

Abstract
Individuals with MECP2 duplication syndrome (MDS) have varying degrees of severity in their mobility, hand use, developmental skills, and susceptibility to infections. In this study, we examine the relationship between duplication size, gene content, and overall phenotype in MDS using a clinical severity scale. Other genes typically duplicated within Xq28 (e.g. GDI1, RAB39B, FLNA) are associated with distinct clinical features independent of MECP2. We additionally compare the phenotype of this cohort (n=48) to other reported cohorts with MDS. Utilizing existing indices of clinical severity in Rett Syndrome, we found that larger duplication size correlates with higher severity in total clinical severity scores (r=.36; p=.02), and in total motor behavioral assessment inventory scores (r=.31; p=.05). Greater severity was associated with having the RAB39B gene duplicated, although most of these participants also had large duplications. Results suggest that developmental delays in the first 6 months of life, hypotonia, vasomotor disturbances, constipation, drooling, and bruxism are common in MDS. This is the first study to show that duplication size is related to clinical severity. Future studies should examine whether large duplications which do not encompass RAB39B also contribute to clinical severity. Results also suggest the need for creating an MDS specific severity scale.
Keywords: MECP2, Genotype, Phenotype, Clinical Severity
Introduction
MECP2 Duplication syndrome (MDS) is a rare X-linked genomic disorder primarily affecting males that is associated with interstitial chromosomal duplications at Xq28 encompassing the MECP2 gene1. This gene encodes methyl CpG binding protein 2 (MeCP2), a critical regulator of neuronal gene transcription that is required for normal brain maturation2,3. Loss of MeCP2 function is the primary cause of Rett Syndrome (RTT), a distinct neurodevelopmental disorder affecting primarily females exhibiting some symptom overlap with MDS. Fewer than 200 cases of MDS have been described worldwide, however, it is likely underdiagnosed and estimated to account for at least 1–2% of all cases of X-linked intellectual disability (ID)1. The core clinical phenotype of MDS involves infantile hypotonia, global developmental delays, choreiform movements, progressive spasticity and recurrent respiratory infections4–6. The shortest duplication sufficient for the core phenotype contains only MECP2 and IRAK1 genes7–9 and the pathogenic role of increased MECP2 dosage is further supported by transgenic mouse models of MDS with overexpression of mouse or human MECP2 that develop impaired coordination, seizures, and impairments in learning and memory10–12. The spectrum of phenotypic features associated with MDS is gradually being defined, with the largest published cohort to date (n=59) reporting high rates of constipation, stereotypic movements, epilepsy, decreased pain sensitivity, scoliosis, motor regression and specific facial dysmorphologies in addition to the aforementioned features13.
Our own prior studies reveal considerable inter-patient differences in severity of several symptom domains within MDS including ambulation, hand function, non-verbal communication and susceptibility to infection14; furthermore, developmental regression occurs in only half of the population with highly variable age of onset15. There is limited published information regarding factors that contribute to this observed variability in symptom severity. Although prior studies have suggested that duplication size is not related to the phenotype4,13, this has not been examined within the context of a severity scale (as opposed to discrete symptoms). Additionally, it is unknown whether specific phenotypic features contribute disproportionately to overall clinical severity and/or changes in severity within this population. This information would aid development of a dynamic index of clinical severity with content validity for MDS as existing scales in use were created and validated for longitudinal studies in RTT16,17. In addition, the degree to which presence/absence of additional salient genes (e.g. SRPK3, L1CAM, FLNA, GDI1, RAB39B) within the duplicated interval contribute to severity has not yet been examined in detail. For example, other studies have noted that increased expression of GDI1 (without duplication of MECP2) is associated with impaired cognition18 as well as autism19. Increased dosage of RAB39B has also been associated with mild to moderate ID and behavioral problems20,21. SRPK3 is involved in muscle growth and homeostasis with overexpression causing muscle degeneration in mice22. A neurological phenotype due to L1CAM overexpression has not been reported, but loss of function mutations are associated with corpus callosum abnormalities on neuroimaging23 that are also seen in MDS24. Duplications in FLNA are associated with bowel dysfunction that is also very common in MDS25,26.
The recent report of phenotypic reversal after antisense oligonucleotide treatment in an animal model of MDS27 suggests disease-modifying therapies may be possible. Clinical trials to test such future therapies will require standardized measures of clinical severity with content validity for MDS that can be applied by investigators across all study sites. Here, we present baseline data from our natural history study of MDS, including how these data compare to previous reports in the literature. We extend the prior studies by utilizing existing standardized indices of clinical severity for RTT while emphasizing relevant phenotypic features in MDS important for improving content validity of these scales. We additionally report whether duplication size or duplication of additional genes within the respective breakpoint intervals contribute to clinical severity. We hypothesize that differences in severity will be noted according to the presence/absence of additional genes within the duplicated interval.
Methods
Participants
This cohort of patients is part of a broader longitudinal, natural history study of Rett Syndrome, and Rett-Related disorders including MDS. The Rett Syndrome and Related Disorders natural history study consists of 14 sites around the United States. The sites are located in centers that have existing clinical and research specialists in these respective syndromes and are geographically spread around the country to facilitate enrollment. 48 participants, including 5 females and 43 males, ranging between the ages of 1–28 years have enrolled in the study to date (Table 1). For each participant, all data is gathered in person via direct clinical exam (by a child neurologist or geneticist), and via parent report. Participants are evaluated on a yearly basis. Given the smaller number of participants who have participated in longitudinal follow-up to date, only baseline data is being reported at this time. The families of all participants provided written informed consent and all procedures performed in the studies were done in accordance with the ethical standards of the respective institutional research committees. Data within the consortium is routinely checked for compliance. The natural history study is registered with Clinicaltrials.gov: NCT02738281.
Table 1:
Comparison of clinical features in the present study vs. prior published studies in the literature
| Present Study | Miguet et al. 2018 | Lim et al. 2017 | Van Esch et al. 2005 | |
|---|---|---|---|---|
| Number of patients | 48 | 59 | 56 | 13 |
| Gender | 43 male/5 female | 59 male | 49 male/7 female | 13 male |
| Age at Diagnosis | 4.16 years (50 months) | 10 years (120 months) | 36 months/male; 24 months/female | NR |
| Age at Exam/self-report | 9.01 years | 11.7 years | 7.9 years | |
| Abnormal development in first 6 months of life | 31/48 (65%) | NR | NR | NR |
| Hypotonia | 42/48 (88%) | 57/58 (98.3%) | 31/48 (64.6%) male; 4/7 (57.1%) female | 12/12 (100%) |
| Recurrent Infections | 27/48 (56%) | 49/55 (89.1%) | 38/49 (77.6%) males; 3/7 (42.9%) females | 5/9 (55.6%) |
| Seizures | 21/48 (43.8%) | 35/59 (59.3%) | 21/49 (42.9%) male; 3/6 (50.0%) female | 4/9 (44.4%) |
| Constipation | 39/48 (81%) | 43/55 (78.2%) | 39/49 (79.6%) | 0/12 (0%) |
| Reflux | 21/48 (44%) | 34/51 (66.7%) | 26/49 (53.1%) male; 3/7 (42.9%) female | NR |
| Drooling | 37/48 (77%) | 40/50 (80.0%) | NR | NR |
| Vasomotor Disturbances | 35/48 (73%) | 15/31 (48.4%) | NR | NR |
| Breathing Disturbances | 12/48 (25%) | NR | NR | NR |
| Scoliosis and/or Kyphosis | 9/48 (19% with scoliosis as a diagnosis) | 23/43 (53.5%) - includes scoliosis and/or kyphosis | 10/46 (21.7%) males; 0/5 (0%) female (scoliosis) | NR |
| Anxiety | 14/48 (29%) | NR | NR | NR |
| Sleep problems | 27/48 (43.8%) | NR | 13/48 (27.1%) male; 1/7 (14,3%) female | NR |
| Bruxism | 34/48 (71%) | 33/46 (71.7%) | 80% | NR |
| Regression | 12/48 (25%) | 12/31 (38.7%) (post seizure onset) | NR | NR |
| High Pain Tolerance | 32/48 (67%) | 29/37 (78.4%) | 25/50 (50.0%) | NR |
| Low activity for age | 29/48 (60%) | NR | NR | NR |
| Irritability | 28/48 (58%) | NR | NR | NR |
| Early Death (before age 25 years) | 2/48 (4%) | 9/59 (15.3%) | NR | 6/11 (54.5%) |
Scales
Clinical Severity Scale (CSS) – clinician rating:
The CSS was developed for use in RTT and has been used to assess almost 2000 children, adolescents, and adults with RTT and related disorders who have been enrolled in the natural history study28. It is a composite score based on thirteen individual ordinal categories measuring common clinical features during an in-person exam (e.g. independent sitting, hand use, scoliosis, language, seizures, autonomic symptoms, onset of stereotypies, regression, head growth, etc.). Individual item scores range from 0 to 4 or 0 to 5 with 0 representing the least severe and 4 or 5 representing increasing severity16. Lower total scores represent milder severity (see supplementary files).
Motor Behavioral Assessment Scale (MBA) – clinician rating:
The MBA has also been used to assess almost 2000 children, adolescents, and adults with RTT and related disorders who have been enrolled in the natural history study28. The current version of this scale consists of 34 items across three subscales: Behavior/Social (irritability, aggression, poor eye gaze, sustained interest, etc.), Orofacial (bruxism, mouthing objects or hands, biting self, breath holding, hyperventilation, etc.), and Motor (bradykinesia, dystonia, ataxia, chorea, etc.), the scores of which are based on current functioning. These are scored during a clinical interview and in-person exam by a specialist once per year. Items are captured on a 5-point Likert scale. Lower total scores indicate milder disease severity (see supplementary files).
Additional clinical features (constipation, reflux, drooling, bruxism, anxiety, sleep problems, irritability, etc.)
Salient clinical features were quantified via parent report and clinician observation according to a scale (none, occasional, frequent, very frequent, constant). For the purposes of this study, these were recoded to determine the presence or absence of these features. The number of infections was quantified over the preceding year at the in-person physician history. The presence of hypotonia, and developmental delays within the first 6 months of life was also quantified via physician exam.
Cytogenetic and Molecular Analyses
Confirmation of duplication was required for entry into the study. Genetic analyses were performed by a variety of laboratories depending upon where the participant was originally diagnosed (e.g. Baylor College of Medicine, University of Chicago, Signature Genomics, Greenwood Genetic Center, Athena, CHOP, Emory, Gene Dx, Duke, etc.). Most participants had their duplications detected via Array Comparative Genomic Hybridization (Array-CGH), while others (mostly older participants) had Multiplex-Ligation-dependent Probe Amplification (MLPA). Some had an earlier version of CMA (version 5), such that clearly delineated breakpoints could not be determined. Genomic positions are based on data from the human genome assembly GRCH37/hg1929.
Results
Demographics
48 participants have enrolled in this study to date. The mean age of participants at evaluation is 9.01 years (SD=6.79 years), and the mean age of diagnosis was 4.16 years (SD=5.38 years). All participants in this study lived in the home; 58% of mothers and 52% of fathers had a bachelor’s or advanced degree. 58% of mothers and 75% of fathers were employed outside of the home.
Clinical Features
Table 1 lists the demographics of participants in this study, along with the frequency of salient clinical features as compared to prior published studies5,13,30,31. When parents were asked to list their chief concern about their child, many reported that lack of effective communication (27%) was their most pressing concern, while 23% reported that seizures were their greatest concern. Of note is that 65% of parents reported that their children had significant delays within the first 6 months of life. In fact, 17 participants were diagnosed with MDS at age 12 months or younger, and 53% of the sample was diagnosed prior to age 2 years. Hypotonia, drooling, vasomotor disturbances (cold hands and/or feet, skin mottling), bruxism, and a high pain tolerance were commonly noted features in this cohort. A diagnosis of epilepsy was only noted in 21/48 participants (44%); and, 27/48 (56%) exhibited recurrent respiratory infections at the time of evaluation (27/48). In addition, only 12 participants had a documented regression at the time of their visit, with the age of those who had regressed being slightly older (M=11.87 years, SD=6.08) as compared to those who had not regressed (M=8.25 years, SD=6.82 years), although this is not statistically significant (p=.11).
Clinical Severity Scale
First, descriptive statistics were conducted to examine CSS scores. Scores on the CSS ranged from 4–38 (M=16.57, SD=7.34). Next, because of the significant differences found between males vs. females with MDS in prior studies32, an ANOVA was used to test for any differences in CSS scores X gender. Significant differences were noted in scores of males vs. females, with males having significantly higher CSS scores (M=17.52; SD=6.95) than females (M=8.60; SD=5.90) (F 1, 47 = 7.55; p<.009). Regardless of gender, the most commonly endorsed items were those that related to developmental functioning (e.g. hand use, ambulation, nonverbal and verbal language, sitting) and stereotypies. Items that were least likely endorsed related to microcephaly and scoliosis.
Motor Behavioral Assessment
A similar approach to analyses was followed as was described for the CSS above. Scores on the MBA ranged from 3–69 (M=33.09; SD=14.03). Significant differences were noted in the scores of males vs. females, with males exhibiting higher (i.e. more severe) scores (M=34.90; SD=13.36) than females (M=17.80; SD=10.38). Most commonly endorsed items related to developmental milestones (as noted with the CSS), bruxism, air/saliva expulsion, pain insensitivity, poor eye gaze, and apraxia/ataxia. Least endorsed items related to self-injury, aggression, breath holding, hyperventilation, myoclonus, and dystonia.
Genetic Breakpoints and Duplication Size
Information on specific breakpoints was available for 40 participants (See Figure 1). Duplication sizes ranged between 210,970–14,461,013bp, with a mean size of 2,375,105bp. Information regarding carrier status was available for 29 participants who chose to have additional genetic testing; n=24 had inherited duplications (with a maternal carrier), while n=5 had de novo duplications. Besides MECP2 and IRAK1, the salient genes within duplication breakpoints with putative functions relevant to clinical aspects of MDS were SRPK3, L1CAM, FLNA, GDI1, and RAB39B.
Figure 1:
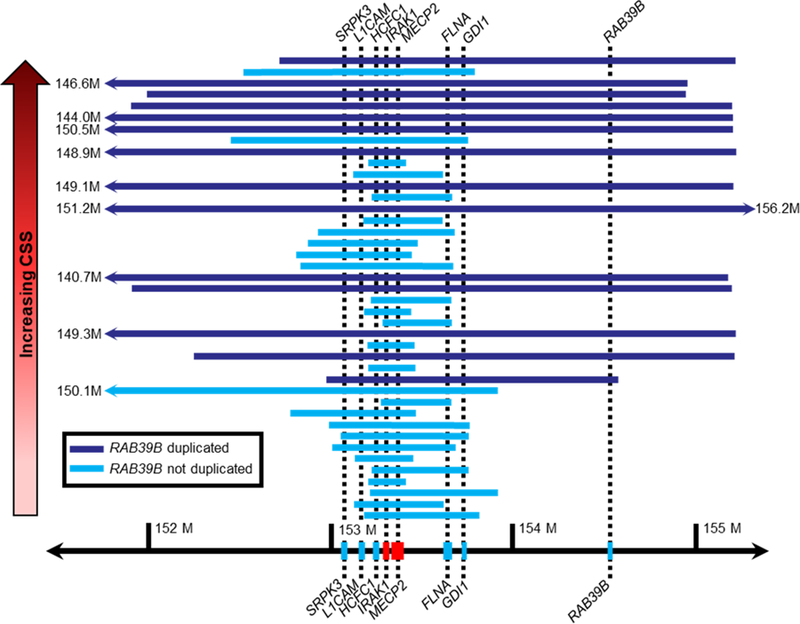
Xq28 Duplication Size X Clinical Severity Score
A larger duplication size significantly correlated with higher severity in total CSS score (r=.36; p=.02), and with greater severity in total MBA score (r=.31; p=.05). Since duplication size is significantly associated with higher CSS and MBA scores, ANCOVA’s (controlling for duplication size) were performed to determine whether the presence/absence of individual genes within the breakpoint interval independently contributes to severity. Testing was done for each of the five genes mentioned above using an ANCOVA. For the CSS, presence of the RAB39B gene in the duplication was associated with greater severity independent of duplication size. A trend toward greater severity was noted when the duplication involved L1CAM. These results (uncorrected values for n=5 statistical ANCOVA tests) are summarized in Table 2. In addition, to address any concerns regarding multiple comparisons, the Benajmini-Hochberg procedure33 was performed, and these results are detailed in the Supplementary Table 3. This test corrects for the false discovery rate. The results remain significant using this correction procedure, that the presence of the RAB39B gene contributes to greater severity independently of duplication size. It is important to note that the false discovery rate was set at .10, based on the literature33 and given the few studies that have been done in this area. This is a conservative rate; however, even if the false discovery rate was set to .05, the results and interpretations of the findings would remain unchanged. The presence/absence of these genes did not have a significant effect on either the total MBA score or its domain specific subscales. These results are summarized in Table 3, and corrected values are presented in Supplementary Table 4 (results were unchanged with regard to significance or interpretation). Follow-up exploratory analyses were conducted to understand better the specific CSS items that seemed to differentiate the severity of participants with and without the RAB39B gene duplication. Follow-up ANOVA’s (including Bonferroni’s correction for multiple comparisons) revealed that microcephaly (partial eta squared = .15), and worse impairment in independent sitting (partial eta squared = .19), ambulation (partial eta squared = .30), and hand use (partial eta squared = .12) all accounted for the differences in severity.
Table 2:
Mean Total CSS scores by the absence/presence of additional gene duplications within the BP interval controlling for duplication size
| SRPK3 | L1CAM | FLNA | GDI1 | RAB39B | |
|---|---|---|---|---|---|
| Absence | 12.33 (4.35) N=15 |
11.75 (4.39) N=12 |
13.64 (4.94) N=11 |
12.72 (4.60) N=18 |
13.08 (5.34) N=26 |
| Presence | 17.88 (7.42) N=25 |
17.54 (7.15) N=28 |
16.62 (7.46) N=29 |
18.32 (7.57) N=22 |
20.86 (6.85) N=14 |
| F value | F=2.78; p=.10 | F=3.13; p=.08 | F=.148; p=.743 | F=2.81; p=.102 | F=8.88; p=.005 |
Table 3:
Mean Grand Total MBA scores by the presence/absence of additional gene duplications within the BP interval controlling for duplication size
| SRPK3 | L1CAM | FLNA | GDI1 | RAB39B | |
|---|---|---|---|---|---|
| Absence | 26.53 (8.86) N=15 |
25.08 (9.25) N=12 |
27.36 (11.17) N=11 |
27.11 (10.19) N=18 |
27.92 (10.71) N=26 |
| Presence | 33.52 (13.52) N=25 |
33.39 (12.80) N=28 |
32.24 (12.69) N=29 |
34.00 (13.30) N=22 |
36.43 (13.65) N=14 |
| F value | F=.885; p=.35 | F=1.77; p=.19 | F=.172; p=.68 | F=.742; p=.395 | F=1.06; p=.31 |
Discussion
The results of this study extend the clinical characterization and genotype-phenotype correlations in MDS and establish the importance of duplication size and gene content as contributing to phenotypic severity. Several large series of patients with MDS have now been described in the literature5,13,30,31. This study examined some previously unreported features in MDS from those cohorts, including delays within the first 6 months of life (common in 65% of this cohort), breathing disturbances (25%), hypoactivity (also quite common at 60%), and behavioral features such as anxiety (29%) and irritability (58%). Aggression and self-injury were not common amongst participants in this study. Consistent with previous cohorts, this study demonstrated that hypotonia, constipation, bruxism, vasomotor disturbances (most often cold hands and/or feet, skin mottling), and high pain tolerance are common in MDS. As found in previous studies32, clinical severity was milder in the females enrolled in this cohort as compared to the males. In contrast, while recurrent respiratory infections are reported to be quite common in MDS, they were seen less frequently in this cohort at the time of evaluation. This may, in part, reflect sampling bias perhaps due to lack of participation by patients perceived by parents to be too fragile to travel to a study site. Alternatively, it is possible that increased awareness of this aspect of the syndrome has resulted in more primary prevention practices including use of airway clearance devices, better hand hygiene, exposure avoidance, and more timely seeking of medical care such that respiratory infections are more likely to be milder or avoided altogether. The rate of epilepsy in this cohort was also lower compared to the recent French cohort13, but was consistent with that of other cohorts5,30. This could reflect the younger age of participants at the time of evaluation and will be important to track over time. In addition, it will be important to track any changes to regression status over time, as this feature was also less frequent in this cohort (also perhaps reflecting a younger age and/or potential sampling bias).
This study is the first to demonstrate that duplication size is related to phenotypic severity; the larger the duplication, the more severely affected the participant was on both indices of clinical severity. In further examining the results and the degree to which the presence/absence of specific genes impacts severity beyond duplication size, individuals with a duplication of RAB39B were more severely affected regarding having microcephaly, and worse motor impairments in terms of hand use, ambulation, and sitting independently. It is important to note that this was independent of duplication size alone, given that duplication size was statistically controlled for in the analyses. This is also the first study to demonstrate that duplications of both MECP2 and RAB39B confer greater clinical severity. RAB39B is strongly expressed in the brain, encoding a small GTPase that regulates neuronal development21,34. Like MECP2, RAB39B appears to be a dosage-sensitive gene and a prior study demonstrated that duplications of RAB39B alone are associated with ID and behavioral problems21, independently of duplication of the MECP2 gene itself. It is not surprising then to see higher severity scores with duplication of RAB39B in our MDS patients. A caveat to these findings, however, is that in this study all but one of the cases with larger duplications and greater severity scores encompassed RAB39B. Only one participant with a large duplication that did not encompass RAB39B had a lower severity score. In future, larger scale studies, to continue to tease apart the degree to which an additional duplication of RAB39B contributes to clinical severity in MDS, it will be important to continue to examine the degree to which large duplications which do not encompass this gene contribute to clinical severity. In other words, if equally large duplications that do not encompass the RAB39B result in lower levels of clinical severity as was seen in one case in this study, then it can be stated with greater confidence that RAB39B independently confers greater severity in MDS. The specific associated features of motor impairments and microcephaly do not recapitulate findings in published reports of RAB39B duplication, however, the neurodevelopmental interactions are likely complex and make it challenging to predict phenotype when both genes are duplicated. Future studies should continue to examine these associations. Over time, it will be important to track whether those with MDS including duplication of RAB39B are more susceptible to developmental regression and whether the regression occurs at a younger age. Although the current study did not detect statistically significant differences in severity for the other genes in question, the relatively small sample size and lack of a disorder specific assessment strategy are limiting factors in interpretation. Thus, future studies should continue to examine the degree to which the other genes contribute to clinical severity and specific clinical features. For example, duplications in FLNA have previously been associated with increased severity of gastrointestinal features25 and while it was not clearly associated with increased overall severity in this study, severity of gastrointestinal issues was not specifically measured (the presence/absence of constipation was noted, but not as part of the severity scale). Additionally, mutations in L1CAM have been associated with cerebral ventricle dilation as well as hypoplasia of the corpus callosum35 both relatively common findings on brain imaging in MDS24 which was not systematically documented in this study. Finally, future studies should examine X-inactivation patterns in females with MDS (this could contribute to differing levels of severity).
It is important to note that the severity scales employed in this study were created for use in RTT, and although some construct validity is evident (e.g. females had milder severity scores than males), future studies should focus on refinement of these scales to include additional items that are more specific to MDS. One of the severity scales employed, the Motor Behavioral Assessment, did not yield any significant results, perhaps due to the nature of and scaling of the items being less specific to MDS. Even within the CSS, some essential phenotypic characteristics specific to MDS (e.g. respiratory infections and hospitalizations, later onset of regression, urinary retention, and bowel obstruction) are not captured, but would be essential for tracking developmental trajectories in MDS and should be part of the future development of a more MDS-specific scale. For example, the age range of regression in RTT is more constricted and confined to younger ages as opposed to those with MDS who tend to regress at later ages36.
There are some important limitations to the present study that should be addressed in future work. Specifically, this cohort still represents a relatively smaller sample size (given the estimated prevalence of MDS) that is skewed toward a younger age range and due to the travel requirement is potentially biased toward individuals who are less clinically severe. In addition, in-person clinical assessments, especially when conducted on a yearly basis, can only provide a snapshot of a child’s functioning. This becomes problematic when assessing medically complex neurodevelopmental disorders such as MDS where patients may have day-to-day fluctuations in functional ability due to their medical comorbidities. For example, temporary loss of ambulation due to an acute seizure or decreased attentiveness due to discomfort from severe constipation could easily bias clinical assessments for the day. Retrospective parental reporting of a patient’s functioning is thus required to fill in the gaps; however, the long intervals of reporting common to the design of natural history studies to minimally inconvenience families creates significant potential for recall bias37. Future efforts employing modern technologies for remote observational data collection and parental reporting through ecological momentary assessment strategies could permit more frequent and timely assessments of patients in their home environment thus helping to minimize the burden of travel while more fully ascertaining the spectrum of severity in neurodevelopmental disorders such as MDS.
In summary, this is the first study of MDS to show that both duplication size and specific gene content play a role in clinical severity. These biomarkers in part explain the observed heterogeneity in clinical presentation. Future studies should refine and expand existing severity scales to include more MDS-specific items, especially given the impending human clinical trials that have been buoyed by the promising pre-clinical studies suggesting MDS could be a reversible disorder27. Such trials will greatly benefit from the biomarkers reported here for purposes of patient stratification; and will also require patient assessment tools that are standardized, dynamic, valid, and reliable to accurately measure outcomes. It is essential that these tools be refined with feedback from all salient stakeholders including clinicians, parents and patient advocacy groups as progress continues toward clinical trial readiness.
Supplementary Material
Acknowledgements:
The investigators gratefully acknowledge the participants and families who took their time to contribute to this study. This project was funded by NIH R01HD084500 to SUP, and NIH U54HD061222 to AKP. The NIH Rare Diseases Clinical Research Network (RDCRN) Rett syndrome, MECP2 Duplication disorder, and Rett-related disorders Consortium (U54HD061222) is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). The RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through a collaboration between NCATS, the NICHD, and NINDS.
Footnotes
Data Availability Statement: This paper adheres to the official RDCRN policy related to natural history studies.
Available data will be released to the repository and will become available to the scientific community one year after publication of planned analyses, or after a period of 5 years from the date when the data were collected, whichever comes first.
Conflict of Interest: There are no conflicts of interest on the part of any investigators involved in this study.
References
- 1.Lupski JR. Genomic disorders ten years on. Genome medicine 2009;1(4):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science May 30 2008;320(5880):1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horvath PM, Monteggia LM. MeCP2 as an Activator of Gene Expression. Trends in neurosciences February 2018;41(2):72–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramocki MB, Peters SU, Tavyev YJ, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Annals of neurology December 2009;66(6):771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Esch H MECP2 Duplication Syndrome. Mol Syndromol April 2012;2(3–5):128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friez MJ, Jones JR, Clarkson K, et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics December 2006;118(6):e1687–1695. [DOI] [PubMed] [Google Scholar]
- 7.del Gaudio D, Fang P, Scaglia F, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med December 2006;8(12):784–792. [DOI] [PubMed] [Google Scholar]
- 8.Bauters M, Van Esch H, Friez MJ, et al. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome research June 2008;18(6):847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lugtenberg D, Kleefstra T, Oudakker AR, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. European journal of human genetics : EJHG April 2009;17(4):444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Human molecular genetics November 1 2004;13(21):2679–2689. [DOI] [PubMed] [Google Scholar]
- 11.Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America April 20 2004;101(16):6033–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Na ES, Nelson ED, Adachi M, et al. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. The Journal of neuroscience : the official journal of the Society for Neuroscience February 29 2012;32(9):3109–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miguet M, Faivre L, Amiel J, et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J Med Genet June 2018;55(6):359–371. [DOI] [PubMed] [Google Scholar]
- 14.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A May 2010;152A(5):1079–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peters SU, Hundley RJ, Wilson AK, Carvalho CM, Lupski JR, Ramocki MB. Brief report: regression timing and associated features in MECP2 duplication syndrome. J Autism Dev Disord October 2013;43(10):2484–2490. [DOI] [PubMed] [Google Scholar]
- 16.Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology April 15 2008;70(16):1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Percy AK, Neul JL, Glaze DG, et al. Rett syndrome diagnostic criteria: Lessons from the Natural History Study. Ann Neurol November 22 2010. [DOI] [PMC free article] [PubMed]
- 18.Vandewalle J, Van Esch H, Govaerts K, et al. Dosage-dependent severity of the phenotype in patients with mental retardation due to a recurrent copy-number gain at Xq28 mediated by an unusual recombination. Am J Hum Genet December 2009;85(6):809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pinto D, Delaby E, Merico D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet May 1 2014;94(5):677–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El-Hattab AW, Fang P, Jin W, et al. Int22h-1/int22h-2-mediated Xq28 rearrangements: intellectual disability associated with duplications and in utero male lethality with deletions. Journal of medical genetics December 2011;48(12):840–850. [DOI] [PubMed] [Google Scholar]
- 21.Vanmarsenille L, Giannandrea M, Fieremans N, et al. Increased dosage of RAB39B affects neuronal development and could explain the cognitive impairment in male patients with distal Xq28 copy number gains. Hum Mutat March 2014;35(3):377–383. [DOI] [PubMed] [Google Scholar]
- 22.Nakagawa O, Arnold M, Nakagawa M, et al. Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes & development September 1 2005;19(17):2066–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weller S, Gartner J. Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): Mutations in the L1CAM gene. Human mutation 2001;18(1):1–12. [DOI] [PubMed] [Google Scholar]
- 24.El Chehadeh S, Faivre L, Mosca-Boidron AL, et al. Large national series of patients with Xq28 duplication involving MECP2: Delineation of brain MRI abnormalities in 30 affected patients. American journal of medical genetics. Part A January 2016;170A(1):116–129. [DOI] [PubMed] [Google Scholar]
- 25.Clayton-Smith J, Walters S, Hobson E, et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. European Journal Of Human Genetics 10/15/online 2008;17:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gargiulo A, Auricchio R, Barone MV, et al. Filamin A is mutated in X-linked chronic idiopathic intestinal pseudo-obstruction with central nervous system involvement. American journal of human genetics April 2007;80(4):751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sztainberg Y, Chen HM, Swann JW, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature December 3 2015;528(7580):123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neul JL, Lane JB, Lee HS, et al. Developmental delay in Rett syndrome: data from the natural history study. J Neurodev Disord 2014;6(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. GRCH37/hg19 2009.
- 30.Lim Z, Downs J, Wong K, Ellaway C, Leonard H. Expanding the clinical picture of the MECP2 Duplication syndrome. Clinical genetics April 2017;91(4):557–563. [DOI] [PubMed] [Google Scholar]
- 31.Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet September 2005;77(3):442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.San Antonio-Arce V, Fenollar-Cortes M, Oancea Ionescu R, et al. MECP2 Duplications in Symptomatic Females: Report on 3 Patients Showing the Broad Phenotypic Spectrum. Child neurology open Jan-Dec 2016;3:2329048X16630673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald JH. Handbook of biological statistics, Third Edition. Baltimore, MD: Sparky House Publishing; 2014. [Google Scholar]
- 34.Gao Y, Wilson GR, Stephenson SEM, Bozaoglu K, Farrer MJ, Lockhart PJ. The emerging role of Rab GTPases in the pathogenesis of Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society February 2018;33(2):196–207. [DOI] [PubMed] [Google Scholar]
- 35.Otter M, Wevers M, Pisters M, et al. A novel mutation in L1CAM causes a mild form of L1 syndrome: a case report. Clinical case reports August 2017;5(8):1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters SU, Hundley RJ, Wilson AK, Carvalho CM, Lupski JR, Ramocki MB. Brief Report: Regression Timing and Associated Features in MECP2 Duplication Syndrome. J Autism Dev Disord March 2 2013. [DOI] [PubMed]
- 37.Ozonoff S, Li D, Deprey L, Hanzel EP, Iosif AM. Reliability of parent recall of symptom onset and timing in autism spectrum disorder. Autism September 1 2017:1362361317710798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.