

Abstract
Elicitation of neutralizing antibody responses against hepatitis C virus (HCV) has been a challenging goal. While the E2 subunit of the HCV envelope glycoprotein complex is a promising target for generating cross-genotype neutralizing antibodies, vaccinations with soluble E2 immunogens generally induce weak neutralizing antibody responses. Here, E2 immunogens (i.e., E2.661 and E2c.661) were loaded into lipid-based nanovaccines and examined for induction of neutralizing antibody responses. Compared with soluble E2 immunogens, E2 nanoparticles elicited 6- to 20-fold higher E2-specific serum IgG titers in mice. Importantly, E2 vaccine nanoparticles analyzed at a single particle level with a flow cytometry-based method revealed interesting dynamics between epitope display on the surfaces of nanoparticles in vitro and induction of neutralizing antibody responses in vivo. E2c.661 nanoparticles that are preferentially bound by a broadly neutralizing antibody, HCV1, in vitro elicit neutralizing antibody responses against both autologous and heterologous HCV virions in vivo. In stark contrast, E2.661 nanoparticles with reduced HCV1-antibody binding in vitro mainly induce autologous neutralizing antibody responses in vivo. These results show that rationale antigen design coupled with interrogation of epitope display on vaccine nanoparticles at a single particle level may aid in vaccine development toward achieving neutralizing antibody responses in vivo.
Keywords: Nanoparticle, vaccine, flow cytometry, antigen, hepatitis C virus
Graphical Abstract
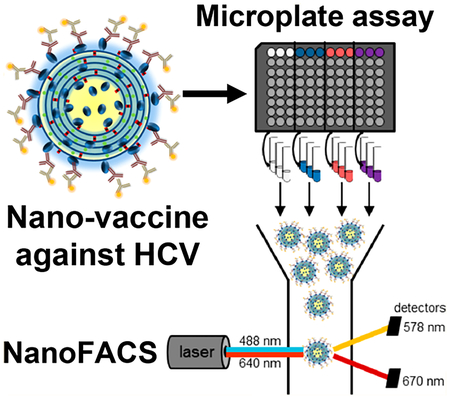
Chronic hepatitis C virus (HCV) infection can lead to liver fibrosis and cirrhosis and is a leading contributor to hepatocellular carcinoma.1,2 Despite the advances of direct-acting antivirals against hepatitis C, the treatment is lengthy (lasting 2−6 months) and requires daily dosing tailored to the diverse genotypes of HCV.3 Furthermore, the escalating crisis of the opioid epidemic and injectable drug use in the United States and elsewhere fuel the spread of HCV.4 An HCV vaccine that can prevent virus transmission or the development of chronic infection will be an important tool for the elimination of this devastating human pathogen.
Analyses of monoclonal antibodies (mAbs) from chronically infected patients have identified cross-genotype neutralizing antibodies recognizing the E2 subunit of the HCV envelope glycoprotein complex,5–9 and extensive research from us and others has led to the development of an engineered E2.661 subunit that retains its conformation and immunogenicity.10–13 Additionally, we developed a new E2 core construct (termed E2c.661) by removing the highly variable region 1 (HVR1) and variable region 2 (VR2) that can skew humoral immune responses away from the E2 neutralizing face, a critical antigenic surface for generating broadly neutralizing antibody responses against HCV.14,15 However, as soluble E2.661 and E2c.661 immunogens lack the multivalent display, concerted orientation, and immunostimulatory danger signals, they induce rather weak neutralizing antibody responses against heterologous HCV virions.
To address these limitations, here we incorporated E2.661 and E2c.661 antigens into interbilayer-cross-linked multi-lamellar vesicles (ICMVs) that can present antigens in a multivalent manner and significantly improve immune responses to protein antigens.16,17 Additionally, we interrogated the display of the antigens on ICMV surfaces and sought to correlate the in vitro epitope recognition of antigen-displaying ICMVs to the in vivo neutralizing antibody responses (Figure 1A). To achieve this, we employed a flow cytometry-based analysis method, termed NanoFACS. Interrogation of ICMVs at a single particle level revealed that E2c.661 ICMVs were preferentially recognized by E2-specific antibodies, including the broadly neutralizing antibody HCV1, compared with E2.661 ICMVs. Mice vaccinated with ICMV formulations generated 6- to 20-fold higher E2-specific serum IgG titers with increased neutralizing capacity against HCV pseudotype particles (HCVpp), compared with soluble controls. Importantly, immune sera from the E2.661 ICMV group preferentially neutralized autologous HCVpp, while immune sera from the E2c.661 ICMV group neutralized both autologous as well as heterologous HCVpp. To the best of our knowledge, this is the first demonstration of correlating in vitro antibody recognition of antigen-displaying nanoparticles at a single particle level to their capacity to generate neutralizing antibody responses in vivo. Our work provides a new analytical approach to characterize and screen nanoparticles for vaccine applications.
Figure 1.

Analysis of E2 antigen display on ICMV vaccines. (A) Schematic of the immunofluorescence staining process. Step 1, ICMVs labeled with DiD were incubated with primary antibodies. Step 2, the samples were incubated with PE-labeled secondary antibodies. Immunofluorescence staining was first measured on a microplate and then analyzed on an individual nanoparticle basis using NanoFACS. (B) Schematic illustration of native HCV E2, E2.661, and E2c.661. (C) Table of percent loading efficiencies of initial antigen (LE %), average diameters by dynamic light scattering (DLS) or nanoparticle tracking analysis (NTA), polydispersity indices (PDI), and zeta potentials (ZP) of ICMVs. (D) Intensity-based size distributions of E2.661 and E2c.661 ICMVs as measured by DLS. (E) Number-based size distributions of E2.661 and E2c.661 ICMVs as measured by NanoSight. Measurements reported as mean ± SD.
Specifically, we derived recombinant HCV antigens E2.661 or E2c.661 from the E2 envelope glycoprotein of the prototypic HCV strain H77 and removed part of the stalk and the entire transmembrane region to improve their solubility (Figure 1B).10,13,14 As the variable regions (VRs) are immunogenic decoys and contribute to immune evasion by HCV,18,19 we removed the N-linked glycans and two VRs in E2.661 to obtain E2c.661 with native-like conformation.14 We encapsulated E2.661 or E2c.661 into ICMVs by the standard lipid film hydration method with the lipid composition of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dio leoyl-sn-glycero-3-phosphoethanolamine-N-[4-(pmaleimidophenyl)butyramide] (MPB) in 1:1 ratio, followed by stapling of apposing lipid layers within multilamellar vesicles with dithiothreitol (DTT) to form ICMVs.16,17 The resulting E2.661 ICMVs and E2c.661 ICMVs exhibited an average diameter of 132 ± 16 and 123 ± 2 nm, respectively, with narrow size distributions of 0.23 ± 0.02 and 0.21 ± 0.02 polydispersity indices (Figures 1C and D) as measured by dynamic light scattering (DLS). In line with these results, tracking individual ICMVs with nanoparticle tracking analysis (NTA) showed a mean diameter of 115 ± 12 and 121 ± 14 nm for E2.661 and E2c.661 ICMVs, respectively (Figures 1C and E). The antigen loading efficiencies of E2.661 ICMVs and E2c.661 ICMVs were 54 ± 8 and 50 ± 7%, respectively, as measured by reducing SDS-PAGE and Coomassie staining (Figure S1).
While these initial characterizations showed comparable antigen loading and particle sizes for both E2.661 ICMVs and E2c.661 ICMVs, these metrics do not provide insights on antigen conformation or orientation on nanoparticle surfaces, which are critical components for induction of neutralizing antibody responses. To evaluate these properties, we developed an indirect immunofluorescence staining assay using E2-specific antibodies recognizing spatially distinct antigenic sites. Specifically, we incubated antigen-loaded ICMVs with antibodies AR1B, AR2A, AR3A, or HCV1 that recognize four different antigenic sites on E2,5,20 followed by incubation with phycoerythrin (PE)-conjugated secondary antibodies and assessing the PE signal. We assessed antibody-mediated recognition of surface-displayed antigens on ICMVs in a population as well as single nanoparticle levels by direct measurement of antibody binding using a microplate-based analysis of bulk samples as well as a flow cytometry-based analysis of individual nanoparticles (Figure 1A).
First, to account for particle loss during the incubation and washing procedures, we added 0.2 molar % DiD, a lipophilic fluorophore, as a nondiscriminate marker of ICMVs and confirmed that the addition of DiD did not change the antigen loading efficiency (Figures S1A−D). The microplate-based assessment of the whole ICMV population showed an average particle recovery rate of 65 ± 5 and 67 ± 4% for E2.661 ICMVs and E2c.661 ICMVs, respectively (Figure 2A). We then quantified binding of E2-specific antibodies on ICMVs by normalizing the PE−antibody signal with the particle recovery rates (Figure 2B). Both E2.661 ICMVs and E2c.661 ICMVs were recognized by all E2-specific antibodies tested. Notably, we observed increased binding of HCV1 antibody on E2c.661 ICMVs, compared with that on E2.661 ICMVs (p < 0.05, Figure 2B). These results indicated maintenance of antigenicity on the surfaces of ICMVs.
Figure 2.

Interrogation of antigen display and antibody recognition on ICMVs. (A and B) Microplate-based analyses of E2.661 ICMVs and E2c.661 ICMVs showing (A) particle recovery after immunofluorescence assay as measured by DiD fluorescence signal and (B) E2-specific antibody binding on ICMVs as measured by PE fluorescence signal normalized by the particle recovery. (C−E) NanoFACS-based analyses of ICMVs showing (C) representative NanoFACS plots from each group tested, (D) particle recovery after immunofluorescence assay as measured by DiD fluorescence signal, and (E) E2-specific antibody binding on ICMVs as measured by PE fluorescence signal normalized by the particle recovery. Measurements reported as mean ± SEM. Statistical analyses performed by two-way ANOVA, followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The same set of ICMV samples from above was subsequently examined with NanoFACS-based analysis. Specifically, we used MoFlo Astrios EQ (Beckman Coulter) equipped with the Dual-PMT (Photomultiplier Tube) Forward Scatter Upgrade with M1 and M2 masks to quantify DiD and PE−antibody signals on individual nanoparticles (Figure 2C). NanoFACS analysis indicated a slight decrease in the DiD signal between the unprocessed and processed samples (Figure 2D). Consistent with the microplate-based method (Figure 2B), we observed strong binding of all E2 epitope-specific antibodies on the surfaces of both E2.661 ICMVs and E2c.661 ICMVs (Figure 2E). Importantly, unlike the results from the microplate-based assay (Figure 2B), NanoFACS analysis of individual nanoparticles revealed significantly enhanced binding of AR1B, AR2A, and HCV1 antibodies on E2c.661 ICMVs (p < 0.01, p < 0.001, and p <0.0001, respectively, Figure 2E), compared with E2.661 ICMVs. This discrepancy suggests technical limitations of the conventional approaches that examine vaccine nano-particles by population-based methods and report the sum of all antibody binding events. We speculate that incubation of vaccine nanoparticles with antibodies may aggregate a subset of nanoparticles because primary and secondary antibodies simultaneously bind to multiple nanoparticles; thus, investigate ing antigen display on individual nanoparticles using a more sensitive approach, such as NanoFACS, can address this issue.
To further interrogate the impact of differential epitope display on humoral immune responses induced by E2.661 ICMVs and E2c.661 ICMVs, we immunized C57BL/6 mice and quantified antibody responses (Figure 3A). We used an immunostimulatory adjuvant, monophosphoryl lipid A (MPLA, an FDA-approved Toll-like receptor 4 agonist), in ICMVs as well as soluble vaccine formulations. Mice were vaccinated subcutaneously at the tail base with the prime dose of 10 μg of antigen and 1 μg of MPLA and two subsequent boost doses of 5 μg of antigen and 0.5 μg of MPLA, followed by ELISA-based measurement of sera IgG specific to the native E1E2 antigen. After the first vaccination, both ICMV formulations achieved seroconversion, and after three rounds of immunizations, animals maintained strong antigen-specific IgG responses throughout 176 days with serum IgG titers at least 6- to 20-fold higher than those of their respective soluble controls (Figure 3B and Figure S2). Notably, the soluble controls required two vaccinations for seroconversion. We did not detect any difference in the magnitude of E2-specific IgG titers between the two ICMV formulations.
Figure 3.

Analyses of humoral immune responses induced by ICMV or soluble vaccine formulations. (A) Vaccination scheme indicating days of subcutaneous vaccinations (black arrows) and serum collection (red arrows). (B) Arithmetic averages of E2-specific serum IgG titers. Measurements reported as mean ± SEM. Statistical analysis was performed on groups with similar antigens (e.g., E2.661 + MPLA vs E2.661 ICMV + MPLA) using two-way ANOVA with matched pairs followed by Tukey’s multiple comparisons test. Statistical significance levels are denoted by asterisks (*) for E2 antigen formulations and pound signs (#) for E2c antigen formulations. (#/*) p < 0.05, (##/**) p < 0.01, (###/***) p <0.001, (####/****) p < 0.0001. (C) Average in vitro pseudotype virus particle neutralization with immune sera collected at various time points.(D) Individual in vitro serum neutralization percentage. LCMV (lymphocytic choriomeningitis virus, negative control), HCV H77 (autologous HCV), and HCV UKN1b12.6 (heterologous strains from genotype 1b).
Strikingly, we detected significant differences in the neutralizing capacities of the immune sera between the E2.661 ICMV and E2c.661 ICMV groups (Figures 3C and D). Specifically, immune sera were used in in vitro neutralization assays performed with HCV pseudotype virus particles (HCVpp) expressing E1E2 glycoproteins from autologous (H77) or heterologous (UKN1b12.6) HCV strains or an unrelated envelope glycoprotein from lymphocytic choriomeningitis virus (LCMV) used as a negative control.
Immune sera from the E2.661 ICMV group exhibited robust neutralizing IgG responses against autologous HCVpp, with 6 out of 7 animals achieving >50% neutralization by day 62, and sustaining strong neutralizing capacity throughout 176 days (Figures 3C and D). In contrast, the soluble E2.661 control group had minimal neutralizing IgG responses (p < 0.0001, Figures 3C and D). Despite the strong autologous neutralizing capacity of the E2.661 ICMV group, their immune sera displayed minimal neutralizing capacity against heterologous HCVpp (Figures 3C and D), with only 3 out of 7 animals exhibiting >25% heterologous neutralization on day 104. In stark contrast, the immune sera from the E2c.661 ICMV group exhibited significantly higher neutralizing activity against both autologous (p < 0.0001) and heterologous HCVpp (p <0.001), compared with the soluble E2c.661 control by day 104 (Figures 3C and D). Although the E2c.661 ICMV group displayed weaker autologous neutralizing activity than the E2.661 ICMV group, 6 out of 7 animals in the E2c.661 ICMV group had immune sera with >25% heterologous neutralization activity on day 104 (Figures 3C and D). Notably, this was sustained up to day 142 at which point 4 out of 7 animals in the E2c.661 ICMV group exhibiting >25% heterologous neutralizing activity, compared with 0 out of 7 animals in the E2.661 ICMV group (p < 0.05, Figures 3C and D). However, the heterologous neutralizing capacity of the E2c.661 ICMV group decreased to the minimal level by day 176. Overall, both ICMV formulations elicited significantly enhanced antigen-specific antibody responses with neutralizing capacity, compared with the soluble groups. In addition, E2c.661 ICMVs, which better-maintained antigenicity of E2 than E2.661 ICMVs as shown by NanoFACS (Figure 2E), achieved robust IgG responses with neutralizing capacities against autologous and heterologous viruses (Figures 3C and D).
Lastly, we examined specific E2 epitopes recognized by the immune sera collected on day 104. Specifically, we assessed immune sera binding to either HVR1 and HCV1 region peptides within E2 using ELISA. We first confirmed that there was no peptide-specific IgG by testing prebleed samples one day before the first immunization (Figure S3). All immune sera from the E2.661 ICMV group on day 104 bound to the HVR1 peptide (Figure 4A), whereas the immune sera from the E2c.661 ICMV group did not bind to the HVR1 peptide since the recombinant E2c.661 construct lacks the HVR1 region (Figures 1B and 4A). Importantly, all immune sera samples (7 of 7) from the E2c.661 ICMV group had HCV1-specific IgG antibodies, compared with only 3 of 7 samples from the E2.661 ICMV group (Figure 4B). Notably, we mainly observed HCV1-specific IgG1 isotype in the E2c.661 ICMV group, whereas balanced IgG1 and IgG2c responses were detected against the native E1E2 antigen in both ICMV groups (Figure S4). These results suggested that the removal of the highly antigenic HVR1 region from E2.661 shifted antibody responses to the adjacent HCV1 region, which may have contributed to the heterologous neutralizing activity elicited by E2c.661 ICMVs (Figures 3C and D).
Figure 4.

Specificity of immune sera to HVR1 and HCV1 peptides. (A and B) Optical densities (OD) of log-fold serum samples after incubating day 104 immune sera on (A) HVR1 peptide-coated (E2 aa 384−411: ETHVTGGNAGRTTAGLVGLLTPGAKQNI) or (B) HCV1 long peptide-coated (E2 aa 407−424: AKQNIQLINTNGSWHINS) ELISA plates.
Interestingly, there was a positive correlation between the HVR1 peptide binding and autologous neutralizing activity of the immune sera for the E2.661 ICMV group (r = 0.58) (Figure S5A). On the other hand, there was a positive correlation between the HCV1 peptide binding and heterologous neutralizing activity of the immune sera for both ICMV groups, with the E2c.661 ICMV group exhibiting a greater correlation between the two factors (r = 0.66), compared with the E2.661 ICMV group (r = 0.32) (Figure S5B). Taken together, these data suggest that the HVR1 region displayed on ICMVs has a positive effect on autologous neutralization, while antibody responses to the HCV1 region, possibly driven by IgG1 antibodies, appear to positively impact heterologous neutralization.
In summary, we developed ICMVs loaded with recombinant HCV E2 antigens and interrogated antigen display and orientation on the surfaces of ICMVs using both whole population- and single-particle-based analyses of antibody− antigen interactions. Our work demonstrates that in vitro assessment of conformational antigen display on nanoparticles can aid in the selection of vaccine formulations for neutralizing antibody responses in vivo. In particular, antigen-loaded ICMVs elicited greater E2-specific IgG titers with superior neutralizing capacities than their respective soluble controls (Figure 3). Immune sera from the E2.661 ICMV group displayed selectivity for autologous neutralization, whereas immune sera from the E2c.661 ICMV group exhibited both autologous and heterologous neutralization (Figure 3C, D). These data correlated with our in vitro NanoFACS analysis in which the broadly neutralizing antibody HCV1 bound with a greater extent to E2c.661 ICMVs (Figure 2E), suggesting a contribution of the HCV1 region in generating heterologous neutralization. Analysis of epitope recognition by immune sera revealed that all mice administered with E2.661 ICMVs generated HVR1-specific antibodies, whereas only 3 of 7 mice elicited antibodies to the HCV1 region (Figure 4). In stark contrast, all mice administered with ICMVs carrying E2c.661 (HVR1 removed) skewed antibody responses to the HCV1 region (Figure 4). These data suggest the importance of rationale antigen design as well as screening for proper antigen display on nanoparticle vaccines, as different orientations may impact the breadth and potency of immune responses in vivo.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by NIH (Grants R01AI079031, R01AI106005, U19AI123861, R01AI127070, R01EB022563, and R01HL125555). J.J.M. is a Young Investigator supported by the Melanoma Research Alliance (Grant 348774), DoD/CDMRP Peer Reviewed Cancer Research Program (Grant W81XWH-16-1-0369), Emerald Foundation, and NSF CAREER Award (1553831). We thank Dr. Jonathan Ball for HCVpp reagents. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the United States Department of the Army or the United States Department of Defense.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nanolett.8b03601.
Additional materials, experimental details, and Supporting Figures S1−S4 (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Martinez-Sierra C; Arizcorreta A; Diaz F; Roldan R; Martin-Herrera L; Perez-Guzman E; Giron-Gonzalez JA Clin. Infect. Dis 2003, 36 (4), 491–8. [DOI] [PubMed] [Google Scholar]
- (2).Ghouri YA; Mian I; Rowe JH J. Carcinog 2017, 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Guidelines for the Screening, Care and Treatment of Persons with Chronic Hpeatits C Infection; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- (4).Liang TJ; Ward JW N. Engl. J. Med 2018, 378 (13), 1169–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Giang E; Dorner M; Prentoe JC; Dreux M; Evans MJ; Bukh J; Rice CM; Ploss A; Burton DR; Law M Proc. Natl. Acad. Sci. U. S. A 2012, 109 (16), 6205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Keck ZY; Li TK; Xia J; Gal-Tanamy M; Olson O; Li SH; Patel AH; Ball JK; Lemon SM; Foung SK J. Virol 2008, 82 (12), 6061–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kong L; Giang E; Nieusma T; Robbins JB; Deller MC; Stanfield RL; Wilson IA; Law MJ Virol 2012, 86 (23), 13085–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Law M; Maruyama T; Lewis J; Giang E; Tarr AW; Stamataki Z; Gastaminza P; Chisari FV; Jones IM; Fox RI; Ball JK; McKeating JA; Kneteman NM; Burton DR Nat. Med 2008, 14 (1), 25–7. [DOI] [PubMed] [Google Scholar]
- (9).Schofield DJ; Bartosch B; Shimizu YK; Allander T; Alter HJ; Emerson SU; Cosset FL; Purcell RH Hepatology 2005, 42(5), 1055–62. [DOI] [PubMed] [Google Scholar]
- (10).Spaete RR; Alexander D; Rugroden ME; Choo QL; Berger K; Crawford K; Kuo C; Leng S; Lee C; Ralston R; et al. Virology 1992, 188 (2), 819–30. [DOI] [PubMed] [Google Scholar]
- (11).Selby MJ; Glazer E; Masiarz F; Houghton M Virology 1994, 204 (1), 114–22. [DOI] [PubMed] [Google Scholar]
- (12).Michalak JP; Wychowski C; Choukhi A; Meunier JC; Ung S; Rice CM; Dubuisson JJ Gen. Virol 1997, 78 (9), 2299–2306. [DOI] [PubMed] [Google Scholar]
- (13).Flint M; Thomas JM; Maidens CM; Shotton C; Levy S; Barclay WS; McKeating JA J. Virol 1999, 73 (8), 6782–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kong L; Giang E; Nieusma T; Kadam RU; Cogburn KE; Hua Y; Dai X; Stanfield RL; Burton DR; Ward AB; Wilson IA; Law M Science 2013, 342 (6162), 1090–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tzarum N; Wilson IA; Law M Front. Immunol 2018, 9, 1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Moon JJ; Suh H; Bershteyn A; Stephan MT; Liu H; Huang B; Sohail M; Luo S; Um SH; Khant H; Goodwin JT; Ramos J; Chiu W; Irvine DJ Nat. Mater 2011, 10 (3), 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Moon JJ; Suh H; Li AV; Ockenhouse CF; Yadava A; Irvine DJ Proc. Natl. Acad. Sci. U. S. A 2012, 109 (4), 1080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Dao Thi VL; Dreux M; Cosset FL Expert Rev. Mol. Med 2011, 13, No. e13. [DOI] [PubMed] [Google Scholar]
- (19).Farci P; Shimoda A; Coiana A; Diaz G; Peddis G; Melpolder JC; Strazzera A; Chien DY; Munoz SJ; Balestrieri A; Purcell RH; Alter HJ Science 2000, 288 (5464), 339–44. [DOI] [PubMed] [Google Scholar]
- (20).Kong L; Giang E; Robbins JB; Stanfield RL; Burton DR; Wilson IA; Law M Proc. Natl. Acad. Sci. U. S. A 2012, 109(24), 9499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.