

Abstract
Mass spectrometry-based protein quantitation is currently used to measure therapeutically relevant protein biomarkers in CAP/CLIA setting to predict likely responses of known therapies. Selected reaction monitoring (SRM) is the method of choice due to its outstanding analytical performance. However, data-independent acquisition (DIA) is now emerging as a proteome-scale clinical assay. We evaluated the ability of DIA to profile the patient-specific proteomes of sample-limited tumor biopsies and to quantify proteins of interest in a targeted fashion using formalin-fixed, paraffin-embedded (FFPE) tumor biopsies (n=12) selected from our clinical laboratory. DIA analysis on the tumor biopsies provided 3,713 quantifiable proteins including actionable biomarkers currently in clinical use, successfully separated two gastric cancers from colorectal cancer specimen solely based on global proteomic profiles, and identified subtype-specific proteins with prognostic or diagnostic value. We demonstrate the potential use of DIA-based quantitation to inform therapeutic decision-making using TUBB3, for which clinical cut-off expression levels have been established by SRM. Comparative analysis of DIA-based proteomic profiles and mRNA expression levels found positively and negatively correlated protein-gene pairs, a finding consistent with previously reported results from fresh-frozen tumor tissues.
Keywords: DIA-Mass Spectrometry, Clinical Proteomics, Protein Biomarkers, Cancer Biomarkers
1. INTRODUCTION
Cellular expression levels of clinically relevant proteins in patient tumor samples can guide selection of cancer therapies. While antibody-based immunohistochemistry (IHC) assays are most widely used in the clinic, mass spectrometry (MS)-based quantitative proteomics has been increasingly applied to clinical samples to objectively measure tumor expression levels of treatment-related protein biomarkers1–2. A targeted strategy using selected reaction monitoring (SRM) has been a method of choice for clinical proteomics due to its high analytical performance including exceptional sensitivity from a fixed quadrupole setting, and inherent selectivity achieved by paired selection of precursor and fragment ions defined as transitions. SRM-based protein detection in archived tissues offers fundamental advantages of robustness as it is less sensitive to pre-analytical variables such as age of tissue sections and fixation time than IHC 3–4. We previously demonstrated that SRM-based quantitation of the targeted therapy marker, human epidermal growth factor receptor 2 (HER2) is superior to IHC scores in predicting outcomes of clinical trial participants treated with anti-HER2 therapy 1, 5.
While SRM is the standard MS-based method for measuring protein concentrations, high-resolution/accurate mass-based parallel reaction monitoring (PRM) benefits from improved signal-to-noise ratio for quantitative analysis and flexibility for assay development. In general, PRM assays can perform better than SRM in more complex samples with high background, which is the case for clinical tumor biopsies 6–7. Both SRM and PRM are used to target specific analytes, often with stable isotope labeled (SIL) internal standards.
Data-independent acquisition (DIA) has recently emerged as a comprehensive solution for protein quantitation8. In contrast to data-dependent acquisition (DDA) where precursor ions are selected sequentially for fragmentation, in DIA mode, all precursor ions within a user-defined m/z window are fragmented in parallel and generate MS2 data with convoluted product ions. As with PRM, DIA benefits from high-resolution mass analyzers to accurately measure product ions that can be used for further quantitative analysis. In general, DIA-based quantitation relies on the MS2 measurement which can perform better than MS1 in complex matrices due to better selectivity and lower background noise 8–9.
Application of DIA to clinical proteomics for precision medicine can offer clear advantages. Its untargeted and systematic sampling process enables acquisition of comprehensive LC-MS data consisting of all precursors and product ions present in the sample within the m/z range measured. Once digitally archived, this proteomic profile can be re-analyzed over and over again with newer clinical hypotheses or analytical strategies which may not have been available at the time of acquisition. For already validated protein biomarkers, DIA data can be analyzed in a targeted fashion by extracting the co-eluting fragment ions corresponding to the peptides of interest for quantitation 8, 10–11. This global proteomic profile in combination with corresponding genomic data from tumor biopsy samples can offer comprehensive characterization of tumor cells for precision medicine.
The analytical performance of DIA for proteomics has been thoroughly demonstrated elsewhere 12–16. In this study, therefore, we sought to evaluate the use of DIA to characterize tumor biology and to measure oncology biomarkers in sample-limited formalin-fixed paraffin-embedded (FFPE) tumor biopsies using 12 archived gastrointestinal (GI) tumor biopsies. We focused to assess if single-shot DIA data from FFPE tumor biopsies using 1 μg protein extracts provide clinically relevant protein targets. DIA peak area of known biomarkers of which tumoral concentrations have been previously established by SRM for clinical use were compared to the SRM-based quantitation acquired from the same samples. Whole genome sequencing results and mRNA expression levels were analyzed, and the correlation results between protein and mRNA were compared with the previously reported correlation data from fresh-frozen tissue samples.
2. METHODS
2.1. Tissue samples
Slides of FFPE tumor tissue and clinical annotations were received in the authors’ clinical laboratory during 2016 (Table S-1 in the Supporting Information). Patients provided consent for research use of anonymized data from their test results.
2.2. Protein extraction and peptide generation
Two consecutive tissue sections per sample were prepared. The first sections were stained with hematoxylin and eosin (H&E), and were used to guide tumor area selection. The second sections were mounted on DIRECTOR® slides (Expression Pathology, Rockville, MD), deparaffinized with xylene, stained with hematoxylin, and subjected to microdissection based on the tumor-specific markup by a board-certified pathologist using MMI CellCut laser microdissection system (Molecular Machines and Industries, Eching, Germany) equipped with UV laser. A set of tissue images before and after laser microdissection of a sample used in this study is included in the Supporting Information (Figure S-1). Collected cells after laser ablation were heated in Liquid Tissue® buffer (Expression Pathology, Rockville, MD) at 95 °C for 1.5 h, and subsequently incubated with trypsin (Promega, Madison, WI) for 16 h at 37 °C. A Micro BCA assay (Thermo Fisher Scientific, Rockford, IL) was performed to determine total protein concentration. For both DIA and SRM analysis, 1 μg tumor protein digest was injected per sample.
2.3. LC-MS/MS experiment for SRM analysis
LC-SRM was performed in NantOmics’ CAP-accredited, CLIA-certified laboratory (Rockville, MD) using a nanoAcquity UPLC system (Waters, Milford, MA) coupled to a TSQ Quantiva mass spectrometer (Thermo Scientific, San Jose, CA)1. In each analysis, 1 μg of sample spiked with 5 fmol SIL internal standard peptides (ThermoFisher Scientific; TUBB3-ISVYYNEASSHK[13C615N2] and EGFR-IPLENL[13C615N1]QIIR) was loaded onto the Waters Symmetry C18 trap column (180 μm x 2 cm, 5 μm) and washed with 100% buffer A (0.1% formic acid in water). After trapping, the sample was loaded onto the Waters HSS T3 analytical column (100 μm x 10 cm, 1.8 μm). Peptides were separated over a 14 min step gradient: 2 min from 1%−9% buffer B (0.1% formic acid in acetonitrile), 6 min from 9%−15% buffer B, 4 min from 15%−25% buffer B, and 2 min from 25%−50% buffer B with a flow rate of 800 nl/min. Eluents entered the mass spectrometer via an electrospray ionization interface with a 2.3 kV spray voltage. The SRM mode used a cycle time of 1 second with Q1/Q3 window 0.7. Details of SRM transitions are found in Supporting Information Table S-2. Data was analyzed using Pinpoint software version 1.3 (Thermo Scientific, San Jose, CA). Protein concentrations were determined by calculating the ratio of endogenous analyte signal to the internal standard signal, and multiplying by the amount of internal standard spiked into the sample (Fig. S-2, S-3 in the Supporting Information).
2.4. LC-MS/MS experiment for DIA analysis
DIA was performed at the University of Washington using a nanoAcquity UPLC system coupled to an Orbitrap Fusion mass spectrometer (Thermo Scientific, Bremen, Germany). A trapping column used for sample cleanup was a fused-silica column (150 μm x 4 cm, 3 μm) packed with C18 packing material (Reprosil Pur 120 C-18-AQ) (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany). The analytical column was a PicoFrit column (75 μm x 30 cm, 3 μm) (New Objective, Woburn, MA) and packed with the same material as the trapping column. In each mass spectrometry run, 1 μg of sample was loaded onto the trapping column and washed with a mixture of 98% buffer A and 2% buffer B. After trapping, the sample was loaded onto the analytical column and separated over a 90 min linear gradient from 2%−35% buffer B. As the peptides were eluted from the column, they entered the mass spectrometer via an electrospray ionization interface with a 2 kV spray voltage.
For DIA with gas-phase fractionation, a pooled sample was analyzed with 13 DIA LC-MS/MS runs collectively covering 350 – 1000 m/z. Each DIA run acquired comprehensive MS/MS data on all precursors in a 50 m/z range. The MS/MS scans used a 2 m/z wide isolation window, acquired with resolving power 30,000 at 200 m/z. The acquisition consists of a cycle of MS/MS scan acquisition with a pair of MS scans acquired every 25 MS/MS scans. For DIA with single-shot acquisition, comprehensive MS/MS data on all precursors between 400 and 800 m/z was acquired with a 20 m/z isolation width. The acquisition consisted of a cycle of MS/MS scan acquisition with an MS scan acquired every 21 MS/MS scans. The MS/MS scans were acquired using an “overlapping window” multiplexing approach in which alternating cycles of MS/MS scans are offset by 10 m/z relative to one another (manuscript in preparation).
2.5. Library generation
The 13 raw DIA files with gas-phase fractionation were converted to mzML using ProteoWizard msconvert17, and submitted to peptide search by the Walnut tool, part of EncyclopeDIA (v. 0.6.3)18. A FASTA file was prepared using the protein sequences from UniProtKB/SwissProt (version 17-Jan, 2018; 20,231 entries)19. This proteome was used as both the “target” and “background” protein sequence database. The former is used to define query peptides after in silico digestion, and the latter to compute statistics for score calibration. The search performed was a tryptic digestion with no fixed or variable modifications, and one missed cleavage allowed. The precursor and fragment mass tolerance were both set to 10 ppm, and fragmentation was modeled using only the y-ion series. The number of quantitative ions was set to 5. Only peptide precursors with charge +2 or +3 were considered in the search. Percolator version 3.01 was selected to calibrate scores and calculate statistics for peptide detection confidence (e.g. q-value)20. After searching, a chromatogram library (.elib) was saved containing peptides with q-value < 0.01 calculated on the combined set of 13 library runs.
2.6. Data query and chromatographic peak integration for quantitation
Prior to data query, the 12 single-shot DIA data were demultiplexed using msconvert (part of ProteoWizard v 3.0.11392) with vendor centroiding enabled and the filter “demultiplex optimization=overlap only” used to enable demultiplexing. The demultiplexed data were output in the .mzML format. The .elib library generated in the previous step was used to query the demultiplexed single-shot DIA data using the EncyclopeDIA tool. The same FASTA was used for the “background” as in the Library Generation step. The Target/Decoy approach was set to “Normal Target/Decoy” and the Data Acquisition type set to “Non-Overlapping DIA” as the data had already been demultiplexed. The minimum and maximum number of quantitative ions were set to 3 and 5, respectively. The RT Align feature was enabled, to use detection of peptide features in common between files to improve detection sensitivity and accuracy as well as the calculation of optimal fragment ions and peak integration boundaries. All other parameters were set to the same values as in the Library Generation step. In both the library generation and wide-window query steps, both MS1 and MS2 data are used for peptide detection, but quantitation is done exclusively with MS2 data. The results from the analysis were exported into a .elib-format library containing peptides detected with an experiment-wide FDR of q < 0.0121.
For quantitation, a Skyline (v. 4.1.1.11756) document was created to visualize the resulting peptide detections and perform chromatographic peak integration and background subtraction21. To generate the document, the .elib file resulting from the data query was added to the document, which causes Skyline to use the peptides and peptide chromatographic peak integration boundaries defined by EncyclopeDIA. Skyline was set to pick the top 5 most intense product ions from the .elib file that are part of the singly charged y-ion series y3 – y(n-1) (ion 3 to last y ion). This effectively causes Skyline to use the top 5 most intense transitions that were determined to be amenable for quantitation by EncyclopeDIA. Any peptides containing fewer than 3 transitions matching these criteria were removed from the document as were peptides containing fewer than 5 or more than 65 amino acids. The centroided, demultiplexed data files were imported into Skyline with precursor and fragment ion mass tolerance set to 10 ppm. Integrated peak areas were exported as a report containing the integrated fragment ion signal intensity for each peptide precursor calculated by Skyline.
For further data analysis, a non-redundant set of 3,713 protein groups was created from a redundant list of 4,239 proteins by grouping proteins containing identical sets of peptides. Protein identifications differing only in Leu/Ile (isobaric) were also grouped. Those proteins associated only with a subset of peptides associated with another protein were removed. Hierarchical clustering was carried out using Perseus software 22, using Euclidean distance, average linkage and pre-processing with k-means (300 cluster, maximum of 10 iterations and 1 restart). Figures were generated using the ggplot2 and cowplot packages in R downloaded from https://github.com/wilkelab/cowplot.
2.7. DNA and RNA analysis
Whole genome sequencing of tumor DNA, germline DNA and tumor RNA from each patient was performed in a NantOmics laboratory using the Illumina platform, as previously described 23. Germline sequencing was from a blood sample; tumor sequencing was from macrodissection of tissue sections mounted on slides according to annotation by a board-certified pathologist. Normal and tumor genomes were sequenced to depths of roughly 30x and 60x respectively. About 300 million RNA reads were sequenced for each tumor. DNA reads were aligned to GRCh37 with BWA24 and mutations and copy number changes were determined using the NantOmics Contraster Pipeline25. Tumor purity was estimated based on the mutant allele frequency in the tumor sample compared to the matched normal sample. RNA reads were aligned with bowtie26 and transcript abundances were quantified with RSEM27.
Protein-mRNA correlation analysis was performed on Log2-scaled protein intensity and mRNA transcripts per million (TPM) values with an offset of 1. Gene set enrichment analysis of the correlation between the protein-mRNA pairs was performed using LIGER (https://github.com/JEFworks/liger). Gene sets used for this analysis came from subcellular localization annotations from UniProt19. and the Human Protein atlas (www.proteinatlas.org)28, as well as gene pathways from KEGG29.
2.8. DIA Data Availability
The Skyline document and raw files for DIA library generation and DIA sample analysis are available at Panorama Public. ProteomeXchange ID: PXD010934. Access URL: https://panoramaweb.org/7nYM9z.url
3. RESULTS AND DISCUSSION
3.1. DIA analysis of therapeutic biomarkers
The overall workflows of proteomic sample preparation from FFPE biopsies and DIA-based mass spectrometric analysis are shown in Fig. 1. A spectral library was generated from a pooled sample analyzed with a narrow isolation window (2 Th; no overlapping) and gas-phase fractionation. The library contained a total of 18,033 peptides amounting to 4,706 human proteins based on UniProtKB/SwissProt sequence database 19. Each of the twelve biopsy samples analyzed with single-shot DIA using a wide isolation window (20 Th; 10 Th overlapped) were searched against this library, resulting in 12,256 quantifiable peptides and 3,713 non-redundant protein groups for downstream analysis (3,600 single protein identifications and 113 protein groups). Detailed list of peptides with their detected retention times and intensities from both precursor ions and fragment ions in 12 samples were included in the Supporting Information (Table S-3 in the Supporting Information). The list of proteins obtained from these biopsy samples represent various cellular sub-locations (Fig. 2A) (Table S-4 and S-5 in the Supporting Information, for 4,239 redundant and 3,713 non-redundant protein identifications, respectively). To assess the depth of coverage of our single-shot DIA quantitation from tumor biopsies, we compared our findings with those of a previous study of FFPE CRC samples that employed multiple proteases, additional biochemical fractionation and DDA mass spectrometry, and reported estimated protein copy numbers per cell30. The copy numbers per cell in the reference [30] were estimated by the “Protein Ruler” method, using MS signal of histones, total MS signal of proteins and measured DNA amount from samples31. Of the common proteins, Fig. 2B shows that our spectral library has good coverage (80%−100%) of high-copy number proteins (>1 × 105 copies per cell), reasonable coverage of medium-copy number proteins (1 × 104 copies per cell; 30%−50%) and weak coverage of low-copy number proteins (1,000 copies and fewer per cell). This is not unexpected given the difference in sample fractionation, sample number and instrument time employed in the comparison study. Our single-shot analysis identified 140 proteins that were not found in the comparison study, likely reflecting tissue- or patient-specific differences. Fig. 2C shows that the copy numbers of our identifications span five orders of magnitude, from approximately 1 × 103 to 1 × 108 copies per cell (Table S-5 in the Supporting Information).
Figure 1.
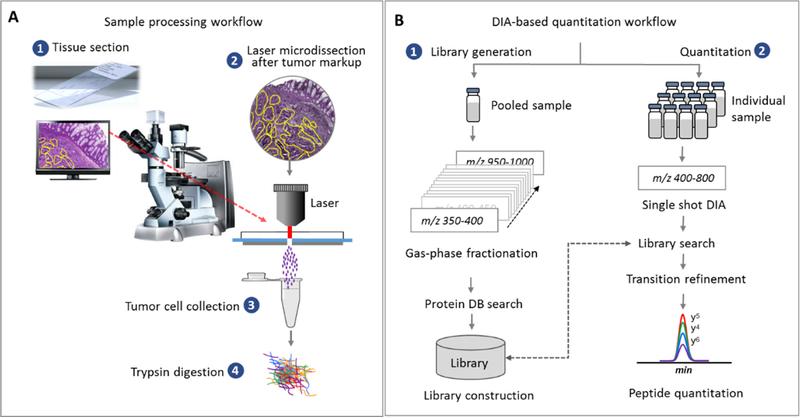
(A) Proteomic analysis of FFPE biopsy samples. Tissue sections mounted on Director® slides were subjected to microdissection (10 μm tissue thickness) after tumor-specific markup. Collected tumor cells were proteolyzed by trypsin after heat treatment. (B) DIA analysis for quantitative analysis. A library containing chromatographic and mass spectrometric attributes (MS1 and MS2 traces) of detected peptides was constructed using the gas-phase-fractionated DIAs in a pooled sample with 2 Th m/z window18. This library was used to match peptides from the DIA data obtained from the individual sample using a wider isolation window (20 Th). Summed AUCs of the fragment ions’ chromatograms per precursor are used to generate quantitative data.
Figure 2.

DIA-based proteomic analysis in tumor biopsies. (A) Detected 4,239 proteins (DIA library) were categorized based on their sub-cellular localization. (B) Binned protein identifications from the chromatogram library (Library) generated using gas phase fractionation and from individual single-shot analysis of samples (SS-DIA) are compared with findings of Wisniewski et al.30–31. (C) Comparison findings [reference 32] are listed by decreasing copies per cell; 95% of our single-shot protein identifications are found between 5 × 106 and 2.5 × 103 copies per cell (n=3,573 matched protein identifications). Eight examples of proteins identified in single-shot analysis spanning the range of copies per cell were indicated. (D) Oncologic biomarkers matched to those cataloged in the Cancer Genome Interpreter database 32. 40 biomarkers at the protein level were detected and quantified across 12 samples.
As evidence of the clinical utility of DIA-based proteomic data from FFPE tumor biopsies, we identified known biomarkers of response to cancer drugs. The 12 biopsies expressed 40 of the 201 non-redundant cancer biomarkers listed in the curated database Cancer Genome Interpreter (CGI)32 (Fig. 2D). Furthermore, among the detected proteins, 131 were targets of FDA-approved therapies; An additional 986 were potential drug targets known to be involved in disease progress and belonging to the same functional classes of approved drug targets (www.drugbank.ca)33 (Table S-4 in the Supporting Information). Recently, a comprehensive analysis of the Cancer Genome Atlas (TCGA) dataset identified 299 cancer driving genes34. In our dataset, 104 proteins corresponding to these genes were quantified (35% of all drivers): this included 76 pan-cancer drivers, and drivers specific to adenocarcinoma of the colon (n=4) and stomach (n=7).
The presence of these biomarkers in patient tumor samples can provide useful information to physicians for therapeutic decision-making. Although cutoff values based on relationships between DIA readouts and therapeutic outcomes have not yet been established, demonstrating the ability of DIA-based proteomic profiling to quantify clinically relevant biomarkers from tumor biopsies is an important first step. Note that the majority of known biomarkers have been developed for genomic testing, including identification of point-mutations as well as gene amplification. We anticipate that protein expression data obtained directly from tumor biopsies by a robust profiling method will identify novel biomarkers and suggest applications for protein-based clinical assays. The ample number of therapeutic targets obtained by a DIA analysis in biopsy samples supports the potential use of such data for discovery of drug targets and therefore new opportunities for developing companion diagnostics models.
3.2. Patient-specific proteomic profiles
The proteomic profiles of the 12 tumor samples were compared with one another to assess the ability of DIA readouts to reflect indication-specific or patient-specific characteristics. The result of unsupervised hierarchical clustering analysis indicated a clear distinction between colorectal cancer (CRC) and the other GI samples (non-CRC; patients #1 and #3) (Fig. S-4 in the Supporting Information). Further comparative analysis between these groups was performed to identify proteins whose expression levels are differentially elevated in CRC or non-CRC samples (Fig. 3A). Requiring a minimum of two peptides per protein identification, we identified a subset of 15 proteins that were significantly overexpressed in CRC as compared to non-CRC samples (Fig. 3B; Table S-6 in the Supporting Information). In a TCGA dataset of multiple human cancers35–36, these 15 targets showed higher mRNA expression level in CRC tissue samples compared to gastric cancer samples. (p=0.0002; two-tailed, paired t-test) (Fig. 3C). Immunohistochemical results from the Human Protein Atlas (www.proteinatlas.org) using verified antibodies37 showed that the same targets were significantly overexpressed in CRC tissue samples compared to gastric tumors (p=0.0039; two-tailed, paired t-test) (Fig. 3D).
Figure 3.
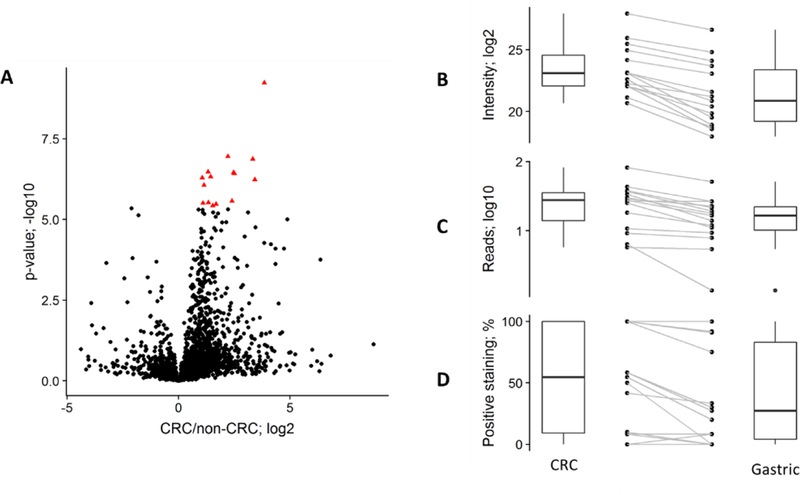
Evaluation of DIA-based differential expression. (A) Volcano plot showing a Welch’s t-test on proteins identified with two or more peptides, comparing CRC (n=10) and non-CRC (n=2) samples. 15 proteins significantly overexpressed in the CRC samples (q-value <0.01; Bonferroni multiple-hypothesis testing correction; red triangles) were selected for further analysis. Boxplots and paired samples are shown for: our proteomic quantification (B; median intensity); median mRNA expression levels obtained in TCGA studies (C); and IHC-based tumor expression levels in the Human Protein Atlas (D).
Among the proteins that were differentially expressed between patients was succinate dehydrogenase B (SDHB). Visual inspection of the peptides inferring SDHB protein (QQYLQSIEER and WMIDSR) confirmed that SDHB was absent only in one biopsy obtained from patient #3. Fig. 4A demonstrates the MS2 traces of the unique SDHB peptide QQYLQSIEER measured in three biopsy samples exhibiting high, moderate, and no expression level, from patients #6, #12, and #3, respectively. No detectable trace of the targeted fragment ions was found at the expected chromatographic space (normalized retention time) in patient #3 while the background traces still remain relatively stable compared to other biopsies (Fig. 4A). Germline DNA analysis found that the patient with missing SDHB protein was heterozygous for a nonsense mutation in the SDHB gene (p.Arg90Ter) while SDHB was intact in the other patients (Fig. 4B).
Figure 4.
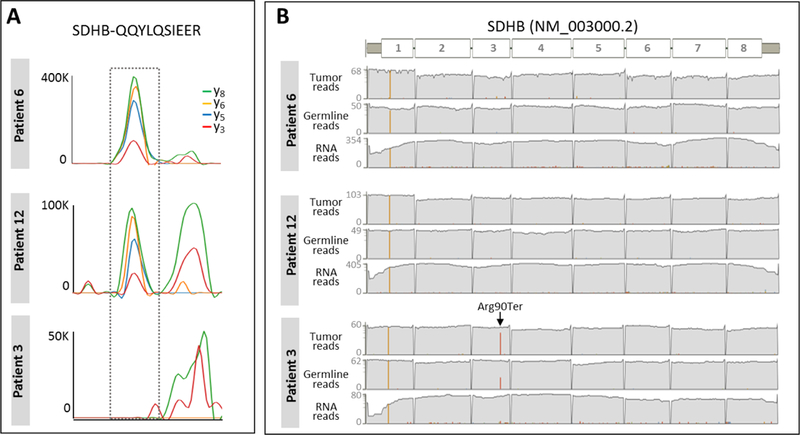
Differential SDHB status. (A) Chromatographic traces of four extracted fragment ions (y3, y5, y6, y8) (± 10 ppm) of a unique SDHB peptide, QQYLQSIEER are shown for three biopsies exhibiting different levels (high, intermediate, absent) of SDHB protein. (B) Depth of tumor DNA, germline DNA, and tumor RNA sequencing reads mapped to SDHB in three biopsies corresponding to the patient samples shown in DIA-MS data left. Red bar indicates the number of reads that contain Arg90ter.
Tumor DNA was significantly enriched for the mutation compared to germline DNA (30/38 reads with the variant in tumor versus 18/50 reads in germline, p=7.6 × 10−6 by Fisher’s exact test). Only 3 out of 35 RNA transcript reads at the locus contained the mutation likely due to degradation of the truncated transcript via nonsense mediated decay. Copy-number analysis indicated loss of heterozygosity for chromosome 1p, including SDHB (1p36.13). The tumor DNA and RNA reads which do not contain the mutation are consistent with the estimate of 70% tumor purity after macrodissection of tumor region for genomic analysis. The absence of SDHB protein in the DIA analysis is consistent with higher tumor purity after microdissection of tumor regions. SDHB deficiency and loss of function is a known tumor driver in certain cancer types38. Approximately 7.5% of gastrointestinal stromal tumors (GIST) are SDH-deficient. As these patients are known to respond poorly to the common targeted therapy for GIST with tyrosine kinase inhibitor imatinib, diagnosis of this sub-type is critical for treatment decision-making38,39–40. This finding demonstrates the potential of DIA-based tumor characterization on FFPE biopsies to identify subtype-specific biomarkers. Seen from the perspective of assay development, the chromatographic and mass spectrometric attributes of the SDHB peptides identified by DIA analysis can be translated directly into the development of a new targeted assay.
Finally, the FDA recently approved an immunotherapy for treatment of tumor with high microsatellite instability (MSI-H) due to mutation or inactivation of genes in the DNA mismatch repair (MMR) pathway. This genetic marker is found in about 15% of CRC. Of the 12 samples analyzed in this study, none were found to be MSI-H by genomic analysis. In proteomic analysis, MMR family proteins including MLH1, MSH2, MSH3, MSH5, and MSH6 were present at high levels across the samples, thus corroborating the genomic profile.
3.3. DIA compared to SRM-based quantitation of predictive biomarkers
We have previously demonstrated that protein concentrations of clinically actionable biomarkers measured by SRM in FFPE tumor biopsies provide objective cutoff values that predict therapeutic outcomes1, 41–42. Those cutoff values can be used to identify eligible patients for relevant therapies or to forgo specific therapies unlikely to benefit the patients. To investigate such utility with DIA-based quantitation, we compared DIA-readouts to the tumoral concentrations (amol/μg) obtained by SRM from the same biopsy samples focusing on TUBB3 (peptide: ISVYYNEASSHK) and EGFR (peptide: IPLENLQIIR). Good agreement between the methods was observed (Fig. 5). An SRM-based clinical cutoff value of 750 amol/μg was previously established for TUBB3 protein as predictive of resistance to taxane-based chemotherapy42. Based on this, biopsies expressing TUBB3 above this cutoff are unlikely to benefit from taxane42–43. If DIA readout of TUBB3 correlates with the SRM results, it would be reasonable to expect that the DIA readout corresponding to the SRM-based cutoff would similarly identify patients unlikely to benefit from taxane. Fig. 5A demonstrates a good correlation between DIA and SRM results on TUBB3 in this study (r2 = 0.80); as an example, a DIA threshold of 4.5E5 would correctly classify 11/12 samples (9/12 as taxane-resistant, 2/12 as taxane-sensitive, 1/12 mis-classified as taxane-resistant). Likewise, EGFR protein expression levels measured by two methods showed a good agreement (r2 = 0.70) (Fig. 5B). From SRM analysis of these samples, two gastric tumors exhibited EGFR expression higher than 320 amol/μg, the median tumoral concentration of EGFR established based on SRM data in our clinical laboratory, and those higher EGFR expressing samples are correlated with the DIA data. High tumor expression of EGFR is an indicator of benefit from cetuximab44. It is worth emphasizing that the experiments of DIA and SRM have been performed with significantly different instrument setups. The chromatography conditions of each method have been optimized independently to meet different purposes (SRM: 14 min separation time up to 50% ACN with 800 nl/min flow rate; DIA: 250 min separation time up to 35% ACN with 300 nl/min flow rate) with different C18 columns as well as different ESI interfaces and instrumentation. Based on previously published results, direct comparison of SRM vs DIA quantification using the same instrument setup should result in a strong correlation (r2 > 0.95)8, 16, 45, while the correlation becomes weaker if the chromatographic conditions are different: Selevescek et al. reported correlations (n=51) between DIA and SRM, with a median correlation of 0.57 r2 46. In this case the weaker correlation is most probably due to the varying transition interferences and/or ion suppression levels, rather than the different acquisition methods of SRM and DIA.
Figure 5.

DIA readouts compared to protein concentrations by SRM. DIA vs SRM-based analysis of the same peptide showed a good agreement between the methods (A) TUBB3 with peptide ISVYYNEASSHK (r2=0.80). The shaded area contains samples with TUBB3 expression below the cutoff indicating likely taxane resistance. (B) EGFR with peptide IPLENLQIIR (r2=0.70). Samples in the shaded area express EGFR above the median concentration by SRM.
This comparison experiment inspires us that DIA-based protein quantification can be used in clinic for therapeutic decision-making, as SRM-based clinical cutoffs could be directly projected to the DIA readouts. Moving forward, however, in order for DIA assays to be applicable in routine clinic, quantitative studies involving dilution curves need to be performed to define the limit of quantification (LOQ) of all peptides, and the standardized intensity normalization strategy needs to be defined for lab-to-lab robustness.
3.4. Protein vs mRNA expression
It is widely accepted that mRNA expression levels of genes are not directly correlated to their cellular protein concentrations. Based on CPTAC’s recent proteogenomic studies conducted on CRC and breast tumor tissues, while overall correlations between mRNA and protein expression were not concordant, subsets of protein-mRNA pairs exhibited strong correlations both positively and negatively47–48. Genes encoding proteins with metabolic functions tended to be correlated with their protein expression levels, and genes encoding ribosome and proteasome compartments tended to be negatively correlated. As DIA data in this study generated expression levels of >3700 proteins, we repeated this analysis by comparing DIA readouts to mRNA levels (TPM values) obtained from the same FFPE tumor biopsies.
The overall correlation of 3429 protein-mRNA pairs across 12 patient samples is shown in Figure 6A with Spearman’s correlation of 0.45. (Fig. 6A) The Spearman’s correlation coefficients calculated for each mRNA-protein pair between the data types ranged from −0.71 to 0.97, and the median of the coefficients across mRNA-protein pairs was 0.31 (Fig. S-5). This result is comparable to the previously reported TCGA studies of fresh-frozen tumor tissues 47–48. We performed gene set enrichment analysis (GSEA)49 of the Spearman’s correlation coefficient between mRNA-protein pairs (Fig. 6B). The significantly enriched pathways with high mRNA-protein correlation (positive GSEA score) are involved in cellular metabolism and immune response. Significantly enriched gene sets with negative mRNA-protein correlation include ribosomes and spliceosomes. This finding also consistent with the previous studies conducted in frozen tissues 47–48.
Figure 6.
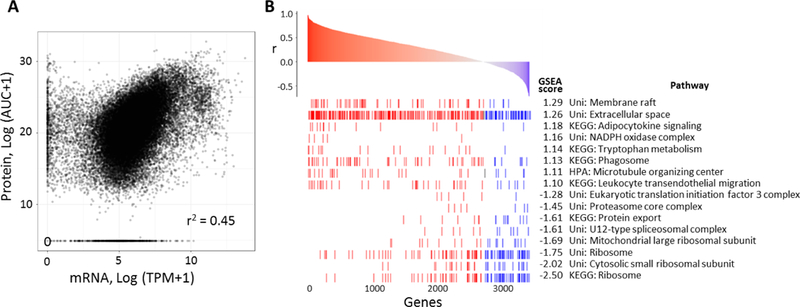
Proteogenomic correlation analysis. (A) Overall correlation of protein and mRNA measurements across 3,429 protein-mRNA pairs from 12 patients (12 × 3,429 points). Spearman’s correlation coefficient is listed. (B) Gene set enrichment analysis of Spearman’s correlation coefficients between each protein-mRNA pair in 12 patients. 8 gene sets with the lowest FDR (<0.05) associated with either high protein-mRNA correlation (high GSEA score) or low protein-mRNA correlation are displayed.
CONCLUSIONS
We anticipate that proteome-wide expression data obtained directly from tumor biopsies by a robust DIA method will provide numerous opportunities in oncology. This includes identifying novel therapeutic targets or biomarkers in discovery mode, and also capitalizing on the high multiplexing capacity of quantitative measurement of protein biomarkers in a clinical setting. In this study, DIA-MS in combination with microdissection of the archived FFPE tumor biopsies achieved good depth of coverage (five orders of magnitude on the estimated protein copy numbers) even without peptide fractionation. Importantly, the quantified proteins include actionable cancer biomarkers, FDA-approved drug targets and known cancer driver genes. Complementary information from genomic and proteomic analyses solidified the conclusion based on molecular profiles, as highlighted by the patient-specific non-detection of SDHB with loss of heterozygosity and nonsense mutation. A comparison of DIA vs SRM for two actionable biomarkers demonstrates the potential of DIA in clinical use for therapeutic decision-making, as SRM-based clinical cutoffs could be directly projected to the DIA-based protein quantitation. We suggest that DIA-based quantification of protein levels and correlation with patient outcomes will permit the development of improved diagnostic tests and extend the utility of known biomarkers beyond genomic testing. In summary, DIA-based analysis of clinical FFPE tumor biopsies represents an attractive diagnostic approach, combining reliable quantitation with broad proteome coverage.
Supplementary Material
ACKNOWLEDGMENTS
Authors acknowledge to Drs. Sheeno Thyparambil, Shankar Sellappan, and Fabiola Cecchi for useful discussions on the clinical applications of this study. This research was supported in part by National Institutes of Health Grants R21 CA192983 and P41 GM103533.
Footnotes
SUPPORTING INFORMATION:
The following supporting information is available free of charge at ACS website http://pubs.acs.org
CONFLICTS OF INTEREST
YJK, SMMS, AJS, SW, WL, CV, RH, TH are employees of NantOmics.
REFERENCES
- 1.An E; Ock CY; Kim TY; Lee KH; Han SW; Im SA; Kim TY; Liao WL; Cecchi F; Blackler A; Thyparambil S; Kim WH; Burrows J; Hembrough T; Catenacci DVT; Oh DY; Bang YJ, Quantitative proteomic analysis of HER2 expression in the selection of gastric cancer patients for trastuzumab treatment. Ann Oncol 2017, 28 (1), 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hembrough T; Liao WL; Hartley CP; Ma PC; Velcheti V; Lanigan C; Thyparambil S; An E; Monga M; Krizman D; Burrows J; Tafe LJ, Quantification of Anaplastic Lymphoma Kinase Protein Expression in Non-Small Cell Lung Cancer Tissues from Patients Treated with Crizotinib. Clin Chem 2016, 62 (1), 252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catenacci DV; Liao WL; Thyparambil S; Henderson L; Xu P; Zhao L; Rambo B; Hart J; Xiao SY; Bengali K; Uzzell J; Darfler M; Krizman DB; Cecchi F; Bottaro DP; Karrison T; Veenstra TD; Hembrough T; Burrows J, Absolute quantitation of Met using mass spectrometry for clinical application: assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PLoS One 2014, 9 (7), e100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoofnagle AN; Wener MH, The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J Immunol Methods 2009, 347 (1–2), 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nuciforo P; Thyparambil S; Aura C; Garrido-Castro A; Vilaro M; Peg V; Jimenez J; Vicario R; Cecchi F; Hoos W; Burrows J; Hembrough T; Ferreres JC; Perez-Garcia J; Arribas J; Cortes J; Scaltriti M, High HER2 protein levels correlate with increased survival in breast cancer patients treated with anti-HER2 therapy. Mol Oncol 2016, 10 (1), 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YJ; Gallien S; van Oostrum J; Domon B, Targeted proteomics strategy applied to biomarker evaluation. Proteomics Clin Appl 2013, 7 (11–12), 739–47. [DOI] [PubMed] [Google Scholar]
- 7.Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ, Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 2012, 11 (11), 1475–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillet LC; Navarro P; Tate S; Rost H; Selevsek N; Reiter L; Bonner R; Aebersold R, Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 2012, 11 (6), O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rardin MJ; Schilling B; Cheng LY; MacLean BX; Sorensen DJ; Sahu AK; MacCoss MJ; Vitek O; Gibson BW, MS1 Peptide Ion Intensity Chromatograms in MS2 (SWATH) Data Independent Acquisitions. Improving Post Acquisition Analysis of Proteomic Experiments. Mol Cell Proteomics 2015, 14 (9), 2405–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberger G; Bludau I; Schmitt U; Heusel M; Hunter CL; Liu Y; MacCoss MJ; MacLean BX; Nesvizhskii AI; Pedrioli PGA; Reiter L; Rost HL; Tate S; Ting YS; Collins BC; Aebersold R, Statistical control of peptide and protein error rates in large-scale targeted data-independent acquisition analyses. Nat Methods 2017, 14 (9), 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruderer R; Sondermann J; Tsou CC; Barrantes-Freer A; Stadelmann C; Nesvizhskii AI; Schmidt M; Reiter L; Gomez-Varela D, New targeted approaches for the quantification of data-independent acquisition mass spectrometry. Proteomics 2017, 17 (9), doi: 10.1002/pmic.201700021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou G; Lou X; Sun Y; Xu S; Zi J; Wang Q; Zhou B; Han B; Wu L; Zhao X; Lin L; Liu S, Biomarker Discovery and Verification of Esophageal Squamous Cell Carcinoma Using Integration of SWATH/MRM. J Proteome Res 2015, 14 (9), 3793–803. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y; Chen J; Sethi A; Li QK; Chen L; Collins B; Gillet LC; Wollscheid B; Zhang H; Aebersold R, Glycoproteomic analysis of prostate cancer tissues by SWATH mass spectrometry discovers N-acylethanolamine acid amidase and protein tyrosine kinase 7 as signatures for tumor aggressiveness. Mol Cell Proteomics 2014, 13 (7), 1753–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ortea I; Rodriguez-Ariza A; Chicano-Galvez E; Arenas Vacas MS; Jurado Gamez B, Discovery of potential protein biomarkers of lung adenocarcinoma in bronchoalveolar lavage fluid by SWATH MS data-independent acquisition and targeted data extraction. J Proteomics 2016, 138, 106–14. [DOI] [PubMed] [Google Scholar]
- 15.Collins BC; Hunter CL; Liu Y; Schilling B; Rosenberger G; Bader SL; Chan DW; Gibson BW; Gingras AC; Held JM; Hirayama-Kurogi M; Hou G; Krisp C; Larsen B; Lin L; Liu S; Molloy MP; Moritz RL; Ohtsuki S; Schlapbach R; Selevsek N; Thomas SN; Tzeng SC; Zhang H; Aebersold R, Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat Commun 2017, 8 (1), 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y; Huttenhain R; Surinova S; Gillet LC; Mouritsen J; Brunner R; Navarro P; Aebersold R, Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 2013, 13 (8), 1247–56. [DOI] [PubMed] [Google Scholar]
- 17.Chambers MC; Maclean B; Burke R; Amodei D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; Hoff K; Kessner D; Tasman N; Shulman N; Frewen B; Baker TA; Brusniak MY; Paulse C; Creasy D; Flashner L; Kani K; Moulding C; Seymour SL; Nuwaysir LM; Lefebvre B; Kuhlmann F; Roark J; Rainer P; Detlev S; Hemenway T; Huhmer A; Langridge J; Connolly B; Chadick T; Holly K; Eckels J; Deutsch EW; Moritz RL; Katz JE; Agus DB; MacCoss M; Tabb DL; Mallick P, A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol 2012, 30 (10), 918–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; Villén J; MacCoss MJ, Comprehensive peptide quantification for data independent acquisition mass spectrometry using chromatogram libraries. Nature Communications 2018, In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The UniProt C, UniProt: the universal protein knowledgebase. Nucleic Acids Res 2017, 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kall L; Canterbury JD; Weston J; Noble WS; MacCoss MJ, Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 2007, 4 (11), 923–5. [DOI] [PubMed] [Google Scholar]
- 21.MacLean B; Tomazela DM; Shulman N; Chambers M; Finney GL; Frewen B; Kern R; Tabb DL; Liebler DC; MacCoss MJ, Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26 (7), 966–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J, The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731. [DOI] [PubMed] [Google Scholar]
- 23.North JP; Golovato J; Vaske CJ; Sanborn JZ; Nguyen A; Wu W; Goode B; Stevers M; McMullen K; Perez White BE; Collisson EA; Bloomer M; Solomon DA; Benz SC; Cho RJ, Cell of origin and mutation pattern define three clinically distinct classes of sebaceous carcinoma. Nature Communications 2018, 9 (1), 1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H; Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25 (14), 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanborn JZ; Chung J; Purdom E; Wang NJ; Kakavand H; Wilmott JS; Butler T; Thompson JF; Mann GJ; Haydu LE; Saw RP; Busam KJ; Lo RS; Collisson EA; Hur JS; Spellman PT; Cleaver JE; Gray JW; Huh N; Murali R; Scolyer RA; Bastian BC; Cho RJ, Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc Natl Acad Sci U S A 2015, 112 (35), 10995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langmead B; Trapnell C; Pop M; Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009, 10 (3), R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li B; Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thul PJ; Akesson L; Wiking M; Mahdessian D; Geladaki A; Ait Blal H; Alm T; Asplund A; Bjork L; Breckels LM; Backstrom A; Danielsson F; Fagerberg L; Fall J; Gatto L; Gnann C; Hober S; Hjelmare M; Johansson F; Lee S; Lindskog C; Mulder J; Mulvey CM; Nilsson P; Oksvold P; Rockberg J; Schutten R; Schwenk JM; Sivertsson A; Sjostedt E; Skogs M; Stadler C; Sullivan DP; Tegel H; Winsnes C; Zhang C; Zwahlen M; Mardinoglu A; Ponten F; von Feilitzen K; Lilley KS; Uhlen M; Lundberg E, A subcellular map of the human proteome. Science 2017, 356 (6340). [DOI] [PubMed] [Google Scholar]
- 29.Kanehisa M; Sato Y; Kawashima M; Furumichi M; Tanabe M, KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016, 44 (D1), D457–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wisniewski JR; Dus-Szachniewicz K; Ostasiewicz P; Ziolkowski P; Rakus D; Mann M, Absolute Proteome Analysis of Colorectal Mucosa, Adenoma, and Cancer Reveals Drastic Changes in Fatty Acid Metabolism and Plasma Membrane Transporters. J Proteome Res 2015, 14 (9), 4005–18. [DOI] [PubMed] [Google Scholar]
- 31.Wisniewski JR; Hein MY; Cox J; Mann M, A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics 2014, 13 (12), 3497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tamborero D; Rubio-Perez C; Deu-Pons J; Schroeder MP; Vivancos A; Rovira A; Tusquets I; Albanell J; Rodon J; Tabernero J; de Torres C; Dienstmann R; Gonzalez-Perez A; Lopez-Bigas N, Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med 2018, 10 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wishart DS; Knox C; Guo AC; Shrivastava S; Hassanali M; Stothard P; Chang Z; Woolsey J, DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 2006, 34 (Database issue), D668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey MH; Tokheim C; Porta-Pardo E; Sengupta S; Bertrand D; Weerasinghe A; Colaprico A; Wendl MC; Kim J; Reardon B; Ng PK; Jeong KJ; Cao S; Wang Z; Gao J; Gao Q; Wang F; Liu EM; Mularoni L; Rubio-Perez C; Nagarajan N; Cortes-Ciriano I; Zhou DC; Liang WW; Hess JM; Yellapantula VD; Tamborero D; Gonzalez-Perez A; Suphavilai C; Ko JY; Khurana E; Park PJ; Van Allen EM; Liang H; Group MCW; Cancer Genome Atlas Research, N.; Lawrence MS; Godzik A; Lopez-Bigas N; Stuart J; Wheeler D; Getz G; Chen K; Lazar AJ; Mills GB; Karchin R; Ding L, Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173 (2), 371–385 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The future of cancer genomics. Nat Med 2015, 21 (2), 99. [DOI] [PubMed] [Google Scholar]
- 36.Tomczak K; Czerwinska P; Wiznerowicz M, The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn) 2015, 19 (1A), A68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlen M; Zhang C; Lee S; Sjostedt E; Fagerberg L; Bidkhori G; Benfeitas R; Arif M; Liu Z; Edfors F; Sanli K; von Feilitzen K; Oksvold P; Lundberg E; Hober S; Nilsson P; Mattsson J; Schwenk JM; Brunnstrom H; Glimelius B; Sjoblom T; Edqvist PH; Djureinovic D; Micke P; Lindskog C; Mardinoglu A; Ponten F, A pathology atlas of the human cancer transcriptome. Science 2017, 357 (6352). [DOI] [PubMed] [Google Scholar]
- 38.Miettinen M; Lasota J, Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) - a review. Int J Biochem Cell Biol 2014, 53, 514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belinsky MG; Cai KQ; Zhou Y; Luo B; Pei J; Rink L; von Mehren M, Succinate dehydrogenase deficiency in a PDGFRA mutated GIST. BMC Cancer 2017, 17 (1), 512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.von Mehren M; Joensuu H, Gastrointestinal Stromal Tumors. J Clin Oncol 2018, 36 (2), 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz S; Cecchi F; Tian Y; Scott K; Bartolomeo MD; Morano F; Fucà G; Martinetti A; Braud FGD; Dominoni F; Milione M; Calegari MA; Orlandi A; Barone C; Pietrantonio F; Hembrough TA, Selecting patients with metastatic colorectal cancer for treatment with temozolomide using proteomic analysis of MGMT. Journal of Clinical Oncology 2017, 35 (15_suppl), 11601–11601. [Google Scholar]
- 42.Cecchi F; Catenacci DVT; Tian Y; Miceli R; Pietrantonio F; Pellegrinelli A; Martinetti A; Bartolomeo MD; Hembrough TA, Quantitative proteomic analysis of TUBB3 to identify gastric cancer patients who may benefit from docetaxel: A reevaluation of the ITACA-S trial. Journal of Clinical Oncology 2017, 35 (4_suppl), 59–59. [Google Scholar]
- 43.Mozzetti S; Ferlini C; Concolino P; Filippetti F; Raspaglio G; Prislei S; Gallo D; Martinelli E; Ranelletti FO; Ferrandina G; Scambia G, Class III beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res 2005, 11 (1), 298–305. [PubMed] [Google Scholar]
- 44.Hutchinson RA; Adams RA; McArt DG; Salto-Tellez M; Jasani B; Hamilton PW, Epidermal growth factor receptor immunohistochemistry: new opportunities in metastatic colorectal cancer. J Transl Med 2015, 13, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faktor J; Sucha R; Paralova V; Liu Y; Bouchal P, Comparison of targeted proteomics approaches for detecting and quantifying proteins derived from human cancer tissues. Proteomics 2017, 17 (5). [DOI] [PubMed] [Google Scholar]
- 46.Selevsek N; Chang CY; Gillet LC; Navarro P; Bernhardt OM; Reiter L; Cheng LY; Vitek O; Aebersold R, Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Mol Cell Proteomics 2015, 14 (3), 739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mertins P; Mani DR; Ruggles KV; Gillette MA; Clauser KR; Wang P; Wang X; Qiao JW; Cao S; Petralia F; Kawaler E; Mundt F; Krug K; Tu Z; Lei JT; Gatza ML; Wilkerson M; Perou CM; Yellapantula V; Huang KL; Lin C; McLellan MD; Yan P; Davies SR; Townsend RR; Skates SJ; Wang J; Zhang B; Kinsinger CR; Mesri M; Rodriguez H; Ding L; Paulovich AG; Fenyo D; Ellis MJ; Carr SA; Nci C, Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534 (7605), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang B; Wang J; Wang X; Zhu J; Liu Q; Shi Z; Chambers MC; Zimmerman LJ; Shaddox KF; Kim S; Davies SR; Wang S; Wang P; Kinsinger CR; Rivers RC; Rodriguez H; Townsend RR; Ellis MJ; Carr SA; Tabb DL; Coffey RJ; Slebos RJ; Liebler DC; Nci C, Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513 (7518), 382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Subramanian A; Tamayo P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES; Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005, 102 (43), 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.