

Abstract
Background
Neuraminidase inhibitors (NIs) are stockpiled and recommended by public health agencies for treating and preventing seasonal and pandemic influenza. They are used clinically worldwide.
Objectives
To describe the potential benefits and harms of NIs for influenza in all age groups by reviewing all clinical study reports of published and unpublished randomised, placebo‐controlled trials and regulatory comments.
Search methods
We searched trial registries, electronic databases (to 22 July 2013) and regulatory archives, and corresponded with manufacturers to identify all trials. We also requested clinical study reports. We focused on the primary data sources of manufacturers but we checked that there were no published randomised controlled trials (RCTs) from non‐manufacturer sources by running electronic searches in the following databases: the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, MEDLINE (Ovid), EMBASE, Embase.com, PubMed (not MEDLINE), the Database of Reviews of Effects, the NHS Economic Evaluation Database and the Health Economic Evaluations Database.
Selection criteria
Randomised, placebo‐controlled trials on adults and children with confirmed or suspected exposure to naturally occurring influenza.
Data collection and analysis
We extracted clinical study reports and assessed risk of bias using purpose‐built instruments. We analysed the effects of zanamivir and oseltamivir on time to first alleviation of symptoms, influenza outcomes, complications, hospitalisations and adverse events in the intention‐to‐treat (ITT) population. All trials were sponsored by the manufacturers.
Main results
We obtained 107 clinical study reports from the European Medicines Agency (EMA), GlaxoSmithKline and Roche. We accessed comments by the US Food and Drug Administration (FDA), EMA and Japanese regulator. We included 53 trials in Stage 1 (a judgement of appropriate study design) and 46 in Stage 2 (formal analysis), including 20 oseltamivir (9623 participants) and 26 zanamivir trials (14,628 participants). Inadequate reporting put most of the zanamivir studies and half of the oseltamivir studies at a high risk of selection bias. There were inadequate measures in place to protect 11 studies of oseltamivir from performance bias due to non‐identical presentation of placebo. Attrition bias was high across the oseltamivir studies and there was also evidence of selective reporting for both the zanamivir and oseltamivir studies. The placebo interventions in both sets of trials may have contained active substances.
Time to first symptom alleviation. For the treatment of adults, oseltamivir reduced the time to first alleviation of symptoms by 16.8 hours (95% confidence interval (CI) 8.4 to 25.1 hours, P < 0.0001). This represents a reduction in the time to first alleviation of symptoms from 7 to 6.3 days. There was no effect in asthmatic children, but in otherwise healthy children there was (reduction by a mean difference of 29 hours, 95% CI 12 to 47 hours, P = 0.001). Zanamivir reduced the time to first alleviation of symptoms in adults by 0.60 days (95% CI 0.39 to 0.81 days, P < 0.00001), equating to a reduction in the mean duration of symptoms from 6.6 to 6.0 days. The effect in children was not significant. In subgroup analysis we found no evidence of a difference in treatment effect for zanamivir on time to first alleviation of symptoms in adults in the influenza‐infected and non‐influenza‐infected subgroups (P = 0.53).
Hospitalisations. Treatment of adults with oseltamivir had no significant effect on hospitalisations: risk difference (RD) 0.15% (95% CI ‐0.78 to 0.91). There was also no significant effect in children or in prophylaxis. Zanamivir hospitalisation data were unreported.
Serious influenza complications or those leading to study withdrawal. In adult treatment trials, oseltamivir did not significantly reduce those complications classified as serious or those which led to study withdrawal (RD 0.07%, 95% CI ‐0.78 to 0.44), nor in child treatment trials; neither did zanamivir in the treatment of adults or in prophylaxis. There were insufficient events to compare this outcome for oseltamivir in prophylaxis or zanamivir in the treatment of children.
Pneumonia. Oseltamivir significantly reduced self reported, investigator‐mediated, unverified pneumonia (RD 1.00%, 95% CI 0.22 to 1.49); number needed to treat to benefit (NNTB) = 100 (95% CI 67 to 451) in the treated population. The effect was not significant in the five trials that used a more detailed diagnostic form for pneumonia. There were no definitions of pneumonia (or other complications) in any trial. No oseltamivir treatment studies reported effects on radiologically confirmed pneumonia. There was no significant effect on unverified pneumonia in children. There was no significant effect of zanamivir on either self reported or radiologically confirmed pneumonia. In prophylaxis, zanamivir significantly reduced the risk of self reported, investigator‐mediated, unverified pneumonia in adults (RD 0.32%, 95% CI 0.09 to 0.41); NNTB = 311 (95% CI 244 to 1086), but not oseltamivir.
Bronchitis, sinusitis and otitis media. Zanamivir significantly reduced the risk of bronchitis in adult treatment trials (RD 1.80%, 95% CI 0.65 to 2.80); NNTB = 56 (36 to 155), but not oseltamivir. Neither NI significantly reduced the risk of otitis media and sinusitis in both adults and children.
Harms of treatment. Oseltamivir in the treatment of adults increased the risk of nausea (RD 3.66%, 95% CI 0.90 to 7.39); number needed to treat to harm (NNTH) = 28 (95% CI 14 to 112) and vomiting (RD 4.56%, 95% CI 2.39 to 7.58); NNTH = 22 (14 to 42). The proportion of participants with four‐fold increases in antibody titre was significantly lower in the treated group compared to the control group (RR 0.92, 95% CI 0.86 to 0.97, I2 statistic = 0%) (5% absolute difference between arms). Oseltamivir significantly decreased the risk of diarrhoea (RD 2.33%, 95% CI 0.14 to 3.81); NNTB = 43 (95% CI 27 to 709) and cardiac events (RD 0.68%, 95% CI 0.04 to 1.0); NNTB = 148 (101 to 2509) compared to placebo during the on‐treatment period. There was a dose‐response effect on psychiatric events in the two oseltamivir "pivotal" treatment trials, WV15670 and WV15671, at 150 mg (standard dose) and 300 mg daily (high dose) (P = 0.038). In the treatment of children, oseltamivir induced vomiting (RD 5.34%, 95% CI 1.75 to 10.29); NNTH = 19 (95% CI 10 to 57). There was a significantly lower proportion of children on oseltamivir with a four‐fold increase in antibodies (RR 0.90, 95% CI 0.80 to 1.00, I2 = 0%).
Prophylaxis. In prophylaxis trials, oseltamivir and zanamivir reduced the risk of symptomatic influenza in individuals (oseltamivir: RD 3.05% (95% CI 1.83 to 3.88); NNTB = 33 (26 to 55); zanamivir: RD 1.98% (95% CI 0.98 to 2.54); NNTB = 51 (40 to 103)) and in households (oseltamivir: RD 13.6% (95% CI 9.52 to 15.47); NNTB = 7 (6 to 11); zanamivir: RD 14.84% (95% CI 12.18 to 16.55); NNTB = 7 (7 to 9)). There was no significant effect on asymptomatic influenza (oseltamivir: RR 1.14 (95% CI 0.39 to 3.33); zanamivir: RR 0.97 (95% CI 0.76 to 1.24)). Non‐influenza, influenza‐like illness could not be assessed due to data not being fully reported. In oseltamivir prophylaxis studies, psychiatric adverse events were increased in the combined on‐ and off‐treatment periods (RD 1.06%, 95% CI 0.07 to 2.76); NNTH = 94 (95% CI 36 to 1538) in the study treatment population. Oseltamivir increased the risk of headaches whilst on treatment (RD 3.15%, 95% CI 0.88 to 5.78); NNTH = 32 (95% CI 18 to 115), renal events whilst on treatment (RD 0.67%, 95% CI ‐2.93 to 0.01); NNTH = 150 (NNTH 35 to NNTB > 1000) and nausea whilst on treatment (RD 4.15%, 95% CI 0.86 to 9.51); NNTH = 25 (95% CI 11 to 116).
Authors' conclusions
Oseltamivir and zanamivir have small, non‐specific effects on reducing the time to alleviation of influenza symptoms in adults, but not in asthmatic children. Using either drug as prophylaxis reduces the risk of developing symptomatic influenza. Treatment trials with oseltamivir or zanamivir do not settle the question of whether the complications of influenza (such as pneumonia) are reduced, because of a lack of diagnostic definitions. The use of oseltamivir increases the risk of adverse effects, such as nausea, vomiting, psychiatric effects and renal events in adults and vomiting in children. The lower bioavailability may explain the lower toxicity of zanamivir compared to oseltamivir. The balance between benefits and harms should be considered when making decisions about use of both NIs for either the prophylaxis or treatment of influenza. The influenza virus‐specific mechanism of action proposed by the producers does not fit the clinical evidence.
Plain language summary
Regulatory information on trials of oseltamivir (Tamiflu) and zanamivir (Relenza) for influenza in adults and children
Oseltamivir and zanamivir have been stockpiled in many countries to treat and prevent seasonal and pandemic influenza, before an influenza vaccine matched to the circulating virus becomes available. Oseltamivir is classified by the World Health Organization as an essential medicine.
How this review has been approached
We have updated and combined our reviews on the antiviral drugs zanamivir and oseltamivir for influenza in adults and children on the basis of the manufacturers' reports to regulators (clinical study reports) and the regulators' comments. We have called these comments and reports 'regulatory information'. Clinical study reports are unpublished, extensive documents with great detail on the trials that formed the basis for market approval. They include the protocols, methods and results. Clinical study reports have until now been confidential, seen only by the manufacturers and regulators.
Why we have taken this approach
In previous versions of this review we identified unresolved discrepancies in the data presented in published trial reports and substantial publication bias. As a consequence, we elected not to use data from journal articles but included the documents generated during licensing processes. We have accessed such data from the UK, USA, European Medicines Agency (EMA), Japanese regulators and clinical study reports from the manufacturers (after a protracted media campaign). This has enabled us to verify information from the randomised, placebo‐controlled trials on adults and children with confirmed or suspected exposure to naturally occurring influenza.
Based on our assessments of the regulatory documents (in excess of 160,000 pages), we came to the conclusion that there were substantial problems with the design, conduct, reporting and availability of information from many of the trials.
What we have found
We have used data from 46 trials (20 oseltamivir and 26 zanamivir studies) in this review. We identified problems in the design of many of the studies that we included, which affects our confidence in their results. We found that both drugs shorten the duration of symptoms of influenza‐like illness (unconfirmed influenza or 'the flu') by less than a day. Oseltamivir did not affect the number of hospitalisations, based on the data from all the people enrolled in treatment trials of oseltamivir. Zanamivir trials did not record this outcome. The effects on pneumonia and other complications of influenza, such as bronchitis, middle ear infection (otitis media) and sinusitis, were unreliably reported, as shown by the case report form in the trial documents. Some forms showed limitations in the diagnostic criteria for pneumonia. Regulatory comments noted problems with missing follow‐up diary cards from participants. In children with asthma there was no clear effect on the time to first alleviation of symptoms.
Prophylaxis trials showed that oseltamivir and zanamivir reduced the risk of symptomatic influenza in individuals and households. There was no evidence of an effect on asymptomatic influenza or on non‐influenza, influenza‐like illness, but trial conduct problems prevent any definitive conclusion.
Oseltamivir use was associated with nausea, vomiting, headaches, renal and psychiatric events; these last three were when it was used to prevent influenza (prophylaxis). Its effect on the heart is unclear: it may reduce cardiac symptoms, but may induce serious heart rhythm problems. In adult treatment trials of zanamivir there was no increased risk of reported adverse events. The evidence on the possible harms associated with the treatment of children with zanamivir was sparse.
Agreement with other findings
The lack of good evidence demonstrating an effect on complications agrees with the conservative conclusions on both drugs drawn by the US Food and Drug Administration (FDA). The FDA only allowed claims of effectiveness of both drugs for the prevention and treatment of symptoms of influenza and not for other effects (including the interruption of person‐to‐person spread of the influenza virus or prevention of pneumonia). The FDA described the overall performance of both drugs as 'modest'.
Mechanism of action for beneficial effects
These findings all suggest that the low immune response with low levels of pro‐inflammatory cytokines, which is induced by the action of oseltamivir carboxylate, may reduce the symptoms of influenza unrelated to an inhibition of influenza virus replication. The potential hypothermic or antipyretic effect of oseltamivir as a central nervous system depressant may also contribute to the apparent reduction of host symptoms. Statements made on the capacity of oseltamivir to interrupt viral transmission and reduce complications are not supported by any data we have been able to access.
The mechanism of action proposed by the producers (influenza virus‐specific) does not fit the clinical evidence which suggests a multi‐system and central action.
Background
This review (known as A159) reports our efforts to get to the bottom of the issue of the effects of NIs by appraising evidence from unpublished clinical study reports (see Glossary, Appendix 1) and regulatory documents containing comments and reviews. We have called the body of clinical studies and regulatory comments 'regulatory information'. For the history and evolution of the review see Appendix 2.
Description of the condition
Influenza is mostly a mild, self limiting infection of the upper airways with local symptoms, including sniffles, nasal discharge, dry cough and sore throat, and systemic symptoms such as fever, headache, aches and pains, malaise and tiredness.
Occasionally patients with influenza develop complications such as pneumonia, otitis media and dehydration or encephalopathy with or without liver failure, which may be due to the effects of the influenza virus itself or associated secondary bacterial infections and/or adverse effects of drugs such as antipyretics (including salicylates and other non‐steroidal anti‐inflammatory drugs) (Hama 2008).
Influenza is not clinically distinguishable from influenza‐like illness (ILI) (Call 2005). Epidemic influenza in humans is caused by influenza A and B viruses. Currently, influenza A/H1N1, influenza A/H3N2 and influenza B cause most influenza infections worldwide (CDC 2013).
Description of the intervention
Neuraminidase inhibitors (NIs) comprise inhaled zanamivir (Relenza, GlaxoSmithKline), oral oseltamivir (Tamiflu, Gilead Sciences and F. Hoffman‐La Roche), parenteral peramivir (BioCryst Ltd), inhaled laninamivir (Daiichi Sankyo Co. Ltd) (Sugaya 2010) and others still under development (Hayden 2009). The use of NIs has increased dramatically since the outbreak of A/H1N1 in April 2009, partly because of the rise in amantadine/rimantadine resistance and, in the early stages of the outbreak, the lack of a vaccine, which meant that NIs became a widespread public health intervention. The World Health Organization (WHO) had previously encouraged member states to stockpile and gain experience of using NIs (WHO 2002a; WHO 2002b; WHO 2004).
How the intervention might work
Although NIs may reduce the ability of the virus to penetrate the mucus in the very early stage of infection (Bhatia 2007; Matrosovich 2004; Moscona 2005; Ohuchi 2006), their main mechanism of action is thought to lie in their ability to inhibit influenza viruses from exiting host cells (Liu 1995; Moscona 2005). The manufacturers state that oseltamivir does not prevent infection, nor affect antibody production (Smith 2006), but it reduces symptom duration probably by reducing viral load, spread and release of cytokines (Hayden 1999; WV15670), diminishing the chance of complications and interrupting person‐to‐person viral spread.
Oseltamivir phosphate (Tamiflu) is the pro‐drug of oseltamivir carboxylate, the effective form. Oseltamivir phosphate dissociates in the gastrointestinal tract to form oseltamivir, which is absorbed and metabolised into oseltamivir carboxylate by hepatic carboxylesterase (h‐CE). Oseltamivir may have a central depressant action (Hama 2008) and may also inhibit human sialidase (Li 2007), causing abnormal behaviour.
Inhaled zanamivir reaches a far lower plasma concentration compared to its intravenous administration (Cass 1999).
Any treatment that reduces the complications of influenza (for example, pneumonia) and the excretion of the virus from infected people might be a useful public health measure to contain an epidemic by limiting the impact and spread of the virus. In addition to symptomatic treatment, prophylactic use for interrupting the spread of disease has informed pandemic planning over the past decade.
Why it is important to do this review
There are three major reasons for conducting this review, in addition to questions of efficacy associated with the clinical use of NIs for influenza:
Influenza antivirals are a commonly used and stockpiled drug against past and future pandemics on the basis of international and national recommendations. These recommendations are based on the claimed and assumed ability of the drug to reduce complications and transmission (HHS 2005; WHO 2007). In theory, containing the spread of influenza allows time for an organised response with longer‐term interventions (such as vaccines), which take time to produce (WHO 2007).
There are legitimate reasons to doubt these claims and the results of previous Cochrane reviews of NIs in adults (Jefferson 2006; Jefferson 2009a) and children (Shun‐Shin 2009), due to the risk of reporting bias, including the certainty of publication bias (Doshi 2012a; Doshi 2012b).
Oseltamivir is now on the list of WHO essential drugs (WHO 2013a; WHO 2013b).
Process
A159 is an amalgamation of two long‐standing Cochrane reviews on the effects of NIs for influenza in healthy adults (Jefferson 2010a, also published as Jefferson 2009a) and children (Matheson 2007), and it is based on the assessment of trials through their clinical study reports and other regulatory information: a decision we made after finding substantial reporting bias in the journal publications of the relevant trials.
For the rationale for this process see Appendix 2.
Examples of discrepancies and reporting bias
We identified that 60% (3145/5267) of patient data from randomised, placebo‐controlled, phase III treatment trials of oseltamivir have never been published. This includes M76001, the biggest treatment trial ever undertaken on oseltamivir (with just over 1400 people of all ages). Exclusion of unpublished data changed our previous findings regarding the ability of oseltamivir to reduce the complications of influenza (Doshi 2009; Jefferson 2009a). In some cases, mistakes in the attribution of adverse events were only discovered through matching summary tables with individual participant listings (Gravenstein 2013; Peters 2001; WV15825).
A modified approach
We have modified the routine Cochrane processes to improve our previous methods, which we now consider inadequate. To resolve inconsistencies and under‐reporting, we changed our approach by no longer including trial data as reported in papers published in biomedical journals. Instead, we treated clinical study reports as our basic unit of analysis. Clinical study reports are often sent to national drug regulators such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) (formerly EMEA), which require far more stringent standards for completeness and accuracy of reporting than biomedical journals. Journal articles can be regarded as a very succinct synthesis of a clinical study report. In addition to seeking clinical study reports, we decided to read and review regulatory documentation. The FDA in particular (and the EMA to a far lesser extent) make many of its scientific reviews available on its website. Unlike Cochrane review authors, regulators can have access to the whole data set and their comments can provide useful insight, helping to achieve a better understanding of trial programmes.
Clinical study reports generally remain hidden from public view and are not readily available for wider scientific scrutiny, despite the wealth of information they contain for those willing and able to spend the time reading them and despite calls to make all relevant trial data public (Doshi 2013; Godlee 2009), as well as the known problems with reporting biases (McGauran 2010; Wieseler 2013).
Implications
This modified approach to a Cochrane review aims to provide patients, clinicians and policy‐makers with the most transparent and independent information possible about NIs for influenza. In addition, it should contribute to improving a European regulatory and pharmacovigilance legal framework, which commentators consider weak (Cohen 2009; Godlee 2009). We believe that as NIs have become public health drugs, recommended and stockpiled globally, independent scrutiny of all the evidence relating to harms and effects on complications is necessary to provide patients, policy‐makers and physicians with a complete and unbiased view of their risks and benefits.
Implication for A/H1N1 (2009) influenza
In response to our 2010 review (Jefferson 2009a; Jefferson 2010a), some have argued that its findings cannot be applied to the 2009 A/H1N1, suggesting that it is a new virus and thus we need new evidence (JAID 2010; Maugh 2009; Nebehay 2009; NHS 2009; NHS 2010). Novel A/H1N1 is a new strain of a subtype that has been circulating since 1977, but it also resembles the A/H1N1 strain that has been circulating since before 1957 (CDC 2009) or before the 1918 pandemic (Itoh 2009). Influenza subtype A/H1N1 was indeed circulating in the clinical trials we have included in our previous reviews. In addition, oseltamivir and zanamivir were approved by regulators worldwide for the treatment and prevention of influenza types A and B, not specific subtypes or strains of influenza A and B. The expectation of regulatory approval is thus that the effects of these drugs demonstrated in clinical trials will apply to future strains of influenza A and B. Use of these drugs during the pandemic was not off‐label. It was approved use because of the assumption that the clinical trial evidence underpinning regulatory approval applied to novel A/H1N1. We reviewed the clinical trial evidence with the expectation that our results, similar to regulators, will apply to all influenza viruses.
Wider implications
The modified approach in this Cochrane review grew out of a realisation that prior methods employed to review NIs were inadequate. There seems to be no compelling reason to think that the lessons learned are limited to these particular drugs (Godlee 2009; Rodgers 2013; Vedula 2009; Vedula 2013; Wieseler 2013). For this reason, our independent scrutiny, using all possible trial information, may inform both the wider debate on the adequacy of existing regulatory frameworks in the adoption of new drugs and the question of whether other systematic reviews should move to this new, more rigorous, approach, which focuses on trial programmes rather than single trials (Eyding 2010; Ioannidis 2010) (see Glossary, Appendix 1). Although there is substantial evidence for the effects of reporting bias in estimates of effectiveness, less is known of its impact on the evidence of harms (Chou 2005). We decided to quantify the additional resources required to follow our modified methodological approach to assess the feasibility of other systematic reviews proceeding in a similar fashion.
See the Differences between protocol and review section for the previous version of the objectives of this review.
Objectives
To describe the potential benefits and harms of NIs for influenza in all age groups by reviewing all clinical study reports of published and unpublished randomised, placebo‐controlled trials and regulatory comments.
Methods
Criteria for considering studies for this review
Types of studies
We included evidence from randomised controlled trials (RCTs) testing the effects of NIs for prophylaxis, post‐exposure prophylaxis (PEP) and treatment of influenza. Prophylaxis is the mode of use of NIs when there is expectation of possible near‐future exposure to influenza. PEP is the use of NIs following probable exposure to influenza but before symptoms develop. Treatment is the use of NIs in persons showing probable signs of influenza.
Due to discrepancies between published and unpublished reports of the same trials, we decided to include only those trials for which we had unabridged clinical study reports (for example, with consecutively numbered pages), even though they may be parts of clinical study reports (i.e. Module 1 only) and information on reports of trials that were considered "pivotal" (i.e. first or second‐line evidence to regulators in support of the registration application).
Types of participants
We included previously healthy people (children and adults). 'Previously healthy' includes people with chronic illness (such as asthma, diabetes, hypertension), but excludes people with illnesses with more significant effects on the immune system (such as malignancy or HIV infection). We included only trials on people exposed to naturally occurring influenza with or without symptoms. We targeted the intention‐to‐treat (ITT) and safety populations as our prior review discovered compelling evidence that the intention‐to‐treat‐influenza‐infected (ITTI), the sub‐population deemed to be influenza‐infected, were not balanced between treatment groups in the Roche oseltamivir trials. In addition, estimates from the ITT population will be more generalisable to clinical practice, where routine testing for influenza is not common in many countries (and even where used, remains of variable accuracy).
Types of interventions
NIs administered by any route compared with placebo during the period in which medication was assumed and during the follow‐up (on‐ and off‐treatment: on‐t and off‐t) periods.
Types of outcome measures
Primary outcomes
Primary outcome measures for treatment studies
Symptom relief
Hospitalisation and complications
Harms
Primary outcome measures for prophylaxis studies
Influenza (symptomatic and asymptomatic, always with laboratory confirmation) and influenza‐like illness (ILI)
Hospitalisation and complications
Interruption of transmission (in its two components, reduction of viral spread from index cases and prevention of onset of influenza in contacts)
Harms
Secondary outcomes
Secondary outcome measures for treatment studies
Symptom relapse after finishing treatment
Drug resistance
Viral excretion
Mortality
Secondary outcome measures for prophylaxis studies
Drug resistance
Viral excretion
Mortality
Whilst overall symptom reduction is well documented, our interest was particularly focused on complications and adverse events, as this is where evidence is currently scarce or inconclusive (Jefferson 2009a; Shun‐Shin 2009). Our preliminary examination of some regulatory documents and some published versions of the studies had identified that some symptoms and sequelae of influenza (such as pneumonia) had been classified as either a 'complication of influenza' or as an 'adverse event of the treatment', or both. This is somewhat confusing and we intended to analyse 'compliharms' (see Glossary, Appendix 1) irrespective of the classification as a 'complication of influenza' or as an 'adverse event of the treatment' (Appendix 3) in oseltamivir trials. Complications of particular interest included pneumonia, bronchitis, otitis media and sinusitis as these were the secondary illnesses often collected in the Roche oseltamivir trials and we agreed that these events are clinically important. Initially we constructed a table to illustrate the design methodology used for each complication by study (Table 1). The table included the following variables: definition of which events are termed complications; where complications are first defined in the clinical study report; diagnosis method; and availability of data. We then stratified our analysis by method of diagnosis with three possible criteria: (1) laboratory‐confirmed diagnosis (e.g. based on radiologically or microbiologically confirmed evidence of infection); (2) clinical diagnosis without laboratory confirmation (diagnosed by a doctor after a clinical examination); (3) other type of diagnosis such as self reported by patient. We conducted analysis of any complication (pneumonia, bronchitis, otitis media and sinusitis) that was classified as serious or led to study withdrawal.
1. Blank case report forms' data capture for secondary illnesses in oseltamivir trials.
| Study | Where in CRF (PDF pg #) | Data captured |
Person reporting (participant/investigator) |
Where reported | Specific field for recording confirmatory assessment (e.g. CXR) | Confirmation (including px) |
| M76001 | 1167 | Yes/no answer to question: "Is this event a secondary illness related to influenza?" Secondary illness is defined: sinusitis, otitis, bronchitis, pneumonia + other chest infections that are not diagnosed as bronchitis and/or pneumonia |
Investigator | In form for "Adverse events or intercurrent illness" | No | Px |
| NV16871 | 361, 389 | Form states: Have there been any changes in the patient’s health including any new conditions or worsening of existing conditions since day 1 (please include secondary illnesses)? Yes/No. If "Yes", please record the details on the "Adverse events or secondary illness" form in the Additional Forms section of the CRF on pg 30.0. All serious adverse events must be reported within 1 working day of occurrence to Roche Pg 30.0 of CRF (PDF pg 389) defines secondary illnesses as sinusitis, otitis media, bronchitis and pneumonia, and asks additional questions such as relationship to test drug and outcome, and leaves space for investigator's comments on the adverse event |
Investigator | Secondary illness not listed as efficacy outcomes Recording of secondary illnesses was to occur in a form titled "Adverse event or secondary illness" |
No | Px |
| WV15670 | 732, 754, 791, 832 | CRF form (PDF pg 732) states: Secondary illness reminder Has the patient reported any sinusitis, otitis, bronchitis, other chest infection or pneumonia since baseline? yes [ ] Complete secondary illness page (not the adverse event page) no [ ] Secondary illness page CRF (PDF pg 754) requests information on date of onset, date resolved, whether treatment was given and, if so, what treatment or medical procedures, total daily dose, and start/end date of treatment or medical procedure In addition, participants could fill in information related to a secondary illness in their diary card in the free‐text box called "Notes" which prompts participants: "Please can you record below any extra information about your flu which may be of interest to us, (for example: did your flu symptoms re‐occur, and if so when?), and have you taken any other treatments. If so please record the treatment name and the dates you took it." (PDF pg 791) |
Participant mediated through Investigator | For investigators, on "Secondary illness" form For participants, on "Notes" section of diary card |
No | |
| WV15671 | 740, 889, 1018 | CRF form (PDF pg 740) states: Secondary illness reminder Has the patient reported any sinusitis, otitis, bronchitis, other chest infection or pneumonia since baseline? yes [ ] Complete secondary illness page (not the adverse event page) no [ ] Secondary illness page CRF (PDF pg 889) requests information on date of onset, date resolved, whether treatment was given and, if so, what treatment or medical procedures, total daily dose and start/end date of treatment or medical procedure Secondary illnesses are listed as sinusitis, otitis, bronchitis, pneumonia and other chest infections that are not diagnosed as bronchitis and/or pneumonia In addition, participants could fill in information related to a secondary illness in their diary card in the free‐text box called "Notes" which prompts participants: "Please can you record below any extra information about your flu which may be of interest to us, (for example: did your flu symptoms re‐occur, and if so when?), and have you taken any other treatments. If so please record the treatment name and the dates you took it." (PDF pg 1018) |
Participant mediated through investigator | Mentioned in M1 and RAP as tertiary outcomes not mentioned in protocol | No | Px |
| WV15673/WV15697 | From 483 | No mention of pneumonia, secondary illness, complications in the CRFs | Unclear | Secondary illnesses not listed in protocol as endpoints. They are listed as safety endpoints in the RAP which states that "pre‐defined" secondary illnesses were "sinusitis, otitis, bronchitis, pneumonia, and other chest infections that are not diagnosed as bronchitis and/or pneumonia, plus recurrence of symptoms from the diary card once alleviation had occurred." (PDF pg 479) | ||
| WV15707 | From 98 | Pg 117 Secondary illness reminder: Has the patient reported any sinusitis, otitis, bronchitis, other chest infection or pneumonia since baseline? yes [] ‐ Complete secondary illness page (not the adverse event page) no [] Pg 131: Diagnostic procedures 1) Were there any diagnostic procedures or tests carried out since day 1 as a result of influenza or secondary illness that were not scheduled as part of protocol? Yes Type of diagnostic procedure or test 1 Chest X‐rays, 2 ECG, 3 Bacterial culture, 4 Bronchoscopy, 5 Pulmonary function test, 6 Viral culture (other than influenza), 7 Blood tests (other than antibody sample), 8 Other specify No Secondary illness page CRF (PDF pg 158) requests information on date of onset, date resolved, whether treatment was given and, if so, what treatment or medical procedures, total daily dose and start/end date of treatment or medical procedure |
Participant mediated through investigator | Mentioned in RAP as tertiary endpoints pg 57‐8 | Yes | Px |
| WV15708 | From 460 | Secondary illness reminder at pg 474: Has the patient reported any new episodes of sinusitis, otitis, bronchitis, other chest infection or pneumonia since screening? yes [] ... Complete adverse event page no [] "Adverse events" CRF collected data on date of onset, initial intensity, test drug adjustment, whether treatment was given (if so, what), most extreme intensity, relationship to test drug, outcome, whether it led to hospitalisation and a free‐text line for recording "Comments on AE" (e.g. PDF pg 479) |
Participant mediated through investigator | Secondary illness not mentioned in protocol | No | Px |
| WV15730 | From 340 | Secondary illness reminder: Has the patient reported any sinusitis, otitis, bronchitis, other chest infection or pneumonia since baseline? yes [] ... Complete secondary illness page (not the adverse event page) no [] The secondary illness page is descriptive of dates and px |
Participant mediated through investigator | Listed as tertiary endpoints in RAP at pg 297 | No | Px |
| WV15758 | From 637 | Has the patient reported any new adverse events or symptoms (including intercurrent illnesses and secondary illnesses)? yes [] record in the adverse events/intercurrent illness section of the case no [] report form Diagnostic confirmation of otitis media from pg 648 onwards |
Participant mediated through investigator | Listed as secondary illnesses in core report Module 1‐2 (pg 36) | Yes | Px |
| WV15759/871 | From 665 | Has the subject reported any adverse events including secondary and intercurrent illnesses? | Participant mediated through investigator | Secondary illnesses not mentioned in protocol, but secondary outcome in core report Note: worth looking at comparisons 1.49 to 1.51 in RM5. No effect but in bronchitis this study has a more conservative effect than NV 16871 which has no definitions and no diagnostics |
Yes | Px |
| WV15799 | From 642 | Secondary illness defined as in M76001. There is a generic physical examination form at pg 704 including "lungs" normal/abnormal specify_______ At pg 709 has the patient reported any new AE including intercurrent or secondary illnesses yes/no. If y record the adverse events/intercurrent illness section of the CRF (noted at pf 746 on to be at the back of the CRF) with FULL HISTORY, PHYSICAL EXAMINATION AND DIAGNOSTIC WORK UP QUESTIONS FOR BRON+PNUM+LRTI+SIN+OM including questions about CXR, MRI, sputum etc. |
Investigator | Proportion of contacts who are classified as having a secondary illness subsequent to a confirmed episode of influenza listed as tertiary endpoints | Yes | Px |
| WV15812/872 | From 285 | Has the patient reported any new adverse events or symptoms (including intercurrent illnesses and secondary illnesses)? yes [] record in the adverse events/intercurrent illness section of the case no [] report form At pg 450‐74 is DIAGNOSIS OF SECONDARY ILLNESS page which is very similar to the one at serial 10 EXHAUSTIVE list of diagnostic procedures |
Participant mediated through investigator | Listed as secondary tertiary in protocol at pg 252 | Yes | Px |
| WV15819/978/876 | From 412 | Pg 437 (adverse event reminder): Has the patient reported any new adverse events or symptoms (including intercurrent illnesses)? yes [] record in the adverse events/intercurrent illness section of the case no [] report form In CRF pg 447 and 443 usual secondary illness reminder From pg 471 DIAGNOSTIC OF SECONDARY ILLNESS. This is a one page list of diagnostics similar to that at serial 10. The question is: "Were there any diagnostic procedures or tests carried out since day 1 as a result of influenza or secondary illness that were not scheduled as part of protocol?" If yes list per serial 10 From pg 486 is a list of diagnostic tests |
Participant mediated through investigator | Secondary illness listed as secondary (required antibiotics) and tertiary outcomes in core report and as an addition in protocol amendment at pg 21 | Yes | Px |
| WV15825 | From 389 | There is a usual note: please go to diagnosis of secondary illness at back of CRF. Pg 487: Is this event a secondary illness related to influenza? DIAGNOSTIC OF SECONDARY ILLNESS From pg 510‐40 with exhaustive list of diagnostics as per serial 10 |
Participant mediated through investigator | Secondary illness listed as secondary outcomes in protocol pg 346 Secondary illnesses recorded on "Adverse events" CRF |
Yes | Px |
| WV16277 | From 415 | Not found | Not found | Secondary illness not listed as efficacy outcomes |
#Events within the first 2 days of the study were excluded *Note that some events are reported as secondary illness and adverse event (AE) but some events are reported as secondary illness only and some events are reported as adverse event only BRON = bronchitis CRF = case report form CXR = chest x‐ray ECG = electrocardiogram LRTI = lower respiratory tract infection MRI = magnetic resonance imaging OM = otitis media PNUM = pneumonia px = prescription ˜RAP = reporting analysis plan SIN = sinusitis
In all cases of influenza complications reporting (pneumonia, bronchitis, sinusitis, otitis media) there is a variable degree of participant self reporting, of investigator mediation (for example, in writing down the details in the case report form) and lack of verification with investigations such as culture or imaging. The 'self reported, investigator‐mediated, unverified' title is relevant to all complications but for brevity we use it as sparingly as possible.
For harms we were limited by the frequency of occurrence of the adverse events collected in the trials. Consequently we meta‐analysed (1) all serious adverse events; (2) all adverse events leading to study withdrawal; (3) all withdrawals; (4) all adverse events within a clinical study report's defined body system; as well as (5) a small group of common adverse events as defined in the FDA drug label for oseltamivir. There were too few events to meta‐analyse (1) deaths; (2) serious adverse events by body system; and (3) any events that had an overall incidence of less than 0.5%. We did not meta‐analyse outcomes with fewer than 10 events in total. We conducted analyses separately for on‐treatment and off‐treatment periods. However, in two cases where (on‐treatment) treatment effects were borderline statistically significant (prophylaxis with oseltamivir: renal body system on‐treatment and psychiatric body system on‐treatment), we conducted additional analysis combining on‐ and off‐treatment periods to maximise statistical power. We conducted dose‐response harms analysis for two treatment trials (WV15670 and WV15671) combined and one prophylaxis study (WV15673/WV15697), as these trials investigated the active agent at multiple doses. These studies included standard‐dose and high‐dose oseltamivir arms. For these analyses we used logistic regression, adjusting for study effects if appropriate (i.e. for the two treatment trials) and testing for trend using a likelihood ratio test. We tested the hypothesis that increased dose of drug leads to increased incidence of adverse effects.
Search methods for identification of studies
To identify trials in the manufacturer‐funded clinical trial programmes for NIs, as well as non‐manufacturer‐funded clinical trials of NIs, we used a variety of methods applied to a variety of sources from the literature, manufacturers and from regulatory bodies. These methods, as well as our methodology for identifying and obtaining relevant clinical study reports, are detailed in Appendix 4, Appendix 5 and Appendix 6.
Electronic searches
We used electronic searches to identify trials not identified by the methods outlined in Appendix 4, particularly for non‐manufacturer‐funded clinical trials. See Appendix 5 for details. For the 2012 review, we updated our searches of the electronic databases of published studies that were previously carried out for the Cochrane reviews on NIs in children (Matheson 2007) and healthy adults (Jefferson 2010a), and then updated the searches again on 22 July 2013.
Searching other resources
For the description of our searches for regulatory information (FDA, EMA, Roche, GlaxoSmithKline (GSK), Japanese Pharmaceuticals and Medical Devices Agency (PMDA)), see Appendix 6.
Data collection and analysis
Collection and inventory of the evidence base was facilitated by the tools specifically developed for the review (Appendix 7). The overall risk of bias is presented graphically in Figure 1 and summarised in Figure 2.
1.
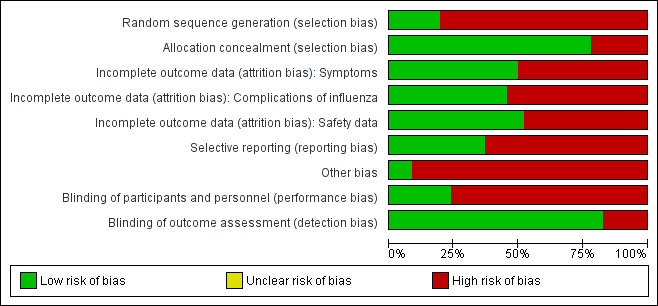
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies. 'Other bias' includes potentially active placebos.
2.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study. 'Other bias' includes potentially active placebos.
Selection of studies
For this 2013 review, two authors (PD, TJ) reapplied the inclusion criteria for the oseltamivir clinical study reports and resolved disagreements by discussion. Two review authors (ES, IO) applied the criteria for the zanamivir clinical study reports while one review author (CH) arbitrated.
For the procedures followed in the 2012 review, see Appendix 8 and Appendix 9.
Data extraction and management
The sizeable quantity of available data led us to subdivide the extraction, appraisal and analysis of the data into a two‐stage exercise. In Stage 1 we assessed the reliability and completeness of the identified trial data. We decided to include in Stage 2 of the review (full analysis following standard Cochrane methods) only data that satisfied the following three criteria.
Completeness. Clinical study reports/unpublished reports include both identifiable CONSORT statement‐specified methods to enable replication of the study. Identifiable CONSORT statement‐specified results (primary outcomes, tables, appendices) must be available.
Internal consistency. All parts (for example, denominators) of the same clinical study reports/unpublished report are broadly consistent.
External consistency. Consistency of data as reported in regulatory documents, other versions of the same clinical study reports/unpublished reports and other references, to be established by cross‐checking.
This was a different approach to that used in the previous version of the current review (Jefferson 2012), since we only had incomplete information at that time and only applied the second and third criteria.
Stage 1
For details of the use of the CONSORT‐based extraction template and the assessment for Stage 1 inclusion in the A159 (Jefferson 2012) review, see Appendix 7. In this review assessment for inclusion in Stage 1 was part of the inclusion procedure.
Stage 2
In Stage 2, one review author extracted data and a second review author checked it. We extracted data onto standard forms, checked and recorded it.
Use of regulatory information
We used regulatory information to assess the possible correlation between citation frequency of oseltamivir treatment trials in the FDA regulatory documents and trial size.
Post‐protocol analyses
After publication of the A159 protocol in December 2010, but before validation of our CONSORT‐based extractions in the Northern Hemisphere spring of 2011, we decided to carry out analyses (which we called post‐protocol analyses) to test five null hypotheses that we had formulated while reading, summarising and reconstructing the clinical study reports. The hypotheses originated from our observations of discrepancies and other unexpected observations in the clinical study reports' data and were informed by reading regulatory information. Appendix 10 reports the rationale, methods to formulate and test, and the results of the hypotheses.
The hypotheses reflect the uncertainty prevailing in the evidence base at a time when full clinical study reports were not available for all studies.
Assessment of risk of bias in included studies
Previous studies comparing regulatory with published or internal company sources of evidence have reported a variety of different biases that affect medical knowledge (Chou 2005; MacLean 2003; McGauran 2010; Wieseler 2013). We will report in detail elsewhere our comments on using the Cochrane 'Risk of bias' tool (Higgins 2011) to appraise clinical study reports and for trial programmes, and our efforts to construct an instrument for assessing risk of bias in complete clinical study reports. A full description of the methods used to quantify biases will be published in another paper.
Measures of treatment effect
To estimate treatment effects we first calculated the risk ratios (RRs) and used the average (mean) control event rate and the pooled RRs reported in the figures to calculate the risk differences (RD). For consistency we adopted this method for both the 'Summary of findings' tables and for the RDs reported in the text. For the analysis we chose to report the RRs as they are more consistent across the studies, and we have reported the heterogeneity for the pooled RR. We reinterpreted the results using the RD as this result is applicable to clinical decision‐making. We calculated mean differences (MDs) for time to first alleviation of symptoms. For time to first alleviation of symptoms we also estimated the treatment effect as the percentage reduction in the average time to first alleviation of symptoms in the placebo group. Most zanamivir clinical study reports only reported treatment effects in terms of medians in each treatment group as well as P values from a hypothesis test comparing the time‐to‐event distributions. These data are insufficient for conducting meta‐analysis. However, often sufficient time‐to‐event data were reported to allow us to estimate restricted means and standard deviations. Restricted means are based on the maximum time reported where alleviation occurred. There were some patients where alleviation was censored at the maximum follow‐up time, therefore restricted means are under‐estimates of the true means. However, the proportion of patients censored was generally low and similar in both treatment arms, hence this limitation is unlikely to have led to bias. The length of follow‐up varied across trials and this has led to high variation in the estimated means and standard deviations (SDs) across trials.
A post hoc analysis was undertaken after we discovered seven zanamivir trials provided data on time to first alleviation of symptoms with and without relief medication. Each patient in the studies may or may not have taken relief medication during the trial. Alleviation of symptoms may have occurred while the patient was taking relief medication and the "standard" comparison was made using this scenario. However, an additional analysis used a stricter definition where alleviation of symptoms could only be achieved without the use of relief medication. For example, a patient may have achieved alleviation using relief medication after five days but took seven days to achieve alleviation without the use of relief medication. The comparison we reported is for all patients where we used the stricter definition for the zanamivir group (alleviation without relief medication) and the less strict definition for the placebo group (alleviation with relief medication).
We planned to use the tri‐dimensional dose‐relatedness, timing and patient susceptibility (DoTS) methodology to assess the likelihood of harms causality (Aronson 2003), but the quality of the data available did not allow for this.
Unit of analysis issues
Problems with unit of analysis are described in the 'Risk of bias' and 'Post‐protocol hypotheses' sections.
Dealing with missing data
We developed a comprehensive strategy for dealing with data that we know are missing at the trial level, i.e. unpublished trials (see Search methods for identification of studies section and Appendix 4, Appendix 5 and Appendix 6) and unreliable published records, which are a very concentrated summary of clinical study reports. For example, in the oseltamivir trial programme, some trials' clinical study reports (e.g. WP16263) consist of 8545 pages. This has a 1000‐fold greater length compared to its published version (Dutkowski 2010), which consists of seven pages. The purpose of this review is to provide as complete a picture as possible of trial programmes, without reliance on the published literature. Appendix 11 reports an example of the content of a typical Roche clinical study report.
Assessment of heterogeneity
We used Tau2 (inverse variance method) and the I2 statistic to estimate between‐study variance as measures of the level of statistical heterogeneity and the Chi2 test to test for heterogeneity.
Assessment of reporting biases
We carried out assessment of reporting biases (comparing clinical study report with the relevant publication) only in the first publication of A159. For this version, as we had complete clinical study reports for the trial programmes of the two drugs, we expected to find all relevant information in these documents and adopted a binary assessment (high risk, low risk or unclear bias).
Data synthesis
We used the random‐effects approach of DerSimonian and Laird based on MDs for analysis of time to first alleviation of symptoms. For all other outcomes we used the random‐effects approach for binary data of DerSimonian and Laird, where Tau2 was estimated using the inverse variance method.
Whilst overall symptom reduction is well documented, our interest was particularly focused on complications and adverse events, as this is where evidence is currently scarce or inconclusive (Jefferson 2009a; Matheson 2007; Shun‐Shin 2009). Our preliminary examination of clinical study reports identified that some symptoms and sequelae of influenza (such as “pneumonia”) had been classified as either a ‘complication of influenza’ or as an 'adverse event of the treatment’ or both. We called this somewhat confusing classification 'compliharms'. We decided to deal with compliharms as follows. We identified complications of particular clinical interest as “pneumonia”, bronchitis, otitis media and sinusitis. We tabulated the type of data capture used for each complication (“secondary illness”) by study including the following variables: definition of what events are termed complications, which part of the clinical study report captured data on complications, who reported and captured the data, which diagnostic method was used, whether and where the diagnostic pathway was (usually a form) and whether prescription for treatment were captured. We then aimed to stratify our analysis by method of diagnosis with three possible criteria: (1) laboratory‐confirmed diagnosis (e.g. based on radiological‐ or microbiologically‐confirmed evidence of infection); (2) clinical diagnosis without laboratory‐confirmation (diagnosed by a doctor/investigator after a clinical examination); (3) other type of diagnosis such as self‐reported by patient. We also conducted analysis of any complication (such as “pneumonia”, bronchitis, otitis media and sinusitis) that was classified as serious or led to study withdrawal.
We tested the effects of oseltamivir in prophylaxis of influenza and influenza‐like illness. However the clinical study reports of prophylaxis trials do not define influenza‐like illness but report eight different definitions for influenza with laboratory‐confirmation (see web extra influenza definitions).
This is a complex and confusing set of definitions where, for example, the definition for Upper Respiratory Tract Infection (URTI) with systemic disturbance is the same as one of the definitions for asymptomatic influenza. After discovering the absence of a definition for influenza‐like illness and the complex and confusing definitions for laboratory‐confirmed influenza, we classified influenza‐like illness as two or more symptoms from the following: nasal congestion, headache, chills/sweats, sore throat, cough, fatigue, myalgia and fever. These were the symptoms reported in the efficacy listing of individual patients in Module 3 of the prophylaxis trials clinical study reports.
In two oseltamivir treatment trials (WV15670; WV15671) and one prophylaxis study (WV15673/WV15697) there were three treatment arms comparing placebo, standard dose and high dose. For time to first alleviation of symptoms we restricted comparison to placebo versus standard dose (as this is how it was reported in the original report). However, for all other outcomes we combined the standard and high‐dose treatment arms. There was little apparent difference in the incidence of outcomes between the standard and high‐dose arms and combining the arms did not appear to cause heterogeneity. However, in two cases there was some evidence of a dose‐response effect. These cases are described more fully in the Results section under 'Analysis of harms'.
The majority of zanamivir trials compared placebo with inhaled zanamivir. However, some trials also included an intranasal zanamivir treatment arm and a combined arm of inhaled and intranasal treatment. The multiple zanamivir arms were generally combined for meta‐analysis as effects appeared similar and did not appear to cause heterogeneity.
Subgroup analysis and investigation of heterogeneity
We investigated the robustness of complications outcomes using subgroup analysis by method of diagnosis. We investigated high estimates of heterogeneity, where possible, using subgroup analysis. For example, we conducted subgroup analysis of time to first alleviation of symptoms in studies of oseltamivir treatment in children by partitioning studies into those of otherwise healthy children and those of children with chronic illness (asthma). Based on a referee's comment, we conducted a subgroup analysis on time to first alleviation of symptoms by infection status for zanamivir. We could not do a similar analysis for oseltamivir because we did not have data on the non‐influenza‐infected patients and we could not correctly identify the patients with influenza infection due to the effect of oseltamivir on antibodies.
In the trial programmes for both oseltamivir and zanamivir there was large variation in treatment effects for pneumonia across the populations studied (i.e. adults and children as well as treatment and prophylaxis), hence we conducted meta‐regression to investigate this heterogeneity. We included all studies that reported pneumonia (32 studies in total) and investigated the four binary factors: age group (adults versus children); drug (oseltamivir versus zanamivir); indication (treatment versus prophylaxis) and method of diagnosis. For oseltamivir studies, the method of diagnosis was either based on data collected on non‐specific adverse events or secondary/intercurrent illness forms or data collected on specific "diagnosis of secondary illness" forms that included objective criteria such as X‐ray confirmation. For zanamivir, two trials included X‐ray confirmation of pneumonia. We conducted meta‐regression in Stata/SE, version 13 for Windows using the metareg command. There were some studies where one treatment group had zero events, therefore we added 0.5 events to all treatment groups for all studies prior to analysis. The dependent variable in the regression was log relative risk. A further post hoc analysis was undertaken after we discovered seven trials provided data on time to first alleviation of symptoms with and without relief medication. Each patient in the studies may or may not have taken relief medication during the trial. Alleviation of symptoms may have occurred while the patient was taking relief medication and the "standard" comparison was made using this scenario. However, an additional analysis used a stricter definition where alleviation of symptoms could only be achieved without the use of relief medication. For example, a patient may have achieved alleviation using relief medication after five days but took seven days to achieve alleviation without the use of relief medication. The comparison we reported is for all patients where we used the stricter definition for the zanamivir group (alleviation without relief medication) and the less strict definition for the placebo group (alleviation with relief medication).
Sensitivity analysis
Sensitivity analyses applicable to our post‐protocol analyses have been covered earlier in the Methods section of this review. We used the fixed‐effect method of Mantel and Haenszel as a sensitivity analysis to supplement our primary analyses using the random‐effects method of DerSimonian and Laird. Random‐effects meta‐analysis is known to be overly conservative with sparse data. Hence we conducted sensitivity analysis using Peto's method on two occasions where we had sparse data and borderline statistically significant results (prophylaxis with oseltamivir: renal body system on‐treatment and psychiatric body system on‐treatment).
Results
Description of studies
We searched trial registries, electronic databases and regulatory archives, corresponded with manufacturers to identify all trials and requested clinical study reports. Although this review focuses on the primary data sources of manufacturers, we checked that there were no published randomised controlled trials (RCTs) from non‐manufacturer sources by running electronic searches in the following databases: the Cochrane Central Register of Controlled Trials (CENTRAL 2013, Issue 6), limited to year published 2010 to 2013 (20 search results); MEDLINE (January 2011 to July week 2, 2013) (56 search results) and MEDLINE (Ovid) from 1 January 2011 to July week 2, 2013 (56 search results); EMBASE (January 2011 to July 2013) (90 search results) and Embase.com from 1 January 2011 to July 2013 (90 search results); and PubMed (not MEDLINE) with no date limit (21 records). We searched PubMed to identify publisher‐submitted records that will never be indexed in MEDLINE and the most recently added records not yet indexed in MEDLINE. To identify reviews that may possibly have referenced further trials we searched: the Database of Reviews of Effects (DARE) (2013, Issue 2 of 4 April) (four search results); the NHS Economic Evaluation Database (NHSEED) (Issue 2 of 4 April 2013) (two search results), both resources part of The Cochrane Library (accessed 22 July 2013), and the Health Economic Evaluations Database (HEED) (searched 22 July 2013) (three search results).
Results of the search
Use of regulatory information
We were able to download 2673 pages from the FDA website. The table of contents is in Table 2, Table 3, Table 4 and Table 5. We used these pages to identify all trials that had been conducted within a drug's trial programme. There was no correlation between citation frequency of oseltamivir treatment trials in the FDA regulatory documents and trial size. The biggest treatment trial (M76001) is cited only four times in three documents, while other contemporary treatment trials are cited far more (WV15670; WV15671; WV15730; WV15812/WV15872; WV15707). WV15670, for example, is cited 46 times in the FDA documents. However, the combined enrolled denominator of the four treatment trials completed at the time (WV15670; WV15671; WV15707; WV15730) was 1442, smaller than M76001 (1459). This suggested that the FDA's regulatory evaluation of Roche's New Drug Application was based predominantly on what Roche had offered them as "pivotal" or trials that best demonstrated the properties of oseltamivir, not the complete evidence base of all oseltamivir trials. One possible alternative explanation for this observation could have been the interval between trial completion, generation of the report and New Drug Applications (NDA) submission. This explanation is supported by the relatively brief interval between completion of the M76001 trial (19 February 1999) and submission (on 30 April 1999) of NDA 021087 to the FDA. However, the core part of the submission (the clinical development programme) contains data from two (at the time) ongoing trials (WV15819/WV15876/WV15978; WV15812/WV15872).
2. Table of contents for studies of zanamivir described in regulatory documentation from the FDA (USA).
| Mentioned study | File name | Pages where study is mentioned (separated by commas) | Note |
| 113502 | |||
| 113625 | |||
| 113678 | |||
| 114045 | |||
| NAI108166 | |||
| 105934 | |||
| NAI106784 | |||
| 107485 | |||
| 108127 | |||
| 112311 | |||
| 112312 | |||
| 113268 | |||
| GCP/95/045 | |||
| NAI10901 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 15.15 | |
| NAI10902 | |||
| NAI30008 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 15 | 7 documents with 14 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin3.pdf | 13 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 19, 19, 20 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 1, 1, 3, 4, 4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview9.pdf | 7.7 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/21036ltr.pdf | 2 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_MEDR.pdf | 33 | ||
| NAI30009 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 1.2 | 7 documents with 110 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P1.pdf | 10, 10, 12, 13, 13, 14, 14, 17, 29, 42, 61, 62, 64, 64, 65, 65, 68 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P2.pdf | 33, 34, 36, 43, 43, 43, 43, 52, 52, 52, 53, 53, 56, 57 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 5, 5, 5, 6, 6, 8, 8 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_MEDR.pdf | 3, 3, 3, 3, 3, 3, 3, 4, 4, 5, 8, 9, 9, 10, 10, 11, 11, 11, 14, 14, 15, 16, 17, 19, 19, 19, 20, 20, 22, 23, 23, 23, 24, 24, 24, 25, 25, 25, 25, 25, 25, 26, 26, 26, 27, 27, 28, 28, 28, 29, 29, 31, 31, 31, 31 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_MICROBR.pdf | 3, 3, 4, 4, 4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_STATR.pdf | 2, 2, 2, 4, 7, 12, 18, 18, 18, 19 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P1.pdf | 31.56 | 1 document with 2 instances | |
| NAI30010 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 1.2 | 6 documents with 65 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P1.pdf | 10, 12, 13, 14, 14, 15, 17, 62, 62, 62, 64 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P2.pdf | 34, 34, 36, 43, 53 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 5, 5, 6, 6 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_MEDR.pdf | 3, 3, 3, 3, 3, 4, 5, 18, 19, 21, 21, 22, 23, 23, 23, 23, 24, 25, 25, 25, 26, 26, 26, 26, 27, 27, 27, 28, 28, 29, 29, 29, 30, 31, 31, 31, 32 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_STATR.pdf | 2, 2, 13, 13, 13, 19 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 6 | 1 document with 1 instance | |
| NAI30012 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 1 | 1 document with 1 instance |
| NAI30015 | |||
| NAI30020 | |||
| NAI30028 | |||
| NAI30034 | |||
| NAI40012 | |||
| NAIA1009 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P1.pdf | 56 | 4 documents with 17 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P2.pdf | 1, 1, 1, 48, 49, 52 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 5, 5, 6 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_MEDR.pdf | 3, 3, 6, 7, 20, 31, 31 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 18 | 5 documents with 5 instances | |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 9 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P2.pdf | 52 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 11 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_STATR.pdf | 2 | ||
| NAIA3002 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin1.pdf | 15 | 13 documents with 122 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 6, 6, 7, 7, 14, 15, 22, 22, 23 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin3.pdf | 1, 4, 4, 12, 12, 12, 12, 17 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 4, 14, 14, 14, 14, 14, 15, 15, 15, 15, 16 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview2.pdf | 1, 2, 3, 4, 4, 5, 6, 6, 6, 8, 8, 9, 9, 9, 12, 12, 15, 16, 16, 16, 17 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview3.pdf | 5, 5, 6, 6, 6, 7, 7, 7, 8, 8, 9, 9, 9, 10, 11, 12, 13, 13, 14, 15, 15, 17, 18, 18, 19, 20, 21 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview4.pdf | 1, 1, 1, 1, 2, 6 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 4, 5, 10, 12 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 1, 1, 2, 2, 2, 2, 3, 3, 4, 4, 5, 5, 7, 8, 10, 11, 12, 14, 16, 16, 16, 16, 16, 17 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 2, 2, 6, 6, 8, 8, 9, 10 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview9.pdf | 10 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐stats.pdf | 7 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 5 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 15 | 1 document with 1 instance | |
| NAIA3003 |
Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 17, 17, 18 | 3 documents with 6 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 4.4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview9.pdf | 22 | ||
| NAIA3004 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin3.pdf | 14 | 4 documents with 8 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 7 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 18, 18, 19 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 4, 4, 4 | ||
| NAIA3005 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin3.pdf | 14 | 5 documents with 12 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 5 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 12, 12, 12, 13, 14, 15, 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 14.15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_ADMINCORRES_P2.pdf | 38 | ||
| NAIB1002 | |||
| NAIB3002 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin1.pdf | 15 | 14 documents with 99 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 14, 15, 15, 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin3.pdf | 11.12 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 4, 14, 14, 14, 14, 14, 14, 14, 14 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview2.pdf | 9, 9, 9, 9, 9, 9, 10, 11, 12, 12, 12, 12, 13, 13, 13, 14, 14, 14, 15, 15, 16, 16, 16, 17 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview3.pdf | 4, 5, 6, 6, 6, 7, 7, 7, 8, 8, 8, 9, 9, 11, 12, 12, 13, 13, 14, 15, 17, 18, 18, 19, 20, 21 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview4.pdf | 1, 1, 1, 1, 2 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 4, 5, 10, 12 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 2, 3, 3, 7, 8, 10, 11, 14, 15, 16, 16, 16 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 7, 8, 8, 8, 9, 9 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview9.pdf | 10.2 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐stats.pdf | 7 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 5.5 | ||
| NAI30011 | |||
| NAIB2007 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin1.pdf | 15 | 7 documents with 18 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 5 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview4.pdf | 14, 15, 15, 16, 16, 17, 17, 17, 18 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 3 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 8, 10, 10, 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 2 | ||
| NAIA2006 | |||
| NAIB2006 | |||
| NAIB1007 | |||
| C94‐009 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 17 | 1 document with 1 instance |
| C94‐085 |
Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 17 | 2 documents with 2 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview9.pdf | 22 | ||
| NAIB1001 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 17 | 1 document with 1 instance |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/20000426_001/21‐036‐S001_RELENZA_BIOPHARMR.pdf | 6 | 1 document with 1 instance | |
| NAIA2005 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin1.pdf | 15 | 10 documents with 44 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 7, 17, 10 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin3.pdf | 2.4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 4.5 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview4.pdf | 2, 2, 3, 3, 3, 3, 5, 6, 6, 6, 6, 8, 8, 8, 9, 11, 12, 12, 13, 14, 14, 14, 14, 14, 15, 18 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 7.7 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 3.4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 2, 5, 9, 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 10 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐microbiology.pdf | 21 | ||
| NAIB2005 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin1.pdf | 15 | 9 documents with 43 instances |
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐admin2.pdf | 17, 20, 20, 22, 23 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview1.pdf | 5.5 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview4.pdf | 3, 3, 3, 7, 8, 8, 8, 9, 10, 11, 11, 11, 11, 11, 12, 12, 12, 13, 14, 14, 14, 14, 14, 14, 14, 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 7.7 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 3.4 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview7.pdf | 2, 9, 15 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview8.pdf | 2 | ||
| Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐microbiology.pdf | 21 | ||
| NAIA/B2008 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview6.pdf | 4 | 1 document with 1 instance |
| NAIA2010 | Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036‐medreview5.pdf | 16 | 1 document with 1 instance |
| NAIA/B2009 | |||
| 167‐02 | |||
| 167‐03 | |||
| 167‐05 | |||
| 167‐04 | |||
| JNAI‐03 | |||
| JNAI‐02 | |||
| JNAI‐01 | |||
| JNAI‐07 | |||
| JNAI‐04 | |||
| PE‐01 | |||
| 167‐101 | |||
| 167T3‐11 |
Zanamivir trials citation by trial ID and source FDA file. Page numbers separated by commas (where applicable) indicate which trial is cited where in which regulatory file. Blank spaces indicate no citation for known trials.
All the studies have been searched in the folder "Tamiflu and Relenza/Relenza/Relenza ‐ NDA 021036/19990726_000/021036". File name is left blank when the study was not present in that folder.
3. Table of contents for studies of oseltamivir described in regulatory documentation from the FDA (USA).
| Referenced study | File name | Pages where study is mentioned (separated by commas) | Note |
| NP15717 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 46.46 | 6 documents with 13 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 14, 15, 15 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 2 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_BioPharmr.pdf | 5, 8, 10, 13, 31 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| NP15718 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 17 | 1 document with 1 instance |
| NP15728 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 16.35 | 3 documents with 6 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 11 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 45, 46, 47 | ||
| NP15757 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 92, 93, 104, 122, 126, 131, 144, 144, 145 | 1 document with 9 instances |
| NP15826 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 47 | 9 documents with 26 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20040624_016/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_ADMINCORRES.pdf | 6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 2 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_BioPharmr.pdf | 4, 5, 5, 8, 8, 8, 10, 17, 29, 30, 30, 30, 30, 30, 31, 31 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 9.1 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 9.1 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20040624_016/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_ADMINCORRES.pdf | 6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| NP15827 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 10.12 | 2 documents with 7 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 16, 16, 17, 17, 17 | ||
| WP15525 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 21, 25, 26, 27, 27, 27, 27, 42, 42, 44 | 3 document with 13 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 2.2 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_BioPharmr.pdf | 29 | ||
| WP15647 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 24, 27, 27 | 2 documents with 4 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 44 | ||
| WP15648 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 39 | 3 documents with 8 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 44.44 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 94, 128, 153, 153, 154 | ||
| WP15676 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 28.33 | 3 documents with 4 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 11 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 45 | ||
| WV15670 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 2, 44, 44 | 6 documents with 45 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 6, 19, 37, 38, 39, 39, 39, 39, 40, 41, 41, 42, 43, 44, 48, 48, 49, 49 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 1, 25, 25, 35, 35, 39, 39, 47 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 3, 3, 4, 4, 5, 5, 5, 8, 9, 10, 17, 17, 21, 22, | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 189 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| WV15671 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 2, 44, 44 | 7 documents with 50 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 6, 16, 19, 24, 24, 25, 25, 26, 27, 27, 28, 32, 34, 35, 36, 37, 38, 39, 39, 39, 40, 41, 46, 49, 49 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 1, 25, 25, 35, 38, 47 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 3, 4, 4, 5, 5, 5, 5, 9, 10, 10, 15, 17, 21 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 189 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| WV15673 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 3 | 3 documents with 50 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 18, 18, 18, 20, 21, 21, 21, 22, 39 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 58, 59, 71, 71, 71, 71, 71, 72, 72, 73, 73, 76, 76, 76, 76, 76, 77, 77, 79, 82, 83, 83, 84, 122, 124, 125, 126, 128, 131, 131, 132, 133, 134, 134, 145, 145, 156, 169, 177, 189 | ||
| WV15697 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 39 | 2 documents with 40 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 58, 59, 71, 71, 71, 71, 71, 72, 72, 73, 73, 76, 76, 76, 76, 76, 77, 77, 79, 82, 83, 83, 84, 122, 126, 128, 131, 131, 131, 132, 133, 134, 145, 145, 152, 153, 156, 162, 189 | ||
| WV15708 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 3 | 3 documents with 39 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 23, 35, 39, 41 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 71, 71, 71, 71, 71, 72, 72, 72, 72, 75, 75, 75, 75, 77, 77, 78, 79, 79, 82, 82, 122, 125, 125, 126, 131, 134, 134, 135, 135, 149, 151, 152, 152, 153 | ||
| WV15708D | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 3 | 2 documents with 3 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 23.35 | ||
| WV15730 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_bior.pdf | 44.44 | 5 documents with 15 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 6, 9, 19, 49, 50, 50 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 1, 1, 25, 25, 27 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 189 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 3 | ||
| WV15731 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 17 | 4 documents with 9 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 5, 30, 37 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Microbr.pdf | 5.6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 5, 30, 37 | ||
| WV15758 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P1.pdf | 12, 19, 19, 36 | 9 documents with 92 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 2, 8, 17, 39, 39, 57, 57 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_BioPharmr.pdf | 3, 4, 5, 5, 5, 8, 10, 17, 27, 30 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Corres.pdf | 6.9 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 5, 5, 9, 9, 10, 11, 12, 12, 16, 18, 18, 18, 19, 19, 31, 31, 31, 33, 33, 35, 36, 37, 37, 37, 37, 37, 37, 37, 40, 43 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Microbr.pdf | 2, 4, 5, 6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 5, 5, 9, 9, 10, 11, 12, 12, 16, 18, 18, 18, 19, 19, 31, 31, 31, 33, 33, 35, 36, 37, 37, 37, 37, 37, 37, 37, 40, 43 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20040624_016/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_ADMINCORRES.pdf | 6, 6, 8 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 2.3 | ||
| WV15759 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P1.pdf | 12.13 | 7 documents with 44 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 39 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 5, 10, 30, 30, 30, 30, 31, 32, 32, 33, 34, 37, 37, 37, 40, 44 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Microbr.pdf | 2, 4, 4, 5, 6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 5, 10, 30, 30, 30, 30, 31, 32, 32, 33, 34, 37, 37, 37, 40, 44 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20040624_016/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_ADMINCORRES.pdf | 6, 6, 9 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 2 | ||
| WV15799 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 28, 28, 28, 28, 28, 29, 29, 30, 30, 30, 30, 30, 31, 31, 31, 31, 32, 32, 32, 32, 32, 33, 33, 34, 34, 35, 35, 35, 36, 37, 37, 37, 37, 37, 38, 38, 38, 39, 39, 40, 40, 40, 40, 40, 58, 60, 71, 71, 71, 71, 71, 72, 72, 73, 76, 76, 76, 77, 78, 79, 84, 85, 122, 125, 125, 126, 126, 128, 131, 140, 140, 140, 143, 147, 149, 156, 162, 169, 175, 187, 203, 208, 208 | 4 documents with 89 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 10.11 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 10.11 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20040624_016/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_ADMINCORRES.pdf | 6.7 | ||
| WV15812 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 3, 6, 10, 12 | 2 documents with 9 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 6, 8, 10, 25, 35 | ||
| WV15819 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P1.pdf | 6, 10, 12, 15 | 2 documents with 8 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/19991027_000/21087_Tamiflu_medr_P2.pdf | 2, 6, 6, 39 | ||
| WV15825 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20001117_002/21‐087SE1‐002_review.pdf | 41, 41, 41, 41, 42, 42, 42, 42, 42, 42, 43, 44, 58, 59, 71, 71, 71, 71, 71, 72, 72, 72, 72, 73, 73, 75, 75, 77, 77, 78, 79, 79, 79, 80, 80, 80, 81, 82, 85, 125, 125, 126, 126, 128, 131, 134, 134, 135, 135, 137, 137, 138, 145, 150, 151, 152, 152, 155, 156, 162, 169, 180, 204, 211 | 1 document with 64 instances |
| WV15871 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P1.pdf | 12.13 | 7 documents with 42 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Admindocs_P2.pdf | 39 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 5, 11, 30, 31, 31, 32, 32, 32, 33, 34, 37, 37, 37, 37, 37, 37, 40 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Microbr.pdf | 2, 5, 6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 5, 11, 30, 31, 31, 32, 32, 32, 33, 34, 37, 37, 37, 37, 37, 37, 40 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021087/20040624_016/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_ADMINCORRES.pdf | 6 | ||
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20040624_010/021087_S016_TAMIFLU CAPSULES ‐ DRY POWDER_BIOPHARMR.pdf | 2 | ||
| WV15872 | Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Medr.pdf | 11.33 | 2 documents with 4 instances |
| Tamiflu and Relenza/Tamiflu/Tamiflu ‐ NDA 021246/20001214_000/21‐246_Tamiflu_Statr.pdf | 11.33 |
Oseltamivir trials citation by trial ID and source FDA file. Page numbers separated by commas (where applicable) indicate which trial is cited where in which regulatory file. Blank spaces indicate no citation for known trials.
Search strategy: WV15758 OR WV 15758 OR Trial 15758 OR Trial15758 OR Trials 15758 OR Trials15758 OR 15758 OR study 15758 OR study15758
4. Table of contents for studies of zanamivir described in regulatory documentation from NICE (UK).
| Mentioned study | File name | Pages where study is mentioned (separated by commas) | Note |
| NAI106784 | |||
| 107485 | |||
| 108127 | |||
| 112311 | |||
| 112312 | |||
| 113268 | |||
| GCP/95/045 | |||
| NAI10901 | |||
| NAI10902 | |||
| NAI30008 | Relenza treatment submission executive summary.pdf | 4 | 3 documents with 10 instances |
| Relenza treatment submission full document.pdf | 5, 26, 26, 26, 146 | ||
| Relenza treatment submission main text.pdf | 5, 26, 26, 26 | ||
| NAI30009 | NAI30010 study report pdf\FINAL NAI30010 for sign‐off.pdf | 102 | 7 documents with 461 instances |
| NAI30009 study report pdf\CSR30009.pdf | |||
| NAI30009 study report pdf\NAI 30009 HO final FSR.pdf | |||
| NAI30009 study report pdf\suptables.pdf | |||
| NAI30009 study report pdf\tables.pdf | |||
| Relenza treatment submission full document.pdf | 16, 16, 17, 18, 18, 18, 18, 19, 27, 30, 31, | ||
| Relenza treatment submission main text.pdf | 16, 16, 17, 18, 18, 18, 18, 19, 27, 30, 31, 76, 128, 130, 132, 134, 144 | ||
| NAI30010 | NAI30010 study report\Final NAI30010 for sign‐off.pdf | 7 documents with 399 instances | |
| NAI30010 study report pdf\NAI30010 HO final FSR.pdf | |||
| NAI30010 study report pdf\suptables.pdf | |||
| NAI30010 study report pdf\tables.pdf | |||
| Relenza prophylaxis submission.pdf | 2, 5, 8, 11, 12, 19, 20, 21, 23, 24 | ||
| Relenza treatment submission full document.pdf | 16, 16, 17, 18, 18, 18, 27, 30, 31, 76, 135, 137, 139, 141, 143, 144 | ||
| Relenza treatment submission main text.pdf | 16, 16, 17, 18, 18, 18, 27, 30, 31 | ||
| NAI30012 | Relenza treatment submission executive summary.pdf | 4 | 3 documents with 8 instances |
| NAI30012 | Relenza treatment submission full document.pdf | 5, 26, 26, 146 | |
| NAI30012 | Relenza treatment submission main text.pdf | 5, 26, 26 | |
| NAI30015 | Relenza treatment submission full document.pdf | 146 | 1 document with 1 instance |
| NAI30020 | |||
| NAI30028 | |||
| NAI30031 | |||
| NAI30034 | |||
| NAI40012 | |||
| NAIA1009 |
NAI30010 study report pdf\FINAL NAI30010 for sign‐off.pdf | 101 | 2 documents with 3 instances |
| NAI30009 study report pdf\CSR30009.pdf | 28.34 | ||
| NAIA3002 |
NAI30010 study report pdf\FINAL NAI30010 for sign‐off.pdf | 102 | 9 documents with 513 instances |
| NAI30009 study report pdf\CSR30009.pdf | 34.95 | ||
| NAI30009 study report pdf\NAI30009 HO final FSR.pdf | 22 | ||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | |||
| NAIA3002 study report pdf\NAIA3002 supporting tables 2.pdf | |||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28, 47, 49 | ||
| Relenza treatment submission full document.pdf | 16, 16, 17, 17, 18, 19, 27, 30, 31, 63, 63, 63, 76, 106, 106, 107, 107, 109, 109, 112, 112, 114, 114, 115, 115, 144 | ||
| Relenza treatment submission main text.pdf | 16, 16, 17, 17, 18, 19, 27, 30, 31 | ||
| NAIA3003 | Relenza prophylaxis submission.pdf | 10 | 1 document with 1 instance |
| NAIA3004 | Relenza prophylaxis submission.pdf | 10 | 1 document with 1 instance |
| NAIA3005 | NAI30010 study report pdf\FINAL NAI30010 for sign‐off.pdf | 36, 94, 94, 94, 95, 96, 96, 101 | 5 documents with 310 instances |
| NAI30010 study report pdf\NAI30010 HO FSR.pdf | 6.18 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | |||
| NAIA3005 study report pdf\TABS.pdf | |||
| Relenza prophylaxis submission.pdf | 2, 5, 6, 12, 13, 13, 15, 15, 16, 16, 17, 17, 18, 18 | ||
| NAIB1002 | |||
| NAIB3001 | NAI30009 study report pdf\CSR30009.pdf | 34, 50, 95 | 11 documents with 374 instances |
| NAI30009 study report pdf\NAI 30009 HO final FSR.pdf | 10.22 | ||
| NAI30010 study report pdf\FINAL NAI30010 for sign‐off.pdf | 102 | ||
| NAI30010 study report pdf \NAI30010 HO FSR.pdf | 17.17 | ||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3001 study report pdf\NAIB3001 full study report.pdf | |||
| NAIB3001 study report pdf\NAIB3001 supporting tables 1.pdf | |||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28 | ||
| Relenza treatment submission full document.pdf | 16, 16, 17, 18, 18, 18, 18, 27, 30, 31, 32, 63, 63, 63, 76, 99, 99, 101, 101, 103, 103, 105, 105, 144, 162 | ||
| Relenza treatment submission main text.pdf | 16, 16, 17, 18, 18, 18, 18, 27, 30, 31, 32 | ||
| NAIB3002 | NAI30009 study report pdf\CSR30009.pdf | 34.95 | 10 documents with 579 instances |
| NAI30009 study report pdf\NAI 30009 HO final FSR.pdf | 22 | ||
| NAI30010 study report pdf\FINAL NAI30010 for sign‐off.pdf | 102 | ||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28, 48, 50 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | |||
| NAIB3002 study report pdf\NAIB3002supporting tables 1.pdf | |||
| NAIB3002 study report pdf\NAIB3002supporting tables 2.pdf | |||
| Relenza treatment submission full document.pdf | 16, 16, 17, 17, 18, 19, 27, 30, 31, 63, 63, 63, 76, 117, 117, 117, 118, 118, 120, 120, 122, 122, 124, 124, 125, 125, 127, 127, 144 | ||
| Relenza treatment submission main text.pdf | 16, 16, 17, 17, 18, 19, 27, 30, 31 | ||
| NAI30011 | Relenza treatment submission full document.pdf | 146 | 1 document with 1 instance |
| NAIB2007 | NAI30009 study report pdf\CSR30009.pdf | 95 | 10 documents with 379 instances |
| NAI30009 study report pdf\NAI 30009 HO final FSR.pdf | 10 | ||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28, 28, 29 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB2007 study report pdf\b2007cr.pdf | |||
| NAIB2007 study report pdf\TABLES.pdf | |||
| NAIB3001 study report pdf\NAIB3001 full study report.pdf | 25.26 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28, 28, 29 | ||
| Relenza treatment submission full document.pdf | 16, 16, 17, 18, 18, 19, 27, 30, 31, 76, 91, 91, 92, 92, 94, 94, 96, 96, 98, 98, 144 | ||
| Relenza treatment submission main text.pdf | 16, 16, 17, 18, 18, 19, 27, 30, 31 | ||
| NAIA2006 | NAIA2005 study report pdf\a2005cr.pdf | 38, 73, 74 | 4 documents with 6 instances |
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28 | ||
| NAIB2006 | NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28 | 3 documents with 3 instances |
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28 | ||
| NAIB1007 | |||
| C94‐009 | |||
| C94‐085 | |||
| NAIB1001 | |||
| NAIB_1001 | |||
| NAIA2005 |
NAI30009 study report pdf\CSR30009.pdf | 95 | 12 documents with 895 instances |
| NAIA2005 study report pdf\a2005cr.pdf | |||
| NAIA2005 study report pdf\APPS_ALL.pdf | |||
| NAIA2005 study report pdf\TBS_ALL.pdf | |||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28, 28, 48, 48 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB2005 study report pdf\b2005cr.pdf | 7, 7, 22, 25, 26, 34, 34, 42, 71, 72, 72 | ||
| NAIB2007 study report pdf\b2007cr.pdf | 76 | ||
| NAIB3001 study report pdf\NAIB3001 full study report.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28, 28, 47, 47 | ||
| Relenza treatment submission full document.pdf | 16, 16, 16, 16, 17, 18, 27, 30, 76, 77, 77, 77, 79, 79, 79, 80, 80, 82, 82, 84, 84, 85, 144, 144 | ||
| Relenza treatment submission main text.pdf | 16, 16, 16, 16, 17, 18, 27, 30 | ||
| NAIB2005 |
NAI30009 study report pdf\CSR30009.pdf | 95 | 12 documents with 838 instances |
| NAIA2005 study report pdf\a2005cr.pdf | 7, 8, 8, 24, 24, 25, 43, 70, 74, 74 | ||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28, 28, 48, 48 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB2005 study report pdf\APPSNEW.pdf | |||
| NAIB2005 study report pdf\b2005cr.pdf | |||
| NAIB2005 study report pdf\TBS_ALL.pdf | |||
| NAIB2007 study report pdf\b2007cr.pdf | 76 | ||
| NAIB3001 study report pdf\NAIB3001 full study report.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28, 28, 47, 47 | ||
| Relenza treatment submission full document.pdf | 16, 16, 16, 16, 17, 18, 27, 30, 76, 77, 79, 79, 85, 85, 85, 86, 86, 88, 88, 90, 90, 144, 144 | ||
| Relenza treatment submission main text.pdf | 16, 16, 16, 16, 17, 18, 27, 30 | ||
| NAIA/B2008 |
NAI30009 study report pdf\CSR30009.pdf | 95 | 6 documents with 16 instances |
| NAI30009 study report pdf\NAI 30009 HO final FSR.pdf | 10 | ||
| NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28, 28, 29, 29 | ||
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3001 study report pdf\NAIB3001 full study report.pdf | 25, 26, 26, 26, 77 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28, 28, 29, 29 | ||
| NAIA2010 | NAIA3005 study report pdf\A3005cr01.pdf | 25 | 1 document with 1 instance |
| NAIA/B2009 | NAIA3002 study report pdf\NAIA3002 full study report.pdf | 28 | 3 documents with 3 instances |
| NAIA3005 study report pdf\A3005cr01.pdf | 25 | ||
| NAIB3002 study report pdf\NAIB3002 full study report.pdf | 28 | ||
| 167‐02 | |||
| 167‐03 | |||
| 167‐05 | |||
| 167‐04 | |||
| JNAI‐03 | |||
| JNAI‐02 | |||
| JNAI‐01 | |||
| JNAI‐07 | |||
| JNAI‐04 | |||
| PE‐01 | |||
| 167‐101 | |||
| 167T3‐11 |
Zanamivir trials citation by trial ID and source NICE file. Page numbers separated by commas (where applicable) indicate which trial is cited where in which file. Blank spaces indicate no citation for known trials
5. Table of contents for studies of oseltamivir described in regulatory documentation from NICE (UK).
| Referenced study | File name volume* | Pages where study is mentioned (separated by commas) | Note |
| 133312 | |||
| GS97‐802 | |||
| 133312 | |||
| GS‐97‐801 | |||
| JP15734 | |||
| JP15735 | |||
| JV15823 | |||
| JV15824 | |||
| JV16284 | |||
| M76001 | 1 | 33, 36, 37, 37, 38, 38, 39, 67, 68, 94, 95, 224 | 1 document with 12 instances |
| M76006 | |||
| ML20910 | |||
| ML22789 | |||
| ML22879 | |||
| MV21118 | |||
| MV22841 | |||
| NCT00298233 | |||
| NCT00555893 | |||
| NCT00707941 | |||
| NCT00799760 | |||
| NCT00830323 | |||
| ML25018 | |||
| NCT00867139 | |||
| NCT00873886 | |||
| NCT01002729 | |||
| NP15717 | 6 | 32, 75, 76, 77 | 2 documents with 5 instances |
| 8 | 68 | ||
| 6 | 73.98 | 1 document with 2 instances | |
| NP15718 | |||
| NP15728 | |||
| NP15757 | 8 | 68 | 1 document with 1 instance |
| NP15826 | 6 | 32, 75, 75, 75, 76, 76, 77, 78, 79, 80, 98 | 1 document with 11 instances |
| NP15827 | 8 | 68 | 1 document with 1 instance |
| NP22770 | |||
| NP25138 | |||
| NP25139 | |||
| NV16871 | |||
| NV20234 | |||
| NV20235 | |||
| NV20236 | |||
| NV20237 | |||
| NV22155 | |||
| NV25118 | |||
| NV25182 | |||
| PP16351 | |||
| WP15517 | 1 | 185.245 | 1 document with 2 instances |
| WP15525 | 1 | 185.245 | 1 document with 2 instances |
| WP15647 | |||
| WP15648 | |||
| WP15676 | |||
| WP15901 | |||
| WP22849 | |||
| WV144181 | |||
| WV15670 | 1 | 33, 36, 37, 37, 38, 38, 39, 47, 48, 48, 49, 49, 50, 53, 54, 54, 55, 163, 171, 188, 207, 209, 224, 245, 245, 252, 253, 253 | 7 documents with 1193 instances |
| 10 | 7, 36, 37, 37 | ||
| 2 | |||
| 3 | |||
| 4 | 90 | ||
| 6 | 35.98 | ||
| 8 | 65 | ||
| 2 | 20, 20, 20, 20, 20 | 1 document with 5 instances | |
| WV15671 |
1 | 33, 36, 37, 37, 38, 38, 39, 47, 48, 49, 49, 50, 53, 54, 54, 55, 163, 171, 188, 207, 209, 224, 245, 245 | 7 documents with 1222 instances |
| 10 | 7, 36, 37, 37 | ||
| 2 | 82 | ||
| 4 | |||
| 5 | |||
| 6 | 35.98 | ||
| 8 | 66 | ||
| WV15673 | 8 | 66 | 1 document with 1 instance |
| WV15673D | 8 | 66 | 1 document with 1 instance |
| WV15697 | 8 | 1 document with 1 instance | |
| WV15697D | 8 | 1 document with 1 instance | |
| WV15707 | 1 | 33, 36, 37, 37, 38, 67, 68, 224, 245, 245, 245, 246 | 1 document with 12 instances |
| WV15708 | |||
| WV15708D | |||
| WV15730 | 1 | 33, 36, 37, 37, 38, 38, 39, 47, 53, 54, 55, 186, 207, 224, 245, 245, 246 | 4 documents with 22 instances |
| 10 | 7, 36, 37 | ||
| 2 | 82 | ||
| 4 | 90 | ||
| WV15731 | 6 | 98 | 1 document with 1 instance |
| WV15758 | 1 | 36, 37, 82, 83, 84, 85, 86, 92, 94, 95, 97, 106, 224, 246 | 4 documents with 424 instances |
| 6 | |||
| 7 | |||
| 8 | 68 | ||
| WV15759 | 1 | 36, 37, 94, 95, 95, 109, 113, 114, 121, 122, 224, 246 | 1 document with 12 instances |
| WV15799 | 1 | 137, 139, 139, 232, 233 | 3 documents with 499 instances |
| 8 | |||
| 9 | |||
| WV15812 | 1 | 36, 37, 37, 38, 38, 39, 67, 68, 68, 107, 107, 107, 108, 108, 121, 121, 122, 123, 224, 246 | 2 documents with 197 instances |
| 10 | |||
| WV15819 | 1 | 33, 36, 37, 37, 38, 58, 58, 59, 59, 60, 61, 62, 62, 65, 65, 67, 68, 224, 246 | 2 documents with 173 instances |
| 10 | |||
| WV15825 | 8 | 66, 66 | 1 document with 2 instances |
| WV15871 | 1 | 109, 246 | 1 document with 2 instances |
| WV15872 | 1 | 36, 37, 37, 38, 38, 39, 67, 68, 68, 107, 107, 108, 108, 121, 121, 122, 123, 224 | 1 document with 18 instances |
| WV15876 | 1 | 246, 246 | 1 document with 2 instances |
| WV15978 | 1 | 67, 70, 175, 246, 246 | 1 document with 5 instances |
| WV16193 | |||
| ML16369 |
Oseltamivir trials citation by trial ID and source NICE file. Page numbers separated by commas (where applicable) indicate which trial is cited where in which file. Blank spaces indicate no citation for known trials.
All the studies have been searched in the folder "Roche submission".
When there is the number of the volume but no pages are mentioned, it means that the code of the study is cited more than 100 times.
*Number of the volume of the Tamiflu NICE submission.
The basis of the selection of trials to regulators is therefore unclear but appears to be dictated by criteria other than availability and size. The importance of trials (to manufacturers and possibly to regulators) may not be based on the same criteria that systematic reviewers would use (i.e. the capability of the trial to answer questions).
Due to the vast size of FDA documents, sometimes hundreds of pages long, it was difficult to determine important emerging themes solely by reading. To identify items of interest in the FDA comments we used word clouds (Feinberg 2009). Word clouds give greater prominence to words that appear more frequently in the source document. The resulting graphic representation showed words such as 'diary' and 'baseline' to be heavily mentioned in the relevant (abridged) text from the FDA's Medical Officer Review (FDA 1999c, PDF page 19). Examining the 'diary' entry in more detail, we found the following FDA comment:
"The majority of subjects participating in the treatment trials had only used the first diary card. The second diary card was issued in 15% to 20% of participants. In response to FDA's request, the applicant provided a summary of diary card dispensing in the 8/6/99 submission. It became apparent that instructions on when to start a second diary card were not uniformly followed in WV15670, WV15671, and WV15730 trials. There were examples of patients who had alleviated symptoms yet also received a second diary card. Conversely, there were also examples of patients who did not alleviate all symptoms but did not receive a second diary card. Thus the second diary card was used inconsistently which is viewed as a flaw of these trials. The lack of consistency in collecting symptom information after alleviation precluded a complete documentation of symptom fluctuation. Also missing second diary cards in subjects who had not alleviated symptoms were responsible for the majority of censored data which may have potentially influenced the results of efficacy analysis. In order to address the impact of censoring, the applicant performed several sensitivity analyses which will be summarized in the Integrated Summary of Efficacy".
This comment highlights problems with the follow‐up procedure of treatments trials, which may have impaired the regulator's ability to draw conclusions on the duration of effect of oseltamivir. It also provides a good example of how graphic methods can help identify crucial comments in vast regulatory files.
Several other experiments with text from the same FDA document showed that the choice of text to be represented as a Word cloud heavily influenced cloud construction, visibility of words and hence our ability to detect important comments. It is for this reason that we decided to adopt a mixed approach: mapping citations while reading FDA comments and integrating such comments in our appraisal of the evidence. Regulatory comments were all the more important, because at the time we developed this method we had few clinical study reports and comments helped to identify the gaps in our knowledge of the trial programmes.
Once the table of contents had been constructed, we postulated that given the huge work involved in reviewing lots of regulatory files, our new instrument could also help us by indicating which parts were more important than others, thus focusing our efforts. We experimented with a variety of methods reported in the Data collection and analysis section.
Clinical study reports
After prolonged correspondence and media pressure (Appendix 2), we were able to access the trial programmes for both oseltamivir and zanamivir without clauses restricting their accessibility to third parties.
Electronic searches
Two review authors (CDM, MT) independently scanned the titles and abstracts of the electronic searches. Three identified studies (NCT00980109; NCT01032837; JPRN‐JapicCTI‐111647) were published versions of trials possibly unknown to us. We wrote to the first trial author to ask for clinical study reports or equivalent on 12 November 2013 who confirmed that the trials had not been completed.
Included studies
The absence of documentation of trial programmes for both drugs, listing all sponsored trials completed or underway, meant we had to rely on a variety of sources for the reconstruction of the trial programmes and identification of relevant clinical study reports. This complexity is reflected in the flowchart presented in Figure 3, illustrating the study selection process for this review. The two main pathways were the spontaneous release of 77 full clinical study reports by Roche (long after our request for 36 of them) and the requests to regulatory authorities and GSK for all the relevant reports.
3.
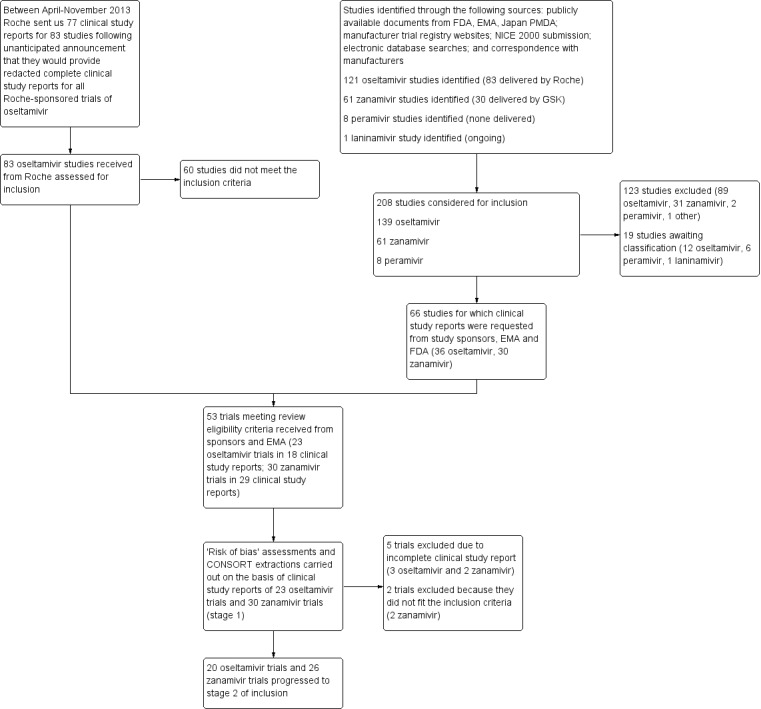
Flow diagram describing the number of studies identified, inclusion, exclusion and progression from identification to stage 1 to stage 2 of the review.
NB Because of the absence of trial programmes for both drugs listing all sponsored trials completed or underway, we had to rely on a variety of sources for the reconstruction of the trial programmes and retrieval of relevant clinical study reports. This complexity is reflected in the flowchart, illustrating the study selection process for this review. The two main pathways were the spontaneous release of 77 clinical full clinical study reports by Roche and the requests to regulatory authorities and GSK for all the relevant reports. There was overlap in trial reports retrieved following the different pathways
We carried out the inclusion into Stage 1 using the clinical study reports, titles, abstracts and any other relevant information. Through this process we identified 208 potentially relevant studies (139 oseltamivir trials, 61 zanamivir trials and eight peramivir trials). We excluded 123 studies (listed in the Characteristics of excluded studies table) as clearly ineligible. A further 19 studies are awaiting classification (see Characteristics of studies awaiting classification). We requested 66 trials from study sponsors, the EMA and the FDA. From these different methods the total number of trials available for assessment for inclusion in our review at Stage 1 was 53.
Twenty three studies of oseltamivir (JV15823; JV15824; M76001; ML16369; NV16871; WV15670; WV15671; WV15673/WV15697; WV15707; WV15708; WV15730; WV15758; WV15759/WV15871; WV15799; WV15812/WV15872; WV15819/WV15876/WV15978; WV15825; WV16277) and 28 of zanamivir (167‐101; JNAI‐01; JNAI‐04; JNAI‐07; NAI30008; NAI30009; NAI30010; NAI30011; NAI30012; NAI30015; NAI30020; NAI30028; NAI30031; NAI30034; NAIA/B2008; NAIA/B2009; NAIA2005; NAIA2006; NAIA3002; NAIA3003; NAIA3004; NAIA3005; NAIB2005; NAIB2006; NAIB2007; NAIB3001; NAIB3002; PE‐01) were included in Stage 1. It was not uncommon for more than one trial to be reported in the same clinical study reports. This was either due to the amalgamation of two or more trials because of low influenza virus circulation and difficulties in recruitment (for example, WV15812/WV15872), or because the trials bore different ID numbers when in reality they followed the same protocol, albeit in two different hemispheres (for example, WV15759/WV15871).
We also identified six completed or ongoing studies of peramivir in dose‐response or placebo‐controlled studies (NCT00419263; NCT00453999; NCT00486980; NCT00610935; NCT00705406; NCT00958776).
The included trials were predominantly conducted in adults during influenza seasons in both hemispheres. A small number of studies were conducted in older people residing in care homes and in people with underlying respiratory diseases. All trials were sponsored by the manufacturers.
Oseltamivir
Of the 23 oseltamivir trials in Stage 1, 15 were multicentre trials conducted in both the Northern and Southern Hemispheres, while eight were done in only one country (USA five, Japan two and China one). In total 9623 participants were included (6574 in treatment trials and 3049 in prophylaxis trials). The age of the participants ranged from 1 to 82 years and the duration of follow‐up varied from 6 to 42 days.
Two of the trials were conducted within nursing homes; 20 were within free‐living populations; one was performed in in‐ and out‐patient departments. Three trials were conducted in children, while participants in 20 trials were adults. In some trials the eligible population included participants at increased risk of influenza complications, or with diagnoses of asthma or chronic obstructive pulmonary disease, but the majority included only otherwise healthy adults. In one trial (WV15730), participants were stratified by smoking status, while those in another trial were stratified by the presence or absence of otitis media (WV15758).
All trials compared orally administered oseltamivir (either as capsules or reconstituted powder) with placebo.
Of the 23 trials, we included 20 RCTs for the analysis examining the use of oseltamivir compared with placebo. Two were excluded from the meta‐analysis because they were only synopsis reports (JV15823; JV15824) and another because it was not a full clinical study report (ML16369).
We finally included 20 oseltamivir trials into Stage 2: 11 on treatment in adults (M76001; WV15670; WV15671; WV15707, WV15730; WV15812/WV15872; WV15819/WV15876/WV15978WV16277), four in children (NV16871; WV15758; WV15759/WV15871), and five on prophylaxis: two in adults (WV15673/WV15697), two in the elderly (WV15708; WV15825) and one in households (WV15799). Of the 15 included treatment trials of oseltamivir only three (M76001; WV15670; WV15758) were successful in recruiting the a priori planned sample size.
Zanamivir
Of the 28 included zanamivir trials, 18 were multicentre trials done in both the Northern and Southern Hemisphere and 10 were done in only one country (Japan five, USA three, Finland and Germany one each). In total 14,628 participants were included (7678 in treatment trials and 6950 in prophylaxis trials). Participants' age ranged from 5 to 12 years to over 65 and duration of follow‐up varied from 5 to 35 days.
Two of the trials were performed within nursing homes; several were within free‐living populations; one was performed within a university student population. In some trials the eligible population included participants at increased risk of influenza complications, or with diagnoses of asthma or chronic obstructive pulmonary disease, but the majority included only adults who were otherwise healthy.
Zanamivir was administered as an intranasal spray, an inhalation or a combination of both and placebos were designed to match. Administration was by the participant in the majority of trials and by nursing staff in the trials within nursing homes. Twenty‐two trials compared inhaled zanamivir with placebo and six trials compared inhaled zanamivir, or intranasal zanamivir, with placebo or usual care.
Of the 28 trials we included 26 RCTs for the analysis examining the use of zanamivir compared with placebo. Two were excluded from the meta‐analysis because one was only a synopsis (NAI30020) and one compared zanamivir to usual care and not placebo (NAIA3003).
We finally included 26 zanamivir trials: 14 on treatment in adults (JNAI‐01; JNAI‐04; JNAI‐07, NAI30008; NAI30011; NAI30012; NAI30015; NAIA/B2008; NAIA2005; NAIA3002; NAIB2005; NAIB2007; NAIB3001; NAIB3002), two in children (NAI30009; NAI30028) and 10 trials in prophylaxis (167‐101; NAI30034; NAIA/B2009; NAIA2006; NAIA3004; NAIA3005; NAIB2006; PE‐01; NAI30010; NAI30031).
Our attempt at collecting sufficient information from regulatory files to reconstruct missing clinical study reports also failed because the information appeared insufficient for a reliable reconstruction.
Excluded studies
We excluded 123 studies from entering Stage 1 for various reasons. Some were pharmacokinetic studies, or had an active comparator, or compared higher‐ versus lower‐dose schedules, or were ongoing trials. A further 19 trials are awaiting assessment (Characteristics of studies awaiting classification).
Risk of bias in included studies
Study level assessments are reported in the 'Risk of bias' tables. To address the problem of reporting bias, we ignored published trial reports and directed our attention to clinical study reports and regulatory information. Our problems in reviewing the copious material at our disposal were how to identify and analyse important details in the midst of thousands of pages of information and how to construct a coherent appraisal of large and complex trial programmes.
In addition, since we gained unrestricted access to the full clinical study reports (apart from personal de‐identifying redactions) we took the view that all information needed to judge risk of bias should be present. Therefore when this information was not available, we judged the corresponding risk of bias element as at 'high' risk of bias. For example, when details of the random sequence generation are missing from journal publications of clinical trials, it is customary to record this as "unknown" risk of bias. This judgment usually carries the assumption that the random sequence generation details are available in more detailed reports. But when these details were still missing in even full clinical study reports, we chose to rate this risk of bias element at "high" risk of bias.
In the following paragraphs we report some of the salient findings using the current Cochrane format but applying the logic of reviewing regulatory data.
Allocation
In 10 of the 20 oseltamivir studies included in Stage 2 the description of random sequence generation is missing. The reporting of all zanamivir trials but one (NAI30028) was biased by the absence of description of random sequence generation.
Blinding
The placebo and active drug capsule cap were not identical in 11 of the 20 trials of oseltamivir. This may have compromised blinding of participants. For all but one of the zanamivir trials we did not have the certificates of analysis to enable us to reconstruct the appearance, taste and texture of the two principles.
Incomplete outcome data
In addition to the missing diary cards in three treatment trials (see Results of the search section), we were unable to identify all data for all outcomes in all oseltamivir trials and in eight of the zanamivir trials. For example, hospitalisations were not reported in zanamivir trials and inconsistently reported in oseltamivir trials. The relevant data in this review come from a table of hospitalisations sent to us by Roche in late 2013. In addition, in some trials we were unable to track individual participants through tables, narratives and individual listings. The issue of compliharms impeded the ascertainment of harms in oseltamivir treatment trials (Appendix 1). We had difficulty in following the logic of compliharms, even with access to full clinical study reports. The definition of adverse events in the RCTs of oseltamivir and zanamivir is different from the ordinary definition of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) E2D guideline, which is as follows: "An adverse event (AE) is any untoward medical occurrence in a patient administered a medicinal product and which does not necessarily have to have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended sign (for example, an abnormal laboratory finding), symptom, or disease temporally associated with the use of a medicinal product, whether or not considered related to this medicinal product". (http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2D/Step4/E2D_Guideline.pdf accessed 27 December 2013).
As an example, the definition of adverse events in the WV15671 study is as follows (PDF page 35): "following the alleviation of influenza‐like symptoms, the recurrence of a single respiratory or constitutional symptom was recorded as an adverse event, however, the reappearance of more than one symptom was recorded as influenza‐like syndrome (i.e. secondary illness) and therefore do not appear as adverse events" and WV15670: "any adverse change from the subject's baseline (pre‐treatment) condition, which occurred during the course of the study after treatment had started, whether considered related to treatment or not". Treatment included all investigational agents (including placebo and comparative agents) administered during the course of the study)" (our emphasis). As a consequence, adverse events that are similar to the symptoms of influenza (such as headache and mild gastrointestinal adverse events) tend to be excluded from the treatment trials.
We identified a report of a site inspection for the adult prophylaxis trial WV15673/WV15697. The FDA carried out the inspection in September 2000 at various trial sites in the US including the West Virginia site (which was responsible for enrolling many hundreds of participants). An FDA official letter reported several violations including failure to report serious harms to the sponsor (Roche) as the protocol required and in addition stated: "... we view the statement in the payment section of the consent form used in the study that subjects '...will receive $300.00 for participating in and completing the study. No payment will be made to you if you withdraw from the study for personal reasons...' to be an improper procedure. When subjects are to be paid for participating in a study, the payment should be prorated for the subject's actual participation in the study in order to avoid the possibility of coercion" (FDA 2000e, PDF page 177). However, the FDA allowed the data (which had been published a year earlier in a prime journal) to stand in support of Roche's application for the prophylaxis indication. We do not know whether the participant contract was standard (i.e. whether the observation of possible improper procedures could be generalised to other sites and other trials), but the document cited by the FDA inspector is the subject of one of our (as yet unfulfilled) Freedom of Information (FOI) requests. The possibility of financial pressure, if confirmed, could seriously confound drop‐out rates because of harms or any other causes in prophylaxis trials.
The significantly higher incidence of diarrhoea in placebo recipients of treatment trial WV15671 was identified by the FDA reviewers who remarked "Diarrhea was reported more frequently among subjects receiving placebo than among subjects receiving Ro 64‐0796 [oseltamivir]. Diarrhoea, although not specified as an inclusion criterion, has been documented to be a clinical manifestation of influenza infection. The reduction in the incidence of diarrhoea for the treatment groups compared with the placebo group could be considered as a possible treatment effect of Ro 64‐0796" (FDA 1999c). However, according to the Japanese Summary Basis for Approval (JSBA) of oseltamivir capsules for prophylaxis, diarrhoea was reported more frequently in the oseltamivir arm (49/986) than in the placebo group (38/973) in the summarised table of adverse events from three trials (WV15673/WV15697; WV15708; WV15825). Our findings are inconsistent with the explanation by the FDA.
Selective reporting
All oseltamivir trials and almost half of the zanamivir trials had selected reporting. The oseltamivir trials showed a consistent trend of missing original protocols (except for M76001), changing outcome definitions while the trial was running, protocol amendments even after the trial had been completed, inconsistent approaches to outcome data collection, missing statistical analysis plans, missing date of unblinding and the use of self reported outcomes such as pneumonia (M76001; WV15670; WV15671; WV15707; WV15730; WV16277). This represent 55% of pneumonia event data. As an example, in trial WV15670, secondary illnesses were patient reported. The body of the clinical study report states that complications requiring antibiotic treatment were specified a priori but even in the final version of the protocol, for which we have the full text, there is no predefined list of secondary illnesses (i.e. no mention of pneumonia, bronchitis, sinusitis or otitis in the protocol), nor did complications have anything to do with antibiotic treatment according to the protocol, nor does the Case Report Form mention specific secondary illnesses by name. Zanamivir trials reported outcomes not specified in the protocol provided.
We found evidence of possible selective reporting bias when we analysed the JSBA data on prophylaxis. The regulatory data reports tables for individual trials as well as 10 pages of summarised tables for three trials of prophylaxis (WV15673/WV15697; WV15708; WV15825). Tables for individual trials include data for high‐dose arms but report few psychiatric adverse events overall. However, the summarised tables list a variety of psychiatric adverse events including psychotic and suicidal adverse events, but not adverse events from the high‐dose group. As a preliminary exploratory analysis, we combined the following suspected serious adverse events collectively: hallucination and delusion that are classified grade 3 (serious) by the National Cancer Institute‐Common Toxicity Criteria Version 2.0 (NCI‐CTC V2.0), psychosis (hallucination and delusion are the two major symptoms of this disease), suicidal attempt that is classified grade 3 (serious) by the Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0 (CTCAE V4.0) and hostility that includes aggression, hostility, violence, murder and commonly considered as serious events though not listed in the NCI‐CTC V2.0 or CTCAE V4.0. Numbers of suspected serious psychotic/suicidal adverse events (including hallucination, psychosis, schizophrenia, paranoia, aggression/hostility and attempted suicide) were five in the oseltamivir group and zero in the placebo group during the on‐treatment period. When the off‐treatment period data are added the total was eight versus one. The prophylaxis programme is crucial in understanding the harms profile of the drug as the potential for harms witnessed to be confounded by the apparently numerous symptoms and signs of influenza infection is far less, as many participants do not become infected with influenza. This makes a causality assessment more straightforward.
Other potential sources of bias
All but three of the oseltamivir treatment trials were under‐recruited. Several of the zanamivir trials were also under‐recruited. We noted the use of different relief medication across different centres within the same trial and in one zanamivir trial (NAI30031), according to the protocol participants receiving antibiotics for bacterial respiratory tract infection should have been excluded but in the trial this did not happen. In the zanamivir trial NAI30034, the definition of "confirmed influenza" was amended after protocol closure.
We also noted several other items that were not included in all full clinical study reports:
Study protocols dated prior to participant enrolment (missing for many oseltamivir trials).
Certificates of analysis for the intervention/placebo preparations.
Patient enrolment dates explicitly reported (only trial inception and cessation dates are given; in zanamivir trials these are partially redacted).
Explicitly reported date of trial unblinding. We frequently noted the statement "the database was authorized on xxxx" to identify the unblinding date but an explicit date is important to report. In some cases, the date of unblinding was reported but the actual date within the month was redacted. This practice also applied to zanamivir protocol amendments.
Authorship and accountability for the writing of the clinical study reports.
Statistical analysis plans in some cases.
Patient consent forms (missing from most zanamivir trials).
Patient information form (missing from most zanamivir trials).
List of randomisation codes (variably included).
Case report form templates in zanamivir trials do not allow for determining who completes the form (patient or clinician).
Core data sheet.
Other important documents that we did not have included:
Study manual of procedures.
Minutes of safety data monitoring committee meetings.
The placebo interventions in both sets of trials may have contained active substances. The placebo for zanamivir trials contained lactose powder, which can potentially cause bronchospasm, while the placebo for oseltamivir trials contained dehydrocholic acid and dibasic calcium phosphate dehydrate, which can cause gastrointestinal symptoms.
Data on participants by influenza‐infected status (in treatment trials) and for participants with influenza‐like illness (in prophylaxis trials) were not reported in the oseltamivir clinical study reports.
Finally, data on the effects of rescue or relief medication (mainly paracetamol/acetaminophen) were incomplete in clinical study reports of oseltamivir trials and not reported separately in all zanamivir trials.
Effects of interventions
Analysis of time to first symptom alleviation
In adult treatment, oseltamivir reduced the time to first alleviation of symptoms by 16.8 hours (95% confidence interval (CI) 8.4 to 25.1 hours, I2 statistic = 0%), representing a 10% reduction from 7 days to 6.3 days (Analysis 1.1; Figure 4). There was no significant effect in asthmatic children: increased by 5.2 hours (95% CI 11.1 hours lower to 21.4 hours higher, I2 statistic = 0%). But there was an effect in otherwise healthy children, based on one trial: 29 hours, 95% CI: 12 to 47 hours, P = 0.001). (Analysis 1.46). Zanamivir reduced time to first alleviation of symptoms in adults by 0.60 days (95% CI 0.39 to 0.81 days, I2 statistic = 9%), which equates to a 14.4 hours (10%) reduction in symptoms from 6.6 days to 6.0 days (Analysis 3.1; Figure 5). There was no significant effect in children: time to first alleviation of symptoms was 1.08 days lower in the zanamivir group (95% CI 2.32 lower to 0.15 days higher, I2 statistic = 72%) (Analysis 3.14).
1.1. Analysis.
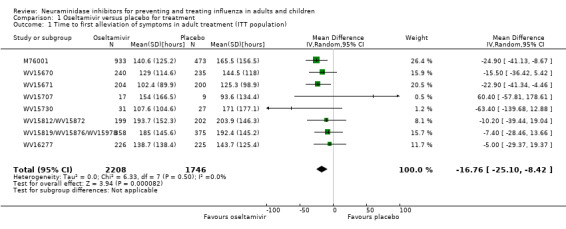
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 1 Time to first alleviation of symptoms in adult treatment (ITT population).
4.
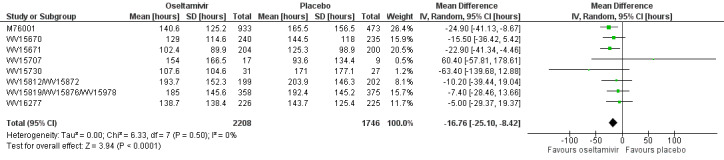
Forest plot of comparison: 1 Oseltamivir versus placebo for treatment, outcome: 1.1 Time to first alleviation of symptoms in adult treatment (ITT population) [hours].
1.46. Analysis.
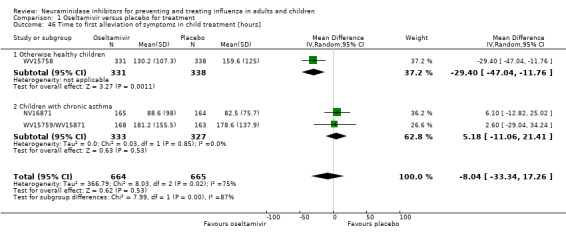
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 46 Time to first alleviation of symptoms in child treatment [hours].
3.1. Analysis.
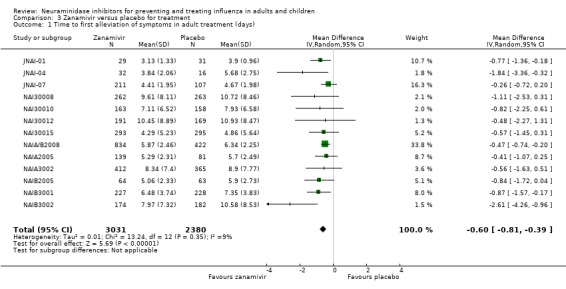
Comparison 3 Zanamivir versus placebo for treatment, Outcome 1 Time to first alleviation of symptoms in adult treatment (days).
5.
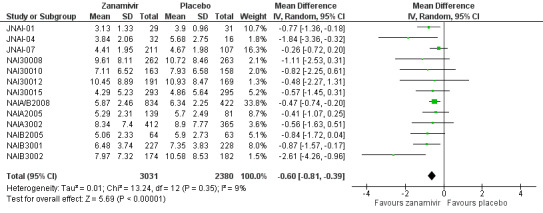
Forest plot of comparison: 3 Zanamivir versus placebo for treatment, outcome: 3.1 Time to first alleviation of symptoms in adult treatment (days).
3.14. Analysis.
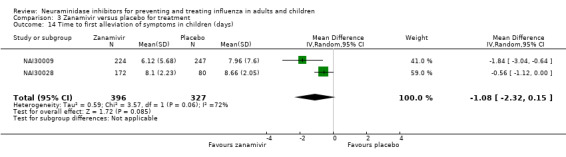
Comparison 3 Zanamivir versus placebo for treatment, Outcome 14 Time to first alleviation of symptoms in children (days).
In eight zanamivir trials that reported on use of relief medication, in all participants the median days to alleviation in both the placebo and the treatment arms was less when compared to those who did not use relief medications (Table 6). In seven zanamivir trials, time to first alleviation of symptoms was also reported with and without rescue medication. Using these data we were able to compare zanamivir without rescue medication with placebo with rescue medication. Overall there was a non‐significant 0.41 day decrease (95% CI 0.47 days lower to 1.29 days higher, I2 statistic = 67%) in time to first alleviation of symptoms in the placebo with rescue medication group, suggesting that zanamivir itself is no better than rescue medication and possibly even less effective, although the varying levels of use of rescue medication in the seven trials did give rise to large heterogeneity (Analysis 3.68; Figure 6).
6. Time to alleviation* of clinically significant symptoms of influenza‐like illness (in all participants and participants with no use of relief medication).
| Study | Sample size | Median days to alleviation for all participants |
Difference in days (P value) |
Median days to alleviation and no use of relief medication | Difference in days (P value) | |||
| Zanamivir (n) | Placebo (n) | Zanamivir | Placebo | Zanamivir | Placebo | |||
| NAI30008 | 262 | 263 | 6.0 | 7.0 | 1.0 (0.123) |
8.0 | 10.0 | 2.0 (0.037) |
| NAI30009 | 224 | 247 | 4.5 | 5.0 | 0.5 (0.011) |
5.0 | 6.0 | 1.0 (0.002) |
| NAI30010 | 76 | 81 | 4.5 | 5.5 | 1.0 (0.033) |
5.5 | 6.75 | 1.25 (0.150) |
| NAI30011 | 237 | 229 | 4.50 | 5.00 | 0.50 (0.495) |
7.0 | 7.0 | 0.0 (0.623) |
| NAI30012 | 191 | 167 | 6.5 | 7.5 | 1.0 (0.159) |
9.0 | 10.0 | 1.0 (0.131) |
| NAI30015 | 293 | 295 | 2.17 | 2.67 | 0.5 (0.166) |
3.17 | 3.83 | 0.66 (0.058) |
| NAIA3002 | 412 | 365 | 5.5 | 6.0 | 0.5 (0.228) |
7.0 | 8.0 | 1.0 (0.054) |
| NAIB3002 | 174 | 182 | 5.0 | 7.5 | 2.5 (< 0.001) |
5.5 | 8.25 | 2.75 (< 0.001) |
*Alleviation defined as no fever (temperature < 37.8 °C), cough recorded as none or mild and muscle/joint aches and pains, sore throat, feverishness/chills and headache recorded as absent/minimal
3.68. Analysis.
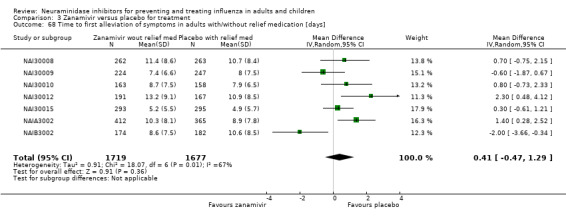
Comparison 3 Zanamivir versus placebo for treatment, Outcome 68 Time to first alleviation of symptoms in adults with/without relief medication [days].
6.
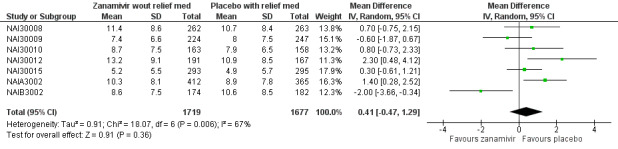
Forest plot of comparison: 3 Zanamivir versus placebo for treatment, outcome: 3.68 Time to first alleviation of symptoms in adults with/without relief medication [days].
In subgroup analysis of time to first alleviation of symptoms in adults by infection status, we found no evidence of a difference in treatment effect for zanamivir on the influenza‐infected subgroup compared to the non‐influenza‐infected subgroup (P = 0.53). The treatment effect was 0.67 days (95% CI 0.35 to 0.99 days, I2 statistic = 17%) for influenza‐infected patients and 0.52 days (95% CI 0.18 to 0.86 days, I2 statistic = 0%) for non‐influenza‐infected patients (Analysis 3.69).
3.69. Analysis.
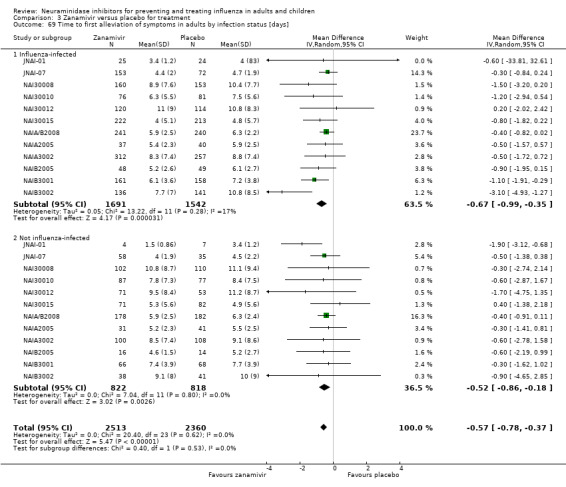
Comparison 3 Zanamivir versus placebo for treatment, Outcome 69 Time to first alleviation of symptoms in adults by infection status [days].
Analysis of hospitalisations
In oseltamivir treatment of adults, there was no significant difference in hospitalisation rate between treatment groups (risk ratio (RR) 0.92, 95% CI 0.57 to 1.50, I2 statistic = 0%) (Analysis 1.2), or in treatment of children (RR 1.92, 95% CI 0.70 to 5.23, I2 statistic = 0%) (Analysis 1.47 ), with wide CIs; or in prophylaxis (RR 1.14, 95% CI 0.66 to 1.94, I2 statistic = 0%) (Analysis 2.7). Data on hospitalisations for the zanamivir studies were not reported.
1.2. Analysis.
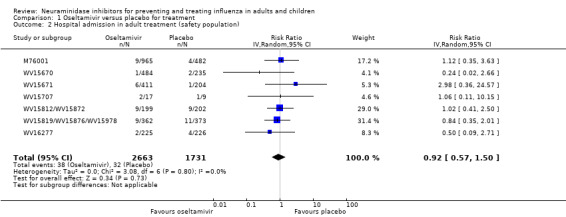
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 2 Hospital admission in adult treatment (safety population).
1.47. Analysis.
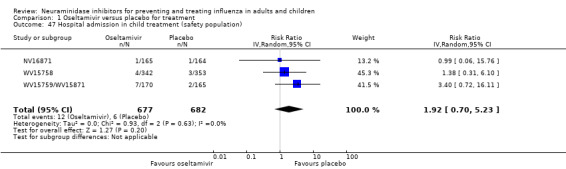
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 47 Hospital admission in child treatment (safety population).
2.7. Analysis.
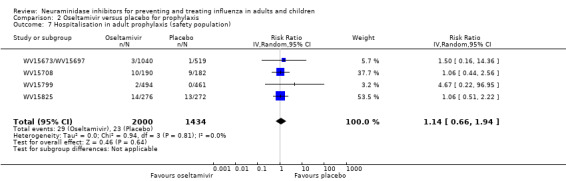
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 7 Hospitalisation in adult prophylaxis (safety population).
Analysis of influenza complications
Pneumonia
In adult treatment trials, oseltamivir significantly reduced self reported, investigator‐mediated, unverified pneumonia (RR 0.55, 95% CI 0.33 to 0.90, I2 statistic = 0%; risk difference (RD) 1.00%, 95% CI 0.22 to 1.49; number needed to treat to benefit (NNTB) = 100, 95% CI 67 to 451) in the treated population. The effect was significant in the six trials that collected data on non‐specific adverse events or secondary/intercurrent illness forms (RR 0.44, 95% CI 0.22 to 0.88, I2 statistic = 0%; RD 0.99%, 95% CI 0.21 to 1.38; NNTB = 101, 95% CI 73 to 470). However, it was not significant in the five trials (two clinical study reports) that used more detailed diagnostic data collection forms, and in no studies that reported on radiological confirmation of pneumonia (Figure 7; Analysis 1.17). There was no significant effect on pneumonia in children (RR 1.06, 95% CI 0.62 to 1.83, I2 statistic = 0%) (Analysis 1.52). In two zanamivir adult trials (NAI30012; NAI30015), pneumonia reporting was based on a stricter definition of X‐ray confirmation and there was also no significant treatment effect (RR 1.02, 95% CI 0.35 to 3.02, I2 = 39%) (Analysis 3.3). In nine zanamivir trials (NAI30008; NAI30010; NAI30011; NAIA/B2008; NAIA2005; NAIA3002; NAIB2007; NAIB3001; NAIB3002), pneumonia was a self reported, investigator‐mediated, unverified outcome (Figure 8; Figure 9). Overall, there was no significant effect of zanamivir on mixed verified and unverified pneumonia in adult treatment (RR 0.90, 95% CI 0.58 to 1.40, I2 statistic = 0%) (Analysis 3.2). Analysis 4.5 shows that in prophylaxis trials, zanamivir reduced the risk of self reported, investigator‐mediated, unverified pneumonia in adults (RR 0.30, 95% CI 0.11 to 0.80, I2 statistic = 0%; RD 0.32%, 95% CI 0.09 to 0.41; NNTB = 311, 95% CI 244 to 1086).
7.
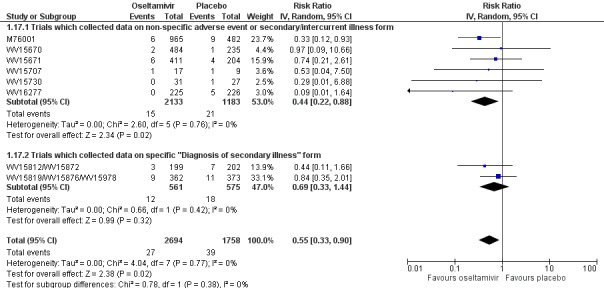
Forest plot of comparison: 1 Oseltamivir versus placebo for treatment, outcome: 1.17 Complications: pneumonia in adult treatment.
1.17. Analysis.
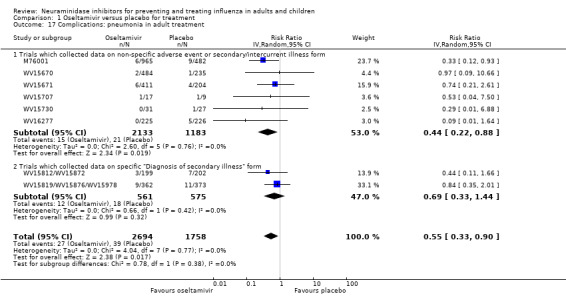
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 17 Complications: pneumonia in adult treatment.
1.52. Analysis.
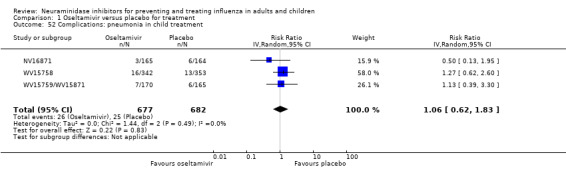
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 52 Complications: pneumonia in child treatment.
3.3. Analysis.
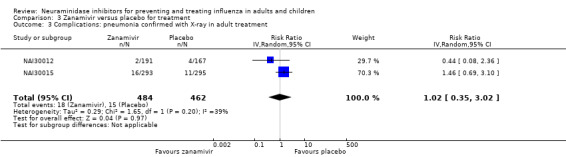
Comparison 3 Zanamivir versus placebo for treatment, Outcome 3 Complications: pneumonia confirmed with X‐ray in adult treatment.
8.

Example Diary card from case‐report form for Zanamivir trial
9.

Example Diary card from case‐report form for Zanamivir trial (cont)
3.2. Analysis.
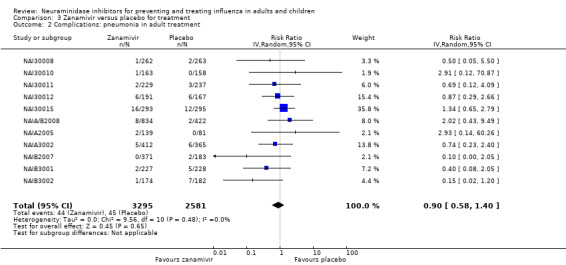
Comparison 3 Zanamivir versus placebo for treatment, Outcome 2 Complications: pneumonia in adult treatment.
4.5. Analysis.
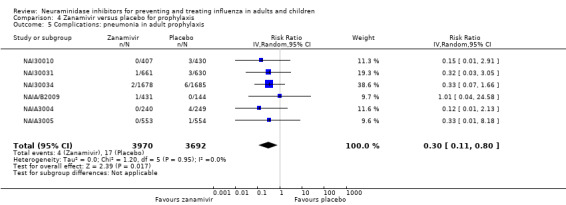
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 5 Complications: pneumonia in adult prophylaxis.
In meta‐regression of 'pneumonia' based on 32 studies, treatment effects were not statistically different by age group (P = 0.22), drug (P = 0.89) or indication (P = 0.14). However, treatment effects were statistically different by method of diagnosis (P = 0.025). For unclear objective diagnosis of pneumonia, the treatment effect was RR 0.51 (95% CI 0.35 to 0.75, I2 statistic = 0%), whereas for objective diagnosis data collection of pneumonia, the treatment effect was 1.01 (95% CI 0.69 to 1.47, I2 statistic = 0%). A subgroup analysis of pneumonia for all 32 studies by method of diagnosis is shown in Analysis 5.1. Please note that estimates in the subgroup analysis are slightly different to those obtained in meta‐regression due to the different methodologies.
5.1. Analysis.
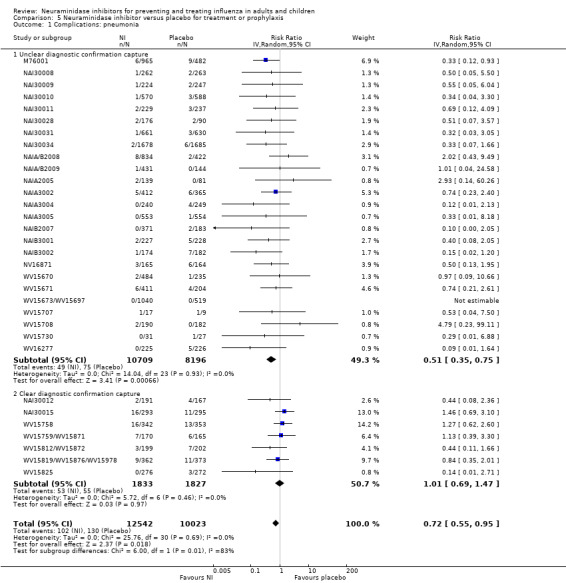
Comparison 5 Neuraminidase inhibitor versus placebo for treatment or prophylaxis, Outcome 1 Complications: pneumonia.
Serious complications and study withdrawals
In oseltamivir trials, treatment did not significantly affect complications classified as serious or those that led to withdrawal from the trial in adults (RR 0.91, 95% CI 0.40 to 2.06, I2 statistic = 0%) (Analysis 1.20) or in children (RR 1.98, 95% CI 0.58 to 6.72, I2 statistic = 0%) (Analysis 1.55). This outcome could not be assessed in oseltamivir prophylaxis due to an insufficient number of events. There was no significant effect of zanamivir, in adult treatment, in reducing the risk of any complication classified as serious or which led to study withdrawal (RR 1.10, 95% CI 0.46 to 2.63, I2 statistic = 0%) (Analysis 3.7) or in prophylaxis (RR 1.09, 95% CI 0.36 to 3.26, I2 statistic = 0%) (Analysis 4.8). This outcome could not be assessed in children due to an insufficient number of events.
1.20. Analysis.
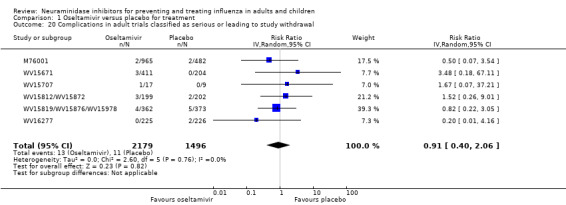
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 20 Complications in adult trials classified as serious or leading to study withdrawal.
1.55. Analysis.
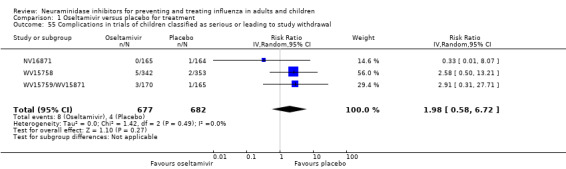
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 55 Complications in trials of children classified as serious or leading to study withdrawal.
3.7. Analysis.
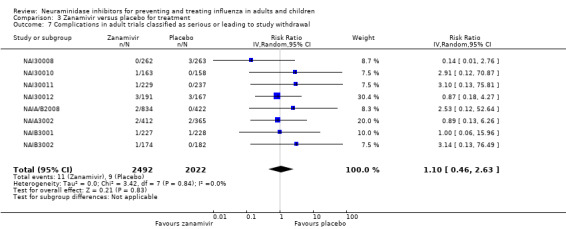
Comparison 3 Zanamivir versus placebo for treatment, Outcome 7 Complications in adult trials classified as serious or leading to study withdrawal.
4.8. Analysis.
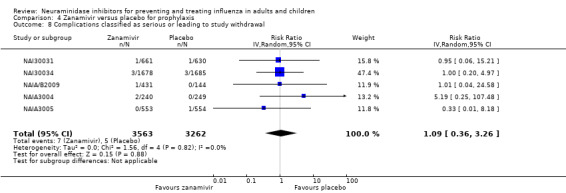
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 8 Complications classified as serious or leading to study withdrawal.
Bronchitis, sinusitis and otitis media
Neither zanamivir (Analysis 4.6; Analysis 4.7) nor oseltamivir (Analysis 2.8; Analysis 2.9) significantly reduced the risk of bronchitis or sinusitis in prophylaxis trials. In adults, treatment with oseltamivir did not significantly reduce the risk of bronchitis (RR 0.75, 95% CI 0.56 to 1.01, I2 statistic = 36%) (Analysis 1.16), sinusitis (RR 1.03, 95% CI 0.76 to 1.40, I2 statistic = 0%) (Analysis 1.18) or otitis media (RR 1.11, 95% CI 0.57 to 2.15, I2 statistic = 0%) (Analysis 1.19). The result for bronchitis was sensitive to the methods used, as a fixed‐effect analysis showed a significant effect (P = 0.02). Oseltamivir did not significantly affect complications in treatment of children (Analysis 1.52 ; Analysis 1.50; Analysis 1.53), including otitis media (RR 0.80, 95% CI 0.62 to 1.02, I2 statistic = 0%) (Analysis 1.51).
4.6. Analysis.
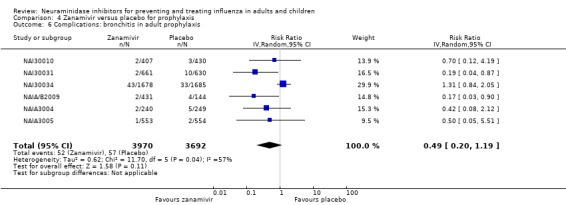
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 6 Complications: bronchitis in adult prophylaxis.
4.7. Analysis.
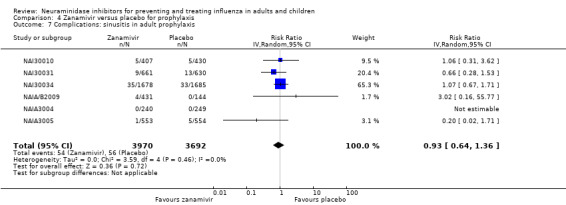
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 7 Complications: sinusitis in adult prophylaxis.
2.8. Analysis.
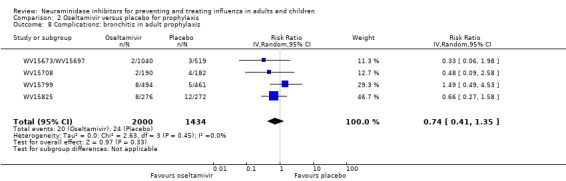
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 8 Complications: bronchitis in adult prophylaxis.
2.9. Analysis.
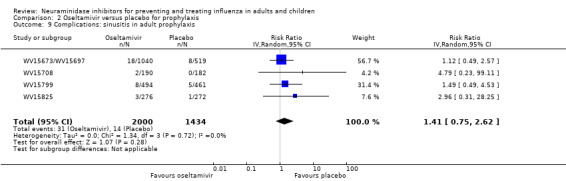
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 9 Complications: sinusitis in adult prophylaxis.
1.16. Analysis.
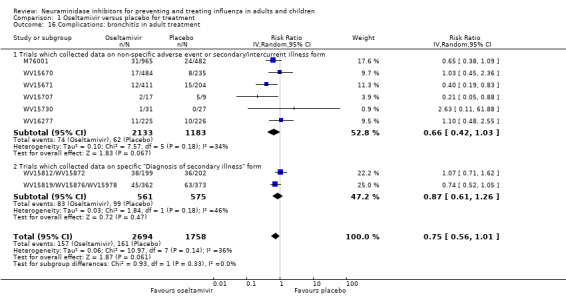
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 16 Complications: bronchitis in adult treatment.
1.18. Analysis.
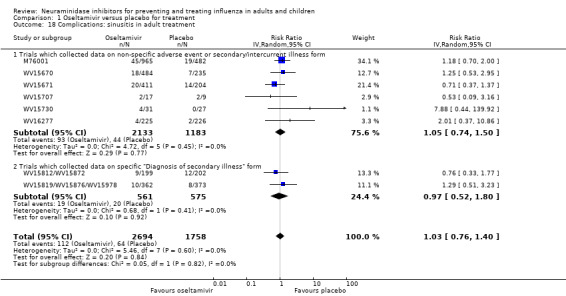
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 18 Complications: sinusitis in adult treatment.
1.19. Analysis.
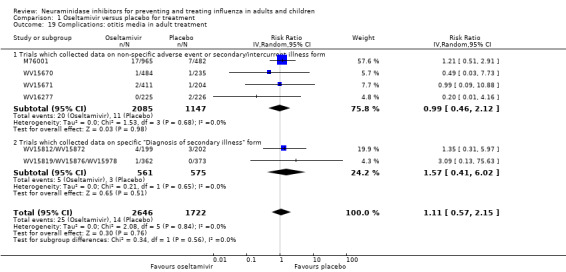
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 19 Complications: otitis media in adult treatment.
1.50. Analysis.
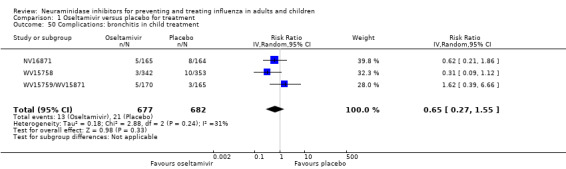
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 50 Complications: bronchitis in child treatment.
1.53. Analysis.
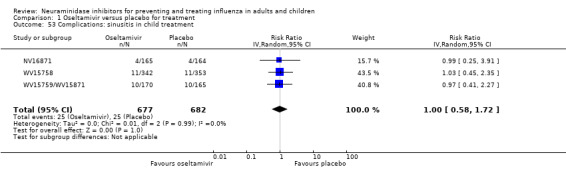
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 53 Complications: sinusitis in child treatment.
1.51. Analysis.
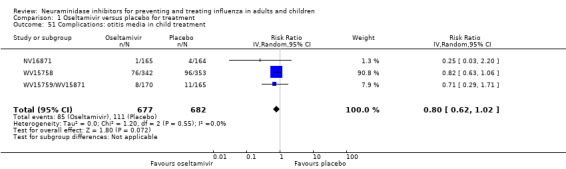
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 51 Complications: otitis media in child treatment.
Treatment with zanamivir significantly reduced the risk of bronchitis in adults (RR 0.75, 95% CI 0.61 to 0.91, I2 statistic = 0%; RD 1.80%, 95% CI 0.65 to 2.80; NNTB = 56, 95% CI 36 to 155) (Analysis 3.4), but did not reduce the risk of sinusitis (Analysis 3.5) or otitis media (Analysis 3.6). In children, zanamivir treatment did not significantly reduce the risk of sinusitis (RR 0.87, 95% CI 0.12 to 6.45, I2 statistic = 40%) (Analysis 3.17) or otitis media (RR 1.00, 95% CI 0.59 to 1.72, I2 statistic = 0%) (Analysis 3.18).
3.4. Analysis.
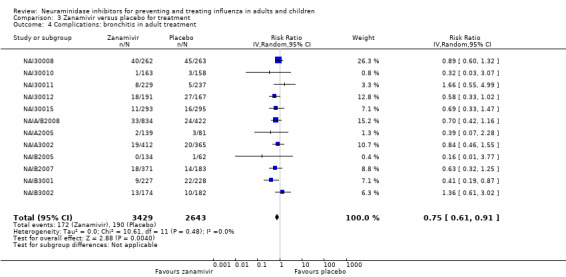
Comparison 3 Zanamivir versus placebo for treatment, Outcome 4 Complications: bronchitis in adult treatment.
3.5. Analysis.
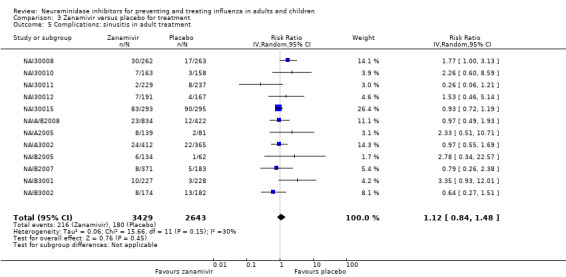
Comparison 3 Zanamivir versus placebo for treatment, Outcome 5 Complications: sinusitis in adult treatment.
3.6. Analysis.
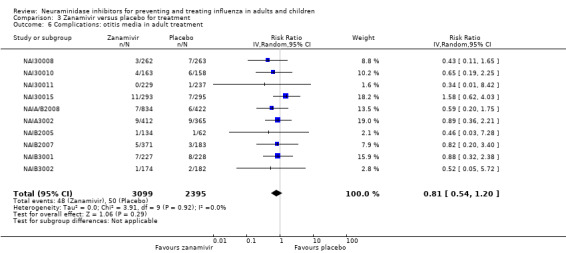
Comparison 3 Zanamivir versus placebo for treatment, Outcome 6 Complications: otitis media in adult treatment.
3.17. Analysis.
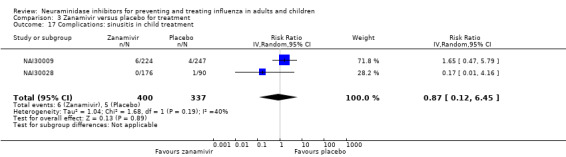
Comparison 3 Zanamivir versus placebo for treatment, Outcome 17 Complications: sinusitis in child treatment.
3.18. Analysis.
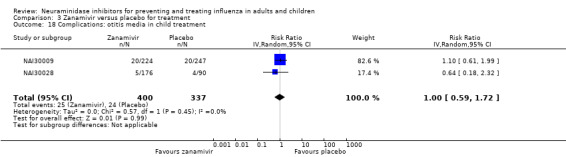
Comparison 3 Zanamivir versus placebo for treatment, Outcome 18 Complications: otitis media in child treatment.
See Table 1 for a summary of the methodology used for collecting and assessing complications in oseltamivir treatment trials. See Table 7 for the overall results for oseltamivir in adults and Table 8 for children. See Table 9 for the overall results for zanamivir in adults and Table 10 for children.
7. Oseltamivir versus placebo for treating influenza in healthy adults.
| Oseltamivir versus placebo for treating influenza in healthy adults | ||||||
| Patient or population: healthy adults with influenza Settings: community, nursing homes Intervention: oseltamivir versus placebo for treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Risk difference (95% CI) | NNTB or NNTH (95% CI) | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oseltamivir versus placebo for treatment | |||||
| Time to first alleviation of symptoms in adult treatment (ITT population) (hours) | The mean time (hours) to first alleviation of symptoms adults in the intervention groups was 16.76 lower (25.1 to 8.42 lower) | 16.8 hours (8.4 to 25.1) | 3954 (8) | N/A | N/A | |
| Adverse events: nausea in adult treatment (on‐treatment) | Study population | RR 1.57 (1.14 to 2.15) | 4452 (8) | ‐3.66% (‐7.39 to ‐0.9) | NNTH = 28 (14 to 112) | |
| 64 per 1000 | 101 per 1000 (73 to 138) | |||||
| Adverse events: vomiting in adult treatment (on‐treatment) | Study population | RR 2.43 (1.75 to 3.38) | 4452 (8) | ‐4.56% (‐7.58 to ‐2.39) | NNTH = 22 (14 to 42) | |
| 32 per 1000 | 77 per 1000 (56 to 108) | |||||
| Adverse events: diarrhoea in adult treatment (on‐treatment) | Study population | RR 0.67 (0.46 to 0.98) | 4452 (8) | 2.33% (0.14 to 3.81) | NNTB = 43 (27 to 709) | |
| 71 per 1000 | 47 per 1000 (32 to 69) | |||||
| Complications: self reported, investigator‐mediated, unverified pneumonia in adult treatment | Study population | RR 0.55 (0.33 to 0.90) | 4452 (8) | 1.00% (0.22 to 1.49) | NNTB = 100 (67 to 451) | |
| 22 per 1000 |
12 per 1000 (7 to 20) |
|||||
| Adverse events: cardiac body system in adult treatment (on‐treatment) | Study population | RR 0.49 (0.25 to 0.97) | 3943 (6) | 0.68% (0.04 to 1.00) | NNTB = 148 (101 to 2509) | |
| 13 per 1000 | 7 per 1000 (3 to 13) | |||||
| Adverse events: hospital admission in adult treatment | Study population |
RR 0.92 (0.57 to 1.50) |
4394 (7) |
0.15% (‐0.78 to 0.91) | NNTB = 687 (NNTB 110 to ∞ to NNTH 128) | |
| 18 per 1000 |
17 per 1000 (11 to 28) |
|||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Negative risk differences indicate harms; positive risk differences indicate benefits. CI: confidence interval; RR: risk ratio; NNTB: number needed to treat to benefit; NNTH: number needed to treat to harm | ||||||
8. Oseltamivir versus placebo for treating influenza in healthy children.
| Oseltamivir versus placebo for treating influenza in healthy children | ||||||
| Patient or population: healthy children with influenza Settings: community Intervention: oseltamivir versus placebo for treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Risk difference (95% CI) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oseltamivir versus placebo for treatment | |||||
| Time to first alleviation of symptoms in child treatment (hours) | The mean time (hours) to first alleviation of symptoms in children in the intervention groups was 8.04 lower (33.34 lower to 17.26 higher) | 1329 (3) | Not significant | |||
| Hospital admission in child treatment (safety population) | Study population | RR 1.92 (0.7 to 5.23) | 1359 (3) | ‐0.81% (‐3.72 to 0.26) | NNTH = 124 (NNTB 379 to ∞ to NNTH 27) | |
| 9 per 1000 | 17 per 1000 (6 to 46) | |||||
| Complications: bronchitis in child treatment | Study population | RR 0.65 (0.27 to 1.55) | 1359 (3) | 1.08% (‐1.69 to 2.25) | NNTB = 93 (NNTB 45 to ∞ to NNTH 59) | |
| 31 per 1000 | 20 per 1000 (8 to 48) | |||||
| Complications: otitis media in child treatment | Study population | RR 0.8 (0.62 to 1.02) | 1359 (3) | 3.26% (‐0.33 to 6.18) | NNTB = 31 (NNTB 17 to ∞ to NNTH 308) | |
| 163 per 1000 | 130 per 1000 (101 to 166) | |||||
| Complications: pneumonia in child treatment | Study population | RR 1.06 (0.62 to 1.83) | 1359 (3) | ‐0.22% (‐3.07 to 1.41) | NNTH = 450 (NNTB 71 to ∞ to NNTH 33) | |
| 37 per 1000 | 39 per 1000 (23 to 68) | |||||
| Adverse events: diarrhoea in child treatment (on‐treatment) | Study population | RR 0.87 (0.58 to 1.28) | 1358 (3) | 0.93% (‐2.01 to 3.02) | NNTB 108 (NNTB 34 to ∞ to NNTH 50) | |
| 72 per 1000 | 63 per 1000 (42 to 92) | |||||
| Adverse events: vomiting in child treatment (on‐treatment) | Study population | RR 1.7 (1.23 to 2.35) | 1358 (3) | 5.34% (1.75 to 10.29) | NNTH = 19 (10 to 57) | |
| 76 per 1000 | 130 per 1000 (94 to 179) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Negative risk differences indicate harm; positive risk differences indicate benefits. CI: confidence interval; RR: risk ratio; NNTB: number needed to treat to benefit; NNTH: number needed to treat to harm | ||||||
9. Zanamivir versus placebo for treating influenza in healthy adults.
| Zanamivir versus placebo for treating influenza in healthy adults | ||||||
| Patient or population: healthy adults with influenza Settings: community, nursing homes Intervention: zanamivir versus placebo for treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Risk difference (95% CI) | NNTB or NNTH (95% CI) | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Zanamivir versus placebo for treatment | |||||
| Time to first alleviation of symptoms in adult treatment (days) | The mean time (days) to first alleviation of symptoms in adults in the intervention groups was 0.60 lower (0.81 to 0.39 lower) | 0.60 days (0.39 to 0.81) | 5411 (13) | N/A | N/A | |
| Complications: pneumonia confirmed with X‐ray in adult treatment | Study population | RR 1.02 (0.35 to 3.02) | 946 (2) | ‐0.06% (‐6.56 to 2.11) | NNTH = 1540 (NNTB 48 to ∞ to NNTH 16) | |
| 32 per 1000 | 33 per 1000 (11 to 98) | |||||
| Adverse events: nausea/vomiting in adult treatment (on‐treatment) | Study population | RR 0.6 (0.39 to 0.94) | 6553 (15) | 1.63% (0.24 to 2.48) | NNTB = 62 (41 to 411) | |
| 41 per 1000 | 24 per 1000 (16 to 38) | |||||
| Adverse events: psychiatric body system in adult treatment (on‐treatment) | Study population | RR 1.16 (0.57 to 2.38) | 4732 (10) | ‐0.09% (‐0.76 to 0.24) | NNTH 1132 (NNTB 421 to ∞ to NNTH 132) | |
| 6 per 1000 | 6 per 1000 (3 to 13) | |||||
| Complications: bronchitis in adult treatment | Study population |
RR 0.75 (0.61 to 0.91) |
6072 (12) |
1.80% (0.65 to 2.80) | NNTB 56 (36 to 155) | |
| 72 per 1000 |
54 per 1000 (44 to 65) |
|||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Negative risk differences indicate harms; positive risk differences indicate benefits. CI: confidence interval; RR: risk ratio; NNTB: number needed to treat to benefit; NNTH: number needed to treat to harm | ||||||
10. Zanamivir versus placebo for treating influenza in healthy children.
| Zanamivir versus placebo for treating influenza in healthy children | ||||||
| Patient or population: healthy children with influenza Settings: community Intervention: zanamivir versus placebo for treatment | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Risk difference (95% CI) | NNTB or NNTH (95% CI) | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Zanamivir versus placebo for treatment | |||||
| Time to first alleviation of symptoms in children (days) | The mean time (days) to first alleviation of symptoms in children in the intervention groups was 1.08 lower (2.32 lower to 0.15 higher) | 723 (2) | NA | NA | ||
| Complications: sinusitis in child treatment | Study population | RR 0.87 (0.12 to 6.45) | 737 (2) | 0.19% (‐8.09 to 1.31) | NNTB = 519 (NNTB 13 to ∞ to NNTH 77) | |
| 15 per 1000 | 13 per 1000 (2 to 96) | |||||
| Complications: otitis media in child treatment | Study population | RR 1.0 (0.59 to 1.72) | 737 (2) | 0.00% (‐5.13 to 2.92) | NNTB = > 1000 (NNTB 35 to ∞ to NNTH 20) | |
| 71 per 1000 | 71 per 1000 (42 to 122) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Negative risk differences indicate harms; positive risk differences indicate benefits. CI: confidence interval; RR: risk ratio; NNTB: number needed to treat to benefit; NNTH: number needed to treat to harm | ||||||
Analysis of influenza outcomes in prophylaxis studies
Symptomatic influenza was lower in the oseltamivir arms compared to placebo in studies of prophylaxis (RR 0.45, 95% CI 0.30 to 0.67, I2 statistic = 0%; RD 3.05%, 95% CI 1.83 to 3.88; NNTB = 33, 95% CI 26 to 55) (Analysis 2.1); but there were no differences for all other influenza outcomes including overall influenza‐like‐illness reported as an adverse event on‐treatment. In household prophylaxis, one small study with missing outcome data and selective reporting, including 405 participants, showed a significant reduction of symptomatic influenza in the oseltamivir arm compared to placebo (RR 0.20, 95% CI 0.09 to 0.44; RD 13.6%, 95% CI 9.52 to 15.47) (Analysis 2.3), but in the same study there was no significant reduction in asymptomatic influenza (RR 1.14, 95% CI 0.39 to 3.33) (Analysis 2.4). Asymptomatic influenza was not significantly reduced and there was no non‐influenza, influenza‐like illness reported throughout the study period.
2.1. Analysis.
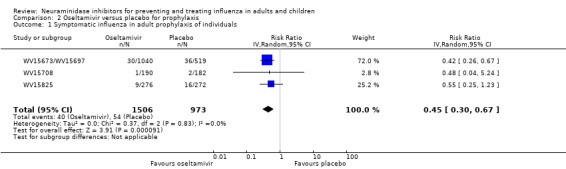
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 1 Symptomatic influenza in adult prophylaxis of individuals.
2.3. Analysis.
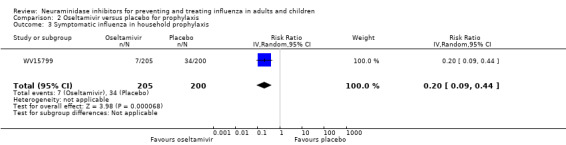
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 3 Symptomatic influenza in household prophylaxis.
2.4. Analysis.
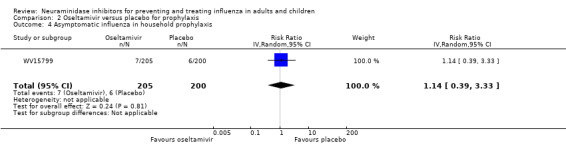
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 4 Asymptomatic influenza in household prophylaxis.
In prophylaxis trials we could not analyse effects on influenza‐like illness because of a lack of definition in the clinical study reports. However, using our definition (see methods), oseltamivir did not reduce influenza‐like illness in participants (RR 0.95, 95% CI 0.86 to 1.06).
The Roche trial programme assessing the effects of oseltamivir in post‐exposure prophylaxis (PEP) submitted to the FDA on 22 May 2000 consisted of two trials: WV15799 and WV16139. We included only trial WV15799 because WV16139 was not placebo‐controlled. WV15799 was a double‐blind, cluster‐randomised trial in which contact clusters of index cases were randomised to oseltamivir 75 mg a day or placebo for seven days. The manufacturer concluded that the trial proved that oseltamivir could prevent influenza in contacts by interrupting transmission from index cases. Interruption of transmission has two components: reduction of viral spread from index cases (measured by nasal shedding of influenza viruses) and prevention of onset of influenza in contacts measured with a mixture of symptoms and signs and 'laboratory confirmation' (i.e. viral culture from the upper airways and/or at least a four‐fold rise in antibody titres measured between baseline and two to three weeks later). The design of the WV15799 is weak. All index cases were left untreated except for a paracetamol rescue pack, making it impossible to assess the effect of oseltamivir on nasal voidance of index cases. Nasal viral voidance was measured only in symptomatic participants thereby missing out on potential asymptomatic infected people.
Zanamivir similarly significantly reduced the risk of symptomatic influenza for individuals (RR 0.39, 95% CI 0.22 to 0.70, I2 statistic = 45%; RD = 1.98%, 95% CI: 0.98 to 2.54; NNTB = 51, 95% CI 40 to 103) (Analysis 4.1), as well as households (RR 0.33, 95% CI 0.18 to 0.58, I2 statistic = 40%; RD = 14.84%, 95% CI 12.18 to 16.55, NNTB = 7, 95% CI 6 to 9) (Analysis 4.3). However, it did not reduce the risk of asymptomatic influenza in the prophylaxis of individuals (RR 0.97, 0.76 to 1.24, I2 statistic = 0%) (Analysis 4.2) or asymptomatic individuals in post‐exposure prophylaxis of households (RR 0.88, 95% CI 0.65 to 1.20, I2 statistic = 0%) (Analysis 4.4). See Table 11 for the overall results for oseltamivir in adults and Table 9 for children. See Table 12 and Table 13 for the overall results for zanamivir in adults and Table 14 for children.
4.1. Analysis.
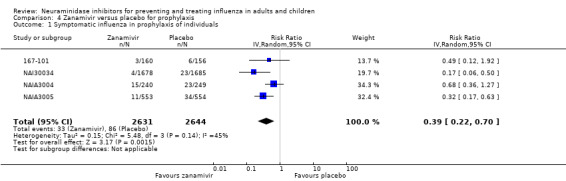
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 1 Symptomatic influenza in prophylaxis of individuals.
4.3. Analysis.
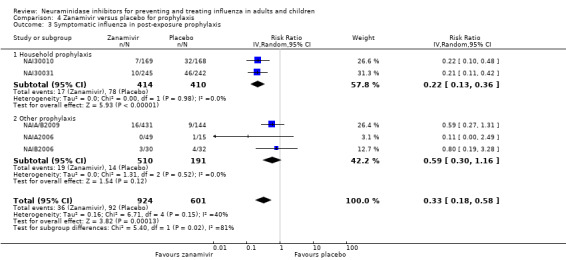
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 3 Symptomatic influenza in post‐exposure prophylaxis.
4.2. Analysis.
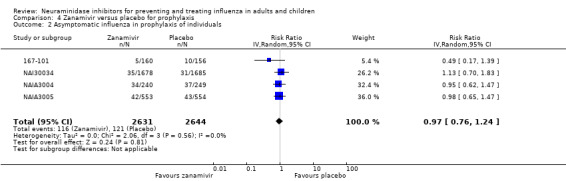
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 2 Asymptomatic influenza in prophylaxis of individuals.
4.4. Analysis.
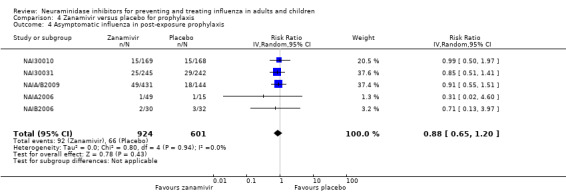
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 4 Asymptomatic influenza in post‐exposure prophylaxis.
11. Oseltamivir versus placebo for preventing influenza in healthy children.
| Oseltamivir versus placebo for preventing influenza in healthy children | |||||
| Patient or population: healthy children without influenza Settings: community Intervention: oseltamivir versus placebo for prophylaxis | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Comments | |
| Assumed risk | Corresponding risk | ||||
| Placebo | Oseltamivir versus placebo for treatment | ||||
| No data | — | — | — | — | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio | |||||
12. Zanamivir versus placebo for preventing influenza in healthy adults.
| Zanamivir versus placebo for preventing influenza in healthy adults | ||||||
| Patient or population: healthy adults without influenza Settings: community, nursing homes Intervention: zanamivir versus placebo for prophylaxis | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Risk difference (95% CI) | NNTB or NNTH (95% CI) | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Zanamivir versus placebo for prophylaxis | |||||
| Symptomatic influenza in prophylaxis of individuals | Study population |
RR 0.39 (0.22 to 0.70) |
5275 (4) |
1.98% (0.98 to 2.54) | NNTB = 51 (40 to 103) | |
| 33 per 1000 | 13 per 1000 (7 to 23) | |||||
| Asymptomatic influenza in prophylaxis of individuals | Study population | RR 0.97 (0.76 to 1.24) | 5275 (4) | 0.14% (‐1.1 to 1.1) | NNTB = 729 (NNTB 91 to ∞ to NNTH 91) | |
| 50 per 1000 | 48 per 1000 (38 to 60) | |||||
| Symptomatic influenza in household prophylaxis | Study population | RR 0.22 (0.13 to 0.36) | 824 (2) | 14.84% (12.18 to 16.55) | NNTB = 7 (6 to 9) | |
| 190 per 1000 | 42 per 1000 (25 to 68) | |||||
| Asymptomatic influenza in household prophylaxis | Study population | RR 0.90 (0.6 to 1.35) | 824 (2) | 1.32% (‐2.2 to 3.84) | NNTB = 76 (NNTB 26 to ∞ to NNTH 46) | |
| 107 per 1000 | 97 per 1000 (64 to 145) | |||||
| Complications: pneumonia in adult prophylaxis | Study population | RR 0.30 (0.11 to 0.8) | 7662 (6) | 0.32% (0.09 to 0.41) | NNTB = 311 (244 to 1086) | |
| 5 per 1000 | 1.5 per 1000 (1 to 4) | |||||
| Complications: bronchitis in adult prophylaxis | Study population |
RR 0.49 (0.02 to 1.19) |
7662 (6) |
0.79% (‐0.29 to 1.24) | NNTB = 127 (to NNTB 81 to ∞ to NNTH 341) | |
| 15 per 1000 |
8 per 1000 (3 to 18) |
|||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Negative risk differences indicate harms; positive risk differences indicate benefits. CI: confidence interval; RR: risk ratio; NNTB: number needed to treat to benefit; NNTH: number needed to treat to harm | ||||||
13. Oseltamivir versus placebo for preventing influenza in healthy adults.
| Oseltamivir versus placebo for preventing influenza in healthy adults | ||||||
| Patient or population: healthy adults without influenza Settings: community, nursing homes Intervention: oseltamivir for prophylaxis Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Risk difference (95% CI) | NNTB or NNTH (95% CI) | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oseltamivir versus placebo for prophylaxis | |||||
| Symptomatic influenza in adult prophylaxis of individuals | Study population | RR 0.45 (0.30 to 0.67) | 2479 (3) | 3.05% (1.83 to 3.88) | NNTB = 33 (26 to 55) | |
| 55 per 1000 | 25 per 1000 (17 to 37) | |||||
| Symptomatic influenza in household prophylaxis | Study population | RR 0.2 (0.09 to 0.44) | 405 (1) | 13.6% (9.52 to 15.47) | NNTB = 7 (6 to 11) | |
| 170 per 1000 | 34 per 1000 (15 to 75) | |||||
| Adverse events: psychiatric body systems in adult prophylaxis (all events on‐ and off‐treatment) | Study population | RR 1.80 (1.05 to 3.08) | 3434 (4 studies) | ‐1.06% (‐2.76 to ‐0.07) | NNTH = 94 (36 to 1538) | |
| 13 per 1000 | 23 per 1000 (14 to 40) | |||||
| Adverse events: headache in adult prophylaxis (on‐treatment) | Study population | RR 1.18 (1.05 to 1.33) | 3434 (4) | ‐3.15% (‐5.78 to ‐0.88) | NNTH = 32 (18 to 115) | |
| 175 per 1000 | 207 per 1000 (184 to 233) | |||||
| Adverse events: nausea in adult prophylaxis (on‐treatment) | Study population | RR 1.96 (1.2 to 3.2) | 3434 (4) | ‐4.15% (‐9.51 to ‐0.86) | NNTH = 25 (11 to 116) | |
| 43 per 1000 | 85 per 1000 (52 to 138) | |||||
| Adverse events: vomiting in adult prophylaxis (on‐treatment) | Study population | RR 1.91 (0.7 to 5.22) | 3434 (4) | ‐0.95% (‐4.41 to 0.31) | NNTH = 106 (NNTB 319 to ∞ to NNTH 23) | |
| 10 per 1000 | 20 per 1000 (7 to 55) | |||||
| Adverse events: headache in adult prophylaxis (off‐treatment) | Study population | RR 0.88 (0.63 to 1.24) | 3434 (4) | 0.44% (‐0.89 to 1.37) | NNTB = 226 (NNTB 74 to ∞ to NNTH 113) | |
| 37 per 1000 | 33 per 1000 (23 to 46) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Negative risk differences indicate harms; positive risk differences indicate benefits. CI: confidence interval; RR: risk ratio; NNTB: number needed to treat to benefit; NNTH: number needed to treat to harm | ||||||
14. Zanamivir versus placebo for preventing influenza in healthy children.
| Zanamivir versus placebo for preventing influenza in healthy children | |||||
| Patient or population: healthy children without influenza Settings: community Intervention: zanamivir versus placebo for prophylaxis | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Comments | |
| Assumed risk | Corresponding risk | ||||
| Placebo | Zanamivir versus placebo for treatment | ||||
| No data | — | — | — | — | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio | |||||
Analysis of harms
Oseltamivir treatment
Nausea, vomiting and diarrhoea
Oseltamivir in the treatment of adults is associated with increased risk of nausea (RR 1.57, 95% CI 1.14 to 2.15, I2 statistic = 43%; RD 3.66%, 95% CI 0.90 to 7.39; number needed to treat to harm (NNTH) = 28, 95% CI 14 to 112) (Analysis 1.5) and vomiting (RR 2.43, 95% CI 1.75 to 3.38, I2 statistic = 12%; RD 4.56%, 95% CI 2.39 to 7.58; NNTH = 22, 95% CI 14 to 42) (Analysis 1.6). It is associated with a decreased risk of diarrhoea (RR 0.67, 95% CI 0.46 to 0.98, I2 statistic = 44%; RD 2.33%, 95% CI 0.14 to 3.81; NNTB = 43, 95% CI 27 to 709) (Analysis 1.7) when compared to placebo during on‐treatment periods. Both nausea and vomiting were associated with significant heterogeneity where treatment effects appeared larger in otherwise healthy adults compared to the elderly and the chronically ill. However, one trial of otherwise healthy adults also showed smaller effects (WV16277). Vomiting was more common in those children on oseltamivir treatment compared to those on placebo (RR 1.70, 95% CI 1.23 to 2.35, I2 statistic = 0%; RD 5.34%, 95% CI 1.75 to 10.29; NNTH = 19, 95% CI 10 to 57) (Analysis 1.63).
1.5. Analysis.
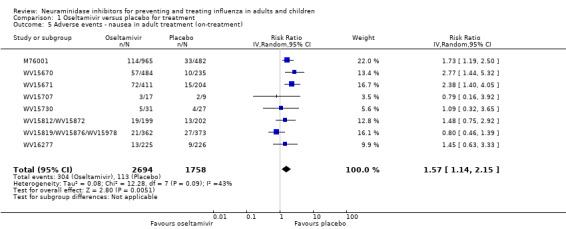
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 5 Adverse events ‐ nausea in adult treatment (on‐treatment).
1.6. Analysis.
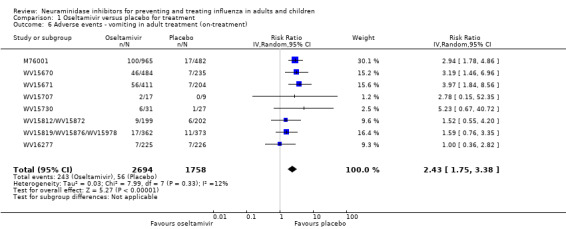
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 6 Adverse events ‐ vomiting in adult treatment (on‐treatment).
1.7. Analysis.
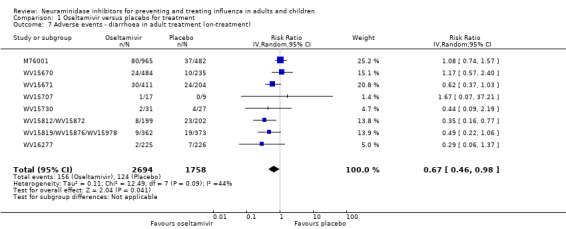
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 7 Adverse events ‐ diarrhoea in adult treatment (on‐treatment).
1.63. Analysis.
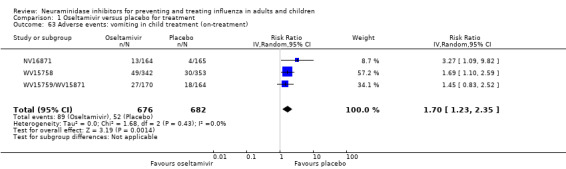
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 63 Adverse events: vomiting in child treatment (on‐treatment).
Cardiac effects
The cardiac effects of oseltamivir are unclear. Exposure to oseltamivir may reduce cardiac general events compared to placebo (RR 0.49, 95% CI 0.25 to 0.97, I2 statistic = 0%; RD 0.68%, 95% CI 0.04 to 1.00; NNTB = 148, 95% CI 101 to 2509), excluding WV16277 in which ECG was included in the safety parameters (Analysis 1.27). However, exposure to oseltamivir may increase QTc prolongation (including borderline) as reported in trial WV16277 (RD 4.0%, 95% CI 0.71 to 7.30; NNTH = 25, 95% CI 14 to 140) compared to placebo during on‐treatment periods.
1.27. Analysis.
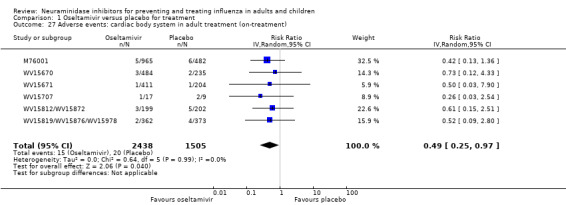
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 27 Adverse events: cardiac body system in adult treatment (on‐treatment).
Psychiatric effects
In treatment trials, there was no significant increase in risk between oseltamivir and on‐treatment psychiatric adverse events overall (Analysis 1.32). However, there was a dose‐response effect in the two "pivotal" treatment trials. In the identically designed trials WV15670 and WV15671 there were two active treatment groups: 150 mg (standard dose) and 300 mg (high dose) oseltamivir per day. In the dose‐response analysis there was an increased risk of psychiatric body system adverse events over the entire follow‐up period (P = 0.038 based on likelihood ratio test). In trial WV15670, the event rates were: 1/204, 1/206 and 4/205 in the placebo, 75 mg and 150 mg arms respectively, whereas trial WV15671 had rates of 2/235, 0/242 and 5/242, respectively.
1.32. Analysis.
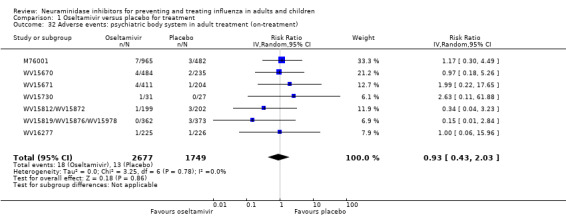
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 32 Adverse events: psychiatric body system in adult treatment (on‐treatment).
Effect on antibodies (post‐protocol hypotheses)
The proportion of patients being diagnosed as influenza‐infected in oseltamivir treatment of adults was significantly lower in the treated compared to the control group (RR 0.95, 95% CI 0.91 to 0.99, I2 statistic = 0%) (Analysis 1.3). The proportion of patients with four‐fold increases in antibody titre was significantly lower in the treated group compared to the control group (RR 0.92, 95% CI 0.86 to 0.97, I2 statistic = 0%) (Analysis 1.4). This represents an absolute difference of 5% between treatment groups. There was a lower proportion of children on oseltamivir with a four‐fold increase in antibodies (RR 0.90, 95% CI 0.80 to 1.00, I2 statistic = 0%) (Analysis 1.49).
1.3. Analysis.
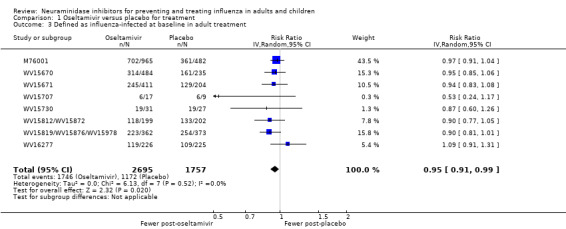
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 3 Defined as influenza‐infected at baseline in adult treatment.
1.4. Analysis.
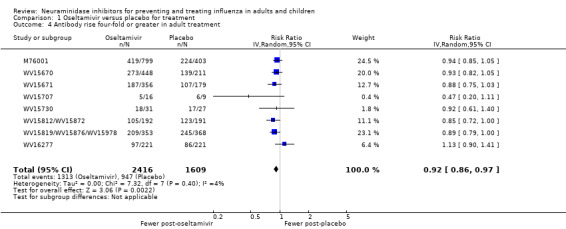
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 4 Antibody rise four‐fold or greater in adult treatment.
1.49. Analysis.
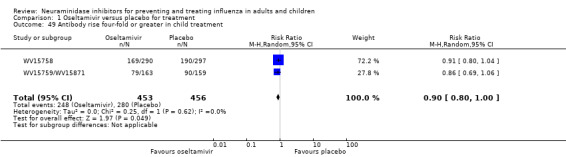
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 49 Antibody rise four‐fold or greater in child treatment.
Oseltamivir prophylaxis
Headaches and nausea
In oseltamivir prophylaxis, there was an increased risk of headaches on‐treatment (RR 1.18, 95% CI 1.05 to 1.33, I2 statistic = 0%; RD 3.15%, 95% CI 0.88 to 5.78; NNTH = 32, (95% CI 18 to 115) (Analysis 2.19; Figure 10) and nausea on‐treatment (RR 1.96, 95% CI 1.20 to 3.20, I2 statistic = 49%; RD 4.15%, 95% CI 0.86 to 9.51; NNTH = 25, 95% CI 11 to 116) (Analysis 2.20). There was also a dose‐response effect for headaches in study WV15673/WV15697 (P = 0.013 based on likelihood ratio test), where on‐treatment rates were: 202/519, 225/520 and 242/520 in the placebo, standard‐dose and high‐dose arms, respectively.
2.19. Analysis.
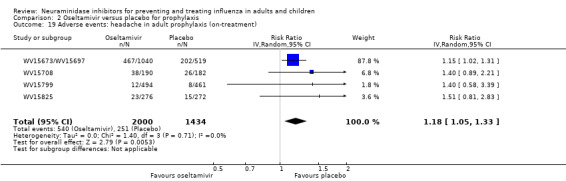
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 19 Adverse events: headache in adult prophylaxis (on‐treatment).
10.
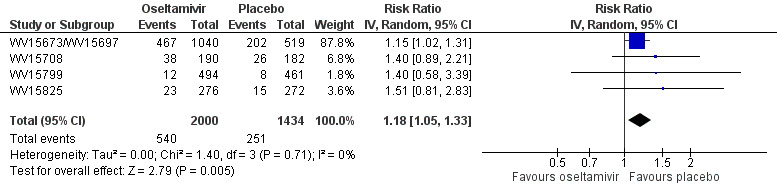
Forest plot of comparison: 2 Oseltamivir versus placebo for prophylaxis, outcome: 2.19 Adverse events: headache in adult prophylaxis (on‐treatment).
2.20. Analysis.
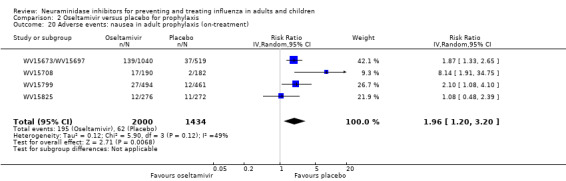
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 20 Adverse events: nausea in adult prophylaxis (on‐treatment).
Psychiatric effects
Figure 11 (Analysis 2.54) shows that in prophylaxis trials of oseltamivir there was a significant increase in patients with psychiatric adverse events over the on‐ and off‐treatment periods (RR 1.80, 95% CI 1.05 to 3.08, I2 statistic = 0%; RD 1.06%, 95% CI 0.07 to 2.76; NNTH = 94, 95% CI 36 to 1538). Initial analysis of patients with psychiatric adverse events in the on‐treatment period showed a borderline statistically significant result (P = 0.06), hence we conducted sensitivity analysis using Peto's method (P = 0.05) as well as the analysis reported in Figure 11.
11.
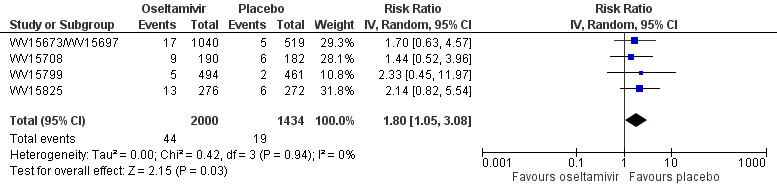
Forest plot of comparison: 2 Oseltamivir versus placebo for prophylaxis, outcome: 2.54 Adverse events: psychiatric body system in adult prophylaxis (on‐ and off‐treatment).
2.54. Analysis.
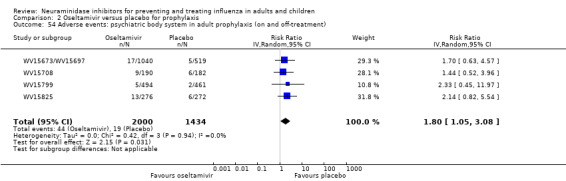
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 54 Adverse events: psychiatric body system in adult prophylaxis (on and off‐treatment).
Table 15 shows a summary of all psychiatric adverse events in oseltamivir prophylaxis trials. Of particular note was an oseltamivir patient in study WV15825 who had severe confusion on day 27 and was hospitalised. On day 28 the patient was taken off medication and the event resolved. On day 29 the patient was discharged from hospital and subsequently resumed medication. However, confusion reappeared on day 32. The initial event was misclassified in the clinical study report as "mental impairment" but has since been corrected in an erratum published in the same journal that published the original trial manuscript (Gravenstein 2013; Peters 2001).
15. Psychiatric adverse events in oseltamivir prophylaxis trials.
| Oseltamivir | Placebo | Total | ||||
| Event type | # Events | % | # Events | % | # Events | % |
| Confusion | 5 | 0.25 | 1 | 0.07 | 6 | 0.17 |
| Depression | 14 | 0.7 | 6 | 0.42 | 20 | 0.58 |
| Hallucinations | 2 | 0.1 | 0 | 0.00 | 2 | 0.06 |
| Anxiety | 7 | 0.35 | 8 | 0.56 | 15 | 0.44 |
| Psychosis | 2 | 0.1 | 1 | 0.07 | 3 | 0.09 |
| Schizophrenia | 1 | 0.05 | 0 | 0.00 | 1 | 0.03 |
| Bipolar disorder | 0 | 0 | 1 | 0.07 | 1 | 0.03 |
| Sleeping disorder | 2 | 0.1 | 0 | 0.00 | 2 | 0.06 |
| Aggression | 1 | 0.05 | 0 | 0.00 | 1 | 0.03 |
| Stress symptoms | 3 | 0.15 | 0 | 0.00 | 3 | 0.09 |
| Restlessness | 1 | 0.05 | 0 | 0.00 | 1 | 0.03 |
| Nervousness | 1 | 0.05 | 0 | 0.00 | 1 | 0.03 |
| Suicide ideation | 1 | 0.05 | 0 | 0.00 | 1 | 0.03 |
| Paranoia | 1 | 0.05 | 0 | 0.00 | 1 | 0.03 |
| Alcohol related | 6 | 0.3 | 2 | 0.14 | 8 | 0.23 |
| Total | 47 | 2.35 | 19 | 1.32 | 66 | 1.92 |
Of the 66 events, 12 were classified as severe intensity (10 oseltamivir, 2 placebo)
Renal effects
There was a non‐significant increase in renal events on‐treatment (RR 3.17, 95% CI 0.96 to 10.49, I2 statistic = 0; RD 0.67%, 95% CI ‐2.93 to 0.01; NNTH = 150, 95% CI NNTH 35 to ∞ to NNTB > 1000) (Analysis 2.38). However, in sensitivity analysis using Peto's method the result for renal events was statistically significant (P = 0.02).
2.38. Analysis.
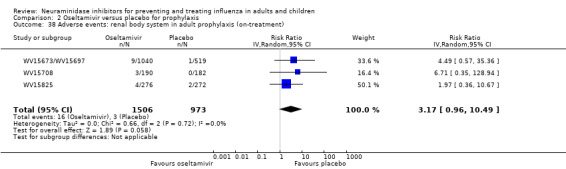
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 38 Adverse events: renal body system in adult prophylaxis (on‐treatment).
Zanamivir
Serious adverse events
There was no significant effect on serious adverse events in adult treatment trials (RR 0.86, 95% CI 0.49 to 1.50, I2 statistic = 0%) (Analysis 3.11).
3.11. Analysis.
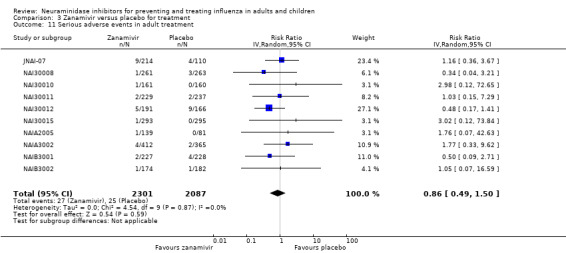
Comparison 3 Zanamivir versus placebo for treatment, Outcome 11 Serious adverse events in adult treatment.
Nausea, vomiting and diarrhoea
In treatment trials, there was no significant effect on diarrhoea in adults (RR 0.87, 95% CI 0.66 to 1.14, I2 statistic = 5%) (Analysis 3.33) or headache (RR 0.84, 95% CI 0.60 to 1.18, I2 statistic = 0) (Analysis 3.35). However, during the on‐treatment phase, nausea and vomiting were significantly less frequent in the zanamivir arm (RR 0.60, 95% CI 0.39 to 0.94, I2 statistic = 0%; RD 1.63%, 95% CI 0.24 to 2.48%; NNTB = 62, 95% CI 41 to 411) (Analysis 3.32).
3.33. Analysis.
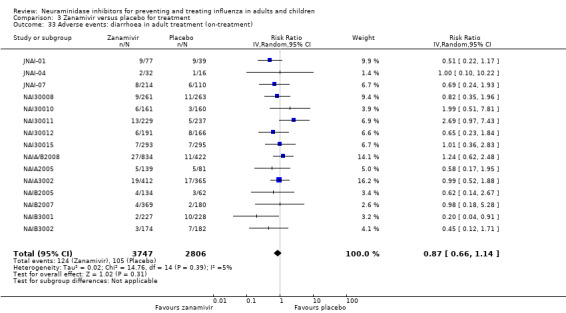
Comparison 3 Zanamivir versus placebo for treatment, Outcome 33 Adverse events: diarrhoea in adult treatment (on‐treatment).
3.35. Analysis.
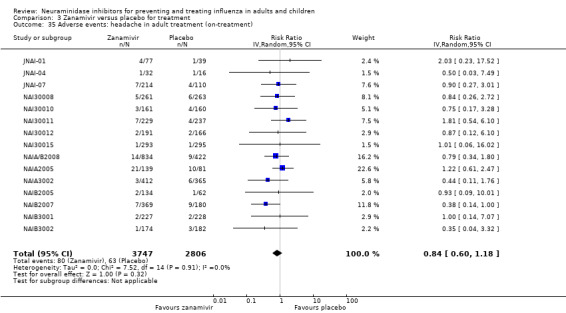
Comparison 3 Zanamivir versus placebo for treatment, Outcome 35 Adverse events: headache in adult treatment (on‐treatment).
3.32. Analysis.
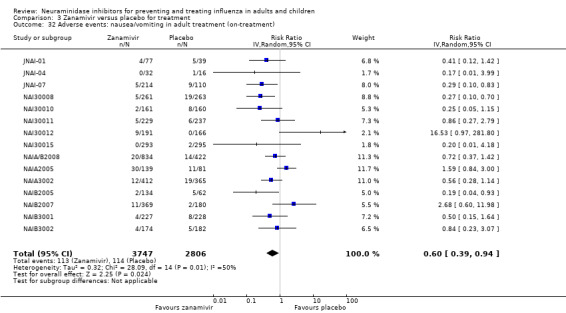
Comparison 3 Zanamivir versus placebo for treatment, Outcome 32 Adverse events: nausea/vomiting in adult treatment (on‐treatment).
Renal, psychiatric and other harms
There was no significant effect observed on the renal system (RR 0.84, 95% CI 0.41 to 1.72, I2 statistic = 0%) (Analysis 3.45), or the psychiatric system (RR 1.16, 95% CI 0.57 to 2.38, I2 statistic = 0%) (Analysis 3.48). In adult treatment trials of zanamivir, there was no significantly increased risk of any other reported adverse events and there was no significant increase in adverse effects observed in prophylaxis trials, including psychiatric (Analysis 4.29) and renal effects (Analysis 4.30) on‐treatment. There was no significant increase in harms associated with zanamivir treatment of children but data were sparse.
3.45. Analysis.
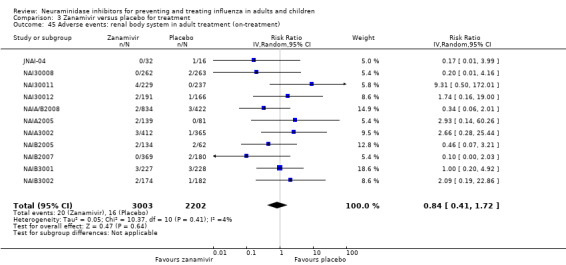
Comparison 3 Zanamivir versus placebo for treatment, Outcome 45 Adverse events: renal body system in adult treatment (on‐treatment).
3.48. Analysis.
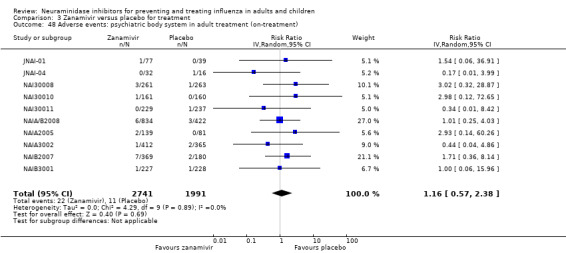
Comparison 3 Zanamivir versus placebo for treatment, Outcome 48 Adverse events: psychiatric body system in adult treatment (on‐treatment).
4.29. Analysis.
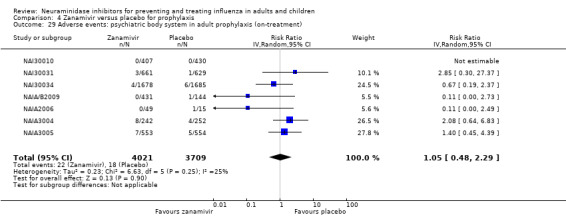
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 29 Adverse events: psychiatric body system in adult prophylaxis (on‐treatment).
4.30. Analysis.
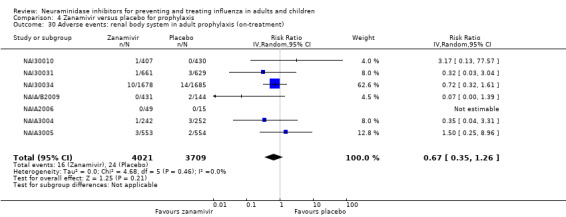
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 30 Adverse events: renal body system in adult prophylaxis (on‐treatment).
Effect on antibodies
There was no significant effect of zanamivir treatment on influenza diagnosis (Analysis 3.8) or probability of a four‐fold increase in antibody titre (Analysis 3.9; Analysis 3.10).
3.8. Analysis.
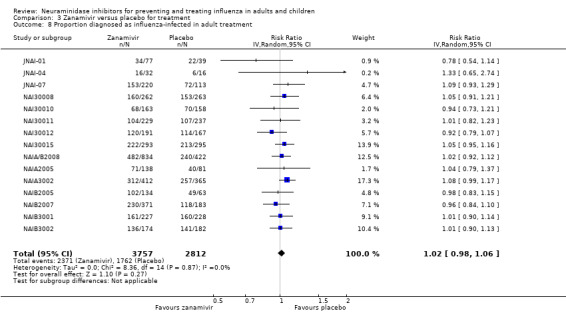
Comparison 3 Zanamivir versus placebo for treatment, Outcome 8 Proportion diagnosed as influenza‐infected in adult treatment.
3.9. Analysis.
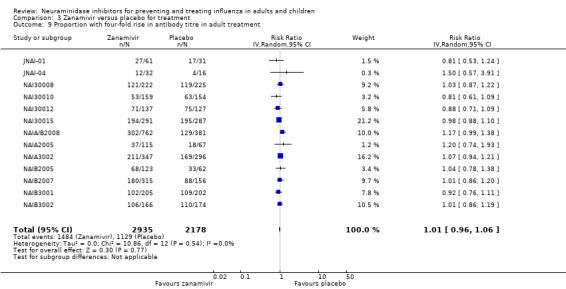
Comparison 3 Zanamivir versus placebo for treatment, Outcome 9 Proportion with four‐fold rise in antibody titre in adult treatment.
3.10. Analysis.
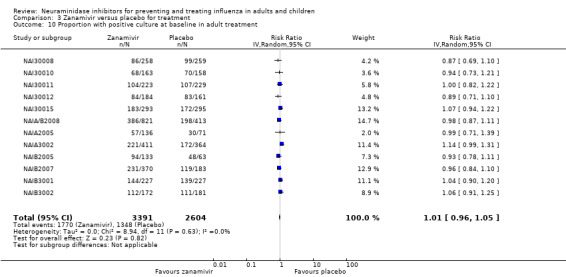
Comparison 3 Zanamivir versus placebo for treatment, Outcome 10 Proportion with positive culture at baseline in adult treatment.
Deaths
In oseltamivir treatment trials, there was one death overall. This event occurred due to acute respiratory syndrome in a placebo patient without influenza in study WV15812/WV15872. In prophylaxis trials, there were four deaths in total, all in elderly patients, with two in the placebo group and two in the oseltamivir group. Causes of death were reported as two cancers, one myocardial infarction and one intestinal perforation. However, for both deaths in the oseltamivir arms the participants experienced acute renal failure on‐treatment prior to death.
There were eight deaths in total in the zanamivir trials. Six of the deaths were caused by neoplasias or cardiovascular events in elderly patients with multiple pathologies. However, two deaths were reported as due to influenza A pneumonia. One participant was on inhaled rimantadine plus placebo and the other on zanamivir.
The results of post‐protocol hypotheses are in Appendix 10.
Discussion
Oseltamivir and zanamivir have small, non‐specific effects on reducing time to alleviation of influenza‐like illness symptoms in adults, but not in asthmatic children. Using either drug as prophylaxis reduces the risk of developing symptomatic influenza. Treatment trials with oseltamivir or zanamivir do not settle the question of whether complications of influenza such as pneumonia are reduced, because of a lack of diagnostic definitions. Use of oseltamivir increases the risk of adverse effects such as nausea, vomiting, psychiatric effects and renal events in adults and the risk of vomiting in children. The lower bioavailability may explain the lower toxicity of zanamivir compared to oseltamivir. The influenza virus‐specific mechanism of action proposed by the producers does not fit the clinical evidence.
Reconstructing trial lists and indexing regulatory comments
Calls for incorporating unpublished data to supplement published trial data in systematic reviews and meta‐analyses highlight deficiencies in the current methods for obtaining the most complete understanding of a drug's effects (Godlee 2010). Our methodological approach entailed comprehensive searching of unpublished sources, with a particular emphasis on obtaining unpublished and internal reports from drug manufacturers intended for regulatory submission and comments from national regulatory bodies. Our decision not to use published evidence as a basis for trial appraisal and data extraction meant that we had to reconcile and synthesise information from multiple unpublished sources. We had to devise a new method of searching, indexing, retrieving and reviewing trial data and to combine this understanding with regulatory comments to produce an informative review. The first step in this process entailed the need to develop our own reconstruction of the trial programme without initial help from outside sources. The reconstructed list of trials and then programmes took a whole‐time‐equivalent (WTE) researcher 20 days to compile. Due to the complexity of the task we suggest that in the future some of the essential phases, such as trial ID checking, be conducted in pairs.
One of the comments received on our protocol suggested that discrepancies between published and unpublished versions of the same data set could be due to mistakes in the non‐peer reviewed, unedited clinical study reports (which may be corrected by the time of publication). Our experience, especially with the non‐reporting of serious adverse events, points to the opposite being the case (Jefferson 2011b). Considering the fact that unintentional errors can occur, we believe the response should not be a resort to published papers as 'most accurate' and best unit of analysis, but rather that clinical study reports ‐ as by far the most comprehensive record of a trial ‐ remain the key unit of analysis, with the expectation that they be amended and kept as accurate as possible over time, with complete documentation of reasons for any amendments. We believed that the results of our review would be undermined without accessing a more complete body of evidence that we knew to be outside the public domain.
In theory, trial registers would be expected to provide a comprehensive picture of a drug's trial programme. However, registers were not our primary instruments to reconstruct zanamivir and oseltamivir trial programmes. Both drugs' programmes were mainly run in the late 1990s, before trial registration became the norm. In addition, registers may suffer from some of the problems that we were trying to address. Bourgeois 2011 audited entries for 546 trials of five major classes of drugs on ClinicalTrials.gov, the biggest prospective register of clinical trials, and found evidence of risk of reporting bias and delay in reporting of results. Another review of 152 trials found that the description of 123 (or 81%) of the trials in the sample had been changed in at least one key element in the time between registration and publication. The most frequent changes regarded outcomes (Huic 2011). Despite the current limits of registers, both specifically to this review and in the way they are run and updated, we believe that registers are an obvious first choice to start reconstruction of trials programmes. Searching for unpublished material has not yet become standard practice in conducting Cochrane reviews (Van Driel 2009), and is currently variably reported (Ghersi 2010).
The indexing and review of regulatory files was also a very laborious task. It took a WTE researcher three days to review the US Food and Drug Administration (FDA) regulator's comments and gain a basic understanding of the content. Four additional days were needed to read and annotate the FDA zanamivir files and 28 days for reading and annotating the oseltamivir files and building the Table of Contents‐Evidence (TOCE). The exercise had to be repeated several times to cross‐check content and expand annotations. Construction of the Table of Contents (TOC) was laborious. A first attempt at electronic mapping the TOC content took 12 and 8 hours respectively for the FDA and National Institute for Health and Clinical Excellence (NICE) regulatory documents. This was carried out using the Adobe Acrobat Optical Character Recognition (OCR) search facility, which enabled mapping of citation counts by document and by trial ID. Initially we used the trial prefix followed by the serial number ('WV15670') as ID. This procedure, however, had one major drawback linked to the nature of regulatory documents. As regulatory documents consist of notes, correspondence and reviews, the same trial is cited in a non‐standardised way. For example, trial WV15670 is cited as 'WV15670' 15 times, as 'WV_15670' 12 times and simply as '15670' 19 times). Thorough searches must be conducted using all the different terms. As this can be very time‐consuming, we decided to compare an Acrobat search with a Boolean string strategy containing all possible citation formats (for example, WV15758 OR WV 15758 OR Trial 15758 OR Trial15758 OR Trials 15758 OR Trials15758 OR 15758 OR study 15758 OR study15758) (this is logically equivalent to 'WV 15758 OR WV 15758') with a term‐by‐term search (i.e. separately searching for WV15758 and then for WV 15758 and so on). We reasoned that if the yield were comparable, the Boolean strategy would have been faster. The yield of citations of the two strategies was the same for six of seven 'tracker' studies but use of a Boolean string was considerably faster (an average of 3 versus 14 hours) than the term‐by‐term strategy. The National Institute of Clinical Excellence (NICE) submission citations took two hours to list in a TOC using a Boolean strategy. We adopted the Boolean search strategy to construct our TOC. Ultimately it is possible that a search with the trial numerals ('15670') may be sufficient to identify the vast majority of citations. To validate this method of searching further our methods should be repeated on other sets of regulatory documents.
Once we had reconstructed the trial programmes we submitted the results to GlaxoSmithKline (GSK) and Roche for their input. We received detailed feedback from both but as late as 2011 Roche's list of trials was incomplete. Despite the laboriousness of the methods, we believe we ended up with a far more comprehensive and less biased set of evidence than that available through the current system of journal‐based publications. This shift in our data synthesis paradigm was made necessary by the numerous and documented discrepancies between regulatory and published evidence and by the sizeable risk of publication bias of the oseltamivir trial programme. The importance of reconstructing the trial programme by first generating a complete trial list was further reinforced upon discovering bias and oversights in regulators' handling of the trial programme. Regulators focus on a few mutually agreed "pivotal" trials whose data analyses are replicated by the FDA but not by the European Medicines Agency (EMA). Both largely ignored trial M76001, the largest oseltamivir treatment trial conducted prior to initial registration of the drug (and still unpublished). While the manufacturer may not have offered it as a "pivotal" trial, far smaller and even ongoing studies were included in the evidence base to support Roche's year 1999 New Drug Application number 021087 (Treatment of uncomplicated acute illness due to influenza infections in adults who have been symptomatic for no more than two days). The depth of the EMA scrutiny is harder to assess as we could find no reports of trial site visits or of data analysis replication, but we identified a pooled analysis of treatment trials, very similar to the Kaiser 2003 analysis which formed the basis for the EMA conclusion that oseltamivir affected complications reported, for example, on EMA's 4 October 2012 Summary of product characteristics (SmPC) (http://www.bmj.com/tamiflu/ema). We requested Modules 3, 4 and 5 (individual listings, demographic data and the statistical analysis report) from the EMA. However, for most oseltamivir trials, the EMA do not have the relevant documents and neither apparently do National Competent Authorities (email from the EMA, 24 May 2011; email from Dutch regulator MEB, 20 July 2011). This means that the Modules do not appear to have been either submitted to or requested by regulators, raising questions as to the extent of scrutiny of the clinical trials during the regulatory review of oseltamivir in Europe.
Our new method
Reviewing huge quantities of complicated data and linked comments is a very difficult and delicate process. The main problem is not so much the appraisal following standard rules and possible synthesis of data (as when we review published information), but the reconstructions and logical threading of a trial programme generating huge amounts of data needing appraisal. Also the manufacturer's full regulatory submission, which may have even more information than a full clinical study report, remains confidential. Most of the essential data required are available in clinical study reports, together with masses of less important data, but as we have explained even in this case there may be important omissions, such as mislaid diary cards for follow‐up. Manufacturers are under obligation to provide regulators with all data requested to enable them to reach a decision: in doing so they produce vast submissions. None of the authors (all experienced systematic reviewers) had any experience of reviewing regulatory information. Given the laborious and painstaking process we tried to identify a quicker and equally reliable way of reviewing regulatory information but could not find any obvious shortcuts. However, we believe that providing a critical overview of a trial programme rather than minute dissection of each trial is necessary. This can be done by identifying the important topics in the trial programme (such as the effects of the drug on symptoms, infection, complications, transmission and well‐being) and following them throughout the programme, knitting the evidence into a coherent narrative. This includes carrying out a high‐level overview of the mode of action of the drug in different populations for different indications. Understanding any drug's mode of action is core to correct reporting of its strengths and limitations. In addition, a large part of the regulatory submission is made up of chemistry, microbiological, animal model pharmacodynamic and pharmacokinetic studies, which are important for shedding light on the trial programme but which seldom feature in systematic reviews. We are unsure as to whether this information could be considered as core information but an exhaustive review of a trial programme should include reviews dedicated to such topics.
These methods were crucial in discovering major concerns in trial conduct and validity, including the lack of comparability between arms induced by subset analysis and by the randomisation‐analysis fork, high positivity rate of influenza, high gastrointestinal events in the placebo arms, possibly active placebo content and possible procedural breaches several trials, which are concerning. Overall, the safest and more conservative option appears to carry out analyses on the basis of the intention‐to‐treat (ITT) population, in which units of randomisation and analysis are the same and many of the potential problems listed are either not present or minimised.
Our novel methodology remains a work in progress.
Regulatory comments
Reviewing regulatory comments was an essential way to deepen our understanding of the trial programme. From early on in our review we hoped that a close reading of regulatory material would allow us to understand the reason for discrepancies between US and European regulators' conclusions regarding the effects of oseltamivir, particularly (but not limited to) their purported effect on complications (Doshi 2009). We were interested in what led the FDA to have far more cautious and conservative statements ‐ as witnessed in the Tamiflu product label and FDA letters ‐ in comparison to European regulators. Our access to huge amounts of FDA regulatory data allowed for many insights but gave us little visibility of manufacturers' responses.
Some of the statements made by the manufacturer in the clinical study reports, and subsequently in contemporaneous publications and advertisements, appeared unsupported by the evidence provided at the time. The FDA drug regulatory reviewers' comments, although laborious to summarise and contextualise (because of the non‐availability of the whole pharmaceutical submission), were confirmed by our reading of the clinical study reports. However, we were unable to find a statement explaining how the FDA reviewed each New Drug Application (NDA). FDA reviewing methods appeared to be a mixture of spot checks, re‐run of statistical analyses and on‐site inspections. An FDA methods volume or standard operational procedure may be among the documents not available from the web but accessible through a Freedom of Information (FOI) request. Neither the FDA nor the EMA have inventories of held documents, making it very difficult to know what to ask for under FOI rules. We concentrated on downloading or asking for specific clinical study reports and related documents or reviewers' comments on a particular NDA. The quantity of information held by regulators is likely to be large. For example, New Drug Application 21‐246, the use of Tamiflu in the treatment of influenza in children submitted to the FDA on 15 June 2000 consisted of 137 volumes of study documents and possibly several electronic files. Although we do not know exactly how long a volume was, we have seen references to up to hundreds of pages in each volume.
Requesting specific documents and packages of information is especially important to allow a more efficient and timely reviewing process when confronted with a large volume of evidence, most of which could be of peripheral value. A request for a specific document is likely to be dealt with far more efficiently than a generic request for "all documentation relating to oseltamivir". This is one of the reasons why developing a TOC for any drug or family of drugs (no matter how time‐consuming) is an absolute prerequisite for any serious attempt at reviewing regulatory evidence. This introduces another very difficult problem: how to handle huge quantities of structured information and the ethics of drawing conclusions from what is still a fragmentary (albeit sizeable) evidence base.
Overall the FDA assessment of the performance of oseltamivir was "modest". This adjective appears six times in a 50‐page review document (FDA 1999c). For example, in the Division Director Memorandum dated 25 October 1999, under the heading "Public health role of antiviral treatment" the FDA states: "The clinical relevance of the modest treatment benefit is a highly subjective question" (FDA 1999c, PDF page 3). The FDA refused to accept claims of oseltamivir's effects on influenza complications as "false or misleading" statements in promotional materials (FDA 2000f). An FDA warning letter seems to imply, for example, that oseltamivir's mode of action is "proposed" or "possibly" [that proposed by the manufacturers] i.e. not certain (FDA 2000f). However, FDA reviewers appear to have missed important problems in Roche's clinical trials (such as the imbalance in the numbers of individuals classified as influenza‐infected in oseltamivir treatment trials). Most of all, no one seems to have questioned the coherence of the evidence with the proposed mode of action of the drug.
Summary of main results
For the first time a Cochrane review is based on all relevant full clinical study reports of a class of drugs integrated by regulatory comments. Also for the first time, all clinical study reports of trials in a manufacturer's programme (regardless of their relevance to the review) are available to readers without any restriction (apart from minimal redactions to protect anonymity further). The role of Roche and GSK in making this possible should be recognised, as well as that of the BMJ, which kept the issue in the public eye until it was resolved.
The evidence we have presented and synthesised shows that both neuraminidase inhibitors (NIs) in this review have symptom‐relieving effects, especially for self reported outcomes. They appear to have symptom‐relieving properties that make people with influenza‐like illness and self reported, investigator‐mediated, unverified pneumonia feel better by shortening symptom duration and reducing the frequency of symptoms such as cough. For oseltamivir, this effect perhaps extends to cardiac symptoms, despite the short duration of treatment (five days). We are unsure what to make of this finding but we think it deserves further investigation.
The issue which triggered our change of evidence‐seeking methods is partly resolved: no definitions of secondary illnesses were given anywhere in the clinical study reports (for example "pneumonia" was defined as "pneumonia" in the case report forms (Table 1) and diagnostic criteria were not given); clinical diagnosis in the absence of criteria and without X‐ray has only a moderate chance of being correct.
We could not decide the level of diagnostic ascertainment of diagnosis of pneumonia and other complications, as it is unclear from the clinical study reports. Definitions of pneumonia were not given and the algorithm for classification of an event as pneumonia was not supplied. In oseltamivir trials, the case report form trigger for recording of adverse events and secondary illness was a question to the participant posed by the investigator. A typical phrasing is as follows: "Secondary illness reminder: Has the patient reported any sinusitis, otitis, bronchitis, other chest infection or pneumonia since baseline?" This was followed by a yes/no box to be ticked and an additional form was to be filled out by the investigator for collecting details on the secondary illness. A record of medications outside trial allocation was elicited in addition to the participant's diary card. The original and Medical Dictionary for Regulatory Activities (MedDRA) terms suggest diagnoses for all secondary illnesses and adverse events but there is no indication how the original and preferred terms were assigned. We therefore considered these outcomes to be "self reported, investigator‐mediated, unverified" outcomes. For a subset of trials, secondary/intercurrent illness and adverse event data were collected on a single, one‐page form. In our meta‐analyses, we called this sub‐analysis "Trials which collected data on non‐specific adverse events or secondary/intercurrent illness form". For a different subset of trials, case report forms contained space to record diagnostic tests such as chest X‐rays, tympanometry and sinus X‐rays for all secondary illness but there was no reporting of such variables in the clinical study reports (Figure 12; Figure 13; Figure 14; Figure 15). In our meta‐analyses, we called this sub‐analysis "Trials which collected data on specific 'Diagnosis of Secondary Illness' form". None of the complications were defined as primary outcomes in any trial, which may explain the poverty of data definition.
12.

Sample "Adverse event or intercurrent illness" form (oseltamivir study M76001)
13.

Sample "Secondary illness" form (oseltamivir study WV15670)
14.

Sample "Diagnosis of secondary illness" form, page 1/2 (oseltamivir study WV15978)
15.

Sample "Diagnosis of secondary illness" form, page 2/2 (oseltamivir study WV15978)
In meta‐regression of all 32 included studies that reported on "pneumonia", we found evidence that treatment effects for pneumonia are statistically different depending on the method of diagnosis. Unclear objective diagnosis was associated with an apparent 46% reduction in pneumonia due to treatment with neuraminidase inhibitors, whereas the use of objective criteria in the data collection showed no evidence of effect, with a risk ratio (RR) of 1.0. Age group (adults versus children), drug (oseltamivir versus zanamivir) and indication (treatment versus prophylaxis) showed no evidence of association with treatment effect.
Meaningful conclusions on the effect of either NI on complications of influenza are difficult to draw based on the trial evidence. In part this was due to the lack of standardised definitions. In addition, meta‐analyses of these outcomes that lacked definitions were based on few events and therefore not robust. Caution is therefore urged in interpreting the meta‐analysis result, which suggests that 100 patients (67 to 451) need to be treated with oseltamivir for one less self reported, investigator‐mediated, unverified pneumonia. The same applies to the zanamivir treatment result, which suggests a reduced risk of self reported, investigator‐mediated, unverified bronchitis in adults (number needed to treat to benefit (NNTB) 56, 95% confidence interval (CI) 36 to 155). The evidence suggested oseltamivir had a similar effect, although the result was non‐significant.
As stated above, there is no evidence that definitions of complications in either paediatric, elderly or adult trials were ever prepared and incorporated in the trials' design. Therefore, the reporting of cases of 'otitis media', 'pneumonia', 'sinusitis' or 'bronchitis' are of unclear significance and importance, making it impossible to attribute a cause and draw conclusions (FDA 2000d). This is probably why the US Food and Drug Administration (FDA)‐approved oseltamivir package insert, since 17 November 2000, has consistently stated: "serious bacterial infections may begin with influenza‐like symptoms or may coexist with or occur as complications during the course of influenza. TAMIFLU has not been shown to prevent such complications." The original product label did not contain such a statement but on 14 April 2000, after oseltamivir was approved for sale in the United States, the FDA sent Roche an untitled letter about "Misleading Efficacy Claims" that the FDA had noted in Roche's promotional materials (FDA 2000a, PDF page 3). One of the statements that Roche made was: "Tamiflu reduces incidence of secondary complications (i.e. bacterial infections) by 45%." The FDA commented: "Further, you have claimed reductions in severity and incidence of secondary infections with Tamiflu that are misleading because they are not supported by substantial evidence" (FDA 2000a, PDF page 3). We do not know how Roche responded to the FDA but in subsequently available Roche promotional material information, Roche's statements were consistent with the FDA's demands (Doshi 2009).
There is uncertainty in the "complications" and "secondary illnesses" outcome definition therefore we carried out an analysis on the data from adult treatment trials on those complications classified as serious or those which led to study withdrawal. For oseltamivir, there was no evidence that treatment affected such complications (risk difference (RD) 0.07%, 95% CI ‐0.78 to 0.44) (Analysis 1.20; Analysis 1.55). This outcome could not be assessed in oseltamivir prophylaxis due to an insufficient number of events. For zanamivir, there was no significant evidence of a treatment effect on such complications (RD ‐0.04%, 95% CI ‐0.64 to 0.24) (Analysis 3.7; Analysis 4.8). This outcome could not be assessed in children due to an insufficient number of events.
Contrary to the FDA, the European Medicines Agency (EMA)'s oseltamivir 'Summary of Product Characteristics' states that oseltamivir significantly reduces the incidence of "specified lower respiratory tract complications (mainly bronchitis) treated with antibiotics" in individuals of 13 years of age and older. This claim is based on "a pooled analysis of all influenza‐positive adults and adolescents (N = 2413) enrolled into treatment studies", of which 1063 were in the placebo group and 1350 were in the oseltamivir‐treated population (EMA 2010). This statement appears in the EMA files as early as 2001 (EMEA 2001). These exact denominators appear in the Kaiser 2003 meta‐analysis.
The design of the trials, as defined in the protocol with amendments, statistical analysis plan and case report forms, does not allow any further inferences. The effect on outcomes that were originally considered of secondary or tertiary importance (such as bronchitis and pneumonia) would have been clarified with better clinical definitions and investigations, as some of the serious adverse events were. These benefited from a paragraph‐length narrative, which reported most of the salient features of the event.
Our previous decision to analyse the effects of oseltamivir and zanamivir on the ITT population has been confirmed for oseltamivir with the demonstration of the effect on antibody responses in participants in treatment trials, although no such effect is discernible for zanamivir. This effect leads to the introduction of selection bias with a significantly reduced probability of being diagnosed with influenza and an imbalance in the two arms if the intention‐to‐treat‐influenza‐infected (ITTI) population is analysed. The effect of oseltamivir on antibodies appears to be carried over to children with influenza‐like illness. Its finding contradicts statements made by the manufacturer.
The seeming incomparability between arms of the influenza‐infected subpopulations in the oseltamivir trials raises the question of how an appropriate analysis should be conducted. If influenza‐infected groups are comparable (as appears to be the case in zanamivir treatment trials) then an appropriate analysis strategy (based on Senn 2004) would be to determine first the effect of treatment in the ITT population. If there is evidence of a treatment effect, then treatment by infected status interaction could be tested. If there was evidence of an interaction, then estimates of treatment effect could be derived separately for the influenza‐infected and non‐influenza‐infected subpopulations. However, this analysis should be conducted on the ITT population using a single appropriate statistical model, obviating the need to conduct separate analysis on the influenza‐infected subpopulation. Roche used geometric mean titres indicating antibody responses in the ITTI population to support their statement that oseltamivir does not affect antibody responses (for example, in Table 16 and linked text of Module 1 of trial WV15799). However, the use of such measures can be misleading. What are required for such an analysis are data on how many ITT population participants responded by arm, at what level of antibody response and how many were tested. Such data could not be identified with certainty. A further effect of choosing a subpopulation analysis (ITTI in treatment trials and ITTIINAB (ITT influenza‐infected index cases who had negative virology at baseline) in prophylaxis trials) as the primary analysis is the restriction of the generalisability of results. This is especially so in the case of design flaws (for example, in the case of the post‐exposure prophylaxis trial WV15799, where all index cases were not treated and around 55% of participants were dropped from the ITTIINAB analysis). In this cluster‐trial design households should be included as random‐effects in the analysis to take account of within‐household correlations.
16. Proportions of contacts with positive serology data (WV15799 ITTIINAB population).
|
Positive serology |
Group | Total | |
|
Placebo N % |
Tamiflu N % |
||
| No | 166 83.0 | 192 93.7 | 358 |
| Yes | 34 17.0 | 13 6.3 | 47 |
| Total | 200 | 205 | 405 |
ITTIINAB population: ITT influenza‐infected index cases who had negative virology at baseline
Chi² P = 0.001
A significant but slight reduction of the proportion with serum antibody (mostly haemagglutination inhibition (HAI) antibody) titre rise by four‐fold or more among those who were tested was shown in this review. This was consistent with the evidence from animal tests using a sub‐clinical dose of oseltamivir in influenza A/H1N1‐infected mice (Takahashi 2010). Takahashi 2010 reported a non‐significant slight reduction of haemagglutinin (HA) specific IgG antibody in serum and spleen, while they reported about an 80% significant reduction of HA specific secretory IgA antibody (s‐IgA Ab) in nasal wash and bronchoalveolar fluids (BALF) on day 12. From this evidence, they warned that the risk of re‐infection may increase in patients showing a low mucosal IgA antibody response following oseltamivir administration. These experiments were done because they had the unexpected finding that paediatric influenza patients treated orally with oseltamivir for five days had significantly low levels (about 60% reduction on day five) of anti‐influenza S‐IgA nasopharyngeal fluids compared to levels in patients not treated with oseltamivir (Sawabuchi 2009). Their findings are consistent with our findings that serum HA inhibition (HAI) antibody response was decreased by oseltamivir administration, though s‐IgG Ab could not be analysed in our study because the data were not reported in the clinical study reports (Sawabuchi 2009; Takahashi 2010). These findings are also consistent with the evidence on the mode of action of oseltamivir from animal models (Module 2 of Mendel 1998; WV15670; WV15671) and from viral challenge, randomised, placebo‐controlled studies in humans (Hayden 1999).
Pro‐inflammatory cytokines, including interleukin 6 (IL‐6), tumour necrosis factor alpha (TNF‐α) and interferon gamma (IFN‐γ), were completely suppressed by oseltamivir administered 28 hours after the experimental inoculation of influenza virus, while the reduction of viral titre in nasal lavages was partial (Hayden 1999).
There is decisive evidence that administration of oseltamivir in animals challenged by respiratory syncytial virus (RSV) that lacks a neuraminidase gene showed a symptom‐relieving effect (decreased weight loss) and inhibition of viral clearance (Moore 2007). These effects were accompanied by a decreased CD+8 T cell surface sialoglycosphingolipid GM1 level, which is regulated by the endogenous sialidase/neuraminidase in response to viral challenge along with suppression of cytokine expression (Moore 2007). They are consistent with those findings from the pharmaceutical company and their investigators. The findings of the study by Moore 2007 suggest a risk of infection and exacerbation of infection by pathogens other than influenza virus in spite of the apparent reduction of symptoms from infection.
Sufficient plasma concentration of oseltamivir carboxylate from orally administered oseltamivir phosphate may act directly on the host endogenous neuraminidase to reduce (or suppress) the immune response even at the dose of 20 mg twice a day for five days. However, the bioavailability of inhaled zanamivir seems to be very broad: about 10% to 70%, as estimated by the area under the curve (AUC) data from the inhalation and intravenous study from the Japanese Summary Basis of Approval (JSBA). The difference in peak concentration (Cmax) was much larger (6 to 37‐fold). This means that inhaled zanamivir could reach a high enough concentration to reduce the immune response, if it is administered at a high dose or a for long period, or if the patient is very susceptible. In fact, a double‐blind, placebo‐controlled trial using healthy volunteers to investigate the effect of zanamivir treatment (20 mg/day for 14 days) on the humoral immune response to influenza vaccine showed that the zanamivir group responded with significantly lower antibody titres to the H1N1 (Cox 2001). Pro‐inflammatory cytokines, including IL‐6, TNF‐α, IFN‐γ and other chemokines, were almost completely suppressed in the viral challenge randomised controlled trial (RCT) using a very high dose (600 mg) of intravenous zanamivir before inoculation of the influenza virus in human adults (Fritz 1999).
These findings all suggest that the low immune response, with a low level of pro‐inflammatory cytokines, induced by the action of oseltamivir carboxylate may reduce the symptoms of influenza irrespective of an inhibition of influenza virus replication, which is widely believed to be the main mode of action of NIs.
In addition, the potential hypothermic or antipyretic effect of oseltamivir (but not zanamivir) as a central nervous system depressant may also contribute to the apparent reduction of host symptoms (Ono 2008; Ono 2013).
Zanamivir had no effect on pneumonia symptoms in treatment trials, even when the diagnosis was supported by a chest X‐ray; nor did it affect antibody responses, but it did affect bronchitis. We think that this shows an undeniable symptom‐relieving effect of both drugs, which also applies to more severe, if undefined, syndromes. Both drugs relieve influenza‐like illness symptoms by around 0.6 to 0.7 of a day, although this is first relief and not necessarily complete relief. In the case of oseltamivir, the mix up with the follow‐up cards does not allow us to draw any conclusions on a possible length of the duration of symptom relief. Also of note is the fact that this important information came to light from the FDA reports and not from the clinical study reports of the relevant trials (WV15670; WV15671). This points to the incomplete nature of reporting in the clinical study reports and the important role of Summary Basis of Approval (SBA) regulatory information.
In a subgroup analysis we found no evidence of a difference in treatment effect for zanamivir on time to first alleviation of symptoms in adults in the influenza‐infected and non‐influenza‐infected subgroups. Both subgroups showed strong evidence of treatment effect of 0.5 to 0.7 days reduction in time to first alleviation of symptoms. This strongly supports our hypothesis that these drugs do not have an influenza‐specific effect.
Oseltamivir relieves symptoms in otherwise healthy children, but no effect was noted with zanamivir, which may be due to the limited power of the two eligible trials with just over 700 children in total. However, oseltamivir does not have any effect on asthmatic children with influenza‐like illness, a population which should benefit most from its use. One explanation for this finding is in the nature of the young asthmatic population, which is well cared for and used to regular powerful medications and close follow‐up. The incremental benefit of oseltamivir is thus likely to be undetectable in such a population. An alternative explanation could be the higher susceptibility of the immune system to suppression by oseltamivir carboxylate in asthmatic children compared with those in the placebo group. The finding that oseltamivir administered to asthmatic children reduces symptoms faster than in placebo recipients at the beginning of the study, but during the off‐treatment period more recovered later than those administered placebo, gives some support to this explanation.
There is no evidence of an effect of oseltamivir on hospitalisations. Hospitalisations are an important but poorly defined outcome in the oseltamivir protocols, inconsistently reported in the clinical study reports and overlooked in the zanamivir protocols and reports.
The oseltamivir trials did not detect any influenza‐related deaths, reflecting the relatively benign nature of influenza in the study populations. The zanamivir trials detected eight deaths, of which only two were likely to be due to influenza and both occurred in the intervention arms. All the trials were likely to be underpowered to detect differential effects on mortality, but the absence of deaths in placebo recipients again underlines the benign nature of influenza. In fact mortality in Japan during the 2009A/H1N1 influenza outbreak was 198 among about 20 million influenza patients (one in 100,000 infected). Early deterioration leading to death was observed more frequently in oseltamivir compared to zanamivir or no antivirals recipients (Hama 2011).
Overall the two drugs have similar benefits but quite different toxicity profiles.
On average, for every 28 (14 to 112) adults treated with oseltamivir there will be one more report of nausea and for every 22 (14 to 42) adults and 19 (10 to 57) children there will be one more report of vomiting. Oseltamivir seems to have an apparent protective effect on diarrhoea, contrary to the other evidence of gastrointestinal disturbance. This finding might be as an effect of a placebo containing dehydrocholic acid or it might be one of the results of the influenza‐like illness symptom‐relieving effects (similar to relief of tachycardia and palpitation). The other apparent gastrointestinal events, such as nausea and vomiting, may be the results of central nervous disorders indicated by "only day 1 increase of vomiting" in treatment trials in children.
For every 62 (41 to 411) adults exposed to zanamivir there will be one less case of nausea and vomiting, but no such effect was visible in children, probably because of a lack of power. Zanamivir does not appear to affect the frequency of bowel movements.
In the prophylaxis data set, "influenza without laboratory confirmation" (i.e. influenza‐like illness) was only partially reported in the oseltamivir clinical study reports and not reported in the zanamivir clinical study reports, except for NAI30034 in which no significant reduction was observed (9% versus 10%). As a consequence we are unable to report on that outcome. The size of the reduction in influenza symptoms in oseltamivir prophylactic trials is inferior in magnitude to that seen in hand washing to prevent severe acute respiratory syndrome (SARS), based on seven case‐control studies (odds ratio (OR) 0.77, 95% CI 0.70 to 0.84, I2 statistic = 68%, RD ‐0.12, ‐0.16 to 0.08, I2 statistic = 26%), the NNTB being approximately 50 for prophylaxis with oseltamivir and eight with hand washing (Jefferson 2011a).
There is a significant reduction in the proportion of patients with symptomatic influenza with both NIs. However, these findings do not reflect the true efficacy for prevention of influenza, because they conceal the positivity of laboratory testing (measured through tests of viral shedding and four‐fold antibody titre rise).
We found an apparent prophylactic effect of zanamivir on pneumonia (which was not defined in case report forms) when it was used for 14 to 28 days. However, we found no evidence of significant effects on other complications and no evidence of an effect of oseltamivir on complications or hospitalisations.
Oseltamivir induced nausea in people undergoing prophylaxis but there was insufficient evidence to show an association with vomiting.
On‐treatment renal adverse events were three times more common in the oseltamivir arms compared to the placebo arms, with 150 treated patients leading to one additional event. The two participants who died in the oseltamivir arms both experienced acute renal failure while on‐treatment, although only one of those events was listed as an adverse event. The unlisted event was in a 91‐year old female who was "withdrawn from the study on Study Day 15 because her estimated creatinine clearance was less than 30 mL/min. The screening laboratory examinations, that were carried out 10 days before the start of study treatment, were normal". Hyperglycaemic adverse events (aggravated diabetes mellitus or hyperglycaemia) were also more common in the oseltamivir arms, with eight events in total (one in WV15673/WV15697, two in WV15708 and five in WV15825) compared to none in the corresponding placebo arms. These data are only presented descriptively as they are too few (< 10) to meta‐analyse formally, as prespecified in our analysis plan.
Finally, oseltamivir caused headaches and psychiatric harms in adult prophylaxis trials. Headaches are one of the most prominent harms of oseltamivir. There is evidence of a dose‐response effect in prophylaxis trials WV15673/WV15697 (P = 0.013), in which headaches were observed in 202/519, 225/520 and 242/520 participants in the placebo, oseltamivir 75 mg once daily and 75 mg twice daily arms, respectively.
In the psychiatric category, several rare and severe single events (nervousness, aggression, hallucinations, psychosis, suicide ideation and paranoia) were reported significantly more frequently in the intervention arm. Added to other more frequently reported but not significantly different events (such as depression and confusion) this gave a large effect and a relatively small number needed to treat to harm of 94 (36 to 1538). The importance of such a finding lies in the distribution of oseltamivir to large numbers of asymptomatic individuals following pandemic plans. There were no prophylaxis trials in children that met our inclusion criteria, therefore we cannot report on prophylaxis harms in this important population.
The question of why oseltamivir treatment trials failed to identify a clear association between oseltamivir and psychiatric harms, although a weak dose‐dependent association was observed, is a moot point. It is possible that influenza‐like illness and influenza symptoms masked the harms in those who were already symptomatic and therefore recruited in the treatment trials (and influenza‐type symptoms were excluded as adverse events to be reported). The reporting issue of compliharms may have helped to mask such events. Alternatively, it could be that these events are rare in the populations studied and that there was insufficient power to detect an association. The CI was wide (0.43 to 2.03) and does not rule out a doubling in risk due to treatment ‐ as was found in the prophylaxis trials. It is also possible that the risk of psychiatric harm increases with increasing dose (as the data from trials WV15670 and WV15671 suggest) and increasing duration of treatment (as the prophylaxis trials suggest).
Toovey 2008 assessed the issue and failed to find an association between neurological and psychiatric adverse events and oseltamivir exposure. The outcomes studied were not based on the a priori definition of psychiatric adverse events as defined in the clinical study reports. The definition was constructed post hoc based on a selected group of adverse events taken from the psychiatric, neurological and injury body systems in the reports. The issues are described fully in Jones 2012 and Toovey's response is in Toovey 2012. Toovey 2008 reviewed only retrospective observational studies and did not review three prospective cohort studies conducted in Japan.
A meta‐analysis of three prospective cohort studies of neuropsychiatric adverse events (NPAEs) in Japanese children show a pooled odds ratio for abnormal behaviours due to oseltamivir exposure of 1.55 (95% CI 1.21 to 1.98; P = 0.0005) without significant heterogeneity (Hama 2010). In one prospective study of several thousand children with influenza carried out to test the hypothesis of a causal relation between oseltamivir and neuropsychiatric events, abnormal behaviour was observed more frequently in oseltamivir recipient children than in controls (RR 1.57, 95% CI 1.34 to 1.83). Abnormal behaviour was observed in 3.4 per 100 person day (or 13.8%) in the oseltamivir group, compared to 2.2 per 100 person‐days (or 8.8%) in the control group (Yorifuji 2009). Reanalysis of this study population, focusing on delirium and unconsciousness, also showed a significant association between oseltamivir and neuropsychiatric events, especially in the very early phase of the illness within a day of commencement of fever (Fuiita 2011). These indicate that prospective and intentional collection with this scale of participants may be necessary in treatment RCTs.
Animal toxicity study results firmly support the effect of oseltamivir on the central nervous system. One of these is the hypothermic effect of oseltamivir (but not zanamivir) administered orally, intraperitoneally (Ono 2008; Ono 2013) and intracerebroventricularly (Ono 2013). The other is that intra‐duodenal or intravenous administration of oseltamivir to mature rats induced respiratory arrest shortly followed by cardiac arrest. These studies clearly show central depressant effects of oseltamivir (Kimura 2012). Moreover in the post‐marketing toxicology phase studies by Roche, many symptoms that the manufacturer considered "item‐related" were observed: alterations in respiration including decreased respiratory rate/gasping and altered mucous membrane/skin colour (pale) prior to death. Although the manufacturer denied the causality (Freichel 2009), symptoms at two hours after administration that showed dose‐related increase were lack of olfactory orientation, lack of cliff aversion and low or very low arousal. Twenty‐four of 52 pups that did not exhibit cliff aversion were later found dead. Fourteen of 17 animals with low or very low arousal died thereafter. These findings are consistent with the clinically observed psychiatric symptoms in the RCTs and post‐marketing spontaneous reports.
Zanamivir was well tolerated. However, a potentially active placebo may have masked the occurrence of bronchospasm in zanamivir trials.
Treatment trials were mostly under‐recruited and often their results pooled post hoc in two or even three trials, and yet they showed very high influenza positivity rates. One possible explanation for this lies in the intensive surveillance carried out in the predefined trial centre areas and the restricted time span of recruitment during high likelihood of positivity periods. This may be why many centres with low levels of recruitment are listed in the clinical study reports; this limits the generalisability of the results to everyday life.
In a primary or secondary prophylaxis indication the postulated central effect of oseltamivir is confined to suppressing symptoms, as infection was not prevented even when oseltamivir was administered prior to the inoculation of influenza virus both in animals (Mendel 1998) and in humans (Hayden 1999) and the prophylaxis trials. However, the central problem remains the incompatibility of the two contrasting claims of its activity against antibody production. If, as reported in many documents, oseltamivir does not interfere with antibody production (see, for example, FDA 2011a; Roche Investigators' Guide), how is it possible that oseltamivir prevents cases of influenza when part of the definition of prevented cases in oseltamivir trials was based on absence of antibody response?
The apparent ability of oseltamivir to interfere with antibody response calls into question the mode of action of the drug and puts in doubt the proposed effects of oseltamivir. One possibility in treatment trials is that oseltamivir administration, by interfering with antibody production, has the effect of selecting the strongest antibody responders in the ITTI subpopulation. These individuals are classified as influenza cases and are included in the oseltamivir arm of the ITTI population. This selected subpopulation probably represents the healthiest or those least likely to experience complications. An alternative consequence could be that interference with antibody production in the oseltamivir arm led to active arm participants being more likely to develop complications due to impaired immune function.
Evidence from prophylaxis and secondary prophylaxis trials suggests that in addition to the apparent similar mode of action as in the treatment studies, suppression of viral shedding in nasal swab may be of importance. In the former, participants who become positive (i.e. who are subsequently classified as cases of influenza) in the oseltamivir arms are the few who mount a strong response despite oseltamivir interference. The remainder (who are significantly more than in the placebo arm) are classified as prevented or avoided cases. However, as prophylaxis clinical study reports do not report antibody responses and viral isolate results for the ITT populations either, it is impossible to tell whether this proposed mode of action fits all the evidence. The effect of oseltamivir on nasal shedding is consistent with the proposed mode of action of NIs in preventing the virus from leaving the host respiratory epithelial cells, which are covered by a mucous layer. Compared with the rather small reduction of symptoms of influenza‐like illness and reduction in antibody rise (up to 10%) by both oseltamivir and zanamivir, the extent of the reduction of symptomatic influenza is almost half. This may be due to reduction of influenza viruses in the nasal swab sample.
In prophylaxis there is no evidence that oseltamivir reduces symptomatic influenza‐like illness. Oseltamivir reduces the number of prophylaxis participants testing positive (based on antibody rise and/or culture test). However, this finding is weakened by oseltamivir’s interference with the viral replication on the swab and effect on antibody production. In addition oseltamivir does not affect asymptomatic influenza and there is no evidence that it interferes with person‐to‐person spread.
Similarly to the FDA (FDA 1999c; FDA 2000c) because of the problems with the design of study WV15799 we could not draw any conclusions on the ability of oseltamivir to interrupt viral transmission.
This is important as the results of trial WV15799 formed part of the WHO3 rationale for use of the drug to interrupt transmission from person to person and allow time before the arrival of vaccines in the event of a pandemic furnishing a seemingly powerful rationale for stockpiling oseltamivir.
This shows the importance of availability of full clinical study reports, something the WHO did not have.
Antibody suppression seems stronger for oseltamivir than zanamivir, probably due to the difference in bioavailability. It may be that evidence of other effects, such as hyperglycaemia and renal impairment (though significance was marginal) in the prophylaxis trials may be due to inhibition of the host's endogenous neuraminidase, which impairs the cell function of various organs (Hama 2008). Overall, the significance of oseltamivir for nasal shedding is unclear but problems with sampling and culture undermine any claims as to its secondary prophylactic properties, as the FDA made clear in its response (FDA 1999c).
The dose‐response increase in psychiatric events in the "pivotal" oseltamivir treatment trials and the increase in vomiting only on day one in treatment trials in children may be due to the sudden onset of the central action of unchanged oseltamivir (Hama 2008). Brain concentration of unchanged oseltamivir increases during the early phase of influenza in juvenile animals (Freichel 2009), due to a reduced or low function of p‐glycoprotein, a major transporter of oseltamivir at the blood‐brain barrier (Hama 2008; Kimura 2012). The likely centrally mediated mode of action of oseltamivir is supported by the finding of adverse events in healthy people in prophylaxis trials. However, these effects may also be derived from a delayed action associated with host endogenous neuraminidase inhibition by oseltamivir (Hama 2008), because this appeared after more than a week's exposure the drug and lasted for more than two weeks. Other effects, such as pain in the limbs, hyperglycaemia or diabetic events, reduction of antibody rise and reduction of cytokine induction, may also be due to the suppression of the host's endogenous neuraminidase by oseltamivir (Moore 2007). Pain in the limbs and metabolic control events (mainly hyperglycaemia) were in excess in the oseltamivir arms, but we did not carry out a formal meta‐analysis, as they were not prespecified in our analysis plan and the number of events was below 10 for metabolic events.
Statements made about the capacity of oseltamivir to interrupt viral transmission and reduce complications are not supported by any data we have been able to access.
We have not reviewed other NIs, such as laninamivir and peramivir, or other antivirals, such as the adamantanes (amantadine and rimantadine), or antipyretic/anti‐inflammatory agents either. Laninamivir and peramivir may be more potent as NIs, because their bioavailability is far higher than zanamivir and may affect the host's endogenous neuraminidase. Adamantanes are well known centrally active agents and may be more harmful than oseltamivir and zanamivir. Anti‐inflammatory antipyretics (except paracetamol) may be more toxic than NIs (Hama 2008). Hence, the other NIs, adamantanes and anti‐inflammatory antipyretics may not be alternatives to oseltamivir and zanamivir.
Overall completeness and applicability of evidence
We used the Cochrane seven‐domain 'Risk of bias' instrument to assess bias. The availability of partial or complete clinical study reports decreased the uncertainty and allowed definitive judgements to be made. Previous unclear risk of bias became certainty of bias or certainty of absence of bias. Certainty or low levels of uncertainty are due to our expectations regarding the complete clinical study reports. We were expecting to have all relevant and consistent information available for our reviews, but when it was not, our judgements changed because we found gaps in the availability of information and inconsistent information. We are still uncertain whether the complete study reports represent an exhaustive and coherent source of trial narrative and data.
In the case of treatment trials, conclusions and generalisations are drawn from a subpopulation in which the two arms do not appear comparable due to the apparent ability of oseltamivir to interfere with influenza antibody production. The effect of oseltamivir on the gastrointestinal tract appears to be notable, although a definitive statement will only be possible once the mode of action and dosage of dibasic calcium phosphate dihydrate and dehydrocholic acid have been clarified. The high percentage of influenza infections appears to be in contrast with the need to pool or delay several trials and the small recruitment size of others because of a lack of influenza circulation. In the case of post‐exposure prophylaxis trials, the selection of the infected population has the effect of excluding from the analysis large percentages (in some cases over 50%) of participants. This brings the generalisability of the results of these trials into question.
Much has been made in the trial programmes of viral nasal voidance as a marker of effect. However, its measurement was unreliable in treatment trials as this verbatim quote from the FDA review shows: "Duration of viral shedding was measured from treatment initiation to the time of the first negative virus culture with no subsequent positive cultures. Upon reviewing a list of viral shedding patterns provided by the applicant on 8/16/99, two problems emerged: (1) the pattern of virus shedding was fluctuating in at least 33 subjects (i.e. pos‐neg‐pos‐neg, with or without a subsequent negative result). (2) In at least 100 subjects, the last virus shedding sample was the first negative sample in sequence, meaning there was not a subsequent negative confirmation. Given the fluctuating pattern of virus shedding, to estimate the duration of viral shedding based on the occurrence of a single first negative data poses a high level of uncertainty" (FDA 1999c).
In all programmes, the effect on complications was based on unclear and potentially unreliable definitions, often at the discretion of local clinicians and confirmation (e.g. radiological confirmation of pneumonia) was not consistently reported when it did occur. In the ITT population, the correct population for analysis, there is no credible effect of oseltamivir against pneumonia as the significance of the term "pneumonia" is not clarified.
In the case of post‐exposure prophylaxis trial WV15799, nasal voidance was measured only in symptomatic subjects as an adjunct to protocol version H. However, this does not prevent the manufacturers from making claims of effect for all these outcomes.
Other general requirements, such as presentation within 36 to 48 hours, raise questions about the generalisability of the research evidence. However, underlying all our doubts is the conflicting evidence on the mode of action of the drug.
Most of the trials were substantially under‐recruited and so had insufficient power individually to answer the research question.
Quality of the evidence
We assessed all full clinical study reports of relevant trials. An example of the kind of detail available in complete clinical study reports and the importance of the trial timeline in assessing the presence of bias is the observation that of the clinical study reports for the included trials, only one contained a protocol that predated the beginning of participant enrolment, only two had statistical analysis plans that clearly predated participant enrolment and three had clearly dated protocol amendments. No oseltamivir clinical study report included a clear date of unblinding.
All reports in our review were sponsored by the manufacturers. It is known that published studies sponsored by the pharmaceutical industry are more likely to have outcomes favouring the sponsor compared to studies which have other sponsors (Lexchin 2003; Lundh 2012). As the evidence relates to published studies, we do not know whether the findings are applicable to clinical study reports.
Potential biases in the review process
The main limitation of our study is our relative inexperience in dealing with large quantities of information and our lack of familiarity with certain trial documents, such as randomisation lists. Randomisation lists appeared to be of two types. The first was a pre‐randomisation list of random codes with which participants' IDs cannot be matched with the participant IDs used within other sections of the clinical study report. The second was a post hoc randomisation list to which individual participants can be matched but the original generated codes are not shown. In both cases the truly random generation of the sequence could not be properly assessed because either the original codes are not provided or original codes cannot be matched to patients.
We have created methods and procedures to address the risk of reporting bias that we identified in published trials, but remain uncertain about the success of these new methods.
Agreements and disagreements with other studies or reviews
Several reviews of NIs are now available (Burch 2009; Cooper 2003; Falagas 2010; Tappenden 2009; Turner 2003), including several separate versions of our previous reviews (Jefferson 2006; Jefferson 2009a; Matheson 2007; Shun‐Shin 2009). All are mainly based on published information and reach similar conclusions to our 2006 review, which sparked the reader's comment and subsequent investigation and change of methods.
Following publication of our review update in December 2009, Roche asked the Harvard‐based academics Hernan and Lipsitch to repeat the Kaiser analysis to confirm or reject Kaiser's conclusions (Hernan 2011). They were not provided with any funding to carry out this analysis and Roche ultimately provided them with patient‐level data sets and Module 1 for the 10 Kaiser trials and one more treatment trial (WV16277). An important methodological difference between Hernan and Lipsitch's analysis and that of Kaiser was Hernan and Lipsitch's decision to privilege a true ITT analysis over the sub‐population analysis featured in the Kaiser analysis. Our Cochrane review also analyses the ITT population.
The Kaiser analysis concluded that oseltamivir provided two statistically significant reductions: in lower respiratory tract complications and in hospitalisations.
Hernan and Lipsitch evaluated lower respiratory tract complications and found a statistically significant, but smaller, reduction in the risk of these complications.
Hernan and Lipsitch omitted evaluating the Kaiser paper's conclusion that oseltamivir reduced the risk of hospitalisation. They wrote, "it was not possible to assess the potential benefit for high‐risk participants who are hospitalised, because the sample size of most studies was too small to consider hospitalisation as an outcome."
Hernan and Lipsitch do not elaborate on or highlight their apparent methodological disagreement with the Kaiser 2003 analysis and it is not reflected in the news article published on the Harvard website entitled "Oseltamivir effect on complications confirmed by reanalysis" (http://ccdd.hsph.harvard.edu/NewsEvents/Oseltamivir‐reanalysis). In fact, Hernan and Lipsitch did not confirm one of the key conclusions of the Kaiser paper (Kaiser 2003).
Unfortunately, the Hernan‐Lipsitch analysis has been cited by influential bodies such as the European Centre for Disease Prevention and Control (ECDC) as "confirmation of the original Kaiser meta‐analysis" (http://ecdc.europa.eu/en/activities/sciadvice/_layouts/forms/Review_DispForm.aspx?ID=561&List=a3216f4c%2Df040%2D4f51%2D9f77%2Da96046dbfd72) despite the fact that Hernan and Lipsitch did not confirm one of the key conclusions of the Kaiser paper (Kaiser 2003).
For complications, while Hernan and Lipsitch clearly produced similar results to Kaiser, we do not think that this means the result is more credible. In view of our findings, we suggest that these results should be interpreted with caution. We have published our preliminary comments (Cochrane Neuraminidase Inhibitors Review Team 2011). The approach Hernan and Lipsitch took in analysing data was insufficient to provide a credible, independent check on validity and reinforces the importance of detailed, critical assessment of entire trial programmes, with access to full‐length study reports. Our analysis questions the coherence between the evidence and the proposed mode of action of oseltamivir.
The Ebell 2012 review concluded that there was "no evidence that oseltamivir reduces the likelihood of hospitalisation, pneumonia or the combined outcome of pneumonia, otitis media and sinusitis in the ITT population". This conclusion was based on Module 1 of the 10 Kaiser trials plus WV16277. These are the same 11 trials as Hernan 2011.
Authors' conclusions
Implications for practice.
On the basis of the findings of this review, clinicians and healthcare policy‐makers should urgently revise current recommendations for use of the neuraminidase inhibitors (NIs) for individuals with influenza. Our findings confirm that both oseltamivir and zanamivir reduce the time to symptomatic improvement in adults (but not asthmatic children) with influenza‐like illness. The size of this effect is small, approximately half a day. It is unclear whether this is superior to treatment with commonly used antipyretic medications. However, we did not find any credible evidence that either oseltamivir or zanamivir reduce the risk of complications of influenza, particularly pneumonia, nor reduce risk of hospitalisation or death. Moreover, even in individuals at higher risk of complications, such as children with asthma or the elderly, we found no evidence of a beneficial effect for reducing risks of complications.
Based on these findings there appears to be no evidence for patients, clinicians or policy‐makers to use these drugs to prevent serious outcomes, both in annual influenza and pandemic influenza outbreaks. Practice recommendations and drug labelling needs to be changed to reflect these findings.
When used as prophylactic agents to prevent the occurrence of influenza in individuals or families, our findings again suggest a minimal effect on prevention. Based on this, there is little support for their use as prophylactic agents, for example, during influenza epidemics. Given that oseltamivir is now recommended as an essential medicine for the treatment of seriously ill patients or those in higher‐risk groups with influenza (H1N1 2009) (WHO 2013a; WHO 2013b), this is of some concern.
Reasons why prophylaxis treatment is not clinically meaningful include: 1) according to modelling studies, for prophylactic treatment to be effective, 80% of the population require at least eight weeks of treatment, which has not been trialled (Longini 2004); 2) models assume the relative risk reduction observed in low risk populations, transfers directly to populations at higher risk (i.e. an absolute treatment effect of 31%, approximately 15‐fold higher than the absolute effects we observed in prophylaxis trials (Longini 2004); 3) A high proportion of people, at least two‐thirds require recognisable influenza symptoms; 4) treatment has to be effective against both asymptomatic and symptomatic infections (which it is not), in modelling studies asymptomatic infections are assumed to be 50% as infectious as symptomatic infections (Longini 2004); 5) because the influenza season can last four to five months, any secondary strategy, such as vaccination, would have to be widely available at the end of the eight week treatment period, and 6) a full understanding of the effect of the treatment in prophylaxis is unknown because symptomatic influenza‐like‐illness without laboratory‐confirmation was only fully reported in one study. This study (NAI30034) showed no difference in proportion of patients with symptomatic influenza‐like‐illness (with or without lab confirmation) (RR 0.90, 95% CI 0.73 to 1.11).
The small benefits we noted in symptomatic improvement and the lack of credible evidence for an effect on serious complications needs to be balanced with the adverse effects found with these drugs in meta‐analyses, especially diabetic/hyperglycaemic, renal and neuropsychiatric effects in all those people for whom the World Health Organization (WHO) recommend use.
Our results do not discount the potential benefit of using zanamivir and oseltamivir in individuals under particular situations, for example in immunocompromised or in compassionate cases, where few other therapeutic options may exist. However, NIs themselves may be immunosuppressants. Our findings do not support the stockpiling of NIs, nor oseltamivir's inclusion in the WHO's list of essential drugs.
The rationale for undertaking the current review and the methods and pressures that needed to be employed to obtain and evaluate the evidence for these drugs has significant implications for the robustness of the scrutiny that new drugs undergo prior to entering widespread clinical use. We believe that several steps now need to be put in place to provide patients, clinicians and policy‐makers with the most transparent assessment of the relative benefits and risks of new drugs.
First, our findings imply that numerous national and international bodies appear willing to accept biased or incomplete trial reports seemingly at face value. This ready willingness is in contrast with the considerable time and effort needed to change their recommendations. Second, published trials are unlikely to provide the level of detail to allow the results of a drug trial to be properly evaluated and risk presenting a partial and potentially biased report of trial conduct and findings. This has implications not only for the reporting of trials but also the weight that can be applied to published studies alone. Third, clinicians and policy‐makers should be cautious in interpreting and using the findings of systematic reviews including only published studies, particularly those that comprise only a portion of an entire drug trial programme, or which contain only a portion of the results of trials. There have been many systematic reviews of NIs, none assessing the full trial programme or full trial results, thus limiting their validity. We suspect a similar situation exists for other drugs. Fourth, clinical policy‐makers at the national and international level should raise the level of scrutiny needed in the cases of drugs that are likely to be used by large numbers of people who are either asymptomatic or have short, self limiting illnesses such as influenza ('public health drugs') and where the potential for benefit (and harm) is vital. Given the weight of these decisions (with serious implications for both correct and incorrect decisions), policy‐makers should not face the barrier of being denied access to what sponsors regard as commercially sensitive information.
Without concerted efforts from multiple stakeholders to put the above measures in place, it is not possible to provide objective assessment of the benefits and risks of new drugs. This risks not only patients potentially missing out on effective treatments, but also patients being exposed to either ineffective (or harmful) treatments.
Implications for research.
Our findings have implications for research on the mechanism of action of NIs, with special regard to any direct central action of oseltamivir and the inhibitory effect of the host endogenous neuraminidase of various organs and systems. We could not reach a consensus on whether further trials are warranted and whether current trials should be discontinued.
The considerable body of evidence from randomised controlled trials (RCTs) included in this review indicates either no effect or a relatively small absolute effect size against the complications of influenza. Such an effect, even if statistically significant, would be too small to warrant treatment with NIs in a primary care setting, especially since effective diagnosis and treatments for rare complications (such as pneumonia) are available. Lack of evidence of an effect on hospitalisations probably indicates lack of severity in the first place. Assuming an influenza incidence rate of 2% (similar to that in the control arms of oseltamivir treatment trials), to detect a 25%, clinically significant reduction in pneumonia, 21,500 participants would have to be enrolled in a clinical trial.
Our calculation is likely to underestimate population size, as the 2% incidence rate was derived from trials that used enhanced ad hoc surveillance systems. Any trial design would have to ensure that the presence of complications is ascertained using objective diagnostic criteria (for example, with confirmation using imaging or laboratory testing for pneumonia). Such trials would also have to consider the ethical implications of conducting studies where the estimate of benefit (based on 11 RCTs) in otherwise healthy people is likely to be small, and would have to be balanced against the apparent risks of adverse effects from NIs. We think research should be aimed at more effective preventive measures and early identification of complications.
Based on the length of time it has taken to provide a definitive answer on the efficacy of the NIs, the challenges in obtaining the full information and the methods that we needed to develop to conduct the evidence synthesis, we believe the main implication of our review is the need for reform of multiple components of the research and development, regulatory and assessment pathway of new drugs.
Pharmaceutical sponsors of drug trials should follow a data access and sharing procedure similar to that of the European Medicines Agency (EMA) and sponsors should make all full clinical study reports available to be downloaded from their websites and shared freely once a regulatory decision has been made. Redactions should be kept to the minimum. Part of this process needs to include a full list of the entire drug development programme, to avoid assessment of an incomplete set of trials. Researchers and industry employees who are listed in trial documents should be considered to have legal responsibility for the conduct and reporting of a trial.
Regulators should post an inventory of their documentary holdings on their websites with a brief description of the main content and size of each file. They should make all information available shortly after making a registration decision on a drug and within a reasonable time period. The information should be in electronic format and anonymised (i.e. participants' details should be removed to prevent each person being identified but no further).
Trial registries have improved the reporting of new trials. However, on their own they will not be adequate to resolve the problems we encountered. The completeness of trial registries needs to tested with a random sampling procedure. Clear instructions for the reporting and updating of their content should be promulgated and penalties imposed on breaches of these procedures. Trial registration should include the original and final versions of a trial protocol, with a full declaration of dated amendments. Procedures for trial unblinding and dates of unblinding should be routinely reported. Registration should be made compulsory for all studies in which human beings are randomly assigned to experimental arms. Ethical and consent procedures for all trials should include obligations of the trial sponsor to ensure results are made public. Failure to report the existence of a trial on humans and to make results available should be considered as an ethical breach of conduct and subject to appropriate penalties.
The methods used to conduct our evidence synthesis need to be repeated across further interventions and by other researchers and may need to be refined further. Given the considerable resources involved in using these methods, a system is needed to prioritise reviews of important drugs so that such methods are reserved for drugs that meet certain conditions. Priority could perhaps be given to first drugs of a new family, drugs considered to be innovative or those that are likely to have a big market impact. Such reviews should be publicly funded and be independent from both regulators and manufacturers. Researchers who conduct these 'high scrutiny' reviews need to be free of recent ties to either government or the pharmaceutical industry. Systematic review groups such as The Cochrane Collaboration should consider both adopting these methods for other drugs and whether perhaps to scrutinise the published reviews of prioritised drugs.
Finally, all documentary evidence relating to a trial on humans (including clinical study reports, regulatory documents, evidence syntheses) should be archived electronically with no statute of limitations.
Authors' note: in reviewing over 2 GB of data there is the possibility of mistakes. The authors would be grateful if readers could identify these. We promise that if we concur the record will be amended accordingly.
Feedback
From Michael Power, Sowerby Centre for Health Informatics at Newcastle, 15 December 2010
Summary
From: Michael Power <michael.power@schin.co.uk> Date: 15 December 2010 18:51 Subject: Neuraminidase inhibitors for influenza ‐ HTA project To: "cdelmar@bond.edu.au" <cdelmar@bond.edu.au>, "jefferson.tom@gmail.com" <jefferson.tom@gmail.com>, Carl Heneghan <carl.heneghan@dphpc.ox.ac.uk> Hi I picked up Carl’s Twitter request for comments on your draft protocol "Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children ‐ a review of unpublished data". So, here are my two comments on the content. The title confused me: I expected it to be a review of unpublished trials to complement your review of published trials. It would be longer but clearer if you could call it "Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children ‐ a review of clinical study reports for published and unpublished trials". The section "How the intervention might work" could be reorganized along the lines of: 0) Metabolism: oseltamivir phosphate (OP), Tamiflu, is the pro‐drug of oseltamivir carboxylate (OC), the effective form. OP dissociates in the gastrointestinal tract to form oseltamivir (OT) which is absorbed and metabolised into OC by hepatic carboxylesterase (h‐CE). 1) Reducing the ability of the virus to penetrate the mucus in the very early stage of infection (Bhatia 2007; Matrosovich 2004; Moscona 2005; Ohuchi 2006). 2) Inhibiting neuraminidase, which enables influenza viruses to exit host cells (Liu 1995; Moscona 2005). 3) Central depression by OT (Hama 2008) may cause hypothermia (Ono 2008). 4) Inhibition by NIs of human sialidase may cause abnormal behaviour (Li 2007).
You have obviously put a huge amount of work and expertise into developing the protocol, and have an even bigger task ahead to complete the review. Congratulations for taking this on. Best wishes Michael
Reply
Thanks for the constructive comments.
We have re‐titled the Protocol to address this concern (and that of feedback from GSK, see below);
We have re‐examined the "How the intervention might work" section but made only small adjustments in the interest of keeping this section short;
We are not sure what problems you might have had printing the pdf file, and hope they are resolved with this new version.
Contributors
Chris Del Mar
From Juan C. Vergara, Intensive Care, Hospital Cruces, 48901 Barakaldo, Spain, 24 February 2011
Summary
From: JUAN CARLOS VERGARA SERRANO <JUANCARLOS.VERGARASERRANO@osakidetza.net> Date: 24 February 2011 12:48 Subject: oseltamivir To: jefferson.tom@gmail.com I've read your Intervention Protocol: Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children ‐ a review of unpublished data. And may be you can be interested in this letter I wrote to de BMJ: http://www.bmj.com/content/340/bmj.c789.extract/reply 1. Early use of oseltamivir does not reduce swine flu mortality, Juan C. Vergara, MD. Intensive Care Unit, Hospital Cruces. 48901 Barakaldo. Spain As you say, in July the National Pandemic Flu Service started providing oseltamivir to anybody who telephoned with a plausible set of symptoms. From 23rd July to 1st December, the National Pandemic Flu Service (NPFS) in the UK, has provided more than one million courses of antiviral medication. By that time the Spanish Health Secretary General, José Martínez Olmos, at the Congress of Deputies, announced that only 6.000 patients (most of them hospitalised) had received oseltamivir in Spain. At the end of January there have been 411 deaths reported due to pandemic (H1N1) 2009 in the UK, and about 300 in Spain. That means 6.7 and 6.5 deaths per million, respectively. These data create serious doubts about the real utility of early use of oseltamivir in preventing deaths from Influenza A H1N1. http://www.nhsdirect.nhs.uk/article.aspx?name=SbSwineflu http://www.congreso.es/public_oficiales/L9/CONG/DS/CO/CO_411.PDF Competing interests: None declared Yours sincerely; J. C. Vergara
Reply
Thank you for your interest.
Contributors
Chris Del Mar
From Dr Helen Steel, GSK, UK, 30 March 2011
Summary
GSK comments on Cochrane Collaboration protocol: neuraminidase inhibitors for preventing and treating influenza in healthy adults and children ‐ a review of unpublished data
General:
The term ‘unpublished data’ is used extensively in the protocol. However, it does not appear to be clearly defined either in the protocol or in Jefferson’s comment in the 15 Jan 2011 edition of the BMJ. Additionally, the term ‘unpublished data’ is misleading. It appears the Cochrane Group use this term interchangeably with Clinical Study Reports, regardless of whether a primary manuscript is available for a given study. We suggest this is clarified or preferably replaced, especially since the term appears extensively in the protocol including the title. Readers are likely to use the terms ‘unpublished data’ and ‘unpublished trials’ (trials for which no primary publication appears in the scientific press) interchangeably. A suggested replacement is ‘Clinical Study Reports’ since this term is not easily misinterpreted and is clearly defined in Jefferson’s BMJ comment.
The ‘scope of clinical trial data’ are defined in Jefferson’s BMJ 15 Jan 2011 comment, as mentioned above (i.e. definitions for clinical study reports, raw data, unpublished trial, published trial, regulatory data). It would seem important that these and any other definitions introduced in the protocol are included in the protocol.
Description of Intervention
This section incorrectly describes Relenza as ‘nebulized zanamivir’. Relenza is formulated in Rotadisks containing foil blisters with a powder mixture of zanamivir and lactose. Relenza is administered by oral inhalation using a breath‐activated device called the Diskhaler. Earlier clinical studies explored several methods of administration, including nebulized and intranasal routes but marketing approval in nearly all countries is currently available only for oral inhalation via Rotadisk/Diskhaler.
Types of Studies
To meet the objective of providing a comprehensive review of neuraminidase inhibitors in preventing and treating influenza, it would seem appropriate that clinical trials from all sources (including sponsors other than industry) be included in this meta‐analysis. Please clarify if this is your intent.
Outcome Measures
More details should be provided on the outcome measures section in the final protocol.
For example, broad outcome measures are stated in the protocol but specific endpoints are not provided. The primary and secondary endpoints of the meta‐analysis should be clearly defined in the final protocol.
e.g.1. A stated primary outcome in the treatment studies is ‘symptom relief’. Does this refer to ‘the time to alleviation of symptoms’ or ‘reduction in symptom score’ or another endpoint? Time to alleviation of clinically significant symptoms was the primary endpoint used in the majority of GSK treatment studies.
e.g.2. Another stated primary outcome is ‘Harms’. Please provide the specific endpoints. Will this refer to ‘incidence of most common AEs’ or ‘incidence of common SAEs’, ‘incidence of complications’ or another endpoint? It is not clear if ‘harms’ are the same as ‘compliharms’. It is not clear what specific events will comprise compliharms.
Prophylaxis studies: Several types of prophylaxis studies were conducted by GSK: household prophylaxis (post‐exposure prophylaxis), community prophylaxis and outbreak control in nursing homes, and as such the designs and/or endpoints are different. It is possible to measure ‘prevention of onset of influenza in contacts’ in these studies but not ‘reduction in viral spread from index cases’ in the majority of prophylaxis studies.
Hospitalisations: As studies were generally conducted in the setting of acute uncomplicated influenza, limited hospitalisation data were collected, and are available only for some studies.
Extracting compliharms: There is a statement that ‘AEs are reported for all participants while complications are only reported for infected subjects’. This statement is not accurate for GSK trials. AEs are reported for all study participants. However, AEs of ILI were not collected in the treatment studies unless the symptoms were considered to be worse than expected for the normal progression of illness. Without knowing the specific safety endpoints, it is unclear whether this will affect the outcome of some of the harms analyses.
Data collection and analysis:
The protocol indicates that clinical study reports will be requested (minus participant identification). In fact many documents for each study will need to be redacted not just to remove participant identification but any personally identifiable information including author and investigator identification.
Missing Data. The protocol states "At the participant level (i.e. within a trial) we will not make any assumptions about missing data." This is not possible, because an analysis of data that is collected in a trial can only be done in the context of assumptions about potential mechanisms that led to data being missing (e.g., missing completely at random, or missing at random).
Meta‐analysis Method. Little detail is given in the protocol. The protocol states that "Whether or not heterogeneity is detected, we will perform a random effects meta‐analysis. Random‐effects methods will be used to compare the dichotomised outcomes (RR and absolute risk reduction (ARR) for efficacy and safety)." There are several different Random Effects methods available (Bayesian or frequentist, DerSimonian & Laird or Maximum‐likelihood or REML), and different approaches to handling rare events (various "corrections" to include trials with zero counts). Furthermore, would random‐effects methods also used to compare the continuous outcomes?
Fixed‐effects Model. The protocol also states that fixed‐effects models will be used in a sensitivity analysis. No details are given with regard to which fixed‐effects models will be used. There are several fixed‐effects models available including Inverse Variance, Mantel‐Haenszel, and Peto’s method. The appropriate method used should also depend on the outcome measures (dichotomous vs. continuous; relative vs. absolute). The approach and choice of models for sparse data and rare events should be provided. Furthermore, various methods in the framework of fixed‐effects model may be explored to evaluate the robustness of the results.
Hazard Ratio. The protocol states "We will convert medians of treatment groups into (log) hazard ratios (estimating the variance of these) to enable meta‐analysis of time to event outcomes." Although hazard ratio (HR) is a standard analysis and widely recommended approach for time‐to‐event data in clinical trials, the HR analysis may not be suitable for the Relenza studies with relatively short follow‐up time because the assumption of proportional hazards required for the proportional hazards model may not hold. GSK did not follow this approach for the original analysis due to the concern stated above. Further the clinical and regulatory interest centred on differences in the time to alleviation not in the relative hazard between treatments. The above issues would be best addressed by using subject level rather than summary data, which GSK have offered to provide to the Cochrane Group.
Analysis Populations. The protocol does not specify which populations will be used for the various analyses, for example, intent‐to‐treat or influenza‐positive or other. We believe that influenza positive population is appropriate, especially for the efficacy analysis using time to alleviation of influenza symptom as a primary endpoint consistent with the prescribing information for Relenza.
Study Duration. No details are given in the protocol with regard to how studies with different follow‐up times will be handled.
Trials with no Events. No details are given in the protocol with regard to how to deal with trials in which there are no events (such as death). By excluding studies with no events will make the event appear more common than it actually is. There are various techniques: Bayesian approach, continuity correction, combining similar trials to avoid having any components of the analysis that have no events.
Sensitivity Analyses. Sensitivity analyses using different outcome measures, statistical models and/or continuity correction factors to assess the robustness of the results are strongly encouraged.
Reply
General:
'unpublished data'. We agree that this term is confusing, and are attracted to the proposal of using 'clinical study reports' instead.
We have attempted to ensure all terms are clear.
Description of intervention
Description of zanamivir (Relenza): we have corrected 'nebulized zanamivir' to 'powder inhalation'.
Types of studies
Yes, we intend to comprehensively review clinical trials from all sources (including sponsors other than industry). This intent is clear from the subsection 'Electronic searching' under the 'Search methods for identification of studies' section.
Outcome measures
Our specified outcomes are those of interest to patients, and their clinicians and policy‐makers. They are therefore likely to be broader than the more specific endpoints selected by trialists. The purpose of Cochrane Reviews are usually to set clinically relevant review questions, and search the literature (or other sources) for answers to them. Sometimes answers to some questions are not available, and this is also documented. Where possible we report outcomes as pre‐specified in the trial protocols, or as pre‐specified in the review protocol, or otherwise reported as a post‐hoc analysis.
e.g. 1. 'symptom relief' may refer to 'the time to alleviation of symptoms' or 'reduction in symptom score', or any other endpoint (including 'area under the curve of symptom score and time').
e.g. 2. 'Harms' include common adverse events (AEs) as well as serious AEs. We agree about the confusion of harms and complications, and have tried to capture the totality of these with the neologism 'compliharms' to avoid classification errors between their different labellings.
Prophylaxis studies: We understand that it is possible to measure 'prevention of onset of influenza in contacts' in some GSK studies but not 'reduction in viral spread from index cases' in others.
Hospitalisations: We understand that hospitalisation data may only be available for some studies. However patient hospitalisation is usually classified as a serious adverse event therefore we expect to identify hospitalisations (not reported separately) in that way.
Extracting compliharms: Your statement that "AEs of ILI were not collected in the treatment studies unless the symptoms were considered to be worse than expected for the normal progression of illness" underlies the complexity of analysing AEs and complications (our 'compliharms'). We have noted in the protocol that the limitation of complications only reported for the infected patients is relevant to the Roche trials only.
Data collection and analysis:
We are interested that not only subject identification would be required to be removed from any documents of clinical study reports but also information personally identifying authors and investigators. We wonder why.
Missing data. We have removed this statement.
Meta‐analysis method. DerSimonian & Laird method will be used. Note that in the case of zero cells (e.g. no events in one group) the RevMan software (which we will use for the analysis) automatically adds 0.5 to each cell of the 2×2 table for any such study. There are no continuous outcomes specified in this review.
Fixed‐effects model. Mantel‐Haenszel method will be used except in the case of sparse data, in which case Peto's method will be used (as recommended in the Cochrane Handbook).
Hazard ratio. We note the concerns with this outcome hence we will also consider analysis of this outcome as a continuous outcome noting that the data are likely to be skewed. We will use the inverse‐variance random‐effects method for this analysis.
Analysis populations. All analysis will be using the intent‐to‐treat population as this is the most methodologically rigorous and clinically relevant.
Study duration. We have specified in the protocol, where appropriate, that we will report outcomes for the on‐treatment and off treatment time periods. If data are not available in the clinical study reports for any time period of the study then we will write to the relevant manufacturer to request the missing data.
Trials with no events. As stated above the RevMan software automatically adds 0.5 to each cell of the 2×2 table for any such study.
Sensitivity analyses. We note this point and agree. Where appropriate, a realistic sensitivity analyses will be conducted.
Contributors
Chris Del Mar
Feedback from Wolfgang Becker‐Brueser, 30 January 2012
Summary
Dear Tom Jefferson,
I read your review about NI for prevention and treating influenza with interest. It's an important work. In the chapter "Why it is important to do this review" I found a small mistake concerning the worldwide stockpiling of oseltamivir which is mentioned to be "CHF 7.6 billion worth of oseltamivir (JACK 2009)". This would be an enormous amount "prior (!) to the emergence of influenza A/H1N1 in 2009". But Andrew JACK wrote in the cited Financial Times (May 13, 2009): "Governments around the world had stockpiled 220m treatments to date, swelling sales since the start of 2003 to SFr7.6bn, largely on the basis of preparation for a pandemic virus that has yet to appear." So 7.6 billion SFr represent sales and not stockpiling.
Wolfgang Becker‐Brueser (physician and pharmacist)
Reply
Thank you. The extent of stockpiling is a closely guarded secret this is why these are estimates. We will probably never know.
Contributors
Tom Jefferson MD
From Frederick G. Hayden, M.D., 2 February 2012
Summary
I am writing to comment on the recently updated meta‐analysis by Jefferson and colleagues published through the Cochrane Collaboration and to request clarifications on several points, as well as to suggest some additional analyses that would be helpful in terms of taking greater advantage of this useful database. While I fully support access of Jefferson and other interested investigators to all of the published and unpublished data from the RCTs of oseltamivir and zanamivir for further analyses, this analysis only focuses on RCTs in ambulatory patients with uncomplicated influenza (the vast majority of whom were previously healthy) and on the period before the 2009 H1N1 pandemic. Consequently, I would urge these investigators to extend their efforts to other populations and datasets examining the risks and benefits of using neuraminidase inhibitors (NAIs) for treatment and prophylaxis. Furthermore, the authors should acknowledge the limitations of their analyses more explicitly and avoid inappropriate extrapolation to populations and influenza events that the RCTs did not adequately address. Differences in disease pathogenesis related to virus and host factors, as well as time to treatment, have important effects on the utility of antiviral agent interventions. My specific comments and recommendations for additional analyses follow:
1. Use of Intention to Treat (ITT) and ITTI‐Infected Groups. The exclusive focus in the current treatment analysis on the ITT population is a readily rectified shortcoming. Outcomes in all three groups of relevance (ITT, ITT‐infected, and ITT‐noninfected) should be presented, so that readers can examine both clinical effectiveness and efficacy for the key endpoints, as well as events in those without documented influenza. Because NAI treatment would not be expected to provide any benefit in non‐influenza illness, not presenting the ITT‐infected outcomes in the analysis underestimates possible beneficial drug effects. Assessment of the non‐infected group provides a valuable control and also enables a determination of whether there was a potential drug‐disease adverse interaction of NAI treatment in non‐influenza patients. Of note, our earlier pooled analysis of physician‐diagnosed lower respiratory tract complications leading to antibiotic use found a significant benefit of oseltamivir in the influenza‐infected patients but not in those enrolled in whom influenza infection was not detected by culture or serology [Kaiser 2003].
2. Sample size considerations. Severe outcomes of influenza infection are sufficiently uncommon in previously healthy people that even large RCTs or combining multiple RCTs would be very unlikely to detect them with confidence. The same point applies to very uncommon endpoints like microbiologically documented bacterial complications and rare adverse effects of treatment. Consequently, conclusions that there is no evidence (from trials) that NAIs reduce the risk of pneumonia, hospitalisations, deaths are overstated, as the evidence considered in this analysis is insufficient to properly address these questions.
The US CDC has estimated age‐related influenza‐related hospitalisation and mortality rates for both seasonal epidemics and the 2009 pandemic [Shrestha 2011]. Jefferson and colleagues should use such event estimates and others to make calculations of the necessary sample sizes to detect reductions in these severe outcomes with NAI therapy in a controlled RCT across a range of clinically relevant effect sizes (e.g., 20%, 35%, 50% reductions). In a related fashion, they should also provide more quantitative estimates for their ability to detect such outcomes with their existing database and comment more precisely on their power to capture particular endpoints.
3. Complications in ambulatory patients. Other clinically relevant endpoints in these previously healthy and at‐risk persons warrant investigation. With regard to influenza‐related complications, the most frequent in previously healthy children and adults are respiratory tract infections (otitis media, bronchitis) leading to antimicrobic use. These are usually not severe and typically not microbiologically documented with respect to etiologies but physician‐diagnosed complications leading to antibiotic use is an outcome that has important clinical and public health implications (i.e., cost, antibiotic resistance, side effects) and also is sufficiently frequent to demonstrate effects of antivirals. We showed such a benefit in adults in our earlier pooled analyses of the then available RCT data on inhaled zanamivir [Kaiser 2000] and oral oseltamivir [Kaiser 2003]. The oseltamivir effect was confirmed in a recent meta‐analysis [Hernan 2011], and another recent Cochrane report confirms an effect on otitis media in children [Wang 2011].
Given the large amount of data available to the investigators, it would be a valuable contribution to also explore the clinical outcomes in greater detail and to clarify the use of terms like severe outcomes. Although uncommon in the populations enrolled in these RCTs, endpoints such as radiographically documented pneumonia, microbiologically documented infections, and hospitalisation or death are clear and should be listed separately in those with or without proven influenza infection. Because of the importance of hospitalisations as an endpoint, it would be helpful to examine not only all‐cause hospitalisations but also relevant subgroups based on likely causation (e.g., events in which influenza was documented or likely implicated including exacerbations of co‐morbidities vs others like accidents, elective surgeries, conditions unlikely to be influenza‐related). In addition to these events, exacerbations of underlying conditions (e.g., asthma, COPD, diabetes, CHF) are of medical importance in influenza outpatients with co‐morbidities and should be examined.
4. Data from observational studies. Typically the patients who are most at risk of severe outcomes (older people, infants and young children, those with underlying chronic conditions) are not included in RCTs. In this regard, the current analysis is limited to placebo‐ or active‐controlled RCTs largely done in previously healthy persons and does not consider the multiple observational studies from different countries that have consistently showed protective effects against severe outcomes like pneumonia and hospitalisation, particularly in those with co‐morbidities, as well as reduced mortality if patients have been hospitalised. A considerable amount of new treatment data was generated in many countries during the 2009 H1N1 pandemic that found timely NAI treatment to be associated with a lower risk for intensive care admission and death (reference list available upon request).
While such data and analyses are weaker than RCT data and subject to bias, these observational studies address key endpoints in at‐risk and seriously ill populations, including patients admitted to a hospital at the time of initiating therapy, that the available RCTs cannot and do not address. Furthermore, the standard of care has evolved such that placebo‐controlled RCT in such patient groups would not be acceptable to investigators or ethics committees. The decision by Jefferson and colleagues not to consider and critically analyse the large amount of observational data with modern techniques means that they are not incorporating key information and many important patient groups in which the available data suggests medically important benefits from early NAI therapy. Such findings from observational data can inform antiviral treatment in more severely ill patients when no other data are available. As discussed above, not to include observational data means that conclusions of no effect on uncommon events or no severe adverse events being detected are almost inevitable. This should be made explicit in the design and the conclusion of the current report.
4. Influenza diagnosis and serologic results. The Jefferson report raises questions about the possible inhibitory effects of oseltamivir therapy on influenza‐specific serologic rises and introduction of bias into the outcomes analysis. Further analyses might help to assess these possibilities. They should compare the primary endpoint of illness alleviation between the oseltamivir and placebo subgroups that were culture‐positive (irrespective of serologic findings) at enrolment, and separately those that were culture‐negative but had serologic evidence of infection.
Of note, one prior study of oseltamivir treatment in pandemic 2009 H1N1 patients, although not in seasonal influenza patients, suggested that early treatment could reduce antibody responses [Cowling 2010]. Jefferson and colleagues should examine the age‐related frequencies of HAI seroconversions and the GMT titre rises in those with influenza‐culture positive illness and separately in those with such HAI rises in absence of culture positivity. Of course, if still available, it would be interesting to test the culture‐negative enrolment samples by RT‐PCR.
The RCT data were generated over multiple seasons in which different influenza A and B viruses were circulating. Influenza B neuraminidases are generally less susceptible to oseltamivir carboxylate and several observational studies indicate that oseltamivir is less effective in influenza B‐ than influenza A‐infected children [Sugaya 2007; Sato 2008]. It would be useful to examine the primary outcome in relation to virus type (A vs. B) and if possible A subtype (H3 vs. H1) in those with documented infections to expand on this point.
5. Other treatment endpoints of interest. Since those enrolled in the RCTs were outpatients, it would be useful to explore other endpoints that reflect patient recovery and impacts on the healthcare system (e.g., nonscheduled return visits for complications or adverse events). Perhaps more important than the time to alleviation endpoint used in the registrational trials might be the times to resumption of usual activities and return to pre‐morbid status.
The authors raise the possibility that oseltamivir might have non‐specific antipyretic effects, and one animal model study has also suggested possible adverse immunomodulatory effects of oseltamivir in RSV infection [Moore 2007]. Consequently, it would be interesting to examine the course of fever resolution (a much earlier event than cough resolution) and of symptoms in oseltamivir‐ and placebo‐treated patients with and without documented influenza infections. In addition, it would be valuable to examine the correspondence (or lack thereof) between influenza virologic measures (e.g., enrolment virus titre, time to culture negativity, change in viral titres over time) and symptom resolution measures in both oseltamivir and placebo groups.
Various cost‐effectiveness analyses on NAI therapy in low‐risk populations have been published with widely divergent outcomes, largely depending on the input assumptions. Using this large database, a more refined analysis that incorporates both the direct and indirect (productivity losses) costs of influenza would be informative.
6. Adverse events with treatment. With regard to drug tolerability, it is important to examine not only the frequencies of reported adverse events but also assess indicators of their severity and interference with compliance (e.g., symptom days, patient reported severity, premature cessation of study drug).
Comparisons of AEs in the placebo groups across zanamivir and oseltamivir studies need to be interpreted with caution, since these studies were performed in different influenza seasons viruses and locations, with different protocols and case record forms, and by different investigators. Only one head‐head RCT of treatment comparing these drugs has been published to date to my knowledge but the design did not include placebo only groups [Duval 2010]. In particular, comparisons in children (page 24) need to be age‐adjusted as there were major differences in those enrolled into the zanamivir (5 years and older) and oseltamivir trials (1 year and older), and the frequencies of gastrointestinal manifestations are much higher in younger children with influenza and other acute illnesses.
7. Prophylaxis endpoints of interest. The analysis of prophylaxis outcomes and the associated discussion requires clarification. The statement on page 5 says: "The FDA has also not allowed an indication for interference of viral transmission within households (the key concept behind post‐exposure prophylaxis)." The key concept behind post‐exposure prophylaxis is prevention of illness in exposed persons, and the primary endpoint in most prophylaxis studies has been symptomatic, laboratory‐confirmed influenza illness. FDA and other regulatory agencies have approved both NAIs for post‐exposure prophylaxis in households and also for longer duration pre‐exposure chemoprophylaxis [reviewed in Khazemi 2009].
The Jefferson analysis seems to focus exclusively on the effect of chemoprophylaxis in "preventing the spread" of influenza, with endpoints presumably determined by evidence of culture or serologically confirmed infection irrespective of illness. While this is one endpoint of interest in such studies, the primary outcome of medical interest is prevention of influenza illness in those exposed. There is abundant RCT data, as well as observational data from the 2009 pandemic, that both inhaled zanamivir and oral oseltamivir have both statistically significant and medically important effects on preventing influenza‐specific illness. Of note, the development of serologic evidence of infection without illness is advantageous in those receiving chemoprophylaxis, as it likely is an immunizing event that protects against future infection and illness by that strain. In addition several oseltamivir RCTs have shown significant but lesser effects on influenza infection in prophylaxis recipients [Welliver 2001; Hayden 1999]. The authors should present all of the relevant endpoints in their analysis of the prophylaxis trials.
8. Adverse effects with prophylaxis. The prophylaxis studies are particularly useful in assessing drug tolerability as symptoms of acute illness present in treatment studies are not confounders and there is a more prolonged duration of drug exposure. However, it is essential to examine not only the frequencies of reported adverse events but also indicators of their severity and possible interference with compliance (e.g., symptom days, patient reported severity, premature cessation of study drug).
For example, the Jefferson posting states that "Similarly, a published prophylaxis trial (Hayden 1999a, known by its trial ID WV15673/WV15697) describes headache as having "occurred in similar proportions of subjects in the three groups (39 to 47 per cent)." but indicates that Japanese regulatory documents reached a different conclusion. My own review of the adverse event tabulations from our 6‐weeks prophylaxis study (tables provided by the sponsor) indicates that the proportions of subjects reporting headache (not otherwise specified) that might have been related to study drug (unrelated reports excluded) during the treatment phase were similar across the placebo (N=116, 22.4%), oseltamivir 75 mg once (N=124, 23.8%), and oseltamivir 75 mg twice (N=132, 25.4%) daily dose groups [Hayden 1999]. Most of these reports indicated mild or moderate intensity and were self‐limited. As indicated in the published paper [Hayden 1999], study withdrawals for AEs or illness occurred infrequently across these same groups (N=10, 1.9%; N= 8, 1.5%; N= 7, 1.3%). Of note, the specified causes for AE‐related withdrawals included three reports of headache associated with other symptoms in the placebo group. In contrast, there were no reports of headache as reason for the withdrawals receiving oseltamivir; gastrointestinal complaints accounted for withdrawals in 4 of 8 oseltamivir 75 mg and 3 of 7 oseltamivir 75 mg twice daily recipients. The total numbers of patients with premature study withdrawal for any reason was 21 (4.0%), 17 (3.3%), and 16 (3.1%) across the three groups, respectively. Overall, severe AEs were reported in 82 (15.8%) of placebo, 75 (14.4%) of oseltamivir 75 mg, and 77 (14.8%) of oseltamivir 75 mg twice daily recipients. We were unable to include these details in the paper because of space limitations but my interpretation remains that no excess of clinically relevant oseltamivir‐related headache occurred during this study. This type of detailed AE analysis incorporating severity measures provides necessary context in interpreting the possible importance of AEs.
9. Peer review. The questions raised and opinions expressed in this and earlier Cochrane reports on NAIs by Jefferson and colleagues have resulted in debate and sometimes confusion among practitioners and policy makers regarding the appropriate use of NAIs in seasonal and pandemic influenza responses. Given the importance of these issues, it would be helpful for any future updates to have proper independent review before posting or publication by the Collaboration, as the Cochrane methodology of publication and then independent peer review is not well understood by many people.
Thank you for the opportunity to provide comments. I look forward to seeing the responses from Dr. Jefferson and his colleagues on these points.
Sincerely,
Frederick G. Hayden, M.D. Stuart S. Richardson Professor of Clinical Virology Professor of Medicine University of Virginia School of Medicine Charlottesville, Virginia, USA
Reference List
1. Cowling BJ, Chan KH, Fang VJ, Lau LL, So HC, Fung RO, Ma ES, Kwong AS, Chan CW, Tsui WW, Ngai HY, Chu DW, Lee PW, Chiu MC, Leung GM, Peiris JS. Comparative epidemiology of pandemic and seasonal influenza A in households. N Engl J Med 2010 June 10;362(23):2175‐84.
2. Duval X, van der WS, Blanchon T, Mosnier A, Bouscambert‐Duchamp M, Tibi A, Enouf V, Charlois‐Ou C, Vincent C, Andreoletti L, Tubach F, Lina B, Mentre F, Leport C. Efficacy of oseltamivir‐zanamivir combination compared to each monotherapy for seasonal influenza: a randomised placebo‐controlled trial. PLoS Med 2010:7(11):e100362.
3. Hayden FG, Atmar RL, Schilling M, Johnson C, Poretz D, Paar D, Huson L, Ward P, Mills RG. Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. N Engl J Med 1999 October 28;341(18):1336‐43.
4. Hernan MA, Lipsitch M. Oseltamivir and risk of lower respiratory tract complications in patients with flu symptoms: a meta‐analysis of eleven randomised clinical trials. Clin Infect Dis 2011 August 1;53(3):277‐9.
5. Kaiser L, Keene ON, Hammond JM, Elliott M, Hayden FG. Impact of zanamivir on antibiotic use for respiratory events following acute influenza in adolescents and adults. Arch Intern Med 2000 November 27;160(21):3234‐40.
6. Kaiser L, Wat C, Mills T, Mahoney P, Ward P, Hayden F. Impact of oseltamivir treatment on influenza‐related lower respiratory tract complications and hospitalisations. Arch Intern Med 2003 July 28;163(14):1667‐72.
7. Khazeni N, Bravata DM, Holty JE, Uyeki TM, Stave CD, Gould MK. Systematic review: safety and efficacy of extended‐duration antiviral chemoprophylaxis against pandemic and seasonal influenza. Ann Intern Med 2009 October 6;151(7):464‐73.
8. Moore ML, Chi MH, Zhou W, Goleniewska K, O'Neal JF, Higginbotham JN, Peebles RS, Jr. Cutting Edge: Oseltamivir decreases T cell GM1 expression and inhibits clearance of respiratory syncytial virus: potential role of endogenous sialidase in antiviral immunity. J Immunol 2007 March 1;178(5):2651‐4.
9. Sato M, Saito R, Sato I, Tanabe N, Shobugawa Y, Sasaki A, Li D, Suzuki Y, Sato M, Sakai T, Oguma T, Tsukada H, Gejyo F, Suzuki H. Effectiveness of oseltamivir treatment among children with influenza A or B virus infections during four successive winters in Niigata City, Japan. Tohoku J Exp Med 2008 February;214(2):113‐20.
10. Shrestha SS, Swerdlow DL, Borse RH, Prabhu VS, Finelli L, Atkins CY, Owusu‐Edusei K, Bell B, Mead PS, Biggerstaff M, Brammer L, Davidson H, Jernigan D, Jhung MA, Kamimoto LA, Merlin TL, Nowell M, Redd SC, Reed C, Schuchat A, Meltzer MI. Estimating the burden of 2009 pandemic influenza A (H1N1) in the United States (April 2009‐April 2010) Clin Infect Dis 2011 January 1;52 Suppl 1:S75‐S82.
11. Sugaya N, Mitamura K, Yamazaki M, Tamura D, Ichikawa M, Kimura K, Kawakami C, Kiso M, Ito M, Hatakeyama S, Kawaoka Y. Lower clinical effectiveness of oseltamivir against influenza B contrasted with influenza A infection in children. Clin Infect Dis 2007 January 15;44(2):197‐202.
12. Wang K, Shun‐Shin M, Gill P, Perera R, Harnden A. Neuraminidase inhibitors for preventing and treating influenza in children. Cochrane Database Syst Rev 2012;1:CD002744.
Welliver R, Monto AS, Carewicz O, Schatteman E, Hassman M, Hedrick J, Jackson HC, Huson L, Ward P, Oxford JS. Effectiveness of oseltamivir in preventing influenza in household contacts: a randomised controlled trial. JAMA 2001 February 4;285(6):748‐54
Submitter has modified conflict of interest statement: Disclosures to BMJ (Updated 4 June 2012)
Dr. Hayden received lecture and/or consulting honoraria from GSK until 2002 and from Roche until 2005. Gilead Sciences from 1996‐1999 and Roche from 1999‐2005 provided grant support to the University of Virginia for oseltamivir studies on which he was PI. Similarly GSK provided grant support to the University of Virginia for zanamivir studies from 1994‐2001. Dr. Hayden served as medical officer in the Global Influenza Programme from 2006‐2008 with funding provided to the University of Virginia through the National Institute of Allergy and Infectious Diseases (NIAID). Since 2008 to present the University of Virginia has received funding from the Wellcome Trust for his part‐time work as influenza research coordinator at the Trust and through NIAID for his work as consultant the Southeast Asia Infectious Diseases Clinical Research Network. From 2008‐11 the University also received honoraria for his participation in the Neuraminidase Inhibitor Susceptibility Network which received funding from Roche and GSK. Since 2008 to present, Dr. Hayden has been an unpaid consultant to multiple companies engaged in the development or marketing of influenza antivirals including Roche and GSK.
Dr. John Treanor reports receiving compensation as a member of the scientific advisory boards of Novartis and Immune Targeting Systems, and has performed consulting work for Pfizer. Within the last 3 years, his group has been funded to perform laboratory assays or conduct clinical trials for Sanofi, GlaxoSmithKline, Protein Sciences Corp, Wyeth, PaxVax, Ligocyte, and Vaxinnate.
Dr. Kaiser reports no financial disclosures.
Frederick G. Hayden
Reply
Response to Dr. Hayden’s comments of 2 February 2012.
We thank Dr. Hayden for his detailed feedback. However nothing he writes allays our basic concerns that:
(1) despite the 16,000 pages we analysed, we currently only have access to a very limited dataset hence cannot carry out many of the analyses Dr. Hayden suggests; (2) analysing the "influenza infected" population in Roche oseltamivir trials, as Dr. Hayden proposes, will lead to misleading results because the treatment groups are not comparable for this population; (3) the observational studies Dr. Hayden urges us to consider are generally of poor quality and only represent the small proportion of patients who are hospitalised with influenza; (4) the Kaiser et al (2003) analysis is seriously flawed; (5) data have been selectively reported.
Below, we provide point‐by‐point responses to Dr. Hayden's concerns. (Please note that point 4 appears twice, to follow the numbering in Dr. Hayden’s letter.)
1. Use of intention to treat (ITT) and ITTI‐infected [sic] groups
We agree, in principle, to conduct analysis using the ITT‐infected (ITTI) sub‐population provided that it is appropriately selected by the results of testing completed before the start of the trial (for example by using only the results of viral culture or rapid testing before randomisation).
However we argue that this is not possible in Roche oseltamivir trials. In these trials, the selection of "infected" or "non‐infected" was dependent on the results of serology that is affected by "use" and "non‐use" of oseltamivir. And the selection of those with "serology‐positive results" appears to have given advantage to the oseltamivir group. Hence the method of selecting the ITT‐Infected population in the trials has fundamental flaws and therefore the results are less reliable than those obtained using the ITT population.
2. Sample size considerations
The Kaiser et al analysis has a number of fundamental problems. First, analyses were performed on the ITT‐infected sub‐population which we have shown to be non‐comparable between treatment groups. Second, the authors analysed an outcome that was different to that pre‐specified in the trials. In the trials, complications included otitis media and sinusitis but in the Kaiser et al paper these were not included. This is an example of selective reporting or "cherry picking". Third, complications were not objectively or consistently measured in the trials. Fourth, outcomes such as pneumonia and bronchitis could be either reported as a complication or as an adverse event according to a classification criteria we do not understand and is not discussed in the Kaiser et al paper. And finally the data from the 10 trials was not meta‐analysed, rather, it was combined as if generated from one single trial.
We could potentially address most of these limitations (except for the third) but we have not been given access to the data despite repeated requests to the manufacturer. However we were able to compare hospitalisations as those data were available to us for the ITT population.
We found no evidence of effect on hospitalisations based on seven studies with a median placebo group event rate of 0.84% (range 0% to 11%): odds ratio (OR) 0.95; 95% CI 0.57 to 1.61, P = 0.86). This result is quite different to that reported by Kaiser et al based on the (non‐comparable) ITT Infected population.
In terms of power analysis, to detect a significant difference at this level of difference of 0.84% (placebo) vs 0.80% (oseltamivir), with alpha of 0.05 and power of 0.8, a RCT with approximately 800,000 participants is required.
3. Complications in ambulatory patients
As we have illustrated above the Kaiser et al (2003) analysis has fundamental flaws that we cannot address because the manufacturer refuses to provide us with the data necessary to conduct a proper analysis.
Analysis of the "population with proven influenza infection" (ITT‐infected population) is not appropriate (see above). Data for the analysis of "population without proven influenza infection" are not available to us.
As we have shown above, the power to detect a difference in all‐cause hospitalisation is very small hence to do a subgroup analysis on this outcome seems unwarranted.
The pharmacological/toxicological adverse effects of oseltamivir can be classified into two major types [3]. One is sudden type occurring during the hypercytokinemic state in the early phase of infection including sudden death [3,4], accidental death after abnormal behaviours and vomiting induced by the central depressing action of unchanged oseltamivir [4]. The second are delayed type of reactions including recurrence or exacerbation of influenza and/or other infection, diabetes, bleeding, renal impairment and delayed type neuropsychiatric reactions related to inhibition of the host’s neuraminidase [3]. Sudden type adverse effects should be collected and analysed only during the early phase of influenza (for example, vomiting was only significantly increased within one day of treatment in the paediatric RCTs). However, delayed type adverse effects should be collected and analysed for a longer period to detect those reactions after a full course of treatment (for example the increase of pneumonia in the off‐treatment period in the paediatric RCTs).
A recently published proportional mortality study has indicated that oseltamivir increases sudden type of death (odds ratio: 5.9) compared with zanamivir users by analysing all death cases among approximately 20 million 2009A/H1N1 influenza patients in Japan. This effect was also true for the comparison of oseltamivir users with non‐users of antivirals [4].
4. Data from observational studies
Observational studies during the 2009 H1N1 influenza outbreak have assessed the effects of oseltamivir on a selected population of hospitalised patients. These represent a very small proportion of the total population who get influenza. While subgroup analyses are important, it is important to not lose sight of the fact that the use and governmental stockpiling of oseltamivir is for its routine use in asymptomatic and symptomatic members of the community. Our review thus considers the evidence base that applies to the vast majority of people.
In addition, the studies Dr. Hayden appears to be referring to are retrospective observational studies in which apparent treatment effects may be the result of an effective treatment but could also be due to confounding effects. Unfortunately there is no way to determine which of these possibilities is true. That is why drug regulators require evidence from RCTs to determine whether or not a drug is approved for use. According to the analysis by Jones and Hama [5], apparent protective effects against severe outcomes like pneumonia, hospitalisation and mortality are possibly derived from survivor treatment selection bias (or immortal time‐bias). This is not an issue for randomised controlled trials because follow up begins at the time of randomisation which is the same for patients allocated to active drug and patients allocated to placebo. However in the case of observational studies treatment can begin at varying times (up to several days) after the onset of symptoms. Therefore a naive comparison that compares a binary outcome, such as death (or other adverse event), or time to an event (survival time) is at high risk of survivor treatment selection bias (also referred to as immortal time bias or simply time dependent bias). This bias can occur, for example, because patients who die early are not given the opportunity to receive treatment. In addition patients who are extremely sick may not be given the opportunity to receive antivirals because other treatments and procedures take priority. This bias can be addressed with an appropriate analysis however this has not been done in any of the observational studies of antiviral use for influenza that we have seen.
4. Influenza diagnosis and serologic results
We do not have access to the data required to conduct all these analyses.
5. Other treatment endpoints of interest
We do not have access to the data required to conduct these analyses (time to resumption of usual activities and return to pre‐morbid status) using the ITT population.
By mentioning the evidence and possible mechanism of action for oseltamivir, we are arguing that fever alleviation and symptom reduction may not be caused by the reduction of viral load but may be the result of inhibition of host’s immune functions including induction of cytokines and antibody production by inhibition of the host’s neuraminidase in addition to central depression by oseltamivir.
Analysis of the population with documented influenza infection (ITT‐Infected population) is not valid (see above). Hence we are unable to conduct a valid analysis in the influenza positive population and data for the influenza negative population has not been provided.
Antibody titre is one of the ways of selecting only subjects infected with influenza. However we have shown that the production of antibodies was consistently lower in the oseltamivir group compared to the placebo group in the treatment trials. Therefore the use of antibody production to confirm influenza in prophylaxis trials is not valid. Moreover comparison of the proportion with confirmed infection between the oseltamivir group(s) and the placebo group will provide misleading results.
Nor are "virus titre", "time to culture negativity" or "change in viral titres over time" a true measure of viral load, because oseltamivir as a neuraminidase inhibitor may conceal positivity by inhibiting the influenza virus from leaving the surface of host respiratory cells (which are covered by a mucous layer on the surface of the cells).
6. Adverse events with treatment
In principle we agree. However, there are many data that show the classification of severity is questionable: for example, we believe that psychosis or hallucinations should be classified as "severe" but this has not always been followed. Therefore, we are planning to propose using new classification methods for the analysis of adverse events in the next update of our review.
We agree that comparisons of adverse events in the placebo groups across zanamivir and oseltamivir studies need to be interpreted with caution.
We agree that the spectrum and severity of adverse events/reactions are different among age groups. Therefore, we propose analysing adverse events/reactions stratified by age, if possible, according to the data in the Clinical Study Reports or individual patients' data in the next step of our systematic review.
7. Prophylaxis endpoints of interest
As described on page 7 of our systematic review, the primary outcome measures for prophylaxis studies are:
influenza (both symptomatic and asymptomatic and laboratory‐confirmed) and influenza‐like illness (ILI);
hospitalisation and complications;
interruption of transmission (in its two components, reduction of viral spread from index cases and prevention of onset of influenza in contacts);
harms.
We did not meta‐analyse data from the prophylaxis trials in this systematic review because the substantial documents for prophylaxis trials were obtained after the time lock of 12 April 2011.
Due to the problems we have illustrated above on using virus titre to confirm influenza infection we plan to amend the primary endpoint for prophylaxis trials to influenza‐like illness (ILI).
There is some fear that those with serologic negative infection without symptoms may be more easily infected with influenza virus in the future, because evidence from animal experiments shows that IgA antibody in the respiratory mucosa is reduced (to about 20% of the control group), while reduction of those of systemic IgG antibody (HI antibody) was slight and not statistically significant [6].
8. Adverse effects with prophylaxis
We agree that the prophylaxis studies are particularly useful in assessing drug tolerability.
As we discussed above ("7. Adverse events with treatment"), there are many data that show the classification of severity is questionable. For example, we believe that psychosis or hallucinations should be classified as "severe" but this has not always been followed. Therefore, we are planning to propose using new classification methods for the analysis of adverse events in the next step of the review.
We mentioned the statement "occurred in similar proportions of subjects in the three groups (39 to 47 per cent)" as an example of reporting bias present in the paper (Dr. Hayden’s reference no. 3; known by its trial ID WV15673/WV15697).
The numbers for headache are 47% (242/520) in high‐dose oseltamivir group, 43% (335/520) in low‐dose oseltamivir group and 39% (202/519) in placebo group. These proportions are not similar and show a significant linear trend of increase with oseltamivir dose (P = 0.013).
In addition, we would be grateful if Dr. Hayden were to supply the definition of "drug related headache among headaches reported as adverse events"? In particular, how was it decided whether a headache was drug‐related or not? We cannot suggest signs or symptoms to distinguish oseltamivir‐induced headache from placebo‐induced headache.
We propose analysing adverse events in clinical study reports, including those for prophylaxis trials.
9. Peer review
We agree that there is confusion among policy‐makers and practitioners but believe this to be justified: the data published and accessible to them appear to have some flaws that need to be resolved. We are encouraged by Dr Hayden’s support for our obtaining all the data necessary to clear the confusion.
Cochrane systematic reviews are stringently peer‐reviewed. Not only are they peer‐reviewed by independent experts prior to publication but the protocols are also peer‐reviewed before being undertaken, to reduce a priori biases. In addition, protocols are available for comment from outside the internal review process – Dr Hayden himself, or employees of Roche the manufacturer of oseltamivir, could have provided input about suggested alterations to the protocol which we would have been glad to receive. To this extent the peer‐review process is more stringent than that employed by most other scientific journals.
RH, MJ, TJ, CDM, PD
References
[1] Moore ML, Chi MH, Zhou W, Goleniewska K, O'Neal JF, Higginbotham JN, Peebles RS Jr. Cutting Edge: Oseltamivir decreases T cell GM1 expression and inhibits clearance of respiratory syncytial virus: potential role of endogenous sialidase in antiviral immunity. J Immunol. 2007 Mar 1;178(5):2651‐4.
[2] Hama R, Jones M et al. An overview of neuraminidase inhibitors on inhibitory effect of immune response including cytokines and antibody production (under submission for publication)
[3] Hama R, Fatal neuropsychiatric adverse reactions to oseltamivir: case series and overview of causal relationships. The International Journal of Risk & Safety in Medicine 20 (2008) 5‐36. Available at http://npojip.org/english/no11.html
[4] Hama R, Jones M, Okushima H, Kitao M, Noda N, Hayashi K, Sakaguchi K. Oseltamivir and early deterioration leading to death: a proportional mortality study for 2009A/H1N1 influenza. Int J Risk Saf Med. 2011;23(4):201‐15. Available at http://iospress.metapress.com/content/5257410g24403m68/fulltext.pdf
[5] Jones M, and Hama R et al. Survivor treatment selection bias in a cohort of 2009A/H1N1 influenza patients from Japan (manuscript in preparation)
[6] Takahashi E, Kataoka K, Fujii K, Chida J, Mizuno D, Fukui M, Hiro‐O Ito, Fujihashi K, Kido H. Attenuation of inducible respiratory immune responses by oseltamivir treatment in mice infected with influenza A virus. Microbes Infect. 2010 Sep;12(10):778‐83.
Contributors
Jefferson T, Jones MA, Doshi P, Del Mar CB, Heneghan CJ, Hama R, Thompson MJ
Additional feedback from Frederick G. Hayden, 10 August 2012
Summary
I am writing to respond to the comments and questions raised by Jefferson and his colleagues to my letter of 2 February 2012 about their report published through the Cochrane Collaboration. While the authors have provided helpful clarifications to many points, I remain concerned about their selective approach to data analysis and presentation. Resolution of these issues is important in anticipation of future analyses by Jefferson and colleagues or by others. Many of their responses indicate that analysis of the cohorts with proven influenza infection (ITT‐infected) are not appropriate but further analyses of patient level data should be able to address their concerns (see below). Also they identify biases that could make oseltamivir look better but not those that could make it look worse than its effectiveness and tolerability likely are in reality. An impartial analysis would identify biases in both directions and attempt to deal with them in a balanced appraisal.
My specific comments and recommendations for additional analyses follow:
1. Use of intention to treat (ITT) and ITTI‐infected groups. One obvious means of addressing the concern about selection bias in defining the ITT‐infected (ITTI) population for analysis is to focus on those who were influenza virus‐positive (irrespective of serologic results) at enrolment. These individuals (ITTI‐virus) represented approximately 70‐85% of those enrolled into the ITTI cohorts across the various RCTs.
In addition, those who were included in the ITTI group solely on the basis of seroconversion could be analysed separately to assess overall comparability in terms of symptom resolution and complications to those who were both virus‐positive (ITTI‐virus) and showed serologic rises. This might also help determine whether inclusion of data from virus‐negative seroconverters would affect overall findings.
In contrast to the Cochrane statement that "And selection of those with "serology‐positive results" appears to have given the advantage to the oseltamivir group", it might alternatively be disadvantageous (bias toward the null) or neutral in effect. If oseltamivir is most beneficial in preventing lower respiratory tract (LRT) complications leading to antibiotic use in those in whom it also prevents seroconversion, as one might expect if its overall treatment effect varies between patients based on timing of administration, individual pharmacokinetics or other factors, then its protective effect on complications will be underestimated because the benefits in those for whom it prevents seroconversion will not be counted. If, on the other hand, treatment works effectively only in those infected who seroconvert and has little or no effect in those in whom it prevents seroconversion, this would increase the apparent benefit. However, the only way in which this sequence seems possible would be if late treatment does not interfere with seroconversion but early treatment does AND late treatment is more effective than early. This is biologically implausible and inconsistent with the observed effects on time to treatment for other outcomes, in which early treatment is associated with greater effects. Alternatively, if oseltamivir treatment has a similar effect on LRT complications in infected who seroconvert and those who do not, this would reduce the numbers in the treated group with and without outcomes in a non‐differential way.
In addition to a possible non‐specific immunomodulatory effect of oseltamivir on serologic responses or possible confounding effect of prior inactivated influenza vaccine which might blunt antibody responses in those with proven influenza (1), one explanation for the apparently lower seroconversion rate in oseltamivir recipients would be that some oseltamivir recipients had low viral replication levels at enrolment that were quickly reduced by treatment and did not stimulate antibody rises, so that in these persons treatment prevented seroconversion. If one assumes that clinical outcomes are linked to viral replication levels as other reports suggest, such individuals would probably have shorter illness duration and also be less likely to develop LRT complications. Consequently, not counting them in the oseltamivir group would bias towards the null and under‐estimate the effect of treatment on both illness resolution and complications. In this regard, comparing outcomes in the ITTI‐virus seroconverters vs non‐seroconverters would be of interest if sufficient numbers are available. Also, as stated previously, analysis of the serologic responses based on time from symptom onset to enrolment, including both frequency of seroconversion and observed titres rises in the ITTI‐virus group compared to placebo, might help address this possibility.
If I have interpreted their report correctly, the post‐hoc analyses by Jefferson and colleagues found an absolute difference of 3.4% in overall infection rates between placebo (68.9%) and oseltamivir (65.5%) groups across the studies they analysed (Figure 5, Table 17). This difference presumably approximates the fraction of virus‐negative, non‐seroconverting but possibly influenza‐infected subjects in oseltamivir group. To what extent this difference might bias outcomes is uncertain but its relatively modest size suggests that misclassification would not be a major confounder in either the ITTI or ITT‐non‐infected groups. Optimally in future studies more sensitive nucleic acid amplification testing will be used to detect infection by influenza and other respiratory viruses and facilitate more clear delineation of the groups of interest.
In summary, further analyses of the RTCs on oseltamivir and zanamivir, the outcomes in all groups of relevance (ITT, ITTI, ITTI‐virus, and ITT‐non‐infected) are important and should be presented as fully as possible. As stated previously, separate assessment of the ITT‐non‐infected group provides a valuable control and also enables a determination of whether there was a potential drug‐disease interaction of NAI treatment in non‐influenza patients. As specific antiviral treatment would not be expected to provide benefit on illness resolution or complications in non‐influenza illness, examining the ITT‐non‐infected groups allows this point to be tested directly. An analysis of 11 oseltamivir RCTs (2) confirmed lack of treatment effect on LRT complications in non‐influenza‐infected subjects compared to placebo. The failure to present outcomes in the ITT‐infected or ITT‐virus cohort underestimates possible beneficial drug effects, whereas full data presentation would enable readers to examine the event rates and magnitude of treatment effect sizes for key outcomes across all relevant groups for themselves.
2. Sample size considerations. The endpoint used in our pooled analysis of oseltamivir RCTs (3) was prospectively defined before the analysis was undertaken and was based on findings in our earlier study of zanamivir treatment effects (4) that indicated inhaled zanamivir reduced LRT illnesses leading to antibiotic prescriptions (RR, 0.60; 95% CI 0.42‐0.85) but not upper respiratory tract ones (RR 0.90; 95% CI 0.63‐1.27). The oseltamivir analysis used all studies available to us at the time, including unpublished clinical study reports, in order to avoid selection bias. The other endpoints of upper respiratory tract complications leading to antibiotic use (6.8% oseltamivir vs 5.9% placebo) and overall antibiotic use (14.0% oseltamivir vs 19.1% placebo; P <.001) were described in our 2003 paper (page 1760). Of note, the reductions in overall antibiotic use in influenza outpatients were similar for zanamivir (28%) and oseltamivir (27%) treatment. The limitations of the clinical diagnoses and retrospective approach used in these studies were described more fully in the earlier zanamivir paper (4). However, the simple pooled analysis we undertook in the oseltamivir paper did not correct for the higher proportion of influenza‐infected, at‐risk individuals in the placebo group, and this was a shortcoming. In any case, we pointed out this difference in the paper (page 1669) and presented the data by each group of interest (previously healthy or at risk) in Tables 3 and 4.
More importantly, our finding that early oseltamivir treatment reduced the likelihood of physician‐diagnosed LRT complications leading to antibiotic use has been confirmed and extended (37% reduction in oseltamivir group; risk ratio 0.63 [95% CI 0.48, 0.82]) in a subsequent meta‐analysis (that controlled for pre‐enrolment risk status and included events from the time of enrolment) of the same 10 RCTs included in our paper and one additional one (2). Furthermore, this analysis found that the unpublished trials for which Jefferson and colleagues apparently do not have data were found to be no more favourable to oseltamivir than the published ones. When only the two published trials in previously healthy persons were considered, the reduction in the 24‐day risk of LRT complications treated with antibiotics was 65% (risk ratio, 0.35; 95% CI 0.15, 0.82) in the oseltamivir arms.
3. Complications in ambulatory patients. Their comments on possible oseltamivir adverse events, including sudden death and neuropsychiatric adverse events (NPAEs), raises important points about the effects of influenza infection itself and possible drug‐disease interactions. A well‐documented relationship exists between NPAEs and influenza infection itself. Differing age‐related patterns of influenza‐associated encephalopathy/encephalitis and NPAEs have been reported in Japanese children and adolescents, and also age‐related differences exist in NAI prescribing patterns in Japan. Consequently, careful analysis is required to assess possible associations. It is important to point out that causal relationships between oseltamivir use and such events remain to be proven. Some analyses have indicated comparable or lower NPAEs rates in oseltamivir‐treated compared to non‐treated influenza patients (reviewed in (5)) and no higher rates of NPAEs have been found in hospitalised infants in the USA (6). Oseltamivir administration to those with influenza‐associated NPAEs does not appear to worsen manifestations (7;8). Of note, the crude reporting rates for possible oseltamivir‐associated NPAEs in Japan and USA were significantly lower during the 2009 pandemic than during preceding influenza seasons (9).
As pointed out by Jefferson and colleagues, the possibility of late‐onset adverse events requires that sufficient follow‐up be incorporated into study design to examine both possible adverse and beneficial effects. However, the low frequencies of such events would likely require much larger numbers of subjects than enrolled in most RCTs. One approach is retrospective examination of large databases that link healthcare visits, clinical diagnoses, and drug administration registries. For example, one cohort study involving over 150,000 subjects (49,238 oseltamivir recipients, 102,692 control patients) reported that oseltamivir treatment of presumed influenza was associated with lower risk of TIA or stroke in the subsequent six months (10). This kind of observational study approach has been undertaken for investigation of outcomes and possible adverse events following influenza immunisation and should also be extended to antivirals.
4. Data from observational studies. Jefferson and colleagues indicate that possible survivor treatment selection bias in observational studies can occur because patients who die early are not given the opportunity to receive treatment. However, there is also the opposite concern that sicker patients, especially in a rapidly evolving illness like influenza, are more likely to initiate therapy at any given time after symptom onset than less ill ones. This would be a conservative bias and reduce the likelihood of observing a treatment effect. Clinical experience during the 2009 H1N1 pandemic indicated that late NAI treatment in critically ill or non‐surviving influenza patients was frequently due to delayed consideration of the diagnosis or failure to appreciate the potential value of starting treatment beyond two days after symptom onset in those with progressive illness or high‐risk conditions. This occurred often despite some of these patients having had prior outpatient contact for their acute illness. Although the published reports indicate that most critically ill patients ultimately received antiviral therapy, delayed treatment commonly led to initiation of NAI administration as part of a salvage effort in a deteriorating patient. In part because of critical care support, even those patients who died in hospital usually survived into the second week of illness or later. Those analysing the large amount of observational data that has been generated in recent years, particularly in the context of the 2009 H1N1 pandemic, need to keep these clinical observations in mind. Of note, a recent analysis of critically ill pandemic H1N1 patients in California compared mortality in untreated patients who survived at least to the day after symptom onset when NAIs were first given to the NAI‐treated ones and found that cases who received NAI up to 4 days after symptom onset were more likely to survive (P < 0.05 for each day 0‐4) (11).
An independent report on the observational studies of influenza antivirals published up to November 2010 (12) conducted a meta‐analyses of the few studies providing effects adjusted for confounders and, while acknowledging the low quality of the evidence based on the GRADE assessment approach, concluded that in high‐risk populations, oral oseltamivir may reduce mortality (odds ratio, 0.23 [95% CI 0.13 to 0.43]) and hospitalisation (odds ratio, 0.75 [95% CI 0.66 to 0.89]). In addition, as reported in multiple studies of hospitalised pandemic 2009 A(H1N1) patients, including high‐risk ones like pregnant women and those admitted with pneumonia, treatment with oseltamivir up to 4 days and in some studies later after illness onset has been associated consistently with better outcomes (11;13‐21). Such observations have served to reinforce US CDC recommendations for using influenza antivirals as early as possible in those with severe or progressive illness, those hospitalised with suspected or proven influenza, and outpatients at higher risk for influenza complications (22). Furthermore, given that the circulating influenza viruses have continued to change, with the pre‐2009 A(H1N1) seasonal viruses being entirely replaced by A(H1N1)pdm09 and now antigenically drifted A(H3N2) and B viruses, ignoring observational data means that only information concerning NAI treatment for influenza viruses that are now no longer circulating is being considered.
5. Other treatment endpoints of interest. The possibility that oseltamivir might have non‐specific antipyretic or immunomodulatory actions unrelated to its antiviral effects has been raised in part on the basis of murine studies (23;24). These possibilities or other symptom‐ modifying effects could be addressed by comparison of the course of fever and individual symptom resolution between oseltamivir and placebo recipients for those enrolled in the RTCs who did not have laboratory evidence for influenza (ITT‐non‐infected). Of note, antipyretics were provided to participants in these trials, so that use of paracetamol (acetaminophen) needs to be included as a confounder in such analyses.
In the published pivotal RCTs of oseltamivir treatment in adults, the fever and symptom reductions observed in oseltamivir recipients were in addition to the effects of paracetamol (acetaminophen). One previous RCT in adults with uncomplicated influenza compared amantadine to aspirin and found faster fever resolution in aspirin recipients but slower resolution of other symptoms and higher rates of adverse effects leading to drug cessation (25). While fever resolution is an objective endpoint of interest, it is generally short‐lived and of limited clinical importance relative to other endpoints like time to symptom alleviation, time to return to usual activities/premorbid status, and complications reductions.
The comment by Jefferson and colleagues on measuring viral loads is confusing. Virologic endpoints like quantitative virus titres (infectious and in recent studies viral RNA), time to culture negativity, and changes in titres over time are essential to determining whether a putative influenza antiviral treatment is exerting an antiviral effect and the magnitude of that effect. Failure to detect an antiviral effect raises questions about issues like compliance, drug absorption and disposition, lack of potency, and resistance emergence. Examining such virologic measures also serves to confirm the likely mechanism of antiviral action of NAIs, inhibiting release from infected cells and spread in respiratory tract secretions to initiate subsequent rounds of replication. Several observational studies during the 2009 pandemic found that early antiviral treatment (<2‐3 days from symptom onset) was associated with reduced duration of viral RNA detection (26‐28). Consequently, in the context of the oseltamivir RCTs, it would be valuable to examine the correspondence between upper respiratory tract influenza virologic measures and symptom resolution and LRT complications in both oseltamivir and placebo groups.
7. Prophylaxis endpoints of interest. As indicated in my initial letter, the key efficacy endpoint for an influenza antiviral used for prophylaxis should be symptomatic, laboratory‐confirmed influenza illness. Given the potential for other respiratory viruses to cause febrile respiratory illness, a focus on ILI as the primary endpoint will inevitably underestimate the protective effects of an influenza‐specific chemoprophylactic agent. Of note, various definitions of symptomatic illness and ILI have been used in the influenza prophylaxis RCTs to date, so that further analyses using standardised definitions would be a helpful contribution. Other secondary endpoints of interest include laboratory documented infection (irrespective of symptoms), ILI, virus‐positive ILI, and laboratory‐confirmed illnesses not meeting the ILI definition. Laboratory confirmation based on both viral culture and in future studies viral RNA detection would take advantage of the greater sensitivity of RNA detection.
8. Adverse effects with prophylaxis. As detailed in the oseltamivir seasonal prophylaxis study protocols and report, the relationship between drug receipt and adverse events, including headache, in these trials (29) was determined by the study staff and investigators during the trial under blinded conditions before data lock. The assessment of causality in adverse events (unrelated, remote, possible, probable) as related to drug administration was made using pre‐specified criteria in the protocol (see Appendix 2) on an individual basis by both interviewing the affected participant and considering various factors including past patterns of headaches, associated symptoms, duration and severity, timing in relation to study drug, and whether the symptom persisted during drug administration. Because of its background frequency in the population, headache is a very common event in longer term studies. When it is mild or transient despite continued drug administration, or when it occurs in context of other events (URI, trauma, stress), headache is unlikely to be drug‐related. Using these criteria and the analysis report provided by the sponsor Roche, we observed headache (not otherwise specified, NOS) that was probably, possibly, or remotely related to study drug administration in 22.4% of placebo, 23.8% of once daily oseltamivir, and 25.4% of twice daily oseltamivir recipients during the 6 weeks of prophylaxis (29). The proportions were 10.2%, 8.7%, and 10.8%, respectively, for headache (NOS) that was possibly or probably related to study drug administration.
Headache is a good example of where it is essential to examine not only the frequencies of reported adverse events but also their severity and functional impact, including premature cessation of study drug. In our 6‐week prophylaxis trial (29), severe headache (NOS) irrespective of relationship to study drug administration was reported in 5.0% of placebo, 3.3% of once daily oseltamivir, and 6.9% of twice daily oseltamivir, respectively. Overall premature study withdrawals were found in 21 (4.4%) of placebo, 17 (3.3%) of once daily oseltamivir, and 16 (3.1%) of twice daily oseltamivir recipients. In three placebo but no oseltamivir recipients, headache was listed as a contributory factor. However, headache was reported to be a factor leading to cessation of oseltamivir prophylaxis in one subject in another prophylaxis study (30) and was also reported at a higher frequency during 6‐weeks prophylaxis in a nursing home‐based RCT (5.5% placebo vs 8.3% oseltamivir)(31), so that further analyses are warranted.
9. Peer review. I thank Jefferson and his colleagues for their clarifications on the Cochrane peer review process, and as indicated above, I have provided my own suggestions on the design of future analyses by them and others. In addition, I have provided a list to the Cochrane Editorial Unit of several dozen potential expert reviewers for future protocols and reports on influenza antivirals.
Thank you for the opportunity to provide these responses and comments.
Sincerely,
Frederick G. Hayden, M.D. Richardson Professor of Clinical Virology Professor of Medicine University of Virginia School of Medicine Charlottesville, Virginia, USA
Reference List
(1) Ohmit SE, Petrie JG, Cross RT, Johnson E, Monto AS. Influenza hemagglutination‐inhibition antibody titres as a correlate of vaccine‐induced protection. J Infect Dis 2011 Dec 15;204(12):1879‐85. (2) Hernan MA, Lipsitch M. Oseltamivir and risk of lower respiratory tract complications in patients with flu symptoms: a meta‐analysis of eleven randomised clinical trials. Clin Infect Dis 2011 Aug 1;53(3):277‐9. (3) Kaiser L, Wat C, Mills T, Mahoney P, Ward P, Hayden F. Impact of oseltamivir treatment on influenza‐related lower respiratory tract complications and hospitalisations. Arch Intern Med 2003 Jul 28;163(14):1667‐72. (4) Kaiser L, Keene ON, Hammond JM, Elliott M, Hayden FG. Impact of zanamivir on antibiotic use for respiratory events following acute influenza in adolescents and adults. Arch Intern Med 2000 Nov 27;160(21):3234‐40. (5) Okamoto E. Is oseltamivir (Tamiflu) safe? Re‐examining the Tamiflu 'ado' from Japan. Expert Rev Pharmacoecon Outcomes Res 2010 Feb;10(1):17‐24. (6) Kimberlin DW, Shalabi M, Abzug MJ, Lang D, Jacobs RF, Storch G, et al. Safety of oseltamivir compared with the adamantanes in children less than 12 months of age. Pediatr Infect Dis J 2010 Mar;29(3):195‐8. (7) Huang YC, Li WC, Tsao KC, Huang CG, Chiu CH, Lin TY. Influenza‐associated central nervous system dysfunction in Taiwanese children: clinical characteristics and outcomes with and without administration of oseltamivir. Pediatr Infect Dis J 2009 Jul;28(7):647‐8. (8) Tanabe T, Hara K, Nakajima M, Shimakawa S, Tamai H. Oseltamivir treatment for children showing abnormal behavior during influenza virus infection. Brain Dev 2010 Jun;32(6):440‐4. (9) Donner B, Bader‐Weder S, Schwarz R, Peng MM, Smith JR, Niranjan V. Safety profile of oseltamivir during the 2009 influenza pandemic. Pharmacoepidemiol Drug Saf 2011 May;20(5):532‐43. (10) Madjid M, Curkendall S, Blumentals WA. The influence of oseltamivir treatment on the risk of stroke after influenza infection. Cardiology 2009;113(2):98‐107. (11) Louie JK, Yang S, Acosta M, Yen C, Samuel MC, Schechter R, et al. Treatment with neuraminidase inhibitors for critically ill patients with influenza A(H1N1)pdm09. Clin Infect Dis 2012 Jul 26. (12) Hsu J, Santesso N, Mustafa R, Brozek J, Chen YL, Hopkins JP, et al. Antivirals for treatment of influenza: a systematic review and meta‐analysis of observational studies. Ann Intern Med 2012 Apr 3;156(7):512‐24. (13) Yang SG, Cao B, Liang LR, Li XL, Xiao YH, Cao ZX, et al. Antiviral therapy and outcomes of patients with pneumonia caused by influenza A pandemic (H1N1) virus. PLoS One 2012;7(1):e29652. (14) Higuera Iglesias AL, Kudo K, Manabe T, Corcho Berdugo AE, Corrales BA, Alfaro RL, et al. Reducing occurrence and severity of pneumonia due to pandemic H1N1 2009 by early oseltamivir administration: a retrospective study in Mexico. PLoS One 2011;6(7):e21838. (15) Mosby LG, Rasmussen SA, Jamieson DJ. 2009 pandemic influenza A (H1N1) in pregnancy: a systematic review of the literature. Am J Obstet Gynecol 2011 Jul;205(1):10‐8. (16) Dubar G, Azria E, Tesniere A, Dupont H, Le RC, Baugnon T, et al. French experience of 2009 A/H1N1v influenza in pregnant women. PLoS One 2010;5(10). (17) Rodriguez A, Zaragoza R, Daz E, Daz JJ, Marques A. Early oseltamivir treatment was associated with improved outcomes in 2009 pandemic influenza A (H1N1)v in Spain. Intensive Care Med 2010;36:S136. (18) Siston AM, Rasmussen SA, Honein MA, Fry AM, Seib K, Callaghan WM, et al. Pandemic 2009 influenza A(H1N1) virus illness among pregnant women in the United States. JAMA 2010 Apr 21;303(15):1517‐25. (19) Sugaya N, Shinjoh M, Mitamura K, Takahashi T. Very low pandemic influenza A (H1N1) 2009 mortality associated with early neuraminidase inhibitor treatment in Japan: analysis of 1000 hospitalised children. J Infect 2011 Oct;63(4):288‐94. (20) Yu H, Feng Z, Uyeki TM, Liao Q, Zhou L, Feng L, et al. Risk factors for severe illness with 2009 pandemic influenza A (H1N1) virus infection in China. Clin Infect Dis 2011 Feb 15;52(4):457‐65. (21) Viasus D, Pano‐Pardo JR, Pachon J, Riera M, Lopez‐Medrano F, Payeras A, et al. Timing of oseltamivir administration and outcomes in hospitalised adults with pandemic 2009 influenza A(H1N1) virus infection. Chest 2011 Oct;140(4):1025‐32. (22) "Have you heard?": CDC recommendations for influenza antiviral medications remain unchanged. http://www.cdc.gov/media/haveyouheard/stories/Influenza_antiviral.html. 2012. (23) Ono H, Nagano Y, Matsunami N, Sugiyama S, Yamamoto S, Tanabe M. Oseltamivir, an anti‐influenza virus drug, produces hypothermia in mice. Biol Pharm Bull 2008 Apr;31(4):638‐42. (24) Wong ZX, Jones JE, Anderson GP, Gualano RC. Oseltamivir treatment of mice before or after mild influenza infection reduced cellular and cytokine inflammation in the lung. Influenza Other Respi Viruses 2011 Sep;5(5):343‐50. (25) Younkin SW, Betts RF, Roth FK, Douglas RG, Jr. Reduction in fever and symptoms in young adults with influenza A/Brazil/78 H1N1 infection after treatment with aspirin or amantadine. Antimicrob Agents Chemother 1983 Apr;23(4):577‐82. (26) Cao B, Li XW, Mao Y, Wang J, Lu HZ, Chen YS, et al. Clinical features of the initial cases of 2009 pandemic influenza A (H1N1) virus infection in China. N Engl J Med 2009 Dec 24;361(26):2507‐17. (27) Ling LM, Chow AL, Lye DC, Tan AS, Krishnan P, Cui L, et al. Effects of early oseltamivir therapy on viral shedding in 2009 pandemic influenza A (H1N1) virus infection. Clin Infect Dis 2010 Apr 1;50(7):963‐9. (28) Li IW, Hung IF, To KK, Chan KH, Wong SS, Chan JF, et al. The natural viral load profile of patients with pandemic 2009 influenza A(H1N1) and the effect of oseltamivir treatment. Chest 2010 Apr;137(4):759‐68. (29) Hayden FG, Atmar RL, Schilling M, Johnson C, Poretz D, Paar D, et al. Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. N Engl J Med 1999 Oct 28;341(18):1336‐43. (30) Welliver R, Monto AS, Carewicz O, Schatteman E, Hassman M, Hedrick J, et al. Effectiveness of oseltamivir in preventing influenza in household contacts: a randomised controlled trial. JAMA 2001 Feb 14;285(6):748‐54. (31) Peters PH, Jr., Gravenstein S, Norwood P, De B, V, Van CA, Gibbens M, et al. Long‐term use of oseltamivir for the prophylaxis of influenza in a vaccinated frail older population. J Am Geriatr Soc 2001 Aug;49(8):1025‐31.
Appendix 2 Definition of Adverse Event Relationship to Treatment Probable This category applies to those adverse events which are considered, with a high degree of certainty, to be related to the test drug. An adverse event may be considered probable if: 1. It follows a reasonable temporal sequence from administration of the study drug. 2. It cannot be reasonably explained by the known characteristics of the subject’s clinical state, environmental or toxic factors, or other modes of therapy administered to the subject. 3. It disappears or decreases on cessation or reduction of dose. (There are important exceptions when an adverse event does not disappear upon discontinuation of the drug, yet drug‐ relatedness clearly exists; e.g., (1) bone marrow depression, (2) tardive dyskinesias). 4. It follows a known pattern of response to the study drug. 5. It reappears upon re‐challenge.
Possible This category applies to those adverse events in which the connection with the test drug administration appears unlikely but cannot be ruled out with certainty. An adverse event may be considered possible if or when: 1. It follows a reasonable temporal sequence from the administration of study drug. 2. It may have been produced by the subject’s clinical state, environmental or toxic factors, or other modes of therapy administered to the subject. 3. It follows a known pattern of response to the study drug.
Remote In general, this category is applicable to an adverse event which meets the following criteria: 1. It does not follow a reasonable temporal sequence from administration of the study drug. 2. It may readily have been produced by the subject’s clinical state, environmental or toxic factors, or other modes of therapy administered to the subject. 3. It does not follow a known pattern of response to the study drug. 4. It does not reappear or worsen when the drug is re‐administered.
Unrelated This category is applicable to those adverse events which are judged to be clearly and incontrovertibly due only to extraneous causes (disease, environment, etc.) and do not meet the criteria for drug relationship listed under remote,possible, or probable.
| Probable | Possible | Remote | Unrelated | |
| Clearly due to extraneous causes | ‐ | ‐ | ‐ | + |
| Reasonable temporal association with drug administration | + | + | ‐ | ‐ |
| May be produced by subjects clinical state | ‐ | + | + | + |
| Known response pattern to suspected drug | + | + | ‐ | ‐ |
| Disappears or decreases on cessation or reduction in dose | + | ‐ | ‐ | ‐ |
| Reappears on re‐challenge | + | ‐ | ‐ | ‐ |
Reply
Reply to Hayden Letter 10 August 2012
Thank you for taking the trouble to provide further feedback to our responses to your first set of feedback comments.
You remain concerned about 1) "…selective approach to data analysis and presentation…", especially with respect to our concern that ITT‐infected (ITTI) criteria are inappropriate; and 2) our identification of biases that may exaggerate the effectiveness of oseltamivir. You detail these concerns in more detail:
1. ITT and ITTI You propose an analysis of ITTI in which patients are categorised not by an immune response (which we regard as potentially flawed because our interpretation of the data suggests the drug may interfere with the immune response) but instead by determining whether patients were seroconverting excreting influenza virus at enrolment.
This sounds sensible, and were the data of symptoms and baseline infectivity (by serology or even virus shedding) available to us in suitable format, we would include this analysis. By this, we would expect the randomisation of patients into the two groups to be independent of the initiation of the drug (that is the "influenza‐positive" or "‐negative") before the drug was administered, in case (as may be with the immune response) the drug interferes with virus excretion (as the manufacturer claims in some of its literature).
You also propose an analysis of those grouped by ITTI from serological conversion with those grouped by virus excretion. This also would be useful, to determine whether or not a bias exists in the current data (in either direction, as you point out – the possible mechanisms you outline are plausible).
However, your hypothesis "If oseltamivir is most beneficial in preventing lower respiratory tract (LRT) complications" IS one of the main issues to be confirmed.
As already described in our review, you reported a reduction of cytokine production in response to influenza infection by oseltamivir in humans:
Hayden FG, Atmar RL, Schilling M, Johnson C, Poretz D, Paar D, et al. Use of the selective oral neuraminidase inhibitor oseltamivir to prevent influenza. New England Journal of Medicine 1999;341(18):1336‐43
These findings suggest that reduction of antibody production cannot simply be assumed to be the result of reduced viral load.
2. Sample sizes You describe in more detail the Kaiser 2003 pooled analysis of complications:
Kaiser L, Wat C, Mills T, Mahoney P, Ward P, Hayden F. Impact of oseltamivir treatment on influenza‐related lower respiratory tract complications and hospitalisations. Arch Intern Med 2003;163:1667‐72
This was central to the start of our unease, after it was pointed out to us (in this Feedback section!) by Hayashi that over half of the data in it were of unpublished trials. You state that the end‐points were established a priori and not post hoc. You admit to shortcomings of the paper but point out that they were declared in the paper itself. You suggest that because the two published trials meta‐analysed had no more favourable drug results than the unpublished, bias is less likely.
We think this is to misunderstand our central concern: we are unable to critically appraise the trials in the usual way because they are not available to us, nor, apparently, any other group unselected by the manufacturer. Incidentally we note that you yourself, even as an author, admit you were unable to locate the data for this paper on request, referring us instead to the sponsoring manufacturer, Roche:
Cohen D. Complications: tracking down the data on oseltamivir. BMJ 2009;339:b5387.
This inability by you (authors) or sponsoring manufacturer to provide data for independent scrutiny is disgraceful, a view shared by others, http://bmj.com/tamiflu.
3. Adverse effects of NIs We find it interesting that you call these adverse events 'complications'. You point to our concerns about neuropsychiatric adverse events (NPAEs), and (correctly) state that any association recorded in the literature "…remains to be proven…" with some references (all were retrospective studies and mostly sponsored by the manufacturer) that suggest that there is no increase over control groups. We have other references suggesting the opposite:
Hama R. Fatal neuropsychiatric adverse reactions to oseltamivir: case series and overview of causal relationship. Int J Risk Safety Med: 20 (2008): 5‐36: http://npojip.org/english/no11.html
Nakamura K, Schwartz BS, Lindegårdh N, Keh C, Guglielmo BJ. Possible neuropsychiatric reaction to high‐dose oseltamivir during acute 2009 H1N1 influenza A infection. Clin Infect Dis. 2010 Apr 1;50:e47‐9.
Kruker AT, Krause M. ["Oseltamivir‐induced delirium"]. Ther Umsch. 2010 Dec;67(12):613‐5. German.
Chung S, Joung YS. Oseltamivir (Tamiflu) induced depressive episode in a female adolescent. Psychiatry Investig. 2010 Dec;7(4):302‐4. Epub 2010 Nov 11.
The following are prospective cohort studies that aimed to analyse the association of NPAEs and administration of NIs, in particular oseltamivir.
Fujiwara F, Ikushima S, Hibi N et al. An analysis of risk factors of abnormal behavior in two seasons (07, 08) of influenza infection. Presentation at the 40th annual meeting of the Japanese Society for Paediatric Infectious Diseases held on 15 and 16 (2008)
Fujita T, Fujii Y, Watanabe Y, Mori M, Yokota S. A pharmacoepidemiological study on the relationship between neuropsychiatric symptoms and therapeutic drugs after influenza infection. Jap J Pharmacoepidemiol 2010; 15: 73‐92.
This preliminary report on the analysis of randomised controlled trials of oseltamivir for prophylaxis contains our response to Roche’s report discussing NPAEs and oseltamivir:
Jones M, Hama R, Jefferson T, Doshi P. Neuropsychiatric adverse events and oseltamivir for prophylaxis (letter). Drug Safety, 2012, 35 (12): 1187‐90.
A proportional mortality study indicates that oseltamivir increases sudden death (odds ratio: 5.9) compared with zanamivir users in an analysis of all deaths among ˜ 20 million 2009A/H1N1 influenza patients in Japan. This effect is also observed for the comparison of oseltamivir users with non‐users.
Hama R, Jones M, Okushima H, Kitao M, Noda N, Hayashi K, Sakaguchi K. Oseltamivir and early deterioration leading to death: a proportional mortality study for 2009A/H1N1 influenza. Int J Risk Saf Med. 2011;23(4):201‐15. http://iospress.metapress.com/content/5257410g24403m68/fulltext.pdf
We have presented many of these studies in our previous reply to you, without response.
Of course the uncertainty about causation is true for many drug adverse events: our duty is to ensure that any such uncertainty is clearly articulated.
Nevertheless we entirely agree that "…observational studies … undertaken for investigation of outcomes and possible adverse events following influenza immunisation … should also be extended to antivirals." However, because this Cochrane review is limited to randomised data, such observational studies would be conducted outside this particular review.
4. Observational data You point to our concerns about observational data in general for answering intervention questions. We acknowledge the plethora of observational data available, and even the meta‐analysis of some of them. This does not detract from our continued concern that the best data for answering these questions are randomised, and to leave most of these data unavailable for independent scrutiny is unforgivable.
Moreover, the observational studies are regarded as poor in quality. A recent systematic review and meta‐analysis of observational data for antivirals for the treatment of influenza concluded, "…therapy with oral oseltamivir and inhaled zanamivir may provide a net benefit over no treatment of influenza. However the confidence in the estimates of the effects for decision making is low to very low."
Hsu J, Santesso N, Mustafa R, Brozek J, Chen YL, Hopkins JP, et al. Ann Intern Med. 2012 Apr 3;156(7):512‐24. doi: 10.1059/0003‐4819‐156‐7‐201204030‐00411. Epub 2012 Feb 27. Antivirals for treatment of influenza: a systematic review and meta‐analysis of observational studies
Incidentally, we are interested in rigorously meta‐analysing these data ourselves, and have put in a protocol to do just that. (Jones M, Hama R. Effect of oseltamivir on mortality in treatment of 2009A/H1N1 influenza patients. PROSPERO 2012:CRD42012002245. Available from: http://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42012002245
The proportional mortality study (above), analysing all influenza deaths in Japan and estimating populations who took antivirals and did not take them as the denominators, provides far more reliable estimates of risk from drug exposures than retrospective analysis of surveillance cases without exposed populations (denominators). Contrary to your suggestion "…there is also the opposite concern that sicker patients, especially in a rapidly evolving illness like influenza, are more likely to initiate therapy at any given time after symptom onset than less ill ones…", no such tendency was detected in this study. Proportions of patients treated with antivirals within 12 hours from the onset of fever were significantly lower in the "not mild" cases (26.5%) than "mild" cases (35.4%) at the time when antiviral was prescribed [Table 2b]. However, no patients who deteriorated before the first presentation at medical facilities were treated with antivirals before deterioration [Table 2a], while 78% of "mild" cases and 55% of "not mild" cases were prescribed antivirals within 48 hours from onset of fever [Tables 2a and 2b]. These may be related to the lower positive results (45%) of rapid testing for influenza virus in the "not mild" cases than that in the "mild" cases (60%) at the first consultation:
Hama R, Jones M, Okushima H, Kitao M, Noda N, Hayashi K, Sakaguchi K. Oseltamivir and early deterioration leading to death: a proportional mortality study for 2009A/H1N1 influenza. Int J Risk Saf Med. 2011;23(4):201‐15. http://iospress.metapress.com/content/5257410g24403m68/fulltext.pdf
5. Other treatment endpoints of interest Does oseltamivir have non‐specific antipyretic or immune‐modulatory actions unrelated to its antiviral effect?
We have already noted the hypothermic and immune‐suppression effect of oseltamivir in humans, some from your own writing.
Hama R. Fatal neuropsychiatric adverse reactions to oseltamivir: case series and overview of causal relationship. Int J Risk Safety Med 2008:20:5‐36
Hayden FG, Treanor JJ, Fritz RS, Lobo M, Betts RF, Miller M, et al. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza: randomised controlled trials for prevention and treatment. JAMA 1999;282:1240‐6.
Your suggestion that antipyretic actions of oseltamivir be tested by comparing those randomised to oseltamivir against those not in the non‐ITTI group is worth consideration (although the results might be difficult to interpret). Again, as mentioned above, it would be good to have access to sufficient data to allow this analysis and others we have outlined in the protocol.
We note your criticism about over‐focusing on fever as a proxy for symptom resolution. We are of course interested in any good measure of the latter that is not only objective but also common to all trials. Nevertheless, despite your criticism, fever is a reasonable marker of 'illness' from infections such as influenza, and probably correlates reasonably well with symptom resolution (especially in the prophylaxis trials) and in the treatment trials (if fever is measured until complete resolution) – it is, after all, a cardinal symptom – and has the great advantage of being clearly measured.
You suggest that we test whether viral excretion correlates with symptoms of influenza. We agree that this would be an interesting analysis, were the data available to us (see above).
7. (Note there was no Point 6) Should we be focusing so much on influenza‐like illness (ILI)?
Of course, if oseltamivir neither reduces antibody production to influenza virus nor conceals testing positivity, selecting only laboratory‐confirmed influenza might be a reasonable end point for prophylaxis trials. However the facts suggest these cannot be assumed.
In any case, the Cochrane Collaboration is dedicated to finding the best available evidence to enable patients and their clinicians to make best‐informed decisions. To that end, ILI is what the vast majority of clinicians and their patients will be facing. Therefore this is an end‐point of direct relevance to them, and we make no apology for including it.
8. Adverse events in prophylactic trials Thanks for this detailed information. Further analyses are indeed what we would like to undertake according to our protocol.
9. Peer review Thanks for offering a list of your own colleagues to act as peer reviewers. We adhere to the principle of ensuring there is methodological expertise as well as content expertise. Your list will be useful to consider when finding peer reviewers.
As you may be aware, because this particular Review Group (Acute Respiratory Infections) has its Co‐ordinating Editor as an Author on this review, the handling of the manuscript is managed by the Central Editorial Unit to minimise any potential conflict of interest.
Contributors
Chris Del Mar, Tom Jefferson, Rokuro Hama, Mark Jones, Peter Doshi, Carl Heneghan, Matthew Thomson.
Feedback from Adam Jacobs, 13 February 2013
Summary
Comment: The selection criteria in the review seem highly unusual. The authors describe a 2‐stage process for including trials.
In the first stage, they require that the trial reports they analyse have "external consistency". As far as I can tell, this means that they must be able to verify the contents of the report from an external source.
This seems an extraordinarily high bar to set. I am not aware that it is part of standard Cochrane methodology. If it were applied across Cochrane reviews more generally, I imagine that very few Cochrane reviews would include any evidence at all, especially given that most Cochrane reviews are done perfectly happily with published papers, whereas this one had the advantage of clinical study reports, which are generally far more reliable and comprehensive than published papers.
It is almost as if the authors have gone out of their way to exclude the evidence, which does not help to answer important questions about the efficacy of neuraminidase inhibitors.
It is also noteworthy that no specific reasons were given for exclusion of studies from stage I of the process: we are only told that "insufficient information was available". In the interests of transparency, it would be better to know specifically what information was lacking.
May I suggest that the authors either explain the reason why they felt the need to use far stricter inclusion criteria than is normal in Cochrane reviews, or revisit their inclusion criteria so that the studies can be analysed.
I agree with the conflict of interest statement below:
I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
Adam Jacobs, Director, Dianthus Medical Limited
Reply
Adam Jacobs writes:
"The selection criteria in the review seem highly unusual. The authors describe a 2‐stage process for including trials. In the first stage, they require that the trial reports they analyse have "external consistency". As far as I can tell, this means that they must be able to verify the contents of the report from an external source."
At page 11 of the review we provide the definition: "External consistency. Consistency of data as reported in regulatory documents, other versions of the same clinical study reports/unpublished reports and other references, to be established by cross‐checking"
"This seems an extraordinarily high bar to set. I am not aware that it is part of standard Cochrane methodology. If it were applied across Cochrane reviews more generally, I imagine that very few Cochrane reviews would include any evidence at all, especially given that most Cochrane reviews are done perfectly happily with published papers, whereas this one had the advantage of clinical study reports, which are generally far more reliable and comprehensive than published papers".
And
"May I suggest that the authors either explain the reason why they felt the need to use far stricter inclusion criteria than is normal in Cochrane reviews, or revisit their inclusion criteria so that the studies can be analysed."
Our review is the first systematic review that we are aware of to be completely based on regulatory information. As our basic element of data synthesis was different, we had to develop new methods which we did transparently and are described in the review. It was a fact that we had received partial clinical study reports for the same trials from both Roche and EMA. We felt the need to ensure these reports were consistent. Whether our methods were an "extraordinarily high bar" or a reasonable bar or too low a bar is a judgement readers can make for themselves.
The background history which informed our methodology is explained in the review itself. At pages 4 and 5 of the review we write:
"In 2009, a reader posted a comment in response to the (then current) 2006 version of this review (Jefferson 2006). He pointed out that the review had endorsed the claim regarding a reduction in complications based on the uncritical inclusion of the Kaiser meta‐analysis (Doshi 2009). The reader pointed out that only two of the 10 ’Kaiser trials’ had been published (Nicholson 2000; Treanor 2000) and the information provided by the Kaiser text about the remaining eight was insufficient for their appraisal. Our subsequent efforts to retrieve and review the eight unpublished trials (representing 2691 patients) were unsuccessful, raising the possibility that the findings of our previous review were not an accurate estimate of the benefits and safety of the drug. In addition, we found clear evidence of possible publication bias (see below) amid concern that some evaluations have not been available to scrutiny by the scientific community (Cohen 2009; Doshi 2009; Freemantle 2009; Godlee 2009)."
"This review is focused on healthy adults and children. It represents the amalgamation of two long‐standing Cochrane reviews on the effects of NIs for influenza in healthy adults (Jefferson 2010a, also published as Jefferson 2009a) and children (Matheson 2007). The reviews were combined to pool our collective expertise and time in extracting and assessing data from clinical study reports, which in the case of some oseltamivir trials, report both adult and paediatric outcomes. Cochrane reviews of NIs in both children and adults generated intense interest from clinicians and media during the influenza outbreak declared a pandemic by the WHO in 2009. The Cochrane review of NIs in healthy adults highlighted the high risk of publication bias (Jefferson 2010a). In 2009, a reader posted a comment in response to the (then current) 2006 version of this review (Jefferson 2006). He pointed out that the review had endorsed the claim regarding a reduction in complications based on the uncritical inclusion of the Kaiser meta‐analysis (Doshi 2009). The reader pointed out that only two of the 10 'Kaiser trials' had been published (Nicholson 2000; Treanor 2000) and the information provided by the Kaiser text about the remaining eight was insufficient for their appraisal. Our subsequent efforts to retrieve and review the eight unpublished trials (representing 2691 patients) were unsuccessful, raising the possibility that the findings of our previous review were not an accurate estimate of the benefits and safety of the drug. In addition, we found clear evidence of possible publication bias (see below) amid concern that some evaluations have not been available to scrutiny by the scientific community (Cohen 2009; Doshi 2009; Freemantle 2009; Godlee 2009).
Our attempts to reconcile published and unpublished evidence by contacting the manufacturer and study authors failed (the latter were unable to provide us with the necessary data; some were not in possession of the data and others may have been restricted by confidentiality agreements). Together with the British Medical Journal (BMJ)we ascertained that ghostwriters had been involved, which means the named authors may not have been in full control of the trial publications (Cohen 2009). We also identified several key differences in licensed indications for oseltamivir between regulatory systems (mainly between the US, Europe and Japan) and under‐reporting of harms. The differences are detailed elsewhere (Doshi 2009) but of particular concern was the insistence of the FDA that oseltamivir has not been shown to reduce complications (FDA 2011a). The FDA has also not allowed an indication for interference of viral transmission within households (the key concept behind post‐exposure prophylaxis). This undermined our confidence in published data and in the findings of our previous Cochrane reviews. In the background of all this were suggestions that NIs may not be as safe as previously assumed, with associations between oseltamivir use and neuropsychiatric adverse reactions of particular concern (Hama 2008)."
Adam Jacobs writes:
"It is almost as if the authors have gone out of their way to exclude the evidence, which does not help to answer important questions about the efficacy of neuraminidase inhibitors."
A page 5 of the review we write:
"During the preparation of the 2010 review and of the current review, we realised that there were multiple sources and different levels of granularity of clinical trial data (see 'The Scope of Clinical Trial Data' table in Jefferson 2011). We decided that clinical study reports and regulatory comments were likely to provide the least biased, most complete and most insightful set of data for our review".
And
"We identified that 60% (3145/5267) of patient data from randomised, placebo‐controlled phase III treatment trials of oseltamivir have never been published. This includes M76001, the biggest treatment trial ever undertaken on oseltamivir (with just over 1400 people of all ages). Exclusion of unpublished data changed our previous findings regarding oseltamivir's ability to reduce complications of influenza (Doshi 2009; Jefferson 2009a)."
Our attempts at identifying and retrieving all available evidence from regulators and manufacturers since 2009 are documented at http://bmj.com/tamiflu.
Adam Jacobs writes:
"It is also noteworthy that no specific reasons were given for exclusion of studies from stage I of the process: we are only told that "insufficient information was available". In the interests of transparency, it would be better to know specifically what information was lacking."
In Table 9 (page 186) we list all studies included in Stage 1 and report details of what data for each were available to us. For, example for trial MV22940 we know that it is likely to be a randomised trial assessing effects of oseltamivir on post exposure prophylaxis but no other data are available to us. In these circumstances we cannot proceed to assessment until the information is available, as explained in the text of the review. However these studies are not excluded but are marked as pending assessment.
We invite Adam Jacobs to read the review and the references which document the history of the review, background and rationale for withdrawing the original review and developing the current version. We also invite Mr Jacobs to clarify what business relation his firm has if any with Roche, GSK and BioCryst Ltd.
It is possible that future Cochrane reviews will include an increasing proportion of regulatory information to minimize the effects of reporting bias. This type of speculation is however beyond the scope of the review.
Contributors
Cochrane Neuraminidase Inhibitors Review Team, 5 March 2013
Prof Chris Del Mar, Coordinating Editor, Acute Respiratory Infections Cochrane Review Group, Australia Dr Peter Doshi, Postdoctoral Fellow, Johns Hopkins University, USA Dr Rokuro Hama, Physician, Pharmaco‐epidemiologist, Japan Institute of Pharmaco‐vigilance, University of Osaka, Japan Dr Carl Heneghan, Clinical Reader, Department of Primary Care Health Sciences, University of Oxford, UK Dr Tom Jefferson, Epidemiologist, Acute Respiratory Infections Cochrane Review Group, Italy Dr Mark Jones, Statistician, University of Queensland, Australia Dr Matthew Thompson, Clinical Reader, Department of Primary Care Health Sciences, University of Oxford, UK
Feedback from Harri Hemilä, 6 May 2013
Summary
Comment: Oseltamivir (Tamiflu) shortens the duration of influenza‐like illness by 13% (95% CI 8% to 18%)
In studies measuring dichotomous outcomes, relative risk (RR) is a standard measure for comparing study groups. The purpose of using RR is to adjust for baseline variability in the occurrence of disease. It is easier to compare two trials on the basis of their RR estimates than on the basis of their absolute effects.
The relative effect should also be calculated for continuous outcomes. Although the duration of disease may vary randomly in placebo groups, there are also biological reasons why diseases in different placebo groups differ in their severity and duration. For example, in Analysis 1.1 of this review, the duration of influenza‐like illness in the placebo group of trial WV15671 is 35% shorter than in the placebo group of trial WV15819/WV15876/WV15978 (Z = 6.5; P = <0.00001; 125h/192h). Such very large baseline differences are not explained by chance. Differences in the study populations, influenza seasons, study protocols, etc. are plausible explanations for the baseline variation. The above‐mentioned baseline difference is much greater than any of those between the oseltamivir (Tamiflu) and placebo groups in the five trials of Analysis 1.1. As for dichotomous outcomes, the baseline variability of continuous outcomes can be adjusted for by calculating the effect in percentages, i.e., the relative effect. Furthermore, the percentage effect is informative for an average reader because the reader may form an opinion on whether, for example, a 10% or 20% average decrease in the duration is worth the cost and effort of the treatment. Separate from the absolute effect in days, the percentage effect shows whether the effect is small or large.
Therefore the effect of oseltamivir should be calculated also as a percentage effect. I calculated the relative effects for the five trials listed in Analysis 1.1, pooled them using the fixed effect inverse variance method of RevMan, and found that the average effect of oseltamivir is a 13% (95% CI 8 to 18%) decrease in the duration of influenza‐like illness.
Furthermore, the relative effect estimate makes it possible to compare the effects of treatments for related conditions. Influenza‐like illness has substantial overlap with the common cold. In our Cochrane review on vitamin C and the common cold we calculated that ≥1 g/day of vitamin C shortens colds in adults by 8% (95% CI 4 to 12%) and in children by 18% (95% CI 9 to 27%) [1]. Another meta‐analysis found that a high dose of zinc (>75 mg/day) as zinc acetate lozenges decreased the duration of colds by 42% (95% CI 35 to 48%) and as zinc lozenges made with other salts by 20% (95% CI 12 to 28%)[2]. The mechanism of the effect of vitamin C and zinc lozenges is not understood; however, there is no reason to assume that their effects are specific, for example, to the rhinovirus. If vitamin C and zinc lozenges have effects on diverse respiratory viruses, they might also have an effect on influenza viruses. In mice, influenza infection decreased vitamin C concentration in bronchoalveolar lavage fluid [3]. In mice, vitamin C deficiency increased lung pathology caused by influenza infection [4]. An early study with influenza patients reported that the occurrence of pneumonia was 80% lower (2 vs. 10 cases) in the vitamin C group, suggesting that vitamin C might also have an effect on influenza in humans [5,6]. If the effects of vitamin C and zinc lozenges on influenza‐like illness are of the same magnitude as their effects on the common cold, then the effects of these treatments compare reasonably with oseltamivir. The comparison of the percentage effects of oseltamivir, vitamin C and zinc lozenges may be useful when considering how future research resources concerning the treatment of respiratory virus infections might be allocated. In this respect, the type of effect measure has a much wider importance than just its use in evaluating the effectiveness of oseltamivir as an issue of its own.
Thus the relative effect estimate adjusts for baseline variations between trials, it is informative for most readers because people are familiar with percentages, and it makes it easier to compare different treatments for related conditions. For these reasons I would like to encourage the authors to calculate and report the relative effect estimates for oseltamivir in the next revision of the review.
References
[1] Hemilä H, Chalker E. Vitamin C for preventing and treating the common cold. Cochrane Database Syst Rev 2013:CD000980. http://dx.doi.org/10.1002/14651858.CD000980.pub4
[2] Hemilä H. Zinc lozenges may shorten the duration of colds: a systematic review. Open Respir Med J 2011;5:51‐8. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3136969/
[3] Buffinton GD, Christen S, Peterhans E, Stocker R. Oxidative stress in lungs of mice infected with influenza A virus. Free Rad Res Commun 1992;16:99‐110 http://www.ncbi.nlm.nih.gov/pubmed/1321077, http://dx.doi.org/10.3109/10715769209049163
[4] Li W, Maeda N, Beck MA. Vitamin C deficiency increases the lung pathology of influenza virus‐infected gulo‐/‐ mice. J Nutr 2006;136:2611‐6. http://www.ncbi.nlm.nih.gov/pubmed/16988135, http://jn.nutrition.org/content/136/10/2611
[5] Kimbarowski JA, Mokrow NJ. Colored precipitation reaction of the urine according to Kimbarowski as an index of the effect of ascorbic acid during treatment of viral influenza [in German]. Deutsche Gesundheitswesen 1967;22:2413‐8. http://www.ncbi.nlm.nih.gov/pubmed/5614915, Translation: http://www.mv.helsinki.fi/home/hemila/T4.pdf
[6] Hemilä H, Louhiala P. Vitamin C for preventing and treating pneumonia. Cochrane Database Syst Rev 2007:CD005532. http://dx.doi.org/10.1002/14651858.CD005532.pub2
I agree with the conflict of interest statement below: I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
Harri Hemilä Department of Public Health, University of Helsinki
Reply
Thank you for your suggestion and comprehensive argument why you think it is important. Indeed in our 2006 and 2009 updates of A047 (the previous review on antivirals for influenza in otherwise healthy adults), we pooled hazard ratios and reported relative effects for time to alleviation of symptoms. However GSK, the manufacturer of zanamivir, made the comment that hazard ratios may not be appropriate due to non‐proportional hazards. Therefore for A159 we reported absolute treatment effects for time to alleviation of symptoms but not relative effects. We agree with your argument and will report absolute and relative effects for time to alleviation of symptoms and other outcomes in the next update of 'Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children' due at the end of 2013.
Contributors
Jefferson T, Jones MA, Doshi P, Del Mar CB, Heneghan CJ, Hama R, Thompson MJ
Review amendments, 16 May 2013
Summary
As reported in the current version of our review, we will complete the review of regulatory information which arrived after our original time lock. We will assess additional evidence from oseltamivir Module 2s, evidence on adverse events following exposure to neuraminidase inhibitors (NIs) and clinically relevant outcomes.
A rationale and description of our methods follows.
Evidence from Module 2s (Ms2) of oseltamivir trials
1. Summary and background
This part of the document will describe our efforts to determine whether the additional information included within Module 2s (Ms2) of clinical study reports (CSRs) would change the risk of bias assessment, identify additional useful or relevant information, and conclusions of the overall body of evidence contained within our existing review. A second aim is to construct and test a tool that could be used to extract, organise and appraise study information contained in such modules.
The items which are most commonly found in the M2 of the oseltamivir trials are: Certificates of Analysis (a report on the colour, composition and content of active and control substance capsules, blank Case Report Forms (case notes for each participant), follow‐up cards/diary cards (on which each participant recorded information such as symptoms), informed consent text and participant contract (to be administered to and signed by each participant), lists of investigators in the trial, investigation review board, ethics committees and study sites' addresses, the Reporting Analysis Plan (Roche's term for the Statistical Analysis Plan or SAP detailing the types of data analyses to be carried out), randomisation list (used to allocate participants and the study Protocol with its amendments when appropriate or available.
1.2 Methods
We received 12 CSR Ms2 from 31 studies requested from EMA by July 2011. Before we reviewed Ms2 we knew they contained protocols, with their amendments, certificate of analyses, blank case report forms, randomisation and participating centres’ lists. However, we had no precise idea whether this was a comprehensive list or whether further items would be identified once we started reviewing. We also noted that the same info was reported elsewhere in the CSRs (for example in the core report) but in a different level of detail. A good example of this is the statistical analysis section of the core report which is a few pages long chapter, compared to the Statistical Analysis Plan (SAP), which is a self contained document included in M2. In addition we were not aware of the existence of any readily available tool to allow us to extract, organise and appraise the information contained in the Ms2.
As consequence we decided to develop our own tool. Our plan is to do this by identifying the types of items contained in the Ms2 available to us and their location in the Ms2. The outline content of all items identified will be checked in the Ms2 because of the potential for differing titles for the same item. For example we have already noticed that Research Analysis Plan (RAP) is sometimes called Data Analysis Plan (DAP) or Statistical Analysis Plan (SAP). Another example are the Protocol Amendment Histories and Protocol Modification History Document. These represented different ways of identifying the same item and need to be given a single identifier. Items such as Data Reporting and Analysis Manual (DRAM) are only cited in one M2. We will also conduct a pilot to identify with certainty which items are present more frequently. We will make a list of what we thought were most present and important items contained in the Ms2 and create a grid based on the sequence of development of the trial design and analysis plan. For example, we want to track whether the reporting of the trial study design in the relevant section of the protocol and its amendments (in M2) is consistent with that described in the core report (in M1). We will also make an initial extraction frame to reconstruct the timeline of the study documents, summarising the number of protocol changes and their dates in sequence. This has the purpose of giving an overview of the main timeline points of the key items of study design and analysis.
We will then pilot our extraction sheet and make changes following discussion with all authors. We will extract the data in the same groups we worked in the original review.
We will define the impact of adding M2 information by measuring the change in risk of bias (ROB) assessment in our review as well as reporting our summary description and appraisal of each trial before and after addition of the data and comparing it with the manufacturer's assessment.
The detailed questions addressed by our analysis are:
Does addition of M2 to M1 change the risk of bias evaluation compared to M1 alone?
Does reading M2 and M1 in CSRs change the risk of bias evaluation compared to using published papers?
Is the current risk of bias tool adequate for assessing trials based on reading M2 then M1 in the CSRs?
Does reading M2 and M1 in the CSRs identify additional useful relevant information for systematically reviewing a trial programme?
We will primarily use descriptive methods to answer the questions. To answer question 1 we will compare the risk of bias in our 2012 review with risk identified after addition of M2 information to our current review using a 3 by 3 contingency table. We will repeat this procedure to answer question 2, by comparing risk of bias in our 2009 BMJ review to our current assessment. This analysis will be based on the subset of trials that were published and included in our 2009 review.
To answer question 3 we will list all the components of other risk of bias in the current review and compared these with previous reviews (2012 and 2009).
To answer the final question we will provide a summary of the items that were identified in our assessment of the trials using the new M2 tool. This will allow us to summarise discrepancies between what was planned in the protocol, what was carried out (RAP, protocol amendments), what was reported in M1, and what was reported in the published papers. The focus would be on the trial programme of research i.e. issues that appeared consistently over the trials.
Adverse events
2. Summary and background
This document outlines how we will conduct the analysis of adverse events as part of the wider Cochrane review of neuraminidase inhibitors (NIs) for prophylaxis and treatment of influenza in healthy adults and children (A159).
We use the term 'adverse events' throughout this document rather than harms or adverse reactions as these latter terms imply causality which may or may not be appropriate.
In keeping with the methods of our previous review we will not use data from journal publications for this proposed analysis. We now have access to multiple clinical study reports (CSRs) for both oseltamivir and zanamivir. To our knowledge this is the first time some of these data have been available outside manufacturers and regulators, and allows for the exploration of events in more detail than is possible using the limited information on safety reported in journal publications. This potentially allows us to address some of the concerns that have arisen in the post marketing period about the possible relationship between neuraminidase inhibitors, oseltamivir in particular, and neuropsychiatric and other harms. The documents available to us contain listings and summaries of adverse events recorded in the trials including narrative summaries of serious adverse events and adverse events leading to study withdrawal.
The adverse events are classified by relationship to the study drug and also, by intensity (mild, moderate, severe, life‐threatening and death). The duration of events is reported and they are also lumped into body systems such as gastrointestinal, neurological, etc.
2.1 Methods
All CSRs of oseltamivir and zanamivir will be included in our analysis. CSRs for prophylaxis, for treatment of adults and for treatment of children will be analysed separately. Adverse events will be initially descriptively compared over the entire treatment and follow‐up period but then potentially stratified by on‐treatment and off‐treatment periods if it appears there may be a difference between treatment groups.
2.2 Adverse events for comparison
2.2.1 Common events
For common events of any intensity with an overall incidence of 2% or more we will compare the incidence between treatment groups. The cut‐off of 2% is based on a power analysis where assuming 4000 patients in total (this is approximately how many patients we have access to in oseltamivir treatment trials of adults as well as in oseltamivir prophylaxis trials of adults), we will have 80% power to detect an odds ratio of 1.75 with 5% level of significance.
2.2.2 Uncommon events
Due to a lack of data to compare uncommon events we will compare events lumped into body systems between treatment groups. If we find evidence of a difference in incidences between groups lumped into a body system we will conduct further analysis if appropriate. This further analysis is to determine whether the difference in incidence is due to any common events included in that body system. For example, in the case of neurological body system, if we found evidence of a difference between treatment groups we would remove all common neurological events such as headaches and repeat the analysis.
2.3 Severe, serious events and events leading to study withdrawal
As well as the analysis described in section 2.2 above we will also conduct a subgroup analysis of just the events with severe intensity, serious events and events leading to study withdrawal. We will use the same definitions of "severe" and "serious" as specified in the CSRs. However we will check the classifications using all the information available in the CSRs including line listings of events, narratives provided for serious events and also for events leading to study withdrawal. Any disagreements with the original classifications will be recorded and any reclassifications will be assessed in a sensitivity analysis. Given it is unlikely there will be sufficient events to conduct separate statistical analysis at the level of body system we will compare the overall distribution of events by body system between treatment groups.
2.4 Incidence of adverse events in the CSRs
As a further check on the validity of the data on adverse events contained in the CSRs we will conduct descriptive comparisons of the incidence of adverse events in the prophylaxis and treatment trials.
This is because of the unclear methods of collecting and classifying adverse events in the trials. A potential adverse event could have been classified as a symptom of influenza, an efficacy outcome (such as complication of influenza) or an adverse event. Hence an informal comparison of the incidence of adverse events in the trials where participants had influenza (or influenza‐like‐illness) and the trials where participants did not have influenza may help show where adverse events could have been under‐reported. We will take into account factors such as age of participants and duration of treatment exposure for these informal analyses. In addition if it is clear that an adverse event was not reported as an adverse event but was included elsewhere in the CSR (e.g. in the efficacy section), we will include that data in our adverse event analyses.
We will also construct a table showing the definitions specified in each CSR for classifying potential adverse events as adverse events, complications or symptoms of influenza.
2.5 Antibody titre
We have already reported that antibody production was lower in the oseltamivir group than in the placebo group in the systematic review of treatment trials of oseltamivir (2012). We will update this analysis by including additional oseltamivir trials as well as assess antibody production in the zanamivir trials.
We will assess antibody production in the prophylaxis trials of oseltamivir and zanamivir by the following methods.
We will first identify the participants who had influenza‐like illness (ILI) or pyrexia. If the proportion is similar between active group and placebo group, the proportion of participants who had four times or higher increase of antibody will be compared between groups.
2.6 Dose‐response analysis
A number of trials included two or more active treatment arms with different doses of study medication given to participants in each of the arms. For these trials we will investigate the dose‐response relationship for common adverse events (as defined above).
2.7. Details of analysis
Initial analysis will be descriptive only where we will report the numbers and percentages of events by treatment group. If there is a potential difference in the pooled percentages between treatment groups (e.g. if there is more than a two standard error difference between percentages) then we will conduct formal meta‐analysis. If indicated we may also conduct additional analyses taking into account event intensity and/or duration.
2.8 Limitation and exploratory analysis
The methods presented above are those that we have pre‐specified prior to formal analysis of the data. A limitation of these methods is that we may fail to detect differences in rare adverse events because these events will be compared along with other types of events within body systems. Therefore in the process of conducting our formal analysis we may generate further hypotheses or conduct additional exploratory analyses. If this is the case then we will clearly label these analyses as exploratory and interpret the findings accordingly.
Types of outcome measures
3. Background
For most people, influenza is a self limiting illness. However the disease can at times lead to serious complications such as pneumonia and hospitalisations, and if treatment with neuraminidase inhibitors can reduce the risk of severe outcomes, this would be an important public health benefit. Another potentially important public health benefit would be the ability of antivirals to interrupt person to person transmission of influenza. Current evidence for these outcomes is scarce or inconclusive. A positive balance of effects on complications and viral spread versus harm profile is the main reason for using NIs in a public health context, especially the orally administered oseltamivir.
All analysis will be based on the intention‐to‐treat (ITT) or safety populations as our prior review discovered compelling evidence that the ITTI (the subpopulation deemed to be influenza‐infected) populations were not balanced between treatment groups in the Roche oseltamivir trials. In addition, estimates from the ITT population will be more generalisable to clinical practice where routine testing for influenza is not common in many countries (and even where used, remains of variable accuracy). Analysis will be conducted separately for prophylaxis trials, treatment trials of adults and treatment trials of children.
The list of outcomes given below includes all potential outcomes that we believe are clinically important. However a number of them may not be formally comparable in this review because there are insufficient numbers of events (e.g. mortality) or they were not adequately measured or reported (e.g. drug resistance).
3.1 Outcome measures for treatment studies
Complications˜ Harms* Symptom relief Hospitalisation Viral excretion Drug resistance Mortality
3.2 Outcome measures for prophylaxis studies
Influenza‐like‐illness^ Complications˜ Harms* Hospitalisation Viral excretion Drug resistance Mortality ˜Complications (secondary illnesses) include pneumonia, bronchitis, otitis media, sinusitis or other respiratory tract infection after influenza‐like illness. Initially we will construct a table to illustrate the design methodology used for each study. The table will include the following variables:
Study/trial ID Where complications are first defined in the CSR (e.g. "as secondary endpoint in 3rd version of protocol six months into trial and two months prior to trial unblinding") Definition of "complication" including types of events, population and time period at risk How complications were measured (see diagnosis methods criteria shown below) Availability of complications data for the ITT population We will then stratify our analysis by method of diagnosis with three possible criteria: a. Lab‐confirmed diagnosis (e.g. based on radiological or microbiologically confirmed evidence of infection). b. Clinical diagnosis without laboratory confirmation (diagnosed by a doctor after a clinical examination). c. Other type of diagnosis such as self‐reported by patient
*A separate section provides the details of our proposed analysis of harms.
^The main outcome of interest is any symptomatic influenza‐like‐illness (ILI). However, we will also conduct separate analyses of influenza (symptomatic and asymptomatic) and non‐influenza ILI.
Reply
TJ
Contributors
Jefferson T, Jones MA, Doshi P, Del Mar CB, Heneghan CJ, Hama R, Thompson MJ
Feedback from Peter Gross, Hackensack University Medical Center, USA, 17 April 2014
Summary
Can Cochrance compare their results on influenza neuraminidase inhibitors with the reduction in symptoms when penicillin is given for strep throat? I think they may be comparable. That would be an important perspective.
I agree with the conflict of interest statement below: I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
Reply
Dear Dr Gross, the Cochrane Collaboration and specifically our ARI Group could certainly design a review looking at the comparative evidence of the effects of neuraminidase inhibitors versus penicillin for sore throat. Both reviews are in the ARI Group's Module, so it would have to be an indirect comparison review. I am not sure there are any of those in the Cochrane Database of Systematic Reviews, but there's always a first time for everything as our regulatory evidence review shows.
It would have to be a new review, with a new protocol, perhaps with new authors. Would you be interested?
All the clinical study reports for the two neuraminidase inhibitors we looked at are at: http://dx.doi.org/10.5061/dryad.77471 and a short introduction at: blog post http://blog.datadryad.org/2014/04/17/tamiflu‐data/
With best wishes, Tom Jefferson on behalf of the authors
Contributors
Peter Gross
Roche feedback on 'Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children', 16 October 2014
Summary
https://editorial‐unit.cochrane.org/cochrane‐review‐neuraminidase‐inhibitors‐influenza
Reply
We have received feedback from Roche and the full document is accessible via the above link, hosted on the Cochrane Editorial Unit's (CEU) website. The review authors submitted their reply to the Roche feedback on 23 March 2015, and this has been posted on the CEU website.
Contributors
Submitted by Barry Clinch,1 James Smith,2 Andy Kenwright,1 Bernadette Surujbally,3 Joanne Harding1 1Roche Products Ltd, Welwyn Garden City, UK; 2F. Hoffmann La‐Roche Ltd, Basel, Switzerland; 3BStats Solutions Ltd, Hertfordshire, UK
Feedback from Ryuko Hatano, 28 August 2017
Summary
Dear Sir/Madam: Greetings from Japan.
We are members of the Association of Victims of Tamiflu‐related Encephalopathy, a group of victims whose children or other family members had serious reactions after taking Tamiflu, including sudden death, accidental death due to abnormal behaviours, and serious sequelae with disability. In this letter, we would like to respectfully request your group to establish the causality between Tamiflu and abnormal behaviours, sudden death or sequelae.
We have learnt that the WHO’s essential medicines list was amended and Tamiflu was moved from the core to the complementary list, and that its use be restricted to severe illness due to confirmed or suspected influenza virus infection in critically ill hospitalized patients. Moreover, the next Expert Committee might consider Tamiflu for deletion, unless new information supporting the use in seasonal and pandemic outbreaks is provided.
We understand that the systematic review your group issued in April 2014 and the opinion you submitted to the WHO have served as the major evidence that promoted the amendment and future possible deletion of Tamiflu from the essential medicines model list. We, all the members of the Association of Victims, deeply appreciate you for conducting such an important systematic review which influenced the policy of WHO.
We know the outline of the results of your systematic review because Dr. Rokuro Hama, a Japanese doctor who is one of the members of the Cochrane team translated some important parts of the review into Japanese and uploaded it on his website: http://www.npojip.org/sokuho/140410.html
According to the translation, the results of the systematic review is summarized as follows:
1) Tamiflu reduced the time to first alleviation of symptoms in adults by 0.7 day (from 7 to 6.3 days). There was no effect in asthmatic children who are the real target of efficacy, although healthy children of Tamiflu group alleviate symptoms one day earlier than placebo group. 2) Tamiflu reduced the production of antibody against influenza. 3) Tamiflu did not decrease hospitalization. 4) There was no evidence that Tamiflu reduce influenza like illness. 5) Tamiflu induced harmful effects such as nausea, vomiting, headache, neurologic symptoms, kidney disorders, diabetic/hyperglycemia and pain in extremities. Zanamivir had no such adverse effects. 6) In particular, neuro‐psychiatric symptoms were significantly more reported in Tamiflu group than in the placebo group in the prophylaxis trials. Dose‐response relationship was also seen in the treatment trials.
“Abnormal behaviour” has been listed as one of the adverse reactions to Tamiflu in the Japanese label of Tamiflu since 2004. Hence, we believe that the accidental deaths due to abnormal behaviours after using Tamiflu should be recognized as death cases caused by side effects of Tamiflu, and that they should be relieved by the government. We submitted the applications for damage relief to the Pharmaceuticals and Medical Devices Agency (PMDA), but they rejected them, stating that “there is no association with Tamiflu use”.
We also believe that the sudden deaths during sleep may be the consequences of side effects of Tamiflu because animals died suddenly after the administration of Tamiflu in animal experiments. Therefore, we applied damage relief for the sudden death cases as well, but PMDA rejected them with the same reason.
We, 14 families filed cases against PMDA to cancel the decision. Three families (one sudden death and two accidental deaths after abnormal behavior) lost the cases at the Supreme Court. The court accepted the claim of PMDA: the causes of both sudden death and accidental death due to abnormal behaviors were caused by influenza related encephalopathy and not by Tamiflu use because Tamiflu has no effect on brain and there is no evidence suggesting causality in animal, clinical and epidemiologic studies.
However, sensory function, cognitive function and consciousness of rats are disturbed and the mortality of rats from sudden death rises as the doses of Tamiflu increases as shown in the review article on the mechanisms of sudden‐onset type of reactions to Tamiflu (Hama et al. 2016). High dose of Tamiflu stops animals’ respiration, followed by cardiac arrest (Kimura and Haji et al. 2013). It induces low body temperature (Ono et al. 2013).
There are some epidemiologic studies which suggest association between Tamiflu use and delirium or unconsciousness (Fujita et al. 2010). There is also an epidemiologic study in which sudden deteriorations leading to death within 12 hours after Tamiflu use were far more frequently reported than after Relenza use (Hama et al. 2011). All these findings suggest close association, but the judges neglected them and concluded that there was “no causal association” based solely on the claims of PMDA.
We ourselves witnessed extremely abnormal course of dying or developing sequelae in our precious children or other family members. Based on these experiences we are convinced that there is no cause for these events other than Tamiflu. However, we are merely lay people, and we have no scientific means to prove it by ourselves. If your group could establish the causality between Tamiflu and abnormal behaviours, sudden death or sequelae, it would be greatly appreciated. If needed, all the members of our group are willing to provide any information about our family cases.
Thank you very much for your understanding and kind assistance in advance. Yours sincerely, All members of the Association of Victims of Tamiflu‐related Encephalopathy
I do not have any affiliation with or involvement in any organisation with a financial interest in the subject matter of my comment
Ms. Ryuko Hatano Affiliation: The Association of Victims of Tamiflu ‐related Encephalopathy Role: Representative
Reply
Dear Ms Hatano,
Thank you for your query. We sympathize with your position. However, unfortunately we do not have enough information to respond to your specific request to “establish the causality between Tamiflu and abnormal behaviours, sudden death or sequelae.” The following explains why we cannot address your specific requests, and offers some thoughts on possibly relevant information that we can speak to based on our Cochrane Review.
Our systematic Review published in 2014 did not conduct a statistical test on every type of adverse event that was recorded in the clinical trials that we studied. Doing so may have led to false positives and false negative as well, undermining the reliability of any associations that we might have otherwise been able to detect. Therefore, as we write in the Review, our approach was to meta‐analyze “(1) all serious adverse events; (2) all adverse events leading to study withdrawal; (3) all withdrawals; (4) all adverse events within a clinical study report’s defined body system; as well as (5) a small group of common adverse events as defined in the FDA drug label for oseltamivir.” (p.7).
Importantly, “There were too few events to meta‐analyse (1) deaths; (2) serious adverse events by body system; and (3) any events that had an overall incidence of less than 0.5%. We did not meta‐analyse outcomes with fewer than 10 events in total” (p.7).
“Abnormal behaviours”, “sudden death” or “sequelae” per se did not have an overall incidence of greater than 0.5%, and therefore we did not conduct statistical tests on them. Our Review however, did conduct statistical tests and make causal inferences regarding other adverse events that may be possibly relevant to your concerns.
We wrote, with respect to oseltamivir being used for the prevention of influenza, that “oseltamivir caused headaches and psychiatric harms in adult prophylaxis trials” (p.38).
The degree to which the risk of psychiatric harms is increased is somewhere between 1 additional psychiatric adverse event for every 36 to 1538 persons that received oseltamivir for the prevention of influenza (p.3).
While our Review “failed to identify a clear association between oseltamivir and psychiatric harms” (p.38) in clinical trials in which oseltamivir was being used for the treatment of influenza, this does not rule out the possibility that a causal relationship exists. It only states that we did not detect one. We wrote: “The question of why oseltamivir treatment trials failed to identify a clear association between oseltamivir and psychiatric harms, although a weak dose‐dependent association was observed, is a moot point. It is possible that influenza‐like illness and influenza symptoms masked the harms in those who were already symptomatic and therefore recruited in the treatment trials (and influenza‐type symptoms were excluded as adverse events to be reported). The reporting issue of compliharms may have helped to mask such events. Alternatively, it could be that these events are rare in the populations studied and that there was insufficient power to detect an association. The CI [confidence interval] was wide (0.43 to 2.03) and does not rule out a doubling in risk due to treatment ‐ as was found in the prophylaxis trials. It is also possible that the risk of psychiatric harm increases with increasing dose (as the data from trials WV15670 and WV15671 suggest) and increasing duration of treatment (as the prophylaxis trials suggest)” (p.38).
In our Review, we discuss other studies, including three prospective cohort studies in Japan, that you may wish to review (p.38), although we did not formally analyze these studies in our systematic review. A careful analysis of these studies, and other studies we did not mention in this reply, would be required to answer your question about the causal link between Tamiflu and deaths.
We hope the above is helpful in responding to your query.
Regards
Contributors
(in alphabetical order) Chris Del Mar Peter Doshi Rokuro Hama Carl Heneghan Jeremy Howick Mark Jones Kamal Mahtani David Nunan Igho Onakpoya Elizabeth Spencer Matthew Thompson
Feedback from Noritoshi Tanida, 14 September 2017
Summary
Dear authors of the Cochrane Acute Respiratory Infections Group, Neuraminidase Inhibitor Review Team.
I have learnt that the Association of Victims of Tamiflu‐related Encephalopathy requested the Cochrane team to establish causality between Tamiflu and abnormal/psychiatric behaviours, sudden death or serious sequelae.
I know the victim's serious situations; the applications for damage relief to the Pharmaceuticals and Medical Devices Agency (PMDA) were turned down and they lost cases in law courts either. Both PMDA and law courts took the same position, saying “there was no serious side effects in Tamiflu.”
Here, the importance of causality establishment is evident as to respond to the request by the victims’ group. I remember that Cochrane collaboration started upon responding lay peoples' query. Hence, I also request the Cochrane team to focus on this issue and to establish causality between Tamiflu and abnormal/psychiatric behaviours, sudden death or sequelae.
In revising the systematic review report considering the establishment of causality between Tamiflu and serious side effects, I hope that the Cochrane Acute Respiratory Infections Group, Neuraminidase Inhibitor Review Team will formally include analyses on events of “injury and poisoning” especially “injury” with Tamiflu treatment.
I believe that all criteria for causal inference by Hill/US surgeon General are satisfied for both abnormal behaviour and sudden death.
I thank you in advance. Sincerely Yours,
I do not have any affiliation with or involvement in any organisation with a financial interest in the subject matter of my comment Noritoshi Tanida (MD, PhD, DTM&H) Affiliation: Former Professor of Department of Medical Humanities, School of Medicine, Yamaguchi University, Japan.
Reply
Dear Professor Tanida,
Thank you for your query. We received a similar request from Ms. Ryuko Hatano on Aug 28, 2017, regarding “abnormal behaviours, sudden death or sequelae.”
As we explain in our response to that query, these events per se did not occur at a frequency greater than 0.5% and therefore we did not meta‐analyze it. The same is true of “injury and poisoning.” This also did not occur at a frequency greater than 0.5% and therefore we did not meta‐analyze it.
Please see our response to her query for more information on our methods and findings.
Regards
Contributors
(in alphabetical order) Chris Del Mar Peter Doshi Carl Heneghan Jeremy Howick Mark Jones Kamal Mahtani David Nunan Igho Onakpoya Elizabeth Spencer Matthew Thompson
What's new
| Date | Event | Description |
|---|---|---|
| 4 January 2018 | Amended | Removed Rokuro Hama's name from the Feedback reply to Noritoshi Tanida's comment, as it had been included by mistake. |
History
Protocol first published: Issue 1, 2011 Review first published: Issue 1, 2012
| Date | Event | Description |
|---|---|---|
| 20 December 2017 | Feedback has been incorporated | Two feedback comments and replies have been added to the review. |
| 27 March 2015 | Feedback has been incorporated | The authors' response has been posted on the CEU website https://editorial‐unit.cochrane.org/cochrane‐review‐neuraminidase‐inhibitors‐influenza |
| 14 January 2015 | Amended | We removed 'healthy' from the review title because the adjective 'healthy' incorrectly describes the review's population. Several included trials are in people with chronic respiratory problems and those in a general population have participants with pre‐existing pathologies as well as healthy people. |
| 1 December 2014 | Feedback has been incorporated | Link to Feedback incorporated. |
| 29 July 2014 | Feedback has been incorporated | Feedback comment and reply added. |
| 22 July 2013 | New search has been performed | Searches updated. We received the last clinical study report in September 2013. Since the first publication of this review in January 2012, we have completed the review of regulatory information that became available after our original time lock. We have assessed additional evidence from oseltamivir Module 2, evidence on adverse events following exposure to neuraminidase inhibitors and clinically relevant outcomes, and cross‐referenced this with the individual listings contained in Modules 3 to 5. We now hold all the relevant full clinical study reports, which we are making publicly accessible with this review. |
| 22 July 2013 | New citation required and conclusions have changed | In this update we found further evidence that the mode of action of oseltamivir is likely to be centrally mediated. We then carried out a toxicity assessment and found that oseltamivir has multi‐system toxicity (renal, neurological, psychiatric, metabolic, gastrointestinal and immune system). The effects of both drugs are modest. Evidence of oseltamivir toxicity is reasonably firm. Zanamivir toxicity is low. Neither drug should be used routinely. The beneficial effects on influenza viruses are slight or difficult to identify. |
| 15 May 2013 | Amended | Amendments to data analyses from oseltamivir trials Module 2; clinical outcomes and adverse events added in the 'Feedback' section and 'Published notes' sections |
| 14 May 2013 | Feedback has been incorporated | Feedback comment and reply added to the review |
| 8 March 2013 | Feedback has been incorporated | Feedback and reply added to the review |
| 28 January 2013 | Amended | New feedback comment and reply posted |
| 7 September 2012 | Amended | Conflict of interest statement updated for a feedback submitter |
| 9 February 2012 | Feedback has been incorporated | Feedback comments added to review |
| 4 May 2011 | Feedback has been incorporated | Feedback from three contributors has been added to the review |
Notes
Since the January 2012 version of A159, we have now completed the review of regulatory information which became available after our original time lock. We have assessed additional evidence from oseltamivir Module 2s, evidence on adverse events following exposure to NIs and clinically relevant outcomes, and cross‐referenced this with individual listings contained in Modules 3 to 5. We now hold all the relevant full clinical study reports, which we are making publicly accessible with this review.
Acknowledgements
Thanks to Jon Deeks, Timothy Aoki, Carlo Di Pietrantonj, Vittorio Demicheli, Janet Wale, John Bartlett, Sree Nair, Tom Fahey, Matthew Shun‐Shin, Anthony Harnden, Nigel Matheson, M Symmonds‐Abrahams and Aziz Sheikh, for input and advice on earlier versions of related reviews. Thanks to Ruth Foxlee, Alex Rivetti and Nia Roberts for helping out with the searches. Peter Collignon and Marcus Muellner helped us with aspects of the review. Thanks to Nicola Ring and Ruth Jepson for advice on the inclusion of qualitative data. We thank Toby Lasserson for providing advice and an independent check of our 'Risk of bias' judgements. The European Medicines Agency (EMA) (formerly EMEA) provided all clinical study reports and reviewers' comments in their archive. Hoffman‐La Roche SA and GlaxoSmithKline provided us with full clinical study reports and answered our queries. Thanks also to the Australian National Health and Medical Research Council (NHMRC) and the UK National Health Service (NHS) Research and Development fund for grants to enable the 2009 healthy adults review update. Philip Carter and Deborah Cohen shared some of their Freedom of Information material; Eliana Ferroni helped develop and cross‐check the TOC. Finally, we wish to thank the following people for commenting on the draft protocol: Maryann Napoli, Janet Wale, Paul Glasziou, David Boltz, Elaine Beller and Anca Zalmanovici Trestioreanu and Marcus Muellner. Thanks to the following people for commenting on the draft 2012 review: Chris Cates, Janet Wale, Paul Glasziou, David Boltz and Robert Ware and the following people for commenting on the draft 2014 review: Chris Cates, Elizabeth Dooley, Janet Wale, David Boltz and Robert Ware.
This project was funded by the NIHR Health Technology Assessment programme and will be published in full in the Health Technology Assessment journal series. Visit the HTA programme website for more details: http://www.nets.nihr.ac.uk/projects/hta/108001. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Department of Health. The National Institute of Health Research (NIHR) School of Primary Care Research (SPCR) provides financial support for Dr Carl Heneghan and funding for an investigators' meeting in Oxford (UK).
Appendices
Appendix 1. Glossary of terms used in this review
Public health drugs: Drugs in which a considerable quantity of public money has been invested and/or are on the WHO essential drugs list.
Clinical study reports: Detailed reports of a clinical trial usually submitted to regulators following a prescribed ICH format. Roche's follow a modular structure (see Appendix 11). Reports can be several hundred pages long and contain details of the planned design, conduct (protocol), analysis (reporting analysis plan or RAP) and results of the trial.
Compliharm: Term describing events defined as either complications or harms according to ambiguous criteria that appeared to include time of analysis (with times either unspecified or inconsistent among trials) and whether participants were infected (by influenza) or not. In oseltamivir treatment trials some potential harms or complications could both be caused by medication or influenza infection (e.g. vomiting), hence our classification as a compliharms.
Time lock: Date (12 April 2011) after which no documentation would be reviewed in the January 2012 version of the review. A cutoff was made necessary by the sheer scale of our data holdings. We were initially funded to review the full clinical study reports of the 10 treatment trials included in the Kaiser et al paper. We were able to access the 10 Module 1s and regulatory comments (approximately 6000 pages in total). As the funder‐stipulated deadline to producing our review progressively shortened and our understanding of the issues evolved we received notification that while the balance of the 10 study reports were unlikely to be accessible by our deadline, we would receive substantial quantities of regulatory documents from the EMA in four tranches. When we held our second face to face meeting in April 2011 we had just received our first tranche of clinical study reports consisting of just over 10 thousand pages, bringing our total holdings to 16,000 pages. We decided that we did not have the resources to review any further documentation within our current funding and imposed a data time lock. Any documentation received after this date would be reviewed if and when we had more resources. The balance of documents (a further 14,000 pages) are included in this review.
TOC: Table of content of regulatory reviews and comments on industry submissions. Our TOC indicates which trial is cited in which document on which page how many times.
TOCE: Annotated version of the TOC. Comments and annotation are preliminary and form the basis for the weaving of the important aspects into the review narrative. (See also Table 2; Table 3; Table 4; Table 5).
Trial ID: Means of identifying a trial. Usually made up of letters and numbers (WV 15799). At times the ID bears a letter suffix indicating the last version of the protocol followed in the trial (e.g. WV 15799H, i.e. trial carried out following amendment H).
Regulatory information: Term comprising clinical study reports (data) and regulatory comments and reviews.
Modules: Basic structure of Roche's trial reports see (Appendix 11). Today, the term 'Modules' refers to the components of a regulatory submission, as set by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) (ICH 2011). Clinical study reports are just one 'Module' of a regulatory submission.
FOI: Freedom of Information. Enshrined by law in the US and EMA policy in Europe. FOI requests in this review have been a means of access to clinical study reports and regulatory comments (regulatory information).
CONSORT‐based extraction: Extraction, synthesis and appraisal method used in this review for data from clinical study reports. Reconstructions were done by pairs of review authors and assessed in the authors' plenary session to decide whether included trials could proceed to stage 2 of the analysis. The structure of the reconstruction follows that of the CONSORT statement.
Protocol: Document reporting the trial's planned design and conduct, with amendments (when relevant). Confusingly also used in submissions and regulatory documents as synonymous with study.
IPD: Individual patient data. Anonymised individual data listings of characteristics and results, which form the basis for the synthetic analyses in clinical study reports.
Trial programme: Series of trials designed and carried out to achieve registration or to answer specific questions. Usually programmes of the same drug or intervention focus on the same indication or the same study population.
Reporting Analysis Plan (RAP): Plan of analysis usually linked to trial protocol explaining what and how the authors intend to analyse.
Japanese Summary Basis for Approval (of a drug) (JSBA): Summary of the application dossiers included as one of the documents prepared and attached by the sponsoring pharmaceutical company. These are submitted to the regulatory body for approval of a new drug.
Appendix 2. The story of A159
The 2009 review: from A047 to A159
In the midst of the A/H1N1 influenza outbreak in June 2009, the Australian and UK governments commissioned an update of our long‐standing Cochrane review on neuraminidase inhibitors (NIs) for influenza in (otherwise) healthy adults (known as A047). Prior to the emergence of influenza A/H1N1 in 2009, governments worldwide stockpiled nearly CHF 7.6 billion worth of oseltamivir (Jack 2009). The World Health Organization (WHO) considered antivirals for influenza important (WHO has recently added oseltamivir to the list of essential medicines (WHO 2011; WHO 2013a; WHO 2013b)). Oseltamivir and zanamivir have been prescribed for the treatment of influenza worldwide since the outbreak of 2009 A/H1N1 influenza. The review (on healthy adults) had first been published in 1999 (as A047) and was updated in 2006 and 2008. At the same time a similar review on children (as Shun‐Shin 2009 or A046) had also been published.
As the review had been updated the previous year, we initially anticipated that the commissioned 2009 update would not require substantial effort and likely reflect only updated pharmacovigilance data and not the incorporation of new trial evidence (Doshi 2009).
In the end, the 2009 update was inconclusive regarding whether or not oseltamivir reduced the risk of complications of influenza (Jefferson 2010a), as we were unable to verify the data underlying manufacturer and government claims to this effect. The claims were based on clinical trial evidence included in a published pooled analysis of 10 manufacturer‐funded clinical trials of oseltamivir for the treatment of influenza in people of 13 years and older (Kaiser 2003). Eight of the 10 trials in the Kaiser et al pooled analysis have never been published (Jefferson 2009a), and their complete data sets were not available from either the authors or the manufacturers. Some of the published trials had been ghost written (Cohen 2009). The largest of the 10 Kaiser trials (M76001), involving over 1400 people, had been briefly reported in conference abstract format but the person whose name appeared on the abstract could not recall ever presenting its results or being involved with the study (Cohen 2009). Our early requests for data from the 10 Kaiser studies were met with an offer from Roche to sign a confidentiality agreement with an embedded secrecy clause preventing us from mentioning the existence of the agreement. The unsigned agreement can be seen at http://www.bmj.com/highwire/filestream/440792/field_highwire_adjunct_files/0.
At the time of publication of the 2009 update and its linked investigation by the BMJ, we were unaware of the size and depth of the oseltamivir evidence development programme. We thought it comprised around 36 trials and we expected that only a proportion of these would fit our inclusion criteria. We also did not realise the size and the level of detail that the clinical study reports contained.
On 31 December 2009, Roche released the core reports (or Module 1s) of the 10 Kaiser trials with no legal agreement signed (only a web‐based agreement that we would not share the documents with other commercial companies). After requesting Roche to provide us with the full study reports, Roche said that the Module 1s were all that were needed for us to complete our job (Doshi 2012a). The missing Modules (or parts), numbered 3 to 4 according to the trial in question, contained protocols, amendments, individual listings and demographic information. It seemed to us that these documents would have contained some important additional material for understanding the trials, their design and interpretation but we were not sure.
In 2010 we started exploring the relationship between the available Module 1s for the only two Kaiser trials that had been published (trials WV15670 or Nicholson 2000 and WV15671 or Treanor 2000).
At about this time we started getting concerned that the oseltamivir trial programme was considerably larger than we first thought. Our interest was awakened by the casual discovery of a confidential 2009 Roche Tamiflu Investigator's Brochure, which was freely accessible on the web. This reported a clinical trial programme of over 60 studies. Searching for an unpublished and hitherto unseen data set requires constructing a reasonably accurate list of all studies of the drug in question. The Roche Investigator's Brochure did not mention some studies we were aware of, and reinforced an idea that we had become clear was essential: the need to develop our own list of trials, because a single, authoritative, up‐to‐date and complete list of all clinical trials conducted on humans using for both drugs did not seem to exist.
When thinking about our next update we decided not to use publications because the majority of treatment trial evidence for Tamiflu remained unpublished, because we had found some discrepancies between CSRs and published equivalents and, mostly, because clinical study reports were so much more detailed and comprehensive than short journal articles, enabling a more thorough critical analysis of the trials. We also decided to expand the scope of A047 by including evidence relating to people of all ages except for immune‐suppressed individuals. This, de facto, amounted to the creation of one new review (A159) with a new protocol subsuming A047 and A046. The protocol for A159 was publicly posted in December 2010.
A new source of evidence for A159
Today the obvious source of information on clinical study reports would be trial registries and company websites, but most trials of both NIs were carried out before inception or wide acceptance of centralised registries and company websites. In 2009‐11, company websites did not and still do not have extensive lists of trials with downloadable clinical study reports. Most people had never heard of clinical study reports before media coverage of our efforts.
We decided to construct our list by using multiple cross‐referencing methods. We constructed a list beginning with clinical trials identified from previous review updates. To this end, we added additional trials in humans from multiple sources, including manufacturer submissions to regulators, drug product information sheets, previous published reviews, Health Technology Assessment (HTA) documents and public and manufacturers' registers (Burch 2009; Cooper 2003; Jefferson 2006; Tappenden 2009; Turner 2003), such as www.ClinicalTrials.gov and www.roche‐trials.com. Regulatory documents also aided the identification of unknown trials. Finally, we also conducted traditional database and grey literature searches (Appendix 5) to identify previously unknown trials.
One of the first things we learned was that to ensure the list did not include duplicate entries, we had to assign to each trial a Unique Trial ID. 'Author' is not a good choice of Unique Trial ID, as different authors can be present across different versions of the same trial (that is, the authors of clinical study reports can be different from publications arising from the same clinical trial). Nor are any other details connected to publications a good option for Unique Trial ID because not all studies are published. Some trials will have company‐specific codes and some will have public clinical trial registry numbers, or both, or neither. To simplify recognition and terminology we used the manufacturer protocol ID as our Unique Trial ID.
Our list was only going to be useful if it had sufficient details to enable us to decide whether it met our inclusion criteria. For each Unique Trial ID, we gathered the following details.
Unique Trial ID
Other IDs
Phase of study
Sponsor
Short description
Official trial title
First authors (name and email)
Type of trial
Comparator
Outcomes assessed
Date of trial
Study period (days)
Population
Number of participants planned
Number of participants enrolled
Number of participants completing
Trial status (for example, completed, ongoing or early termination)
Publication status (a citation or understanding of why it was not published)
How identified (to record how the trial was discovered)
Notes
Once we had as complete a list of trials as possible, we contacted manufacturers and sent them our draft list, asking them to check the accuracy and completeness of our list. Roche, GlaxoSmithKline (GSK) and BioCryst all did so, and in doing so we learned of hitherto unknown trials.
Occasionally, the existence of further unknown trials was detected weeks and months after we thought we had a 'complete' list. This may be inevitable given that trial identification often takes place in unpredictable ways, for example while reading through detailed regulatory reports.
We engaged in prolonged correspondence with Roche and GSK and requested a series of regulatory documents under freedom of information policies from both the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). No substantial comments were made by Roche on the protocol of 159 which has been publicly available in one form or another since December 2010.
Specifically, we applied to EMA under their new release policy for 26 clinical study reports in their holdings relating to oseltamivir and GSK's zanamivir. The result was the delivery (starting in late March 2011) of 16 clinical study reports, all containing Modules 1 and 2 plus one complete report for oseltamivir (trial WP16263). (See table 1 in Doshi 2012b). None were available for zanamivir, as the EMA had not played a part in its market authorisation. These formed the basis for the 2012 version of A159.
At the date of completion of data searches for A159 (12 April 2011), Roche had only provided us with partial clinical study reports despite five requests for full clinical study reports. The material obtained from Roche included the first section (or so‐called 'Module 1' or 'Core Report') of a full clinical study report, each of which contain four to five Modules (Appendix 11) for the 10 oseltamivir treatment trials included in the Kaiser 2003 meta‐analysis. Not contained in the provided Module 1s are trial protocols with the list of amendments and original reporting analysis plans. These Module 1s comprise 3195 pages. Roche had not made available any further material and indicated it did not intend to answer our requests for clarification on aspects of trials and for availability of the missing parts of complete clinical study reports. In addition we had a 53‐page report in English of the treatment trial ML16369, sponsored by Shanghai Roche Pharmaceutical Ltd. Regardless of success with our requests to obtain full clinical study reports, we decided to update our review with available material and subsequently update it as and when additional data became available.
Our searches of publication databases did not add any significant information.
Following a change of policy at the EMA prompted by similar efforts of the Nordic Cochrane Centre (Gotzsche 2011), we received an additional eight clinical study reports (10,737 pages) in response to a freedom of information request. An additional 14,700 pages of further clinical study reports and 33 pages of regulators' comments arrived after our search deadline. All of the materials received from the EMA are related to oseltamivir. The EMA has no access to information for zanamivir, as it is a nationally authorised product in Europe (correspondence with Xavier Luria, 23 March 2011 and David Mackay 20 July 2011). At present we hold all Module 1s and 2 of oseltamivir trials we have requested. From GSK we have received the promise of individual patient data. Many of the clinical study reports used in this review were obtained via freedom of information requests.
We still await an FDA decision regarding similar requests sent to FDA in January 2011.
We were able to download 2673 pages of Summary Basis for Approval (SBA) documents from the FDA website, 31 from EMA and 508 pages of Japanese SBA. We indexed the content and then constructed an extended table of contents, giving a summary of each file, thereby facilitating navigation of these complex documents. Once the table of contents had been constructed, we postulated that given the huge work involved in reviewing lots of regulatory files, including clinical study reports, we needed new instruments to indicate which parts were more important than others, thus focusing our efforts. We experimented with a variety of methods reported in the earlier version of the review. We have now devised and used what for us is a satisfactory instrument for critically assessing clinical study reports in their entirety. We intend publishing this separately.
The EMA releases, coupled with documents from the FDA SBA for both drugs, formed the basis for the version of A159 that we eventually published in January 2012. The review reported our efforts to get to the bottom of the issue of the effects of NIs by appraising evidence from unpublished clinical study reports (see Glossary (Appendix 1) and regulatory documents containing comments and reviews. We called the body of clinical studies and regulatory comments 'regulatory information' as all of these documents are either created for or by regulators. To our knowledge this was the first Cochrane review ever conducted on the basis of regulatory documents only.
Due to funding timelines and the sizeable amount of regulatory information already available to us we decided to review material available as at 12 April 2011. This meant reviewing FDA SBA material and core reports (Module 1s) in our possession from EMA data releases received by this time.
Study selection and extraction in 2011
In 2011 our methods were a mixture of established and novel, reflecting the size of the task, our lack of experience in dealing with large amounts of very detailed regulatory information and the lack of a complete set of clinical study reports. For example, scanning of titles and abstracts was done in double, but selection of studies for inclusion from the list constructed during our search was done in quadruple with disagreements resolved by discussion.
For many studies we only had titles and in some cases a very brief description of content, thus we assigned three categories to our trials:
definitely included;
definitely excluded; and
trials for which we needed further information.
We excluded studies definitely not meeting the inclusion criteria on the basis of the available information (e.g. the title described the trial as a pharmacokinetic study). Where appropriate we requested further information from the trials' sponsor, usually copies of the clinical study reports (minus participant identification) for each trial that was definitely included or for which we needed further information. We did not contact first/corresponding authors of published versions of the trials on the basis of our experience with the 2009 review.
Data extraction and management reflected the lack of established methods for reviewing regulatory material. We subdivided the extraction, appraisal and analysis of the data into a two‐stage exercise, including studies in the analysis phase only if we judged their reports reliable and complete. To help structure the information we used CONSORT statement‐based extraction forms aimed at assembling a concise version of the clinical study reports, which included all important methods as well as defined and extracted all relevant outcomes. We colour‐coded the original text to flag up uncertainty or lack of clarity or need for more information from other (then inaccessible) parts of the clinical study report.
During this process we excluded all six peramivir trials as we were informed by the manufacturers that no clinical study reports would be available.The 2012 A159 review was based on 15 oseltamivir clinical study core reports and 10 very brief zanamivir study reports. The former came from EMA, while the latter had been part of a GSK submission to the UK's National Institute for Health and Care Excellence (NICE) which the BMJ had passed onto us together with the SBA material. Our 2010 protocol for A159 was not very detailed on how we would handle this mass of information for the simple reason that no one had ever done it before. We quite literally were learning as we were going along and our understanding of the structure and content of clinical study reports evolved. We created and tested five post‐protocol hypotheses (see Appendix 10), which had originated from the findings in the reports. The best example was our finding of an unnaturally high (up to 80%) influenza positivity rate in treatment trials, which in some cases had been pooled because of lack of viral circulation. We hypothesised that screening for influenza positivity had been carried out prior to enrolment of people with influenza‐like illness. This was not borne out by the evidence available to us. We know now that a far simpler explanation is more likely: careful selection of the time period for trial participant enrolment based on when surveillance data suggested high influenza activity led to the recruitment of small numbers of participants from each of many centres with a high likelihood of influenza positivity, but at the time we had limited information available. The 201
2 A159 review analyses were based on the ITT population, which we had found to be the only reliable analysis unit as oseltamivir appeared to have an effect on antibody production in people with influenza, leading to an imbalance in numbers of subjects in the influenza‐infected (so‐called ITTI) sub‐population. This in effect introduced confounding in what otherwise should have been well‐designed double‐blind trials. The only effect that was clearly identifiable was a modest shortening of influenza‐like illness symptoms by less than a day. This led us to believe that oseltamivir had an aspecific powerful effect on symptoms not mediated by any action on influenza viruses but possibly via an anti‐inflammatory mechanism. The trials had been clearly designed with a commercial focus in mind and some of the claims made by the manufacturer (especially on the effect on interruption of transmission and on pneumonia) were not supported by the evidence in our possession.
2012 to the present day
Soon after the publication of the review, the BMJ agreed to publish our correspondence with Roche, GSK, EMA, Centers for Disease Control and Prevention (CDC) and WHO, recording our attempts at retrieving the full reports without any conditions attached and to understand the basis for promotion of the drugs (especially oseltamivir) by public health bodies. The correspondence (which is hundreds of pages long) formed the basis for what then became the BMJ Open Data campaign and a stimulus for the later AllTrials campaign. Public exposure of our efforts and copious media coverage had the direct effect of ensuring the unconditional release of 77 reports of oseltamivir of 82 studies sponsored by Roche and the equivalent of the 30 studies we had requested from GSK. For the full correspondence see http://www.bmj.com/tamiflu and http://www.bmj.com/relenza. The reports (amounting to over 100,000 pages) are made available with this review for the first time at [insert URL when available], marking a small but significant victory for open science.
Before receiving the full reports, we resumed reviewing the remainder of the material we had received in 2011. This mainly consisted of Module 2s (Roche terminology for pre‐study documents). Module 2s contained the information originally denied to us by Roche: study protocols with their amendments, randomisation lists, blank case report forms (CRFs), certificates of analysis describing appearance and content of active and control capsules and, at times, statistical analysis plans (SAPs). CRFs are containers for the rawest form of recorded data at the individual participant level.
We had no tools for reviewing and synthesising this information, so again we had to create our own. The instrument is made up of three parts each with a separate function. In the first part there is a template for a brief summary description of the trial. The format fits into the RevMan Characteristics of Included Studies (CIST) table and is mainly descriptive. The second part is the appraisal of the trial following the Cochrane 'Risk of bias' format. The third part reconstructs the trial timeline and provides a checklist and position in the document of the various elements such as protocol, protocol amendments and study period with dates. The last part aims to check the internal consistency and coherence of each element (such as numbers screened, numbers randomised, interventions, comparators) across all the different documents, starting from the earliest available version of the protocol. The form also contains suggestions on where to look for the information. This is based on our experience gained in this review and in a descriptive review of 78 clinical study reports of 14 different drugs and biologicals (Doshi 2013).
While designing the tool we also asked ourselves whether access to Module 2 information (and later the full study reports) changed our perception of the trial and specifically our 'Risk of bias' assessment. We found that access to what are supposed to be full study reports should provide clarity and remove the rationale for 'unclear' risk of bias judgements and ideally remove the concept of risk leaving just 'bias', at least for certain study design elements such as attrition bias. Either a design element introduces bias or it does not. In the case of the 15 full oseltamivir clinical study reports we reviewed when constructing our tool, only one contained a protocol which predated the beginning of participant enrolment, only two had statistical analysis plans that clearly predated participants' enrolment and three had clearly dated protocol amendments. No clinical study report reported a clear date of unblinding.
During the latter part of 2013, we received from the manufacturers tens of thousands of pages of full clinical study reports for both programmes combined.
The history and conclusions form the backdrop to this version of A159.
Oseltamivir studies received from Hoffman La Roche SA in CSR format:
PV15616 (= GS 97‐801)
PV15615 (= GS 97‐802)
ML17279 – publication only
ML19340 – publication only
JV15823 – English translation of Gaiyo summary
JV15824 – English translation of Gaiyo summary
ML17279 (=WV17052)
ML19340 (= COSMOS Study)
Zanamivir studies received from GSK in CSR format:
Appendix 3. Compliharms: events alternatively recorded as complications or harms
Roche clinical study report of oseltamivir treatment trial: "The following symptoms, signs and common sequelae associated with influenza were excluded from specific adverse event reporting if they occurred during the period of drug treatment provided their appearance was in conjunction with one or more other influenza‐related symptoms. The recrudescence of single discrete signs/symptoms associated with influenza syndrome were recorded as adverse events."
[Event by body system]
Respiratory Cough Pneumonia Bronchitis/tracheitis Sinusitis Dyspnoea/difficulty breathing
Cardiovascular Tachycardia
Eyes, ears, nose and throat Sore throat Nasal obstruction Earache Otitis Coryza Conjunctivitis
Central nervous system Headache Fatigue
Musculoskeletal Myalgia
Other Fever Rigor Malaise/asthenia Chills
Source: "Appendix 1. Events Associated with Influenza Syndrome". Roche Clinical Study Report No. W‐144117, Protocol WV15707, Module I‐43
A 1999 FDA medical review of oseltamivir: "As symptoms and common sequelae of influenza were collected as endpoint data, these symptoms, signs and common complications were specifically excluded from reporting as adverse events. The following table [above] lists events associated with influenza syndrome which were excluded from adverse event reporting. … In addition, following the alleviation of influenza‐like symptoms, the recurrence of a single respiratory or constitutional symptom was recorded as an adverse event; however, the reappearance of more than one symptom was recorded as influenza‐like syndrome (i.e. secondary illness). Comment: As the applicant [Hoffman‐La Roche] stated in a written response dated 6/11/99, some sites incorrectly reported symptoms occurring prior to the cessation of the primary illness as secondary illness."
Emphasis in the original. Oseltamivir Medical Review. US FDA Center for Drug Evaluation and Research, Application No. 021087, 25 October 1999, page 15. www.accessdata.fda.gov/drugsatfda_docs/nda/99/21087_Tamiflu_medr_P1.pdf
Appendix 4. Searches for clinical study reports
Searching an unpublished and hitherto unseen data set requires constructing a reasonably accurate list of all studies of the drug in question. The obvious source of such information would be trial registries but most trials of both NIs were carried out before inception or wide acceptance of centralised registries. As single, authoritative, up‐to‐date and complete lists of all clinical trials conducted on humans using a given drug are rarely available in the public domain, there was no alternative to constructing our own. We decided to do so by using multiple, cross‐referencing methods. We constructed a list beginning with clinical trials identified from previous review updates. To this end, we added additional trials in humans from multiple sources, including manufacturer submissions to regulators, drug product information sheets, previous published reviews, Health Technology Assessment (HTA) documents and public and manufacturers' registers (Burch 2009; Cooper 2003; Jefferson 2006; Tappenden 2009; Turner 2003), such as www.ClinicalTrials.gov and www.roche‐trials.com. Regulatory documents also aided the identification of unknown trials (see also Searching other resources). Finally, we also conducted traditional database searches (Appendix 5) and searches of grey literature to identify previously unknown trials.
To ensure the list did not include duplicate entries, we assigned each trial a Unique Trial ID. 'Author' is not a good choice of Unique Trial ID, as different authors can be present across different versions of the same trial (that is, the authors of clinical study reports can be different from publications arising from the same clinical trial). Nor are any other details connected to publications a good option for Unique Trial ID because not all studies are published. Some trials will have company‐specific codes and some will have public clinical trial registry numbers, or both or neither.
The majority of trials cited in this review are manufacturer‐funded (with corresponding manufacturer protocol IDs) and to simplify recognition and terminology we have used the manufacturer protocol ID as our Unique Trial ID.
A list is only helpful so long as it has sufficient details to enable us to decide whether it meets our inclusion criteria. For each Unique Trial ID, we gathered the following details.
Unique Trial ID
Other IDs
Phase of study
Sponsor
Short description
Official trial title
First authors (name and email)
Type of trial
Comparator
Outcomes assessed
Date of trial
Study period (days)
Population
Number of participants planned
Number of participants enrolled
Number of participants completing
Trial status (for example, completed, ongoing or early termination)
Publication status (a citation or understanding of why it was not published)
How identified (to record how the trial was discovered)
Notes
Once we had as complete a list of trials as possible, we contacted manufacturers and sent them our draft list, asking them to check accuracy and completeness of our list. Roche, GSK and BioCryst all did so, and in doing so we learned of hitherto unknown trials.
Occasionally, the existence of other hitherto unknown trials was detected weeks and months after we thought we had a 'complete' list. We feel this is inevitable given that trial identification often takes place in unpredictable ways, for example while reading through detailed regulatory reports. We engaged in prolonged correspondence with both manufacturers and requested a series of regulatory documents under FOI law from both the FDA and EMA.
Appendix 5. Searches of the electronic databases
Although this review focuses on the primary data sources of manufacturers, to check that there were no published randomised controlled trials (RCTs) from non‐manufacturer sources, we ran electronic searches in the following databases:
the Cochrane Central Register of Controlled Trials (CENTRAL 2013, Issue 6) limited to year published 2010‐2013 (20 search results);
MEDLINE (January 2011 to July week 2, 2013) (56 search results) and MEDLINE (Ovid) from 1 January 2011 to July week 2, 2013 (56 search results);
EMBASE (January 2011 to July 2013) (90 search results) and Embase.com from 1 January 2011 to July 2013 (90 search results);
PubMed (NOT MEDLINE) no date limit (21 records). We searched PubMed to identify publisher submitted records that will never be indexed in MEDLINE and the most recently added records not yet indexed in MEDLINE.
To identify reviews that may possibly have referenced further trials we searched:
the Database of Reviews of Effect (DARE) (2013 Issue 2 of 4 April) (four search results);
the NHS Economic Evaluation Database (NHSEED) Issue 2 of 4 April 2013 (two search results) ‐ both resources are part of The Cochrane Library, www.thecochranelibrary.com (accessed 22 July 2013);
the Health Economic Evaluations Database (HEED) (searched 22 July 2013) (three search results).
Previously we had searched the Cochrane Central Register of Controlled Trials (CENTRAL) (eight search results); MEDLINE (Ovid) from 1 May 2009 to 12 April 2011 (31 search results); EMBASE from 1 January 2010 to 12 April 2011 (54 search results); DARE (five search results) and NHSEED (five search results). CENTRAL, DARE and NHSEED are part of The Cochrane Library, www.thecochranelibrary.com (Issue 2, 2011, accessed 1 June 2011). All search results were loaded to an electronic library (EndNote).
We used the following search strategy to search MEDLINE and CENTRAL. We combined the MEDLINE search with the Cochrane Highly Sensitive Search Strategy for identifying randomised trials in MEDLINE: sensitivity‐ and precision‐maximising version (2008 revision); Ovid format (Lefebvre 2011). We adapted the search strategy for EMBASE. We imposed no publication or language restrictions.
MEDLINE (Ovid)
1 Influenza, Human/ 2 exp Influenzavirus A/ 3 exp Influenzavirus B/ 4 (influenza* or flu).tw. 5 or/1‐4 6 Oseltamivir/ 7 Zanamivir/ 8 neuraminidase inhibitor*.tw. 9 (oseltamivir or zanamivir or tamiflu or relenza or peramivir or gs4071).tw,nm. 10 or/6‐9 11 5 and 10
EMBASE.com
17 #13 AND #16 16 #14 OR #15 833616 15 random*:ab,ti OR placebo*:ab,ti OR factorial*:ab,ti OR crossover*:ab,ti OR 'cross over':ab,ti OR 'cross‐over':ab,ti OR volunteer*:ab,ti OR assign*:ab,ti OR allocat*:ab,ti OR ((singl* OR doubl*) NEAR/1 blind*):ab,ti 14 'randomised controlled trial'/exp OR 'single blind procedure'/exp OR 'double blind procedure'/exp OR 'crossover procedure'/exp 13 #4 AND #12 12 #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 11 oseltamivir:ab,ti OR zanamivir:ab,ti OR tamiflu:ab,ti OR relenza:ab,ti OR peramivir:ab,ti OR laninamivir:ab,ti OR gs4071:ab,ti 10 'sialidase inhibitor':ab,ti OR 'sialidase inhibitors':ab,ti 9 'neuraminidase inhibitor':ab,ti OR 'neuraminidase inhibitors':ab,ti 8 'sialidase inhibitor'/exp 7 'peramivir'/de 6 'zanamivir'/de 5 'oseltamivir'/de 4 #1 OR #2 OR #3 3 influenza*:ab,ti OR flu:ab,ti 2 'influenza virus a'/exp OR 'influenza virus b'/de 1 'influenza'/exp
Appendix 6. Searches for regulatory information
We searched the following sources:
the US Food and Drug Administration (FDA);
the European Medicines Agency (EMA) (formerly EMEA);
Roche;
the Japanese regulator (Pharmaceuticals and Medical Devices Agency (PMDA)) Summary Basis for Approval (SBA).
We conducted a search of the FDA regulatory documentation of the New Drug Applications (NDA) and supplementary New Drug Applications (sNDA) of both drugs (FDA 2011b). The FDA NDA documentation includes medical, statistical, microbiological and other reviews, product labels, reports of site inspections, meetings with manufacturers and records of the decision‐making leading to registration and post‐marketing requirements. We also searched 'Warning Letters' dispatched by the FDA (FDA 2011c).
To organise receipt of FDA materials, we created a Table of Contents (TOC) listing all the regulatory and pharmaceuticals documents accessible to us. The TOC's function was that of an index, searchable quick reference guide and research tool to enable us to carry out quantitative (e.g. citation density analysis) and qualitative analyses (e.g. theme summaries) of the content. We also needed a rapid aide memoir with brief summaries of the evidence contained in each regulatory document listed in the TOC. We called this aide memoir the TOCE (Table of Contents ‐ Evidence). As the TOCE contains copious working personal notes aimed to understand the regulatory narrative, we have not reproduced it here but its content is woven into the narrative of this review.
Due to the length and format of regulatory documents, we realised in building the TOC that there was a need to formalise the search and identification methods of trials referenced in the FDA documentation. We concentrated on where each trial is mentioned in the documentation by its pharmaceutical code. So, for example, if trial WV15670 is mentioned 60 times by that code in a particular file, then the TOC will report the page numbers in which it is cited, which could be any number up to 60. The unit of search was the file, as a FDA PDF file can contain many different types of documents scanned into the same file. TOC and TOCE are among the tools we specifically constructed for the review (Appendix 1).
We wanted to validate our new methods, therefore we compared the yield of Optical Character Recognition (OCR) searching and handsearching of the PDF files of the FDA regulatory material using the same trial ID as a working example.
We also searched the material sent to us by Roche for our 2009 update.
We searched the website of the Japanese PMDA (http://www.info.pmda.go.jp/shinyaku/shinyaku_previous_index.html) for data relating to NIs approved in 1999 and 2000 and http://www.info.pmda.go.jp/approvalSrch/PharmacySrchInit for NIs approved since 2001. We identified 1575 pages of documents relating to the regulatory review by the PMDA and the Japanese Ministry of Health, Labor and Welfare (JMHLW) and the Japanese SBA of oseltamivir capsules for treatment (2000), and prophylaxis of oseltamivir dry syrup for children (2002) and oseltamivir capsules for prophylaxis of influenza (2004) and their re‐examination results. The Japanese regulatory body introduced a system to disclose their examination results and SBA in 1999 instead of the prior system, 'full disclosure requirement system', which had been introduced in 1967. Although these documents included preclinical, methodological, clinical (pharmacological, toxicity and pharmacokinetics with metabolism) data and clinical (phase I to phase III) studies and contain more precise data than the published papers, no complete clinical study reports were publicly available. Therefore, one review author (RH) asked the JMHLW on 29 July 2010 to disclose all documents reporting the evidence base for the approval of oseltamivir for these indications. The JMHLW sent RH a letter of refusal dated 2 September 2010, with the explanation "because the disclosure of such documents might hurt the right, position or other fair benefit in the competition of the corporation concerned". We waited six months to take further action hoping that the required clinical study reports would be forthcoming from the manufacturers. When this did not happen, RH filed a suit to overturn the JMHLW decision with the Osaka (Japan) District Court on 28 February 2011. The District Court petition was rejected on 19 April 2013 and the Osaka High Court rejected it on 29 November 2013. No appeal to the Supreme Court was made because substantial clinical study reports had already disclosed from various sources.
Appendix 7. Modified CONSORT statement‐based extraction template for clinical study reports
|
Title and drug name Include source documents used: | ||
| Modified CONSORT extraction template http://www.consort‐statement.org/ | ||
| Introduction CONSORT number | ||
| Background and objectives | 2a | Scientific background and explanation of rationale |
| 2b | Specific objectives or hypotheses | |
| Insert text: | ||
| Methods | ||
| Trial design | 3a | Description of trial design (such as parallel, factorial) including allocation ratio |
| 3b | Important changes to methods after trial commencement (such as eligibility criteria), with reasons | |
| Insert text: | ||
| Participants | 4a | Eligibility criteria for participants |
| 4b | Settings and locations where the data were collected | |
| Insert text: | ||
| Interventions | 5 | The interventions for each group with sufficient details to allow replication, including how and when they were actually administered |
| Insert text: | ||
| Outcomes | 6a | Completely defined prespecified primary and secondary outcome measures, including how and when they were assessed |
| 6b | Any changes to trial outcomes after the trial commenced, with reasons | |
| Insert text: | ||
| Sample size | 7a | How sample size was determined |
| 7b | When applicable, explanation of any interim analyses and stopping guidelines | |
| Randomisation: | ||
| Sequence generation | 8a | Method used to generate the random allocation sequence |
| 8b | Type of randomisation; details of any restriction (such as blocking and block size) | |
| Allocation concealment mechanism | 9 | Mechanism used to implement the random allocation sequence (such as sequentially numbered containers), describing any steps taken to conceal the sequence until interventions were assigned |
| Implementation | 10 | Who generated the random allocation sequence, who enrolled participants and who assigned participants to interventions |
| Insert text: | ||
| Blinding | 11a | If done, who was blinded after assignment to interventions (for example, participants, care providers, those assessing outcomes) and how |
| 11b | If relevant, description of the similarity of interventions | |
| Statistical methods | 12a | Statistical methods used to compare groups for primary and secondary outcomes |
| 12b | Methods for additional analyses, such as subgroup analyses and adjusted analyses | |
| Insert text: | ||
| Results | ||
| Participant flow (a diagram is strongly recommended) | 13a | For each group, the numbers of participants who were randomly assigned, received intended treatment and were analysed for the primary outcome |
| 13b | For each group, losses and exclusions after randomisation, together with reasons | |
| Recruitment | 14a | Dates defining the periods of recruitment and follow‐up |
| 14b | Why the trial ended or was stopped | |
| Insert text: | ||
| Baseline data | 15 | A table showing baseline demographic and clinical characteristics for each group |
| Numbers analysed | 16 | For each group, number of participants (denominator) included in each analysis and whether the analysis was by original assigned groups |
| Insert text: | ||
| Outcomes and estimation | 17a | For each primary and secondary outcome, results for each group and the estimated effect size and its precision (such as 95% confidence interval) |
| 17b | For binary outcomes, presentation of both absolute and relative effect sizes is recommended | |
| Insert text: | ||
| Ancillary analyses | 18 | Results of any other analyses performed, including subgroup analyses and adjusted analyses, distinguishing prespecified from exploratory |
| Insert text: | ||
| Harms | 19 | All important harms or unintended effects in each group (for specific guidance see CONSORT for harms) |
| Insert text: | ||
| Other information | ||
| Registration | 23 | Registration number and name of trial registry |
| Protocol | 24 | Where the full trial protocol can be accessed, if available |
| Funding | 25 | Sources of funding and other support (such as supply of drugs), role of funders |
| Insert text: | ||
| First author Date of completion Conflicts of interest Second author check Date of check Conflicts of interest | ||
Appendix 8. Stage 1 of the 2012 A159 review
Two review authors assessed each study (with studies allocated randomly to three pairs of review authors). The lists of included studies (33 for oseltamivir, 30 for zanamivir, six for peramivir) were randomly created by the program Edgar II (Brown 2011). Every study was openly allocated to each group according to its number.
We initially included six peramivir trials in the randomisation/allocation sequence but subsequently decided not to proceed further, as we were informed by the manufacturers that no clinical study reports would be available until after registration with the US Food and Drug Administration (FDA) (correspondence with Bill Sheridan, 20 August 2010). One review author (TJ) was assigned to the attempted reconstruction of clinical study reports from the FDA documents.
Two weeks before 'time lock' (see Glossary in Appendix 1) we received the first batch of clinical study reports from the EMA (formerly EMEA), containing an additional four clinical study reports (including one complete four‐module clinical study report) of studies we wanted to include. This time random allocation was achieved by writing trial IDs on one set of tickets and asking an external researcher to allocate them to groups, the names of which had been written on another set of tickets.
Authors in pairs separately extracted data from the same clinical study reports of studies included in Stage 1 of the review. When we had more than one copy of the same clinical study reports from different sources (for example, clinical study reports submitted to a regulatory body and clinical study reports from a pharmaceutical company), we independently extracted data from each of the copies and then compared the results. We aimed to record and tabulate disagreements between data extracted from the same source and between different sources. We extracted data using a modified CONSORT statement‐based extraction template (Appendix 7).
The modified CONSORT‐based extraction template aimed to assemble a concise version of the clinical study reports, which would include all important methods as well as define and extract all relevant outcomes. The CONSORT‐based extraction template includes the features that would be expected to be found in a published trial report but in far greater detail. Our reconstructions do not include introduction or discussion sections. We extracted the following for each trial.
Background and objectives.
Methods: including trial design, important changes to methods after trial commencement (such as eligibility criteria), with reasons.
Participants: including eligibility criteria for participants and settings and locations where the data were collected.
Interventions: the interventions for each group with sufficient details to allow replication, including how and when they were actually administered.
Outcomes: prespecified primary and secondary outcome measures, including how and when they were assessed and changes to trial outcomes after the trial commenced, with reasons.
Sample size: how it was determined and explanation of any interim analyses and stopping guidelines.
Randomisation: including sequence generation and method used to generate the random allocation sequence.
Blinding: who was blinded after assignment to treatment groups.
Statistical methods: methods used to compare groups for primary and secondary outcomes and methods for additional analyses, such as subgroup analyses and adjusted analyses.
Results: participant flow, numbers of participants randomly assigned, losses and exclusions after randomisation, together with reasons. Baseline demographic and clinical characteristics for each group.
Outcomes: primary and secondary outcome results for each group.
Ancillary analyses: results of any other analyses performed, including subgroup analyses and adjusted analyses, distinguishing prespecified from exploratory.
Harms: all important harms or unintended effects in each group.
One review author completed the CONSORT‐based extraction on the template in full (Appendix 7), with the name and date of completion and a statement of conflict of interests. A second review author checked the extraction. We extracted data, text, tables and figures directly from the relevant sections of the clinical study reports into the appropriate section of the template. We did not change the text in any way apart from clarifying abbreviations or spellings, but we highlighted some text. We used three types of text highlighting in the document.
Yellow: where text, figures or tables need to be checked with further information (for example, if an adverse event is referred to in appendices or a further clinical study reports Module). Red: where text or comments were inserted by one or both review authors but required an additional opinion due to concerns that there is the potential for discrepancies in the clinical study reports. Green: any text or tables added by us to the template (for example, a reconstructed table of adverse events).
Two review authors (CH, MT) independently piloted the reconstruction method on oseltamivir trial WV15671 with data from Module 1 of the clinical study report from Roche and data submitted to the UK National Institute for Health and Care Excellence (NICE). We discussed the pilot reconstruction amongst the whole review team for clarification. At a face‐to‐face meeting we discussed the reliability and completeness of each reconstructed trial in the light of comments and other information from regulatory sources with a view to inclusion of the trial in Stage 2. We resolved all differences in opinion by consensus. We reached decisions on whether a trial moved to Stage 2 by consensus. We planned to record dissent when consensus was not possible.
Appendix 9. Applying inclusion criteria for the A159 2012 review
For the 2012 A159 review two review authors (CDM, MT) independently scanned the titles and abstracts identified from the searches of the published literature. None of the identified items were published versions of trials unknown to us. Four review authors (TJ, CH, MJ, RH) independently read all data relating to the studies on the list constructed during our search and selected studies that seemingly fulfilled our inclusion criteria. One review author (PD) compiled the assessments into a single sheet for another review author (CDM). One review author (CDM) resolved disagreements by discussion.
We assigned three categories to identified trials from our complete list:
definitely included;
definitely excluded; and
trials for which we needed further information.
We excluded studies definitely not meeting inclusion criteria on the basis of available information (e.g. the title described the trial as a pharmacokinetic study). Where appropriate we requested further information from the trials' sponsor, usually copies of the clinical study reports (minus participant identification) for each trial that was definitely included or for which we needed further information. We did not contact first/corresponding authors of published versions of the trials on the basis of our experience with the 2009 A047 review.
Appendix 10. Post‐protocol hypotheses ‐ methods and results
This text is carried over from the 2012 version of this review and is provided for record completeness
Methods
The hypotheses (expressed as null hypotheses) are listed below, in order of their generation (not necessarily of importance). Their rationale is explained further down the text.
Hypothesis 1. Incidence of certain harms is not associated with placebo content. Hypothesis 2. Oseltamivir (or zanamivir) does not affect antibody production in treatment trials. Hypothesis 3. Oseltamivir does not affect antibody production in post‐exposure (or secondary prophylaxis) trials. Hypothesis 4. The number of trial centres and centre withdrawals does not affect the proportion of placebo patients subsequently diagnosed with influenza infection (originally the outcome was effect size). Hypothesis 5. In oseltamivir treatment trials there is no association between the order of randomisations and naso‐pharyngeal swabbing (i.e. randomising participants first and then swabbing or swabbing first and then randomising) and the proportion of placebo patients subsequently diagnosed with influenza infection.
Hypothesis 1. Incidence of certain harms is not associated with placebo content. Rationale. While reviewing the US Food and Drug Administration (FDA) critique of zanamivir, we noted the regulators' concern over the apparent drop in forced expiratory volume (FEV) following zanamivir inhalation (FDA 1999a), which appeared to be enhanced by the lactose powder excipient content of the active blister (FDA 1999b). The powder, which causes bronchospasm in susceptible individuals, was contained in both the active and the placebo blisters. This principle of using a matching placebo is of course correct but may have had the effect of increasing the incidence of bronchospasm (or asthma‐related episodes) in both arms. This is clearly reported as a warning in the 1999 FDA label "Because the placebo consisted of inhaled lactose powder, which is also the vehicle for the active drug, some adverse events occurring at similar frequencies in different treatment groups could be related to lactose vehicle inhalation" (FDA 2000b p.10).
We reasoned by analogy and reviewed the medication content of the available clinical study reports of oseltamivir trials. The detailed information comparing content and physical characteristics and batch numbers is in Table 22. Roche's use of the word 'matching' is not strictly correct as two principles present in the placebo capsules (dehydrocholic acid and dibasic calcium phosphate dihydrate) are not listed as being present in the active oseltamivir capsules. We could not locate the reason for such a choice in the clinical study reports but both substances may have gastrointestinal action if consumed in large enough quantities.
17. Oseltamivir and placebo intervention contents by trial.
| Trial ID | Description oseltamivir/batch no. | Description placebo/batch no. |
Certified content (oseltamivir) |
Certified content (placebo) |
Ref (PDF page number) |
| M76001 | Size 2 capsules containing 75 mg oseltamivir/V01‐00 (GS 4104), batch number GMZ 0082 | Size 2 placebo capsules for oseltamivir/V02‐00 (GS 4104), batch number GMZ 0083 | Unknown (certificate of analysis not provided) | Unknown (certificate of analysis not provided) | 20 |
| WP16263 | Grey opaque body, light yellow opaque cap/PT2247C01 | Grey opaque body, ivory opaque cap/GMZ 0163 |
Oseltamivir 97.5 mg | Dehydrocholic acid | 19 and 422 |
| WV15670 | Size 2 capsules containing 75 mg Ro 64‐0796/V01‐00 (GS 4104), batch number GMZ 0067; caramel opaque body, caramel opaque cap | Size 2 placebo capsules for Ro 64‐0796/V02‐00 (GS 4104), batch number GMZ 0066; caramel opaque body, caramel opaque cap | Oseltamivir 94.3 mg | Dehydrocholic acid 6.13 mg | 13, 834‐5 |
| WV15671 | Capsules (size 2) containing 75 mg Ro 64‐0796 (GS 4104)/V01; batch number GMZ 0067/GMZ 0065. Capsules are caramel opaque body, caramel opaque cap. Also used batch GMZ 0067 capsules caramel opaque body, caramel opaque cap (for oseltamivir 94.3 mg) | Matching placebo capsules (size 2) for Ro 64‐0796 (GS 4104)/V02; batch number GMZ 0066. Capsules are caramel opaque body, caramel opaque cap | Oseltamivir 93.1 mg and oseltamivir 94.3 mg | Dehydrocholic acid 6.13 mg | 13, 764‐7 |
| WV15673/WV15697 | GS 4104 (Ro 64‐0796) provided as size 2 caramel‐coloured capsules containing 75 mg of active drug and packaging material consisting of dehydrocholic acid, dibasic calcium phosphate dihydrate, pregelatinised starch, povidone, talc and sodium stearyl fumarate." Ro 64‐0796/V01‐00 batch GMZ 0067; caramel opaque body, caramel opaque cap | Placebo provided as size 2 caramel‐coloured capsules, containing dehydrocholic acid, dibasic calcium phosphate dihydrate, pregelatinised starch, povidone, talc and sodium stearyl fumarate Ro64‐0796/V02‐00 batch GMZ 0066; caramel opaque body, caramel opaque cap | 94.3 mg | Dehydrocholic acid 6.13 mg | 385, 540‐2 |
| WV15707 | Ro 64‐0796 was provided as a size 2 capsule containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. Ro 64‐0796 (GS4104)/V01‐00 batch number GMZ 0082 | Placebo was provided as a size 2 capsule containing dehydrocholic acid, dibasic calcium phosphate dihydrate and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. Placebo Ro 64‐0796/V02‐00 batch number GMZ 0066 | Ro 64‐0796/002 100.5 mg | Dehydrocholic acid | 3, 517‐9 |
| WV15708 | Size 2 capsules of 75 mg; Ro 64‐0796/V01‐00 batch no. GMZ 0082; caramel opaque body, caramel opaque cap | Matching size 2 placebo capsules Ro 64‐0796/V02‐00 batch no. GMZ 0083; caramel opaque body, caramel opaque cap | Oseltamivir 100.5 mg | Dehydrocholic acid | 21‐2, 517‐9 |
| WV15730 | Ro 64‐0796 was provided as a caramel, opaque, size 2 capsule containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. Ro 64‐0796 (GS4104)/V01‐00 batch number GMZ 0082 | Placebo was provided as a caramel, opaque, size 2 capsule containing dehydrocholic acid, dibasic (calcium phosphate dihydrate and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. Placebo Ro 64‐0796/V02‐00 batch number GMZ 0083 | Oseltamivir 100.5 mg | Dehydrocholic acid | 24, 504‐5 |
| WV15758 | 2 batches of the paediatric formulation were used in the present study: 1. Ro 64‐0796/V20‐01 (0.6% syrup); batch no. G HK 0180/05 2. Ro 64‐0796/V20‐01 (0.6% syrup); batch no. G HK 0180/06 |
2 batches of the corresponding placebo formulation were used: 1. Ro 64‐0796/V19‐01; batch no. G HK 0179/04 2. Ro 64‐0796/V19‐01; batch no. G HK 0179/05 |
Oseltamivir 0.768 g (G HK 0180/05), 0.763 g (G HK 0180/06) | Dehydrocholic acid | 27, 1043‐5 |
| WV15759/15871 | Ro 64‐0796 was to be provided as a dry powder for reconstitution with water. The powdered formulation contains the active ingredient, sorbitol and saccharin sodium (sweeteners), betacarotene (colouring agent), permageal 31 tutti frutti (flavour), cellulose, xanthan gum and methylhydroxy/propylhydroxybenzoate | Unknown (certificates of analysis not in our possession) | Unknown (certificates of analysis not in our possession) | 36 | |
| WV15799 | Ro 64‐0796 was provided as ivory, opaque, size 2 capsule containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. Ro 64‐0796 (GS4104)/V14‐00 batch numbers GMZ 0124/03 and GMZ 0129/03 | Placebo was provided as an ivory, opaque, size 2 capsule containing dehydrocholic acid, dibasic calcium phosphate dihydrate and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. Placebo Ro 64‐0796/V16‐00 batch number GMZ 0136 | Unknown (certificates of analysis not in our possession) | Unknown (certificates of analysis not in our possession) | 24 |
| WV15812/WV15872 | Ro 64‐0976 was provided as size 2 capsules containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate | Matching placebo was provided as size 2 capsules containing dehydrocholic acid, dibasic calcium phosphate dihydrate, pregelatinised starch, povidone, talc, sodium stearyl fumarate | Unknown (certificate of analysis not provided) | Unknown (certificate of analysis not provided) | 18 |
| WV15876/WV15819/WV15978 | Capsules (size 2) containing 95.8 mg oseltamivir phosphate, equivalent to 75 mg oseltamivir: formulation V14; batch numbers GMZ 0124/03, GMZ 0129/03. Both batches: grey opaque body, ivory opaque cap The following statement appears after the description of the placebo; whether it applies to oseltamivir capsules is unclear: "Excipients for each capsule consisted of dehydrocholic acid, dibasic calcium diphosphate dihydrate, pregelatinized starch, povidone, talc, sodium stearyl fumarate." |
Matching placebo capsules (size 2) for oseltamivir: formulation V16; batch numbers GMZ 0136, GMZ 0163. Both batches: grey opaque body, ivory opaque cap The following statement appears after the description of the placebo; whether it applies to oseltamivir capsules is unclear: "Excipients for each capsule consisted of dehydrocholic acid, dibasic calcium diphosphate dihydrate, pregelatinized starch, povidone, talc, sodium stearyl fumarate." |
Oseltamivir 95.7 mg (GMZ 0124/03); 97.9 mg (GMZ 0129/03) | Dehydrocholic acid | 21, 944‐9 |
| WV16277 | Oseltamivir was provided as size 2 capsules containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate.. Oseltamivir: 75 mg capsules, Ro 64‐0796/V14, batch number PT2247C01 Capsules are caramel opaque body, caramel opaque cap |
Matching placebo was provided as size 2 capsules, containing dehydrocholic acid, dibasic calcium phosphate dihydrate, pregelatinised starch, povidone, talc, sodium stearyl fumarate Matching placebo capsules: Ro 64‐0796/V16, batch number GMZ 0163. Capsules are caramel opaque body, caramel opaque cap | Unknown (certificate of analysis not provided) | Unknown (certificate of analysis not provided) | 20 |
| NV16871 | Capsules containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate. All participants over the age of 13 or who weighed > 40 kg received this dosage form. 2. A paediatric suspension containing 12 mg oseltamivir per ml of reconstituted solution and the following excipients: sorbitol, titanium dioxide, sodium benzoate, xanthan gum, monosodium citrate, saccharin sodium and Permaseal 11900‐31 tutti frutti (flavour). All participants of 12 years and under or who weighed ≤ 40 kg received this dosage form or 10 doses Ro 64‐0796/V14 (oseltamivir 75 mg capsules), batch PT2247C01 Ro 64‐0796/V37 (oseltamivir powder for oral suspension), batch B1023 |
Matching placebo was to be provided as capsules and as suspension Ro 64‐0796/V16 (placebo capsules), batch GMZ 0163 Ro 64‐0796/VF01 (placebo powder for oral suspension), batch C0318A001 |
Unknown (certificate of analysis not provided) | Unknown (certificate of analysis not provided) | 13‐14, 24 |
NB Most content dosage unavailable at review time‐lock.
On this basis we formulated two hypotheses: 1a. There is no association between incidence of gastrointestinal harms and a placebo containing dehydrocholic acid in oseltamivir trials. 1b. There is no association between incidence of asthma‐related events and a placebo containing lactose powder in zanamivir trials.
To test hypothesis 1a we assessed the oseltamivir trials for which we had clinical study reports Module 1 (M76001; WV15670; WV15671; WV15707; WV15812/WV15872; WV15730; WV15819/WV15876/WV15978; WV15758; WV15799) for gastrointestinal tract (GIT) harms including nausea, vomiting and diarrhoea as well as participants withdrawing from the studies due to adverse events. We meta‐analysed the results from these studies using the inverse variance random‐effects method. We assessed heterogeneity using the Chi2 test and used Tau2 to estimate between‐study variance. To investigate whether placebo containing dehydrocholic acid may be associated with gastrointestinal harms we compared adverse event rates in placebo groups from the oseltamivir trials (where placebo contained dehydrocholic acid) with adverse event rates in the placebo groups from the zanamivir trials (where placebo did not contain dehydrocholic acid). This comparison was done informally using 1) data obtained from the FDA labels of oseltamivir and zanamivir (FDA 2000b; FDA 2011a) as well as 2) the trials for which we have clinical study reports. As a sensitivity analysis we assumed a similar gastrointestinal adverse event rate in the placebo groups of the oseltamivir trials as was observed in the placebo groups of the zanamivir trials and then repeated the meta‐analysis (as described above). We also speculated that withdrawals in the placebo groups due to gastrointestinal adverse events were possibly related to dehydrocholic acid and removed these for the sensitivity analysis.
For hypothesis 1b we assessed asthma‐related events in nine zanamivir trials for which we had clinical study reports (NAIA3002; NAIB3002; NAIA2005; NAIB2005; NAIB2007; NAIB3001; NAIA3005; NAI30010; NAI30009). We meta‐analysed the results from these studies using the inverse variance random‐effects method. We assessed heterogeneity using the Chi2 test and used Tau2 to estimate between‐study variance. To investigate whether placebo containing lactose powder may be associated with asthma‐related events we informally compared event rates in placebo groups from the zanamivir trials (where placebo contained lactose powder) with event rates in the placebo groups from the oseltamivir trials (where placebo did not contain lactose powder). As a sensitivity analysis we assumed a similar asthma‐related event rate in the placebo groups of the zanamivir trials as was observed in the placebo groups of the oseltamivir trials and then repeated the meta‐analysis (as described above).
Hypothesis 2. Oseltamivir (or zanamivir) does not affect antibody production in treatment trials. Rationale. All oseltamivir influenza treatment trials specify the primary efficacy analysis population as the influenza‐infected population, not the randomised intention‐to‐treat (ITT) base population. The influenza‐infected population (known as ITTI, or intention‐to‐treat‐infected in clinical study reports) is determined post‐randomisation based on the results of laboratory testing by culture and/or antibody rise (comparing paired sera from the same participant). The sample for culture and the first sample of sera are taken before commencement of trial product but the second or the third sera are taken after patients are treated with trial medication. It is vital that placebo and active groups of patients have the same odds of being classified as influenza‐infected, otherwise any comparison between influenza‐infected groups will be potentially affected by bias and will essentially be a non‐randomised comparison. If trial medication affects the production of antibodies, the selection of the influenza‐infected population (which is partly based on antibody production) is confounded by taking the trial medication.
Roche have stated on multiple occasions (Smith 2006; Ward 2005; section 3.2.4.2 Serology WV15799) that ingestion of oseltamivir does not affect antibody production and the FDA supports this, stating that "In studies of naturally acquired and experimental influenza, treatment with TAMIFLU did not impair normal humoral antibody response to infection" (FDA 2011a).
However, we noticed unequal numbers of individuals in the influenza‐infected population subgroup in numerous trials. In addition, Takahashi et al reported that oseltamivir significantly suppressed respiratory mucosal secretory immunoglobulin (Ig) A responses to antigen (Ag)‐specific antibody (Ab) production and also the induction of Ag‐specific IgA Ab‐forming cells in an animal experiment (Takahashi 2010). If taking oseltamivir affects the production of IgG antibody as well, it may affect the selection of the influenza‐infected population.
We are also unsure of the implication for immunisation with influenza vaccine. According to the FDA, no influenza vaccine interaction study has been conducted with oseltamivir (FDA 2011a).
To test the hypothesis we compared: (1) the odds of participants in the ITT population subsequently classified as influenza‐infected; and (2) the odds of participants in the ITT population with a four‐fold or more rise of antibody between the placebo and active arms of the trials. If ingestion of oseltamivir does not affect antibody production then we expect the odds of being classified as influenza‐infected to be the same for the placebo and active arms. Therefore, we tested a null hypothesis that the odds of having a four‐fold or more rise of antibody was the same for the placebo and active arms. We meta‐analysed the results from these studies using the inverse variance random‐effects method. We assessed heterogeneity using the Chi2 test and used Tau2 to estimate between‐study variance. The trials included in this analysis were the 10 oseltamivir treatment trials analysed by Kaiser 2003 plus WV15758 for oseltamivir and NAIA3002, NAIB3002, NAIA2005, NAIB2005, NAIB2007, NAIB3001, NAI30009 for zanamivir. These are all the treatment trials for which we have clinical study reports Module 1. In an additional analysis we also assessed the oseltamivir trial conducted in China by Shanghai Roche Pharmaceutical Ltd for which we have a partial clinical study report (ML16369).
Hypothesis 3. Oseltamivir does not affect antibody production in post‐exposure (or secondary prophylaxis) trials. Rationale. According to the clinical study report of WV15799, the trial programme assessing the effects of oseltamivir in post‐exposure prophylaxis (PEP) consisted of two trials: WV15799 and WV16139. The Module 1s of both trials together with copious FDA notes on trial WV15799 were available to us at 'time lock'. However the PEP trial WV16139 was not standard care or placebo‐controlled and so we excluded it from the review.
WV15799 was a double‐blind, cluster‐randomised trial in which contact clusters of index cases were randomised to oseltamivir 75 mg a day or placebo for seven days. The trial formed an integral part of the "pivotal" trials package for the supplementary application and review for prophylaxis use of oseltamivir 75 mg in people aged more than 13 years of age, submitted to the FDA on 22 May 2000, approved on 20 November 2000 (FDA 2000c). In the clinical study report Module 1 the manufacturer claimed that the trial provided evidence of the drug's capacity to prevent influenza in contacts by interrupting its transmission from index cases. Since all index cases were left untreated except for a paracetamol rescue pack, it is hard to see how such a claim can be made. The interruption of transmission claim has two components: reduction of viral spread from index cases (measured by nasal shedding of influenza viruses) and prevention of onset of influenza in contacts. This latter claim was based on the definition of (prevented) influenza cases: a mixture of symptoms signs and 'laboratory confirmation' (i.e. viral culture from the upper airways and/or at least a four‐fold rise in antibody titres measured between baseline and two to three weeks later). The results of the trial later formed the basis for claims of the drug's effectiveness in interrupting transmission from person to person (WHO 2007) and allow time before the arrival of vaccines in the event of a pandemic. The interruption of transmission claim provided a powerful rationale for stockpiling oseltamivir (see for example vol 8, p.61‐62 NICE 2000: "Ro 64‐0796 successfully interrupts the transmission of influenza within households ... and suggests that Ro 64‐0796 [oseltamivir] would control the spread of influenza in other closed communities associated with high risk of transmission, such as nursing homes" … "Ro 64‐0796 also effectively interrupted virus transmission within households.")
The interruption of transmission indication was accepted by agencies such as the World Health Organization (WHO) and the US Centers for Disease Control and Prevention (CDC), but the US FDA refused to register and allow publicity based on any further indication beyond treatment and prophylactic effects on symptoms (FDA 2000f). Review of the evidence from the study protocol and Module 1 together with the FDA criticism explains the rationale for the FDA not supporting the manufacturers' claims. The design of the trial did not allow for comparison of the effects of treating index cases with oseltamivir versus placebo (as all index cases were not medicated) and a repeat viral culture was not performed for all participants. Viral culture was performed at baseline for all participants and thereafter only in participants with influenza‐like illness symptoms (see Schedule of assessment for the contact case, WV15799, and the FDA Medical Officer report (FDA 1999c)). Any participants presenting at follow‐up with symptoms of influenza had throat and nasal swabs taken in order to confirm the presence or absence of influenza infection (FDA 2000c), thereby missing out on potential asymptomatic infected people. However, a recent review of transmission studies has found no convincing evidence of spread from pre‐symptomatic or asymptomatic subjects (Patrozou 2009), which might explain the FDA's caution in sanctioning any such claim for oseltamivir.
Our review of the clinical study report's Module 1 identified further problems with the conduct and reporting of the trial and discrepancies both within the clinical study reports and between the study and its protocol. In the protocol (version H) there is no mention of viral shedding measurement. This appears to be a post‐protocol addition, which would explain the unsystematic nature of the viral excretion measurement remarked on by the FDA (i.e. taken from symptomatic contacts only). The primary population of analysis is the so called ITTIINAB population (contacts of ITT influenza‐infected index cases who had negative virology at baseline). Although defined in the protocol, the selection and presentation of results for the intention‐to‐treat contacts of the influenza‐infected index case not infected at baseline (ITTIINAB) population has the effect of excluding 57% of the placebo (200/456) and 59% of the oseltamivir (205/497) participants. The effect of selection on the clustering was not formally tested in a sensitivity analysis. Nor is the potential weakness of such a choice discussed in the WV15799 clinical study report. We carried out an analysis using Fisher's exact test, which showed that there was no statistical evidence that the placebo and oseltamivir groups' cluster sizes were distributed differently based on households with an infected index case (P = 0.56) (Table 2). By analysing the population by influenza status of the index case, instead of unit of randomisations (all index cases), the beneficial effects of the cluster‐randomisations are potentially lost, introducing unknown biases into the analysis. In addition, the generalisability of the conclusions may not be easily applied to clinical practice where testing of suspected influenza cases is often not practical. Cross‐checking the definition of ITTIINAB with that reported in the protocol of the other PEP trial, WV16193 (excluded from this review) yields a different definition (PDF page 589) "The primary outcome in this study (WV15799) was the incidence of influenza occurring among contacts of influenza‐infected index cases (the intent‐to‐treat‐index‐infected population)".
Throughout the clinical study report of trial WV15799 there are many other apparently contradictory statements on important aspects of the trial, for example, on how many viral swabs and paired sera tests were carried out. The text at page 50 of the Module 1 reports that "For 21 of the 26 contacts with laboratory‐confirmed clinical influenza in the ITTIINAB population the diagnosis was confirmed by culture" but Table 19 shows the 26 contacts as shedding virus at days two to eight. The same table reports that 178 placebo contacts and 201 oseltamivir contacts were negative for virology (which suggests that they were tested) at days two and eight. However, viral testing only took place at baseline and thereafter only in symptomatic participants. The number of contacts in which influenza was diagnosed only by serology is unclear but it appears to be five (26 minus 21). These inconsistencies highlight one of the fundamental conceptual problems in understanding the whole oseltamivir prophylaxis trial programme: the mode of action of the drug. Our interpretation of the text suggests that oseltamivir does not prevent infection and does not affect influenza antibody response. As stated above, the claim that oseltamivir does not affect antibody responses has been made by the manufacturers. However, an antibody response is part of the definition of influenza. We are unsure how it is possible that oseltamivir could prevent influenza by stopping symptoms appearing and antibodies rising while at the same time leaving antibody production unaffected.
It is for this reason that we decided to test whether administration of oseltamivir for PEP affected the production of antibodies to influenza viruses. The distribution of change in antibodies from baseline to follow‐up was compared between the arms of the trials for contacts of the index cases. Analysis was performed using Wilcoxon two‐sample test separately for each type of antibody in each trial. An additional analysis of proportion of contacts having a four‐fold or greater rise in influenza‐specific antibody titre in antibodies was compared between groups using the Chi2 test. Antibody data were not available for index cases, who were left untreated. In WV15799, antibody testing may have been undertaken at day 1, day 8 and at day 21 ± 4 days for all contacts. Day 8 blood samples for influenza antibody analysis were stored to measure influenza antibody levels only in those contacts who did not attend the follow‐up visit (day 17 to 25). Analysis was based on data from the ITTIINAB population at pages 59‐60 and Appendix 60 of the clinical study report's Module 1.
Hypothesis 4. The number of trial centres and centre withdrawals does not affect the proportion of placebo patients subsequently diagnosed with influenza infection (originally the outcome was effect size) and Hypothesis 5. In oseltamivir treatment trials there is no association between the order of randomisations and naso‐pharyngeal swabbing (i.e. randomising participants first and then swabbing or swabbing first and then randomising) and the proportion of placebo patients subsequently diagnosed with influenza infection (originally the outcome was effect size). Rationale. The proportion of ITT population in the treatment trials of NIs that are subsequently diagnosed as infected with influenza is higher (˜ 50% to 80%) than is usually seen in the course of the winter season in routine clinical care, although high peaks can occur for a very limited period. We know that in some treatment trials, such as WV15670 and WV15671, centres were activated to "recruit subjects during an influenza outbreak in the locality, detected using standardised surveillance techniques." We postulated that unreported procedures may also have been used in the trials to obtain these high proportions of influenza to ILI cases. Two procedures that may have been used are: 1) use of rapid influenza tests to screen out patients based on negative results; 2) dropping of centres that recruited low proportions of infected patients. The use of rapid testing of patients prior to randomisation has been reported in at least one of the zanamivir treatment trials (NAIB3001), in oseltamivir trial WV15670 as a means of excluding infection with H5N1 in the Hong Kong Centre, as a pilot surveillance in suburban London during the 1998 to 1999 winter (NICE 2000 vol.1) and in most oseltamivir paediatric trials to exclude respiratory syncytial virus (RSV) infection. In addition, the schedule of testing varies by trial for the oseltamivir trials with swabbing performed either before randomisation or after randomisation. In at least one oseltamivir treatment trial (WV15730) it was reported that no viral culture was performed at centres from South America (FDA 1999c). As a result of these observations we reformulated Hypothesis 4 as follows: the number of centres and centre withdrawals does not affect the proportion of placebo patients subsequently diagnosed with influenza infection (originally the outcome was primary outcome effect size) in oseltamivir treatment trials and Hypothesis 5 as in oseltamivir treatment trials there is no association between the order of randomisations and naso‐pharyngeal swabbing (i.e. randomising participants first and then swabbing or swabbing first and then randomising) and the proportion of placebo patients subsequently diagnosed with influenza infection.
To test hypothesis 4, we used Spearman's rank method to estimate the correlation between average number of patients recruited per centre and the proportion of placebo patients subsequently diagnosed with influenza infection. The placebo patients were used for the proportion of patients subsequently diagnosed with influenza infection because, as we show later in the review, there is evidence that oseltamivir interferes with antibody production and antibody response was used to diagnose influenza infection. We did not analyse the number of centres dropped from studies because information on this variable was not available in Module 1s of the clinical study reports for the included trials (information on patients recruited to each centre is reported in Module 2 which we do not currently have access to).
Hypothesis 5 was generated to attempt to explain the seemingly high proportion of influenza‐infected influenza‐like illness cases in treatment trials. However, we did not formally test this hypothesis as there was only one clinical study report reporting randomisation first then swabbing second (WV15819/WV15876/WV15978) (see also Appendix 10).
Results
The results of our post‐protocol analyses are also reported in Figure and/or Table format.
Hypothesis 1a tested in a sensitivity analysis whether the incidence of gastrointestinal harms may be associated with exposure of participants to a placebo containing dehydrocholic acid. The data obtained from the oseltamivir trials clinical study reports is shown in Table 15.
Overall, the crude adverse event incidence in the placebo groups of the oseltamivir trials was 5.5% for nausea, 3.6% for vomiting and 7.0% for diarrhoea. This compares with crude incidence in the nine zanamivir treatment trials' placebo groups of 4.1% for nausea and vomiting (reported as a combined outcome in the clinical study reports) and 2.8% for diarrhoea. Two studies (WV15670; WV15671) compared three treatment groups: oseltamivir 150 mg bid, oseltamivir 75 mg bid and placebo. To maintain the blinding in these trials, each participant took two pills twice daily. Therefore the participants in the oseltamivir 75 mg bid group took one placebo tablet twice daily. We note that in trial WV15671 there was evidence of a dose‐response effect of placebo on incidence of diarrhoea: oseltamivir 150 mg bid (5.9%), oseltamivir 75 mg bid (8.7%) and placebo (11.8%) (P = 0.036). However, there was no evidence found of a similar trend in trial WV15670 (P = 0.88). We were unable to carry out a similar analysis for paediatric treatment trial WV15758 because a detailed content of the placebo preparations is not available (see Table 22).
Random‐effects meta‐analysis of the data in Table 15 provided the following results.
Nausea: increased odds of adverse events due to oseltamivir (OR 1.62, 95% CI 1.17 to 2.26, P = 0.004). Vomiting: increased odds of adverse events due to oseltamivir (OR 2.32, 95% CI 1.62 to 3.31, P < 0.001). Diarrhoea: decreased odds of adverse events due to oseltamivir (OR 0.72, 95% CI 0.53 to 0.97, P = 0.03). Withdrawal from treatment due to adverse events: no evidence of a difference between treatment groups (OR 1.08, 95% CI 0.66 to 1.76, P = 0.75).
We carried out a sensitivity analysis by assuming placebo rates of gastrointestinal adverse events in oseltamivir trials based on those observed in placebo groups of similar zanamivir trials. Overall rates of nausea, vomiting and diarrhoea in placebo groups of zanamivir treatment trials for adults and adolescents were 3%, 2% and 4% compared to oseltamivir treatment trials for adults and adolescents where rates were 6%, 3% and 10% respectively based on FDA‐reported data (FDA 2000b; FDA 2011a). Conversely, other common adverse events such as headaches, cough and dizziness had similar incidences of 2% to 3% in the placebo groups of zanamivir and oseltamivir treatment trials (FDA 2000b; FDA 2011a). In the treatment trials of children the rates of nausea, vomiting and diarrhoea in placebo groups of zanamivir treatment trials were 2%, 3% and 2% compared to oseltamivir treatment trials of children where rates were 4%, 9% and 11% respectively. Our conservative estimate is that the oseltamivir placebo increased rates of nausea two‐fold (risk ratio (RR) = 2), vomiting (RR 1.5) and diarrhoea (RR 2.5) compared to the placebo arms in zanamivir trials. Based on the adult and adolescent trials we could conservatively speculate that the substances in the oseltamivir trials placebo increase nausea, vomiting and diarrhoea by 100% (6%/3%), 50% (3%/2%) and 150% (10%/4%) respectively. This could also be considered a conservative assumption because it is plausible that the lactose powder used as the placebo in the zanamivir trials also induced gastrointestinal symptoms, especially in patients that were lactose intolerant. Adjusting the actual rates of these events in the oseltamivir trials placebo groups to be consistent with the zanamivir trials placebo group rates (as reported by the FDA (FDA 2000b; FDA 2011a) and re‐running the random‐effects meta‐analysis we obtained the following results.
Nausea: increased odds of adverse events due to oseltamivir (OR 3.33, 95% CI 2.44 to 4.54, P < 0.001; test for heterogeneity P = 0.33). Vomiting: increased odds of adverse events due to oseltamivir (OR 3.46, 95% CI 2.51 to 4.78, P < 0.001; test for heterogeneity P = 0.37). Diarrhoea: increased odds of adverse events due to oseltamivir (OR 1.86, 95% CI 1.39 to 2.50, P < 0.001; test for heterogeneity P = 0.50).
The estimated effect sizes for nausea and vomiting have increased based on the sensitivity analysis. The effect on diarrhoea has reversed, indicating oseltamivir is possibly associated with increased odds of this adverse event. The results of our analysis support an alternative interpretation to that of the FDA.
Finally, we carried out a sensitivity analysis of withdrawal from treatment due to adverse events by assuming no withdrawals due to gastrointestinal events in the placebo group. In total there were nine patients in the oseltamivir trials' placebo groups that withdrew due to gastrointestinal events. When these withdrawals are not included the following result is obtained based on random‐effects meta‐analysis:
Withdrawal from treatment due to adverse events: no evidence of a difference between treatment groups (OR 1.48, 95% CI 0.87 to 2.51, P = 0.15; test for heterogeneity P = 0.40).
We conclude that participants in placebo arms of oseltamivir treatment trials experience a higher rate of gastrointestinal adverse events compared to their zanamivir counterparts. As the zanamivir trials' inclusion criteria were similar to the oseltamivir trials (fever and two additional symptoms of influenza‐like illness (ILI)) this observation cannot plausibly be explained by an incremental role of influenza infection in the genesis of such heterogeneity. It is possible that the difference in reported gastrointestinal adverse events in the placebo groups of zanamivir and oseltamivir trials is due to differences in the collection of these events. However, other common adverse events such as headaches, cough and dizziness had very similar rates in the placebo groups of zanamivir and oseltamivir trials. Despite the results of this sensitivity analysis it is impossible without a clear statement of dosage and rationale of use to assess the role of dehydrocholic acid and possibly calcium phosphate in the causation of such a high incidence of gastrointestinal adverse events.
For hypothesis 1b the data obtained from the zanamivir treatment trials clinical study reports are shown in Table 16.
Over all the nine zanamivir trials the incidence of asthma (including asthma exacerbation) in the placebo groups was 2.1% compared to 0.9% in the placebo groups of the oseltamivir trials. Random‐effects meta‐analysis of the data in Table 16 provided the following results for the combined outcome of any asthma event:
Asthma: decreased odds of adverse events due to zanamivir (OR 0.54, 95% CI 0.34 to 0.86, P = 0.01).
We carried out a sensitivity analysis by assuming placebo rates of asthma‐related adverse events in zanamivir trials based on those observed in similar oseltamivir trials. If we assume a rate of asthma events in the placebo groups of the nine zanamivir trials similar to that observed in the oseltamivir trials we obtain the following result based on random‐effects meta‐analysis:
Asthma: no evidence of a difference between treatment groups (OR 1.27, 95% CI 0.71 to 2.26, P = 0.42; test for heterogeneity P = 0.68).
We conclude that zanamivir trial placebo recipients appear to have a higher incidence of asthma‐related events than their oseltamivir counterparts. Again, as the inclusion criteria were similar for both trial programmes this finding is not likely to be due to severity of influenza infections but associated with exposure to lactose powder and possibly to the active principle. This is a point remarked on by the FDA.
For hypothesis 2 (oseltamivir (or zanamivir) does not affect antibody production in treatment trials) the relevant trials showed strong and consistent evidence that patients randomised to active treatment had reduced odds of being classified as influenza‐infected (OR 0.83, 95% CI 0.73 to 0.94, P = 0.003) with no evidence of heterogeneity (heterogeneity Chi2 test = 2.80 (df = 7) P = 0.90; estimate of between‐study variance Tau2 = 0.00) (see Table 17). There was also strong evidence that patients randomised to active treatment had reduced odds of having four‐fold or higher rise in antibody titres (OR 0.79, 95% CI 0.70 to 0.90, P < 0.001) with no evidence of heterogeneity (heterogeneity Chi2 test = 4.61 (df = 7) P = 0.71; estimate of between‐study variance Tau2 = 0.00) (see Table 17).
In contrast, the zanamivir trials showed no evidence that patients randomised to active treatment had reduced odds of being classified as influenza‐infected (OR 1.05, 95% CI 0.90 to 1.24, P = 0.52) with no evidence of heterogeneity (heterogeneity Chi2 test = 3.03 (df = 6) P = 0.81; estimate of between‐study variance Tau2 = 0.00) (see Table 18).
These results have important implications for the oseltamivir treatment trials programme and for all ongoing trials. All influenza‐infected populations are selected post‐randomisation and post‐trial termination on the basis of laboratory findings (all ITT participants being symptomatic at entry, with aetiology unknown). However, as oseltamivir appears to affect antibody production (or perhaps testing, or both), there may be some participants in the oseltamivir group who were infected with influenza but not diagnosed by the antibody rise and were therefore not counted in the influenza‐infected population. These may have subsequently been excluded from the efficacy analysis. It is also possible that the strength of the antibody production limit to qualify for an influenza infection‐induced antibody rise (four‐four fold and above from baseline) had the effect of selecting the 'stronger' responders into the influenza‐infected subgroup of the oseltamivir arm. This would mean that the best antibody producers were selected and this may have led to inflated treatment estimates of efficacy in influenza‐infected populations.
To investigate this possibility we calculated the correlation between the odds of being classified as infected in the oseltamivir group compared to the placebo group and the size of the primary treatment effect (time to alleviation of symptoms in the ITTI population). In treatment trials all participants are recruited on the basis of symptoms of influenza‐like illness. According to the mechanism of action proposed by the manufacturer, infected participants given oseltamivir up to 48 hours from symptom onset should have an antibody response which, given the effects of randomisation, should be similar to that of placebo recipients. Non‐responders or weak responders should be spread evenly across the trial arms. All treatment trials of oseltamivir showing evidence of a treatment effect on the primary outcome of the study were included in the analysis. This included two trials for which we did not have clinical full study reports (ML16369; JV15823). We included these trials to increase variation in the two variables used for the analysis. In addition, two trials were excluded: WV15707 which had a total ITTI sample size of 12 participants; and WV15812/WV15872, which was a treatment trial in chronically ill adults that showed no evidence of a treatment effect. Results showed strong evidence of a correlation (Spearman rank correlation = ‐0.83, P = 0.01) (Table 19). The correlation was highly negative, indicating that lower odds of being classified as ITTI in the oseltamivir group compared to the placebo group is associated with larger treatment effects for the primary outcome of the studies. In contrast, there was no evidence of a correlation between the odds of being classified as infected in the oseltamivir group compared to the placebo group (Table 19) and the size of the treatment effect in the ITT population (Spearman rank correlation = ‐0.23, P = 0.66). A limitation of this analysis is that data for the ITT population for two trials were not available (WV15730; JV15823) (Table 19).
Thus, all influenza‐infected comparisons are potentially confounded by the action of the drug (oseltamivir but probably not zanamivir) and are essentially non‐randomised comparisons. Any analyses should be based on ITT populations in oseltamivir treatment trials. Analyses and data considered for inclusion in systematic reviews should be based on the ITT (or safety) populations only.
Our analysis of Hypothesis 3 shows that the odds of having a four‐fold rise in antibodies is 0.33 (95% CI 0.16 to 0.67) for the oseltamivir group compared to placebo (hence a much bigger effect compared to the treatment trials). Due to insufficient information provided in the clinical study report we were unable to take account of the clustering in this analysis, hence the confidence intervals are possibly under‐estimated; however an analysis that takes into account clustering is unlikely to change the conclusions. These results show that oseltamivir prophylaxis is associated with lower odds of a four‐fold rise in antibodies and this appears to be due to a difference in the distribution of antibody rise in HIAAH3 antibodies but not HIAAH1 or HIB antibodies (see Table 16, Table 21, Table 22 and Table 23). In summary no conclusions can be drawn from the available evidence on the effects of the drug on viral transmission. The mode of action in prophylaxis appears mainly to be ascribed to symptom suppression or control. There is uncertainty around other possible effects of the drug especially given its interaction with the production of antibodies.
We rejected Hypothesis 4 and are currently unable to test Hypothesis 5
We rejected Hypothesis 4 as there was no evidence of correlation between average recruited subjects per centre and the proportion of placebo patients subsequently diagnosed with influenza infection (Spearman correlation = 0.26; P = 0.53). Table 24 shows that the average recruited participants per centre ranged from 2 to 11 which appears very low for international, multicentre trials. Two studies failed to reach their recruitment target (WV15707 and WV15730) and two clinical study reports were made up of multiple trials due to the original trial's poor recruitment (WV15819/WV15876/WV15978 and WV15812/WV15872) (Table 24). In addition the proportion of placebo patients subsequently diagnosed with influenza infection ranged from 63% to 75%, implying little between‐trial variation.
We are currently unable to test Hypothesis 5 as only one oseltamivir clinical study report (of three trials) reported randomisation first then swabbing second (WV15819/WV15876/WV15978). In this study the proportion of placebo patients that were confirmed as influenza‐infected was 68.1%. This compares with the other seven clinical study reports where swabbing was carried out first and randomisation second and the proportion of placebo patients that were confirmed as influenza‐infected ranged from 63.2% to 74.9% with mean 68.1%. Hence it seems that swabbing after randomisation made no difference in the treatment trial programme where this practice is reported. However, with only one clinical treatment study report randomising prior to swabbing available to us, the power to detect a difference in the proportion of placebo patients subsequently diagnosed with influenza infection is low. We hope to be able to retest this hypothesis as more data become available.
Appendix 11. Example of contents of a Clinical Study Report (from page 1 of WV15670 report)
Final study report modules
This report consists of five modules. Those not supplied in this submission were obtainable from the sponsor on request.
MODULE I: CORE REPORT AND STUDY PUBLICATIONS
Introduction Rationale Objectives Methodology Efficacy results Safety results Discussion/conclusions Appendices
MODULE II: PRESTUDY DOCUMENTS AND STUDY METHODOLOGY
Protocol and amendment history Blank CRF Subject information sheet Glossary of original and preferred terms Randomisation list Reporting analysis plan (RAP) Certificates of analysis List of investigators List of responsible ethics committees
MODULE III: INDIVIDUAL SUBJECT LISTINGS OF DEMOGRAPHIC AND EFFICACY DATA
Demographic data listings Previous and concomitant diseases Previous and concomitant medications Efficacy listings
MODULE IV: INDIVIDUAL SUBJECT LISTINGS OF SAFETY DATA
Laboratory parameters Vital signs data
MODULE V: STATISTICAL REPORT
Data and analyses
Comparison 1. Oseltamivir versus placebo for treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Time to first alleviation of symptoms in adult treatment (ITT population) | 8 | 3954 | Mean Difference (IV, Random, 95% CI) | ‐16.76 [‐25.10, ‐8.42] |
| 2 Hospital admission in adult treatment (safety population) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 0.92 [0.57, 1.50] |
| 3 Defined as influenza‐infected at baseline in adult treatment | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.95 [0.91, 0.99] |
| 4 Antibody rise four‐fold or greater in adult treatment | 8 | 4025 | Risk Ratio (IV, Random, 95% CI) | 0.92 [0.86, 0.97] |
| 5 Adverse events ‐ nausea in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.57 [1.14, 2.15] |
| 6 Adverse events ‐ vomiting in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 2.43 [1.75, 3.38] |
| 7 Adverse events ‐ diarrhoea in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.67 [0.46, 0.98] |
| 8 Withdrawal from adult treatment trial due to adverse events | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.56, 1.48] |
| 9 All withdrawals from adult treatment | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.02 [0.73, 1.41] |
| 10 Adverse events ‐ cough in adult treatment (on‐treatment) | 6 | 3943 | Risk Ratio (IV, Random, 95% CI) | 0.63 [0.41, 0.96] |
| 11 Adverse events ‐ abdominal pain in adult treatment (on‐treatment) | 6 | 4368 | Risk Ratio (IV, Random, 95% CI) | 1.00 [0.64, 1.55] |
| 12 Adverse events: dizziness in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.77 [0.51, 1.18] |
| 13 Adverse events: headache in adult treatment (on‐treatment) | 7 | 4426 | Risk Ratio (IV, Random, 95% CI) | 1.17 [0.72, 1.90] |
| 14 Serious adverse events: overall in adult treatment (on‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 0.96 [0.51, 1.80] |
| 15 Serious adverse events: overall in adult treatment (off‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 0.73 [0.39, 1.37] |
| 16 Complications: bronchitis in adult treatment | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.75 [0.56, 1.01] |
| 16.1 Trials which collected data on non‐specific adverse event or secondary/intercurrent illness form | 6 | 3316 | Risk Ratio (IV, Random, 95% CI) | 0.66 [0.42, 1.03] |
| 16.2 Trials which collected data on specific "Diagnosis of secondary illness" form | 2 | 1136 | Risk Ratio (IV, Random, 95% CI) | 0.87 [0.61, 1.26] |
| 17 Complications: pneumonia in adult treatment | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.55 [0.33, 0.90] |
| 17.1 Trials which collected data on non‐specific adverse event or secondary/intercurrent illness form | 6 | 3316 | Risk Ratio (IV, Random, 95% CI) | 0.44 [0.22, 0.88] |
| 17.2 Trials which collected data on specific "Diagnosis of secondary illness" form | 2 | 1136 | Risk Ratio (IV, Random, 95% CI) | 0.69 [0.33, 1.44] |
| 18 Complications: sinusitis in adult treatment | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.03 [0.76, 1.40] |
| 18.1 Trials which collected data on non‐specific adverse event or secondary/intercurrent illness form | 6 | 3316 | Risk Ratio (IV, Random, 95% CI) | 1.05 [0.74, 1.50] |
| 18.2 Trials which collected data on specific "Diagnosis of secondary illness" form | 2 | 1136 | Risk Ratio (IV, Random, 95% CI) | 0.97 [0.52, 1.80] |
| 19 Complications: otitis media in adult treatment | 6 | 4368 | Risk Ratio (IV, Random, 95% CI) | 1.11 [0.57, 2.15] |
| 19.1 Trials which collected data on non‐specific adverse event or secondary/intercurrent illness form | 4 | 3232 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.46, 2.12] |
| 19.2 Trials which collected data on specific "Diagnosis of secondary illness" form | 2 | 1136 | Risk Ratio (IV, Random, 95% CI) | 1.57 [0.41, 6.02] |
| 20 Complications in adult trials classified as serious or leading to study withdrawal | 6 | 3675 | Risk Ratio (IV, Random, 95% CI) | 0.91 [0.40, 2.06] |
| 21 Culture‐positive at baseline in adult treatment | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.01 [0.95, 1.07] |
| 22 Adverse events: general body system in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.88 [0.67, 1.17] |
| 23 Adverse events: neurological body system in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.05 [0.80, 1.38] |
| 24 Adverse events: respiratory body system in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.81 [0.65, 1.00] |
| 25 Adverse events: infection body system in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.85 [0.71, 1.01] |
| 26 Adverse events: gastrointestinal body system in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.25 [1.08, 1.45] |
| 27 Adverse events: cardiac body system in adult treatment (on‐treatment) | 6 | 3943 | Risk Ratio (IV, Random, 95% CI) | 0.49 [0.25, 0.97] |
| 28 Adverse events: ear body system in adult treatment (on‐treatment) | 7 | 4426 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.61, 1.60] |
| 29 Adverse events: eye body system in adult treatment (on‐treatment) | 7 | 4426 | Risk Ratio (IV, Random, 95% CI) | 1.00 [0.52, 1.92] |
| 30 Adverse events: metabolism body system in adult treatment (on‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 0.81 [0.46, 1.43] |
| 31 Adverse events: musculoskeletal body system in adult treatment (on‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.01 [0.59, 1.73] |
| 32 Adverse events: psychiatric body system in adult treatment (on‐treatment) | 7 | 4426 | Risk Ratio (IV, Random, 95% CI) | 0.93 [0.43, 2.03] |
| 33 Adverse events: skin body system in adult treatment (on‐treatment) | 7 | 4426 | Risk Ratio (IV, Random, 95% CI) | 1.14 [0.63, 2.06] |
| 34 Adverse events: cardiac body system in adult treatment (off‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 1.21 [0.55, 2.64] |
| 35 Adverse events: ear body system in adult treatment (off‐treatment) | 6 | 4368 | Risk Ratio (IV, Random, 95% CI) | 1.17 [0.57, 2.42] |
| 36 Adverse events: gastrointestinal body system in adult treatment (off‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 1.08 [0.74, 1.58] |
| 37 Adverse events: general body system in adult treatment (off‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 0.90 [0.50, 1.62] |
| 38 Adverse events: infection body system in adult treatment (off‐treatment) | 7 | 4426 | Risk Ratio (IV, Random, 95% CI) | 0.80 [0.61, 1.03] |
| 39 Adverse events: musculoskeletal body system in adult treatment (off‐treatment) | 7 | 4394 | Risk Ratio (IV, Random, 95% CI) | 0.84 [0.54, 1.30] |
| 40 Adverse events: neurological body system in adult treatment (off‐treatment) | 6 | 4368 | Risk Ratio (IV, Random, 95% CI) | 1.29 [0.87, 1.91] |
| 41 Adverse events: respiratory body system in adult treatment (off‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.94 [0.71, 1.24] |
| 42 Adverse events: skin body system in adult treatment (off‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 0.81 [0.42, 1.56] |
| 43 Adverse events: cough in adult treatment (off‐treatment) | 8 | 4452 | Risk Ratio (IV, Random, 95% CI) | 1.01 [0.55, 1.85] |
| 44 Adverse events: headache in adult treatment (off‐treatment) | 6 | 4368 | Risk Ratio (IV, Random, 95% CI) | 1.34 [0.83, 2.15] |
| 45 Adverse events: nausea in adult treatment (off‐treatment) | 6 | 4368 | Risk Ratio (IV, Random, 95% CI) | 1.05 [0.50, 2.23] |
| 46 Time to first alleviation of symptoms in child treatment [hours] | 3 | 1329 | Mean Difference (IV, Random, 95% CI) | ‐8.04 [‐33.34, 17.26] |
| 46.1 Otherwise healthy children | 1 | 669 | Mean Difference (IV, Random, 95% CI) | ‐29.40 [‐47.04, ‐11.76] |
| 46.2 Children with chronic asthma | 2 | 660 | Mean Difference (IV, Random, 95% CI) | 5.18 [‐11.06, 21.41] |
| 47 Hospital admission in child treatment (safety population) | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 1.92 [0.70, 5.23] |
| 48 Defined as influenza‐infected at baseline in child treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 0.92 [0.84, 1.01] |
| 49 Antibody rise four‐fold or greater in child treatment | 2 | 909 | Risk Ratio (M‐H, Random, 95% CI) | 0.90 [0.80, 1.00] |
| 50 Complications: bronchitis in child treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 0.65 [0.27, 1.55] |
| 51 Complications: otitis media in child treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 0.80 [0.62, 1.02] |
| 52 Complications: pneumonia in child treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 1.06 [0.62, 1.83] |
| 53 Complications: sinusitis in child treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 1.00 [0.58, 1.72] |
| 54 Complications: pneumonia in child treatment by on‐ and off‐treatment | 3 | Risk Ratio (IV, Random, 95% CI) | Subtotals only | |
| 54.1 On‐treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 0.87 [0.48, 1.60] |
| 54.2 Off‐treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 2.83 [0.52, 15.31] |
| 55 Complications in trials of children classified as serious or leading to study withdrawal | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 1.98 [0.58, 6.72] |
| 56 Withdrawal from child treatment trial due to adverse events | 2 | 1029 | Risk Ratio (IV, Random, 95% CI) | 0.99 [0.33, 3.01] |
| 57 All withdrawals from child treatment | 2 | 1029 | Risk Ratio (IV, Random, 95% CI) | 0.94 [0.56, 1.60] |
| 58 Serious adverse events: overall in child treatment (on‐treatment) | 2 | 1029 | Risk Ratio (IV, Random, 95% CI) | 1.97 [0.59, 6.56] |
| 59 Serious adverse events: overall in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.38, 8.46] |
| 60 Adverse events: abdominal pain in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.10 [0.62, 1.95] |
| 61 Adverse events: diarrhoea in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 0.87 [0.58, 1.28] |
| 62 Adverse events: nausea in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 0.87 [0.50, 1.51] |
| 63 Adverse events: vomiting in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.70 [1.23, 2.35] |
| 64 Adverse events: abdominal pain in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.39, 2.11] |
| 65 Adverse events: cough in child treatment (off‐treatment) | 2 | 1029 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.27, 1.85] |
| 66 Adverse events: diarrhoea in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.36, 1.40] |
| 67 Adverse events: headache in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.13 [0.55, 2.34] |
| 68 Adverse events: vomiting in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.57, 2.02] |
| 69 Adverse events: ear body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.18 [0.30, 4.56] |
| 70 Adverse events: gastrointestinal body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.18 [0.96, 1.44] |
| 71 Adverse events: general body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.95 [0.47, 1.92] |
| 72 Adverse events: infection body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 0.75 [0.59, 0.95] |
| 73 Adverse events: neurological body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 0.66 [0.17, 2.62] |
| 74 Adverse events: respiratory body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.02 [0.73, 1.43] |
| 75 Adverse events: skin body system in child treatment (on‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.26 [0.71, 2.22] |
| 76 Adverse events: ear body system in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.10 [0.52, 2.32] |
| 77 Adverse events: gastrointestinal body system in child treatment (off‐treatment) | 2 | 1029 | Risk Ratio (IV, Random, 95% CI) | 1.15 [0.69, 1.91] |
| 78 Adverse events: general body system in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.00 [0.54, 1.86] |
| 79 Adverse events: infection body system in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.14 [0.82, 1.58] |
| 80 Adverse events: neurological body system in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 1.07 [0.51, 2.26] |
| 81 Adverse events: respiratory body system in child treatment (off‐treatment) | 3 | 1358 | Risk Ratio (IV, Random, 95% CI) | 0.94 [0.65, 1.35] |
| 82 Culture‐positive at baseline in child treatment | 3 | 1359 | Risk Ratio (IV, Random, 95% CI) | 0.93 [0.83, 1.04] |
1.8. Analysis.
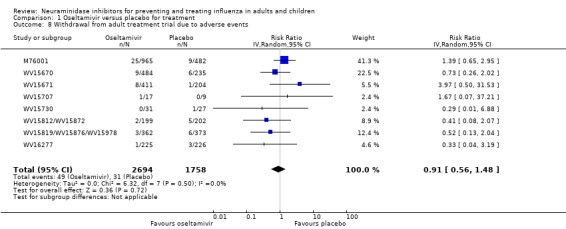
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 8 Withdrawal from adult treatment trial due to adverse events.
1.9. Analysis.
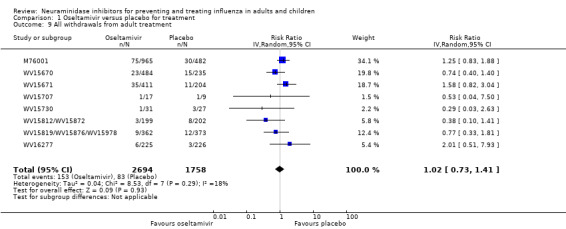
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 9 All withdrawals from adult treatment.
1.10. Analysis.
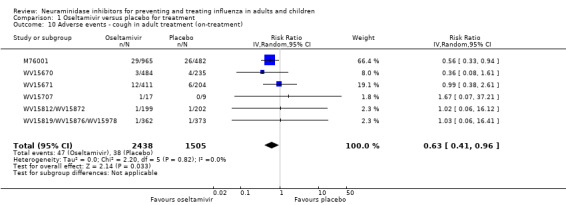
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 10 Adverse events ‐ cough in adult treatment (on‐treatment).
1.11. Analysis.
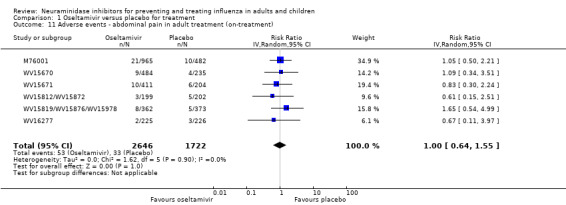
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 11 Adverse events ‐ abdominal pain in adult treatment (on‐treatment).
1.12. Analysis.
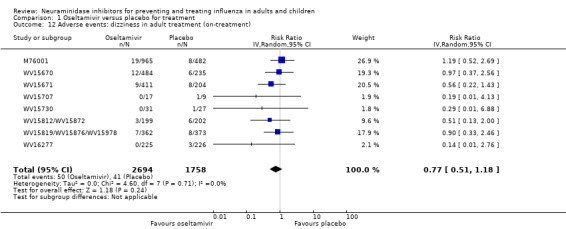
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 12 Adverse events: dizziness in adult treatment (on‐treatment).
1.13. Analysis.
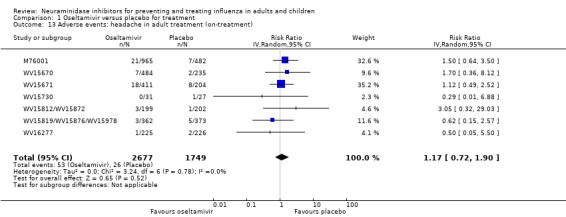
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 13 Adverse events: headache in adult treatment (on‐treatment).
1.14. Analysis.
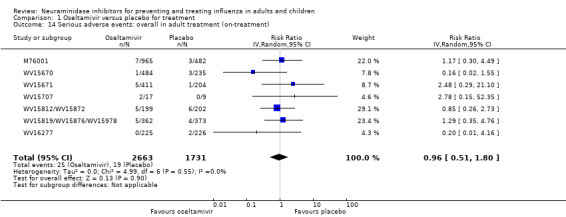
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 14 Serious adverse events: overall in adult treatment (on‐treatment).
1.15. Analysis.
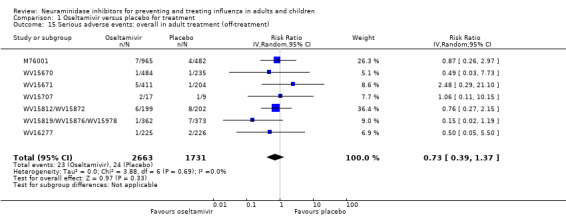
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 15 Serious adverse events: overall in adult treatment (off‐treatment).
1.21. Analysis.
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 21 Culture‐positive at baseline in adult treatment.
1.22. Analysis.
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 22 Adverse events: general body system in adult treatment (on‐treatment).
1.23. Analysis.
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 23 Adverse events: neurological body system in adult treatment (on‐treatment).
1.24. Analysis.
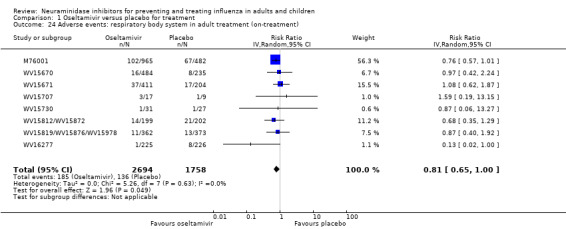
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 24 Adverse events: respiratory body system in adult treatment (on‐treatment).
1.25. Analysis.
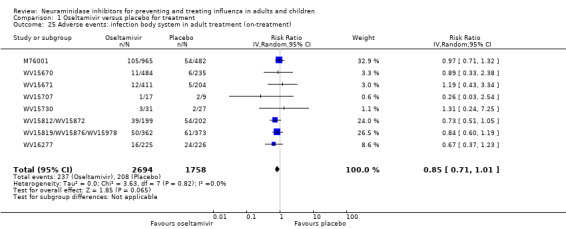
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 25 Adverse events: infection body system in adult treatment (on‐treatment).
1.26. Analysis.
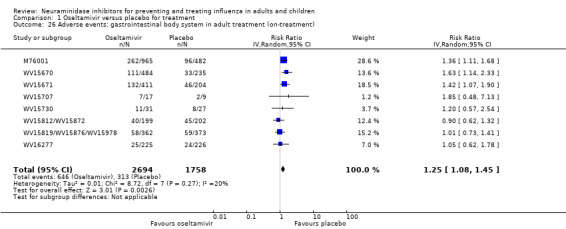
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 26 Adverse events: gastrointestinal body system in adult treatment (on‐treatment).
1.28. Analysis.
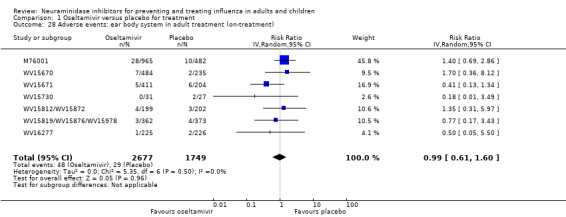
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 28 Adverse events: ear body system in adult treatment (on‐treatment).
1.29. Analysis.
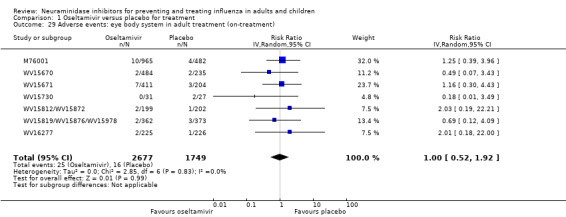
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 29 Adverse events: eye body system in adult treatment (on‐treatment).
1.30. Analysis.
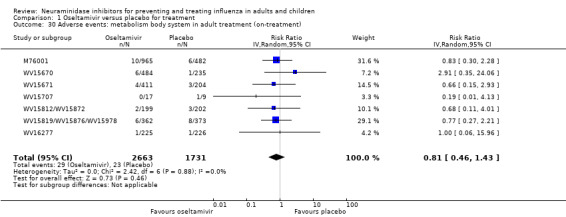
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 30 Adverse events: metabolism body system in adult treatment (on‐treatment).
1.31. Analysis.
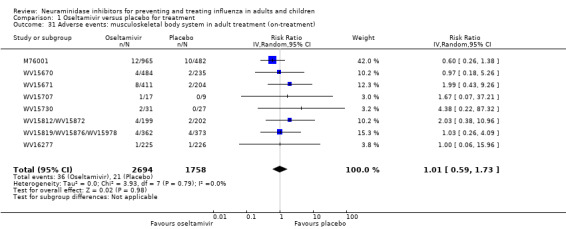
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 31 Adverse events: musculoskeletal body system in adult treatment (on‐treatment).
1.33. Analysis.
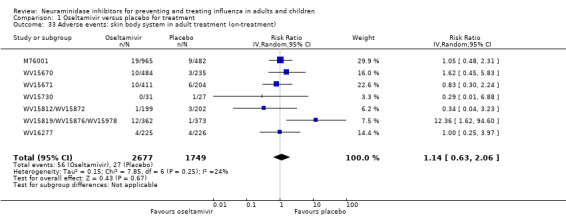
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 33 Adverse events: skin body system in adult treatment (on‐treatment).
1.34. Analysis.
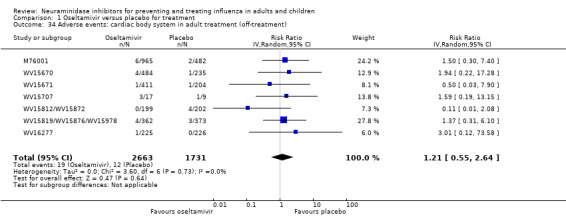
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 34 Adverse events: cardiac body system in adult treatment (off‐treatment).
1.35. Analysis.
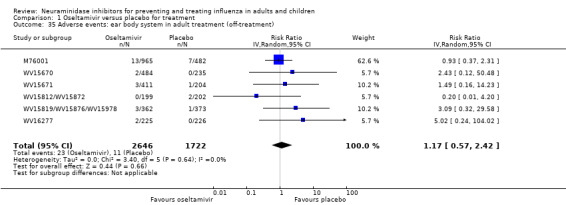
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 35 Adverse events: ear body system in adult treatment (off‐treatment).
1.36. Analysis.
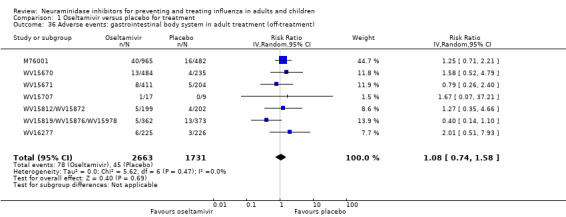
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 36 Adverse events: gastrointestinal body system in adult treatment (off‐treatment).
1.37. Analysis.
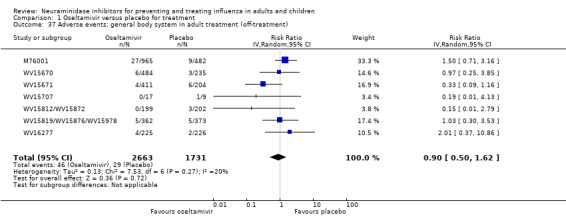
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 37 Adverse events: general body system in adult treatment (off‐treatment).
1.38. Analysis.
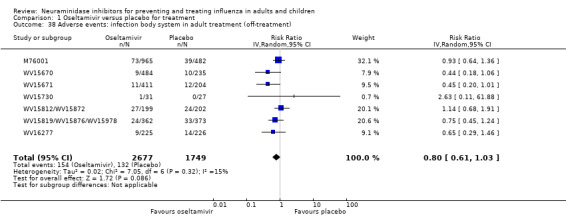
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 38 Adverse events: infection body system in adult treatment (off‐treatment).
1.39. Analysis.
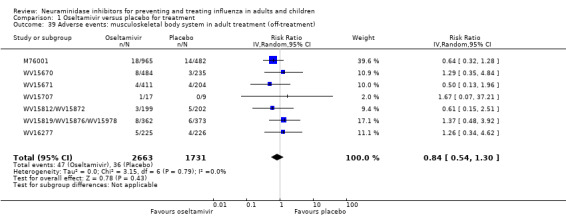
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 39 Adverse events: musculoskeletal body system in adult treatment (off‐treatment).
1.40. Analysis.
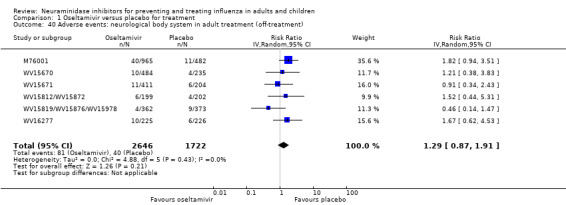
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 40 Adverse events: neurological body system in adult treatment (off‐treatment).
1.41. Analysis.
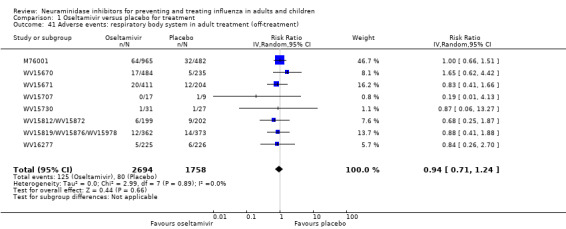
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 41 Adverse events: respiratory body system in adult treatment (off‐treatment).
1.42. Analysis.
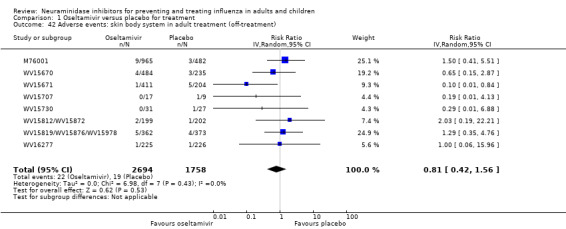
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 42 Adverse events: skin body system in adult treatment (off‐treatment).
1.43. Analysis.
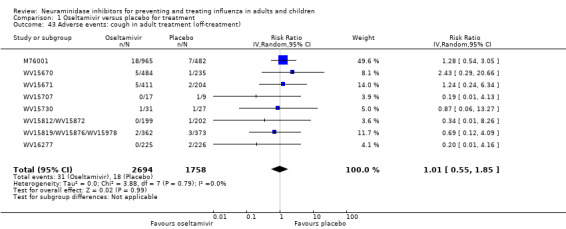
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 43 Adverse events: cough in adult treatment (off‐treatment).
1.44. Analysis.
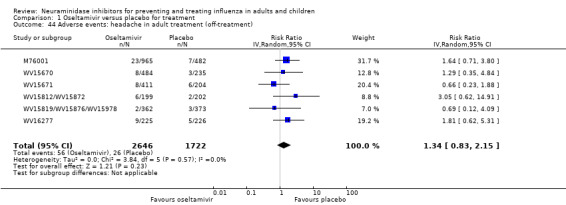
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 44 Adverse events: headache in adult treatment (off‐treatment).
1.45. Analysis.
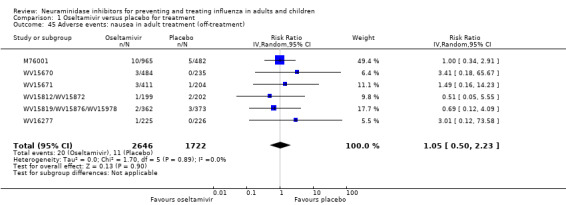
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 45 Adverse events: nausea in adult treatment (off‐treatment).
1.48. Analysis.
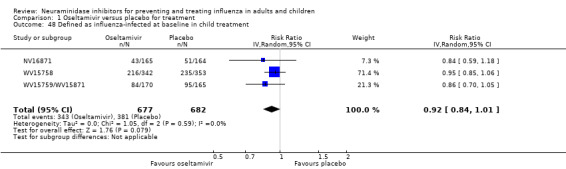
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 48 Defined as influenza‐infected at baseline in child treatment.
1.54. Analysis.
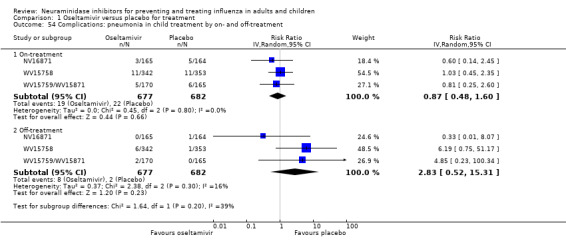
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 54 Complications: pneumonia in child treatment by on‐ and off‐treatment.
1.56. Analysis.
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 56 Withdrawal from child treatment trial due to adverse events.
1.57. Analysis.
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 57 All withdrawals from child treatment.
1.58. Analysis.
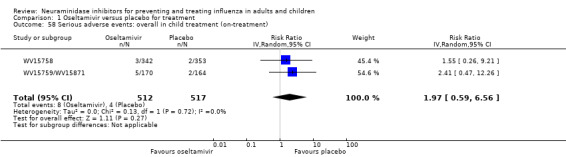
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 58 Serious adverse events: overall in child treatment (on‐treatment).
1.59. Analysis.
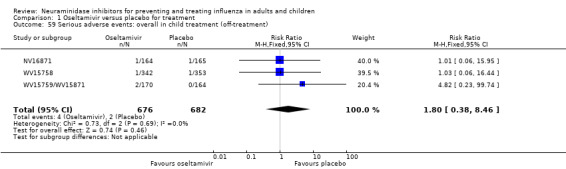
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 59 Serious adverse events: overall in child treatment (off‐treatment).
1.60. Analysis.
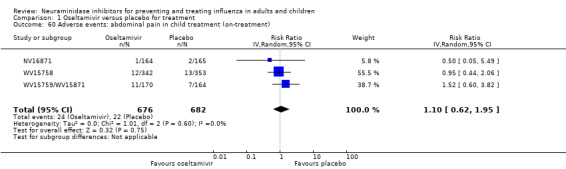
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 60 Adverse events: abdominal pain in child treatment (on‐treatment).
1.61. Analysis.
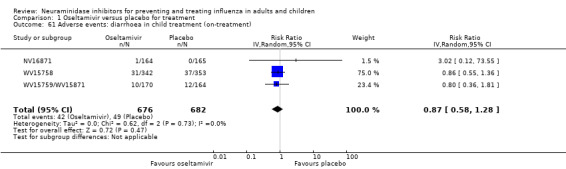
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 61 Adverse events: diarrhoea in child treatment (on‐treatment).
1.62. Analysis.
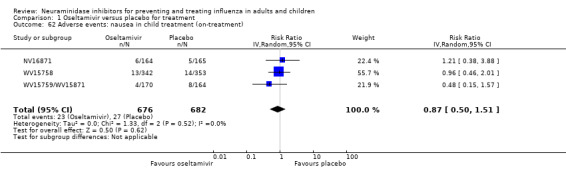
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 62 Adverse events: nausea in child treatment (on‐treatment).
1.64. Analysis.
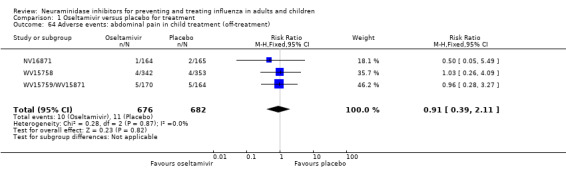
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 64 Adverse events: abdominal pain in child treatment (off‐treatment).
1.65. Analysis.
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 65 Adverse events: cough in child treatment (off‐treatment).
1.66. Analysis.
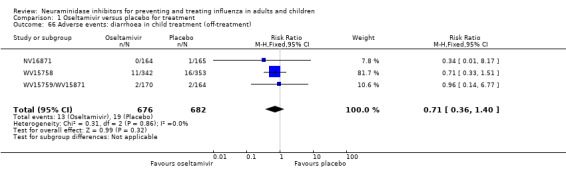
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 66 Adverse events: diarrhoea in child treatment (off‐treatment).
1.67. Analysis.
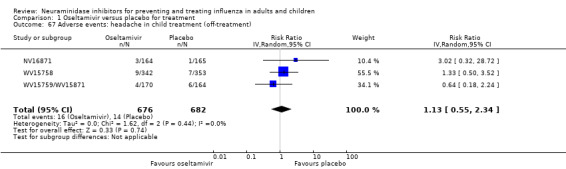
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 67 Adverse events: headache in child treatment (off‐treatment).
1.68. Analysis.
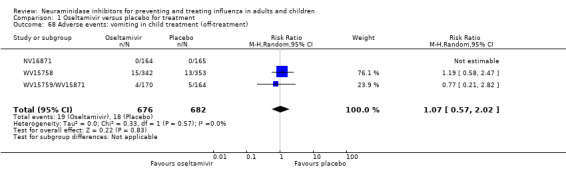
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 68 Adverse events: vomiting in child treatment (off‐treatment).
1.69. Analysis.
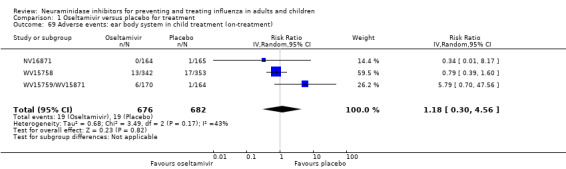
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 69 Adverse events: ear body system in child treatment (on‐treatment).
1.70. Analysis.
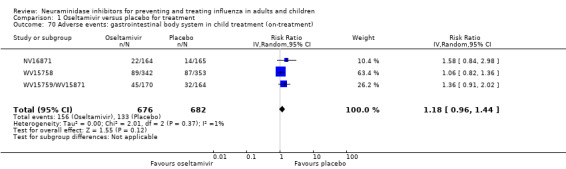
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 70 Adverse events: gastrointestinal body system in child treatment (on‐treatment).
1.71. Analysis.
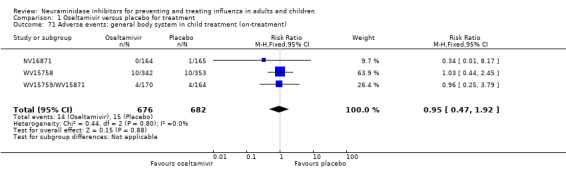
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 71 Adverse events: general body system in child treatment (on‐treatment).
1.72. Analysis.
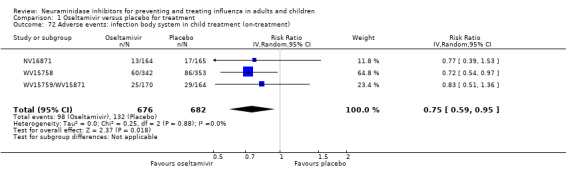
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 72 Adverse events: infection body system in child treatment (on‐treatment).
1.73. Analysis.
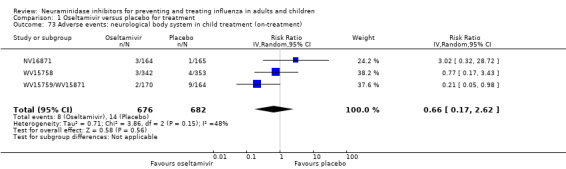
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 73 Adverse events: neurological body system in child treatment (on‐treatment).
1.74. Analysis.
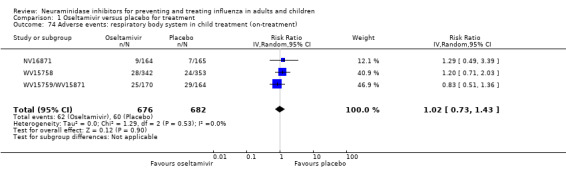
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 74 Adverse events: respiratory body system in child treatment (on‐treatment).
1.75. Analysis.
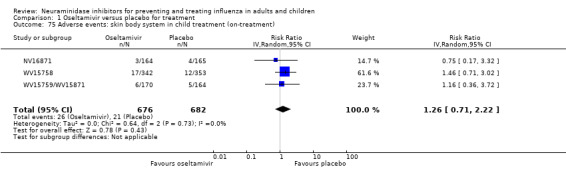
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 75 Adverse events: skin body system in child treatment (on‐treatment).
1.76. Analysis.
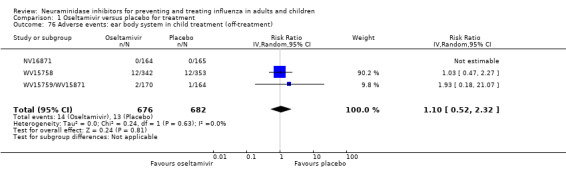
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 76 Adverse events: ear body system in child treatment (off‐treatment).
1.77. Analysis.
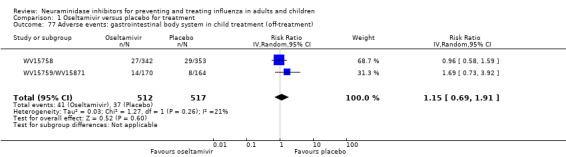
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 77 Adverse events: gastrointestinal body system in child treatment (off‐treatment).
1.78. Analysis.
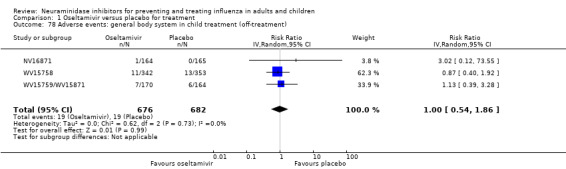
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 78 Adverse events: general body system in child treatment (off‐treatment).
1.79. Analysis.
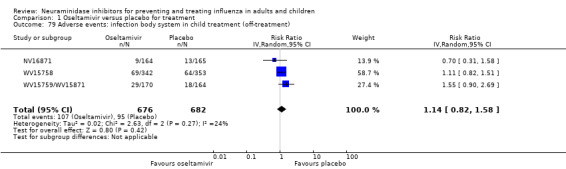
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 79 Adverse events: infection body system in child treatment (off‐treatment).
1.80. Analysis.
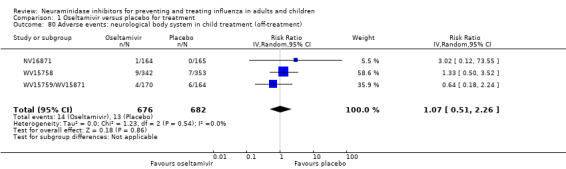
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 80 Adverse events: neurological body system in child treatment (off‐treatment).
1.81. Analysis.
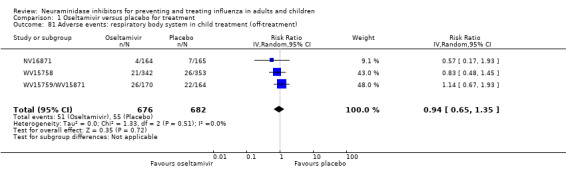
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 81 Adverse events: respiratory body system in child treatment (off‐treatment).
1.82. Analysis.
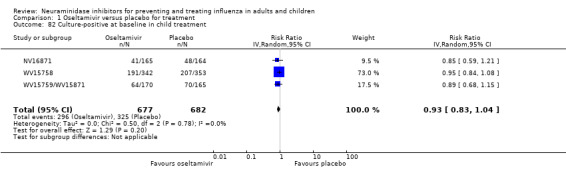
Comparison 1 Oseltamivir versus placebo for treatment, Outcome 82 Culture‐positive at baseline in child treatment.
Comparison 2. Oseltamivir versus placebo for prophylaxis.
2.2. Analysis.
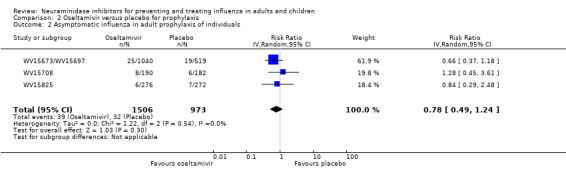
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 2 Asymptomatic influenza in adult prophylaxis of individuals.
2.5. Analysis.
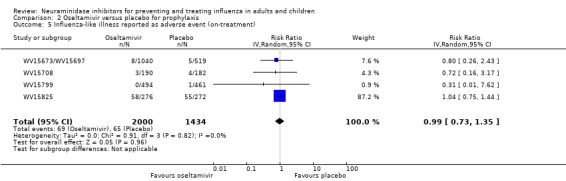
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 5 Influenza‐like illness reported as adverse event (on‐treatment).
2.6. Analysis.
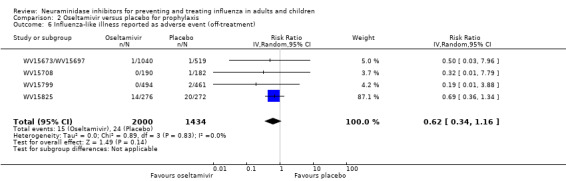
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 6 Influenza‐like illness reported as adverse event (off‐treatment).
2.10. Analysis.
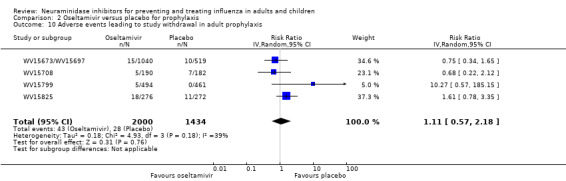
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 10 Adverse events leading to study withdrawal in adult prophylaxis.
2.11. Analysis.
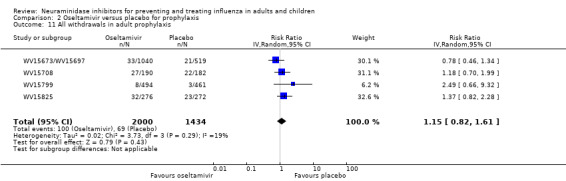
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 11 All withdrawals in adult prophylaxis.
2.12. Analysis.
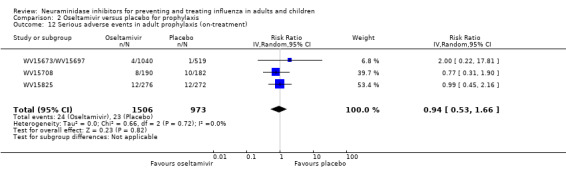
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 12 Serious adverse events in adult prophylaxis (on‐treatment).
2.13. Analysis.
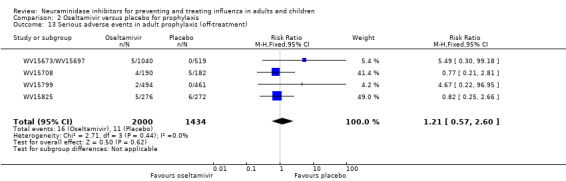
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 13 Serious adverse events in adult prophylaxis (off‐treatment).
2.14. Analysis.
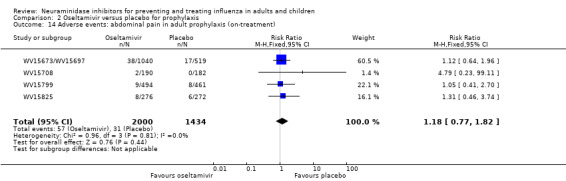
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 14 Adverse events: abdominal pain in adult prophylaxis (on‐treatment).
2.15. Analysis.
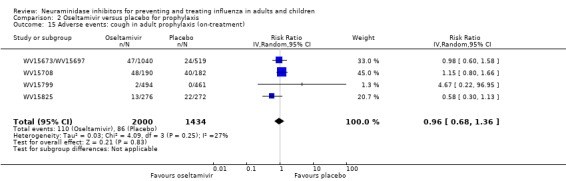
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 15 Adverse events: cough in adult prophylaxis (on‐treatment).
2.16. Analysis.
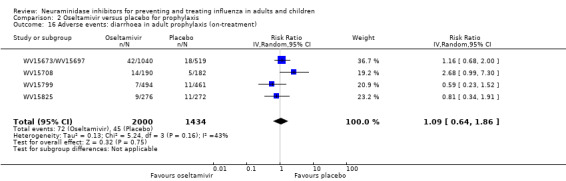
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 16 Adverse events: diarrhoea in adult prophylaxis (on‐treatment).
2.17. Analysis.
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 17 Adverse events: dizziness in adult prophylaxis (on‐treatment).
2.18. Analysis.
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 18 Adverse events: fatigue in adult prophylaxis (on‐treatment).
2.21. Analysis.
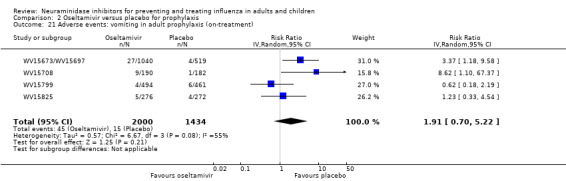
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 21 Adverse events: vomiting in adult prophylaxis (on‐treatment).
2.22. Analysis.
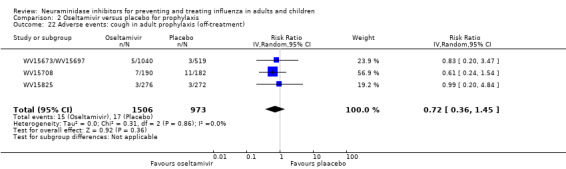
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 22 Adverse events: cough in adult prophylaxis (off‐treatment).
2.23. Analysis.
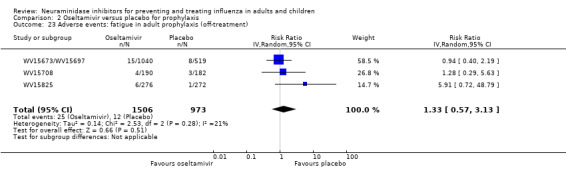
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 23 Adverse events: fatigue in adult prophylaxis (off‐treatment).
2.24. Analysis.
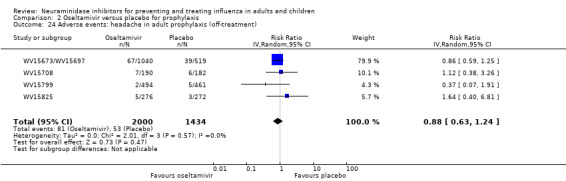
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 24 Adverse events: headache in adult prophylaxis (off‐treatment).
2.25. Analysis.
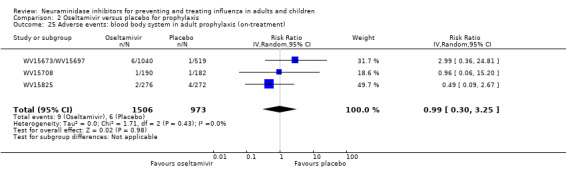
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 25 Adverse events: blood body system in adult prophylaxis (on‐treatment).
2.26. Analysis.
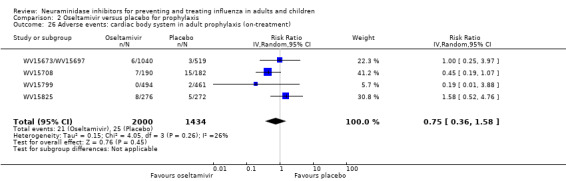
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 26 Adverse events: cardiac body system in adult prophylaxis (on‐treatment).
2.27. Analysis.
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 27 Adverse events: ear body system in adult prophylaxis (on treatment).
2.28. Analysis.
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 28 Adverse events: eye body system in adult prophylaxis (on‐treatment).
2.29. Analysis.
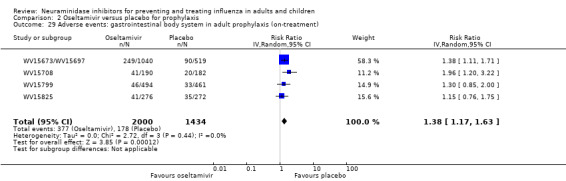
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 29 Adverse events: gastrointestinal body system in adult prophylaxis (on‐treatment).
2.30. Analysis.
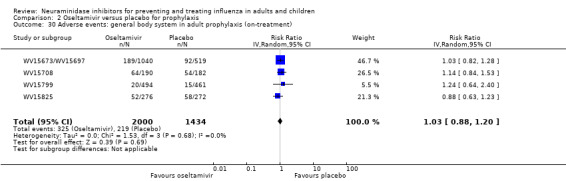
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 30 Adverse events: general body system in adult prophylaxis (on‐treatment).
2.31. Analysis.
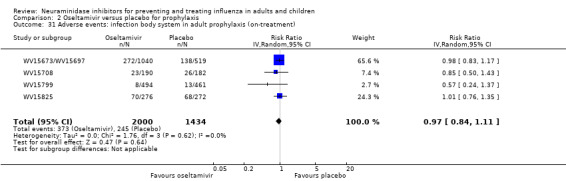
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 31 Adverse events: infection body system in adult prophylaxis (on‐treatment).
2.32. Analysis.
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 32 Adverse events: immune body system in adult prophylaxis (on‐treatment).
2.33. Analysis.
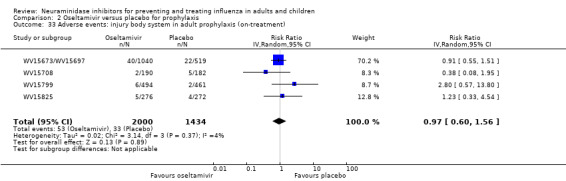
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 33 Adverse events: injury body system in adult prophylaxis (on‐treatment).
2.34. Analysis.
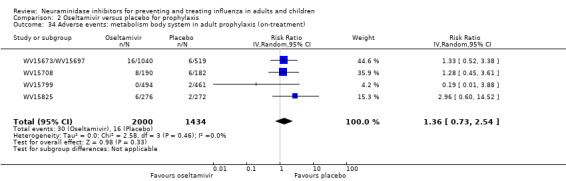
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 34 Adverse events: metabolism body system in adult prophylaxis (on‐treatment).
2.35. Analysis.
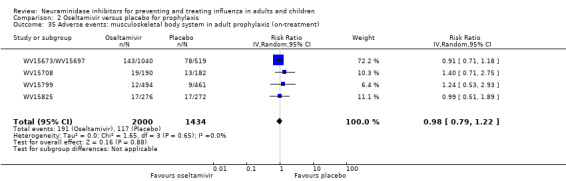
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 35 Adverse events: musculoskeletal body system in adult prophylaxis (on‐treatment).
2.36. Analysis.
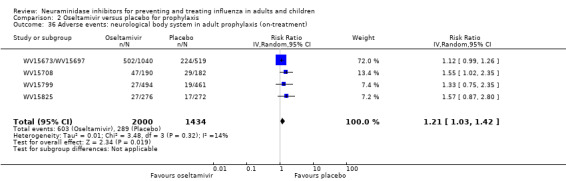
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 36 Adverse events: neurological body system in adult prophylaxis (on‐treatment).
2.37. Analysis.
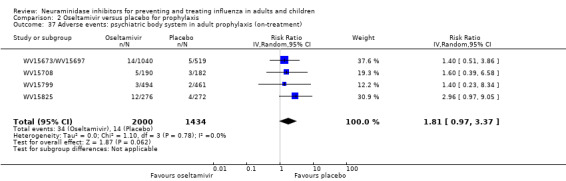
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 37 Adverse events: psychiatric body system in adult prophylaxis (on‐treatment).
2.39. Analysis.
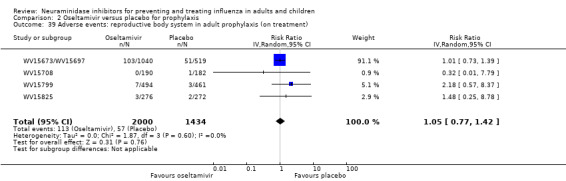
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 39 Adverse events: reproductive body system in adult prophylaxis (on treatment).
2.40. Analysis.
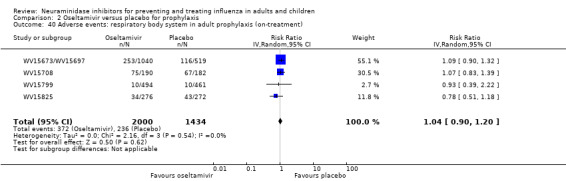
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 40 Adverse events: respiratory body system in adult prophylaxis (on‐treatment).
2.41. Analysis.
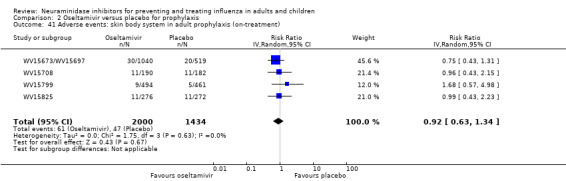
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 41 Adverse events: skin body system in adult prophylaxis (on‐treatment).
2.42. Analysis.
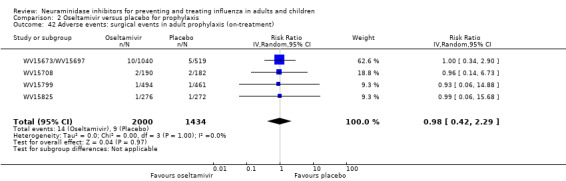
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 42 Adverse events: surgical events in adult prophylaxis (on‐treatment).
2.43. Analysis.
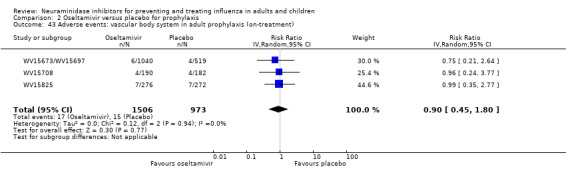
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 43 Adverse events: vascular body system in adult prophylaxis (on‐treatment).
2.44. Analysis.
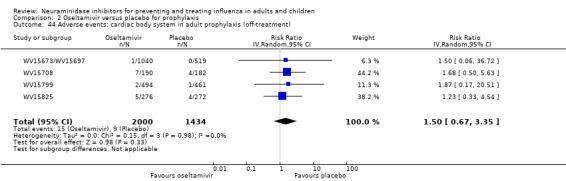
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 44 Adverse events: cardiac body system in adult prophylaxis (off‐treatment).
2.45. Analysis.
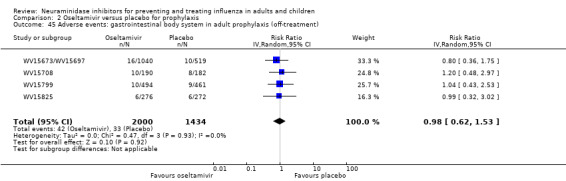
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 45 Adverse events: gastrointestinal body system in adult prophylaxis (off‐treatment).
2.46. Analysis.
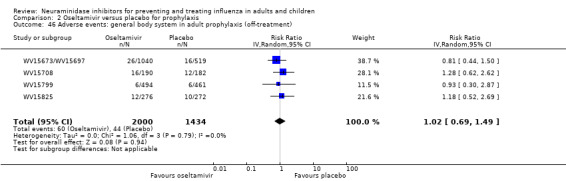
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 46 Adverse events: general body system in adult prophylaxis (off‐treatment).
2.47. Analysis.
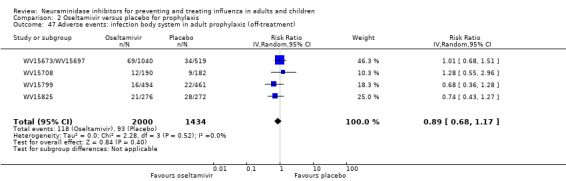
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 47 Adverse events: infection body system in adult prophylaxis (off‐treatment).
2.48. Analysis.
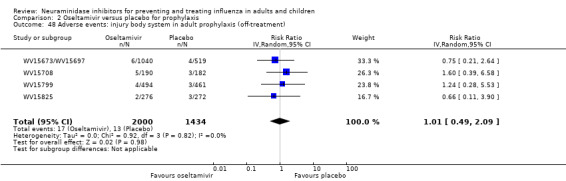
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 48 Adverse events: injury body system in adult prophylaxis (off‐treatment).
2.49. Analysis.
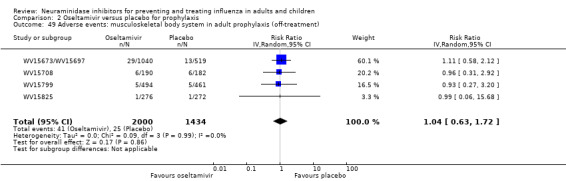
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 49 Adverse events: musculoskeletal body system in adult prophylaxis (off‐treatment).
2.50. Analysis.
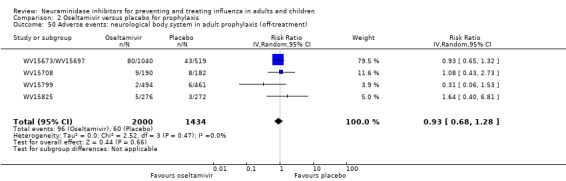
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 50 Adverse events: neurological body system in adult prophylaxis (off‐treatment).
2.51. Analysis.
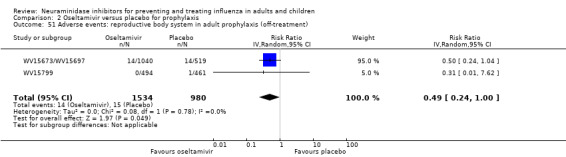
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 51 Adverse events: reproductive body system in adult prophylaxis (off‐treatment).
2.52. Analysis.
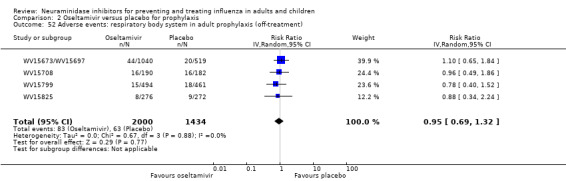
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 52 Adverse events: respiratory body system in adult prophylaxis (off‐treatment).
2.53. Analysis.
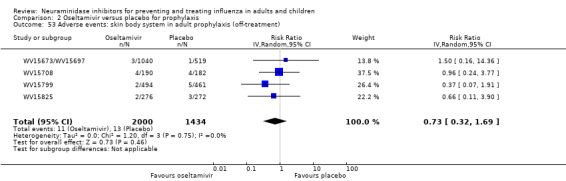
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 53 Adverse events: skin body system in adult prophylaxis (off‐treatment).
2.55. Analysis.
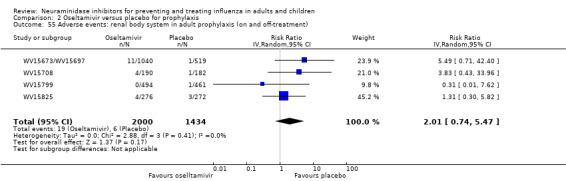
Comparison 2 Oseltamivir versus placebo for prophylaxis, Outcome 55 Adverse events: renal body system in adult prophylaxis (on and off‐treatment).
Comparison 3. Zanamivir versus placebo for treatment.
3.12. Analysis.
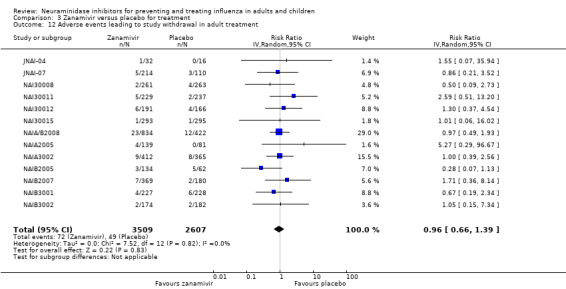
Comparison 3 Zanamivir versus placebo for treatment, Outcome 12 Adverse events leading to study withdrawal in adult treatment.
3.13. Analysis.
Comparison 3 Zanamivir versus placebo for treatment, Outcome 13 All withdrawals in adult treatment.
3.15. Analysis.
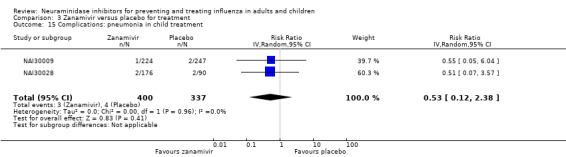
Comparison 3 Zanamivir versus placebo for treatment, Outcome 15 Complications: pneumonia in child treatment.
3.16. Analysis.
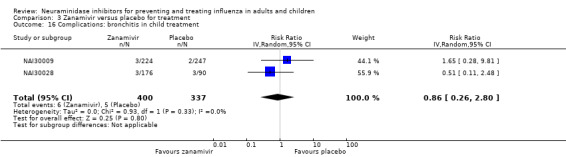
Comparison 3 Zanamivir versus placebo for treatment, Outcome 16 Complications: bronchitis in child treatment.
3.19. Analysis.
Comparison 3 Zanamivir versus placebo for treatment, Outcome 19 Proportion diagnosed as influenza‐infected in child treatment.
3.20. Analysis.
Comparison 3 Zanamivir versus placebo for treatment, Outcome 20 Proportion with four‐fold increase in antibodies in child treatment.
3.21. Analysis.
Comparison 3 Zanamivir versus placebo for treatment, Outcome 21 Proportion with positive culture at baseline in child treatment.
3.22. Analysis.
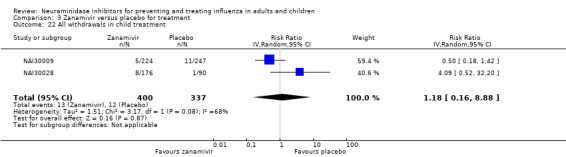
Comparison 3 Zanamivir versus placebo for treatment, Outcome 22 All withdrawals in child treatment.
3.23. Analysis.
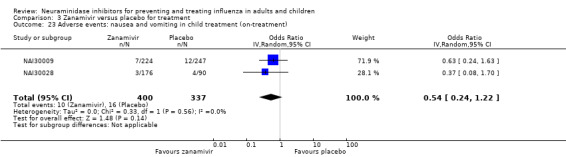
Comparison 3 Zanamivir versus placebo for treatment, Outcome 23 Adverse events: nausea and vomiting in child treatment (on‐treatment).
3.24. Analysis.
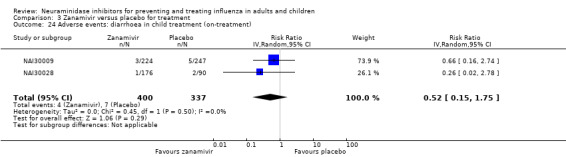
Comparison 3 Zanamivir versus placebo for treatment, Outcome 24 Adverse events: diarrhoea in child treatment (on‐treatment).
3.25. Analysis.
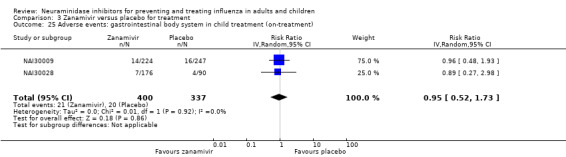
Comparison 3 Zanamivir versus placebo for treatment, Outcome 25 Adverse events: gastrointestinal body system in child treatment (on‐treatment).
3.26. Analysis.
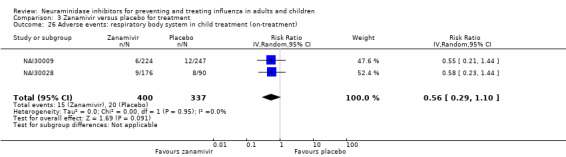
Comparison 3 Zanamivir versus placebo for treatment, Outcome 26 Adverse events: respiratory body system in child treatment (on‐treatment).
3.27. Analysis.
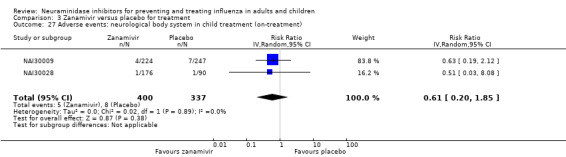
Comparison 3 Zanamivir versus placebo for treatment, Outcome 27 Adverse events: neurological body system in child treatment (on‐treatment).
3.28. Analysis.
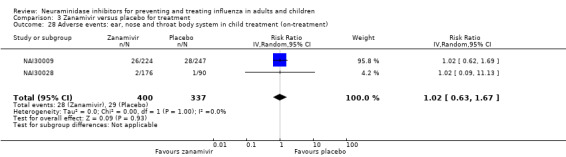
Comparison 3 Zanamivir versus placebo for treatment, Outcome 28 Adverse events: ear, nose and throat body system in child treatment (on‐treatment).
3.29. Analysis.
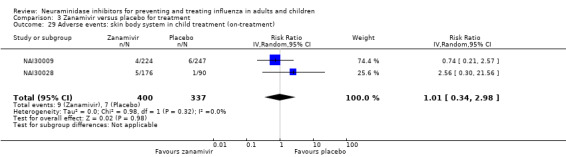
Comparison 3 Zanamivir versus placebo for treatment, Outcome 29 Adverse events: skin body system in child treatment (on‐treatment).
3.30. Analysis.
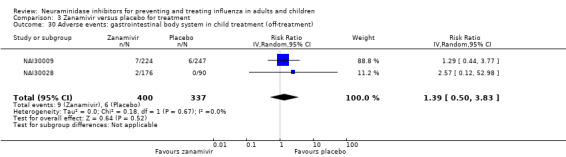
Comparison 3 Zanamivir versus placebo for treatment, Outcome 30 Adverse events: gastrointestinal body system in child treatment (off‐treatment).
3.31. Analysis.
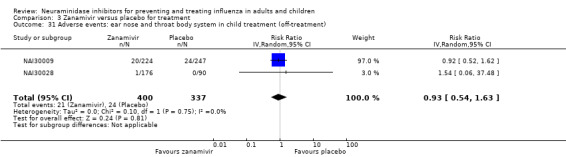
Comparison 3 Zanamivir versus placebo for treatment, Outcome 31 Adverse events: ear nose and throat body system in child treatment (off‐treatment).
3.34. Analysis.
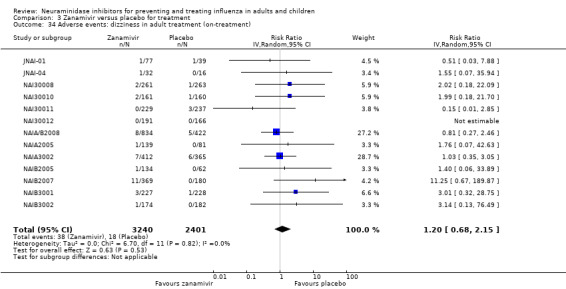
Comparison 3 Zanamivir versus placebo for treatment, Outcome 34 Adverse events: dizziness in adult treatment (on‐treatment).
3.36. Analysis.
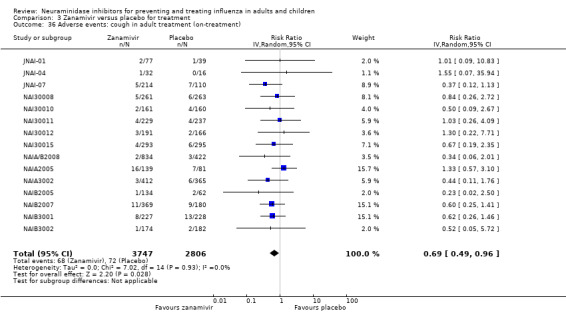
Comparison 3 Zanamivir versus placebo for treatment, Outcome 36 Adverse events: cough in adult treatment (on‐treatment).
3.37. Analysis.
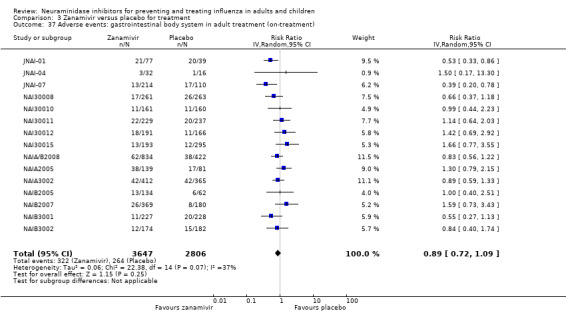
Comparison 3 Zanamivir versus placebo for treatment, Outcome 37 Adverse events: gastrointestinal body system in adult treatment (on‐treatment).
3.38. Analysis.
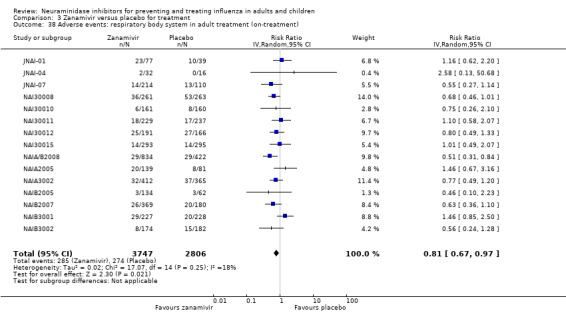
Comparison 3 Zanamivir versus placebo for treatment, Outcome 38 Adverse events: respiratory body system in adult treatment (on‐treatment).
3.39. Analysis.
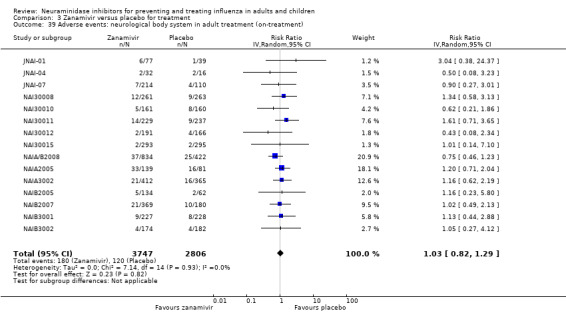
Comparison 3 Zanamivir versus placebo for treatment, Outcome 39 Adverse events: neurological body system in adult treatment (on‐treatment).
3.40. Analysis.
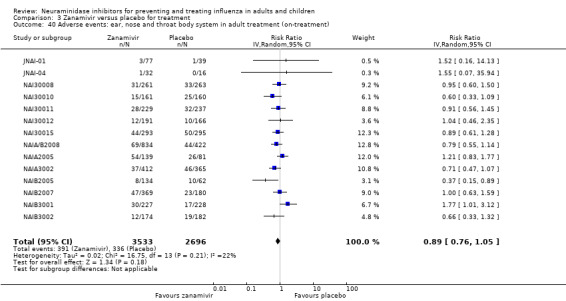
Comparison 3 Zanamivir versus placebo for treatment, Outcome 40 Adverse events: ear, nose and throat body system in adult treatment (on‐treatment).
3.41. Analysis.
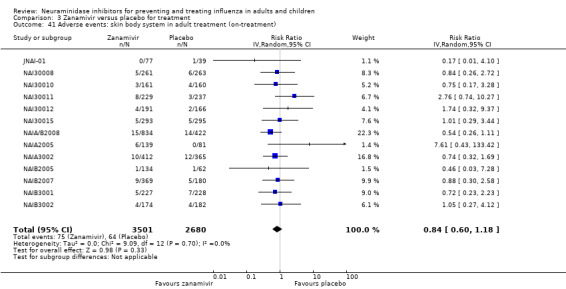
Comparison 3 Zanamivir versus placebo for treatment, Outcome 41 Adverse events: skin body system in adult treatment (on‐treatment).
3.42. Analysis.
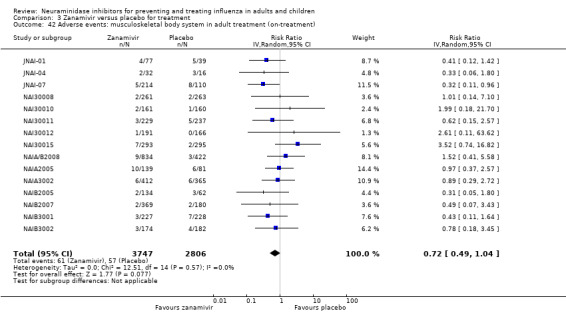
Comparison 3 Zanamivir versus placebo for treatment, Outcome 42 Adverse events: musculoskeletal body system in adult treatment (on‐treatment).
3.43. Analysis.
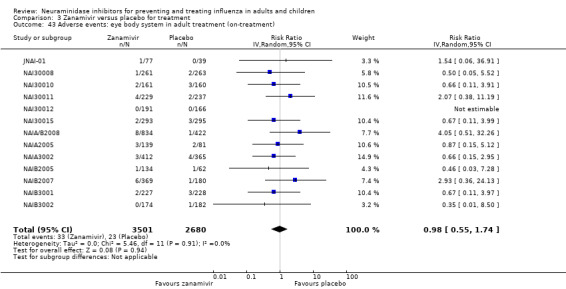
Comparison 3 Zanamivir versus placebo for treatment, Outcome 43 Adverse events: eye body system in adult treatment (on‐treatment).
3.44. Analysis.
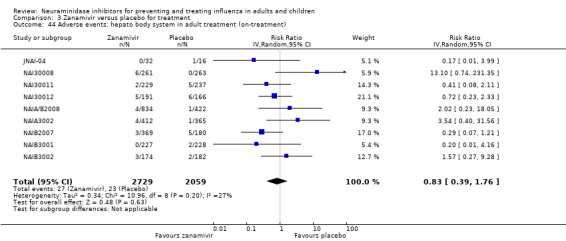
Comparison 3 Zanamivir versus placebo for treatment, Outcome 44 Adverse events: hepato body system in adult treatment (on‐treatment).
3.46. Analysis.
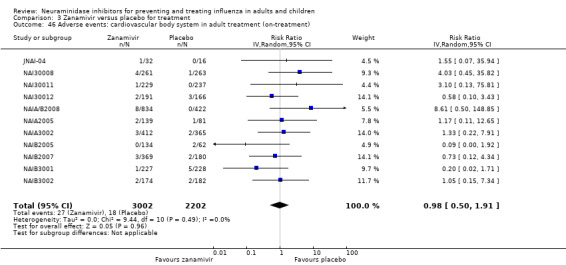
Comparison 3 Zanamivir versus placebo for treatment, Outcome 46 Adverse events: cardiovascular body system in adult treatment (on‐treatment).
3.47. Analysis.
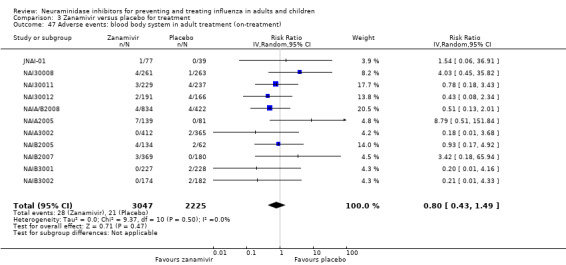
Comparison 3 Zanamivir versus placebo for treatment, Outcome 47 Adverse events: blood body system in adult treatment (on‐treatment).
3.49. Analysis.
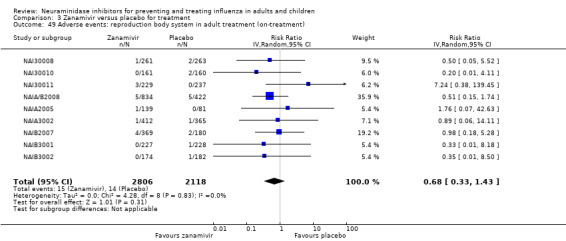
Comparison 3 Zanamivir versus placebo for treatment, Outcome 49 Adverse events: reproduction body system in adult treatment (on‐treatment).
3.50. Analysis.
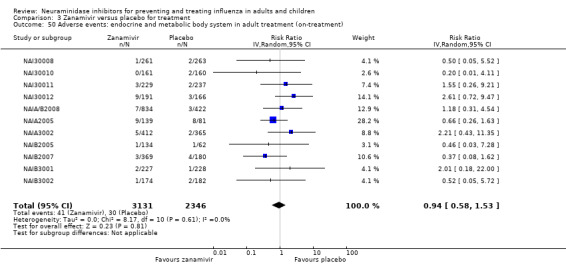
Comparison 3 Zanamivir versus placebo for treatment, Outcome 50 Adverse events: endocrine and metabolic body system in adult treatment (on‐treatment).
3.51. Analysis.
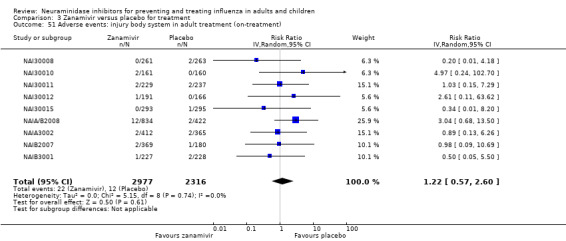
Comparison 3 Zanamivir versus placebo for treatment, Outcome 51 Adverse events: injury body system in adult treatment (on‐treatment).
3.52. Analysis.
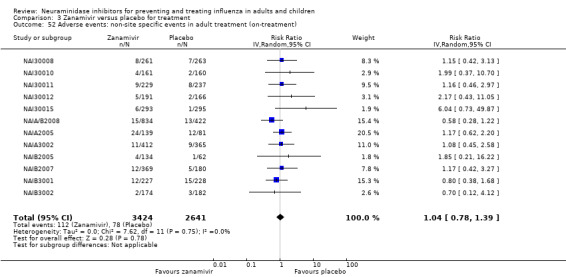
Comparison 3 Zanamivir versus placebo for treatment, Outcome 52 Adverse events: non‐site specific events in adult treatment (on‐treatment).
3.53. Analysis.
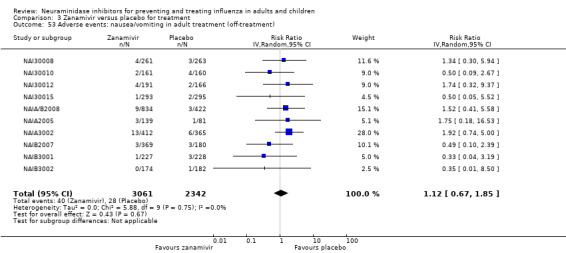
Comparison 3 Zanamivir versus placebo for treatment, Outcome 53 Adverse events: nausea/vomiting in adult treatment (off‐treatment).
3.54. Analysis.
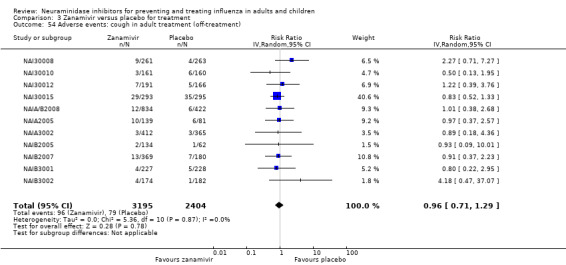
Comparison 3 Zanamivir versus placebo for treatment, Outcome 54 Adverse events: cough in adult treatment (off‐treatment).
3.55. Analysis.
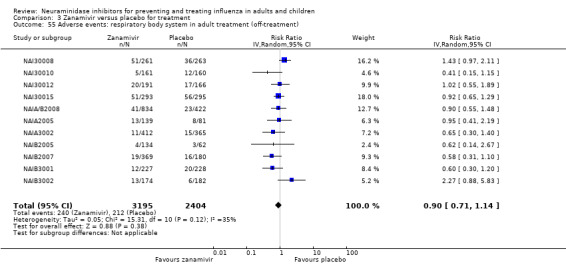
Comparison 3 Zanamivir versus placebo for treatment, Outcome 55 Adverse events: respiratory body system in adult treatment (off‐treatment).
3.56. Analysis.
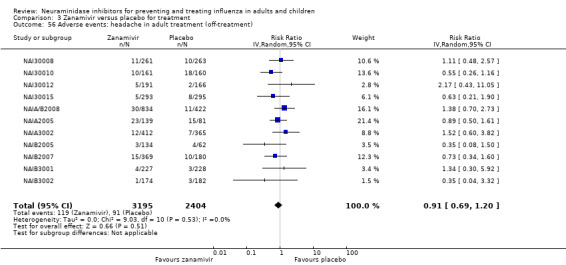
Comparison 3 Zanamivir versus placebo for treatment, Outcome 56 Adverse events: headache in adult treatment (off‐treatment).
3.57. Analysis.
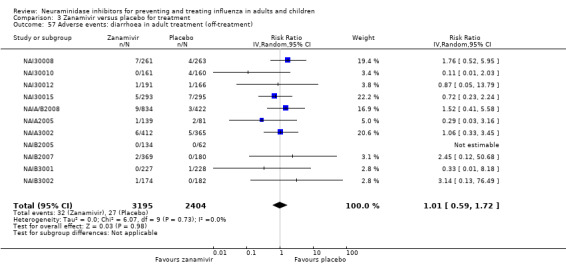
Comparison 3 Zanamivir versus placebo for treatment, Outcome 57 Adverse events: diarrhoea in adult treatment (off‐treatment).
3.58. Analysis.
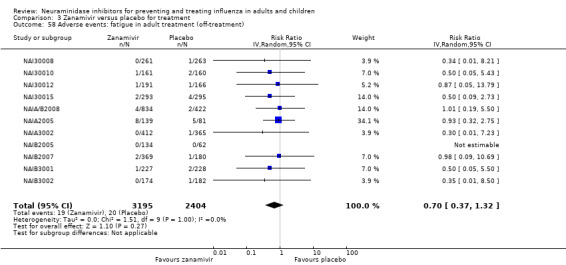
Comparison 3 Zanamivir versus placebo for treatment, Outcome 58 Adverse events: fatigue in adult treatment (off‐treatment).
3.59. Analysis.
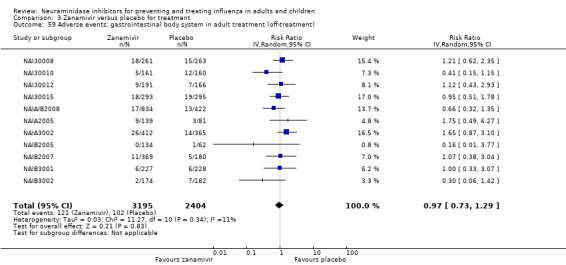
Comparison 3 Zanamivir versus placebo for treatment, Outcome 59 Adverse events: gastrointestinal body system in adult treatment (off‐treatment).
3.60. Analysis.
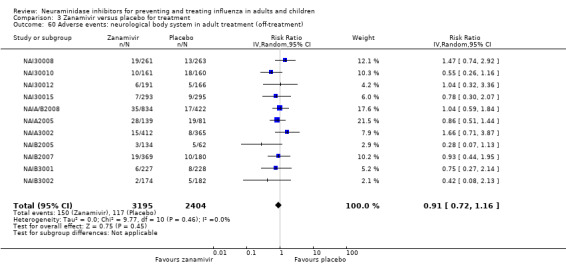
Comparison 3 Zanamivir versus placebo for treatment, Outcome 60 Adverse events: neurological body system in adult treatment (off‐treatment).
3.61. Analysis.
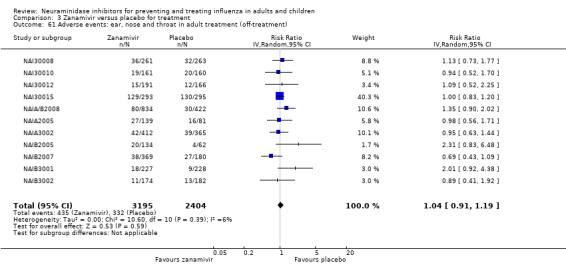
Comparison 3 Zanamivir versus placebo for treatment, Outcome 61 Adverse events: ear, nose and throat in adult treatment (off‐treatment).
3.62. Analysis.
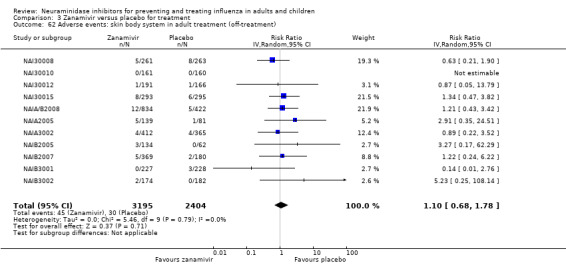
Comparison 3 Zanamivir versus placebo for treatment, Outcome 62 Adverse events: skin body system in adult treatment (off‐treatment).
3.63. Analysis.
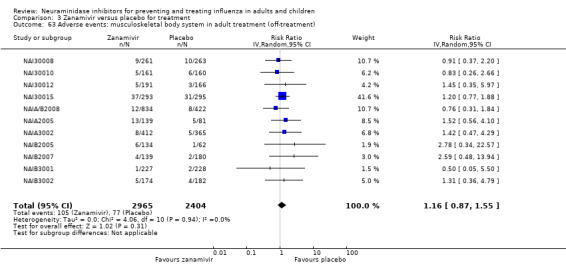
Comparison 3 Zanamivir versus placebo for treatment, Outcome 63 Adverse events: musculoskeletal body system in adult treatment (off‐treatment).
3.64. Analysis.
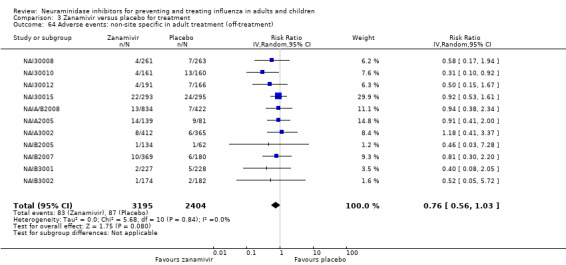
Comparison 3 Zanamivir versus placebo for treatment, Outcome 64 Adverse events: non‐site specific in adult treatment (off‐treatment).
3.65. Analysis.
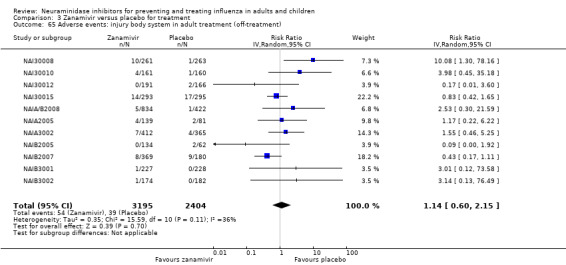
Comparison 3 Zanamivir versus placebo for treatment, Outcome 65 Adverse events: injury body system in adult treatment (off‐treatment).
3.66. Analysis.
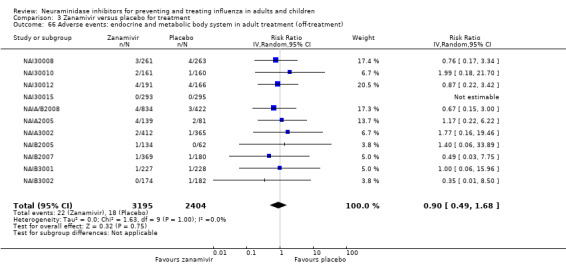
Comparison 3 Zanamivir versus placebo for treatment, Outcome 66 Adverse events: endocrine and metabolic body system in adult treatment (off‐treatment).
3.67. Analysis.
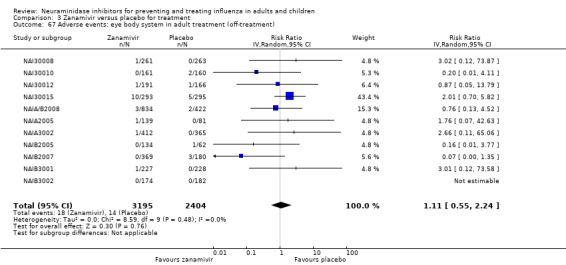
Comparison 3 Zanamivir versus placebo for treatment, Outcome 67 Adverse events: eye body system in adult treatment (off‐treatment).
Comparison 4. Zanamivir versus placebo for prophylaxis.
4.9. Analysis.
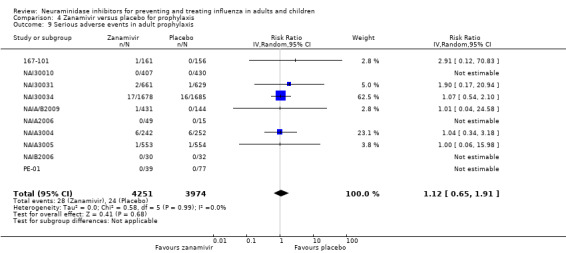
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 9 Serious adverse events in adult prophylaxis.
4.10. Analysis.
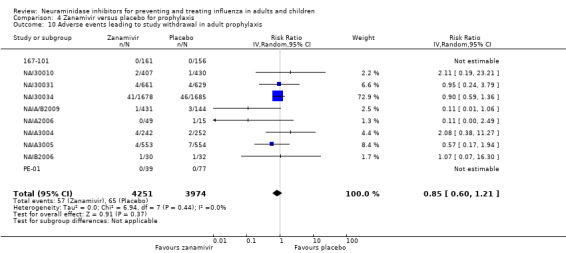
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 10 Adverse events leading to study withdrawal in adult prophylaxis.
4.11. Analysis.
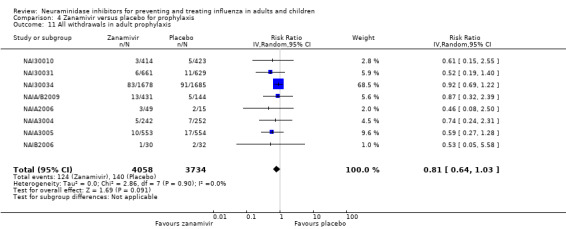
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 11 All withdrawals in adult prophylaxis.
4.12. Analysis.
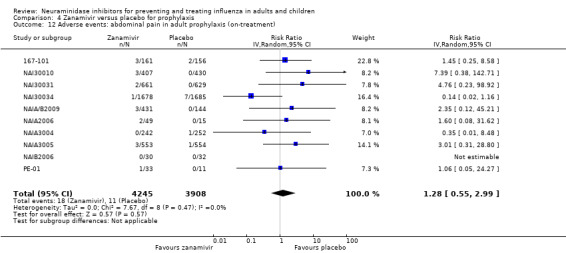
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 12 Adverse events: abdominal pain in adult prophylaxis (on‐treatment).
4.13. Analysis.
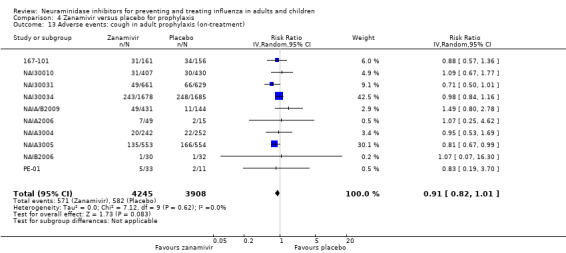
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 13 Adverse events: cough in adult prophylaxis (on‐treatment).
4.14. Analysis.
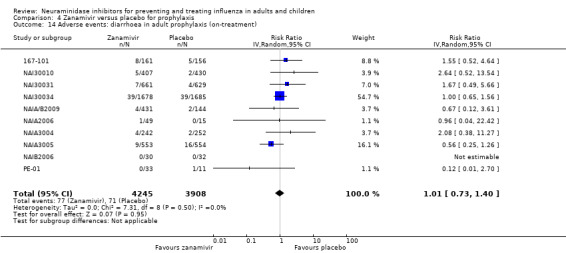
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 14 Adverse events: diarrhoea in adult prophylaxis (on‐treatment).
4.15. Analysis.
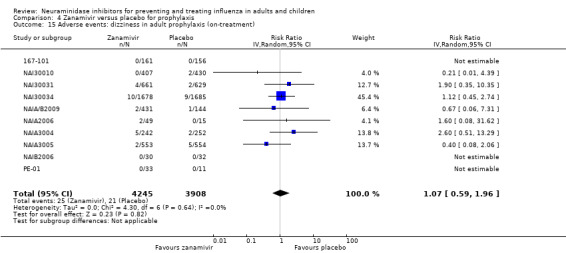
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 15 Adverse events: dizziness in adult prophylaxis (on‐treatment).
4.16. Analysis.
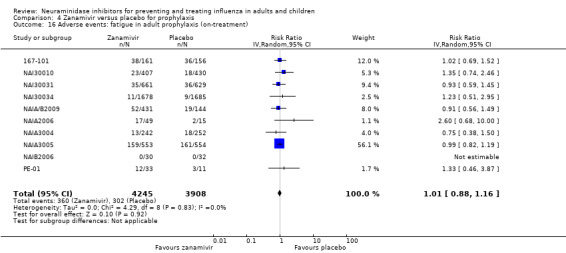
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 16 Adverse events: fatigue in adult prophylaxis (on‐treatment).
4.17. Analysis.
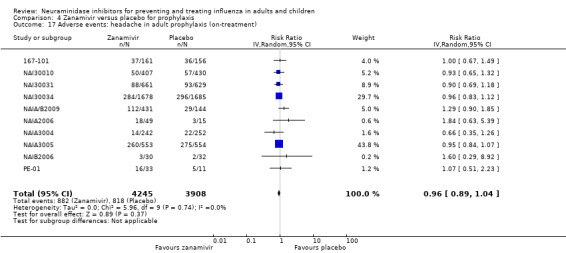
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 17 Adverse events: headache in adult prophylaxis (on‐treatment).
4.18. Analysis.
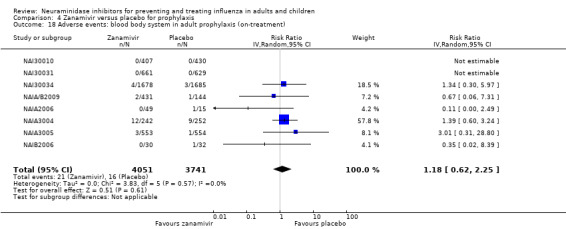
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 18 Adverse events: blood body system in adult prophylaxis (on‐treatment).
4.19. Analysis.
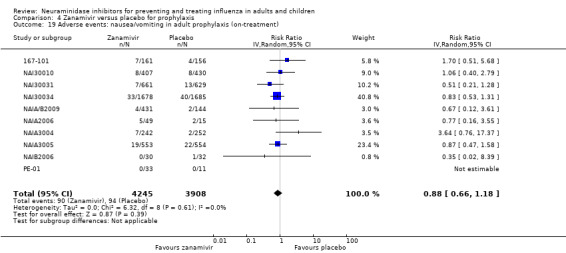
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 19 Adverse events: nausea/vomiting in adult prophylaxis (on‐treatment).
4.20. Analysis.
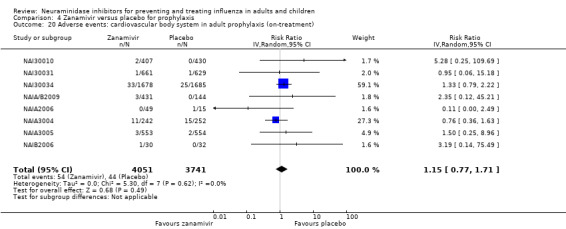
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 20 Adverse events: cardiovascular body system in adult prophylaxis (on‐treatment).
4.21. Analysis.
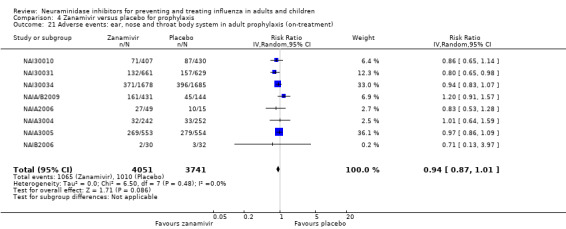
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 21 Adverse events: ear, nose and throat body system in adult prophylaxis (on‐treatment).
4.22. Analysis.
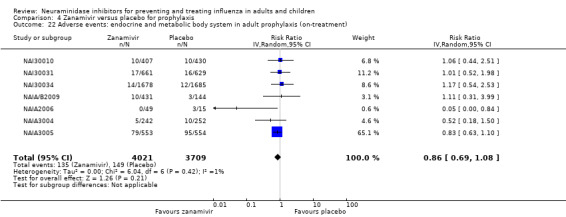
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 22 Adverse events: endocrine and metabolic body system in adult prophylaxis (on‐treatment).
4.23. Analysis.
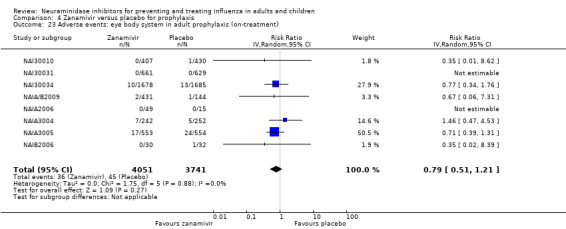
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 23 Adverse events: eye body system in adult prophylaxis (on‐treatment).
4.24. Analysis.
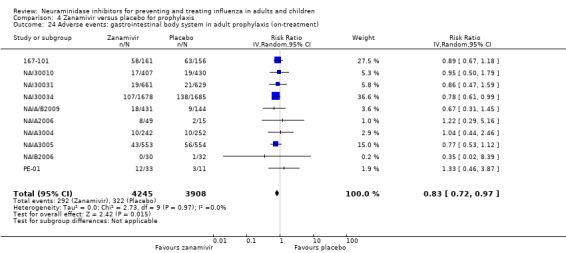
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 24 Adverse events: gastrointestinal body system in adult prophylaxis (on‐treatment).
4.25. Analysis.
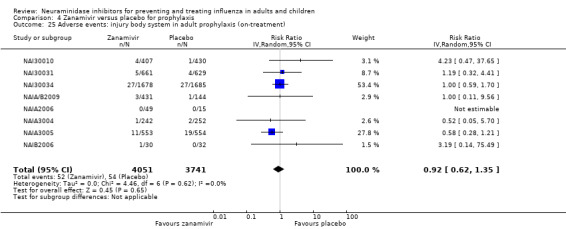
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 25 Adverse events: injury body system in adult prophylaxis (on‐treatment).
4.26. Analysis.
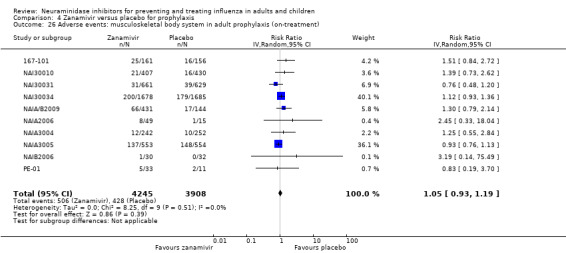
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 26 Adverse events: musculoskeletal body system in adult prophylaxis (on‐treatment).
4.27. Analysis.
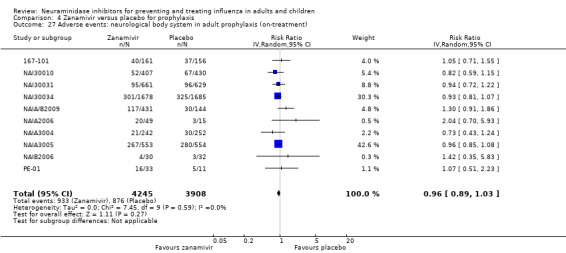
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 27 Adverse events: neurological body system in adult prophylaxis (on‐treatment).
4.28. Analysis.
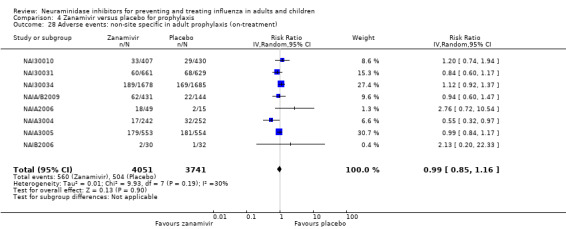
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 28 Adverse events: non‐site specific in adult prophylaxis (on‐treatment).
4.31. Analysis.
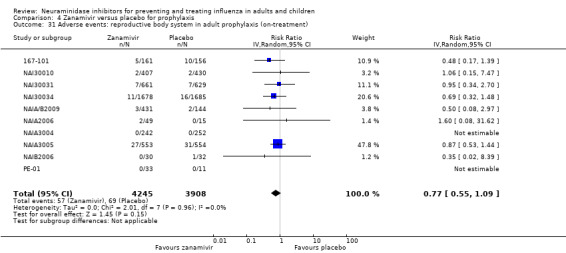
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 31 Adverse events: reproductive body system in adult prophylaxis (on‐treatment).
4.32. Analysis.
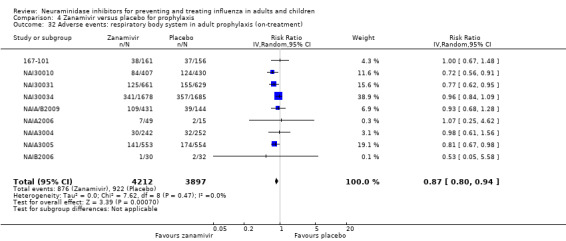
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 32 Adverse events: respiratory body system in adult prophylaxis (on‐treatment).
4.33. Analysis.
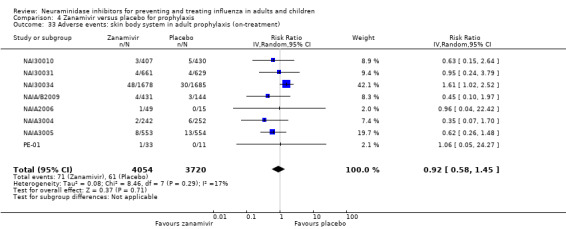
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 33 Adverse events: skin body system in adult prophylaxis (on‐treatment).
4.34. Analysis.
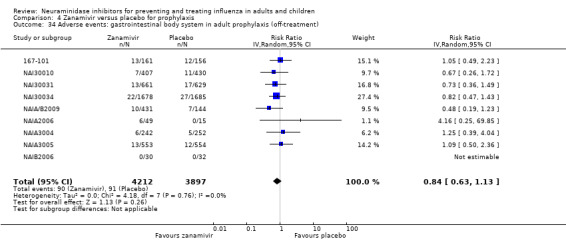
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 34 Adverse events: gastrointestinal body system in adult prophylaxis (off‐treatment).
4.35. Analysis.
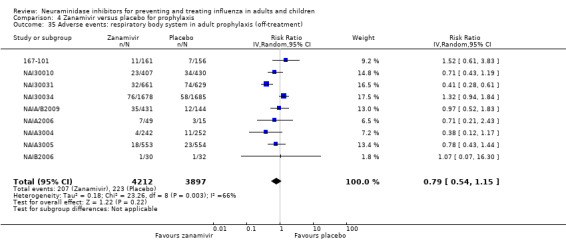
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 35 Adverse events: respiratory body system in adult prophylaxis (off‐treatment).
4.36. Analysis.
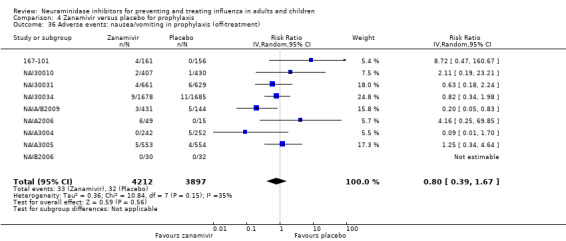
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 36 Adverse events: nausea/vomiting in prophylaxis (off‐treatment).
4.37. Analysis.
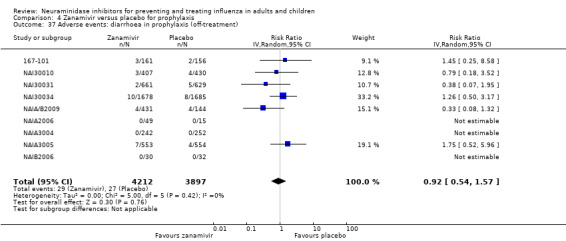
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 37 Adverse events: diarrhoea in prophylaxis (off‐treatment).
4.38. Analysis.
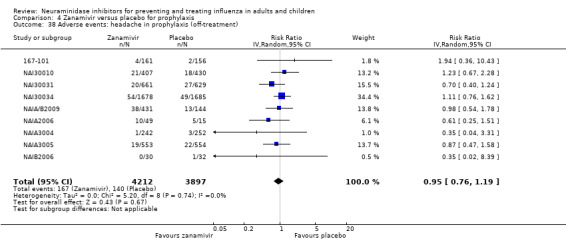
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 38 Adverse events: headache in prophylaxis (off‐treatment).
4.39. Analysis.
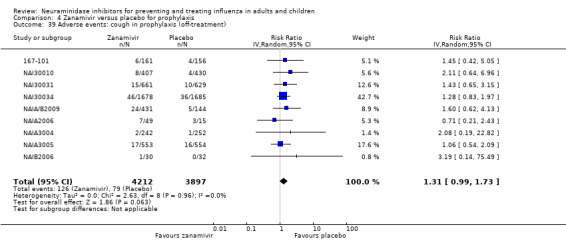
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 39 Adverse events: cough in prophylaxis (off‐treatment).
4.40. Analysis.
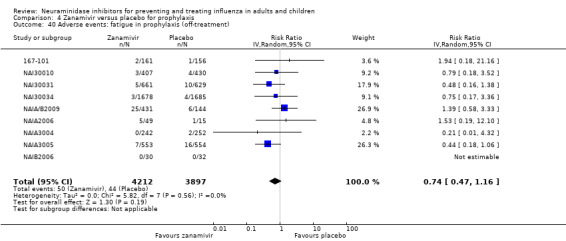
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 40 Adverse events: fatigue in prophylaxis (off‐treatment).
4.41. Analysis.
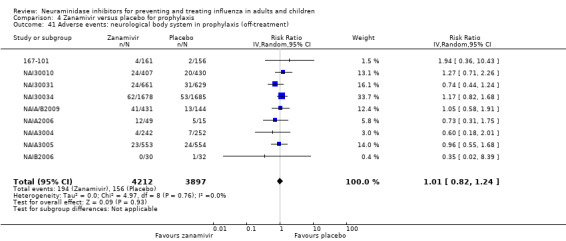
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 41 Adverse events: neurological body system in prophylaxis (off‐treatment).
4.42. Analysis.
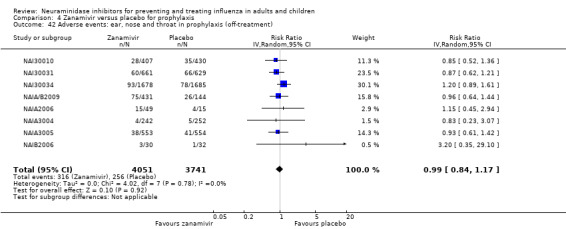
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 42 Adverse events: ear, nose and throat in prophylaxis (off‐treatment).
4.43. Analysis.
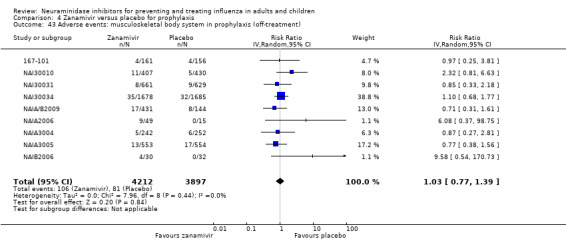
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 43 Adverse events: musculoskeletal body system in prophylaxis (off‐treatment).
4.44. Analysis.
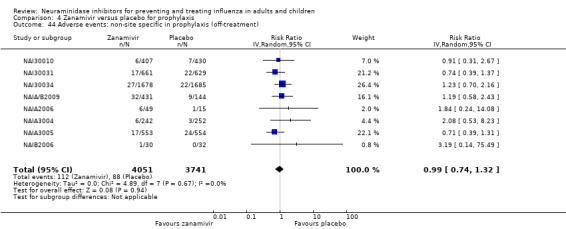
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 44 Adverse events: non‐site specific in prophylaxis (off‐treatment).
4.45. Analysis.
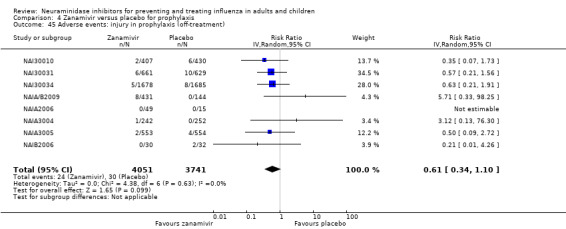
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 45 Adverse events: injury in prophylaxis (off‐treatment).
4.46. Analysis.
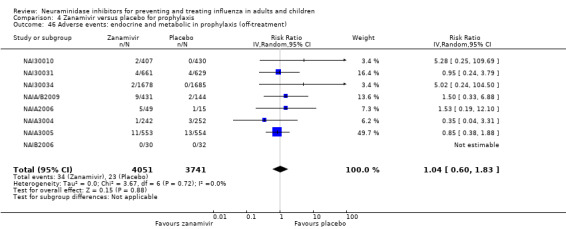
Comparison 4 Zanamivir versus placebo for prophylaxis, Outcome 46 Adverse events: endocrine and metabolic in prophylaxis (off‐treatment).
Comparison 5. Neuraminidase inhibitor versus placebo for treatment or prophylaxis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Complications: pneumonia | 32 | 22565 | Risk Ratio (IV, Random, 95% CI) | 0.72 [0.55, 0.95] |
| 1.1 Unclear diagnostic confirmation capture | 25 | 18905 | Risk Ratio (IV, Random, 95% CI) | 0.51 [0.35, 0.75] |
| 1.2 Clear diagnostic confirmation capture | 7 | 3660 | Risk Ratio (IV, Random, 95% CI) | 1.01 [0.69, 1.47] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
167‐101.
| Methods | A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study of zanamivir for the prophylaxis of influenza infection. Phase III study Location, number of centres: Japan, 5 centres Duration of study: 36 days (duration of administration: 28 days) |
|
| Participants | Number screened: not available Number randomised: 319 (zanamivir: 161; placebo: 158) Number completed (participants for efficacy analysis): 317 (zanamivir: 161; placebo: 156) M = 26.5%, F = 73.5% among 317 Mean age: 33.6 years Baseline details: all (at least most) Japanese; 1 (0.6%) vaccinated in zanamivir group, 0 in placebo group (within 8 months) Inclusion criteria: 1. Male or female healthcare workers aged 18 years and above at the time of the study 2. Participants who were able to take the first dose of study medication following notification of an influenza outbreak by the investigator 3. Participants who were willing and able to give written informed consent to participate in the study (if the subject is aged less than 20 years old, written informed consent is necessary from the subject itself and from the parental authority) 4. Able to use the diskhaler properly 5. Participants were willing and able to adhere to the protocol and the instructions of the investigator 6. Participants who, in the opinion of the investigator, could complete the symptom‐diary card Exclusion criteria: 1. Participants who have influenza‐like illness (one or more of fever 37.5 °C or more, headache, sore throat, feverishness, muscle or joint pain, or cough) prior to the study during this influenza season 2. Participants who have any underlying illness which could influence the efficacy and safety assessment 3. Participants who were known or suspected to be hypersensitive to any component of the study medication (GG167, lactose) and relief medications (acetaminophen and dextromethorphan) 4. Participants who had received an investigational drug in the previous 3 months 5. Participants who had received any influenza antiviral therapy in the previous 7 days, e.g. amantadine 6. Participants who were pregnant, breast‐feeding or intending to become pregnant during the study 7. Evidence of, or history of, such things as alcoholism, drug abuse, psychiatric disorders 8. Other conditions which the investigator decide inappropriate for participants of the study Definition of patient populations for analysis Intention‐to‐treat population: N = 319 (zanamivir: 161; placebo: 158): number randomised irrespective of the study medication This is a secondary population for the analysis of efficacy data Population for efficacy analysis: Full analysis set (FAS): N = 317 (zanamivir: 161; placebo: 156) All randomised participants except those who did not take at least 1 dose of study agent or whose efficacy data were not available. This is the secondary population for the analysis of efficacy Non‐vaccinated set (NVS) (N = 316) (zanamivir: 160; placebo: 156) All non‐vaccinated randomised participants who took at least 1 dose of study drug. This is the primary population for the analysis of efficacy Per‐protocol set (PPS) (N = 315) (zanamivir: 159; placebo: 156) [needs translation] Safety population (N = 317) (zanamivir: 161; placebo: 156) All randomised participants except those who met the exclusion criteria and did not take at least 1 dose of study agent. Primary population for analysis of safety data. This is the secondary population for the analysis of efficacy |
|
| Interventions | Zanamivir 10 mg once daily Placebo |
|
| Outcomes |
The primary efficacy endpoint was the proportion of subjects who, during prophylaxis (d1 to d28), developed symptomatic, laboratory‐confirmed influenza A or B infection (1) "Symptomatic influenza" was defined as the presence of at least 2 of the following influenza symptoms: fever of 37.5 °C or more, headache, sore throat, feverishness, myalgia, cough. Symptoms must have been present concurrently for 3 consecutive diary card entries (approximately 1.5 days) (2) Laboratory confirmation of influenza infection was a positive result by any of the following methods: culture or seroconversion The secondary efficacy endpoint 1. Proportion of subjects who, during prophylaxis (d 3 to d 28), developed symptomatic, laboratory‐confirmed influenza A or B infection that satisfied the 2 conditions above (1) and (2) 2. Proportion of subjects who, during prophylaxis (d 1 to d 28), developed laboratory‐confirmed influenza infection 3. Proportion of subjects who, during prophylaxis (d 1 to d 28), developed laboratory‐confirmed influenza infection and developed fever of 37.5 °C or more 4. Proportion of subjects who, during prophylaxis (d 1 to d 28), developed fever of 37.5 °C or more irrespective of the laboratory test results 5. Number of days, out of 28, the subject recorded use of relief medications (acetaminophen and cough suppressant) 6. Maximum recorded score on diary card 7. Development of secondary complications of influenza 8. Other outcomes to explore unique aspects of zanamivir based on the results Safety outcome: The safety measure was adverse events. Adverse events were defined as any unwanted or unexpected events. However, symptoms that were observed or did not worsen were not included as adverse events (pg 69 to 71/122; protocol) |
|
| Notes | Study period: 20 December 1999 to 3 March 2000 There was notable inconsistency in the results across different outcomes (see 'Risk of bias' section). The time period for assessment of antibody rise may have been inappropriate and increased the risk of bias in observed effects on outcomes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | 1. Participants were medical workers. There may have been selection bias prior to randomisation. pg 32/96 (CSR‐PTC) and pg 36/122 (protocol) 2. The protocol says (pg 43/122, protocol), "study agents were randomly allocated" before distribution of the block (kumi) of study agents. However the random sequence generation method is not described. Randomisation was done before the distribution of the block of the study agents. Each block (kumi) contained 6 packages of study agents (3 packages for zanamivir and 3 packages for placebo) for 6 participants |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Unequal proportion of drop‐outs between groups |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Unequal proportion of drop‐outs between groups |
| Incomplete outcome data (attrition bias) Safety data | High risk | Unequal proportion of drop‐outs between groups |
| Selective reporting (reporting bias) | High risk | The placebo contained lactose, which may cause bronchospasm in some participants. Hence the placebo used in this study may be a good control for the efficacy analysis but is not a good control for the safety analysis, because it increases the probability of adverse events in the control (lactose) group |
| Other bias | High risk | Use of healthcare workers could have resulted in under‐reporting of adverse events |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both treatments were identical in colour |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No evidence of code‐breaking |
JNAI‐01.
| Methods | Double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre trial to investigate the efficacy, safety and route of administration of zanamivir in the treatment of influenza A and B | |
| Participants |
Inclusion criteria: Subjects who met the following criteria and were judged to have influenza A or B viral infection by the investigator when influenza A or B was circulating in the region: 1. Otherwise healthy males or females aged 16 to 65 years old. Inpatient or outpatient 2. Have influenza‐like illness (fever > 37.5 °C and at least 2 of the following symptoms: headache, myalgia, cough, sore throat) 3. Presenting within 24 hours (at most 36 hours) from influenza‐like illness symptom onset 4. Willing to co‐operate for the study and able to follow the investigator's instructions Exclusion criteria: 1. Suspected bacterial infection 2. Patients with chronic respiratory disease (including bronchial asthma), cardiovascular disease (excluding hypertension without complications), chronic metabolic disease (including diabetes), hepatic or renal impairment, anaemia or immunosuppression 3. Unstable chronic illness 4. Were receiving other intranasal or oral drug for topical application 5. Females who were pregnant, of child‐bearing potential, breast‐feeding or trying to become pregnant during the study period 6. Had received an investigational drug in the previous 3 months 7. Judged to be inappropriate for the study by the investigator Populations for efficacy analysis Randomised and treated population: 116 cases (A37, B40, C 39). 24 cases (8 cases each) were excluded due to GCP non‐compliance (1) Intention‐to‐treat population: N = 92 (A29, B32, C31) (116 randomised and treated minus 24 non‐compliant with GCP were considered "intention‐to‐treat population". This is a secondary population for the analysis of efficacy data (2) Per‐protocol population: N = 68 (A25, B19, C24) (3) Influenza‐positive population: N = 56 (A16, B18, C22) (4) Efficacy population: N = 49 (A15, B14, C20) Populations for safety analysis: 116 cases (A37, B40, C 39) |
|
| Interventions | GG167 10 mg inhaled GG167 inhaled plus intranasal 6.4 mg administered twice daily for 5 days Placebo |
|
| Outcomes |
Primary: time to alleviation of the 3 major and 5 major influenza symptoms (3 systemic symptoms typical of influenza: fever, headache and myalgia + 2 upper respiratory symptoms: cough and sore throat) Secondary: time to alleviation of individual symptoms (fever, headache, myalgia, cough and sore throat) Safety: incidence of drug‐related abnormal symptoms/abnormal laboratory changes |
|
| Notes | Random allocation was done prior to the distribution and registration of participants | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method used for sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Centrally controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | High risk | The trial was terminated prematurely |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | The trial was terminated prematurely |
| Incomplete outcome data (attrition bias) Safety data | High risk | The trial was terminated prematurely |
| Selective reporting (reporting bias) | High risk | Methods of primary outcomes were not predefined in the protocol |
| Other bias | High risk | Corrected data were used for analysis. Endpoints were not described in the protocol; under‐recruitment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Treatments were identical in colour |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No breaking of the code allowed |
JNAI‐04.
| Methods | Double‐blind, placebo‐controlled trial of zanamivir in the treatment of influenza viral infection A and B. Late phase II study for dose comparison | |
| Participants | 16 years or older with influenza‐like symptoms Inclusion criteria:
Exclusion criteria:
Populations for efficacy analysis Number screened N = 53 (3 were not eligible) Randomised N = 50 (1 was not treated and 1 was excluded due to incomplete informed consent) Randomised and treated population: 48 cases Low‐dose active group (LA): 19, high‐dose active group (HA): 13 Low‐dose placebo group (LP): 10, high‐dose placebo group (HP): 6 (1) Intention‐to‐treat population: 48 cases (LA19, HA13, LP10, HP6) (2) Per‐protocol population: 38 cases (LA15, HA10, LP8, HP5) (3) Influenza‐positive population: 22 cases (LA11, HA5, LP2, HP4) (4) Efficacy population: 18 cases (LA9, HA4, LP2, HP3) |
|
| Interventions | Zanamivir 10 mg twice daily for 5 days Zanamivir 20 mg twice daily for 5 days "Low" placebo twice daily for 5 days "High" placebo twice daily for 5 days |
|
| Outcomes |
Primary: time to alleviation of the 3 major influenza symptoms (fever, headache and myalgia) Secondary: ‐ Time to alleviation of 5 major influenza symptoms (fever, headache and myalgia + cough and sore throat) ‐ Proportion of subjects with individual influenza‐like symptoms (fever, headache, myalgia, feverishness, cough, sore throat, arthralgia, lumbago, chillness, fatigue, nasal symptoms, diarrhoea and poor appetite) Safety: incidence of abnormal symptoms/abnormal laboratory changes, which were not denied as having causality by the investigator |
|
| Notes | The primary and secondary outcomes were not defined in the protocol | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation |
| Incomplete outcome data (attrition bias) Symptoms | High risk | The study was terminated prematurely |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | The study was terminated prematurely |
| Incomplete outcome data (attrition bias) Safety data | High risk | The study was terminated prematurely |
| Selective reporting (reporting bias) | High risk | Populations of analysis were not predefined |
| Other bias | High risk | The primary and secondary outcomes were not defined in the protocol |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Low and high dose differed in colour |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Investigators were informed of the randomisation codes before unblinding |
JNAI‐07.
| Methods | A randomised, placebo‐controlled trial of zanamivir in the treatment of influenza viral infection A and B. Late phase II study for dose comparison | |
| Participants | Individuals visiting a participating outpatient centre where influenza A and B were circulating, who were diagnosed as having influenza A and B based on satisfying the following inclusion criteria:
Exclusion criteria:
Populations for efficacy analysis Number screened N = 336 (among those 4 were not eligible) Randomised with double‐blinded state N = 333 (among those 4 were withdrawn consent) Randomised and treated population under double‐blinded state (ITT): 329 cases Low‐dose active group (LA): 107, high‐dose active group (HA): 111 Low‐dose placebo group (LP): 54, high‐dose placebo group (HP): 57 (1) ITT (intention‐to‐treat) population: 329 cases (LA107, HA111, LP54, HP57) (2) FAS (full analysis set: ITT ‐ 5 (consent withdrawn) ‐ 4 (incomplete case record): 318 cases (LA101, HA110, LP51, HP56) (3) Per‐protocol population (irrespective of influenza positivity): 244 cases (LA81, HA84, LP38, HP41) (4) Influenza‐positive population (irrespective of protocol adherence): 225 cases (LA71, HA82, LP34, HP38) (5) Efficacy population (per‐protocol and influenza‐positive): 172 cases (LA55, HA63, LP26, HP28) Populations for safety analysis: 329 cases (LA107, HA111, LP54, HP57) Safety outcome: The safety measure was the incidence of abnormal symptoms/abnormal laboratory changes which include or exclude those "causality denied". "Abnormal symptoms or abnormal laboratory findings" are the adverse events defined as newly observed or remarkably worsened symptoms after treatment started or abnormal or worsened laboratory findings compared with those before treatment irrespective of the causal assessment |
|
| Interventions | Zanamivir 10 mg twice daily for 5 days Zanamivir 20 mg twice daily for 5 days "Low" placebo twice daily for 5 days "High" placebo twice daily for 5 days |
|
| Outcomes |
Primary: time to alleviation of the 3 major influenza symptoms (influenza, fever, headache) Secondary: time to alleviation of 5 major influenza symptoms Change in antibody titre |
|
| Notes | The interventions were distinguishable in appearance | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method used for sequence generation not specified |
| Allocation concealment (selection bias) | High risk | Low and high doses of intervention differed in colour |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Case narratives not fully provided |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Only the data for hospitalisation reported |
| Incomplete outcome data (attrition bias) Safety data | High risk | Symptoms that were part of the subject's influenza‐like illness (ILl) according to the opinion of the investigator did not need to be reported as AEs. Only new or worsened symptoms (abnormal symptoms) and laboratory test results (abnormal laboratory tests results) were required to be reported |
| Selective reporting (reporting bias) | High risk | Populations of analysis were not predefined |
| Other bias | High risk | Baseline temperatures were almost significantly lower in the zanamivir group than the placebo group |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Low and high doses of intervention differed in colour |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Low and high doses of intervention differed in colour |
JV15823.
| Methods | Randomised, placebo‐controlled, multicentre study of oseltamivir (Ro 64‐0796) in the treatment of influenza in Japanese participants | |
| Participants | Over 16 years of age; present within 36 hours of onset of symptoms of fever > 38.0 °C, plus 2 influenza symptoms | |
| Interventions | 75 mg capsules, Ro 64‐0796/V14 batch no. GMZ 0129/03 Matching placebo capsules: Ro 64‐0796/V16 batch no. GMZ 0136 |
|
| Outcomes |
Primary outcome The time to alleviation of all symptoms Secondary outcomes Total symptom score AUC Change in virus titre Time to return to the afebrile state Adverse events |
|
| Notes | The available study report is a 29‐page document in Japanese. The design and methods are similar to those of WV 15670 and 15671 (the "pivotal" treatment trials in adults) and the trial was intended as a "bridge" with the Western trial programme. The report does not contain any supporting data (i.e. statistical analysis plan, protocol, amendments, certificates of analysis and audit, randomisation lists, lists of investigators, IRB clearance and individual listings) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Not specified as protocol and methods section were not available |
| Allocation concealment (selection bias) | High risk | Not specified as protocol and methods section were not available |
| Incomplete outcome data (attrition bias) Symptoms | High risk | 92 participants excluded in efficacy analysis because infection with influenza virus could not be verified in the efficacy analysis |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | 92 participants excluded in efficacy analysis because infection with influenza virus could not be verified in the efficacy analysis |
| Incomplete outcome data (attrition bias) Safety data | High risk | No drop‐outs because of harms were reported but no breakdown by on‐ and off‐treatment status reported |
| Selective reporting (reporting bias) | High risk | Not specified as protocol and methods section were not available |
| Other bias | High risk | Not specified as protocol and methods section were not available |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not specified as protocol and methods section were not available |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not specified as protocol and methods section were not available |
JV15824.
| Methods | Randomised, double‐blind, placebo‐controlled trial to evaluate the efficacy and safety of Ro64‐0796 for the prophylaxis of influenza A and B | |
| Participants | Participants ≥ 16 years of age and without influenza‐like symptoms | |
| Interventions | Ro64‐0796/V14 75 mg capsule Ro64‐0796/V16 placebo capsule |
|
| Outcomes |
Primary outcome Rate of occurrence of influenza Secondary outcomes Rate of occurrence for patients infected with non‐clinical influenza Rate of occurrence for patients infected with non‐symptomatic influenza Rate of occurrence for patients infected with influenza‐like disease Safety |
|
| Notes | The available clinical study report is a 15‐page document translated into English from the Japanese original. The design and methods are similar to those of WV 15697/15673 (the "pivotal" prophylaxis trials in adults) and the trial was meant as a "bridge" with the Western trial programme. The report does not contain any supporting data (i.e. statistical analysis plan, protocol, amendments, certificates of analysis and audit, randomisation lists, lists of investigators, IRB clearance and individual listings) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Not specified as protocol and methods section were not available: method of sequence generation not reported |
| Allocation concealment (selection bias) | High risk | Not specified as protocol and methods section were not available |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT analysis included all randomised participants; all participants who took at least 1 dose of medication were included in the safety population (CSR G‐146); no systematic differences in drop‐outs |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | ITT analysis included all randomised participants |
| Incomplete outcome data (attrition bias) Safety data | High risk | All participants who took at least 1 dose of medication were included in the safety population (CSR G‐146); no systematic differences in drop‐outs but no breakdown by on‐ and off‐treatment status reported, only aggregate in treatment summary table |
| Selective reporting (reporting bias) | High risk | Not specified as protocol and methods section were not available; unclear if there were any amendments |
| Other bias | High risk | No reporting of methods; no protocol; 4 participants not registered received the clinical trial drug |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not specified as protocol and methods section were not available |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not specified as protocol and methods section were not available |
M76001.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study stratified by onset of influenza symptoms. Influenza surveillance programme set up to track outbreak of virus across the United States Location, number of centres: USA (164 centres) Duration of study: 21 (+/‐4) days |
|
| Participants | Number screened: not available Number randomised: 1459 (oseltamivir: 965; placebo: 482. N randomised but did not receive study drug: 12) Number completed: 1344 M = 44% F = 56% Mean age: 35 years Baseline details: 81% Caucasian Inclusion criteria
Exclusion criteria
Definition of patient populations for analysis Intention‐to‐treat (ITT) infected population (N = 1063) This population was the primary analysis population and was used for summaries and analyses of efficacy parameters and consisted of the same patients as the ITT population but excluded patients who did not have laboratory‐confirmed infections. Patients were analysed according to the groups to which they were randomised ITT population (N = 1447) The ITT population consisted of all patients who took at least 1 dose of study medication and had at least 1 efficacy measurement. Patients with protocol violations or deviations were retained in the ITT population. Patients were analysed according to the groups to which they were randomised Safety population (N = 1447) Not defined Standard population (N = 932) This population was used for summaries of selected efficacy parameters. It included all patients who were randomised, who had no major protocol violations or deviations, who had laboratory‐confirmed influenza and who received at least the first 6 scheduled doses |
|
| Interventions | Intervention: oseltamivir (size 2 capsules) 75 mg bid Control: matching placebo (size 2 capsules) bid For each treatment arm, patients were provided with a blister pack containing 12 capsules for 10 doses (2 extra capsules in case of damage or mishandling) Treatment period: 5 days Follow‐up period: 12 to 18 days post‐treatment Co‐interventions: paracetamol 500 mg was also provided for symptomatic relief, if necessary |
|
| Outcomes |
Primary outcome Duration of illness Length of time until alleviation of the symptoms of influenza (nasal congestion, sore throat, cough, aches and pains, fatigue, headaches and chills/sweats). The time to alleviation of all 7 symptoms corresponds to the duration over which subsequent area under the curve calculations were made Secondary outcomes
|
|
| Notes | The final protocol is dated 2 October 1998. There were no amendments to the protocol. The first patients received treatment on 24 December 1998. The Module 2 does not contain the statistical analysis plan, amendments or protocol amendments or certificates of analysis. As a consequence no additional information to further develop the M1 extraction was obtained | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list shows random sequence and centralised phone driven system was used |
| Allocation concealment (selection bias) | Low risk | Centralised phone driven system: "The randomisation numbers were allocated by a central randomisation service, ICTI (Interactive Clinical Technologies Inc., Princeton, NJ)." "The investigator or study coordinator telephoned the Randomization Center to report their centre's identification number, the patient's initials, date of birth and time from the onset of flu symptoms. The Randomization Center then assigned a unique patient identification number and a corresponding medication number for each patient. The investigator or coordinator entered these numbers in the appropriate place on the case report form." |
| Incomplete outcome data (attrition bias) Symptoms | High risk | In the absence of IPD and CRFs we cannot account for all participants |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Unclear how complications of influenza were defined clinically |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events (AEs) could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting was inconsistent |
| Selective reporting (reporting bias) | High risk | Certificate of analysis is missing |
| Other bias | High risk | Potentially active placebo |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Capsule size is reported but no details of colour or taste or contents |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Randomisation code was unavailable to monitors or statisticians |
ML16369.
| Methods | Randomised, double‐blind, placebo‐controlled trial conducted during the influenza epidemic season Location, number of centres: Beijing and Shanghai, China; 7 study centres Duration of study: 21 days |
|
| Participants | Number screened: not available Number randomised: 478 (baseline data on ITTI population only: 273 (oseltamivir: 134; placebo: 139)) Number completed: 451 M = (ITTI) 50% F = (ITTI) 50% Mean age: 31 years Baseline details: baseline information only available for the ITTI population Smoking history: 20%; influenza virus: A (62%); B (36.5%); unknown (1.5%) Inclusion criteria
Exclusion criteria
Definition of patient populations for analysis ITT infected population (N = 273) The population for primary efficacy analyses was the intention‐to‐treat‐infected (ITTI) population comprising randomised participants who received at least 1 dose of study drug and had laboratory‐confirmed influenza (a positive culture on day 1 and/or ≥ 4‐fold increase in HAI antibody between baseline and day 21 of the study) ITT population (N = 451) The ITT population consisted of all participants who took at least 1 dose of study medication. The safety population included all participants who received at least 1 dose of study medication and who had at least 1 safety follow‐up, whether or not withdrawn prematurely Safety population (N = 459) Not defined |
|
| Interventions | Intervention: oseltamivir 75 mg (Ro 64‐0796) bid Control: matching placebo bid Treatment period: 5 days Follow‐up period: up to day 21 Co‐interventions: contents of rescue pack of medication provided to study participants not reported |
|
| Outcomes |
Primary outcome Duration of illness: the median duration was presented for each treatment group together with 95% confidence intervals. Kaplan‐Meier graphs of duration of symptoms according to treatment group were provided. Although the primary analysis for the primary parameter was done using the ITTI population, an additional analysis for the primary parameter was done using the ITT population Secondary outcomes
|
|
| Notes | Conducted during influenza season from January to April 2001. No further information available in 2011 to 2013 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Described as randomised; procedure generating randomisation schedule not available |
| Allocation concealment (selection bias) | Low risk | Randomisation was pharmacy‐controlled |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT population outcome data reported |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Based on all participants irrespective of compliance with treatment or infection status |
| Selective reporting (reporting bias) | Low risk | Symptoms and outcomes of primary interest were available for ITT population |
| Other bias | High risk | No information available on placebo contents; under‐recruitment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Participants and staff remained blinded to allocation status throughout the study. The investigator received a sealed envelope for each subject in the trial, for use in emergencies. Each envelope contained the identity of a subject's treatment." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Inadequate information available to ascertain whether outcome assessors were aware of treatment group assignment |
NAI30008.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered twice daily for 5 days in the treatment of influenza in patients aged 12 ≥ years diagnosed with asthma or COPD Location, number of centres: USA (46 centres); UK (36); France (23); South Africa (11); Norway (10); Canada (9); Australia (6); Germany (5); Slovakia (3); Austria (2); Belgium (2); Denmark (2); Sweden (2) Chile (1); Israel (1) Duration of study: 4 weeks |
|
| Participants | Males or females aged ≥ 12 years with influenza‐like illness | |
| Interventions | Zanamivir (5 mg per inhalation) 2 inhalations twice a day, via Rotadisks/modified Diskhaler Placebo 2 inhalations twice a day, via Rotadisks/modified Diskhaler |
|
| Outcomes |
Primary outcome: Time to alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to alleviation of clinically significant symptoms of influenza and no use of relief medication Time until the subject returned to normal activities Sleep disturbance measured by the number of nights during the treatment period and during the study period for which the subject recorded Maximum daily temperature Total number of tablets of supplied paracetamol taken over the treatment period Total number of spoonfuls of supplied cough mixture taken over the treatment period Incidence of complications of influenza and the incidence of associated antibiotic use Mean PEFR as recorded on the diary card over the treatment period FEV1 and PEFR as recorded in the clinic on days 6 and 28 Viral titres from throat swabs Global assessment of symptoms as recorded by the physician on day 6 |
|
| Notes | Study period: June 1998 to April 2000. Asthmatic patients were excluded after trial commencement; discrepancies in rescue medications were reported and different allocation concealment routines occurred in different centres | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method used in sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Unblocked randomisation schedule provided by sponsor |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT analysis included all randomised participants |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | The authors display data for safety and incidence of complications of influenza based on the ITT population |
| Incomplete outcome data (attrition bias) Safety data | Low risk | The authors display data for safety and incidence of complications of influenza based on the ITT population |
| Selective reporting (reporting bias) | Low risk | Outcomes reported as listed in protocol |
| Other bias | Low risk | Reasons for all protocol amendments were clearly reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Sealed envelopes; only in the case of an emergency, when knowledge of the study drug was essential for the clinical management or welfare of the subject, the investigator could unblind a subject's treatment assignment |
NAI30009.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group multicentre study to investigate the efficacy and safety of zanamivir (GG167) 10 mg administered by inhalation twice daily for 5 days in the treatment of symptomatic influenza A and B viral infections in children ages 5 to 12
Location, number of centres: USA (36 centres); Canada (6); France (7); Germany (6); Belgium (2); Finland (2); Spain (2); Russia (2); Sweden (2); Israel (1) United Kingdom (1) Duration of study: 14 to 28 days |
|
| Participants | Children aged 5 to 12 years with influenza‐like illness (ILI) | |
| Interventions | Zanamivir (5 mg per inhalation), 2 inhalations twice daily via Rotadisk/Diskhaler Placebo, 2 inhalations twice daily via Rotadisk/Diskhaler |
|
| Outcomes |
Primary outcome: time to alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to alleviation of clinically significant symptoms of influenza and no use of relief medication Time until the participant returned to normal activities Incidence of complications of influenza Antibiotic use for complications Mean overall assessment of diary card symptom score over post‐treatment Number of days out of study days 2 to 5 where cough was recorded Maximum daily diary card temperature Number of days out of study days 2 to 5 where the participant's parent recorded use of any relief medication Total number of 12‐hour periods during which supplied paracetamol was taken over the treatment period Total number of 12‐hour periods during which supplied dextromethorphan cough mixture (pholcodine in Europe) was taken over the treatment period Investigator Global Assessment of Symptoms at the study day 3 visit and at the post‐treatment visit Day 3 viral titre from throat swab Temperature as measured at the clinic visit on study day 3 for those participants with this assessment |
|
| Notes | Study conducted: 11 January 1999 to 19 April 1999. Different methods were used for allocation concealment in different countries | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method used for sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | An unblocked randomisation schedule was used. Eligible participants were allocated the next (i.e. lowest) sequential participant treatment number available at each centre |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT population included data for patients with missing diary cards |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | ITT population included data for patients with missing diary cards |
| Incomplete outcome data (attrition bias) Safety data | Low risk | ITT population included data for patients with missing diary cards |
| Selective reporting (reporting bias) | High risk | Per‐protocol population not defined in the protocol |
| Other bias | High risk | Use of different relief medications across different centres |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Only in the case of an emergency, when knowledge of the study drug was essential for the clinical management or welfare of the participant, could the investigator unblind a participant's treatment assignment. If the investigator broke the blind for an individual participant, the date and reason was recorded on the "status of treatment blind" page in the CRF. The investigator did not reveal the blind to the monitor |
NAI30010.
| Methods | A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir (GG167) 10 mg administered once a day for 10 days in the prevention of transmission of symptomatic influenza A and B viral infections within families
Location, number of centres: USA (11 centres); Canada (2); Finland (1); UK (1) Duration of study: 11 days |
|
| Participants | 2 to 5 family members with at least 1 adult and 1 child with presence of influenza‐like illness Definition of patient populations for analysis ITT population (N = 1158) Index cases ≥ 5 years and contact cases ≥ 5 years randomised to treatment. Index cases and contact cases < 5 years of age who did not receive treatment, were excluded from the ITT analysis. The family was included if at least 1 randomised family member was in the population Safety population (N = 1158) Index cases and contact cases who took at least 1 dose of study medication. Randomised participants excluded if there was clear evidence of failure to take study medication |
|
| Interventions | Zanamivir powder (5 mg per inhalation) via Rotadisk/Diskhaler Placebo 2 inhalations via Rotadisk/Diskhaler Co‐interventions Relief medication pack (contents not specified) |
|
| Outcomes |
Primary outcome: proportion of randomised families in whom at least 1 randomised contact developed symptomatic, laboratory‐confirmed influenza A or B infection Secondary outcomes: Proportion of randomised families in whom at least 1 randomised contact developed laboratory‐confirmed influenza A or B infection Proportion of randomised families in whom at least 1 randomised contact developed laboratory‐confirmed influenza A or B infection and where symptoms began anytime from start of treatment to day 11 Proportion of randomised families in whom at least 1 randomised contact developed a febrile illness during days 1 to 11 Proportion of randomised families in whom at least 1 contact case developed laboratory‐confirmed influenza infection Time to alleviation of clinically significant symptoms for randomised index cases Time to alleviation of clinically significant symptoms and no use of relief medication for randomised index cases Number of days unable to perform normal activities Number of days of use of relief medication Proportion of randomised families in whom at least 1 randomised contact developed a secondary infection Temperature of randomised index case at clinic visit on study day 5 |
|
| Notes | In Europe, it was discovered that clinical supplies were incorrectly packed [CSF pg 9]; recruitment was stopped and procedures were put in place to correct this error; all randomised family members received the same treatment as the index case [CSF pg 10] Study period: October 1998 to May 1999 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method used to generate sequence unspecified |
| Allocation concealment (selection bias) | Low risk | Blocked randomisation provided by sponsor |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | All randomised participants included in ITT population for efficacy; low number of drop‐outs |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | All randomised participants included in ITT population for efficacy; low number of drop‐outs |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Safety population based on randomised participants |
| Selective reporting (reporting bias) | Low risk | Outcomes reported as specified in protocol and statistical analyses plan |
| Other bias | High risk | Protocol amendment before participant recruitment Protocol amendments were undertaken during the study, which involved the changing of endpoints and removal of laboratory tests, as well as including new exclusion criteria. In Europe, it was discovered that clinical supplies were incorrectly packed; recruitment was stopped and procedures were put in place to correct this error; all randomised family members received the same treatment as the index case |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Incorrect packing of supplies; use of different colours for index and contact cases; randomising all families to same treatment. No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Breaking of the randomisation code was forbidden |
NAI30011.
| Methods | Randomised, double‐blind, placebo‐controlled study to evaluate the impact of inhaled zanamivir treatment on workplace attendance and healthcare outcomes due to influenza A and B infections | |
| Participants | Males or females aged 18 years and above with influenza‐like illness | |
| Interventions | 4 x 5 mg doses of micronised zanamivir (as dry powder, lactose blend) Matching placebo (lactose only) |
|
| Outcomes |
Primary outcome: time to alleviation of influenza symptoms Secondary outcomes: Time absent from work due to influenza symptoms Time to perception of influenza symptom improvement |
|
| Notes | Protocol amendments were undertaken during the study which involved the changing of endpoints and removal of laboratory tests, as well as including new exclusion criteria | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | The ITT population is included in the safety data |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | The ITT population is included in the safety data |
| Incomplete outcome data (attrition bias) Safety data | Low risk | The ITT population is included in the safety data |
| Selective reporting (reporting bias) | Low risk | Outcomes reported as specified in the protocol |
| Other bias | High risk | 2 protocol amendments occurred during trial; in 1 of these, there was re‐defining of endpoints and removal of a laboratory test for antibody detection |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Only in the case of an emergency, when knowledge of the study drug was essential for the clinical management or welfare of the participant, could the investigator unblind a participant's treatment assignment. The investigator was not to reveal the blind to the monitor |
NAI30012.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered twice daily for 5 days in the treatment of symptomatic influenza A and B viral infections in participants aged > 65 years | |
| Participants | Male or female participants aged > 65 years (with or without underlying medical conditions) with ILI | |
| Interventions | Zanamivir (GG167) powder (5 mg inhalations), 2 inhalations, twice daily, for 5 days, via Rotadisk/Diskhaler Placebo, 2 inhalations, twice daily, for 5 days, via Rotadisk/Diskhaler |
|
| Outcomes |
Primary outcome: time to alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to alleviation of clinically significant symptoms and no use of relief medication Complications of influenza Complications of influenza requiring antibiotic use Time to alleviation of individual symptoms Time to afebrile status Use of supplied paracetamol Global assessment of symptoms by the investigator Time to return to 'how I felt before influenza illness' Time to return to normal activities Viral titre levels at day 2/3, day 6 and day 14 |
|
| Notes | More participants in the placebo group had a high‐risk medical condition compared with the zanamivir group, in both the ITT and influenza‐positive populations. 2 deaths reported; neither were considered to be related to the drugs but case narratives are blanked out | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Randomisation code provided by sponsor; unblocked randomisation schedule and sequential participant treatment number |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | All randomised participants were analysed in the ITT sample for efficacy |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | All randomised participants were analysed in the ITT sample for efficacy |
| Incomplete outcome data (attrition bias) Safety data | Low risk | The safety population included all participants randomised to treatment and who took at least 1 dose of study medication |
| Selective reporting (reporting bias) | Low risk | All outcomes specified in the protocol reported |
| Other bias | High risk | More participants in the placebo group had a high‐risk medical condition compared with the zanamivir group, in both the ITT and influenza‐positive populations; protocol was amended 8 times during the trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Breaking of the code by opening the hidden portion of the detachable drug label or hidden entry envelope was expressly forbidden. Only in the case of an emergency, when knowledge of the study drug was essential for the clinical management or welfare of the participant, could the investigator unblind a participant's treatment assignment. The investigator was not to reveal the blind to the monitor |
NAI30015.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered twice daily for 5 days in the treatment of symptomatic influenza A and B infections in armed service personnel | |
| Participants | Participants were conscripts of the Finnish Army, living in residential units with influenza‐like illness | |
| Interventions | Inhaled zanamivir 10 mg (2 x 5 mg blisters), twice daily, for 5 days, via Rotadisk/Diskhaler Placebo (2 blisters), twice daily, for 5 days, via Rotadisk/Diskhaler |
|
| Outcomes |
Primary outcome: time to alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to alleviation of clinically significant symptoms of influenza and no relief medication Time to afebrile status Total symptom score Viral load data Complications Maximum daily diary temperature during treatment Total number of 12‐hour periods during which supplied paracetamol was taken over the treatment period Global assessment of symptoms Time to alleviation of individual symptoms |
|
| Notes | No explicit report on randomisation and blinding procedures provided | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; no information on method of sequence generation |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | All randomised participants were analysed in the ITT sample for efficacy |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | All randomised participants were analysed in the ITT sample for efficacy |
| Incomplete outcome data (attrition bias) Safety data | Low risk | The safety population comprised all randomised participants |
| Selective reporting (reporting bias) | High risk | Protocol listed "MEP abnormalities (in a sub‐set of participants)" as a secondary endpoint but it was not reported in the CSR |
| Other bias | Low risk | Only 1 protocol amendment comprising typographical corrections |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators received hidden entry envelopes that contained the code break for each treatment number. Breaking of the code by opening the hidden entry envelopes was expressly forbidden except in the event of a medical emergency where the identity of the drug was necessary in order to treat the participant properly. In the event of such an emergency, it was requested that the investigator make every effort to contact the study monitor or designee prior to breaking the code |
NAI30020.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre study in 2 parallel groups, to investigate the efficacy and safety of inhaled zanamivir (10 mg bid via Diskhaler), for 5 days, in high‐risk patients with symptomatic influenza A and/or B infection | |
| Participants | Eligible participants were adults aged ≥ 18 years, with symptomatic influenza A and/or B infection: feverishness (> 37.8 °C) and at least 2 of the 4 following symptoms: headache, muscle pains, myalgia, sore throat and cough, also with influenza‐quick‐test positive and categorised as "high risk" High risk was defined as: males and females aged > 60 years, participants living in an old peoples' home, participants with respiratory disorders including COPD or asthma, participants with diabetes mellitus, chronic renal failure or cardiovascular disorders Exclusions included pregnant women or women at risk of becoming pregnant during the study; participants with a suspected bacterial respiratory infection; participants with a known sensitivity to components of the intervention or placebo treatments or the emergency medications |
|
| Interventions | Zanamivir 2 inhalations (5 mg per inhalation) twice daily for 5 days Placebo 2 inhalations (5 mg per inhalation) twice daily for 5 days Participants were additionally provided with paracetamol and cough syrup as symptomatic medication, with guidance to take these only if there was "an acute need" |
|
| Outcomes |
Primary outcome: time to alleviation of fever (time to first measurement with a temperature < 37.8 °C, maintained for the following 24 hours) Secondary outcomes: Time to alleviation of the other clinically relevant symptoms Time to return to normal activity Incidence of complications Loss of productivity assessed by number of days taken off work Use of medical services |
|
| Notes | Only a synopsis was provided for this trial; a full clinical study report was not available The total number of patients analysed was not clearly reported It appears that intention‐to‐treat analysis was not performed 43/329 randomised participants took the study drug but were excluded from the analysis according to prespecified exclusion criteria (temperature too low, no diary available, fever stopped before medication taken ‐ Clinical Research Associate findings at investigator's site) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method used for sequence generation not specified |
| Allocation concealment (selection bias) | High risk | Concealment of allocation was not reported |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Blinding of participants and personnel was not reported |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Blinding of outcome assessors was not reported |
| Incomplete outcome data (attrition bias) Safety data | High risk | Blinding of participants, personnel and assessors was not reported |
| Selective reporting (reporting bias) | High risk | Statistical analysis plan was not available and it is not possible to know the pre specification of outcomes |
| Other bias | High risk | The trial appeared to under‐recruit but the numbers of participants are not clearly reported |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of participants and personnel was not reported |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors was not reported |
NAI30028.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre, comparative study in parallel groups to investigate the efficacy and tolerability of inhaled zanamivir, administered 2 x daily for 5 days | |
| Participants | Children aged from 5 to 12 years with symptomatic influenza A and/or influenza B infection | |
| Interventions | Zanamivir (dry powder for oral inhalation) in a circular foil pack (Rotadisks®) with 4 regularly distributed blisters each containing 5 mg zanamivir blended with 20 mg lactose Placebo Rotadisks® containing 25 mg lactose |
|
| Outcomes |
Primary outcome: time to alleviation of all the clinically significant symptoms of influenza Secondary outcomes: Time to return to normal activities Complications |
|
| Notes | The 2 SAEs (febrile convulsion and stomach ache) were considered by the investigators to be zanamivir‐related | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random blocks |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled |
| Incomplete outcome data (attrition bias) Symptoms | High risk | 5 participants were excluded from the analysis of efficacy due to insufficient diary card data |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | 5 participants were excluded from the analysis of efficacy due to insufficient diary card data |
| Incomplete outcome data (attrition bias) Safety data | High risk | 5 participants were excluded from the analysis of efficacy due to insufficient diary card data |
| Selective reporting (reporting bias) | High risk | The primary endpoint was originally the time to alleviation of the main signs/symptoms of influenza but this was later adjusted to the time to alleviation of fever. The presence of fever was indicated for all participants but the value was not recorded for 11 participants; data for these participants was, therefore, excluded from the per‐protocol analysis |
| Other bias | Low risk | Low number of drop‐outs in both groups; reasons reported. All participants included in safety population. There were no protocol amendments |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Identical Rotadisks used in both groups; placebo was reported to be lactose. No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were only allowed to break the study blind in an emergency, where knowledge of the participant's study medication was essential on clinical grounds |
NAI30031.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered once a day for 10 days in the prevention of transmission of symptomatic influenza A and B viral infections within households | |
| Participants | The index case was the first household member for whom a clinical diagnosis of ILI could be made | |
| Interventions | Zanamivir (GG167) powder (5 mg per inhalation) 2 inhalations via Rotadisk/Diskhaler Placebo, 2 inhalations via Rotadisk/Diskhaler |
|
| Outcomes |
Primary outcome: the proportion of randomised households in which at least 1 randomised contact case developed symptomatic, laboratory‐confirmed influenza A or B infection Secondary outcomes: The proportion of randomised households in which at least 1 randomised contact case developed laboratory‐confirmed influenza infection The proportion of randomised households for which at least 1 randomised contact case developed symptomatic influenza‐like illness (irrespective of laboratory confirmation). The proportion of randomised households in which at least 1 randomised contact case developed symptomatic, laboratory‐confirmed influenza infection, excluding any failures that occurred within 1 day of the start of prophylaxis The proportion of randomised households in which at least 1 randomised contact case developed laboratory‐confirmed influenza and a febrile illness The number of days out of 28 at least 1 randomised contact case was unable to perform all their normal activities The number of days out of 28 at least 1 randomised contact case recorded the use of relief medication The proportion of randomised households in which at least 1 randomised contact case developed any secondary complication of influenza The proportions of randomised households in which at least 1 randomised contact case develops symptomatic, laboratory‐confirmed influenza and the flu type of the index case matches that of all the contact cases who develop influenza Time to alleviation of clinically significant symptoms of influenza in contact cases developing symptomatic, laboratory‐confirmed influenza during prophylaxis |
|
| Notes | Some outcomes in the clinical study reports were not listed in the protocol | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided, but sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Blocked randomisation schedule was used. Each randomised household contact case was allocated the next (i.e. lowest) sequential treatment number available for that household |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT population available |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | ITT population available |
| Incomplete outcome data (attrition bias) Safety data | Low risk | The safety population was defined as all contact cases randomised to treatment who took at least 1 dose of study medication |
| Selective reporting (reporting bias) | High risk | Several outcomes reported in clinical study reports were not listed in the protocol |
| Other bias | High risk | Patients receiving antibiotics for bacterial RTI were excluded in the protocol but not in the clinical study reports |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Breaking of code only permitted in emergency |
NAI30034.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered once a day for 28 days in the prevention of symptomatic influenza A and B viral infections in community‐dwelling, high‐risk participants aged ≥ 12 years | |
| Participants | Community‐dwelling males and females aged ≥ 12 years at greatest risk for developing complications from influenza | |
| Interventions | Zanamivir powder (5 mg per inhalation) 2 inhalations, once daily for 28 days, via Rotadisk/Diskhaler Placebo, 2 inhalations, once daily for 28 days, via Rotadisk/Diskhaler |
|
| Outcomes |
Primary outcome: the proportion of randomised participants who, during prophylaxis (days 1 to 28), developed symptomatic, influenza A or B infection confirmed by culture/serology Secondary outcomes: The proportion of randomised participants who developed influenza confirmed by culture/serology The proportion of randomised participants who, during prophylaxis, developed influenza‐like illness (ILI); developed a febrile illness |
|
| Notes | Protocol states several other secondary outcome measures; these modifications are not listed in the protocol amendment section: the total number of over‐the‐counter medications used; the proportion of randomised participants who require a prescription medication; the total number of prescription medications used; the proportion of randomised participants who have an unscheduled healthcare contact; and the total number of unscheduled healthcare contacts | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation; a blocked randomisation schedule was used. Eligible participants were always allocated the next (i.e. lowest) sequential treatment number available |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Low number of drop‐outs, reasons reported. There were the same number of participants in the safety population and the ITT population |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Low number of drop‐outs, reasons reported. There were the same number of participants in the safety population and the ITT population |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Low number of drop‐outs, reasons reported. There were the same number of participants in the safety population and the ITT population |
| Selective reporting (reporting bias) | Low risk | Outcomes reported as specified in protocol |
| Other bias | High risk | 5 protocol amendments, with 1 after trial commencement; amended the definition of confirmed influenza |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators, participants and study monitors were unaware of which treatment a participant was randomly assigned to receive. Only in the case of an emergency, when knowledge of the study drug was essential for the clinical management or welfare of the participant, was the investigator permitted to unblind a participant's treatment assignment |
NAIA/B2008.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre, parallel‐group study to investigate the efficacy and safety of zanamivir administered twice or 4 times a day for the treatment of influenza A and B viral infections | |
| Participants | Males and females aged ≥ 13 years with influenza‐like illness; ≥ 18 years in some centres | |
| Interventions | Zanamivir was provided as a solution for intranasal administration at a concentration of 16 mg/ml in saline and as a dry powder for inhalation Placebo was normal saline (for intranasal spray) and dry lactose powder to match zanamivir (for inhalation) |
|
| Outcomes |
Primary outcome:
Time until the alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to eradication of major influenza symptoms Time to alleviation of each of the symptoms in the diary card Combined symptom analysis Time until the patient returned to normal activities Number of days that the overall symptom assessment was recorded Number of days that at least 1 symptom was recorded Number of days that sleep disturbance was recorded as 'not at all' or 'slightly' over the whole period covered by the diary card Maximum daily temperature Mean daily number of administrations of supplied paracetamol (acetaminophen) over the treatment period Mean daily number of administrations of supplied cough mixture over the treatment period Investigator‐rated global assessment of symptoms Number of cases of hospitalisation resulting from influenza infection |
|
| Notes | Combined symptom analysis as a secondary endpoint was added to the final analysis plan after the trial commenced (14 May 1996). The symptoms of feverishness, headache, myalgia, cough and sore throat were summarised by the mean symptom score to provide a measure of overall severity | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation codes provided; but method used for sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Generated using GWRD program (PACT) |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Similar number of withdrawals in both groups (6%) |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Similar number of withdrawals in both groups (6%) |
| Incomplete outcome data (attrition bias) Safety data | Low risk | ITT population was safety population |
| Selective reporting (reporting bias) | High risk | Several secondary endpoints were changed; 1 was added to the clinical study reports that was not in the protocol |
| Other bias | High risk | Protocol amendments do not list changes to secondary endpoints; variation in minimum age for inclusion |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Each investigator was provided with a sealed envelope containing the individual code‐break envelopes for patients in their centre. These were only to be opened in a medical emergency, where knowledge of the study treatment was essential for further management of the patient |
NAIA/B2009.
| Methods | Multicentre, randomised, double‐blind, placebo‐controlled trial was designed to investigate the efficacy and safety of zanamivir (inhaled, intranasal, inhaled combined with intranasal versus placebo) in the prevention of and/or reduction in progression of influenza (post‐exposure prophylaxis) | |
| Participants | Individuals without flu symptoms aged ≥ 13 years (aged ≥ 18 years in 3 European countries) at risk of developing influenza having been in close contact with an index case | |
| Interventions | Zanamivir (16 mg/mL), 2 intranasal sprays per nostril (0.1 mL per spray) twice daily plus placebo 2 inhalations twice daily Zanamivir (5 mg per inhalation), 2 inhalations twice daily plus placebo 2 sprays per nostril twice daily Zanamivir (5 mg per inhalation), 2 inhalations twice daily plus zanamivir (16 mg/mL), 2 intranasal sprays per nostril (0.1 mL per spray) twice daily Placebo 2 inhalations twice daily plus placebo 2 sprays per nostril twice daily |
|
| Outcomes |
Primary: the proportion with symptomatic influenza, with laboratory confirmation, during treatment period Secondary: The proportion of patients with a fever (temperature 37.8 °C) during the treatment period Number of days over the study period (day 1 to 5) that the patient recorded any symptom on the diary card as 'none' or 'mild' Number of days over the study period (day 1 to 5) that the patient recorded the overall symptom assessment on the diary card as 'none' or 'mild' Investigator‐rated global assessment of symptoms (GAS) |
|
| Notes | The study aimed to recruit 840 but randomised 575 participants; the protocol was amended 6 times | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Symptom outcomes analysed with intention‐to‐treat population (all randomised) |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Outcomes analysed with intention‐to‐treat population (all randomised) |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Safety outcomes analysed with safety population (all randomised who took at least 1 dose of study treatment) |
| Selective reporting (reporting bias) | Low risk | Primary and secondary outcomes reported as stated in protocol |
| Other bias | High risk | Patient‐rated assessment not reported as outcome; lack of statistically significant power |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The monitors were blinded as to the study treatment administered. No investigator revealed the blind for study medication prior to trial completion |
NAIA2005.
| Methods | Randomised, double‐blind, placebo‐controlled multicentre trial, designed to evaluate the efficacy and safety of inhaled and intranasal zanamivir in the treatment of influenza A and B viral infections | |
| Participants | Eligible participants were individuals aged ≥ 13 years generally in good health, enrolled within 48 hours of the onset of influenza‐like illness | |
| Interventions | Zanamivir 2 inhalations (5 mg per inhalation) twice a day plus placebo 2 sprays per nostril twice a day Zanamivir 2 inhalations (5 mg per inhalation) twice a day plus zanamivir (16 mg/mL) 2 intranasal sprays per nostril (0.1 mL per spray) twice a day Co‐interventions Paracetamol, dextromethorphan hydrobromide and pseudoephedrine hydrochloride |
|
| Outcomes |
Primary outcome: time to alleviation of major influenza symptoms Secondary outcomes: Time (in days from initiation of treatment) to alleviation of feverishness, headache and myalgia Time (in days from initiation of treatment) to eradication of major signs and symptoms of influenza Time (in days from initiation of treatment) to alleviation and eradication of individual symptoms of influenza Combined symptom analysis Mean daily temperature over the study treatment period Number of days (from initiation of treatment) until the patient was able to return to normal activities Number of days out of days 1 to 10 that the patient recorded any symptom as 'moderate' or 'severe' Number of days out of days 1 to 10 that the patient recorded their overall symptom assessment (OSA) as 'moderate' or 'severe' Number of days out of days 2 to 10 that the patient recorded sleep disturbance as 'moderate' or 'severe' Mean supportive drug use (e.g. paracetamol, cough mixtures and decongestants) over the study treatment period Investigator global assessment of symptoms Day at which viral shedding fell below limit of quantitation (core centres only). Area under the viral shedding curve (core centres only) |
|
| Notes | A protocol amendment changed 1 exclusion criterion from patients with influenza vaccines administered since August 1993 to patients with influenza vaccines administered since 1 October 1994 during the recruitment period Major protocol amendments
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Described as randomised; procedure generating randomisation schedule not available |
| Allocation concealment (selection bias) | Low risk | Centrally generated, pharmacy‐controlled randomisation "Each investigator was provided with a sealed envelope containing the individual code break envelopes for patients in their centre." |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Withdrawals included in ITT analyses for both efficacy and safety. Data from infected and non‐infected participants were available |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Insufficient evidence to indicate that administration of zanamivir affects antibody response in similar way to oseltamivir. The influenza‐positive population is less likely to reflect a non‐randomised comparison |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Safety population based on randomised participants |
| Selective reporting (reporting bias) | Low risk | Change in SAP before trial commencement; outcomes reported as outlined in protocol |
| Other bias | High risk | Exposure to lactose in test drugs may have underestimated true risk of asthma events |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigator, all study staff, patients and the monitors were blinded as to the study treatment (zanamivir or placebo) administered; each investigator was provided with a sealed envelope containing the individual code‐break envelopes for patients in their centres. These were to be opened in a medical emergency only, where knowledge of the study treatment was essential for further management of the patient |
NAIA2006.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre trial investigating efficacy and safety of zanamivir in preventing progression of influenza A and B from asymptomatic to symptomatic, among cases exposed to suspected index cases | |
| Participants | Eligible participants were aged ≥ 13 years, with no signs or symptoms of influenza | |
| Interventions | Zanamivir (16 mg/mL) 2 intranasal sprays per nostril (0.1 mL per spray) twice a day plus placebo 2 inhalations twice a day Zanamivir 2 inhalations (5 mg per inhalation) twice a day plus placebo 2 sprays per nostril twice a day Zanamivir 2 inhalations (5 mg per inhalation) twice a day plus zanamivir (16 mg/mL) 2 intranasal sprays per nostril (0.1 mL per spray) twice a day Placebo 2 inhalations twice a day plus placebo 2 sprays per nostril twice a day |
|
| Outcomes |
Primary outcome: the proportion of patients with laboratory‐confirmed influenza during treatment plus at least 2 clinically significant symptoms of influenza of 'moderate' or 'severe' severity during the study treatment period Secondary outcomes: The proportion of patients with influenza The proportion of patients with a fever (temperature 37.8 °C) during the treatment period Number of days over the study period (day 1 to 5) that the patient recorded any symptom on the diary card as 'moderate' or 'severe' Number of days over the study period (day 1 to 5) that the patient recorded the overall symptom assessment on the diary card as 'moderate' or 'severe' Number of days over the study period (day 1 to 5) that the patient recorded 'moderate' or 'severe' sleep disturbance Number of days over the study period (day 1 to 5) that the patient recorded that they were unable to perform normal activities |
|
| Notes | The aim was to recruit 380 participants. Only 64 contact case participants were recruited and randomised | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled; codes were in sealed envelopes. No investigator revealed the blind for study medication prior to trial completion |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT and safety population included all randomised and all who took at least 1 dose of medication respectively |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | ITT and safety population included all randomised and all who took at least 1 dose of medication respectively |
| Incomplete outcome data (attrition bias) Safety data | Low risk | ITT and safety population included all randomised and all who took at least 1 dose of medication respectively |
| Selective reporting (reporting bias) | Low risk | Outcomes were reported as stated in the protocol |
| Other bias | High risk | Inability to recruit adequate numbers, as initially planned |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigator, all study staff, patients and the monitors were blinded as to the study treatment administered (zanamivir or placebo). Codes were to be opened in a medical emergency only, where knowledge of the study treatment was essential for further management of the patient |
NAIA3002.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre trial to investigate the safety and efficacy of inhaled zanamivir in the treatment of symptomatic influenza A and B in adolescents and adults Location, number of centres: USA (72 centres); Canada (12) Duration of study: 28 days |
|
| Participants | Individuals aged ≥ 12 years, with ILI, in locations with influenza circulating in the community | |
| Interventions | Zanamivir (5 mg per inhalation), 2 inhalations twice daily via Rotadisk/Diskhaler Placebo, 2 inhalations twice daily, via Rotadisk/Diskhaler Treatment period: 5 days Follow‐up period: 23 days post‐treatment Co‐interventions: relief pack of medication (paracetamol and cough mixture) |
|
| Outcomes |
Primary outcome: time until alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to alleviation of clinically significant symptoms of influenza and no use of relief medication Maximum daily temperature summarised by area under the curve (AUC) over the treatment period Time until the participant returned to normal activities Time to alleviation of each individual symptom Mean overall influenza score Mean symptom score for each of the individual symptoms collected on the diary card Total number of tablets of supplied paracetamol taken Total number of spoonfuls of supplied cough mixture taken Global assessment of symptoms at the post‐treatment visit Incidence of complications of influenza Day 3 viral titre from throat swab Productivity and healthcare resource utilisation outcomes |
|
| Notes | Major protocol amendments
Study period: October 1997 to April 1998 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | "...packs containing zanamivir or matching placebo were provided by the Pharmaceutical Supplies Department of Glaxo Wellcome Research and Development to Glaxo Wellcome Inc. The supplies were labelled and packed in Clinical Supply Operations at GWI for distribution to the study centres by Simirex, Inc., Mt. Laurel, NJ." |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Data for ITT and IP populations available |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | All randomised participants included in ITT for efficacy; and all who took at least 1 dose of medication included in ITT for safety |
| Incomplete outcome data (attrition bias) Safety data | Low risk | All who took at least 1 dose of medication included in safety population |
| Selective reporting (reporting bias) | Low risk | All outcomes reported as specified in protocol |
| Other bias | Low risk | Protocol amendments were explained and do not appear to have any influence on the outcomes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigator, all staff, participants and study monitors were blinded as to the study treatment administered. Breaking of the code by opening the hidden portion of the detachable drug label was expressly forbidden except in the event of a medical emergency where the identity of the drug had to be known in order to treat the participant properly |
NAIA3003.
| Methods | Randomised, double‐blind, placebo‐controlled, multicentre trial investigating the efficacy and safety of inhaled zanamivir compared with standard care in controlling outbreaks of influenza in nursing homes | |
| Participants | Participants in nursing homes were randomised when healthy and followed until an outbreak of influenza was declared in that nursing home | |
| Interventions | Zanamivir 2 inhalations (5 mg per inhalation) once a day plus 1 placebo tablet once a day Placebo 2 inhalations once a day plus 1 rimantadine tablet (100 mg) once a day |
|
| Outcomes |
Primary outcome: the proportion of randomised participants who, during prophylaxis, developed a new sign or symptom and had laboratory confirmation of influenza infection Secondary outcomes: The proportion of randomised participants who, during prophylaxis, developed febrile illness The proportion of randomised participants who, during prophylaxis (days 1 to 15) or anytime during the study (days 1 to 28), developed complications of influenza and had subsequent associated laboratory confirmation of influenza infection The proportion of randomised participants who, during prophylaxis (days 1 to 15) or anytime during the study (days 1 to 28), took an antibiotic due to complications of influenza and had subsequent associated laboratory confirmation of influenza infection The proportion of randomised participants who, during days 3 to 15 of prophylaxis, developed a new sign or symptom with subsequent associated laboratory confirmation of influenza infection The proportion of randomised participants who, during prophylaxis, had laboratory‐confirmed influenza infection |
|
| Notes | 4 protocol amendments were made and do not appear to have any influence on the outcomes. Adverse event data were not extracted from this study because of exposure to rimantadine | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | All randomised participants included in ITT for efficacy |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | All randomised participants included in ITT for efficacy |
| Incomplete outcome data (attrition bias) Safety data | Low risk | All who took at least 1 dose of medication included in ITT for safety |
| Selective reporting (reporting bias) | Low risk | Outcomes were reported as specified in the protocol and analysis plan |
| Other bias | High risk | 2 protocol amendments for interim safety analysis; some re‐randomisation of patients occurred |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blind. As such, the investigators, participants and study monitors were unaware of which treatment a participant was randomly assigned to receive. Breaking of the code by opening the hidden portion of the detachable drug label was expressly forbidden except in the event of a medical emergency where the identity of the drug was necessary in order to treat the participant properly |
NAIA3004.
| Methods | Randomised, placebo‐controlled, multicentre trial performed in 12 nursing homes in Lithuania, the Netherlands and Israel | |
| Participants | Participants without ILI, living in nursing homes | |
| Interventions | Zanamivir 2 inhalations (5 mg per inhalation) once a day Placebo 2 inhalations once a day |
|
| Outcomes |
Primary: the proportion of randomised participants who, during prophylaxis, developed a new sign or symptom and had laboratory confirmation of influenza infection Secondary: The proportion of randomised participants who, during prophylaxis, developed febrile illness (defined as a temperature of > 99.0 °F or > 37.2 °C) and had laboratory confirmation of influenza infection The proportion of randomised participants who, during prophylaxis (days 1 to 15) or anytime during the study (days 1 to 28), developed complications of influenza and had laboratory confirmation of influenza infection The proportion of randomised participants who, during prophylaxis (days 1 to 15) or anytime during the study (days 1 to 28), took an antibiotic due to complications of influenza and had laboratory confirmation of influenza infection The proportion of randomised participants who, during days 3 to 15 of prophylaxis, developed a new sign or symptom with laboratory confirmation of influenza infection The proportion of randomised participants who, during prophylaxis, had laboratory‐confirmed influenza infection |
|
| Notes | There were 12 SAEs during the study. There were 3 deaths, causes as follows: MI during post‐prophylaxis follow‐up; liver cirrhosis and congestive heart failure (6 months after study completion); pleural effusion and probable lung cancer (3 months after the study). Some re‐randomisation of patients occurred | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Symptom outcomes were reported using the intention‐to‐treat population (all randomised) as specified in the protocol and analysis plan |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Outcomes were reported using the intention‐to‐treat population (all randomised) as specified in the protocol and analysis plan |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Safety outcomes were reported using the safety population (all randomised who took at least 1 dose of study medication) as specified in the protocol and analysis plan |
| Selective reporting (reporting bias) | Low risk | Outcomes were reported as specified in the protocol and analysis plan |
| Other bias | High risk | The protocol was amended 4 times; twice to allow for interim safety analysis and once to allow for extra recruitment. Some re‐randomisation of patients occurred |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators, participants and study monitors were unaware of which treatment a participant was randomly assigned to receive. Breaking of the code by opening the hidden portion of the detachable drug label was expressly forbidden except in the event of a medical emergency where the identity of the drug was necessary in order to treat the participant properly |
NAIA3005.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study designed to investigate the efficacy and safety of zanamivir given by the inhaled route in the prevention of symptomatic disease caused by influenza A and B viral infections in community‐dwelling adults
Location, number of centres: USA, 2 centres Duration of study: 35 days |
|
| Participants | Males or females 18 years or greater from a university community without symptoms indicative of influenza prior to the prophylaxis phase of the study Definition of patient populations for analysis Intention‐to‐treat population (N = 1107) Not specified. Safety (N = 1107) All randomised participants who took 1 dose of study drug. Primary population for analysis of safety data Non‐vaccinated population (N = 948) All non‐vaccinated randomised participants who took at least 1 dose of study drug. Primary population for the analysis of efficacy Per‐protocol (N = 891) Not specified |
|
| Interventions | Zanamivir (5 mg per inhalation), 2 inhalations once a day via Rotadisk/Diskhaler Placebo, 2 inhalations once a day, via Rotadisk/Diskhaler Treatment period: 28 days Follow‐up period: 7 days post‐treatment Co‐interventions: relief medication provided as paracetamol and cough mixture |
|
| Outcomes |
Primary outcome: the proportion of non‐vaccinated randomised participants who, during prophylaxis, developed symptomatic, laboratory‐confirmed influenza A or B infection Secondary outcomes: The proportion of randomised participants who developed laboratory‐confirmed influenza infection The proportion of randomised participants who, during days 3 to 28 of prophylaxis, developed symptomatic, laboratory‐confirmed influenza infection The proportion of randomised participants who, during prophylaxis, developed a febrile illness with laboratory confirmation of influenza infection. A febrile illness was defined as a temperature of 37.8 °C The proportion of randomised participants who during prophylaxis developed a febrile illness irrespective of laboratory confirmation of influenza The maximum recorded score during days 1 to 28 for each of the symptoms recorded on the diary card The number of days out of 28 the participant was unable to perform all their normal activities The number of days out of 28 the participant recorded use of relief medication The proportion of randomised participants who, during prophylaxis, developed a secondary complication of influenza and had subsequent associated laboratory confirmation of influenza infection The proportion of randomised participants who, during prophylaxis, developed a secondary complication of influenza, irrespective of laboratory confirmation of influenza The proportion of randomised participants who required antibiotics The proportion of randomised participants who required an OTC medication The proportion of randomised participants who required a prescribed medication The proportion of randomised participants who had an unscheduled healthcare contact plus the mean number of unscheduled healthcare contacts The proportion of randomised participants confined to bed/incapacitated plus the mean duration of incapacity because of influenza The proportion of randomised participants who missed at least 1/2 day from work/school because of influenza and the mean duration missed from work/school |
|
| Notes | ITT population comprised all randomised participants. However, patients who had no diary card data from days 1 to 5 or were withdrawn prematurely from study treatment were not included Patients who were at risk of developing complications were excluded from the study and the investigators did not list complications as an outcome Inconsistencies occurred in the Protocol Summary, Study Plan and Study Procedures (Protocol 2 Amendment); cough and sore throat were listed as primary symptoms in clinical study reports but not in the protocol and the investigator global assessment was not listed in the protocol but was listed in the clinical study reports Study performed prior to influenza season in 1997 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; but method used to generate randomisation sequence not stated in clinical study reports |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Similar number of participant adverse events in zanamivir and placebo |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Similar number of participant adverse events in zanamivir and placebo |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Similar number of participant adverse events in zanamivir and placebo |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in protocol reported |
| Other bias | High risk | Exposure to lactose in test drugs may have underestimated true risk of asthma events. The protocol was amended twice during the course of the study; case narratives and CRFs blank |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Placebo described as "matching"; no certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Breaking of the code by opening the hidden portion of the detachable drug label was expressly forbidden except in the event of a medical emergency where the identity of the drug had to be known in order to treat the participant properly |
NAIB2005.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled and intranasal zanamivir in the treatment of influenza A and B viral infections Location, number of centres: Belgium (3 centres); Finland (1); France (5); Germany (1); Italy (2); Netherlands (1); Norway (6); Spain (5); Sweden (4); UK (4) Duration of study: 28 days |
|
| Participants | Male or female aged ≥18 years with influenza‐like illness for < 48 hours Number screened: not available Number randomised: 197 (zanamivir (inhaled): 64; zanamivir (inhaled and intranasal): 70; placebo: 63) Number completed: 185 M = 53 F = 47 Mean age: 34 years Baseline details: 96% Caucasian; 24% smokers Inclusion criteria
Exclusion criteria
Definition of patient populations for analysis Influenza‐positive population (N = 151) All participants in the ITT population with laboratory‐confirmed influenza determined either from pre‐treatment diagnostic sample or a positive serology result. If diagnostic sample and serology were both positive but indicated different influenza types, influenza type was assigned according to diagnostic sample result. Secondary population for assessment of efficacy Intention‐to‐treat population (N = 197) All randomised participants included in the treatment group to which they were assigned even if no medication was taken. Primary population for assessment of efficacy Safety (N = 196) Participants randomised to treatment who took at least 1 dose of study medication. Participants excluded if there was clear evidence of failure to take any study medication. Used for safety data |
|
| Interventions | Zanamivir (5 mg per inhalation) 2 inhalations twice a day plus placebo 2 sprays per nostril (0.1 mL per spray) twice a day Zanamivir (5 mg per inhalation) 2 inhalations twice a day plus zanamivir (16 mg/mL) 2 sprays per nostril (0.1 mL per spray, equivalent to 1.6 mg zanamivir) twice a day Placebo 2 inhalations twice a day plus placebo 2 sprays per nostril (0.1 mL per spray) twice a day Treatment period: 5 days Follow‐up period: 23 days post‐treatment Co‐interventions: relief medication described and measured as an outcome but not clear whether this was administered as co‐intervention |
|
| Outcomes |
Primary outcome: time to alleviation of major influenza symptoms Secondary outcomes: Time (in days from initiation of treatment) to eradication of major signs and symptoms of influenza Combined symptom analysis Mean daily temperature over the study treatment period (days 1 to 5) Number of days (from initiation of treatment) until the patient was able to return to normal activities Number of days out of days 1 to 10 that the patient recorded any symptom as 'moderate' or 'severe' Number of days out of days 1 to 10 that the patient recorded their overall symptom assessment (OSA) as 'moderate' or 'severe' Number of days out of days 2 to 10 that the patient recorded sleep disturbance as 'moderate' or 'severe' Mean supportive drug use Investigator global assessment of symptoms Day at which viral shedding fell below limit of quantitation (core centres only) Area under the viral shedding curve (core centres only) |
|
| Notes | The original intention was for 273 patients (91 patients per treatment group) to be recruited but due to the relatively low incidence of influenza, this target was not achieved. The protocol for this study was amended twice. Both of these amendments were implemented prior to the commencement of recruitment. As a result of both internal discussions and external discussions with regulatory authorities, the statistical methods employed were substantially different from those documented in the protocol. Cough and sore throat were reported as major symptom in clinical study reports but not in the protocol and discrepancies occurred between the protocol and clinical study reports regarding evaluation of viral shedding Study period: November 1994 to April 1995 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; but method used to generate randomisation sequence not stated in clinical study reports |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Data from ITT and IP populations available |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | No systematic differences in drop‐out |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Based on randomised participants |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in protocol reported |
| Other bias | High risk | As a result of both internal discussions and external discussions with regulatory authorities, the statistical methods employed were substantially different from those documented in the protocol |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The blind was maintained by ensuring that trial medication and placebo had an identical appearance, shape, administration schedule, smell and taste; no certificates of analysis were provided. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigator, all study staff, patients and the monitors were blinded as to the study treatment administered. The blind was maintained by ensuring that trial medication and placebo had an identical appearance, shape, administration schedule, smell and taste. 1 envelope was opened for a patient 3 months after the conclusion of their treatment but as this was some time after treatment, it was not seen to affect the trial. The blind was not broken for any other patients entered into this study |
NAIB2006.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study designed to evaluate the efficacy and safety of inhaled zanamivir in the prevention of progression of influenza A and B viral infections | |
| Participants | Male or female ≥ 18 years exposed to an index case with symptoms of influenza‐like illness within the previous 48 hours | |
| Interventions | Zanamivir (5 mg per inhalation), 2 inhalations twice a day by Rotadisk/Diskhaler Placebo to match zanamivir, 2 inhalations twice a day by Rotadisk/Diskhaler |
|
| Outcomes |
Primary outcome: the proportion of patients with laboratory‐confirmed influenza during treatment plus at least 2 clinically significant symptoms of influenza of 'moderate' or 'severe' severity during the study treatment period Secondary outcomes: Proportion of patients with a documented fever during the study (temperature > 37.8 °C) Number of days with any 'moderate' or 'severe' symptoms Number of days overall assessment of symptoms as 'moderate' or 'severe' Number of days with moderate or severe sleep disturbance Number of days patient was unable to perform normal activities Investigator global assessment |
|
| Notes | Statistical methods section were revised to allow for stratification of vaccinated participants In the efficacy evaluations, nasal symptoms (nasal congestion, rhinorrhoea) were changed to nasal congestion (blocked, runny nose) Safety and non‐vaccinated populations were not defined in the protocol |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided, but method used for sequence generation not specified in clinical study reports |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Patients who had no diary card data from days 1 to 5 were withdrawn prematurely from study treatment. Page numbers in clinical study reports blanked |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Patients who were at risk of developing complications were excluded from the study and the investigators did not list complications as an outcome. Page numbers in clinical study reports blanked |
| Incomplete outcome data (attrition bias) Safety data | High risk | Patients were removed from the safety population if there was documented evidence of failure to take any study medication. Page numbers in clinical study reports blanked |
| Selective reporting (reporting bias) | Low risk | All outcomes listed in the protocol were reported in the clinical study reports. Pages number blanked |
| Other bias | High risk | Inconsistencies in the protocol summary, study plan and study procedures. Clinical study report pages for this information were censored |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Investigators and participants reported to be blinded but the colour and appearance of the inhalers not described [clinical study reports page blanked out]; no certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No investigator revealed the blind for study medication prior to trial completion [clinical study reports page blanked out] |
NAIB2007.
| Methods | Placebo‐controlled, double‐blind trial to evaluate the efficacy and safety of inhaled zanamivir and the combination of inhaled plus intranasal zanamivir in the treatment of influenza A and B viral infections Location, number of centres: Australia, New Zealand, South Africa (24 centres) Duration of study: 28 days |
|
| Participants | Male or females, aged 13 years and above (aged 16 and above, or aged 18 and above in some centres) with laboratory‐confirmed influenza or influenza‐like illness as defined by symptoms of feverishness and at least 2 of the following: headache, myalgia, cough, sore throat of less than or equal to 48 hours duration Number screened: not available Number randomised: 554 (inhaled zanamivir: 188; inhaled and intranasal zanamivir: 183; placebo: 183) Number completed: 456 M = 52% F = 48% Mean age: 30 years |
|
| Interventions | Zanamivir (5 mg per inhalation) 2 inhalations twice daily plus placebo 2 sprays per nostril (0.5 mL per spray) twice daily Zanamivir (5 mg per inhalation) 2 inhalations twice daily plus zanamivir (1 mg/mL) 2 sprays per nostril (0.5 mL per spray) twice daily Placebo 2 inhalations twice daily plus placebo 2 sprays per nostril (0.5 mL per spray) twice daily Treatment period: 5 days Follow‐up period: 23 days post‐treatment Co‐interventions: paracetamol was provided for symptomatic relief |
|
| Outcomes |
Primary outcome: time to alleviation of clinically significant symptoms of influenza Secondary outcomes: Time (in days from initiation of treatment) to alleviation of individual symptoms of influenza (feverishness, headache, myalgia, cough, sore throat, nasal congestion, weakness and loss of appetite). Alleviation of an influenza symptom was defined as a score of 'none' or 'mild', which had to be maintained over the next 24 hours Time (in days from initiation of treatment) until the patient returned to normal activities. This had to be maintained for the following 24 hours Time (in days from initiation of treatment) until the patient was able to return to usual daily activities and perform these as well as normal Mean symptom score. The 5 symptoms of feverishness, headache, myalgia, cough and sore throat were summarised by the mean symptom score over days 1 to 5 of treatment Number of days over the study period (days 1 to 5) that the patient recorded their overall symptom assessment (OSA) as 'moderate' or 'severe' Number of days over the study period (days 1 to 5) that at least 1 symptom was rated as 'moderate' or 'severe' Number of days over the study period (days 2 to 5) that sleep disturbance was recorded as 'moderate' or 'severe' Maximum daily temperature summarised over the study treatment period (days 1 to 5) Mean daily paracetamol use over the study treatment period (days 1 to 5) Investigator‐rated global assessment of symptoms (GAS). The influenza infection status of patients rated by the investigator at the post‐treatment visit as 'none', 'mild, 'moderate' or 'severe' Incidence of secondary infections |
|
| Notes | Due to logistic difficulties in receiving the results of influenza diagnostic tests within 48 hours of influenza symptom onset, some patients were recruited on the basis of a clinical diagnosis of influenza Cough and sore throat were reported as major symptoms in clinical study reports but not in the protocol Protocol amendment 3 necessitated revising the definition of serious adverse events and time line changes for the reporting adverse events. Amendments were also made to definitions for primary and secondary efficacy parameters and statistical analyses were modified Protocol amendment 4 was made to the age range included (13 to 65 years, 16 to 65 years and 18 to 65 years, respectively) and inclusion or exclusion of patients with asthma, to meet local regulatory and ethics committee requirements Study period: May 1995 to May 1996 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; method of sequence generation not described in clinical study reports |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled allocation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | ITT and IP population data available for symptom relief |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Attrition does not appear to be significantly different between groups; reasons for attrition reported |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Based on randomised participants |
| Selective reporting (reporting bias) | High risk | Protocol amendment 3, which necessitated revising the definition of serious adverse events and timeline changes for reporting adverse events. Cumulative day of alleviation used to report primary outcome rather than mean or median; this was not specified in the protocol |
| Other bias | High risk | Exposure to lactose in test drugs may have underestimated true risk of asthma events. Amendments were also made to definitions for primary and secondary efficacy parameters, inclusion/exclusion of participants; and statistical analyses were modified |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Each investigator was provided with a sealed envelope containing the individual code‐break envelopes for patients in their centres but colour and appearance were not reported; no certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "...the monitors were blinded as to the study treatment administered." No investigator broke the blind for study medication prior to trial completion |
NAIB3001.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir administered twice daily in the treatment of influenza A and B viral infections in adults Location, number of centres: Australia (6 centres); New Zealand (4); South Africa (3) Duration of study: 28 days |
|
| Participants | Males or females aged ≥ 12 years (age mean 37) with laboratory‐confirmed influenza or influenza‐like illness Number screened: not specified Number randomised: 455 (zanamivir: 227; placebo: 228) Number completed: 428 Definition of patient populations for analysis Influenza‐positive population (N = 321) Secondary population for assessing efficacy. Defined as all participants the safety population who had confirmed influenza. Participants were included in this population if baseline culture test was positive or if rapid diagnostic test was positive or if serology results confirmed influenza infection (≥ 4‐fold increase in influenza antibody from day 1 to day 28) Sensitivity analysis also performed for primary endpoint on population of patients confirmed as influenza‐positive by either culture or serology ITT population (N = 455) Primary population for assessing efficacy. All randomised patients, regardless of whether or not the study drug was actually taken or completion of study. Participants analysed according to assigned treatment group irrespective of which medication they took during the study Safety population (N = 455) Primary population for the analysis of safety data. Defined as all participants randomised to treatment who took at least 1 dose of study medication. Randomised patients were excluded if there was clear evidence of failure to take study medication. Participants were analysed according to treatment group of the actual medication they took the majority of the time High risk population (N = 76) All patients in safety population at greater risk of complications if they became infected with influenza. Analysis of 'high risk' population restricted to primary endpoint, complications, adverse event incidence and serious adverse event incidence All participants ≥ 65 years were in this population. In addition, conditions thought to predispose patients to greater risk of complications from influenza included concurrent cardiovascular conditions (excluding hypertension), concurrent respiratory conditions (asthmatics excluded if unmedicated), concurrent metabolic conditions and those who were immunocompromised |
|
| Interventions | Zanamivir (5 mg per inhalation) 2 inhalations twice a day Placebo 2 inhalations twice a day Treatment period: 5 days Follow‐up period: 23 days post‐treatment Co‐interventions: paracetamol and cough mixture were provided for symptomatic relief |
|
| Outcomes |
Primary outcome: time until alleviation of major signs and symptoms of influenza Secondary outcomes: Time to eradication of major signs and symptoms of influenza. Major signs and symptoms of symptoms of influenza were defined as fever, headache, myalgia, sore throat and cough. Eradication was defined as a score of 'none' for all symptoms, which had to be maintained over the next 24 hours Time to alleviation of each of the diary card symptoms calculated separately Time until the patient returned to normal activities. This was defined as the first day on which the patient recorded that they were able to carry out all their usual daily activities. This was required to be maintained for 24 hours (2 consecutive diary card entries) Mean symptom score over post‐treatment assessments on days 1 to 5 and on days 1 to 14 Maximum daily temperature, the maximum of the temperatures was obtained for each day. The weighted mean of these values was calculated over the study treatment period. The AUC was calculated using trapezoidal methods Sleep disturbance. The definition of sleep disturbance described in the protocol (i.e. mean number of days when sleep was disturbed 'not at all' or 'slightly') was changed to number of days out of days 2 to 14 for which the patient recorded 'moderate', 'quite a bit' or 'severe' sleep disturbance. This change was made prior to blinding the study and described in the data analysis plan (DAP) Number of tablets of paracetamol taken over the treatment period (days 1 to 5) and days 1 to 14 Number of spoonfuls of cough mixture taken over the treatment period (days 1 to 5) and days 1 to 14 Incidence of complications of influenza and associated antibiotic use Investigator global assessment of symptoms |
|
| Notes | The study was conducted by 22 investigators in 3 countries (Australia, New Zealand and South Africa); only 13 of these investigators recruited patients The protocol defined the influenza‐positive population as a subset of the intention‐to‐treat population. This was changed in the data analysis plan to be a subset of the safety population. For this study, both populations were the same so the change was irrelevant Investigator global assessment of symptoms was not described in the protocol but planned prior to blinding the study Protocol amendment: At 3 Australian centres an additional study protocol was designed to collect pharmacoeconomic data was instigated. This involved interviews with influenza‐positive patients after their day 28 visit Study period: recruitment commenced in May 1997 and rolled over to 1998 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; but method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Based on ITT and IP populations |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Insufficient evidence to indicate that administration of zanamivir affects antibody response in similar way to oseltamivir. The influenza‐positive population is less likely to reflect a non‐randomised comparison |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Based on randomised participants. The safety population included all patients randomised to treatment who took at least 1 dose of study medication |
| Selective reporting (reporting bias) | Low risk | Outcomes listed in protocol reported in clinical study reports |
| Other bias | High risk | Exposure to lactose in test drugs may have underestimated true risk of asthma events. Investigator global assessment of symptoms was not defined in the protocol but was planned prior to unblinding the study and specified in the DAP. It is not clear why the patient assessment was not included to assess concordance |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Unclear if Rotadisks were identical in shape and appearance; no certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | No code‐break envelopes were opened in this study |
NAIB3002.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir (GG167) 10 mg administered twice a day for 5 days in the treatment of symptomatic influenza A and B viral infections in adolescents and adults Location, number of centres: multicentre study in Europe: Belgium (2 centres); Denmark (3); Finland (2); France (10); Germany (5); Holland (1); Italy (2); Norway (7); Spain (1); Sweden (6); UK (3) Duration of study: 28 days |
|
| Participants | Males or females aged ≥ 12 years (mean age: 37 years) with influenza‐like illness (ILI) Number screened: not available Number randomised: 356 (zanamivir: 174; placebo: 182) Number completed: 349 Definition of patient populations for analysis Influenza‐positive population (N = 277) Primary population for assessing efficacy. Defined as all participants in safety population with confirmed influenza. Participants considered influenza‐positive if positive result obtained from any of: baseline culture or polymerase chain reaction (PCR) assay, or if participants showed seroconversion (≥ 4‐fold increase in convalescent antibody titres compared with baseline demonstrated by haemagglutination inhibition) ITT population (N = 356) All randomised participants, regardless of whether study drug was taken or study completion. Participants who did not take medication to which they were randomised included in treatment group assigned. This was the secondary population for assessing efficacy Safety population (N = 356) All participants who took at least 1 dose of study medication. Participants only excluded from safety population if clear evidence of failure to take study medication. Participants who did not take medication to which they were randomised would have been included in the treatment group of the actual medication they took the majority of the time. This was the primary population for the analysis of safety data High risk (N = 32) Defined as those who could experience more prolonged or severe illness, or suffer complications from influenza due to age or underlying medical condition |
|
| Interventions | Four 5 mg doses of micronized zanamivir (as dry powder, lactose blend) or matching placebo (lactose only) Treatment period: 5 days Follow‐up period: 23 days post‐treatment Co‐interventions: paracetamol and cough mixture were provided for symptomatic relief |
|
| Outcomes |
Primary outcome: time until alleviation of clinically significant symptoms of influenza Secondary outcomes: Time to alleviation of clinically significant symptoms and no use of relief medication Time until the participant returns to normal activities Time to alleviation of each individual symptom score Mean overall influenza score Mean symptom score for each of the individual symptoms collected on the diary card Maximum daily temperature Total number of tablets of supplied paracetamol Total number of spoonfuls of supplied cough mixture Global assessment of symptoms at the post‐treatment visit Incidence of complications of influenza and associated antibiotic use |
|
| Notes | Protocol amendments
Study period: recruitment planned for between October 1997 and April 1998 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation list provided; but method of sequence generation not specified |
| Allocation concealment (selection bias) | Low risk | Pharmacy‐controlled randomisation |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | Based on ITT and IP populations |
| Incomplete outcome data (attrition bias) Complications of influenza | Low risk | Insufficient evidence to indicate that administration of zanamivir affects antibody response in similar way to oseltamivir. The influenza‐positive population is less likely to reflect a non‐randomised comparison |
| Incomplete outcome data (attrition bias) Safety data | Low risk | All 356 participants randomised to treatment were included in the safety population; missing diary cards were accounted for in analysis |
| Selective reporting (reporting bias) | Low risk | Primary and secondary endpoints reported as specified in protocol |
| Other bias | High risk | 3 protocol amendments in the course of the study |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Unclear if Rotadisks were identical in shape and appearance; no certificates of analysis were provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "...study monitors were blinded as to the study treatment administered." The code‐break envelopes were to be opened only in the event of a medical emergency where knowledge of the treatment received was absolutely necessary for management of the participant. Prior to breaking the blind the investigator was asked to discuss the matter with the study monitor or designee |
NV16871.
| Methods | Randomised, double‐blinded, placebo‐controlled, parallel‐group trial of the effect of treatment with oseltamivir on improving rate of recovery from influenza‐mediated asthma symptoms and exacerbations in children (6 to 17 years) with asthma Location, number of centres: 50 centres in 10 countries mostly in Eastern Europe | |
| Participants | A total of 329 randomised of whom 28.6% (i.e. less than estimated rate of 50%) had laboratory‐confirmed influenza; ITTI population had 51 placebo and 43 oseltamivir patients | |
| Interventions | Oseltamivir by weight dosing bid for 5 days or placebo | |
| Outcomes | Primary efficacy outcome was % change in FEV1 from days 1 to 2 to day 6 measured by spirometry. Study stratified for asthma severity at baseline (mild or moderate/severe) and time from influenza symptoms onset to first study drug dose (< 24 hours or ≥ 24 hours) | |
| Notes | There was no significant difference in outcomes between the 2 groups, though a trend in favour of oseltamivir was reported. Secondary outcomes based on diary symptoms appeared to show faster recovery, reduced rate of complications and reduced use of antibiotics in the oseltamivir group. In terms of safety the overall incidence of adverse events was higher in the oseltamivir group than the placebo group (24% versus 21%). This was mainly due to a higher incidence of gastrointestinal events (10% versus 6%), of which vomiting (8% versus 2%) was most significant The definition of the M2 Module itself was not clear in the 614 page PDF reviewed and despite page by page review some things, e.g. the certificates of analysis, were not found and protocol amendments or statistical analysis plan amendments were also not reported. No additional information from M2 changes the original summary and risk of bias |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list shows random sequence and centralised phone‐driven system was used |
| Allocation concealment (selection bias) | Low risk | Centralised phone system |
| Incomplete outcome data (attrition bias) Symptoms | High risk | In the absence of IPD and CRFs we cannot account for all participants |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Unclear how complications of influenza were defined clinically |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified either as symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same participant in different categories |
| Selective reporting (reporting bias) | High risk | SAP and certificates of analysis and amendments are missing. GCP infringements noted in 2 centres |
| Other bias | High risk | Placebo content unclear |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Placebo colour and taste not clear |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Centrally randomised scheme |
PE‐01.
| Methods | A double‐blind, double‐dummy, randomised, placebo‐controlled, parallel group, multicentre study to investigate the efficacy and safety of oral and inhaled GG167 in reducing the development of influenza symptoms | |
| Participants |
Inclusion criteria: Index case: Individuals who the investigator diagnosed as having symptoms of influenza A and B by the following criteria: individuals who satisfied the following conditions for influenza infection between the commencement of the symptoms and the first presentation to the participating centre, were willing to participate in the study and could be expected to follow the investigator's orders:
Contact: Person who had any following chance of contact with an index case within 4 days after the commencement of influenza symptoms of the index case and was diagnosed that he/she may be infected with influenza:
Participants had to be at least 16 years old, understand how to use the investigational agents and intend to co‐operate with the study and could be expected to follow the investigator's orders Exclusion criteria:
Definition of patient populations for analysis ITT population N = 44 (IH 11, IN 11, IH + IN 11 and PL 11) Protocol compatible population: N = 40 (IH9, IN 11, IH + IN 10, PL 10) Safety population: N = 44 (IH 11, IN 11, IH + IN 11 and PL 11) Safety outcome The safety measure was the incidence of abnormal symptoms/abnormal laboratory changes in which causality determinations were not completely denied. "Abnormal symptoms or abnormal laboratory findings" are the adverse events defined as newly observed or remarkably worsened symptoms after treatment started or clinically untoward abnormal or abnormally changed laboratory findings after treatment started irrespective of the causal assessment |
|
| Interventions | Orally inhaled zanamivir 10 mg + intranasally nebulised placebo Orally inhaled placebo + intranasally nebulised zanamivir 6.4 mg Orally inhaled zanamivir 10 mg + intranasally nebulised zanamivir 6.4 mg Orally inhaled placebo + intranasally nebulised placebo |
|
| Outcomes |
Primary: the proportion of subjects who, during prophylaxis (day 1 to day 5), developed symptomatic influenza Secondary:
|
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Method of sequence generation not specified |
| Allocation concealment (selection bias) | High risk | Not described |
| Incomplete outcome data (attrition bias) Symptoms | High risk | The trial was terminated prematurely |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | The trial was terminated prematurely |
| Incomplete outcome data (attrition bias) Safety data | High risk | The trial was terminated prematurely |
| Selective reporting (reporting bias) | High risk | Analysis, including primary outcomes, was planned after study unblinding |
| Other bias | High risk | Under‐recruitment |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificate of analysis |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Not described |
WV15670.
| Methods | Randomised, double‐blind, placebo‐controlled study in people with symptoms of influenza. Centres were activated to recruit participants during an influenza outbreak in the locality, detected using standardised surveillance techniques Location, number of centres: 51 centres in Europe, 11 in Canada and 1 in Hong Kong Duration of study: 21 (+/‐ 4 days) |
|
| Participants | Number screened: not available Number randomised: 719 (oseltamivir 75 mg: 242; oseltamivir 150 mg: 242; placebo: 235) Number completed: 688 M = 51% F = 49% Mean age: 37.4 Baseline details Inclusion criteria:
For the purposes of analysis and definition of the study populations, the criteria were adjusted to accept a baseline temperature of 37.8 °C and entry into the studies up to 40 hours post onset of illness, thereby accounting for differences between criteria evaluated at time of entry and criteria at time of first dose Exclusion criteria:
Definition of patient populations for analysis ITTI population (N = 425) Participants who were discovered to have been infected with laboratory‐confirmed influenza ITT population (N = 726) All randomised participants irrespective of influenza status |
|
| Interventions | Intervention:
Control: placebo size 2 capsules Treatment period: 5 days Follow‐up period: 12 to 20 days post‐treatment Co‐interventions: participants were provided with a rescue pack of paracetamol (500 mg) for symptomatic relief. The amount of medication was noted on the participant's diary card. Participants were requested not to use any other medication for the relief of symptoms during the study treatment period. However, if any other medication was taken, this was to be recorded |
|
| Outcomes |
Primary outcome: duration of illness, defined as the length of time to first alleviation of the symptoms of influenza (nasal congestion, sore throat, cough, aches and pains, fatigue, headaches and chills/sweats). This was calculated from 'time 0' (study drug initiation) to the time at which all 7 symptoms were alleviated Secondary outcomes:
|
|
| Notes | Protocol amendments
Study period: December 1997 to April 1998 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Some discrepancies noted from the Module 2 summary regarding the treatment group assignment of participants experiencing adverse events. This does not necessarily affect the method for generating the randomisation sequence but could affect ITT and safety populations |
| Allocation concealment (selection bias) | Low risk | "The randomisation numbers were generated by a central randomisation service, ICTI (Interactive Clinical Technologies inc., Princeton, NJ, USA)." "The investigator telephoned the centre to report the participant's initials, date of birth and smoking history. The randomisation number was then supplied by the centre in the form of a message on an interactive voice response system (IVRS). The investigator entered the randomisation number in the appropriate place on the case report form." |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Appendix 19 (time to alleviation of all symptoms (ITT population)) has summary info for ITT. However, the extent of missing data is unknown for this outcome |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Incomplete outcome data on complications is very likely given the inadequacies present in the way info on complications was collected. In summary, this was to ask patients to self report complications at day 8 and day 21 (follow‐up visit at end of the study) |
| Incomplete outcome data (attrition bias) Safety data | Low risk | AEs could be classified as either symptoms of influenza, complications of influenza and AEs. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Secondary illnesses were patient‐reported The body of the clinical study reports states that complications requiring antibiotic treatment were specified a priori, but even in the final version of the protocol for which we have the full text, there is no predefined list of secondary illnesses (i.e. no mention of pneumonia, bronchitis, sinusitis or otitis in the protocol), nor did complications have anything to do with antibiotic treatment according to the protocol, nor does the CRF mention specific secondary illnesses by name In addition, the protocol does not pre‐specify any secondary illnesses. They first appear in the RAP and are in the core report (Module 1). The protocol indicates that the data would be collected, so the absence of mention of secondary illnesses as an outcome (even tertiary) is not easily explicable In addition, the decision of ITTI selection for primary outcome for reporting itself may be the selective reporting bias at the start, because this could break the balance between both groups. This bias may have both factor of patient selection (selection bias) and reporting selection (reporting bias), although they were prespecified |
| Other bias | High risk | The protocol reports that dehydrocholic acid and dibasic calcium phosphate dihydrate were in the oseltamivir as well as placebo capsules. It is not clear why it is reported in another clinical study report that the dehydrocholic acid was added to the placebo to match the bitter taste of oseltamivir The certificate of analysis reports capsule No 1 (placebo) contains 6.13 mg of dehydrocholic acid. Placebo contained dehydrocholic acid but the dosage was not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "In order to maintain blinding, each participant had 2 bottles of medication for each dose interval. 1 capsule was administered from each bottle twice per day at approximately 12 hour intervals. The first dose was administered during the first (day 1) visit Each bottle was labelled with the participant number and contained identical capsules of either active compound or placebo. Those participants receiving 75 mg bid received 1 capsule containing 75 mg from 1 bottle and a matching capsule containing placebo from the other bottle at each dosing. Participants receiving doses of 150 mg bid received 1 capsule containing 75 mg active drug from each bottle at each dosing." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key to the randomisation code was available at the Study centre, to the Roche Monitors, Statisticians or at Roche Headquarters. In the event of a medical emergency the blind could be broken, if this was considered absolutely necessary to properly manage the participant, by contacting the randomisation centre. The blinding was not required to be broken for any participant during the study." |
WV15671.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study design in participants presenting with influenza‐like illness Location, number of centres: USA, 57 centres Duration of study: 12 days (+/‐ 4 days) |
|
| Participants | Number screened: not described Number randomised: 627 (oseltamivir 75 mg bid: 209; oseltamivir 150 mg bid: 210; placebo: 208) Number completed: 581 M = 49% F = 51% Mean age: 32.6 Baseline details Inclusion criteria: Fever ≥ 100 °F plus
Exclusion criteria:
Definition of patient populations for analysis Intention‐to‐treat infected population (N = 375) All participants who took 1 dose of the study drug and were subsequently discovered to have laboratory‐confirmed influenza Standard population (N not presented) As for the ITTI population, except that this was further restricted to those who took at least 5 doses of the study drug ITT population (N = 615) All participants who took at least 1 dose of the study drug. Following a request from the regulators this population was included in hypothesis testing for the primary efficacy endpoint |
|
| Interventions | Intervention:
Control: matching placebo capsules (2) for Ro 64‐0796 (GS 4104) orally bid for 5 days Treatment period: 5 days Follow‐up period: 12 to 20 days post‐treatment Co‐interventions: rescue pack consisting of paracetamol (500 mg) for symptomatic relief |
|
| Outcomes |
Primary outcome: time to alleviation of symptoms (nasal congestion, sore throat, cough, aches and pains, fatigue, headache and chills/sweats) as derived from participant symptom questionnaire. Calculated from time 0 (study drug initiation) to the time at which all 7 symptoms were alleviated. Participants who withdrew prior to the alleviation of symptoms were censored at the time of withdrawal Secondary outcomes:
|
|
| Notes | Protocol amendments Protocol amendment D (16 February 1998) revised the analyses and definition of secondary and tertiary parameters in the study. Virus titre was removed as a secondary outcome following the detection of significant variability between 2 virology laboratories with respect to these assays. The major virology parameter in these studies thus became the duration of virus shedding following inclusion into the trial. It was also believed that peak virus titre might have occurred prior to baseline for a significant number of participants entering the trial and hence this particular parameter was not analysed Study period: December 1997 to April 1998 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Telephone access available |
| Allocation concealment (selection bias) | High risk | "Randomisation was conducted by a central randomisation service by telephone. The investigator/study coordinator telephoned the randomisation centre giving the participants initials, date of birth and smoking history and the treatment number was then supplied by the centre. The randomisation number was entered in the appropriate place on the participant's Case Report Form by the investigator." |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Missing data imputed but number missing not provided. Higher attrition in treatment groups for fever |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Diagnosis of complication not standardised and based on objective criteria. Method of diagnosis was based on local centre definitions. Unknown what effect this could have on classification of outcome |
| Incomplete outcome data (attrition bias) Safety data | High risk | AEs could be classified as either symptoms of influenza, complications of influenza and AEs. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Missing summary table of hospitalisations. ITT data provided in Modules 3 to 5 |
| Other bias | High risk | Unknown what effect additional substances in placebo could have on AEs. Number of centres in Module 1 not consistent with Module 2 (60 versus 57) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matching placebo used "In order to maintain the double blind nature of the study, participants received 2 capsules twice daily for all treatments." "The identification number was added by the investigator at the time of randomisation" "No open key to the code was available at the Study centre..." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The identification number was added by the investigator at the time of randomisation." "No open key to the code was available at the Study centre, to the Monitors, Statisticians or at Gilead/Roche Headquarters" |
WV15673/WV15697.
| Methods | Combined analysis of 2 randomised, double‐blind, placebo‐controlled trials. Participants were requested to return to the clinic when investigators determined that influenza was present in the community Location, number of centres: USA; 6 centres Duration of study: 8 weeks |
|
| Participants | Number screened: not specified Number randomised: 1562 (oseltamivir 75 mg: 520; oseltamivir 150 mg: 521; placebo: 521) Number completed: 1505 M = 37% F = 63% Mean age: 34 years. Baseline details: 80% Caucasian; 11% African‐American; 3% Hispanic Inclusion criteria:
Exclusion criteria:
Definition of patient populations for analysis ITTI population Not applicable ITT population (N = 1559) All participants randomised to treatment and who took at least 1 dose of study medication |
|
| Interventions | Intervention: 1. Oseltamivir 75 mg once daily plus placebo (total daily dose: 75 mg) 2. Oseltamivir 75 mg twice daily (total daily dose: 150 mg) Control: placebo twice daily Treatment period: 6 weeks Follow‐up period: 2 weeks post‐treatment Co‐interventions: none specified |
|
| Outcomes |
Primary outcome: laboratory‐confirmed clinical influenza during the 6‐week treatment period Secondary outcomes:
|
|
| Notes | Study period not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Absence of information on randomisation procedure |
| Allocation concealment (selection bias) | High risk | The Module 1 (PDF page 23) reports: "The participant's randomisation numbers were generated by Roche and incorporated into double‐blind labeling. No open key was available at any of the study sites or to any Roche personnel involved with the study". We presume this means that Roche generated the codes and stuck them on the packaging and ICTI allocated through phone in. |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Effect of oseltamivir on antibody response impacting on diagnosis |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Original protocol missing |
| Other bias | High risk | The data set for the study is aggregated from 2 separate trials. The placebo event rates for influenza infection are very different and their aggregation conceals the variation between the results of the different studies. We tried separating estimates of effect for individual participants in the 2 trials. We failed because although participants' ID codes and centres were known, centre codes were redacted from individual participant listings, which meant that we could not disaggregate results by trial for use in meta‐analysis |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Capsule size but no details of colour, taste or contents was reported |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key to the code was available" |
WV15707.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study. Stratification by vaccination status (current season or not) and chronic obstructive airways disease (present/absent) Location, number of centres: Australia, South Africa and South America, 13 centres Duration of study: 21 +/‐4 days |
|
| Participants | Number screened: not described Number randomised: 26 (oseltamivir: 17; placebo: 9) Number completed: 25 M = 59% F = 41% Mean age: 71.5 years Baseline details Inclusion criteria:
Exclusion criteria: Not described Definition of patient populations for analysis ITTI population (N = 12) Analysis of participants according to the groups to which they were randomised, having received at least 1 dose of study treatment and laboratory‐confirmed influenza virus infection ITT population (N = 26) Analysis of participants according to the groups to which they were randomised, having received at least 1 dose of study treatment, irrespective of influenza infection status Standard population (N not reported) Population with no major protocol violations or deviations and laboratory‐confirmed influenza, who received at least the first 6 scheduled doses within 72 hours/first 5 doses within 72 hours and went on to take 9 or 10 doses. Analysis according to treatment received |
|
| Interventions | Intervention: oseltamivir 75 mg bid (total daily dose 150 mg) Control: placebo (provided as size 2 capsule containing dehydrocholic acid, dibasic calcium phosphate dihydrate and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate) Treatment period: 10 days Follow‐up period: 7 to 15 days post‐treatment Co‐interventions: not specified |
|
| Outcomes |
Primary outcome: duration of illness (time to alleviation of symptoms) Secondary outcomes:
|
|
| Notes | Study period not specified No viral swab data were collected on South American patients. This population was therefore excluded from the analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation service by phone |
| Allocation concealment (selection bias) | Low risk | Central randomisation service |
| Incomplete outcome data (attrition bias) Symptoms | High risk | In the absence of IPD and CRFs we cannot account for all participants |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Outcomes of primary interest for the ITT population not made available to the review authors |
| Other bias | High risk | Placebo contained dehydrocholic acid at a dose of 6.3 mg. Placebo contained dehydrocholic acid at a dose of 6.3 mg |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Presentation of placebo described as identical |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Centrally generated randomisation code |
WV15708.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group, multicentre study carried out in elderly persons in residential homes. Participants were randomised to treatment when a local outbreak was detected. Stratification factors were vaccine status and presence or absence of COPD Location, number of centres: Australia, New Zealand, South Africa, Brazil (14 centres) Duration of study: 8 weeks |
|
| Participants | Number screened: not described Number randomised: 372 (oseltamivir: 190; placebo: 182) Number completed: 335 M = 41% F = 59% Mean age: 79 years Baseline details: 99% Caucasian; 69% vaccinated against influenza; 12% had COPD. 90% participants had other pre‐existing diseases, of which diabetes was more common in oseltamivir than placebo (17.4% versus 8.8% respectively) Inclusion criteria: Resident in care home Exclusion criteria: Not listed Definition of patient populations for analysis ITT population Not described. Incidence of influenza was low |
|
| Interventions | Intervention: oseltamivir 75 mg od (total daily dose: 75 mg) Control: matching placebo Treatment period: 6 weeks Follow‐up period: 2 weeks post‐treatment Co‐interventions: not specified |
|
| Outcomes |
Primary outcome: laboratory‐confirmed clinical influenza, defined as: fever (temperature > 99 °F) plus 1 respiratory symptom (cough, sore throat, nasal symptoms) and 1 constitutional symptom (headache, myalgia, sweats/chills, fatigue). Laboratory confirmation by either virus shedding within 2 days of symptom onset or 4‐fold increase in influenza antibody Secondary outcomes:
|
|
| Notes | Study period not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Randomisation numbers generated by Roche but more details of method and randomisation schedule not available |
| Allocation concealment (selection bias) | High risk | Insufficient details given |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups. Low rates of attrition from treatment groups were noted |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Patient‐reported outcomes |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Postdate changes to protocol after closure of enrolment. Outcome changes should be consistent with data collected |
| Other bias | High risk | Placebo contents and colour and similarity to active drug not described; very low rates of influenza or ILI in trial, so could not analyse for primary outcome of efficacy |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Information on appearance of placebo capsules not available |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Centrally generated randomisation code |
WV15730.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study. Participants were stratified by current smoking behaviour (smoker/non‐smoker). Centres activated to recruit participants during an influenza outbreak in the locality, detected using standardised surveillance techniques Location, number of centres: Australia and South Africa, 12 centres Duration of study: 21 +/‐ 4 days |
|
| Participants | Number screened: not described Number randomised: 58 (oseltamivir: 31; placebo: 27) Number completed: 56 M = 52% F = 48% Mean age: 35 years Baseline details: 93% Caucasian; 21% smoking history Inclusion criteria:
Exclusion criteria:
Definition of patient populations for analysis ITTI population (N = 38) Participants analysed according to groups to which they were randomised providing they had received at least 1 dose of study treatment and had laboratory‐confirmed influenza virus infection ITT population (N = 58) The ITT population consisted of the same participants as the ITTI population; also included participants who did not have laboratory‐confirmed influenza but took at least 1 dose of study medication. Participants analysed by groups to which they were randomised Safety population (N = 58) All participants randomised, who received at least 1 dose of study medication and at least 1 safety follow‐up, whether or not they had withdrawn prematurely. Participants who received therapy other than intended were analysed according to therapy received Standard population (N = 38) All randomised participants without major protocol violations or deviations, with laboratory‐confirmed influenza and who received at least the first 6 scheduled doses within 72 hours or who received the first 5 doses within 72 hours and went on to take 9 or 10 doses. Participants analysed according to treatment received |
|
| Interventions | Intervention: oseltamivir 75 mg bid (total daily dose: 150 mg), given as size 2 capsule Control: placebo, given as size 2 capsule Treatment period: 5 days Follow‐up period: between 12 and 20 days post‐treatment Co‐interventions: rescue medication pack |
|
| Outcomes |
Primary outcome: time to alleviation of symptoms. Assessed as alleviation of nasal congestion, sore throat, cough, aches and pains, fatigue, headache and feeling feverish. Time to alleviation of symptoms calculated from study drug initiation to time at which all symptoms were alleviated. Participants withdrawing prior to alleviation of all symptoms were censored at the time of withdrawal Secondary outcomes:
|
|
| Notes | Study period not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Original randomisation list not provided |
| Allocation concealment (selection bias) | Low risk | Central randomisation service |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Missing data imputed; number missing not reported |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Diagnosis of complication not standardised and based on objective criteria. Method of diagnosis was based on local centre definitions. Unknown what effect this could have on classification of outcome |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | 70% under‐recruitment |
| Other bias | High risk | Placebo capsule contained dehydrocholic acid. Unknown what effect this could have had on AEs Mentioned in protocol amendment that South American (SA) sites could not diagnose influenza by culture due to delays in processing, however there is no mention in M1 or rest of M2 that SA sites were included |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Certificates of analysis show identical colour and size |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key to the code was available at the study centre, to the monitors, statistician or at Roche Headquarters. In the event of a medical emergency the blinding was to be broken if considered absolutely mandatory to properly manage the patient by contacting the randomisations centre. The blinding was not broken for any participant during the study." |
WV15758.
| Methods | Randomised, double‐blind, placebo‐controlled study stratified for the presence of acute otitis media Location, number of centres: USA and Canada, 80 centres Duration of study: 28 +/‐4 days |
|
| Participants | Number screened: not described Number randomised: 698 (oseltamivir: 342; placebo: 356) Number completed: 655 M = 50% F = 50% Mean age: 5.34 years Baseline details: 65% Caucasian; 18% otitis media Inclusion criteria:
Exclusion criteria:
Definition of patient populations for analysis ITTI infected population (N = 452) Participants analysed according assigned treatment provided they received at least 1 dose of study treatment with some follow‐up efficacy data and had laboratory‐confirmed influenza virus infection as determined post entry into the study ITT population (N = 695) All participants who took at least 1 dose of study medication with some follow‐up efficacy data irrespective of influenza virus infection. Participants analysed according to the groups to which they were randomised Safety population (N = 695) Participants who received at least 1 dose of study medication and who received at least 1 safety follow‐up, whether or not withdrawn prematurely. Participants analysed according to treatment received Standard population (N = 396) Used for summaries of efficacy parameters. All participants who had no major protocol violations or deviations, who had laboratory‐confirmed influenza and who received at least the first 6 scheduled doses within 72 hours or who received the first 5 doses within 72 hours but went on to take 9/10 doses. Participants analysed according to the treatment received |
|
| Interventions | Intervention: oseltamivir 2 mg/kg (not exceeding a maximum of 100 mg/dose) bid Control: placebo bid Study drugs administered as dry powder to be reconstituted with water Treatment period: 5 days Follow‐up period: 19 to 27 days post‐treatment Co‐interventions: relief medication was provided but details not specified |
|
| Outcomes |
Primary outcome: Time to freedom from illness: defined as the length of time taken from the start of treatment to the point at which all of the following criteria were met
The duration of the event was calculated from 'time 0' (study drug initiation) to the time at which all the above 3 conditions were simultaneously met and remained true for a minimum of 24 hours Secondary outcomes:
|
|
| Notes | Protocol amendments
Study period: December 1998 to April 1999 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Original randomisations not provided in randomisation list |
| Allocation concealment (selection bias) | Low risk | "Randomization was conducted by a central randomisations service, ICTI (Interactive Clinical Technologies Inc., Princeton, NJ). The investigator telephoned the centre to report the participant's date of birth, sex and weight. The randomisations number was then supplied by the centre in the form of a message on an interactive voice response system (IVRS). The investigator entered the randomisations number in the appropriate place on the case report form. The participant randomisation numbers were allocated sequentially within a stratum in the order in which participants were enrolled." |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Primary outcome changed during trial. Missing data imputed, number missing not reported |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Diagnosis of complications not standardised and based on objective criteria. Method of diagnosis was based on local centre definitions. Unknown what effect this may have had on classification of outcome |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Original protocol missing |
| Other bias | High risk | Dehydrocholic acid in placebo. Unknown what impact this could have had on adverse events |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Certificates of analysis available |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key to the code was available (...) to the Roche monitors, statisticians or at Roche Headquarters." |
WV15759/WV15871.
| Methods | Randomised, double‐blind, stratified placebo‐controlled study. Stratification by asthma severity Location, number of centres: not available Duration of study: not available |
|
| Participants | Number screened: not provided Number randomised: not provided (oseltamivir: NA; placebo: NA) Number completed: not provided M = NA F = NA Mean age: NA Baseline details: NA Inclusion criteria:
Exclusion criteria: None specified Definition of patient populations for analysis ITTI population (N = NA) Not specified ITT population (N = NA) Not specified |
|
| Interventions | Intervention: oseltamivir: 2.0 mg/kg bid Control: matching placebo bid Study drugs administered as dry powder Treatment period: 5 days Follow‐up period: not specified Co‐interventions: not specified |
|
| Outcomes |
Primary outcome: Composite of all of the following:
Secondary outcomes:
|
|
| Notes | Study period not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Only treatment received provided on randomisation lists |
| Allocation concealment (selection bias) | Low risk | Central randomisation service provided by telephone |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Missing data imputed for primary outcome but numbers missing not provided |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Diagnosis of complications not standardised and based on objective criteria. Method of diagnosis was based on local centre definitions. Unknown what effect this may have had on classification of outcome |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Missing data for hospitalisations ITT population |
| Other bias | High risk | Placebo capsule contained dehydrocholic acid. Unknown what effect this may have had on adverse events |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Certificates of analysis show identical colour and size |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key to the code was available (...) to the Roche monitors, statisticians or at Roche Headquarters." |
WV15799.
| Methods | Randomised, double‐blind, placebo‐controlled cluster trial recruiting families of 3 to 8 members. Households recruited if any member developed an influenza‐like illness during an influenza outbreak within the community (index case) Location, number of centres: USA (35 centres); Canada (11 centres); Denmark (1 centre); Finland (6 centres); Germany (6 centres); Netherlands (3 centres); Norway (2 centres); Switzerland (1 centre); UK (8 centres) Duration of study: 21 +/‐ 4 days |
|
| Participants | Number screened: not described Number randomised: 962 (oseltamivir: 498; placebo: 464) Number completed: 944 M = not reported F = not reported Mean age: range from 1 to 76 years Baseline details: 13% contacts had received influenza vaccination in the same season. 40% contacts had pre‐existing diseases (most frequently reported: asthma (3.0%), hypertension (5.7%), drug hypersensitivity (3.9%) and depression (2.9%)) Inclusion criteria:
Exclusion criteria: Not specified Definition of patient populations for analysis ITT (contacts: N = 550; index cases: 370) People residing in the same house as an index case (someone with ILI, irrespective of baseline infection status) ITTI (contacts: N = 405; index cases: 163) People residing in the same house as a positive index case (somebody with confirmed influenza at baseline) Standard population: N = unclear Mentioned but not described |
|
| Interventions | Intervention: oseltamivir 75 mg od (total daily dose: 75 mg) Control: placebo Treatment period: 7 days Follow‐up period: 10 to 18 days post‐treatment Co‐interventions: index case received paracetamol |
|
| Outcomes |
Primary outcome: Incidence of laboratory‐confirmed clinical influenza in contacts of the index case. Defined as fever plus at least 1 respiratory symptom (cough, sore throat, nasal congestion) and 1 constitutional symptom (fatigue, aches and pains, headache, feeling feverish), all recorded on the same day (either by the investigator as an illness visit report on the CRF, or by the participant on their diary card) plus laboratory confirmation of influenza infection Secondary outcomes:
|
|
| Notes | Study period not specified | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Described as randomised; procedure generating randomisation described |
| Allocation concealment (selection bias) | Low risk | Adequate information to ascertain concealment of allocation available |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the intervention groups. Only symptomatic participants were swabbed. Low rates of attrition from treatment groups were noted |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Original protocol missing |
| Other bias | High risk | Diagnosis breakdown by culture and/or antibody titre rises not reported |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Certificates of analysis show identical colour |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Centrally generated randomisation code. Randomisation key only available to the clinical trial supplies group for packaging purposes. Some participant numbers in M1 could not be found on the randomisation list (e.g. 3913 and 3921) |
WV15812/WV15872.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group design. Stratification performed by presence of chronic obstructive airways disease (COAD) Location, number of centres: northern hemisphere (80 centres) and southern hemisphere (20 centres) during influenza seasons Duration of study: 21 +/‐ 4 days |
|
| Participants | Number screened: not reported Number randomised: 404 (oseltamivir: 200; placebo: 204) Number completed: 393 M = 44% F = 56% Mean age: 52 years Baseline details: COAD 76%; vaccination: 28%; smoking: 22% Inclusion criteria:
Exclusion criteria:
Definition of patient populations for analysis ITTI population (N = 231) All patients who had at least 1 dose of study medication and who had a laboratory‐confirmed influenza virus infection. Data were analysed according to treatment assignment at randomisation ITT population (N = 402) All randomised patients who received at least 1 dose of study medication Safety population (N = 401) Randomised participants who received at least 1 dose of study medication and had at least 1 post‐baseline safety assessment Standard population (N = 236) Participants from ITTI population without major protocol violations and who received at least the first 6 scheduled doses within 72 hours, or received the first 5 doses within 72 hours and went on to take 9 out of the 10 doses |
|
| Interventions | Intervention: oseltamivir 75 mg bid (total daily dose 150 mg) Control: placebo bid Treatment period: 10 days Follow‐up period: 7 to 15 days Co‐interventions: pack of paracetamol (500 mg) |
|
| Outcomes |
Primary outcome: time to alleviation of illness (derived from a patient‐rated symptom questionnaire). The 7 symptoms assessed in the questionnaire were:
Secondary outcomes:
|
|
| Notes | Study period: WV15812: January to April 1999; WV15872: June to October 1999 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation service, block randomisation (block size = 4) |
| Allocation concealment (selection bias) | Low risk | Central randomisation service |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Missing symptoms data not reported. Available data analysed by ITTI population and not ITT |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Missing symptoms data not reported. Available data analysed by ITTI population and not ITT |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Original protocol for WV15812 missing |
| Other bias | High risk | Placebo contained dehydrocholic acid. Dosage not available. Active drug Ro 64‐0796 batches GMZ 0124/03 and GMZ 0129/03; matching placebo GMZ 0136 |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No certificate of analysis to confirm same shape/size/colour |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | From Module 1: "No open key to the code was available at the Study Centre, to the Monitors, Statisticians or at Roche Headquarters. The blind was to be broken only in the event of a medical emergency if considered absolutely necessary to manage the patient." |
WV15819/WV15876/WV15978.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group design. Participants were stratified according to vaccination status (vaccinated in the current influenza season or not) and coexistence or not of chronic obstructive airways disease (COAD) Location, number of centres: France, the Netherlands, Belgium, Germany, Switzerland, United Kingdom, Norway, Sweden, Denmark, Israel, Lithuania, Estonia, Poland, Canada USA, Canada, South Africa, New Zealand, Australia; 169 centres Duration of study: 21 +/‐ 4 days |
|
| Participants | Number screened: not reported Number randomised: 726 (oseltamivir: 362; placebo: 374) Number completed: 715 M = 43% F = 57% Mean age: 73 years Baseline details: 98% Caucasian; COAD: 8%; vaccination: 43% Inclusion criteria:
Exclusion criteria:
Definition of patient populations for analysis ITTI population (N = 477) Primary analysis population for efficacy. Participants analysed according to the groups to which they were randomised, provided they received at least 1 dose of study treatment and had laboratory‐confirmed influenza virus infection. Participants with protocol violations or deviations were retained in the ITTI population ITT population (N = 735) All participants who took at least 1 dose of study medication. Participants analysed according to the groups to which they were randomised Safety population (N = 736) All randomised participants who received at least 1 dose of study medication and who had at least 1 safety follow‐up, whether or not withdrawn prematurely. Data from participants were analysed according to therapy they received Standard population (N = 445) All randomised participants who had no major protocol violations or deviations, laboratory‐confirmed influenza virus infection and who received at least the first 6 scheduled doses within 72 hours or who received the first 5 doses within 72 hours but went on to take 9 out of 10 total doses. Participants were analysed according to treatment received |
|
| Interventions | Intervention: oseltamivir 75 mg bid (total daily dose 150 mg) given as size 2 capsules Control: matching placebo size 2 capsules Treatment period: 5 days Follow‐up period: 12 to 20 days post‐treatment Co‐interventions: rescue pack of paracetamol |
|
| Outcomes |
Primary outcome: Duration of illness given as summary measures from Kaplan‐Meier survival curves Secondary outcomes:
|
|
| Notes | Protocol amendments
Study period: Northern Hemisphere centres recruited during flu seasons in 1998 and 1999; Southern Hemisphere centres recruited during flu seasons in 1999 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation list provided |
| Allocation concealment (selection bias) | Low risk | Central randomisation service provided by telephone |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Missing data imputed but number missing unknown |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups. Also diagnosis not based on standardised objective criteria but based on local centre definitions. Unknown what effect this may have had on classification of outcome |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | Selection of symptom reporting after major protocol amendment |
| Other bias | High risk | Module 1 implies active contains dehydrocholic acid but certificate of analysis suggests otherwise. Unknown what effect this may have had on adverse events |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Certificates of analysis show identical colour and size |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key to the code was available at the study centres, to the monitors, statisticians or at Roche headquarters. In the event of a medical emergency the blind could be broken, if considered absolutely mandatory to properly manage the subject, by contacting the randomisations centre." |
WV15825.
| Methods | Randomised, double‐blind, placebo‐controlled trial in residential homes for elderly people. Participants were recruited when a local outbreak was detected, defined as 2 cases in immediate vicinity within 7 days or 1 case in the home itself Location, number of centres: USA (16 centres), UK (1 centre), France (4 centres), Belgium (2 centres) and the Netherlands (3 centres) Duration of study: 8 weeks |
|
| Participants | Number screened: not reported Number randomised: 548 (oseltamivir: 276; placebo: 272) Number completed: 493 M = 31% F = 69% Mean age: 82 years Baseline details: 92% Caucasian; 4% Black; 4% Hispanic. 80% vaccinated; 14% COAD Inclusion criteria: No inclusion criteria detailed. Study conducted in residential homes for the elderly Exclusion criteria: Not specified Definition of patient populations for analysis Prophylaxis study, differentiation between populations at baseline not undertaken |
|
| Interventions | Intervention: oseltamivir 75 mg (frequency of administration not specified) Control: placebo Treatment period: not specified Follow‐up period: 8 weeks Co‐interventions: not specified |
|
| Outcomes |
Primary outcome: Laboratory‐confirmed clinical influenza. Defined as fever (temperature > 99 °F) plus 1 respiratory symptom (cough, sore throat, nasal symptoms) plus 1 constitutional symptom (headache, myalgia, sweats/chills, fatigue) confirmed by either virus shedding within 2 days of symptom onset or 4‐fold increase in influenza antibody Secondary outcomes: Adverse events |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Centrally generated randomisation code. No details reported. Randomisation key only available to the clinical trial supplies group for packaging purposes. Some participant numbers in M1 could not be found on the randomisation list (e.g. #3913 and #3921) |
| Allocation concealment (selection bias) | High risk | In the Module 1 at page 21: "The subject randomisation numbers were generated by Roche and incorporated into double‐blind labeling. No open key was available at any of the study sites or to any Roche personnel involved with the study". Roche generated the codes and stuck them on the packaging and ICTI allocated through phone‐in system |
| Incomplete outcome data (attrition bias) Symptoms | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the intervention groups. Only symptomatic participants were swabbed. Low rates of attrition from treatment groups were noted |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Possible effect of oseltamivir on antibody production makes the assessment of influenza status and associated complications in the infected subpopulation non‐comparable between the treatment groups |
| Incomplete outcome data (attrition bias) Safety data | High risk | Adverse events could be classified as either symptoms of influenza, complications of influenza or adverse events. Reporting is inconsistent and some trials reported the same outcome in the same patient in different categories |
| Selective reporting (reporting bias) | High risk | No definitions given – see pg 69 and protocol 367. Events after positive swab are 3 in placebo group and 1 in oseltamivir group (pg 70 also pg 330‐1) Participants with ILI in adverse events table are not included in efficacy analysis |
| Other bias | High risk | Retrospective changes to protocol and reporting analysis plan |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Certificate of analysis available. Placebo contained dehydrocholic acid. Dosage not available. More information needed on whether patients and personnel could distinguish between the 2 treatments |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "No open key will be available" |
WV16277.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study. Patients were stratified by age, gender, vaccination status (vaccinated in current season or not) and coexistence of chronic obstructive airways disease (present/absent). The objectives of the study were: ‐ To investigate the clinical efficacy of oseltamivir in patients with influenza ‐ To investigate the safety and tolerability of oseltamivir in patients with influenza ‐ To investigate the effect of oseltamivir on well being and daily disruption of patients with influenza ‐ To investigate the antiviral efficacy of oseltamivir in patients with influenza (in selected patients) |
|
| Participants | Patients aged 13 or more years (or 18 or more in countries with local IRB requirements) presenting with symptoms of influenza (fever 37.8 °C or more with at least 2 of the following symptoms: cough, sore throat, nasal congestion, chills/sweats, headache, myalgia (aches and pains), fatigue) | |
| Interventions | Oseltamivir was provided as size 2 capsules containing 75 mg of active drug and packaging material consisting of pregelatinised starch, povidone, talc and sodium stearyl fumarate Matching placebo was provided as size 2 capsules, containing dehydrocholic acid, dibasic calcium phosphate dihydrate, pregelatinised starch, povidone, talc and sodium stearyl fumarate The following batch numbers of drugs were used in all centres: Oseltamivir: 75 mg capsules, Ro 64‐0796/V14, batch number PT2247C01 Matching placebo capsules: Ro 64‐0796/V16, batch number GMZ 0163 The medication was to be taken within 36 hours of first symptom onset at the clinic |
|
| Outcomes | This study was designed to investigate whether oseltamivir suppressed the main clinical symptoms (fever and cough) of patients with laboratory‐confirmed influenza. For all time‐to‐event analyses, time zero was defined as coinciding with the initiation of treatment. For all endpoints defined in this section that involve 'alleviation' of symptoms, alleviation occurred at the start of the 24‐hour period in which the symptom was less than or equal to 1 (mild) and remained less than or equal to 1 for at least 24 hours Primary outcome: The primary efficacy parameter was the proportion of patients alleviated of cough (none or mild for at least 24 hours) and without fever (37.2 °C or less) at 36 hours after initiation of study medication Secondary outcomes: Symptoms 1. The efficacy parameter to reflect the duration of illness was the length of time until alleviation of all symptoms. This was calculated from Time 0 (study drug initiation) to the length of time over which the area under the curve (AUC) for symptoms was to be calculated. Participants withdrawing prior to alleviation of symptoms were censored at last assessment 2. The efficacy parameter to reflect the extent and severity of illness was the AUC for symptoms. This was calculated from Time 0 (study drug initiation) to the time at which all symptoms were alleviated. Scores were calculated twice daily by totaling the separate symptoms that form the symptom scale. The AUC of these scores was then calculated for each patient using the trapezoidal rule. The baseline for each symptom was defined as the assessment prior to first study drug intake even though this might be before Time 0 3. Proportion of participants who were afebrile (oral temperature 37.2 °C or less) at 24 hours, 36 hours and 48 hours 4. Proportion of patients with fever each day (fever was defined as an oral temperature of 37.8 °C or higher at any assessment during that day) 5. At 12‐hourly intervals over the first 2 days, the proportion of patients with alleviation of cough and no fever (37.2 °C or less), with a reduction in ≥ 1 symptom score, with alleviation of cough, with alleviation of myalgia and with alleviation of cough and no fever (38 °C or less) |
|
| Notes | ECGs were collected for a subset of (intensive) patients only, on day 1, day 3 and day 5. Further measurements were to be taken at day 10 or day 21 if abnormal results were observed at the previous visit. ECG tests were not part of the protocol but are mentioned as part of a "sub‐study" at PDF pg 63 Blank CRFs, searches under bronchitis, secondary identified only cases of bronchitis classified as harms or pre‐existing conditions. Modules 3 and 4 table of content pages (PDF pages 660 and 3011) do not report listings of complications. H&L meta‐analysis shows confidence interval without recognisable point estimate |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ICTI‐run central randomisation scheme |
| Allocation concealment (selection bias) | High risk | Different coloured caps in size 2 capsules. Content of capsules stated, certificate of analysis missing perhaps because the August 2000 certificates from WP16263 are sufficient, but are not referenced. Concentration of dehydrocholic acid and CaPO4 unreported |
| Incomplete outcome data (attrition bias) Symptoms | Low risk | No systematic differences in drop‐out rates between intervention groups; outcomes reported as specified in the protocol |
| Incomplete outcome data (attrition bias) Complications of influenza | High risk | Great difficulty in reconciling rationale with focus on cardiac complications of influenza with design and reporting of trial |
| Incomplete outcome data (attrition bias) Safety data | Low risk | Narratives for serious adverse events and drop‐outs are reported with centre ID redacted |
| Selective reporting (reporting bias) | High risk | Undated amendment to text indicates sub‐study of selected "intensive" population on the basis of viral isolates at baseline reported in the CRF: PDF page 388 "In selected patients (qualified as intensive patients in the Case Report Form), ECG's will be taken at baseline, day 3 and day 5. In these intensive patients, nose and throat swabs will be taken for virus culture at screening and on study days 3 and 5. In these patients also, a blood sample for laboratory safety measurements will be taken on days 1, 3 and 5 (and day 10/21 if indicated by the day 5/10 results)." Reason for change: "The sampling for the virology at baseline has been restricted to the selected intensive patients and the collection of the pharma‐economic data in the questionnaire has been updated" |
| Other bias | High risk | Certificate of analysis missing. Grossly underpowered trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Different coloured capsules (identified through batch numbers and certificates of analysis of other study reports) |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Different coloured capsules unknown concentration of content |
AEs: adverse events AUC: area under the curve bid: twice daily CSR: clinical study report CARIF: severity score CDC: US Centers for Disease Control and Prevention COAD: chronic obstructive airways disease CONSORT: Consolidated Standards of Reporting Trials COPD: chronic obstructive pulmonary disease CPK: inflammation marker CRF: clinical report form d: day DAP: data analysis plan ECG: electrocardiogram EMA: European Medicines Agency EMEA: formerly EMA f: female FAS: full analysis set FEV1: forced expiratory volume (at interval 1 in spirometry testing) FDA: US Food and Drug Administration GCP: good clinical practice h: hour HAI: anti‐haemagglutinin antibody IH: inhaled ILI: influenza‐like illness IN: intranasal IP: infection‐positive IPD: individual participant data IRB: institutional review board ITT: intention‐to‐treat (population) ITTI: intention‐to‐treat (influenza)‐infected (population) LRTI: lower respiratory tract infection m: male MEP: middle ear pressure MI: myocardial infarction N: number NA: not applicable od: once daily OTC: over‐the‐counter PCR: polymerase chain reaction PEFR: peak expiratory flow rate P‐R:1 of the segments of the ECG trace QRS: 1 of the segments of the ECG trace QT: 1 of the segments of the ECG trace QTc: 1 of the segments of the ECG trace RAP: reporting analysis plans R‐R: risk ratio RSV: respiratory syncytial virus RTI: respiratory tract infection SAE: serious adverse event SAP: statistical analysis plan URTI: upper respiratory tract infection WBC: white blood cell WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| 105934 | Post‐marketing study |
| 107485 | Dose‐ranging study |
| 108127 | Non‐randomised study |
| 112311 | Pharmaco‐availability study |
| 112312 | Pharmaco‐availability study |
| 113268 | Pharmaco‐availability study |
| 113502 | Non‐comparative study |
| 113625 | Pharmacokinetics study |
| 113678 | Non‐comparative study |
| 114045 | Survey |
| 114373 | Not placebo/do nothing controlled |
| 167‐02 | Dose ranging phase 1 in volunteers, no influenza exposure |
| 167‐03 | Dose ranging phase 1 in volunteers, no influenza exposure |
| 167‐04 | Dose ranging phase 1 in volunteers, no influenza exposure |
| 167‐05 | Dose ranging phase 1 in volunteers, no influenza exposure |
| 167T3‐11 | An open‐label trial of 20 mg CG167 (zanamivir) in the treatment of influenza viral infection in children aged ≤ 5 and < 15 years old (open‐label study). Non‐randomised; the intervention group was compared to a survey group. 18 page summary available with no title |
| ADS‐TCAD‐PO206 | Not placebo/do nothing controlled |
| BP21288 | Pharmacokinetics study |
| C94‐009 | Pharmacokinetics study |
| C94‐085 | Pharmacokinetics study |
| GCP/95/045 | Pharmacokinetics study |
| JNAI‐02 | Unknown study. Only ID traced |
| JNAI‐03 | Unknown study. Only ID traced |
| JP15734 | Pharmacokinetics non‐comparative study |
| JP15735 | Does not test treatment, prophylaxis or PEP and there was no exposure to influenza |
| JV16284 | Open‐label, no control |
| JV21490 | No influenza circulation, phase IV study with unusual oseltamivir dosages |
| M76006 | Not placebo/do nothing controlled |
| ML17279 | CSR bears no title. Study of community pharmacist availability |
| ML17713 | Non‐comparative study |
| ML19340 | Text in French. Community pharmacist availability study |
| ML20542 | Not placebo/do nothing controlled |
| ML21954 | Not placebo/do nothing controlled |
| ML22789 | Not placebo/do nothing controlled |
| ML22872 | Not placebo/do nothing controlled |
| ML22879 | Not placebo/do nothing controlled |
| ML25018 | Bioavailability study |
| ML25087 | Not placebo/do nothing controlled |
| ML25094 | Non‐comparative study |
| ML25157 | Pharmacokinetics study |
| ML25176 | Pharmacokinetics study |
| ML25179 | Not placebo/do nothing controlled |
| ML25265 | Non‐comparative observational study |
| ML25266 | Pharmacokinetics study |
| MP20691 | Pharmacokinetics study |
| MV20043 | Transmission study |
| MV20050 | Dose‐ranging study |
| MV22926 | Non‐comparative study |
| MV22949 | Pharmacokinetics study |
| MV22951 | Pharmacokinetics study |
| MV22963 | Pharmacokinetics study |
| MV22970 | Pharmacokinetics study |
| NAI106784 | Pharmacokinetics study |
| NAI108166 | Pharmacokinetics study |
| NAI10901 | Comparator is vaccine |
| NAI10902 | Pharmacokinetics study |
| NAI40012 | Instructional leaflet study |
| NAIA1009 | Pharmacokinetics study |
| NAIA2010 | Open‐label, rimantadine‐controlled, cluster‐randomised trial |
| NAIB1001 | Pharmacokinetics study |
| NAIB1002 | Pharmacokinetics study |
| NAIB1007 | Pharmacokinetics study |
| NCT00297050 | Dose‐ranging study |
| NCT00416962 | Not placebo/do nothing controlled |
| NCT00867139 | Not placebo/do nothing controlled in immunocompromised people |
| NCT00957996 | Peramivir study ‐ does not have placebo/do nothing comparator |
| NCT01063933 | Pharmacokinetics study |
| Not applicable (registry) | Unknown study. Only ID traced. Identified from Reddy D. J Antimicrob Chemother 2010; 65 Suppl 2: ii35–40 (doi:10.1093/jac/dkq014) Table 2. http://jac.oxfordjournals.org/cgi/content/full/65/suppl_2/ii35/DKQ014TB2 |
| NP15525 | Pharmacokinetics study |
| NP15717 | Pharmacokinetics study |
| NP15718 | Pharmacokinetics study |
| NP15719 | Pharmacokinetics study |
| NP15728 | Pharmacokinetics study |
| NP15729 | Pharmacokinetics study |
| NP15743 | Palatability study, open‐label |
| NP15757 | Pharmacokinetics study |
| NP15810 | Pharmacokinetics study |
| NP15826 | Pharmacokinetics study |
| NP15827 | Pharmacodynamics study |
| NP15881 | Palatability study in children |
| NP15901 | Pharmacokinetics study |
| NP15912 | Palatability study in children |
| NP16472 | Not placebo/do nothing controlled |
| NP22770 | Pharmacokinetics study |
| NP25138 | Not placebo/do nothing controlled |
| NP25139 | Not placebo/do nothing controlled |
| NP25140 | Pharmacokinetics study |
| NV20234 | Immunocompromised participants |
| NV20235 | Immunocompromised participants |
| NV20237 | Resistance study |
| NV22155 | Not placebo/do nothing controlled |
| NV22158 | Registry study |
| NV25118 | Pharmacokinetics study |
| NV25182 | Not placebo/do nothing controlled |
| NV25655 | Open‐label pharmacokinetics study |
| PP15974 | Pharmacokinetics study |
| PP16351 | Pharmacokinetics study |
| PP16361 | Pharmacokinetics study |
| PV15615 | Viral challenge study |
| PV15616 | Viral challenge study |
| WP15517 | Pharmacokinetics study |
| WP15525 | Pharmacokinetics study |
| WP15647 | Pharmacokinetics study |
| WP15648 | Pharmacokinetics study |
| WP15676 | Pharmacokinetics study |
| WP15979 | Bioavailability study |
| WP16094 | Pharmacokinetics study |
| WP16134 | Bioequivalence study |
| WP16137 | Bioequivalence study |
| WP16225 | Bioequivalence study |
| WP16226 | Pharmacokinetics study |
| WP16254 | Pharmacokinetics study |
| WP16263 | No influenza circulation, phase IV study |
| WP16295 | Open‐label absorption study |
| WP17721 | Pharmacokinetics study |
| WP18308 | Pharmacokinetics study |
| WP20727 | Pharmacokinetics study |
| WP20749 | Not placebo/do nothing controlled |
| WP21272 | Pharmacokinetics study |
| WP22849 | Pharmacokinetics study |
| WV15731 | No placebo arm |
| WV16139 | Unknown study. Only ID traced. ID could be a typo |
| WV16193 | Not placebo/do nothing controlled |
CSR: clinical study report ID: identity number PEP: post‐exposure prophylaxis
Characteristics of studies awaiting assessment [ordered by study ID]
JPRN‐JapicCTI‐111647.
| Methods | A randomised, double‐blind, placebo‐controlled study to confirm the efficacy in the prevention of influenza virus infection (Phase 3 study) Duration of the study 2011‐10‐1 to 2012‐6‐30 Sponsor: Daiichi Sankyo Co., Ltd |
| Participants | — |
| Interventions | Laninamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
ML20589.
| Methods | Economic and social benefits of treating and preventing influenza in aged care facilities Sponsor: The University of Sydney, Australia anzctr.org.au number ACTRN12606000278538 |
| Participants | — |
| Interventions | Oseltamivir, 3 different regimens |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
ML20910.
| Methods | A study of Tamiflu (oseltamivir) treatment in laboratory‐confirmed influenza Sponsor: Hoffmann‐La Roche NCT00436124 |
| Participants | — |
| Interventions | Oseltamivir |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
ML21776.
| Methods | Study to evaluate nosocomial transmission of influenza Sponsor: University Hospitals, Leicester NCT00798421 |
| Participants | — |
| Interventions | — |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
MV21118.
| Methods | Early oseltamivir treatment of influenza in children 1 to 3 years of age Sponsor: Hospital District of Southwestern Finland NCT00593502 |
| Participants | — |
| Interventions | Oseltamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
MV21737.
| Methods | Long‐term influenza prophylaxis with inhaled zanamivir or oral oseltamivir Sponsor: University of Oxford NCT00980109 |
| Participants | — |
| Interventions | Oseltamivir, zanamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
MV21879.
| Methods | Oseltamivir randomised controlled efficacy trial Sponsor: International Centre for Diarrhoeal Disease Research, Bangladesh NCT00707941 |
| Participants | — |
| Interventions | Oseltamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
MV22841.
| Methods | An observational clinical trial of influenza A/H1N1 2009 resistance under standard duration oseltamivir treatment Sponsor: not known |
| Participants | — |
| Interventions | Oseltamivir |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
MV22940.
| Methods | A randomised controlled trial on the effect of post‐exposure oseltamivir prophylaxis on influenza transmission in nursing homes Sponsor: National Institute for Public Health and the Environment (RIVM), The Netherlands NCT01053377 |
| Participants | — |
| Interventions | Oseltamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00419263.
| Methods | Evaluation of the efficacy and safety of peramivir in subjects with uncomplicated acute influenza Sponsor: BioCryst Pharmaceuticals |
| Participants | — |
| Interventions | Peramivir |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00453999.
| Methods | Evaluation of the efficacy and safety of peramivir in adults with acute serious or potentially life‐threatening influenza Sponsor: BioCryst Pharmaceuticals |
| Participants | — |
| Interventions | Peramivir |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00486980.
| Methods | Intramuscular peramivir for the treatment of uncomplicated influenza Sponsor: BioCryst Pharmaceuticals |
| Participants | — |
| Interventions | Peramivir |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00555893.
| Methods | Efficacy study of early versus late oseltamivir administration for treating and preventing influenza |
| Participants | — |
| Interventions | Oseltamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00610935.
| Methods | Intramuscular peramivir in subjects with uncomplicated acute influenza Sponsor: Marshfield Clinic Research Foundation |
| Participants | — |
| Interventions | Peramivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00705406.
| Methods | A phase II, multicentre, randomised, placebo‐controlled, study to evaluate the efficacy and safety of intramuscular peramivir 600 mg in subjects with uncomplicated acute influenza Sponsor: BioCryst Pharmaceuticals |
| Participants | — |
| Interventions | Peramivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00958776.
| Methods | A study to evaluate the efficacy and safety of IV peramivir in addition to standard of care compared to standard of care alone in adults and adolescents who are hospitalised due to influenza Sponsor: BioCryst Pharmaceuticals |
| Participants | Peramivir |
| Interventions | — |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT00980109.
| Methods | Long‐term influenza prophylaxis with inhaled zanamivir or oral oseltamivir Sponsor: University of Oxford |
| Participants | — |
| Interventions | Oseltamivir, zanamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NCT01032837.
| Methods | A study of Tamiflu (oseltamivir) for treatment of influenza with a focus on (H1N1) 2009 flu strain Sponsor: Hoffmann‐La Roche |
| Participants | — |
| Interventions | Oseltamivir, placebo |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
NV20236.
| Methods | A study of Tamiflu (oseltamivir) for seasonal prophylaxis of influenza in children Sponsor: Hoffmann‐La Roche NCT00412555 |
| Participants | — |
| Interventions | Oseltamivir |
| Outcomes | — |
| Notes | Awaiting assessment as we do not yet have the clinical study reports for this study |
Differences between protocol and review
We have made a number of changes to the text of A159 during the process of turning the protocol into the review. This reflects our evolving understanding of the issues, during the relatively long period when work on the review was underway.
We have changed the review title to reflect the nature of the evidence. The old title was: Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children ‐ a review of clinical study reports.
We have also re‐written the objective twice, tightening up the text to bring it in line with our initial intentions and clarifying its meaning. The old objectives were: "To review clinical study reports (CSRs) identified from published and unpublished randomised controlled trials (RCTs) and relevant regulatory data on effectiveness and harms of NIs for influenza in all age groups" and "To review published and unpublished clinical study reports and other relevant regulatory data on effectiveness and harms of NIs for influenza in all age groups (and compare them with our published review)."
We changed the emphasis of the objectives on unpublished study reports as we had decided from the start to concentrate on regulatory information. Similarly, comparison of published versus unpublished data is an important and worthwhile effort, but the original objective possibly misled readers as to its importance in our work. We had always conceptualised it as a low‐priority task we could carry out only if we had time following our review of unpublished data. We have also avoided using acronyms, which we thought cumbersome and confusing to the reader.
Our initial intention was to review clinical study reports and regulatory comments making up what we have subsequently called 'regulatory information'. The edits do not reflect a change in intent but our slowly evolving understanding of the problems we faced and our solutions to address these problems. As one of many examples, the transition from a world in which studies were identified by names and years (Nicholson 2000), to one in which the same trial is identified by a series of letters and numbers (WV15670), was not easy.
While the review was underway, we identified several unforeseen issues, such as placebo content and the effect of oseltamivir on antibodies. To test the relevant hypotheses we carried out post‐protocol analyses, which had not been present in the original protocol but were derived from our protocol‐stated intention to assess programmes and not single trials. These are now reported in their entirety in Appendix 10.
In May 2013, we added amendments to the review for: data analyses from oseltamivir trials Module 2s, clinical outcomes and adverse events added in the Feedback section. In the text we explain the rationale and methods applying to regulatory information received after our 2011 time lock, which could not be implemented in time for the current review (see also Appendix 2). For the 16 May 2013 amendments see Feedback.
Contributions of authors
TJ, PD, CDM, MT, MJ and CH were authors of the separate relevant Cochrane reviews. The protocol for the 2012 review was written by TJ, PD and CDM. All authors contributed to the writing of this protocol and devised the approach strategies to the data sources. CH provided logistical support. For the 2012 review, all authors reconstructed clinical trials using the CONSORT statement‐based extraction template, TJ reviewed regulatory material and TJ, MJ, CH, RH and CDM applied the inclusion criteria. CDM supervised the process and arbitrated when necessary. MJ carried out the statistical analyses. RH reviewed the Japanese data together with MJ and PD. TJ reviewed the FDA files. CDM and MT screened the electronic searches. TJ prepared the final text and all authors contributed to the final draft. Toby Lasserson contributed editorial support. For the 2014 review TJ, PD, CDM, MT, RH, MJ and CH amended the protocol. TJ and PD applied the inclusion criteria to the oseltamivir clinical study reports. CH and IO applied the inclusion criteria to the zanamivir clinical study reports. MJ supervised the process and arbitrated when necessary. MJ carried out the statistical analyses. RH reviewed the Japanese data together with MJ and PD. TJ, PD, CH, IO, ES, DN and JH extracted the clinical study reports. CDM and MT screened the electronic search updates. TJ prepared the final text and all authors contributed to the final draft.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research (NIHR), UK.
The review has been prepared with support from a NIHR (UK) grant 10/80/01
Declarations of interest
All review authors have applied for and received competitive research grants. TJ, PD, CDM, MT, RH, MJ and CH are co‐recipients of the NIHR grant to carry out this review ( http://www.nets.nihr.ac.uk/projects/hta/108001 ). In addition:
Prof Jefferson receives royalties from his books published by Blackwell and Il Pensiero Scientifico Editore, Rome. Dr Jefferson is occasionally interviewed by market research companies for anonymous interviews about Phase 1 or 2 pharmaceutical products. In 2011‐2013 Dr Jefferson acted as an expert witness in a litigation case related to oseltamivir phosphate; Tamiflu [Roche] and in a labour case on influenza vaccines in healthcare workers in Canada. In 1997‐99 Dr Jefferson acted as consultant for Roche, in 2001‐2 for GSK and in 2003 for Sanofi‐Synthelabo for pleconaril (an anti‐rhinoviral which did not get approval from FDA). Dr Jefferson is a consultant for IMS Health.
Dr Doshi received EUR 1500 from the European Respiratory Society in support of his travel to the society's September 2012 annual congress in Vienna, where he gave an invited talk on oseltamivir.
Prof Del Mar was a Board member of two companies to commercialise research at Bond University, part of his responsibilities as Pro‐Vice Chancellor (Research) until 2010, receives fees for editorial and guideline developmental work and royalties from books, and is in receipt of institutional grants from NHMRC (Aus), NIHR (UK) and HTA (UK) and from a private donor (for support of the editorial base of the Cochrane ARI Group).
Dr Hama receives royalties from two books published in 2008 titled "Tamiflu: harmful as was afraid" and "In order to escape from drug‐induced encephalopathy". Dr Hama provided scientific opinions and expert testimony on 11 adverse reaction cases related to oseltamivir and gefitinib.
Dr Howick has received expenses and payments from Johns Hopkins and the American Society for Neurophysiological Monitoring as an EBM consultant. Dr Howick has received funding from the Wellcome Trust, the Medical Research Council of the UK, the Economics and Social Science Research Council of the UK and he is currently a National Institute for Health Research non‐clinical research fellow. He has received payment from the Canadian Medical Association Journal for writing a book review and receives royalties from the publication of his book from Blackwell/Wiley.
Dr Heneghan receives payment for running educational courses at the University of Oxford and University of Oxford ISIS consulting services for external teaching and training. He also receives royalties for books (Evidence Based Toolkit series by Blackwell BMJ Books).
Dr Onakpoya has no additional interests to disclose. Dr Thompson has no additional interests to disclose. Dr Jones has no additional interests to disclose. Dr Spencer has no additional interests to disclose. Dr Nunan has no additional interests to disclose. Dr Mahtani has no additional interests to disclose.
Edited (no change to conclusions)
References
References to studies included in this review
167‐101 {published data only}
- GSK. Investigation of the efficacy of GG167 (zanamivir) in the treatment of influenza viral infections (late Phase II study: dose comparison study). Data on file.
JNAI‐01 {published data only}
- GSK. Investigation of the efficacy of CG167 in the treatment of influenza viral infections (phase II study) (Protocol NoJNAI‐01). Double blind, double dummy, randomized, placebo controlled, parallel group, multicenter study to investigate safety and route of administration of CG167 when inhaled, CG167 10 mg, or the combination of inhaled CG167 10 mg plus intranasal CG167 6.4 mg, administered twice daily for 5 days in the treatment of influenza A and B viral infections. Data on file.
JNAI‐04 {published data only}
- GSK. A multicenter two way layout randomized placebo‐controlled double‐blind trial parallel group comparative trial on the efficacy and safety of GG167 (zanamivir) 10 mg twice a day and 20 mg twice a day in the treatment of influenza type A and type B infections (late Phase II study: dose comparison study) (Protocol No. JNAI‐04). Data on file.
JNAI‐07 {published data only}
- GSK. A multicenter two way layout randomized placebo‐controlled double‐blind trial parallel group comparative trial on the efficacy and safety of GG167 (zanamivir) 10 mg twice a day and 20 mg twice a day in the treatment of influenza type A and type B infections (late Phase II study: dose comparison study) (Protocol No.JNAI‐07). Data on file.
JV15823 {published data only}
- No authors listed. A randomized, placebo‐controlled, multicenter study of oseltamivir (Ro 64‐0796) in the treatment of influenza in Japanese subjects (Translation of summary Japanese report ‐ of 29 pages). Data on file.
JV15824 {published data only}
- No authors listed. Phase 3 study for prophylaxis of influenza with Ro64‐0796 (15 page summary from Japanese). Data on file.
M76001 {published data only}
- McGarty T. A randomized, double‐blind, placebo‐controlled, multicenter study of efficacy based on the time to treatment of influenza infection with the neuraminidase inhibitor Ro 64‐0796 (also known as GS 4104). Data on file.
ML16369 {published data only}
NAI30008 {published data only}
- No authors listed. A double‐blind, randomized, placebo‐controlled, parallel group, multi‐center study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered twice daily for five days in the treatment of influenza in patients 12 years or over diagnosed with asthma or chronic obstructive pulmonary disease. Data on file.
NAI30009 {published data only}
- Alfors S, Keene O, Grice R, Hammond J, Hendricks V, Martin N, et al. A double‐blind, randomized, placebo‐controlled, parallel‐group, multicenter study to investigate the efficacy and safety of zanamivir (GG167) 10 mg administered by inhalation twice daily for five days in the treatment of symptomatic influenza A and B viral infections in children ages 5‐12. Data on file.
NAI30010 {published data only}
- Hunter S, Reilly L, Sharp S, West M, Alfors S, Hammond J, et al. A double‐blind, randomized, placebo‐controlled, parallel‐group, multicenter study to investigate the efficacy and safety of inhaled zanamivir (GG167) 10 mg administered once a day for 10 days in the prevention of transmission of symptomatic influenza A and B viral infections within families. Data on file.
NAI30011 {published data only}
- No authors listed. A randomised, double‐blind, placebo‐controlled study to evaluate the impact of inhaled zanamivir treatment on workplace attendance due to influenza A and B infections. Data on file.
NAI30012 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered twice daily for five days in the treatment of symptomatic influenza A and B viral infections in subjects aged over 65 years. Data on file.
NAI30015 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered twice daily for five days in the treatment of symptomatic influenza A and B viral infections in armed services personnel. Data on file.
NAI30020 {unpublished data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, multicenter study in 2 parallel groups, to investigate the efficacy and safety of inhaled zanamivir (10 mg bd. via Diskhaler), for 5 days, in high risk patients with symptomatic influenza A and / or B infection. Data on file Synopsis only available.
NAI30028 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, multicenter study in 2 parallel groups, to investigate the efficacy and safety of inhaled zanamivir (10 mg bd via Diskhaler), for 5 days, in children aged 5 to 12 years with symptomatic influenza A and / or B infection. Data on file.
NAI30031 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered once a day for 10 days in the prevention of transmission of symptomatic influenza A and B viral infections within households. Data on file.
NAI30034 {published data only}
- No authors listed. A double‐blind, randomized, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered once a day for 28 days in the prevention of symptomatic influenza A and B viral infections in community‐dwelling high‐risk populations. Data on file.
NAIA/B2008 {published data only}
- No authors listed. A double‐blind, randomized, placebo‐controlled, multicenter, parallel‐group study to investigate the efficacy and safety of zanamivir administered twice or four times a day for the treatment of influenza A and B viral infections. Data on file.
NAIA/B2009 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, multicentre, parallel‐group study to demonstrate the efficacy and safety of zanamivir in the prevention and/or progression of influenza A and B viral infections. Data on file.
NAIA2005 {published data only}
- MacLeod A, Gummer M, Raniga K, Hirst H, Keene O, Ossi M, et al. A double‐blind, randomised, placebo‐controlled multi‐centre study to investigate the efficacy and safety of inhaled and intranasal zanamivir in the treatment of influenza A and B viral infections. Data on file.
NAIA2006 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled multicentre study to investigate the efficacy and safety of zanamivir therapy in the prevention of progression of influenza A and B viral infections. Data on file.
NAIA3002 {published data only}
- Elliott M, Flack N, Keene O, Szymborski P, Vega R (PharmaResearch, Inc). A double‐blind, randomized, placebo‐controlled, parallel‐group, multicenter study to investigate the efficacy and safety of inhaled zanamivir (GG167) 10 mg administered twice a day for five days in the treatment of symptomatic influenza A and B viral infections in adolescents and adults. Data on file.
NAIA3003 {published data only}
- No authors listed. A double‐blind, randomized, parallel‐group, multi‐center study to investigate the efficacy and safety of inhaled zanamivir 10 mg administered once a day compared to the standard of care in controlling nursing home influenza outbreaks. Data on file.
NAIA3004 {published data only}
- No authors listed. A double‐blind, randomized, placebo‐controlled, parallel‐group, multi‐center study to investigate the efficacy and safety of inhaled zanamivir 10 mg once a day in controlling nursing home influenza outbreaks. Data on file.
NAIA3005 {published data only}
- Elliott M, Hunter S, Flack N, Crisp A, Szymborski P, Vega R. A double‐blind, randomized, placebo‐controlled, parallel‐group, multicenter study to investigate the efficacy and safety of zanamivir (GG167) 10 mg administered once a day for 28 days in the prevention of symptomatic influenza A and B viral infections in community dwelling adults. Data on file.
NAIB2005 {published data only}
- Leong J, Brennan J, Gummer M, Keene O, Wightman K. A double‐blind, randomised, placebo‐controlled, parallel‐group, multi‐centre study to investigate the efficacy and safety of inhaled plus intranasal zanamivir in the treatment of influenza A and B viral infections. Data on file.
NAIB2006 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir in preventing progression of influenza A and B viral infections. Data on file.
NAIB2007 {published data only}
- Perich R, Solterbeck A, Keene O, Leong J, Raniga K, MacLeod A. A double‐blind, randomised, placebo‐controlled, parallel‐group, multi‐centre study to investigate the efficacy and safety of inhaled and inhaled plus intranasal zanamivir in the treatment of influenza A and B viral infections. Data on file.
NAIB3001 {published data only}
- Campion K, Gummer M, Keene O. A double‐blind, randomized, placebo‐controlled, parallel‐group, multicenter study to investigate the efficacy and safety of zanamivir administered twice daily in the treatment of influenza A and B viral infections in adults. Data on file.
NAIB3002 {published data only}
- Man CY, Keene ON, Challoner T (Challoner Associates). A double‐blind, randomised, placebo‐controlled, parallel‐group, multicentre study to investigate the efficacy and safety of inhaled zanamivir (GG167) 10 mg administered twice a day for five days in the treatment of symptomatic influenza A and B viral infections in adolescents and adults. Data on file.
NV16871 {published data only}
- No authors listed. A double‐blind, randomized, stratified, placebo‐controlled study of oseltamivir in the treatment of influenza in children with asthma. Data on file.
PE‐01 {unpublished data only}
- No authors listed. A trial to investigate efficacy to reduce development of influenza symptoms in influenza infected patients treated with GG167 (protocol number PE‐01 phase II clinical trial). A double‐blind double‐dummy, randomized, placebo‐controlled, parallel group, multicenter study to investigate the efficacy reducing development of influenza symptoms and, safety and rout of administration of GG 157 when orally inhaled GG 167 10 mg, 6.4 mg nebulized intranasally or the combination of inhaled GG167 10 mg plus intranasal GG167 6.4 mg was administered twice day for 5 days in the treatment of influenza A and B viral infections. Data on file.
WV15670 {published data only}
- Dorkings J. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomized controlled trial. Data on file.
WV15671 {published data only}
WV15673/WV15697 {published data only}
- No authors listed. Efficacy of Ro 64‐0796 when used as chemoprophylaxis against natural influenza infection. Data on file.
WV15707 {published data only}
- Grosse M. A multi‐center, randomized, double‐blind, placebo‐controlled, parallel group study of oseltamivir treatment in elderly patients with influenza. Data on file.
WV15708 {published data only}
- No authors listed. A double‐blind randomised placebo controlled study of Ro 64‐0796 (also known as GS4104) used in elderly subjects for the prevention of clinical influenza during influenza season. Data on file.
WV15730 {published data only}
- Dorkings J. A double‐blind, stratified, randomized, placebo controlled study of Ro 64‐0796 (GS4104) in the treatment of influenza infection in adults. Data on file.
WV15758 {published data only}
- No authors listed. A double‐blind, randomized, stratified, placebo‐controlled study of Ro 64‐0796 (also known as GS 4104) in the treatment of children with influenza. Data on file.
WV15759/WV15871 {published data only}
- Gerster T. A double‐blind, randomized, stratified, placebo‐controlled study of oseltamivir phosphate (Ro 64‐0796, also known as GS 4104) in the treatment of influenza in children with chronic asthma. Data on file.
WV15799 {published data only}
- No authors listed. A double‐blind randomised placebo controlled study of Ro 64‐0796 (also known as GS4104) for the prevention of clinical influenza post exposure in families. Data on file.
WV15812/WV15872 {published data only}
- McCarvil M. A double‐blind, stratified, randomised, placebo controlled study of Ro 64‐0796 (also known as GS4104) in the treatment of influenza in chronically ill adults. Data on file.
WV15819/WV15876/WV15978 {published data only}
- No authors listed. A double‐blind, randomized, stratified, placebo‐controlled study of Ro 64‐0796 (also known as GS4104) in the treatment of influenza infection in elderly patients. Data on file.
WV15825 {published data only}
- No authors listed. A double‐blind, randomised, placebo‐controlled study of Ro 64‐0796 (also known as GS4104) used in elderly subjects for the prevention of clinical influenza during the influenza season. Data on file.
WV16277 {published data only}
- No authors listed. A double‐blind, randomised, stratified, placebo‐controlled study of oseltamivir in the treatment of influenza infection in patients. Data on file.
References to studies excluded from this review
105934 {published data only}
- A post‐marketing surveillance to monitor the safety of RELENZA (zanamivir) administered in Korean subjects according to the prescribing information. Data on file.
107485 {published data only}
- An open label, single‐dose, five‐way crossover study examining relative oral bioavailability of zanamivir with bioenhancing excipients following direct release into mid‐small intestine using gamma scintigraphy and the InteliSite Companion Capsule in healthy subjects. Data on file.
108127 {published data only}
- An open‐label, non‐randomized, single‐dose study to evaluate serum zanamivir pharmacokinetics following intravenous administration to human subjects with renal impairment compared to subjects without renal impairment. Data on file.
112311 {published data only}
- Special drug use investigation for Relenza (resistant appearance). Data on file.
112312 {published data only}
- Special drug use investigation for Relenza (efficacy). Data on file.
113268 {published data only}
- Drug use investigation for Relenza. Data on file.
113502 {published data only}
- Prophylactic efficacy of Relenza against influenza A and B. Data on file.
113625 {published data only}
- A randomized, placebo controlled, 3‐way crossover study to investigate the safety, tolerability, and pharmacokinetics of repeat dose zanamivir/placebo 10 mg administered twice daily for 5 days by a rotahaler compared to the diskhaler in healthy subjects. Data on file.
113678 {published data only}
- An open‐label, multi‐center, single arm study to evaluate the safety and tolerability of intravenous zanamivir in the treatment of hospitalized adult, adolescent and pediatric subjects with confirmed influenza infection. Data on file.
114045 {published data only}
- Collection of patients' background information Relenza® sentinel site monitoring program in Japan. Data on file.
114373 {published data only}
- A phase III international, randomized, double‐blind, double‐dummy study to evaluate the efficacy and safety of 300 mg or 600 mg of intravenous zanamivir twice daily compared to 75 mg of oral oseltamivir twice daily in the treatment of hospitalized adults and adolescents with influenza. Data on file.
167‐02 {published data only}
- A study of GG167 single blind, single administration ‐ phase I. Data on file.
167‐03 {published data only}
- A study of GG167 single blind, single administration ‐ phase I. Data on file.
167‐04 {published data only}
- A study of GG167 single blind, single administration ‐ phase I. Data on file.
167‐05 {published data only}
- A study of GG167 single blind, single administration ‐ phase I. Data on file.
167T3‐11 {published data only}
- (Zanamivir trial. Title unknown). Data on file.
ADS‐TCAD‐PO206 {published data only}
- A randomized open label study comparing the efficacy, safety, and tolerability of oral administration of amantadine and ribavirin with oseltamivir versus oseltamivir to influenza A virus infected immunocompromised subjects. Data on file.
BP21288 {published data only}
- A single‐center, open‐label, single dose, exploratory study in Caucasian and Japanese healthy subjects to investigate the pharmacokinetics of oseltamivir and its metabolite in plasma and cerebrospinal fluid. Data on file.
C94‐009 {published data only}
C94‐085 {published data only}
GCP/95/045 {published data only}
- A study to investigate the pharmacokinetics of GG167 in subjects with impaired renal function. Data on file.
JNAI‐02 {published data only}
- (Zanamivir trial. Title unknown). Data on file.
JNAI‐03 {published data only}
- (Zanamivir trial. Title unknown). Data on file.
JP15734 {published data only}
- Single ascending oral dose study of tolerability, safety and pharmacokinetics (including effect of food) of the neuraminidase inhibitor Ro 64‐0796 in healthy male volunteers. Data on file.
JP15735 {published data only}
- Multiple oral dose study of the tolerability, safety and PK of the neuraminidase inhibitor Ro64‐0796: direct PK comparison between Japanese and Caucasian subjects. Data on file.
JV16284 {published data only}
- Phase II clinical study of oseltamivir phosphate (Ro64‐0796) for the treatment of influenza in children. Data on file.
JV21490 {unpublished data only}
- Post‐marketing clinical study of oseltamivir phosphate on nighttime ECG in healthy adult male subjects. Data on file 2008.
M76006 {published data only}
ML17279 {unpublished data only}
- An observational study to assess the accuracy of diagnosis of influenza in community pharmacies. Data on file.
ML17713 {published data only}
- Phase IV study on Tamiflu® capsule 75 in the elderly aged 80 years or older (a single dose oral administration study for assessing pharmacokinetics in the elderly not infected with influenza virus). Data on file.
ML19340 {unpublished data only}
- Impact of oseltamivir (Tamiflu ®) in post‐exposure prophylaxis influenza on mortality and morbidity in institutionalised elderly people. [Impact de l'oseltamivir (Tamiflu®) en prophylaxie antigrippale post‐exposition, sur la mortalité et la morbidité des personnes âgées institutionnalisées.]. Data on file.
ML20542 {published data only}
- Evaluation of combination therapy with oseltamivir and zanamivir versus monotherapy in the treatment of virologically confirmed influenza in primary care a randomised double blind controlled trial study. Data on file.
ML21954 {published data only}
- Efficacy and safety of combination therapies with oseltamivir & zanamivir or oseltamivir & amantadine versus oseltamivir monotherapy in the treatment of seasonal influenza A infection. Data on file.
ML22789 {published data only}
- An unblinded, comparative, randomized study of influenza A/H1N1 2009 resistance in patients with standard and double dose oseltamivir treatment. Data on file.
ML22872 {published data only}
- Viral shedding/resistance with double duration oseltamivir in infected patients (New Zealand). Data on file.
ML22879 {published data only}
- Viral shedding/resistance with standard dose/duration oseltamivir in infected patients (UK). Data on file.
ML25018 {published data only}
- A study of the relative oral bioavailability of the antiflu medicine oseltamivir (Tamiflu®) in patients in the intensive care unit. Data on file.
ML25087 {published data only}
- Viral shedding/resistance with double dose oseltamivir in infected patients (Australia). Data on file.
ML25094 {published data only}
- Nasogastric administration of OP in infected patients with respiratory failure. Data on file.
ML25157 {published data only}
ML25176 {published data only}
- Open‐label pharmacokinetic of oseltamivir in healthy obese Thai adult subjects. Data on file.
ML25179 {published data only}
- A randomized, double‐blinded controlled trial comparing high vs standard dose oseltamivir in severe, influenza infection in ICU. "ROSII Study". Data on file.
ML25265 {published data only}
- Probing the functional expression of carboxyl esterase in preterm neonates using oseltamivir: a pragmatic observational study. Data on file.
ML25266 {published data only}
- Plasma levels of oseltamivir in H1N1 infected patients supported by extracorporeal membrane oxygenation: a single‐centre cohort study. Data on file.
MP20691 {published data only}
- Effect of probenecid on the pharmacokinetics of oseltamivir. Data on file.
MV20043 {published data only}
- A prospective study to assess household transmission of influenza and emergence and transmissibility of drug resistance to oseltamivir following treatment of children with influenza A and B. Data on file.
MV20050 {published data only}
- High‐dose versus standard‐dose oseltamivir for the treatment of severe influenza and avian influenza: a phase II double‐blind, randomized clinical trial. Data on file.
MV22926 {published data only}
MV22949 {published data only}
- A study of the pharmacology of oseltamivir (Tamiflu) in pregnancy. Data on file.
MV22951 {published data only}
- Pharmacokinetics of Tamiflu® (oseltamivir) in patients receiving extracorporeal membrane oxygenation (ECMO) and or continuous venovenous hemodialysis (CVVHD). Data on file.
MV22963 {published data only}
- Pharmacokinetics of oseltamivir in critically ill adult patients. Data on file.
MV22970 {published data only}
- Observational study on the pharmacokinetics of oseltamivir in the treatment of influenza during lactation. Data on file.
NAI106784 {published data only}
- Phase I, open‐label study to evaluate steady‐state serum and pulmonary pharmacokinetics following intravenous administration of zanamivir in healthy adult subjects. Data on file.
NAI108166 {published data only}
- Phase 1, open‐label study to evaluate potential pharmacokinetic interactions between orally‐administered oseltamivir and intravenous zanamivir in healthy Thai adult subjects. Data on file. [DOI] [PMC free article] [PubMed]
NAI10901 {published data only}
- A double‐blind, randomised, placebo‐controlled study to evaluate the effect of inhaled zanamivir 10 mg od for 28 days on anti‐haemagglutinin antibody production (HAI titre) following co‐administration with Fluvirin™ trivalent influenza vaccine in healthy adult subjects. Data on file.
NAI10902 {published data only}
- An open label, randomized evaluation of the direct measurement of zanamivir concentrations in respiratory secretions following a single dose inhalation of 10 mg RELENZA™ via DISKHALER in health volunteers. Data on file.
NAI40012 {published data only}
- An open‐label, multi‐center study of the patient instructional leaflet for RELENZA DISKHALER. Data on file.
NAIA1009 {published data only}
- Pharmacokinetics of zanamivir (GG167) following inhaled administration in pediatric subjects with signs and symptoms of respiratory illness. Data on file.
NAIA2010 {published data only}
- Pilot, cluster randomised, open, single centre, parallel group study to evaluate the efficacy and safety of zanamivir in controlling influenza outbreaks and preventing the development of resistant influenza cases in a high risk nursing home population, compared with usual care. Data on file.
NAIB1001 {published data only}
NAIB1002 {published data only}
- A study to evaluate the effect of repeat doses of GG167 dry powder on pulmonary function and bronchial hyper‐responsiveness in asthmatic subjects. Data on file.
NAIB1007 {published data only}
- A GG167 Pharmacokinetic Study to Select a Regimen for Prophylaxis. Data on file.
NCT00297050 {published data only}
- A phase I double‐blind, placebo‐controlled, dose‐escalating study to evaluate the safety and tolerability of intravenous peramivir in healthy subjects. Data on file.
NCT00416962 {published data only}
- An open‐label, multiple dose, randomized, three‐period crossover study in healthy volunteers to evaluate the effect of co‐administration of amantadine 100 mg BID and oseltamivir 75 mg BID on the pharmacokinetic properties of amantadine and oseltamivir. Data on file.
NCT00867139 {published data only}
- TCAD vs. monotherapy for influenza A in immunocompromised patients. Data on file.
NCT00957996 {published data only}
- A phase 3, open‐label, randomized study of the antiviral activity, safety, and tolerability of intravenous peramivir in hospitalized subjects with confirmed or suspected influenza infection. Data on file.
NCT01063933 {published data only}
- A pharmacokinetic/pharmacodynamic and safety evaluation of investigational intravenous peramivir in children with influenza disease (CASG 117). Data on file.
Not applicable (registry) {published data only}
- (Oseltamivir trial. Title unknown). Data on file.
NP15525 {published data only}
- Multiple ascending oral dose study of the tolerability, safety and pharmacokinetics of the neuraminidase inhibitor Ro 64‐0796 in healthy volunteers. Data on file.
NP15717 {published data only}
- Study of the PD and PK of the neuraminidase inhibitor Ro 64‐0796 (GS4104) in the treatment of volunteers experimentally infected with human influenza B virus. Data on file.
NP15718 {published data only}
- An excretion balance and pharmacokinetic study of Ro 64‐0796 after a single oral dose of 14C‐labelled Ro 64‐0796 and an intravenous dose of 14C‐labelled Ro 64‐0802 in healthy male subjects. Data on file.
NP15719 {published data only}
- Study of the pharmacokinetics and absolute bioavailability of the neuraminidase inhibitor Ro 64‐0796. Data on file.
NP15728 {published data only}
- An open‐label study of the effect of cimetidine and probenecid on the pharmacokinetics of Ro 64‐0796/GS4104 in healthy subjects. Data on file.
NP15729 {published data only}
- An open‐label bioequivalence and food effect study of the clinical trial and market formulations of Ro 64‐0796 in healthy subjects. Data on file.
NP15743 {unpublished data only}
- No authors listed. A palatability study of the neuraminidase inhibitor (Ro 64‐0796), formulated as an oral formulation. Data on file.
NP15757 {published data only}
- No authors listed. Study of the pharmacokinetics and pharmacodynamics of the neuraminidase inhibitor Ro 64‐0796 (GS4104) in the prophylaxis of experimental infection of volunteers with the human influenza B virus. Data on file.
NP15810 {published data only}
- An open‐label bioequivalence and food effect study of the clinical trial and market formulations of Ro 64‐0796 in healthy subjects. Data on file.
NP15826 {published data only}
- An open‐label study of pharmacokinetics of Ro 64‐0796/GS4104 in children. Data on file.
NP15827 {published data only}
- Study of the pharmacodynamics of the neuraminidase inhibitor in the treatment of subjects experimentally infected with the human influenza B virus. Data on file.
NP15881 {published data only}
- Palatability testing of the neuraminidase inhibitor Ro 64‐0796 in children. Research Report No. W‐144154/27 October 1999. Data on file.
NP15901 {published data only}
- An open‐label, two‐way crossover pharmacokinetic drug interaction study of neuraminidase inhibitor Ro 64‐0796/GS4104 and amoxicillin in healthy volunteers. Data on file.
NP15912 {published data only}
- Palatability testing of the neuraminidase inhibitor Ro 64‐0796 in children. Data on file.
NP16472 {published data only}
- A single center, open label, multiple dose oral oseltamivir suspension study in end‐stage‐renal disease (ESRD) patients on hemodialysis (HD) and continuous ambulatory peritoneal dialysis (CAPD). Data on file.
NP22770 {published data only}
- An open‐label, multiple dose, randomized, three‐period crossover study in healthy subjects to evaluate the effect of co‐administration of oseltamivir (RO0640796) 75 mg twice daily and rimantadine 100 mg twice daily on the pharmacokinetic properties of oseltamivir and rimantadine. Data on file.
NP25138 {published data only}
- A study of intravenous oseltamivir [Tamiflu] in infants with influenza. Data on file.
NP25139 {published data only}
- A study of intravenous Tamiflu (oseltamivir) in children with influenza. Data on file.
NP25140 {published data only}
- PK and safety of multiple ascending doses of iv oseltamivir in healthy adults. Data on file.
NV20234 {published data only}
- A randomized, double‐blind trial evaluating conventional and high dose Tamiflu in the treatment of influenza in immunocompromised patients. Data on file.
NV20235 {published data only}
- A double‐blind, randomized, placebo controlled multicenter trial of oseltamivir for the seasonal prophylaxis of influenza in immunocompromised patients. Data on file.
NV20237 {published data only}
- An influenza resistance information study (IRIS). Data on file.
NV22155 {published data only}
- A randomized, multicenter trial of oseltamivir [Tamiflu] doses of 75 mg for 5 or 10 days versus 150 mg for 5 or 10 days to evaluate the effect on the duration of viral shedding in influenza patients with pandemic (H1N1) 2009. Data on file.
NV22158 {published data only}
- Avian/pandemic influenza registry final report, 30 August 2012. Data on file.
NV25118 {published data only}
- A randomized, multicenter, parallel study of the safety, pharmacokinetics and the effect on viral activity of intravenously administered Tamiflu [oseltamivir] in patients with influenza over 13 years of age. Data on file.
NV25182 {published data only}
- A prospective, observational safety study in children ≤ 24 months of age receiving oseltamivir for the treatment or prophylaxis of influenza infection. Data on file.
NV25655 {published data only}
- An open‐label, prospective, single oral dose study evaluating the pharmacokinetics, safety and tolerability of oseltamivir (Tamiflu) in adult subjects on peritoneal dialysis (PD) using a rapid cycle regimen to simulate automated peritoneal dialysis (APD) and in adult subjects with creatinine clearance from 10‐30 mL/min not on dialysis. Data on file.
PP15974 {published data only}
- A single oral dose, multi‐center, open label study of the pharmacokinetics, safety and tolerability of Ro 04‐0796/GS4104 in ESRD subjects on hemodialysis and peritoneal dialysis. Data on file.
PP16351 {published data only}
- An open label study of the pharmacokinetics of oseltamivir (Ro 64‐0796) in children aged 0 ‐ 5 years old after a single dose. Data on file.
PP16361 {published data only}
- Double‐blind, randomized, placebo controlled, single ascending i.v. dose study of the tolerability (with emphasis of nausea and vomiting), safety, pharmacokinetics of oseltamivir (Ro 64‐0796) and its active metabolite oseltamivir carboxylate (Ro 64‐0802) in healthy male volunteers. Data on file.
PV15615 {published data only}
- GS97802 ‐ challenge flu A treatment. Data on file.
PV15616 {published data only}
- GS‐97801 challenge flu A treatment. Data on file.
WP15517 {published data only}
- Single ascending oral dose study of the tolerability, safety and pharmacokinetics (including effect of food) of the neuraminidase inhibitor GS4104 in healthy volunteers. Data on file.
WP15525 {published data only}
- Multiple ascending oral dose study of the tolerability, safety and pharmacokinetics of the neuraminidase inhibitor, GS4104 in healthy volunteers. Data on file.
WP15647 {published data only}
- Multiple ascending oral dose study of the tolerability, safety and pharmacokinetics of the neuraminidase inhibitor Ro 64‐0796 in healthy elderly volunteers. Data on file.
WP15648 {published data only}
- Multiple oral dose study of the pharmacokinetics, tolerability and safety of the neuraminidase inhibitor Ro 64‐0796 in patients with renal impairment. Data on file.
WP15676 {published data only}
- Study of the safety and pharmacokinetics of the neuraminidase inhibitor Ro 64‐0796 in healthy volunteers when administered concomitantly with paracetamol (acetaminophen). Data on file.
WP15979 {published data only}
- An open‐label, relative bioavailability study of the phase III pediatric clinical trial and market formulations of Ro 64‐0796 in healthy volunteers. Data on file.
WP16094 {published data only}
- An open‐label, three‐way crossover, pharmacokinetic drug interaction study of neuraminidase inhibitor Ro 64‐0796 and aspirin in healthy subjects. Data on file.
WP16134 {published data only}
- An open label bioequivalence and food effect study of the enteric coated and immediate release formulations of oseltamivir in healthy subjects. Data on file.
WP16137 {published data only}
- An open‐label, bioequivalence study of the phase III pediatric clinical trial and market oral suspension formulations of Ro 64‐0796 in healthy volunteers. Data on file.
WP16225 {published data only}
- An open‐label, relative bioavailability study of the market suspension (with improved process), the clinical trial suspension and market capsule formulation of Ro 64‐0796 (Tamiflu, oseltamivir) in healthy subjects. Data on file.
WP16226 {published data only}
- A study of the pharmacokinetics of oseltamivir (Ro 64‐796) and its active metabolite Ro 64‐0802 following single oral dosing of Ro 64‐0796 to healthy volunteers and patients with moderate hepatic impairment. Data on file.
WP16254 {published data only}
- A pharmacokinetic drug interaction study of oseltamivir (Ro 64‐0796) and antacid in healthy volunteers. Data on file.
WP16263 {published data only}
- A randomized, double blind, parallel group, placebo controlled study of the effect of oseltamivir on ECG intervals in healthy subjects. Data on file.
WP16295 {unpublished data only}
- A randomized, open label study of the site of absorption of oseltamivir in healthy subjects using an Enterion capsule. Data on file.
WP17721 {published data only}
- Clinical pharmacokinetics of cyclosporine or mycophenolate with and without a concurrent single dose of oseltamivir phosphate in patients with a renal transplant. Data on file.
WP18308 {published data only}
- Comparison of the pharmacokinetics of Ro 64‐0802 following a single dose of oseltamivir phosphate either in a capsule or a drinking solution. Data on file.
WP20727 {published data only}
- A combined single ascending dose, multiple ascending dose and exploratory bioavailability study to investigate the safety, tolerability and pharmacokinetics of intravenous Ro 64‐0796 in healthy volunteers. Data on file.
WP20749 {published data only}
- Oseltamivir treatment for children less than 24 months of age with influenza. Data on file.
WP21272 {published data only}
- An open‐label, randomized 2‐period crossover study to investigate the pharmacodynamics, pharmacokinetics, safety and tolerability of warfarin, and the pharmacokinetics, safety and tolerability of oseltamivir, when given in combination. Data on file.
WP22849 {published data only}
- An open label, prospective, pharmacokinetic/pharmacodynamic and safety evaluation of oseltamivir (Tamiflu®) in the treatment of infants 0 to < 12 months of age with confirmed influenza infection. Data on file.
WV15731 {published data only}
- A double‐blind, randomized, stratified pilot study of Ro 64‐0796 (also known as GS4104) in children with influenza. Data on file.
WV16139 {published data only}
- (Oseltamivir trial. Title unknown). Data on file. [Mentioned in EMA EPAR dated 23 March 2006 pdf page 14 but possibly same trial as WV 16193]
WV16193 {published data only}
- A randomized, open‐label, parallel group study of oseltamivir used for management of influenza in households. Data on file.
References to studies awaiting assessment
JPRN‐JapicCTI‐111647 {published data only (unpublished sought but not used)}
- Kashiwagi S, Watanabe A, Ikematsu H, Awamura S, Okamoto T, Uemori M, et al. Laninamivir octanoate for post‐exposure prophylaxis of influenza in household contacts: a randomized double blind placebo controlled trial. Journal of Infection and Chemotherapy 2013;19:740‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
ML20589 {published data only}
- Economic and social benefits of treating and preventing influenza in aged care facilities. Data on file.
ML20910 {published data only}
- A randomized, open label study to evaluate the effect of Tamiflu on viral shedding and on serum and cytoplasmic inflammatory cytokine concentrations in patients with laboratory‐confirmed influenza. Data on file.
ML21776 {published data only}
- Pilot study to develop a model to evaluate nosocomial transmission of influenza. Data on file.
MV21118 {published data only}
- A double‐blind, randomized, placebo‐controlled study of early oseltamivir treatment of influenza in children 1‐3 years of age. Data on file.
MV21737 {published data only}
- A phase 4, multi‐center, randomized, double blind, placebo controlled study, to evaluate the safety of inhaled zanamivir 10 mg versus placebo and oral oseltamivir 75 mg versus placebo for influenza prophylaxis in healthy volunteers for 16 weeks. Data on file.
MV21879 {published data only}
- Efficacy of oseltamivir in reducing the duration of clinical illness, viral shedding, and transmissibility reduction within households among participants in an influenza disease burden surveillance cohort in urban Dhaka, Bangladesh. Data on file.
MV22841 {published data only}
- Viral shedding/resistance with standard dose/duration oseltamivir in infected patients (South Africa). Data on file.
MV22940 {published data only}
- A randomised controlled trial on the effect of post‐exposure oseltamivir prophylaxis on influenza transmission in nursing homes (PEPpIE). Data on file.
NCT00419263 {published data only}
- A phase II, multicenter, randomized, double‐mask, placebo‐controlled study to evaluate the efficacy and safety of intramuscular peramivir in subjects with uncomplicated acute influenza. Data on file.
NCT00453999 {published data only}
- A phase II, multicenter, randomized, double‐mask, double‐dummy study comparing the efficacy and safety of peramivir administered intravenously once daily versus oseltamivir administered orally twice daily in adults with acute serious or potentially life‐threatening influenza. Data on file.
NCT00486980 {published data only}
- A phase 3 multicenter, randomized, double blind, placebo‐controlled study to evaluate the efficacy and safety of intramuscular peramivir in subjects with uncomplicated acute influenza. Data on file.
NCT00555893 {published data only}
- Monitoring influenza severity and transmission on Tamiflu (MISTT). Data on file.
NCT00610935 {published data only}
- A phase 3 multicenter, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy and safety of intramuscular peramivir in subjects with uncomplicated acute influenza. Data on file.
NCT00705406 {published data only}
- A phase II, multicenter, randomized, placebo‐controlled, study to evaluate the efficacy and safety of intramuscular peramivir 600 mg in subjects with uncomplicated acute influenza. Data on file.
NCT00958776 {published data only}
- A phase 3, multicenter, randomized, double‐blind, controlled study to evaluate the efficacy and safety of peramivir administered intravenously in addition to standard of care compared to standard of care alone in adults and adolescents who are hospitalized due to serious influenza. Data on file.
NCT00980109 {published data only (unpublished sought but not used)}
- Anekthananon T, Pukritayakamee S, Ratanasuwan W, Jittamala P, Werarak P, Charunwatthana P, et al. Oseltamivir and inhaled zanamivir as influenza prophylaxis in Thai health workers: A randomized, double‐blind, placebo‐controlled safety trial over 16 weeks. Journal of Antimicrobial Chemotherapy 2013;68:697‐707. [NCT00980109 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01032837 {published data only (unpublished sought but not used)}
- Dharan NJ, Fry AM, Kieke BA, Coleman L, Meece J, Vandermause M, et al. Clinical and virologic outcomes in patients with oseltamivir‐resistant seasonal influenza A (H1N1) infections: results from a clinical trial. Influenza and Other Respiratory Viruses 2012;6:153‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
NV20236 {published data only}
- An open label, multicenter trial of oseltamivir prophylaxis of seasonal influenza in children. Data on file.
Additional references
Aronson 2003
- Aronson JK, Ferner RE. Joining the DoTS: new approach to classifying adverse drug reactions. BMJ 2003;327(7425):1222‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhatia 2007
- Bhatia A, Kast RE. How influenza's neuraminidase promotes virulence and creates localized lung mucosa immunodeficiency (Letter). Cellular and Molecular Biology 2007;12(1):111‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bourgeois 2011
- Bourgeois FT, Murthy S, Mandl KD. Outcome reporting among drug trials registered in ClinicalTrials.gov. Annals of Internal Medicine 2010;153:158‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brown 2011
- Brown JKM. Experimental design generator and randomiser. http://www.edgarweb.org.uk/ 2011 (accessed 24 August 2011).
Burch 2009
- Burch J, Corbett M, Stock C, Nicholson K, Elliot AJ, Duffy S, et al. Prescription of anti‐influenza drugs for healthy adults: a systematic review and meta‐analysis. Lancet 2009;9(9):537‐45. [DOI] [PubMed] [Google Scholar]
Call 2005
- Call SA, Vollenweider MA, Hornung CA, Simel DL, McKinney WP. Does this patient have influenza?. JAMA 2005;8:987‐97. [DOI] [PubMed] [Google Scholar]
Cass 1999
- Cass LM, Efthymiopoulos C, Bye A. Pharmacokinetics of zanamivir after intravenous, oral, inhaled or intranasal administration to healthy volunteers. Clinical Pharmacokinetics 1999;36(Suppl 1):1‐11. [DOI] [PubMed] [Google Scholar]
CDC 2009
- U.S. Centers for Disease Control and Prevention. Serum cross‐reactive antibody response to a novel influenza A (H1N1) virus after vaccination with seasonal influenza vaccine. Morbidity and Mortality Weekly Report 2009;58(19):521‐4. [PubMed] [Google Scholar]
CDC 2013
- U.S. Centers for Disease Control and Prevention. Influenza activity — United States, 2012–13 season and composition of the 2013–14 influenza vaccine. Morbidity and Mortality Weekly Report 2013;62(23):473‐9. [PMC free article] [PubMed] [Google Scholar]
Chou 2005
- Chou R, Helfand M. Challenges in systematic reviews that assess treatment harms. Annals of Internal Medicine 2005;142(2 Pt 2):1090‐9. [DOI] [PubMed] [Google Scholar]
Cochrane Neuraminidase Inhibitors Review Team 2011
- Cochrane Neuraminidase Inhibitors Review Team. Does oseltamivir really reduce complications of influenza?. Clinical Infectious Diseases 2011;53(12):1302–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cohen 2009
- Cohen D. Complications: tracking down the data on oseltamivir. BMJ 2009;339:b5387. [DOI] [PubMed] [Google Scholar]
Cooper 2003
- Cooper NJ, Sutton AJ, Abrams KR, Wailoo A, Turner D, Nicholson KG. Effectiveness of neuraminidase inhibitors in treatment and prevention of influenza A and B: systematic review and meta‐analyses of randomised controlled trials. BMJ 2003;326(7401):1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cox 2001
- Cox RJ, Mykkeltvedt E, Sjursen H, Haaheim LR. The effect of zanamivir treatment on the early immune response to influenza vaccination. Vaccine 2001;19(32):4743‐9. [DOI] [PubMed] [Google Scholar]
Doshi 2009
- Doshi P. Neuraminidase inhibitors ‐ the story behind the Cochrane review. BMJ 2009;339:b5164. [DOI] [PubMed] [Google Scholar]
Doshi 2012a
- Doshi P, Jefferson T, Mar C. The imperative to share clinical study reports: recommendations from the Tamiflu experience. PLoS Med 2012;9(4):e1001201. [DOI: 10.1371/journal.pmed.1001201] [DOI] [PMC free article] [PubMed] [Google Scholar]
Doshi 2012b
- Doshi P, Jones MA, Jefferson T. Rethinking credible evidence synthesis. BMJ 2012;344:d7898. [DOI: 10.1136/bmj.d7898] [DOI] [PubMed] [Google Scholar]
Doshi 2013
- Doshi P, Jefferson T. Clinical study reports of randomised controlled trials: an exploratory review of previously confidential industry reports. BMJ Open 2013;3:e002496. [DOI: 10.1136/bmjopen-2012-002496] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dutkowski 2010
- Dutkowski R, Smith JR, Davies BE. Safety and pharmacokinetics of oseltamivir at standard and high dosages. International Journal of Antimicrobial Agents 2010;35:461–7. [DOI] [PubMed] [Google Scholar]
Ebell 2012
- Ebell MH, Call M, Shinholser J. Effectiveness of oseltamivir in adults: a meta‐analysis of published and unpublished clinical trials. Family Practice 2012;30(2):125‐33. [DOI: 10.1093/fampra/cms059] [DOI] [PubMed] [Google Scholar]
EMA 2010
- European Medicines Agency. Annex I. Summary of product characteristics (Tamiflu 30 mg hard capsule). http://www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500033106 (accessed 21 January 2010).
EMEA 2001
- European Agency for the Evaluation of Medicinal Products. Tamiflu. Oseltamivir phosphate. (EMEA/H/C/402) CPMP recommendation & scientific discussion consolidated list of questions (June 28, 2001). http://www.ema.europa.eu/.
Eyding 2010
- Eyding D, Lelgemann M, Grouven U, Harter M, Kromp M, Kaiser T, et al. Reboxetine for acute treatment of major depression: systematic review and meta‐analysis of published and unpublished placebo and selective serotonin reuptake inhibitor controlled trials. BMJ 2010;341:c4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Falagas 2010
- Falagas ME, Koletsi PK, Vouloumanou EK, Rafailidis PI, Kapaskelis AM, Rello J. Effectiveness and safety of neuraminidase inhibitors in reducing influenza complications: a meta‐analysis of randomized controlled trials. Journal of Antimicrobial Chemotherapy 2010;65:1330‐46. [DOI: 10.1093/jac/dkq158] [DOI] [PubMed] [Google Scholar]
FDA 1999a
- Food, Drug Administration. Administrative/correspondence documents part 2. Relenza (Zanamivir). Application No.: 21036. http://www.accessdata.fda.gov/drugsatfda_docs/nda/99/021036‐admin2.pdf 2009 (accessed 26 August 2009).
FDA 1999b
- Food, Drug Administration. Medical review part 6. Relenza (zanamivir). Application No.: 21036. http://www.accessdata.fda.gov/drugsatfda_docs/nda/99/021036‐medreview6.pdf 2009 (accessed 26 August 2009).
FDA 1999c
- Food, Drug Administration. Tamiflu (Oseltamivir Phosphate) Capsule. Medical Review Part 2 (Application No.: 021087). http://www.accessdata.fda.gov/drugsatfda_docs/nda/99/21087_Tamiflu_medr_P1.pdf 1999 (accessed 26 August 2009).
FDA 2000a
- Food, Drug Administration. Letter from FDA to Hoffman‐La Roche Inc. re "NDA 21‐087 TAMIFLU (oseltamivir phosphate) MACMIS ID#8675. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM166329.pdf 2000 (accessed 14 April 2000).
FDA 2000b
- Food, Drug Administration. Drug approval package. Relenza (zanamivir). Application No.: 021036S001. Label. http://www.accessdata.fda.gov/drugsatfda_docs/label/2000/21036S1LBL.PDF 2000 (accessed 26 August 2009).
FDA 2000c
- Food, Drug Administration. Drug approval package. Tamiflu (oseltamivir). Application No.: 021087‐SE1‐002. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21‐087SE1‐002_review.pdf 2000 (accessed 27 August 2009).
FDA 2000d
- Food, Drug Administration. Review. Tamiflu (oseltamivir). NDA 021087 Supplement 002. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21‐087SE1‐002_review.pdf 2000 (accessed 26 August 2009).
FDA 2000e
- Food, Drug Administration. Site inspection report in Review. Tamiflu (oseltamivir). NDA 021087 Supplement 002. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21‐087SE1‐002_review.pdf 2000 (accessed 26 August 2009):177.
FDA 2000f
- Food, Drug Administration. Faxed letter to Roche (file UCM166329) [NDA 21‐087TAMIFLU (oseltamivir phosphate) MACMIS ID#8675]. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM166329.pdf 2000 (accessed 19 October 2010).
FDA 2011a
- F. Hoffman‐La Roche. Tamiflu label (for FDA NDA no. 021087). http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/021087s057lbl.pdf 2011 (accessed 7 February 2011).
FDA 2011b
- Food, Drug Administration. Drugs@FDA. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/ 2011 (accessed 5 July 2011).
FDA 2011c
- Food, Drug Administration. Warning letters. http://www.fda.gov/ICECI/EnforcementActions/WarningLetters/default.htm 2011 (accessed 5 July 2011).
Feinberg 2009
- Feinberg J. Wordle. http://www.wordle.net/ 2009 (accessed 15 September 2010).
Freichel 2009
- Freichel C, Prinssen E, Hoffmann G, Gand L, Beck M, Weiser T, et al. Oseltamivir is devoid of specific behavioral and other central nervous system effects in juvenile rats at supratherapeutic oral doses. International Journal of Virology 2009;5(3):119‐30. [Google Scholar]
Fritz 1999
- Fritz RS, Hayden FG, Calfee DP, Cass LM, Peng AW, Alvord WG, et al. Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo‐controlled trial of intravenous zanamivir treatment. Journal of Infectious Diseases 1999;180(3):586‐93. [DOI] [PubMed] [Google Scholar]
Fuiita 2011
- Fujita T, Fujii Y, Watanabe Y, Mori M, Yokota S. A pharmacoepidemiological study on the relationship between neuropsychiatric symptoms and therapeutic drugs after influenza infection. Japanese Journal of Pharmacoepidemiology 2010;15:73‐92. [Google Scholar]
Ghersi 2010
- Ghersi D, Clarke MJ, Reveiz L. Do Cochrane reviews search databases of ongoing trials, and how well do they report these searches? Oral presentation at the Joint Cochrane and Campbell Collaboration. http://www.cochrane.org/sites/default/files/uploads/abstract_book_keystone_2010.pdf. 2010, issue Suppl CD000002:45‐6. [DOI: 10.1002/14651858] [DOI]
Godlee 2009
- Godlee F, Clarke M. Why don't we have all the evidence on oseltamivir?. BMJ 2009;339:b5351. [DOI] [PubMed] [Google Scholar]
Godlee 2010
Gotzsche 2011
Gravenstein 2013
- Gravenstein S, Peters P. Erratum. Journal of the American Geriatrics Society 2013;61:478. [Google Scholar]
Hama 2008
- Hama R. Fatal neuropsychiatric adverse reactions to oseltamivir: case series and overview of causal relationships. International Journal of Risk and Safety in Medicine 2008;20:5‐36. [Google Scholar]
Hama 2010
- Hama R, Jones M, Hayashi K, Yanagi K, Sakaguchi K. Oseltamivir: a systematic review and meta‐analysis of adverse effects in prospective cohort studies. Presentation at the 16th Japanese Society for Pharmaco‐epidemiology (JSPE) and 5th Activities and Co‐operation for Drug Safety in Asia (ACPE) Joint Meeting. Tokyo, 2010.
Hama 2011
- Hama R. Jones M, Hayashi K, Sakaguchi K. Oseltamivir and early deterioration leading to death. International Journal of Risk & Safety in Medicine 2011;23:201‐15. [http://iospress.metapress.com/content/5257410g24403m68/fulltext.pdf] [DOI] [PubMed] [Google Scholar]
Hayden 1999
- Hayden FG, Treanor JJ, Fritz RS, Lobo M, Betts RF, Miller M, et al. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza: randomized controlled trials for prevention and treatment. JAMA 1999;282(13):1240‐6. [DOI] [PubMed] [Google Scholar]
Hayden 2009
- Hayden F. Developing new antiviral agents for influenza treatment: what does the future hold?. Clinical Infectious Diseases 2009;48(Suppl 1):3‐13. [DOI] [PubMed] [Google Scholar]
Hernan 2011
- Hernan MA, Lipsitch M. Oseltamivir and risk of lower respiratory tract complications in patients with flu symptoms: a meta‐analysis of eleven randomized clinical trials. Clinical Infectious Diseases 2011;53(3):277‐9. [PUBMED: 21677258] [DOI] [PMC free article] [PubMed] [Google Scholar]
HHS 2005
- U.S. Department of Health and Human Services. HHS pandemic influenza plan. http://www.hhs.gov/pandemicflu/plan/pdf/HHSPandemicInfluenzaPlan.pdf 2005 (accessed 9 June 2009).
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from: http://www.cochrane‐handbook.org.
Huic 2011
- Huic M, Marusic M, Marusic A. Completeness and changes in registered data and reporting bias of randomized controlled trials in ICMJE journals after trial registration policy. PLoS ONE 2011;6(9):e25258. [DOI: 10.1371/journal.pone.0025258] [DOI] [PMC free article] [PubMed] [Google Scholar]
ICH 2011
- The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). M4: the common technical document. http://www.ich.org/products/ctd.html 2011 (accessed 13 July 2011).
Ioannidis 2010
- Ioannidis JPA, Karassa FB. The need to consider the wider agenda in systematic reviews and meta‐analyses: breadth, timing, and depth of the evidence. BMJ 2010;341:c4875. [DOI] [PubMed] [Google Scholar]
Itoh 2009
- Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, et al. In vitro and in vivo characterization of new swine‐origin H1N1 influenza viruses. Nature 2009;460(7258):1021‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jack 2009
- Jack A. Roche steps up production of Tamiflu after virus scare. Financial Times 13 May 2009:19.
JAID 2010
- Japanese Association for Infectious Diseases. Question and answers on the Japanese Association for Infectious Diseases urgent recommendation "Handling pandemic influenza in routine care institutions". http://www.kansensho.or.jp/topics/090525influenza_qanda.html 2010 (accessed 9 May 2010).
Jefferson 2006
- Jefferson TO, Demicheli V, Pietrantonj C, Jones M, Rivetti D. Neuraminidase inhibitors for preventing and treating influenza in healthy adults. Cochrane Database of Systematic Reviews 2006, Issue 3. [DOI: 10.1002/14651858.CD001265.pub2] [DOI] [PubMed] [Google Scholar]
Jefferson 2009a
- Jefferson T, Jones M, Doshi P, Mar C. Neuraminidase inhibitors for preventing and treating influenza in healthy adults: systematic review and meta‐analysis. BMJ 2009;339:b5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jefferson 2010a
- Jefferson T, Jones M, Doshi P, Mar C, Dooley L, Foxlee R. Neuraminidase inhibitors for preventing and treating influenza in healthy adults. Cochrane Database of Systematic Reviews 2010, Issue 2. [DOI: 10.1002/14651858.CD001265.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jefferson 2011a
- Jefferson T, Mar CB, Dooley L, Ferroni E, Al‐Ansary LA, Bawazeer GA, et al. Physical interventions to interrupt or reduce the spread of respiratory viruses. Cochrane Database of Systematic Reviews 2011, Issue 7. [DOI: 10.1002/14651858.CD006207.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jefferson 2011b
- Jefferson T, Doshi P, Thompson M, Heneghan C. Ensuring safe and effective drugs: who can do what it takes?. BMJ 2011;342:c7258. [PUBMED: 21224325] [DOI] [PubMed] [Google Scholar]
Jones 2012
- Jones M, Hama R, Jefferson T, Doshi P. Neuropsychiatric adverse events and oseltamivir for prophylaxis. Drug Safety 2012;35(12):1187‐8. [DOI] [PubMed] [Google Scholar]
Kaiser 2003
- Kaiser L, Wat C, Mills T, Mahoney P, Ward P, Hayden F. Impact of oseltamivir treatment on influenza‐related lower respiratory tract complications and hospitalizations. Archives of Internal Medicine 2003;163:1667‐72. [DOI] [PubMed] [Google Scholar]
Kimura 2012
- Kimura S, Niwa Y, Iwajima Y, Nagano Y, Yamamoto S, Ohi Y, et al. High doses of oseltamivir phosphate induce acute respiratory arrest in anaesthetized rats. Basic & Clinical Pharmacology & toxicology 2012;111(4):232‐9. [DOI] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326(7400):1167‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2007
- Li C, Yu Q, Ye Z, Sun Y, He Q, Li X, et al. A nonsynonymous SNP in human cytosolic sialidase in a small Asian population results in reduced enzyme activity: potential link with severe adverse reactions to oseltamivir. Cell Research 2007;17(4):357‐62. [DOI] [PubMed] [Google Scholar]
Liu 1995
- Liu C, Eichelberger MC, Compans RW, Air GM. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. Journal of Virology 69;2:1099‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Longini 2004
- Longini IM Jr, Halloran ME, Nizam A, Yang Y. Containing pandemic influenza with antiviral agents. Am J Epidemiology 2004;159(7):623‐33. [DOI] [PubMed] [Google Scholar]
Lundh 2012
- Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.MR000033.pub2] [DOI] [PubMed] [Google Scholar]
MacLean 2003
- MacLean CH, Morton SC, Ofman JJ, Roth EA, Shekelle PG. How useful are unpublished data from the Food and Drug Administration in meta‐analysis?. Journal of Clinical Epidemiology 2003;56(1):44‐51. [DOI] [PubMed] [Google Scholar]
Matheson 2007
- Matheson NJ, Harnden AR, Perera R, Sheikh A, Symmonds‐Abrahams M. Neuraminidase inhibitors for preventing and treating influenza in children. Cochrane Database of Systematic Reviews 2007, Issue 1. [DOI: 10.1002/14651858.CD002744.pub2] [DOI] [PubMed] [Google Scholar]
Matrosovich 2004
- Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk H. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. Journal of Virology 2004;78(22):12665‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Maugh 2009
- Maugh TH II. British Medical Journal questions efficacy of Tamiflu for swine flu ‐ or any flu. http://latimesblogs.latimes.com/booster_shots/2009/12/british‐medical‐journal‐questions‐efficacy‐of‐tamiflu‐for‐swine‐flu‐or‐any‐flu.html 2009 (accessed 8 December 2009).
McGauran 2010
- McGauran N, Wieseler B, Kreis J, Schuler Y, Kolsch H, Kaiser T. Reporting bias in medical research ‐ a narrative review. Trials 2010;11(1):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mendel 1998
- Mendel DB, Tai CY, Escarpe PA, Li W, Sidwell RW, Huffman JH, et al. Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrobial Agents and Chemotherapy 1998;42(3):640‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2007
- Moore ML, Chi MH, Zhou W, Goleniewska K, O'Neal JF, Higginbotham JN, et al. Cutting edge: oseltamivir decreases T cell GM1 expression and inhibits clearance of respiratory syncytial virus: potential role of endogenous sialidase in antiviral immunity. Journal of Immunology 2007;178(5):2651‐4. [DOI] [PubMed] [Google Scholar]
Moscona 2005
- Moscona A. Neuraminidase inhibitors for influenza. New England Journal of Medicine 2005;353(13):1363‐73. [DOI] [PubMed] [Google Scholar]
Nebehay 2009
- Nebehay S. WHO backs findings on Tamiflu for seasonal flu. http://www.reuters.com/article/idUSGEE5BA0UY20091211?type=marketsNews 2009 (accessed 11 December 2009).
NHS 2009
- National Health Service. NHS choices. Antivirals and swine flu. http://www.nhs.uk/news/2009/12December/Pages/Antivirals‐and‐swine‐flu.aspx 2009 (accessed 17 May 2010).
NHS 2010
- National Health Service. NHS Choices. Swine flu ‐ questions and answers. http://www.nhs.uk/Conditions/Pandemic‐flu/Pages/QA.aspx 2010 (accessed 17 May 2010).
NICE 2000
- Roche. Tamiflu (oseltamivir phosphate) NICE submission. (leaked document) 1 May 2000.
Nicholson 2000
- Nicholson KG, Aoki FY, Osterhaus AD, Trottier S, Carewicz O, Mercier CH, et al. Efficacy and safety of oseltamivir in treatment of acute influenza: a randomised controlled trial. Neuraminidase Inhibitor Flu Treatment Investigator Group. Lancet 2000;355(9218):1845‐50. [DOI] [PubMed] [Google Scholar]
Ohuchi 2006
- Ohuchi M, Asaoka N, Sakai T, Ohuchi R. Roles of neuraminidase in the initial stage of influenza virus infection. Microbes and Infection 2006;8(5):1287‐93. [DOI] [PubMed] [Google Scholar]
Ono 2008
- Ono H, Nagano Y, Matsunami N, Sugiyama S, Yamamoto S, Tanabe M. Oseltamivir, an anti‐influenza virus drug, produces hypothermia in mice. Biological and Pharmaceutical Bulletin 2008;31(4):638‐42. [DOI] [PubMed] [Google Scholar]
Ono 2013
- Ono H, Iwajima Y, Nagano Y, Chazono K, Maeda Y, Ohsawa M, et al. Reduction in sympathetic nerve activity as a possible mechanism for the hypothermic effect of oseltamivir, an anti‐influenza virus drug, in normal mice. Basic & Clinical Pharmacology & Toxicology 2013;113(1):25‐30. [DOI] [PubMed] [Google Scholar]
Patrozou 2009
- Patrozou E, Mermel LA. Does influenza transmission occur from asymptomatic infection or prior to symptom onset?. Public Health Reports 2009;124:193‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Peters 2001
- Peters PH Jr, Gravenstein S, Norwood P, Bock V, Couter A, Gibbens M, et al. Long‐term use of oseltamivir for the prophylaxis of influenza in a vaccinated frail older population. Journal of the American Geriatrics Society 2001;49(8):1025‐31. [PUBMED: 11555062] [DOI] [PubMed] [Google Scholar]
Roche Investigators' Guide
- Roche. Investigators Guide. http://www.roche.be/fmfiles/re7189007/CU056/10_Investigators_brochure.pdf.
Rodgers 2013
- Rodgers MA, Brown JVE, Heirs MK, Higgins JPT, Mannion RJ, Simmonds MC, et al. Reporting of industry funded study outcome data: comparison of confidential and published data on the safety and effectiveness of rhBMP‐2 for spinal fusion. BMJ 2013;346:f3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sawabuchi 2009
- Sawabuchi T, Suzuki S, Iwase K, Ito C, Mizuno D, Togari H, et al. Boost of mucosal secretory immunoglobulin A response by clarithromycin in paediatric influenza. Respirology 2009;14(8):1173‐9. [DOI] [PubMed] [Google Scholar]
Senn 2004
- Senn SJ. Added values: controversies concerning randomization and additivity in clinical trials. Statistics in Medicine 2004;23:3729‐53. [DOI] [PubMed] [Google Scholar]
Shun‐Shin 2009
- Shun‐Shin M, Thompson M, Heneghan C, Perera R, Harnden A, Mant D. Neuraminidase inhibitors for treatment and prophylaxis of influenza in children: systematic review and meta‐analysis of randomised controlled trials. BMJ 2009;339:b3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2006
- Smith J, Dutkowski R, Ward P. Antivirals for influenza in healthy adults. Lancet 2006;367(9522):1571; author reply 1573. [PUBMED: 16698402] [DOI] [PubMed] [Google Scholar]
Sugaya 2010
- Sugaya N, Ohashi Y. Long‐acting neuraminidase inhibitor laninamivir octanoate (CS‐8958) versus oseltamivir as treatment for children with influenza virus infection. Antimicrobial Agents and Chemotherapy 2010;54(6):2575‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Takahashi 2010
- Takahashi E, Kataoka K, Fujii K, Chida J, Mizuno D, Fukui M, et al. Attenuation of inducible respiratory immune responses by oseltamivir treatment in mice infected with influenza A virus. Microbes and Infection / Institut Pasteur 2010;12(10):778‐83. [PUBMED: 20452454] [DOI] [PubMed] [Google Scholar]
Tappenden 2009
- Tappenden P, Jackson R, Cooper K, Rees A, Simpson E, Read R, et al. Amantadine, oseltamivir and zanamivir for the prophylaxis of influenza (including a review of existing guidance no. 67): a systematic review and economic evaluation. Health Technology Assessment 2009;11(iii, ix‐xii):1‐246. [DOI] [PubMed] [Google Scholar]
Toovey 2008
- Toovey S, Rayner C, Prinssen E. Assessment of neuropsychiatric adverse events in influenza patients treated with oseltamivir. A comprehensive review. Drug Safety 2008;31(12):1097‐114. [DOI] [PubMed] [Google Scholar]
Toovey 2012
- Toovey S. The author's reply. Drug Safety 2012;35(12):1188‐90. [Google Scholar]
Treanor 2000
- Treanor JJ, Hayden FG, Vrooman PS, Barbarash R, Bettis R, Riff D, et al. Efficacy and safety of the oral neuraminidase inhibitor oseltamivir in treating acute influenza: a randomized controlled trial. US Oral Neuraminidase Study Group. JAMA 2000;283(8):1016‐24. [DOI] [PubMed] [Google Scholar]
Turner 2003
- Turner D, Wailoo A, Nicholson K, Cooper N, Sutton A, Abrams K. Systematic review and economic decision modelling for the prevention and treatment of influenza A and B. Health Technology Assessment 2003;7(35):iii‐iv, xi‐xiii, 1‐170. [DOI] [PubMed] [Google Scholar]
Van Driel 2009
- Driel ML, Sutter A, Maeseneer J, Christiaens T. Searching for unpublished trials in Cochrane reviews may not be worth the effort. Journal of Clinical Epidemiology 2009;62:838‐44. [DOI] [PubMed] [Google Scholar]
Vedula 2009
- Vedula SS, Bero L, Scherer RW, Dickersin K. Outcome reporting in industry‐sponsored trials of gabapentin for off‐label use. New England Journal of Medicine 2009;361(20):1963–71. [DOI] [PubMed] [Google Scholar]
Vedula 2013
- Vedula SS, Li T, Dickersin K. Differences in reporting of analyses in internal company documents versus published trial reports: comparisons in industry‐sponsored trials in off‐label uses of gabapentin. PLoS Medicine 2013;10(1):e1001378. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ward 2005
- Ward P, Small I, Smith J, Suter P, Dutkowski R. Oseltamivir (Tamiflu) and its potential for use in the event of an influenza pandemic. Journal of Antimicrobial Chemotherapy 2005;55(Suppl 1):5‐21. [PUBMED: 15709056] [DOI] [PubMed] [Google Scholar]
WHO 2002a
- World Health Organization. Global agenda on influenza‐‐adopted version. Part I. Releve Epidemiologique Hebdomadaire/Section d'Hygiene du Secretariat de la Societé des Nations = Weekly Epidemiological Record/Health Section of the Secretariat of the League of Nations 2002;77(22):179‐82. [PUBMED: 12061030] [PubMed] [Google Scholar]
WHO 2002b
- World Health Organization. Adoption of global agenda on influenza ‐ Part II. Releve Epidemiologique Hebdomadaire/Section d'Hygiene du Secretariat de la Societé des Nations = Weekly Epidemiological Record/Health Section of the Secretariat of the League of Nations 2002;77(23):191‐5. [PUBMED: 12073536] [PubMed] [Google Scholar]
WHO 2004
- World Health Organization. WHO guidelines on the use of vaccines and antivirals during influenza pandemics. http://www.who.int/csr/resources/publications/influenza/11_29_01_A.pdf 2009 (accessed 30 November 2009).
WHO 2007
- World Health Organization. WHO interim protocol: rapid operations to contain the initial emergence of pandemic influenza. http://www.who.int/influenza/resources/documents/RapidContProtOct15.pdf October 2007 (accessed 17 May 2011).
WHO 2011
- World Health Organization. UNEDITED REPORT of the 18th Expert Committee on the selection and use of essential medicines. http://www.who.int/entity/selection_medicines/Complete_UNEDITED_TRS_18th.pdf 2011 (accessed 7 September 2011).
WHO 2013a
- World Health Organization. WHO Model List of Essential Medicines. ADULTS, 18th edition (April 2013). Rev. Oct 2013. http://www.who.int/medicines/publications/essentialmedicines/18th_EML_Final_web_8Jul13.pdf 2013 (accessed 25 November 2013).
WHO 2013b
- World Health Organization. WHO Model List of Essential Medicines for Children. 4th list (April 2013). Rev. Oct 2013. http://apps.who.int/iris/bitstream/10665/93143/1/EMLc_4_eng.pdf 2013 (accessed 25 November 2013).
Wieseler 2013
- Wieseler B, Wolfram N, McGauran N, Kerekes MF, Vervölgyi V, Kohlepp P, et al. Completeness of reporting of patient‐relevant clinical trial outcomes: comparison of unpublished clinical study reports with publicly available data. PLoS Medicine 2013;10(10):e1001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yorifuji 2009
- Yorifuji T, Suzuki E, Tsuda T. Oseltamivir and abnormal behaviors: true or not?. Epidemiology 2009;20(4):619‐21. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Jefferson 2012
- Jefferson T, Jones MA, Doshi P, Mar CB, Heneghan CJ, Hama R, et al. Neuraminidase inhibitors for preventing and treating influenza in healthy adults and children. Cochrane Database of Systematic Reviews 2012, Issue 1. [DOI: 10.1002/14651858.CD008965.pub3] [DOI] [PubMed] [Google Scholar]