

Abstract
Post-translationally spliced peptides have recently garnered significant interest as potential targets for cancer immunotherapy and as contributors to autoimmune diseases such as type 1 diabetes, yet feasible identification methods for spliced peptides have yet to be developed. Here we present Neo-Fusion, a search program for discovering spliced peptides in tandem mass spectrometry data. Neo-Fusion utilizes two separated ion database searches to identify the two halves of each spliced peptide, and then it infers the full spliced sequence. This strategy allows for the identification of spliced peptides without peptide length constraints, providing a broadly applicable tool suitable for identification of spliced peptides in a variety of systems, such as the HLA-I and HLA-II immunopeptidomes and in vitro digested protein samples obtained from organelles, cells, or tissues of interest. Using simulated spliced peptides to benchmark Neo-Fusion, 25% of all simulated spliced peptides were identified at a measured false-discovery rate of 5% for HLA-I. Neo-Fusion provides the research community with a powerful new tool to aid in the study of the prevalence and biological significance of post-translationally spliced peptides.
Keywords: bioinformatics searching, false discovery rate, human leukocyte antigen, immunopeptidomics, mass spectrometry, proteasome-spliced peptides, spliced peptides
Graphical Abstract
INTRODUCTION
Neoantigens are in vivo-expressed antigen peptides to which the immune system has not previously been exposed. The presentation of neoantigens on cell surfaces allows the immune system to recognize and destroy the neoantigen-presenting cells. These processes have important consequences in cancer immunotherapy,1 as well as in autoimmune diseases like type 1 diabetes.2 The ability to identify neoantigens is thus of paramount importance for both understanding and controlling immune responses. The two main approaches to identify neoantigens are nucleic acid sequencing and mass spectrometry. While nucleic acid sequencing is able to reveal genetic variations from which possible neoantigens may be inferred, it does not provide direct information on the molecules that are actually expressed, the levels at which they are presented, nor the nature of post-translational modifications (PTMs) that may be present, all of which contribute to immunogenicity.3 Mass spectrometry (MS), in contrast, is able to provide such information, but is limited in its current capabilities.4 In particular, recent developments have highlighted the possible role of proteasome-generated spliced peptides (PSPs) in immune recognition, which are challenging to identify by MS. It has been reported that the proteasome, widely known for digesting proteins into peptide fragments, can ligate such fragments together to form novel peptide sequences not otherwise found in the proteome (i.e., neoantigens).5 These PSPs can occur as either cis- or trans-PSPs, where the two halves originate from either the same protein or from two distinct proteins, respectively.6 To date, several such post-translationally spliced peptides have been identified in vivo, and many were reported to be immunogenic.2,5,7–16
New tools are needed to comprehensively identify such post-translationally spliced peptides in complex samples. Mass spectrometry is currently the premier strategy for peptide identification, but it is largely reliant on prior knowledge of the proteome, such as canonical databases or spectral libraries. No such resources exist for post-translationally spliced peptides, making their detection and identification diffcult. Nonspliced peptides reflect the linear sequence of their respective parent protein, but post-translationally spliced peptides result from the fusion of two distinct sequences, thereby blinding traditional proteomic database searches to their existence. De novo proteomics can be employed to obtain a putative peptide sequence, but de novo identification significantly underperforms in comparison to database searches when applied to complex mixtures such as the immunopeptidome.17 Liepe et al. sought to determine the prevalence of PSPs in the human leukocyte antigen class I immunopeptidome (HLA-I-Ip) by identifying cis-PSPs. In their approach, a massive database of theoretical cis-PSPs was generated through in silico splicing of all known human canonical protein sequences. This strategy suffered from several limitations. The large size of the cis-PSP database necessitated substantial search times and possibly an underestimated false discovery rate (FDR) arising from sequence similarities between nonspliced peptides and PSPs.18 The approach was unable to identify trans-PSPs, as the combinatorics of generating all possible trans-PSPs are even more substantial than those for generating all possible cis-PSPs. To reduce the issue of database inflation in the cis-PSP database, Liepe et al. also limited their search to only peptides 9−12 residues in length, which excluded a substantial fraction of the HLA-I-Ip and prevented the extension of the study to HLA-II antigens, which are typically longer than HLA-I antigens and have a broader distribution of lengths.
Here we present Neo-Fusion, an open-source, publicly available program for the detection and identification of spliced peptides such as PSPs. Neo-Fusion rapidly discovers spliced peptides using a traditional database size and a confidence threshold obtained through comparisons with nonspliced peptides, allowing for the identification of cis-spliced peptides without any length constraints. Neo-Fusion works by using separate ion-type database searches of MS spectra to identify both halves of a spliced peptide and then splicing the two halves together in silico. Using simulated PSPs generated from real data, Neo-Fusion was able to identify approximately one-quarter of all simulated cis-PSPs at a 5% FDR. Analysis of MS data from an HLA-I-Ip, an HLA-II-Ip, and trypsin-digested proteins from mouse islets indicates that an upper limit of 1− 4% of all peptides in these samples are potentially spliced peptides. However, it is not generally possible to confirm that these identifications truly correspond to spliced peptides, as these identified sequences could derive from other sources such as contaminants, genetic variants, protein isoforms, and PTMs. An essential part of any search for spliced peptides is the careful evaluation of all candidates and the consideration of alternative explanations for their presence in the sample.
METHODS
Data Sets
Neo-Fusion was used to search for spliced peptides in three publicly available data sets representing three unique sample types: an HLA-I-Ip, an HLA-II-Ip, and a fractionated tryptic digest of whole tissue lysate. Specifically, the datafiles were the HLA-I-Ip of primary fibroblast cells originally published by Bassani-Sternberg et al.19 and later analyzed by Liepe et al.,6 a single replicate of an HLA-II-Ip for mantle-cell lymphoma,20 and nine MS files for a fractionated tryptic digest of B6 mouse pancreatic islet lysate.21 The HLA-I and HLA-II associated antigens were previously isolated through immunoaffinity purification of HLA-I and HLA-II followed by antigen elution. The tryptic peptide sample was previously prepared by in vitro enzymatic digestion of proteins. In each sample, the peptides were analyzed via high pressure liquid chromatography and tandem mass spectrometry (HPLC-MS/MS). Additional information on the sample preparations and analyses of these datafiles can be found in their original manuscripts. HLA-I-Ip and HLA-II-Ip files were searched against a UniProtKB/Swiss-Prot.xml protein database of Homo sapiens accessed December 12, 2017. The tryptic files were searched against a UniProtKB/ Swiss-Prot.xml protein database of Mus musculus accessed November 1, 2016. All mass spectrometry files were calibrated prior to analysis using MetaMorpheus (version 0.0.276).22 Global-PTM-Discovery23 in MetaMorpheus was conducted to inform the database of common PTMs and prevent modified nonspliced peptides from being erroneously assigned as spliced peptides.
Neo-Fusion
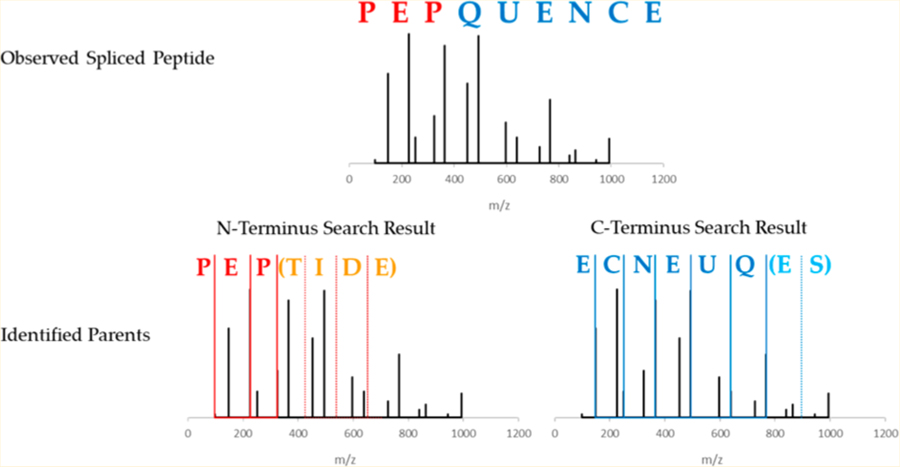
Neo-Fusion (https://github.com/zrolfs/MetaMorpheus/tree/Neo-Fusion) employs a two-part algorithm comprised of (a) two separate database searches to identify the parent peptides followed by (b) in silico splicing of these parent peptides to generate spliced peptide candidates. For higher-energy collisional dissociation (HCD), the N-terminal ion database consists of b-ions, while the C-terminal ion database consists of y-ions. Traditional database searches compare the experimental spectra with theoretical spectra, wherein theoretical b- and y-ions are generated in silico from a database of expected peptide sequences. In contrast, Neo-Fusion utilizes two separate databases, one for each terminus, containing only the ion series originating from the given terminus (e.g., b- or y-ions, Figure 1A). Each database serves to produce a single parent peptide for its respective terminus (Figure 1B). Furthermore, Neo-Fusion employs open mass searches that ignore precursor masses, in contrast to traditional searches that require theoretical and experimental precursor masses to match within a narrow tolerance. Open mass searches are necessary for the Neo-Fusion search algorithm, because the two parent peptides giving rise to a spliced peptide rarely, if ever, have the same precursor mass as the spliced peptide itself. Recent advances in indexed search methods have made open mass searches feasible on a reasonable time scale.24 Note that the aim of these searches is strictly to identify the parents of spliced peptides. Spectra that match to nonspliced sequences are still considered as possible spliced peptide spectra and are not discarded. The indexed, open mass, separate ion database searches are conducted in Neo-Fusion using the Morpheus scoring algorithm.25 To aid in the identification of each terminus, complementary ions are added to the experimental spectra, and peaks with a mass below 600 Da are doubly weighted to more significantly impact the score near the termini. The highest scoring peptide from each separate ion database search for each spectrum is saved, such that each spectrum now possesses two parent peptides.
Figure 1.
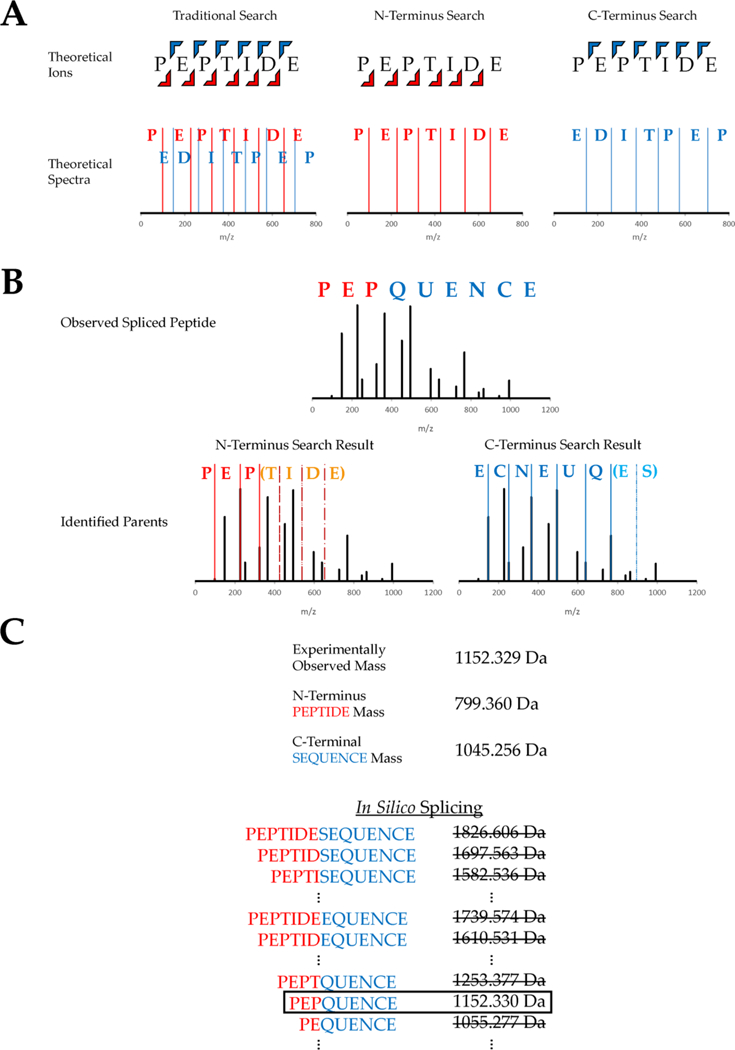
Workflow of the database search algorithm used to identify spliced peptides. (A) Two separate databases are generated containing only N-terminal ions or C-terminal ions. (B) Experimental spectra are compared against both databases in an open mass tolerance search, and the highest scoring match from each search is recorded. This example shows an experimental spectrum for the spliced peptide “PEP-QUENCE, derived from parent peptides “PEPTIDE” and “SEQUENCE”. Both parents are identified from the separate ion database searches. (C) The two parent peptide assignments have different precursor masses than the experimental spectrum, and in silico splicing is used to identify the full spliced sequence. This is accomplished through a narrow mass tolerance comparison between the observed precursor mass and all theoretical spliced precursor masses that can be obtained from the two parents.
After the separate ion database searches have identified the two parents of a spliced peptide, Neo-Fusion attempts to determine the complete spliced sequence through in silico splicing of the two parents. Every theoretical spliced product of the two parent peptides is evaluated as to whether its total mass matches that of the observed precursor mass within a reasonable mass tolerance (Figure 1C). Every spliced product that possesses the same precursor mass as the observed spectrum is recorded as a possible spliced candidate. Experimental spectra where the precursor mass cannot be obtained from in silico splicing are deemed nonspliced peptides. The surviving spliced peptide-spectrum matches (PSMs) are inspected for missing experimental fragment ion peaks that may cause sequence ambiguity. Spliced PSMs that lack both ion series at any peptide bond are considered ambiguous, and therefore additional spliced sequences are generated that could be equally likely to explain the observed spectra. For example, if “PEPQUENCE” does not contain a b1 or a y8 product ion, then “EPPQUENCE” will also be generated, provided it can be formed through splicing, because both sequences have the same Morpheus score.25 These spliced peptides are considered ambiguous, because Neo-Fusion is unable to differentiate the sequence. The generation of every ambiguous sequence is achieved by using all possible combinations of the 20 amino acids that can both account for the missing fragment(s) and be formed by the splicing of any protein(s) in the specified database. Finally, all of the resulting spliced candidate sequences are exported in a .FASTA file and searched in a second-pass, traditional database search to obtain a Morpheus score25 that indicates how well the spectrum matches the spliced peptide sequence as a whole.
Confidence Measurements
Traditional proteomics is largely dependent on the target-decoy approach to FDR determination. The target-decoy approach assumes that any spectrum that is incorrectly assigned has an equal probability of being assigned as a target or decoy peptide. This enables the count of decoy matches at any score threshold to serve as a proxy for the count of false-positive target assignments. However, this target-decoy strategy is challenging to employ in the analysis of spliced peptides for two reasons. First, peptide splicing is not yet predictable. Therefore, attempts to construct a target database of spliced peptides are prone to missing actual spliced peptides and to containing spliced peptides not present in the sample. This stands in contrast to traditional target protein databases, which are constructed from previously curated and validated protein sequences. Second, we have found that strategies for producing spliced peptide databases yield many sequences that have similar precursor masses and fragmentation patterns to existing nonspliced sequences. Target spliced peptides are derived from the splicing of nonspliced sequences and are thus often very similar to one of their nonspliced parents. These sequence similarities arise through a variety of isobaric amino acid permutations and substitutions (Table 1). The target-decoy approach struggles to distinguish between such similar sequences, which are hundreds of times more frequent when searching for spliced peptides than nonspliced peptides alone (Table 1). It has been observed that over 95% of nonspliced PSMs can be assigned a similar, incorrect sequence that scores greater than or equal to the correct sequence, and this was found to be independent of the mass spectrometer or search algorithm used.26 The large number of sequence similarities introduced into the search space by spliced peptides results in a substantial problem of nonspliced peptide spectra being incorrectly assigned as spliced peptides with comparable or better scores. This is further compounded by nonspliced peptide spectra that are absent from the search space, such as peptides containing PTMs, single amino acid variants, or peptides outside of the specified amino acid length constraints. When such variant peptides are present in the sample but not in the search space, they provide likely targets for misidentification (i.e., false positives).
Table 1.
Common Sequence Similarity Types and Their Frequencies in Nonspliced and Cis-Spliced Searches
| Observed Peptide | Similar Sequencea | Nonspliced Frequencyb | Cis-Spliced Frequencyb | |
|---|---|---|---|---|
| Permutation | CQMMQNPRACLEMS | QCMMQNPRAGLEMS | 1.42E-3 | 0.660 |
| Replacement | CQMMQNPRACLEMS | CQMMQNPRAGLDMT | 2.57E-4 | 0.384 |
| Insertion | CQMMQNPRAGLEMS | CQMMQGGPRAGLEMS | 2.56E-5 | 0.106 |
| Deletion | CQMMQNPRAGLEMS | CQMMQN PRQLEMS | Ί.76Ε−5 | 0.054 |
| Substitution | CQMMQNPRAGLEMS | CQMMQNPRAGLEME | 1.69E-4 | 0.265 |
| Acetyl |
These sequences have identical precursor masses and similar fragmentation patterns, making the correct sequence diffcult to distinguish.
Frequencies are the average number of similar sequences per theoretical peptide. This was calculated for 9-mer, nonspliced peptides from the human.xml database. The computational power required for this calculation limited the similarity comparisons to peptides generated from the same protein. The database-wide frequencies are thus expected to be much higher.
The sequence similarity problem, as we have just described, is a challenge to the correct identification of spliced peptides in complex samples. This problem grows by orders of magnitude when analyzing immunopeptidomes. HLA-I/II antigens are generated by proteasomal or lysosomal degradation of intact proteins, respectively,27 and neither process is known to possess specific digestion motifs. Without established methods to predict digestion, proteins can be cleaved at any available residue. The lengths of these peptides vary, but they are typically 8−16 amino acids for HLA-I-associated antigens and 8−25 amino acids for HLA-II-associated antigens. This is an extraordinary diversity of sequences, which are diffcult to navigate even when one does not consider splicing. Once splicing is introduced, there is an additional ∼300-fold increase in search space when constraining the search to only cis-spliced peptides possessing intervening sequences of 1−25 amino acids.6 The resulting database expansion makes such searches particularly diffcult.
Neo-Fusion avoids the problems that arise from a target-decoy based FDR by using a set of heuristic thresholds that reduces the number of reported false-positive spliced peptide discoveries. This is achieved through a recursive loop that aims to maximize the number of reported spliced peptides at an estimated false-positive rate (Figure 2). The heuristic considers four parameters: a minimum required score difference (delta score) between the highest scoring spliced and nonspliced sequence for each spectrum, a maximum nonspliced q-value required for spectra to be assigned as gold standard nonspliced spectra, a minimum allowed spliced peptide score, and a maximum allowed false-positive rate. In the first round of the computation, the minimum delta score and maximum nonspliced q-value are arbitrarily chosen. Each spectrum that has a delta score lower than the chosen minimum is considered nonspliced and ignored for the remainder of the round. This helps to mitigate incorrect spliced peptide assignments by increasing the score necessary for a spliced sequence to compete with a nonspliced sequence. Every spectrum with a nonspliced q-value lower than the chosen maximum q-value has its spliced sequence marked as a false positive (Figure 3). The assumption is that spliced sequences matching to confident nonspliced peptide spectra must be incorrect spliced assignments. The minimum allowed spliced peptide score for the round is simply the lowest gold standard nonspliced peptide score. Having now removed nonspliced spectra and assigning incorrect sequences as false positives, all spectra are sorted in decreasing order by the spliced peptide score for each spectrum. Moving down the list of spectra, the ratio of marked false positives to true positives is calculated. This calculation stops when either the percentage of false positives to true positives reaches the maximum allowed false-positive rate (e.g., 1%) or the spliced score dips below the minimum allowed spliced score threshold. The number of putative true positives is then tallied. The entire process is repeated with alternative values for the minimum allowed delta score and the maximum nonspliced q-value for gold standard assignments until the number of true positives is maximized. This optimization is achieved by iterating through the discrete q-values, between 0 and 0.05, observed for the nonspliced PSMs. Each discrete q-value is tested with every minimum allowed delta score between 1 and 5, with allowed score intervals of 0.5 (1.0, 1.5, 2.0, …, 4.5, 5.0). The combination of q-value and delta score that produces the most putative true positive spliced peptide at a 1% false-positive rate is selected to yield the optimized parameters. The optimized parameters for all reported runs can be found in Table S1.
Figure 2.
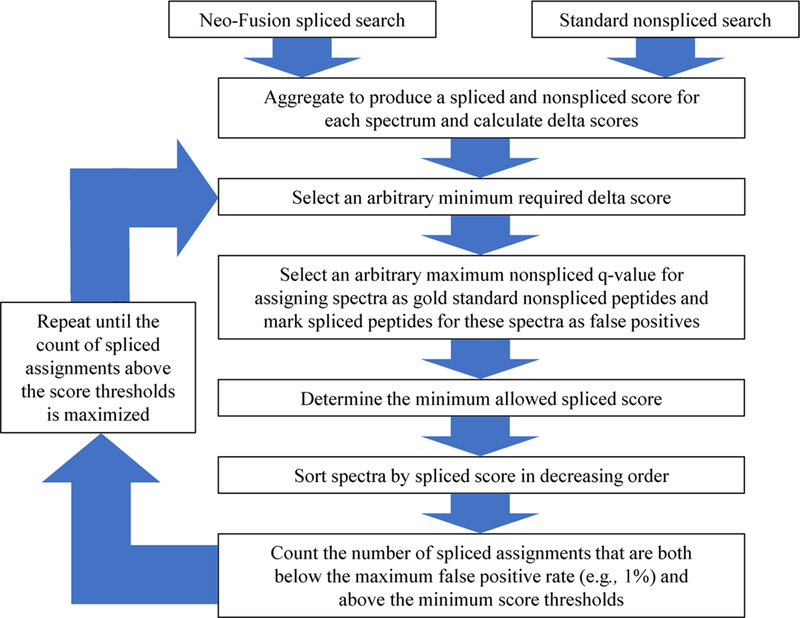
Flowchart showing the heuristic determination of confidence thresholds for spliced peptide discoveries. The inputs required for this heuristic determination are the highest scoring spliced and nonspliced sequences for each spectrum, which are identified by Neo-Fusion through separate spliced and nonspliced searches.
Figure 3.
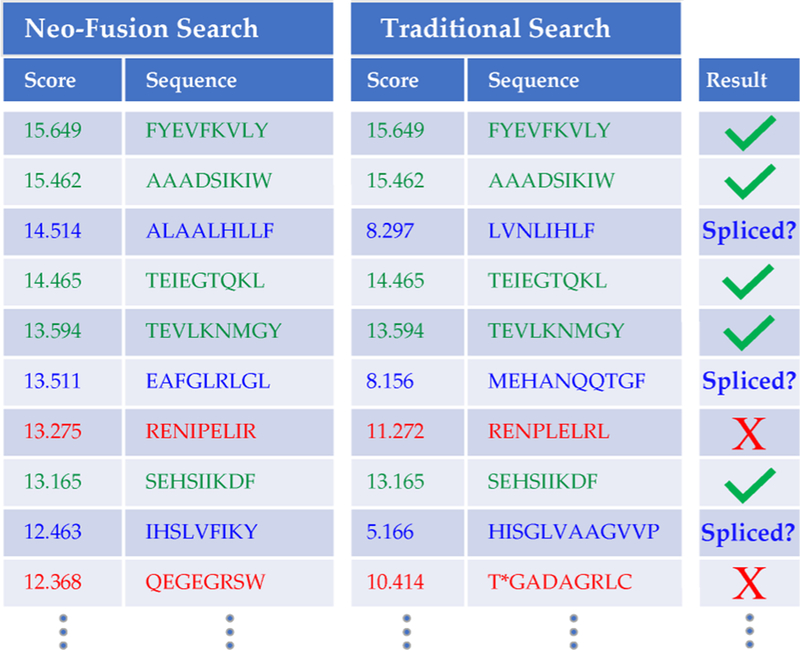
Example comparison of Neo-Fusion and nonspliced results. Green rows indicate spectra where the delta score was below an arbitrary threshold, so a nonspliced sequence was assigned. Blue rows show where Neo-Fusion generated a high scoring spliced sequence that better explained the spectrum than a low-confidence nonspliced peptide. Red rows are false positives where a spliced sequence outscored a gold standard nonspliced sequence. The minimum delta score and minimum gold standard score for this example are 1.5 and 10, respectively. *The nonspliced sequence “TGADAGRLC” contains a formylated threonine.
Neo-Fusion required 11 h to complete all Neo-Fusion tasks for the HLA-I file (44075 MS/MS spectra) when running on a 4-core, 16 GB RAM workstation. The uncompressed Neo-Fusion software requires 17 MB of disk space, and the output generated from Neo-Fusion for the HLA-I file required 1.8 GB of disk space for the results and 6.8 GB for the theoretical fragment ion indexes used to decrease subsequent search times. The indexes are partitioned, such that the entire database does not have to be in active memory at once. This enables Neo-Fusion to be useable on low RAM workstations.
RESULTS AND DISCUSSION
Neo-Fusion’s ability to confidently discover spliced peptides in complex HLA-I-Ip, HLA-II-Ip, and tryptic samples was benchmarked by simulating spliced peptides in silico within the reference database for each sample. These simulations were accomplished by using a traditional database search to confidently identify nonspliced peptides in each sample. A subset of these nonspliced sequences were then randomly selected, cleaved in silico at a random residue specific to each peptide, and a resulting half of each sequence was shifted a random number of residues (1−25) within the original protein sequences in the reference database (Figure S1). This process effectively removed the linear nonspliced peptide sequences from the database and generated proteins containing the two halves of each spliced peptide. The use of such simulated spliced peptides allowed us to search real experimental spectra, instead of simulated spectra, and know a priori which spectra correspond to “spliced peptides” of known sequences. MetaMorpheus (version 0.0.276) was used to identify nonspliced peptides at a 1% FDR. We replaced 5% of these confidently identified nonspliced peptides at random with simulated spliced peptides, based upon a reported upper limit of 2−6% of the HLA-I-Ip being comprised of cis-spliced peptides.18 This process was repeated 20 times with different subsets of confident peptides to produce simulation replicates spanning the breadth of all identified peptides. These spliced peptide simulations were conducted for each of the three sample types, effectively generating 20 replicates per sample wherein a subset of observed spectra corresponded to “spliced peptides” and whose correct sequences were known.
The results of the benchmark simulation are shown in Table 2. For the HLA-I-Ip datafile, 4450 unique nonspliced peptides were identified by MetaMorpheus at a 1% FDR and used in the simulation. Overlapping peptide sequences were not included in the simulations, such that the actual number of unique nonspliced peptides identified is higher than 4450. 222 unique spliced sequences (5%) were simulated for each of the 20 replicates. Across all replicates in the HLA-I-Ip simulation, an average of 55.5 simulated cis-spliced peptides were correctly identified in each replicate, indicating that Neo-Fusion has a true-positive rate of 25% for HLA-I cis-spliced peptides. Using the known sequences of the simulated spliced peptides, we were able to evaluate Neo-Fusion’s false-positive rate. Roughly 5% of the 25% of spliced peptides found were assigned an incorrect sequence, indicating a reasonable FDR of 5% for cis-spliced peptide identification. This means that if a spliced peptide exists in the sample and Neo-Fusion identifies it as spliced, then there is a 5% chance that an incorrect spliced sequence will be assigned to it. The benchmark FDR is not a measure of how many nonspliced peptides were incorrectly assigned as spliced, as we do not know a priori which peptides were spliced in vivo. The FDR measured for HLA-II cis-spliced peptides was found to be 0%, which we attribute to the low number of simulated spliced peptides introduced into the sample. If the number of peptides present in the sample had been similar to that of HLA-I or the trypsin fraction, then we would expect an observed cis-spliced FDR closer to 5%.
Table 2.
Neo-Fusion Results for Simulated and Real Spliced Peptide Searches
| HLA-1 | HLA-II | T rypsina | |
| Search Parametersb | |||
| Peptide Length Limitation | 8–16 | 8–25 | 7–50 |
| Precursor Mass Tolerance (PPM) | 6.6 | 5.2 | 4.5 |
| Product Mass Tolerance (PPM) | 15.2 | 23.6 | 21.0 |
| Benchmark Results Using Simulated PSPs | (Fraction #5 of 9) | ||
| Total Simulated PSPs | 4450 | 440 | 3608 |
| Simulated PSPs per Replicate | 222 | 22 | 180 |
| Average Number of Simulated Cis-PSPs Found ± SD | 55.5 ± 10.4 | 4.6 ±2.4 | 76.0 ±8.1 |
| Average Percent of Simulated Cis-PSPs Found ± SD | 25.0% ±4.7 | 20.7% ± 10.7 | 42.2% ±4.5 |
| Average Simulated Cis-PSP FDR ± SD | 5.3% ±2.6 | 0.0% ± 0.0c | 3.5% ± 1.8 |
| Average Number of Simulated Cis- and Trans-PSPs Found ± SD | 48.0 ±8.4 | 5.3 ±2.4 | 89.9 ±8.7 |
| Average Percent of Simulated Cis- and Trans-PSPs Found ± SD | 21.6% ±3.8 | 24.3% ± 10.9 | 50.0% ± 4.8 |
| Average Simulated Cis- and Trans-PSPs FDR ± SD | 31.7% ±6.7 | 23.1% ±20.5c | 30.3% ± 4.4 |
| Real Spliced Peptide Sequences Discovered | (All 9 fractions) | ||
| Real Cis-Spliced (“1%” FDR) | 62 | 13 | 487 |
| Real Cis-Spliced Without Ambiguityd (“ 1 %” FDR) | 47 | 9 | 340 |
| Real Cis- and Trans-Spliccd (“1%” FDR) | 156 | 81 | 2490 |
| Real Cis- and Trans-Spliced Without Ambiguityd (“1%” FDR) | 29 | 10 | 496 |
The trypsin sample was offine fractionated, while the HLA samples were not. A single trypsin fraction (#5 of 9) was used for the benchmark simulation, but all fractions were used for the discovery of real spliced peptides.
All files were searched using b- and y-ions from HCD fragmentation, fixed carbamidomethylation of cysteine, variable oxidation of methionine, a maximum of 4096 isoforms, and a maximum of 2 modifications per peptide. Trypsin was allotted two missed cleavages of lysine and arginine. Mass tolerances were selected based on MetaMorpheus calibration suggestions.
The low FDRs and high variance reported for the HLA-II sample are due to the scarcity of nonspliced identifications; no richer HLA-II datafiles are available to our knowledge.
Ambiguity is defined as two or more PSP sequences sharing the highest score for a single spectrum.
Table 2 additionally displays simulated spliced peptide results from Neo-Fusion searches allowing both cis- and trans-spliced peptides. Note that when searching for trans-spliced peptides, Neo-Fusion does not differentiate between cis and trans. Therefore, we were able to use the same simulated cis-spliced peptides to benchmark both cis- and trans-spliced peptides. The difference when searching for both cis and trans, compared to cis only, is a greatly increased search space, because the two parent peptides can now originate from multiple unique proteins. A consequence of this greater search space is a substantial increase in FDR compared to searching for cis only. While cis-spliced peptides had an observed FDR of 5%, trans-spliced peptides had substantially higher and more varied FDRs. Thus, searching for both types of spliced peptides can decrease the number of spliced discoveries when applying a confidence threshold, as observed for the HLA-I-Ip datafile. The extremely large number of possible trans-spliced sequences yields false-positive conditions similar to those observed for de novo peptide sequencing, making trans-spliced FDRs particularly diffcult to estimate.17 The high FDRs observed across all sample types for trans-spliced peptides indicate that Neo-Fusion is largely unable to identify trans-spliced peptides. The roughly 5% FDR observed for cis-spliced peptides is more manageable, and true positives can be confirmed using additional heuristics such as retention time prediction28 and fragment ion intensity prediction models.29 Therefore, the Neo-Fusion algorithm and software reported herein provide a broadly applicable method for the identification of cis-spliced peptides.
We compared Neo-Fusion’s ability to identify cis-spliced peptides with the state-of-the-art de novo peptide sequencing tool PEAKS 8.5.30 In the HLA-I sample, Neo-Fusion was able to identify 25% of simulated cis-spliced peptides at a roughly 5% FDR. PEAKS auto de novo sequencing software was used to generate five candidate sequences for each spectrum in the HLA-I sample using the following parameters: “Parent Mass Error Tolerance”: 10 ppm, “Fragment Mass Error Tolerance”: 0.02 Da, “Enzyme”: None, “Fixed Modifications”: Carbami-domethylation 57.02, “Variable Modifications”: Oxidation (M) 15.99. Only sequences with a minimum local confidence score over 80 for every amino acid position were kept.18 PEAKS results for spectra that were not used in the simulation were discarded. PSMs were assigned as correct if any of the five sequences matched the simulated spliced peptide and otherwise were marked as incorrect. Isoleucine and leucine were considered identical for this comparison. The PSMs were then sorted by average local confidence (ALC) from highest to lowest, and an FDR cutoff was drawn where 5% of results did not agree with the known simulated sequences (ALC = 93.89%).17 Using this method, PEAKS identified 20.1% of all simulated spliced peptides at a 5% FDR, whereas Neo-Fusion was able to identify 25.0% of all simulated spliced peptides at a similar FDR.
Comparison of the Neo-Fusion and PEAKS results showed a 41% overlap in the identified sequences; thus using both programs together yielded a total of 37% of the simulated spliced peptides present. The different results between these two methods can be largely attributed to the product ions used for scoring. Neo-Fusion uses the Morpheus score, which only counts b- and y-ions for HCD fragmentation.25 Although PEAKS searches for b- and y-ions, it also considers additionally ion types (a, b-H2O, b-NH3) for its scoring function.30 The current dependence of Neo-Fusion on the Morpheus score heavily favors spliced peptide spectra with more b- and y-ions for HCD or c- and z-ions for electron-transfer dissociation (Figure 4). Indeed, only 4% of the Neo-Fusion identified simulated spliced peptides had fewer than 12 combined b- and y-ions. When comparing the Neo-Fusion and PEAKS results for spectra that had 12 or more combined b- and y-ions, there is a better agreement with a 72% overlap in the identified sequences.
Figure 4.
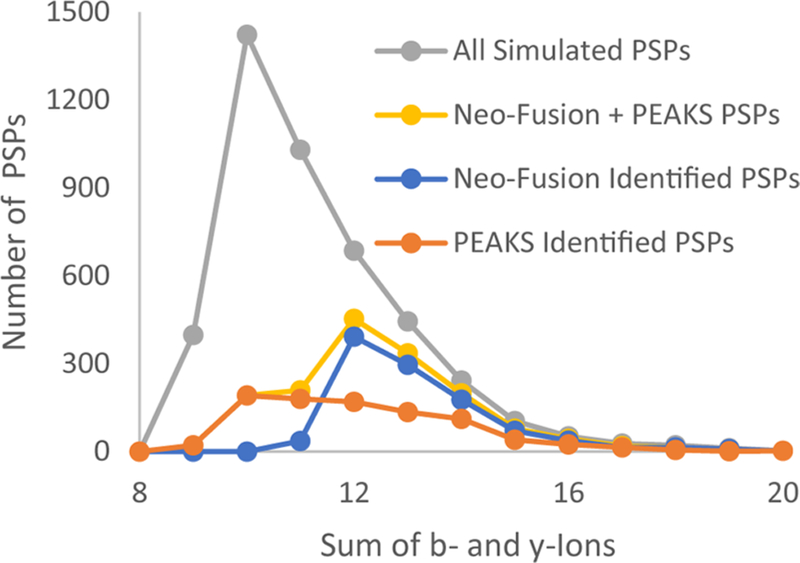
Distribution of Morpheus scores (the sum of b- and y-ions) for all simulated spliced peptides from the HLA-I datafile and for simulated spliced peptides identified by Neo-Fusion and PEAKS.
Having shown that Neo-Fusion is effective at identifying cis-spliced peptides in complex samples, we searched for real spliced peptides in the HLA-I-Ip fibroblast data. This file was previously reported by Liepe et al. to have over one-fourth of its immunopeptidome comprised of PSPs,6 and the list of previously identified PSP PSMs (LM_PSPs) was kindly provided by the authors (Table S2). Using Neo-Fusion, we were largely unable to reidentify the previously reported LM_PSPs. To ensure this was not an issue with Neo-Fusion, we appended the LM_PSPs directly into the reference database and conducted traditional database searches. Using MetaMorpheus22 and X! Tandem31 searches, we were still unable to reidentify the LM_PSPs. The observation that multiple other search engines were unable to reidentify these with similar search spaces suggests that many of these reported PSPs are likely incorrect.18,32,33 The inability to reidentify these LM_PSPs has been previously reported for the HLA-I fibroblast data.18
We further investigated the LM_PSPs using the SSRCalc web-based retention time prediction software (version Q) to observe whether the predicted hydrophobicity indexdees (HIs) for the reported PSPs correlate well with their observed retention times (RT).28 Reverse-phase HPLC, which separates peptides based on their affinity to a hydrophobic stationary phase, was used prior to MS/MS.19 We can therefore expect that the predicted HIs should strongly correlate with the observed RTs, and we find that the reported nonspliced peptides confirm such a correlation (Figure 5A). However, the LM_PSPs appear to largely deviate from the narrow range observed for the nonspliced peptides (Figure 5B). Rather, the LM_PSP distribution resembles the distribution of known false-positive decoy peptides (Figure 5C), suggesting that the LM_PSPs themselves are largely false-positive PSPs. A fraction of the reported PSPs do possess expected HIs for their RTs, but these can largely be explained as close sequences with similar HIs and fragmentation spectra. As additional evidence against the LM_PSPs, the HLA-I binding motifs34 observed between the LM_PSPs and the reported nonspliced peptides appear to disagree (Figure 5D−F).18 HLA-I antigens are well-known to bind to HLA-I at the N2 and C1 anchor residues (Figure 5D).35 The LM-PSPs display a weaker C1 specificity and a complete absence of N2 specificity compared to the nonspliced peptides. Finally, we were able to reassign nearly half of the LM_PSPs to nonspliced sequences at a 1% FDR (Table S2).22 Most of these nonspliced sequences were missed in the Liepe et al. analysis because of a smaller search space compared to that used here.33 Specifically, we both allowed a greater distribution of peptide lengths (8−16 amino acids instead of 9−12) and included oxidized methionine and other common PTMs identified in the samples with Global-PTM-Discovery.23 Many of the reassignments are similar sequences that are present in the reference database and can be easily misassigned.26 The reassigned PSMs possess superior HI fidelity and better fit the HLA-I binding motifs observed for the previously reported nonspliced identifications, suggesting that these are correct reassignments (Figure S2).
Figure 5.
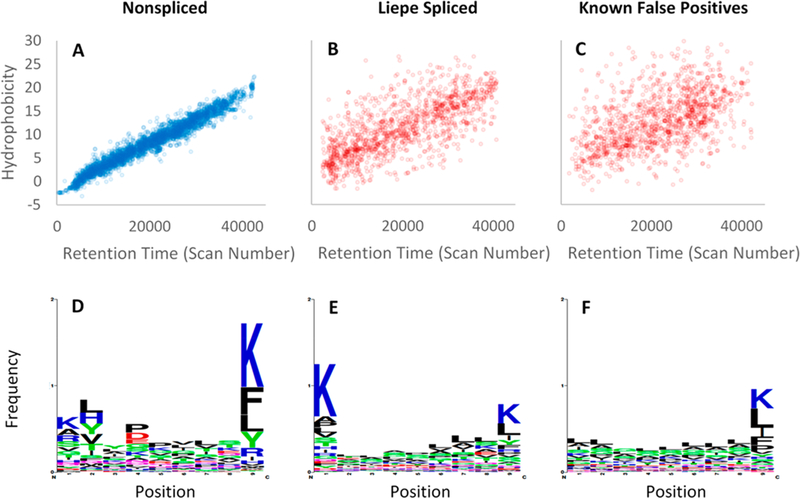
Comparison of peptide identifications previously reported by Liepe et al. for HLA-I-Ip of primary fibroblasts. (A) Nonspliced identifications show tight correlations between SSRCalc predicted HI and observed RT. (B) LM_PSPs show a weak correlation between HI and RT, indicating an underestimated FDR. (C) Known false positive PSMs are displayed to visualize the poor correlation between HI and RT for incorrect identifications. The false positives PSMs are the top scoring decoys from a MetaMorpheus search, where the number of decoys plotted is equal to the number of LM_PSPs. (D) The nonspliced sequences have common residues at the N2 (#2) and C1 (#9) anchor positions where the antigens bind to HLA-I. The LM_PSPs (E) and decoy identifications (F) display a weaker C1 specificity and an absence of N2 specificity.
Neo-Fusion identified a modest number of peptides that could be explained by cis-splicing in each of the three sample types (Table 2 and Tables S3−S5). These Neo-Fusion identified cis-spliced peptides have well-behaved retention times for their predicted hydrophobicities (Figure S3), with an expected number of outliers when considering the FDRs found in the benchmark experiment (Table 2). The number of identified cis-spliced peptides, when accounting for the false-negative rate found in the benchmark simulation experiment, is consistent with a previous upper limit estimate that 2−6% of the HLA-I-Ip is comprised of cis-PSPs.18 A larger number of trans-spliced peptides were identified by Neo-Fusion, but these were generally more ambiguous than the cis-spliced peptides and presumably suffer from the high FDRs observed in the benchmark simulation experiment (Table 2 and Tables S6− S8). Nevertheless, the RTs observed for these trans-spliced sequences are remarkably close to those expected for their predicted HIs (Figure S3).
In the tryptic digest of proteins from mouse islets, Neo-Fusion identified roughly 2% of all observed peptides as being explainable through cis-splicing events. This seems to be an unreasonable result, as the specific sample had been prepared using the filter-aided sample preparation protocol,36 in which small, endogenous peptides are removed by passing through a 30 kDa molecular weight cutoff filter prior to tryptic digestion. Therefore, if 2% of all identified peptides were indeed spliced peptides, then that would suggest that an unreasonable number of intact proteins, instead of peptides, are spliced in vivo. In pursuit of other explanations for these possibly spliced sequences, we expanded our nonspliced search space using the unreviewed UniProtKB/TrEMBL database, some common contaminant proteins, and Global-PTM-Discovery.23 Addi-tionally, liberal search conditions were used that allowed for semi-tryptic digestion, three missed cleavages, any peptide length greater than five amino acids, and an expanded precursor mass tolerance of 10 ppm. With this increased search space, we found that over 60% of the supposedly spliced sequences could be explained as less well-documented nonspliced peptides (Table S5 and Table S8). This illustrates how Neo-Fusion, while able to identify putative spliced sequences, cannot determine sequence origin definitively. The Neo-Fusion reported sequences may be proteasome-generated spliced peptides, but other possible explanations for these sequences include proteins that are unannotated in UniProtKB/Swiss-Prot, alternative RNA-splicing, chromoso-mal rearrangements, and single amino acid variants.37 This uncertainty necessitates that putative spliced peptide sequences be confirmed by follow-up validation experiments to be certain of their origins. One such method might be through in vitro proteasomal degradation assays.38 In the absence of such validation, putative spliced sequences identified by Neo-Fusion or other strategies should be regarded merely as hypotheses rather than as unambiguous findings.
At present, given the wide variety of possible sources for apparently spliced sequences in complex proteomic samples, it is not possible to definitively determine the prevalence of spliced peptides in biology. Rather, as indicated previously by Mylonas et al.,18 we can only place an upper limit, in the range of 1−4%, on their frequency. Future work will focus on automated methods to validate the putative spliced sequences discovered by Neo-Fusion. The use of predicted hydro-phobicity in comparison to retention time,39 predicted binding affinity for HLA-I antigens,35 spectral libraries,40 and machine learning predicted fragmention intensities29 are all promising developments to reduce the false-positive rate and improve the sensitivity of spliced peptide discovery.
CONCLUSIONS
The identification of spliced peptides in complex samples is a fascinating challenge with important implications in funda-mental biology, autoimmune responses, and cancer immuno-therapy. Standard database search strategies used for peptide identification are ineffective for spliced peptide analysis, because of the combinatorial explosion in search space size caused by the relatively unconstrained nature of the spliced peptide sequences considered. We present an algorithm for the identification of spliced peptides in MS data obtained from complex biological samples. This algorithm was implemented in the software program Neo-Fusion, and the software’s performance was benchmarked through the detection of simulated spliced peptides in real-world data. We were able to detect 25% of such simulated cis-spliced peptides at an FDR of roughly 5%. In the selected HLA-I-Ip sample, Neo-Fusion found that approximately 1% of identified peptide sequences could be explained as cis-spliced peptides. Neo-Fusion is written in the C# programming language, is open-source, and is publicly available at https://github.com/zrolfs/MetaMorpheus/tree/Neo-Fusion.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Cancer Institute of the National Institutes of Health (NIH) under Award Numbers U24CA199347 and R01CA193481. The content is solely the responsibility of the authors and does not necessarily represent the offcial views of the NIH. We thank A. D. Attie for originally introducing us to this interesting problem.
ABBREVIATIONS
- PTM
post-translational modification
- MS
mass spectrometry
- PSP
proteasome-generated spliced peptide
- HLA-I/II-Ip
human leukocyte antigen class I or class II immunopeptidome
- FDR
false discovery rate
- HPLC-MS/MS
high pressure liquid chromatography and tandem mass spectrometry
- HCD
higher-energy collisional dissociation
- PSM
peptide-spectrum match
- ALC
average local confidence
- LM_PSPs
spliced peptides reported by Liepe et al
- HI
hydrophobicity index
- RT
retention time
Footnotes
Accession Codes
The mass spectrometry data have been previously deposited in the PRIDE41 Archive at www.ebi.ac.uk/pride/archive under accession numbers PXD000394 for HLA-I (20130504_EX-Q3_MiBa_SA_Fib-2.raw) and PXD004746 for HLA-II (e001323.raw). All nine trypsin files are available via FTP from the PeptideAtlas42 data repository by accessing the following link: http://www.peptideatlas.org/PASS/PASS00470.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Wirth TC; Kühnel F Neoantigen Targeting−Dawn of a New Era in Cancer Immunotherapy? Front. Immunol 2017, 8, 1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Delong T; Wiles TA; Baker RL; Bradley B; Barbour G; Reisdorph R; Armstrong M; Powell RL; Reisdorph N; Kumar N; et al. Pathogenic CD4 T Cells in Type 1 Diabetes Recognize Epitopes Formed by Peptide Fusion. Science (Washington, DC, U. S.) 2016, 351 (6274), 711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ward JP; Gubin MM; Schreiber RD The Role of Neoantigens in Naturally Occurring and Therapeutically Induced Immune Responses to Cancer. Adv. Immunol 2016, 130, 25–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Creech AL; Ting YS; Goulding SP; Sauld JFK; Barthelme D; Rooney MS; Addona TA; Abelin JG The Role of Mass Spectrometry and Proteogenomics in the Advancement of HLA Epitope Prediction. Proteomics 2018, 18 (12), 1700259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vigneron N; Stroobant V; Chapiro J; Ooms A; Degiovanni G; Morel S; van der Bruggen P; Boon T; Van den Eynde BJ An Antigenic Peptide Produced by Peptide Splicing in the Proteasome. Science (Washington, DC, U. S.) 2004, 304 (5670), 587–590. [DOI] [PubMed] [Google Scholar]
- (6).Liepe J; Marino F; Sidney J; Jeko A; Bunting DE; Sette A; Kloetzel PM; Stumpf MPH; Heck AJR; Mishto M A Large Fraction of HLA Class I Ligands Are Proteasome-Generated Spliced Peptides. Science 2016, 354 (6310), 354–358. [DOI] [PubMed] [Google Scholar]
- (7).Wiles TA; Delong T; Baker RL; Bradley B; Barbour G; Powell RL; Reisdorph N; Haskins K An Insulin-IAPP Hybrid Peptide Is an Endogenous Antigen for CD4 T Cells in the Non-Obese Diabetic Mouse. J. Autoimmun 2017, 78, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mishto M; Liepe J Post-Translational Peptide Splicing and T Cell Responses. Trends Immunol 2017, 38 (12), 904–915. [DOI] [PubMed] [Google Scholar]
- (9).Hanada K; Yewdell JW; Yang JC Immune Recognition of a Human Renal Cancer Antigen through Post-Translational Protein Splicing. Nature 2004, 427 (6971), 252–256. [DOI] [PubMed] [Google Scholar]
- (10).Warren EH; Vigneron NJ; Gavin MA; Coulie PG; Stroobant V; Dalet A; Tykodi SS; Xuereb SM; Mito JK; Riddell SR; et al. An Antigen Produced by Splicing of Noncontiguous Peptides in the Reverse Order. Science (Washington, DC, U. S.) 2006, 313 (5792), 1444–1447. [DOI] [PubMed] [Google Scholar]
- (11).Dalet A; Vigneron N; Stroobant V; Hanada K-I; Van den Eynde BJ Splicing of Distant Peptide Fragments Occurs in the Proteasome by Transpeptidation and Produces the Spliced Antigenic Peptide Derived from Fibroblast Growth Factor-5. J. Immunol 2010, 184 (6), 3016–3024. [DOI] [PubMed] [Google Scholar]
- (12).Dalet A; Robbins PF; Stroobant V; Vigneron N; Li YF; El-Gamil M; Hanada K. -i.; Yang JC; Rosenberg SA; Van den Eynde BJ An Antigenic Peptide Produced by Reverse Splicing and Double Asparagine Deamidation. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (29), E323–E331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Michaux A; Larrieu P; Stroobant V; Fonteneau J-F; Jotereau F; Van den Eynde BJ; Moreau-Aubry A; Vigneron N A Spliced Antigenic Peptide Comprising a Single Spliced Amino Acid Is Produced in the Proteasome by Reverse Splicing of a Longer Peptide Fragment Followed by Trimming. J. Immunol 2014, 192 (4), 1962–1971. [DOI] [PubMed] [Google Scholar]
- (14).Berkers CR; de Jong A; Schuurman KG; Linnemann C; Meiring HD; Janssen L; Neefjes JJ; Schumacher TNM; Rodenko B; Ovaa H Definition of Proteasomal Peptide Splicing Rules for High-Efficiency Spliced Peptide Presentation by MHC Class I Molecules. J. Immunol 2015, 195 (9), 4085–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Berkers CR; de Jong A; Schuurman KG; Linnemann C; Geenevasen JAJ; Schumacher TNM; Rodenko B; Ovaa H Peptide Splicing in the Proteasome Creates a Novel Type of Antigen with an Isopeptide Linkage. J. Immunol 2015, 195 (9), 4075–4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ebstein F; Textoris-Taube K; Keller C; Golnik R; Vigneron N; Van den Eynde BJ; Schuler-Thurner B; Schadendorf D; Lorenz FKM; Uckert W; et al. Proteasomes Generate Spliced Epitopes by Two Different Mechanisms and as Efficiently as Non-Spliced Epitopes. Sci. Rep 2016, 6 (1), 24032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Devabhaktuni A; Elias JE Application of de Novo Sequencing to Large-Scale Complex Proteomics Data Sets. J. Proteome Res 2016, 15 (3), 732–742. [DOI] [PubMed] [Google Scholar]
- (18).Mylonas R; Beer I; Iseli C; Chong C; Pak H-S; Gfeller D; Coukos G; Xenarios I; Muller M; Bassani-Sternberg M Estimating the Contribution of Proteasomal Spliced Peptides to the HLA-I Ligandome. Mol. Cell. Proteomics 2018, mcp.RA118.000877. [DOI] [PMC free article] [PubMed]
- (19).Bassani-Sternberg M; Pletscher-Frankild S; Jensen LJ; Mann M Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation. Mol. Cell. Proteomics 2015, 14 (3), 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Khodadoust MS; Olsson N; Wagar LE; Haabeth OAW; Chen B; Swaminathan K; Rawson K; Liu CL; Steiner D; Lund P; et al. Antigen Presentation Profiling Reveals Recognition of Lymphoma Immunoglobulin Neoantigens. Nature 2017, 543 (7647), 723–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shortreed MR; Wenger CD; Frey BL; Sheynkman GM; Scalf M; Keller MP; Attie AD; Smith LM Global Identification of Protein Post-Translational Modifications in a Single-Pass Database Search. J. Proteome Res 2015, 14 (11), 4714–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Solntsev SK; Shortreed MR; Frey BL; Smith LM Enhanced Global Post-Translational Modification Discovery with MetaMorpheus. J. Proteome Res 2018, 17 (5), 1844–1851. [DOI] [PubMed] [Google Scholar]
- (23).Li Q; Shortreed MR; Wenger CD; Frey BL; Schaffer LV; Scalf M; Smith LM Global Post-Translational Modification Discovery. J. Proteome Res 2017, 16 (4), 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry−based Proteomics. Nat. Methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wenger CD; Coon JJ A Proteomics Search Algorithm Specifically Designed for High-Resolution Tandem Mass Spectra. J. Proteome Res 2013, 12 (3), 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Colaert N; Degroeve S; Helsens K; Martens L Analysis of the Resolution Limitations of Peptide Identification Algorithms. J. Proteome Res 2011, 10 (12), 5555–5561. [DOI] [PubMed] [Google Scholar]
- (27).Blum JS; Wearsch PA; Cresswell P Pathways of Antigen Processing. Annu. Rev. Immunol 2013, 31, 443–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krokhin OV Sequence-Specific Retention Calculator. Algorithm for Peptide Retention Prediction in Ion-Pair RP-HPLC: Application to 300- and 100-Å Pore Size C18 Sorbents. Anal. Chem 2006, 78 (22), 7785–7795. [DOI] [PubMed] [Google Scholar]
- (29).Zhou X-X; Zeng W-F; Chi H; Luo C; Liu C; Zhan J; He S-M; Zhang Z PDeep: Predicting MS/MS Spectra of Peptides with Deep Learning. Anal. Chem 2017, 89 (23), 12690–12697. [DOI] [PubMed] [Google Scholar]
- (30).Ma B; Zhang K; Hendrie C; Liang C; Li M; Doherty-Kirby A; Lajoie G PEAKS: Powerful Software for Peptidede Novo Sequencing by Tandem Mass Spectrometry. Rapid Commun. Mass Spectrom 2003, 17 (20), 2337–2342. [DOI] [PubMed] [Google Scholar]
- (31).Craig R; Beavis RC A Method for Reducing the Time Required to Match Protein Sequences with Tandem Mass Spectra. Rapid Commun. Mass Spectrom 2003, 17 (20), 2310–2316. [DOI] [PubMed] [Google Scholar]
- (32).Shteynberg D; Nesvizhskii AI; Moritz RL; Deutsch EW Combining Results of Multiple Search Engines in Proteomics. Mol. Cell. Proteomics 2013, 12 (9), 2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tessier D; Lollier V; Larre C; Rogniaux H Origin of Disagreements in Tandem Mass Spectra Interpretation by Search Engines. J. Proteome Res 2016, 15 (10), 3481–3488. [DOI] [PubMed] [Google Scholar]
- (34).Crooks GE; Hon G; Chandonia J-M; Brenner SE WebLogo: A Sequence Logo Generator. Genome Res 2004, 14 (6), 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jurtz V; Paul S; Andreatta M; Marcatili P; Peters B; Nielsen M NetMHCpan-4.0: Improved Peptide−MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol 2017, 199 (9), 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wisniewski JR; Zougman A; Nagaraj N; Mann M Universal Sample Preparation Method for Proteome Analysis. Nat. Methods 2009, 6 (5), 359–362. [DOI] [PubMed] [Google Scholar]
- (37).Pearson H; Daouda T; Granados DP; Durette C; Bonneil E; Courcelles M; Rodenbrock A; Laverdure J-P; Côte C; Mader S; et al. MHC Class I−associated Peptides Derive from Selective Regions of the Human Genome. J. Clin. Invest 2016, 126 (12), 4690–4701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liepe J; Mishto M; Textoris-Taube K; Janek K; Keller C; Henklein P; Kloetzel PM; Zaikin A The 20S Proteasome Splicing Activity Discovered by SpliceMet. PLoS Comput. Biol 2010, 6 (6), e1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Dorfer V; Maltsev S; Winkler S; Mechtler K CharmeRT: Boosting Peptide Identifications by Chimeric Spectra Identification and Retention Time Prediction. J. Proteome Res 2018, 17, 2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shao W; Pedrioli PGA; Wolski W; Scurtescu C; Schmid E; Vizcaíno JA; Courcelles M; Schuster H; Kowalewski D; Marino F; et al. The SysteMHC Atlas Project. Nucleic Acids Res 2018, 46 (D1), D1237–D1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Vizcaíno JA; Cote RG; Csordas A; Dianes JA; Fabregat A; Foster JM; Griss J; Alpi E; Birim M; Contell J; et al. The Proteomics Identifications (PRIDE) Database and Associated Tools: Status in 2013. Nucleic Acids Res 2012, 41, D1063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Desiere F; Deutsch EW; King NL; Nesvizhskii AI; Mallick P; Eng J; Chen S; Eddes J; Loevenich SN; Aebersold R The PeptideAtlas Project. Nucleic Acids Res 2006, 34 (90001), D655–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.