monitoring boundaries were not drawn. The Z‐curve (blue line) crossed the conventional boundaries (dotted green line) favouring azathioprine for azathioprine versus no intervention, but did not cross the conventional boundaries for colchicine versus no intervention. This indicates that there is a high risk of random errors in both these comparisons.
5.
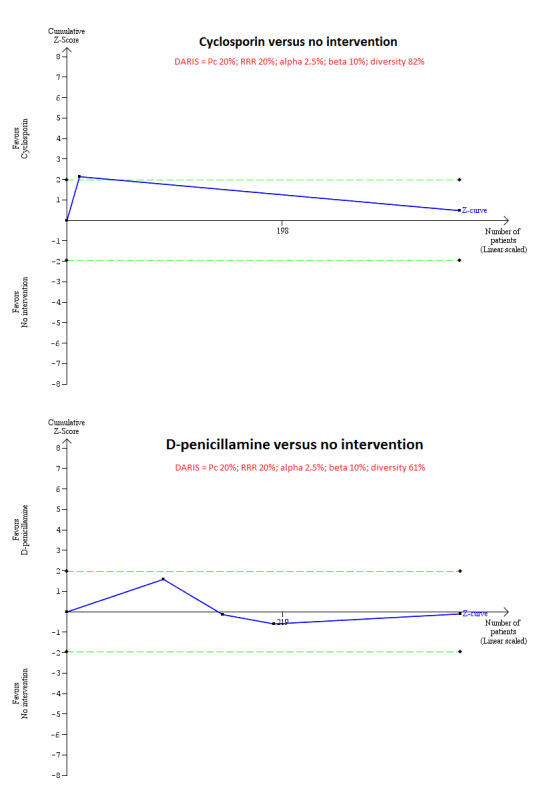
Trial Sequential Analysis of mortality at maximal follow‐up: ciclosporin versus no intervention and D‐penicillamine versus no intervention.
Based on an alpha error of 2.5%, power of 90% (beta error of 10%), a relative risk reduction (RRR) of 20%, a control group proportion observed in the trials (Pc = 20%), and diversity observed in the analyses (82% for ciclosporin versus no intervention and 61% for D‐penicillamine versus no intervention), the accrued sample size (394 for ciclosporin versus no intervention and 423 for D‐penicillamine versus no intervention) was only a small fraction of the diversity adjusted required information size (DARIS) (25,098 for ciclosporin versus no intervention and 11,623 for D‐penicillamine versus no intervention); therefore, the trial sequential monitoring boundaries were not drawn. The Z‐curve (blue line) crossed the conventional boundaries (dotted green line) favouring ciclosporin for ciclosporin versus no intervention, but did not cross the conventional boundaries for D‐penicillamine versus no intervention. This indicates that there is a high risk of random errors in both these comparisons.
6.
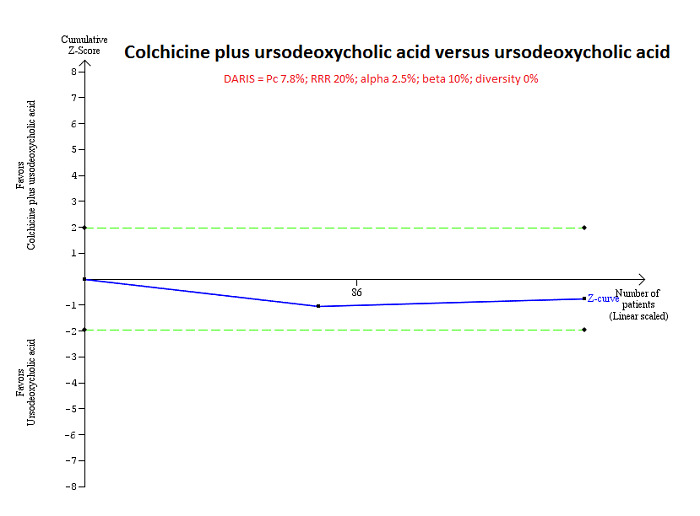
Trial Sequential Analysis of mortality at maximal follow‐up: colchicine plus UDCA versus UDCA.
Based on an alpha error of 2.5%, power of 90% (beta error of 10%), a relative risk reduction (RRR) of 20%, a control group proportion observed in the trials (Pc = 7.8%), and diversity observed in the analyses (0%), the accrued sample size (160 participants) was only a small fraction of the diversity adjusted required information size (DARIS) (13,316); therefore, the trial sequential monitoring boundaries were not drawn. The Z‐curve (blue line) did not cross the conventional boundaries (green dotted line). This indicates that there is a high risk of random errors in both this comparison.
Discussion
Summary of main results
We included 74 trials (5902 participants) in this review, and included 4274 participants from 46 trials in one or more outcomes in this review. We did not perform the planned network meta‐analysis because there was no closed loop (i.e. outcomes for which direct and indirect estimates were available to allow us estimation of inconsistency). Therefore, we reported only the direct comparisons. We reported the results from frequentist meta‐analysis only as it is more familiar to people.
Although mortality at maximal follow‐up was lower in people who received azathioprine versus no intervention, there was no evidence of any reduction in mortality by any intervention, either at less than one year or between one and five years. However, this evidence is unreliable because the Christensen 1985 trial excluded a large proportion of participants (25%) (i.e. only 185/224 participants were included in the meta‐analysis). The Trial Sequential Analysis showed that only a small proportion of the required information size was reached and the risk of random errors was high. In addition to the risk of systematic errors and random errors, the proportion of people who died was high (71.3%) in the no intervention group of the Christensen 1985 trial compared to the other trials (the overall mortality at maximal follow‐up was 20.8%). Although this difference could be due to the shorter follow‐up periods in some of the other trials, the mortality observed in this trial was much higher than that observed in the other trials with similar or longer follow‐up such as Epstein 1979; Hendrickse 1999; and Papatheodoridis 2002. The general care of people with cirrhosis is likely to have improved since the 1980s and it is unlikely to be as high as that mortality observed in Christensen 1985. This is another reason why there is no need to recommend azathioprine routinely in people with primary biliary cholangitis.
There was no evidence of a decrease in liver transplantation, decompensated liver disease, or cirrhosis in any of the interventions compared with no intervention. However, several interventions increased the number of people with, and total number of, adverse events. Although the Trial Sequential Analysis revealed that only a small proportion of the required information size was reached, the risk of random errors was high. Thus, concluding that there were more adverse events in some of these comparisons is only of academic interest because none of the interventions appeared to result in clinical benefit.
However, it has to be pointed out that the periods of follow‐up in the trials were sufficiently long to identify any differences in clinical outcomes because primary biliary cholangitis is a slowly progressive disease. Trials with sufficient follow‐up (e.g. five or 10 years) are required to detect any differences in clinically important outcomes.
Overall completeness and applicability of evidence
The trials included symptomatic and asymptomatic primary biliary cholangitis, AMA‐positive and AMA‐negative primary biliary cholangitis, treatment‐naive people, and people regardless of the treatments that they had received previously. However, majority of the trials excluded people with advanced liver cirrhosis and primary biliary cholangitis in people with other liver diseases. Therefore, this review is applicable to people with primary biliary cholangitis without advanced liver cirrhosis or with coexisting other liver diseases.
Quality of the evidence
The overall quality of evidence was very low for all the outcomes. The major reasons for this were the high risk of bias in the trials, in particular, exclusion of participants from the analysis after randomisation, small sample size, and imprecision. Overall, there were serious concerns about whether the effect estimates observed were the true effect estimates.
Potential biases in the review process
We followed the guidance of the Cochrane Handbook for Systematic Reviews of Interventions with two review authors independently selecting trials and extracting data (Higgins 2011). We performed a thorough search of the literature. However, the search period includes the premandatory trial registration era and it is possible that some trials on treatments that were not effective or were harmful were not reported at all.
We excluded studies which compared variations in the different treatments. Hence, this review does not provide information on whether one variation is better than another.
We only included randomised clinical trials which are known to focus mostly on benefits and do not collect and report harms in a detailed manner. Therefore, we might have missed a large number of studies that addressed the reporting of harms. Accordingly, this review is biased towards benefits ignoring harms. We did not search for interventions and trials registered at regulatory authorities (e.g. US Food and Drug Administration; European Medicines Agency, etc.). This may have overlooked trials and as such trials usually are unpublished, the lack of inclusion of such trials may make our comparisons look more advantageous than they really are. However, this is of academic interest only because there is no evidence of benefit of any treatment in people with primary biliary cholangitis (i.e. there is no reason to suggest that any of the treatments should be used in routine clinical practice regardless of the adverse event profile of the intervention).
We planned to perform a network meta‐analysis. However, it was not possible to assess whether the potential effect modifiers were similar across different comparisons. Performing a network meta‐analysis in this scenario can be misleading. Therefore, we did not perform the network meta‐analysis, and assessed the comparative benefits and harms of different interventions using standard Cochrane methodology.
Agreements and disagreements with other studies or reviews
We identified three network meta‐analyses on this topic (Zhu 2015a; Zhu 2015b; Zhu 2015c). We disagreed with the authors of these reviews that UDCA in combination with corticosteroids or methotrexate are effective interventions in the treatment of primary biliary cholangitis. The disagreements were probably due to considering mortality and liver transplantation separately in this review compared to Zhu 2015a and Zhu 2015b and only including evidence prior to cross‐over in our review. In particular, the cross‐over was not true cross‐over where the interventions were swapped but all the participants belonging to the 'no intervention' were switched over to the intervention. Therefore, it was not possible to obtain the effect estimate adjusted for intra‐participant correlation either. It should be also noted that the decision to switch the no intervention to intervention was based on improvement of some laboratory parameters which are invalidated surrogate outcomes. This can only be considered as observational evidence. We disagree with current EASL and AASLD guideline recommendations that UDCA should be used for the management of primary biliary cholangitis (EASL 2009; Lindor 2009). Again, these recommendations were based on observational evidence and invalidated surrogate outcomes (Gluud 2007).
We agreed with several systematic reviews that showed that none of the interventions are effective in improving survival or other major clinical outcomes such as cirrhosis or liver transplantation (Giljaca 2010; Rudic 2012a; Rudic 2012b; Yin 2015; Zhang 2015).
Authors' conclusions
Implications for practice.
Based on very low quality evidence, there is currently no evidence that any intervention is beneficial for primary biliary cholangitis.
Implications for research.
Randomised clinical trials need to be conducted and reported according to the SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) statement (Chan 2013) and the CONSORT statement (Schulz 2010). Future randomised clinical trials ought to be adequately powered, performed in people who are generally seen in the clinic rather than in highly selected participants, employ blinding, avoid post‐randomisation dropouts or planned cross‐overs, should have sufficient follow‐up period (e.g. five to 10 years or more), and use clinically important outcomes such as mortality, health‐related quality of life, cirrhosis, decompensated cirrhosis, and liver transplantation.
What's new
| Date | Event | Description |
|---|---|---|
| 12 April 2017 | Amended | The Cochrane Central Editorial Unit requested removal of the 'attempted network meta‐analysis' phrase from the end of the review title, as this further description of the review might create confusion in the reader. Although we followed the planned methodology for network meta‐analysis, it was not possible to assess whether the potential effect modifiers were similar across different comparisons. Therefore, we did not perform the network meta‐analysis and instead assessed the comparative benefits and harms of different interventions using standard Cochrane methodology. |
Notes
There is considerable overlap between the 'Methods' of this review and those of several other reviews written by the same group of authors.
Acknowledgements
We thank the Cochrane Comparing of Multiple Interventions Methods Group and the Cochrane Hepato‐Biliary for their support and advice.
Peer reviewers: Frederik Nevens, Belgium; Annarosa Floreani, Italy. Contact editor: Christian Gluud, Denmark. Sign‐off editor: Christian Gluud, Denmark.
Cochrane Review Group funding acknowledgement: the Danish State is the largest single funder of the Cochrane Hepato‐Biliary Group through its investment in The Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Copenhagen University Hospital, Denmark.
Disclaimer: the views and opinions expressed in this review are those of the review authors and do not necessarily reflect those of the Danish State or The Copenhagen Trial Unit.
Appendices
Appendix 1. Methods for network meta‐analysis if we find this is possible in future
Measures of treatment effect
Relative treatment effects
For dichotomous variables (e.g. proportion of participants with serious adverse events or any adverse events), we will calculate the odds ratio with 95% credible interval (or Bayesian confidence interval) (Severini 1993). For continuous variables (e.g. quality of life reported on the same scale), we will calculate the mean difference with 95% credible interval. We will use standardised mean difference values with 95% credible interval for quality of life if included trials use different scales. For count outcomes (e.g. number of adverse events and serious adverse events), we will calculate the rate ratio with 95% credible interval. For time‐to‐event data (e.g. mortality at maximal follow‐up), we will calculate hazard ratio with 95% credible interval.
Relative ranking
We will estimate the ranking probabilities for all treatments of being at each possible rank for each intervention. Then, we will obtain the surface under the cumulative ranking curve (SUCRA) (cumulative probability) and rankogram (Salanti 2011; Chaimani 2013).
Unit of analysis issues
We will collect data for all trial treatment groups that meet the inclusion criteria. The codes for analysis, that we will use, accounts for the correlation between the effect sizes from trials with more than two groups.
Assessment of heterogeneity
We will assess clinical and methodological heterogeneity by carefully examining the characteristics and design of included trials. We will assess the presence of clinical heterogeneity by comparing effect estimates under different categories of potential effect modifiers. Different study designs and risk of bias may contribute to methodological heterogeneity.
We will assess the statistical heterogeneity by comparing the results of the fixed‐effect model meta‐analysis and the random‐effects model meta‐analysis, between‐study standard deviation (tau2 and comparing this with values reported in the study of the distribution of between‐study heterogeneity (Turner 2012)), and by calculating the I2 statistic (using Stata/SE 14.2). If we identify substantial heterogeneity, clinical, methodological, or statistical, we will explore and address heterogeneity in a subgroup analysis (see 'Subgroup analysis and investigation of heterogeneity for network meta‐analysis' section).
Assessment of transitivity across treatment comparisons
We will evaluate the plausibility of transitivity assumption (the assumption that the participants included in the different studies with different immunosuppressive regimens can be considered to be a part of a multi‐arm randomised clinical trial and could potentially have been randomised to any of the treatments) (Salanti 2012). In other words, any participant that meets the inclusion criteria is, in principle, equally likely to be randomised to any of the above eligible interventions. If there was any concern that the clinical safety and effectiveness are dependent upon the effect modifiers, we will not perform a network meta‐analysis on all participant subgroups.
Assessment of reporting biases
For the network meta‐analysis, we will judge the reporting bias by the completeness of the search (i.e. searching various databases and including conference abstracts), as we do not currently find any meaningful order to perform a comparison‐adjusted funnel plot as suggested by Chaimani 2012. However, if we find any meaningful order, for example, the control group used depended upon the year of conduct of the trial, we will use comparison‐adjusted funnel plot as suggested by Chaimani 2012.
Data synthesis
Methods for indirect and mixed comparisons
We will conduct network meta‐analyses to compare multiple interventions simultaneously for each of the primary and secondary outcomes. Network meta‐analysis combines direct evidence within trials and indirect evidence across trials (Mills 2012). We will obtain a network plot to ensure that the trials were connected by treatments using Stata/SE 14.2 (Chaimani 2013). We will exclude any trials that were not connected to the network. We will conduct a Bayesian network meta‐analysis using the Markov chain Monte Carlo method in OpenBUGS 3.2.3 as per the guidance from the National Institute for Health and Care Excellence (NICE) Decision Support Unit (DSU) documents (Dias 2014a). We will model the treatment contrast (i.e. log odds ratio for binary outcomes, mean difference or standardised mean difference for continuous outcomes, log rate ratio for count outcomes, and log hazard ratio for time‐to‐event outcomes) for any two interventions ('functional parameters') as a function of comparisons between each individual intervention and an arbitrarily selected reference group ('basic parameters') (Lu 2006) using appropriate likelihood functions and links. We will use binomial likelihood and logit link for binary outcomes, Poisson likelihood and log link for count outcomes, binomial likelihood and complementary log‐log link for time‐to‐event outcomes, and normal likelihood and identity link for continuous outcomes. We will perform a fixed‐effect model and random‐effects model for the network meta‐analysis. We will report both models for comparison with the reference group in a forest plot. For pairwise comparison, we will report the fixed‐effect model if the two models report similar results; otherwise, we will report the more conservative model.
We will use a hierarchical Bayesian model using three different initial values using codes provided by NICE DSU (Dias 2014a). We will use a normal distribution with large variance (10,000) for treatment effect priors (vague or flat priors). For the random‐effects model, we will use a prior distributed uniformly (limits: 0 to 5) for between‐trial standard deviation but assume similar between‐trial standard deviation across treatment comparisons (Dias 2014a). We will use a 'burn‐in' of 5000 simulations, check for convergence visually, and run the models for another 10,000 simulations to obtain effect estimates. If we do not obtain convergence, we will increase the number of simulations for 'burn‐in'. If we do not obtain convergence still, we will use alternate initial values and priors using methods suggested by Van Valkenhoef 2012. We will also estimate the probability that each intervention ranks at one of the possible positions using the NICE DSU codes (Dias 2014a).
Assessment of inconsistency
We will assess inconsistency (statistical evidence of the violation of transitivity assumption) by fitting both an inconsistency model and a consistency model. We will use the inconsistency models used in the NICE DSU manual, as we plan to use a common between‐study deviation for the comparisons (Dias 2014b). In addition, we will use the design‐by‐treatment full interaction model (Higgins 2012) and IF (inconsistency factor) plots (Chaimani 2013) to assess inconsistency. In the presence of inconsistency, we will assess whether the inconsistency is because of clinical or methodological heterogeneity by performing separate analyses for each of the different subgroups mentioned in the ‘Subgroup analysis and investigation of heterogeneity for network meta‐analysis’ section.
If there was evidence of inconsistency, we will identify areas in the network where substantial inconsistency might be present in terms of clinical and methodological diversities between trials and, when appropriate, limit network meta‐analysis to a more compatible subset of trials.
Direct comparison
We will perform the direct comparisons using the same codes and the same technical details.
Sample size calculations
To control for the risk of random errors, we will interpret the information with caution when the accrued sample size in the network meta‐analysis (i.e. across all treatment comparisons) is less than the required sample size (required information size). For calculation of the required information size, see Appendix 3.
Subgroup analysis and investigation of heterogeneity for network meta‐analysis
We will assess the differences in the effect estimates between the subgroups listed in Subgroup analysis' and 'Investigation of heterogeneity' sections using meta‐regression with the help of the OpenBUGS code (Dias 2012a) if we include a sufficient number of trials. We will use the potential modifiers as study level co‐variates for meta‐regression. We will calculate a single common interaction term (Dias 2012a). If the 95% credible intervals of the interaction term do not overlap zero, we will consider this as evidence of difference in subgroups.
Presentation of results
We will present the effect estimates with 95% credible interval for each pairwise comparisons calculated from the direct comparisons and network meta‐analysis. We will also present the cumulative probability of the treatment ranks (i.e. the probability that the treatment is within the top two, the probability that the treatment is within the top three, etc.) in graphs (SUCRA) (Salanti 2011). We will also plot the probability that each treatment is best, second best, third best, etc. for each of the different outcomes (rankograms), which are generally considered more informative (Salanti 2011; Dias 2012b).
We will present the 'Summary of findings' tables for mortality. In the 'Summary of findings', we will follow the approach suggested by Puhan and colleagues (Puhan 2014). First, we will calculate the direct and indirect effect estimates and 95% credible intervals using the node‐splitting approach (Dias 2010), that is calculate the direct estimate for each comparison by including only trials in which there was direct comparison of treatments and the indirect estimate for each comparison by excluding the trials in which there was direct comparison of treatments. Then we will rate the quality of direct and indirect effect estimates using GRADE which takes into account the risk of bias, inconsistency, directness of evidence, imprecision, and publication bias (Guyatt 2011). Then, we will present the estimates of the network meta‐analysis and rated the quality of network meta‐analysis effect estimates as the best quality of evidence between the direct and indirect estimates (Puhan 2014). In addition, in the same table, we will present illustrations and information on the number of trials and participants as per the standard 'Summary of findings' table.
Appendix 2. Search strategies
| Database | Time span | Search strategy |
| Cochrane Central Register of Controlled Trials (CENTRAL) (Wiley) | Issue 2, 2017. | #1 MeSH descriptor: [Liver Cirrhosis, Biliary] explode all trees #2 (primary biliary cholangitis or PBC) #3 #1 or #2 |
| MEDLINE (OvidSP) | January 1947 to February 2017. | 1. exp Liver Cirrhosis, Biliary/ 2. (primary biliary cholangitis or PBC).ti,ab. 3. 1 or 2 4. randomized controlled trial.pt. 5. controlled clinical trial.pt. 6. randomized.ab. 7. placebo.ab. 8. drug therapy.fs. 9. randomly.ab. 10. trial.ab. 11. groups.ab. 12. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 13. exp animals/ not humans.sh. 14. 12 not 13 15. 3 and 14 |
| Embase (OvidSP) | January 1974 to February 2017. | 1. exp primary biliary cholangitis/ 2. (primary biliary cholangitis or PBC).ti,ab. 3. 1 or 2 4. exp crossover‐procedure/ or exp double‐blind procedure/ or exp randomized controlled trial/ or single‐blind procedure/ 5. (((((random* or factorial* or crossover* or cross over* or cross‐over* or placebo* or double*) adj blind*) or single*) adj blind*) or assign* or allocat* or volunteer*).af. 6. 4 or 5 7. 3 and 6 |
| Science Citation Index Expanded (Web of Knowledge) | January 1945 to February 2017. | #1 TS=(primary biliary cholangitis or PBC) #2 TS=(random* OR rct* OR crossover OR masked OR blind* OR placebo* OR meta‐analysis OR systematic review* OR meta‐analys*) #3 #1 AND #2 |
| World Health Organization International Clinical Trials Registry Platform Search Portal (apps.who.int/trialsearch/Default.aspx) | February 2017. | Condition: "primary biliary cholangitis" or PBC |
| ClinicalTrials.gov | February 2017. | Interventional Studies | "primary biliary cholangitis" OR PBC | Phase 2, 3, 4 |
Appendix 3. Sample size calculation
The five‐year mortality in people with primary biliary cholangitis is 20% (Kim 2000). The required information size based on a control group proportion of 20%, a relative risk reduction of 20% in the intervention group, type I error of 5%, and type II error of 20% is 2894 participants. Network analyses are more prone to the risk of random errors than direct comparisons (Del Re 2013). Accordingly, a greater sample size is required in indirect comparisons than direct comparisons (Thorlund 2012). The power and precision in indirect comparisons depends upon various factors, such as the number of participants included under each comparison and the heterogeneity between the trials (Thorlund 2012). If there is no heterogeneity across the trials, the sample size in indirect comparisons would be equivalent to the sample size in direct comparisons. The effective indirect sample size can be calculated using the number of participants included in each direct comparison (Thorlund 2012). For example, a sample size of 2500 participants in the direct comparison A versus C (nAC) and a sample size of 7500 participants in the direct comparison B versus C (nBC) results in an effective indirect sample size of 1876 participants. However, in the presence of heterogeneity within the comparisons, the sample size required is higher. In the above scenario, for an I2 statistic for each of the comparisons A versus C (IAC2) and B versus C (IBC2) of 25%, the effective indirect sample size is 1407 participants. For an I2 statistic for each of the comparisons A versus C and B versus C of 50%, the effective indirect sample size is 938 participants (Thorlund 2012). If there were only three groups and the sample size in the trials is more than the required information size, we planned to calculate the effective indirect sample size using the following generic formula (Thorlund 2012):
((nAC × (1 ‐ IAC2)) × (nBC × (1 ‐ IBC2))/((nAC × (1 ‐ IAC2)) + (nBC × (1 ‐ IBC2)).
There is currently no method to calculate the effective indirect sample size for a network analysis involving more than three intervention groups.
Data and analyses
Comparison 1. Main analysis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality at maximal follow‐up | 28 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Azathioprine versus no intervention | 2 | 224 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.32, 0.98] |
| 1.2 Chlorambucil versus no intervention | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 3.28] |
| 1.3 Colchicine versus no intervention | 2 | 122 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.32, 1.85] |
| 1.4 Cyclosporin versus no intervention | 3 | 390 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.51, 1.50] |
| 1.5 D‐Penicillamine versus no intervention | 5 | 423 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.57, 1.44] |
| 1.6 Glucocorticosteroids versus no intervention | 1 | 36 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.14, 2.92] |
| 1.7 Malotilate versus no intervention | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.47, 8.48] |
| 1.8 Methotrexate versus no intervention | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.83 [1.01, 76.96] |
| 1.9 UDCA versus no intervention | 6 | 734 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.60, 1.64] |
| 1.10 Bezafibrate plus UDCA versus UDCA | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 9.67 [0.45, 207.78] |
| 1.11 Colchicine plus UDCA versus UDCA | 2 | 158 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.84 [0.38, 8.91] |
| 1.12 Methotrexate plus UDCA versus UDCA | 2 | 290 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.55, 2.51] |
| 1.13 Obeticholic acid plus UDCA versus UDCA | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.06, 38.46] |
| 2 Mortality (< 1 year) | 8 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Azathioprine versus no intervention | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.16, 2.10] |
| 2.2 Colchicine versus no intervention | 1 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.22, 3.33] |
| 2.3 Cyclosporin versus no intervention | 1 | 12 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 3.63] |
| 2.4 D‐Penicillamine versus no intervention | 1 | 189 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.35, 1.42] |
| 2.5 Ursodeoxycholic acid (UDCA) versus no intervention | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.6 Colchicine plus UDCA versus UDCA | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.13, 7.45] |
| 2.7 Methotrexate plus UDCA versus UDCA | 1 | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.8 Obeticholic acid plus UDCA versus UDCA | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.06, 38.46] |
| 3 Mortality (1 to 5 years) | 20 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Azathioprine versus no intervention | 1 | 185 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.30, 1.04] |
| 3.2 Chlorambucil versus no intervention | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 3.28] |
| 3.3 Colchicine versus no intervention | 1 | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.22, 2.25] |
| 3.4 Cyclosporin versus no intervention | 2 | 378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.54, 1.64] |
| 3.5 D‐Penicillamine versus no intervention | 4 | 234 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.59, 2.08] |
| 3.6 Glucocorticosteroids versus no intervention | 1 | 36 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.14, 2.92] |
| 3.7 Malotilate versus no intervention | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.47, 8.48] |
| 3.8 Methotrexate versus no intervention | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.83 [1.01, 76.96] |
| 3.9 UDCA versus no intervention | 5 | 716 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.60, 1.64] |
| 3.10 Bezafibrate plus UDCA versus UDCA | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 9.67 [0.45, 207.78] |
| 3.11 Colchicine plus UDCA versus UDCA | 1 | 74 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.28 [0.24, 113.87] |
| 3.12 Methotrexate plus UDCA versus UDCA | 1 | 265 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.55, 2.51] |
| 4 Serious adverse events (proportion) | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Colchicine versus no intervention | 1 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 D‐Penicillamine versus no intervention | 1 | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 28.77 [1.57, 526.67] |
| 4.3 Obeticholic acid versus no intervention | 1 | 165 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.21, 15.73] |
| 4.4 UDCA versus no intervention | 3 | 380 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.5 UDCA versus bezafibrate | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.6 Bezafibrate plus UDCA versus UDCA | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.7 Colchicine plus UDCA versus UDCA | 1 | 74 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.08 [0.12, 78.14] |
| 4.8 Lamivudine plus zidovudine plus UDCA versus UDCA | 1 | 59 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.04, 5.43] |
| 4.9 Obeticholic acid plus UDCA versus UDCA | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.58 [1.02, 12.51] |
| 5 Serious adverse events (number of events) | 1 | Rate Ratio (Fixed, 95% CI) | Subtotals only | |
| 5.1 Obeticholic acid plus UDCA versus UDCA | 1 | 216 | Rate Ratio (Fixed, 95% CI) | 1.66 [0.75, 3.66] |
| 6 Adverse events (proportion) | 19 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Cyclosporin versus no intervention | 3 | 390 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.04 [1.98, 4.68] |
| 6.2 D‐Penicillamine versus no intervention | 2 | 287 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.51 [2.56, 7.93] |
| 6.3 Malotilate versus no intervention | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 11.43 [1.40, 93.04] |
| 6.4 Obeticholic acid versus no intervention | 1 | 165 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.58 [1.31, 15.95] |
| 6.5 UDCA versus no intervention | 3 | 380 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.50, 4.25] |
| 6.6 Azathioprine plus UDCA versus UDCA | 1 | 42 | Odds Ratio (M‐H, Fixed, 95% CI) | 19.67 [0.94, 413.50] |
| 6.7 Bezafibrate versus UDCA | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.8 Bezafibrate plus UDCA versus UDCA | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.29 [0.12, 89.81] |
| 6.9 Colchicine plus UDCA versus UDCA | 2 | 42 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.20 [0.63, 60.80] |
| 6.10 Colestilan plus UDCA versus UDCA | 1 | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.11 Glucocorticosteroids plus UDCA versus UDCA | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.54 [1.35, 22.84] |
| 6.12 Methotrexate plus UDCA versus UDCA | 1 | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 115.0 [4.98, 2657.48] |
| 6.13 TauroUDCA versus UDCA | 1 | 30 | Odds Ratio (M‐H, Fixed, 95% CI) | 21.0 [2.16, 204.61] |
| 6.14 Glucocorticosteroids plus UDCA versus azathioprine plus UDCA | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.08, 2.12] |
| 7 Adverse events (number) | 14 | Rate Ratio (Random, 95% CI) | Subtotals only | |
| 7.1 Chlorambucil versus no intervention | 1 | 24 | Rate Ratio (Random, 95% CI) | 3.67 [1.04, 12.87] |
| 7.2 Cyclosporin versus no intervention | 3 | 390 | Rate Ratio (Random, 95% CI) | 2.58 [1.26, 5.31] |
| 7.3 D‐Penicillamine versus no intervention | 3 | 303 | Rate Ratio (Random, 95% CI) | 2.99 [1.04, 8.63] |
| 7.4 Malotilate versus no intervention | 1 | 101 | Rate Ratio (Random, 95% CI) | 6.13 [1.38, 27.14] |
| 7.5 Obeticholic acid versus no intervention | 1 | 76 | Rate Ratio (Random, 95% CI) | 1.41 [1.13, 1.75] |
| 7.6 Azathioprine plus glucocorticosteroids plus UDCA versus UDCA | 1 | 50 | Rate Ratio (Random, 95% CI) | 1.32 [0.88, 1.97] |
| 7.7 Bezafibrate plus UDCA versus UDCA | 1 | 29 | Rate Ratio (Random, 95% CI) | 11.79 [0.65, 213.14] |
| 7.8 Colchicine plus UDCA versus UDCA | 1 | 24 | Rate Ratio (Random, 95% CI) | 5.91 [0.28, 123.08] |
| 7.9 Methotrexate plus UDCA versus UDCA | 1 | 27 | Rate Ratio (Random, 95% CI) | 30.64 [1.84, 510.76] |
| 7.10 TauroUDCA versus UDCA | 1 | 191 | Rate Ratio (Random, 95% CI) | 1.17 [0.81, 1.71] |
| 8 Liver transplantation | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Cyclosporin versus no intervention | 2 | 378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.43, 1.72] |
| 8.2 D‐Penicillamine versus no intervention | 1 | 189 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.06, 15.05] |
| 8.3 Methotrexate versus no intervention | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.58] |
| 8.4 UDCA versus no intervention | 5 | 640 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.48, 1.68] |
| 8.5 Bezafibrate plus UDCA versus UDCA | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.6 Methotrexate plus UDCA versus UDCA | 1 | 265 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.35, 1.39] |
| 9 Decompensated liver disease | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 D‐Penicillamine versus no active treatment | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 UDCA versus no intervention | 2 | 237 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.60 [0.86, 2.98] |
| 9.3 Azathioprine plus UDCA versus UDCA | 1 | 42 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.05, 5.18] |
| 9.4 Colchicine plus UDCA versus UDCA | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.04, 1.07] |
| 9.5 Glucocorticosteroids plus UDCA versus UDCA | 1 | 66 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.11, 2.69] |
| 9.6 Methotrexate plus UDCA versus UDCA | 1 | 265 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.77, 2.33] |
| 9.7 Obeticholic acid plus UDCA versus UDCA | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.06, 38.46] |
| 9.8 Glucocorticosteroids plus UDCA versus azathioprine plus UDCA | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.10, 11.18] |
| 10 Cirrhosis | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Azathioprine versus no intervention | 1 | 31 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.18, 3.41] |
| 10.2 UDCA versus no intervention | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 1.53] |
| 10.3 Azathioprine plus glucocorticosteroids plus UDCA versus UDCA | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.03, 2.90] |
Comparison 2. Stratified by dose.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality at maximal follow‐up | 29 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Azathioprine versus no intervention | 2 | 224 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.32, 0.98] |
| 1.2 Chlorambucil versus no intervention | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 3.28] |
| 1.3 Colchicine versus no intervention | 2 | 122 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.77 [0.32, 1.85] |
| 1.4 Cyclosporin versus no intervention | 3 | 390 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.51, 1.50] |
| 1.5 D‐Penicillamine versus no intervention | 5 | 423 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.57, 1.44] |
| 1.6 Glucocorticosteroids versus no intervention | 1 | 36 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.14, 2.92] |
| 1.7 Malotilate versus no intervention | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.47, 8.48] |
| 1.8 Methotrexate versus no intervention | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.83 [1.01, 76.96] |
| 1.9 UDCA (low) versus no intervention | 2 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.03, 3.47] |
| 1.10 UDCA (moderate) versus no intervention | 4 | 670 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.62, 1.77] |
| 1.11 UDCA (low) versus UDCA (high) | 1 | 106 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.06, 17.06] |
| 1.12 UDCA (moderate) versus UDCA (high) | 1 | 103 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.01, 9.05] |
| 1.13 UDCA (low) plus colchicine versus UDCA (low) | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.13, 7.45] |
| 1.14 UDCA (low) plus methotrexate versus UDCA (low) | 1 | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 1.15 UDCA (moderate) versus UDCA (low) | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.72] |
| 1.16 Bezafibrate plus UDCA (moderate) versus UDCA (moderate) | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 9.67 [0.45, 207.78] |
| 1.17 Colchicine plus UDCA (moderate) versus UDCA (moderate) | 1 | 74 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.28 [0.24, 113.87] |
| 1.18 Methotrexate plus UDCA (moderate) versus UDCA (moderate) | 1 | 265 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.55, 2.51] |
| 1.19 Obeticholic acid (low) plus UDCA (moderate) versus UDCA (moderate) | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.06, 38.46] |
| 2 Mortality (< 1 year) | 9 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 Azathioprine versus no intervention | 1 | 39 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.58 [0.16, 2.10] |
| 2.2 Colchicine versus no intervention | 1 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.22, 3.33] |
| 2.3 Cyclosporin versus no intervention | 1 | 12 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 3.63] |
| 2.4 D‐Penicillamine versus no intervention | 1 | 189 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.35, 1.42] |
| 2.5 UDCA (low) versus no intervention | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.6 UDCA (low) versus UDCA (high) | 1 | 106 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.04 [0.06, 17.06] |
| 2.7 UDCA (moderate) versus UDCA (high) | 1 | 103 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.01, 9.05] |
| 2.8 Obeticholic acid (low) plus UDCA (moderate) versus UDCA (moderate) | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.06, 38.46] |
| 2.9 UDCA (low) plus colchicine versus UDCA (low) | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.13, 7.45] |
| 2.10 UDCA (low) plus methotrexate versus UDCA (low) | 1 | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.11 UDCA (moderate) versus UDCA (low) | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.35 [0.01, 8.72] |
| 3 Mortality (1 to 5 years) | 20 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 Azathioprine versus no intervention | 1 | 185 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.30, 1.04] |
| 3.2 Chlorambucil versus no intervention | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 3.28] |
| 3.3 Colchicine versus no intervention | 1 | 58 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.22, 2.25] |
| 3.4 Cyclosporin versus no intervention | 2 | 378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.94 [0.54, 1.64] |
| 3.5 D‐Penicillamine versus no intervention | 4 | 234 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.59, 2.08] |
| 3.6 Glucocorticosteroids versus no intervention | 1 | 36 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.14, 2.92] |
| 3.7 Malotilate versus no intervention | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.47, 8.48] |
| 3.8 Methotrexate versus no intervention | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.83 [1.01, 76.96] |
| 3.9 UDCA (low) versus no intervention | 1 | 46 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.03, 3.47] |
| 3.10 UDCA (moderate) versus no intervention | 4 | 670 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.62, 1.77] |
| 3.11 Bezafibrate plus UDCA (moderate) versus UDCA (moderate) | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 9.67 [0.45, 207.78] |
| 3.12 Colchicine plus UDCA (moderate) versus UDCA (moderate) | 1 | 74 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.28 [0.24, 113.87] |
| 3.13 Methotrexate plus UDCA (moderate) versus UDCA (moderate) | 1 | 265 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.55, 2.51] |
| 4 Serious adverse events (proportion) | 12 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 Colchicine versus no intervention | 1 | 64 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 D‐Penicillamine versus no intervention | 1 | 52 | Odds Ratio (M‐H, Fixed, 95% CI) | 28.77 [1.57, 526.67] |
| 4.3 Obeticholic acid (high) versus no intervention | 1 | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.14 [0.57, 46.17] |
| 4.4 Obeticholic acid (low) versus no intervention | 1 | 76 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 8.22] |
| 4.5 Obeticholic acid (moderate) versus no intervention | 1 | 86 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.05, 13.01] |
| 4.6 UDCA (low) versus no intervention | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.7 UDCA (moderate) versus no intervention | 2 | 362 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.8 UDCA (low) versus bezafibrate | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.9 Obeticholic acid (low) versus obeticholic acid (high) | 1 | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.00, 1.61] |
| 4.10 Obeticholic acid (moderate) versus obeticholic acid (high) | 1 | 89 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.02, 1.37] |
| 4.11 Obeticholic acid (moderate) versus obeticholic acid (low) | 1 | 86 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.43 [0.10, 61.39] |
| 4.12 Lamivudine plus zidovudine plus UDCA (moderate) versus UDCA (moderate) | 1 | 59 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.04, 5.43] |
| 4.13 UDCA (moderate) versus obeticholic acid (low) plus UDCA (moderate) | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.08, 0.98] |
| 4.14 Bezafibrate plus UDCA (low) versus UDCA (low) | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.15 UDCA (moderate) versus UDCA (low) | 1 | 59 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.16 Colchicine plus UDCA (moderate) versus UDCA (moderate) | 1 | 74 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.08 [0.12, 78.14] |
| 5 Serious adverse events (number of events) | 1 | Rate Ratio (Fixed, 95% CI) | Subtotals only | |
| 5.1 Obeticholic acid (low) plus UDCA (moderate) versus UDCA (moderate) | 1 | Rate Ratio (Fixed, 95% CI) | 1.66 [0.75, 3.66] | |
| 6 Adverse events (proportion) | 20 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 Cyclosporin versus no intervention | 3 | 390 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.04 [1.98, 4.68] |
| 6.2 D‐Penicillamine versus no intervention | 2 | 287 | Odds Ratio (M‐H, Fixed, 95% CI) | 4.51 [2.56, 7.93] |
| 6.3 Malotilate versus no intervention | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 11.43 [1.40, 93.04] |
| 6.4 Obeticholic acid (high) versus no intervention | 1 | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 16.6 [0.90, 305.59] |
| 6.5 Obeticholic acid (low) versus no intervention | 1 | 76 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.59 [0.41, 6.17] |
| 6.6 Obeticholic acid (moderate) versus no intervention | 1 | 86 | Odds Ratio (M‐H, Fixed, 95% CI) | 8.81 [1.01, 76.73] |
| 6.7 UDCA (low) versus no intervention | 1 | 18 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.8 UDCA (moderate) versus no intervention | 2 | 362 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.45 [0.50, 4.25] |
| 6.9 Glucocorticosteroids plus UDCA (moderate) versus azathioprine plus UDCA (moderate) | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.08, 2.12] |
| 6.10 UDCA (low) versus bezafibrate | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.11 Obeticholic acid (low) versus obeticholic acid (high) | 1 | 79 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.09 [0.00, 1.78] |
| 6.12 Obeticholic acid (moderate) versus obeticholic acid (high) | 1 | 89 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.02, 9.62] |
| 6.13 Obeticholic acid (moderate) versus obeticholic acid (low) | 1 | 86 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.53 [0.59, 51.70] |
| 6.14 Bezafibrate plus UDCA (low) versus UDCA (low) | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.29 [0.12, 89.81] |
| 6.15 Colestilan plus UDCA (low) versus UDCA (low) | 1 | 11 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.16 Methotrexate plus UDCA (low) versus UDCA (low) | 1 | 25 | Odds Ratio (M‐H, Fixed, 95% CI) | 115.0 [4.98, 2657.48] |
| 6.17 UDCA (moderate) versus UDCA (low) | 1 | 59 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 6.18 Azathioprine plus UDCA (moderate) versus UDCA (moderate) | 1 | 42 | Odds Ratio (M‐H, Fixed, 95% CI) | 19.67 [0.94, 413.50] |
| 6.19 Colchicine plus UDCA (moderate) versus UDCA (moderate) | 2 | 42 | Odds Ratio (M‐H, Fixed, 95% CI) | 6.20 [0.63, 60.80] |
| 6.20 Glucocorticosteroids plus UDCA (moderate) versus UDCA (moderate) | 2 | 135 | Odds Ratio (M‐H, Fixed, 95% CI) | 5.54 [1.35, 22.84] |
| 6.21 TauroUDCA (moderate) versus UDCA (moderate) | 1 | 30 | Odds Ratio (M‐H, Fixed, 95% CI) | 21.0 [2.16, 204.61] |
| 7 Adverse events (number) | 15 | Rate Ratio (Fixed, 95% CI) | Subtotals only | |
| 7.1 Chlorambucil versus no intervention | 1 | Rate Ratio (Fixed, 95% CI) | 3.67 [1.04, 12.87] | |
| 7.2 Cyclosporin versus no intervention | 3 | Rate Ratio (Fixed, 95% CI) | 1.87 [1.51, 2.32] | |
| 7.3 D‐Penicillamine versus no intervention | 3 | Rate Ratio (Fixed, 95% CI) | 2.64 [1.78, 3.91] | |
| 7.4 Malotilate versus no intervention | 1 | Rate Ratio (Fixed, 95% CI) | 6.13 [1.38, 27.14] | |
| 7.5 Obeticholic acid (high) versus no intervention | 1 | Rate Ratio (Fixed, 95% CI) | 1.91 [1.50, 2.44] | |
| 7.6 Obeticholic acid (low) versus no intervention | 1 | Rate Ratio (Fixed, 95% CI) | 1.05 [0.80, 1.39] | |
| 7.7 Obeticholic acid (moderate) versus no intervention | 1 | Rate Ratio (Fixed, 95% CI) | 1.25 [0.97, 1.62] | |
| 7.8 Obeticholic acid (low) versus obeticholic acid (high) | 1 | Rate Ratio (Fixed, 95% CI) | 0.55 [0.43, 0.70] | |
| 7.9 Obeticholic acid (moderate) versus obeticholic acid (high) | 1 | Rate Ratio (Fixed, 95% CI) | 0.66 [0.53, 0.81] | |
| 7.10 Obeticholic acid (moderate) versus obeticholic acid (low) | 1 | Rate Ratio (Fixed, 95% CI) | 1.19 [0.93, 1.53] | |
| 7.11 UDCA (low) versus UDCA (high) | 1 | Rate Ratio (Fixed, 95% CI) | 2.08 [0.78, 5.53] | |
| 7.12 UDCA (moderate) versus UDCA (high) | 1 | Rate Ratio (Fixed, 95% CI) | 0.73 [0.21, 2.60] | |
| 7.13 UDCA (low) plus methotrexate versus UDCA (low) | 1 | Rate Ratio (Fixed, 95% CI) | 30.64 [1.84, 510.76] | |
| 7.14 UDCA (moderate) versus UDCA (low) | 1 | Rate Ratio (Fixed, 95% CI) | 0.35 [0.11, 1.10] | |
| 7.15 Azathioprine plus glucocorticosteroids plus UDCA (moderate) versus UDCA (moderate) | 1 | Rate Ratio (Fixed, 95% CI) | 1.32 [0.88, 1.97] | |
| 7.16 Bezafibrate plus UDCA (moderate) versus UDCA (moderate) | 1 | Rate Ratio (Fixed, 95% CI) | 11.79 [0.65, 213.14] | |
| 7.17 Colchicine plus UDCA (moderate) versus UDCA (moderate) | 1 | Rate Ratio (Fixed, 95% CI) | 5.91 [0.28, 123.08] | |
| 7.18 TauroUDCA (moderate) versus UDCA (moderate) | 1 | Rate Ratio (Fixed, 95% CI) | 1.17 [0.81, 1.71] | |
| 8 Liver transplantation | 12 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 Cyclosporin versus no intervention | 2 | 378 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.43, 1.72] |
| 8.2 D‐Penicillamine versus no intervention | 1 | 189 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.06, 15.05] |
| 8.3 Methotrexate versus no intervention | 1 | 60 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.17 [0.02, 1.58] |
| 8.4 UDCA (low) versus no intervention | 2 | 162 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.24, 4.06] |
| 8.5 UDCA (moderate) versus no intervention | 3 | 478 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.88 [0.44, 1.76] |
| 8.6 UDCA (low) versus UDCA (high) | 1 | 106 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.17 [0.13, 79.71] |
| 8.7 UDCA (moderate) versus UDCA (high) | 1 | 103 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.37 [0.13, 84.70] |
| 8.8 UDCA (moderate) versus UDCA (low) | 1 | 101 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.06, 17.47] |
| 8.9 Bezafibrate plus UDCA (moderate) versus UDCA (moderate) | 1 | 27 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8.10 Methotrexate plus UDCA (moderate) versus UDCA (moderate) | 1 | 265 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.70 [0.35, 1.39] |
| 9 Decompensated liver disease | 7 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 D‐Penicillamine versus no intervention | 1 | 24 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 9.2 UDCA (moderate) versus no intervention | 2 | 351 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.84, 2.12] |
| 9.3 Obeticholic acid (low) plus UDCA (moderate) versus UDCA (moderate) | 1 | 216 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.55 [0.06, 38.46] |
| 9.4 UDCA (low) plus colchicine versus UDCA (low) | 1 | 84 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.21 [0.04, 1.07] |
| 9.5 Azathioprine plus UDCA (moderate) versus UDCA (moderate) | 1 | 42 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.52 [0.05, 5.18] |
| 9.6 Glucocorticosteroids plus UDCA (moderate) versus UDCA (moderate) | 1 | 66 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.11, 2.69] |
| 9.7 Methotrexate plus UDCA (moderate) versus UDCA (moderate) | 1 | 151 | Odds Ratio (M‐H, Fixed, 95% CI) | 2.00 [0.79, 5.04] |
| 9.8 Glucocorticosteroids plus UDCA (moderate) versus azathioprine plus UDCA (moderate) | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.10, 11.18] |
| 10 Cirrhosis | 3 | Odds Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 Azathioprine versus no intervention | 1 | 31 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.79 [0.18, 3.41] |
| 10.2 UDCA (low) versus no intervention | 1 | 22 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.15 [0.01, 1.53] |
| 10.3 Azathioprine plus glucocorticosteroids plus UDCA (moderate) versus UDCA (moderate) | 1 | 50 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.28 [0.03, 2.90] |
2.1. Analysis.
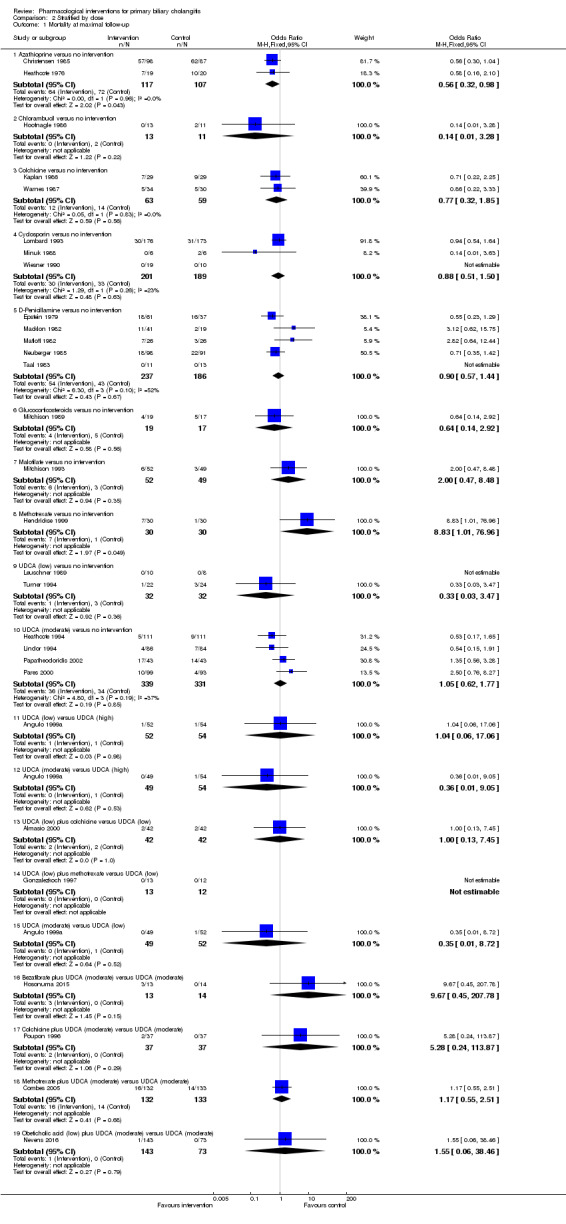
Comparison 2 Stratified by dose, Outcome 1 Mortality at maximal follow‐up.
2.2. Analysis.
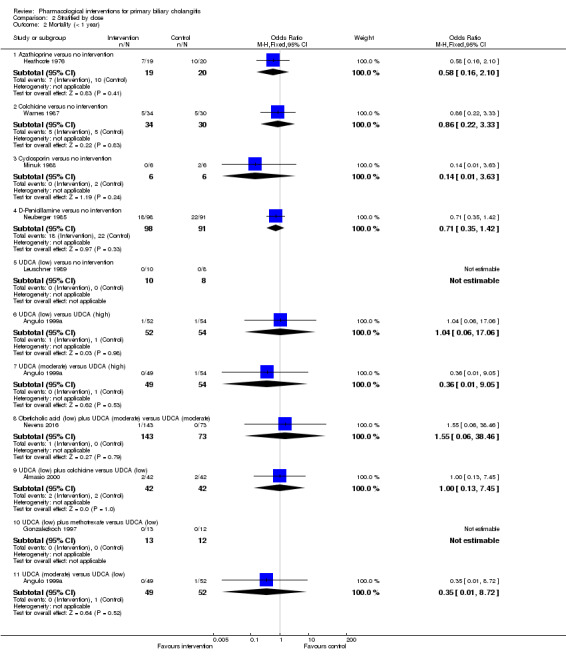
Comparison 2 Stratified by dose, Outcome 2 Mortality (< 1 year).
2.3. Analysis.
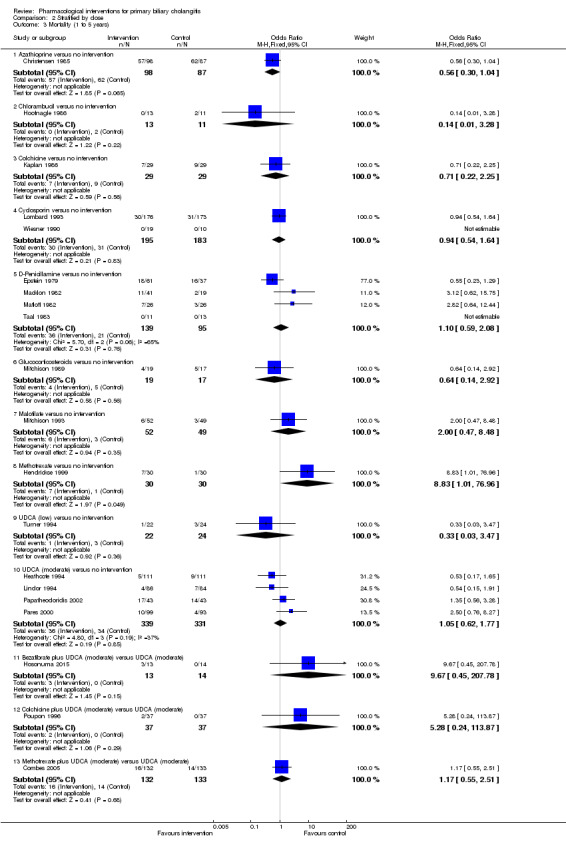
Comparison 2 Stratified by dose, Outcome 3 Mortality (1 to 5 years).
2.4. Analysis.
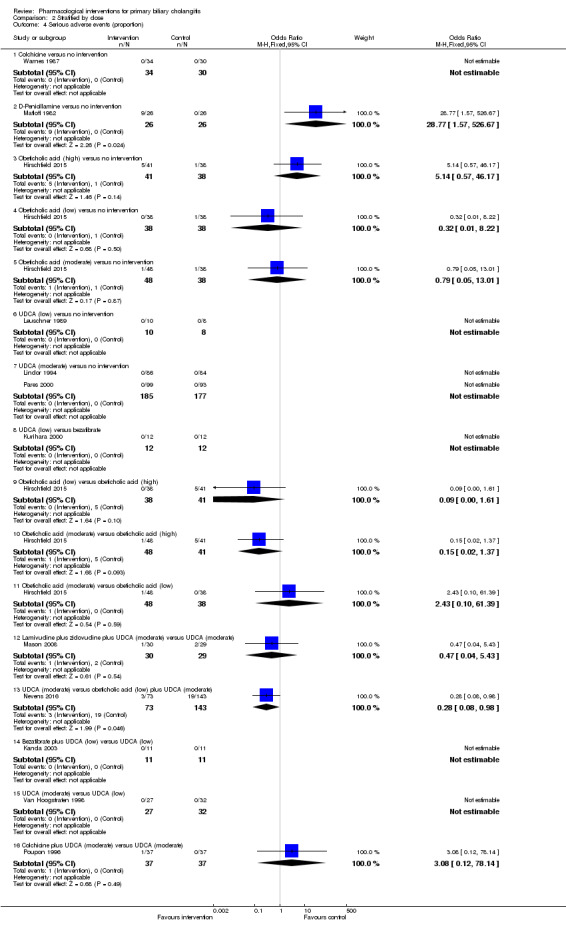
Comparison 2 Stratified by dose, Outcome 4 Serious adverse events (proportion).
2.5. Analysis.
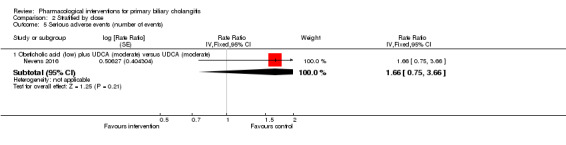
Comparison 2 Stratified by dose, Outcome 5 Serious adverse events (number of events).
2.6. Analysis.
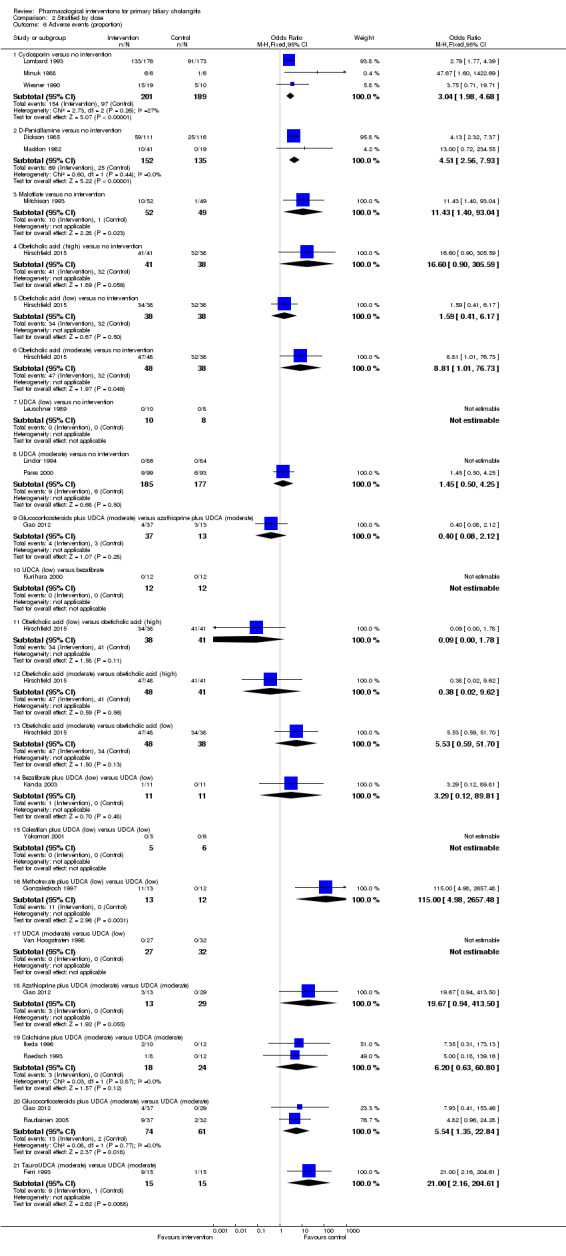
Comparison 2 Stratified by dose, Outcome 6 Adverse events (proportion).
2.7. Analysis.
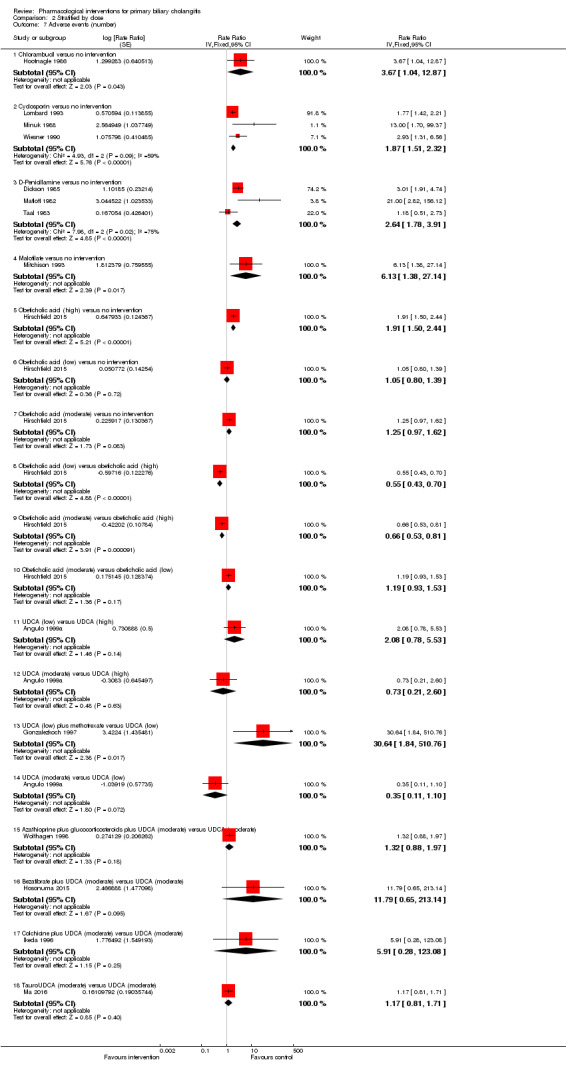
Comparison 2 Stratified by dose, Outcome 7 Adverse events (number).
2.8. Analysis.
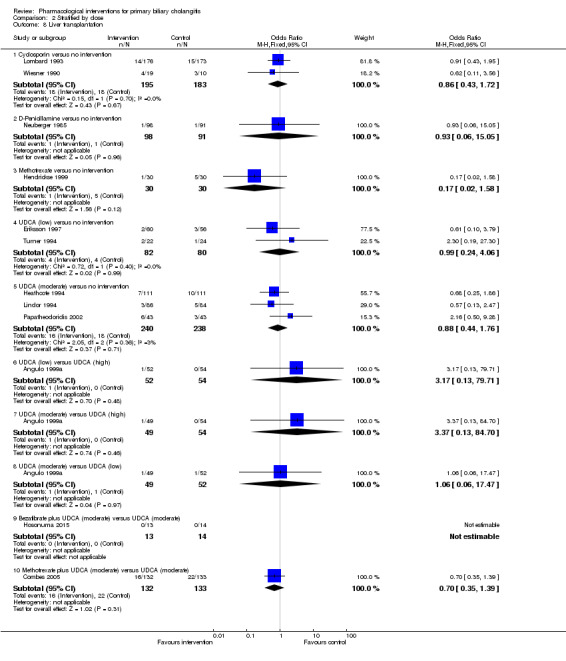
Comparison 2 Stratified by dose, Outcome 8 Liver transplantation.
2.9. Analysis.
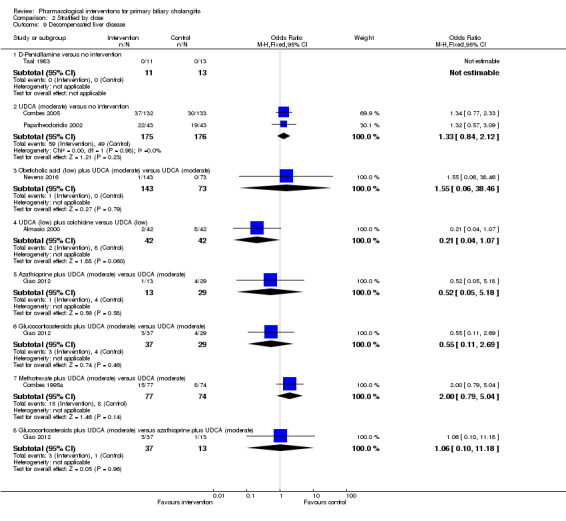
Comparison 2 Stratified by dose, Outcome 9 Decompensated liver disease.
2.10. Analysis.
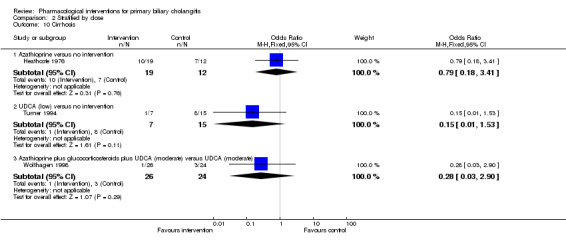
Comparison 2 Stratified by dose, Outcome 10 Cirrhosis.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Almasio 2000.
| Methods | Randomised clinical trial. | |
| Participants | Country: Italy. Number randomised: 90. Post‐randomisation dropouts: 6 (6.7%). Revised sample size: 84. Mean age: 54 years. Females: 81 (96.4%). Symptomatic participants: 84 (100%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + colchicine (n = 42). Further details: UDCA: 250 mg BD for 3 years + colchicine: 1 mg/day for 3 years. Group 2: UDCA (low) (n = 42). Further details: UDCA: 250 mg BD for 3 years. |
|
| Outcomes | Mortality, decompensated liver disease. | |
| Notes | Reasons for post‐randomisation dropouts: adverse effects and low compliance. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Colchicine, 1 mg daily, or an indistinguishable placebo were randomly assigned to patients according to a computer‐generated list developed separately for each centre". |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was performed by a central study unit…". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Low risk | Comment: no money received for the trial; the drug was provided by Abc Farmaceutici S.p.a (author's reply). |
| Other bias | Low risk | Comment: no other bias noted. |
Angulo 1999a.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 155. Post‐randomisation dropouts: not stated. Revised sample size: 155. Mean age: 53 years. Females: 130 (83.9%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 3 groups. Group 1: UDCA (low) (n = 52). Further details: UDCA: 5 mg/kg/day to 7 mg/kg/day; duration: 1 to 2 years. Group 2: UDCA (moderate) (n = 49). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day; duration: 1 to 2 years. Group 3: UDCA (high) (n = 54). Further details: UDCA: 23 mg/kg/day to 25 mg/kg/day; duration: 1 to 2 years. |
|
| Outcomes | Mortality, adverse events, liver transplantation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was carried out separately for each of the eight strata with a computer‐generated, blocked, randomized drug assignment schedule". |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomized by a statistician (D.W.M.), and the drug was provided by a pharmacist who was not involved in the patient's clinical evaluation or follow‐up". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients, physicians, nurses, and study coordinator were unaware throughout the study which dose was being administered. To assure blindness patients received the same number of tablets by mixing UDCA‐tablets with placebo‐tablets in a ratio defined by their assigned dose; therefore, the number of tablets taken per day according to the body weight was exactly the same regardless of the dose assigned". Comment: identical placebo used and authors stated double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: unclear whether all participants randomised were included in the analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other risk of bias. |
Arora 1990.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 9. Post‐randomisation dropouts: not stated. Revised sample size: 9. Mean age: not stated. Females: not stated. Symptomatic participants: 9 (100%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 5 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 5). Further details: UDCA: 10 mg/kg/day for 5 months. Group 2: placebo (n = 4). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: placebo used and authors stated double blind; however, unclear whether identical placebo used. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: placebo used and authors stated double blind; however, unclear whether identical placebo used. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other risk of bias. |
Askari 2010.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 28. Post‐randomisation dropouts: 0 (0%). Revised sample size: 28. Mean age: 54 years. Females: 26 (92.9%). Symptomatic participants: not stated. AMA positive: 27 (96.4%). Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: tetrathiomolybdate (n = 13). Further details: tetrathiomolybdate: 10 mg/day to 120 mg/day based on serum ceruloplasmin levels; duration: not stated. Group 2: placebo (n = 15). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The patients were assigned to the placebo arm or the tetrathiomolybdate arm using a table of random numbers". |
| Allocation concealment (selection bias) | Low risk | Quote: "Central allocation by pharmacy" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "There were not post‐randomisation drop‐outs" (author's reply). |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Low risk | Quote: "Supported by Grant FD‐02590‐02 from the U.S. Food and Drug Administration's Orphan Products Office, the General Clinical Research Center of the University of Michigan Hospitals, Grant MO1‐ RR000042 from the National Institutes of Health, and Grant Ul1‐ RR024986 Clinical and Translational Science Awards". |
| Other bias | High risk | Comment: unclear whether the participants continued to take UDCA in both groups. |
Battezzati 1993.
| Methods | Randomised clinical trial. | |
| Participants | Country: Italy. Number randomised: 88. Post‐randomisation dropouts: 2 (2.3%). Revised sample size: 86. Mean age: 55 years. Females: 78 (90.7%). Symptomatic participants: 86 (100%). AMA positive: 77 (89.5%). Responders: not stated. Mean follow‐up period (for all groups): minimum 6 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 42). Further details: UDCA: 250 mg BD for 6 months. Group 2: placebo (n = 44). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: lost to follow‐up. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization of treatment assignments was performed separately for each centre: patients were consecutively given indistinguishable medications, which had been assigned by the central pharmacy according to a computer‐ generated list. UDCA and an identical‐appearing placebo were obtained through the courtesy of ABC Farmaceutici, Torino, Italy". |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization of treatment assignments was performed separately for each centre: patients were consecutively given indistinguishable medications, which had been assigned by the central pharmacy according to a computer‐ generated list. UDCA and an identical‐appearing placebo were obtained through the courtesy of ABC Farmaceutici, Torino, Italy". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Randomization of treatment assignments was performed separately for each centre: patients were consecutively given indistinguishable medications, which had been assigned by the central pharmacy according to a computer‐ generated list. UDCA and an identical‐appearing placebo were obtained through the courtesy of ABC Farmaceutici, Torino, Italy". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Randomization of treatment assignments was performed separately for each centre: patients were consecutively given indistinguishable medications, which had been assigned by the central pharmacy according to a computer‐ generated list. UDCA and an identical‐appearing placebo were obtained through the courtesy of ABC Farmaceutici, Torino, Italy". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other risk of bias. |
Bobadilla 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: Mexico. Number randomised: 40. Post‐randomisation dropouts: not stated. Revised sample size: 40. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + colchicine (n = 21). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day for 1 year + colchicine: 1 mg/day for 5 days in a week for 1 year. Group 2: placebo (n = 19). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo used in double‐blind trial, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo used in double‐blind trial, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Bodenheimer 1988.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 57. Post‐randomisation dropouts: 10 (17.5%). Revised sample size: 47. Mean age: 52 years. Females: not stated. Symptomatic participants: 45 (95.7%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): 33 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: colchicine (n = 28). Further details: colchicine: 0.6 mg BD orally for 5 years. Group 2: placebo (n = 29). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: non‐compliance. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The design of our trial was that of a double‐blind, randomized evaluation of colchicine (0.6 mg) twice daily compared with an identically appearing placebo". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The design of our trial was that of a double‐blind, randomized evaluation of colchicine (0.6 mg) twice daily compared with an identically appearing placebo". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "The colchicine and placebo tablets were prepared and generously supplied by Eli Lilly and Company, Indianapolis, Ind". |
| Other bias | Low risk | Comment: no other source of bias. |
Bowlus 2014.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 216. Post‐randomisation dropouts: not stated. Revised sample size: 216. Mean age: 56 years. Females: 197 (91.2%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 3 groups. Group 1: obeticholic acid (low) (n = 73). Further details: obeticholic acid (low): 5 mg orally for 12 months; frequency not stated. Group 2: obeticholic acid (low) (n = 73). Further details: obeticholic acid (low): 10 mg orally for 12 months; frequency not stated. Group 3: placebo (n = 70). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used in double‐blind trial, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used in double‐blind trial, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Comment: Several authors had advised pharmaceutical companies or were employees of pharmaceutical company. |
| Other bias | Low risk | Comment: no other source of bias. |
Cash 2013.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 21. Post‐randomisation dropouts: 8 (38.1%). Revised sample size: 13. Mean age: 55 years. Females: not stated. Symptomatic participants: not stated. AMA positive: 13 (100%). Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: simvastatin (n = 7). Further details: simvastatin: 20 mg/day orally for 12 months. Group 2: placebo (n = 6). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: adverse effects. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "Patient treatment randomization and allocation was performed independently by the Department of Research Pharmacology in the Royal Victoria Hospital at the initial baseline visit". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The patients were blinded but the healthcare providers were not" (author's reply). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Outcome assessors were not blinded" (author's reply). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Low risk | Quote: "Financial support: The Royal Victoria Hospital Liver Support Group". |
| Other bias | High risk | Quote: "Patients were allowed to continue previous prescriptions for primary biliary cholangitis. It was not clear whether this was balanced across groups". |
Christensen 1985.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 248. Post‐randomisation dropouts: 63 (25.4%). Revised sample size: 185. Mean age: 55 years. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Mean follow‐up period (for all groups): minimum 63 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: azathioprine (n = 98). Further details: azathioprine: escalating doses up to a maximum of 100 mg/day; duration: not stated. Group 2: placebo (n = 87). |
|
| Outcomes | Mortality. | |
| Notes | Reasons for post‐randomisation dropouts: lost to follow‐up. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were randomized to azathioprine or placebo separately for each centre and for each sex by the sealed envelope technique". Comment: further details of sealed envelope technique were not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | High risk | Quote: "This work was also supported by the Wellcome Foundation. J.N. was supported by Ciba‐Geigy Ltd". |
| Other bias | Low risk | Comment: no other source of bias. |
Combes 1995a.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 151. Post‐randomisation dropouts: 0 (0%). Revised sample size: 151. Mean age: 49 years. Females: 134 (88.7%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Other exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 77). Further details: UDCA: 10 mg/kg/day to 12 mg/kg/day for 2 years. Group 2: placebo (n = 74). |
|
| Outcomes | Decompensated liver disease. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "Supported in part by a research grant from Ciba‐Geigy". |
| Other bias | Low risk | Comment: no other risk of bias. |
Combes 2005.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 265. Post‐randomisation dropouts: 0 (0%). Revised sample size: 265. Mean age: 51 years. Females: 245 (92.5%). Symptomatic participants: not stated. AMA positive: 265 (100%). Responders: not stated. Median follow‐up period (for all groups): 91 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + methotrexate (n = 132). Further details: UDCA: 15 mg/kg/day for 2 years + methotrexate: 2.5 mg orally once a week. Group 2: UDCA (moderate) (n = 133). Further details: UDCA: 15 mg/kg/day for 2 years. |
|
| Outcomes | Mortality, liver transplantation, decompensated liver disease. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | High risk | Quote: "By provision of UDCA by Ciba‐Geigy Corporation, and subsequently Novartis; by provision of methotrexate and its placebo by Lederle Laboratories, and subsequently Wyeth‐Ayerst Laboratories". |
| Other bias | Low risk | Comment: no other source of bias. |
Dickson 1985.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 309. Post‐randomisation dropouts: 82 (26.5%). Revised sample size: 227. Mean age: not stated. Females: 200 (88.1%). Symptomatic participants: 182 (80.2%). AMA positive: not stated. Responders: not stated. Median follow‐up period (for all groups): 60 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = 111). Further details: D‐penicillamine: 1000 mg/day; duration: not stated. Group 2: placebo (n = 116). |
|
| Outcomes | Adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: histological stages < 3; alcoholism. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to drug or placebo groups according to a table of random numbers". |
| Allocation concealment (selection bias) | Low risk | Quote: "Penicillamine and placebo (furnished to us through the courtesy of Merck Sharp and Dohme, West Point, Pa.) were dispensed in identical yellow capsules by a central pharmacist". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | High risk | Quote: "Penicillamine and placebo (furnished to us through the courtesy of Merck Sharp and Dohme, West Point, Pa.) were dispensed in identical yellow capsules by a central pharmacist". |
| Other bias | High risk | Comment: authors presented the results of only a subgroup of participants without explaining the reason for this. |
Epstein 1979.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 98. Post‐randomisation dropouts: not stated. Revised sample size: 98. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): mean: 66 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = 61). Further details: D‐penicillamine: 600 mg/day to 900 mg/day for 12 months. Group 2: placebo (n = 37). |
|
| Outcomes | Mortality. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The original double‐blind design of the trial was discontinued after a year because both major and minor side effects identified treated patients". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "The original double‐blind design of the trial was discontinued after a year because both major and minor side effects identified treated patients". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Eriksson 1997.
| Methods | Randomised clinical trial. | |
| Participants | Country: Sweden. Number randomised: 116. Post‐randomisation dropouts: 15 (12.9%). Revised sample size: 101. Mean age: 57 years. Females: 99 (98%). Symptomatic participants: 39 (38.6%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 60). Further details: UDCA: 500 mg/day for 24 months. Group 2: placebo (n = 56). |
|
| Outcomes | Liver transplantation. | |
| Notes | Reasons for post‐randomisation dropouts: adverse effects, alcoholic hepatitis, liver transplantation, death. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "We acknowledge the financial support from Meda AB, Searle AB, and the Swedish Medical Research Council (03x‐4793)". |
| Other bias | Low risk | Comment: no other source of bias. |
Ferri 1993.
| Methods | Randomised clinical trial. | |
| Participants | Country: Italy. Number randomised: 30. Post‐randomisation dropouts: 0 (0%). Revised sample size: 30. Mean age: 53 years. Females: 27 (90%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 6 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: TUDCA (moderate) (n = 15). Further details: TUDCA: 13 mg/day to 15 mg/day for 6 months. Group 2: UDCA (moderate) (n = 15). Further details: UDCA: 13 mg/day to 15 mg/day for 6 months. |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: adverse events, the only outcome of interest reported in this study were available from all randomised participants. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Gao 2012.
| Methods | Randomised clinical trial. | |
| Participants | Country: China. Number randomised: 79. Post‐randomisation dropouts: not stated. Revised sample size: 79. Mean age: 53 years. Females: 77 (97.5%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Other inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 3 groups. Group 1: UDCA (moderate) (n = 29). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day; duration: not stated. Group 2: UDCA (moderate) + glucocorticosteroids (n = 37). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day; duration: not stated + prednisolone: 7.5 mg/day; duration: not stated. Group 3: UDCA (moderate) + azathioprine (n = 13). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day; duration: not stated + azathioprine: 1 mg/kg/day; duration: not stated. |
|
| Outcomes | Adverse events, decompensated liver disease. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Goddard 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 57. Post‐randomisation dropouts: not stated. Revised sample size: 57. Mean age: not stated. Females: not stated. Symptomatic participants: 30 (52.6%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): 15 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 4 groups. Group 1: UDCA (low) (n = not stated). Further details: UDCA: 10 mg/kg/day; duration: not stated. Group 2: colchicine (n = not stated). Further details: colchicine: 1 mg/day; duration: not stated. Group 3: UDCA (low) + colchicine (n = not stated). Further details: UDCA: 10 mg/kg/day; duration: not stated + colchicine: 1 mg/day; duration: not stated. Group 4: placebo (n = not stated). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: placebo used; however, the authors did not mention blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: placebo used; however, the authors did not mention blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Gonzalezkoch 1997.
| Methods | Randomised clinical trial. | |
| Participants | Country: Chile. Number randomised: 25. Post‐randomisation dropouts: not stated. Revised sample size: 25. Mean age: 50 years. Females: 25 (100%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 11 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + methotrexate (n = 13). Further details: UDCA: 250 mg BD for 48 weeks + methotrexate: 10 mg/week given over 48 hours each week for 48 months. Group 2: UDCA (low) (n = 12). Further details: UDCA: 250 mg BD for 48 weeks. |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "A physician who was blinded to the treatment, followed them up clinically, evaluated clinical symptoms, adverse side effects, complications and compliance". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Heathcote 1976.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 45. Post‐randomisation dropouts: 6 (13.3%). Revised sample size: 39. Mean age: 51 years. Females: not stated. Symptomatic participants: 39 (100%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: azathioprine (n = 19). Further details: azathioprine: 2 mg/kg; frequency and duration: not stated. Group 2: control (n = 20). |
|
| Outcomes | Mortality, cirrhosis. | |
| Notes | Reasons for post‐randomisation dropouts: adverse events, wrong diagnosis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were randomly allocated to the treatment or control group, using the sealed envelope technique". Comment: further details of sealed envelope technique not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "No placebo was given to the control patients". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "No placebo was given to the control patients". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Low risk | Quote: "This work was supported by the Medical Research Council and the Ingram Fund". |
| Other bias | Low risk | Comment: no other source of bias. |
Heathcote 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: Canada. Number randomised: 222. Post‐randomisation dropouts: not stated. Revised sample size: 222. Mean age: 56 years. Females: 206 (92.8%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 111). Further details: UDCA: 14 mg/kg/day for 2 years. Group 2: placebo (n = 111). |
|
| Outcomes | Mortality, liver transplantation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was done separately at each centre by the study pharmacist using consecutive identification numbers, and patients were stratified according to whether they were symptomatic or asymptomatic". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Once informed consent was obtained from the patients, double‐blind randomization to UDCA or an identical placebo (1 : 1) was conducted". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Once informed consent was obtained from the patients, double‐blind randomization to UDCA or an identical placebo (1 : 1) was conducted". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: unclear whether the authors have reported the outcomes on all randomised participants. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | High risk | Quote: "Study medications kindly provided by Interfalk Canada and Jouveinal Inc., Quebec, Canada". |
| Other bias | Low risk | Comment: no other source of bias. |
Hendrickse 1999.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 60. Post‐randomisation dropouts: not stated. Revised sample size: 60. Mean age: 57 years. Females: 55 (91.7%). Symptomatic participants: 57 (95%). AMA positive: 51 (85%). Responders: not stated. Mean follow‐up period (for all groups): minimum 68 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: methotrexate (n = 30). Further details: methotrexate: 2.5 mg 3 times weekly for 6 years. Group 2: placebo (n = 30). Further details: placebo: 3 times weekly for 6 years. |
|
| Outcomes | Mortality, liver transplantation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized in groups of 10 by a random‐number technique, operated by the hospital pharmacy, to receive 2.5 mg MTX [methotrexate] or identical placebo tablets, both supplied by Lederle Laboratories, on Friday, Saturday, and Sunday of each week for up to 6 years". |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomized in groups of 10 by a random‐number technique, operated by the hospital pharmacy, to receive 2.5 mg MTX or identical placebo tablets, both supplied by Lederle Laboratories, on Friday, Saturday, and Sunday of each week for up to 6 years". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: placebo used in this double‐blind trial; however, the authors did not state whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor morbidity reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Hirschfield 2015.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 165. Post‐randomisation dropouts: 0 (0%). Revised sample size: 165. Mean age: 55 years. Females: 157 (95.2%). Symptomatic participants: not stated. AMA positive: 134 (81.2%). Responders: 0 (0%). Mean follow‐up period (for all groups): all participants followed up for 3 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 3 groups. Group 1: obeticholic acid (low) (n = 38). Further details: obeticholic acid (low): 10 mg for 85 days; frequency not stated. Group 2: obeticholic acid (moderate) (n = 48). Further details: obeticholic acid (moderate): 25 mg for 85 days; frequency not stated. Group 3: obeticholic acid (high) (n = 41). Further details: obeticholic acid (high): 50 mg for 85 days; frequency not stated. Group 4: placebo (n = 38). |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The computerized randomization schedule used a block size of 4 at each center". |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | High risk | Quote: "Patients received varying doses of UDCA". |
Hoofnagle 1986.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 24. Post‐randomisation dropouts: 0 (0%). Revised sample size: 24. Mean age: 47 years. Females: 23 (95.8%). Symptomatic participants: 24 (100%). AMA positive: 22 (91.7%). Responders: not stated. Mean follow‐up period (for all groups): 52 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: chlorambucil (n = 13). Further details: chlorambucil: 2 mg OD; duration: not stated. Group 2: control (n = 11). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomized by random numbers (generated by pharmacy) to either chlorambucil or no therapy. Pre‐computer age. They were kept in envelopes" (author's reply). |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomized by random numbers (generated by pharmacy) to either chlorambucil or no therapy. Pre‐computer age. They were kept in envelopes" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Not a blinded study" (author's reply). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "The outcomes were not blinded" (author's reply). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | Low risk | Quote: "The study was funded by the NIH intramural program" (author's reply). |
| Other bias | Low risk | Comment: no other source of bias. |
Hosonuma 2015.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 27. Post‐randomisation dropouts: 0 (0%). Revised sample size: 27. Mean age: 64 years. Females: 22 (81.5%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): minimum: 96 months. Inclusion criteria
Other inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + bezafibrate (n = 13). Further details: UDCA: 12 mg/kg/day to 15 mg/kg/day; duration: not stated + bezafibrate: 400 mg/day; duration: not stated. Group 2: UDCA (moderate) (n = 14). Further details: UDCA: 12 mg/kg/day to 15 mg/kg/day; duration: not stated. |
|
| Outcomes | Mortality, adverse events, liver transplantation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Sealed opaque envelopes" (author's reply). |
| Allocation concealment (selection bias) | Low risk | Quote: "These patients were randomly allocated to treatment with either UDCA alone (the UDCA group) or with the combination therapy (the UDCA+BF [bezafibrate] group), according to sequential sealed envelopes in blocks of four to ensure equal randomization for the duration of the study". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "However, our study was an unblinded, open trial and was therefore not free from bias". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "However, our study was an unblinded, open trial and was therefore not free from bias". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and adverse events reported. |
| For‐profit bias | Low risk | Quote: "This study was supported by the authors' own research funds". |
| Other bias | Low risk | Comment: no other source of bias. |
Ikeda 1996.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 22. Post‐randomisation dropouts: 0 (0%). Revised sample size: 22. Mean age: 61 years. Females: 19 (86.4%). Symptomatic participants: 7 (31.8%). AMA positive: 22 (100%). Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + colchicine (n = 10). Further details: UDCA: 9 mg/kg/day to 15 mg/kg/day for 2 years + colchicine: 1 mg/day for 2 years. Group 2: UDCA (moderate) (n = 12). Further details: UDCA: 9 mg/kg/day to 15 mg/kg/day for 2 years. |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Quote: "Part of the present study was supported by a grant from the Intractable Liver Diseases Research Committee, the Ministry of Health and Welfare, Japan". Comment: unclear how the remaining part of the funds were obtained. |
| Other bias | High risk | Comment: dose range for UDCA was very wide. |
Iwasaki 2008a.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 45. Post‐randomisation dropouts: not stated. Revised sample size: 45. Mean age: 56 years. Females: 37 (82.2%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: bezafibrate (n = 20). Further details: bezafibrate: 400 mg/day for 52 weeks. Group 2: UDCA (low) (n = 25). Further details: UDCA: 600 mg/day for 52 weeks. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "Consecutive patients from these hospitals were randomized centrally at the Kanagawa Dental University and were enrolled into the study if they met the following criteria". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "A randomized, open study design was used because there was no suitable placebo for bezafibrate available". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "A randomized, open study design was used because there was no suitable placebo for bezafibrate available". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Low risk | Quote: "The Ministry of Health, Labour and Welfare of Japan supported this study from 2002 to 2004 with a Health Science Research Grant on a Specific Disease (Study of Intractable Liver Diseases) to chief scientist Gotaro Toda". |
| Other bias | Low risk | Comment: no other bias. |
Iwasaki 2008b.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 22. Post‐randomisation dropouts: not stated. Revised sample size: 22. Mean age: 54 years. Females: 19 (86.4%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + bezafibrate (n = 10). Further details: UDCA: 600 mg/day for 52 weeks + bezafibrate: 400 mg/day for 52 weeks. Group 2: UDCA (low) (n = 12). Further details: UDCA: 600 mg/day for 52 weeks. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "Consecutive patients from these hospitals were randomized centrally at the Kanagawa Dental University and were enrolled into the study if they met the following criteria". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "A randomized, open study design was used because there was no suitable placebo for bezafibrate available". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "A randomized, open study design was used because there was no suitable placebo for bezafibrate available". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Low risk | Quote: "The Ministry of Health, Labour and Welfare of Japan supported this study from 2002 to 2004 with a Health Science Research Grant on a Specific Disease (Study of Intractable Liver Diseases) to chief scientist Gotaro Toda". |
| Other bias | Low risk | Comment: no other bias. |
Kanda 2003.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 22. Post‐randomisation dropouts: 0 (0%). Revised sample size: 22. Mean age: 56 years. Females: 19 (86.4%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): minimum 7 months. Inclusion criteria
Other inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + bezafibrate (n = 11). Further details: UDCA: 600 mg/day for 6 months + bezafibrate: 400 mg/day for 52 weeks. Group 2: UDCA (low) (n = 11). Further details: UDCA: 600 mg/day for 6 months. |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: mortality was not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Kaplan 1986.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 60. Post‐randomisation dropouts: 3 (5%). Revised sample size: 57. Mean age: not stated. Females: 57 (100%). Symptomatic participants: 45 (78.9%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: colchicine (n = 28). Further details: colchicine: 0.6 mg BD for ≥ 2 years. Group 2: placebo (n = 29). |
|
| Outcomes | Mortality. | |
| Notes | Reasons for post‐randomisation dropouts: adverse effects, despondent about treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: authors stated that this was a double‐blind trial and used a placebo; however, they did not state whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: authors stated that this was a double‐blind trial and used a placebo; however, they did not state whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Kaplan 1999.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 87. Post‐randomisation dropouts: 2 (2.3%). Revised sample size: 85. Mean age: 51 years. Females: 82 (96.5%). Symptomatic participants: 71 (83.5%). AMA positive: 77 (90.6%). Responders: not stated. Mean follow‐up period (for all groups): minimum 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: colchicine (n = 43). Further details: colchicine: 0.6 mg BD for 2 years. Group 2: methotrexate (n = 42). Further details: methotrexate: 15 mg/week orally. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: withdrawal from study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both the patients and investigators were blinded to the treatment assignments". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Both the patients and investigators were blinded to the treatment assignments". Comment: authors stated that this was a double‐blind trial and used a placebo; however, they did not state whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Kowdley 2011.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 59. Post‐randomisation dropouts: not stated. Revised sample size: 59. Mean age: 55 years. Females: 50 (84.7%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 3 groups. Group 1: obeticholic acid (low) (n = 20). Further details: obeticholic acid (low): 10 mg OD for 12 weeks. Group 2: obeticholic acid (high) (n = 16). Further details: obeticholic acid (high): 50 mg OD for 12 weeks. Group 3: placebo (n = 23). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although this was a double‐blind trial and placebo was used, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although this was a double‐blind trial and placebo was used, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Comment: some of the coauthors were from the pharmaceutical industry. |
| Other bias | Low risk | Comment: no other source of bias. |
Kurihara 2000.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 24. Post‐randomisation dropouts: not stated. Revised sample size: 24. Mean age: 60 years. Females: 23 (95.8%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: bezafibrate (n = 12). Further details: bezafibrate: 400 mg/day for 1 year. Group 2: UDCA (low) (n = 12). Further details: UDCA: 600 mg/day for 1 year. |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Leuschner 1989.
| Methods | Randomised clinical trial. | |
| Participants | Country: Germany. Number randomised: 20. Post‐randomisation dropouts: 2 (10%). Revised sample size: 18. Mean age: not stated. Females: 18 (100%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 10). Further details: UDCA: 10 mg/kg/day for 9 months. Group 2: placebo (n = 8). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, unclear whether identical placebo used. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, unclear whether identical placebo used. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Leuschner 1999.
| Methods | Randomised clinical trial. | |
| Participants | Country: Germany. Number randomised: 40. Post‐randomisation dropouts: 1 (2.5%). Revised sample size: 39. Mean age: 58 years. Females: 37 (94.9%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + corticosteroids (n = 20). Further details: UDCA: 10 mg/kg/day to 15 mg/kg/day for 2 years + budesonide: 3 mg 3 times daily for 2 years. Group 2: UDCA (moderate) (n = 19). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: personal reasons. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Complete randomization was done according to Rancode + (version 3.1; IDV‐Co., Marsaglia and Bray, Gauting, Germany)". |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, unclear whether identical placebo used. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, unclear whether identical placebo used. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "UDCA, budesonide, and placebo were provided by Dr. Falk Pharma GmbH (Freiburg, Germany)". |
| Other bias | Low risk | Comment: no other source of bias. |
Liberopoulos 2010.
| Methods | Randomised clinical trial. | |
| Participants | Country: Greece. Number randomised: 10. Post‐randomisation dropouts: not stated. Revised sample size: 10. Mean age: 57 years. Females: 8 (80%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + fenofibrate (n = 6). Further details: UDCA: 600 mg/day for 8 weeks + fenofibrate: 200 mg/day for 8 weeks. Group 2: UDCA (low) (n = 4). Further details: UDCA: 600 mg/day for 8 weeks. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Continue open‐label UDCA". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Continue open‐label UDCA". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Quote: "This study was conducted independently; no company or institution supported it financially. Some of the authors have given talks, attended conferences and participated in trials and advisory boards sponsored by various pharmaceutical companies". Comment: unclear whether the authors were in the advisory board of related pharmaceutical companies. |
| Other bias | Low risk | Comment: no other source of bias. |
Lim 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 32. Post‐randomisation dropouts: not stated. Revised sample size: 32. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = not stated). Further details: UDCA: 10 mg/kg/day to 12 mg/kg/day; duration: not stated. Group 2: placebo (n = not stated). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although a placebo was used in this double‐blind trial, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although a placebo was used in this double‐blind trial, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Lindor 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 180. Post‐randomisation dropouts: 10 (5.6%). Revised sample size: 170. Mean age: 53 years. Females: 160 (94.1%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 86). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day; duration: not stated. Group 2: placebo (n = 84). |
|
| Outcomes | Mortality, adverse events, liver transplantation. | |
| Notes | Reasons for post‐randomisation dropouts: not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients, physicians, nurses, and study coordinators were blinded as to whether active drug or placebo was being administered". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The patients, physicians, nurses, and study coordinators were blinded as to whether active drug or placebo was being administered". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "Supported by Falk Pharma and Interfalk". |
| Other bias | Low risk | Comment: no other risk of bias. |
Lindor 1997.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 150. Post‐randomisation dropouts: not stated. Revised sample size: 150. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 3 groups. Group 1: UDCA (low) (n = not stated). Further details: UDCA: 5 mg/kg/day to 7 mg/kg/day; duration: not stated. Group 2: UDCA (moderate) (n = not stated). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day; duration: not stated. Group 3: UDCA (high) (n = not stated). Further details: UDCA: 22 mg/kg/day to 25 mg/kg/day; duration: not stated. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other risk of bias. |
Lombard 1993.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 349. Post‐randomisation dropouts: 0 (0%). Revised sample size: 349. Mean age: 54 years. Females: 298 (85.4%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Median follow‐up period (for all groups): 31 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: ciclosporin (n = 176). Further details: ciclosporin: 3 mg/kg/day to maintain levels at 150 ng/mL by polyclonal radioimmunoassay or 75 ng/mL by monoclonal radioimmunoassay. Group 2: placebo (n = 173). |
|
| Outcomes | Mortality, adverse events, liver transplantation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Sealed envelopes" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "The authors are grateful to Sandoz Pharmaceuticals, Basle, Switzerland and their international sub‐offices for supplying Sandimmune and placebo for this study and for their support throughout. The authors are grateful to Sandoz Pharmaceuticals, Basle, Zerland and their international sub‐offices for supplying Sandimmune and placebo for this study and for their support throughout". |
| Other bias | Low risk | Comment: no other source of bias. |
Ma 2016.
| Methods | Randomised clinical trial. | |
| Participants | Country: China. Number randomised: 199. Post‐randomisation dropouts: 8 (4.0%). Revised sample size: 191. Mean age: 51 years. Females: 167 (83.9%). Symptomatic participants: 38 (19.9%). AMA positive: 187 (97.9%). Responders: not stated. Mean follow‐up period (for all groups): all participants: 6 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 66). Further details: UDCA: 250 mg 3 times daily for 24 weeks. Group 2: TUDCA (moderate) (n = 125). Further details: TUDCA: 250 mg 3 times daily for 24 weeks. |
|
| Outcomes | Adverse events. | |
| Notes | Reason for drop‐outs: not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A centralized telecommunication‐based interactive voice response system was used for patient randomization after patient eligibility was determined through clinical and laboratory screening assessments". |
| Allocation concealment (selection bias) | Low risk | Quote: "A centralized telecommunication‐based interactive voice response system was used for patient randomization after patient eligibility was determined through clinical and laboratory screening assessments". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: unclear whether all participants were included in the analysis. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | High risk | Quote: "This study was sponsored by the Beijing Trendful Kangjian Medical Information Consulting Co., Ltd. and the Major Science and Technology Special Project of China Twelfth Five‐year Plan (2012ZX10002003). Registration Number: NCT01829698". |
| Other bias | Low risk | Comment: no other source of bias. |
Macklon 1982.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 60. Post‐randomisation dropouts: 0 (0%). Revised sample size: 60. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): 37 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = 41). Further details: D‐penicillamine: 250 mg/day or 1 g/day; duration: not stated. Group 2: placebo (n = 19). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used, there is no mention of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used, there is no mention of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Manzillo 1993a.
| Methods | Randomised clinical trial. | |
| Participants | Country: Italy. Number randomised: 32. Post‐randomisation dropouts: not stated. Revised sample size: 32. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 1 month. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: SAMe (n = 16). Further details: SAMe: 800 mg/day IV for 2 weeks. Group 2: placebo (n = 16). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used, there was no mention of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used, there was no mention of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Manzillo 1993b.
| Methods | Randomised clinical trial. | |
| Participants | Country: Italy. Number randomised: 6. Post‐randomisation dropouts: not stated. Revised sample size: 6. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 2 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: SAMe (n = 3). Further details: SAMe: 1600 mg/day orally for 8 weeks. Group 2: placebo (n = 3). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used, there was no mention of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used, there was no mention of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Mason 2008.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 59. Post‐randomisation dropouts: 0 (0%). Revised sample size: 59. Mean age: 56 years. Females: 58 (98.3%). Symptomatic participants: not stated. AMA positive: 59 (100%). Responders: 0 (0%). Mean follow‐up period (for all groups): all participants followed up for 6 months. Inclusion criteria
Other exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: lamivudine + zidovudine + UDCA (moderate) (n = 30). Further details: lamivudine: 150 mg BD for 6 months + zidovudine: 300 mg BD for 6 months + UDCA: 13 mg/kg/day to 15 mg/kg/day for 6 months. Group 2: UDCA (moderate) (n = 29). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day for 6 months. |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomisation was performed centrally at the University of Alberta by a dynamic randomization" (author's reply). |
| Allocation concealment (selection bias) | Low risk | Quote: "Sealed opaque envelopes" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Dilan Clinical Packaging Ltd (Mississauga, ON, Canada) coded samples ensuring that the investigators and patients were blinded to the treatment". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Dilan Clinical Packaging Ltd (Mississauga, ON, Canada) coded samples ensuring that the investigators and patients were blinded to the treatment". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | High risk | Quote: "This study was funded in full by GlaxoSmithKline and Axcan Pharma". |
| Other bias | Low risk | Comment: no other bias. |
Matloff 1982.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 52. Post‐randomisation dropouts: 0 (0%). Revised sample size: 52. Mean age: 52 years. Females: 48 (92.3%). Symptomatic participants: not stated. AMA positive: 42 (80.8%). Responders: not stated. Mean follow‐up period (for all groups): minimum 24 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = 26). Further details: D‐penicillamine: 1 g/day; duration: not stated. Group 2: placebo (n = 26). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, there was no mention about identical placebo. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, there was no mention about identical placebo. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "We are indebted to Merck, Sharp and Dohme Research Laboratories for providing the D‐penicillamine and placebo tablets". |
| Other bias | Low risk | Comment: no other bias. |
Mayo 2015.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 45. Post‐randomisation dropouts: 3 (6.7%). Revised sample size: 42. Mean age: 56 years. Females: 38 (90.5%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: NGM282 (n = 27). Further details: NGM282: 0.3 mg/day or 3 mg/day SC for 28 days. Group 2: placebo (n = 15). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although placebo was used in this double‐blind trial, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "Grant/Research Support: Intercept, Salix, NGM, Lumena, Gilead". |
| Other bias | Low risk | Comment: no other bias. |
Mazzarella 2002.
| Methods | Randomised clinical trial. | |
| Participants | Country: Italy. Number randomised: 42. Post‐randomisation dropouts: not stated. Revised sample size: 42. Mean age: not stated. Females: 37 (88.1%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 72 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (high) (n = 21). Further details: UDCA (high): 30 mg/kg/day for 6 years. Group 2: UDCA (moderate) (n = 21). Further details: UDCA (moderate): 10.5 mg/kg/day for 6 years. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
McCormick 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 18. Post‐randomisation dropouts: 0 (0%). Revised sample size: 18. Mean age: 60 years. Females: 14 (77.8%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: thalidomide (n = 10). Further details: thalidomide: 100 mg/day for 6 months. Group 2: placebo (n = 8). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "Thalidomide and identical placebo tablets were supplied by Penn Pharmaceuticals Ltd". |
| Other bias | Low risk | Comment: no other bias. |
Minuk 1988.
| Methods | Randomised clinical trial. | |
| Participants | Country: Canada. Number randomised: 12. Post‐randomisation dropouts: 0 (0%). Revised sample size: 12. Mean age: 55 years. Females: 11 (91.7%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: ciclosporin (n = 6). Further details: ciclosporin: maintain serum radioimmunoassay dosage between 100 ng/mL and 200 ng/mL for 12 months. Group 2: placebo (n = 6). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "…patients were randomized by sealed envelope to receive either cyclosporin A or placebo". Comment: further details of the sealed envelope method not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although a placebo was used, there was no mention about blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: although a placebo was used, there was no mention about blinding. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Comment: the drugs were provided by the pharmaceutical company. |
| Other bias | Low risk | Comment: no other bias. |
Mitchison 1989.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 36. Post‐randomisation dropouts: 0 (0%). Revised sample size: 36. Mean age: 55 years. Females: 33 (91.7%). Symptomatic participants: 35 (97.2%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 36 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: glucocorticosteroids (n = 19). Further details: prednisolone: 10 mg/day for 36 months (loading dose was used). Group 2: placebo (n = 17). |
|
| Outcomes | Mortality. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were paired according to the presence or absence of cirrhosis, their age by decade, menopausal status (for women) and their serum bilirubin (greater or less than 30 µmoles per litre)". Comment: minimisation used. |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were paired according to the presence or absence of cirrhosis, their age by decade, menopausal status (for women) and their serum bilirubin (greater or less than 30 µmoles per litre)". Comment: minimisation used. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Study was double‐blind for the first year, single blind thereafter (patients were blinded)". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Study was double‐blind for the first year, single blind thereafter (patients were blinded)". |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other bias. |
Mitchison 1993.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 104. Post‐randomisation dropouts: 3 (2.9%). Revised sample size: 101. Mean age: 54 years. Females: 93 (92.1%). Symptomatic participants: 101 (100%). AMA positive: not stated. Responders: not stated. Median follow‐up period (for all groups): 25 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: malotilate (n = 52). Further details: malotilate: 500 mg 3 times daily; mean duration: 23 months. Group 2: placebo (n = 49). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: elementary data not available. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Random sequence was generated by the trial statistician with tables with random numbers" (author's reply). |
| Allocation concealment (selection bias) | Low risk | Quote: "Sequentially numbered identical containers" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Both patients and doctors were unaware of the nature of the tablets". Comment: placebo used to achieve blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Both patients and doctors were unaware of the nature of the tablets". Comment: placebo used to achieve blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "The study was supported in part by Zyma S.A., Nyon, Switzerland, and by Nihon Nohyaku, Tokyo, Japan". |
| Other bias | Low risk | Comment: no other source of bias. |
Nakai 2000.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 23. Post‐randomisation dropouts: not stated. Revised sample size: 23. Mean age: 57 years. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + bezafibrate (n = 13). Further details: UDCA: 600 mg/day; duration: not stated + bezafibrate: 400 mg/day; duration: not stated. Group 2: UDCA (low) (n = 10). Further details: UDCA: 600 mg/day; duration: not stated. |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Low risk | Quote: "This work was supported by a Grant‐in‐Aid for Scientific Research from the Ministry of Education, Science and Culture of Japan". |
| Other bias | Low risk | Comment: no other source of bias. |
Neuberger 1985.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 189. Post‐randomisation dropouts: not stated. Revised sample size: 189. Mean age: not stated. Females: 174 (92.1%). Symptomatic participants: 172 (91%). AMA positive: 163 (86.2%). Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = 98). Further details: D‐penicillamine: 1200 mg/day; duration: not stated. Group 2: placebo (n = 91). |
|
| Outcomes | Mortality, liver transplantation. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "Opaque sealed envelopes" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind trial, identical appearing placebo" (author's reply). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Assessors were blinded, identical placebo" (author's reply). |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Unclear risk | Quote: "Not pharmaceutical funding" (author's reply). |
| Other bias | Low risk | Comment: no other source of bias. |
Nevens 2016.
| Methods | Randomised clinical trial. | |
| Participants | Country: multicentric; international. Number randomised: 217. Post‐randomisation dropouts: 1. Revised sample size: 216. Mean age: 56 years. Females: 196 (90.7%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: obeticholic acid (low) + UDCA (moderate) (n = 143). Further details: obeticholic acid: 5 mg to 10 mg for 1 year + UDCA: 13 mg/kg/day to 15 mg/kg/day for 1 year. Group 2: UDCA (moderate) (n = 73). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day for 1 year. |
|
| Outcomes | Mortality, adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: withdrawal from study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "On a predefined randomization code (generated by the Sponsor or designee) using an IWRS". |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomization number will be recorded in the CRF and will serve for patient identification and for assignment of appropriate study medication and bottle number(s) by the IWRS". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used and authors stated double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "Supported by Intercept Pharmaceuticals". Comment: trial funded by industrial sources which might benefit by the nature of the results. |
| Other bias | Low risk | Comment: no other source of bias. |
Oka 1990.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 52. Post‐randomisation dropouts: 7 (13.5%). Revised sample size: 45. Mean age: 59 years. Females: 41 (91.1%). Symptomatic participants: 17 (37.8%). AMA positive: 41 (91.1%). Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 22). Further details: UDCA: 600 mg/day for 24 weeks. Group 2: placebo (n = 23). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: worsening liver disease, lack of compliance. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Low risk | Quote: "The patients were allocated to two groups, a UDCA group and a placebo group, by a single monitor according to a randomization scheme in which the number of patients allocated to two groups tended to be equal". |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "UDCA and placebo tablets were generously furnished by Tokyo Tanabe Pharmaceutical Company". |
| Other bias | Low risk | Comment: no other source of bias. |
Papatheodoridis 2002.
| Methods | Randomised clinical trial. | |
| Participants | Country: Greece. Number randomised: 92. Post‐randomisation dropouts: 6 (6.5%). Revised sample size: 86. Mean age: 54 years. Females: 77 (89.5%). Symptomatic participants: 86 (100%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): 89 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 43). Further details: UDCA: 12 mg/kg//day to 15 mg/kg//day for ≥ 2 years. Group 2: control (n = 43). |
|
| Outcomes | Mortality, liver transplantation, decompensated liver disease. | |
| Notes | Reasons for post‐randomisation dropouts: not stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization was carried out by serially numbered sealed envelopes containing random table numbers 14 patients crossed over from placebo to UDCA". |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization was carried out by serially numbered sealed envelopes containing random table numbers 14 patients crossed over from placebo to UDCA". |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Patients and healthcare providers were not blinded" (author's reply). |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "The outcome assessors were not blinded" (author's reply). |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts in the initial report. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | High risk | Quote: "Support for this work was provided during the first 2 years of the study by a research grant the pharmaceutical company Galenica Hellas and by the Greek Ministry of Health and Welfare". |
| Other bias | High risk | Comment: 14 participants crossed over from placebo to UDCA. |
Pares 2000.
| Methods | Randomised clinical trial. | |
| Participants | Country: Spain. Number randomised: 192. Post‐randomisation dropouts: 0 (0%). Revised sample size: 192. Mean age: 54 years. Females: 179 (93.2%). Symptomatic participants: not stated. AMA positive: 172 (89.6%). Responders: not stated. Median follow‐up period (for all groups): 41 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 99). Further details: UDCA 14 mg/kg/day to 16 mg/kg/day; duration: 25 to 73 months. Group 2: placebo (n = 93). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: unclear whether all participants were included in the analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "We are indebted to Zambon S. A., Laboratorio Farmaceutico for supplying the UDCA and placebo capsules, and for the invaluable administrative support". |
| Other bias | Low risk | Comment: no other risk of bias. |
Poupon 1991a.
| Methods | Randomised clinical trial. | |
| Participants | Country: France. Number randomised: 149. Post‐randomisation dropouts: 3 (2%). Revised sample size: 146. Mean age: 56 years. Females: 134 (91.8%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) (n = 73). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day for 2 years. Group 2: placebo (n = 73). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: bilirubin > 300 μmol/L, ascites, other coexisting disease. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "This work was supported in part by Synthélabo‐Recherche and in Canada by Jouveinal and Interfalk". |
| Other bias | Low risk | Comment: no other source of bias. |
Poupon 1996.
| Methods | Randomised clinical trial. | |
| Participants | Country: France. Number randomised: 74. Post‐randomisation dropouts: not stated. Revised sample size: 74. Mean age: 54 years. Females: 63 (85.1%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + colchicine (n = 37). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day for 2 years + colchicine: 1 mg/day for 5 days in a week for 2 years. Group 2: UDCA (moderate) (n = 37). Further details: UDCA: 13 mg/kg/day to 15 mg/kg/day for 2 years. |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: unclear whether all randomised participants were included for analysis. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "Supported in part by Laboratoires Houde (France) and Jouveinal (Canada)". |
| Other bias | Low risk | Comment: no other source of bias. |
Raedsch 1993.
| Methods | Randomised clinical trial. | |
| Participants | Country: Germany. Number randomised: 28. Post‐randomisation dropouts: 8 (28.6%). Revised sample size: 20. Mean age: 54 years. Females: 20 (100%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + colchicine (n = 8). Further details: UDCA: 10 mg/kg/day to 12 mg/kg/day for 24 months + colchicine: 1 mg/day for 24 months. Group 2: UDCA (moderate) (n = 12). Further details: UDCA: 10 mg/kg/day to 12 mg/kg/day for 24 months. |
|
| Outcomes | Adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: adverse events, lost to follow‐up. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although a placebo used in this double‐blind trial, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although a placebo used in this double‐blind trial, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Rautiainen 2005.
| Methods | Randomised clinical trial. | |
| Participants | Country: Finland. Number randomised: 77. Post‐randomisation dropouts: 8 (10.4%). Revised sample size: 69. Mean age: 53 years. Females: 60 (87%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 36 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + glucocorticosteroids (n = 37). Further details: UDCA: 15 mg/kg/day for 3 years + budesonide: 6 mg/day for 3 years. Group 2: UDCA (moderate) (n = 32). Further details: UDCA: 15 mg/kg/day for 3 years. |
|
| Outcomes | Adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: adverse effects, death, refused follow‐up biopsy. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Randomization was done centrally at Helsinki University Hospital with sealed envelopes in a block of 10". Comment: further details of sealed envelope technique not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Study design was randomized but open because placebo for budesonide was not available for us". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "Study design was randomized but open because placebo for budesonide was not available for us". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | High risk | Quote: "Medication was supplied free of charge by AstraZeneca Finland (budesonide, Entocort) and Leiras Finland (UDCA, Adursal)". |
| Other bias | Low risk | Comment: no other source of bias. |
Senior 1991.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 20. Post‐randomisation dropouts: 1 (5%). Revised sample size: 19. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 18 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 9). Further details: UDCA (low): 8 mg/kg/day to 12 mg/kg/day for 6 months. Group 2: placebo (n = 10). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | Reasons for post‐randomisation dropouts: had coexisting gallstones. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although a placebo used in this double‐blind trial, unclear whether the placebo was identical. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although a placebo used in this double‐blind trial, unclear whether the placebo was identical. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: there were post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | High risk | Quote: "and ursodiol supplies provided by Ciba‐Geigy Corporation". |
| Other bias | Low risk | Comment: no other source of bias. |
Smart 1990.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 20. Post‐randomisation dropouts: not stated. Revised sample size: 20. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: antioxidants (n = not stated). Further details: antioxidant: cocktail of vitamin E 100 mg, zinc 135 mg, and selenium 100 μg daily; duration: not stated. Group 2: placebo (n = not stated). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: although a placebo was used, no mention of blinding made. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: although a placebo was used, no mention of blinding made. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Steenbergen 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: Belgium. Number randomised: 14. Post‐randomisation dropouts: not stated. Revised sample size: 14. Mean age: 51 years. Females: 12 (85.7%). Symptomatic participants: not stated. AMA positive: 13 (92.9%). Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + methotrexate (n = 8). Further details: UDCA: 500 mg/day; duration: not stated + methotrexate: 15 mg/week; duration: not stated. Group 2: control (n = 6). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Using a random number table…". |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: information not available. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: information not available. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Taal 1983.
| Methods | Randomised clinical trial. | |
| Participants | Country: Netherlands. Number randomised: 24. Post‐randomisation dropouts: not stated. Revised sample size: 24. Mean age: 49 years. Females: 23 (95.8%). Symptomatic participants: 24 (100%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 18 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = 11). Further details: D‐penicillamine: 250 mg/day to 1000 mg/day (escalating dose) and then 500 mg/day: total duration: 1 year. Group 2: placebo (n = 13). |
|
| Outcomes | Mortality, adverse events, decompensated liver disease. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Triger 1980.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 35. Post‐randomisation dropouts: not stated. Revised sample size: 35. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: D‐penicillamine (n = not stated). Further details: D‐penicillamine: 250 mg to 875 mg (escalating dose). Group 2: placebo (n = not stated). |
|
| Outcomes | Mortality. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: in this double‐blind trial, unclear whether the placebo was identical to active treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: in this double‐blind trial, unclear whether the placebo was identical to active treatment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | High risk | Quote: "The UDCA and placebo tablets were generously donated by Thames Laboratories, Wrexham, Wales". |
| Other bias | Low risk | Comment: no other source of bias. |
Turner 1994.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 46. Post‐randomisation dropouts: 0 (0%). Revised sample size: 46. Mean age: 58 years. Females: 44 (95.7%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): all participants followed up for 24 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 22). Further details: UDCA: 10 mg/kg/day for 2 years. Group 2: placebo (n = 24). |
|
| Outcomes | Mortality, liver transplantation, cirrhosis. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no post‐randomisation dropouts. |
| Selective reporting (reporting bias) | High risk | Comment: adverse events not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Ueno 2005.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 20. Post‐randomisation dropouts: not stated. Revised sample size: 20. Mean age: not stated. Females: 16 (80%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: lamivudine (n = not stated). Further details: lamivudine: 100 mg/day for 3 months. Group 2: placebo (n = not stated). |
|
| Outcomes | None of the outcomes of interest reported. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: neither mortality nor adverse events reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Van Hoogstraten 1998.
| Methods | Randomised clinical trial. | |
| Participants | Country: Netherlands. Number randomised: 61. Post‐randomisation dropouts: 2 (3.3%). Revised sample size: 59. Mean age: 57 years. Females: 55 (93.2%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) (n = 32). Further details: UDCA (low): 10 mg/kg/day for 6 months. Group 2: UDCA (moderate) (n = 27). Further details: UDCA (moderate): 20 mg/kg/day for 6 months. |
|
| Outcomes | Adverse events. | |
| Notes | Reasons for post‐randomisation dropouts: developed liver failure, lost to follow‐up. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Tables with random numbers" (author's reply). |
| Allocation concealment (selection bias) | Low risk | Quote: "Opaque closed envelopes" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "randomised open controlled trial". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "randomised open controlled trial". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | High risk | Quote: "This study was supported in part by Zambon Nederland BV, Amersfoort, the Netherlands". |
| Other bias | Low risk | Comment: no other source of bias. |
Warnes 1987.
| Methods | Randomised clinical trial. | |
| Participants | Country: UK. Number randomised: 64. Post‐randomisation dropouts: not stated. Revised sample size: 64. Mean age: not stated. Females: not stated. Symptomatic participants: not stated. AMA positive: 64 (100%). Responders: not stated. Median follow‐up period (for all groups): 19 months. Inclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: colchicine (n = 34). Further details: colchicine: 0.5 mg BD; duration: not stated. Group 2: placebo (n = 30). |
|
| Outcomes | Mortality, adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "To ensure that treatment groups were comparable, patients were stratified according to serum bilirubin level at entry (A, < 19/µmol/1; B, 20‐34/ µmol/L; C, 35‐102/µmol/L; D, >102/µmol/1). The first patient in any pair was allocated by the staff pharmacist to the treatment or placebo group by reference to random tables. The pair was completed when another patient, in the same bilirubin group and with an age within 5 years of the first patient, was entered into the study. The second member of the pair was allocated to the alternative treatment group. The study was double‐blind". Comment: minimisation method used. |
| Allocation concealment (selection bias) | Low risk | Quote: "To ensure that treatment groups were comparable, patients were stratified according to serum bilirubin level at entry (A, < 19/µmol/1; B, 20‐34/ µmol/L; C, 35‐102/µmol/L; D, >102/µmol/1). The first patient in any pair was allocated by the staff pharmacist to the treatment or placebo group by reference to random tables. The pair was completed when another patient, in the same bilirubin group and with an age within 5 years of the first patient, was entered into the study. The second member of the pair was allocated to the alternative treatment group. The study was double‐blind". Comment: minimisation method used. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
Wiesner 1990.
| Methods | Randomised clinical trial. | |
| Participants | Country: USA. Number randomised: 40. Post‐randomisation dropouts: 11 (27.5%). Revised sample size: 29. Mean age: 46 years. Females: 28 (96.6%). Symptomatic participants: not stated. AMA positive: not stated. Responders: not stated. Median follow‐up period (for all groups): 35 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: ciclosporin (n = 19). Further details: ciclosporin: 4 mg/kg/day. Group 2: placebo (n = 10). |
|
| Outcomes | Mortality, adverse events, liver transplantation. | |
| Notes | Reasons for post‐randomisation dropouts: follow‐up < 1 year. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | Low risk | Comment: mortality and morbidity reported. |
| For‐profit bias | High risk | Quote: "Supported by a grant from Sandoz and by the Mayo foundation". |
| Other bias | Low risk | Comment: no other source of bias. |
Wolfhagen 1998.
| Methods | Randomised clinical trial. | |
| Participants | Country: Netherlands. Number randomised: 50. Post‐randomisation dropouts: not stated. Revised sample size: 50. Mean age: 52 years. Females: 45 (90%). Symptomatic participants: not stated. AMA positive: not stated. Responders: 0 (0%). Mean follow‐up period (for all groups): all participants followed up for 12 months. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (moderate) + azathioprine + glucocorticosteroids (n = 26). Further details: UDCA: 10 mg/kg/day for 6 months + azathioprine: 50 mg/day for 6 months + prednisolone: 10 mg/day for 6 months. Group 2: UDCA (moderate) (n = 24). Further details: UDCA: 10 mg/kg/day for 6 months. |
|
| Outcomes | Adverse events, cirrhosis. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Tables with random numbers" (author's reply). |
| Allocation concealment (selection bias) | Low risk | Quote: "Opaque closed envelopes" (author's reply). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: identical placebo used in this double‐blind trial. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | High risk | Quote: "Supported by…Zambon Nederland B.v. and Glaxo Wellcome Research and Development Ltd". |
| Other bias | Low risk | Comment: no other source of bias. |
Yokomori 2001.
| Methods | Randomised clinical trial. | |
| Participants | Country: Japan. Number randomised: 11. Post‐randomisation dropouts: not stated. Revised sample size: 11. Mean age: 54 years. Females: 9 (81.8%). Symptomatic participants: 11 (100%). AMA positive: not stated. Responders: not stated. Mean follow‐up period (for all groups): not stated. Inclusion criteria
Exclusion criteria
|
|
| Interventions | Participants were randomly assigned to 2 groups. Group 1: UDCA (low) + colestilan (n = 5). Further details: UDCA: 600 mg/day for 8 weeks + colestilan: 6.42 mg/day for 4 weeks. Group 2: UDCA (low) (n = 6). Further details: UDCA: 600 mg/day for 8 weeks. |
|
| Outcomes | Adverse events. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: information not available. |
| Allocation concealment (selection bias) | Unclear risk | Comment: information not available. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "open‐label". |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "open‐label". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: information not available. |
| Selective reporting (reporting bias) | High risk | Comment: mortality not reported. |
| For‐profit bias | Unclear risk | Comment: information not available. |
| Other bias | Low risk | Comment: no other source of bias. |
AMA: antimitochondrial antibody; BD: twice daily; IV: intravenous; OD: once daily; SAMe: S‐adenosyl methionine; SC: subcutaneous; TUDCA: taurodeoxycholic acid; UDCA: ursodeoxycholic acid.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Angulo 1999b | Long‐term follow‐up of participants included in an included trial (Lindor 1994), but the randomisation was not maintained. |
| Angulo 1999c | Long‐term follow‐up of participants included in an included trial (Lindor 1994), but the randomisation was not maintained. |
| Angulo 2002 | Comparison of different administration schedules of the same dose of UDCA. |
| Attili 1994 | Not in people with primary biliary cholangitis. |
| Avezov 2004a | Not a randomised clinical trial. |
| Avezov 2004b | Not a randomised clinical trial. |
| Bach 2003 | Not a randomised clinical trial. |
| Batta 1989 | Not a randomised clinical trial. |
| Beukers 1988 | Not a randomised clinical trial. |
| Blanche 1994 | Not a randomised clinical trial. |
| Bonis 2006 | Not a randomised clinical trial. |
| Borum 1990 | Not a primary study (editorial). |
| Bray 1991 | Cross‐over RCT; no results presented before cross‐over. |
| Carbone 2016 | Long‐term follow‐up of Nevens 2016, but excluded because randomisation not maintained. |
| Chazouilleres 1995 | Not a randomised clinical trial. |
| Christensen 1986 | Not a primary study (letter to editor). |
| Combes 1989 | Not a primary study (editorial). |
| Combes 2004 | Not a primary study (editorial). |
| Combes 2005b | Long‐term follow‐up of participants in an included RCT (Combes 1995a); however, all participants received the intervention after the end of the initial study. |
| Copaci 2001 | Not a randomised clinical trial. |
| Corpechot 2000 | Long‐term follow‐up of an included trial (Poupon 1991a); however, all participants received the active intervention at the end of the trial period. |
| Corpechot 2001 | Not a primary study (editorial). |
| Crosignani 1996a | Cross‐over RCT; no outcomes of interest reported before cross‐over. |
| Crosignani 1996b | In this RCT of different doses of TUDCA, participants who were intolerant to the drug were replaced. This affected the randomisation. |
| De la Mora 1994 | No separate data on people who were randomised (included non‐randomised participants in the results). |
| Degott 1999 | Long‐term follow‐up of an included trial (Poupon 1991a); however, all participants received the active intervention at the end of the trial period. |
| Dickson 1991 | No separate data on people who were randomised (included non‐randomised participants in the results). |
| Emond 1996 | Long‐term follow‐up of an included trial (Combes 1995a); however, all participants received the active intervention at the end of the trial period. |
| Fischer 1967 | Not a randomised clinical trial. |
| Golovanova 2010 | Not a randomised clinical trial. |
| Heathcote 1993 | Not a randomised clinical trial. |
| Heathcote 1995 | Not a primary study. |
| Hirschfield 2011 | Not a randomised clinical trial. |
| Hishon 1982 | Not a randomised clinical trial. |
| Howat 1966 | Not a randomised clinical trial. |
| Hwang 1993 | Cross‐over RCT; none of the outcomes of interest reported prior to cross‐over. |
| Invernizzi 1996 | Cross‐over RCT, no results presented before cross‐over. |
| Invernizzi 2015 | Not a primary study. |
| Itakura 2004 | Not a primary study. |
| Jazrawi 1999 | Not a randomised clinical trial. |
| Jones 2006 | Not a primary study (letter to editor). |
| Jorgensen 2002 | Long‐term follow‐up of an included trial (Lindor 1994); however, all participants received the active intervention at the end of the trial period. |
| Joshi 2002 | Not a primary study. |
| Kaplan 1993 | Not a primary study (editorial). |
| Kaplan 1998 | Not a primary study (letter to editor). |
| Kaplan 2004 | Long‐term follow‐up of an included trial (Kaplan 1999), but the treatment was changed at the completion of the RCT. |
| Kaplan 2009 | Not a primary study (letter to editor). |
| Kisand 1996 | Quasi‐randomised study (allocation by case numbers). |
| Kisand 1998 | Quasi‐randomised study (allocation by case numbers). |
| Kowdley 2014a | Not a randomised clinical trial. |
| Kowdley 2014b | Not a randomised clinical trial. |
| Kowdley 2015 | Long‐term follow‐up of Kowdley 2011, but excluded because randomisation was not maintained. |
| Kugler 1991 | Not a primary study (commentary). |
| Kurihara 2002 | Not a randomised clinical trial. |
| Lampe 1972 | Not a randomised clinical trial. |
| Larghi 1997 | Cross‐over RCT; no results presented before cross‐over. |
| Lee 2003 | Not a randomised clinical trial. |
| Leung 2010 | Long‐term follow‐up of a subgroup of participants in an included trial (Kaplan 1999), where additional interventions were added after completion of the trial period. |
| Leung 2011 | Long‐term follow‐up of a subgroup of participants in an included trial (Kaplan 1999), where additional interventions were added after completion of the trial period. |
| Leuschner 1990 | Not a primary study (review). |
| Leuschner 1993a | Not a primary study (review). |
| Leuschner 1993b | Quasi‐randomised study (allocation by alternation). |
| Leuschner 1996a | Quasi‐randomised study (allocation by alternation). |
| Leuschner 1996b | Quasi‐randomised study (allocation by alternation). |
| Leuschner 1997 | Not a primary study (review). |
| Leuschner 1998 | Not a primary study (review). |
| Levy 2004 | Not a primary study (editorial). |
| Licinio 2015 | Not a primary study (letter to editor). |
| Lim 2000 | Not a randomised clinical trial. |
| Lindor 1994a | Not a randomised clinical trial |
| Lindor 1995a | Long‐term follow‐up an included RCT (Lindor 1994); however, all participants received the intervention after the completion of the RCT. |
| Lindor 1995b | Not a randomised clinical trial. |
| Lindor 1995c | Not a primary study (review). |
| Lindor 1996 | Long‐term follow‐up an included RCT (Lindor 1994); however, all participants received the intervention after the completion of the RCT. |
| Lindor 2000 | Not a primary study (letter to editor). |
| Lindor 2005 | Not a primary study (review). |
| Lindor 2007 | Not a primary study (review). |
| Lytvyak 2015 | Cross‐over RCT; no outcomes reported prior to cross‐over. |
| Lytvyak 2016 | Not a randomised clinical trial |
| Miettinen 1993 | Quasi‐randomised study (allocation by case numbers). |
| Miettinen 1995 | Quasi‐randomised study (allocation by case numbers). |
| Muntoni 2010 | Only 4 participants in this trial had primary biliary cholangitis and separate data not available for these 4 participants. |
| Nikolaidis 2006 | Only 5 participants had primary biliary cholangitis and only 1 of them received placebo. Separate data not available on these participants. |
| Ohmoto 2001 | Not a randomised clinical trial. |
| Ohmoto 2006 | Not a randomised clinical trial. |
| Pan 2013 | Only 5 participants had primary biliary cholangitis. Separate data not available for these participants. |
| Pares 2009 | Not a randomised clinical trial. |
| Podda 1989 | Cross‐over study of different doses of UDCA; outcomes not reported at the end of first treatment. |
| Poupon 1989 | Not a primary study (review). |
| Poupon 1990 | Not a primary study (review). |
| Poupon 1991b | Not a primary study (commentary). |
| Poupon 1994 | Long‐term follow‐up of an included trial (Poupon 1991a); however, all participants received the active intervention at the end of the trial period. |
| Poupon 1997 | Not a primary study. |
| Poupon 1999 | Not a randomised clinical trial. |
| Poupon 2003 | Not a primary study. |
| Raedsch 1989 | Not a randomised clinical trial. |
| Reed 1982 | Not a primary study (editorial). |
| Robson 1994 | Not a randomised clinical trial. |
| Roda 2002 | Not a randomised clinical trial. |
| Savolainen 1983 | Unclear whether this was a randomised clinical trial. |
| Schaffner 1982 | Comparison of 2 doses of D‐penicillamine with no other treatment as comparator. |
| Setchell 1994 | In this RCT of different doses of TUDCA, participants who were intolerant to the drug were replaced. This affected the randomisation. |
| Setchell 1996 | In this RCT of different doses of TUDCA, participants who were intolerant to the drug were replaced. This affected the randomisation. |
| Stellaard 1979 | Not a randomised clinical trial. |
| Taal 1985 | Not a randomised clinical trial. |
| Tang 2008 | Not a pharmacological agent. |
| Tong 2012 | Not a pharmacological agent. |
| Verma 1999 | Cross‐over RCT; no results presented before cross‐over. |
| Verma 2000 | Not a primary study (review). |
| Vogel 1988 | Not a randomised clinical trial. |
| Vuoristo 1995 | Quasi‐randomised study (allocation by case numbers). |
| Vuoristo 1997 | Quasi‐randomised study (allocation by case numbers). |
| Wiesner 1994 | Not a randomised clinical trial. |
| Wolfhagen 1995 | Not a randomised clinical trial. |
| Yan 2007 | Not a primary study (letter to editor). |
| Yano 2002 | Not a randomised clinical trial. |
| Zuin 1991 | Symptomatic treatment of dyslipidaemia associated with primary biliary cholangitis. |
TUDCA: taurodeoxycholic acid; UDCA: ursodeoxycholic acid.
Characteristics of studies awaiting assessment [ordered by study ID]
O'Brian 1990.
| Methods | Full text not available. |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes |
Zaman 2006.
| Methods | Full text not available. |
| Participants | |
| Interventions | |
| Outcomes | |
| Notes |
Characteristics of ongoing studies [ordered by study ID]
ChiCTR‐IPR‐16008935.
| Trial name or title | Biochemical Response of PBC‐AIH Overlap Syndrome Induced by Ursodeoxycholic Acid Only or Combination Therapy of Immunosuppressive Agents |
| Methods | Randomised parallel clinical trial |
| Participants | People with primary biliary cholangitis and autoimmune hepatitis overlap syndrome. |
| Interventions | Group 1: UDCA + immunosuppression Further details: not provided. Group 2: UDCA. Further details: not provided. |
| Outcomes | Adverse events |
| Starting date | Not stated. |
| Contact information | yangli_hx@scu.edu.cn |
| Notes | Status: recruiting. |
EUCTR2015‐002698‐39‐GB.
| Trial name or title | A 12‐Week, Double‐Blind, Randomized, Placebo‐Controlled, Phase 2 Study to Evaluate the Effects of Two Doses of MBX‐8025 in Subjects with Primary Biliary Cirrhosis (PBC) and an Inadequate Response to Ursodeoxycholic Acid (UDCA). |
| Methods | Randomised, placebo‐controlled, double‐blind clinical trial. |
| Participants | People with primary biliary cholangitis (non‐responders). |
| Interventions | Group 1: MBX‐8025. Further details: not provided. Group 2: placebo. |
| Outcomes | None of the outcomes of interest for this review measured in this trial. |
| Starting date | Not stated. |
| Contact information | KRosemark@cymabay.com |
| Notes | Status: recruiting. |
NCT02308111.
| Trial name or title | A Phase 3b, Double‐Blind, Randomized, Placebo‐Controlled, Multicenter Study Evaluating the Effect of Obeticholic Acid on Clinical Outcomes in Patients With Primary Biliary Cirrhosis |
| Methods | Phase 3, double‐blind, randomised, placebo‐controlled, multicentre study. |
| Participants | People with primary biliary cholangitis. |
| Interventions | Group 1: obeticholic acid. Further details: obeticholic acid 5 mg to 10 mg tablets once daily for the duration of the study based on tolerability at 3 months. Group 2: placebo. Further details: 1 tablet daily for the remainder of the study. |
| Outcomes | Mortality, liver transplantation, liver decompensation, hepatocellular carcinoma. |
| Starting date | December 2014. |
| Contact information | dshapiro@interceptpharma.com |
| Notes | Status: recruiting. |
NCT02701166.
| Trial name or title | The Effect of Bezafibrate on Cholestatic Itch |
| Methods | Double‐blind, randomised, placebo‐controlled clinical trial. |
| Participants | People with primary biliary cholangitis. |
| Interventions | Group 1: bezafibrate. Further details: bezafibrate 400 mg/day. Group 2: placebo. |
| Outcomes | None of the outcomes of interest for this review are measured in this trial. |
| Starting date | February 2016. |
| Contact information | u.h.beuers@amc.uva.nl |
| Notes | Status: recruiting. |
NCT02823353.
| Trial name or title | Fenofibrate in Combination with Ursodeoxycholic Acid in Primary Biliary Cirrhosis: a Randomized Control Study |
| Methods | Phase 3, open‐label, randomised clinical trial. |
| Participants | People with primary biliary cholangitis. |
| Interventions | Group 1: UDCA + fenofibrate. Further details: not provided. Group 2: UDCA. Further details: not provided. |
| Outcomes | None of the outcomes of interest for this review are measured in this trial. |
| Starting date | January 2016. |
| Contact information | hanying@fmmu.edu.cn |
| Notes | Status: recruiting. |
NCT02823366.
| Trial name or title | Fenofibrate for Patients with Primary Biliary Cirrhosis who had an Inadequate Response to Ursodeoxycholic Acid |
| Methods | Phase 3, open‐label, randomised clinical trial. |
| Participants | People with primary biliary cholangitis. |
| Interventions | Group 1: UDCA + fenofibrate Further details: not provided. Group 2: UDCA. Further details: not provided. |
| Outcomes | None of the outcomes of interest for this review measured in this trial. |
| Starting date | January 2016. |
| Contact information | hanying@fmmu.edu.cn |
| Notes | Status: recruiting. May be the same as NCT02823353. |
NCT02937012.
| Trial name or title | Efficacy and Security of Bezafibrate in Patients with Primary Biliary Cirrhosis without Biochemical Response to Ursodeoxycholic Acid: a Randomized, Double‐blind, Placebo‐controlled Trial |
| Methods | Randomised, double‐blind, placebo‐controlled clinical trial. |
| Participants | People with primary biliary cholangitis (non‐responders). |
| Interventions | Group 1: UDCA + bezafibrate. Further details: bezafibrate 200 mg capsule every 12 hours + UDCA 13 mg/kg/day to 15 mg/kg/day for 12 months. Group 2: UDCA + placebo. Further details: placebo capsule (for bezafibrate 200 mg capsule) every 12 hours + UDCA 13 mg/kg/day to 15 mg/kg/day for 12 months. |
| Outcomes | Quality of life. |
| Starting date | October 2016. |
| Contact information | ericlopezmendez@yahoo.com.mx sergio_sg@hotmail.com |
| Notes | Status: recruiting. |
NCT02943447.
| Trial name or title | A Phase 2, Randomized, Double‐Blind, Placebo Controlled Study Evaluating the Safety, Tolerability, and Efficacy of GS‐9674 in Subjects with Primary Biliary Cholangitis without Cirrhosis |
| Methods | Randomised, double‐blind, placebo‐controlled clinical trial. |
| Participants | People with primary biliary cholangitis. |
| Interventions | Group 1: GS‐9674. Further details: GS‐9674 30 mg for 12 weeks. Group 2: placebo. |
| Outcomes | Adverse events. |
| Starting date | December 2016. |
| Contact information | GS‐US‐427‐4024@Gilead.com |
| Notes | Status: recruiting. |
NCT02965911.
| Trial name or title | A Randomized Controlled Clinical Trial on the Efficacy and Safety of Fenofibrate Combined with Ursodeoxycholic Acid in PBC Patients with an Incomplete Biochemical Response to UDCA |
| Methods | Open‐label, randomised clinical trial. |
| Participants | People with primary biliary cholangitis. |
| Interventions | Group 1: fenofibrate + UDCA. Further details: UDCA 13 mg/kg/day to 15 mg/kg/day + fenofibrate 200 mg once daily for 12 months. Group 2: UDCA. Further details: UDCA 13 mg/kg/day to 15 mg/kg/day. |
| Outcomes | None of the outcomes of interest for this review measured in this trial. |
| Starting date | January 2016. |
| Contact information | zszou302@163.com |
| Notes | Status: recruiting. |
UDCA: ursodeoxycholic acid.
Differences between protocol and review
It was not possible to assess whether the potential effect modifiers were similar across different comparisons. Therefore, we did not perform the network meta‐analysis and assessed the comparative benefits and harms of different interventions using standard Cochrane methodology. The methodology that we plan to use if we conduct a network meta‐analysis in the future is available in Appendix 1.
We performed Trial Sequential Analysis in addition to conventional method of assessing the risk of random errors using P values.
Contributions of authors
FS identified the trials, extracted the data for half the trials, and completed the characteristics tables. KG identified the trials, extracted the data, performed the analysis, and wrote the review. LHE extracted the data for half the trials. ET, BD, and DT critically commented on the review.
Sources of support
Internal sources
University College London, UK.
External sources
National Institute for Health Research, UK.
Declarations of interest
This report is independent research funded by the National Institute for Health Research (NIHR Cochrane Programme Grants, 13/89/03 ‐ Evidence‐based diagnosis and management of upper digestive, hepato‐biliary, and pancreatic disorders). The views expressed in this publication are those of the review authors and not necessarily those of the National Health System (NHS), the NIHR, or the Department of Health.
FS has no financial disclosures. KG has no financial disclosures. LHE has no financial disclosures. ET has participated in advisory boards for Astra Zeneca and ViiV healthcare. BD has no financial disclosures. DT was funded by Astellas for his attendance at the International Liver Transplantation Society meeting in 2014. He received GBP 25,000 from Boston Scientific to fund a clinical research fellow in 2013.
Edited (no change to conclusions)
References
References to studies included in this review
Almasio 2000 {published data only}
- Almasio P, Provenzano G, Battezzati PM, Podda M, Todros L, Rosina F, et al. The Italian multi‐centre randomized controlled trial of UDCA vs colchicine plus UDCA in symptomatic primary biliary cholangitis. Hepatology 1994;20(4 (Pt 2)):73. [Google Scholar]
- Almasio PL, Floreani A, Chiaramonte M, Provenzano G, Battezzati P, Crosignani A, et al. Multicentre randomized placebo‐controlled trial of UDCA with or without colchicine in symptomatic primary biliary cholangitis. Alimentary Pharmacology & Therapeutics 2000;14(12):1645‐52. [DOI] [PubMed] [Google Scholar]
- Battezzati PM, Zuin M, Crosignani A, Allocca M, Invernizzi P, Selmi C, et al. Ten‐year combination treatment with colchicine and UDCA for primary biliary cholangitis: a double‐blind, placebo‐controlled trial on symptomatic patients. Alimentary Pharmacology & Therapeutics 2001;15(9):1427‐34. [DOI] [PubMed] [Google Scholar]
- Podda M, Almasio P, Battezzati PM, Crosignani A. Long‐term effect of the administration of UDCA alone or with colchicine in patients with primary biliary cholangitis: a double‐blind multicentre study. 68th Falk Symposium; 1992 Oct 12‐14; Basel, Switzerland. 1993:310‐5.
Angulo 1999a {published data only}
- Angulo P, Dickson ER, Therneau TM, Jorgensen RA, Smith C, DeSotel CK, et al. Comparison of three doses of UDCA in the treatment of primary biliary cholangitis: a randomized trial. Journal of Hepatology 1999;30(5):830‐5. [DOI] [PubMed] [Google Scholar]
Arora 1990 {published data only}
- Arora R, Batta AK, Salen G, O'Brien C, Senior JR. Effect of ursodiol on bile acid conjugation in patients with primary biliary cholangitis. Hepatology 1990;12(4 (Pt 2)):994. [Google Scholar]
- Batta AK, Arora R, Salen G, Katz S. UDCA improves liver function and reduces serum and urinary endogenous bile acids in primary biliary cholangitis. Hepatology 1988;8(5):1221. [Google Scholar]
- Batta AK, Arora R, Salen G, O'Brien C, Senior JR. Effect or ursodiol on biliary bile acid composition and conjugation in patients with primary biliary cholangitis. Gastroenterology 1990;98(2 (Pt 2)):A567. [Google Scholar]
Askari 2010 {published data only}
- Askari F, Innis D, Dick RB, Hou GQ, Marrero J, Greenson J, et al. Treatment of primary biliary cholangitis with tetrathiomolybdate: results of a double‐blind trial. Translational Research 2010;155(3):123‐30. [DOI] [PubMed] [Google Scholar]
Battezzati 1993 {published data only}
- Anonymous. UDCA (UDCA) for symptomatic primary biliary cholangitis (PBC): a double‐blind multicenter trial. Journal of Hepatology 1989;9(Suppl 1):S44. [Google Scholar]
- Battezzati PM, Podda M, Bianchi FB, Naccarato R, Orlandi F, Surrenti C, et al. UDCA for symptomatic primary biliary cholangitis. Preliminary analysis of a double‐blind multicenter trial. Italian multicenter group for the study of UDCA in PBC. Journal of Hepatology 1993;17(3):332‐8. [DOI] [PubMed] [Google Scholar]
- Podda M, Battezzati PM, Crosignani A, Bianchi FB, Fusconi M, Chiaramonte M, et al. UDCA (UDCA) for symptomatic primary biliary cholangitis (PBC): a double‐blind multicenter trial. Hepatology 1989;10(4):639. [Google Scholar]
Bobadilla 1994 {published data only}
- Bobadilla J, Vargas F, Dehesa M, Zapata L, Kaplan M, Nava R, et al. Colchicine and ursodiol in the treatment of primary biliary cholangitis. Hepatology 1994;20(4 (Pt 2)):332a. [Google Scholar]
Bodenheimer 1988 {published data only}
- Bodenheimer H Jr, Schaffner F, Pezzullo J. Evaluation of colchicine therapy in primary biliary cholangitis. Gastroenterology 1988;95(1):124‐9. [DOI] [PubMed] [Google Scholar]
- Bodenheimer H, Schaffner F, Pezzullo J. A randomized double‐blind controlled trial of colchicine in primary biliary cholangitis. Hepatology 1985;5(5):968. [Google Scholar]
- Bodenheimer H, Schaffner F, Pezzullo J. Colchicine therapy in primary biliary cholangitis. Hepatology 1986;6(5):1172. [Google Scholar]
- Zifroni A, Schaffner F. Long‐term follow‐up of patients with primary biliary cholangitis on colchicine therapy. Hepatology 1991;14(6):990‐3. [PubMed] [Google Scholar]
Bowlus 2014 {published data only}
- Bowlus CL, Pockros PJ, Drenth J, Floreani A, Vincent C, Luketic VA, et al. Obeticholic acid in PBC patients: the utility of titration based on therapeutic response and tolerability. Hepatology 2014;60:353a. [Google Scholar]
Cash 2013 {published data only}
- Cash WJ, O'Neill S, O'Donnell ME, McCance DR, Young IS, McEneny J, et al. Randomized controlled trial assessing the effect of simvastatin in primary biliary cholangitis. Liver International 2013;33(8):1166‐74. [DOI] [PubMed] [Google Scholar]
Christensen 1985 {published data only}
- Christensen E, Neuberger J, Crowe J. Beneficial effect of azathioprine and prediction of prognosis in primary biliary cholangitis. Final results of an international trial. Gastroenterology 1985;89(5):1084‐91. [DOI] [PubMed] [Google Scholar]
- Christensen E, Neuberger J, Crowe J, Popper H, Portmann B, Deniach D, et al. Azathioprine in primary biliary cholangitis: late results of an international trial. Liver 1984;4(1):81. [Google Scholar]
- Christensen E, Neuberger J, Crowe J, Popper H, Portmann B, Doniach D, et al. Azathioprine in primary biliary cholangitis: late results of an international trial. Gut 1983;24(10):A995‐6. [Google Scholar]
- Crowe J, Christensen E, Smith M. Azathioprine in primary biliary cholangitis: a preliminary report of an international trial. Gastroenterology 1980;78(5 I):1005‐10. [PubMed] [Google Scholar]
Combes 1995a {published data only}
- Carithers RL, Luketic VA, Peters M, Zetterman RK, Garcia‐Tsao G, Munoz SJ. Extended follow‐up of patients in the US multicenter trial of UDCA for primary biliary cholangitis. Gastroenterology 1996;110(Suppl 4):A1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes B, Carithers RL Jr, Maddrey WC, Lin D, McDonald MF, Wheeler DE, et al. A randomized, double‐blind, placebo‐controlled trial of UDCA in primary biliary cholangitis. Hepatology 1995;22(3):759‐66. [PubMed] [Google Scholar]
- Combes B, Carithers RL Jr, Maddrey WC, Munoz S, Garcia‐Tsao G, et al. Biliary bile acids in primary biliary cholangitis: effect of UDCA. Hepatology 1999;29(6):1649‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combes B, Carithers RL Jr, McDonald MF, Maddrey WC, Munoz SJ, Boyer JL, et al. UDCA therapy in patients with primary biliary cholangitis. Hepatology 1991;14(4 (Pt 2)):91a. [Google Scholar]
- Combes B, Carithers RL, Maddrey WC, Munoz SJ, McDonald MF, Garcia‐Tsao G. A randomized, double‐blind, placebo‐controlled trial of UDCA in primary biliary cholangitis. 68th Falk Symposium; 1992 Oct 12‐14; Basel, Switzerland. 1993:289‐91.
- Combes B, Carithers RL, Maddrey WC, Munoz SJ, McDonald MF, Garcia‐Tsao G, et al. A randomized, double‐blind, placebo‐controlled trial of UDCA (UDCA) in primary biliary cholangitis. Hepatology 1993;18(4 (Pt 2)):175a. [PubMed] [Google Scholar]
- Combes B, Markin RS, Wheeler DE, Rubin R, West AB, Mills AS, et al. The effect of UDCA on the florid duct lesion of primary biliary cholangitis. Hepatology 1999;30(3):602‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Combes 2005 {published data only}
- Combes B, Emerson SS, Flye NL. The primary biliary cholangitis (PBC) ursodiol (UDCA) plus methotrexate (MTX) or its placebo study (PUMPS) ‐ a multicenter randomized trial. Hepatology 2003;38(4):210A‐1A. [Google Scholar]
- Combes B, Emerson SS, Flye NL, Munoz SJ, Luketic VA, Mayo MJ, et al. Methotrexate (MTX) plus UDCA (UDCA) in the treatment of primary biliary cholangitis. Hepatology 2005;42(5):1184‐93. [DOI] [PubMed] [Google Scholar]
- Munoz S, Carithers R, Emerson SS, Flye N, Kowdley K, Combes B. Absence of pulmonary toxicity in primary biliary cholangitis (PBC) treated with methotrexate and ursodiol. Hepatology 1998;28(Suppl 4):392a. [Google Scholar]
- NCT00004784. Phase III randomized study of ursodiol with vs without methotrexate for primary biliary cholangitis. clinicaltrials.gov/ct2/show/NCT00004784 (date first received: 24 February 2000).
Dickson 1985 {published data only}
- Deering TB, Dickson ER, Fleming CR. Effect of D penicillamine on copper retention in patients with primary biliary cholangitis. Gastroenterology 1977;72(6):1208‐12. [PubMed] [Google Scholar]
- Dickson ER. The syndrome of primary biliary cholangitis. Journal of Rheumatology ‐ Supplement 1981;7:121‐3. [PubMed] [Google Scholar]
- Dickson ER, Fleming CR, Geall MG. A double blind controlled study using D‐penicillamine in chronic cholangiolitic hepatitis (primary biliary cholangitis). Gastroenterology 1977;72(5 II):A‐26. [Google Scholar]
- Dickson ER, Fleming TR, Wiesner RH. Trial of penicillamine in advanced primary biliary cholangitis. New England Journal of Medicine 1985;312(16):1011‐5. [DOI] [PubMed] [Google Scholar]
- Dickson ER, Wiesner RH, Baldus WP, Fleming CR, Ludwig JL. D‐penicillamine improves survival and retards histologic progression in primary biliary‐cirrhosis. Gastroenterology 1982;82(5 Part 2):1225. [Google Scholar]
- Fleming CR, Ludwig J, Dickson ER. Asymptomatic primary biliary cholangitis. Presentation, histology, and results with D‐penicillamine. Mayo Clinic Proceedings 1978;53(9):587‐93. [PubMed] [Google Scholar]
- Locke GR 3rd, Therneau TM, Ludwig J, Dickson ER, Lindor KD. Time course of histological progression in primary biliary cholangitis. Hepatology 1996;23(1):52‐6. [DOI] [PubMed] [Google Scholar]
- Powell FC, Rogers RS, Dickson ER. Primary biliary cholangitis and lichen planus. Journal of the American Academy of Dermatology 1983;9(4):540‐5. [DOI] [PubMed] [Google Scholar]
Epstein 1979 {published data only}
- Epstein G, Williers D, Jain S, Potter BJ, Thomas HC, Sherlock S. Effect of penicillamine on immune complexes and immunoglobulins in primary biliary cholangitis (PBC). Gut 1978;19(Suppl 3):A994. [Google Scholar]
- Epstein O, Cook D, Jain S, Sherlick S. D‐Penicillamine in primary biliary cholangitis (PBC) ‐ an untested (and untestable?) treatment. Gut 1984;25:A1134. [Google Scholar]
- Epstein O, Cook DG, Jain S, McIntyre N, Sherlock S. D‐Penicillamine and clinical‐trials in PBC. Hepatology 1984;4(5):1032. [Google Scholar]
- Epstein O, Cook DG, Jain S, Sherlock S. D‐Penicillamine in PBC ‐ an untested (and untestable?) treatment. Journal of Hepatology 1985;1(Suppl 1):S49. [Google Scholar]
- Epstein O, Villiers D, Jain S. Reduction of immune complexes and immunoglobulins induced by D‐penicillamine in primary biliary cholangitis. New England Journal of Medicine 1979;300(6):274‐8. [DOI] [PubMed] [Google Scholar]
- Epstein O, Jain S, Lee RG. D‐Penicillamine treatment improves survival in primary biliary cholangitis. Lancet 1981;1(8233):1275‐7. [DOI] [PubMed] [Google Scholar]
- Epstein O, Jain S, Lee RG, Cook DG, Boss AM, Scheuer PJ, et al. D‐Penicillamine treatment improves survival in primary biliary‐cirrhosis. Gut 1981;22(5):A433‐A. [DOI] [PubMed] [Google Scholar]
- Jain S, McGee JD, Scheuer PJ, Samourian S, Sherlock S. A controlled trial of D‐penicillamine therapy in primary biliary cholangitis and chronic active hepatitis. Digestion 1976;14:523. [Google Scholar]
- Jain S, Scheuer PJ, Samourian S. A controlled trial of D‐penicillamine therapy in primary biliary cholangitis. Lancet 1977;1(8016):831‐4. [DOI] [PubMed] [Google Scholar]
- Jain S, Scheur PJ, Samourian S, McGee JD, Sherlock S. A controlled trial of D‐penicillamine therapy in primary biliary cholangitis. Gut 1976;17(Suppl 2):822. [Google Scholar]
Eriksson 1997 {published data only}
- Eriksson LS, Olsson R, Glauman H, Prytz H, Befrits H, Lindgren S, et al. UDCA (UDCA) in patients with primary biliary cholangitis (PBC): results of a two‐year randomized placebo‐controlled study (abstract). Scandinavian Journal of Gastroenterology 1995;30(Suppl 209):35. [Google Scholar]
- Eriksson LS, Olsson R, Glauman H, Prytz H, Befrits R, Ryden BO, et al. UDCA treatment in patients with primary biliary cholangitis. A Swedish multicentre, double‐blind, randomized controlled study. Scandinavian Journal of Gastroenterology 1997;32(2):179‐86. [DOI] [PubMed] [Google Scholar]
Ferri 1993 {published data only}
- Ferri F, Bernocchi P, Fedeli S. [Taurodeoxycholic acid in the treatment of primary biliary cholangitis. A controlled study in comparison to UDCA]. Clinica Terapeutica 1993;143(4):321‐6. [PubMed] [Google Scholar]
Gao 2012 {published data only}
- Gao LX, Zhang FC, Wang L, Zhang X, Liu B. The clinical observation of different therapeutic strategies in combined primary biliary cholangitis and Sjogren syndrome. Chung‐Hua Nei Ko Tsa Chih 2012;51(11):851‐4. [PubMed] [Google Scholar]
Goddard 1994 {published data only}
- Goddard C, Smith A, Hunt L, Halder T, Hillier V, Rowan B, et al. Surrogate markers of response in a trial of UDCA (UDCA) and colchicine in primary biliary cholangitis (PBC). Gut 1995;36(Suppl 1):A30. [Google Scholar]
- Goddard CJR, Hunt L, Smith A, Followfield G, Rowan B, Warnes TW. A trial of UDCA (UDCA) and colchicine in primary biliary cholangitis (PBC). Hepatology 1994;20(4 (Pt 2)):151a. [Google Scholar]
Gonzalezkoch 1997 {published data only}
- Gonzalezkoch A, Brahm J, Antezana C, Smok G, Cumsille MA. The combination of UDCA and methotrexate for primary biliary cholangitis is not better than UDCA alone. Journal of Hepatology 1997;27(1):143‐9. [DOI] [PubMed] [Google Scholar]
Heathcote 1976 {published data only}
- Heathcote J, Ross A, Sherlock S. A prospective controlled trial of azathioprine in primary biliary cholangitis. Gastroenterology 1976;70(5 PT.1):656‐60. [PubMed] [Google Scholar]
- Ross A, Sherlock S. A controlled trial of azathioprine in primary biliary cholangitis. Gut 1971;12(2):770. [Google Scholar]
- Ross A, Sherlock S. A trial of azathioprine in primary biliary cholangitis. Gut 1970;11(12):1058. [PubMed] [Google Scholar]
Heathcote 1994 {published data only}
- Heathcote EJ, Cauch‐Dudek K, Walker V, Bailey RJ, Blendis LM, Ghent CN, et al. The Canadian multicenter double‐blind randomized controlled trial of UDCA in primary biliary cholangitis. Hepatology 1994;19(5):1149‐56. [PubMed] [Google Scholar]
- Heathcote EJL, Cauch K, Walker V, Bailey RJ, Blendis LM, Ghent CN, et al. The Canadian multicenter double‐blind randomized controlled trial of UDCA in primary biliary‐cirrhosis. Hepatology 1992;16(4):A91. [PubMed] [Google Scholar]
- Heathcote EJL, Cauch K, Walker V, Blendis LM, Pappas SC, Wanless IR. A double‐blind randomized controlled multicentre trial of UDCA in primary biliary cholangitis: results from a 1991 interim analysis. 68th Falk Symposium; 1992 Oct 12‐14; Basel, Switzerland. 1993:294‐8.
- Neuman MG, Cameron RG, Haber JA, Katz GG, Blendis LM. An electron microscopic and morphometric study of ursodeoxycholic effect in primary biliary cholangitis. Liver 2002;22(3):235‐44. [DOI] [PubMed] [Google Scholar]
- Neuman MG, Cameron RG, Shear NH, Blendis LM. Electron microscopic study on antifibrotic effects of ursodeoxycholate treatment in primary biliary cholangitis. Bile Acids and Cholestasis 1999;108:254‐69. [Google Scholar]
- Worobetz LJ, Cauch‐Dudek K, Heathcote EJ. The effect of UDCA (UDCA) on anti‐mitochondrial antibody (AMA) titre in primary biliary cholangitis (PBC). Gastroenterology 1995;108(4 Suppl 3):A1200. [Google Scholar]
Hendrickse 1999 {published data only}
- Giaffer MH, Hendrickse M, Soomoro I, Triger DR, Underwood JCE, Gleeson D. Low‐dose methotrexate in treatment of primary biliary cholangitis. Gut 1995;36(Suppl 1):A30. [Google Scholar]
- Hendrickse M, Rigney E, Giaffer MH, Soomoro I, Triger DR, Underwood JC, et al. Low‐dose methotrexate in primary biliary cholangitis: long‐term results of a placebo‐controlled trial. Hepatology 1997;26(4):479. [DOI] [PubMed] [Google Scholar]
- Hendrickse MT, Rigney E, Giaffer MH, Soomro I, Triger DR, Underwood JC, et al. Low‐dose methotrexate is ineffective in primary biliary cholangitis: long‐term results of a placebo‐controlled trial. Gastroenterology 1999;117(2):400‐7. [DOI] [PubMed] [Google Scholar]
Hirschfield 2015 {published data only}
- Hirschfield G, Kowdley K, Mason A, Luketic V, Gordon S, Vincent C, et al. Long‐term (LT) therapy of a farnesoid X receptor (FXR) agonist obeticholic acid (OCA) maintains biochemical response in primary biliary cholangitis (PBC). Journal of Hepatology 2012;56:S372. [Google Scholar]
- Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cholangitis and inadequate response to UDCA. Gastroenterology 2015;148(4):751‐61.e8. [DOI] [PubMed] [Google Scholar]
- Luketic V, Gordon SG, Vincent C, Chapman R, Mayo M, Kowdley K, et al. The FXR agonist obeticholic acid improves a transplant free survival‐proven biochemical response criterion in placebo controlled primary biliary cholangitis studies. Journal of Hepatology 2014;60(1 (Suppl 1)):S193‐4. [Google Scholar]
- Luketic VA, Invernizzi P, Trauner M, Regula J, Mazzella G, Strasser SI, et al. Efficacy of obeticholic acid in primary biliary cholangitis as assessed by response criteria associated with clinical outcome: a poise analysis. Hepatology 2014;60:355a‐6a. [Google Scholar]
- Marschall H, Luketic VA, Mason AL, Lindor KD, Hirschfield GM, Gordon SC, et al. The farnesoid X receptor (FXR) agonist obeticholic acid (INT‐747, 6α‐ethyl chenodeoxycholic acid) in combination with UDCA (UDCA) increases plasma FGF‐19 concentrations but not bile acid concentration or profile in primary biliary cholangitis (PBC). Hepatology 2010;52(Suppl S1):355a. [Google Scholar]
- Mason AL, Lindor KD, Bacon BR, Vincent C, Neuberger JM, Wasilenko ST. Multi‐center, double blind, randomized controlled trial of zidovudine and lamivudine (Combivir) therapy for patients with primary biliary cholangitis. Hepatology 2007;46(4):264A. [Google Scholar]
Hoofnagle 1986 {published data only}
- Hoofnagle JH, Davis GL, Schafer DF, Peters M, Avigan MI, Pappas SC, et al. Randomized trial of chlorambucil for primary biliary cholangitis. Gastroenterology 1986;91(6):1327‐34. [DOI] [PubMed] [Google Scholar]
- Hoofnagle JH, Davis GL, Schafer DF, Peters MG, Avigan MI, Hanson RG, et al. Randomized trial of chlorambucil for primary biliary‐cirrhosis. Hepatology 1984;4(5):1062. [DOI] [PubMed] [Google Scholar]
Hosonuma 2015 {published data only}
- Hosonuma K, Sato K, Yamazaki Y, Yanagisawa M, Hashizume H, Horiguchi N, et al. A prospective randomized controlled study of long‐term combination therapy using UDCA and bezafibrate in patients with primary biliary cholangitis and dyslipidaemia. American Journal of Gastroenterology 2015;110(3):423‐31. [DOI] [PubMed] [Google Scholar]
- Sato K, Hosonuma K, Yamazaki Y, Yanagisawa M, Hashizume H, Horiguchi N, et al. Long‐term prognosis of combination therapy with UDCA and bezafibrate for primary biliary cholangitis: a prospective, multicenter, randomized controlled study. Hepatology International 2014;8(1 (Suppl 1)):S15. [Google Scholar]
Ikeda 1996 {published data only}
- Ikeda T, Tozuka S, Noguchi O, Kobayashi F, Sakamoto S, Marumo F, et al. Effects of additional administration of colchicine in UDCA‐treated patients with primary biliary cholangitis: a prospective randomized study. Journal of Hepatology 1996;24(1):88‐94. [DOI] [PubMed] [Google Scholar]
Iwasaki 2008a {published data only}
- Iwasaki S, Ohira H, Nishiguchi S, Zeniya M, Kaneko S, Onji M, et al. The efficacy of UDCA and bezafibrate combination therapy for primary biliary cholangitis: a prospective, multicenter study. Hepatology Research 2008;38(6):557‐64. [DOI] [PubMed] [Google Scholar]
Iwasaki 2008b {published data only}
- Iwasaki S, Ohira H, Nishiguchi S, Zeniya M, Kaneko S, Onji M, et al. The efficacy of UDCA and bezafibrate combination therapy for primary biliary cholangitis: a prospective, multicenter study. Hepatology Research 2008;38(6):557‐64. [DOI] [PubMed] [Google Scholar]
Kanda 2003 {published data only}
- Kanda T, Yokosuka O, Imazeki F, Saisho H. Bezafibrate treatment: a new medical approach for PBC patients?. Journal of Gastroenterology 2003;38(6):573‐8. [DOI] [PubMed] [Google Scholar]
Kaplan 1986 {published data only}
- Johnston DE, Kaplan MM, Miller KB, Connors CM, Milford EL. Histocompatibility antigens in primary biliary cholangitis. American Journal of Gastroenterology 1987;82(11):1127‐9. [PubMed] [Google Scholar]
- Kaplan MM, Alling DW, Wolfe HJ, Zimmerman HJ. Colchicine is effective in the treatment of primary biliary cholangitis. Hepatology 1985;5(5):967. [Google Scholar]
- Kaplan MM, Alling DW, Zimmerman HJ, Wolfe HJ, Sepersky RA, Hirsch GE, et al. A prospective trial of colchicine for primary biliary cholangitis. (abstract). Acta Gastroenterologica Belgica 1987;50(3):382. [Google Scholar]
- Kaplan MM, Alling DW, Zimmerman HJ, Wolfe HJ, Sepersky RA, Hirsch GS, et al. A prospective trial of colchicine for primary biliary cholangitis. New England Journal of Medicine 1986;315(23):1448‐54. [DOI] [PubMed] [Google Scholar]
- Miller LC, Kaplan MM. Serum interleukin‐2 and tumor necrosis factor‐alpha in primary biliary cholangitis: decrease by colchicine and relationship to HLA‐DR4. American Journal of Gastroenterology 1992;87(4):465‐70. [PubMed] [Google Scholar]
Kaplan 1999 {published data only}
- Kaplan M, Schmid C, McKusick A, Provenzale D, Sharma A, Sepe T. Double blind trial of methotrexate (MTX) versus colchicine (COLCH) in primary biliary cholangitis (PBC). Hepatology 1993;18(4 (Pt 2)):176a. [Google Scholar]
- Kaplan MM, Dickstein G, Schmid C. Methotrexate (MTX) improves histology in primary biliary cholangitis (PBC). Hepatology 1994;20(4 (Pt 2)):152a. [Google Scholar]
- Kaplan MM, Schmid C, Provenzale D, Sharma A, Dickstein G, McKusick A. A prospective trial of colchicine and methotrexate in the treatment of primary biliary cholangitis. Gastroenterology 1999;117(5):1173‐80. [DOI] [PubMed] [Google Scholar]
- Miller LC, Sharma A, McKusick AF, Tassoni JP, Dinarello CA, Kaplan MM. Synthesis of interleukin‐1 beta in primary biliary cholangitis: relationship to treatment with methotrexate or colchicine and disease progression. Hepatology 1995;22(2):518‐24. [PubMed] [Google Scholar]
- NCT00004748. Phase III randomized, double‐blind, placebo‐controlled study of low‐dose oral methotrexate vs colchicine for primary biliary cholangitis. www.clinicaltrials.gov/ct2/show/NCT00004748 Date first received: 24 February 2000.
- Sharma A, Provenzale D, McKusick A, Kaplan MM. Interstitial pneumonitis after low‐dose methotrexate therapy in primary biliary cholangitis. Gastroenterology 1994;107(1):266‐70. [DOI] [PubMed] [Google Scholar]
Kowdley 2011 {published data only}
- Kowdley K, Jones D, Luketic V, Chapman R, Burroughs A, Hirschfield G, et al. An international study evaluating the farnesoid X receptor agonist obeticholic acid as monotherapy in PBC. Journal of Hepatology 2011;54:S13. [Google Scholar]
Kurihara 2000 {published data only}
- Kurihara T, Niimi A, Maeda A, Shigemoto M, Yamashita K. Bezafibrate in the treatment of primary biliary cholangitis: comparison with UDCA. American Journal of Gastroenterology 2000;95(10):2990‐2. [DOI] [PubMed] [Google Scholar]
Leuschner 1989 {published data only}
- Guldutuna S, Leuschner U, Imhof M, Zimmer G. Treatment of chronic active hepatitis and primary biliary cholangitis with UDCA. Zeitschrift Fur Gastroenterologie 1992;30 Suppl 1:49‐54. [PubMed] [Google Scholar]
- Leuschner U, Fischer H, Guldutuna S, Kurtz W, Gatzen M, Hellstern A. Does UDCA (UDCA) influence cell membrane architecture in patients with primary biliary cholangitis (PBC)?. Gastroenterology 1989;96(5 (Pt 2)):A621. [DOI] [PubMed] [Google Scholar]
- Leuschner U, Fischer H, Hubner K. [UDCA in the treatment of primary biliary cholangitis: results of a controlled study]. Zeitschrift fur Gastroenterologie ‐ Verhandlungsband 1989;24:133. [PubMed] [Google Scholar]
- Leuschner U, Fischer H, Kurtz W, Guldutuna S, Hubner K, Hellstern A, et al. UDCA in primary biliary cholangitis: results of a controlled double‐blind trial. Gastroenterology 1989;97(5):1268‐74. [DOI] [PubMed] [Google Scholar]
- Leuschner U, Fisher H, Hübner K, Güldütuna S, Gatzen M, Hellstern A. UDCA (UDCA) treatment of primary biliary cholangitis: clinical and histological results of a controlled study. 52nd Falk Symposium; 1988 Jun 9‐11; Freiburg, Germany 1989;41:355‐8. [Google Scholar]
Leuschner 1999 {published data only}
- Leuschner M, Maier KP, Schlichting J, Strahl R, Herrmann G, Dahm HH, et al. Combination of UDCA (UDCA) with budesonide (BUD) is superior to UDCA‐mono‐therapy in primary biliary cholangitis (PBC). Journal of Hepatology 1999;30(Suppl 1):57. [Google Scholar]
- Leuschner M, Maier KP, Schlichting J, Strahl R, Herrmann G, Dahm HH, et al. UDCA (UDCA) and budesonide (BUD) in the treatment of primary biliary cholangitis (PBC): a prospective double‐blind trial. Hepatology 1999;30(4):471a. [DOI] [PubMed] [Google Scholar]
- Leuschner M, Maier KP, Schlichting J, Strahl R, Herrmann G, Dahm HH, et al. [UDCA (UDCA)‐placebo versus UDCA‐budesonide by primary biliary cholangitis (PBC). A double‐blind study]. Zeitschrift fur Gastroenterologie 1999;37(9):897. [Google Scholar]
- Leuschner M, Maier KP, Schlichting J, Strahl S, Herrmann G, Dahm HH, et al. Oral budesonide and UDCA for treatment of primary biliary cholangitis: results of a prospective double‐blind trial. Gastroenterology 1999;117(4):918‐25. [DOI] [PubMed] [Google Scholar]
Liberopoulos 2010 {published data only}
- Liberopoulos EN, Florentin M, Elisaf MS, Mikhailidis DP, Tsianos E. Fenofibrate in primary biliary cholangitis: a pilot study. Open Cardiovascular Medicine Journal 2010;4:120‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lim 1994 {published data only}
- Lim AG, Jazrawi RP, Maxwell JD, Northfield TC. UDCA (UDCA) improves hepatic excretion in primary biliary cholangitis (PBC). Gut 1994;35(S5):S11. [Google Scholar]
- Lim AG, Jazrawi RP, Petroni ML, Pereira S, Maxwell JD, Northfield TC. T‐lymphocyte activation in primary biliary cholangitis: effects of UDCA. Falk Symposium XIII International Bile Acid Meeting 1994;80:147. [Google Scholar]
Lindor 1994 {published data only}
- Balan V, Dickson ER, Jorgensen RA, Lindor KD. Effect of UDCA on serum lipids of patients with primary biliary cholangitis. Mayo Clinic Proceedings 1994;69(10):923‐9. [DOI] [PubMed] [Google Scholar]
- Batts KP, Jorgensen RA, Dickson ER, Hofmann AF, Rossi SS, Ludwig J, et al. The effects of UDCA on hepatic inflammation and histologic stage in patients with primary biliary cholangitis. Hepatology 1993;18(4 (Pt 2)):175a. [Google Scholar]
- Batts KP, Jorgensen RA, Dickson ER, Lindor KD. Effects of UDCA on hepatic inflammation and histological stage in patients with primary biliary cholangitis. American Journal of Gastroenterology 1996;91(11):2314‐7. [PubMed] [Google Scholar]
- Dickson ER, Lindor KD, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA. Ursodiol is effective therapy for patients with primary biliary cholangitis. 68th Falk Symposium; 1992 Oct 12‐14; Basel, Switzerland. 1993:292‐3.
- Jorgensen RA, Dickson ER, Hofmann AF, Rossi SS, Lindor KD. Characterisation of patients with a complete biochemical response to UDCA. Gut 1995;36(6):935‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacerda MA, Lindor KD, Jorgensen RA, Rossi SS, Hofmann AF, Salen GR, et al. Dissimilar patterns of serum and biliary bile‐acids in primary biliary‐cirrhosis (PBC) patients treated with UDCA (UDCA). Hepatology 1993;18(4):A174. [Google Scholar]
- Laurin JM, DeSotel CK, Jorgensen RA, Dickson ER, Lindor KD. The natural history of abdominal pain associated with primary biliary cholangitis. American Journal of Gastroenterology 1994;89(10):1840‐3. [PubMed] [Google Scholar]
- Lindor KD, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA, Dickson ER. UDCA (UDCA) is beneficial therapy for patients with primary biliary cholangitis (PBC). Hepatology 1992;16(2 (Pt 2)):91a. [Google Scholar]
- Lindor KD, Dickson ER, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA, et al. UDCA in the treatment of primary biliary cholangitis. Gastroenterology 1994;106(5):1284‐90. [DOI] [PubMed] [Google Scholar]
- Lindor KD, Janes CH, Crippin JS, Jorgensen RA, Dickson ER. Bone disease in primary biliary cholangitis: does UDCA make a difference?. Hepatology 1995;21(2):389‐92. [PubMed] [Google Scholar]
- Lindor KD, Jorgensen RA, Dickson ER. UDCA delays the onset of esophageal varices in primary biliary cholangitis. Hepatology 1995;22(4 (Pt 2)):125a. [DOI] [PubMed] [Google Scholar]
- Lindor KD, Jorgensen RA, Therneau TM, Malinchoc M, Dickson ER. UDCA delays the onset of esophageal varices in primary biliary cholangitis. Mayo Clinic Proceedings 1997;72(12):1137‐40. [DOI] [PubMed] [Google Scholar]
- Lindor KD, Lacerda MA, Jorgensen RA, DeSotel CK, Batta AK, Salen G, et al. Relationship between biliary and serum bile acids and response to UDCA in patients with primary biliary cholangitis. American Journal of Gastroenterology 1998;93(9):1498‐504. [DOI] [PubMed] [Google Scholar]
- Siegel JL, Jorgensen R, Angulo P, Lindor KD. Treatment with UDCA is associated with weight gain in patients with primary biliary cholangitis. Journal of Clinical Gastroenterology 2003;37(2):183‐5. [DOI] [PubMed] [Google Scholar]
- Zukowski TH, Jorgensen RA, Dickson ER, Lindor KD. Autoimmune conditions associated with primary biliary cholangitis: response to UDCA therapy. American Journal of Gastroenterology 1998;93(6):958‐61. [DOI] [PubMed] [Google Scholar]
Lindor 1997 {published data only}
- Lindor KD, Jorgensen R, Therneau TM, Smith C, Mahoney DW, Dickson ER. Comparison of three different doses of UDCA in the treatment of primary biliary cholangitis: a randomized trial. Hepatology 1997;26(4):1240. [Google Scholar]
Lombard 1993 {published data only}
- Guanabens N, Pares A, Navasa M, Martinez de Osaba MJ, Hernandez ME, Munoz J, et al. Cyclosporin a increases the biochemical markers of bone remodeling in primary biliary cholangitis. Journal of Hepatology 1994;21(1):24‐8. [DOI] [PubMed] [Google Scholar]
- Guañabens N, Parés A, Navasa M, Rivera F, Muñoz J, Rodés J. Influence of cyclosporin a in bone metabolism in primary biliary cholangitis. Revista Española de Reumatología 1990;17(Suppl 1):14‐5. [Google Scholar]
- Lombard M, Portmann B, Neuberger J, Williams R, Tygstrup N, Ranek L, et al. Cyclosporin a treatment in primary biliary cholangitis: results of a long‐term placebo controlled trial. Gastroenterology 1993;104(2):519‐26. [DOI] [PubMed] [Google Scholar]
- Lombard M, Portmann BP, Tygstrup N, Ranek L, Larsen HR, Trepo C, et al. Cyclosporin a in primary biliary cholangitis: results of a long‐term placebo controlled trial and effect on survival. Hepatology 1990;12(4 (Pt 2)):872. [Google Scholar]
Ma 2016 {published data only}
- Ma H, Zeng M, Han Y, Yan H, Tang H, Sheng J, et al. A multicenter, randomized, double‐blind trial comparing the efficacy and safety of TUDCA and UDCA in Chinese patients with primary biliary cholangitis. Medicine 2016;95(47):e5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Macklon 1982 {published data only}
- Bassendine M, Macklon A, Mulcahy R, James O. Controlled trial of high and low dose D‐penicillamine (DP) in primary biliary cholangitis (PBC): results at three years. Gut 1982;23:A909. [Google Scholar]
- Macklon AF, Bassendine MF, James OFW. Controlled trial of D‐penicillamine in primary biliary cholangitis: incidence of side effects and relation to dose. Hepatology 1982;2(1):166. [Google Scholar]
Manzillo 1993a {published data only}
- Manzillo G, Piccinino F, Surrenti C, Frezza M, Grazie C. Double‐blind, placebo‐controlled study with parenteral and oral S‐adenosyl‐L‐methionine (SAMe) in primary biliary cholangitis. II United European Gastroenterology Week 1993:A337. [Google Scholar]
Manzillo 1993b {published data only}
- Manzillo G, Piccinino F, Surrenti C, Frezza M, Grazie C. Double‐blind, placebo‐controlled study with parenteral and oral S‐adenosyl‐L‐methionine (SAMe) in primary biliary cholangitis. II United European Gastroenterology Week 1993:A337. [Google Scholar]
Mason 2008 {published data only}
- Mason A, Luketic V, Lindor K, Hirschfield G, Gordon S, Mayo M. Farnesoid‐x receptor agonists: a new class of drugs for the treatment of PBC? An international study evaluating the addition of INT‐747 to UDCA. Journal of Hepatology 2010;52(Suppl 1):S1‐S2. [Google Scholar]
- Mason AL, Lindor KD, Bacon BR, Vincent C, Neuberger JM, Vvasilenko ST. Clinical trial: randomized controlled study of zidovudine and lamivudine for patients with primary biliary cholangitis stabilized on ursodiol. Alimentary Pharmacology & Therapeutics 2008;28(7):886‐94. [DOI] [PubMed] [Google Scholar]
Matloff 1982 {published data only}
- Matloff D, Resnick R, Alpert E, Kaplan M. D‐Penicillamine does not alter the course of primary biliary cholangitis. Clinical Research 1979;27:579A. [Google Scholar]
- Matloff DS, Alpert E, Resnick RH, Kaplan MM. A prospective trial of D‐penicillamine in primary biliary cholangitis. New England Journal of Medicine 1982;306(6):319‐26. [DOI] [PubMed] [Google Scholar]
Mayo 2015 {published data only}
- Mayo MJ, Wigg AJ, Roberts SK, Arnold H, Hassanein TI, Leggett BA, et al. NGM282, a novel variant of FGF‐19, demonstrates biologic activity in primary biliary cholangitis patients with an incomplete response to UDCA: results of a phase 2 multicenter, randomized, double blinded, placebo controlled trial. Hepatology 2015;62:263A‐4A. [Google Scholar]
Mazzarella 2002 {published data only}
- Enrico R, Federica B, Andrea L, Patrizia S, Roda A, Mazzella G. Treatment of early (I‐II stage) primary biliary cholangitis (PBC): high versus standard UDCA doses after 15 years follow‐up. A randomized open controlled trial. Hepatology 2011;54(4 (Suppl)):1209a. [Google Scholar]
- Mazzarella G, Azzolini F, Casanova S, Giovanelli S, Ferrara F, Liva S, et al. Standard vs high dose UDCA in PBC: efficacy on liver function tests and histology after six years of treatment in a randomised controlled trial. Gastroenterology 2002;123(1):66. [Google Scholar]
McCormick 1994 {published data only}
- McCormick PA, Scott F, Epstein O, Burroughs AK, Scheuer PJ, McIntyre N. Thalidomide as therapy for primary biliary cholangitis: a double‐blind placebo controlled pilot study. Journal of Hepatology 1994;21(4):496‐9. [DOI] [PubMed] [Google Scholar]
Minuk 1988 {published data only}
- Hanley DA, Ayer LM, Gundberg CM, Minuk GY. Parameters of calcium metabolism during a pilot study of cyclosporin a in patients with symptomatic primary biliary cholangitis. Clinical & Investigative Medicine ‐ Medecine Clinique et Experimentale 1991;14(4):282‐7. [PubMed] [Google Scholar]
- Minuk G, Bohme C, Burgess E, Hershfield N, Kelly J, Shaffer E, et al. A prospective, double‐blind, randomized, controlled trial of cyclosporine a in primary biliary‐cirrhosis. Hepatology 1987;7(5):1119. [Google Scholar]
- Minuk G, Bohme C, Burgess E, Hershfield N, Kelly J, Shaffer E, et al. A prospective, randomized, placebo‐controlled study of cyclosporine a in primary biliary‐cirrhosis. Clinical and Investigative Medicine ‐ Medecine Clinique et Experimentale 1987;10(4):B131. [Google Scholar]
- Minuk GY, Bohme CE, Burgess E, Hershfield NB, Kelly JK, Shaffer EA, et al. Pilot study of cyclosporin a in patients with symptomatic primary biliary cholangitis. Gastroenterology 1988;95(5):1356‐63. [DOI] [PubMed] [Google Scholar]
- Parsons HG, Thirsk JE, Frohlich J, Dias V, Minuk GY. Effect of cyclosporin a on serum lipids in primary biliary cholangitis patients. Clinical & Investigative Medicine ‐ Medecine Clinique et Experimentale 1989;12(6):386‐91. [PubMed] [Google Scholar]
Mitchison 1989 {published data only}
- Mitchison HC, Bassendine MF, Malcolm AJ, Watson AJ, Record CO, James OF. A pilot, double‐blind, controlled 1‐year trial of prednisolone treatment in primary biliary cholangitis: hepatic improvement but greater bone loss. Hepatology 1989;10(4):420‐9. [DOI] [PubMed] [Google Scholar]
- Mitchison HC, Bassendine MF, Watson AJ, Record CO, James OFW. Double blind placebo‐controlled trial of prednisolone treatment in primary biliary cholangitis (PBC): a 3 year update. Journal of Hepatology 1989;9(1):P4. [Google Scholar]
- Mitchison HC, Palmer JM, Bassendine MF, Watson AJ, Record CO, James OF. A controlled trial of prednisolone treatment in primary biliary cholangitis. Three‐year results. Journal of Hepatology 1992;15(3):336‐44. [DOI] [PubMed] [Google Scholar]
- Mitchison HC, Watson AJ, Bassendine MF, Record CO, James OFW. A pilot double blind controlled trial of prednisolone treatment in primary biliary cholangitis (PBC). Journal of Hepatology 1986;3(Suppl 1):S28. [DOI] [PubMed] [Google Scholar]
Mitchison 1993 {published data only}
- Buuren HR, Schalm SW. Beneficial effect of malotilate on primary biliary cholangitis (PBC): results of a multicentre controlled trial. Journal of Hepatology 1989;9(Suppl 1):S28. [Google Scholar]
- Mitchison HC, Mutimer DJ, James OFW, Triger DR, Moller B, Hopf U, et al. The results of a randomized double blind controlled trial evaluating malotilate in primary biliary cholangitis. Journal of Hepatology 1993;17(2):227‐35. [DOI] [PubMed] [Google Scholar]
Nakai 2000 {published data only}
- Nakai S, Masaki T, Kurokohchi K, Deguchi A, Nishioka M. Combination therapy of bezafibrate and UDCA in primary biliary cholangitis: a preliminary study. American Journal of Gastroenterology 2000;95(1):326‐7. [DOI] [PubMed] [Google Scholar]
- Nakai S, Masaki T, Morita T, Deguchi A, Nishioka M. The effect of bezafibrate in patients with primary biliary cholangitis. Hepatology 1999;30(4 Suppl):566a. [Google Scholar]
Neuberger 1985 {published data only}
- Neuberger J, Christensen E, Popper H, Portmann B, Caballeri J, Rodes J, et al. D‐Penicillamine in primary biliary cholangitis: preliminary results of an international trial. Gut 1983;24(10):A968. [Google Scholar]
- Neuberger J, Christensen E, Popper H, Portmann B, Caballeri J, Rodes J, et al. D‐Penicillamine in primary biliary cholangitis: preliminary results of an international trial. Liver 1984;4(1):74. [Google Scholar]
- Neuberger J, Christensen E, Portmann B, Caballeria J, Rodes J, Ranek L, et al. Double blind controlled trial of D‐penicillamine in patients with primary biliary cholangitis. Gut 1985;26(2):114‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nevens 2016 {published data only}
- Andreone P, Mazzella G, Invernizzi P, Floreani A, Picaro LA, Adorini L. Efficacy and safety of obeticholic acid in patients with primary biliary cirrhosis: an analysis of the Italian patients from a phase 3, randomized, placebo controlled study. Digestive and Liver Disease 2016;48:e81. [Google Scholar]
- Andreone P, Mazzella G, Strasser SI, Bowlus CL, Invernizzi P, Drenth J, et al. The FXR agonist obeticholic acid (OCA) improves liver biochemistry parameters correlated with clinical benefit across a range of patient characteristics. Hepatology 2014;60:360a. [Google Scholar]
- Hirschfield GM, Floreani A, Trivedi PJ, Pencek R, Liberman A, Marmon T, et al. Long‐term effect of obeticholic acid on transient elastography and AST to platelet ratio index in patients with PBC. Hepatology 2017;64(1 Suppl S1):110A‐1A. [Google Scholar]
- Mayo M, Kremer AE, Beuers U, Marmon T, Hooshmand‐Rad R, Pencek R, et al. Mitigation of pruritus during obeticholic acid treatment in patients with primary biliary cirrhosis: strategies and successes. Gastroenterology 2016;150(4 Suppl):S1072. [Google Scholar]
- Nevens F, Andreone P, Mazzella G, Strasser S, Bowlus C, Invernizzi P, et al. The first primary biliary cholangitis (PBC) phase 3 trial in two decades‐an international study of the FXR agonist obeticholic acid in PBC patients. Journal of Hepatology 2014;60(1 Suppl 1):S525‐6. [Google Scholar]
- Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. New England Journal of Medicine 2016;375(7):631‐43. [DOI] [PubMed] [Google Scholar]
- Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus CL, Invernizzi P, et al. An international phase 3 study of the FXR agonist obeticholic acid in PBC patients: effects on markers of cholestasis associated with clinical outcomes and hepatocellular damage. Hepatology 2014;60:347a‐8a. [Google Scholar]
- Pares A, Pencek R, Peters Y, Marmon T, MacConell L, Adorini L, et al. FXR agonism with obeticholic acid may attenuate bone mineral density decrease in subjects with primary biliary cirrhosis. Journal of hepatology 2015;62:S786. [Google Scholar]
- Pencek R, Lutz K, Marmon T, MacConell L. Evaluation of the posology of obeticholic acid (OCA) in patients with PBC. Hepatology 2015;62:525a‐6a. [Google Scholar]
- Peters Y, Hooshmand‐Rad R, Pencek R, Owens‐Grillo J, Marmon T, MacConell L, et al. Long‐term safety of OCA in patients with PBC. Hepatology 2015;62:530a. [Google Scholar]
- Peters Y, Hooshmand‐Rad R, Pencek R, Owens‐Grillo J, Marmon T, Macconell L, et al. Long‐term safety of obeticholic acid in patients with primary biliary cirrhosis. Digestive and Liver Disease 2016;48:e117‐8. [Google Scholar]
- Pockros PJ, Reddy KG, Owens‐Grillo J, Marmon T, MacConell L. Efficacy of obeticholic acid in patients with primary biliary cholangitis and renal impairment. Hepatology 2017;64(1 Suppl S1):205A. [Google Scholar]
- Trauner M, Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus CL, et al. Sustained improvement in the markers of cholestasis in an open label long term safety extension study of obeticholic acid in primary biliary cirrhosis patients. Hepatology 2015;62:511a‐2a. [Google Scholar]
- Vierling JM, Hirschfield GM, Jones D, Groszmann RJ, Kowdley KV, Pencek R, et al. Efficacy of obeticholic acid treatment in patients with primary biliary cholangitis with cirrhosis. Hepatology 2017;64(1 Suppl S1):187A. [Google Scholar]
Oka 1990 {published data only}
- Oka H, Toda G, Ikeda Y, Hashimoto N, Hasumura Y, Kamimura T, et al. A multi‐center double‐blind controlled trial of UDCA for primary biliary cholangitis. Gastroenterologia Japonica 1990;25(6):774‐80. [DOI] [PubMed] [Google Scholar]
- Toda G, Oka H, Hasumura Y, Kamimura T, Ohat Y, Tsuji T, et al. A multicenter double‐blind controlled trial of UDCA for primary biliary cholangitis in Japan. XI International Bile Acid Meeting Bile Acids as Therapeutic Agents ‐ From Basic Science to Clinical Practice 1990:76. [Google Scholar]
Papatheodoridis 2002 {published data only}
- Hadziyannis S. Long‐term treatment of primary biliary cholangitis with UDCA: the third year of a controlled trial. XI International Bile Acid Meeting Bile Acids as Therapeutic Agents ‐ From Basic Science to Clinical Practice 1990:57‐8. [Google Scholar]
- Hadziyannis S, Hadziyannis E. A randomised controlled trial of UDCA (UDCA) in primary biliary cholangitis (PBC). Hepatology 1988;8(5):1421. [Google Scholar]
- Hadziyannis S, Hadziyannis E, Lianidou E, Makris A. Long‐term treatment of primary biliary cholangitis with UDCA: the third year of a controlled trial. Bile Acids as Therapeutic Agents From Basic Science to Clinical Practice Falk Symposium 58 1990:287‐96. [Google Scholar]
- Hadziyannis SJ, Hadziyannis ES, Makris A. A randomized controlled trial of UDCA (UDCA) in primary biliary cholangitis (PBC). European Journal of Clinical Investigation 1989;19(Pt 2):A15. [Google Scholar]
- Hadziyannis SJ, Hadziyannis ES, Makris A. A randomized controlled trial of UDCA (UDCA) in primary biliary cholangitis (PBC). Hepatology 1989;10(4):580. [Google Scholar]
- Papatheodoridis GV, Deutsch M, Hadziyannis E, Tzakou A, Hadzivannis SJ. Ursodeoxycholic‐acid for primary biliary cholangitis: final results of a 12‐year prospective, randomised, controlled trial. Journal of Hepatology 2000;32:40. [DOI] [PubMed] [Google Scholar]
- Papatheodoridis GV, Hadziyannis ES, Deutsch M, Hadziyannis SJ. UDCA for primary biliary cholangitis: final results of a 12‐year, prospective, randomized, controlled trial. American Journal of Gastroenterology 2002;97(8):2063‐70. [DOI] [PubMed] [Google Scholar]
Pares 2000 {published data only}
- Pares A. Long‐term treatment of primary biliary cholangitis with UDCA: results of a randomized, double‐blind, placebo‐controlled trial. Journal of Hepatology 1997;26(Suppl 1):S166. [Google Scholar]
- Pares A, Caballeria L, Bruguera M, Rodes J. Factors influencing historical progression of early primary biliary cholangitis effect of UDCA (abstract). Journal of Hepatology 2001;34(1):189‐90. [Google Scholar]
- Pares A, Caballeria L, Rodes J. Long‐term UDCA treatment delays progression of mild primary biliary cholangitis (abstract). Journal of Hepatology 2001;34(1):187‐8. [Google Scholar]
- Pares A, Caballeria L, Rodes J, Bruguera M, Rodrigo L, Garcia‐Plaza A, et al. Long‐term effects of UDCA in primary biliary cholangitis: results of a double‐blind controlled multicentric trial. UDCA‐cooperative group from the Spanish Association for the Study of the Liver. Journal of Hepatology 2000;32(4):561‐6. [DOI] [PubMed] [Google Scholar]
Poupon 1991a {published data only}
- Calmus Y, Poupon R. UDCA (UDCA) in the treatment of chronic cholestatic diseases. Biochimie 1991;73(10):1335‐8. [DOI] [PubMed] [Google Scholar]
- Huet PM, Huet J, Hotte S. Long term effect of UDCA (UDCA) on hepatic function and portal hypertension in primary biliary cholangitis (PBC). Hepatology 1994;20(4 (Pt 2)):202a. [Google Scholar]
- Huet PM, Willems B, Huet J, Poupon R. Effects of UDCA (UDCA) on hepatic function and portal hypertension in primary biliary cholangitis (PBC). Hepatology 1990;12(4 (Pt 2)):907. [Google Scholar]
- Poupon R, Chazouilleres O, Balkau B, Poupon RE. Clinical and biochemical expression of the histopathological lesions of primary biliary cholangitis. UDCA‐PBC Group. Journal of Hepatology 1999;30(3):408‐12. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Balkau B, Eschwege E, Poupon R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cholangitis. New England Journal of Medicine 1991;324(22):1548‐54. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Balkau B, Guechot J, Heintzmann F. Predictive factors in UDCA‐treated patients with primary biliary cholangitis: role of serum markers of connective tissue. Hepatology 1994;19(3):635‐40. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Balkau B, Poupon R. Beneficial effect of UDCA (UDCA) in primary biliary cholangitis (PBC) final results of the French Canadian trial. Hepatology 1990;12(4 (Pt 2)):872. [Google Scholar]
- Poupon RE, Chretien Y, Balkau B, Niard AM, Poupon R. Ursodeoxycholic therapy for primary biliary cholangitis: a four year controlled study. Hepatology 1992;16(2 (Pt 2)):91a. [Google Scholar]
- Poupon RE, Chretien Y, Poupon R, Paumgartner G. Serum bile acids in primary biliary cholangitis: effect of UDCA therapy. Hepatology 1993;17(4):599‐604. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Eschwege E, Poupon R. UDCA for the treatment of primary biliary cholangitis. Interim analysis of a double‐blind multicentre randomized trial. The UDCA‐PBC Study Group. Journal of Hepatology 1990;11(1):16‐21. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Ouguerram K, Chretien Y, Verneau C, Eschwege E, Magot T, et al. Cholesterol‐lowering effect of UDCA in patients with primary biliary cholangitis. Hepatology 1993;17(4):577‐82. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Ouguerram K, Chretien Y, Verneau C, Eschwege E, Magot T, et al. [Hypocholesterolemic properties of UDCA in patients with primary biliary cholangitis]. Medecine & Chirurgie Digestives 1995;24(3):163‐4. [Google Scholar]
- Poupon RE, Poupon R. UDCA (UDCA) for treatment of primary biliary cholangitis (PBC): interim analysis of a double‐blind multicenter randomized trial. Hepatology 1989;10(4):639. [Google Scholar]
Poupon 1996 {published data only}
- Huet P, Willems B, Huet J, Poupon R. Effects of UDCA (UDCA) on hepatic function and portal hypertension in primary biliary cholangitis (PBC). XII International Bile Acid Meeting Bile Acids and the Hepatobiliary System from Basic Science to Clinical Practice Falk Symposium No 68 1992:118. [Google Scholar]
- Huet PM, Huet J, Deslauriers J. Long‐term UDCA (UDCA) and colchicine (C) treatment in primary biliary cholangitis (PBC): effect on hepatic function and portal hypertension. Canadian Journal of Gastroenterology 1996;10(Suppl A):S47. [Google Scholar]
- Huet PM, Huet J, Poupon RE, Deslauriers J. The combination of UDCA (UDCA) and colchicine (C) for patients with primary biliary cholangitis (PBC): effect on hepatic function and portal hypertension. Hepatology 1996;23(1):I‐49. [Google Scholar]
- Poupon RE, Huet PM, Poupon R, Bonnand AM, Nhieu JT, Zafrani ES. A randomized trial comparing colchicine and UDCA combination to UDCA in primary biliary cholangitis. UDCA‐PBC Study Group. Hepatology 1996;24(5):1098‐103. [DOI] [PubMed] [Google Scholar]
- Poupon RE, Niard AM, Huet PM, Miguet JP, Mathieuchandelier C, Doffoel M, et al. A randomized trial comparing the combination UDCA (UDCA) and colchicine to UDCA alone in primary biliary‐cirrhosis. Hepatology 1994;20(4):A151. [Google Scholar]
Raedsch 1993 {published data only}
- Raedsch R, Stiehl A, Walker S, Rudi J, Schlenker T, Gerteis C. Controlled study on the effects of a combined colchicine plus UDCA treatment in primary biliary cholangitis. 68th Falk Symposium; 1992 Oct 12‐14; Basel, Switzerland. 1993:303‐9.
- Raedsch R, Stiehl A, Walker S, Scherrmann JM, Kommerell B. [Combined colchicine plus UDCA‐treatment of primary biliary cholangitis: results of a placebo‐controlled double‐blind study]. Zeitschrift Fur Gastroenterologie 1992;30(Suppl 1):55‐7. [PubMed] [Google Scholar]
- Raedsch R, Stiehl A, Walker S, Theilmann L, Kommerell B. Effects of UDCA and colchicine plus UDCA in primary biliary cholangitis: a double‐blind pilot study. Bile Acids as Therapeutic Agents From Basic Science to Clinical Practice Falk Symposium 58 1991, (36):301‐4. [Google Scholar]
- Raedsch R, Stiehl A, Walker S, Theilmann L, Kommerell B. Influence of UDCA and URSO plus colchicine on primary biliary cholangitis: a double‐blind controlled pilot study. Klinische Wochenschrift 1991;69(Suppl 23):84. [Google Scholar]
Rautiainen 2005 {published data only}
- Rautiainen H, Farkkila M, Neuvonen M, Sane T, Karvonen AL, Nurm H, et al. Pharmacokinetics and bone effects of budesonide in primary biliary cholangitis. Alimentary Pharmacology & Therapeutics 2006;24(11‐12):1545‐52. [DOI] [PubMed] [Google Scholar]
- Rautiainen H, Karkkainen P, Karvonen AL, Nurmi H, Pikkarainen P, Nuutinen H, et al. Budesonide combined with UDCA to improve liver histology in primary biliary cholangitis: a three‐year randomized trial. Hepatology 2005;41(4):747‐52. [DOI] [PubMed] [Google Scholar]
Senior 1991 {published data only}
- O'Brien CB, Senior JR, Sternlieb JM, Sample M, Saul SM, Arora R. Ursodiol treatment of primary biliary cholangitis. Gastroenterology 1990;98(2 (Pt 2)):A617. [Google Scholar]
- Senior JR, O'Brien C. Mortality risk indices as outcome measures of the effectiveness of UDCA treatment of cholestatic liver diseases. Bile Acids as Therapeutic Agents From Basic Science to Clinical Practice Falk Symposium 58 1991:273‐85. [Google Scholar]
- Senior JR, O'Brien CB, Dickson ER. Effect of oral ursodiol treatment on the predicted probability of mortality in primary biliary cholangitis. Hepatology 1990;12(2):438. [Google Scholar]
Smart 1990 {published data only}
- Smart HL, Cann PA, Thuluvath PJ, Triger DR. A double blind placebo controlled study of antioxidants in primary biliary cholangitis (PBC). Gut 1990;31(10):A1184. [Google Scholar]
Steenbergen 1994 {published data only}
- Steenbergen W, Sciot R, Eycken P, Desmet V, Fevery J. Combined treatment with methotrexate and UDCA in primary biliary cholangitis (PBC). Journal of Hepatology 1994;21(Suppl 1):S89. [Google Scholar]
- Steenbergen W, Sciot R, Eyken P, Desmet V, Fevery J. Methotrexate alone or in combination with UDCA as possible treatment in primary biliary cholangitis. Cholestatic Liver diseases: New Strategies for Prevention and Treatment of Hepatobiliary and Cholestatic Liver Diseases Falk Symposium 75 1994:246‐54. [Google Scholar]
- Steenbergen W, Sciot R, Eyken P, Desmet V, Fevery J. Combined treatment with methotrexate and UDCA in non‐cirrhotic primary biliary cholangitis. Acta Clinica Belgica 1996;51(1):8‐18. [PubMed] [Google Scholar]
Taal 1983 {published data only}
- Taal BG, Schalm SW. Prednisone plus D‐penicillamine, D‐penicillamine and placebo compared in primary biliary‐cirrhosis syndrome. Gastroenterology 1981;80(5 Part 2):1351. [Google Scholar]
- Taal BG, Schalm SW, Kate FWJ. Double‐blind controlled study of penicillamine in primary biliary cholangitis: dose‐dependent effects. Nederlands Tijdschrift voor Geneeskunde 1982;126(12):547. [Google Scholar]
- Taal BG, Schalm SW, Kate FW, Berge Henegouwen GP, Brandt KH. Low therapeutic value of D‐penicillamine in a short‐term prospective trial in primary biliary cholangitis. Liver 1983;3(6):345‐52. [DOI] [PubMed] [Google Scholar]
Triger 1980 {published data only}
- Triger DR, Manifold IH, Cloke P, Underwood JCE. D‐Penicillamine in primary biliary cholangitis: two year results of a single centre, doubled‐blind controlled trial. Gut 1980;21(10):A919‐20. [Google Scholar]
Turner 1994 {published data only}
- Myszor M, Turner I, Mitshison H, Bennett M, Burt AD, James OFW. No symptomatic or histological benefit from UDCA treatment in PBC after 1 year controlled pilot study. Hepatology 1990;12(2):415. [Google Scholar]
- Turner IB, Myszor M, Mitchison HC, Bennett MK, Burt AD, James OF. A two year controlled trial examining the effectiveness of UDCA in primary biliary cholangitis. Journal of Gastroenterology & Hepatology 1994;9(2):162‐8. [DOI] [PubMed] [Google Scholar]
Ueno 2005 {published data only}
- Ueno Y, Moritoki Y, Kanno N, Fukushima K, Yamagiwa Y, Kogure T, et al. Randomized double blind control trial of reverse transcriptase inhibitor for the treatment of UDCA‐resistant PBC. Gastroenterology 2005;128(4):A775‐A. [Google Scholar]
Van Hoogstraten 1998 {published data only}
- Hoogstraten HJF, Smet MBM, Renooij W, Hop WCJ, Berge‐Henegouwen GP, Schalm SW. A randomized controlled trial evaluating therapy with UDCA in daily doses of 10 mg/kg versus 20 mg/kg in primary biliary cholangitis. European Journal of Gastroenterology & Hepatology 1998;10(12):A7. [Google Scholar]
- Hoogstraten HJF, Smet MBM, Renooij W, Breed JGS, Engels L, Ouden‐Muller JW, et al. A randomized trial in primary biliary cholangitis comparing UDCA in daily doses of either 10 mg/kg or 20 mg/kg. Alimentary Pharmacology & Therapeutics 1998;12(10):965‐71. [DOI] [PubMed] [Google Scholar]
- Hoogstraten HJF, Smet MBM, Renooij W, Hop WCJ, Buuren HR, VanBerge‐Henegouwen GP. A randomized controlled trial evaluating therapy with UDCA in daily doses of 10 mg/kg versus 20 mg/kg in primary biliary cholangitis. Gastroenterology 1998;114(4):A1358‐A. [Google Scholar]
Warnes 1987 {published data only}
- Warnes T, Babbs C, Smith A, Lee F, Haboubi NY, Johnson PJ, et al. A controlled trial of colchicine in primary biliary cholangitis (PBC). Journal of Hepatology 1985;1(Suppl 2):S348. [DOI] [PubMed] [Google Scholar]
- Warnes TW, Goddard CJR, Smith A, Rowan BP, Hunt L. Liver function and prognosis in primary biliary cholangitis: 'sharp' and 'blunt' tests and the influence of colchicine treatment on survival. Hepatology 1996;23(1):I‐82. [Google Scholar]
- Warnes TW, Smith A, Lee F, Haboubi NY, Johnson PJ, Hunt L. A controlled trial of colchicine in primary biliary cholangitis. Hepatology 1984;4(5):1022. [DOI] [PubMed] [Google Scholar]
- Warnes TW, Smith A, Lee FI, Haboubi NY, Johnson PJ, Hunt L. A controlled trial of colchicine in primary biliary cholangitis. Trial design and preliminary report. Journal of Hepatology 1987;5(1):1‐7. [DOI] [PubMed] [Google Scholar]
Wiesner 1990 {published data only}
- Wiesner RH, Dickson ER, Lindor KD, Jørgensen R, LaRusso NF, Baldus W. A controlled clinical trial evaluating cyclosporin in the treatment of primary biliary cholangitis: a preliminary report. Hepatology 1987;7(5):1025. [Google Scholar]
- Wiesner RH, Ludwig J, Lindor KD, Jorgensen RA, Baldus WP, Homburger HA, et al. A controlled trial of cyclosporine in the treatment of primary biliary cholangitis. New England Journal of Medicine 1990;322(20):1419‐24. [DOI] [PubMed] [Google Scholar]
Wolfhagen 1998 {published data only}
- Hoogstraten H, Wolfhagen FHJ, Berge Henegouwen GP, Schalm SW, Kate FJW, Hop WCJ, et al. Combined bile acid‐immunosuppressive therapy for primary biliary cholangitis. Results of a 1‐year multi centre, placebo controlled trial. European Journal of Gastroenterology & Hepatology 1996;8(Suppl 12):A 41. [Google Scholar]
- Lim AG, Wolfhagen FH, Verma A, Buuren HR, Jazrawi RP, Levy JH, et al. Soluble intercellular adhesion molecule‐1 in primary biliary cholangitis: effect of UDCA and immunosuppressive therapy. European Journal of Gastroenterology & Hepatology 1997;9(2):155‐61. [DOI] [PubMed] [Google Scholar]
- Lim AG, Wolfhagen FHJ, Verma A, Buuren HR, Jazrawi RP, Levy JH, et al. Soluble intercellular adhesion molecule 1 in primary biliary cholangitis: effect of UDCA and immunosuppressive therapy. Hepatology 1995;22(4 (Pt 2)):124a. [Google Scholar]
- Lim AG, Wolfhagen FHJ, Verma A, Buuren HR, Jazrawi RP, Northfield TC. Soluble intercellular adhesion molecule‐1 in primary biliary cholangitis: effect of UDCA, prednisone and azathioprine. Falk Symposium Bile Acids and Immunology 1995;86:9. [Google Scholar]
- Lim AG, Wolfhagen FHJ, Verma A, Jazrawi RP, Bururen HR, Levy JH, et al. Combination UDCA and immunosuppressive therapy for the treatment of primary biliary cholangitis. Gut 1995;37(Suppl 2):A27. [Google Scholar]
- Hoogstraten HJF, Wolfhagen FHJ, Henegouwen GPB, Schalm SW, tenKate FJW, Hop WCJ. Combined bile acid ‐ immunosuppressive therapy for primary biliary cholangitis. Results of a 1‐year multi centre, placebo controlled trial. Hepatology 1996;24(4 Suppl):167. [Google Scholar]
- Wolfhagen FHJ, Buuren HR, Berge Henegouwen GP, Hattum J, Ouden JW, Kerbert MJ, et al. A randomized placebo‐controlled trial with prednisone/azathioprine in addition to UDCA in primary biliary cholangitis. Journal of Hepatology 1994;21(Suppl 1):S49. [Google Scholar]
- Wolfhagen FHJ, Buuren HR, Berge‐Henegouwen GP, Hattum J, Ouden JW, Kerbert MJ. Prednisone/azathioprine treatment in primary biliary cholangitis (PBC) a randomized, placebo‐controlled trial. Netherlands Journal of Surgery 1995;46:A10. [Google Scholar]
- Wolfhagen FHJ, Lim AG, Verma A, Buuren HR, Jazrawi RP, Northfield TC, et al. Soluble ICAM‐1 in primary biliary cholangitis (PBC) during combined treatment with UDCA, prednisone and azathioprine. Netherlands Journal of Surgery 1995;47:A29‐30. [Google Scholar]
- Wolfhagen FHJ, Hoogstraten HJF, Buuren HR, Berge‐Henegouwen GP, Kate FJW, Hop WCJ, et al. Triple therapy with UDCA, prednisone and azathioprine in primary biliary cholangitis: A 1‐year randomized, placebo‐controlled study. Journal of Hepatology 1998;29(5):736‐42. [DOI] [PubMed] [Google Scholar]
Yokomori 2001 {published data only}
- Yokomori H, Oda M, Ishii H. Effects of UDCA and colestilan versus UDCA alone on serum bile acids and pruritus: a randomized, open‐label study. Current Therapeutic Research ‐ Clinical and Experimental 2001;62(3):221‐9. [Google Scholar]
References to studies excluded from this review
Angulo 1999b {published data only}
- Angulo P, Batts KP, Therneau TM, Jorgensen RA, Dickson ER, Lindor KD. Long‐term UDCA delays histological progression in primary biliary cholangitis. Hepatology 1999;29(3):644‐7. [DOI] [PubMed] [Google Scholar]
Angulo 1999c {published data only}
- Angulo P, Lindor KD, Therneau TM, Jorgensen RA, Malinchoc M, Kamath PS, et al. Utilization of the Mayo risk score in patients with primary biliary cholangitis receiving UDCA. Liver 1999;19(2):115‐21. [DOI] [PubMed] [Google Scholar]
Angulo 2002 {published data only}
- Angulo P, Petz JL, Jorgensen RA, Lindor KD. A randomized, cross‐over study evaluating single‐ and multiple‐daily dosage schedules of UDCA in primary biliary cholangitis. Gastroenterology 2002;122(4):A629. [Google Scholar]
Attili 1994 {published data only}
- Attili AF, Rusticali A, Varriale M, Carli L, Repice AM, Callea F. Effect of UDCA on serum enzymes and liver histology in patients with chronic active hepatitis ‐ a 12 month double‐blind, placebo‐controlled trial. Journal of Hepatology 1994;20(3):315‐20. [DOI] [PubMed] [Google Scholar]
Avezov 2004a {published data only}
- Avezov SA, Mansurova F. [Efficacy of combined use of UDCA and heptral in the treatment of primary biliary cholangitis]. Klinicheskaia Meditsina 2004;82(2):46‐9. [PubMed] [Google Scholar]
Avezov 2004b {published data only}
- Avezov SA, Mansurov F. [Efficacy of combined administration of UDCA and hepthral in the treatment of primary biliary cholangitis]. Klinicheskaia Meditsina 2004;82(3):55‐8. [PubMed] [Google Scholar]
Bach 2003 {published data only}
- Bach N, Bodian C, Bodenheimer H, Croen E, Berk PD, Thung SN, et al. Methotrexate therapy for primary biliary cholangitis. American Journal of Gastroenterology 2003;98(1):187‐93. [DOI] [PubMed] [Google Scholar]
Batta 1989 {published data only}
- Batta AK, Salen G, Arora R, Shefer S, Tint GS, Abroon J, et al. Effect of UDCA on bile acid metabolism in primary biliary cholangitis. Hepatology 1989;10(4):414‐9. [DOI] [PubMed] [Google Scholar]
Beukers 1988 {published data only}
- Beukers R, Schalm SW. Effect of cyclosporine and cyclosporine plus prednisone in primary biliary cholangitis. Transplantation Proceedings 1988;20(3 Suppl 4):340‐3. [PubMed] [Google Scholar]
Blanche 1994 {published data only}
- Blanche P, Sicard D. Methotrexate for polymyositis associated with primary biliary cholangitis. Clinical & Experimental Rheumatology 1994;12(6):694. [PubMed] [Google Scholar]
Bonis 2006 {published data only}
- Bonis PA, Kaplan M. Methotrexate for treatment of primary biliary cholangitis. Hepatology 2006;43(3):632; author reply 633. [DOI] [PubMed] [Google Scholar]
Borum 1990 {published data only}
- Borum M, Fromm H. UDCA in the treatment of primary biliary cholangitis: first controlled data. Hepatology 1990;12(1):172‐3. [DOI] [PubMed] [Google Scholar]
Bray 1991 {published data only}
- Bray GP, Padova C, Tredger JM, Williams R. A comparison of S‐adenosylmethionine (SAMe), rifampicin (R) and UDCA (UDCA) in primary biliary cholangitis (PBC): interim results. Journal of Hepatology 1991;13(Suppl 2):S101. [Google Scholar]
Carbone 2016 {published data only}
- Carbone M, Jones D, Mells GF, Pencek R, Marmon T, Shapiro D. Predicted risk of end stage liver disease with continued standard of care and subsequent addition of obeticholic acid in patients with PBC. Hepatology 2016;63 (1 Suppl 1):184A‐5A. [Google Scholar]
Chazouilleres 1995 {published data only}
- Chazouilleres O, Legendre C, StMaur P, Ismail PR, Poupon R. Histological course of primary biliary cholangitis (PBC) treated with UDCA (UDCA). Hepatology 1995;22(4 (Pt 2)):125a. [Google Scholar]
Christensen 1986 {published data only}
- Christensen E, Neuberger J, Crowe J, Portmann B, Williams R, Altman DG, et al. Azathioprine and prognosis in primary biliary cholangitis. Gastroenterology 1986;90(2):508‐9. [DOI] [PubMed] [Google Scholar]
Combes 1989 {published data only}
- Combes B. Prednisolone for primary biliary cholangitis ‐ good news, bad news. Hepatology 1989;10(4):511‐3. [DOI] [PubMed] [Google Scholar]
Combes 2004 {published data only}
- Combes B, Luketic VA, Peters MG, Zetterman RK, Garcia‐Tsao G, Munoz SJ, et al. Prolonged follow‐up of patients in the U.S. Multicenter trial of UDCA for primary biliary cholangitis. American Journal of Gastroenterology 2004;99(2):264‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Combes 2005b {published data only}
- Combes B. Reflections on therapeutic trials in primary biliary cholangitis. Hepatology 2005;42(5):1009. [DOI] [PubMed] [Google Scholar]
Copaci 2001 {published data only}
- Copaci I, Micu L, Cojocaru L. UDCA and methotrexate for primary biliary cholangitis. Journal of Hepatology 2001;34(1):59. [Google Scholar]
Corpechot 2000 {published data only}
- Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of UDCA therapy on liver fibrosis progression in primary biliary cholangitis. Hepatology 2000;32(6):1196‐9. [DOI] [PubMed] [Google Scholar]
Corpechot 2001 {published data only}
- Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of UDCA therapy on liver fibrosis progression in primary biliary cholangitis. European Journal of Gastroenterology & Hepatology 2001;13(1):90. [Google Scholar]
Crosignani 1996a {published data only}
- Crosignani A, Larghi A, Invernizzi P, Battezzati PM, DeValle G, Zuin M, et al. Tauroursodeoxycholic and UDCAs for the treatment of primary biliary cholangitis: a crossover study. Hepatology 1996;23(1):P111. [Google Scholar]
Crosignani 1996b {published data only}
- Crosignani A, Battezzati PM, Setchell KDR, Invernizzi P, Covini G, Zuin M, et al. TauroUDCA for treatment of primary biliary cholangitis ‐ a dose‐response study. Digestive Diseases and Sciences 1996;41(4):809‐15. [DOI] [PubMed] [Google Scholar]
Degott 1999 {published data only}
- Degott C, Zafrani ES, Callard P, Balkau B, Poupon RE, Poupon R. Histopathological study of primary biliary cholangitis and the effect of UDCA treatment on histology progression. Hepatology 1999;29(4):1007‐12. [DOI] [PubMed] [Google Scholar]
De la Mora 1994 {published data only}
- Mora G, Bobadilla J, Romero P, Rodríguez‐Leal G, Morán S, Kershenobich D, et al. Does treatment with UDCA (UDCA) really diminish cholesterol serum levels in primary biliary cholangitis (PBC)?. Hepatology 1994;19:571. [Google Scholar]
Dickson 1991 {published data only}
- Dickson ER, Lindor KD. Beneficial effects of UDCA in an open trial of patients with primary biliary cholangitis. Bile Acids as Therapeutic Agents From Basic Science to Clinical Practice Falk Symposium 58 1991, (32):271‐2. [Google Scholar]
Emond 1996 {published data only}
- Emond M, Carithers RL, Luketic VA, Peters M, Zetterman RK, Garcia‐Tsao G. Does UDCA improve survival in patients with primary biliary cholangitis? Comparison of outcome in the US multicenter trial to expected survival using the Mayo Clinic prognostic model. Hepatology 1996;24(4 (Pt 2)):168a. [Google Scholar]
Fischer 1967 {published data only}
- Fischer JA, Schmid M. Treatment of primary biliary cholangitis with azathioprine. Lancet 1967;1(7487):421‐4. [DOI] [PubMed] [Google Scholar]
Golovanova 2010 {published data only}
- Golovanova EV, Khomeriki SG, Petrakov AV, Serova TI. Budesonide in treatment of patients with cross primary biliary cholangitis and autoimmune hepatitis. Eksperimental'Naia i Klinicheskaia Gastroenterologiia 2010, (8):113‐7. [PubMed] [Google Scholar]
Heathcote 1993 {published data only}
- Heathcote EJL, Cauch K, Walker V, Blendism LM, Ghent CN, Pappas SC. A four‐year follow‐up study of UDCA therapy for primary biliary cholangitis. Gastroenterology 1993;104(4 (Pt 2)):A914. [Google Scholar]
Heathcote 1995 {published data only}
- Heathcote EJ, Lindor KD, Poupon R, Cauchdudek K, Dickson ER, Trout R, et al. Combined analysis of French, American and Canadian randomized controlled trials of UDCA therapy in primary biliary cholangitis. Gastroenterology 1995;108(4 Suppl 3):A1082. [Google Scholar]
Hirschfield 2011 {published data only}
- Hirschfield GM, Mason AL, Gordon SC, Luketic VA, Mayo M, Vincent C, et al. A long term safety extension trial of the farnesoid x receptor (FXR) agonist obeticholic acid (OCA) and UDCA in primary biliary cholangitis (PBC). Hepatology 2011;54(S1):429a. [Google Scholar]
Hishon 1982 {published data only}
- Hishon S, Tobin G, Ciclitira PJ. A clinical trial of levamisole in primary biliary cholangitis. Postgraduate Medical Journal 1982;58(685):701‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Howat 1966 {published data only}
- Howat HT, Ralston AJ, Varley H, Wilson JA. The late results of long‐term treatment of primary biliary cholangitis by corticosteroids. Revue Internationale d' Hepatologie 1966;16(2):227‐38. [PubMed] [Google Scholar]
Hwang 1993 {published data only}
- Hwang SJ, Chan CY, Lee SD, Wu JC, Tsay SH, Lo KJ. UDCA in the treatment of primary biliary cholangitis: a short‐term, randomized, double‐blind controlled, cross‐over study with long‐term follow up. Journal of Gastroenterology & Hepatology 1993;8(3):217‐23. [DOI] [PubMed] [Google Scholar]
Invernizzi 1996 {published data only}
- Invernizzi A, Setchell DDR, Crosignani A, Larghi A, Battezzatti PM, O'Connell N, et al. Comparison between tauroursodeoxycholic and UDCAs in patients with primary biliary cholangitis: a cross over study. Hepatology 1996;24(4 (Pt 2)):168a. [Google Scholar]
Invernizzi 2015 {published data only}
- Invernizzi P, Pencek R, Marmon T, MacConell L, Shapiro D. Integrated efficacy summary for obeticholic acid in subjects with primary biliary cholangitis. Journal of Hepatology 2015;62:S778. [Google Scholar]
Itakura 2004 {published data only}
- Itakura J, Izumi N, Nishimura Y, Inoue K, Ueda K, Nakanishi H, et al. Prospective randomized crossover trial of combination therapy with bezafibrate and UDCA for primary biliary cholangitis. Hepatology Research 2004;29(4):216‐22. [DOI] [PubMed] [Google Scholar]
Jazrawi 1999 {published data only}
- Jazrawi RP, Verma A, Ahmed HA, Northfield TC. Effect of UDCA on cholestatic features and complications of primary biliary cholangitis. Bile Acids and Cholestasis 1999;108:231‐46. [Google Scholar]
Jones 2006 {published data only}
- Jones DE, Bhala N, Newton JL. Reflections on therapeutic trials in primary biliary cholangitis: a quality of life oriented counter‐view. Hepatology 2006;43(3):633; Author reply 634. [DOI] [PubMed] [Google Scholar]
Jorgensen 2002 {published data only}
- Jorgensen R, Angulo P, Dickson ER, Lindor KD. Results of long‐term ursodiol treatment for patients with primary biliary cholangitis. American Journal of Gastroenterology 2002;97(10):2647‐50. [DOI] [PubMed] [Google Scholar]
Joshi 2002 {published data only}
- Joshi S, Cauch‐Dudek K, Wanless IR, Lindor KD, Jorgensen R, Batts K, et al. Primary biliary cholangitis with additional features of autoimmune hepatitis: response to therapy with UDCA. Hepatology 2002;35(2):409‐13. [DOI] [PubMed] [Google Scholar]
Kaplan 1993 {published data only}
- Kaplan MM. New strategies needed for treatment of primary biliary cholangitis?. Gastroenterology 1993;104(2):651‐3. [DOI] [PubMed] [Google Scholar]
Kaplan 1998 {published data only}
- Kaplan M. Primary biliary cholangitis. Lancet 1998;351(9097):216. [DOI] [PubMed] [Google Scholar]
Kaplan 2004 {published data only}
- Kaplan MM, Cheng S, Price LL, Bonis PA. A randomized controlled trial of colchicine plus ursodiol versus methotrexate plus ursodiol in primary biliary cholangitis: ten‐year results. Hepatology 2004;39(4):915‐23. [DOI] [PubMed] [Google Scholar]
Kaplan 2009 {published data only}
- Kaplan MM, Poupon R. Treatment with immunosuppressives in patients with primary biliary cholangitis who fail to respond to ursodiol. Hepatology 2009;50(2):652. [DOI] [PubMed] [Google Scholar]
Kisand 1996 {published data only}
- Kisand KE, Karvonen AL, Vuoristo M, Farkkila M, Lehtola J, Inkovaara J, et al. UDCA treatment lowers the serum level of antibodies against pyruvate dehydrogenase and influences their inhibitory capacity for the enzyme complex in patients with primary biliary cholangitis. Journal of Molecular Medicine 1996;74(5):269‐72. [DOI] [PubMed] [Google Scholar]
Kisand 1998 {published data only}
- Kisand KE, Kisand KV, Karvonen AL, Vuoristo M, Mattila J, Makinen J, et al. Antibodies to pyruvate dehydrogenase in primary biliary cholangitis: correlation with histology. APMIS 1998;106(9):884‐92. [DOI] [PubMed] [Google Scholar]
Kowdley 2014a {published data only}
- Kowdley K, Hirschfield G, Chapman R, Vincent C, Jones D, Pares A, et al. Long‐term treatment of primary biliary cholangitis with the FXR agonist obeticholic acid shows durable efficacy. Journal of Hepatology 2014;60(1 Suppl 1):S192‐3. [Google Scholar]
Kowdley 2014b {published data only}
- Kowdley KV, Pencek R, Marmon T, Shapiro D, Hooshmand‐Rad R. FXR agonist obeticholic acid: sustained improvement in markers of cholestasis and long‐term safety in patients with primary biliary cholangitis through 4 years. Hepatology 2014;60:361a. [Google Scholar]
Kowdley 2015 {published data only}
- Kowdley KV, Shah H, Mason A, Luketic VA, Pencek R, Marmon T, et al. Long‐term safety and efficacy of obeticholic acid treatment in primary biliary cirrhosis after more than 4 years of treatment. Hepatology 2015;62:521a‐2a. [Google Scholar]
Kugler 1991 {published data only}
- Kugler CF, Fleig WE. [Placebo‐controlled double‐blind study of cyclosporin a in primary biliary cholangitis]. Zeitschrift Fur Gastroenterologie 1991;29(12):663‐4. [PubMed] [Google Scholar]
Kurihara 2002 {published data only}
- Kurihara T, Maeda A, Shigemoto M, Yamashita K, Hashimoto E. Investigation into the efficacy of bezafibrate against primary biliary cholangitis, with histological references from cases receiving long term monotherapy. American Journal of Gastroenterology 2002;97(1):212‐4. [DOI] [PubMed] [Google Scholar]
Lampe 1972 {published data only}
- Lampe K, Hudepohl M, Schopen RD. [Immunosuppressive therapy of chronic aggressive hepatitis and primary biliary cholangitis]. Medizinische Klinik 1972;67(15):527‐34. [PubMed] [Google Scholar]
Larghi 1997 {published data only}
- Larghi A, Crosignani A, Battezzati PM, Valle G, Allocca M, Invernizzi P, et al. Ursodeoxycholic and tauro‐UDCAs for the treatment of primary biliary cholangitis: a pilot crossover study. Alimentary Pharmacology & Therapeutics 1997;11(2):409‐14. [DOI] [PubMed] [Google Scholar]
Lee 2003 {published data only}
- Lee YM, Kaplan MM. Efficacy of colchicine in patients with primary biliary cholangitis poorly responsive to ursodiol and methotrexate. American Journal of Gastroenterology 2003;98(1):205‐8. [DOI] [PubMed] [Google Scholar]
Leung 2010 {published data only}
- Leung J, Bonder A, Sasson M, Bonis P, Kaplan M. 19‐year follow‐up of patients in a double‐blind trial of colchicine plus ursodiol versus methotrexate plus ursodiol in the treatment of primary biliary cholangitis. Gastroenterology 2010;1:S218. [Google Scholar]
Leung 2011 {published data only}
- Leung J, Bonis PA, Kaplan MM. Colchicine or methotrexate, with ursodiol, are effective after 20 years in a subset of patients with primary biliary cholangitis. Clinical Gastroenterology & Hepatology 2011;9(9):776‐80. [DOI] [PubMed] [Google Scholar]
Leuschner 1990 {published data only}
- Leuschner U, Güldütuna S, Fischer H, Hellstern A, Hübner K. UDCA in the treatment of primary biliary cholangitis: the Frankfurt experience. Strategies for the treatment of hepatobiliary diseases Falk symposium 53 1990, (9):83‐90. [Google Scholar]
Leuschner 1993a {published data only}
- Leuschner M, Güldütuna S, Imhof M, Bhati S, You T, Leuschner U. UDCA therapy in primary biliary cholangitis. 68th Falk Symposium; 1992 Oct 12‐14; Basel, Switzerland 1993:299‐302. [Google Scholar]
Leuschner 1993b {published data only}
- Leuschner M, Guldutuna S, Benjaminov A, Hubner K, Leuschner U. Interim evaluation of a prospective double‐blind trial of UDCA (UDCA) versus UDCA plus prednisolone in primary biliary cholangitis (PBC). Gastroenterology 1993;104(4):A938. [Google Scholar]
Leuschner 1996a {published data only}
- Leuschner M, Guldutuna S, You T, Hubner K, Bhatti S, Leuschner U. UDCA and prednisolone versus UDCA and placebo in the treatment of early stages of primary biliary cholangitis. Journal of Hepatology 1996;25(1):49‐57. [DOI] [PubMed] [Google Scholar]
Leuschner 1996b {published data only}
- Leuschner M, Guldutuna S, Hubner K, You T, Leuschner U. UDCA (UDCA) and prednisolone in the treatment of primary biliary cholangitis (PBC). Results of a controlled double‐blind trial. Gastroenterology 1996;110(4):A1250. [Google Scholar]
Leuschner 1997 {published data only}
- Leuschner U, Maier KP, Guldutuna S, Parte‐Peterhans S, Leuschner M. UDCA in combination with prednisolone or budesonide in the therapy of primary biliary cholangitis. Bile Acids in Hepatobiliary Diseases: Basic Research and Clinical Application 1997;93:299‐302. [Google Scholar]
Leuschner 1998 {published data only}
- Leuschner U, Maier P, Schitling J, Strahl R, Herrmann G, Leuschner M. UDCA and budesonide in the treatment of primary biliary cholangitis. XV International Bile Acid Meeting Bile Acids and Cholestasis Vol 1998;Falk Symposium 108:60‐1. [Google Scholar]
Levy 2004 {published data only}
- Levy C, Angulo P. UDCA and long‐term survival in primary biliary cholangitis. American Journal of Gastroenterology 2004;99(2):269‐70. [PubMed] [Google Scholar]
Licinio 2015 {published data only}
- Licinio R, Facciorusso A, Castellaneta NM, Leo A. Combination therapy of UDCA and bezafibrate in patients with primary biliary cholangitis: the end of the steroid era in autoimmune liver diseases?. American Journal of Gastroenterology 2015;110(7):1086. [DOI] [PubMed] [Google Scholar]
Lim 2000 {published data only}
- Lim AG, Jazrawi RP, Ahmed HA, Northfield TC. UDCA ‐ an immunomodulator in primary biliary cholangitis?. Bile Acids in Hepatobiliary Disease 2000;110B:30‐5. [Google Scholar]
Lindor 1994a {published data only}
- Lindor KD, Jorgensen RA, Anderson ML, Gores GJ, Baldus WP, Dickson ER. The combination of UDCA (UDCA) and methotrexate (MTX) for patients with primary biliary cholangitis (PBC): the results of a pilot study. Hepatology 1994;20(4 (Pt 2)):202. [DOI] [PubMed] [Google Scholar]
Lindor 1995a {published data only}
- Lindor KD, Therneau TM, Jorgensen RA, Malinochoc M, Dickson ER. Effects of UDCA (UDCA) on survival in patients with primary biliary‐cirrhosis (PBC). Gastroenterology 1995;108(4 Suppl 3):A1111. [DOI] [PubMed] [Google Scholar]
Lindor 1995b {published data only}
- Lindor KD, Dickson ER, Jorgensen RA, Anderson ML, Wiesner RH, Gores GJ, et al. The combination of UDCA and methotrexate for patients with primary biliary cholangitis: the results of a pilot study. Hepatology 1995;22(4 I):1158‐62. [DOI] [PubMed] [Google Scholar]
Lindor 1995c {published data only}
- Lindor K. Long‐term experience with UDCA for patients with primary biliary cholangitis and primary sclerosing cholangitis. International Falk Workshop Bile Acids in Liver Diseases 1995:141‐5. [Google Scholar]
Lindor 1996 {published data only}
- Lindor KD, Therneau TM, Jorgensen RA, Malinchoc M, Dickson ER. Effects of UDCA on survival in patients with primary biliary cholangitis. Gastroenterology 1996;110(5):1515‐8. [DOI] [PubMed] [Google Scholar]
Lindor 2000 {published data only}
- Lindor KD, Poupon R, Poupon R, Heathcote EJ, Therneau T. UDCA for primary biliary cholangitis. Lancet 2000;355(9204):657‐8. [DOI] [PubMed] [Google Scholar]
Lindor 2005 {published data only}
- Lindor KD. Dose effect of UDCA used in the treatment of primary biliary cholangitis and primary sclerosing cholangitis. Bile Acid Biology and Its Therapeutic Implications 2005;141:225‐9. [Google Scholar]
Lindor 2007 {published data only}
- Lindor K. UDCA for the treatment of primary biliary cholangitis. New England Journal of Medicine 2007;357(15):1524‐9. [DOI] [PubMed] [Google Scholar]
Lytvyak 2015 {published data only}
- Lytvyak E, Montano‐Loza AJ, Saxinger L, Mason A. Combination anti‐retroviral therapy provides reduction in human betaretrovirus load and durable biochemical responses in patients with primary biliary cholangitis. Hepatology 2015;62:528A. [Google Scholar]
Lytvyak 2016 {published data only}
- Lytvyak E, Montano‐Loza AJ, Mason AL. Combination antiretroviral studies for patients with primary biliary cirrhosis. World Journal of Gastroenterology 2016;22(1):349‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Miettinen 1993 {published data only}
- Miettinen TA, Farkkila M, Vuoristo M, Karvonen AL, Leino R, Lehtola J. Improvement of serum noncholesterol sterols may indicate retarded progression of primary biliary cholangitis (PBC) in a randomized placebo controlled two‐year trial with colchicine and UDCA. Gastroenterology 1993;104(4 (Pt 2)):A954. [Google Scholar]
Miettinen 1995 {published data only}
- Miettinen TA, Farkkila M, Vuoristo M, Karvonen AL, Leino R, Lehtola J, et al. Serum cholestanol, cholesterol precursors, and plant sterols during placebo‐controlled treatment of primary biliary cholangitis with UDCA or colchicine. Hepatology 1995;21(5):1261‐8. [PubMed] [Google Scholar]
Muntoni 2010 {published data only}
- Muntoni S, Rojkind M, Muntoni S. Colchicine reduces procollagen III and increases pseudocholinesterase in chronic liver disease. World Journal of Gastroenterology 2010;16(23):2889‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nikolaidis 2006 {published data only}
- Nikolaidis N, Kountouras J, Giouleme O, Tzarou V, Chatzizisi O, Patsiaoura K, et al. Colchicine treatment of liver fibrosis. Hepato‐Gastroenterology 2006;53(68):281‐5. [PubMed] [Google Scholar]
Ohmoto 2001 {published data only}
- Ohmoto K, Mitsui Y, Yamamoto S. Effect of bezafibrate in primary biliary cholangitis: a pilot study. Liver 2001;21(3):223‐4. [DOI] [PubMed] [Google Scholar]
Ohmoto 2006 {published data only}
- Ohmoto K, Yoshioka N, Yamamoto S. Long‐term effect of bezafibrate on parameters of hepatic fibrosis in primary biliary cholangitis. Journal of Gastroenterology 2006;41(5):502‐3. [DOI] [PubMed] [Google Scholar]
Pan 2013 {published data only}
- Pan XL, Zhao L, Li L, Li AH, Ye J, Yang L, et al. Efficacy and safety of TUDCA in the treatment of liver cirrhosis: a double‐blind randomized controlled trial. Journal of Huazhong University of Science and Technology‐Medical Sciences 2013;33(2):189‐94. [DOI] [PubMed] [Google Scholar]
Pares 2009 {published data only}
- Pares A. Excellent long‐term survival in patients with primary biliary cholangitis treated with UDCA. Bile Acid Biology and Therapeutic Actions 2009;165:259‐69. [Google Scholar]
Podda 1989 {published data only}
- Podda M, Ghezzi C, Battezzati PM, Bertolini E, Crosignani A, Petroni ML, et al. Effect of different doses of UDCA in chronic liver disease. Digestive Diseases & Sciences 1989;34(12 Suppl):59S‐65S. [DOI] [PubMed] [Google Scholar]
Poupon 1989 {published data only}
- Poupon R, Poupon R, the UDCA‐PBCG. UDCA for primary biliary cholangitis. International Lugano Symposium on Biliary Physiology and Diseases: Strategies for the Treatment of Hepatobiliary Diseases Falk Symposium No 53 1989, issue 22.
Poupon 1990 {published data only}
- Poupon R, Poupon R, The UDCA‐PBCG. UDCA in the treatment of primary biliary cholangitis. Strategies for the treatment of hepatobiliary diseases. Falk Symposium 53 1990:79‐81. [Google Scholar]
Poupon 1991b {published data only}
- Poupon RE, Balkau B, Eschwege E, Poupon R, Kaplan MM. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cholangitis. Annals of Internal Medicine 1991;115(6 Suppl 2):48. [DOI] [PubMed] [Google Scholar]
Poupon 1994 {published data only}
- Poupon RE, Poupon R, Balkau B. Ursodiol for the long‐term treatment of primary biliary cholangitis. The UDCA‐PBC study group. New England Journal of Medicine 1994;330(19):1342‐7. [DOI] [PubMed] [Google Scholar]
Poupon 1997 {published data only}
- Poupon RE, Lindor KD, CauchDudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of UDCA in primary biliary cholangitis. Gastroenterology 1997;113(3):884‐90. [DOI] [PubMed] [Google Scholar]
Poupon 1999 {published data only}
- Poupon RE, Bonnand AM, Chretien Y, Poupon R. Ten‐year survival in UDCA‐treated patients with primary biliary cholangitis. The UDCA‐PBC study group. Hepatology 1999;29(6):1668‐71. [DOI] [PubMed] [Google Scholar]
Poupon 2003 {published data only}
- Poupon RE, Lindor KD, Pares A, Chazouilleres O, Poupon R, Heathcote EJ. Combined analysis of the effect of treatment with UDCA on histologic progression in primary biliary cholangitis. Journal of Hepatology 2003;39(1):12‐6. [DOI] [PubMed] [Google Scholar]
Raedsch 1989 {published data only}
- Raedsch R, Stiehl A, Hopf U, Moller B. [Effect of UDCA treatment on primary biliary cholangitis]. Zeitschrift fur Gastroenterologie ‐ Verhandlungsband 1989;24:125‐7. [PubMed] [Google Scholar]
Reed 1982 {published data only}
- Reed M. Penicillamine therapy 'encouraging' in primary biliary cholangitis study. JAMA 1982;248(1):11‐2. [PubMed] [Google Scholar]
Robson 1994 {published data only}
- Robson SC, Neuberger JM, Williams R. The influence of cyclosporine a therapy on sex hormone levels in pre‐ and post‐menopausal women with primary biliary cholangitis. Journal of Hepatology 1994;21(3):412‐6. [DOI] [PubMed] [Google Scholar]
Roda 2002 {published data only}
- Roda E, Azzaroli F, Nigro G, Piazza F, Jaboli F, Ferrara F, et al. Improved liver tests and greater biliary enrichment with high dose UDCA in early stage primary biliary cholangitis. Digestive & Liver Disease 2002;34(7):523‐7. [DOI] [PubMed] [Google Scholar]
Savolainen 1983 {published data only}
- Savolainen ER, Miettinen TA, Pikkarainen P, Salaspuro MP, Kivirikko KI. Enzymes of collagen synthesis and type III procollagen aminopropeptide in the evaluation of D‐penicillamine and medroxyprogesterone treatments of primary biliary cholangitis. Gut 1983;24(2):136‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schaffner 1982 {published data only}
- Schaffner F, Sternlieb I, Sachs H. A 2 dose level randomized double‐blind controlled trial of penicillamine in primary biliary cholangitis ‐ the 1st 2 years. Hepatology 1982;2(5):714. [Google Scholar]
Setchell 1994 {published data only}
- Setchell KDR, Rodrigues C, Podda M, Crosignani A. TauroUDCA (TUDCA) appears more effective than UDCA at displacing hydrophobic bile acids from the bile acid pool of patients with primary biliary cholangitis (PBC). Hepatology 1994;20(4 (Pt 2)):150a. [Google Scholar]
Setchell 1996 {published data only}
- Setchell KDR, Rodrigues CMP, Podda M, Crosignani A. Metabolism of orally administered TUDCA in patients with primary biliary cholangitis. Gut 1996;38(3):439‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stellaard 1979 {published data only}
- Stellaard F, Bolt MG, Boyer JL, Klein PD. Phenobarbital treatment in primary biliary cholangitis. Differences in bile acid composition between responders and nonresponders. Journal of Laboratory & Clinical Medicine 1979;94(6):853‐61. [PubMed] [Google Scholar]
Taal 1985 {published data only}
- Taal BG, Schalm SW. Cryoglobulins in primary biliary cholangitis: prevalence and modulation by immunosuppressive therapy. Zeitschrift Fur Gastroenterologie 1985;23(5):228‐34. [PubMed] [Google Scholar]
Tang 2008 {published data only}
- Tang HH, Chen YJ, Tong GD, Zhou DQ, He JS, Zhou XZ, et al. [Efficacy of UDCA combined with Tongdan Decoction in treatment of patients with primary biliary cholangitis]. World Chinese Journal of Digestology 2008;16(13):1417‐24. [Google Scholar]
Tong 2012 {published data only}
- Tong GD, Tang HH, Wei CS, Chen YJ, He JS, Zhou XZ, et al. Efficacy of UDCA combined with Tongdan Decoction on immunological indices and histopathological changes in primary biliary cholangitis patients. Chinese Journal of Integrative Medicine 2012;18(1):16‐22. [DOI] [PubMed] [Google Scholar]
Verma 1999 {published data only}
- Verma A, Jazrawi RP, Ahmed HA, Davis T, Bland JM, Benson M, et al. Optimum dose of UDCA in primary biliary cholangitis. European Journal of Gastroenterology & Hepatology 1999;11(10):1069‐76. [DOI] [PubMed] [Google Scholar]
Verma 2000 {published data only}
- Verma A, Jazrawi RP, Ahmed HA, Northfield TC. The optimum dose of UDCA in primary biliary cholangitis. Bile Acids in Hepatobiliary Disease 2000;110B:25‐9. [Google Scholar]
Vogel 1988 {published data only}
- Vogel W, Kathrein H, Judmaier G, Braunsteiner H. Deterioration of primary biliary cholangitis during treatment with UDCA. Lancet 1988;1(8595):1163. [DOI] [PubMed] [Google Scholar]
Vuoristo 1995 {published data only}
- Vuoristo M, Farkkila M, Karvonen AL, Leino R, Lehtola J, Makinen J, et al. A placebo‐controlled trial of primary biliary cholangitis treatment with colchicine and UDCA. Gastroenterology 1995;108(5):1470‐8. [DOI] [PubMed] [Google Scholar]
Vuoristo 1997 {published data only}
- Vuoristo M, Farkkila M, Gylling H, Karvonen AL, Leino R, Lehtola J, et al. Expression and therapeutic response related to apolipoprotein e polymorphism in primary biliary cholangitis. Journal of Hepatology 1997;27(1):136‐42. [DOI] [PubMed] [Google Scholar]
Wiesner 1994 {published data only}
- Wiesner RH. Progression of primary biliary cholangitis on UDCA. Gastroenterology 1994;106(2):555. [PubMed] [Google Scholar]
Wolfhagen 1995 {published data only}
- Wolfhagen FH, Buuren HR, Schalm SW, Kate FJ, Hattum J, Eskens FA, et al. Can UDCA induce disease remission in primary biliary cholangitis? The Dutch Multicentre PBC Study Group. Journal of Hepatology 1995;22(3):381. [DOI] [PubMed] [Google Scholar]
Yan 2007 {published data only}
- Yan G, Erik C, Gluud C. The long‐term beneficial effects of UDCA in primary biliary cholangitis are highly questionable. American Journal of Gastroenterology 2007;102(2):464‐5. [DOI] [PubMed] [Google Scholar]
Yano 2002 {published data only}
- Yano K, Kato H, Morita S, Takahara O, Ishibashi H, Furukawa R. Is bezafibrate histologically effective for primary biliary cholangitis?. American Journal of Gastroenterology 2002;97(4):1075‐7. [DOI] [PubMed] [Google Scholar]
Zuin 1991 {published data only}
- Zuin M, Grandinetti G, Camisasca M, Boga E, Ravizza L, Molteni P. A comparison of cholestyramine and diethylaminoethyl‐dextran for the treatment of hyperlipidemia and pruritus of primary biliary cholangitis. Current Therapeutic Research, Clinical and Experimental 1991;49(4):659‐65. [Google Scholar]
References to studies awaiting assessment
O'Brian 1990 {published data only}
- O'Brian C, Senior J, Sternlieb J, Saul S. Caution: not all patients with primary biliary cholangitis may successfully be treated by ursodiol. Second International Meeting on Pathochemistry, Pathophysiology and Pathomechanisms of the Biliary System and New Strategies for the Treatment of Hepato‐biliary Diseases 1990:208. [Google Scholar]
Zaman 2006 {published data only}
- Zaman A. Methotrexate in combination with UDCA is ineffective in the treatment of primary biliary cholangitis: commentary. Evidence‐Based Gastroenterology 2006;7(1):21‐2. [Google Scholar]
References to ongoing studies
ChiCTR‐IPR‐16008935 {unpublished data only}
- Biochemical Response of PBC‐AIH Overlap Syndrome Induced by Ursodeoxycholic Acid Only or Combination Therapy of Immunosuppressive Agents. Ongoing study Not stated..
EUCTR2015‐002698‐39‐GB {unpublished data only}
- A 12‐Week, Double‐Blind, Randomized, Placebo‐Controlled, Phase 2 Study to Evaluate the Effects of Two Doses of MBX‐8025 in Subjects with Primary Biliary Cirrhosis (PBC) and an Inadequate Response to Ursodeoxycholic Acid (UDCA).. Ongoing study Not stated..
NCT02308111 {published and unpublished data}
- Lindor K, Hansen B, Pencek R, Hooshmand‐Rad R, Marmon T, MacConell L, et al. A phase 3b, double blind, placebo controlled study evaluating the effect of obeticholic acid on clinical outcomes in subjects with primary biliary cholangitis at elevated risk of progression to liver transplant or death. Journal of Hepatology 2015;62:S850‐1. [Google Scholar]
NCT02701166 {unpublished data only}
- The Effect of Bezafibrate on Cholestatic Itch. Ongoing study February 2016..
NCT02823353 {unpublished data only}
- Fenofibrate in Combination with Ursodeoxycholic Acid in Primary Biliary Cirrhosis: a Randomized Control Study. Ongoing study January 2016..
NCT02823366 {unpublished data only}
- Fenofibrate for Patients with Primary Biliary Cirrhosis who had an Inadequate Response to Ursodeoxycholic Acid. Ongoing study January 2016..
NCT02937012 {unpublished data only}
- Efficacy and Security of Bezafibrate in Patients with Primary Biliary Cirrhosis without Biochemical Response to Ursodeoxycholic Acid: a Randomized, Double‐blind, Placebo‐controlled Trial. Ongoing study October 2016..
NCT02943447 {unpublished data only}
- A Phase 2, Randomized, Double‐Blind, Placebo Controlled Study Evaluating the Safety, Tolerability, and Efficacy of GS‐9674 in Subjects with Primary Biliary Cholangitis without Cirrhosis. Ongoing study December 2016..
NCT02965911 {unpublished data only}
- A Randomized Controlled Clinical Trial on the Efficacy and Safety of Fenofibrate Combined with Ursodeoxycholic Acid in PBC Patients with an Incomplete Biochemical Response to UDCA. Ongoing study January 2016..
Additional references
Baldursdottir 2012
- Baldursdottir TR, Bergmann OM, Jonasson JG, Ludviksson BR, Axelsson TA, Bjornsson ES. The epidemiology and natural history of primary biliary cholangitis: a nationwide population‐based study. European Journal of Gastroenterology and Hepatology 2012;24(7):824‐30. [DOI] [PubMed] [Google Scholar]
Bergasa 2000
- Bergasa NV, Mehlman JK, Jones EA. Pruritus and fatigue in primary biliary cholangitis. Bailliere's Best Practice & Research: Clinical Gastroenterology 2000;14(4):643‐55. [DOI] [PubMed] [Google Scholar]
Boberg 1998
- Boberg KM, Aadland E, Jahnsen J, Raknerud N, Stiris M, Bell H. Incidence and prevalence of primary biliary cholangitis, primary sclerosing cholangitis, and autoimmune hepatitis in a Norwegian population. Scandinavian Journal of Gastroenterology 1998;33(1):99‐103. [DOI] [PubMed] [Google Scholar]
Boonstra 2014
- Boonstra K, Kunst AE, Stadhouders PH, Tuynman HA, Poen AC, Nieuwkerk KM, et al. Rising incidence and prevalence of primary biliary cholangitis: a large population‐based study. Liver International 2014;34(6):e31‐8. [DOI] [PubMed] [Google Scholar]
Chaimani 2012
- Chaimani A, Salanti G. Using network meta‐analysis to evaluate the existence of small‐study effects in a network of interventions. Research Synthesis Methods 2012;3(2):161‐76. [DOI] [PubMed] [Google Scholar]
Chaimani 2013
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta‐analysis in STATA. PLoS One 2013;8(10):e76654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chan 2013
- Chan AW, Tetzlaff JM, Altman DG, Laupacis A, Gotzsche PC, Krleza‐Jeric K, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Annals of Internal Medicine 2013;158(3):200‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Del Re 2013
- Re AC, Spielmans GI, Flückiger C, Wampold BE. Efficacy of new generation antidepressants: differences seem illusory. PLoS One 2013;8(6):e63509. [DOI] [PMC free article] [PubMed] [Google Scholar]
DeMets 1987
- DeMets DL. Methods for combining randomized clinical trials: strengths and limitations. Statistics in Medicine 1987;6(3):341‐50. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Dias 2010
- Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta‐analysis. Statistics in Medicine 2010;29(7‐8):932‐44. [DOI] [PubMed] [Google Scholar]
Dias 2012a
- Dias S, Sutton AJ, Welton NJ, Ades AE. NICE DSU Technical Support Document 3: heterogeneity: subgroups, meta‐regression, bias and bias‐adjustment, September 2011 (last updated April 2012). www.nicedsu.org.uk/TSD3%20Heterogeneity.final%20report.08.05.12.pdf (accessed 27 March 2014).
Dias 2012b
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 1: introduction to evidence synthesis for decision making, April 2011 (last updated April 2012). www.nicedsu.org.uk/TSD1%20Introduction.final.08.05.12.pdf (accessed 27 March 2014).
Dias 2014a
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: a generalised linear modelling framework for pairwise and network meta‐analysis of randomised controlled trials, August 2011 (last updated April 2014). www.nicedsu.org.uk/TSD2%20General%20meta%20analysis%20corrected%2015April2014.pdf (accessed 8 October 2014). [PubMed]
Dias 2014b
- Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: inconsistency in networks of evidence based on randomised controlled trials, May 2011 (last updated April 2014). www.nicedsu.org.uk/TSD4%20Inconsistency.final.15April2014.pdf (accessed 8 October 2014). [PubMed]
Dronamraju 2010
- Dronamraju D, Odin J, Bach N. Primary biliary cholangitis: environmental risk factors. Disease Markers 2010;29(6):323‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
EASL 2009
- European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases. Journal of Hepatology 2009;51(2):237‐67. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ (Clinical Research Ed.) 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
EuroQol 2014
- EuroQol. About EQ‐5D, 2014. www.euroqol.org/about‐eq‐5d.html (accessed 8 October 2014).
Floreani 2011
- Floreani A, Caroli D, Variola A, Rizzotto ER, Antoniazzi S, Chiaramonte M, et al. A 35‐year follow‐up of a large cohort of patients with primary biliary cholangitis seen at a single centre. Liver International 2011;31(3):361‐8. [DOI] [PubMed] [Google Scholar]
Gershwin 2005
- Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cholangitis: a controlled interview‐based study of 1032 patients. Hepatology 2005;42(5):1194‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Giljaca 2010
- Giljaca V, Poropat G, Stimac D, Gluud C. Methotrexate for primary biliary cholangitis. Cochrane Database of Systematic Reviews 2010, Issue 5. [DOI: 10.1002/14651858.CD004385.pub3] [DOI] [PubMed] [Google Scholar]
Gluud 2007
- Gluud C, Brok J, Gong Y, Koretz RL. Hepatology may have problems with putative surrogate outcome measures. Journal of hepatology 2007;46(4):734‐42. [PUBMED: 17316871] [DOI] [PubMed] [Google Scholar]
Gluud 2017
- Gluud C, Nikolova D, Klingenberg SL. Cochrane Hepato‐Biliary Group. About The Cochrane Collaboration (Cochrane Review Groups (CRGs)) 2017, Issue 2. Art. No.: LIVER.
Gong 2004a
- Gong Y, Gluud C. Colchicine for primary biliary cholangitis. Cochrane Database of Systematic Reviews 2004, Issue 2. [DOI: 10.1002/14651858.CD004481.pub2] [DOI] [PubMed] [Google Scholar]
Gong 2004b
- Gong Y, Klingenberg SL, Gluud C. D‐Penicillamine for primary biliary cholangitis. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD004789.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gong 2007a
- Gong Y, Christensen E, Gluud C. Azathioprine for primary biliary cholangitis. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD006000.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gong 2007b
- Gong Y, Christensen E, Gluud C. Cyclosporin A for primary biliary cholangitis. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD005526.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guanabens 2013
- Guanabens N, Monegal A, Cerda D, Muxi A, Gifre L, Peris P, et al. Randomized trial comparing monthly ibandronate and weekly alendronate for osteoporosis in patients with primary biliary cholangitis. Hepatology 2013;58(6):2070‐8. [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, et al. GRADE guidelines: 1. Introduction ‐ GRADE evidence profiles and summary of findings tables. Journal of Clinical Epidemiology 2011;64(4):383‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2012
- Higgins JPT, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta‐analysis: concepts and models for multi‐arm studies. Research Synthesis Methods 2012;3(2):98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH‐GCP 1997
- International Conference on Harmonisation Expert Working Group. International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline. Guideline for good clinical practice CFR & ICH Guidelines. Vol. 1, Pennsylvania: Barnett International/PAREXEL, 1997. [Google Scholar]
Jakobsen 2014
- Jakobsen JC, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta‐analytic methods. BMC Medical Research Methodology 2014;14(1):120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jones 1999
- Jones EA, Kate FJ, Ter Borg F, Houben M, Reesink HW, Chamuleau RA. Combination therapy with mycophenolate mofetil and UDCA for primary biliary cholangitis. European Journal of Gastroenterology & Hepatology 1999;11(10):1165‐9. [PubMed] [Google Scholar]
Kim 2000
- Kim WR, Lindor KD, Locke GR 3rd, Therneau TM, Homburger HA, Batts KP, et al. Epidemiology and natural history of primary biliary cholangitis in a US community. Gastroenterology 2000;119(6):1631‐6. [DOI] [PubMed] [Google Scholar]
Kjaergard 2001
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta‐analyses. Annals of Internal Medicine 2001;135(11):982‐9. [DOI] [PubMed] [Google Scholar]
Lammert 2013
- Lammert C, Nguyen DL, Juran BD, Schlicht E, Larson JJ, Atkinson EJ, et al. Questionnaire based assessment of risk factors for primary biliary cholangitis. Digestive and Liver Disease 2013;45(7):589‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lazaridis 2007
- Lazaridis KN, Talwalkar JA. Clinical epidemiology of primary biliary cholangitis: incidence, prevalence, and impact of therapy. Journal of Clinical Gastroenterology 2007;41(5):494‐500. [DOI] [PubMed] [Google Scholar]
Leung 2005
- Leung PS, Coppel RL, Gershwin ME. Etiology of primary biliary cholangitis: the search for the culprit. Seminars in Liver Disease 2005;25(3):327‐36. [DOI] [PubMed] [Google Scholar]
Li Wei 2012
- Li Wei X, Yan X, Shi Chun R, Zhang Ai P. Chlorambucil for patients with primary biliary cirrhosis. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD008714.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lindor 2009
- Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. Primary biliary cholangitis. Hepatology 2009;50(1):291‐308. [DOI] [PubMed] [Google Scholar]
Lu 2006
- Lu G, Ades AE. Assessing evidence inconsistency in mixed treatment comparisons. Journal of the American Statistical Association 2006;101(474):447‐59. [Google Scholar]
Lundh 2017
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2017, Issue 2. [DOI: 10.1002/14651858.MR000033.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Macaskill 2001
- Macaskill P, Walter SD, Irwig L. A comparison of methods to detect publication bias in meta‐analysis. Statistics in Medicine 2001;20(4):641‐54. [DOI] [PubMed] [Google Scholar]
Metcalf 1997
- Metcalf JV, Bhopal RS, Gray J, Howel D, James OF. Incidence and prevalence of primary biliary cholangitis in the city of Newcastle upon Tyne, England. International Journal of Epidemiology 1997;26(4):830‐6. [DOI] [PubMed] [Google Scholar]
Mills 2012
- Mills EJ, Ioannidis JP, Thorlund K, Schünemann HJ, Puhan MA, Guyatt GH. How to use an article reporting a multiple treatment comparison meta‐analysis. JAMA 2012;308(12):1246‐53. [DOI] [PubMed] [Google Scholar]
Moher 1998
- Moher D, Pham B, Jones A, Cook DJ, Jadad AR, Moher M, et al. Does quality of reports of randomised trials affect estimates of intervention efficacy reported in meta‐analyses?. Lancet 1998;352(9128):609‐13. [DOI] [PubMed] [Google Scholar]
Myers 2009
- Myers RP, Shaheen AA, Fong A, Burak KW, Wan A, Swain MG, et al. Epidemiology and natural history of primary biliary cholangitis in a Canadian health region: a population‐based study. Hepatology 2009;50(6):1884‐92. [DOI] [PubMed] [Google Scholar]
NCBI 2014
- NCBI. Liver cirrhosis, biliary, 2014. www.ncbi.nlm.nih.gov/mesh/68008105 (accessed 19 October 2014).
Newell 1992
- Newell DJ. Intention‐to‐treat analysis: implications for quantitative and qualitative research. International Journal of Epidemiology 1992;21(5):837‐41. [DOI] [PubMed] [Google Scholar]
OpenBUGS 3.2.3 [Computer program]
- Members of OpenBUGS Project Management Group. OpenBUGS. Version 3.2.3. Members of OpenBUGS Project Management Group, 2014.
Ormarsdottir 2004
- Ormarsdottir S, Mallmin H, Naessen T, Petren‐Mallmin M, Broome U, Hultcrantz R, et al. An open, randomized, controlled study of transdermal hormone replacement therapy on the rate of bone loss in primary biliary cholangitis. Journal of Internal Medicine 2004;256(1):63‐9. [DOI] [PubMed] [Google Scholar]
Parikh‐Patel 2001
- Parikh‐Patel A, Gold EB, Worman H, Krivy KE, Gershwin ME. Risk factors for primary biliary cholangitis in a cohort of patients from the United States. Hepatology 2001;33(1):16‐21. [DOI] [PubMed] [Google Scholar]
Paumgartner 2002
- Paumgartner G, Beuers U. UDCA in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36(3):525‐31. [DOI] [PubMed] [Google Scholar]
Perez 2009
- Perez MJ, Briz O. Bile‐acid‐induced cell injury and protection. World Journal of Gastroenterology 2009;15(14):1677‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pla 2007
- Pla X, Vergara M, Gil M, Dalmau B, Cistero B, Bella RM, et al. Incidence, prevalence and clinical course of primary biliary cholangitis in a Spanish community. European Journal of Gastroenterology and Hepatology 2007;19(10):859‐64. [DOI] [PubMed] [Google Scholar]
Prince 2005
- Prince M, Christensen E, Gluud C. Glucocorticosteroids for primary biliary cholangitis. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD003778.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Prince 2010
- Prince MI, Ducker SJ, James OF. Case‐control studies of risk factors for primary biliary cholangitis in two United Kingdom populations. Gut 2010;59(4):508‐12. [DOI] [PubMed] [Google Scholar]
Puhan 2014
- Puhan MA, Schünemann HJ, Murad MH, Li T, Brignardello‐Petersen R, Singh JA, et al. A GRADE Working Group approach for rating the quality of treatment effect estimates from network meta‐analysis. BMJ (Clinical Research Ed.) 2014;349:g5630. [DOI] [PubMed] [Google Scholar]
Rautiainen 2007
- Rautiainen H, Salomaa V, Niemela S, Karvonen AL, Nurmi H, Isoniemi H, et al. Prevalence and incidence of primary biliary cholangitis are increasing in Finland. Scandinavian Journal of Gastroenterology 2007;42(11):1347‐53. [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Royle 2003
- Royle P, Milne R. Literature searching for randomized controlled trials used in Cochrane reviews: rapid versus exhaustive searches. International Journal of Technology Assessment in Health Care 2003;19(4):591‐603. [DOI] [PubMed] [Google Scholar]
Rudic 2011a
- Rudic JS, Giljaca V, Krstic MN, Bjelakovic G, Gluud C. Bisphosphonates for osteoporosis in primary biliary cholangitis. Cochrane Database of Systematic Reviews 2011, Issue 12. [DOI: 10.1002/14651858.CD009144.pub2] [DOI] [PubMed] [Google Scholar]
Rudic 2011b
- Rudic JS, Poropat G, Krstic MN, Bjelakovic G, Gluud C. Hormone replacement for osteoporosis in women with primary biliary cholangitis. Cochrane Database of Systematic Reviews 2011, Issue 12. [DOI: 10.1002/14651858.CD009146.pub2] [DOI] [PubMed] [Google Scholar]
Rudic 2012a
- Rudic JS, Poropat G, Krstic MN, Bjelakovic G, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD000551.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rudic 2012b
- Rudic JS Poropat G, Krstic MN, Bjelakovic G, Gluud C. Bezafibrate for primary biliary cirrhosis. Cochrane Database of Systematic Reviews 2012, Issue 1. [DOI: 10.1002/14651858.CD009145.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Salanti 2011
- Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple‐treatment meta‐analysis: an overview and tutorial. Journal of Clinical Epidemiology 2011;64(2):163‐71. [DOI] [PubMed] [Google Scholar]
Salanti 2012
- Salanti G. Indirect and mixed‐treatment comparison, network, or multiple‐treatments meta‐analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Research Synthesis Methods 2012;3(2):80‐97. [DOI] [PubMed] [Google Scholar]
Savović 2012a
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials: combined analysis of meta‐epidemiological studies. Health Technology Assessment 2012;16(35):1‐82. [DOI] [PubMed] [Google Scholar]
Savović 2012b
- Savović J, Jones HE, Altman DG, Harris RJ, Jüni P, Pildal J, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials. Annals of Internal Medicine 2012;157(6):429‐38. [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [DOI] [PubMed] [Google Scholar]
Schulz 2010
- Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ (Clinical Research Ed.) 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Selmi 2010
- Selmi C, Gershwin ME. The etiology mystery in primary biliary cholangitis. Digestive Diseases 2010;28(1):105‐15. [DOI] [PubMed] [Google Scholar]
Severini 1993
- Severini TA. Bayesian interval estimates which are also confidence intervals. Journal of the Royal Statistical Society. Series B (Methodological) 1993;55(2):533‐40. [Google Scholar]
Song 2008
- Song M, Song Z, Barve S, Zhang J, Chen T, Liu M, et al. Tetrathiomolybdate protects against bile duct ligation‐induced cholestatic liver injury and fibrosis. Journal of Pharmacology and Experimental Therapeutics 2008;325(2):409‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sood 2004
- Sood S, Gow PJ, Christie JM, Angus PW. Epidemiology of primary biliary cholangitis in Victoria, Australia: high prevalence in migrant populations. Gastroenterology 2004;127(2):470‐5. [DOI] [PubMed] [Google Scholar]
Stata/SE 14.2 [Computer program]
- StataCorp LP. Stata/SE 14.2 for Windows[64‐bit x86‐64]. Version 14. College Station: StataCorp LP, 2017.
Talwalkar 2005
- Talwalkar JA, Angulo P, Keach JC, Petz JL, Jorgensen RA, Lindor KD. Mycophenolate mofetil for the treatment of primary biliary cholangitis in patients with an incomplete response to UDCA. Journal of Clinical Gastroenterology 2005;39(2):168‐71. [PubMed] [Google Scholar]
Talwalkar 2006
- Talwalkar JA, Donlinger JJ, Gossard AA, Keach JC, Jorgensen RA, Petz JC, et al. Fluoxetine for the treatment of fatigue in primary biliary cholangitis: a randomized, double‐blind controlled trial. Digestive Diseases and Sciences 2006;51(11):1985‐91. [DOI] [PubMed] [Google Scholar]
Ter Borg 2004
- Ter Borg PC, Os E, Broek WW, Hansen BE, Buuren HR. Fluvoxamine for fatigue in primary biliary cholangitis and primary sclerosing cholangitis: a randomised controlled trial [ISRCTN88246634]. BMC Gastroenterology 2004;4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thorlund 2011
- Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for Trial Sequential Analysis (TSA). Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, Denmark. Available from www.ctu.dk/tsa 2011:1‐115.
Thorlund 2012
- Thorlund K, Mills EJ. Sample size and power considerations in network meta‐analysis. Systematic Reviews 2012;1:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
TSA 2011 [Computer program]
- Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen. TSA version 0.9. Copenhagen Trial Unit, Centre for Clinical Intervention Research, Copenhagen, 2011.
Turner 2012
- Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JP. Predicting the extent of heterogeneity in meta‐analysis, using empirical data from the Cochrane Database of Systematic Reviews. International Journal of Epidemiology 2012;41(3):818‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Van Valkenhoef 2012
- Valkenhoef G, Lu G, Brock B, Hillege H, Ades AE, Welton NJ. Automating network meta‐analysis. Research Synthesis Methods 2012;3(4):285‐99. [DOI] [PubMed] [Google Scholar]
Ware 2014
- Ware JE. SF‐36® health survey update, 2014. www.sf‐36.org/tools/sf36.shtml (accessed on 8 October 2014).
Wetterslev 2008
- Wetterslev J, Thorlund K, Brok J, Gluud C. Trial Sequential Analysis may establish when firm evidence is reached in cumulative meta‐analysis. Journal of Clinical Epidemiology 2008;61(1):64‐75. [DOI] [PubMed] [Google Scholar]
Wetterslev 2017
- Wetterslev J, Jakobsen JC, Gluud C. Trial Sequential Analysis in systematic reviews with meta‐analysis. BMC Medical Research Methodology 2017;17(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wood 2008
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta‐epidemiological study. BMJ (Clinical Research Ed.) 2008;336(7644):601‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yin 2015
- Yin Q, Li J, Xia Y, Zhang R, Wang J, Lu W, et al. Systematic review and meta‐analysis: bezafibrate in patients with primary biliary cholangitis. Drug Design, Development and Therapy 2015;9:5407‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2015
- Zhang Y, Li S, He L, Wang F, Chen K, Li J, et al. Combination therapy of fenofibrate and UDCA in patients with primary biliary cholangitis who respond incompletely to UDCA monotherapy: a meta‐analysis. Drug Design, Development and Therapy 2015;9:2757‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhu 2015a
- Zhu GQ, Huang S, Huang GQ, Wang LR, Lin YQ, Wu YM, et al. Optimal drug regimens for primary biliary cholangitis: a systematic review and network meta‐analysis. Oncotarget 2015;6(27):24533‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhu 2015b
- Zhu GQ, Shi KQ, Huang S, Huang GQ, Lin YQ, Zhou ZR, et al. Network meta‐analysis of randomized controlled trials: efficacy and safety of UDCA‐based therapies in primary biliary cholangitis. Medicine 2015;94(11):e609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhu 2015c
- Zhu GQ, Shi KQ, Huang GQ, Wang LR, Lin YQ, Braddock M, et al. A network meta‐analysis of the efficacy and side effects of UDCA‐based therapies for primary sclerosing cholangitis. Oncotarget 2015;6(29):26757‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]