

Introduction
Pediatric drug-drug interactions (DDIs) can be life threatening, and a plan for assessing the interaction potential should be part of every pediatric drug development program. However, DDI studies are rarely performed in the pediatric population for ethical and practical reasons. Consequently, there is a paucity of information available regarding pediatric DDIs. This article will propose strategies for evaluating pharmacokinetic/pharmacodynamic DDIs throughout pediatric drug development and clinical use.
THE IMPORTANCE OF EVALUATING PEDIATRIC DDIs
Pediatric Patients Are at Risk for DDIs
Children are exposed to multiple medications throughout hospitalization which increases their likelihood of experiencing DDIs. A retrospective cohort study using the Pediatric Health Information System database reported that out of 54,549 admissions to the pediatric intensive care unit, children were exposed on average, to 10 distinct medications daily and to 20 medications cumulatively during hospitalization.1 The most common medications in this study included acetaminophen, fentanyl, midazolam, ranitidine, heparin, morphine, potassium chloride, furosemide, lidocaine, and epinephrine.1 In another retrospective cohort study using the Pediatric Health Information System database, approximately half of hospitalized children were associated with a potential DDI based upon 498,956 pediatric hospitalizations within 42 United States Children’s Hospitals.2 Additionally, 41% of these potential DDIs were considered “major” based on the Micromedex® DRUG-REAX® classification system.2 Among the most common major DDIs was fentanyl in combination with morphine or midazolam, with the potential additive effects on respiratory depression.2 These studies affirm the need to determine DDI potential and clinical management of DDIs in the pediatric population.
Adult DDI Data Cannot Simply Be Extrapolated to Pediatric Populations
Pharmacokinetic (PK) DDIs are evaluated in healthy adult volunteers during drug development, are communicated in the label, and are monitored through post-marketing surveillance following drug approval. However, limited studies have characterized PK alterations due to DDIs in pediatric patients, particularly infants where blood sampling and recruitment are most challenging.3 Per 21 CFR 50 subpart D regarding clinical investigations in pediatric subjects which are associated with a more than minimal risk, children cannot be enrolled in clinical trials unless there is an anticipated benefit for the research subjects.4 Consequently, a DDI assessment would have to be conducted in pediatric patients requiring the drug combination as part of therapy.
As a result, recommendations from adult DDI studies are often extrapolated to the pediatric population to guide clinical management of pediatric DDIs. However, extrapolating adult DDI data to pediatric patients can under or over predict the magnitude of DDIs. In a systematic literature review, fold interactions were compared between 31 pediatric studies and 33 adult studies for 24 drug pairs using clearance, steady state plasma concentrations, or area under the concentration versus time curve (AUC).3 Fold interactions were higher (>1.25-fold) or lower (<0.8-fold) in pediatric patients compared to adults for 15 and 8, respectively, out of these 33 cases.3 By example, digoxin plus amiodarone and lamotrigine plus valproate resulted in a 2.18 fold higher and 0.58 fold lower exposure, respectively, in pediatrics compared to adults.3 Although these results may be impacted by the study design used, variability in pharmacokinetics, and the specific pediatric age groups evaluated, this systematic review highlighted that differences beween adult and pediatric DDI potential may exist and that additional studies are needed. Furthermore, a physiologically-based pharmacokinetic (PBPK) model demonstrated that the magnitude of metabolic DDIs depended on the ontogeny profiles of the relevant drug metabolizing enzymes as well as the fractional elimination pathway of the drug being inhibited.5
Additional variables that can influence DDI potential in pediatric patients relative to adults may also be important. For example, other physiological changes besides ontogeny of drug metabolizing enzymes may need to be considered, including changes in intragastric pH, gastric emptying, intestinal motility, protein binding, and transporter ontogeny. Differences in diet, drug combinations, formulations, and dosing may also play a role in dictating pediatric DDI potential. Additionally, the exposure-response relationship may differ between adults and pediatric patients because of altered expression and function of proteins mediating drug effect, as well as age related physiological factors including changes in CD4 T-cell count with age.6 In fact, the majority of potential DDIs identified retrospectively in hospitalized pediatric patients were associated with pharmacodynamic interactions such as additive respiratory depression and gastrointestinal toxicity.2 Moreover, age-related changes in disease progression and safety can occur due to organ development and altered tissue distribution.3
OPPORTUNITIES FOR EVALUATING PEDIATRIC DDIs
Modeling and Simulation May Predict and Evaluate Pediatric DDIs
Quantitative approaches, such as population PK (PopPK) and PBPK modeling and simulation, can be used to evaluate pediatric DDIs during drug development. PopPK modeling combined with sparse sampling is widely used to characterize drug disposition in neonates and children.7 Implementing allometric scaling for size in addition to a sigmoidal function accounting for organ maturation may be able to distinguish age and size effects on clearance from other patient specific factors including DDIs.8 If a concomitant drug is identified as a significant predictor of variability in a PK parameter, simulations can be performed to optimize dosing for children receiving the drug combination. PBPK modeling, which integrates physiological information along with drug-specific properties to predict drug-disposition throughout the body, can facilitate the evaluation of potential pediatric DDIs. A major advantage is that PBPK modeling can predict a-priori exposure and can be extrapolated to different populations and pediatric age groups. However, there are some gaps in knowledge regarding age related changes in drug absorption, distribution, excretion, and ontogeny of metabolizing enzymes and transporters which still require further exploration. Although PBPK models are widely used during drug development for providing therapeutic management in adults experiencing DDIs, this strategy has infrequently been applied to the pediatric population. Additional opportunities exist for leveraging PBPK modeling to account for age related changes and to predict pediatric DDI potential during drug development (Figure 1).
Figure 1: Application of physiologically-based pharmacokinetic (PBPK) modeling and simulation to predict drug-drug interaction (DDI) potential in pediatric patients.

Adult PBPK models can be developed incorporating drug-specific, system-specific, and study protocol and formulation properties, and then evaluated and further refined using adult clinical data. Next, DDI potential can be evaluated by incorporating in-vitro induction or inhibition parameters and then further refined using adult DDI data. Adult PBPK models can be scaled to pediatric patients including anthropomorphic and ontogeny functions, and then model performance and scaling can be evaluated using available pediatric data. Next, DDIs can be simulated in pediatric patients in order to provide therapeutic recommendations across pediatric ages likely to receive the drug. Finally, dosing recommendations can be evaluated using opportunistic pharmacokinetic data or using prospectively captured data from an adaptive trial. During adapative trials, efficacy and safety of the dosages and drug combinations can be monitored in pediatric patients throughout the trial at pre-specified times and the dosing regimen can be modified according to these interim study results.
Adaptive Trials May Mitigate the Risks of Performing Prospective Pediatric DDI Studies
The risks associated with prospectively evaluating pediatric DDIs may be mitigated using adaptive trials, in which modifications to the trial or statistical procedure are modified at pre-specified times without diminishing the validity of the study. One study in healthy adults used an adaptive 2-cohort strategy to mitigate tolerability concerns associated with evaluating the DDI potential between GSK239512 and the strong cytochrome P450 3A inhibitor ketoconazole.9 PBPK modeling predicted a 4-fold increase in GSK239512 exposure after coadministration with ketoconazole, which informed the dose selected for subjects in cohort 1.9 The safety and PK data from cohort 1 justified providing a higher dose of GSK239512 for subjects in cohort 2 receiving the drug combination.9 A similar approach can be applied for pediatric patients where dose adjustments are first based upon scaling an adult PBPK model to pediatric populations, followed by prospectively evaluating the drug combination in pediatric patients with safety monitored throughout the trial at pre-specified times.
Opportunistic Clinical Data May Facilitate Pediatric DDI Evaluation
Some of the challenges associated with evaluating pediatric DDIs as post-marketing requirements or for marketed drugs may be overcome by leveraging opportunistic data, which are data collected in pediatric patients reciving medications per standard of care. Drug concentration measurements from remaining blood samples collected at times of routine laboratory draws can be used for PopPK and PBPK model development and evaluation. Opportunistic clinical data that are documented in the patient chart, such as vital signs (e.g., heart rate, blood pressure), laboratory values (e.g., serum creatinine, liver transaminases) and clinical symptoms (e.g., sedation, pain), can be used for pharmacodynamic assessment. Consent rates are likely higher because of the minimal risk that these studies pose to pediatric patients. However, limitations with opportunistic data include the random nature of sample collection and measurement of drug responses, as well as the fact that many confounding variables may affect the interpretation of the patient data (e.g., organ dysfunction, other concomitant medications, and comorbidities). As a result, use of opportunistic data would likely require collecting data from a sufficiently large sample size while also controlling for confounding factors.
Leveraging “Real World” Data for Pediatric DDI Evaluation Can Overcome Ethical Barriers
Although several retrospective cohort studies have reported potential DDIs in children, limitations in the electronic health record (EHR) database precluded determination of clinical outcomes associated with these potential DDIs.2 Consequently, opportunities exist to relate potential DDIs in children with clinical outcomes using EHR data. In addition, PK DDIs could be prospectively assessed for drugs undergoing therapeutic drug monitoring, such as gentamicin, vancomycin, phenobarbital, carbamazepine, phenytoin, tacrolimus, cyclosporine, and caffeine. Recently, a pediatric PopPK model was developed for posaconazole utilizing therapeutic drug monitoring data which was able to detect a 42% reduction in bioavailability in the presence of proton pump inhibitors.10 Additionally, pharmacodynamic analyses utilizing clinical or laboratory adverse events can be performed with or without PK data to assess pediatric DDIs. Controlling for confounding variables will likely be required given the retrospective study design and complexity of the patient population. Additional limitations include suboptimal PK sampling times (e.g., only troughs or peaks may be measured) plus laboratory or adverse events are restricted to those that are measured and recorded in the EHR. However, advantages of this approach are that institutional review boards generally consider these studies to be exempt or have expedited approval, and information from a large pediatric population can be evaluated.
CONCLUSIONS
Limited pediatric DDI data are currently available, and virtually no PK DDI studies have been conducted in neonates and infants. However, considering DDI potential should be part of every pediatric drug development program. This is particularly true when physiological changes, such as the ontogeny of enzymes and transporters, are expected to affect DDI potential in pediatric patients receiving or likely to receive multiple drugs concurrently. Prior to drug approval, PBPK modeling can investigate pediatric DDI potential and inform dose adjustments that can be evaluated prospectively in pediatric studies where multiple drugs are administered together. When pediatric PK studies are performed and pediatric DDI data are available, the impact of DDIs can be evaluated for coadministered drugs using PopPK modeling. After drug approval, pediatric DDIs should continue to be monitored when appropriate, and opportunistic and EHR data can be particulary useful during post-marketing surveillance (Figure 2).
Figure 2: Approaches to evaluate pediatric pharmacokinetic (PK) drug-drug interactions (DDIs) throughout pediatric drug development and post-marketing.
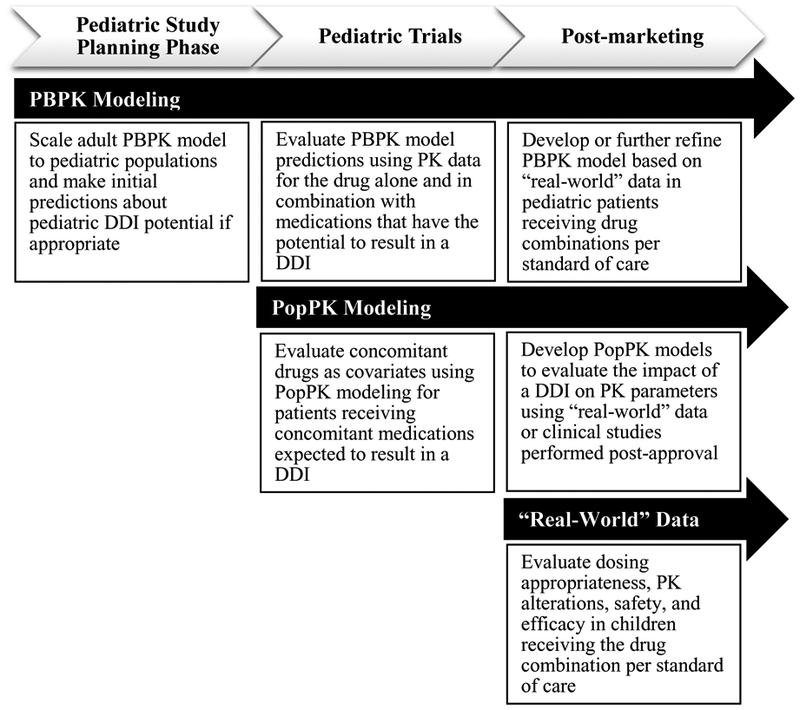
Prior to pediatric drug approval, physiologically-based pharmacokinetic (PBPK) modeling and simulation can investigate pediatric DDI potential and inform dose adjustments that can be evaluated through prospective pediatric trials, such as an adaptive trial. PBPK models can also be developed or further refined using “real-world” pediatric DDI data for marketed drugs. Once pediatric PK and coadministered drug data are available, PopPK models can be developed and concomitant drugs can be evaluated as predictors of inter-individual variability in PK parameters. Dosing simulations can then be performed based on the final population PK (PopPK) model to optimize dosing for children receiving the drug combination of interest. After drug approval, studies leveraging “real-world” data can be performed to evaluate dosing appropriateness, PK alterations, safety, and efficacy in children receiving the drug combination of interest per standard of care.
Supplementary Material
Additional references
Acknowledgments
Funding:
S.N.S. receives financial support from the National Institutes of General Medical Sciences (NIGMS) under award T32GM086330. D.G. receives support for research from National Institute of Child Health and Human Development (NICHD) under award K23HD083465.
Footnotes
Conflict of Interest:
As an Associate Editor for Clinical Pharmacology & Therapeutics, Shiew-Mei Huang was not involved in the review or decision process for this paper.
Disclaimer:
The opinions expressed in this manuscript are those of the authors and should not be interpreted as the position of the U.S. Food and Drug Administration or the National Institutes of Health.
References:
- 1.Dai D, Feinstein JA, Morrison W, Zuppa AF & Feudtner C Epidemiology of Polypharmacy and Potential Drug–Drug Interactions Among Pediatric Patients in ICUs of U.S. Children’s Hospitals. Pediatr. Crit. Care Med 17, e218–e228 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feinstein J, Dai D, Zhong W, Freedman J & Feudtner C Potential Drug−Drug Interactions in Infant, Child, and Adolescent Patients in Children’s Hospitals. Pediatrics 135, 3–12 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Salem F, Rostami-Hodjegan A & Johnson TN Do Children Have the Same Vulnerability to Metabolic Drug-Drug Interactions as Adults? A Critical Analysis of the Literature. J. Clin. Pharmacol 53, 559–566 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Protection of Human Subjects. Additional Safeguards for Children in Clinical Investigations. 21 C.F.R. § 50.52 (2017).
- 5.Salem F, Johnson TN, Barter ZE, Leeder JS & Rostami-Hodjegan A Age Related Changes in Fractional Elimination Pathways for Drugs: Assessing the Impact of Variable Ontogeny on Metabolic Drug-Drug Interactions. J. Clin. Pharmacol 53, 857–865 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Hoare RL, Veys P, Klein N, Callard R & Standing JF Predicting CD4 T-Cell Reconstitution Following Pediatric Hematopoietic Stem Cell Transplantation. Clin. Pharmacol. Ther 102, 349–357 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Standing JF et al. Dosing of Ceftriaxone and Metronidazole for Children With Severe Acute Malnutrition. Clin. Pharmacol. Ther (2018). March 25 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Germovsek E, Barker CIS, Sharland M & Standing JF Pharmacokinetic–Pharmacodynamic Modeling in Pediatric Drug Development, and the Importance of Standardized Scaling of Clearance. Clin. Pharmacokinet (2018). April 19 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J et al. An Adaptive Design to Investigate the Effect of Ketoconazole on Pharmacokinetics of GSK239512 in Healthy Male Volunteers. J. Clin. Pharmacol 55, 505–511 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Boonsathorn S et al. Clinical Pharmacokinetics and Dose Recommendations for Posaconazole in Infants and Children. Clin. Pharmacokinet (2018). April 20 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional references