

Abstract
We have performed deep proteomic profiling down to as few as 9 Panc-1 cells using sample fractionation, TMT multiplexing, and a carrier/reference strategy. Off line fractionation of the TMT-labeled sample pooled with TMT-labeled carrier Panc-1 whole cell proteome was achieved using alkaline reversed phase spin columns. The fractionation in conjunction with the carrier/reference (C/R) proteome allowed us to detect 47 414 unique peptides derived from 6261 proteins, which provided a sufficient coverage to search for single amino acid variants (SAAVs) related to cancer. This high sample coverage is essential in order to detect a significant number of SAAVs. In order to verify genuine SAAVs versus false SAAVs, we used the SAVControl pipeline and found a total of 79 SAAVs from the 9-cell Panc-1 sample and 174 SAAVs from the 5000-cell Panc-1 C/R proteome. The SAAVs as sorted into high confidence and low confidence SAAVs were checked manually. All the high confidence SAAVs were found to be genuine SAAVs, while half of the low confidence SAAVs were found to be false SAAVs mainly related to PTMs. We identified several cancer-related SAAVs including KRAS, which is an important oncoprotein in pancreatic cancer. In addition, we were able to detect sites involved in loss or gain of glycosylation due to the enhanced coverage available in these experiments where we can detect both sites of loss and gain of glycosylation.
Keywords: small number of cells, sample fractionation, LC−MS/MS, single amino acid variants (SAAVs), cancer related SAAVs
Graphical Abstract
INTRODUCTION
Protein mutations such as single amino acid variants (SAAVs) can result in a change of protein abundance, folding, stability, function, its interactions with other proteins, and the proteins subcellular localization.1–3 SAAVs have been shown to play an important role in diseases including cancer, where the SAAVs can lead to activated oncogene expression or inactivation of tumor suppressor gene expression.4 SAAVs may also have a central role in tumorigenesis and subsequent cancer progression.5 A systematic discovery of variants in protein amino acid sequences is crucial for our understanding of the mechanisms for tumor progression and would offer a potential for future therapy and treatment of cancer.6,7
Cellular heterogeneity may derive from mutations in the genome as cells divide and different clones of the cells may be generated.8 The different clones can result in different phenotypes including in cancer with increased aggressiveness. A small population of these cells with altered phenotype may eventually determine the progression of the tumor.9 Thus, analysis of small numbers of cells even down to a single cell is an important goal, as current methods are mainly applicable to analysis of bulk populations. The data from bulk populations often results in the loss of information on those unique cell subpopulations that may be drivers of important cellular processes.10,11 There has been a significant advance in the area of analysis of small numbers of cells for genomic and transcriptomic profiling of cells recently.12,13 Single cell profiling is essential for rare subpopulations in cancer such as cancer stem cells (CSCs), where there is a limited number of cells available for analysis.14,15
Proteomic analysis of a small number of cells is currently challenging due to the lack of sensitive methods to process the extremely low amounts of cellular proteins for liquid chromatography/tandem mass spectrometry (LC–MS/MS).16 This becomes even more difficult for detection of protein variants where relatively high peptide coverage for the protein is required in order to observe the unique peptides that define the different isoforms. We have thus adopted a method for sample preparation and analysis in which processing of small numbers of cells is performed with minimal steps to prevent protein losses. We have also used a carrier/reference strategy based on TMT (Tandem Mass Tags) labeling, where the MS/MS spectra from a larger number of cells are used to trigger detection of those from a limited number of cells. This strategy has resulted in detection of up to 6000 proteins in a sample of 9 Panc-1 cells with sufficient coverage to detect a significant number of single amino acid protein variants, many of which are known to be cancer related.
In the current work, we use carrier/reference LC–MS/MS detection to demonstrate the analysis of the proteomes from small numbers of cells for identification of single amino acid variants. However, an important issue is that for the detection of SAAVs based on mass spectrometry analysis and database searching, there is a risk for the presence of a high false positive detection rate that results from misidentified PTMs or other false assignments. In order to deal with false positives, most studies have used a special custom database for database searching.7,17–22 Few studies have focused on false discovery rate (FDR) to reduce false positive results in these variant identifications.23–25 Herein, we have addressed the challenge of false positives using software called SAVControl, which can sort SAAVs into categories of genuine variants versus likely false positives. This software has been utilized with data from as few as nine cells analyzed using a Panc-1 carrier/reference and incorporating a detailed manual check to distinguish true SAAVs versus false positives.26 Those SAAVs identified as genuine SAAVs are identified with high probability, whereas those identified as likely false positives are indeed found to be so in at least half of these cases. The methods are applied to analysis of cells from Panc-1 cell lines for bulk conditions, 5000 cells, down to a small number of cells, i.e., nine cells.
MATERIALS AND METHODS
Cell Culture and Counting
The Panc-1 cell line was obtained from the American Type Culture Collection (ATCC). These cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) antibiotic-antimycotic. Panc-1 cells were cultured at 37°C in a humidified atmosphere with 5% CO2. The cells were harvested when the cells covered approximately 60% of the dish surface. The supernatant was removed after the harvested cells were standing for 40 min in PBS solution. This procedure was repeated 4 times, and the harvested cells were counted with trypan blue staining using a hemocytometer under a microscope (TRM-301, Bresser Science) where approximately 10k cells were obtained. The trypan blue indicates that these are live cells and these cells were able to be cultured.27 The cell suspension was diluted with PBS into around 3 cells per 1 μL. The cell number in a 0.5 mL LoBind Eppendorf tube was counted under the microscope carefully to determine the exact number of Panc-1 cells. Samples with different numbers of Panc-1 cells, including 31 and 9 cells were obtained after carefully counting of the cells in the tubes under the microscope.
Protein Extraction and Sample Preparation
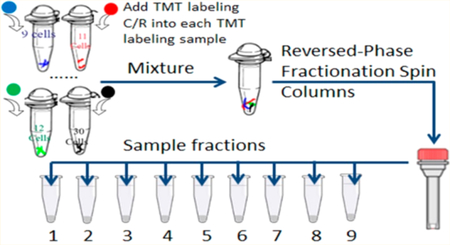
Because of the amount of protein in 9–31 cells, precautions were followed to minimize protein losses. Once obtained the samples were not transferred until the carrier/reference proteome was added to the TMT-labeled sample. Cells that were archived in 200 μL PCR tubes were suspended in 10 μL of 0.05% ProteaseMax (Promega) 100 mM TEAB (pH = 8.5) and 0.5 mM TCEP. Lysis was accomplished by sonication in a cup-horn sonicator (Q-Sonica). Cysteine residues were then alkylated with 2 mM IAA for 30 min under dark conditions. The proteins were digested by addition of 20 ng sequencing grade Trypsin (Promega) overnight at 37 °C. After the digestion, the samples were labeled with the appropriate TMT-11plex reagent by adding sufficient reagent for labeling 5 μg of protein. The samples were quenched by adding 10 mM hydroxylamine for 15 min at room temperature.28,29 The sample set information is presented in Table S1. TMT-labeled C/R proteome after quenching was always added to the sample tube as part of the pooling procedure. After pooling all samples with the full amount of C/R proteome the pool was fractionated using the Pierce high pH reversed-phase peptide fractionation kit (Thermo Fisher Scientific). As a control, the C/R proteome was also fractionated independently using the same kit. Thus, any losses from the small numbers of cells are minimized during further handling since they are a small fraction of the total population.
LC–MS/MS Analysis
Each of 9 fractions was analyzed on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, CA). The entire amount of TMT11-plex labeled sample in each fraction was analyzed using a 90 min gradient from 4% to 30% acetonitrile with 0.1% formic acid. We used the Easy 1000 nano UHPLC system (Thermo Fisher Scientific, CA) and Acclaim PepMap 100, 75 μm × 2 cm trap with Acclaim PepMap RSLC, 75 μm × 25 cm column (Dionex). The column effluent is analyzed directly by LC–MS/MS where all material is injected into a single run because of the limited amount of sample. The mass spectral data acquisition was performed using the software Xcalibur v2.3 (Thermo Fisher Scientific, CA) in data dependent mode. The ESI spray voltage was set at 2500 V. A full mass scan (m/z 400–1400) was performed, and using the auto normal scan the most intense ions in the full scan were chosen for MS/MS analysis. The normalized collision energy was set at 39% for high-energy collision-induced dissociation (HCD) fragmentation. The maximum injection time was set 150 ms. The Orbitrap resolution was set at 60 k for both MS1 and MS2 spectra. Filter dynamic exclusion settings were set as follows: repeat count 1 and exclusion duration 60 s. Both mass tolerances low and high were set at 10 ppm.
Database Searching
The protein sequence database, CanProVar (v2.0),30 was downloaded freely from http://canprovar2.zhang-lab.org/ for database searching. The CanProVar (v2.0) database includes 65 963 distinct human cancer protein variants collected and integrated from COSMIC, HPI, OMIM, TCGA, BioMart, and 825 106 coding SNPs from dbSNP. Corresponding nonvariant proteins come from the Ensembl database (Homo sapiens, v53). The reversed sequences of the same size were combined in the protein sequence database as decoys for FDR estimation.
Approximately 90% of the proteins in CanProVar 2.0 contain SAAVs derived from known single-nucleotide polymorphism (SNPs) and cancer-related variants. Database searching was conducted using the Mascot algorithm (v2.5.1, Matrix Science Inc.). The searching parameters were set as follows: (1) oxidation (+15.995 Da) at Methionine was set as a dynamic modification; (2) carbamidomethylation (+57.021 Da) at Cysteine and TMT Tags labeling at N-termini and Lysine (+229.163 Da) were set as static modifications; (3) the tolerance of precursor ions and fragment ions was set as 10 ppm and 0.05 Da, respectively; (4) the maximum missed trypsin cleavage sites allowed was 2; (5) the minimum peptide length was set at 5 amino acids and unrestricted peptide-level false discovery rate (FDR) was enabled; (6) charge status was set as +2, +3 and +4. Unrestricted peptide-level FDR setting can obtain extremely exaggerated high coverage of peptide identification mixing with real SAAVs and false positive SAAVs. The protein profile database searching was performed using Proteome Discoverer (V1.4, Thermo Fisher Scientific, CA) with the same parameter settings except using FDR 1% and Swiss-Prot Homo sapiens database (reviewed, downloaded in April 2014 with 26 152 entries). After database searching based on Mascot, the resulting data were applied for advanced site-level quality control of variant peptide identifications based on SAVControl software, which has proven to be effective for removal of false positive SAAVs sites.26 For advance data processing, SAVControl first filters out false peptide identifications using transfer FDR control, and then evaluates the reliability of the SAAV sites by unrestricted mass shift relocation and introduction of alternative interpretations such as modifications.26 All resulting variant sites are finally classified into three levels: Level I (reliable results), Level II (ambiguous results), and Level III (unreliable results). All level II SAAVs were further confirmed by manual check based on mass spectra.
In order to compare with the high quality control results provided by SAVControl, we also performed the SAAVs database search using Mascot with the same parameters except using FDR 1% peptide level. The Gene Ontology (GO) for these proteins with SAAV sites were performed using QIAGEN’S Ingenuity Pathway Analysis (IPA) online software (http://www.ingenuity.com/products/ipa). As for cancer-related SAAVs analysis, each SAAV was manually checked via the open source CanProVar 2.0 online search. The potential N-glycosylation occurs at the consensus sequence N-X-S/T/C, where X can be any amino acid except proline.31 We manually checked the potential site and counted the number of gain or lost potential N-glycan sites. The mass spectrometry data have been deposited to the ProteomeXchange32 Consortium via the PRIDE33 partner repository with the data set identifier PXD011471.
RESULTS AND DISCUSSION
Proteomic Profiles in Different Numbers of Panc-1 Cells via the Carrier/Reference Method
We have prepared and analyzed small numbers of Panc-1 cell samples by mass spectrometry. This included 9 and 31 cell samples with fractionation and TMT labeling as described above using the carrier/reference method. A total of 47 414 unique peptides derived from 6261 proteins from 9 cells were detected as compared to the 5000 cell reference where 65 901 unique peptides derived from 7435 proteins can be detected over a 90 min gradient for each fraction on an Orbitrap Fusion mass spectrometer using an FDR of 1% (Table S2). The number of identified proteins is much higher than in a previous study of 50 000 cells due to the strategies below.34
Three strategies were used to achieve the deep proteome analysis from small numbers of cells. One factor is the application of the carrier/reference method using protein samples derived from 5000 Panc-1 cells. The signal from the carrier/reference channels triggers the data acquisition for selected ions and allows signals from channels with much less protein to be identified as being the same ion as that in the carrier/reference channels. This method has been applied by Budnik et al. using 100–200 carrier cells where 583 proteins were quantified.35 A second strategy is the use of sample fractionation to reduce the peptide complexity and obtain the deepest possible coverage. Each sample was fractioned into 9 aliquots. Multiple fractionations can improve signal-to-noise, proteome coverage and reduce interference between peptides in quantitative proteomics analysis.36 Without this fractionation only a limited number of proteins/peptides can be detected. The third strategy involves minimizing steps to avoid sample loss such as using only 2 μL PBS buffer for single cell suspension, minimum transfer sample steps to reduce the proteins/peptides sticking on the surface of the LoBind tube and tip, and the use of cell disruption by ultrasonication.37
The extremely high protein (84%) and peptide (72%) overlap shows that the use of extensive fractionation and other methods used herein provide high efficiency for detection of large numbers of peptides for the reference 5000 cells, which increases the detection of the same precursor ions in the 9-cell sample by mass spectrometry (see Figure 1). These methods are essential for achieving the coverage required for detection of SAAVs. It should be noted that these methods for achieving this deep proteome detection were performed using off the shelf components.
Figure 1.
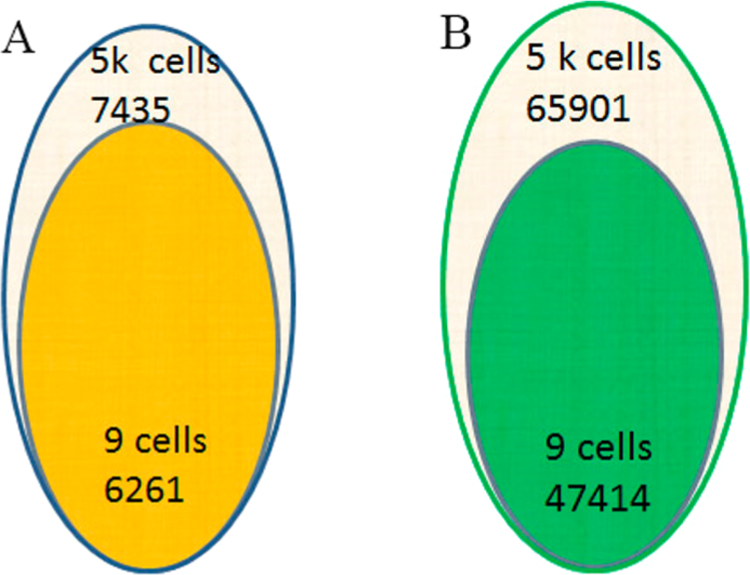
Overlap of identified proteins (A) and peptides (B) in 9 cells and 5000 cells sample.
It should be noted that in recent work using a nanodroplet processing in one pot for trace samples (nanoPOTS) technology for studying small numbers of cells that a proteome coverage of between 1500 proteins by MS/MS alone and 3100 proteins by MS/MS plus Match Between Runs (MBR) was achieved. However, the peptide coverage obtained was more limited at between 7.3k and 18k without and with MBR, where higher numbers of peptide identifications are required for SAAVs analysis.38
We conducted GO analysis of the detected proteins using the online software IPA. The subcellular distribution and protein groups in the 9-cell sample are shown in Figure 2. Among the total of 6261 proteins, nearly half were assigned to the cytoplasm, one-third derived from the nucleus, and the remaining proteins were assigned to the plasma membrane and extracellular space. As for the protein function cluster, approximately half of the proteins are not assigned a detailed function. Approximately one-fourth of the proteins are enzymes. There are also 8%, 7%, and 5% proteins that are transcription regulators, transporters, and kinases. A small percentage of proteins are peptidases, phosphatases, translation regulators, transmembrane receptors, and ion channels.
Figure 2.
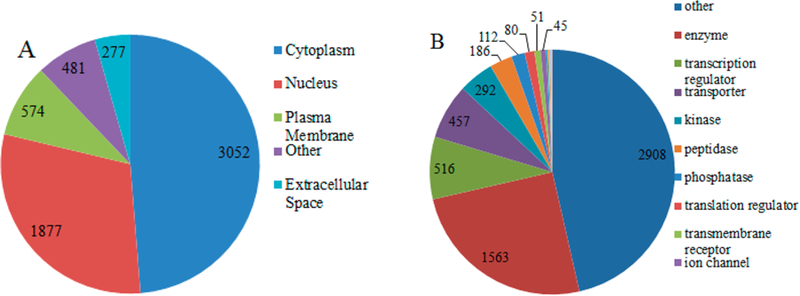
Gene Ontology (GO) analysis of 6261 detected proteins derived from 9 cells. Subcellular distribution of detected proteins (A). Types of detected proteins (B).
SAAVs Quality Control
An important issue is filtering the potential SAAVs detected for false positives.18,20 We performed database searching using the Mascot algorithm and obtained the results shown in Table 1 from 4 sample sets. The number of SAAVs with unrestricted FDR is very large, and most are false positives. The average number of SAAVs are 4278 from 4 sample sets. Only one tenth of the SAAVs succeeded to filter after setting the global FDR 1%. If the results derived from unrestricted FDR were filtered with stringent transfer FDR of 1%, then only 3% of potential true SAAVs were successfully filtered. However, there is still a high false positive rate for SAAVs. After the use of SAVControl quality control, 98% of SAAVs were found to be false positive SAAVs derived from unrestricted FDR database searching. If we applied a global FDR of 1%, a common target-decoy strategy on all peptide identification, to control the quality, there are still approximately 2-fold false positive SAAVs compared to the strategy using transfer FDR 1%, a more stringent FDR control method.26 There also still remain one-third false positive SAAVs compared to database searching using SAVControl to filter the false positive SAAVs. The SAVControl software controls variant peptide identifications by first filtering false positive SAAVs using transfer FDR control and then relocating and introducing alternative interpretations such as modifications, which remove false positive SAAVs very effectively.
Table 1.
Identification of the Number of SAAVs Based on Different FDR Strategies for Database Searching
| unrestricted FDR | global FDR (1%) | transfer FDR (1%) | SAVControl | |
|---|---|---|---|---|
| Sample Set_1 | 3626 | 358 | 99 | 78 |
| Sample Set_2 | 2987 | 326 | 83 | 66 |
| Sample Set_3 | 5348 | 474 | 173 | 123 |
| Sample Set_4 | 5154 | 465 | 166 | 115 |
| Average (Mean ± SD) | 4278 ± 1000 | 405 ± 65 | 130 ± 40 | 95 ± 25 |
| Filter ratio | N/A | 91% | 97% | 98% |
An important issue in SAAVs identification is the quality control involved in SAAVs detection where some percentage will be false matches to the databases.18 These may be due to the presence of PTMs or other forms of false assignments.17,26 FDR is essentially a statistical and computational measure to establish the certainty of the peptide identification in the data derived from tandem mass spectrometry.39 It is mandatory to set the FDR, usually as 1%, for target-decoy database searching strategy.40 In general, it works well for common database searching; however, it is unreliable for identification of peptides with variants. The reason for this has been discussed in previous studies.26 For a given score threshold, the FDR of potential peptides identification with SAAVs may be significantly different from the common global FDR of all peptides identification due to the FDR heterogeneity between peptides with SAAVs and nonvariant peptides.26 Some researchers have overcome this issue by applying a separate FDR for target-decoy FDR estimation and peptides with SAAVs, or a refined separate FDR, or more accurate transfer FDR strategy.25,41 In addition, amino acid modification also increases the incorrect identification of variant sites.42 Variants and modifications are two of the main obstacles to achieve highly reliable SAAVs identification.26
For SAAVs quality control using the SAVControl software, the method assigns scores for the SAAVs based on the highest scores to level I (reliable results), an intermediate score to Level II (ambiguous results) and a low score to Level III (unreliable results).26 This is shown in Figure 3 for four different sample sets where the SAAVs results are sorted into these different categories. The 4 sample sets information is presented in Table S1. Each sample was aliquoted to 9 fractions prior to analyzing on an Orbitrap Fusion mass spectrometer.
Figure 3.
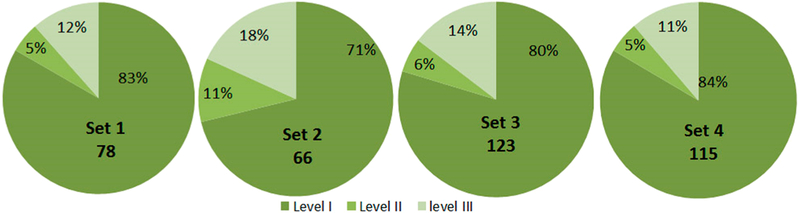
Proportions of SAAVs assigned into three levels by SAVControl software in 4 sample sets 1–4.
SAAVs Manual Validation
We randomly selected several of the SAAVs identified from SAVControl as being level I with high confidence and checked the fragment ions to determine if they matched the theoretical fragment ions. One such representative level I SAAV is shown in Figure 4. In this peptide the fragment ions matched the expected fragment ions. We found this to be true for all level I SAAVs detected. For level II from the database searching results, we checked all these potential SAAVs based on mass spectra. All potential SAAVs were validated manually by checking all fragment ions individually. Only those SAAVs where the observed fragment ions matched the expected fragment ions m/z were considered as genuine SAAVs. Representative SAAVs from level II and level III are shown below (Figure 5 and Figure 6). For the 9-cell Panc-1 sample we found 2 level II SAAVs that were genuine after manual check; however, 3 level III SAAVs were false positives (Table S3). This strategy illustrates the importance of this procedure for correct identification of SAAVs.
Figure 4.

Representative level I SAAV with fragment ions matched as expected. Amino acid Methionine at the 12th position was changed into amino acid Valine (M558V) in protein Septin-9.
Figure 5.
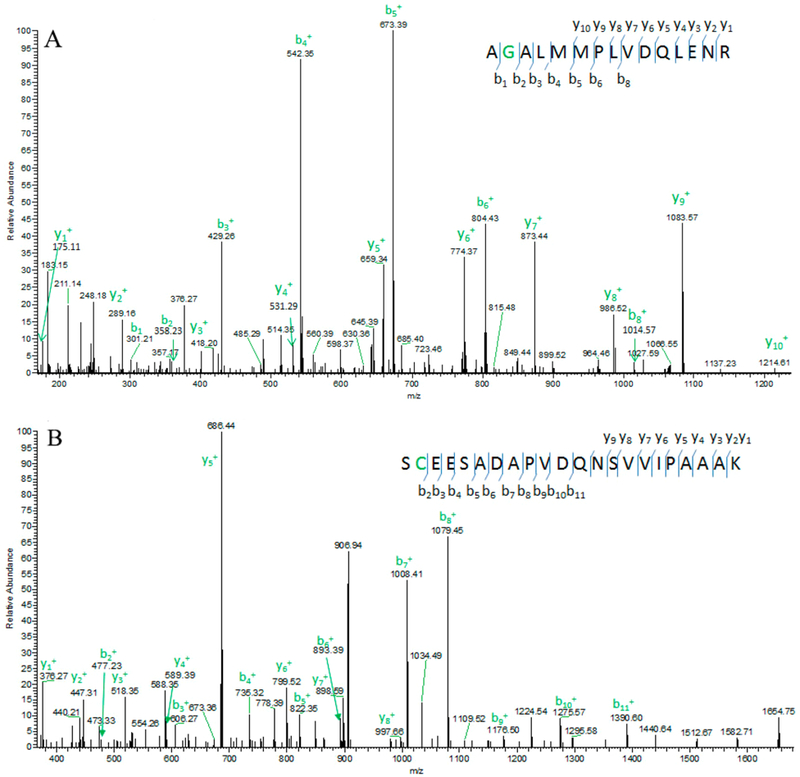
Representative level II SAAV with fragment ions matched as expected. Amino acid Glutamate the second position was changed into amino acid Glycine (E1936G) in protein HEATR1 (HEAT repeat-containing protein 1) (A). False positive level II SAAV where some fragment ions matched; however, the observed precursor ions do not match. If N changed to D, the observed precursor ions match the theoretical precursor ions (B).
Figure 6.
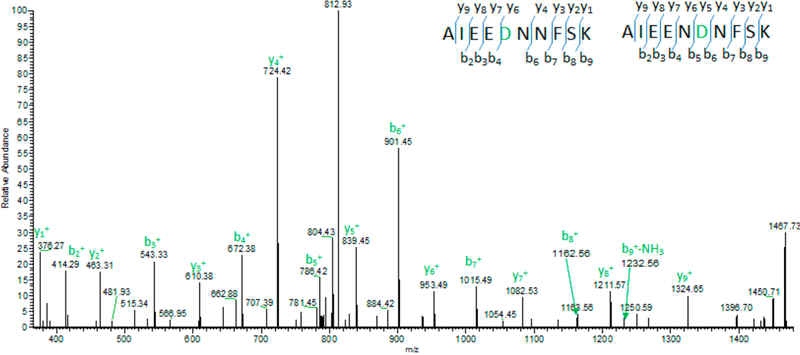
On the basis of a manual check, the fragment ions match the peptide AIEENDNFSK better than that of AIEEDNNFSK. Thus, SAVControl assigned the mass spectra to AIEEDNNFSK in level III, indicating a false positive SAAV.
Comparison of the SAAVs in Different Numbers of Panc-1 Cells
The use of the carrier reference method allows us the ability to analyze small numbers of cells and detect a significant number of SAAVs (see Table 2). We can assign the different number of SAAVs in different cell samples due to the labeling of the samples with TMT before mixing. The number of SAAVs detected were 79 and 174 derived from 9 cells and 5000 cells, respectively. There were 151 SAAVs detected from 5000 cells in the four samples and 174 SAAVs detected when combined with the 5000 cells of the C/R when analyzed separately (Table 2). These numbers are based on the sample fractionation method used, which provides relatively high peptide coverage. In the current analysis, we have detected over 79 SAAVs for the 9 cell sample (Table 2, Table S3).
Table 2.
Identification of the Number of SAAVs in 9–31 Cells and 5000 Cells Samples
| cell number | SAAVs number in carrier/reference | SAAVs in all small number of cells | SAAVs in 9 cells |
|---|---|---|---|
| 9–31 | 151 | 106 | 79 |
| 5000 | 174 | N/A | N/A |
Cancer Related SAAVs
We searched for cancer related SAAVs in the CanProVar 2.0 online resource (http://canprovar2.zhang-lab.org/) and found 8 cancer-related SAAVs derived from 8 proteins. Among them, KRAS mutation (G12D, G12 V, G12R and G12C) has been investigated as the initiating genetic event for pancreatic cancer of approximately 95% of pancreatic intraepithelial neoplasia (PanINs) patients (Table 3).43 TFRC is revealed as a marker of malignant phenotype in human pancreatic cancer but without any report on site G142S related to pancreatic cancer.44 The R521K variant has been associated with cancer severity in EGFR-expressing tumors, such as gliomas, lung cancer and colorectal carcinoma based on DNA sequence analysis.45,46 SEPT9 has mutations in liver and stomach cancer, but there are still a significant number of mutations in pancreatic cancer.47
Table 3.
Cancer Related SAAVs Detected in 9 Panc-1 Cells
| protein name | gene name | peptide | variant site in protein | level | related cancer |
|---|---|---|---|---|---|
| GTPase KRAS | KRAS | LVWGADGVGK | G12D | Level I | Pancreatic Cancer |
| Transferrin receptor protein 1 | TFRC | LDSTDFTSTIK | G142S | Level I | Intestines cancer |
| Epidermal growth factor receptor | EGFR | ATGQVC HALC SPEGC WGPEPK | R521K | Level I | Intestines cancer |
| Septin-9 | SEPT9 | LNEGSSAMANGVEEK | M558V | Level I | Thyroid carcinoma |
| Epidermal growth factor receptor substrate 15 | EPS IS | EKDPEMFCDPFTSATTTTNK | I668M | Level I | Skin cancer |
| Calumenin | CALU | YIYDNVENQWQDFDMNQPGLISWDEYR | E115D | Level I | Hepatocellular carcinoma |
| ATP synthase subunit d, mitochondrial | ATPSH | NLIPFDQMTIENLNEAFPETK | D135N | Level I | Skin cancer |
| 60 kDa heat shock protein, mitochondrial | HSPDl | DMAIATGGAVFGEQGLTLNLEDVQPHDLGK | E328Q | Level II | Breast cancer |
Gain or Lost Potential N-Glycosylation Sites Due to SAAVs in 9-Cell and 5000-Cell Samples
Ma et al. recently investigated the novel phosphorylation sites created by SAAVs.22 Here we manually checked the N-glycosylation consensus sites in peptides with detected SAAVs and identified 9 potential N-glycosylation sites that were affected (see Table 4). Three sites involve potential gain of glycosylation, whereas six sites involve potential loss of glycosylation. We further checked the mass spectra of N-glycan (N-GlcNAc) oxonium ions. The typical N-linked glycan backbone fragmentation can be identified by the oxonium ions at m/z 126.06, 138.06, 168.09, 186.07, 204.09.33 Two of the gain of N-glycosylation sites were shown to have an actual gain in glycosylation due to the presence of the oxonium ions. For the loss of N-glycosylation sites, we currently cannot show an actual loss of glycosylation.
Table 4.
N-Glycosylation Gain or Lost Due to Variant
| protein name | peptide | variant | N-X-S/T/C | gain or lost | detected in sample |
|---|---|---|---|---|---|
| GC-rich sequence DNA-binding factor 2 | DIDLSCGSGSSK | N211S | yes | lost | 9 cells |
| NSFL1 cofactor p47 | ASSSILINESEPTTNIQ1R | D290N | yes | gain | 9 cells, 5000 cells |
| Keratin | VIDDTDITR | N193D | yes | lost | 9 cells, 5000 cells |
| MKI67 FHA domain-interacting nucleolar phosphoprotein | YENESLQSGR | N104S | yes | lost | 5000 cells |
| GC-rich sequence DNA-binding factor 2 | DIDLSCGSGSSK | N211S | yes | lost | 5000 cells |
| Mab-21 domain containing 1 | GAPMDPNESPAAPEAALPK | T35N | yes | gain | 5000 cells |
| Polyribonucleotide nucleotidyltransferase 1 | EILQIMDK | N590D | yes | lost | 5000 cells |
| annexin A1 | GGPGSAVSPYPTFDPSSDVAALHK | N43D | yes | lost | 5000 cells |
| Inner membrane protein, mitochondrial | QKGDTPASATAPTEAAQNISAAGDTLSVPAPAVQPEESLK | I148N | yes | gain | 5000 cells |
CONCLUSION
In this work we were able to perform deep proteomic profiling down to 9 Panc-1 cells using a combination of fractionation and a carrier/reference method. This deep profiling allowed us to detect 47 414 unique peptides derived from 6261 proteins, which provided a sufficient coverage to search for SAAVs related to cancer. This high sample coverage is essential in order to detect a significant number of SAAVs. Without this coverage only a very small number of SAAVs can be detected. In order to verify genuine SAAVs versus false SAAVs, we ran our data through the SAVControl pipeline and found a total of 79 SAAVs from the 9-cell Panc-1 sample and 174 SAAVs from the 5000-cell Panc-1 sample. The SAAVs as sorted into high confidence and low confidence SAAVs were checked manually, where all the high confidence SAAVs were found to be genuine SAAVs, while half of the low confidence SAAVs as categorized by SAVControl were found to be false SAAVs. We also were able to identify 8 cancer-related SAAVs including KRAS, which is an important oncoprotein in pancreatic cancer.43 In addition, we were able to detect sites involved in loss or gain of glycosylation. These sites had been predicted in previous work, but are difficult to detect without sufficient coverage. In the current work we detect both sites of loss and gain of glycosylation. Future work will extend this work down to single cell detection with enhanced fractionation and using improved slow flow separations. Also, new C/R reference strategies will be designed to investigate SAAVs in circulating tumor cells from patient blood and cancer stem cells from tissues.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Song Nie from Catalent, Inc. for critical reading of the manuscript. We acknowledge partial support of this work under NIH R01GM49500 (DML). Also the assistance of the Wayne State University Proteomics Core, which is supported through NIH grants P30 ES020957, P30 CA 022453 and S10 OD010700.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00694.
Notes
The authors declare no competing financial interest.
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD011471.
REFERENCES
- (1).Araya CL; Fowler DM; Chen WT; Muniez I; Kelly JW; Fields S A fundamental protein property, thermodynamic stability, revealed solely from large-scale measurements of protein function. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (42), 16858–16863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lori C; Lantella A; Pasquo A; Alexander LT; Knapp S; Chiaraluce R; Consalvi V Effect of single amino acid substitution observed in cancer on Pim-1 kinase thermodynamic stability and structure. PLoS One 2013, 8 (6), e64824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang CX; Pallan PS; Zhang W; Lei L; Yoshimoto FK; Waterman MR; Egli M; Guengerich FP Functional analysis of human cytochrome P450 21A2 variants involved in congenital adrenal hyperplasia. J. Biol. Chem 2017, 292 (26), 10767–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Olivier M; Hollstein M; Hainaut P TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect. Biol 2010, 2 (1), a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gu X; Xing L; Shi G; Liu Z; Wang X; Qu Z; Wu X; Dong Z; Gao X; Liu G; Yang L; Xu Y The circadian mutation PER2(S662G) is linked to cell cycle progression and tumorigenesis. Cell Death Differ 2012, 19 (3), 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nie S; Yin H; Tan Z; Anderson MA; Ruffin MT; Simeone DM; Lubman DM Quantitative analysis of single amino acid variant peptides associated with pancreatic cancer in serum by an isobaric labeling quantitative method. J. Proteome Res 2014, 13 (12), 6058–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Vegvari A Mutant Proteogenomics. Adv. Exp. Med. Biol 2016, 926, 77–91. [DOI] [PubMed] [Google Scholar]
- (8).Meacham CE; Morrison SJ Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501 (7467), 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Magee JA; Piskounova E; Morrison SJ Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21 (3), 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gavasso S; Gullaksen SE; Skavland J; Gjertsen BT Single-cell proteomics: potential implications for cancer diagnostics. Expert Rev. Mol. Diagn 2016, 16 (5), 579–589. [DOI] [PubMed] [Google Scholar]
- (11).Spiller DG; Wood CD; Rand DA; White MRH Measurement of single-cell dynamics. Nature 2010, 465 (7299), 736–745. [DOI] [PubMed] [Google Scholar]
- (12).Chung W; Eum HH; Lee HO; Lee KM; Lee HB; Kim KT; Ryu HS; Kim S; Lee JE; Park YH; Kan ZY; Han W; Park WY Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun 2017, 8, 15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chen CY; Xing D; Tan LZ; Li H; Zhou GY; Huang L; Xie XS Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356 (6334), 189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Visvader JE; Lindeman GJ Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8 (10), 755–68. [DOI] [PubMed] [Google Scholar]
- (15).Nie S; McDermott SP; Deol Y; Tan Z; Wicha MS; Lubman DM A quantitative proteomics analysis of MCF7 breast cancer stem and progenitor cell populations. Proteomics 2015, 15 (22), 3772–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tian RJ; Wang SA; Elisma F; Li L; Zhou H; Wang LS; Figeys D Rare cell proteomic reactor applied to stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative proteomics study of human embryonic stem cell differentiation. Mol. Cell. Proteomics 2011, 10 (2), M110.000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Alfaro JA; Ignatchenko A; Ignatchenko V; Sinha A; Boutros PC; Kislinger T Detecting protein variants by mass spectrometry: a comprehensive study in cancer cell-lines. Genome Med 2017, 9 (1), 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sheynkman GM; Shortreed MR; Frey BL; Scalf M; Smith LM Large-scale mass spectrometric detection of variant peptides resulting from nonsynonymous nucleotide differences. J. Proteome Res 2014, 13 (1), 228–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lichti CF; Mostovenko E; Wadsworth PA; Lynch GC; Pettitt BM; Sulman EP; Wang Q; Lang FF; Rezeli M; Marko-Varga G; Vegvari A; Nilsson CL Systematic identification of single amino acid variants in glioma stem-cell-derived chromosome 19 proteins. J. Proteome Res 2015, 14 (2), 778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mostovenko E; Vegvari A; Rezeli M; Lichti CF; Fenyo D; Wang QH; Lang FF; Sulman EP; Sahlin KB; Marko-Varga G; Nilsson CL Large scale identification of variant proteins in glioma stem cells. ACS Chem. Neurosci 2018, 9 (1), 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Garin-Muga A; Corrales FJ; Segura V Proteogenomic analysis of single amino acid polymorphisms in cancer research. Adv. Exp. Med. Biol 2016, 926, 93–113. [DOI] [PubMed] [Google Scholar]
- (22).Ma SY; Menon R; Poulos RC; Wong JWH Proteogenomic analysis prioritises functional single nucleotide variants in cancer samples. Oncotarget 2017, 8 (56), 95841–95852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cao RF; Shi Y; Chen SG; Ma YM; Chen JJ; Yang J; Chen G; Shi TL dbSAP: single amino-acid polymorphism database for protein variation detection. Nucleic Acids Res 2017, 45, D827–D832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ivanov MV; Lobas AA; Karpov DS; Moshkovskii SA; Gorshkov MV Comparison of false discovery rate control strategies for variant peptide identifications in shotgun proteogenomics. J. Proteome Res 2017, 16 (5), 1936–1943. [DOI] [PubMed] [Google Scholar]
- (25).Li J; Su ZL; Ma ZQ; Slebos RJC; Halvey P; Tabb DL; Liebler DC; Pao W; Zhang B A Bioinformatics workflow for variant peptide detection in shotgun proteomics. Mol. Cell. Proteomics 2011, 10 (5), M110.006536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xinpei Yi BW; Zhiwu A; Fuzhou G; Jing L; Yan F Quality control of single amino acid variations detected by tandem mass spectrometry. J. Proteomics 2018, 187, 144–151. [DOI] [PubMed] [Google Scholar]
- (27).Strober W Trypan blue exclusion test of cell viability. Curr. Protoc Immunol 2015, 111, A3.B.1–A3.B.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nie S; Lo A; Wu J; Zhu JH; Tan ZJ; Simeone DM; Anderson MA; Shedden KA; Ruffin MT; Lubman DM Glycoprotein biomarker panel for pancreatic cancer discovered by quantitative proteomics analysis. J. Proteome Res 2014, 13 (4), 18731–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yin H; Tan Z; Wu J; Zhu J; Shedden KA; Marrero J; Lubman DM Mass-selected site-specific core-fucosylation of serum proteins in hepatocellular carcinoma. J. Proteome Res 2015, 14 (11), 4876–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang M; Wang B; Xu J; Wang X; Xie L; Zhang B; Li Y; Li J CanProVar 2.0: An Updated Database of Human Cancer Proteome Variation. J. Proteome Res 2017, 16 (2), 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tan ZJ; Yin HD; Nie S; Lin ZX; Zhu JH; Ruffin MT; Anderson MA; Simeone DM; Lubman DM Large-scale ddentification of core-fucosylated glycopeptide sites in pancreatic cancer serum using mass spectrometry. J. Proteome Res 2015, 14 (4), 1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Deutsch EW; Csordas A; Sun Z; Jarnuczak A; Perez-Riverol Y; Ternent T; Campbell DS; Bernal-Llinares M; Okuda S; Kawano S; Moritz RL; Carver JJ; Wang MX; Ishihama Y; Bandeira N; Hermjakob H; Vizcaino JA The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res 2017, 45 (D1), D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vizcaino JA; Csordas A; del-Toro N; Dianes JA; Griss J; Lavidas I; Mayer G; Perez-Riverol Y; Reisinger F; Ternent T; Xu QW; Wang R; Hermjakob H 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 2016, 44 (D1), D447–D456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tan Z; Nie S; McDermott SP; Wicha MS; Lubman DM Single amino acid variant profiles of subpopulations in the MCF-7 breast cancer cell Line. J. Proteome Res 2017, 16 (2), 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Slavov N; Budnik B; Levy E Mass-spectrometry of single mammalian cells quantifies proteome heterogeneity during lineage specification. bioRxiv 2017, 102681. [DOI] [PMC free article] [PubMed]
- (36).Cao ZJ; Tang HY; Wang H; Liu Q; Speicher DW Systematic comparison of fractionation methods for in-depth analysis of plasma proteomes. J. Proteome Res 2012, 11 (6), 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Goebel-Stengel M; Stengel A; Tache Y; Reeve JR The importance of using the optimal plasticware and glassware in studies involving peptides. Anal. Biochem 2011, 414 (1), 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen YF; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian WJ; Kelly RT Nanodroplet processing platform for deep and quantitative proteome profiling of 10−100 mammalian cells. Nat. Commun 2018, 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Burger T Gentle introduction to the statistical foundations offalse discovery rate in quantitative proteomics. J. Proteome Res 2018, 17 (1), 12–22. [DOI] [PubMed] [Google Scholar]
- (40).Reiter L; Claassen M; Schrimpf SP; Jovanovic M; Schmidt A; Buhmann JM; Hengartner MO; Aebersold R Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell. Proteomics 2009, 8 (11), 2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fu Y; Qian XH Transferred subgroup false discovery rate for rare post-translational modifications detected by mass spectrometry. Mol. Cell. Proteomics 2014, 13 (5), 1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Choong WK; Lih TSM; Chen YJ; Sung TY Decoding the effect of isobaric substitutions on identifying missing proteins and variant peptides in human proteome. J. Proteome Res 2017, 16 (12), 4415–4424. [DOI] [PubMed] [Google Scholar]
- (43).Kanda M; Matthaei H; Wu J; Hong SM; Yu J; Borges M; Hruban RH; Maitra A; Kinzler K; Vogelstein B; Goggins M Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142 (4), 730–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ryschich E; Huszty G; Knaebel HP; Hartel M; Buchler MW; Schmidt J Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer 2004, 40 (9), 1418–1422. [DOI] [PubMed] [Google Scholar]
- (45).Hsieh YY; Tzeng CH; Chen MH; Chen PM; Wang WS Epidermal growth factor receptor R521K polymorphism shows favorable outcomes in KRAS wild-type colorectal cancer patients treated with cetuximab-based chemotherapy. Cancer Science 2012, 103 (4), 791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lassman AB; Rossi MR; Razier JR; Abrey LE; Lieberman FS; Grefe CN; Lamborn K; Pao W; Shih AH; Kuhn JG; Wilson R; Nowak NJ; Cowell JK; DeAngelis LM; Wen P; Gilbert MR; Chang S; Yung WA; Prados M; Holland EC Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: Tissue analysis from North American Brain Tumor Consortium Trials 01−03 and 00−01. Clin. Cancer Res 2005, 11 (21), 7841–7850. [DOI] [PubMed] [Google Scholar]
- (47).Angelis D; Spiliotis ET Septin mutations in human cancers. Front. Cell Dev. Biol 2016, 4, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.