

Electron cryo-microscopy (cryo-EM), is a rapidly emerging method for the determination of macromolecular complexes with an expanding presence in both the scientific literature and public data archives. Indeed, its role in moving biochemistry into a new era was recognized by the 2017 Nobel Prize in Chemistry to Jacques Dubochet, Joachim Frank, and Richard Henderson (nobelprize.org/prizes/chemistry/2017). EM Data Bank (EMDB) now holds more than 6500 cryo-EM density maps, and Protein Data Bank (PDB) holds more than 2500 structure coordinates, contributed by the growing cryo-EM community (Patwardhan and Lawson, 2016).
A typical cryo-EM study begins by imaging many thousands of individual particles of a large molecular machine, such as a virus, ribosome, enzyme complex, or membrane receptor, embedded in a thin film of vitreous ice. The structural features of particles are generally invisible to the naked eye, but through complex series of computational processing steps that correct, sort, align and average individual images, the assembly’s overall 3-dimensional shape is revealed. Recognizable features such as helices and sheets--and in optimal cases, individual amino acid side-chains and nucleic acid bases--can then guide assembly of an atomic coordinate model, carried out either de novo or based on previous structures of its molecular components or related assemblies. The atomic model can then in turn be analyzed to reveal important molecular features.
How can cryo-EM scientists have confidence that they have produced the best possible, most correct structure given their image data? In addition, what key indicators can the larger scientific community use to evaluate cryo-EM structure quality for independent analyses? Several tests have been developed to ensure that a reconstruction is on the right track (Chen et al., 2013; Henderson et al., 2011), and one metric, Fourier Shell Correlation (FSC), is now commonly used to estimate a density map’s resolution (Henderson et al., 2012; Patwardhan et al., 2012; van Heel and Schatz, 2017). However, the field has yet to agree on standards for evaluation of map resolvability and reliability as well as quality of fit of map-derived models. This state of affairs is not unusual for a relatively young experimental method, as was the case for macromolecular X-ray crystallography some years ago (Read et al., 2011). However, given the rapidly increasing impact of cryo-EM, it is becoming more and more urgent to develop criteria that can be used to evaluate the quality of a given structure and to rank its relative quality among other experimentally-derived structures.
Following in the spirit of previous community-based activities (Ludtke et al., 2012; Marabini et al., 2015; Zhu et al., 2004), the EMDataBank Project (Lawson et al., 2016) recently sponsored two challenges in order to highlight the need for map and model validation standards, and to expedite development of quantitative tools for cryo-EM structure comparison and assessment with community participation (challenges.emdatabank.org). Additional goals were to develop benchmark datasets, encourage development of best practices, and to compare and contrast different approaches.
The 2016 Cryo-EM Map and Model Challenges were overseen by expert committees charged with developing reconstruction and modelling challenge tasks suitable for all levels of expertise, promoting worldwide participation, evaluating results, and producing final reports. Benchmarks of varying size and complexity were selected based on current state-of-the-art detectors and processing methods, consisting of molecular machines recently elucidated in the 2–5 Å resolution range (Fig. 1). The map challenge data consisted of raw micrograph images archived in the Electron Microscopy Public Image Archive (EMPIAR; Iudin et al., 2016). Model challenge data consisted of density maps archived in EMDB.
Figure 1.

October 2017 Joint Challenges Workshop participants.
The scientific community responded enthusiastically with more than 90 scientists worldwide participating as data contributors, challengers, assessors and committee members. The challenges provided software developers the opportunity to tune and demonstrate their packages, and provided novice users a chance to learn reconstruction and modelling methodology using established benchmarks. A total of 66 maps and 142 models were contributed, each with supporting details about the workflow from benchmark to final result ([dataset] Lawson et al., 2017; [dataset] Lawson et al., 2018). Following internal review, all of the submitted maps and models were released, blinded to software and submitter identity, for assessment by volunteer expert teams.
In order to share and fully explore the results and analyses of both challenges with the community, a workshop was held October 6–8, 2017 at Stanford University/SLAC National Accelerator Laboratory, with one full day devoted to each challenge plus a final half-day for wrap-up discussion. More than 60 participants travelled to present and discuss their findings face-to-face, providing a unique opportunity for two somewhat separate communities (reconstruction and molecular modelling) to come together to review the challenge results, to address the need for robust validation procedures for both density maps and models, and to make recommendations for future challenge events (Figure 1). The following sections summarize some key results and recommendations gleaned from the workshop discussions and also introduce the articles contributed to this special issue.
The Map Challenge
The 27 Map Challenge participant groups represented a broad spectrum of expertise, from novices to software developers. In the first set of articles of this issue, five participating developer groups describe their methods for creating maps from the benchmark raw images (see in this issue Bell et al., 2018; Donati et al., 2018; Heymann, 2018b; Sorzano et al., 2018; Stagg and Mendez, 2018b).
Overall, the majority of the maps submitted were qualitatively similar to maps reported in original benchmark studies, and all eleven reconstruction packages represented in the Challenge were able to produce maps of equivalent quality. For just one of the seven benchmark targets (Figure 2), challengers were able to improve upon the original published result.
Figure 2.
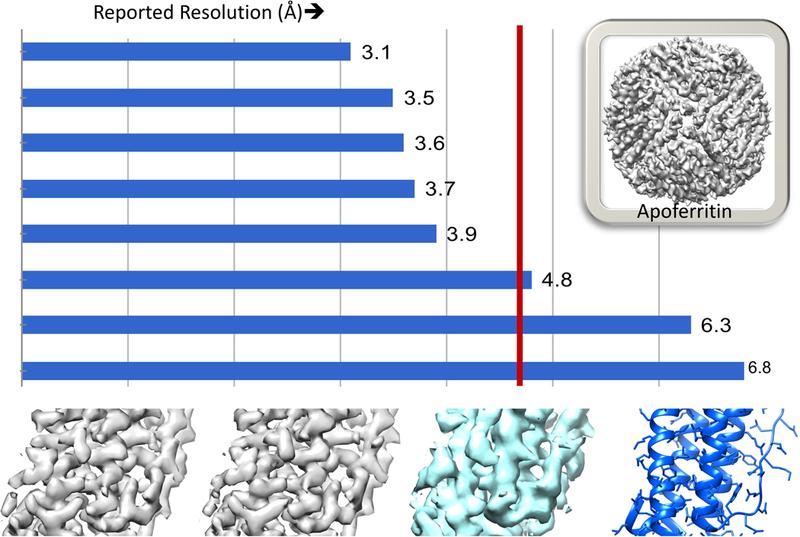
Map Challenge results for the Apoferritin benchmark. Top: Distribution of reported resolutions. Red vertical line indicates the resolution of the original study by Russo and Passmore (Russo and Passmore, 2014). Bottom, left to right: Representative sections of the 3.1 Å and 3.5 Å resolution maps, original study’s reconstruction (EMD-2788), and reference model (PDB 4v1w).
At the workshop it was noted that map quality could vary considerably for the same software package/benchmark target in the hands of different users. Workshop participants noted that there are still pitfalls to mastering use of reconstruction packages and voiced a need for bullet-proof reconstruction workflows, especially in light of the rapidly growing community of new practitioners.
Map Challenge Assessors compared the participants’ reported resolution, determined in most cases using the FSC criterion that has become the standard (if imperfect) metric reported in literature and data archives (Scheres and Chen, 2012). Map quality was judged by visual inspection and additional computational measures of similarity and interpretability, presenting several novel algorithms for comparisons (see in this issue Heymann, 2018a; Jonic, 2018; Marabini et al., 2018; Pintilie and Chiu, 2018; Stagg and Mendez, 2018a).
Most strikingly, resolvability, as judged by model-ability (capability to accurately trace a structural model), was shown to vary substantially among maps with similar reported resolution. It is clear from comparison of the map challenge submissions that current practices for determining resolution via FSC, especially with regards to masking, are inconsistent and therefore limit the value of reported resolution for ranking/comparing independently produced maps. Best-practice standards for post-reconstruction processing and FSC-based resolution evaluation are therefore needed. The full assessor team has produced a combined summary of all assessments along with their recommendations (Heymann et al., 2018, this issue).
The Model Challenge
In both ab initio and model improvement categories, the 16 participant groups in the Model Challenge demonstrated that the field has made great strides in developing software for building models into cryo-EM maps. Many excellent modelling tools are now publicly available. Challengers demonstrated that they were able to correctly trace significant portions of the benchmark map targets in a fully automatic manner, and in some cases make substantive improvements relative to original benchmark models (Figure 3). Five developer groups (see in this issue Chen and Baker, 2018; Donati et al., 2018; Terashi and Kihara, 2018; Terwilliger et al., 2018; Wang et al., 2018) plus a group of students newly introduced to Cryo-EM-based modelling (Yu et al., 2018, this issue) have contributed detailed descriptions of their methods.
Figure 3.

Model Challenge results for the TrpV1 Channel benchmark. Five submissions in the model improvement category (light colors) are compared with the original model (dark brown, PDB 3j5p; Liao et al. (2013)). The Map-vs-Model FSC curves show the resolution-dependent correlation of each model-derived map with the experimental map. In the bottom inset, EMRinger scores of submitted models are compared. Higher scores indicate better overall Cγ placement within map density (Barad et al., 2015).
Model assessment was supported by the creation of an analysis pipeline built on a prototype of the Critical Assessment of protein Structure Prediction (CASP) evaluation system (Kryshtafovych et al., 2018, this issue). Multiple global and local measures for model geometry, reference model similarity, and fit-to-map were compiled using fifteen different software tools. Assessors were then able to focus on comparing and contrasting the different measures of the submitted models. Model accuracy largely depended on the model building category (ab initio or optimization; see [dataset] Kryshtafovych et al. (2018b)). Using different measures provided different perspectives on the model assessment and showed that different scores are often non-correlated. For instance, EMRinger score, a novel fit-to-map metric that measures overall model Cγ placement within map density (Barad et al., 2015), was shown to be largely independent of two other commonly used measures for global fit, Map-vs-Model FSC and real-space correlation coefficient (examples shown in Fig. 3). Assessors also looked directly at models superimposed with maps to identify specific issues such as out-of-register tracing and suboptimal local stereochemistry. It was agreed among meeting participants that further review of global fit metrics is needed to determine which combinations are the most useful. Residue-level metrics that properly take the electron scattering properties of charged residues into account also need to be developed.
The quality of a model ultimately depends upon resolvability of the map being traced. However, as demonstrated by the Map Challenge, map resolvability does not fully correlate with reported resolution, and is difficult to assess from density alone. With both reconstruction and modelling software experts present, the joint workshop sparked lively discussions about the potential of model-based metrics to estimate not only model quality but also map resolvability, e.g. using model-map correlation tools to report specifically on protein backbone and side-chain placement such as EMRinger (Barad et al., 2015), CaBlam (Williams et al., 2017) and amino acid Z-scores (Pintilie and Chiu, 2018, this issue). Regions of uncertainty in a map can be readily flagged through atom or residue-level displacement parameters and/or local map-model density correlation. The final article in this issue recommends a number of additional strategies for improving cryoEM-based models (Richardson et al., 2018, this issue).
Empowering the next generation of cryo-EM scientists will require strong focus on creating best practices in reconstruction and model building, as well as robust community-approved structure validation methods, similar to what the X-ray crystallography community has done (Read et al., 2011). This round of Cryo-EM Challenges has stimulated important discussions that will help propel this field forward to meet these ends. We intend to continue the conversation and spur further method development by sponsoring additional challenges in the coming years.
Acknowledgements
The 2016 Cryo-EM Challenges and 2017 Workshop were supported by the National Institutes of Health [grant number R01-GM079429]. We are grateful to all of the participants of the challenges and the joint workshop discussions. We extend special thanks to both expert committees for their guidance. Maps: Bridget Carragher (chair), Jose-Maria Carazo, Wen Jiang, John Rubinstein, Peter Rosenthal, Fei Sun, Janet Vonck, and Ardan Patwardhan. Models: Paul Adams (chair), Axel Brunger, Randy Read, Torsten Schwede, Maya Topf, Gerard Kleywegt, Ardan Patwardhan and Andriy Kryshtafovych. We also thank Michael Norman for a discretionary award that enabled map challenge participants to perform calculations at the UCSD Supercomputer Center, and the Agouron Institute and ThermoFisher Scientific for co-sponsoring the Workshop.
References
- [dataset] Kryshtafovych A, Adams PD, Lawson CL, Chiu W, 2018b. Distribution of evaluation scores for the models submitted to the second cryo-EM model challenge, Data in Brief, doi: 10.1016/j.dib.2018.08.214 [DOI] [PMC free article] [PubMed]
- [dataset] Lawson CL, Chiu W, Carragher B, Carazo J-M, Jiang W, Patwardhan A, Rubinstein J, Rosenthal P, Sun F, Vonck J, Bai X, Bell J, Caputo N, Chakraborty A, Chen D-H, Chen J, Diaz-Avalos R, Donati L, Estrozi L, Galaz Montoya J, Gati C, Gomez-Blanco J, Grigorieff N, Gros P, Heymann B, Leith A, Li F, Ludtke S, Nans A, Nilchian M, Punjabi A, Sixma T, Tegunov D, Yang K, Yu G, Zhang J, Sala R, 2017. CryoEM Maps and Associated Data Submitted to the 2015/2016 EMDataBank Map Challenge 10.5281/zenodo.1185426 [DOI]
- [dataset] Lawson CL, Kryshtafovych A, Chiu W, Adams P, Brünger A, Kleywegt G, Patwardhan A, Read R, Schwede T, Topf M, Afonine P, Avaylon J, Baker M, Braun T, Cao W, Chittori S, Croll T, DiMaio F, Frenz B, Grudinin S, Hoffmann A, Hryc C, Joseph AP, Kawabata T, Kihara D, Mao B, Matthies D, McGreevy R, Nakamura H, Nakamura S, Nguyen L, Schroeder G, Shekhar M, Shimizu K, Singharoy A, Sobolev O, Tajkhorshid E, Teo I, Terashi G, Terwilliger T, Wang K, Yu I, Zhou H, Sala R, 2018. CryoEM Models and Associated Data Submitted to the 2015/2016 EMDataBank Model Challenge 10.5281/zenodo.1165999 [DOI]
- Barad BA, Echols N, Wang RY, Cheng Y, DiMaio F, Adams PD, Fraser JS, 2015. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JM, Fluty AC, Durmaz T, Chen M, Ludtke SJ, 2018. New software tools in EMAN2 inspired by EMDatabank map challenge. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Chen M, Baker ML, 2018. Automation And Assessment Of De Novo Modeling With Pathwalking In Near Atomic Resolution CryoEM Density Maps. J. Struct. Biol, this issue. [DOI] [PubMed]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R, 2013. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135C, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati L, Nilchian M, Sorzano COS, Unser M, 2018. Fast Multiresolution Reconstruction for Cryo-EM. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Henderson R, Chen S, Chen JZ, Grigorieff N, Passmore LA, Ciccarelli L, Rubinstein JL, Crowther RA, Stewart PL, Rosenthal PB, 2011. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J. Mol. Biol 413, 1028–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, Jiang W, Ludtke SJ, Medalia O, Penczek PA, Rosenthal PB, Rossmann MG, Schmid MF, Schroder GF, Steven AC, Stokes DL, Westbrook JD, Wriggers W, Yang H, Young J, Berman HM, Chiu W, Kleywegt GJ, Lawson CL, 2012. Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann B, 2018a. Map Challenge Assessment: Fair comparison of single particle cryoEM reconstructions. J. Struct. Biol, this issue. [DOI] [PubMed]
- Heymann B, 2018b. Single Particle Reconstruction and Validation using Bsoft for the Map Challenge. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Heymann B, Marabini R, Kazemi M, Sorzano COS, Holmdahl M, Mendez JH, Stagg SM, Jonic S, Palovcak E, Armache J-P, Zhao J, Cheng Y, Pintilie G, Chiu W, Patwardhan A, Carazo J-M, 2018. The First Single Particle Analysis Map Challenge: A Summary of the Assessments. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Iudin A, Korir PK, Salavert-Torres J, Kleywegt GJ, Patwardhan A, 2016. EMPIAR: a public archive for raw electron microscopy image data. Nat. Methods 13, 387–388. [DOI] [PubMed] [Google Scholar]
- Jonic S, 2018. A methodology using Gaussian-based density map approximation to assess sets of cryo-electron microscopy density maps. J. Struct. Biol, this issue. [DOI] [PubMed]
- Kryshtafovych A, Adams PD, Lawson CL, Chiu W, 2018. Evaluation System and Web Infrastructure for the Second Cryo-EM Model Challenge. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Lawson CL, Patwardhan A, Baker ML, Hryc C, Garcia ES, Hudson BP, Lagerstedt I, Ludtke SJ, Pintilie G, Sala R, Westbrook JD, Berman HM, Kleywegt GJ, Chiu W, 2016. EMDataBank unified data resource for 3DEM. Nucleic Acids Res 44, D396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, Cheng Y, 2013. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Lawson CL, Kleywegt GJ, Berman H, Chiu W, 2012. The 2010 cryo-em modeling challenge. Biopolymers 97, 651–654. [DOI] [PubMed] [Google Scholar]
- Marabini R, Kazemi M, Sorzano COS, Carazo J-M, 2018. Map Challenge: Analysis using a Pair Comparison Method based on Fourier Shell Correlation. J. Struct. Biol, this issue. [DOI] [PubMed]
- Marabini R, Carragher B, Chen S, Chen J, Cheng A, Downing KH, Frank J, Grassucci RA, Bernard Heymann J, Jiang W, Jonic S, Liao HY, Ludtke SJ, Patwari S, Piotrowski AL, Quintana A, Sorzano CO, Stahlberg H, Vargas J, Voss NR, Chiu W, Carazo JM, 2015. CTF Challenge: Result summary. J. Struct. Biol 190, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan A, Lawson CL, 2016. Databases and Archiving for CryoEM. Methods Enzymol 579, 393–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan A, Carazo JM, Carragher B, Henderson R, Heymann JB, Hill E, Jensen GJ, Lagerstedt I, Lawson CL, Ludtke SJ, Mastronarde D, Moore WJ, Roseman A, Rosenthal P, Sorzano CO, Sanz-Garcia E, Scheres SH, Subramaniam S, Westbrook J, Winn M, Swedlow JR, Kleywegt GJ, 2012. Data management challenges in three-dimensional EM. Nat. Struct. Mol. Biol 19, 1203–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintilie G, Chiu W, 2018. Assessment of Structural Features in Cryo-EM Density Maps using SSE and Side Chain Z-Scores. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Read RJ, Adams PD, Arendall WB 3rd, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, Perrakis A, Richardson JS, Sheffler WH, Smith JL, Tickle IJ, Vriend G, Zwart PH, 2011. A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19, 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JS, Williams CJ, Videau LL, Chen VB, 2018. Assessment of detailed conformations suggests strategies for improving cryoEM models: helix at lower resolution, ensembles, pre-refinement fixups, and validation at multi-residue length scale. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Russo CJ, Passmore LA, 2014. Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science 346, 1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S, 2012. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorzano COS, Vargas J, Trevin JMR, Jiménez A, Melero R, Martinez M, Ramírez-Aportela E, Conesa P, Vilas JL, Marabini R, Carazo JM, 2018. A new algorithm for high-resolution reconstruction of Single Particles by Electron Microscopy. J. Struct. Biol, this issue. [DOI] [PubMed]
- Stagg SM, Mendez JH, 2018a. Assessing the quality of single particle reconstructions by atomic model building. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Stagg SM, Mendez JH, 2018b. Processing Apoferritin with the Appion Pipeline. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Terashi G, Kihara D, 2018. De novo main-chain modeling with MAINMAST in 2015/2016 EM Model Challenge. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Terwilliger T, Adams PD, Afonine PV, Sobolev OV, 2018. Map segmentation, automated model-building and their application to the Cryo-EM Model Challenge. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- van Heel M, Schatz M, 2017. Reassessing the Revolution’s Resolutions (preprint). bioRxiv
- Wang Y, Shekhar M, Thifault D, Williams CJ, McGreevey R, Richardson JS, Singharoy A, Tajkhorshid E, 2018. Constructing Atomic Structural Models into Cryo-EM densities using Molecular Dynamics - Pros and Cons. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, Jain S, Lewis SM, Arendall WB 3rd, Snoeyink J, Adams PD, Lovell SC, Richardson JS, Richardson DC, 2017. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci [DOI] [PMC free article] [PubMed]
- Yu I, Nguyen L, Avaylon J, Wang K, Zhou ZH, 2018. Building Atomic Models Based on Near Atomic Resolution cryoEM Maps with Existing Tools. J. Struct. Biol, this issue. [DOI] [PMC free article] [PubMed]
- Zhu Y, Carragher B, Glaeser RM, Fellmann D, Bajaj C, Bern M, Mouche F, de Haas F, Hall RJ, Kriegman DJ, Ludtke SJ, Mallick SP, Penczek PA, Roseman AM, Sigworth FJ, Volkmann N, Potter CS, 2004. Automatic particle selection: results of a comparative study. J. Struct. Biol 145, 3–14. [DOI] [PubMed] [Google Scholar]