

Abstract
Polypeptides are promising carriers for chemotherapeutics: they have minimal toxicity, can be recombinantly synthesized with precise control over molecular weight, and enhance drug pharmacokinetics as self-assembled nanoparticles. Polypeptide-based systems also provide the ability to achieve active targeting with genetically encoded targeting ligands. While passive targeting promotes accumulation of nanocarriers in solid tumors, active targeting provides an additional layer of tunable control and widens the therapeutic window. However, fusion of most targeting proteins to polypeptide carriers exposes the limitations of this approach: the residues that are used for drug attachment are also promiscuously distributed on protein surfaces. We present here a universal methodology to solve this problem by the site-specific attachment of extrinsic moieties to polypeptide drug delivery systems without cross-reactivity to fused targeting domains. We incorporate an unnatural amino acid, p-acetylphenylalanine, to provide a biorthogonal ketone for attachment of doxorubicin in the presence of reactive amino acids in a nanobody-targeted, elastin-like polypeptide nanoparticle. These nanoparticles exhibit significantly greater cytotoxicity than nontargeted controls in multiple cancer cell lines.
Keywords: Unnatural amino acids, active targeting, cancer drug delivery, polypeptides, self-assembly, nanoparticles
GRAPHICAL ABSTRACT:
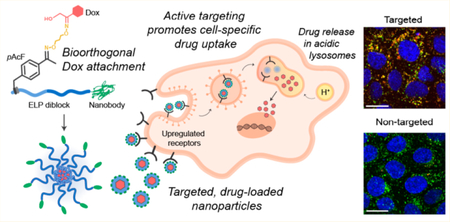
While small molecule drugs are highly potent cytotoxic agents, their poor solubility, short half-life, and insufficient bioavailability often require the use of delivery strategies, such as polymeric nanoscale drug carriers, to improve their delivery to solid tumors.1 Recent work has demonstrated the importance of three key elements for effective tumor treatment by nanoparticle drug carriers: (1) long circulation time,2,3 (2) active targeting,4,5 and (3) site-specific attachment of cytotoxic drugs.6,7 Long blood circulation is advantageous for regional accumulation in tumors via the enhanced permeability and retention effect, also called passive targeting.8 Active targeting of nanoparticles provides the second stage of tumor specific delivery as it enables tumor cell-selective uptake, complementary to the regional accumulation provided by passive targeting, widening the therapeutic window of the drug.9 Finally, site-specific conjugation of small molecule drugs to these carriers with tunable chemistries ensures a homogeneous drug carrier population and predictable release.10
Recombinant peptide polymers are attractive for the design of nanoparticle delivery systems because they can be produced recombinantly in high yield in E. coli as monodisperse macromolecules and their sequence and chain length are easily manipulated at the gene level.11,12 They are also nontoxic and biodegradable.13 We have pioneered the development of elastin-like polypeptides (ELPs) as a class of peptide polymers that self-assemble into nanoparticles for drug delivery.14–16
ELPs consist of the sequence (VPGXG)n, where the guest residue X is any amino acid except proline, and n is the number of repeats.17 ELPs are thermally responsive; they phase separate in aqueous solution when heated above their cloud point temperature, also called the inverse transition temperature (Tt), and resolubilize completely upon cooling below their Tt.18 Diblock copolymer ELPs, with blocks possessing distinct Tts as individual segments, can be designed to self-assemble into nanoscale micelles at physiological temperatures.19 ELPs can also be fused to other proteins and peptides at their N-or C-termini, or both, without loss of thermal responsivity.20 Self-assembling ELPs have been used as nanoparticle drug carriers to increase the half-life of small molecule drugs for potent tumor regression.21–23
Genetically encoded synthesis provides exquisite control over the design and tunability of ELP nanoparticles for drug delivery, as it enables the size and shape of the nanoparticle to be controlled,19,24 as well as the site and stoichiometry of small molecule drug conjugation,14,25 and allows introduction of targeting protein or peptide domains.26 However, because of the limited chemical diversity available with the naturally occurring amino acids, we cannot simultaneously control all of these parameters in the same nanoparticle. Chemically reactive lysine and cysteine residues are typically used for site-specific attachment of small molecule drugs and imaging agents;27,28 this approach only works well with polypeptide nanoparticles or carrier proteins that do not include these residues in their native sequence. Unfortunately, the inclusion of most targeting proteins exposes the limitations of this approach because cysteine and lysine residues are often integral to the bioactivity of these domains and found in solvent-accessible, and hence highly reactive, locations.29,30
To develop a universal approach for the site-specific attachment of extrinsic moieties to ELP nanoparticle-based drug delivery systems without cross-reactivity with a protein targeting domain, we chose to investigate the co-translational incorporation of an unnatural amino acid (UAA) into ELPs that provides a unique, bio-orthogonal reactive moiety in its side-chain for site specific attachment of small molecule drugs or imaging agents. The novel technology we describe herein represents, to the best of our knowledge, the first actively targeted, polypeptide-based nanoparticle with site-specific drug conjugation against a complex background of reactive amino acids that is made possible only through the introduction of bioorthogonal attachment chemistry.
We chose the approach of genetic code expansion pioneered by Schultz and co-workers to incorporate a bio-orthogonal residue, p-acetylphenylalanine (pAcF), as the site of drug attachment.31 The incorporation of pAcF introduces a novel reactive ketone that can participate in bioorthogonal conjugation reactions. Our approach uses a specifically evolved M. jannaschii tRNATyr that recognizes the amber stop codon in E. coli, and its cognate, evolved M. jannaschii aminoacyl tRNA synthetase (aaRSTyr), allowing site-specific incorporation of Tyr analogs at the stop codon with high yield and >95% efficiency.32 We also used a recoded strain of E. coli that has recently been developed for UAA incorporation (C321.ΔA) as the expression host.33,34 This strain has all 321 amber stop codons in its genome recoded to the ochre stop codon and the corresponding release factor 1 deleted, which greatly improves the yield of UAA incorporation into proteins.35
We chose the EgA1 nanobody, a small antibody fragment derived from camelid single-chain antibodies, as the targeting domain for our system.36 Nanobodies are compact and are therefore unlikely to perturb self-assembly of the ELP into nanoparticles when appended on the corona, and unlike full-length antibodies, they can be expressed recombinantly in E. coli alone or as fusions.37 Importantly, the EgA1 nanobody contains four lysine residues and a pair of cysteine residues, thereby representing a good candidate to validate the generality of our approach. The EgA1 nanobody recognizes and binds to human epidermal growth factor receptor (EGFR) with an affinity of 276 nM.38 We chose EGFR as the target, as it is highly upregulated or aberrantly expressed in multiple cancer types including breast, ovarian, brain, lung, and as many as 30% of all carcinomas.39–41
As the cytotoxic payload for our system, we chose a small molecule chemotherapeutic, Doxorubicin (Dox). Dox stabilizes an intermediate covalent complex between topoisomerase II and genomic DNA, causing damaging double-stranded breaks and exit from the cell cycle,42 and is widely used for the treatment of hematopoietic malignancies, breast, lung, ovary, stomach, and thyroid carcinomas, and bone and soft tissue sarcomas.16,25,43,44 However, Dox exhibits dose-limiting myelosuppression, mucositis, and cardiotoxicity, and a targeted nanoparticle formulation of this drug greatly improves its in vivo performance and reduces off-target toxicity.45,46
The design of our Dox-loaded nanoparticles is illustrated in Figure 1A. We designed and synthesized a gene that encodes a self-assembling diblock ELP fused to a peptide leader at the N-terminus of the hydrophobic block that incorporates the site of Dox attachment, pAcF, and the EgA1 nanobody at the C-terminus of the hydrophilic block (pAcF-ELPBC-EgA1, Table S1). We designed the diblock ELP in this construct so that it has a critical micellization temperature (CMT) that lies between room temperature and body temperature. Upon heating to a temperature above the CMT, the N-terminal hydrophobic ELP block desolvates, while the C-terminal hydrophilic ELP block remains solvated, creating an amphiphile that self-assembles into nanoscale micelles that are stable at physiological temperatures. The EgA1 nanobody is displayed on the corona of the micelle, while Dox is sequestered in the core of and is covalently attached to the ketone group of pAcF via a linker that creates a pH-sensitive oxime bond between the drug and the ELP,47,48 allowing for release of Dox in the acidic lysosomal compartments of cells after uptake by receptor-mediated endocytosis.
Figure 1.
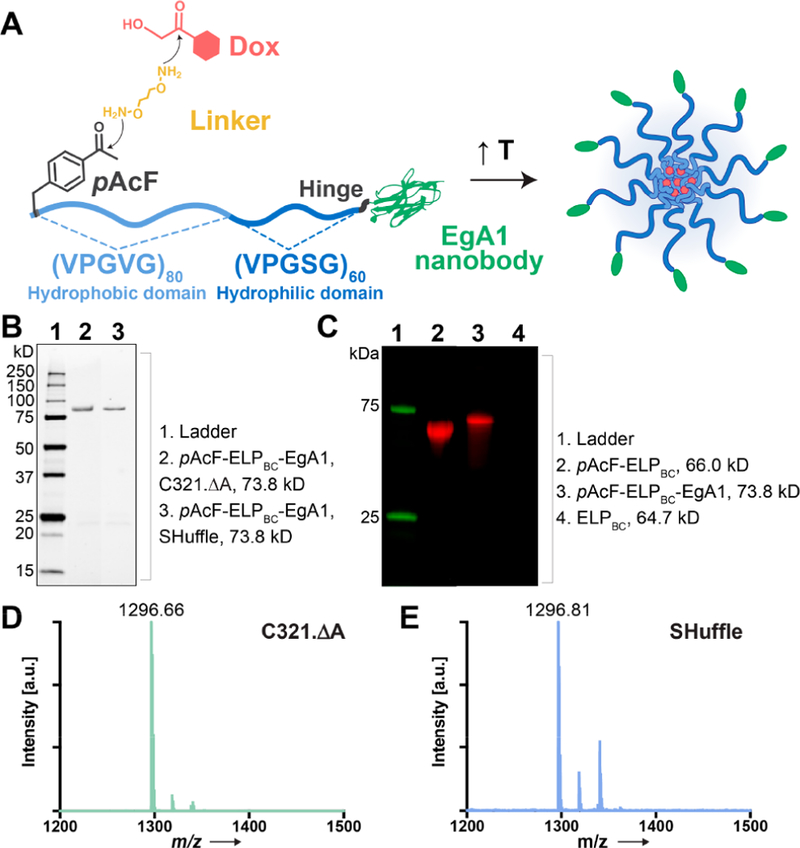
Design and expression of pAcF-ELPBC-EgA1. (A) Schematic showing design and assembly of Dox-pAcF-ELPBC-EgA1 nanoparticles. Dox (red) is conjugated to the pAcF residue (dark gray) at the N-terminus of the amphiphilic ELPBC chain (blue) by a telechelic hydroxylamine linker (yellow). The hydrophilic ELP block is fused to the EgA1 nanobody (green) by a flexible hinge (black). (B) SDS-PAGE of pAcF-ELPBC-EgA1 in C321.ΔA (lane 2) and SHuffle E. coli (lane 3). (C) Fluorescence imaging of SDS-PAGE qualitatively confirms reactivity of the ketone group on the pAcF residue in pAcF-ELPBC (lane 2) and pAcF-ELPBC-EgA1 (lane 3) with a hydroxylamine dye. ELPBC without pAcF incorporated is not labeled (lane 4). Tryptic digest of these constructs followed by MALDI-TOF-MS shows a single peak that is consistent with the incorporation of a pAcF residue in pAcF-ELPBC-EgA1 of protein expressed in (D) C321.ΔA (1296.66 m/z) and (E) SHuffle E. coli (1296.81 m/z).
We expressed a library of constructs in E. coli from a plasmid-borne gene for these studies (Tables S1 and S2). In addition to our primary construct of interest, pAcF-ELPBC-EgA1, our library includes the base diblock ELP (ELPBC), a pAcF-containing ELPBC without the EgA1 nanobody that serves as a negative control for targeting (pAcF-ELPBC), and a targeted ELPBC without a pAcF residue that serves as negative control for site-specific Dox conjugation (ELPBC-EgA1). We expressed these proteins in one of three E. coli expression hosts; the base diblock ELPBC was solubly expressed in high yield (160 mg L−1) in the standard protein expression cell line, BL21(DE3). The introduction of the pAcF residue and EgA1 nanobody directed us to investigate the use of two different E.coli lines previously reported for expression of constructs containing these elements.35,49 The constructs containing pAcF, pAcF-ELPBC and pAcF-ELPBC-EgA1, were expressed in the genomically recoded C321.ΔA cell line with yields of 30 mg L−1 and 15 mg L−1, respectively. For constructs including the EgA1 nanobody, ELPBC-EgA1 and pAcF-ELPBC-EgA1, which introduces a disulfide bond into the protein, we used the SHuffle T7 Express E. coli, as this line is optimized to enhance disulfide bond formation and protein solubility. We were able to express these two constructs as soluble proteins in high yield (50 mg L−1 and 12 mg L−1, respectively). We explored expression of our final product, pAcF-ELPBC-EgA1, in both SHuffle and C321.ΔA E. coli as each provides a unique advantage for expression. The engineered SHuffle strain expresses heterologous chaperones to enhance the production of functional, soluble disulfide-bonded protein under the control of a T7 promotor. Conversely, the recoded C321.ΔA strain is ideally suited for unnatural amino acid incorporation with an unassigned stop codon that can be reassigned exclusively for an unnatural residue of interest as well as the corresponding release factor deleted to eliminate any competition with a termination signal.
We purified all constructs by inverse transition cycling (ITC), a nonchromatographic method for the purification of ELPs and their fusions that exploits the LCST phase behavior of ELPs.50,51 SDS-PAGE of ITC-purified pAcF-ELPBC-EgA1 from C321.ΔA and SHuffle T7 Express E. coli showed that four cycles of ITC provided protein with >95% purity (Figure 1B). The SDS-PAGE of all other constructs is shown in Figure S1.
We investigated the incorporation of pAcF with a labeling experiment wherein pAcF-ELPBC and pAcF-ELPBC-EgA1 were reacted with ketone-reactive Alexa697-hydroxylamine dye (Figure 1C). Fluorescence imaging of SDS-PAGE of the reaction product qualitatively confirms reactivity of the pAcF-ELPBC (lane 2) and pAcF-ELPBC-EgA1 (lane 3) with the ketone-reactive dye and hence indicated the successful incorporation of pAcF in these constructs.
We next confirmed the incorporation of pAcF in pAcF-ELPBC-EgA1 expressed in the SHuffle and C321.ΔA cell lines by mass spectrometry. The leader peptide containing the pAcF residue was cleaved by trypsin and analyzed by matrix assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS). The observed mass of the N-terminal peptide generated by trypsin cleavage of pAcF-ELPBC-EgA1 is 1296.66 Da for protein expressed in C321.ΔA and 1296.81 Da expressed in Shuffle, both of which are in excellent agreement with the theoretical mass of 1296.73 Da, confirming the successful incorporation of pAcF (Figure 1D). While we determined that either E. coli line would be a suitable expression host for simultaneously encoding pAcF and the structured EgA1 nanobody, we selected C321.ΔA for pAcF-ELPBC-EgA1 expression for all subsequent experiments as this line provided a modestly greater yield compared to the SHuffle strain.
We further quantified the incorporation efficiency of pAcF with ESI-LC−MS and found >98% incorporation efficiency of pAcF in both pAcF-ELPBC and pAcF-ELPBC-EgA1 (Figure S2, Table S3). While the C321.ΔA and tRNA/synthetase pair we employed for protein expression have been optimized for greatest incorporation fidelity of pAcF, there is a minor population, < 2%, of natural amino acids misincorporated at the pAcF site. The misincorporated residues we detected include the aromatic Tyr, Trp, and Phe, consistent with literature reports for this cell line.34,52–54
We next validated the bioactivity of the EgA1 nanobody by flow cytometry. We examined the binding of ELPBC-EgA1 for EGFR and cellular uptake in a panel of eight cell lines with a range of EGFR expression levels to identify candidate cell lines for in vitro testing of our conjugates (Figure 2, Figures S3 and S4). To perform flow cytometry analysis, we fluorescently labeled the N-termini of both ELPBC-EgA1 and ELPBC with AlexaFluor488-NHS ester, incubated the labeled proteins with the different cell lines, and analyzed the cells by flow cytometry. In a mouse NIH3T3 fibroblast line transfected with human EGFR (NIH3T3 EGFR+) that has a high level of EGFR expression (1.5 × 106 receptors per cell),55 ELPBC-EgA1 exhibited 18-fold higher uptake as compared to the non-targeted ELPBC (Figure 2A,D). Two cell lines with a range of reported sensitivity to Dox56,57 also exhibited significantly higher uptake as compared to ELPBC. These are the squamous carcinoma line A431, that showed a 13-fold higher uptake as compared to the nontargeted ELPBC (Figure 2B,D), and the ovarian adenocarcinoma line SKOV-3 that exhibited a five-fold higher uptake of ELPBC-EgA1 than ELPBC (Figure 2C,D). Flow cytometry experiments with a negative control, the untransfected fibroblast cell line (NIH3T3 EGFR-) showed no significant difference between uptake of the targeted and nontargeted constructs, confirming the specificity of the EgA1 nanobody for EGFR (Figure S3E,F).
Figure 2.
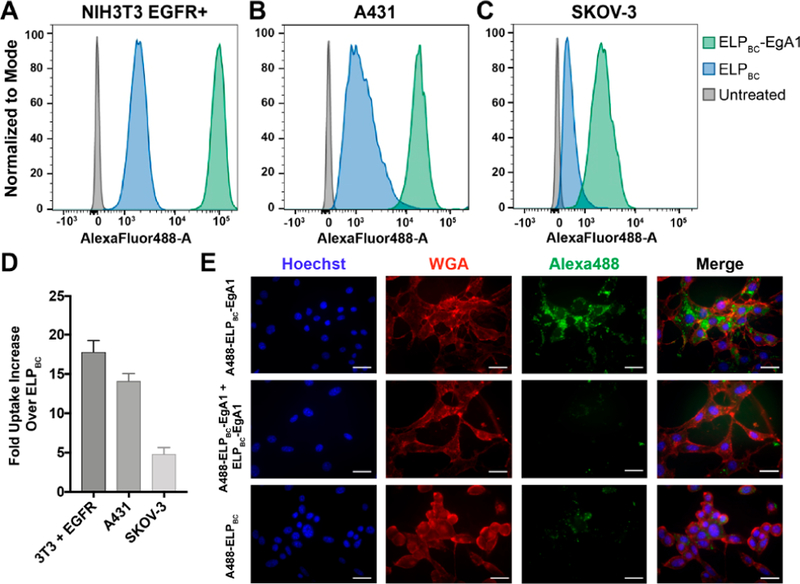
Flow cytometry confirms specificity of the EgA1 nanobody for human epidermal growth factor receptor (EGFR). Fluorescently labeled ELPBC-EgA1 or ELPBC were incubated with various cell lines and analyzed by flow cytometry. (A) NIH3T3 murine fibroblasts transfected with human EGFR (NIH3T3 EGFR+); (B) A431 squamous carcinoma cells; and (C) SKOV-3 ovarian adenocarcinoma cells all indicate enhanced uptake of the ELPBC-EgA1 as compared to ELPBC. (D) Geometric mean fluorescent intensities (gMFI) of the cell populations were used to quantify the fold uptake of ELPBC-EgA1 over ELPBC and shows the range of nanobody-mediated targeting of EGFR across the cell lines. (E) Fluorescence microscopy images of NIH3T3 EGFR+ cells incubated with AlexaFluor488-ELPBC-EgA1, AlexaFluor488-ELPBC-EgA1 with 10-fold excess unlabeled ELPBC-EgA1, and AlexaFluor488-ELPBC shows increased uptake of the AlexaFluor488-ELPBC-EgA1 construct, while the cells incubated with 10-fold excess unlabeled ELPBC-EgA1 demonstrate the specificity of the EgA1 nanobody. The samples incubated with AlexaFluor488-ELPBC show the basal level of nanoparticle uptake. Nuclei stained with Hoechst (blue), cell membranes stained with AlexaFluor594-wheat germ agglutinin (red), and all ELP constructs with AlexaFluor488 (green). Scale bars 25 μm.
We next visualized the intracellular uptake of fluorescently labeled ELPBC-EgA1 in the EGFR-transfected fibroblast line by fluorescence microscopy (Figure 2E). After 24 h of incubation of NIH3T3 EGFR+ cells with Alexa488-ELPBC-EgA1, high levels of intracellular accumulation of the labeled protein were observed. We confirmed specificity of the EgA1 nanobody by coincubating Alexa488-ELPBC-EgA1 with a 10-fold molar excess of unlabeled ELPBC-EgA1 to compete with the Alexa488-ELPBC-EgA1 for binding to EGFR; these cells show little intracellular fluorescence, as do cells incubated with the negative control, Alexa488-ELPBC. We further confirmed the specificity of EgA1 for EGFR by preincubating cells with excess, fluorescently labeled EGF followed by Alexa647-ELPBC-EgA1. We observed simultaneously high levels of intracellular EGF and little Alexa647-ELPBC-EgA1, whereas uptake of Alexa647-ELPBC-EgA1 was significant in a control experiment in which cells were not preincubated with EGF (Figure S5). Together, the flow cytometry and fluorescence microscopy results demonstrate that the EgA1 nanobody: (1) maintains specificity for EGFR when fused to ELPBC, (2) enhances intracellular uptake of an ELPBC fusion by cells that overexpress EGFR, and (3) can be used for targeting the ELPBC to a panel of human cancer cell lines that overexpress EGFR.
We next used the bioorthogonal ketone group on the pAcF residue as the site of conjugation for our drug payload, Dox. To conjugate Dox to pAcF, we employed a two-step reaction scheme (Figure 3A, Table S4). First, we activated pAcF in 1 with an excess of the telechelic linker 2. While the reaction with pAcF-ELPBC proceeds at pH 4.0 with high efficiency (74.5%), we found the presence of aniline catalyst58 allowed the reaction to proceed nearly to completion (90.9%) at pH 6.2, a pH that is more optimal for the stability of pAcF-ELPBC-EgA1 (see eq S1 for calculation of Dox labeling efficiency). We removed unreacted linker by centrifugal ultrafiltration and then reacted the intermediate construct 3 with an excess of Dox 4 in the presence of aniline and removed unreacted Dox using ultrafiltration from the final product 5. We confirmed each individual reaction step by digesting the products 1, 3, and 5 with trypsin and analyzing the digested peptide fragments by MALDI-TOF-MS. The spectra of the liberated peptides showed one major peak for each product, which increased in molecular weight by the expected amount after each reaction step (Figure 3B, full spectra Figure S6). We further validated linker and Dox attachment after each reaction step with ESI-LC/MS and analyzed the composition of the reaction products (Figure S7, Table S3). We found each reaction step had >85% of the desired product and minor populations of starting material and dimer reaction products. We confirmed purity of our final reaction product using size exclusion chromatography (SEC) and the characteristic absorbance of Dox at 488 nm (Figure 3C, Figure S8). We validated the mechanism of Dox release, acid-catalyzed oxime hydrolysis, by incubating solutions of Dox-pAcF-ELPBC-EgA1 conjugates in either pH 4.0 or pH 7.4 buffer and quantifying the amount of Dox released at various time points with SEC (Figure 3D). We observed steady-state release of 58.2 ± 1.4% after incubation for 48 h in pH 4.0 buffer and negligible release at pH 7.4, indicating these micelles will release drug only after receptor-mediated endocytosis into acidic lysosomal compartments.
Figure 3.
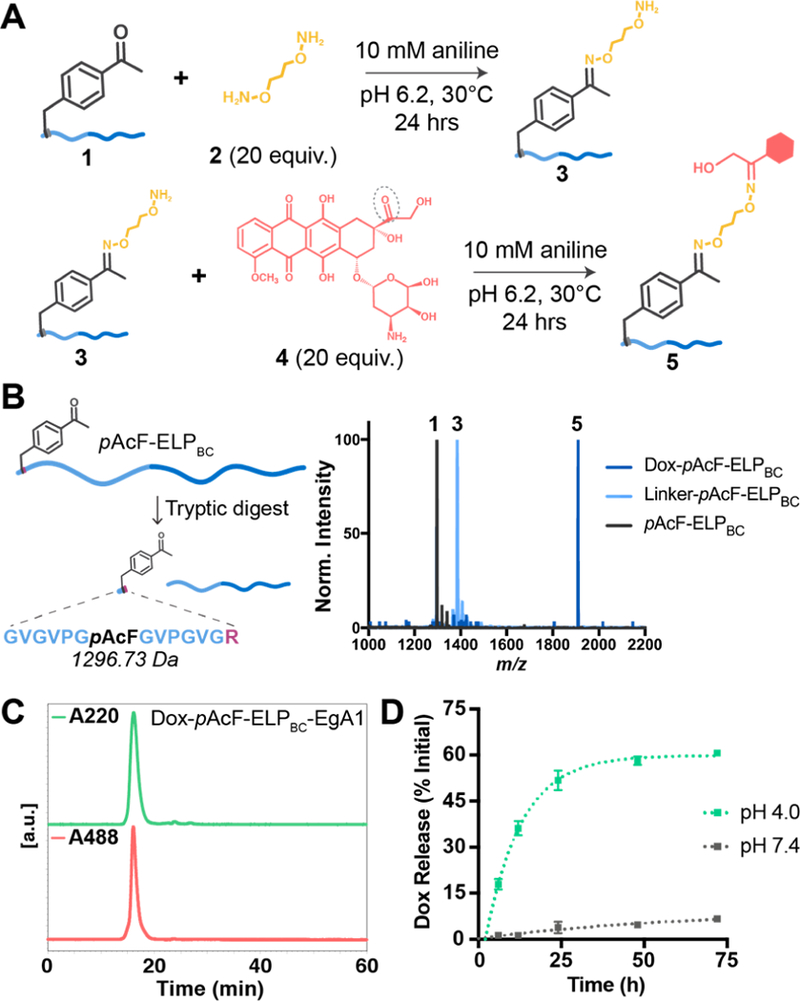
Confirming Dox attachment and acid-catalyzed release from pAcF-ELPBC and pAcF-ELPBC-EgA1. (A) Schematic of the two-step Dox conjugation to pAcF. First, pAcF-ELPBC 1 is reacted with the hydroxylamine linker 2 in the presence of 10 mM aniline catalyst to form the intermediate 3. This product is purified and reacted excess doxorubicin 4 under the same conditions to form the final conjugate 5. (B) The leader peptide containing pAcF can be liberated by tryptic digest from pAcF-ELPBC to confirm successful reaction steps via matrix assisted laser desorption ionization mass spectroscopy (MALDI-TOF): first, incorporation of pAcF in 1 (expected MW 1296.73 Da, observed 1296.65 Da), then modification with linker in 3 (expected MW 1384.84 Da, observed 1384.78 Da), and finally conjugation of Dox onto 5 (expected MW 1911.41 Da, observed 1910.15 Da). (C) Attachment of Dox to pAcF-ELPBC-EgA1 and purity of the final conjugate is confirmed using size exclusion chromatography (SEC), analyzing the spectrum at both A220 nm, to monitor the elution time of the conjugate, and A488 nm, the characteristic absorbance of Dox, to confirm attachment and purity. (D) pH-dependent release of Dox from Dox-pAcF-ELPBC-EgA1 is assessed by SEC at pH 4.0 (green) and pH 7.4 (gray).
We characterized the size and shape of both the Dox-pAcF-ELPBC-EgA1 and nontargeted control Dox-pAcF-ELPBC nano-particles by light scattering and cryogenic transmission electron microscopy (cryo-TEM). Dynamic light scattering (DLS), performed in dilute solution as a function of temperature, showed the critical micellization temperature (CMT) of both pAcF-ELPBC-EgA1 and pAcF-ELPBC to be ∼32 °C and unchanged upon conjugation of Dox (Figure 4A). The Dox-pAcF-ELPBC-EgA1 construct assembles into monodisperse nanoparticles with a hydrodynamic radius (Rh) of 44.7 ± 4.4 nm, while the Dox-pAcF-ELPBC construct forms smaller particles with a Rh of 23.7 ± 0.9 nm (Figure 4B). A dilution series of Dox-pAcF-ELPBC-EgA1 analyzed by DLS revealed the critical micellization concentration (CMC) to be between 1 and 5 μM, consistent with findings for other ELPBC nanoparticles (Figure S9).26 Static light scattering (SLS) revealed the Dox-pAcF-ELPBC-EgA1 particles to contain approximately 22 chains per particle, while the Dox-pAcF-ELPBC particles contain approximately 28 chains per particle (Table 1, Figure S10). The positively charged EgA1 nanobody (pI 9.08) likely increases Dox-pAcF-ELPBC-EgA1 particle size while reducing the Nagg by charge repulsion as compared to Dox-pAcF-ELPBC. Together with the Dox conjugation efficiency, this indicates each EgA1-targeted nanoparticle is loaded with 17–18 Dox molecules.
Figure 4.
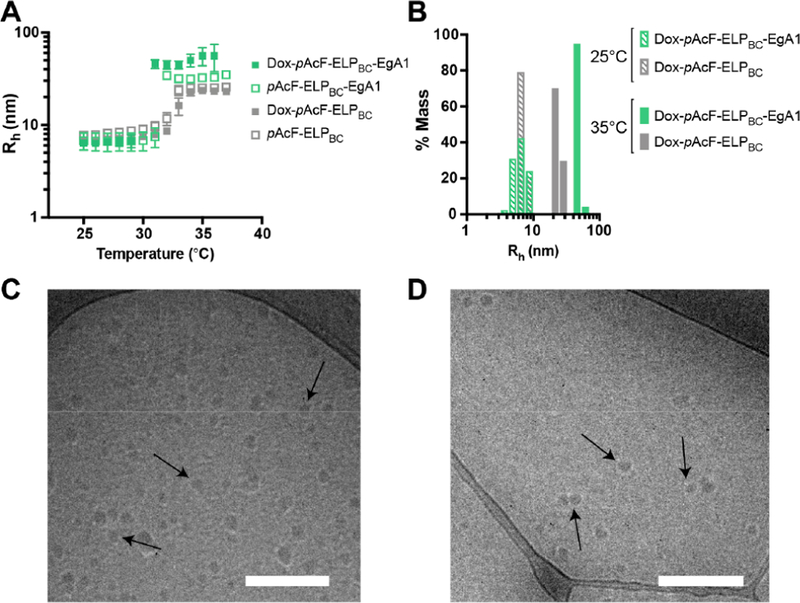
Dox conjugates form uniform nanoparticles at physiologic temperatures. (A) Characterization of the particle self-assembly by dynamic light scattering (DLS) shows hydrodynamic radius (Rh) as a function of temperature for pAcF-ELPBC-EgA1 (green) and pAcF-ELPBC (gray) before and after Dox conjugation. (B) Polydispersity of the samples by % mass at temperatures below (25 °C, unimers) and above (35 °C, micelles) the critical micellization temperature (CMT). Visualization of the micelles by cryogenic transmission electron microscopy (cryo-TEM) illustrates the monodispersity of the nanoscale assemblies of (C) Dox-pAcF-ELPBC-EgA1 and (D) Dox-pAcF-ELPBC. Scale bars 200 nm.
Table 1.
Static Light Scattering of Dox-pAcF-ELPBC and Dox-pAcF-ELPBC-EgA1 Performed at 35 °C, above the CMT
| Rga (nm) | Rhb (nm) | ρc | MWmicelle (g mol−1) | Naggd | |
|---|---|---|---|---|---|
| Dox-pAcF-ELPBC | 20.3 | 18.4 | 1.1 | 1.85 × 106 | 27.8 |
| Dox-pAcF-ELPBC-EgA1 | 37.2 | 41.8 | 0.9 | 1.63 × 106 | 21.8 |
Rg, radius of gyration.
Rh, hydrodynamic radius.
p, form factor.
Nagg, number of chains per micelle.
We visualized both particles with cryo-TEM to further confirm their spherical shape and size (Figure 4C, 4D, Figure S11). Core sizing analysis of these particles indicated roughly equivalent core diameters of 24.7 ± 4.4 nm for the Dox-pAcF-ELPBC particles and 29.4 ± 6.2 nm for the Dox-pAcF-ELPBC-EgA1 nanoparticles. This further indicates the increased size of the Dox-pAcF-ELPBC-EgA1 particles is due to size differences in the corona, where the EgA1 nanobody is presented. These studies confirm that the Dox-loaded particles are spherical, stable above their CMT at physiological temperature (37 °C), and in the ideal sub-100 nm size range for tumor penetration,59 making them suitable for the delivery of drugs to solid tumors.
We next evaluated the cytotoxicity of the Dox-pAcF-ELPBC-EgA1 and Dox-pAcF-ELPBC particles in two EGFR positive cell lines: A431 and SKOV-3.60,61 We chose these two tumor lines, as they express different level of the EGFR, with A431 expressing higher levels of EGFR than SKOV-3. Examining the response of tumor cells that have different levels of receptor expression is critical to demonstrate the clinical utility of our platform, as EGFR expression level varies widely between cancer types,62 within individual tumors,63 and at different time points of treatment.64 These two cell lines also differ in their relative sensitivity to Dox, with the A431 line being more sensitive than SKOV-3. After treatment with increasing concentrations of free drug and assessing cell viability, we determined the IC50 of Dox in A431 cells is 1.38 μM, while that of SKOV-3 cells is 31.8 μM (Figure 5A, 5B). While A431 represents the ideal tumor for targeted drug treatment as it is a highly receptor-positive, drug-sensitive line, SKOV-3 represents a more clinically relevant subset of tumor cells, those with intermediate receptor expression and a lower sensitivity to Dox.57
Figure 5.
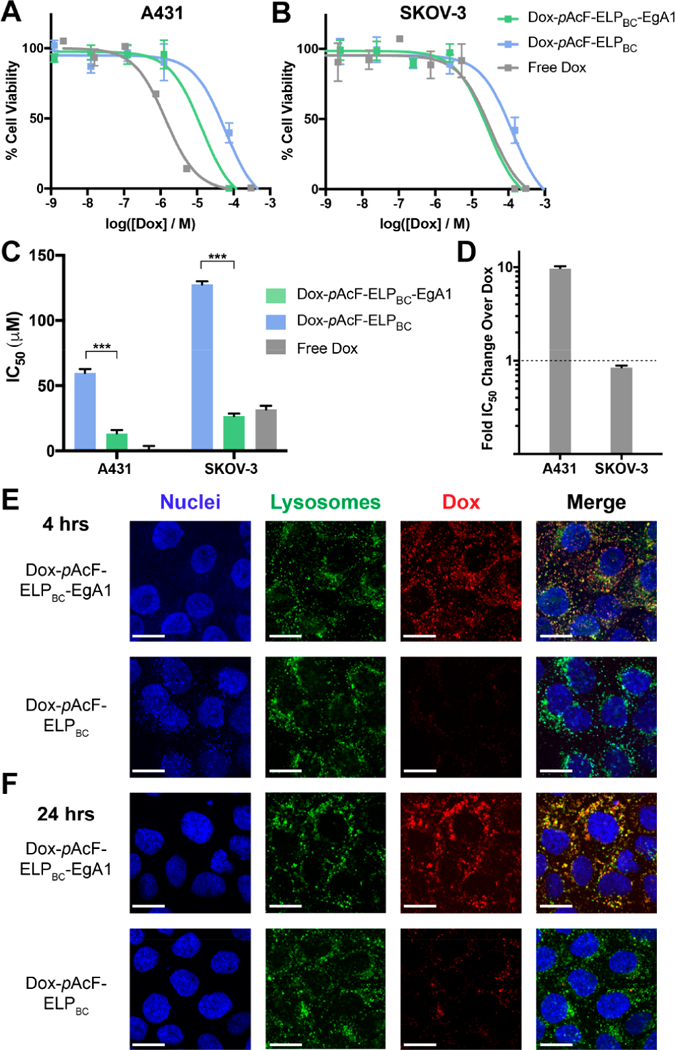
EgA1-targeted Dox-pAcF-ELPBC-EgA1 is significantly more potent in cancer cells via lysosomal-mediated release. Cytotoxicity of Dox-pAcF-ELPBC-EgA1 (green), Dox-pAcF-ELPBC (blue), and free Dox (gray) in (A) A431 and (B) SKOV-3 cells. (C) IC50 values of these cell viability assays confirms the targeted Dox-pAcF-ELPBC-EgA1 is more cytotoxic than the nontargeted control, Dox-pAcF-ELPBC. p < 0.001. (D) Normalized IC50 of Dox-pAcF-ELPBC-EgA1 to IC50 of free Dox indicates the effect of active targeting on the cytotoxicity of Dox. Spinning disk confocal imaging of A431 cells incubated with Dox-pAcF-ELPBC-EgA1 or Dox-pAcF-ELPBC for (E) 4 h or (F) 24 h. Hoechst (blue) staining shows nuclei, CytoPainter (green) indicates low pH endolysosomal compartments, and Dox (red) shows subcellular localization of Dox conjugates. Merged images show colocalization (yellow) between lysosomes and Dox. Scale bars 20 μm.
We treated the two cell lines with increasing concentrations of the targeted and nontargeted Dox conjugates and measured cell viability after treatment. In both cell lines, the Dox-pAcF-ELPBC-EgA1 particles have significantly lower IC50 values than the nontargeted control Dox-pAcF-ELPBC particles, confirming the therapeutic utility of the targeting domain in an in vitro setting (Figure 5C). We further analyzed the data by normalizing the IC50 values of the targeted Dox-pAcF-ELPBC-EgA1 to that of free Dox, as it illustrates the effect of targeting on treatment efficacy (Figure 5D). Importantly, in the SKOV-3 line with lower Dox sensitivity, the presence of the targeting domain drives the IC50 to be lower than that of free drug (normalized value 0.84), strongly highlighting the ability of this targeting domain to enhance the cytotoxicity of Dox. Because the A431 line is already highly sensitive to free drug, it is therefore difficult to affect the cytotoxic threshold, and we observe a modest increase in IC50 of the targeted therapy as compared to free Dox. Nevertheless, in both cell lines, the decrease in IC50 over an order of magnitude with the targeted as compared to nontargeted control indicates a significant widening of the therapeutic window. This attribute of the targeted construct, along with the potential benefits for improved pharmacokinetics and tumor accumulation3 by delivering a drug in a nanoparticle formulation in vivo is likely to far outweigh the observed higher in vitro cytotoxicity of free drug. The fusions without attached Dox exhibited no cytotoxicity, indicating that neither EgA1 nor the ELP contributed to cytotoxicity observed for the Dox-conjugated constructs (Figure S12).
To investigate the subcellular trafficking of these particles after uptake, we imaged the Dox-loaded particles using spinning-disk confocal microscopy in the A431 cell line. We incubated adherent A431 cells with Dox-pAcF-ELPBC-EgA1 and Dox-pAcF-ELPBC for 4 h (Figure 5E, Figure S13A,B) and 24 h (Figure 5F, Figure S13C,D). We then imaged colocalization of Dox with endolysosomes by staining cells with a lysosomal tracking dye, CytoPainter LysoDeep Red, that selectively accumulates and fluoresces in the acidic late endosomal and lysosomal compartments (pH 4.5–4.8). In cells stained with Cytopainter, we found significantly greater intracellular accumulation of Dox delivered by Dox-pAcF-ELPBC-EgA1 compared to Dox-pAcF-ELPBC. We also found colocalization of Dox and Cytopainter at both 4 and 24 h of incubation, with the effect being more pronounced at 24 h. We evaluated the degree of colocalization with Pearson’s R coefficient. This metric is a measure of the linear correlation between two variables, herein the fluorescence intensities for Dox and Cytopainter at each pixel location in the confocal images. R = 1.0 indicates a perfect linear relationship and hence exact colocalization of the drug with endolyosomes, and R = −1.0 indicates a negative linear correlation between the two variables, and in this context indicates a complete lack of colocalization of Dox with endolysosomes (Figure S13E).65,66 The Dox-pAcF-ELPBC-EgA1 samples increased in endolysosomal colocalization from R = 0.66 to R = 0.76 from 4 to 24 h incubation. Conversely, the nontargeted control Dox-pAcF-ELPBC had R < 0.5 at both time points, indicating little colocalization and suggesting a longer time required for uptake of these particles, which results in the lower efficacy observed in the in vitro cytotoxicity studies.
We also confirmed the intensity of lysosomal dye was similar between all four regions of interest analyzed to ensure that the observed differences in colocalization were not due to a different number of, or intensity of, lysosomes analyzed in the different images (Figure S13F). These colocalization analyses revealed both (1) greater levels of and (2) a time-dependent increase in endolysosomal colocalization with Dox-pAcF-ELPBC-EgA1 as compared to the nontargeted Dox-pAcF-ELPBC. This demonstrates that the receptor-mediated endocytosis of the EgA1-containing construct concurrently promotes both more rapid entry into the cell and pH-mediated drug release. Taken together, these in vitro experiments validate that the active targeting of Dox-pAcF-ELPBC-EgA1 directly translates into the exceptional tumor cytotoxicity of this construct.
The platform we have developed combines a bioorthogonal reactive group with high-affinity targeting in a single protein-based particle and has three distinct advantages over existing approaches: (1) our biopolymer vehicle ensures monodisperse, nontoxic nanoparticles with starting materials that are easily produced recombinantly by overexpression in E. coli and purified at high yield via its LCST phase behavior; (2) the location of drug-loading is precisely specified, and the orthogonal UAA-mediated drug conjugation chemistry permits the inclusion of the cysteine-containing targeting domain into the construct; (3) the EgA1 nanobody delivers the targeted drug-loaded nanoparticles to EGFR-positive cancer cells via receptor-mediated endocytosis.67,68 While previous approaches to cancer nanotherapeutics have directed targeting of nanoparticles with scaffold proteins,69,70 utilized UAAs for biorthogonal drug attachment,71–73 and formulated biopolymer nanoparticles as small molecule drug carriers,74,75 our platform is the first to combine these three elements into one powerful therapeutic agent.
In conclusion, the platform described herein reveals a promising strategy for simultaneously genetically encoding both a drug conjugation site and structured bioactive domain into a biopolymer-based nanoparticle, an approach previously inaccessible with existing techniques. The use of an unnatural amino acid as the site of drug attachment allows for the incorporation of a nanobody as the targeting element, which requires a disulfide bond for its stability. Our platform maintains site-specificity of drug attachment against the complex chemical background of proteins that have a distribution of reactive amino acids. This study is the first example of using molecular engineering to decouple the drug reactivity from any polypeptide or protein of interest; our highly tunable system can be modified and expanded upon to incorporate a variety of cytotoxic drugs or active targeting domains. We anticipate future work will validate the efficacy of these carriers in a range of tumor types in vivo to elucidate the full therapeutic potential of our multifunctional targeted, drug-loaded biopolymer nanoparticles.
Supplementary Material
Acknowledgments
Funding
This work was supported by NIH R01 EB00188 to A.C.
ABBREVIATIONS
- ELP
elastin-like polypeptide
- ELPBC
ELP diblock copolymer
- UAA
unnatural amino acid
- pAcF
p-acetylphenylalanine
- EGFR
epidermal growth factor receptor
- Dox
Doxorubicin
- CMT
critical micellization temperature
- CMC
critical micellization concentration
- MALDI-TOF-MS
matrix assisted laser desorption ionization-time-of-flight mass spectroscopy
- DLS
dynamic light scattering
- SLS
static light scattering
- SEC
size exclusion chromatography
- cryoTEM
cryogenic transmission electron microscopy
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Cho K; Wang X; Nie S; Chen ZG; Shin DM Clin. Cancer Res 2008, 14 (5), 1310–6. [DOI] [PubMed] [Google Scholar]
- (2).Yoo J-W; Chambers E; Mitragotri S Curr. Pharm. Des 2010, 16 (21), 2298–2307. [DOI] [PubMed] [Google Scholar]
- (3).Blanco E; Shen H; Ferrari M Nat. Biotechnol 2015, 33 (9), 941–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bazak R; Houri M; El Achy S; Kamel S; Refaat TJ Cancer Res. Clin. Oncol 2015, 141 (5), 769–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Bertrand N; Wu J; Xu X; Kamaly N; Farokhzad OC Adv. Drug Delivery Rev 2014, 66, 2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hao Y; Zheng C; Wang L; Hu Y; Guo H; Song Q; Zhang H; Zhang Z; Zhang YJ Mater. Chem. B 2017, 5 (11), 2133–2144. [DOI] [PubMed] [Google Scholar]
- (7).Werengowska-Ciećwierz K; Wiśniewski M; Terzyk AP; Furmaniak S Adv. Condens. Matter Phys 2015, 2015, 1–27. [DOI] [PubMed] [Google Scholar]
- (8).Matsumura Y; Maeda H Cancer Res 1986, 46 (12), 6387–6392. [PubMed] [Google Scholar]
- (9).Dreher MR; Simnick AJ; Fischer K; Smith RJ; Patel A; Schmidt M; Chilkoti AJ Am. Chem. Soc 2008, 130 (2), 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Torchilin VP; Lukyanov AN Drug Discovery Today 2003, 8 (6), 259–266. [DOI] [PubMed] [Google Scholar]
- (11).Meyer DE; Chilkoti A Biomacromolecules 2002, 3 (2), 357–367. [DOI] [PubMed] [Google Scholar]
- (12).Navon Y; Bitton R Isr. J. Chem 2016, 56 (8), 581–589. [Google Scholar]
- (13).Nettles DL; Chilkoti A; Setton LA Adv. Drug Delivery Rev 2010, 62 (15), 1479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bhattacharyya J; Bellucci JJ; Weitzhandler I; McDaniel JR; Spasojevic I; Li X; Lin CC; Chi JT; Chilkoti A Nat. Commun 2015, 6, 7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu W; Dreher MR; Furgeson DY; Peixoto KV; Yuan H; Zalutsky MR; Chilkoti AJ Controlled Release 2006, 116 (2), 170–8. [DOI] [PubMed] [Google Scholar]
- (16).Mastria EM; Chen M; McDaniel JR; Li X; Hyun J; Dewhirst MW; Chilkoti AJ Controlled Release 2015, 208, 52–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Urry DW; Hugel T; Seitz M; Gaub HE; Sheiba L; Dea J; Xu J; Parker T Philos. Trans. R. Soc., B 2002, 357 (1418), 169–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chilkoti A; Christensen T; MacKay JA Curr. Opin. Chem. Biol 2006, 10 (6), 652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).MacEwan SR; Weitzhandler I; Hoffmann I; Genzer J; Gradzielski M; Chilkoti A Biomacromolecules 2017, 18 (2), 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Trabbic-Carlson K; Meyer DE; Liu L; Piervincenzi R; Nath N; LaBean T; Chilkoti A Protein Eng., Des. Sel 2004, 17 (1), 57–66. [DOI] [PubMed] [Google Scholar]
- (21).Callahan DJ; Liu W; Li X; Dreher MR; Hassouneh W; Kim M; Marszalek P; Chilkoti A Nano Lett 2012, 12 (4), 2165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).MacKay JA; Chen M; McDaniel JR; Liu W; Simnick AJ; Chilkoti A Nat. Mater 2009, 8 (12), 993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).McDaniel JR; MacEwan SR; Li X; Radford DC; Landon CD; Dewhirst M; Chilkoti A Nano Lett 2014, 14 (5), 2890–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Weitzhandler I; Dzuricky M; Hoffmann I; Garcia Quiroz F; Gradzielski M; Chilkoti A Biomacromolecules 2017, 18 (8), 2419–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).McDaniel JR; Macewan SR; Dewhirst M; Chilkoti AJ Controlled Release 2012, 159 (3), 362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang J; Dzuricky M; Chilkoti A Nano Lett 2017, 17 (10), 5995–6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Raphel J; Parisi-Amon A; Heilshorn SJ Mater. Chem 2012, 22 (37), 19429–19437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lim DW; Nettles DL; Setton LA; Chilkoti A Biomacromolecules 2007, 8 (5), 1463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Boohaker RJ; Lee MW; Vishnubhotla P; Perez JM; Khaled AR Curr. Med. Chem 2012, 19 (22), 3794–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shehu A; Kavraki LE; Clementi C Protein Sci 2008, 17 (3), 482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang L; Zhang Z; Brock A; Schultz PG Proc. Natl. Acad. Sci. U. S. A 2003, 100 (1), 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu CC; Schultz PG Annu. Rev. Biochem 2010, 79, 413–44. [DOI] [PubMed] [Google Scholar]
- (33).Isaacs FJ; Carr PA; Wang HH; Lajoie MJ; Sterling B; Kraal L; Tolonen AC; Gianoulis TA; Goodman DB; Reppas NB; Emig CJ; Bang D; Hwang SJ; Jewett MC; Jacobson JM; Church GM Science 2011, 333 (6040), 348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lajoie MJ; Rovner AJ; Goodman DB; Aerni HR; Haimovich AD; Kuznetsov G; Mercer JA; Wang HH; Carr PA; Mosberg JA; Rohland N; Schultz PG; Jacobson JM; Rinehart J; Church GM; Isaacs FJ Science 2013, 342 (6156), 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Amiram M; Haimovich AD; Fan C; Wang Y-S; Aerni H-R; Ntai I; Moonan DW; Ma NJ; Rovner AJ; Hong SH; Kelleher NL; Goodman AL; Jewett MC; Soll D; Rinehart J; Isaacs FJ Nat. Biotechnol 2015, 33 (12), 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Muyldermans S Annu. Rev. Biochem 2013, 82, 775–97. [DOI] [PubMed] [Google Scholar]
- (37).Roovers RC; Laeremans T; Huang L; De Taeye S; Verkleij AJ; Revets H; de Haard HJ; van Bergen en Henegouwen PMP Cancer Immunol. Immunother 2007, 56 (3), 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Hofman EG; Ruonala MO; Bader AN; van den Heuvel D; Voortman J; Roovers RC; Verkleij AJ; Gerritsen HC; van Bergen en Henegouwen PMP J. Cell Sci 2008, 121 (15), 2519–2528. [DOI] [PubMed] [Google Scholar]
- (39).Yewale C; Baradia D; Vhora I; Patil S; Misra A Biomaterials 2013, 34 (34), 8690–707. [DOI] [PubMed] [Google Scholar]
- (40).Cerami E; Gao J; Dogrusoz U; Gross BE; Sumer SO; Aksoy BA; Jacobsen A; Byrne CJ; Heuer ML; Larsson E; Antipin Y; Reva B; Goldberg AP; Sander C; Schultz N Cancer Discovery 2012, 2 (5), 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Gao J; Aksoy BA; Dogrusoz U; Dresdner G; Gross B; Sumer SO; Sun Y; Jacobsen A; Sinha R; Larsson E; Cerami E; Sander C; Schultz N Sci. Signaling 2013, 6 (269), pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Rivankar SJ Cancer Res. Ther 2014, 10 (4), 853–8. [DOI] [PubMed] [Google Scholar]
- (43).Dreher MR; Raucher D; Balu N; Michael Colvin O; Ludeman SM; Chilkoti AJ Controlled Release 2003, 91 (1–2), 31–43. [DOI] [PubMed] [Google Scholar]
- (44).Furgeson DY; Dreher MR; Chilkoti AJ Controlled Release 2006, 110 (2), 362–9. [DOI] [PubMed] [Google Scholar]
- (45).Tacar O; Sriamornsak P; Dass CR J. Pharm. Pharmacol. 2013, 65 (2), 157–70. [DOI] [PubMed] [Google Scholar]
- (46).Bi D; Zhao L; Yu R; Li H; Guo Y; Wang X; Han M Drug Delivery 2018, 25 (1), 564–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Jin Y; Song L; Su Y; Zhu L; Pang Y; Qiu F; Tong G; Yan D; Zhu B; Zhu X Biomacromolecules 2011, 12 (10), 3460–8. [DOI] [PubMed] [Google Scholar]
- (48).Kalia J; Raines RT Angew. Chem., Int. Ed 2008, 47 (39), 7523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Zarschler K; Witecy S; Kapplusch F; Foerster C; Stephan H Microb. Cell Fact 2013, 12, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Meyer DE; Chilkoti A Protein purification by inverse transition cycling. In Protein Interactions; Cold Spring Harbor Laboratory Press, 2002; pp 329–343. [Google Scholar]
- (51).Meyer DE; Chilkoti A Nat. Biotechnol 1999, 17 (11), 1112–1115. [DOI] [PubMed] [Google Scholar]
- (52).Pirman NL; Barber KW; Aerni HR; Ma NJ; Haimovich AD; Rogulina S; Isaacs FJ; Rinehart J Nat. Commun 2015, 6, 8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).O’Donoghue P; Prat L; Heinemann IU; Ling J; Odoi K; Liu WR; Soll D FEBS Lett 2012, 586 (21), 3931–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Aerni HR; Shifman MA; Rogulina S; O’Donoghue P; Rinehart J Nucleic Acids Res 2015, 43 (2), No. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pedersen MW; Tkach V; Pedersen N; Berezin V; Poulsen HS Int. J. Cancer 2004, 108, 643–653. [DOI] [PubMed] [Google Scholar]
- (56).Kibria G; Hatakeyama H; Akiyama K; Hida K; Harashima H Biol. Pharm. Bull 2014, 37 (12), 1926. [DOI] [PubMed] [Google Scholar]
- (57).Yang W; Soares J; Greninger P; Edelman EJ; Lightfoot H; Forbes S; Bindal N; Beare D; Smith JA; Thompson IR; Ramaswamy S; Futreal PA; Haber DA; Stratton MR; Benes C; McDermott U; Garnett MJ Nucleic Acids Res 2012, 41 (Database issue), D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Dirksen A; Hackeng TM; Dawson PE Angew. Chem., Int. Ed 2006, 45 (45), 7581–4. [DOI] [PubMed] [Google Scholar]
- (59).Wittrup KD; Thurber GM; Schmidt MM; Rhoden JJ Methods Enzymol 2012, 503, 255–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wang K; Li D; Sun L OncoTargets Ther 2016, 9, 377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhang F; Wang S; Yin L; Yang Y; Guan Y; Wang W; Xu H; Tao N Anal. Chem 2015, 87 (19), 9960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ponten F; Jirstrom K; Uhlen MJ Pathol 2008, 216 (4), 387–93. [DOI] [PubMed] [Google Scholar]
- (63).Algars A; Avoranta T; Osterlund P; Lintunen M; Sundstrom J; Jokilehto T; Ristimaki A; Ristamaki R; Carpen O PLoS One 2014, 9 (6), No. e99590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bozic I; Reiter JG; Allen B; Antal T; Chatterjee K; Shah P; Moon YS; Yaqubie A; Kelly N; Le DT; Lipson EJ; Chapman PB; Diaz LA Jr.; Vogelstein B; Nowak MA eLife 2013, 2, No. e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Adler J; Parmryd I Cytometry, Part A 2010, 77 (8), 733–42. [DOI] [PubMed] [Google Scholar]
- (66).Bolte S; Cordelieres FP J. Microsc 2006, 224 (3), 213–32. [DOI] [PubMed] [Google Scholar]
- (67).Oliveira S; Schiffelers RM; van der Veeken J; van der Meel R; Vongpromek R; van Bergen En Henegouwen PM; Storm G; Roovers RC J. Controlled Release 2010, 145 (2), 165–75. [DOI] [PubMed] [Google Scholar]
- (68).Bareford L; Swaan P Adv. Drug Delivery Rev 2007, 59 (8), 748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wang M; Thanou M Pharmacol. Res 2010, 62 (2), 90–99. [DOI] [PubMed] [Google Scholar]
- (70).Friedman A; Claypool S; Liu R Curr. Pharm. Des 2013, 19 (35), 6315–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Kularatne SA; Deshmukh V; Ma J; Tardif V; Lim RK; Pugh HM; Sun Y; Manibusan A; Sellers AJ; Barnett RS; Srinagesh S; Forsyth JS; Hassenpflug W; Tian F; Javahishvili T; Felding-Habermann B; Lawson BR; Kazane SA; Schultz PG Angew. Chem., Int. Ed 2014, 53 (44), 11863–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Hutchins BM; Kazane SA; Staflin K; Forsyth JS; Felding-Habermann B; Schultz PG; Smider VV J. Mol. Biol 2011, 406 (4), 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Oller-Salvia B; Kym G; Chin JW Angew. Chem., Int. Ed 2018, 57 (11), 2831–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Nitta S; Numata K Int. J. Mol. Sci 2013, 14 (1), 1629–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Hudson D; Margaritis A Crit. Rev. Biotechnol 2014, 34 (2), 161–179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.