

Abstract
During oxidative stress, K63-linked polyubiquitin chains accumulate in the cell and modify a variety of proteins including ribosomes. Knowledge of the precise sites of K63 ubiquitination is key to understand its function during the response to stress. To identify the sites of K63 ubiquitin, we developed a new mass-spectrometry based method that quantified >1,100 K63 ubiquitination sites in yeast responding to oxidative stress induced by H2O2. We determined that under stress, K63 ubiquitin modified proteins are involved in several cellular functions including ion transport, protein trafficking, and translation. The most abundant ubiquitination sites localized to the head of the 40S subunit of the ribosome, modified assembled polysomes, and affected the binding of translation factors. The results suggested a new pathway of post-initiation control of translation during oxidative stress and illustrated the importance of high-resolution mapping of noncanonical ubiquitination events.
Keywords: K63 ubiquitin, oxidative stress, ribosome, translational control, proteomics
Introduction
Ubiquitination is one of the most prevalent post-translational modifications in eukaryotes, modifying thousands of proteins and involving hundreds of enzymes1–3. It is primarily known as the signal for protein degradation in the nucleus and the cytosol3. However, a variety of ubiquitin chain types can be conjugated to a given substrate, and their functions expand to protein trafficking, protein-protein interaction, and other signaling processes4,5. This diversity in ubiquitin chain types is determined by the internal lysine residues linking the ubiquitin molecules. K48 and K11 ubiquitin chains are the main signal for protein degradation while K63 ubiquitin is the only linkage not related to protein degradation by the proteasome 4,6–8.
The abundance and the dynamics of the different chains and signals can vary according to the cellular environment. In response to oxidative stress, multiple ubiquitin chain types, including K63 and K48 linkages, rapidly and strongly accumulate in the cells9. We have recently shown that K63 polyubiquitin chains modified ribosomes, retained polysomes, and supported cellular survival during oxidative stress.9 However, the exact sites of K63 ubiquitin and its role in ribosome function remained unknown. Finding the modification sites is a crucial next step to understand how ubiquitin regulates ribosome activity and protein expression.
Given the multitude of different ubiquitination types that affect the ribosome and other proteins10–12, we need tools to identify, map, and disentangle the distinct ubiquitin signals to understand their physiological role in vivo. Mass spectrometry is the current tool of choice to investigate protein ubiquitination and its physiological roles13. Mass spectrometry analysis typically identifies ubiquitinated lysines via the di-glycyl remnant (GG) left by tryptic digest of the target protein14,17–23. Importantly though, this tryptic digest also processes the ubiquitin chain, removing information of the linkage type that modified the protein.
Therefore, we developed a new method, called Ub-DiGGer, which comprises a sequential enrichment strategy that preserves information on the linkage type, in this case K63, but also extracts modification sites with high specificity and proteome coverage. Using Ub-DiGGer, we identified ~1,100 K63 ubiquitination sites in the yeast proteome responding to stress. The modification affected several pathways, such as translation, amino acid transport, and the Golgi apparatus. We investigated the modification of the ribosome in more detail and present results that suggest a role of K63 ubiquitin in translation shutdown upon oxidative stress in a mechanism independent of the canonical inhibition of initiation via reduced availability of the ternary complex.
Materials and Methods
Yeast strains and growth.
All yeast Saccharomyces cerevisiae strains used in this study are described in Supplementary Table S3. Standard recombination methods were used to delete and tag genes, which were confirmed by PCR and western blot, respectively.
Sensitivity assay.
Cells were grown to logarithmic phase in minimal medium containing 0.67% yeast nitrogen base, 2% dextrose, and required amino acids at 30°C. Cellular density was normalized to OD600 of 0.2. Cells were then sequentially diluted at 1:5 ratio and plated in YPD rich medium plates containing: 50 ng/ml rapamycin, 20 ug/ul anisomycin, 0.6 or 1.2 mM H2O2.
Isolation of K63 ubiquitinated peptides.
The sequential ubiquitin enrichment was applied to identify and quantify the lysine residues modified by K63 ubiquitin in response to oxidative stress. For each biological replicate, 2.4 liters of each strain (WT and K63R) were cultured to mid-log phase into synthetic SILAC media supplemented with light and heavy isotopes of lysine and arginine, respectively (L-Arg6 13C; L-Lys8 13C, 15N; Cambridge Isotopes). Cells were grown for at least 10 generations in the SILAC medium before being treated with 0.6 mM H2O2 for 30 min at 30 °C. Equal amounts of WT and K63R cells were collected by centrifugation, mixed, and disrupted by glass-bead agitation at 4 °C in lysis buffer: 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 mM EDTA, 20 mM chloroacetamide, 50 nM FLAG K63-TUBE peptide (LifeSensors), 1X EMD Millipore protease inhibitor cocktail set I. Approximately 350 mg of protein were loaded onto a column containing M2 anti-FLAG resin (Sigma). Sample was eluted with 0.1 M glycine pH 2.5, neutralized with NaOH and buffered with 100 mM Tris-HCl pH 8.0, 1 mM CaCl2 prior to overnight digestion with Trypsin/Lys-C (Promega). Samples were desalted using C18 Hypersep Spin Column (Thermo), dried completely and resuspended in IAP buffer from PTMScan Ubiquitin K-ε-GG kit (Cell Signaling). For the enrichment of diglycyl-lysine peptides (GG), tryptic peptides were incubated for 4h at 4 °C with anti-GG beads and eluted with 0.15% formic acid according to the manufacturer’s protocol. Samples were desalted once again with Hypersep tips and resuspended in 0.1% FA, prior to LC-MS/MS analysis.
Polysome proteomics.
For the quantitative analysis of the composition of polysomes and monosomes, two biological replicates were prepared from cells with and without 30 min of 0.6 mM H2O2 treatment. Cell disruption was performed as described below and approximately 400 μg of RNA from each yeast strain wild-type and K63R grown in heavy and light SILAC medium, respectively, were sedimented in a sucrose gradient. The monosome fraction (40S, 60S and 80S) was separated from the polysome fractions, proteins were precipitated with 10% trichloroacetic acid (TCA), protein pellet was washed twice with ice-cold acetone, dried out, and resuspended in 8M urea buffered in 50 mM Tris-HCl pH 8.0, 1 mM CaCl2. Samples were digested in a two-step fashion by incubating with Trypsin/Lys-C mix (Promega) for 3h followed by dilution of urea to 1M with Tris-buffer pH 8.0 for overnight tryptic digestion at 37 °C. Samples were desalted using Hypersep spin tips (Thermo) prior to LC-MS/MS analysis.
Mass spectrometry analysis.
Protein preparation for proteomics analysis was performed as previously described9. Briefly, tryptically digested proteins isolated by polysome profiling were separated on a 25-cm Thermo Acclaim PepMap C18 column (75 μm ID, 2 μm particle size, 100 Å pore size, cat # 164734) by reverse-phase chromatography using a gradient of 5–60% acetonitrile over 2.5 h, performed on an Eksigent NanoLC 2DPlus liquid chromatography system. The eluted peptides were injected in-line into an LTQ Orbitrap Elite mass spectrometer (Thermo Scientific). Data-dependent analysis was performed at a resolution of 120,000 at the MS level, AGC at 1e6, maximum injection time of 100 ms, with the top 20 most intense ions selected from each MS full scan, and dynamic exclusion set to 30 seconds if m/z acquisition was repeated within a 30-second interval. MS2 was acquired at a resolution of 15,000, AGC at 5e4 and maximum injection time of 100ms. The K63 ubiquitinomics samples were loaded onto a 50 cm Easy Spray PepMap C18 column (75 μm ID, 2 μm particle, 100 Å pore size, cat. No ES803) in-line with a Q-Exactive (Thermo Scientific) mass spectrometer using a 75-min gradient (0–40% ACN). Data dependent analysis was performed at resolution of 70,000 at the MS level, AGC at 1e6 and maximum injection time of 120 ms. Top 20 most intense ions selected from each MS full scan, with dynamic exclusion set to 30-second if m/z acquisition was repeated within a 30-second interval. MS2 was acquired at a resolution of 17,500, AGC at 5e4 and maximum injection time of 120ms. The RAW data files were processed using MaxQuant to identify and quantify protein and peptide abundances. The spectra were matched against the yeast Saccharomyces cerevisiae Uniprot database. Protein identification was performed using 10 ppm tolerance with a posterior global FDR of 1% based on the reverse sequence of the yeast FASTA file. Up to two missed trypsin cleavages were allowed, and oxidation of methionine and N-terminal acetylation were searched as variable post-translational modification and cysteine carbamidomethylation as fixed. For K63 ubiquitinated peptides, diglycyl-lysine (GlyGly(K)) was also searched as variable modification.
Polysome analysis.
Polysome profiling was performed as described previously9. Briefly, a total of 400 μg of RNA was sedimented by ultracentrifugation for 150 min at 38,000 rpm (Beckman SW41Ti rotor) at 4 °C. MgCl2 concentration varied from 1 to 3 mM in the lysis buffer and from 1 to 30 mM in the sucrose buffer. Polysome traces were collected with PeakChart software coupled to a Density Gradient Fractionation System (Brandel).
Western blot.
Proteins were separated by standard 10% SDS-PAGE, with samples loaded in Laemmli buffer, transferred for 2h to a PVDF membrane, and immunoblotted using the following antibodies: anti-K63 ubiquitin (1:4,000; EMD Millipore, cat. No. 05–1308, clone apu3); anti-GAPDH (1:4,000; Abcam, cat. No. ab9485); anti-Rps10 (1:6,000, Sigma, cat no. WH0004736M1); anti-Rps6 (1:4,000; Abcam cat no. ab40820), anti-puromycin (1,2:500, EMD Millipore MABE343, clone 12D10), anti HA (1:2,500; Invitrogen, cat no. 71–5500), anti-K48 ubiquitin (1:8,000; Cell Signaling, cat. No 8081).
Translation activity.
Puromycin incorporation into newly synthesized proteins was performed to measure translation activity. Cells were treated in the presence or absence of 0.6 mM H2O2 for 15 min prior to 30 min incubation with 0.9 mM puromycin. Cells were collected by centrifugation and lysate was prepared as described above. Twenty micrograms of protein were loaded onto a 10% SDS-PAGE gel for immunoblotting.
Results
In-depth characterization of the K63 ubiquitin-modified yeast proteome
We developed a mass spectrometry-based method that maps the sites of ubiquitin modification for a specific linkage type (Figure 1A). The method, called Ub-DiGGer, first isolates the targets of a selective linkage with a highly specific K63 TUBE (LifeSensors) enrichment tool. K63 TUBE exhibits 1,000–10,000X higher affinity for K63 ubiquitin compared to other linkages and the low concentration used in this study (50 nM) prevents the isolation of other linkages9,14. The isolated proteins are then digested with trypsin and subjected to a second enrichment step that isolates the di-glycyl (GG) remnant on the modified lysine. Finally, the isolated peptides are identified by mass spectrometry. To quantify K63 ubiquitin changes in response to oxidative stress and to monitor the specificity of the method, we combined Ub-DiGGer with Stable Isotopic Labeling in Cell culture (SILAC)15 of wild-type cells compared to the K63R ubiquitin mutant, which is incapable of assembling K63 ubiquitin chains (Figure S1A-S1B). Our final dataset is comprised of K63 homotypical and chains branched or mixed with K63 linkages, which we consider global K63 ubiquitin targets. Fold-change of all signature peptides for the distinct ubiquitin linkages is presented in Figure S1D. Using the K63R ubiquitin mutant strain as a control to account for false positive identifications, we mapped 1,113 K63 ubiquitin sites at 1% peptide based false discovery rate. We selected K63 ubiquitinated peptides whose SILAC ratios changed at least 1.6-fold compared to the K63R mutant. The cutoff of 1.6 was defined based on a 5% false discovery rate determined previously using the K63 TUBE system9. Results were consistent across both replicates illustrating the high quality and reproducibility of the data (Supplementary Table S1, Figure S1A). The peptides mapped to 490 distinct proteins (Supplementary Table S1).
Figure 1.
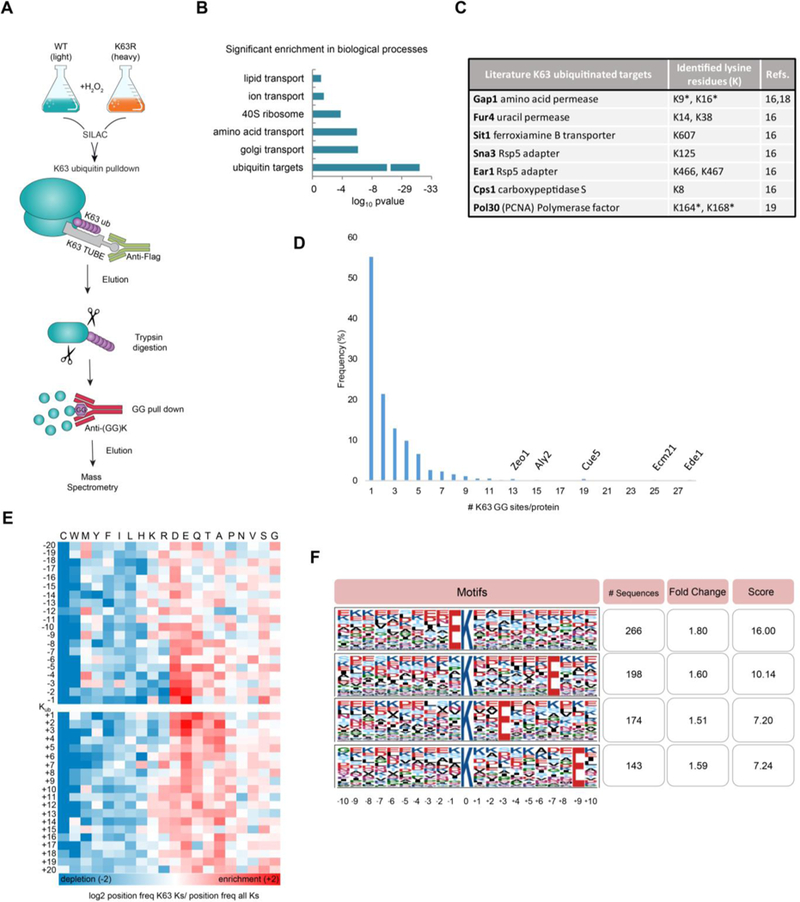
K63 ubiquitin-modified proteome in response to oxidative stress. (A) Scheme describing the Ub-DiGGer method to identify and quantify K63 ubiquitinated lysine residues. (B) Histogram of Gene Ontology analysis for K63 target proteins using DAVID functional annotation tool. p-values are Bonferroni-corrected for multiple hypotheses testing. (C) Sites identified for known K63 ubiquitinated proteins. *K63 ubiquitin sites previously identified and confirmed by this study. (D) Frequency distribution of the number of K63 GG sites per protein. Targets containing multiple K63 ubiquitin sites are annotated. (E) Heatmap shows log 2 ratio of the frequency of the amino acid position relative to the K63 ubiquitin modified lysine (Kub) over the frequency of the amino acids at the respective position on all lysine residues in the yeast proteome. The order of the amino acids (columns) was based on hierarchical clustering of their ratio profiles. Fold change enrichment is shown in red and depletion in blue. (F) K63 ubiquitin motif analysis determined by Motif-X44. Top four score motifs are displayed centered on the ubiquitinated lysine and a window of ± 10 residues.
The identified peptides showed a significant enrichment for ubiquitinated proteins (Figure 1B, p-value <e-30) further validating our approach. In addition, the peptides arose from proteins from the Golgi apparatus and amino acid transport (p-value < e-6, Figure 1B). Other targets functioned in autophagy, ion transport, carbohydrate metabolism, endocytosis, and nutritional response (Figure S1E). These results were in agreement with reports that many proteins involved in trafficking, metabolism, and translation are regulated by K63 ubiquitin16,17.
To-date, a handful of K63 targets have been confirmed for yeast, which we used for further validation of our dataset. Indeed, our dataset reported K63 ubiquitin sites for the permeases Fur4 (K14, K38), the transporter Sit1 (K607), the Rsp5 adapters Sna3 (K125) and Ear1 (K466, K467) and for the carboxypeptidase Cps1 (K8). All these proteins have been previously characterized as targets of K63 ubiquitin16. Moreover, our data retrieved the known K63 modified lysine residues in Gap1, i.e. K9 and K1618 and also identified K164 in PCNA, a well-characterized site of K63 ubiquitin important for DNA repair signaling19. Moreover, we found that most targets contained three or fewer K63 ubiquitin sites (Figure 1D), but some proteins involved in endocytosis or cell wall organization were highly decorated with K63 ubiquitin chains, having more than ten GG sites (e,g, Ede1, Ecm21, and Zeo1, Figure 1D).
Our results also corroborated the hypothesis of the absence of a sequence motif signaling for ubiquitination 20. Importantly, our data arose from a very specific subset of ubiquitination events: K63 ubiquitin changes in response to oxidative stress. Despite this highly focused dataset, we did not identify a sequence motif surrounding the K63 ubiquitin sites (Figure 1E and 1F). However, we identified physicochemical properties surrounding the modified sites: i) an enrichment of acidic amino acids (glutamate and aspartate); ii) an enrichment in glutamine and alanine in the positions immediately following the modified lysine; and iii) a major depletion of cysteine and tryptophan surrounding the modified lysine (Figure 1E). Therefore, our results support the school of thought that ubiquitination does not occur exclusively via a recognition motif20. Instead, structural components associated with amino acid sequence might be recognized by ubiquitination enzymes.
Many of the K63 ubiquitination events in response to stress mapped to the ribosome, and we investigated these events in more detail. First, many of these ribosome GG sites have been previously identified in yeast 1,2,9,21 but without distinction of the associated chain type (Supplementary Table S1). These sites were significantly enriched in components of the 40S ribosome (Figure 1B, p-value < e-3.5). In total, 78 K63 ubiquitin sites mapped to 37 ribosomal proteins, and amongst them, 45 sites mapped to 40S proteins and three additional sites mapped to Asc1 (RACK1), a 40S-associated protein (Supplementary Table S1). This preference is substantial given that the 40S subunit is much smaller and has fewer lysine residues than the 60S subunit (539 and 834, respectively). Further, the SILAC ratios for the 40S subunit sites were significantly higher than those for the 60S (Figure S1F, t-test, p < e-6).
When mapping K63 ubiquitin sites to a publicly available X-ray structure of the yeast 80S ribosome22, we found that most sites were located at the solvent exposed surface of the ribosomes (Figure 2A and 2B). Moreover, the majority of the sites, including those that changed most drastically under stress, clustered at the head of the 40S subunit (Figure 2 and Figure S2A).
Figure 2.

K63 ubiquitin modifies the head of the 40S ribosome. Mapping of the K63 ubiquitinated sites (orange) to the 3D structure of the yeast ribosome (PDB id:4v88)22. Figures highlight K63 modification at (A) the 80S ribosome and (B) at the head of the 40S ribosome showing the most abundantly K63 ubiquitinated subunits colored in yellow (Rps2/uS5), green (Rps3/uS3), magenta (Rps12/eS12), blue (Rps20/uS10), and teal (Rps21/eS21). Ribosome associated protein Asc1 (RACK1) is shown in red. (C) Anti-K63 and anti-K48 ubiquitin western blot of polysome profiling fractions from cells treated with H2O2 for the designated times. Loading controls are presented in Figure S2B. (D,E) Sucrose sedimentation profiles of polysome from wild-type cells in which translation was inhibited (D) at initiation after cells were transferred to glucose depleted medium or (E) at elongation after treatment with 150 μg/ml cycloheximide. Anti-K63 ubiquitin blot was performed for cells cultivated in the presence or absence of H2O2. WT, wild-type SUB280 yeast strain. K63R, ubiquitin K63R mutant SUB413 yeast strain.
K63 ubiquitin regulates translation post initiation
First, we showed that K63 ubiquitin rapidly affected free subunits (40S and 60S), monosomes (80S), and polysomes after stress induction by H2O2 (Figure 2C and Figure S2B). K63 ubiquitin accumulated heavily in the monosome and polysome fractions while K48 ubiquitin accumulated mostly in the lighter fraction of the profile (Figure 2C). Next, we tested if modification of assembled 80S ribosomes arose from the unassembled subunits being modified prior to their assembly or if K63 ubiquitin affected mono- and polysomes directly. We assayed for this question by measuring K63 ubiquitination of polysomes when translation was inhibited and therefore no assembly or progression of monosomes occurred. Using glucose deprivation or cycloheximide to inhibit initiation and elongation, respectively, K63 ubiquitin modified both free subunits and mono- and polysomes (Figure 2D and 2E), supporting our hypothesis that K63 ubiquitin directly targets assembled ribosomes and polysomes.
The entire head of the 40S ribosome is a flexible and dynamic structure that harbors the decoding center, it surrounds the mRNA entry and exit site, engages tRNAs, and is heavily regulated during the different stages of translation 23,24. Therefore, we conducted additional experiments to test for the impact of extensive K63 ubiquitination of the 40S subunit on ribosome activity during oxidative stress.
First, we tested for the specific stage of translation that was affected by K63 ubiquitination. During oxidative stress, translation initiation is rapidly inhibited by the Gcn2 kinase that phosphorylates the initiation factor eIF2α, thereby reducing the levels of translation-competent ribosomes 25,26. To test whether K63 ubiquitin interfered with inhibition of translation initiation by Gcn2, we analyzed polysome levels by sucrose sedimentation profiling. The K63R ubiquitin mutant strain displayed lower levels of polysomes when compared to the wild-type under stress (Figure 3A and 3B). Also, deletion of GCN2 did not affect the accumulation of K63 ubiquitin (Figure S2C). However, both strains maintained the polysome levels when GCN2 was deleted and initiation was fully operational under stress (Figure 3C and 3D). This finding suggested that K63 ubiquitin’s function was independent of the inhibition of initiation. Supporting this conclusion, cells deleted for GCN2 in the K63R background (K63R-gcn2Δ) did not rescue the K63R cell’s sensitivity to translation inhibitors (Figure S2D). Second, we showed that ribosomes for the K63R strain were less stable than the wild-type. At low MgCl2 concentration, which destabilizes 80S particles, samples from K63R cells under stress were devoid of both mono- and polysomes and consisted mostly of free 40S and 60S subunits. In contrast, 80S, disomes, and trisomes were still observed in the wild-type strain under stress (Figure 3E).
Figure 3.

K63R ribosomes are more sensitive to stress than wild-type, independently of GCN2. Sucrose sedimentation profiles of polysome from (A) wild-type, (B) K63R, (C) gcn2Δ, and (D) K63R-gcn2Δ. Cells were incubated in the presence or the absence of 0.6 mM H2O2 for 30 min. Anti-K63 ubiquitin blots of the same experiment are present in Figure S2C. (E) Polysomes from wild-type and K63R under H2O2 were extracted at low magnesium chloride concentration (1 mM). Western blot of sedimentation fractions was performed for 60S large and 40S small subunit proteins Rpl10 and Rps6, respectively. WT, wild-type SUB280 yeast strain. K63R, ubiquitin K63R mutant SUB413 yeast strain. (F,G) Western blot against puromycin was used to evaluate de novo protein synthesis. Cells were treated with H2O2 for 15 or 60 min prior to treatment with 0.9 mM puromycin for additional 30 min. Anti-GAPDH blot was used as loading control.
Considering the 40S location of the K63 ubiquitin sites, the low stability of K63R ribosomes, and the independence from translation initiation, our data suggested that K63 ubiquitin might regulate ribosome activity post initiation. Inhibition of translation at elongation had been proposed during oxidative stress without a known mechanism25–28. To test for post-initiation regulation mediated by K63 ubiquitin, we measured translation output by monitoring the incorporation of puromycin into newly synthesized proteins. The wild-type cells showed low puromycin incorporation in response to H2O2, confirming strong translation repression (Figure 3F). In agreement with earlier findings25,28, deletion of the GCN2 was insufficient to restore translation output under stress, suggesting additional inhibition of translation beyond initiation (Figure 3F). In the absence of stress, K63R mutant cells had similar puromycin incorporation as the wild-type indicating normal translation levels. However, translation output remained mostly unaltered in the K63R mutant in response to H2O2 suggesting that K63 ubiquitin was involved in translation halt, independently of initiation (Figure 3F).
In the K63R mutant, normal translation output could be achieved either by constant production of protein that is unaffected by stress or by elongating ribosomes that remain on the mRNAs that still translate during the early phases of the stress response. In the latter scenario, we would expect puromycin incorporation to decrease over time, as there would be little to no de novo translation initiation due to Gcn2 activity during stress. To test this hypothesis, we used short (15min) and long (1h) H2O2 incubation and monitored protein synthesis. We found that incorporation of puromycin occurred at normal levels in the K63R mutant compared to the repressed wild-type regardless of the duration of the stress (Figure 3G). These findings provided another line of evidence for K63 ubiquitin acting via a mechanism independent from the inhibition of translation initiation.
K63 ubiquitin alters the ribosome’s interaction with translation factors
K63 ubiquitin can act as a scaffold, recruit ubiquitin-binding proteins, or prevent binding of additional proteins, thus affecting a variety of signaling pathways independently of proteasomal degradation6. Since our results indicated that K63 ubiquitin modifies assembled monosomes and polysomes (Figure 2C-E) and affects their stability and activity (Figure 3), we tested if these roles arose from K63 ubiquitin impacting the binding of regulatory factors to the ribosome.
We examined two candidate factors which are known to regulate ribosome function: the ribosome associated factor Asc1 (RACK1)11 and the E3 ubiquitin enzyme Hel212. Asc1 (RACK1) was modified by K63 ubiquitin in our experimental system (Supplementary Table S1), and localized next to K63 ubiquitin sites on the 40S ribosome (Figure 2B). We therefore tested the effect of ASC1 deletion on ribosome ubiquitination, stability, and stress response. Although deletion of ASC1 rendered yeast cells more sensitive to oxidative stress and translation inhibitors (Figure S3A), ASC1 deletion did not result in active translation (Figure S3B), did not show reduced abundance of polysomes (Figure S3C), or diminished K63 ubiquitin levels (Figure S3C) – the molecular phenotypes observed in the K63R mutant strain. ASC1 therefore appeared to work independently of K63 ubiquitination. Similarly, we showed that the ubiquitin ligase Hel2 was not responsible for the accumulation of K63 ubiquitin in response to oxidative stress (Figure S3D). Interestingly, we observed a substantial decrease in polysome levels of hel2Δ under stress (Figure S3D), but the inhibition of translation assessed by puromycin incorporation was similar to the wild-type (Figure S3E). In agreement with our previous work9, rad6Δ shows no accumulation of K63 ubiquitin in response to stress and reduced levels of polysomes supporting its key role in the modification of ribosomes (Figure S3D).
To systematically screen for other factors whose binding to the ribosome could be affected by K63 ubiquitin during oxidative stress, we used mass spectrometry to analyze samples from the sucrose sedimentation profile (Figure 4A, Figure S4A, Supplementary Table S2). While the abundance of ribosomal proteins was mostly unchanged, many translation factors were more abundant in the wild-type compared to the K63R mutant. This effect was observed in the monosomal (Figure 4B) and also in the polysomal fraction (Figure S4B). Amongst these factors, we identified several components of the eukaryotic initiation factor eIF3 complex, e.g. Nip1 (eIF3a) and Tif35 (eIF3g)(Figure 4B, Figure S4B). eIF3 is a key translation factor which binds to the 40S subunit, stabilizes the ternary complex, recruits capped mRNA, and assists translation initiation 29,30.
Figure 4.
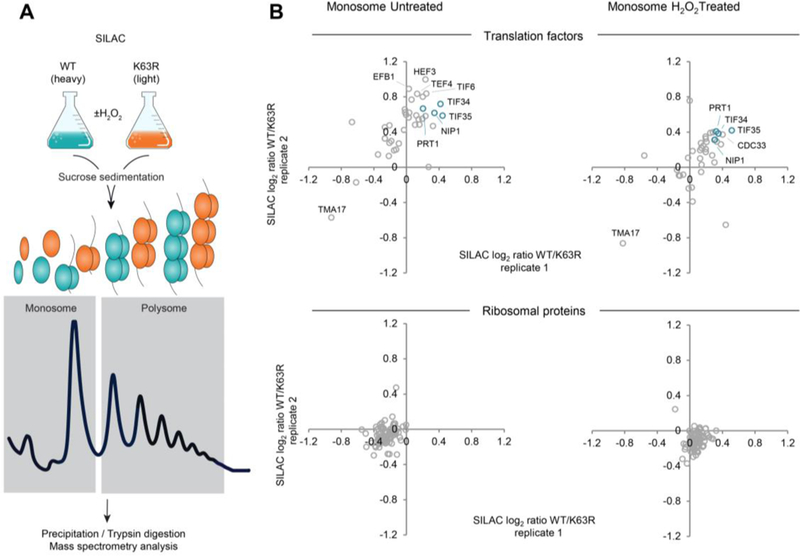
Ribosome proteomics reveals K63 ubiquitin-dependent binding of translation factors. (A) Scheme of polysome proteomics method following sucrose sedimentation profiling. (B) Scatter plot of log2 SILAC WT/K63R ratios of protein intensity from two independent replicates in the monosome fraction. (top) SILAC ratio of translation factors, (bottom) SILAC ratio of ribosomal proteins. Subunits of the eIF3 translation initiation factor are labeled in teal. WT, wild-type GMS280 yeast strain. K63R, ubiquitin K63R mutant GMS413 yeast strain.
We tested for both Nip1 and Tif35 if their binding to the ribosome depended on K63 ubiquitin. The abundance of Nip1 bound to ribosomes remained similar in both the wild-type and the K63R strain (Figure S5A) and could suggest a more transient interaction. Conversely, Tif35’s presence is not detected in polysomes from untreated K63R ubiquitin mutant strain compared to wild-type (Figure 5A) and Tif35 total abundance is decreased in the mutant in response to stress (Figure 5B). These results for both, Nip1 and Tif35, were robust to changes in blotting exposure (Figure S5B and S5C). Consistently, reduced expression of TIF35 in the DAmP strain31 sensitized cells to oxidative stress and to translation inhibitors (Figure S5D), and slightly increased translation as measured by puromycin incorporation (Figure S5E). Tif35 association with the ribosome might therefore be affected by K63 ubiquitin.
Figure 5.
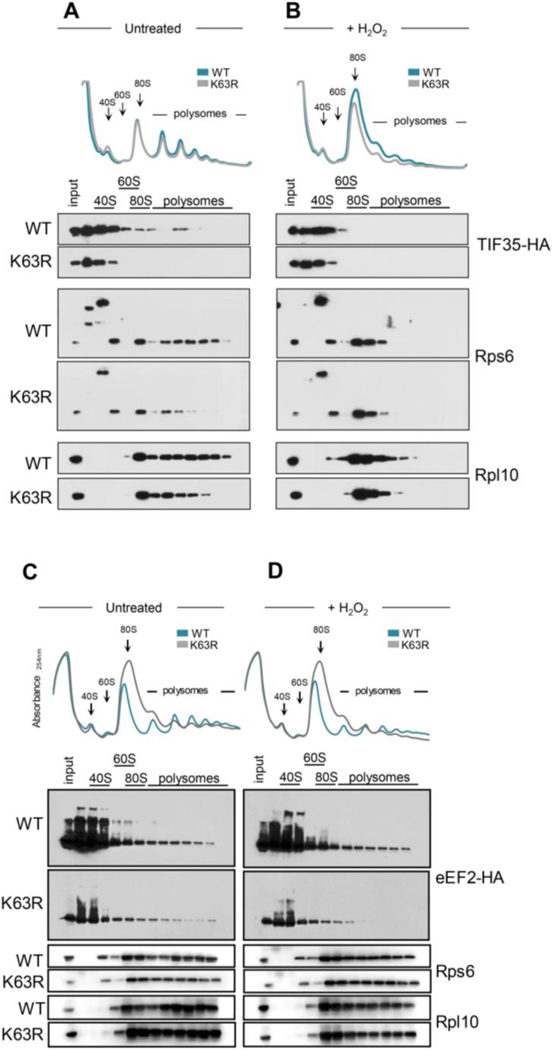
Tif35 and eEF2 are depleted in K63R ubiquitin polysomes. (A,B) Sucrose sedimentation profiles of polysomes from wild-type and K63R mutant strain expressing HA-tagged Tif35 (A) untreated or (B) subjected to 0.6 mM H2O2 for 30 min. (C,D) Sucrose sedimentation profiles of polysomes from wild-type and K63R mutant strain expressing HA-tagged eEF2 (EFT1) (C) untreated or (D) subjected to 0.6mM H2O2 for 30 min. Western blot of sedimentation fractions were performed for anti-HA, 60S large protein Rpl10, and 40S small protein Rps6.
Finally, we demonstrated that the K63R ubiquitin mutation also affected the binding of the elongation factor eEF2, but not eEF3. eEF2 is the elongation factor that interacts with the ribosome during the translocation stage of elongation32, while eEF3 is a yeast-specific elongation factor involved in the post-translocation step of translation, aiding the release of tRNAs from the E-site33. We found that in wild-type cells, a larger amount of eEF2 was present and proportionally bound to the ribosome compared to the K63R mutant (Figure 5C, 5D, S6A). This result indicated that K63 ubiquitin could stabilize eEF2 interaction with the ribosomes. In comparison, eEF3 binding to ribosomes did not change (Figure S6B and S6C). Our results suggested that the accumulation of K63 ubiquitin on the 40S ribosome affected either the dynamics of binding and releasing of translation factors or the stability of these interactions, ultimately hindering the progression of the elongation cycle in response to oxidative stress.
Conclusions
We developed a new mass spectrometry-based method, Ub-DiGGer, to monitor the K63 ubiquitin-modified proteome in response to oxidative stress. The novelty of the method lies in its ability to identify the targets of a specific ubiquitin linkage (K63) and the modified lysine residues simultaneously. Previous methods successfully characterized either the ubiquitin modified proteome20,34 or quantified the cellular abundances of different ubiquitin linkages7,35.
Our method selectively isolated the K63 ubiquitinated proteins prior to tryptic digest and hence enabled large-scale and quantitative site analysis (Figure 1A, Supplementary Table S1). The data provided in this study is unbiased, large scale, quantitative, linkage-specific, and acquired in response to defined conditions – all aspects that render the information highly useful for biological interpretation. Given the emergence of new enrichment methods of sufficient sensitivity and specificity, Ub-DiGGer can be expanded to other ubiquitin linkage types.
Our experimental set up identified >1,100 K63 GG sites, providing the largest resource on global K63 ubiquitin to date, fostering our understanding of K63 ubiquitin targets and functions in the cell. K63 ubiquitin targeted proteins from Golgi-mediated, ion, and amino acid transport, endocytosis, and carbohydrate and lipid metabolism (Supplementary Table S1). We illustrated the wealth in information gained from our data at the example of translation control. Finally we showed that our method was able to retrieve proteins and ubiquitin sites that were previously identified in yeast (Supplementary Table S1) or confirmed as K63 ubiquitin substrates16,18,19 (Figure 1C), supporting the importance and validity of the approach. In the future, combined analysis of changes in ubiquitination levels and protein abundance will provide further insights in the quantification and role of individual ubiquitin sites during stress response. Individual mutation of ribosome ubiquitin sites have produced partial responses in translation and cell viability in human cell lines 10,11. Although sequences are conserved at the protein level, further investigation will be required to understand the role and evolutionary conservation of each K63 site (Figure S6D).
We further illustrated the significance of this dataset by investigating the functional role of K63 ubiquitin in translation regulation. We first showed that a large cluster of K63 modified sites are located at the head of the 40S subunit (Figure 2), which is ribosome’s regulatory center23,24,30. K63 ubiquitin acts independently of GCN2 and therefore, independently of initiation control (Figure 3C, 3D and 3F). Although translation initiation is largely inhibited under oxidative stress, it has been shown that elongation transit time and ribosome occupancy of mRNA at the beginning of the open reading frame increase27,36,37, and that de-repression of translation initiation (gcn2Δ) is insufficient to recover protein synthesis25,27,28,36,37. Temporary translation repression is an essential part of environmental stress response26, and our results consistently suggested that K63 ubiquitin affects ribosome activity post initiation.
Our results also indicated that this role resulted from differential effects of K63 ubiquitin on translation factors and their interaction with the ribosome (Figure 5). While a detailed mechanism remained to be revealed, K63 ubiquitin was essential for proper association of these factors with the ribosome (Figure 4 and 5). Dysregulated binding of elongation factors (e.g. eEF2) could severely impact protein production and cell resistance to stress. Conversely, dysregulated binding of initiation factors (e.g. eIF3) could affect mRNA recruitment, 40S-60S association, or 40S scanning, acting as fail-safe mechanisms in response to stress.
Almost two decades after the initial discovery of K63 ubiquitin modification of a single ribosomal protein, Rpl28 (uL15)38, only few large-scale studies of ribosome ubiquitination exist to date. One such recent study described ubiquitinated sites at the 40S subunit during the unfolded protein response induced by stress of the endoplasmic reticulum 10. The residues were modified primarily by mono-ubiquitin or K48 di-ubiquitin. Additional work have highlighted the role of the ZNF598 ubiquitin ligase - which is a homologue of yeast HEL2 - in quality control to resolve stalled ribosomes 11,12,39. Although these enzymes are important in quality control, the ubiquitin role in this system remains elusive. K63 ubiquitin had been previously implicated in the regulation of quality control during No-Go surveillance40 however, our data suggest that Hel2 is not the E3 responsible for K63 ubiquitination of ribosomes under oxidative stress (Fig. S3D). Furthermore, heterogeneous populations of ribosomes are required to effectively translate specific subpools of transcripts but the molecular mechanism responsible for their recognition remains yet to be elucidated41. Finally, our group is investigating post-initiation mechanisms of translational control mediated by K63 ubiquitin in response to oxidative stress and the use of proteomics combined with targeted molecular approaches provides excellent tools to study this system. Ubiquitination is a widespread post-translational modification of ribosomes and collectively, we are building the knowledge required to solve this complex regulatory code that will lead to the characterization of the physiological roles of ubiquitin in cellular health. Given the large number of ubiquitin enzymes encoded in the genome, the diversity of ubiquitin chains, and the number of modifiable lysine residues in the ribosome, the putative regulatory network governing ubiquitination is extremely complex, and perhaps as intricate as that of histone modifications 42. The tools and data presented here are important in elucidating the regulatory network of ubiquitination.
Supplementary Material
Acknowledgements
We thank D. Finley for providing yeast strains, J.R. Cussiol for the yeast deletion templates and support in yeast genetics. We are indebted to Evan Baugh for the assistance with the structural mapping.
The K63 ubiquitin mass spectrometric experiments were conducted in the NYU Proteomics Resource Center (Dr. Ueberheide), which is in part supported by the NYU School of Medicine.
We thank V. Simoes, M. Beck, P. Kastritis, M. Gorospe, and H. D. Ryoo for the scientific discussions and comments on the manuscript.
This work was supported in part by the US National Institutes of Health K99/R00 award ES025835 (GMS). CV acknowledges funding by the NIH/NIGMS (R01 GM113237 and 1R35GM127089–01), and the Zegar Family Foundation Fund for Genomics Research at New York University.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publication website
Competing financial interests
The authors declare no competing financial interests.
The authors declare no financial interest. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Starita LM, Lo RS, Eng JK, von Haller PD & Fields S Sites of ubiquitin attachment in Saccharomyces cerevisiae. Proteomics 12, 236–240, doi: 10.1002/pmic.201100166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peng J et al. A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21, 921–926, doi: 10.1038/nbt849 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Finley D, Ulrich HD, Sommer T & Kaiser P The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360, doi: 10.1534/genetics.112.140467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yau R & Rape M The increasing complexity of the ubiquitin code. Nat Cell Biol 18, 579–586, doi: 10.1038/ncb3358 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Komander D The emerging complexity of protein ubiquitination. Biochem Soc Trans 37, 937–953, doi: 10.1042/BST0370937 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Tomanov K, Luschnig C & Bachmair A Ubiquitin Lys 63 chains - second-most abundant, but poorly understood in plants. Frontiers in plant science 5, 15, doi: 10.3389/fpls.2014.00015 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu P et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145, doi: 10.1016/j.cell.2009.01.041 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nathan JA, Kim HT, Ting L, Gygi SP & Goldberg AL Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J 32, 552–565, doi: 10.1038/emboj.2012.354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva GM, Finley D & Vogel C K63 polyubiquitination is a new modulator of the oxidative stress response. Nat Struct Mol Biol 22, 116–123, doi: 10.1038/nsmb.2955 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Higgins R et al. The Unfolded Protein Response Triggers Site-Specific Regulatory Ubiquitylation of 40S Ribosomal Proteins. Mol Cell, doi: 10.1016/j.molcel.2015.04.026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sundaramoorthy E et al. ZNF598 and RACK1 Regulate Mammalian Ribosome-Associated Quality Control Function by Mediating Regulatory 40S Ribosomal Ubiquitylation. Mol Cell 65, 751–760 e754, doi: 10.1016/j.molcel.2016.12.026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsuo Y et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nature communications 8, 159, doi: 10.1038/s41467-017-00188-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sylvestersen KB, Young C & Nielsen ML Advances in characterizing ubiquitylation sites by mass spectrometry. Current opinion in chemical biology 17, 49–58, doi: 10.1016/j.cbpa.2012.12.009 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Sims JJ et al. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat Methods 9, 303–309, doi: 10.1038/nmeth.1888 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ong SE et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1, 376–386 (2002). [DOI] [PubMed] [Google Scholar]
- 16.Erpapazoglou Z, Walker O & Haguenauer-Tsapis R Versatile roles of k63-linked ubiquitin chains in trafficking. Cells 3, 1027–1088, doi: 10.3390/cells3041027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romero-Barrios N & Vert G Proteasome-independent functions of lysine-63 polyubiquitination in plants. The New phytologist 217, 995–1011, doi: 10.1111/nph.14915 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Lauwers E, Jacob C & Andre B K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J Cell Biol 185, 493–502, doi: 10.1083/jcb.200810114 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G & Jentsch S RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141, doi: 10.1038/nature00991 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Kim W et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell 44, 325–340, doi: 10.1016/j.molcel.2011.08.025 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hitchcock AL, Auld K, Gygi SP & Silver PA A subset of membrane-associated proteins is ubiquitinated in response to mutations in the endoplasmic reticulum degradation machinery. Proc Natl Acad Sci U S A 100, 12735–12740, doi: 10.1073/pnas.2135500100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ben-Shem A et al. The structure of the eukaryotic ribosome at 3.0 A resolution. Science 334, 1524–1529, doi: 10.1126/science.1212642 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Llacer JL et al. Conformational Differences between Open and Closed States of the Eukaryotic Translation Initiation Complex. Mol Cell 59, 399–412, doi: 10.1016/j.molcel.2015.06.033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson RJ, Hellen CU & Pestova TV The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11, 113–127, doi: 10.1038/nrm2838 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shenton D et al. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J Biol Chem 281, 29011–29021, doi: 10.1074/jbc.M601545200 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Grant CM Regulation of translation by hydrogen peroxide. Antioxid Redox Signal 15, 191–203, doi: 10.1089/ars.2010.3699 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Gerashchenko MV, Lobanov AV & Gladyshev VN Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc Natl Acad Sci U S A 109, 17394–17399, doi: 10.1073/pnas.1120799109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knutsen JH et al. Stress-induced inhibition of translation independently of eIF2alpha phosphorylation. Journal of cell science 128, 4420–4427, doi: 10.1242/jcs.176545 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrmannova A et al. Structural analysis of an eIF3 subcomplex reveals conserved interactions required for a stable and proper translation pre-initiation complex assembly. Nucleic Acids Res 40, 2294–2311, doi: 10.1093/nar/gkr765 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erzberger JP et al. Molecular architecture of the 40SeIF1eIF3 translation initiation complex. Cell 158, 1123–1135, doi: 10.1016/j.cell.2014.07.044 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuldiner M et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123, 507–519, doi: 10.1016/j.cell.2005.08.031 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Taylor DJ et al. Structures of modified eEF2 80S ribosome complexes reveal the role of GTP hydrolysis in translocation. EMBO J 26, 2421–2431, doi: 10.1038/sj.emboj.7601677 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersen CB et al. Structure of eEF3 and the mechanism of transfer RNA release from the E-site. Nature 443, 663–668, doi: 10.1038/nature05126 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Xu G, Paige JS & Jaffrey SR Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat Biotechnol 28, 868–873, doi: 10.1038/nbt.1654 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirkpatrick DS et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol 8, 700–710, doi: 10.1038/ncb1436 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Liu B, Han Y & Qian SB Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell 49, 453–463, doi: 10.1016/j.molcel.2012.12.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shalgi R et al. Widespread regulation of translation by elongation pausing in heat shock. Mol Cell 49, 439–452, doi: 10.1016/j.molcel.2012.11.028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spence J et al. Cell cycle-regulated modification of the ribosome by a variant multiubiquitin chain. Cell 102, 67–76 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Juszkiewicz S & Hegde RS Initiation of Quality Control during Poly(A) Translation Requires Site-Specific Ribosome Ubiquitination. Mol Cell, doi: 10.1016/j.molcel.2016.11.039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saito K, Horikawa W & Ito K Inhibiting K63 polyubiquitination abolishes no-go type stalled translation surveillance in Saccharomyces cerevisiae. PLoS genetics 11, e1005197, doi: 10.1371/journal.pgen.1005197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi Z et al. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol Cell 67, 71–83 e77, doi: 10.1016/j.molcel.2017.05.021 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jenuwein T & Allis CD Translating the histone code. Science 293, 1074–1080, doi: 10.1126/science.1063127 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Vizcaino JA et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456, doi: 10.1093/nar/gkv1145 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou MF & Schwartz D Biological sequence motif discovery using motif-x. Current protocols in bioinformatics / editoral board, Baxevanis Andreas D. ... [et al. ] Chapter 13, Unit 13 15–24, doi: 10.1002/0471250953.bi1315s35 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.