

Abstract
A complementary process to the Pauson–Khand annulation is described that is well suited to forging densely substituted/oxygenated cyclopentenone products (including fully substituted variants). The reaction is thought to proceed through a sequence of metallacycle-mediated bond-forming events that engages an internal alkyne and a β-keto ester in an annulation process that forges two C–C bonds. A variant of this annulation process has also been established that delivers deoxygenated cyclopentenones that lack the allylic tertiary alcohol.
TOC Graphic:

Carbocyclic systems bearing dense patterns of oxygenation present lingering challenges for efficient chemical synthesis.1 While diverse polycyclic systems can be forged with a great variety of available ring-forming processes, even the most powerful of modern methods are not well equipped to simultaneously establish numerous and/or contiguous fully substituted sp3 centers (quaternary centers and tertiary alcohols) within the newly formed ring. This widely appreciated characteristic of available methods has resulted in the conception of synthesis strategies to highly substituted and oxygenated carbocycles that proceed in two distinct phases, where ring formation is strategically decoupled from the challenge of establishing densely substituted and/or highly oxygenated motifs.1d Despite impressive innovations that enable hydrocarbon functionalization after ring-formation,2 carbocyclic systems that contain numerous/contiguous fully substituted sp3 carbons remain quite difficult to prepare. Of the many annulation processes routinely employed in target-oriented synthesis campaigns, Pauson–Khand and Pauson–Khand-like reactions3 stand out as particularly powerful for establishing cyclopentenones, especially when conducted in an intramolecular fashion (Figure 1A). Despite the great history of successfully harnessing the power of such annulation processes in complex molecule synthesis,4 this class of reactions is most effective with sparsely substituted alkenes (Figure 1B).3 Here, we describe a mode of reactivity that can be employed for the formation of densely substituted cyclopentenones that are not directly accessible with Pauson–Khand-type reactions, enabling direct formation of fully substituted five-membered ring-containing products (Figure 1C).
Figure 1.

Introduction.
As illustrated in Figure 2A, we have recently described a metallacycle-mediated annulation reaction that proceeds through intramolecular reaction of an alkyne with a β-diketone,5 and have been investigating the potential value of this annulation process in studies targeting a total synthesis of ryanodol.6 This previous success served as a foundation to the current investigations that are based on the recognition that arelated process may be quite useful for generating densely substituted cyclopentenones that are not accessible with Pauson–Khand-like annulation reactions. Proceeding through unique bond-forming events (Figure 2B), it was anticipated that formation of a metallacyclopropene7 (A) would be followed by intramolecular reaction with the proximal ketone to generate an oxametallacyclopentene intermediate (B). Subsequent engagement of the metal–carbon σ-bond of this intermediate in an intramolecular addition reaction to the tethered ester could result in formation of a bridged polycylic metal-alkoxide (C), the rearrangement and hydrolysis of which would deliver a fully substituted and oxygenated cyclopentenone product.
Figure 2.

An approach to cyclopentenone synthesis by intramolecular metallacycle-mediated coupling of an alkyne to a β-keto ester.
Investigations began as illustrated in eqs. 1–3 of Figure 3, and demonstrate that the planned annulation process was feasible. These initial experiments revealed that treatment of simple unsaturated β-keto ester substrates with the combination of Ti(Oi-Pr)4 (2 equiv) and c-C5H9MgCl (4 equiv) results in formation of the expected cyclopentenone product in good yield (up to 77%),8 noting that the nature of the ester has some impact on efficiency of the process. Perhaps not surprisingly, the t-Bu ester 3 was less effective in the annulation reaction, delivering the product 4 in a depressed yield of 50% – it is believed that this decreased efficiency is due primarily to steric effects imposed by the t-Bu substituent. Moving on, minor modification of the quaternary center between the two reactive carbonyls had little impact on the efficiency of the process. As depicted in eqs. 4 and 5, substrates 5 and 7 were smoothly converted to the tricyclic products 6 and 8 in 71 and 83% yield, respectively.
Figure 3.

Intramolecular annulation reactions between alkynes and β-keto esters: initial exploration.
The substitution on the alkynyl carbon distal to the β-keto ester appeared to have a significant impact on efficiency of the annulation process. Moving from substrates that contain a methyl group at this position to others that contain a phenyl (eq. 6), substituted allyl (eq. 7), and trimethylsilyl substituent (eq. 8), all resulted in more complex reaction mixtures from which only modest quantities of the expected products could be obtained (45, 42, and 37%, respectively).
Disappointingly, the effectiveness of this organometallic transformation was also negatively impacted by tether length (eq. 9). It is certainly appreciated that tether length plays an important role in the efficiency and viability of all intramolecular Pauson–Khand-like reactions, and it is indeed typical that substrates with tethers that result in the formation of a five-membered ring are most effective in a great variety of intramolecular reactions.9 While it was understood that strategic substitution of the tether in 15 may certainly result in enhanced efficiency for cyclization due to the impact of conformation on rate of cyclization (i.e. Thorpe-Ingold effect),10 attention was directed to other concerns regarding the potential utility of this new annulation reaction in stereoselective synthesis.11
As illustrated in Figure 3B, the annulation process can be conducted in a highly diastereoselective manner. Notably, with a substrate containing an α-chiral center (17), the annulation process proceeds with exquisite levels of stereoselection. Here, the bicyclic product 18 was isolated in 62% yield and with ≥20:1 dr. When the stereocenter is relocated to the β-position (symmetrically disposed about the alkyne and the ketone), as in 19 (eq. 11), the annulation reaction also proceeds with acceptable efficiency (68%), albeit without observable levels of diastereoselection. In more conformationally constrained systems (i.e. 21 and 23, eqs 12–13), the annulation also proceeds to deliver fused tricyclic products 22 and 24 as single isomers.12
While these latter annulation reactions proceeded in a manner that indicates the great stereocontrol possible in this new carbocycle-forming reaction, the diminished efficiencies (43% and 40%, respectively) were concerning. As illustrated in Figure 4, additional studies have revealed that the initial products of this metallacycle-mediated annulation process are not stable to the reaction conditions. For example, treatment of the tricyclic enone 22 with the combination of Ti(Oi-Pr)4 and c-C5H9MgCl, resulted in a relatively clean conversion to the deoxygenated product 25. As such, competing rates of initial cyclization and subsequent deoxygenation can complicate efforts to utilize this annulation process in diverse molecular environments. If the deoxygenated product is desired, it is possible to conduct a tandem annulation/deoxygenation with synthetically useful efficiency. As illustrated in eqs. 15 and 16, the β-keto ester substrates 2 and 21 are converted directly to the deoxygenated bi- and tricyclic products 26 and 25 in 47% and 43% yield, simply by employing additional quantities of Ti(Oi-Pr)4 (~4 equiv.) and Grignard reagent (~8 equiv.).
Figure 4.

Products of the annulation are not stable under the reaction conditions; modified reaction conditions realize sequential cyclization and deoxygenation.
While providing direct access to densely substituted cyclopentenones not readily available from other annulation methods, it was discovered that the products of this annulation process are rather resistant to otherwise straightforward functional group manipulations. That said, redox manipulations that engage the alkene of the enone were straightforward, and provided a means to generate additional stereodefined carbocyclic systems. As illustrated in Figure 5, directed epoxidation, conjugate reduction, and heterogeneous hydrogenation were all effective for stereoselectively generating the bicyclic cyclopentanones 27, 28, and 30 (in all cases, no evidence was found for the production of stereoisomeric products).
Figure 5.
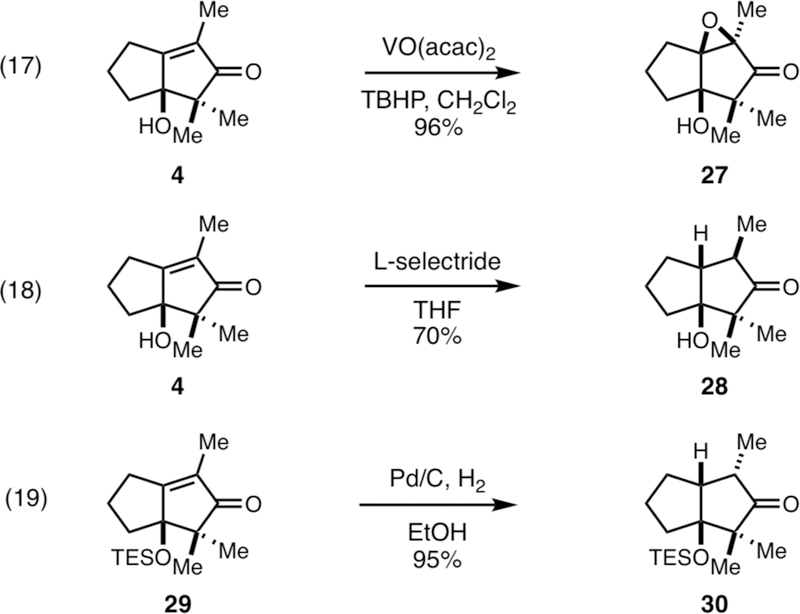
Some stereoselective transformations of the cyclopentenone products.
In conclusion, we report a general annulation process for the synthesis of densely functionalized and highly oxygenated carbocycles that proceeds through the union of alkynes with β-keto esters. The reactions that have emerged through these studies are complementary to classic carbocycle-forming processes in organic chemistry, delivering products not readily available from Pauson–Khand and Pauson–Khand-like annulation reactions. The reactivity realized is thought to proceed through a metallacycle-centered reaction cascade that engages each metal–carbon bond of a metallacyclopropene in reactions with two different carbonyl systems. In addition to establishing the basic coupling process and reporting preliminary scope substitution (ester and alkyne) and tether length, our studies have also shed light on structural features that lead to stereoselection and led to the discovery of a competing reaction process that results in the formation of deoxygenated products. While complex, and certainly appearing to be sensitive to substrate structure, we look forward to exploring key features of this annulation reaction in ongoing methods development projects and target-oriented synthesis campaigns.
Supplementary Material
ACKNOWLEDGMENT:
We gratefully acknowledge financial support of this work by the National Institutes of Health – NIGMS (GM124004).
Footnotes
SUPPORTING INFORMATION PARAGRAPH:
Experimental procedures and tabulated spectroscopic data for new compounds (PDF) are available free of charge via the Internet at http://pubs.acs.org/page/jacsat/submission/authors.html. This includes the preparation of each substrate for the Ti-mediated annulation reactions described.
REFERENCES:
- (1).(a) Wender PA Nat. Prod. Rep 2014, 31, 433–440. [DOI] [PubMed] [Google Scholar]; (b) Ishihara Y; Baran PS Synlett 2010, 1733–1745. [Google Scholar]; (c) Burns NZ; Baran PS; Hoffmann RW Angew. Chem. Int. Ed 2009, 48, 2854–2867. [DOI] [PubMed] [Google Scholar]; (d) Chen K; Baran PS Nature 2009, 459, 824–828. [DOI] [PubMed] [Google Scholar]
- (2).For recent reviews, see: (a) Hartwig JF J. Am. Chem. Soc 2016, 138, 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Qiu Y; Gao S Nat. Prod. Rep 2016, 33, 562–581. [DOI] [PubMed] [Google Scholar]; (c) Nakamura A; Nakada M Synthesis 2013, 45, 1421–1451. [Google Scholar]; (d) Chen DY-K; Youn SW Chem. Eur. J 2012, 18, 9452–9474. [DOI] [PubMed] [Google Scholar]; (e) Gutekunst WR; Baran PS Chem. Soc. Rev 2011, 40, 1976–1991. [DOI] [PubMed] [Google Scholar]
- (3).(a) Brummond KM; Kent JL Tetrahedron, 2000, 56, 3263–3283. [Google Scholar]; Schore NE Org. React 1991, 40, 1–90. For examples with more substituted enyne substrates, see: [Google Scholar]; (b) Knudsen MJ; Schore NE J. Org. Chem 1984, 49, 5025–5026. [Google Scholar]; (c) Schore NE; Knudsen J Org. Chem 1987, 52, 569–580. [Google Scholar]; (d) Schore NE; Rowley EG J. Am. Chem. Soc 1988, 110, 5224–5225. [Google Scholar]; (e) Rowley EG; Schore NE J. Org. Chem 1992, 57, 6853–6861. [Google Scholar]; (f) Stolle A; Becker H; Salaün J; de Meijere, Tetrahedron Lett 1994, 35, 3517–3520. [Google Scholar]; (g) Tormo J; Verdaguer X; Moyano A; Pericás MA; Riera A Tetrahedron 1996, 52, 14021–14040. [Google Scholar]; (h) Tormo J; Moyano A; Pericás MA; Riera AJ Org. Chem 1997, 62, 4851–4856. [DOI] [PubMed] [Google Scholar]; (i) Ishizaki M; Iwahara K; Kyoumura K; Hoshino O Synlett 1999, 587–589. [Google Scholar]; (j) Kerr WJ; McLaughlin M; Morrison AJ; Pauson PL Org. Lett 2001, 3, 2945–2948. [DOI] [PubMed] [Google Scholar]; (k) Ishizaki M; Iwahara K; Nimi Y; Satoh H; Osamu H Tetrahedron 2001, 57, 2729–2738. [Google Scholar]; (l) Son SU; Park KH; chung YK J. Am. Chem. Soc 2002, 124, 6838–6839. [DOI] [PubMed] [Google Scholar]; (m) de Meijere A; Becker H; Stolle A; Kozhushkov SI; Bes MT; Salaün J; Noltemeyer M Chem. Eur. J 2005, 11, 2471–2482. [DOI] [PubMed] [Google Scholar]
- (4).(a) Jamison TF; Shambayati S; Crowe WE; Schreiber SL J. Am. Chem. Soc 1997, 119, 4353–4363. [Google Scholar]; (b) Chuang KV; Xu C; Reisman SE Science, 2016, 353, 912–915. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jørgensen L; McKerrall SJ; Kuttruff CA; Ungeheuer F; Felding J; Baran PS Science, 2013, 341, 878–882. [DOI] [PubMed] [Google Scholar]
- (5).Kier MJ; Leon RM; O’Rourke NF; Rheingold AL; Micalizio GC J. Am. Chem. Soc 2017, 139, 12374–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Du K; Kier MJ; Rheingold AL; Micalizio GC Org. Lett 2018, 20, 6457–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For reviews of Ti(Oi-Pr)4-based metallacycle-mediated coupling chemistry see: (a) Wolan A; Six Y Tetrahedron 2010, 66, 15–61. [Google Scholar]; (b) Kulinkovich OG; de Meijere A Chem. Rev 2000, 100, 2789–2834. [DOI] [PubMed] [Google Scholar]; (c) Sato F; Urabe H; Okamoto S Chem. Rev 2000, 100, 2835–2886. [DOI] [PubMed] [Google Scholar]; (d) Reichard HA; McLaughlin M; Chen MZ; Micalizio GC Eur. J. Org. Chem 2010, 391–409. [DOI] [PMC free article] [PubMed]; (e) Reichard HA; Micalizio GC Chem. Sci 2011, 2, 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Micalizio GC Early Transition Metal Mediated Reductive Coupling Reactions. In: Molander GA, Knochel P (eds.), Comprehensive Organic Synthesis, 2nd edition, Vol. 5, Oxford: Elsevier, 2014, pp. 1660–1737. For a review of the use of metallacycle-mediated cross-coupling in natural product synthesis, see: [Google Scholar]; (g) O’Rourke NF; Kier MJ; Micalizio GC Tetrahedron 2016, 72, 7093–7123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).A variety of Grignard reagents are known to be useful for converting Ti(Oi-Pr)4 to a reactive organometallic intermediate in the context of metallacycle-mediated bond-forming processes. The choice to employ c-C5H9MgCl instead of i-PrMgCl (see ref. #5) here is viewed at this point in time as being arbitrary.
- (9).For example, see: Bird R; Knipe AC; Stirling CJM J. Chem. Soc. Perkin Trans 2, 1973, 1215–1220. [Google Scholar]
- (10).(a) Beesley RM; Ingold CK; Thorpe JF J. Chem. Soc. Trans 1915, 107, 1080–1106. [Google Scholar]; (b) Jung ME; Piizzi G Chem. Rev 2005, 105, 1735–1766. [DOI] [PubMed] [Google Scholar]
- (11).Substrates explored for this annulation uniformly contained a quaternary center between the two ketones due to anticipated problems associated with competitive enolization under the reaction conditions. Attempts to employ an α-monosubstituted β-keto imide were unsuccessful. For an interesting selective enolization of such a β-keto imide, see: Evans DA; Clark JS; Metternich R; Novack VJ; Sheppard GS J. Am. Chem. Soc 1990, 112, 866–868. [Google Scholar]
- (12).Switching the nature of the ethereal solvent used for these annulation reactions was not found to have a significant impact on efficiency (Et2O vs. THF). Also, the competing process of deoxygenation, as seen in Figure 4, represents an ever-present obstacle for optimization of the annulation reaction to deliver tertiary alcohol-containing products due to what appears to be substrate-dependent variable rates of reaction for the subsequent deoxygenation. Over the course of our initial study, benefits were sometimes observed by minor modification of the reaction conditions or experimental setup (see Supporting Information for experimental details).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.