

Abstract
Background
Abatacept inhibits the co‐stimulation of T cells and disrupts the inflammatory chain of events that leads to joint inflammation, pain, and damage in rheumatoid arthritis.
Objectives
To assess the efficacy and safety of abatacept in reducing disease activity, pain, and improving function in people with rheumatoid arthritis.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2007, Issue 1), MEDLINE (from 1966), EMBASE (from 1980), ACP Journal Club (from 2000), and Biosis Previews (from 1990) in March 2007 and December 2008. We contacted authors of included studies and the abatacept manufacturer.
Selection criteria
Randomized controlled trials comparing abatacept alone, or in combination with disease‐modifying anti‐rheumatic drugs (DMARDs) or biologics, to placebo or other DMARDs or biologics in patients with moderate to severe rheumatoid arthritis.
Data collection and analysis
Two authors independently assessed search results and risk of bias, and extracted data. We obtained adverse event data from trials, long‐term extension studies, and regulatory agencies.
Main results
Seven trials with 2908 patients were included. Compared with placebo, patients in the abatacept group were 2.2 times more likely to achieve an ACR 50 response at one year (RR 2.21, 95% confidence interval (CI) 1.73 to 2.82) with a 21% (95% CI 16% to 27%) absolute risk difference between groups. The number needed to treat to achieve an ACR 50 response was 5 (95% CI 4 to 7). Significant improvements in physical function and a reduction in disease activity and pain were found in abatacept‐treated patients compared to placebo. One RCT found abatacept significantly slowed the radiographic progression of joint damage at 12 months compared to placebo, although it is not clear what the clinical relevance of this difference may be. There may be a risk of attrition bias. Total adverse events were greater in the abatacept group (RR 1.05, 95% CI 1.01 to 1.08). Other harm outcomes were not significant with the exception of a greater number of serious infections at 12 months in the abatacept group (Peto odds ratio 1.91 (95% CI 1.07 to 3.42). Serious adverse events were increased when abatacept was given in combination with other biologics (RR 2.30, 95% CI 1.15 to 4.62).
Authors' conclusions
There is moderate‐level evidence that abatacept is efficacious and safe in the treatment of rheumatoid arthritis. Abatacept should not be used in combination with other biologics to treat rheumatoid arthritis. The withdrawal and toxicity profile appears acceptable at the present time but further long‐term studies and post‐marketing surveillance are required to assess harms and sustained efficacy.
Plain language summary
Abatacept for rheumatoid arthritis
This summary of a Cochrane review presents what we know from research about the effect of abatacept on rheumatoid arthritis. Although expensive, if supported by the overall body of evidence, the claims of their benefit upon both symptoms and radiographic progression, and their low rate of short term side effects make them of great interest to patients with RA.
The review shows that in people with rheumatoid arthritis:
‐ Abatacept probably reduces joint damage as seen on the x‐ray. ‐ Abatacept probably improves pain, function and other symptoms of rheumatoid arthritis. ‐ Abatacept probably reduces disease activity.
We do not have precise information about side effects and complications. This is particularly true for rare but serious side effects. Possible side effects may include a serious infection or upper respiratory infection. Rare complications may include certain types of cancer.
What is rheumatoid arthritis and what is abatacept? When you have rheumatoid arthritis, your immune system, which normally fights infection, attacks the lining of your joints. This makes your joints swollen, stiff and painful. The small joints of your hands and feet are usually affected first. There is no cure for rheumatoid arthritis at present, so the treatments aim to relieve pain and stiffness and improve your ability to move.
Abatacept is one of a group of medications called selective costimulation modulators (immunomodulators). It works by blocking the activity of T‐cells, a type of immune cell in the body that causes swelling and joint damage in people who have rheumatoid arthritis.
Best estimate of what happens to people with rheumatoid arthritis who take abatacept:
X‐rays of the joints
‐There was no damage to joints of people who took abatacept after 12 months. ‐The damage to joints of people who took a placebo was 0.27 units on a scale of 0 to 145 units.
Pain (higher scores mean worse or more severe pain)
‐ People who took abatacept rated their pain to be 12 points lower on a scale of 0 to 100 after 12 months with abatacept (12% absolute improvement).
‐People who took abatacept rated their pain to be 37 on a scale of 0 to 100 after 12 months. ‐People who took a placebo rated their pain to be 49 on a scale of 0 to 100.
ACR 50 (number of tender or swollen joints and other outcomes such as pain and disability)
‐20 more people out of 100 experienced improvement in the symptoms of their rheumatoid arthritis after 12 months with abatacept (20% absolute improvement).
‐37 people out of 100 experienced improvement in the symptoms of their rheumatoid arthritis. ‐17 people out of 100 who took a placebo experienced improvement.
Physical Function
‐25 more people out of 100 had better physical function after 12 months with abatacept (25% absolute improvement). ‐64 people out of 100 had better physical function. ‐39 people out of 100 who took a placebo had better physical function.
Disease activity
‐32 more people out of 100 were considered to have low disease activity of their rheumatoid arthritis after 12 months with abatacept (32% absolute improvement). ‐42 people out of 100 were considered to have low disease activity of their rheumatoid arthritis. ‐10 people out of 100 who took a placebo were considered to have low disease activity of their rheumatoid arthritis.
Summary of findings
Summary of findings for the main comparison. Abatacept (2 and 10mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic for rheumatoid arthritis.
| Abatacept (2 and 10mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic for rheumatoid arthritis | ||||||
| Patient or population: patients with rheumatoid arthritis Settings: Intervention: Abatacept (2 and 10mg/kg) + DMARDs/biologic Comparison: Placebo + DMARDs/biologic | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo + DMARDs/biologic | Abatacept (2 and 10mg/kg) + DMARDs/biologic | |||||
| ACR 50% improvement Follow‐up: 12 months | RR 2.21 (1.73 to 2.82) | 993 (3 studies) | ⊕⊕⊕⊝ moderate1,2,3 | Absolute risk difference= 21% (16% to 27%). Relative percent change=121% (73% to 182%). NNT=5 (4 to 7)4 | ||
| 168 per 1000 | 371 per 1000 (291 to 474) | |||||
| Pain measured at end of study on a 100 mm visual analog scale. Scale from: 0 (better) to 100 (worse). Follow‐up: 12 months | The mean pain in the control groups was 49.24 mm | The mean pain in the intervention groups was 10.71 lower (12.97 to 8.45 lower) | 1425 (1 study5) | ⊕⊕⊕⊝ moderate2 | Absolute risk difference=‐11% (‐13% to ‐8.5%). Relative percent change=‐18% (‐22% to ‐14%). NNT=5 (4 to 6)4 | |
| Improvement in physical function (HAQ: greater than 0.3 increase from baseline, 0‐3 scale) Follow‐up: 12 months | RR 1.62 (1.35 to 1.95) | 638 (1 study6) | ⊕⊕⊕⊝ moderate1 | Absolute risk difference= 24% (16% to 32%). Relative percent change= 62% (35% to 95%). NNT=5 (4 to 7)4 | ||
| 393 per 1000 | 637 per 1000 (531 to 766) | |||||
| Achievement of low disease activity state (DAS 28 less than 3.2, scale 0‐10) Follow‐up: 12 months | RR 4.33 (2.84 to 6.59) | 638 (1 study6) | ⊕⊕⊕⊝ moderate1 | Absolute risk difference=33% (26% to 39%). Relative percent change=333% (184% to 559%). NNT=4 (3 to 5)4 | ||
| 98 per 1000 | 424 per 1000 (278 to 646) | |||||
| Total serious adverse events Follow‐up: 6 to 12 months | RR 1.05 (0.87 to 1.28) | 3151 (6 studies) | ⊕⊕⊕⊝ moderate1,2,3,7 | Absolute risk difference=1% (‐2% to 3%). Relative percent change=5% (‐14% to 29%). NNT=n/a4 | ||
| 121 per 1000 | 127 per 1000 (105 to 155) | |||||
| Change in radiographic progression measured by Genant‐modifed Sharp erosion score (increase in score means more joint damage). Scale from: 0 to 145. Follow‐up: 12 months | The median change in radiographic progression in the control group was 0.27 units | The median change in radiographic progression in the intervention group was 0 units | 586 (1 study6) | ⊕⊕⊕⊝ moderate1,8 | Note there was no change in the abatacept group. MD ‐0.27 (‐0.42, ‐0.12). Absolute RD=‐0.2% (‐0.3% to ‐0.08%). Relative percent change=‐1.2% (‐1.9% to ‐0.6%). 9 | |
| Long‐term serious adverse events Follow‐up: 2 years | See comment | See comment | Not estimable | 950 (2 studies11) | ⊕⊕⊝⊝ low10 | Number of patients with SAE: Genovese 2005: 103/357; 23.4 SAE/100 patient‐years; 70% completed the LTE. Kremer 2006: 149/593; 16.3 SAE/100 patient‐years; 90.5% completed the LTE. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Kremer 2006: Intention to treat analysis not performed. 9 patients in abatacept group and 5 in placebo group excluded from analysis. 2 Weinblatt 2006: 15 people randomized were not treated and not included in analysis 3 Kremer 2003: Risk of attrition bias ‐ less than 80% completion rate in treatment group at 12 months 4 NOTE: Number needed to treat (NNT)=n/a when result is not statistically significant. NNT for dichotomous outcomes calculated using Cates NNT calculator (http://www.nntonline.net/visualrx/). NNT for continuous outcomes calculated using Wells Calculator (CMSG editorial office). 5 Outcome based on Weinblatt 2006 6 Outcome based on Kremer 2006 7 Weinblatt 2007: Risk of attrition bias ‐ less than 80% completion rate in the treatment group at 12 months 8 Radiographic data obtained for 90% of study participants 9 RD=risk difference 10 Long‐term serious adverse events based on observational data. Two RCTs had a long‐term extension (LTE) phase in which people in the placebo group during the RCT switched to abatacept for the LTE. 11 Based on 2 long‐term extension studies (LTE) of RCTs. Participants on placebo in the RCT switched to abatacept treatment.
Background
Rheumatoid arthritis is a chronic auto‐immune disease which affects the synovial lining of many joints and tendon sheaths resulting in persistent inflammation (Schumacher 1993). It is associated with significant morbidity, disability, and impaired quality of life (Badley 2003). Rheumatoid arthritis incidence is estimated to be 13 to 36 per 100,000 for females and less for males, with a prevalence in the UK as high as 0.8% (Woolf 2004). In terms of costs to society, the loss of 9.4 million working days to rheumatoid arthritis in 1999‐2000 has been estimated to represent an annual loss in productivity of 833 million British pounds (ARC 2005). In the US, the prevalence of rheumatoid arthritis in white adults over 18 years old was estimated to be 0.6% (Helmick 2008).
Disease‐modifying anti‐rheumatic drugs (DMARDs), such as methotrexate (Suarez‐Almazor 1998), leflunomide (Osiri 2002), hydroxychloroquine (Suarez‐Almazor 2000), and sulfasalazine (Suarez‐Almazor 1998b), have been shown to reduce disease activity, to slow disease progression (i.e. reduce the rate of new joint erosions) and to improve patients' quality of life. However, a significant proportion of rheumatoid arthritis patients are unable to tolerate these agents for long periods of time or only experience a partial benefit from these traditional DMARDs, or both. Another class of drugs called 'biologics' have been developed over the past ten years. These drugs mimic substances that occur in the immune system during an inflammatory reaction and are able to specifically target parts of the immune system to reduce inflammation, which in turn reduces the symptoms of rheumatoid arthritis.
Tumour necrosis factor (TNF)‐alpha is a protein that the body produces during the inflammatory response. The following biologic agents that target TNF‐alpha are currently available: infliximab (Remicade) is a chimeric (mouse/human) monoclonal antibody, golimumab (Simponi) is a fully human monoclonal antibody, etanercept (Enbrel) is a receptor fusion protein that binds to TNF‐alpha, adalimumab (Humira) is a recombinant human IgG1 monoclonal antibody specific for human TNF‐alpha, and and certolizumab pegol (Cimzia) is a recombinant, humanized antibody Fab' fragment specific for human TNF‐alpha. Infliximab, etanercept, and adalimumab have been shown to substantially and rapidly improve rheumatoid arthritis symptoms and to slow radiographic progression (Blumenauer 2002; Blumenauer 2003; Navarro‐Sarabia 2005). Golimumab and certolizumab pegol have recently received licensing approval and a Cochrane review is underway to assess the effects of certolizumab pegol on rheumatoid arthritis (Ruiz 2009).
Despite their effectiveness, not all patients respond to TNF‐alpha blockade and therefore other therapeutic options are needed. Abatacept (brand name Orencia) was approved by the US Food and Drug Administration (FDA) in December 2005 for use in adult patients with moderate to severe rheumatoid arthritis who have not responded adequately either to oral DMARDS (such as methotrexate) or to the TNF‐alpha antagonists. It is a selective costimulation modulator, inhibiting T‐cell (T lymphocyte) activation by binding to CD80 and CD86 (the costimulatory antigens), thereby blocking interaction with CD28 (the costimulatory receptor). It is the first biologic to work by disrupting T‐cell activation. Activated T‐cells occur early in the inflammatory reaction so by preventing their activation, the chain of events that leads to joint inflammation, pain, and damage is prevented. Abatacept is administered intravenously over approximately 30 minutes and after the first dose additional doses are given at two and four weeks and then every four weeks (Orencia 2007).
The use of biologics is limited by their high cost and uncertainty about adverse events. Although estimates vary by country, the annual cost of etanercept treatment is estimated at $17,160 CDN and $21,385 CDN for infliximab (PMPRB 2004). The cost for one year of abatacept treatment is approximately $22,000 USD (ACR 2007). Although expensive, if supported by the overall body of evidence, the claims of their benefit upon both symptoms and radiographic progression, and their low rate of short term side effects make them of great interest to patients with rheumatoid arthritis. At this time it is appropriate to conduct a systematic review of randomized controlled trials of abatacept to quantify the benefits and potential harms of its use.
Objectives
To assess the efficacy and safety of abatacept in reducing disease activity and pain, and improving function in people with rheumatoid arthritis.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs) were included. To be eligible for inclusion, the generation of the allocation sequence had to be truly random; for example, generation of the sequence by a computer or random numbers table. Trials had to be a minimum of three months duration. Trials of less than six months duration were used to investigate short‐term efficacy and safety while studies longer than six months addressed longer‐term efficacy and safety. We considered data from published and unpublished RCTs for inclusion. We checked websites of regulatory agencies for reported adverse effects.
Types of participants
Patients at least 16 years of age meeting the ACR 1987 revised criteria for rheumatoid arthritis (Arnett 1988).
Types of interventions
RCTs comparing abatacept alone or in combination with DMARDs or biologics to placebo or other DMARDs or biologics. There were no restrictions with regard to dosage or duration of intervention.
Types of outcome measures
Major outcomes
Efficacy
The primary outcome is the ACR 50 response rate to treatment with abatacept as defined by the American College of Rheumatology (ACR) (Felson 1995). The variables included in this definition are:
tender joint count;
swollen joint count;
patient's assessment of pain (visual analogue scale (VAS) or Likert scale);
patient and physician assessment of disease activity (VAS or Likert scale);
patient assessment of functional ability (Health Assessment Questionnaire (HAQ), Arthritis Impact Measurement Scales (AIMS), McMaster Toronto Arthritis (MACTAR)); and
laboratory parameters (i.e. acute phase reactants, such as erythrocyte sedimentation rate (ESR) or C‐reactive protein (CRP).
An ACR 20/50/70 response is defined as a 20%/50%/70% improvement in tender and swollen joint counts and the same level of improvement in three of the five following variables: patient and physician global assessments, pain, HAQ, and acute phase reactants.
Adverse events
Since RCTs are usually of limited duration, mainly short‐term adverse events were assessed. However, regulatory agency websites and long‐term extensions of included RCTs were also reviewed for potential longer‐term adverse events.
Specific adverse event outcomes of interest were:
adverse events, including allergic reactions, and infections;
serious adverse events, including serious infections, and lymphoma; and
withdrawals due to lack of efficacy, and adverse events.
Secondary outcomes
Individual ACR criteria and ACR 20 and 70 response criteria as outlined above.
Radiographic progression, as measured by the Sharp, modified Sharp or Larsen methods (also considered a primary outcome for studies longer than one year in duration).
European League Against Rheumatism (EULAR) criteria (Van Gestel 1996), which define response (good, moderate and none) according to certain cut‐offs for both the absolute values and relative changes in the Disease Activity Score (DAS) (Van der Heijde 1993). The DAS is a composite index that includes the combination of the values of tender and swollen joint counts, patient's global assessment of disease activity, and erythrocyte sedimentation rate (ESR) value. When a 28‐joint count is used the index is reported as DAS 28. The DAS28 is scored on a scale from 0 to 10 to indicate the current activity of rheumatoid arthritis; a higher number indicates higher disease activity. According to the DAS‐Score website, “A DAS28 above 5.1 means high disease activity whereas a DAS28 below 3.2 indicates low disease activity. Remission is achieved by a DAS28 lower than 2.6.”(DAS 2009). A 'good' EULAR response is defined as a decrease in the DAS or DAS 28 of more than 1.2 points from baseline with a final DAS less than 2.4 (or DAS 28 less than 3.2). A EULAR response of 'None' is defined as a decrease in DAS or DAS 28 less than 0.6 or a decrease greater than 0.6 and less than 1.2 with a final DAS greater than 3.7 (or DAS 28 greater than 5.1). Any other scores are regarded as 'moderate' response.
Health‐related quality of life (HRQOL) as measured by the SF‐36 or other instruments.
Search methods for identification of studies
Electronic searches
The original search strategy developed for MEDLINE in the protocol is reported in Appendix 1. After further discussions with the Trials Search Co‐ordinator and assessment of the results of this search, we further refined the search strategy to the strategy listed in Appendix 2. We screened 492 records from the original search in MEDLINE and compared these to the results of the new search strategy. All the records of interest retrieved using the original strategy were contained in the new search results. The MeSH headings of 'Immunosuppressive Agents' and 'Antirheumatic Agents' in the original search strategy were removed. In addition, since abatacept has a different mechanism of action from the tumour necrosis factor (TNF) biologics, references to TNF were removed from the original strategy. The MeSH headings of 'Immunoconjugates' and 'Antigens, Differentiation' were retained in the revised strategy. The new search strategy for MEDLINE was adapted for the other electronic databases as shown in the appendices.
We searched the following electronic databases initially up to March 2007: the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library, 2007 Issue 1) Appendix 3, MEDLINE Appendix 2, EMBASE Appendix 4, ACP Journal Club Appendix 5, and ISI Web of Science (Biosis Previews) Appendix 6. We searched the FDA website for references to trials of abatacept. We searched abstracts from ACR and EULAR conferences using Biosis Previews.
The search was not limited by language, year of publication or type of publication.
We ran updated search in January 2009 to capture publications between 1 January 2007 and 31 December 2008.
Searching other resources
We also searched reference lists from comprehensive reviews and identified clinical trials. We contacted content experts and the pharmaceutical company that manufactures abatacept to obtain clarification and any relevant additional unpublished data.
We searched websites of the following regulatory agencies for reported adverse events using the terms 'rheumatoid arthritis', 'abatacept' and 'orencia' on 1 April 2009.
'Current Problems in Pharmacovigilance' (http://www.mhra.gov.uk/Publications/Safetyguidance/CurrentProblemsinPharmacovigilance/index.htm) (this was superseded by 'Drug Safety Update' in July 2007. Both databases were searched under 'drug alerts').
Australian Adverse Drug Reactions Bulletin (http://www.tga.gov.au/adr/aadrb.htm).
Food and Drug Administration FDA Medwatch (US) ‐ Adverse Event Reporting System (AERS) FDA Medwatch ( http://www.fda.gov/medwatch/safety.htm).
European Public Assessment Reports from the European Medicines Evaluation Agency (http://www.emea.europa.eu/).
Data collection and analysis
Selection of studies
We used Reference Manager 11 software to manage the records retrieved from the searches of the electronic databases. We tracked results from handsearches on paper. We created the data extraction form in Word and captured all article information except outcome results in this form. We tracked outcome results in an Excel spreadsheet for easier entry into RevMan (RevMan 2008).
Two authors (LM, JS) independently reviewed the results of the various searches. We reviewed titles and abstracts and when more information was required to determine whether the trial met the inclusion criteria, we obtained the full text. We kept a record of reasons for excluding studies. We resolved disagreement by consensus and there was no need to contact a third party for a decision. Two German language articles were summaries of included studies so no further translation was required.
Data extraction and management
Two authors (LM, JS) independently extracted data from the included trials and entered these into RevMan 5. Variance measures were missing for many continuous outcomes (only P values were reported in the published articles) so we obtained additional data from Bristol‐Myers Squibb.
We decided a priori that the following data from each trial would be extracted.
General study information, such as title, authors, contact address, publication source, publication year, country, study sponsor.
Characteristics of the study: design, study setting, inclusion/exclusion criteria, quality criteria (e.g. randomization method, allocation procedure, blinding of patients, caregivers and outcome assessors, withdrawals and drop‐outs, intention‐to‐treat (ITT) analysis).
Characteristics of the study population and baseline characteristics of the intervention and control groups (age, sex, duration of disease, treatment history, presence of co‐morbidity and peripheral disease, concurrent treatments) and numbers in each group.
Characteristics of the intervention, such as treatment comparators, dose, method of administration, frequency of administration, and duration of treatment.
Outcomes measures as noted above (changes in disease outcome, adverse events, withdrawal from treatment).
Results for the intention‐to‐treat population (if reported), outcome measures at the end of the placebo phase, and any summary measures with standard deviations, confidence intervals, and P values where given, drop‐out rate, and reasons for withdrawal.
Assessment of risk of bias in included studies
Two independent authors (LM, JS) assessed the risk of bias of the included studies. As recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008), the following methodological domains were assessed.
Sequence generation ‐ was the method used to generate the allocation sequence appropriate to produce comparable groups?
Allocation sequence concealment ‐ was the method used to conceal the allocation sequence appropriate to prevent the allocation being known in advance of, or during, enrolment?
Blinding of participants, personnel and outcome assessors ‐ were measures used to blind study participants, personnel, and outcome assessors from knowledge of which intervention a participant received?
Incomplete outcome data ‐ how complete were the outcome data for the primary outcomes? Were drop‐out rates and reasons for withdrawal reported? Were missing data imputed appropriately? We considered an overall completion rate of 80% or higher as a low risk of bias. If completion rates were only provided by group, a less than 80% completion rate in the treatment group was considered a high risk of bias.
Selective outcome reporting ‐ were appropriate outcomes reported and were any key outcomes missing?
Other potential threats to validity (considering external validity, e.g. relevant use of co‐interventions) ‐ what was the funding source of each of the studies?
We explicitly judged each of these criteria using: Yes = low risk of bias; No = high risk of bias; and Unclear = either lack of information or uncertainty over the potential for bias.
Measures of treatment effect
We analyzed the results of the studies using RevMan 5.0 (RevMan 2008). We summarized data in a meta‐analysis if they were sufficiently homogeneous, both clinically and statistically. We expressed continuous data as mean difference (MD) or standardized mean difference (SMD), depending on the similarity of scales measuring an outcome. We expressed dichotomous data as relative risk (RR) or in the case of rare events (< 10%), such as death, we used the Peto odds ratio (Peto OR).
Some transformations were necessary to enter continuous data into RevMan. For Kremer 2006, standard error (SE) was converted to standard deviation (SD) using the formula, SD= SE x sqrt(N). The mean percent improvement from baseline and standard error were provided for Kremer 2003. Mean percent improvement was used to calculate the end of study score using the formula e‐b/b x 100=% improvement from baseline and the standard deviation at baseline was assumed for the standard deviation at end of study.
Assessment of heterogeneity
In addition to reviewing forest plots, we formally tested heterogeneity of the data using the Chi2 with a P value < 0.10 indicating significant heterogeneity. We also assessed the I2 statistic (Higgins 2003). A value greater than 50% may indicate substantial heterogeneity. In the case of substantial heterogeneity, we explored the data further, including subgroup analyses, in an attempt explain the heterogeneity.
Assessment of reporting biases
A funnel plot was performed to assess the possibility of publication bias.
Data synthesis
Since this is a recent drug on the market, it was expected that the trials would be performed in similar populations and that there would be little 'between‐study' variation. Thus, we specified a fixed‐effect model a priori. However, if significant heterogeneity was found and could not be explained, we decided that a random‐effects model would be used to assess the results.
Subgroup analysis and investigation of heterogeneity
We planned the following subgroup analyses a priori in order to explore possible effect size differences.
Intervention ‐ different dosage, duration of treatment.
Characteristics of participants ‐ severity of baseline disease; age; disease duration; sex; disease with or without peripheral joint involvement.
For this review, we assessed results separately at three, six and 12 months, by two dosages (2 mg/kg and 10 mg/kg), by duration of disease (average of less than eight years and greater than eight years), and by study eligibility criteria (anti‐TNF failures or DMARD failures).
Sensitivity analysis
We planned the following sensitivity analyses a priori in order to explore effect size differences and the robustness of conclusions.
Effect of study quality ‐ defined as adequate allocation concealment and outcome assessor blinding.
Effect of imputation of missing data or statistical transformations.
Summary of findings table
We completed 'Summary of findings' tables included in RevMan 5 in order to communicate the key outcomes of the review. We also determined the absolute risk difference and relative percent change and entered these into the comments column of the 'Summary of findings' table. For dichotomous data, the absolute risk difference is calculated by using RevMan to generate the Risk Difference analysis and then reporting the result as a percentage. The relative percent change is calculated by finding the relative risk (RR) from RevMan and then applying the formula RR‐1 equals the relative percent change. The number needed to treat (NNT) was calculated from the control group event rate (unless the population event rate was known) and the relative risk using the Visual Rx NNT calculator (Cates 2004).
For continuous outcomes, the absolute risk difference is the mean difference expressed as a percentage. The relative percent change is the absolute change divided by the baseline mean of the control group. The NNT was calculated using the Wells calculator software available at the Cochrane Musculoskeletal Group editorial office. The minimal clinically important difference (MCID) for pain was 20%, based on Tubach 2007, for input into the calculator. We also carried out a sensitivity analysis for 30%, based on Farrar 2001.
We used GRADE software to provide an overall grading of the quality of the evidence.
Additional data
We contacted trial authors and Bristol‐Myers Squibb, the manufacturer of abatacept, additional information about risk of bias aspects of the trials (e.g. allocation concealment and blinding) and variance and other outcomes not reported in the published reports, which was provided.
Results
Description of studies
See Additional Figure 1 for a flow diagram of the search results. The 'Characteristics of included studies' table provides further details about each included trial.
1.
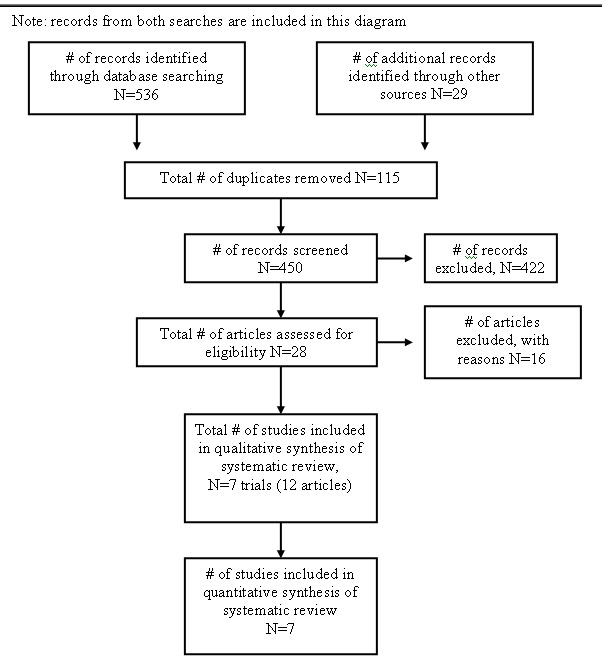
Search result flow diagram
Results of the search
The search of electronic databases in March 2007 resulted in 173 records. Handsearching of the American College of Rheumatology 2005 and 2006 conference abstracts resulted in 29 additional records. After duplicates were removed, there were a total of 172 records. We excluded 155 based on title or abstract. We retrieved seventeen full text articles. We excluded seven articles after reviewing the full text (see 'Characteristics of excluded studies' for further details). A total of ten articles met the inclusion criteria (Emery 2006; Genovese 2005; Kremer 2003; Kremer 2005; Kremer 2006; Moreland 2002; Russell 2007; Weinblatt 2006; Weinblatt 2007; Westhovens 2006). These ten articles correspond to six clinical trials. Three trials had multiple publications. The multiple publications reported different outcomes from the main trial publication. Emery 2006, Kremer 2003, and Kremer 2005 all referred to one trial; for the purpose of this review, Kremer 2003 is considered the primary publication. Kremer 2006 and Russell 2007 referred to one trial and for the purpose of this review, Kremer 2006 is considered the primary publication. Genovese 2005 and Westhovens 2006 referred to a single trial and Genovese 2005 is considered the primary publication.
An updated search of electronic databases in January 2009 from the period January 2007 to December 2008 (with the exception of Biosis Previews which was searched back to 1980) resulted in 363 records. After removing duplicates, there were a total of 278 records to review. Nine articles were excluded. We identified one new RCT (Schiff 2008). Cole 2008 is a publication of health related quality of life data from the Genovese 2005 and Kremer 2006 trials.
We searched pharmacovigilance websites for information on adverse events. 'Current Problems in Pharmacovigilance' (http://www.mhra.gov.uk/Publications/Safetyguidance/ CurrentProblemsinPharmacovigilance/index.htm), which was superseded by 'Drug Safety Update' in July 2007, did not report any adverse event data. The Australian Adverse Drug Reactions Bulletin also did not report any adverse event data. There were 70 hits from the FDA Medwatch site search. The Drug Product Label found on the website (FDA 2007, dated 13 March 2007) reported adverse events and these are described in the Discussion of this review. There were 22 hits on the European Medicines Agency site under 'Human Medicines'. The European Public Assessment Report on Orencia (http://www.emea.europa.eu/humandocs/Humans/EPAR/orencia/orencia.htm) contained the Scientific Discussion document (EMEA 2007) which listed adverse events from controlled and open label studies. These are further described in the Discussion of this review.
Design
All included trials were reported to be randomized, double‐blind, placebo‐controlled trials. All trials except Moreland 2002 and Schiff 2008 reported a randomization ratio of 2:1 for treatment to control. Moreland 2002 had six treatment arms and one placebo. Schiff 2008 had two treatment arms (abatacept or infliximab) and one placebo (randomised 3:3:2 to abatacept, infliximab and placebo. On day 198 of the trial, placebo‐treated patients were reallocated to abatacept (with blinding maintained).
Sample sizes
Sample sizes ranged from 121 in Weinblatt 2007 to 1441 in the Weinblatt 2006 trial (where safety was the primary outcome).
Setting
All seven trials were reported as 'multicenter' trials, but no specific information (except in Schiff 2008) was provided regarding the setting. All trials except Genovese 2005 and Weinblatt 2007 reported that they were multinational studies. No further information was provided regarding which countries participated in the study. Weinblatt 2007 reported that the study was conducted at 40 centers in the US. Schiff 2008 was undertaken at a total of 86 sites (US 20 sites, Europe 18 sites: five in Poland, four in Spain, four in Sweden, two in Russia, two in Denmark and one in Switzerland, Canada 11 sites, Australia six sites, Mexico 10 sites, Argentina five sites, Brazil eight sites, Peru five sites, and South Africa three sites).
Participants
Seven trials with 2908 patients were included in this analysis; 1863 were randomized to abatacept and 1045 to placebo. The majority of patients were white women. The average age of participants in all the trials was early to mid‐50s, with a range of 48.3 years in the control group of the Moreland 2002 trial to 55.8 years in the control group of Kremer 2003. To be eligible for the trials, patients had to have had active disease despite treatment with disease‐modifying anti‐rheumatic drugs (DMARDs) in Kremer 2003; Kremer 2006; Moreland 2002; and Schiff 2008. Genovese 2005 required that eligible patients had an inadequate response to three months of anti‐TNF therapy. For Weinblatt 2007, eligible patients must have received etanercept for more than three months and still have active disease. Patients with an inadequate response to DMARDs or biologics were eligible for Weinblatt 2006. The average disease duration in most trials was between eight and 13 years, except in Moreland 2002 in which the average duration of disease was much shorter: only 3.4 years.
Intervention
Most trials used a dosage of abatacept of 10 mg/kg and patients continued to use a DMARD in addition to abatacept for the duration of the study (Genovese 2005; Kremer 2003; Kremer 2006; Schiff 2008). In Weinblatt 2006, a trial designed to assess safety, patients remained on DMARDS or biologics throughout the trial. Kremer 2003 had a treatment arm where patients received 2 mg/kg of abatacept. In Weinblatt 2007 patients received 2 mg/kg of abatacept and also received etanercept (25 mg/kg twice weekly) until the open‐label long‐term extension period after which all patients received 10 mg/kg. Moreland 2002 was the only study in which no concurrent DMARD use was allowed. Moreland had three arms with three different dosages 0.5, 2 and 10 mg/kg, but for the purpose of this review, only the patients that received 10 mg/kg were analyzed, since this corresponds to the standard recommended dose. Schiff 2008 had two treatment arms (abatacept or infliximab) and one placebo. Patients were randomised 3:3:2 to abatacept (approximately 10 mg/kg), infliximab (3 mg/kg) and placebo. Patients were on a background of methotrexate therapy. On day 198 of the trial, placebo‐treated patients were reallocated to abatacept (with blinding maintained) and those patients initially randomized to abatacept or infliximab continued their treatment.
Abatacept was administered intravenously in all trials. A similar dosing schedule was followed in all trials with three treatments in the first month and then every month until the end of the study. The duration of trials ranged from 85 days (Moreland 2002) to six months (Genovese 2005; Kremer 2003) to 12 months (Kremer 2003; Kremer 2006; Schiff 2008; Weinblatt 2006; Weinblatt 2007).
Outcomes
All trials reported a primary outcome measure. In most trials, the primary outcome was the proportion of patients meeting the ACR 20 response criteria (Genovese 2005; Kremer 2003; Kremer 2006; Moreland 2002). Kremer 2006 also listed radiographic progression of joint erosions and HAQ‐DI as primary outcomes. Weinblatt 2007 used a modified ACR 20 in that the laboratory measure of C‐reactive protein (CRP) was excluded from the definition. The reason provided for this modification is that etanercept (which was administered concurrently to abatacept) normalized CRP levels. The primary objective of Weinblatt 2006 was to evaluate the safety of abatacept in patients with active rheumatoid arthritis. Thus, outcome measures in this trial included occurrence of adverse events, serious adverse events, discontinuations due to adverse events, death, and clinically significant changes. In Schiff 2008, the primary outcome measure was the reduction in Disease Activity Score (DAS) 28 (based on erythrocyte sedimentation rate levels; DAS28 (ESR)) with abatacept versus placebo at six months.
Secondary outcomes were also similar across trials and included ACR 50, ACR 70, individual ACR criteria components, DAS28, health related quality of life, and adverse events. Based on 12 months results from the Kremer 2003 trial, Emery 2006 reported the percentage of patients whose SF‐36 physical or mental component score was "better", "the same", or "worse" than the baseline score, using the definition of two standard error of the mean (SEM) as the minimal clinically important difference.
Most trials reported the timing of the primary outcome at six months (Genovese 2005; Kremer 2003; Kremer 2006; Weinblatt 2007) except Moreland 2002, which measured the primary outcome at 85 days and Weinblatt 2006, which reported adverse events at one year. Kremer 2005 reported the results of the Kremer 2003 trial at one year and Emery 2006 reported the health related quality of life measures of this trial at the one‐year mark. Schiff 2008 was a one‐year trial and they reported efficacy and safety results for abatacept, infliximab and placebo at day 197. Since the placebo group was switched to abatacept after this date, the patients who were reallocated were excluded from the one‐year abatacept group results.
Funding
All trials were sponsored by Bristol‐Myers Squibb, the manufacturer of abatacept.
Excluded studies
We excluded 16 records after retrieving the publication. Three were excluded because they were review articles (NHS 2004; Taylor 2006; Teng 2005); two were German language summaries of included trials (Alten 2006; Kruger 2005); one was a document provided to the FDA as supporting documentation for market approval (FDA 2005); four were reports of long‐term extensions to included RCTs (Genant 2008; Genovese 2008b; Haggerty 2007; Kremer 2008); two were meeting abstracts that were later published in full (Genovese 2004; Genovese 2005a); two did not have an outcome of interest (Li 2008; Weisman 2006); and two were post hoc analyses of included studies (Hassett 2007; Wells 2008).
Risk of bias in included studies
We contacted trial authors and Bristol‐Myers Squibb and asked them to provide further details about the methods of concealing allocation and blinding in those trials where this was not clear from the published study report.
Allocation
One trial clearly reported adequate allocation concealment in that each patient was assigned a unique, sequential patient number using a central (interactive voice) randomization system, and the randomization schedules were generated and kept sealed until the unblinding of the study (Weinblatt 2007). A central randomization procedure was reported in three studies (Genovese 2005; Kremer 2003; Kremer 2006) and this was taken to mean that adequate allocation concealment was performed. The authors of Weinblatt 2006 provided information detailing adequate allocation concealment. Bristol‐Myers Squibb (BMS) provided additional information confirming that Moreland 2002 was adequately concealed. Schiff 2008 did not mention allocation concealment in the published article and additional information was not available from Bristol‐Myers Squibb. However, given that all previous studies had adequate allocation concealment, it is likely that Schiff 2008 was adequately concealed as well.
Blinding
All trials were reported as 'double‐blind'. One trial did not give any further details (Moreland 2002) but further information was obtained from Bristol‐Myers Squibb to clarify that blinding of patients and outcome assessors was adequate. Genovese 2005 and Kremer 2006 reported that patients, study personnel, and clinical assessors were blinded. Patients and assessors were reported as blinded in Kremer 2003. Authors of Weinblatt 2006 and Weinblatt 2007 provided additional information clarifying that the investigators, infusion nurses and pharmacy and study personnel were blinded. Schiff 2008 reported that assessors, physicians and patients were blinded to the treatment group assignment for one year.
Incomplete outcome data
In judging the risk of bias for this item, we considered a less than 80% completion rate in the treatment group as a high risk of bias. We also assessed whether missing data were imputed appropriately and whether an intention‐to‐treat analysis was reported for the primary outcome.
For the primary efficacy measures, three trials (Kremer 2003; Schiff 2008; Weinblatt 2007) reported a proper intention‐to‐treat analysis. The other trials did not perform a proper intention‐to‐treat analysis; that is, one in which all randomized patients were assessed according to the group they were randomized to. Genovese 2005; Kremer 2006; Moreland 2002 and Weinblatt 2006 reported an intention‐to‐treat analysis on those subjects who received at least one infusion of study medication. In each of these studies there were a few patients (less than 1% of those randomized) who were randomized but not included in the analysis. The articles referred to this as a 'modified intention‐to‐treat' analysis. Two studies (Kremer 2006 and Weinblatt 2006) excluded patients from efficacy analyses due to protocol violations but included them in safety analyses. We judged these studies to have a higher risk of bias for efficacy outcomes compared to safety outcomes.
The completion rates in the abatacept‐treated group ranged from 68% in Weinblatt 2007 to 94.2% in the six‐month Schiff 2008 trial. In all trials, fewer patients in the placebo‐treated group completed the trial compared to the treatment arm. More patients who were treated with placebo withdrew due to lack of efficacy. The percentage of those completing the trial in the placebo group ranged from 60% in the 12‐month results of Kremer 2003 to 97% in the six‐month Schiff 2008 trial. The Weinblatt 2007 trial had much lower completion rates than the other trials (68% and 61% in treatment and placebo groups, respectively). Two studies (Kremer 2003 and Weinblatt 2007) were judged as having a high risk of bias due to low completion rates.
Missing data were imputed using last observation carried forward in most trials. For ACR response, those who discontinued were considered to be non‐responders in most trials. In Kremer 2003, "patients who discontinued the study because of worsening disease were considered to have had no response; for those who discontinued the study for other reasons the values for the last efficacy observation were carried forward". Using two separate criteria for imputing data may not be appropriate; for instance, a participant may not advise investigators that the reason they are no longer attending follow‐up visits is due to worsening disease and thus the last observation may be carried forward instead of assigning the patient a status of 'no response'. Additionally, some patients may have multiple reasons for withdrawal that they may or may not share with study staff.
Selective reporting
All trials reported outcome measures as recommended by the Outcome Measures in Rheumatology (OMERACT) group. The primary outcome in the Weinblatt 2007 trial was a modification of the ACR 20 in that it excluded the laboratory measure of CRP due to the normalizing effect of etanercept, which was given in combination with abatacept during the study. The modification to the outcome was done early in the study and was judged to have not contributed any bias to the study. The time points reported in all of the studies were reasonable. There is little risk of bias due to selective reporting in these trials.
Bias due to funding source
All trials were sponsored by the manufacturer of abatacept, Bristol‐Myers Squibb. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004).
Summary assessment of risk of bias
Figure 2 and Figure 3 provide a graphical summary of the results of risk of bias for the seven included studies.
2.
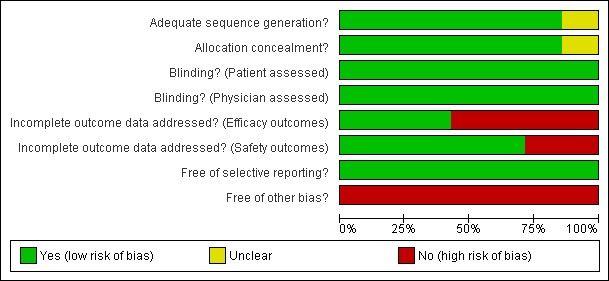
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across all included studies.
3.

Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
For the primary outcome ACR 50 response at 12 months, the studies included in the meta‐analysis rate well in terms of adequate allocation concealment and blinding and reporting of appropriate outcomes. However, there is a concern about bias in terms of incomplete outcome data due to the high drop‐out rate in two of the four studies and the fact that two studies excluded participants from efficacy analyses, but included them in safety analyses. Another concern is that all studies were sponsored by the manufacturer of abatacept.
Effects of interventions
See: Table 1
Seven trials with 2908 patients were included in this analysis; 1863 were randomized to abatacept and 1045 to placebo. Results for efficacy and harms will be presented separately.
Efficacy
Abatacept (10 mg/kg and 2 mg/kg combined) + DMARDs or biologics versus placebo + DMARDs or biologics
The primary outcome variable for this review was the ACR 50 response.
An ACR 20 response was achieved in significantly more abatacept‐treated patients compared to control at three, six and 12 months. At three months, the relative risk (RR) was 1.70 (95% confidence interval (CI) 0.93 to 3.12) (Moreland 2002). The RR was 1.79 (95% CI 1.59 to 2.02) in favor of abatacept at six months (Genovese 2005; Kremer 2003; Kremer 2006; Schiff 2008; Weinblatt 2007) and 1.79 (95% CI 1.55 to 2.07) at 12 months (Kremer 2003; Kremer 2006; Weinblatt 2007). These same trials were included in the results below for the ACR 50 and 70 results.
At three months, abatacept‐treated patients did not achieve a statistically significant higher ACR 50 response compared to control group (RR 2.50, 95% CI 0.52 to 11.96) (Moreland 2002). A statistically significant ACR 50 response was achieved in more abatacept‐treated patients compared to control at six and 12 months. The RR at six months was 2.47 (95% CI 2.00 to 3.07) and 2.21 (95% CI 1.73 to 2.82) at 12 months in favor of abatacept. The result at six months had evidence of moderate heterogeneity (P = 0.13, I2 = 44%). This can be explained by the inclusion of the Weinblatt 2007 trial which was not statistically significant (RR 1.33, 95% CI 0.63 to 2.83) and was the only trial to use a dosage of 2 mg/kg and in combination with etanercept. Removing Weinblatt 2007 from the six‐month pooled analysis reduced the heterogeneity and resulted in a pooled RR of 2.59 (95% CI 2.07 to 3.25). However, this is not a great difference from the RR which included Weinblatt 2007. For the ACR 50 response, there was an absolute difference of 21% (95% CI 16% to 27%). The number needed to treat in order to achieve an ACR 50 response at one year was 5 (95% CI 4 to 7).
A statistically significant higher ACR 70 response was found at six and 12 months in abatacept‐treated patients versus control group. The RR at six months was 3.53 (95% CI 2.41 to 5.16) and 4.02 (95% CI 2.62 to 6.18) at 12 months in favor of abatacept.
Trials reporting results for patient‐reported pain did not provide any measures of variance in Kremer 2003, Moreland 2002 and Weinblatt 2006. Weinblatt 2007 did report variance in the published article and Kremer 2006 provided the mean and standard error in an appendix. Table 2 provides the results for pain that were reported in the trials. We contacted trial authors and Bristol‐Myers Squibb to obtain the missing information. Variance measures were obtained for Kremer 2003 and Weinblatt 2006. In Kremer 2003 there was a statistically significant reduction in pain on a 100 mm visual analogue scale (VAS) (lower score means less pain) in the abatacept group compared to placebo at both six and 12 months (MD ‐22.49, 95% CI ‐28.00 to ‐16.98 at 12 months). In Kremer 2006, the abatacept group had significantly less pain at 12 months compared to placebo (MD ‐12.60, 95% CI ‐16.82 to 8.39). In Weinblatt 2006, there was also a statistically significant reduction in patient‐reported pain between groups at 12 months (mean difference (MD) ‐10.71, 95% CI ‐12.97 to ‐8.45). When pooling these three studies, the overall MD was ‐12.45, 95% CI ‐14.33 to ‐10.57) but there was high heterogeneity: I²=87%. Weinblatt 2006 was chosen to calculate the following two statistics because this was a large study with a wide variety of participants. The relative percent change from baseline was ‐18% (95% CI ‐22% to ‐14%). The NNT was 5 (95% CI 4 to 6) when a minimal clinically important difference (MCID) of 20% was assumed and 8 (95% CI 6 to 10) when an MCID of 30% was assumed. Results for pain in the two trials were not pooled at 12 months due to significant heterogeneity. The mean difference in pain scores between abatacept + etanercept and placebo + etanercept groups was not statistically significant in Weinblatt 2007. Genovese 2005 and Schiff 2008 did not report results for pain in the published articles.
1. Patient ‐reported pain results.
| Study ID | Pain scale | Baseline mean | End of study mean | % improvement, mean | Mean change from baseline | Variance (P value or CI) |
| Moreland 2002 ‐ abatacept; 85 days | 1‐5 | 3.47 | 2.43 | 28.1 | ‐ | NR |
| Moreland 2002 ‐ placebo; 85 days | 1‐5 | 3.55 | 3.24 | 4.6 | ‐ | NR |
| Kremer 2003 ‐ abatacept; 6 months | 100mm VAS | NR | NR | ‐ | ‐46.4 | P < 0.05 (between groups) |
| Kremer 2003 ‐ placebo; 6 months | 100mm VAS | NR | NR | ‐ | ‐8.4 | ‐ |
| Weinblatt 2006 ‐ abatacept; 1 yr | VAS | NR | NR | ‐ | ‐26.3 | P < 0.001 (within group) |
| Weinblatt 2006 ‐ placebo; 1 yr | VAS | NR | NR | ‐ | ‐16.4 | P < 0.001 (within group) |
| Weinblatt 2007 ‐ abatacept; 1 yr | NR | 65.5 | 43.6 | 33.4 | ‐22.0 | P < 0.001 (within group) |
| Weinblatt 2007 ‐ placebo; 1 yr | NR | 53.2 | 47.4 | 10.9 | ‐7.1 | P < 0.001 (within group) |
| Kremer 2006 ‐ abatacept; 1 yr | 100mm VAS | NR | NR | ‐ | ‐35.8 | ‐12.6 95% CI (‐16.9 to ‐8.39) |
| Kremer 2006 ‐ placebo; 1 yr | 100mm VAS | NR | NR | ‐ | ‐23.2 |
NR = not reported
VAS = visual analogue scale
*calculated as the average of the changes in the individual patient data
Clinically meaningful improvement in physical function was defined as a > 0.3 increase from baseline on the HAQ (0 to 3 scale) in Genovese 2005,Kremer 2006, Schiff 2008, and Weinblatt 2007. In Kremer 2003 it was defined as a > 0.22 increase from baseline on the HAQ. Genovese 2005 measured this outcome at six months and Kremer 2006 at 12 months. The results were similar regardless of the definition used. The pooled relative risk of clinically meaningful improvement in HAQ was 1.69 (95% CI 1.51 to 1.90) in favor of abatacept. In terms of the absolute risk difference between treated and control groups, the HAQ (> 0.3) at 12 months had an absolute difference of 24% (95% CI 16% to 32%). The number needed to treat in order to achieve an HAQ > 0.3 response at one year was 5 (95% CI 4 to 7).
The ACR core components ‐ patient global assessment, physician global assessment, physical function, tender joint count, and swollen joint count ‐ were all statistically significant in favor of abatacept. High heterogeneity was found when pooling the results of patient and physician global assessment and physical function, but the individual studies (Kremer 2003; Kremer 2006; Weinblatt 2006) all found a statistically significant improvement in the abatacept group compared to placebo. There was a significant reduction in the number of tender and swollen joints in the abatacept group compared to placebo (pooled results; MD ‐7.30, 95% CI ‐8.79 to ‐5.80 for tender joints and MD ‐4.81, 95% CI ‐5.79 to ‐3.83 for swollen joints).
The abatacept group was significantly more likely to reach a low disease activity state at six months (pooled RR 3.36, 95% CI 2.28 to 4.96) and at 12 months (RR 4.33, 95% CI 2.84 to 6.59). Achievement of a low disease activity state was defined as a DAS28 < 3.2 and at 12 months there was an absolute difference of 33% (95% CI 26% to 39%). The number needed to treat in order to achieve an DAS28 < 3.2 response at one year was 4 (95% CI 3 to 5). Those in the abatacept group were significantly more likely to achieve disease remission (defined as a DAS28<2.6) at twelve months (relative risk 12.74 (95% CI, 4.76 to 34.15). Schiff 2008 reported the DAS28 ESR as their primary outcome measure. At six months, there was a statistically significant reduction in DAS28 ESR in favor of abatacept (MD ‐0.95, 95% CI ‐1.20 to ‐0.70).
Health related quality of life (QoL) outcomes were measured at six and 12 months and these outcomes from Kremer 2003 (published in Emery 2006) and Genovese 2005 (published in Westhovens 2006) were pooled together for the dichotomous results presented. Scoring 'better' on the physical component score was statistically significantly higher in the abatacept group (RR 1.90, 95% CI 1.52 to 2.39). There was no statistically significant difference at six or 12 months in terms of the percent of patients scoring their SF‐36 physical component score as 'worse' from baseline (pooled RR 0.71, 95% CI 0.44 to 1.14). The relative risk was 0.66 (95% CI 0.56 to 0.78) for scoring the physical component score as 'the same' in favor of placebo. Genovese 2005 found a significant increase in the abatacept‐treated group in the number of patients achieving the population norm of the SF‐36 PCS at six months (RR 2.36, 95% CI 1.34 to 4.14). The SF‐36 physical component score was also presented as continuous data; possible scores are between 0 and 100, with a higher score indicating better health. The pooled mean difference of three trials (Kremer 2006; Schiff 2008; Genovese 2005) was statistically significant in favor of abatacept (MD 4.29, 95% CI 3.22 to 5.35).
The percent of patients scoring as 'worse' on the SF‐36 mental component score (MCS) was statistically significantly reduced in abatacept‐treated patients (RR 0.64, 95% CI 0.44 to 0.94) in a pooled analysis of two trials (Kremer 2003; Genovese 2005) that included results measured at six and 12 months. The relative risk was not statistically significant for scoring the mental component score as 'the same'. Scoring 'better' on the mental component score was statistically significantly higher in the abatacept group (RR 1.42, 95% CI 1.14 to 1.76). The SF‐36 mental component score was also presented as continuous data; possible scores are between 0 and 100, with a higher score indicating better health. The pooled mean difference of three trials (Kremer 2006; Schiff 2008; Genovese 2005) was statistically significant in favor of abatacept (MD 2.72, 95% CI 1.57 to 3.87).
Cole 2008 re‐examined QoL outcomes from Genovese 2005 and Kremer 2006 to estimate the outcomes of medical expenditure and likelihood of job loss. Using formulae based on QoL outcomes measured in the trial, they estimated that monthly medical expenditures decreased significantly in the abatacept group versus placebo at the endpoints. In Kremer 2006 (MTX‐failures) by day 365 the abatacept group's expenditures decreased by $220 (from $614 to $394), while the placebo group's expenditures were reduced by $152 ($614 to $462). The between‐group difference was statistically significant. In Genovese 2005 (anti‐TNF failures) by day 169 the abatacept group's expenditures decreased by $169 (from $696 to $527), while the placebo group's expenditures were reduced by $24 ($696 to $672). The between group difference was statistically significant. Likelihood of job loss was found to be statistically significantly different in the abatacept arm in both the MTX‐failure and anti‐TNF failure studies compared to placebo. There was a 25% to 64% greater likelihood of job loss from six months to two years.
Abatacept (2 mg/kg) + etanercept (25mg) versus placebo + etanercept
Statistically significant differences were not achieved for an ACR 20, ACR 50, or ACR 70 response or patient‐reported pain between the treatment and control groups at either six or 12 months (Weinblatt 2007).
Radiographic progression
Abatacept (10 mg/kg and 2 mg/kg combined) + DMARDs or biologics versus placebo + DMARDs or biologics
The only RCT that reported results of structural joint change was the Kremer 2006 AIM trial. The study found that compared to placebo, abatacept statistically significantly reduced the progression of joint damage after twelve months, though the progression was minimal in both groups.
They measured the change in progression of erosions in hands, wrists, and feet using the Genant‐modified Sharp score and found an approximately 50% reduction in change from baseline values in the abatacept group compared to placebo at 12 months. This was one of three primary outcome measures specified for this study. The mean increase in score from baseline was 0.63 for abatacept versus 1.14 for placebo in erosion score (no variance provided). An increase in erosion score indicates worsening joint damage; thus the abatacept group performed better because they had a smaller increase in erosion score than the placebo group.
In the published report on the AIM trial (Kremer 2006), the median change score from baseline and interquartile range (IQR) were provided and this was used to approximate the mean and SD (assuming SD = IQR/1.35) for entry into RevMan. The report states that there was no change from baseline in the abatacept group and a 0.27 change from baseline in the placebo group for the erosion score. Using the above approximation, there was a statistically significant mean difference in favor of abatacept (MD ‐0.27, 95% CI ‐0.42 to ‐0.12). The absolute risk difference is ‐0.2% (95% CI ‐0.3% to ‐0.08%) and the relative percent change is ‐1.2% (95% CI ‐1.9% to ‐0.6%). This difference in radiographic progression of the erosion score between groups is statistically significant, although the clinical significance of this difference is not clear to us (maximum normalized erosion score is 145). After the one‐year double‐blind period, participants were eligible to enter an open‐label, long‐term extension (LTE) and receive abatacept therapy. The results from this LTE are described in the Discussion of this review.
Subgroup analyses
Eligibility criteria
Trials were grouped according to whether eligibility criteria for the trial required patients to be inadequate responders to methotrexate (Kremer 2003; Kremer 2006; Schiff 2008) or inadequate responders to anti‐TNF alpha drugs (Genovese 2005; Weinblatt 2007). In Weinblatt 2006, the large safety trial, and Moreland 2002 participants were either DMARD or biologic failures.
At six months, those who were inadequate responders to biologic therapy were at a slightly larger relative risk for achieving an ACR 20 response (RR 2.27, 95% CI 1.67 to 3.07) compared to those who were methotrexate failures (RR 1.68, 95% CI 1.48 to 1.91). Similar results were found for the ACR 50 and ACR 70 responses. The relative risk for an ACR 50 response was lower in methotrexate failures (RR 2.38, 95% CI 1.89 to 3.00) compared to biologic failures (RR 2.96, 95% CI 1.67 to 5.25). The relative risk for an ACR 70 response was lower in methotrexate failures (RR 3.16, 95% CI 2.12 to 4.71) compared to biologic failures (RR 7.05, 95% CI 1.98 to 25.14). There was significant heterogeneity in the anti‐TNF failure group analysis and we think this is due to the fact that the interventions were different in Weinblatt 2007 (abatacept plus etanercept) and Genovese 2005 (abatacept plus DMARD). The six‐month results in the anti‐TNF failure group are heavily influence by the large relative risks of the Genovese study which included people with long disease duration, high activity, and who were anti‐TNF failures.
At twelve months, a pooled analysis of the methotrexate‐failure populations (Kremer 2003; Kremer 2006) demonstrated statistically significant results in favour of abatacept for ACR 20 (RR 1.81, 95% CI 1.56 to 2.10), ACR 50 (RR 2.27, 95% CI 1.76 to 2.93), and ACR70 (RR 4.29 (95% CI 2.73 to 6.73). The anti‐TNF failure population study with twelve month data (Weinblatt 2007) used a lower dose of abatacept in combination with etanercept and did not find a significant difference in the ACR20/50/70 responses.
Dosage
We carried out a subgroup analysis to look at the effect of the 2 mg/kg dose on ACR 20, 50, and 70 improvement compared to those trials using 10 mg/kg. As noted above, the 2 mg/kg dose was given in combination with etanercept and it was not statistically significant at any time point. There were no major changes to the relative risks once the 2 mg/kg dose was removed from the analysis of the combined dose (comparison 4).
Disease duration
All studies except Moreland 2002 enrolled patients with a disease duration greater than eight years. As Moreland 2002 was a pilot study that only provided three‐month data, this subgroup analysis was not undertaken.
Sensitivity analyses
Effect of study quality
This was defined as adequate allocation concealment and outcome assessor blinding. All studies except Schiff 2008 (unclear allocation concealment) reported adequate allocation concealment and blinding. Excluding Schiff 2008 from the ACR 50 response at six months did not significantly change the result (with Schiff 2008, ACR 50 RR 2.47 (95% CI 2.00 to 3.07); excluding Schiff 2008 ACR 50 RR 2.62 (95% CI 2.05 to 3.37)).
Harms
Total adverse events were significantly greater in the abatacept group compared to placebo but the relative risk was low (RR 1.05, 95% CI 1.01 to 1.08). Total serious adverse events, withdrawals due to adverse events, serious infections, upper respiratory infections, malignancies, and mortality were not statistically significantly different between the treatment and control groups, based on pooled results at six and 12 months. However, there were a greater number of serious infections at 12 months in the abatacept‐treated group (Peto odds ratio (OR) 1.91, 95% CI 1.07 to 3.42). This analysis included the Weinblatt 2007 trial in which abatacept was given in combination with etanercept. Removing this study resulted in a lower odds ratio which was just statistically significant (Peto OR 1.82, 95% CI 1.00 to 3.32). Total withdrawals favored the abatacept‐treated group (RR 0.60, 95% CI 0.52 to 0.70).
Other adverse events such as cough, nausea, dizziness, and diarrhea were not statistically significant between the abatacept and placebo groups, although for each of these outcomes there was a trend in favor of placebo. There was a greater number of headaches reported in the abatacept group compared to placebo for the pooled results at three, six, and 12 months (RR 1.45, 95% CI 1.20 to 1.74). There was also a higher relative risk of an infusion reaction (defined as a reaction within 24 hours after administration of treatment) in the abatacept group (RR 1.30, 95% CI 1.13 to 1.50).
Weinblatt 2006 was a large RCT (n = 1441) designed to assess safety over a 12‐month period. The article reports that "no formal tests were planned to compare AE (adverse event) incidence rates between treatment groups." Data was entered into RevMan to obtain the relative risks provided below. Patients were on a background of either traditional non‐biologic, biologic DMARDs, or both. The abatacept and placebo groups had similar frequencies of total adverse events (90% and 87% respectively; RR 1.04, 95% CI 1.00 to 1.08). Serious adverse events, withdrawals due to adverse events, and serious infections were not statistically different between the abatacept and placebo groups. Total withdrawals favored the abatacept group (RR 0.71, 95% CI 0.55 to 0.91). A post hoc analysis evaluated the incidence of adverse events in patients on a background of biologic therapy compared to non‐biologic therapy. Total adverse events, serious adverse events and withdrawals due to adverse events were similar in the abatacept plus non‐biologic subgroup compared to the placebo plus non‐biologic subgroups. However, in the group receiving abatacept plus a biologic DMARD, total adverse events were found to occur more frequently (95.1%) compared to the placebo plus a biologic DMARD group (89.1%). Withdrawals due to adverse events, total serious adverse events, and withdrawals due to serious adverse events all occurred more frequently in the abatacept plus biologic group. Weinblatt 2007 was designed to assess the safety and efficacy of abatacept in combination with etanercept. As noted above, the efficacy outcomes were not statistically significant. No safety outcomes were statistically significant, with the exception of total serious adverse events (Peto odds ratio 3.49, 95% CI 1.08 to 11.34) (see Analysis 2.11).
Given the concerns in the Weinblatt 2006 trial report, we undertook a post hoc analysis to assess the effect of harms in patients on a background therapy of biologic treatment compared to placebo. We included and pooled 288 patients and dosages of 2 mg/kg and 10 mg/kg in this analysis. The relative risk of total serious adverse events in the abatacept group was statistically significantly more than the placebo group (RR 2.30, 95% CI 1.15 to 4.62) as well as withdrawals due to adverse events (Peto odds ratio 2.68 95% CI 1.07 to 6.72). Total adverse events were not more significant (RR 1.06, 95% CI 0.98 to 1.14). The following outcomes were not statistically significant, but there was a trend towards favoring the placebo group: serious infections (Peto odds ratio 3.20, 95% CI 0.86 to 11.97) and upper respiratory tract infections (Peto odds ratio 1.79, 95% CI 0.75 to 4.26).
There were no deaths reported in Kremer 2003 at six and 12 months and in Weinblatt 2007. At 12 months, there was no significant difference between the abatacept and placebo groups in terms of mortality (Peto odds ratio 0.58, 95% CI 0.17 to 2.04) (Kremer 2003; Kremer 2006; Weinblatt 2006; Weinblatt 2007). Given that there were few events in this analysis, we used the Peto odds ratio. No deaths were reported in patients on a background of biologic therapy and abatacept (Analysis 3.7).
Publication bias
We assessed publication bias using a funnel plot of the ACR 50 response at six months. Additional Figure 4 shows the resulting funnel plot. Although there are only five trials (Moreland 2002 does not provide six‐month data and Weinblatt 2006 does not measure ACR 50) included in this assessment, there is clear symmetry in the plot. Thus, it does not appear that there is evidence of publication bias in this review.
4.
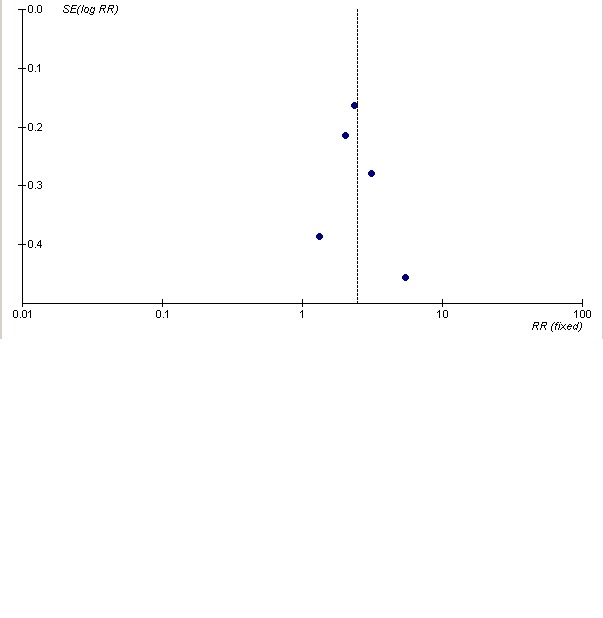
Assessment of publication bias
Discussion
Summary of main results
The primary efficacy outcome, a 50% improvement in tender and swollen joint counts and the same level of improvement in three of the five following variables: patient and physician global assessments, pain, HAQ, and acute phase reactants, was statistically significant in favor of abatacept compared to placebo at six and 12 months. Patients in the abatacept group were between 2 and 2.5 times more likely to achieve these responses compared to those in the placebo group. In addition, significant improvements in physical function and a reduction in disease activity was found in abatacept‐treated patients. A significant decrease in patient‐reported pain in the abatacept group compared to placebo was found in three trials at both six and 12 months. In terms of the absolute risk difference between treated and control groups, the ACR 50 at 12 months had a 21% absolute difference (95% CI 16% to 27%). The number needed to treat in order to achieve an ACR 50 response at one year was 5 (95% CI 4 to 7). This NNT is similar to those found in a systematic review of another biologic agent, adalimumab, an anti‐TNF alpha inhibitor, in patients with moderate to severe disease and failure with previous disease‐modifying anti‐rheumatic drugs. The NNT for an ACR 50 response with adalimumab was 3.0 (95% CI 2.0 to 6.0) (Navarro‐Sarabia 2005). A subgroup analysis based on eligibility criteria of an inadequate response to disease‐modifying anti‐rheumatic drugs (DMARD) therapy versus an inadequate response to anti‐TNF therapy found that in both groups abatacept produced a statistically significant ACR50 response compared to placebo at six months. Therefore, based on placebo‐controlled trials of up to one year duration, it appears that abatacept is efficacious in improving signs and symptoms of patients with active, moderate to severe, rheumatoid arthritis who have failed either DMARD or anti‐TNF therapy.
Kremer 2006 demonstrated that at 12 months abatacept statistically significantly slowed the progression of structural joint damage compared with placebo. A significant reduction in structural damage progression was also observed in a two‐year open‐label study. Out of 547 patients who completed the double‐blind phase of the trial, 538 started the open label phase (98.5%) and 90.5% of those patients completed one year of therapy. Genant 2008 reported the results of the radiographic findings after two‐year, open‐label follow up of the AIM study. Treatment with abatacept for two years was found to statistically significantly reduce erosion scores compared to treatment of one year with placebo followed by one year of abatacept. The mean change in erosion score was 0.84 in patients treated with abatacept for two years compared to 0.62 in patients treated with placebo for 12 months prior to entering the abatacept‐treated open‐label portion of the study. The mean change scores were significantly lower from years one to two, compared to baseline to year one, and 79% of those patients with no radiographic progression at year one continued to have no progression at year two, indicating a continuing maintenance effect of abatacept. As with the results of the radiographic progression in the RCT, although the difference in radiographic score is statistically significant, it is not clear what the clinical significance of this difference may be.
Two studies (Genovese 2005; Kremer 2006) had long‐term extension phases after the double‐blind RCT phase. Kremer 2008 reported the two‐year results of the AIM (Kremer 2006) study after a one‐year, long‐term extension in which patients on placebo for the RCT were switched to abatacept and those randomized to abatacept continued taking it. In the abatacept group 89% finished the one‐year RCT and 74% in the placebo group. All but seven patients in the abatacept group and one in the placebo group started the long‐term extension study. Of those patients, 90.5% completed one year of the long‐term extension. The ACR 20, 50, and 70 responses in the original abatacept group at two years were similar to those at one year: ACR 20 responses were 81.9% and 80.3%, ACR 50 responses were 54.0% and 55.6%, and ACR 70 responses were 32.4% and 34.3%, at one and two years respectively. Disease activity (measured by DAS28), physical function (measured by HAQ‐DI) and health related quality of life (measured by SF‐36) outcome responses were also maintained at two years. In the group originally receiving placebo during the RCT period, ACR 20, 50, and 70 responses at the end of the long‐term extension were similar to the year two response of the original abatacept group. Thus, it appears that response to abatacept therapy is well‐maintained.
Genovese 2008b reported the efficacy and safety of the two‐year, long‐term extension of the Genovese 2005 trial. In the abatacept group 86.4% finished the six‐month double‐blind phase of the study and 74.4% in the placebo group. All but five patients enrolled in the 18 month long‐term extension phase and 70% completed the 18 months of treatment with abatacept. At 24 months, the improvements in ACR 20, 50, and 70 that were observed at six months were maintained. For example, the percentage of patients achieving an ACR 50 response for patients originally randomized to abatacept was 23.5% and 33.2% at six and 24 months, respectively. In those patients originally randomized to placebo, their response was similar to the original abatacept group after 24 months of therapy, as demonstrated by 32.3% of this group achieving an ACR 50 response at two years. A similar maintenance of improvement in physical function using the HAQ‐DI was also reported. The percentage of abatacept‐treated patients experiencing a low‐disease activity state (LDAS, definition DAS28‐CRP <= 3.2) improved from 18.3% at six months to 32% at 24 months, but this was an 'as observed', post hoc analysis. The authors conclude that abatacept maintains a response over two years in those patients who have failed anti‐TNF therapy.
Results of efficacy analysis must be balanced by assessment of harms. In this review, total adverse events were significantly greater in the abatacept group compared to placebo but the relative risk was low (RR1.05, 95% CI 1.01 to 1.08). Total serious adverse events, withdrawals due to adverse events, upper respiratory infections, malignancies, and mortality were not statistically significantly different between the treatment and control groups when the six and 12‐month results were pooled together. However, there were a greater number of serious infections at 12 months in the abatacept‐treated group (Peto odds ratio 1.91, 95% CI 1.07 to 3.42). However, most randomized trials are of short duration and can only provide data on short‐term safety in populations of reasonable size. RCTs are not designed to adequately assess safety in the intermediate or long term, or to detect uncommon, rare adverse events, which are best monitored and studied in observational studies and post‐marketing surveillance studies.
One RCT (Weinblatt 2006) was designed to assess safety over a one‐year period, although they did not plan formal statistical tests to compare adverse event rates between treatment groups. While adverse events in the abatacept‐treated group occurred with similar frequency compared to placebo, a post hoc analysis of abatacept in combination with biologic background therapies was associated with an increase in the rate of serious adverse events. The study authors concluded that "abatacept is not recommended for use in combination with biologic therapy." We conducted a post hoc analysis in RevMan to compare outcomes on harms in those patients on a biologic background therapy (anakinra or anti‐TNF) compared to placebo. To increase the sample size for this analysis, we pooled the trials with 2 mg/kg and 10 mg/kg dosages of abatacept. By including the lower dosage, we may expect that there are fewer adverse events than had only the 10 mg/kg dosage been included. We found that there was a significant increase in the total number of serious adverse events (RR 2.30, 95% CI 1.15 to 4.62) in the abatacept group compared to placebo. Lack of statistically significantly differences in total number of adverse events, serious infections, upper respiratory tract infections, and withdrawals due to adverse events between groups may be due to small sample size and short follow‐up duration. However, there was a strong trend towards a higher relative risk of these harmful outcomes in the abatacept group, so we concur with the authors of the Weinblatt 2006 and Weinblatt 2007 studies that recommend that abatacept not be given in combination with other biologic treatments.
Genovese 2008b and Kremer 2008 published data on the safety of abatacept after the two‐year, long‐term extension phase of Genovese 2005 and Kremer 2006, respectively. Genovese 2008b reported that after two years, the cumulative adverse events were 'comparable' to those observed in the six‐month, double‐blind phase. Out of 357, patients 103 reported a serious adverse event. There were 25.6 serious adverse events per 100 patient‐years in the double‐blind phase and 23.4 over the cumulative two‐year period, and 5.3 serious infections per 100 patient‐years in the double blind and 5.0 in the cumulative period. 3.9% and 7.0% of patients withdrew due to adverse events in the double‐blind phase and cumulative phase, respectively. Two patients died over the cumulative period, one during the double‐blind phase. Eleven malignancies were reported but it was unclear if they were related to use of abatacept.
In Kremer 2008, the number of serious adverse events per 100 patient‐years was 17.7 in the one‐year, double‐blind phase and 16.3 over the two‐year cumulative period. Out of 593 patients, 149 reported a serious adverse event. There were 4.2 and 4.3 serious infections per 100 patient‐years in the double‐blind and two‐year cumulative period, respectively. 4.4% and 6.4% of patients withdrew due to adverse events in the double‐blind phase and cumulative phase, respectively. There were three deaths over the two years, one in the double‐blind phase. Fourteen malignancies were reported over the two years.
The safety of abatacept was recently summarized, based on the two Phase IIb and three Phase III studies included in this review as well as unpublished data from open‐label follow‐up studies (Sibilia 2007). Over the double‐blind and open‐label phases, a cumulative exposure of 4764 patient–years of treatment was assessed. As was described in the open‐label studies mentioned above, the incidence of serious infections or malignancies did not appear to increase in the open‐label phase and was similar to that found in the double‐blind phase. Editorial support for the Sibilia article was provided by Bristol‐Myers Squibb.
The abatacept label information published on the FDA website (FDA 2007) highlighted the increase in serious infections, lymphoma and lung cancer in those patients taking abatacept compared to placebo. The label also included a warning about using abatacept concurrently with anti‐TNF therapy. No significant benefit in terms of efficacy was found with concurrent therapy and the concerns about an increase in infections and serious infections warranted a warning that abatacept should not be administered to patients on concurrent anti‐TNF therapy.
The European Public Assessment Report's Scientific Discussion document on abatacept (EMEA 2007) assessed adverse events in controlled trials and the results were similar to those found in this review. The report highlighted signals of a possible increased risk of infection and autoimmune disorders that will need to be further investigated, as well as malignancies, given abatacept's mechanism of action. The report concluded that ongoing pharmacovigilance is needed to address concerns about 'potential rare and unexpected severe adverse effects of abatacept'.
At this time, the lack of long‐term data on potential harms of abatacept use means that firm conclusions cannot be drawn. However, it does appear that the withdrawal and toxicity profile is appropriate to allow further monitoring of adverse events.
'Summary of findings table 1' summarizes key efficacy and safety outcomes discussed above.
Overall completeness and applicability of evidence
Seven published trials addressed the use of abatacept for rheumatoid arthritis. Participants in the majority of the included studies had moderate to severe rheumatoid arthritis of at least eight years duration. However, the high levels of disease activity seen in the patients included in these trials may not be typical of patients seen in daily clinical practice. Patients selected for RCTs generally have few major comorbidities, though the published exclusion criteria in these trials did not list specific exclusions, other than pregnant and nursing women. Of note, the Weinblatt 2006 study was designed to assess the safety of abatacept in patients expected to be encountered in general practice. Patients with conditions such as congestive heart failure (CHF), asthma, chronic obstructive pulmonary disease (COPD), and diabetes mellitus were included. The prevalence of these diseases in the study population was: CHF 1% to 2%, asthma 6%, COPD 4%, and diabetes mellitus 6%.
Included trials generally did not enrol patients with early disease so it is not clear how efficacious abatacept may be in people with early disease. Most trials used a standard dosage of approximately 10 mg/kg. One trial did use a lower dosage in combination with etanercept and concluded that given the lack of efficacy and increase in adverse events, abatacept in combination with another biologic is not recommended.
An editorial on the Kremer 2006 study (Boers 2006) highlights the desire of clinicians for active comparator trials once efficacy of a treatment has been established against placebo. Boers suggests that Kremer 2006 provides a clinically useful comparison of methotrexate against abatacept plus methotrexate because the trial probably enrolled patients who were partial methotrexate responders rather than methotrexate failures.
Appropriate outcomes based on OMERACT recommendations were assessed to establish short‐term efficacy. Further studies are needed to assess long‐term efficacy and safety.
Quality of the evidence
Seven trials with 2908 patients were included in this analysis; 1863 were randomized to abatacept and 1045 to placebo.
The Outcome Measures in Rheumatology (OMERACT) group has had great success in standardizing outcomes that should be measured in trials of interventions for rheumatoid arthritis and other arthritic conditions. The trials included in this systematic review reported outcome measures as recommended by OMERACT for trials on rheumatoid arthritis patients. There is little risk of bias due to selective reporting in these trials.
When combining studies it is important that the outcome measures are comparable. Of note, different definitions of serious adverse events were used in the assessment of these events across the trials. Some trials used MedDRA version 7, others used version 8, some trial reports provided a long list of specific criteria for the definition of a serious adverse event and others were not specific. We assumed for the purpose of this review that the definitions were similar enough to warrant combining.
Adequate allocation concealment can avoid selection bias in controlled trials and there is evidence that inadequate allocation concealment leads to an overestimation of the treatment effect (Schultz 1995). Additional information was obtained from the manufacturer and authors for three studies to clarify the method of allocation. All studies included in this review, with the exception of Schiff 2008, reported adequate allocation concealment. Although not reported in the published article and the information was not available from Bristol‐Myers Squibb, we think it is likely that there was adequate allocation concealment given that previous trials used a central randomization system managed by Bristol‐Myers Squibb, who also sponsored the Schiff 2008 trial. Additional information was also obtained regarding clarification on blinding of study participants, investigators, and outcome assessors. After this information was obtained, all included studies were deemed to be adequately blinded for patient assessed and physician assessed outcomes. Given that the primary outcome measured in most trials was the percentage of patients meeting a 20 or 50 ACR response and that this composite measure includes subjective measures such as tender and swollen joint counts and physician assessment of disease activity, the reporting of blinding of participants and outcome assessors is necessary to ensure detection bias has not been introduced in these studies.
Completion rates were greater than 80% in the treatment group in all but two studies (Kremer 2003; Weinblatt 2007). All the trials reported the numbers of patients who dropped out in the treatment and placebo groups. The drop‐out rates were higher in the placebo group than the treatment group in all trials and there was a much higher rate of withdrawal due to lack of efficacy in the placebo groups. In most trials the missing data were imputed using last observation carried forward analysis and for dichotomous outcomes like ACR responses, missing data were considered non‐responders. However, only three out of seven trials reported a proper intention‐to‐treat analysis. The other trials reported an intention‐to‐treat analysis as one defined by those subjects who received at least one infusion of study medication. Although fewer than 1% of participants were affected, it is of interest to know why patients who were randomized did not receive the study drug.
For the primary outcome ACR 50 response at 12 months, the four studies included in the meta‐analysis rated well in terms of adequate allocation concealment and blinding and reporting of appropriate outcomes. However, there is a concern over attrition bias given the high drop‐out rate in two of the four studies and that two studies excluded participants from efficacy analyses but included them in safety analyses. Another concern is that all studies were sponsored by the manufacturer of abatacept and there is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004).
A funnel plot indicated that there was no evidence of publication bias, though this result should be interpreted with caution as only five studies that measured the ACR 50 outcome at six months were included in the plot.
With regards to detecting adverse events in RCTs, an interesting paper (Yazici 2008) recently highlighted the fact that an inadequate sample size (Type II error) is a possible reason that a significant difference in the number of adverse events between treatment and placebo groups is often not observed. All the included studies except for the main safety study of abatacept (Weinblatt 2006) termed themselves 'efficacy and safety' studies. But in most trials there was no discussion of necessary sample sizes to detect adverse events. Weinblatt 2006 noted that it was powered to find adverse events occurring at a rate of 0.2%, but no formal statistical tests between groups were planned for this trial.
We concluded that there is 'moderate' level evidence for the short‐term efficacy outcomes of ACR 50 response, physical function, and disease activity. Longer‐term adverse event data was judged to be of 'low' level quality as it was based on observational data (extensions of RCTs).
Potential biases in the review process
We undertook a systematic, thorough search of the literature to identify all studies meeting the inclusion criteria for this review. The manufacturer of abatacept was also contacted to ensure no trials were missing. We are confident that all trials meeting the inclusion criteria were included in this review. A trial published in January 2009 was identified and will be included in a update of this review.
Study selection and data extraction were done in duplicate and independently and we reached consensus by discussing any discrepancies.
Published trial reports did not provide enough details to adequately assess risk of bias and some variance measures necessary for meta‐analysis were missing from the report. We contacted authors and Bristol‐Myers Squibb for further information. The majority of requested data was provided but it is a limitation of this review that not all the data were available.
A protocol was published for this review. All analyses were specified a priori, with the exception of a post hoc analysis of the efficacy and safety of abatacept in combination with biologics, after included studies demonstrated concern over an increase in adverse events with limited efficacy.
Agreements and disagreements with other studies or reviews
Cochrane Reviews of other approved biologics, including anti‐TNF agents, infliximab (Blumenauer 2002), etanercept (Blumenauer 2003), and adalimumab (Navarro‐Sarabia 2005), have also demonstrated clinical efficacy in patients with rheumatoid arthritis.
There was one comparative study between anti‐TNF agents and abatacept – the Schiff 2008 RCT of abatacept versus infliximab versus placebo, sponsored by Bristol‐Myers Squibb. This study was designed to have power to detect differences between abatacept and placebo and infliximab and placebo, but not powered to detect differences between abatacept and infliximab. The first six months was placebo‐controlled and then those patients treated with placebo were re‐allocated to abatacept (with blinding maintained) for the next six months. The efficacy of abatacept compared to infliximab was a secondary outcome of the study. Both biologics were significantly better than placebo in efficacy outcomes and similar responses were found with the two biologics at six months. However, by twelve months, the abatacept‐treated group had a statistically significantly higher ACR20 response, achievement of a low disease activity state, DAS28 (ESR) and good EULAR response. The point estimates of other outcomes such as physical function and ACR 50 and 70 responses favoured abatacept, though there was no statistical difference between the two groups. Of note is that the infliximab study dosage was 3mg/kg and physicians may increase this dosage by as much as 30% to achieve a response in their patients. At twelve months, the infliximab‐treated group had more acute infusion events, serious adverse events (SAE), serious infections, and discontinuations due to SAE than abatacept. The authors concluded that abatacept appeared to be better at maintaining efficacy at twelve months and had a better safety profile at twelve months than infliximab.
Given that there are similar efficacy profiles against placebo with these agents, it is of interest to see how patients might respond in other head‐to‐head comparison trials.
A recent review of trials of abatacept for rheumatoid arthritis summarized the development of abatacept and the results of each Phase II and III trials (Buch 2008) but no meta‐analysis was undertaken. The article was part of a supplement entitled 'Co‐stimulation blockade: from bench to bedside' and publication of that supplement was sponsored by Bristol‐Myers Squibb, the manufacturer of abatacept. A similar review summarized the individual results of clinical and patient‐reported outcomes of six Phase II and III trials for the use of abatacept for rheumatoid arthritis Massarotti 2008. All the studies included in the Buch and Massarotti articles were included in this systematic review. Both reviews concluded that abatacept resulted in significant improvements in efficacy outcomes in patients with rheumatoid arthritis.
Salliot 2009 performed a recent meta‐analysis of RCTs to investigate the risk of serious infections in rituximab, anakinra, and abatacept for rheumatoid arthritis. They found no significant increase in risk of serious infection with the use of abatacept in five trials (pooled Mantel‐Haenszel OR 1.35, 95% CI 0.78 to 2.32). For this review, we used the Peto odds ratio given the low event rate in the treatment and placebo groups. We also found that when the six and 12‐month results were pooled, there was no significant increase in serious infections (Peto OR 1.56, 95% CI 0.93 to 2.61). However, the pooled 12‐month results of three trials did result in a statistically significant increase in serious infections in the abatacept group compared to placebo (Peto OR 1.91, 95% CI 1.07 to 3.42). Additional trials and post‐marketing surveillance studies are needed to determine the long‐term safety profile of this agent and whether the clinical benefits of abatacept found in the current clinical trials will be sustained.
Authors' conclusions
Implications for practice.
Abatacept has been shown to be an efficacious biologic for the treatment of rheumatoid arthritis, although this systematic review does raise concerns about the risk of bias of included studies. Although long‐term studies are lacking, current data suggest that it is well tolerated. On‐going post‐marketing surveillance is required to determine the incidence of adverse events and the sustainability of treatment response.
Implications for research.
Published trials should follow the CONSORT statement (Consort 2001) for reporting controlled trials. Uncertanties regarding the methodology employed in the trials in this review may have been avoided had the CONSORT checklist been adhered to. Efficacy of different biologics against placebo in treating moderate to severe rheumatoid arthritis has been established and it would now be useful to investigate the efficacy of different biologics in head‐to‐head trials. Further trials are needed to determine the long‐term safety profile of abatacept and whether the level of efficacy found in the trials included in this review is sustained over time.
What's new
| Date | Event | Description |
|---|---|---|
| 10 August 2009 | Amended | CMSG ID: C103‐R |
History
Protocol first published: Issue 3, 2008 Review first published: Issue 4, 2009
| Date | Event | Description |
|---|---|---|
| 19 February 2009 | Amended | Converted to new review format. |
Acknowledgements
Thanks to Louise Falzon, Trials Search Co‐ordinator of the Cochrane Musculoskeletal Group, and Nancy Santesso for providing comments on the search strategy. This systematic review was conducted as part of a course on systematic review methodology, taught by Dean Fergusson and David Moher, in the Masters of Epidemiology and Community Medicine program at the University of Ottawa.
Appendices
Appendix 1. Original search strategy
| MEDLINE |
| The following search strategy was the original strategy developed for MEDLINE for this review: 1. exp arthritis, rheumatoid/ or *arthritis, juvenile rheumatoid/ or *caplan's syndrome/ or *felty's syndrome/ or *rheumatoid nodule/ or *sjogren's syndrome/ or *spondylitis, ankylosing/ or *still's disease, adult‐onset/ 2. (felty$ adj2 syndrome).tw. 3. (caplan$ adj2 syndrome).tw. 4. rheumatoid nodule.tw. 5. (sjogren$ adj2 syndrome).tw. 6. (sicca adj2 syndrome).tw. 7. still$ disease.tw. 8. (spondylitis adj2 ankylosing).tw. 9. (arthritis adj2 rheumat$).tw. 10. 10 or/1‐9 11. abatacept.tw. 12. orencia.tw. 13. anti‐interleukin$.tw. 14. anti‐tumor necrosis factor$.tw. 15. anti‐tumour necrosis factor$.tw. 16. anti‐tnf.tw. 17. exp Tumor Necrosis Factor 18. exp Immunosuppressive Agents 19. exp Immunoconjugates 20. exp Antirheumatic Agents 21. exp Antigens, Differentiation 22. or/11‐21 23. 10 and 22 24. clinical trial.pt. 25. randomized.ab. 26. placebo.ab. 27. dt.fs. 28. clinical trials/ 29. randomly.ab. 30.trial.ti. 31.groups.ab. 32. or/24‐29 33. animals/ 34. humans/ 35. 33 and 34 36. 33 not 35 37. 32 not 36 |
Appendix 2. MEDLINE search strategy
| Database: Ovid MEDLINE(R) 1950 to March Week 2 2007 |
| 1 exp arthritis, rheumatoid/ (82649) 2 (arthritis adj2 rheumat$).tw. (51835) 3 (felty$ adj2 syndrome).tw. (604) 4 (caplan$ adj2 syndrome).tw. (100) 5 rheumatoid nodule.tw. (179) 6 (sjogren$ adj2 syndrome).tw. (7824) 7 still$ disease.tw. (1027) 8 (spondylitis adj2 ankylosing).tw. (5833) 9 or/1‐8 (94331) 10 exp Immunoconjugates/tu (1645) 11 exp Antigens, Differentiation/tu (524) 12 abatacept.tw. (48) 13 abatacept.rn. (1547) 14 orencia.tw. (5) 15 ctla4Ig.tw. (342) 16 CTLA‐4IG.tw. (78) 17 CTLA4‐Ig.tw. (256) 18 or/10‐17 (3696) 19 clinical trial.pt. (433538) 20 randomized.ab. (146315) 21 placebo.ab. (97069) 22 dt.fs. (1154061) 23 clinical trials/ (130516) 24 randomly.ab. (107387) 25 trial.ti. (65869) 26 groups.ab. (763970) 27 or/19‐26 (2143950) 28 animals/ (4012600) 29 humans/ (9623654) 30 28 and 29 (964920) 31 28 not 30 (3047680) 32 27 not 30 (2026221) 33 9 and 18 and 32 (89) |
Appendix 3. CENTRAL search strategy
| CENTRAL, DARE, HTA |
| #1 MeSH descriptor Arthritis, Rheumatoid explode all trees in MeSH products #2 felty near/2 syndrome in All Fields in all products #3 caplan near/2 syndrome in All Fields in all products #4 rheumatoid nodule in All Fields in all products #5 sjogren* near/2 syndrome in All Fields in all products #6 still* next disease in All Fields in all products #7 arthritis near/2 rheumat* in All Fields in all products #8 spondylitis near/2 ankylosing #9 (#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8) #10 abatacept #11 orencia #12 ctla4Ig #13 CTLA‐4IG #14 CTLA4‐Ig #15 (#10 OR #11 OR #12 OR #13 OR #14) #16 (#9 AND #15) |
Appendix 4. EMBASE search strategy
| EMBASE 1980 to 2007 Week 12 |
| 1 exp arthritis, rheumatoid/ (55465) 2 (arthritis adj2 rheumat$).tw. (41958) 3 (felty$ adj2 syndrome).tw. (375) 4 (caplan$ adj2 syndrome).tw. (34) 5 rheumatoid nodule.tw. (146) 6 (sjogren$ adj2 syndrome).tw. (6148) 7 still$ disease.tw. (805) 8 (spondylitis adj2 ankylosing).tw. (4720) 9 or/1‐8 (70216) 10 abatacept.tw. (67) 11 exp abatacept/ (340) 12 orencia.tw. (38) 13 ctla4Ig.tw. (316) 14 CTLA‐4IG.tw. (69) 15 CTLA4‐Ig.tw. (243) 16 CTLA‐4‐Ig.tw. (85) 17 or/10‐16 (986) 18 random$.ti,ab. (330681) 19 factorial$.ti,ab. (6740) 20 (crossover$ or cross over$ or cross‐over$).ti,ab. (35382) 21 placebo$.ti,ab. (97190) 22 (doubl$ adj blind$).ti,ab. (76436) 23 (singl$ adj blind$).ti,ab. (6561) 24 assign$.ti,ab. (92926) 25 allocat$.ti,ab. (29201) 26 volunteer$.ti,ab. (88947) 27 crossover procedure.sh. (18389) 28 double blind procedure.sh. (63195) 29 randomized controlled trial.sh. (115217) 30 single blind procedure.sh. (6449) 31 or/18‐30 (564943) 32 exp animal/ or nonhuman/ or exp animal experiment/ (3097329) 33 exp human/ (5675468) 34 32 and 33 (441828) 35 32 not 34 (2655501) 36 31 or 35 (3148641) 37 9 and 17 and 36 (34) |
Appendix 5. ACP Journal Club search strategy
| ACP Journal Club |
| Keyword search: CTLA4‐Ig OR abatacept OR orencia OR ctla4Ig OR CTLA‐4IG |
Appendix 6. Biosis Previews search strategy
| Biosis Previews |
| #8 #7 AND #1 #7 #6 OR #5 OR #4 OR#3 OR #2 #6 TI=CTLA‐4Ig #5 TI=CTLA4‐Ig #4 TI=CTLA4Ig #3 TI=Orencia #2 TI=abatacept #1 DS=rheumatoid arthritis DocType=All document types; LitType=All literature types; Language=All languages; Taxa Notes=All Taxa Notes; Database=BIOSIS Previews; Timespan=1990‐2007 |
Data and analyses
Comparison 1. Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ACR 20% improvement | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 3 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.7 [0.93, 3.12] |
| 1.2 6 months | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.59, 2.02] |
| 1.3 12 months | 3 | 993 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.55, 2.07] |
| 2 ACR 50% improvement | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 3 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.52, 11.96] |
| 2.2 6 months | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [2.00, 3.07] |
| 2.3 12 months | 3 | 993 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.73, 2.82] |
| 3 ACR 70% improvement | 6 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 3 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.25, 100.20] |
| 3.2 6 months | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.53 [2.41, 5.16] |
| 3.3 12 months | 3 | 993 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.02 [2.62, 6.18] |
| 4 Improvement in physical function (HAQ: >0.22 or >0.3 increase from baseline, 0‐3 scale) | 4 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 6 months (>0.22) | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [1.29, 2.33] |
| 4.2 6 months (>0.3) | 2 | 655 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.73 [1.41, 2.13] |
| 4.3 12 months (0.22) | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.27, 2.52] |
| 4.4 12 months (>0.3) | 1 | 638 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.62 [1.35, 1.95] |
| 5 Achievement of low disease activity state (DAS 28<3.2, scale 0‐10) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 6 months | 2 | 1027 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.36 [2.28, 4.96] |
| 5.2 12 months | 1 | 638 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.33 [2.84, 6.59] |
| 6 Achievement of remission (DAS 28 <2.6, scale 0‐10) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 6 months | 2 | 1027 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [0.57, 11.03] |
| 6.2 12 months | 1 | 638 | Risk Ratio (M‐H, Random, 95% CI) | 12.74 [4.76, 34.15] |
| 7 DAS‐28 ESR | 1 | 266 | Mean Difference (IV, Fixed, 95% CI) | ‐0.95 [‐1.20, ‐0.70] |
| 8 SF‐36 physical component score ‐ % same | 2 | 623 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.56, 0.78] |
| 8.1 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.61, 0.88] |
| 8.2 12 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.55 [0.41, 0.75] |
| 9 SF‐36 physical component score ‐ % better | 2 | 623 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.90 [1.52, 2.39] |
| 9.1 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.38, 2.70] |
| 9.2 12 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.88 [1.39, 2.54] |
| 10 SF‐36 physical component score ‐ % worse | 2 | 623 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.44, 1.14] |
| 10.1 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.41, 1.27] |
| 10.2 12 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.29, 1.63] |
| 11 SF‐36 mental component score ‐ % same | 2 | 623 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.86 [0.73, 1.03] |
| 11.1 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.71, 1.14] |
| 11.2 12 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.64, 1.05] |
| 12 SF‐36 mental component score ‐ % better | 2 | 623 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [1.15, 1.76] |
| 12.1 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.37 [1.03, 1.83] |
| 12.2 12 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.50 [1.08, 2.07] |
| 13 SF‐36 mental component score ‐ % worse | 2 | 623 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.64 [0.44, 0.94] |
| 13.1 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.72 [0.48, 1.09] |
| 13.2 12 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.15, 1.08] |
| 14 SF‐36 mental component score | 3 | 1293 | Mean Difference (IV, Fixed, 95% CI) | 2.72 [1.57, 3.87] |
| 15 SF‐36 physical component score | 3 | 1293 | Mean Difference (IV, Fixed, 95% CI) | 4.29 [3.22, 5.35] |
| 16 No. achieving population norm SF‐36 physical component score ‐ 6 months | 1 | 389 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.36 [1.34, 4.14] |
| 17 Patient reported pain (100 mm VAS) | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 17.1 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 17.2 12 months | 3 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 18 Patient global assessment | 3 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 18.1 6 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 18.2 12 months | 3 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 19 Physician global assessment | 3 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 19.1 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 19.2 12 months | 3 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 20 Physical function (HAQ‐DI & MHAQ) | 3 | Std. Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 20.1 6 months | 1 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 20.2 12 months | 3 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 21 Tender joint count | 2 | 1011 | Mean Difference (IV, Fixed, 95% CI) | ‐7.30 [‐8.79, ‐5.80] |
| 21.1 6 months | 1 | 232 | Mean Difference (IV, Fixed, 95% CI) | ‐7.36 [‐10.58, ‐4.14] |
| 21.2 12 months | 2 | 779 | Mean Difference (IV, Fixed, 95% CI) | ‐7.28 [‐8.97, ‐5.59] |
| 22 Swollen joint count | 2 | 1011 | Mean Difference (IV, Fixed, 95% CI) | ‐4.81 [‐5.79, ‐3.83] |
| 22.1 6 months | 1 | 232 | Mean Difference (IV, Fixed, 95% CI) | ‐4.93 [‐7.14, ‐2.72] |
| 22.2 12 months | 2 | 779 | Mean Difference (IV, Fixed, 95% CI) | ‐4.78 [‐5.87, ‐3.68] |
| 23 Radiographic progression (maximum erosion score 145) | 1 | 586 | Mean Difference (IV, Fixed, 95% CI) | ‐0.27 [‐0.42, ‐0.12] |
| 24 Withdrawals due to adverse events | 6 | 3105 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.30 [0.91, 1.85] |
| 24.1 6 months | 2 | 657 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.11 [0.42, 2.96] |
| 24.2 12 months | 4 | 2448 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.33 [0.90, 1.95] |
| 25 All withdrawals | 7 | 3169 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.60 [0.52, 0.70] |
| 25.1 3 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.12, 0.92] |
| 25.2 6 months | 2 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.65 [0.44, 0.96] |
| 25.3 12 months | 4 | 2448 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.61 [0.51, 0.72] |
| 26 Serious infections | 5 | 2871 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.56 [0.93, 2.61] |
| 26.1 6 months | 2 | 657 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.76 [0.25, 2.28] |
| 26.2 12 months | 3 | 2214 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.91 [1.07, 3.42] |
| 27 Upper respiratory infections | 5 | 2839 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.22 [0.84, 1.76] |
| 27.1 6 months | 1 | 391 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.75 [0.32, 1.77] |
| 27.2 12 months | 4 | 2448 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.36 [0.90, 2.04] |
| 28 Total adverse events | 5 | 2871 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [1.01, 1.08] |
| 28.1 6 months | 2 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.97, 1.15] |
| 28.2 12 months | 3 | 2214 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.04 [1.01, 1.08] |
| 29 Total serious adverse events | 6 | 3151 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.86, 1.29] |
| 29.1 6 months | 2 | 703 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.80 [0.49, 1.31] |
| 29.2 12 months | 4 | 2448 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.89, 1.39] |
| 30 Death | 6 | 3105 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.82 [0.26, 2.60] |
| 30.1 6 months | 2 | 657 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 5.02 [0.29, 88.42] |
| 30.2 12 months | 4 | 2448 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.58 [0.17, 2.04] |
| 31 Malignancies | 5 | 2710 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.00 [0.59, 1.71] |
| 31.1 6 months | 1 | 266 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.70 [0.04, 11.72] |
| 31.2 12 months | 4 | 2444 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.02 [0.59, 1.75] |
| 32 Cough | 3 | 1241 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.16 [0.80, 1.70] |
| 32.1 6 months | 1 | 234 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.56, 2.76] |
| 32.2 12 months | 3 | 1007 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.14 [0.74, 1.75] |
| 33 Nausea | 5 | 1696 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.89, 1.58] |
| 33.1 3 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.52, 11.96] |
| 33.2 6 months | 2 | 625 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.09 [0.65, 1.80] |
| 33.3 12 months | 3 | 1007 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.18 [0.82, 1.69] |
| 34 Headache | 6 | 3137 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.45 [1.20, 1.74] |
| 34.1 3 months | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.0 [1.06, 60.32] |
| 34.2 6 months | 2 | 625 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.42 [0.85, 2.38] |
| 34.3 12 months | 4 | 2448 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.41 [1.15, 1.72] |
| 35 Dizziness | 3 | 1164 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.52 [0.92, 2.53] |
| 35.1 6 months | 1 | 391 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.66 [0.25, 85.84] |
| 35.2 12 months | 2 | 773 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.44 [0.86, 2.41] |
| 36 Diarrhea | 4 | 1632 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.94, 1.85] |
| 36.1 6 months | 2 | 625 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.71, 2.49] |
| 36.2 12 months | 3 | 1007 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.31 [0.88, 1.96] |
| 37 Infusion reaction (within 24 hours after infusion) | 4 | 2750 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.30 [1.13, 1.50] |
| 37.1 6 months | 2 | 657 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.44, 1.64] |
| 37.2 12 months | 2 | 2093 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [1.16, 1.55] |
1.1. Analysis.
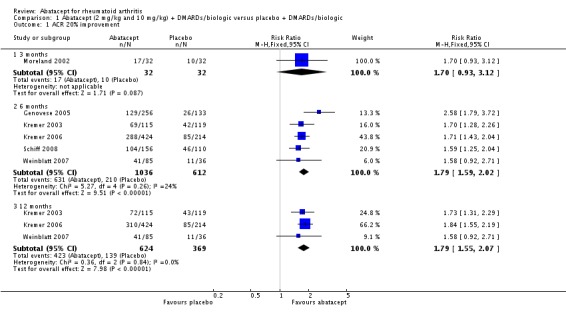
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 1 ACR 20% improvement.
1.2. Analysis.
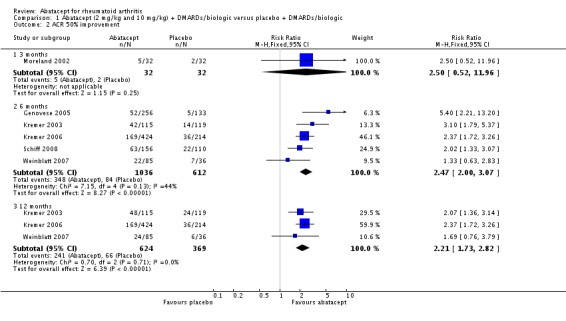
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 2 ACR 50% improvement.
1.3. Analysis.
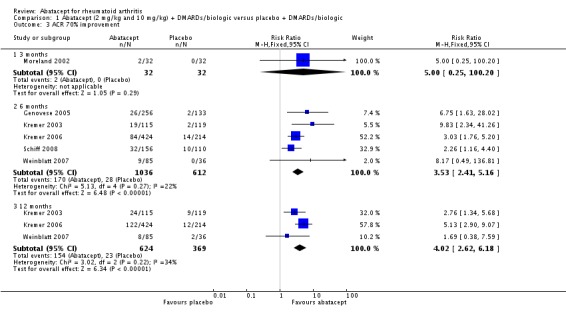
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 3 ACR 70% improvement.
1.4. Analysis.
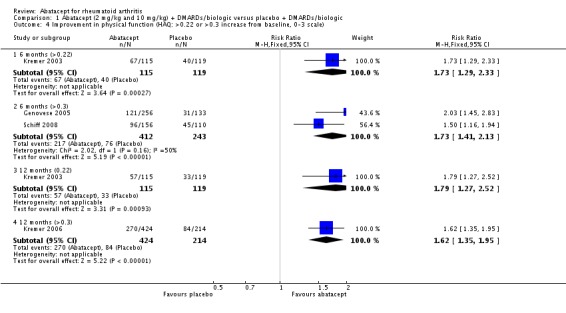
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 4 Improvement in physical function (HAQ: >0.22 or >0.3 increase from baseline, 0‐3 scale).
1.5. Analysis.
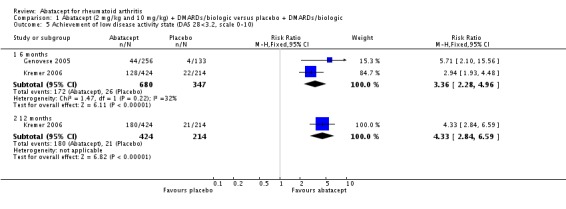
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 5 Achievement of low disease activity state (DAS 28<3.2, scale 0‐10).
1.6. Analysis.
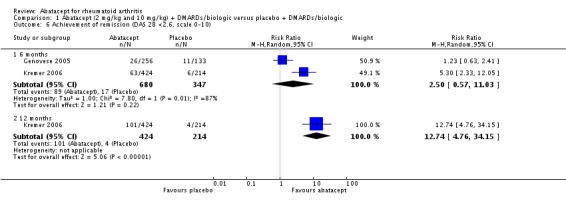
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 6 Achievement of remission (DAS 28 <2.6, scale 0‐10).
1.7. Analysis.
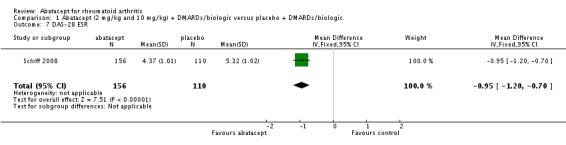
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 7 DAS‐28 ESR.
1.8. Analysis.
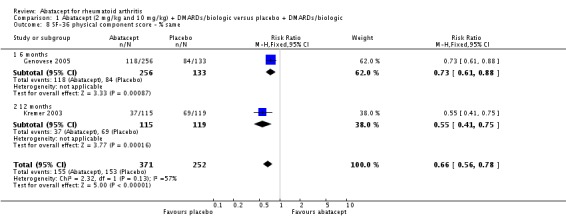
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 8 SF‐36 physical component score ‐ % same.
1.9. Analysis.
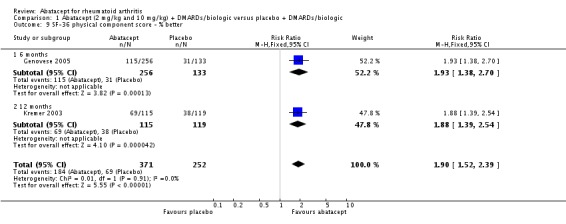
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 9 SF‐36 physical component score ‐ % better.
1.10. Analysis.
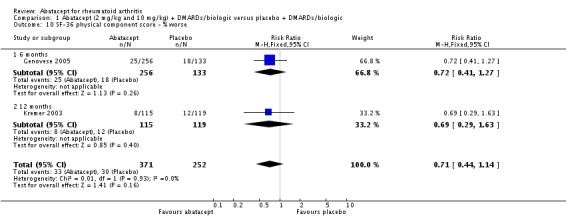
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 10 SF‐36 physical component score ‐ % worse.
1.11. Analysis.
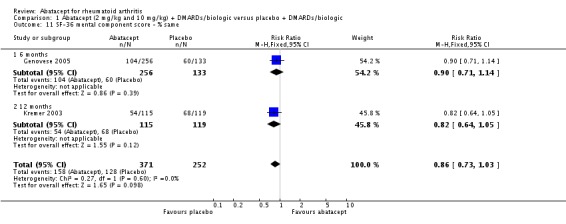
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 11 SF‐36 mental component score ‐ % same.
1.12. Analysis.
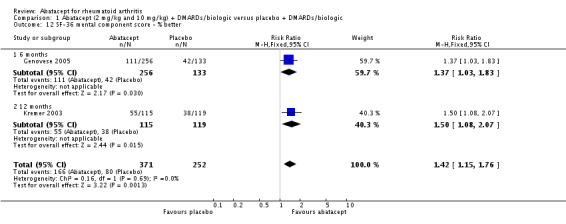
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 12 SF‐36 mental component score ‐ % better.
1.13. Analysis.
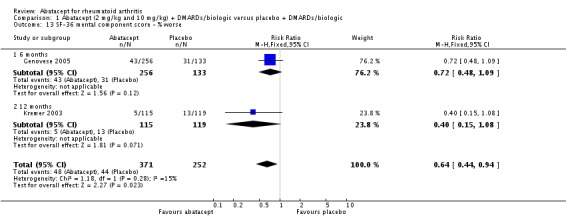
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 13 SF‐36 mental component score ‐ % worse.
1.14. Analysis.
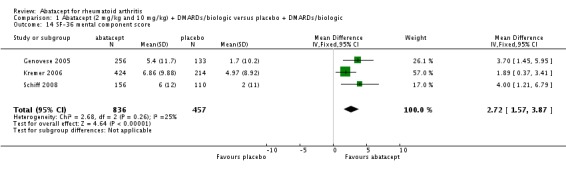
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 14 SF‐36 mental component score.
1.15. Analysis.
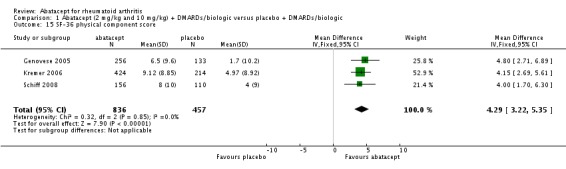
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 15 SF‐36 physical component score.
1.16. Analysis.
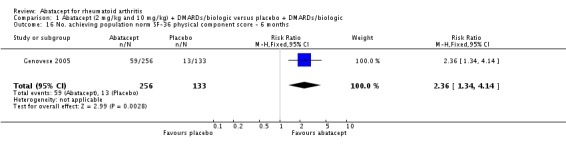
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 16 No. achieving population norm SF‐36 physical component score ‐ 6 months.
1.17. Analysis.
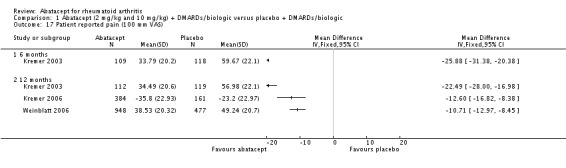
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 17 Patient reported pain (100 mm VAS).
1.18. Analysis.
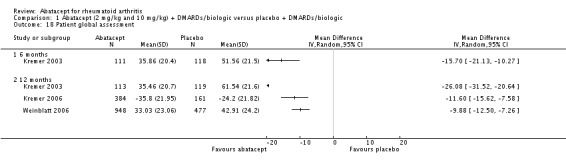
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 18 Patient global assessment.
1.19. Analysis.
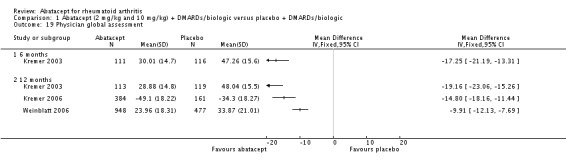
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 19 Physician global assessment.
1.20. Analysis.
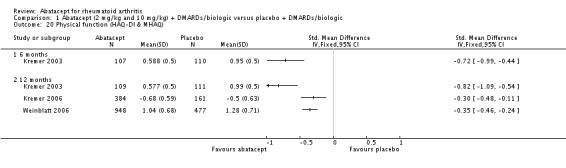
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 20 Physical function (HAQ‐DI & MHAQ).
1.21. Analysis.
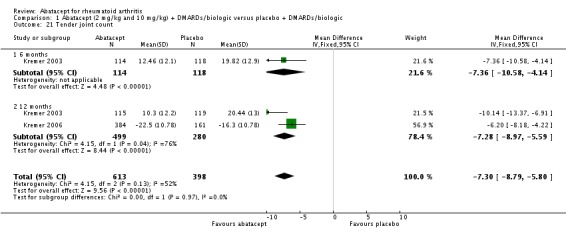
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 21 Tender joint count.
1.22. Analysis.
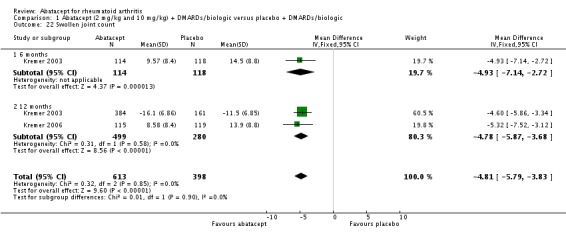
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 22 Swollen joint count.
1.23. Analysis.
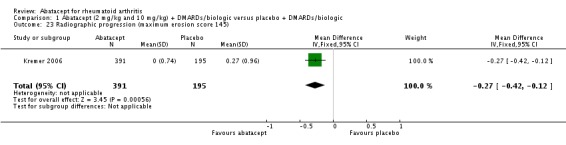
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 23 Radiographic progression (maximum erosion score 145).
1.24. Analysis.
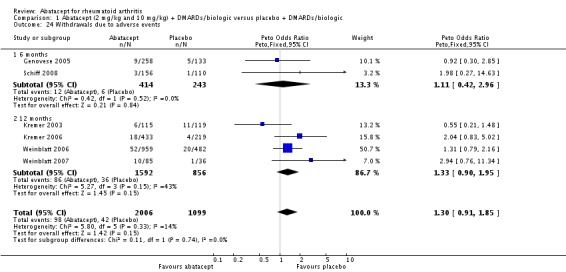
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 24 Withdrawals due to adverse events.
1.25. Analysis.
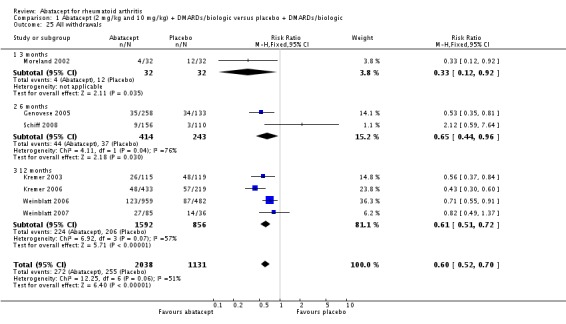
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 25 All withdrawals.
1.26. Analysis.
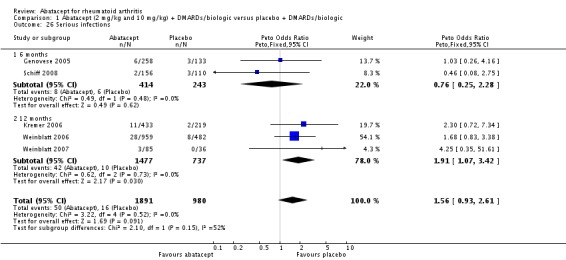
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 26 Serious infections.
1.27. Analysis.
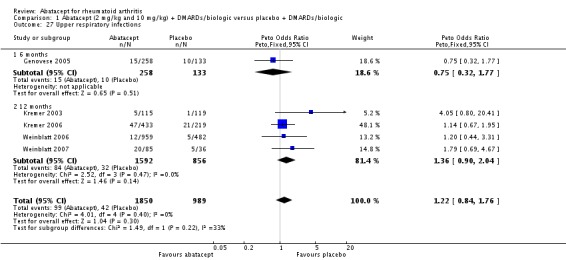
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 27 Upper respiratory infections.
1.28. Analysis.
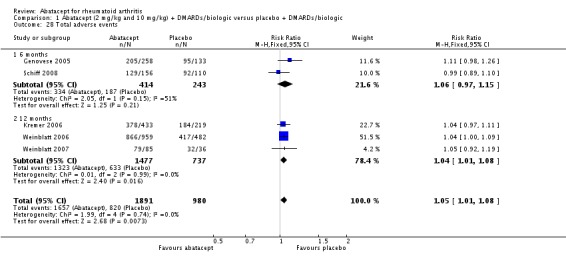
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 28 Total adverse events.
1.29. Analysis.
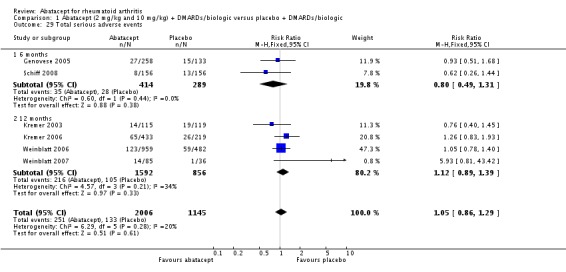
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 29 Total serious adverse events.
1.30. Analysis.
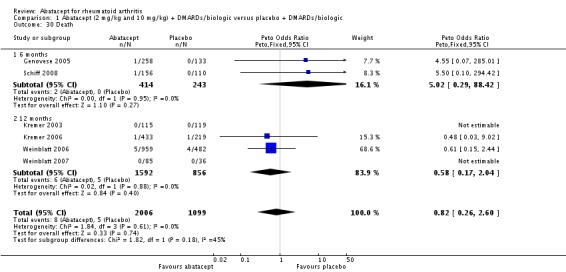
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 30 Death.
1.31. Analysis.
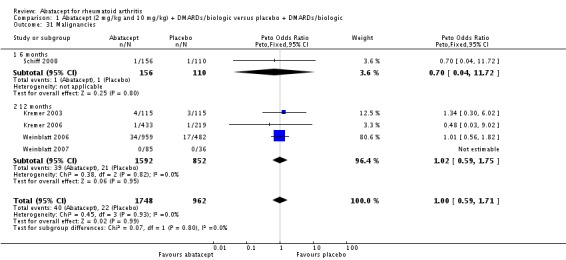
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 31 Malignancies.
1.32. Analysis.
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 32 Cough.
1.33. Analysis.
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 33 Nausea.
1.34. Analysis.
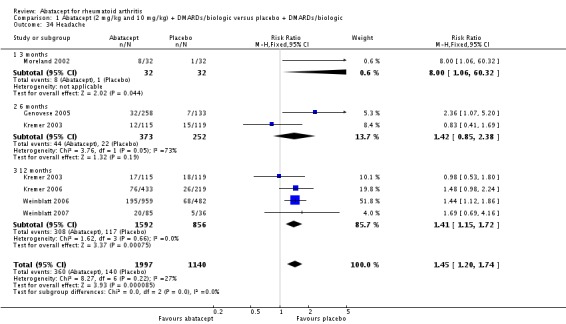
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 34 Headache.
1.35. Analysis.
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 35 Dizziness.
1.36. Analysis.
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 36 Diarrhea.
1.37. Analysis.
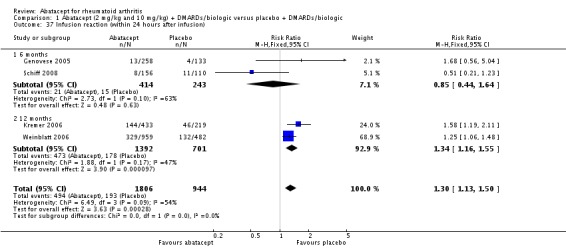
Comparison 1 Abatacept (2 mg/kg and 10 mg/kg) + DMARDs/biologic versus placebo + DMARDs/biologic, Outcome 37 Infusion reaction (within 24 hours after infusion).
Comparison 2. Abatacept (2 mg/kg) + etanercept versus placebo + etanercept.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ACR 20% improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 6 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.92, 2.71] |
| 1.2 12 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.92, 2.71] |
| 2 ACR 50% improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 6 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.63, 2.83] |
| 2.2 12 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.76, 3.79] |
| 3 ACR 70% improvement | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 6 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.17 [0.49, 136.81] |
| 3.2 12 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.38, 7.59] |
| 4 Physical function (mHAQ, 0‐3 scale) | 1 | 121 | Mean Difference (IV, Fixed, 95% CI) | ‐0.10 [‐0.27, 0.07] |
| 5 Patient assessment of pain (100 mm VAS) | 1 | 121 | Mean Difference (IV, Fixed, 95% CI) | ‐3.40 [‐14.86, 8.06] |
| 6 Withdrawals due to adverse events ‐ 12 months | 1 | 121 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.94 [0.76, 11.34] |
| 7 All withdrawals ‐ 12 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.49, 1.37] |
| 8 Serious infections ‐ 12 months | 1 | 121 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.25 [0.35, 51.61] |
| 9 Upper respiratory infections ‐ 12 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.69 [0.69, 4.16] |
| 10 Total adverse events ‐ 12 months | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.05 [0.92, 1.19] |
| 11 Total serious adverse events ‐ 12 months | 1 | 121 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.49 [1.08, 11.34] |
| 12 Death ‐ 12 months | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only |
2.1. Analysis.
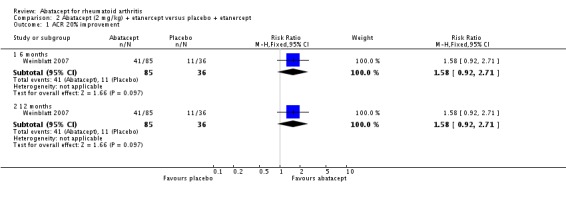
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 1 ACR 20% improvement.
2.2. Analysis.
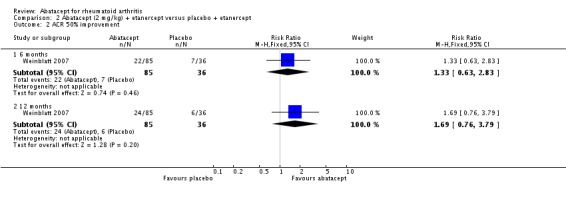
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 2 ACR 50% improvement.
2.3. Analysis.
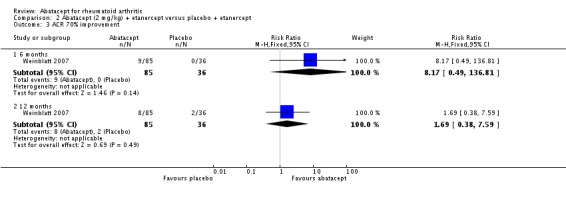
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 3 ACR 70% improvement.
2.4. Analysis.
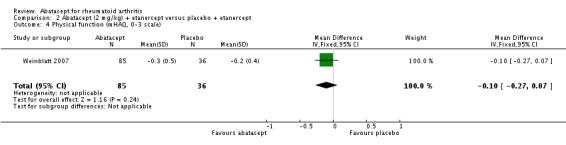
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 4 Physical function (mHAQ, 0‐3 scale).
2.5. Analysis.
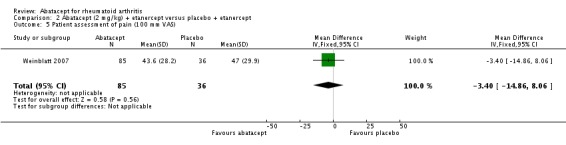
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 5 Patient assessment of pain (100 mm VAS).
2.6. Analysis.
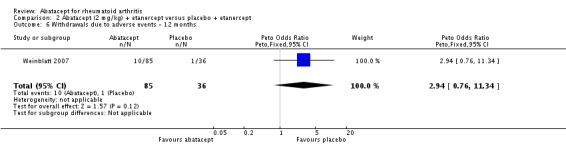
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 6 Withdrawals due to adverse events ‐ 12 months.
2.7. Analysis.
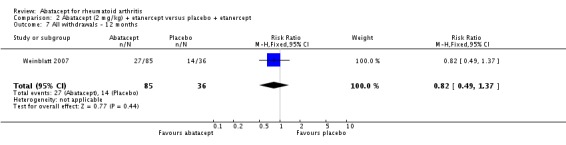
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 7 All withdrawals ‐ 12 months.
2.8. Analysis.
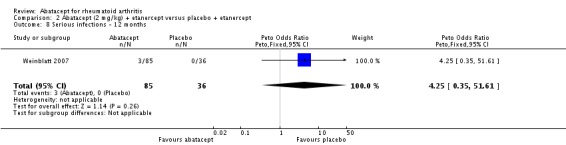
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 8 Serious infections ‐ 12 months.
2.9. Analysis.
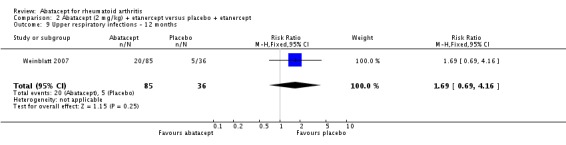
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 9 Upper respiratory infections ‐ 12 months.
2.10. Analysis.
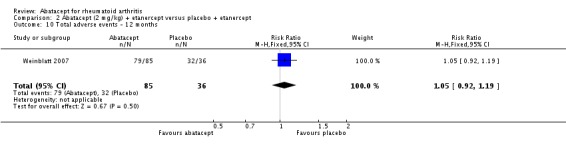
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 10 Total adverse events ‐ 12 months.
2.11. Analysis.
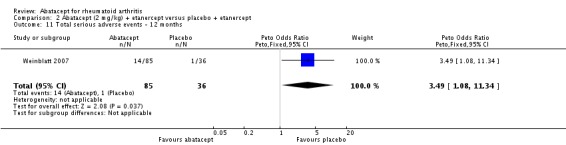
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 11 Total serious adverse events ‐ 12 months.
2.12. Analysis.
Comparison 2 Abatacept (2 mg/kg) + etanercept versus placebo + etanercept, Outcome 12 Death ‐ 12 months.
Comparison 3. Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawals due to adverse events ‐ 12 months | 2 | 288 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.68 [1.07, 6.72] |
| 2 All withdrawals ‐ 12 months | 2 | 706 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.00, 37.14] |
| 3 Serious infections ‐ 12 months | 2 | 288 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.20 [0.86, 11.97] |
| 4 Upper respiratory infections ‐ 12 months | 2 | 288 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.79 [0.75, 4.26] |
| 5 Total adverse events ‐ 12 months | 2 | 288 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.06 [0.98, 1.14] |
| 6 Total serious adverse events ‐ 12 months | 2 | 288 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.30 [1.15, 4.62] |
| 7 Death ‐ 12 months | 2 | 288 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
3.1. Analysis.
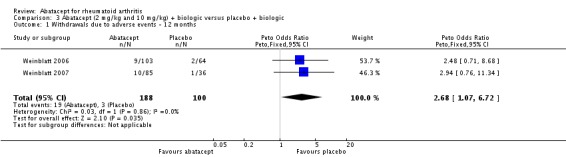
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 1 Withdrawals due to adverse events ‐ 12 months.
3.2. Analysis.
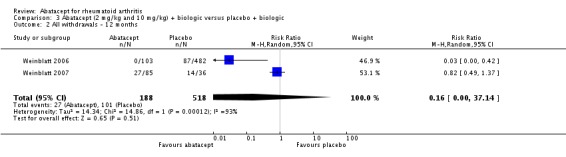
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 2 All withdrawals ‐ 12 months.
3.3. Analysis.
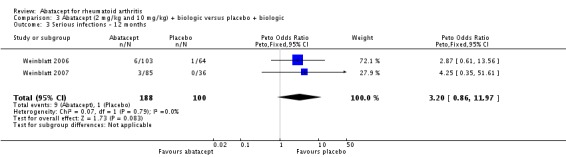
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 3 Serious infections ‐ 12 months.
3.4. Analysis.
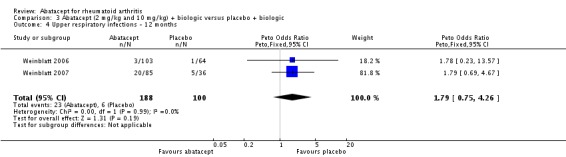
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 4 Upper respiratory infections ‐ 12 months.
3.5. Analysis.
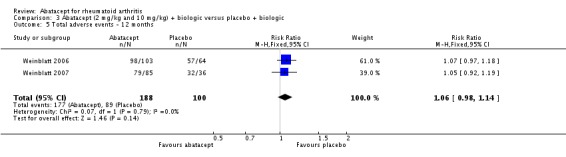
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 5 Total adverse events ‐ 12 months.
3.6. Analysis.
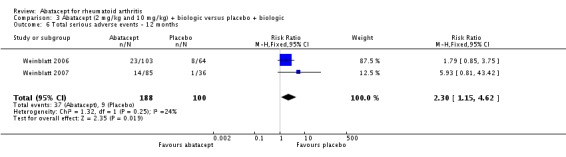
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 6 Total serious adverse events ‐ 12 months.
3.7. Analysis.
Comparison 3 Abatacept (2 mg/kg and 10 mg/kg) + biologic versus placebo + biologic, Outcome 7 Death ‐ 12 months.
Comparison 4. Abatacept versus placebo (by dosage).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ACR 20% improvement | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 6 months ‐ 2 mg/kg | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.58 [0.92, 2.71] |
| 1.2 6 months ‐ 10 mg/kg | 4 | 1527 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.81 [1.60, 2.04] |
| 1.3 6 months ‐ combined dosage | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.59, 2.02] |
| 2 ACR 50% improvement | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 6 months ‐ 2 mg/kg | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.63, 2.83] |
| 2.2 6 months ‐ 10 mg/kg | 4 | 1527 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.59 [2.07, 3.25] |
| 2.3 6 months ‐ combined dosage | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [2.00, 3.07] |
| 3 ACR 70% improvement | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 6 months ‐ 2 mg/kg | 1 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.17 [0.49, 136.81] |
| 3.2 6 months ‐ 10 mg/kg | 4 | 1527 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.43 [2.34, 5.04] |
| 3.3 6 months ‐ combined dosage | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.53 [2.41, 5.16] |
4.1. Analysis.
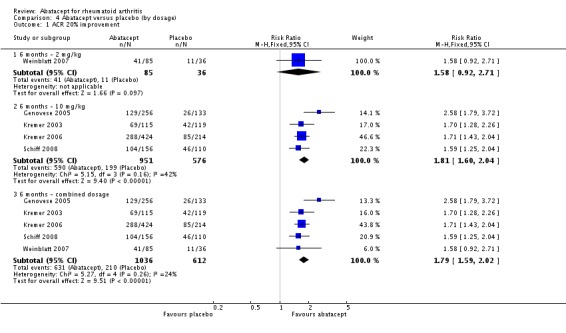
Comparison 4 Abatacept versus placebo (by dosage), Outcome 1 ACR 20% improvement.
4.2. Analysis.
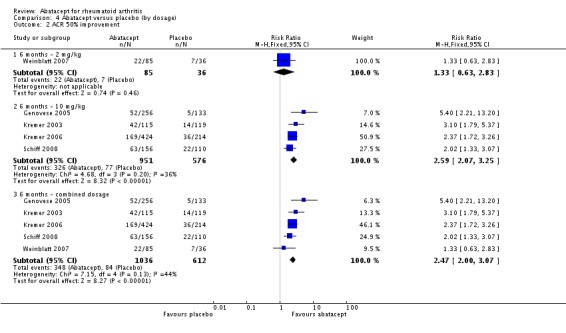
Comparison 4 Abatacept versus placebo (by dosage), Outcome 2 ACR 50% improvement.
4.3. Analysis.
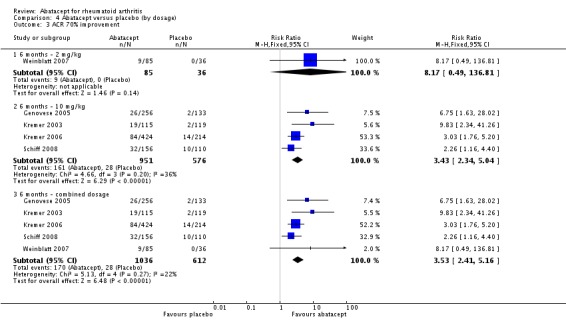
Comparison 4 Abatacept versus placebo (by dosage), Outcome 3 ACR 70% improvement.
Comparison 5. Abatacept versus placebo (by study eligibility criteria).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ACR 20% improvement | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 MTX failures | 3 | 1138 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.68 [1.48, 1.91] |
| 1.2 Biologic failures | 2 | 510 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.27 [1.67, 3.07] |
| 2 ACR 50% improvement | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 MTX failures | 3 | 1138 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.38 [1.89, 3.00] |
| 2.2 Biologic failures | 2 | 510 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.96 [1.67, 5.25] |
| 3 ACR 70% improvement | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 MTX failures | 3 | 1138 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.16 [2.12, 4.71] |
| 3.2 Biologic failures | 2 | 510 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.05 [1.98, 25.14] |
5.1. Analysis.
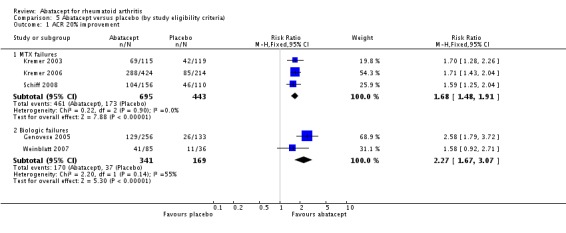
Comparison 5 Abatacept versus placebo (by study eligibility criteria), Outcome 1 ACR 20% improvement.
5.2. Analysis.
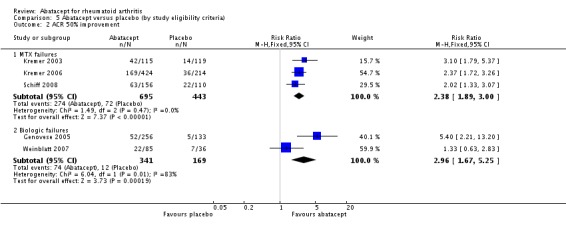
Comparison 5 Abatacept versus placebo (by study eligibility criteria), Outcome 2 ACR 50% improvement.
5.3. Analysis.
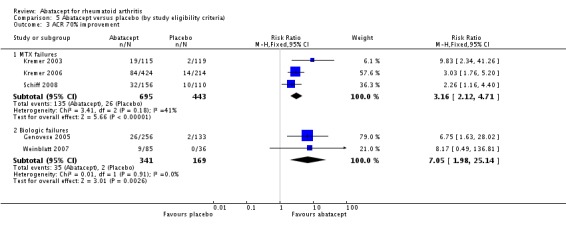
Comparison 5 Abatacept versus placebo (by study eligibility criteria), Outcome 3 ACR 70% improvement.
Comparison 6. Abatacept versus placebo (funnel plot).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ACR 20% improvement ‐ 6 months | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.59, 2.02] |
| 1.1 6 months ‐ for funnel plot | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.79 [1.59, 2.02] |
| 2 ACR 50% improvement ‐ 6 months | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [2.00, 3.07] |
| 2.1 6 months | 5 | 1648 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.47 [2.00, 3.07] |
| 3 ACR 50% ‐ 1 year | 3 | 993 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.73, 2.82] |
| 3.1 12 months | 3 | 993 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.21 [1.73, 2.82] |
6.1. Analysis.
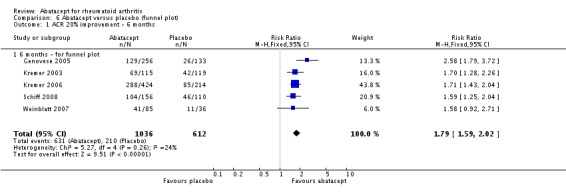
Comparison 6 Abatacept versus placebo (funnel plot), Outcome 1 ACR 20% improvement ‐ 6 months.
6.2. Analysis.
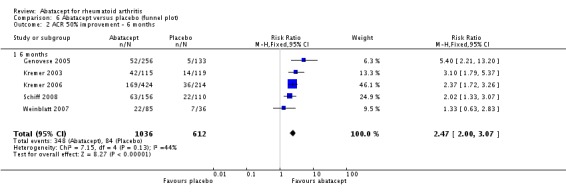
Comparison 6 Abatacept versus placebo (funnel plot), Outcome 2 ACR 50% improvement ‐ 6 months.
6.3. Analysis.
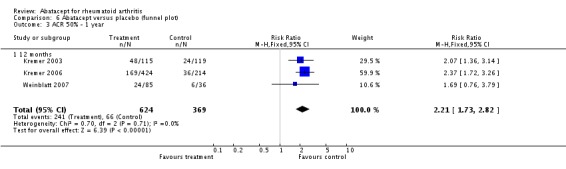
Comparison 6 Abatacept versus placebo (funnel plot), Outcome 3 ACR 50% ‐ 1 year.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Genovese 2005.
| Methods | Multicenter (89 sites). Randomized, double‐blind, phase III trial. 2:1 abatacept to placebo ratio of random assignment. 6‐month study. Stratification by former vs. current users of anti‐TNF therapy. | |
| Participants | Abatacept N = 258, age (mean, SD) 53.4 +/‐ 12.4, % female = 77.1, duration of RA in years (mean, SD) = 12.2 +/‐ 8.5
Placebo N = 133, age (mean, SD) 52.7 +/‐ 11.3, % female = 79.7, duration of RA in years (mean, SD) = 11.4 +/‐ 8.9 Eligibility: ACR criteria for RA, > 18 years old, had RA for at least 1 year and had an inadequate response to anti–TNF therapy with etanercept, infliximab, or both at the approved dose after at least 3 months of treatment. Two groups of patients were enrolled: those receiving anti–TNFtherapy at the time of screening (current users) and those who had previously received such therapy (former users). All users were required to stop taking etanercept or infliximab for at least 28 or 60 days, respectively, before undergoing randomization. |
|
| Interventions | Abatacept (10 mg per kg) + DMARD or placebo + DMARD, administered in a 30‐minute intravenous infusion on days 1, 15, and 29 and every 28 days thereafter, up to and including day 141 | |
| Outcomes | Two primary:
ACR 20 response and the proportion of patients with an improvement of at least 0.3 from baseline in the HAQ (exceeding MCID of 0.22) at 6 months Secondary: ACR 50 and ACR 70 at six months DAS28 Health‐related quality of life (SF‐36) Adverse events |
|
| Notes | ATTAIN ‐ Abatacept Trial in Treatment of Anti‐TNF Inadequate Responders Study was also reported in Westhovens 2006 Study supported by Bristol‐Myers Squibb and a grant (5 M01 RR000070) from the National Center for Research Resources, National Institutes of Health | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | "central randomization" |
| Allocation concealment? | Low risk | "central randomization" |
| Blinding? Patient assessed | Low risk | "The drug was prepared by pharmacists or other qualified personnel who had no interaction with the patients. Medication was administered intravenously in a blinded fashion by qualified personnel." |
| Blinding? Physician assessed | Low risk | "All clinical assessments of response were performed in a blinded fashion by the same trained assessors throughout the study." |
| Incomplete outcome data addressed? Efficacy outcomes | Low risk | 86% in treatment group and 74% in placebo group completed 6 months. Used imputation to account for missing data in the analysis. All withdrawals accounted for except that according to the flow diagram, 2 did not meet the eligibility criteria after randomization. "All efficacy analyses included all randomized patients who received at least one dose of study medication". Also, "Two patients in the abatacept group were excluded from the efficacy analysis because of a protocol violation" Judged a low risk of bias given the > 80% completion rate in the treatment group. |
| Incomplete outcome data addressed? Safety outcomes | Low risk | 86% in treatment group and 74% in placebo group completed 6 months. Used imputation to account for missing data in the analysis. All withdrawals accounted for except that according to the flow diagram, 2 did not meet the eligibility criteria after randomization. Judged a low risk of bias given the > 80% completion rate in the treatment group. |
| Free of selective reporting? | Low risk | "There were two primary end points: the proportion of patients with an ACR 20 response and the proportion of patients with an improvement of at least 0.3 from baseline in the Health Assessment Questionnaire (HAQ) disability index (exceeding the minimal clinically important change of 0.22) at 6 months. |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
Kremer 2003.
| Methods | Six‐month, double‐blind, randomized, placebo‐controlled trial. Phase II. Multicenter, multinational. 2:1 abatacept to placebo ratio of random assignment. | |
| Participants | Abatacept N = 220, age (mean, SD) 54.7 range 23 to 80 years, % female = 66, duration of RA in years (mean, SD) = 8.9 +/‐ 8.3
Placebo N = 119, age (mean, SD) 55.8 range 17 to 83 years, % female = 75.0, duration of RA in years (mean, SD) = 9.7 +/‐ 9.8
Patients with rheumatoid arthritis who had an inadequate response to methotrexate Inclusion criteria: ACR criteria for RA & functional class I, II, or III with active disease. Treated with MTX (10 to 30 mg weekly) for at least 6 months and to have received a stable dose for 28 days before enrolment. Concomitant medications: All patients continued to receive methotrexate. All other DMARDs were discontinued. Stable low‐dose corticosteroids and NSAIDs were permitted. Exclusion: nursing or pregnant |
|
| Interventions | Abatacept 2 mg/kg + MTX (N = 105); Abatacept 10 mg/kg + MTX (N = 115); Placebo + MTX (N = 119) Abatacept or placebo was infused intravenously over a 30‐minute period on days 1, 15, and 30 and monthly thereafter for a total of 6 months. Only 10 mg/kg arm reported for this review. | |
| Outcomes | Primary: ACR 20 response at 6months Secondary outcomes: ACR 50 & 70 Health‐related quality of life (SF‐36) Adverse events |
|
| Notes | Study was also reported in Kremer 2003 and Emery 2006. The study sponsor (Bristol Myers Squibb) was involved in the design of the study, collection of the data, and analysis of the data. The academic investigators had access to the data and were responsible for interpreting the data. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | "central randomization" |
| Allocation concealment? | Low risk | "central randomization" |
| Blinding? Patient assessed | Low risk | "Double blind". Additional info provided by BMS: subjects and clinical assessor(s) were blinded to treatment assignment |
| Blinding? Physician assessed | Low risk | "Double blind". "Assessments were performed by rheumatologists or trained professional staff members who were unaware of patients’ treatment assignments and were not involved in the infusion of CTLA4Ig or placebo." |
| Incomplete outcome data addressed? Efficacy outcomes | High risk | 86% of abatacept group and 66% of placebo completed 6 months of treatment. At 12 months (results reported in Kremer 2005), 78% of abatacept group and 60% of placebo completed the study. Missing data were imputed for analysis. "Patients who discontinued the study because of worsening disease were considered to have had no response; for those who discontinued the study for other reasons the values for the last efficacy observation were carried forward." All withdrawals accounted for. Efficacy outcomes reported for total number of randomized patients. Judged a high risk of bias due to > 20% drop‐out rate at 12 months in the treatment group. |
| Incomplete outcome data addressed? Safety outcomes | High risk | 86% of abatacept group and 66% of placebo completed 6 months of treatment. At 12 months (results reported in Kremer 2005), 78% of abatacept group and 60% of placebo completed the study. Missing data were imputed for analysis. "Patients who discontinued the study because of worsening disease were considered to have had no response; for those who discontinued the study for other reasons the values for the last efficacy observation were carried forward." All withdrawals accounted for. Safety outcomes reported for total number of randomized patients. Judged a high risk of bias due to > 20% drop‐out rate at 12 months in the treatment group. |
| Free of selective reporting? | Low risk | "The primary efficacy variable was the percentage of patients who had a 20 percent improvement according to ACR criteria (an ACR 20 response) at six months." |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
Kremer 2006.
| Methods | 1‐year, multicenter, multinational, randomized, double‐blind, placebo‐controlled study. Phase III. 2:1 abatacept to placebo ratio of random assignment. | |
| Participants | Abatacept N = 433, age (mean, SD) 51.5 +/‐ 12.9, % female = 77.8, duration of RA in years (mean, SD) = 8.5 +/‐ 7.3 Placebo N = 219, age (mean, SD) 50.4 +/‐ 12.4, % female = 81.7, duration of RA in years (mean, SD) = 8.9 +/‐ 7.1 Eligible patients were at least 18 years of age, had RA for at least 1 year, and met the ARA criteria for RA. 2:1 randomization ratio. Abatacept N = 258 and placebo N = 133. RA was persistent and active despite MTX. All patients must have been treated with MTX for 3 months or longer, with a stable dose for 28 days before enrolment. Washout of all DMARDs at least 28 days before randomization. Concomitant medications: NSAIDs, corticosteroids with dosages equal to 10 mg of prednisone or less per day, stabilized for 25 days before randomization. Active disease at randomization. Exclusion: positive tuberculin skin test result unless they had completed treatment for latent tuberculosis before enrolment |
|
| Interventions | Abatacept (10 mg/kg) (N = 258) + MTX or placebo + MTX (N = 133). Study medication given by 30‐minute intravenous infusion on days 1,15, and 29 and then every 28 days up to and including day 337. No premedication was required. All patients received MTX, 15 mg or more per week, although MTX at 10 mg per week was acceptable if the patient had a history of toxicity | |
| Outcomes | Three primary:
ACR 20 response at 6 months The proportion of patients in each group with clinically significant improvement (>= 0.3 unit) in the HAQ‐DI score at 1 year The radiographic progression of joint erosions (assessed by comparing changes from baseline in the Genant‐modified Sharp score) at 1 year Secondary: ACR 50 and ACR 70 at 6 months and all ACR responses at 1 year DAS28 HAQ‐DI Health related quality of life (SF‐36) Adverse events |
|
| Notes | AIM ‐ Abatacept in Inadequate Responders to Methotrexate study. This trial is also reported in Russell 2007. This trial was sponsored by Bristol‐Myers Squibb. The funding source helped design the study in consultation with the authors and provided statistical support for data analysis. Interpretation of the data was aided by the funding biostatisticians, with input from the authors. The funding source was not involved in the decision to submit the article for publication. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | "'central randomization" |
| Allocation concealment? | Low risk | "'central randomization'" |
| Blinding? Patient assessed | Low risk | "Double"; stated patient and "investigators were blinded to treatment group assignment throughout the 1‐year study." |
| Blinding? Physician assessed | Low risk | "Physicians blinded to treatment group assignment performed assessments at enrolment and at every visit before treatment administration" |
| Incomplete outcome data addressed? Efficacy outcomes | High risk | 89% of treatment group and 74% of placebo group completed the 1 year study. All withdrawals accounted for except for four patients that were randomized but not treated (Figure 1). "We performed all efficacy and safety analyses on a modified intention‐to‐treat population, defined as all randomly assigned patients who received at least 1 dose of study medication." However, "Nine abatacept‐treated patients and 5 placebo recipients from 1 site were excluded from all efficacy analyses before unblinding due to nonadherence but were included in all safety analyses." Judged a high risk of bias due to exclusion of patients from efficacy analysis. |
| Incomplete outcome data addressed? Safety outcomes | Low risk | 89% of treatment group and 74% of placebo group completed the 1 year study. All withdrawals accounted for except for 4 patients that were randomized but not treated. "We performed all efficacy and safety analyses on a modified intention‐to‐treat population, defined as all randomly assigned patients who received at least 1 dose of study medication." |
| Free of selective reporting? | Low risk | "Our 3 primary objectives were to evaluate the proportion of patients in each group with a 20% improvement in American College of Rheumatology (ACR) response criteria (ACR 20) at 6 months, the proportion of patients in each group with clinically significant improvement (0.3 unit) in the Health Assessment Questionnaire Disability Index (HAQ‐DI) score at 1 year, and the radiographic progression of joint erosions (assessed by comparing changes from baseline in the Genant‐modified Sharp score) at 1 year." |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
Moreland 2002.
| Methods | Multicenter, multi‐national, randomized, double‐blind, placebo‐controlled trial. Phase II. Trial duration = 85 days. | |
| Participants | Abatacept N = 214, age (mean, SD) 51.5 +/‐ 11.5, % female = 69, duration of RA in years (mean, SD) = 3.4+/‐2.1, 94% white, 3% black Placebo N = 32, age (mean, SD) 48.3 +/‐ 11.7, % female = 81, duration of RA in years (mean, SD) = 3.2 +/‐ 2.0, 94% white, 4% black Mean duration of RA: 3.4 yrs +/‐ 2 yrs (SD) Inclusion criteria: ACR criteria for RA and functional class I, II, or III. Age 18 to 65 years with a disease duration < 7 years. Treated unsuccessfully with at least 1 DMARD or etanercept. Patients had to agree to discontinue any DMARD or etanercept treatment from 28 days prior to the day 1 dose of the study medication through study day 85. Treatment with low‐dose corticosteroids or NSAIDs could be continued provided the prescribed dosage remained stable. Negative tuberculin skin test within the last 6 months or documentation of course of adequate chemoprophylaxis of tuberculosis. Exclusion: pregnancy, nursing | |
| Interventions | Patients were randomized to 1 of 7 treatment groups: abatacept at 0.5 mg/kg, 2 mg/kg, or 10 mg/kg; LEA29Y at 0.5 mg/kg, 2 mg/kg, or 10 mg/kg; or placebo. Study medication was administered on days 1, 15, 29, and 57. No concurrent DMARDs were allowed. Days 1 to 85 were considered to be the treatment period; follow up continued through day 169 For this review, abatacept 10 mg/kg (N = 32) and placebo (N = 32) were considered | |
| Outcomes | Primary: ACR 20% on day 85 Secondary: ACR 50, ACR 70, core set measures Adverse events. |
|
| Notes | Funding source not clearly stated. Employees of Bristol Myers Squibb are listed as co‐authors. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | "Each subject was assigned a unique, sequential subject number beginning with 001, 002, 003, etc., for identification throughout the study. The subject number was not to be used for any other participant at the site. Treated subjects who did not complete the study were not replaced. Randomization schedules were generated and kept by the Biostatistics and Data Management Department of BMS." (Additional info obtained from BMS) |
| Allocation concealment? | Low risk | "The Centralized Randomization Center (CRC) assigned each subject a unique, non‐sequential, randomization number corresponding with the treatment kit number. The Investigator Project Notebook contained instructions on the Centralized Enrolment and Randomization System. Randomization schedules were generated and kept by the Biostatistics and Data Management Department of BMS." (Additional info obtained from BMS). |
| Blinding? Patient assessed | Low risk | "Subjects and clinical assessor(s) were not aware of the treatment being administered. Study drug was required to be supplied to study personnel in a manner such that neither study personnel nor subjects were aware of whether they were receiving active drug or placebo." (Additional information provided by BMS). |
| Blinding? Physician assessed | Low risk | "Subjects and clinical assessor(s) were not aware of the treatment being administered. Study drug was required to be supplied to study personnel in a manner such that neither study personnel nor subjects were aware of whether they were receiving active drug or placebo." (Additional information provided by BMS). |
| Incomplete outcome data addressed? Efficacy outcomes | Low risk | 81% of total study participants completed the 85 days of the study. 22% of abatacept group (all doses) discontinued. Missing data were imputed using LOCF or non‐responders for ACR response. All withdrawals accounted for. "All efficacy analyses were carried out on the intent‐to‐treat population, defined as all randomized subjects who received at least 1 infusion of study medication." (2 people were dropped from the study after randomization). Judged a low risk of bias because the overall trial completion rate was > 80%. |
| Incomplete outcome data addressed? Safety outcomes | Low risk | 81% of total study participants completed the 85 days of the study. 22% of abatacept group (all doses) discontinued. All withdrawals accounted for. Adverse events described for the population of randomized subjects who received at least 1 infusion of study medication (2 people were dropped from the study after randomization). Judged a low risk of bias because the overall trial completion rate was > 80%. |
| Free of selective reporting? | Low risk | ACR 20 specified as primary outcome and is appropriate |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
Schiff 2008.
| Methods | ATTEST was a randomized, double‐blind, double‐dummy, placebo‐ and active (infliximab)‐controlled, 12‐month global trial | |
| Participants | Abatacept, n = 156, infliximab, n = 165, placebo, n = 110. Eligible patients met the American College of Rheumatology (ACR) criteria for RA, were at least 18 years of age, had RA for at least 1 year and had an inadequate response to MTX. Abatacept group: Mean age (SD) = 49 yrs (12.5); gender = 83.3% female; disease duration (yrs) (SD) = 7.9 (8.5) Placebo group: Mean age (SD) = 49.4 yrs (11.5); gender = 87.3% female; disease duration (yrs) (SD) = 8.4 (8.6) |
|
| Interventions | Adult patients with active RA and an inadequate response to MTX were randomised by centre in a 3:3:2 ratio to 6 months of abatacept (approximating 10 mg/kg, n = 156), infliximab (3 mg/kg, n = 165), or placebo (n = 110) treatment by intravenous (IV) infusion, on a background of MTX | |
| Outcomes | Primary outcome: DAS28 (ESR) Secondary: EULAR criteria were used to assess good responses ACR 20, 50 and 70 Physical function (HAQ‐DI) HRQoL(SF‐36) Adverse events |
|
| Notes | ATTEST: "Abatacept or infliximab vs placebo, a Trial for Tolerability, Efficacy and Safety in Treating rheumatoid arthritis’’ On day 198, placebo treated patients were reallocated to abatacept (with blinding maintained). Patients initially randomized to abatacept or infliximab continued their treatment. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Not stated in published article |
| Allocation concealment? | Unclear risk | Not stated in published article |
| Blinding? Patient assessed | Low risk | Additional info from BMS: "Subjects and clinical assessor(s) were blinded to treatment assignment". "In order to maintain the blind, study drug was administered intravenously (IV) on Days 1, 15, 29, 43, 57, 85, 113, 141, 169, 197, 225, 253, 281, 309, and 337 as described below. Because the dosing regimens for abatacept and infliximab were different, subjects received normal saline (NS) at some dosing visits to maintain the integrity of the blind." |
| Blinding? Physician assessed | Low risk | Additional info from BMS: "Subjects and clinical assessor(s) were blinded to treatment assignment". |
| Incomplete outcome data addressed? Efficacy outcomes | Low risk | 94.2% in abatacept group and 97.3% in placebo group completed treatment at 6 months. Missing data were imputed using LOCF or non‐responders for ACR response. All withdrawals accounted for. "All patients who received at least one dose of study medication were assessed for efficacy and safety". Judged a low risk of bias given the > 80% completion rate in the treatment group. |
| Incomplete outcome data addressed? Safety outcomes | Low risk | 94.2% in abatacept group and 97.3% in placebo group completed treatment at 6 months. All withdrawals accounted for. "All patients who received at least one dose of study medication were assessed for efficacy and safety". Judged a low risk of bias given the > 80% completion rate in the treatment group. |
| Free of selective reporting? | Low risk | "The primary endpoint was to evaluate a reduction in disease activity, measured by Disease Activity Score 28 (based on erythrocyte sedimentation rate levels; DAS28 (ESR)) with abatacept vs placebo at 6 months." |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
Weinblatt 2006.
| Methods | 1‐year, multicenter, randomized, double‐blind, placebo‐controlled trial. 2:1 abatacept to placebo ratio of random assignment | |
| Participants | Abatacept N = 959, Placebo N = 482; overall, age: 52.3 +/‐ 11.8 yrs (mean, SD); duration of RA: 9.7 +/‐ 8.9 years (mean, SD) Inclusion: ACR criteria for the diagnosis of RA and functional classes I, II, III, or IV. Patients had to have active disease despite receiving background DMARDs and/or biologic therapy, warranting additional therapy at the discretion of the investigator. Patient’s global assessment of disease activity, VAS >= 20 mm. Had to have been receiving >= 1 biologic and/or non biologic DMARD for at least 3 months, and at a stable dose for at least 28 days prior to day 1 of the trial. Exclusion: unstable or uncontrolled renal, endocrine, hepatic, hematologic, gastrointestinal, pulmonary, cardiac, or neurologic diseases, or any autoimmune disorder other than RA as the main diagnosis. Also, active or chronic recurrent bacterial infections unless treated and resolved, active herpes zoster infection within the previous 2 months, hepatitis B or hepatitis C virus infection, and active or latent TB. |
|
| Interventions | Abatacept (10 mg/kg) (N = 959) or placebo (N = 482) by intravenous infusion. Medication (abatacept or placebo) was administered via a 30‐minute intravenous infusion on days 1, 15, and 29, and every 4 weeks thereafter, for a total of 14 doses. All patients were required to continue to receive their background RA therapies (biologic DMARDs, non biologic DMARDs, or a combination of both) at study entry. Stable, low‐dose oral corticosteroids (10 mg/day or less) and/or stable doses of NSAIDs were allowed. | |
| Outcomes | Primary objective: the ASSURE trial was to evaluate the safety of abatacept in patients with active RA, including those with comorbid conditions. Outcomes measured included: occurrence of AEs, SAEs, discontinuations
due to AEs, death, clinically significant changes Secondary: Disability Index of the HAQ; patient’s global assessment of disease activity, patient’s global assessment of pain, and physician’s global assessment of disease activity using a 100 mm VAS Results were provided both overall and separately for non‐biologic background and biologic background therapy |
|
| Notes | ASSURE ‐ Abatacept Study of Safety in Use with Other RA Therapies Funding: supported by Bristol‐Myers Squibb | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Additional information from author: "'the randomization was done by computer allocation and was maintained by a central independent organization which was not affiliated with the sponsor or the investigators" |
| Allocation concealment? | Low risk | Additional information from author: "'the randomization was done by computer allocation and was maintained by a central independent organization which was not affiliated with the sponsor or the investigators" |
| Blinding? Patient assessed | Low risk | "Double"; additional information from author: "the patients, investigators, infusion nurses and pharmacy and study personnel were blinded" |
| Blinding? Physician assessed | Low risk | "Double"; additional information from author: "the patients, investigators, infusion nurses and pharmacy and study personnel were blinded" |
| Incomplete outcome data addressed? Efficacy outcomes | High risk | 87% of patients in the treatment group and 82% in the placebo group completed 12 months of treatment. 15 people were randomized but not treated with either abatacept or placebo (Figure 1). "Sixteen patients were excluded from the efficacy analysis due to compliance issues at one center." Primary outcome of this study is safety. Judged a high risk of bias due to exclusion of patients from efficacy analysis. |
| Incomplete outcome data addressed? Safety outcomes | Low risk | 87% of patients in the treatment group and 82% in the placebo group completed 12 months of treatment. 15 people were randomized but not treated with either abatacept or placebo (Figure 1). Judged a low risk of bias as safety analysis included all patients treated. |
| Free of selective reporting? | Low risk | The primary objective of the ASSURE trial was to evaluate the safety of abatacept in patients with active RA..." "All patients who received at least 1 dose of study medication were evaluated for the occurrence of adverse events (AEs), serious adverse events (SAEs), discontinuations due to AEs, death, clinically significant changes in vital signs, physical examination abnormalities, and clinical laboratory test abnormalities." |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
Weinblatt 2007.
| Methods | Multicenter, randomized, double‐blind, placebo‐controlled trial with an open‐label long‐term extension (LTE) phase, conducted at 40 centres in the US between 26 February 2001 and 13 October 2004. | |
| Participants | Abatacept (N = 85): mean (SD) age = 49.8+/‐23.7, % female = 78; duration of RA in years (mean, SD) = 13+/‐10; Placebo (N = 36): mean (SD) age = 54.3+/‐28.7, % female = 72; duration of RA in years (mean, SD) = 12.8+/‐8.6 Eligible patients were > 18 years of age and met the ACR criteria for RA and were in functional class I, II or III. Patients must have received etanercept 25 mg twice weekly for > 3 months and have > 8 swollen joints (66‐joint count) and > 10 tender joints (68‐joint count). The original protocol definition of required C reactive protein (CRP) concentration at entry was > 2 mg/dl; however, owing to the effect of etanercept on normalising CRP levels, there was a high initial rate of screen failures. Therefore, the protocol was modified so that CRP elevation was not required for entry and the CRP threshold of > 2 mg/dl was never executed. Important exclusion criteria included active or latent infection, recent opportunist infection, tuberculosis requiring treatment within the previous 3 years, history of cancer within the previous 5 years or history of drug or alcohol misuse. Pregnant and nursing women were excluded. |
|
| Interventions | Abatacept (2 mg/kg) and etanercept (25 mg twice weekly) (N = 85) or placebo and etanercept (25 mg twice weekly) (N = 36). 2:1 ratio for randomisation. Etanercept (25 mg, twice weekly) was continued in all patients for the duration of the study. Abatacept was administered intravenously on days 1, 15, and 30, and every 4 weeks thereafter. MTX and other DMARDs were stopped at least 28 days before randomization, with the exception of leflunomide, which was stopped > 60 days before randomization. Low‐dose corticosteroids (10 mg/day) or NSAIDs were allowed, provided the dose remained stable during the study. Analgesics were also permitted at all times except (12 hours before a joint evaluation. Addition of hydroxychloroquine, sulfasalazine, leflunomide or MTX was allowed after 6 months of double‐blind treatment, as considered appropriate by the investigator according to the patient’s condition. Patients completing double‐blind treatment were eligible to enter the LTE. All patients entering the LTE were switched to receive abatacept at a fixed dose approximating 10 mg/kg (according to weight range). During the LTE, patients were permitted to increase, decrease or discontinue corticosteroids (to a maximum maintenance dose of 10 mg prednisone equivalent daily), etanercept (to a maximum of 25 mg twice weekly) and NSAIDs according to their condition. | |
| Outcomes | Primary endpoint of the double‐blind phase was a modified ACR 20 (defined as >20% improvement in tender and swollen joints and > 20% improvement in 2 of the remaining four core measures (pain, physical function, modified Health Assessment Questionnaire, and patient and physician global assessments. CRP values were excluded from the definition) response rate at 6 months. The CRP values were excluded due to the normalizing effect of etanercept on CRP levels. The secondary endpoint of the double‐blind phase was the proportion of patients achieving a modified ACR 50 response at 6 months. The primary objective of the LTE was to assess the safety and tolerability of abatacept in combination with etanercept during long‐term administration in patients with active rheumatoid arthritis. Secondary efficacy measures included the modified ACR 50 and ACR 70 criteria at 6 months, standard ACR 20, ACR 50 and ACR 70 responses, and improvements in individual ACR criteria components |
|
| Notes | Study was funded and sponsored by Bristol‐Myers Squibb which was involved in the design of the study, the collection and analysis of the data, the writing of the report and the decision to submit the report. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | "central randomization" |
| Allocation concealment? | Low risk | "central randomization" |
| Blinding? Patient assessed | Low risk | "Double"; "the patients, investigators, infusion nurses and pharmacy and study personnel were blinded" (additional information from author) |
| Blinding? Physician assessed | Low risk | "Double"; "the patients, investigators, infusion nurses and pharmacy and study personnel were blinded" (additional information from author) |
| Incomplete outcome data addressed? Efficacy outcomes | High risk | 68% in abatacept group and 61% in placebo group completed 12 months. Missing data was imputed appropriately. All withdrawals accounted for. "One additional patient was randomised but did not receive the study drug and was not included in any analyses." Judged a high risk of bias since drop‐out rate is > 80%. |
| Incomplete outcome data addressed? Safety outcomes | High risk | 68% in abatacept group and 61% in placebo group completed 12 months. "One additional patient was randomised but did not receive the study drug and was not included in any analyses. Judged a high risk of bias since drop‐out rate is > 80%. |
| Free of selective reporting? | Low risk | "The original protocol definition of required C reactive protein (CRP) concentration at entry was > 2 mg/dl; however, owing to the effect of etanercept on normalising CRP levels, there was a high initial rate of screen failures. Therefore, the protocol was modified so that CRP elevation was not required for entry and the CRP threshold of > 2 mg/dl was never executed. The primary end point (ACR 20) was also modified early in the study to accommodate this finding." "Owing to the effect of etanercept on normalising CRP levels in this population, the primary and secondary end points were based on modified ACR 20 criteria, defined as >20% improvement in tender and swollen joints and >20% improvement in two of the remaining four core measures (pain, physical function, modified Health Assessment Questionnaire, and patient and physician global assessments). CRP values were excluded from the definition. Secondary efficacy measures included the modified ACR 50 and ACR 70 criteria at 6 months, standard ACR 20, ACR 50 and ACR 70 responses, and improvements in individual ACR criteria components." Judged a low risk of bias as the modification to the primary outcome occurred early in the study and was due to an unexpected finding that the drug being given in combination with abatacept had the effect of normalising CRP levels. |
| Free of other bias? | High risk | Funded by drug company. There is evidence that industry‐sponsored trials may overestimate the treatment effect (Bhandari 2004) |
ACR = American College of Rheumatology AE = adverse event BMS = Bristol‐Myers Squibb CRP = C reactive protein DMARD = disease‐modifying anti‐rheumatic drugs HAQ‐DI = Health Assessment Questionnaire Disability Index IV = intravenous LOCF = last observation carried forward MTX = methotrexate RA = rheumatoid arthritis SAE = serious adverse event SD = standard deviation VAS = visual analogue scale vs = versus yrs = years
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alten 2006 | German summary of Kremer 2003 trial |
| FDA 2005 | Review |
| Genant 2008 | Long term extension of Kremer 2006 |
| Genovese 2004 | ACR meeting abstract. Full text of trial was published in 2005. |
| Genovese 2005a | ACR meeting abstract. RCT data provided in Genovese 2005; the rest is long‐term extension data |
| Genovese 2008b | Long‐term extension of Genovese 2005 |
| Haggerty 2007 | Long‐term extension of Kremer 2006 |
| Hassett 2007 | Based on only 20 patients from the ATTAIN RCT |
| Kremer 2008 | Long‐term extension of Kremer 2006 trial |
| Kruger 2005 | German summary of Kremer 2003 trial |
| Li 2008 | Not an outcome of interest |
| NHS 2004 | Not a RCT; a review of recent trials |
| Taylor 2006 | Not a RCT; a review |
| Teng 2005 | Not a RCT; a review |
| Weisman 2006 | Outcome not of interest to systematic review |
| Wells 2008 | Not a RCT |
ACR = American College of Rheumatology RCT = randomized controlled trial
Differences between protocol and review
We undertook a subgroup analysis of adverse events in patients on a background therapy of biologics. This analysis was not prespecified in the protocol but arose out of a concern from one of the included studies which analysed their results by background therapy (non‐biologic versus biologic).
Contributions of authors
LM drafted the protocol, JAS provided comments
Review of abstracts ‐ LM, JAS
Data abstraction ‐ LM, JAS
Assessment of study quality ‐ LM, JAS
Review draft ‐ LM
Draft revision ‐ LM, JAS
Sources of support
Internal sources
University of Ottawa, Canada.
Minneapolis VA Medical Center, USA.
External sources
No sources of support supplied
Declarations of interest
None known. This systematic review did not receive specific funding.
Edited (no change to conclusions)
References
References to studies included in this review
Genovese 2005 {published data only}
- Cole J, Li T, Lin P, MacLean R, Wallenstein GV. Treatment impact on estimated medical expenditure and job loss likelihood in rheumatoid arthritis: re‐examining quality of life outcomes from a randomized placebo‐controlled clinical trial with abatacept. Rheumatology 2008;47(7):1044‐50. [DOI] [PubMed] [Google Scholar]
- Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor alpha inhibition. New England Journal of Medicine 2005;353(11):1114‐23 [erratum appears in New England Journal of Medicine 2005;353(21):2311]. [DOI] [PubMed] [Google Scholar]
- Westhovens R, Cole JC, Li T, Martin M, Maclean R, Lin P, et al. Improved health‐related quality of life for rheumatoid arthritis patients treated with abatacept who have inadequate response to anti‐TNF therapy in a double‐blind, placebo‐controlled, multicentre randomized clinical trial. Rheumatology 2006;45(10):1238‐46. [DOI] [PubMed] [Google Scholar]
Kremer 2003 {published and unpublished data}
- Emery P, Kosinski M, Li T, Martin M, Williams GR, Becker JC, et al. Treatment of rheumatoid arthritis patients with abatacept and methotrexate significantly improved health‐related quality of life. Journal of Rheumatology 2006;33(4):681‐9. [PubMed] [Google Scholar]
- Kremer JM, Dougados M, Emery P, Durez P, Sibilia J, Shergy W, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve‐month results of a phase IIb, double‐blind, randomized, placebo‐controlled trial. Arthritis & Rheumatism 2005;52(8):2263‐71 [erratum appears in Arthritis & Rheumatism 2005;52(10):3321]. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Westhovens R, Leon M, Giorgio E, Alten R, Steinfeld S, et al. Treatment of rheumatoid arthritis by selective inhibition of T‐cell activation with fusion protein CTLA4Ig. New England Journal of Medicine 2003;349(20):1907‐15. [DOI] [PubMed] [Google Scholar]
Kremer 2006 {published and unpublished data}
- Kremer J, Genant H, Moreland L, Russel A, Emery P, Abud‐Mendoza C, et al. Effects of abatacept in patients with methotrexate‐resistant active rheumatoid arthritis. Annals of Internal Medicine 2006;144:865‐76. [DOI] [PubMed] [Google Scholar]
- Russell AS, Wallenstein GV, Li T, Martin MC, Maclean R, Blaisdell B, et al. Abatacept improves both the physical and mental health of patients with rheumatoid arthritis who have inadequate response to methotrexate treatment. Annals of the Rheumatic Diseases 2007;66:189‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moreland 2002 {published and unpublished data}
- Moreland LW, Alten R, BF, Appelboom T, Leon M, Emery P, et al. Costimulatory blockade in patients with rheumatoid arthritis: a pilot, dose‐finding, double‐blind, placebo‐controlled clinical trial evaluating CTLA‐4Ig and LEA29Y eighty‐five days after the first infusion. Arthritis & Rheumatism 2002;46(6):1470‐9. [DOI] [PubMed] [Google Scholar]
Schiff 2008 {published and unpublished data}
- Schiff M, Keiserman M, Codding C, Songcharoen S, Berman A, Nayiager S, Saldate C, et al. Efficacy and safety of abatacept or infliximab vs placebo in ATTEST: a phase III, multi‐centre, randomised, double‐blind, placebo‐controlled study in patients with rheumatoid arthritis and an inadequate response to methotrexate. Annals of the Rheumatic Diseases 2008;67(8):1096‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weinblatt 2006 {published and unpublished data}
- Weinblatt M, Combe B, Covucci A, Aranda R, Becker JC, Keystone E. Safety of the selective costimulation modulator abatacept in rheumatoid arthritis patients receiving background biologic and nonbiologic disease‐modifying antirheumatic drugs: a one‐year randomized, placebo‐controlled study. Arthritis & Rheumatism 2006;54(9):2807‐16. [DOI] [PubMed] [Google Scholar]
Weinblatt 2007 {published and unpublished data}
- Weinblatt M, Schiff M, Goldman A, Luggen M, Li T, Chen D, et al. Selective costimulation modulation using abatacept in patients with active RA while receiving etanercept: a randomised clinical trial. Annals of the Rheumatic Diseases 2007;66:228‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Alten 2006 {published data only}
- Alten R. Long‐term therapy with the selective costimulation modulator CTLA4lg in rheumatoid arthritis [German]. Zeitschrift fur Rheumatologie 2006;65(3):252‐3. [DOI] [PubMed] [Google Scholar]
FDA 2005 {published data only}
- FDA Briefing document: ORENCIA (abatacept) BLA 125118/0. FDA website (http://www.fda.gov/OHRMS/DOCKETS/ac/05/briefing/2005‐4170B1_02_01‐FDA‐Abatacept.pdf.) (accessed 22 February 2007).
Genant 2008 {published data only}
- Genant HK, Peterfy CG, Westhovens R, Becker JC, Aranda R, Vratsanos G, et al. Abatacept inhibits progression of structural damage in rheumatoid arthritis: results from the long‐term extension of the AIM trial. Annals of the Rheumatic Diseases 2008;67(8):1084‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Genovese 2004 {published data only}
- Genovese M, Luggen M, et al. Efficacy and safety of abatacept (CTLA41g), a selective co‐stimulation modulator, in rheumatoid arthritis patients not responding adequately to anti‐TNF‐alpha therapy: results of the phase III ATTAIN (Abatacept trial in treatment of Anti‐TNF INadequate responders). Arthritis & Rheumatism 2004;50(12):4103. [Google Scholar]
Genovese 2005a {published data only}
- Genovese M, Luggen M, et al. Sustained improvements through 18 months with abatacept in rheumatoid arthritis patients with an inadequate response to anti‐TNF therapy. Arthritis & Rheumatism 2005;52(12):4064. [Google Scholar]
Genovese 2008b {published data only}
- Genovese M, Schiff M, et al. Efficacy and safety of the selective co‐stimulation modulator abatacept following 2 years of treatment in patients with rheumatoid arthritis and an inadequate response to anti‐tumour necrosis factor therapy. Annals of the Rheumatic Diseases 2008;67(4):547‐54. [DOI] [PubMed] [Google Scholar]
Haggerty 2007 {published data only}
- Haggerty HG, Abbott MA, et al. Evaluation of immunogenicity of the T cell costimulation modulator abatacept in patients treated for rheumatoid arthritis. Journal of Rheumatology 2007;34(12):2365‐73. [PubMed] [Google Scholar]
Hassett 2007 {published data only}
- Hassett A, Maclean J, et al. An observational evaluation of psychological well‐being in rheumatoid arthritis in the attain trial for ABATACEPT. Annals of the Rheumatic Diseases 2007;66 (Suppl.2):278. [Google Scholar]
Kremer 2008 {published data only}
- Kremer JM, Genant HK, et al. Results of a two‐year follow up study of patients with rheumatoid arthritis who received a combination of abatacept and methotrexate. Arthritis & Rheumatism 2008;58(4):953‐63. [DOI] [PubMed] [Google Scholar]
Kruger 2005 {published data only}
- Kruger K. Selective inhibition of T‐cell activation with fusion protein CTLA4lg as a new treatment option for rheumatoid arthritis [German]. Zeitschrift fur Rheumatologie 2005;64(1):40‐1. [DOI] [PubMed] [Google Scholar]
Li 2008 {published data only}
- Li T, Gignac M. Decreased external home help use with improved clinical status in rheumatoid arthritis: an exploratory analysis of the abatacept in inadequate responders to methotrexate (AIM) trial. Clinical Therapeutics 2008;30(4):734‐48. [DOI] [PubMed] [Google Scholar]
NHS 2004 {published data only}
- Abatacept (CTLA4Ig) for rheumatoid arthritis unresponsive to current therapies: new and emerging technology briefing. National Horizon Scanning Centre, University of Birmingham (http://www.pcpoh.bham.ac.uk/publichealth/horizon/PDF_files/2004reports/Abatacept.pdf) (accessed 2 March 2007) 2004.
Taylor 2006 {published data only}
- Taylor PC. Is abatacept an effective treatment for patients with RA who do not respond to other anti‐TNF treatments?. Nature Clinical Practice Rheumatology 2006;2(3):128‐9. [DOI] [PubMed] [Google Scholar]
Teng 2005 {published data only}
- Teng GG, Turkiewicz AM, Moreland LW. Abatacept: a costimulatory inhibitor for treatment of rheumatoid arthritis. Expert Opinion on Biological Therapy 2005;5(9):1245‐54. [DOI] [PubMed] [Google Scholar]
Weisman 2006 {published data only}
- Weisman MH, Durez P, Hallegua D, Aranda R, Becker JC, Nuamah I, et al. Reduction of inflammatory biomarker response by abatacept in treatment of rheumatoid arthritis. Journal of Rheumatology 2006;33(11):2162‐6. [PubMed] [Google Scholar]
Wells 2008 {published data only}
- Wells G, Li T, et al. Responsiveness of patient reported outcomes including fatigue, sleep quality, activity limitation, and quality of life following treatment with abatacept for rheumatoid arthritis. Annals of the Rheumatic Diseases 2008;67(2):260‐5. [DOI] [PubMed] [Google Scholar]
Additional references
ACR 2007
- ACR Hotline. http://www.rheumatology.org/publications/hotline/0506newdrugs.asp (accessed 3 February 2007).
ARC 2005
- ARC Epidemiology Unit. http://www.arc.org.uk/about_arth/bigpic.htm#3 ‐ARC Epidemiology Unit (accessed 3 February 2007).
Arnett 1988
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association revised criteria for the classification of rheumatoid arthritis. Arthritis & Rheumatism 1988;31:315‐24. [DOI] [PubMed] [Google Scholar]
Badley 2003
- Badley E, DesMeules M, eds. Arthritis in Canada. An ongoing challenge. Ottawa: Health Canada, 2003. [Google Scholar]
Bhandari 2004
- Bhandari M, Busse JW, Jackowski D, Montori VM, Schunemann H, Sprague S, et al. Association between industry funding and statistically significant pro‐industry findings in medical and surgical randomized trials. Canadian Medical Association Journal 2004;170(4):477‐80. [PMC free article] [PubMed] [Google Scholar]
Blumenauer 2002
- Blumenauer B, Judd M, Wells G, Burls A, Cranney A, Hochberg M, et al. Infliximab for the treatment of rheumatoid arthritis. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD003785] [DOI] [PMC free article] [PubMed] [Google Scholar]
Blumenauer 2003
- Blumenauer B, Judd M, Cranney A, Burls A, Coyle D, Hochberg M, et al. Etanercept for the treatment of rheumatoid arthritis. Cochrane Database of Systematic Reviews 2003, Issue 3. [DOI: 10.1002/14651858.CD004525.] [DOI] [PubMed] [Google Scholar]
Boers 2006
- Boers M. Abatacept in rheumatoid arthritis: a new branch on the “biologics” tree (editorial). Annals of the Rheumatic Diseases 2006;144:933‐5. [DOI] [PubMed] [Google Scholar]
Buch 2008
- Buch MH, Vital EM, Emery P. Review: abatacept in the treatment of rheumatoid arthritis. Arthritis Research & Therapy 2008;10(Suppl 1):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cates 2004 [Computer program]
- Cates C. Visual Rx 2.0 NNT Calculator. Dr Chris Cates EBM Website www.nntonline.net, 2004.
Consort 2001
- Moher D, Schulz K, Altman DG, the CONSORT Group. Revised recommendations for improving the quality of reports of parallel group randomized trials. Lancet 2001;357:1191‐4. [PubMed] [Google Scholar]
DAS 2009
- DAS‐Score.nl. Home of the DAS. Disease Activity Score in rheumatoid arthritis. http://www.das‐score.nl/www.das‐score.nl/ (accessed 24 July 2009).
EMEA 2007
- Orencia Scientific Discussion document ‐ European Medicines Agency, European Public Assessment Report. http://www.emea.europa.eu/humandocs/PDFs/EPAR/orencia/H‐701‐en6.pdf 2007.
Farrar 2001
- Farrar J, Young Jr. JP, LaMoreauxb L, Werthb JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11‐point numerical pain rating scale. Pain 2001;94:149‐58. [DOI] [PubMed] [Google Scholar]
FDA 2007
- FDA Orencia Label. FDA Center for Drug Evaluation and Research http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#apphist 13 March 2007 (accessed 1 April 2009).
Felson 1995
- Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith D, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis & Rheumatism 1995;38:727‐35. [DOI] [PubMed] [Google Scholar]
Helmick 2008
- Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh Kent C, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis and Rheumatism 2008;58(1):15‐25. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins J, Thompson S, Deeks J, Altman D. Measuring inconsistency in meta‐analysis. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Massarotti 2008
- Massarotti E. Clinical and patient‐reported outcomes in clinical trials of abatacept in the treatment of rheumatoid arthritis. Clinical Therapeutics 2008;30(3):429‐42. [DOI] [PubMed] [Google Scholar]
Navarro‐Sarabia 2005
- Navarro‐Sarabia F, Ariza‐Ariza R, Hernandez‐Cruz B, Villanueva I. Adalimumab for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005113.pub2.] [DOI] [PubMed] [Google Scholar]
Orencia 2007
- Orencia website. http://www.orencia.com/orencia/channels/content.jsp?BV_UseBVCookie=Yes&channelName=Left_Navigation/200@How_Orencia_different (accessed 1 February 2007).
Osiri 2002
- Osiri M, Shea B, Robinson V, Suarez‐Almazor M, Strand V, Tugwell P, et al. Leflunomide for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD002047] [DOI] [PMC free article] [PubMed] [Google Scholar]
PMPRB 2004
- Report on New Patented Drugs ‐ Humira. Patented Medicines Prices Review Board (http://www.pmprb‐cepmb.gc.ca/english/view.asp?x=478&pf=1) (accessed 24 July 2009) 2004.
RevMan 2008 [Computer program]
- The Nordic Cochrane Centre. The Cochrane Collaboration.. Review Manager (RevMan). Version 5.0. Copenhagen: The Nordic Cochrane Centre. The Cochrane Collaboration., 2008.
Ruiz 2009
- Ruiz Garcia V, Burls A, Cabello López JCL, Fry‐Smith AFS, Gálvez Muñoz JG, Jobanputra P, et al. Certolizumab pegol (CDP870) for rheumatoid arthritis in adults (Protocol). Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD007649] [DOI] [PubMed] [Google Scholar]
Salliot 2009
- Salliot C, Dougados M, Gossec L. Risk of serious infections during rituximab, abatacept and anakinra treatments for rheumatoid arthritis: meta‐analyses of randomised placebo‐controlled trials. Annals of the Rheumatic Diseases 2009;68:25‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schultz 1995
- Schultz K, Chalmers I, Hayes R, Altman D. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schumacher 1993
- Schumacher HR Jr, Klippel JH, Koopman WJ. Chapter 10. Rheumatoid Arthritis. Primer on the rheumatic diseases. 10th Edition. Arthritis Foundation, 1993. [Google Scholar]
Sibilia 2007
- Sibilia J, Westhovens R. Safety of T‐cell co‐stimulation modulation with abatacept in patients with rheumatoid arthritis. Clinical Experimental Rheumatology 2007;25 (Suppl. 46):S46‐S56. [PubMed] [Google Scholar]
Suarez‐Almazor 1998
- Suarez‐Almazor ME, Belseck E, Shea BJ, Tugwell P, Wells G. Methotrexate for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 1998, Issue 2. [DOI: 10.1002/14651858.CD000957.] [Google Scholar]
Suarez‐Almazor 1998b
- Suarez‐Almazor ME, Belseck E, Shea BJ, Tugwell P, Wells G. Sulfasalazine for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 1998, Issue 2. [DOI: 10.1002/14651858.CD000958] [Google Scholar]
Suarez‐Almazor 2000
- Suarez‐Almazor ME, Belseck E, Shea BJ, Homik JJEH, Wells G, Tugwell P. Antimalarials for treating rheumatoid arthritis. Cochrane Database of Systematic Reviews 2000, Issue 4. [DOI: 10.1002/14651858.CD000959] [DOI] [PubMed] [Google Scholar]
Tubach 2007
- Tubach F, Ravaud P, Beaton D, Boers M, Bombarier C, Felson DT, et al. Minimal clinically important improvement and patient acceptable symptom state for subjective outcome measures in rheumatic disorders. Journal of Rheumatology 2007;34:1188‐93. [PMC free article] [PubMed] [Google Scholar]
Van der Heijde 1993
- Heijde DM, Hof M, Riel PL, Putte LB. Development of a disease activity score based on judgment in clinical practice by rheumatologists. Journal of Rheumatology 1993;20:579‐81. [PubMed] [Google Scholar]
Van Gestel 1996
- Gestel AM, Prevoo ML, Hof MA, Rijswijk MH, Putte LB, Riel PL. Development and validation of the European League Against Rheumatism response criteria for rheumatoid arthritis. Comparison with the preliminary American College of Rheumatology and the World Health Organisation/International League Against Rheumatism criteria. Arthritis & Rheumatism 1996;39:34‐40. [DOI] [PubMed] [Google Scholar]
Woolf 2004
- Woolf A, Åkesson K, Compston J, et al. European action towards better musculoskeletal health ‐ a public health strategy to reduce the burden of musculoskeletal conditions; the Bone and Joint Decade, 2004. The Bone & Joint Decade, Department of Orthopedics, University Hospital, SE‐221 85 LUND, Sweden, 2004. [Google Scholar]
Yazici 2008
- Yazici Y, Adler NM, Yazici H. Most tumour necrosis factor inhibitor trials in rheumatology are undeservedly called 'efficacy and safety' trials: a survey of power considerations. Rheumatology 2008;47(7):1054‐7. [DOI] [PubMed] [Google Scholar]