

Abstract
SETD2, a histone H3 lysine trimethyltransferase, is frequently inactivated and associated with recurrence of clear cell renal cell carcinoma (ccRCC). However, the impact of SETD2 loss on metabolic alterations in ccRCC is still unclear. In this study, SETD2 null isogenic 38E/38F clones derived from 786-O cells were generated by zinc finger nucleases, and subsequent metabolic, genomic, and cellular phenotypic changes were analyzed by targeted metabolomics, RNA-sequencing, and biological methods, respectively. Our results showed that, compared to parental 786-O cells, 38E/38F cells had elevated levels of MTT/Alamar blue levels, ATP, glycolytic/mitochondrial respiratory capacity, citrate synthase (CS) activity, and TCA metabolites such as aspartate, malate, succinate, fumarate, and α-ketoglutarate. The 38E/38F cells also utilized alternative sources beyond pyruvate to generate acetyl-CoA for the TCA cycle. Moreover, 38E/38F cells showed disturbed gene networks mainly related to mitochondrial metabolism and the oxidation of fatty acids and glucose, which was associated with increased PGC1α, mitochondrial mass, and cellular size/complexity. Our results indicate that SETD2 deficiency induces a metabolic switch toward enhanced oxidative phosphorylation in ccRCC, which can be related to PGC1α-mediated metabolic networks. Therefore, this current study lays the foundation for the further development of a global metabolic analysis of cancer cells in individual patients, which ultimately will have significant potential for the discovery of novel therapeutics and precision medicine in SETD2 inactivated ccRCC.
Keywords: clear cell renal cell carcinoma (ccRCC), SETD2, mitochondria, oxidative phosphorylation, PGC1α
Graphical Abstract
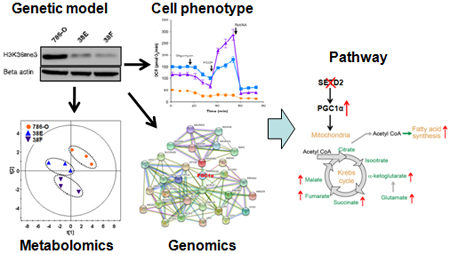
Introduction
Kidney and renal pelvis cancer is the 10th most common cancer in the United States, with an estimated 63,340 new cases and 14,970 associated deaths in 2018.1 Renal cell carcinoma (RCC) is the predominant type, accounting for up to 85% of all kidney cancers. Of those RCC patients, 70–75% are sub-typed as clear cell RCC (ccRCC).2 Although surgery remains the standard of care for patients with early-stage ccRCC (stages I-III), roughly 30% of these patients will progress to distant metastases following resection of localized tumors. Despite recent advances in systemic therapy, median survival drops to ~2 years after development of metastatic disease.3
A common molecular feature of ccRCC is the inactivation of the von Hippel-Lindau (VHL) tumor suppressor protein through a variety of mechanisms.2 VHL, an E3 ubiquitin ligase, regulates the cellular response to hypoxia through its interaction with hypoxia inducible factor-1 (HIF-1). Under normoxic conditions, VHL mediates ubiquitylation and proteasomal degradation of HIF-1 via interaction with the oxygen-dependent degradation domain core of HIF-1.4 The inactivation of VHL in ccRCC cell leads to elevated activity of HIF-1 and a number of downstream genes essential for tumor metabolism and angiogenesis, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and GLUT1.2, 4 However, the loss of VHL has been shown to be insufficient for tumorigenesis in mice, 5 suggesting that other factors are also involved in ccRCC onset and progression.
In addition to functional alterations of VHL, several genes regulating chromatin remodeling in close genomic proximity to VHL are frequently mutated in RCC, including those of the SWI/SNF chromatin remodeling complex (PBRM1 and BAP-1) and histone lysine 36 trimethylase SETD2.6, 7 SETD2, the histone methyltransferase responsible for the trimethylation of H3K36, is inactivated in approximately 10–32% of ccRCC cases.6, 8 SETD2 promotes transcriptional elongation and plays important roles in double-stranded break repair, DNA methylation, and RNA splicing.9, 10 In ccRCC, the homozygous loss of SETD2 activity leads to a global decline of H3K36me3 histone markers and is associated with reduced survival and increased risk of recurrence.10 Moreover, metastatic ccRCC exhibits markedly reduced H3K36me3 compared to matched primary ccRCCs.11 These reports strongly suggest that SETD2 mutations drive the ccRCC progression, but the specific mechanism remains unclear.
Increasingly, kidney cancer is being recognized as a metabolic disease, and many mutated genes in RCC, such as VHL, fumarate hydratase (FH) and succinate dehydrogenase (SDH), are involved in cellular respiration and energy metabolism.12 HIF-1-induced genetic reprogramming promotes a classic Warburg phenotype in RCC through VHL or mechanisms dependent on metabolic enzymes.12–14 For instance, loss of VHL in RCC leads to a HIF-1-dependent reprogramming of energy metabolism that includes elevated glucose uptake, glycolysis, and lactate production accompanied by a reciprocal decrease in respiration under aerobic conditions.13 Inactivation of FH, a tricarboxylic acid (TCA) cycle enzyme, in RCC results in glucose-mediated generation of cellular reactive oxygen species (ROS) and ROS-dependent HIF-1 stabilization, which consequently enhances aerobic glycolysis and reduces reliance on mitochondrial respiration in RCC.14 However, the impact of SETD2 inactivation on the metabolic phenotype of RCC is rarely reported and remains unknown.
In this study, we explored metabolic alterations in SETD2 deficient cell lines. Two independent SETD2 null isogenic clones were generated from 786-O cells by zinc finger nucleases, and ensuing changes in metabolism, genes and cellular phenotypes were analyzed using targeted metabolomics, RNA-seq and molecular biological methods, respectively.
Methods and Materials
Cell culture
The 786-O ccRCC cell line (ATCC, Manassas, VA, USA) was cultured in RPMI 1640 medium (Corning, Corning, NY, USA) supplemented with 10% FBS (HyClone, South Logan, UT, USA), 1% penicillin/streptomycin (Gibco, Grand Island, NY, USA), and 2 mM L-glutamine (Gibco, Grand Island, NY, USA). SETD2 null isogenic cell lines including 38E and 38F were generated by the zinc finger nuclease method as previously described,10 and were cultured under identical conditions to those of 786-O cells.
Cell proliferation assay
The proliferation rates of each cell were determined by MTT (Sigma, St. Louis, MO, USA) and Alamar Blue (Invitrogen, Carlsbad, CA, USA). For the MTT test, 100 μL of MTT (1 mg/ml) solution was added into 96-well plates and incubated at 37 °C for 2 h. After removing media, 100 μL of isopropanol/1N HCl (v:v = 24:1) was added into each well to dissolve formazan crystals, and the absorbance at 570 nm was measured by a microplate reader (BioTek Epoch, Winooski, VT, USA). For the Alamar Blue assay, 100 μL of Alamar Blue (10% in medium) was added into 96-well plates and incubated at 37 °C for 2 h. Then, the absorbance of Alamar Blue solution was measured by a microplate reader (BioTek Epoch) at 600 nm.
Measurement of cellular ATP
Cellular ATP levels were measured using the CellTiter-Glo ATP assay kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, 25 μL of CellTiter-Glo reagent was added into each well of a 384-well plate. After mixing contents for 2 min on a shaker to induce cell lysis, the plate was then incubated at room temperature for 10 min. The luminescence was measured by a microplate luminometer (BioTek, VT, USA).
Seahorse assay
Mitochondrial and glycolytic rates were measured by a Seahorse XF96 extracellular flux analyzer (Seahorse Biosciences, Billerica, MA, USA) with Mitochondrial Stress Test (MST) and Glycolysis Stress Test (GST) assay kits. Cells (1.8×104 per well) were plated in quadruplicate, using XF96 extracellular flux assay plates in 200 μL XF base medium equilibrated to a pH of 7.4. After adherence for 2 hours at 37 °C, media were changed to GST or MST buffer. For MST, compounds were added in the following order: oligomycin (1 μM), FCCP (1 μM), and rotenone/antimycin A (0.5 μM). For GST, compounds were added in the following order: glucose (10 mM), oligomycin (1 μM), and 2-DG (50 mM). Values for each measurement were averaged across quadruplicate wells and displayed as oxygen consumption rate (OCR, pmol O2/min) for MST and extracellular acidification rate (ECAR, mpH/min) for GST.
Metabolic enzymatic activity assay
The activity of citrate synthase (CS) was measured by commercial kits (Cayman, Ann Arbor, MI, USA) according to the manufacturer’s instructions. In brief, the absorbance of cell samples was recorded at 412 nm and then normalized to cell mass following the addition of acetyl-coA and oxaloacetate reagents for 10 min.
Targeted metabolomics analysis with GC-MS
Intracellular metabolites were extracted by methanol/H2O (v:v = 80:20) and analyzed using GC-MS. Briefly, extracted samples were incubated with 30 μL O-methylhydroxylamine hydrochloride solution in pyridine (Sigma-Aldrich, St. Louis, MO, USA) at 60 °C for 90 min. Next, 70 μL of N-tert-Butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA, Sigma, St. Louis, MO, USA) was added and placed at 60 °C for 30 min. GC-MS spectral acquisition was performed on an Agilent 7890 GC-5975C MSD system (Agilent Technologies, Inc., Santa Clara, CA, USA). For analysis, 1 μL derivatized sample was injected using the splitless mode, and helium (purity > 99.999%) was used as the carrier gas. The flow rate of carrier gas was 0.5 mL/min. The temperature of injection port and transfer line was 250°C and 290°C, respectively. Chromatographic separation was performed on a Zorbax DB5-MS + 10 m Duragard Capillary Column (30 m x 250 μm x 0.25 μm). The column temperature was maintained at 60 °C for 1 min, then increased at a rate of 10 °C/min to 325 °C and held at this temperature for 10 min. The solvent delay was 9.69 min. Mass spectral signals were recorded in full scan mode using electron ionization (EI, 70 eV), ion source temperature was 230°C, EM voltage was 1225 V, and mass range was 50–600 m/z with a scan frequency of 2.7 scans/second.
The external calibration curves were used for quantification, and different concentrations of external standard solutions (encompassing 3–5 orders of magnitude) were prepared and run together with study samples. While the GC-MS data were collected in the scanning mode, the raw GC-MS data were extracted in a targeted way to monitor 11 compounds of our strong interest (mainly in the TCA cycle), using Agilent MassHunter software (Version B.09.00). The parameters used to extract the data were all developed and optimized using pure standards, and the targeted m/z values were listed in Table S1. The metabolite levels were then further normalized to the protein concentration of each sample.
Measurement of oxygen consumption rates
The oxygen consumption rates of cells were detected by an oxygen electrode assay with 10 mM glucose. The basal oxygen consumption rate, the non-glycolytic rate after addition of sodium fluoride (NaF, pyruvate dehydrogenase phosphatase inhibitor, Sigma), and the ability of cells to oxidize lactate to form pyruvate for mitochondrial respiration were determined.
RNA-Seq analysis
The RNA-Seq analysis was performed as previously described.10 Briefly, the genomic RNA was extracted from cells using the RNease Mini Kit (Qiagen, Hilden, Germany), and residual DNA was removed by DNAse I. DNA libraries were prepared with Illumina’s TruSeq RNA sample preparation V2 kit, and final amplification was performed using TruSeq bar-coded primers. Amplified DNAs (DNA libraries) were sequenced on the Illumina Genome Analyzer II (Illumina, San Diego, CA, USA). We performed analysis of RNA sequencing data sets using cufflinks41 and ChIPRNAseq-PRO42 software packages to generate prediction models of transcript splice isoforms. RNA abundance was predicted by calculating the fragments per kilobase of exon per million fragments mapped (FPKM) of each transcript prediction. Changes in FPKM were converted to log2 values and were visualized using a heatmap. The gene ontology (GO) analysis was performed using STRING software.
Western Blotting
Cells were harvested and lysed in ice-cold radio immunoprecipitation assay (RIPA) buffer supplemented with a protease inhibitor cocktail and phosphatase inhibitors (Sigma, MO, USA). Protein concentrations were determined by BCA kit (Bio-Rad, Hercules, CA, USA). Briefly, 10 μg of protein were loaded on 12% tris-glycine polyacrylamide gel prior to being transferred to a PVDF membrane on ice. After transfer, membranes were blocked with 5% milk in tris-buffered saline with 0.1% Tween-20 (TBST) for 30 min. Membranes were then incubated with primary antibodies: anti-H3K36me3 (1:1000, Active Motif, Carlsbad, CA), anti-CS (1:1000, Cell Signaling, Danvers, MS), and anti-PGC1α (1:1000, Cell Signaling) at 4 °C overnight. After washing with TBST, membranes were incubated with goat anti-rabbit/mouse HRP (1:10,000, Jackson Immunoresearch, West Grove, PA) for 1 h at room temperature. After washing with TBST, membranes were observed by chemiluminescent kit (Thermo Scientific, Carlsbad, CA, USA).
MitoTracker Red staining
Mitochondria were labeled with MitoTracker Red (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Cells were stained with 100 nM MitoTraker Red at 37 °C for 30 min. After washing with PBS, stained cells were observed by confocal microscopy (LSM 510, Zeiss, Germany).
Statistical analysis
All data, expressed as mean ± SEM, were analyzed by one-way ANOVA with Tukey’s post hoc analysis to control for multiple comparisons using SPSS software (Version 22.0, IBM Corporation, USA); the significance threshold (α) was set to 0.05. Principal component analysis (PCA) of metabolites data was performed using SIMCA-P 11.5 software (Umetrics, Umea, Sweden) as previously described.15
Results
Loss of SETD2 increases overall metabolic activity of ccRCC cells
We first performed proliferation assays to measure the growth rates of SETD2 deficient cells. MTT luminescence was significantly higher in 38E and 38F cells at 3 hours after plating (Figure 1A, S1), and this finding was also corroborated by comparable results obtained from the Alamar Blue assay (Figure 1B), suggesting a higher intracellular oxidase activity in SETD2 deficient ccRCC cells. Moreover, we observed higher ATP levels in 38E and 38F cells than those of 786-O cells (Figure 1C). In addition, loss of SETD2 led to a global decline of H3K36me3 (Figure 1D–E), and increased cell size and intercellular complexity as indicated by forward scatter (FSC) and side scatter (SSC) histograms from 38E and 38F cells (Figure S2).
Figure 1.

Loss of SETD2 increases the metabolic rate in ccRCC cells. The cell metabolic rate was measured by (A) MTT (n=3) and (B) Alamar Blue assays (n=3) at 3 hours after cell plating. (C) Cellular ATP production was measured by CellTiter-Glo reagent (n=3). (D-E) Western blot and quantitative analysis for H3K36me3 histone protein (*p<0.05).
Loss of SETD2 enhances mitochondrial and glycolytic metabolism in ccRCC cells
Seahorse results showed that SETD2 deficient 38E/38F cells had both higher mitochondrial (Figure 2A) and glycolytic (Figure 2B) metabolic rates than those of 786-O cells. For MST (Figure 2C), compared to 786-O cells, 38E and 38F cells had 153.9% and 121.0% of the basal mitochondrial OCR, 251.3% and 147.1% of the proton leak-derived OCR, and 104.8% and 107.9% of the ATP synthesis-derived OCR, respectively. The OCR associated with maximal respiration, spare respiratory capacity, and non-mitochondrial oxygen consumption in 38E and 38F cells were 601.5% and 1322.4%, 1516.6% and 3438.9%, and 320.2% and 167.9% higher than those of 786-O cells, respectively. For GST (Figure 2D), 38E and 38F cells had 167.8% and 133.1% of ECAR, and 79.5% and 102.5% of glycolytic capacity compared to 786-O cells, respectively. Glycolytic reserves for 38E and 38F were 2.6- and 2.4-fold higher than 786-O cells, respectively. Non-glycolytic acidification in 38E and 38F cells was more than 2-fold higher than that of 786-O cells. Additionally, 786-O cells consumed oxygen at a rate of 50.7 pmol O2/min with an acidification rate of 58.2 mpH /min, while 38E and 38F cells had OCRs 2.98- and 2.33-folds higher, as well as ECARs 1.81- and 2.01-folds higher than those of 786-O cells, respectively (Figure 2E).
Figure 2.

Loss of SETD2 enhances glycolytic and mitochondrial respiration in ccRCC cells. (A) Mitochondrial stress test (MST) and (B) glycolysis stress test (GST) were performed on Seahorse (n=3). ECAR and OCR were measured by addition of glucose fuel source and different metabolic inhibitors. (C) Mitochondrial activity and (D) glycolytic activity metabolic parameters were calculated from MST and GST, respectively. (E) OCR vs. ECAR of SETD2 isogenic cell lines under conditions of basal respiration (*p<0.05).
Loss of SETD2 enhances TCA cycle metabolic enzyme activity in ccRCC cells
We further analyzed the activity of CS, the first rate-limiting enzyme in the TCA cycle, in SETD2 deficient 38E/38F cells. Our data showed significantly elevated CS activity in 38E and 38F cells compared to 786-O cells (Figure 3A), but no significant difference in CS protein was observed between 786-O and 38F cells (Figure 3B), suggesting the elevated CS activity to be independent of CS protein level.
Figure 3.

Loss of SETD2 enhances the TCA cycle metabolic enzyme in ccRCC cells. (A) Evaluation of citrate synthase (CS) activity in 786-O and isogenic SETD2-deficient 38E/38F cells (n=3, *p<0.05). (B) Western blot for CS protein expression in ccRCC cells.
Loss of SETD2 increases TCA cycle metabolite production in ccRCC cells
To further validate observed metabolic changes of the TCA cycle, GC-MS-based targeted metabolomics was performed. The PCA score plot showed clear separation between 786-O and 38E/38F groups, and TCA metabolites had similar patterns of alterations in 38E/38F cells (Figure 4A–C). Further analysis showed 38E/38F cells had significantly higher levels of aspartate, malate, succinate, fumarate, and α-ketoglutarate, and lower levels of lactate than 786-O cells (Figure 4C). Furthermore, levels of citrate, isocitrate, cis-aconitic acid, and glutamate in 38E/38F cells were slightly increased, but no significant difference between 786-O and 38E/38F groups was observed (Figure S3).
Figure 4.

Targeted GC-MS analysis of 786-O and isogenic SETD2-deficient 38E/38F cells. (A) PCA score plot and (B) scatter plot based on TCA cycle metabolites from 786-O, 38E, and 38F cells. (C) Quantitative analysis of aspartate, malate, succinate, fumarate, α-ketoglutarate, and lactate concentration in each cell type (n=3, *p<0.05).
SETD2-deficient ccRCC cells utilize alternative sources of acetyl-CoA beyond pyruvate for maintenance of TCA cycle activity
Base oxygen consumption of isogenic SETD2-deficient 38F cells was 41.2% higher than 786-O cells (Figure S4). After NaF addition, the oxygen consumption of 786-O cells was reduced by 30.2%, but remained unchanged for 38F cells, suggesting that SETD2-deficient ccRCC cells could utilize additional sources of acetyl-CoA beyond pyruvate for maintaining TCA cycle activity. The addition of lactate increased oxygen consumption in both 38F and 786-O cells, indicating the oxidation of lactate to form pyruvate. Furthermore, results from the TCGA-KIRC dataset showed SETD2 homozygous deficient ccRCC tumors to have lower levels of PDP1 and higher levels of SHMT2 (both involved in pyruvate metabolism) compared to WT tumors (Figure S5).
Loss of SETD2 disturbs gene networks of mitochondrial and fatty acid metabolism
RNAseq data showed SETD2 deficiency to cause dysregulation of many genes in ccRCC cells, and gene enrichment analysis showed the main biological processes affected by SETD2 to be mitochondrial, lipid (fatty acids), glucose, coenzyme, and purine metabolism (Figure 5A–B). In addition, PGC1α and its related gene networks were altered by SETD2 mutations (Figure 5C). For instance, gene networks related to AMPK signaling such as AMPKa2, glucose metabolism such as SLC2A4 (GLUT4) and glycogen synthase 2 (GYS2), fatty acid metabolism such as Acetyl-CoA carboxylase 2 (ACC2) and carnitine palmitoyltransferase 1 (CPT1), mitochondrial function such as PPARGC1A (PGC1α) and ETC subunit composition genes (complex I to V) were significantly enhanced in isogenic SETD2-deficient 38F cells (Figure 6A–B). Moreover, genes involved in fatty acid metabolism such as hepatic lipase (LIPC), CPT1B, acyl-CoA synthases 2 and 5 (ACSL2 / ACSL5), and acyl-CoA dehydrogenase (ACADL) were also up-regulated in SETD2-deficient 38F cells (Figure S6).
Figure 5.

RNA-seq analysis of 786-O and isogenic SETD2-deficient 38E and 38F cells. (A) Visualization of the gene alterations between 786-O and 38E/38F cells (red indicates up-regulated genes and green indicates down-regulated genes compared to 786-O cells). (B) Gene enrichment analysis revealed the cellular biological processes affected by SETD2 inactivation. (C) Identification of disturbed gene regulation networks in SETD2-deficient 38E/38F cells.
Figure 6.

Gene network analysis of isogenic SETD2-deficient ccRCC cell lines. (A) Activation of pathways that promote glucose uptake, mitochondrial biogenesis, and fatty acid oxidation in SETD2-deficient cells. (B) Genes involved in ETC subunit composition were increased in 38F cells (red indicates up-regulated genes and blue indicates down-regulated genes compared to 786-O cells).
Loss of SETD2 increases PGC1α and mitochondrial mass in ccRCC cells
Since PGC1α is a central regulator of mitochondrial oxidative phosphorylation and fatty acid metabolism,16 we hypothesized that the elevated metabolic activity in SETD2-deficient ccRCC cells is associated with increased PGC1α expression. Indeed, we found that isogenic SETD2-deficient 38F cells had a significant increase of the PGC1α protein (2.5-folds) compared to 786-O cells (Figure 7A). In addition, staining of the living cells with a mitochondrial stain revealed higher mitochondrial mass in isogenic SETD2-deficient 38E/38F cells than that in 786-O cells (Figure 7B–C). Although PGC1α protein expression is not available in public genomic datasets such as The Cancer Genome Atlas, we examined the mRNA expression of PPARGC1A, the gene that encodes PGC1α, in SETD2 wild-type and mutant tumors. In our analysis, we found that the SETD2 mutant group produced a slightly higher expression although the difference was not significant (Figure S7, p˃0.05).
Figure 7.

Loss of SETD2 increases PGC1α and mitochondrial mass in ccRCC cells. (A) Western blot for PGC1α protein expression in 786-O and isogenic SETD2-deficient 38F cells (**p<0.01). (B) MitoTracker Red staining for 786-O, 38E, and 38F cells (400x magnification). (C) Relative mitochondrial mass in each cell type as determined by MitoTracker Red staining (n=3, *p<0.05).
Discussion
Although the connection between RCC and dysregulated metabolism has been previously reported,12 the effects of SETD2 inactivation on ccRCC metabolic phenotype and its contribution to tumor metastasis are still unknown. In this study, we observed higher luminescence of MTT and Alamar Blue in SETD2 deficient 38E/38F cells than 786-O cells. MTT is reduced by a variety of cytosolic and mitochondrial oxidases such as NAD(P)H, FADH, FMNH, and NADH.17 Alamar Blue is reduced by the same cohort of enzymes as that for MTT as well as cytochromes involved in the respiratory chain.18 Thus, these results suggest that loss of SETD2 leads to increased overall metabolic activity in ccRCC cells. Moreover, the elevated ATP levels also suggest increased mitochondrial respiration in SETD2-deficient cells. Further, Seahorse results showed that SETD2 inactivation induced a significant increase in the glycolytic and mitochondrial respiration rates in RCC cells. SETD2-deficient cells had higher basal glycolytic rates and a shut-down of ATP synthesis in the electron transport chain (ETC) by oligomycin led to increased ECAR. There was very little increase in ECAR of 786-O cells after oligomycin addition, suggesting that 786-O cells were operating at the near maximal glycolytic rate under basal conditions and thus had a limited ability to adapt to increased energy demand. Moreover, SETD2-inactivated cells showed more pronounced effects on mitochondrial respiration compared to 786-O cells. The addition of FCCP (an ETC uncoupler) revealed that SETD2-deficient cells have an enormous capacity to meet the energetic demand. In contrast, the OCRs in response to FCCP in 786-O cells were lower than their initial rates, creating an overall negative spare respiratory capacity. Although many cancers exhibit the classic Warburg effect and rely less on mitochondrial respiration for their energetic demand, a similar metabolic switch observed in this study has been reported in prostate cancer and was associated with poor overall survival.19 Thus, our results indicate that SETD2 inactivation can enhance both glycolytic and mitochondrial respiration with greater capacity for mitochondrial oxidative metabolism in ccRCC cells.
The Seahorse results further supported the elevated activities of TCA enzymes such as CS in SETD2-deficient cells. The activity of CS, responsible for the conversion of citrate from acetyl-CoA and oxaloacetate, was significantly increased in SETD2-deficient cells, although there was no apparent increase in CS protein. In addition, we observed increased levels of TCA cycle metabolites such as aspartate, malate, succinate, fumarate, and α-ketoglutarate in SETD2-deficient 38E/38F cells. Since pyruvate to acetyl-CoAs conversion is essential for maintenance of the TCA cycle, and since pyruvate dehydrogenase (PDH) is usually inactivated in tumor cells,20 our results suggest a potential link between SETD2 deficiency and pyruvate metabolism in ccRCC cells. Interestingly, we found several key genes that regulate pyruvate metabolism, such as SHMT2 and PDP1, to be altered in homozygous SETD2-mutated RCC from the TCGA-KIRC dataset.21, 22 SHMT2 is involved in glycine-serine metabolism as an alternative route to generate pyruvate 21 and, thus, its up-regulation might divert pyruvate for the production of acetyl-CoA in SETD2-deficient RCCs. PDP1, an activator of PDH to generate acetyl-CoA,22 was reduced in SETD2-deficient tumors, suggesting a decreased reliance on pyruvate as a source of acetyl-CoA for the TCA cycle in SETD2-deficient RCCs. This hypothesis is partly supported by the reduced oxygen consumption in 786-O cells but not SETD2-deficient 38F cells after treatment with NaF (a PDP1 inhibitor).22 These results suggest that pyruvate is a major source of acetyl-CoA in 786-O cells, but SETD2-deficient cells may use other fuel sources to generate acetyl-CoA for the TCA cycle.
To further explore the underlying molecular mechanisms, RNAseq and gene network analysis was performed in 786-O and 38E/38F cells. We found that loss of SETD2 disturbed networks related to glucose, mitochondrial, and fatty acid metabolism. For instance, GLUT4, a principal transporter for cellular glucose uptake,23 was significantly increased in SETD2-deficient cells. PGC1α, a master regulator for cellular energy homeostasis,16 was also elevated by SETD2 inactivation. Additionally, both mitochondrial uncoupling genes (UCP2, UCP3, and SLC25A27) and ETC subunit composition genes (NDUSF4, SDHA, COX5b, and ATP5A1) were significantly up-regulated in SETD2-deficient cells. Indeed, the up-regulation of mitochondrial biogenesis has been reported in cancers, and some cancers have mutations in mitochondrial TCA cycle enzymes.24 PGC1α has been recognized as a key stimulator for mitochondrial biogenesis, which can induce oxidative phosphorylation and TCA cycle gene expression.16,25 Moreover, PGC1α has also been shown to promote colon and liver carcinogenesis growth via enhancing mitochondrial and fatty acid metabolism.26 Thus, our results indicate that enhanced mitochondrial oxidative metabolism in SETD2-deficient ccRCC cells may be influence by PGC1α expression. This hypothesis is supported by the observed elevation in PGC1α protein, and its associated increase in mitochondrial mass, cell size, and intercellular complexity in SETD2-deficient cells. As previously reported, PGC1α mRNA has multiple isoforms,27 and its mRNA levels may not correlate with PGC1α protein expression. Moreover, there are likely other factors in tissue that could influence PGC1 expression such as VHL and HIF1-α,28 while our SETD2 isogenic cells have the same VHL/HIF phenotype. Thus, comparison of PGC1 isoforms and protein expression in SETD2 WT vs mutation tumor of ccRCC patients is needed in future studies.
Although we examined independently generated SETD2 deficient cell lines, our results in cultured cells may not accurately reflect the in vivo tumor microenvironment.29 First, cultured cells are limited to selection of rapidly proliferating clones under nonphysiological conditions and our results should be confirmed in murine models in vivo to better recapitulate the tissue microenvironment. Second, although we detected increased levels of TCA metabolites, we did not see a correlation with RNA sequencing or Western blot of key enzymes. Similarly, a prior study did not see a correlation in human RCC tumors between RNA sequencing and direct measurement of metabolites.30 Third, there are likely other factors involved in mitochondrial biogenesis beyond PGC1α,24 thus future studies should examine the influence of the tumor microenvironment on biogenesis.
Conclusion
Taken together, our study observed that loss of SETD2 is associated with a metabolic switch in ccRCC cell lines toward enhanced oxidative phosphorylation and lipogenesis, and its mechanism can be potentially related to PGC1α-mediated metabolic networks (Figure 8). Moreover, our results suggest a need for a comprehensive metabolomics analysis of cancer cells with SETD2 inactivation in vivo to specifically identify pathways involved in this metabolic switch, which provides a number of opportunities to identify novel therapeutic targets in kidney cancer.
Figure 8.

Schematic diagram of PGC1α overexpression and enhanced mitochondrial oxidative metabolism induced by SETD2 inactivation in ccRCC cells. Elevated TCA metabolites (green) in SETD2-deficient cells may be shunted toward increased fatty acid synthesis, leading to cancer metastasis.
Supplementary Material
Acknowledgements
This work was supported in part by the China Scholarship Council, Arizona State University (ASU), and the Gloria A. and Thomas J. Dutson Jr. Kidney Research Endowment. THH is supported by National Cancer Institute (R01CA224917) and the Department of Defense (W81XWH-17-1-0546). Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. The funding agencies had no role in the study design.
Footnotes
Conflict of Interest Disclosure
The authors declare no competing financial interest.
References
- 1.Siegel RL; Miller KD; Jemal A Cancer statistics, 2018. CA Cancer J Clin 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Barata PC; Rini BI Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J Clin 2017, 67, 507–524. [DOI] [PubMed] [Google Scholar]
- 3.Cohen HT; McGovern FJ Renal-cell carcinoma. N Engl J Med 2005, 353, 2477–90. [DOI] [PubMed] [Google Scholar]
- 4.Wiesener MS; Munchenhagen PM; Berger I; Morgan NV; Roigas J; Schwiertz A; Jurgensen JS; Gruber G; Maxwell PH; Loning SA; Frei U; Maher ER; Grone HJ; Eckardt KU Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res 2001, 61, 5215–5222. [PubMed] [Google Scholar]
- 5.Haase VH; Glickman JN; Socolovsky M; Jaenisch R Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A 2001, 98, 1583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piva F; Santoni M; Matrana MR; Satti S; Giulietti M; Occhipinti G; Massari F; Cheng L; Lopez-Beltran A; Scarpelli M; Principato G; Cascinu S; Montironi R BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert Rev Mol Diagn 2015, 15, 1201–1210. [DOI] [PubMed] [Google Scholar]
- 7.Su D; Singer EA.; Srinivasan R. Molecular pathways in renal cell carcinoma: recent advances in genetics and molecular biology. Curr Opin Oncol 2015, 27, 217–223. [DOI] [PubMed] [Google Scholar]
- 8.Ho TH; Choueiri TK; Wang K; Karam JA; Chalmers Z; Frampton G; Elvin JA; Johnson A; Liu X; Lin Y; Joseph RW; Stanton ML; Miller VA; Stephens PJ; Ross JS; Ali SM; Pal SK Correlation Between Molecular Subclassifications of Clear Cell Renal Cell Carcinoma and Targeted Therapy Response. Eur Urol Focus 2016, 2, 204–209. [DOI] [PubMed] [Google Scholar]
- 9.Li J; Duns G; Westers H; Sijmons R; van den Berg A; Kok K SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget 2016, 7, 50719–50734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiedemann RL; Hlady RA; Hanavan PD; Lake DF; Tibes R; Lee JH; Choi JH; Ho TH Robertson KD. Dynamic reprogramming of DNA methylation in SETD2-deregulated renal cell carcinoma. Oncotarget 2016, 7, 1927–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho TH; Park IY; Zhao H; Tong P; Champion MD; Yan H; Monzon FA; Hoang A; Tamboli P Parker AS; Joseph RW; Qiao W; Dykema K; Tannir NM; Castle EP; Nunez-Nateras R et al. High-resolution profiling of histone h3 lysine 36 trimethylation in metastatic renal cell carcinoma. Oncogene 2016; 35, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linehan WM; Srinivasan R; Schmidt LS The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol 2010, 7, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Semenza GL HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr 2007, 39, 231–4 [DOI] [PubMed] [Google Scholar]
- 14.Sudarshan S; Sourbier C; Kong HS; Block K; Valera Romero VA; Yang Y; Galindo C; Mollapour M; Scroggins B; Goode N; Lee MJ; Gourlay CW; Trepel J; Linehan WM; Neckers L Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 2009, 29, 4080–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J; Wang C; Liu F; Lu Y; Cheng J Metabonomics revealed xanthine oxidase-induced oxidative stress and inflammation in the pathogenesis of diabetic nephropathy. Anal Bioanal Chem 2015, 407, 2569–79. [DOI] [PubMed] [Google Scholar]
- 16.Austin S; St-Pierre J PGC1alpha and mitochondrial metabolism-emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 2012, 125, 4963–4971. [DOI] [PubMed] [Google Scholar]
- 17.Berridge MV; Tan AS Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys 1993, 303, 474–482. [DOI] [PubMed] [Google Scholar]
- 18.Rampersad SN Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors (Basel) 2012, 12, 12347–12360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zadra G; Photopoulos C; Loda M The fat side of prostate cancer. Biochim Biophys Acta 2013, 1831, 1518–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feron O Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol 2009, 92, 329–33. [DOI] [PubMed] [Google Scholar]
- 21.Kim D; Fiske BP; Birsoy K; Freinkman E; Kami K; Possemato RL; Chudnovsky Y; Pacold ME; Chen WW; Cantor JR; Shelton LM; Gui DY; Kwon M; Ramkissoon SH; Ligon KL; Kang SW; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean P; Kunjara S; Greenbaum AL; Gumaa K; Lopez-Prados J; Martin-Lomas M; Rademacher TW Reciprocal control of pyruvate dehydrogenase kinase and phosphatase by inositol phosphoglycans. Dynamic state set by “push-pull” system. J Biol Chem 2008, 283, 33428–33436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Govers R Cellular regulation of glucose uptake by glucose transporter GLUT4. Adv Clin Chem 2014, 66, 173–240. [DOI] [PubMed] [Google Scholar]
- 24.Zong WX; Rabinowitz JD; White E Mitochondria and Cancer. Mol Cell 2016, 61, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Austin S; Klimcakova E; St-Pierre J Impact of PGC-1alpha on the topology and rate of superoxide production by the mitochondrial electron transport chain. Free Radic Biol Med 2011, 51, 2243–2248. [DOI] [PubMed] [Google Scholar]
- 26.Bhalla K; Hwang BJ; Dewi RE; Ou L; Twaddel W; Fang HB; Vafai SB; Vazquez F; Puigserver P; Boros L; Girnun GD PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res 2011, 71, 6888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruas JL; White JP; Rao RR; Kleiner S; Brannan KT; Harrison BC; Greene NP; Wu J; Estall JL; Irving BA; Lanza IR; Rasbach KA; Okutsu M; Nair KS; Yan Z; Leinwand LA; Spiegelman BM A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 2012; 151, 1319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H; Gao P; Fukuda R; Kumar G; Krishnamachary B; Zeller KI; Dang CV; Semenza GL HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 2007, 11, 407–20. [DOI] [PubMed] [Google Scholar]
- 29.Davidson SM; Papagiannakopoulos T; Olenchock BA; Heyman JE; Keibler MA; Luengo A; Bauer MR; Jha AK; O’Brien JP; Pierce KA; Gui DY; Sullivan LB; Wasylenko TM; Subbaraj L; Chin CR; Stephanopolous G; Mott BT; Jacks T; Clish CB; Vander Heiden MG Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 2016, 23, 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakimi AA; Reznik E; Lee CH; Creighton CJ; Brannon AR; Luna A; Aksoy BA; Liu EM; Shen R; Lee W; Chen Y; Stirdivant SM; Russo P; Chen YB; Tickoo SK; Reuter VE; Cheng EH; Sander C; Hsieh JJ An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.