

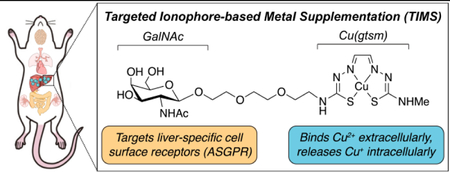
Abstract
Copper deficiency is implicated in a variety of genetic, neurological, cardiovascular, and metabolic diseases. Current approaches for addressing copper deficiency rely on generic copper supplementation, which can potentially lead to detrimental off-target metal accumulation in unwanted tissues and subsequently trigger oxidative stress and damage cascades. Here we present a new modular platform for delivering metal ions in a tissue-specific manner and demonstrate liver-targeted copper supplementation as a proof of concept of this strategy. Specifically, we designed and synthesized a N-acetylgalactosamine-functionalized ionophore, Gal-Cu(gtsm), to serve as a copper-carrying “Trojan Horse” that targets liver-localized asialoglycoprotein receptors (ASGPRs) and releases copper only after being taken up by cells, where the reducing intracellular environment triggers copper release from the ionophore. We utilized a combination of bioluminescence imaging and inductively-coupled plasma mass spectrometry assays to establish ASGPR-dependent copper accumulation with this reagent in both liver cell culture and mouse models with minimal toxicity. The modular nature of our synthetic approach presages that this platform can be expanded to deliver a broader range of metals to specific cells, tissues, and organs in a more directed manner to treat metal deficiency in disease.
Graphical Abstract
INTRODUCTION
Copper is a required nutrient for all living organisms1, enabling fundamental life processes spanning respiration,2 antioxidant defense,3,4 neurotransmitter synthesis,5,6 metabolism,7,8 and cell signaling.9–12 Biological copper deficiency hinders these essential functions and correlates with various pathologies including Menkes disease,13 familial amyotrophic lateral sclerosis,14,15 neurodegenerative disorders,16–18 cardiovascular disease,19,20 and metabolic disorders.21–27 Current approaches for addressing copper deficiency rely on metal supplementation, often through the administration of copper ionophores, which are molecules that are distinct from copper chelators in that they deliver and release metals into cells rather than sequester and remove metals from cells.28,29 Conventional copper ionophores include copper complexes with histidine,30 bisthiosemicarbazones,31–33 8-hydroxyquinolines,16,34 and others.29 The small and hydrophobic nature of these ionophores, which typically chelate copper in the Cu2+ oxidation state, enables their passive diffusion through the cell membrane, after which the reducing intracellular environment acts as a redox trigger for reduction of the metal complex and subsequent Cu+ release to copper-binding molecules and proteins (Figure 1A).
Figure 1.
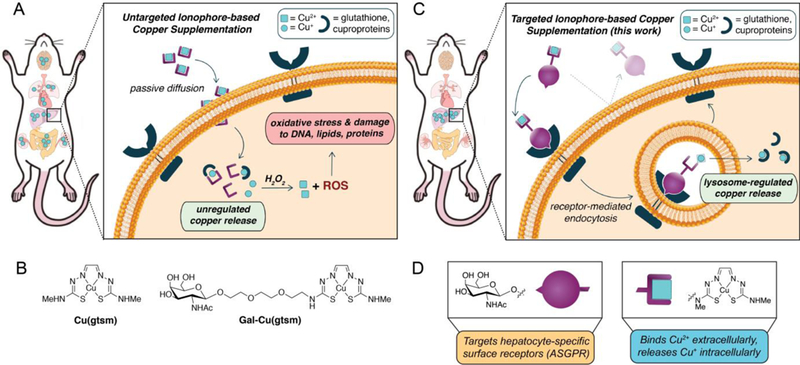
Schematic diagram comparing delivery methods for ionophore-based copper supplementation. (A) Conventional ionophores increase copper levels in many organs due to the non-specific nature of passive diffusion. For example, Cu(gtsm) releases copper intracellularly following reduction in the cytosolic medium. If too much copper is released such that it exceeds the cell’s copper buffering capacity, excess Cu+ can generate reactive oxygen species (ROS) through Fenton-like reactions to induce oxidative stress and damage to the cell. (B) Molecular structures for the untargeted Cu(gtsm) and targeted Gal-Cu(gtsm) ionophores studied here. (C) The targeted ionophore-based metal supplementation (TIMS) strategy presented here enables receptor-mediated metal accumulation with minimal off-target delivery, as shown by liver-selective copper supplementation. The hydrophilicity of the targeted ionophore precludes passive diffusion; ionophore internalization only occurs upon ligand-receptor recognition and endocytosis. Copper release is likely controlled by homeostatic cues at the level of the lysosome to enable regulated copper delivery. (D) The bifunctional ionophore design for Gal-Cu(gtsm) uses a triethyleneglycol linker to join the Cu(gtsm) moiety, which binds copper selectively and releases it upon intracellular reduction, with a GalNAc targeting group that is a specific ligand for ASGPR proteins expressed on the cell membranes of hepatocytes.
Whereas ionophores offer improved cellular copper supplementation relative to the inorganic copper salts typically administered in vitamins, several inherent drawbacks arise from the generic, untargeted nature of copper delivery. First, because copper delivery occurs mostly through a passive transport mechanism, copper delivery into cells is not regulated: for example, if copper supplementation exceeds the buffering capacity of the cytosol, oxidative damage to biomolecules such as DNA, RNA, lipids, and proteins may occur via Fenton-like chemistry.35 Indeed, copper ionophore supplementation routinely leads to oxidative stress and damage.36,37 Second, traditional untargeted copper delivery approaches lack tissue specificity.38,39 As such, if the intent is to treat copper deficiency in a specific organ of interest, such indiscriminate copper delivery will lead to aberrant ROS generation and oxidative damage in off-target tissues (Figure 1A). To address these challenges, we now present a new, versatile approach to site-specific metal delivery, termed “Targeted Ionophore-based Metal Supplementation (TIMS)”, which relies on pairing metal-delivering ionophores with targeting ligands that recognize specific cell surface receptors localized to cells and tissues of interest.
We demonstrate the viability of this TIMS approach through the development of a targeted copper supplement that enables selective delivery of copper to the liver in a non-toxic manner owing to interest in addressing hepatic copper deficiency connected to the pathogenesis of non-alcoholic fatty liver disease (NAFLD),21–23,26,27 which is estimated to afflict 24% of the global population (or 1.8 billion people).40 NAFLD often leads to severe liver diseases and shows a strong association with cardiovascular diseases, obesity, diabetes, and cancer.41–43 Indeed, recent work from our laboratories utilized a caged copper luciferin (CCL-1) probe to longitudinally monitor copper levels over the course of NAFLD progression in mice, revealing that hepatic copper deficiency manifests in a diet-induced NAFLD model prior to the formal onset of fatty liver disease. Along these lines, we prepared targeted bifunctional ionophore Gal-Cu(gtsm) and benchmarked its copper delivery properties in cell culture and mice against the untargeted Cu(gtsm) ionophore (Figure 1B). We utilized ICP-MS and bioluminescent imaging techniques to characterize copper supplementation across tissues, observing that the targeted Gal-Cu(gtsm) enables selective hepatic copper supplementation. Furthermore, we utilized a variety of toxicity and histological staining assays to establish that Cu delivery via the TIMS compound is non-toxic whereas untargeted delivery via passive diffusion incurs significant damage to the liver as well as off-target renal toxicity. Finally, we performed SDS-PAGE and Western blot analysis to characterize expression of copper storage and trafficking proteins, finding that both copper import/export as well as storage proteins are elevated with TIMS treatment. Taken together, this study provides a new type of liver-specific copper supplement and establishes a starting point for the broader use of TIMS reagents as tool compounds to probe the functional roles that metals play in specific cell, tissue, and/or organ types in normal physiology and as directed therapeutic supplements to address metal deficiency-based pathologies.
RESULTS AND DISCUSSION
Design and Synthesis of a Liver-Targeted Copper Supplement.
Figure 1C outlines the TIMS strategy, where (i) ligand-receptor recognition triggers endocytosis and internalization of the receptor-ionophore complex, (ii) the complex releases the metal in the endolysosomal pathway through either intracellular reduction or degradation, and (iii) the metal ion binds to metal storage or trafficking proteins, which can then utilize the metal as needed. In support of this approach, recent studies have shown that the lysosome is a key organelle in dynamic metal regulation,44–46 and mediating metal delivery through this organelle’s homeostatic cues may offer a safer alternative for increasing bioavailable metal ion pools compared to unregulated cytosolic metal release. We note that for this strategy to be effective, the targeted ionophore must be either large or hydrophilic enough to hinder passive diffusion such that metal supplementation is strictly receptor-mediated.
With these design principles in mind, we synthesized a bifunctional ionophore that targets the asialoglycoprotein receptor (ASPGR), a C-type lectin membrane receptor that is specifically expressed in the liver at high levels (e.g., ca. 500,000 ASGPR copies per primary hepatocyte47) as shown in Figure 1D. We utilized an N-acetylgalactosamine (GalNAc) ligand as our targeting group, which binds ASGPR with a Kd value of ca. 10−5 M−1.48 Liver targeting via ASGPR/galactose recognition is a robust strategy that has been used to deliver drugs,49,50 proteins,51 siRNA,52,53 and CRISPR-Cas9;54 related work has employed metal chelators55,56 to address copper excess-based disorders such as Wilson’s disease. Although multivalent47 galactose conjugates offer higher-affinity binding over their monovalent48,57 counterparts, we decided on the monovalent targeting group in our first-generation design owing to improved atom economy for our payload, a small organometallic complex, as well as for scalable synthesis for longitudinal animal studies.
The design for our TIMS agent is conceptually related to the approach taken for site-specific metal removal, with the key difference being that we attach a targeting group to a metal ionophore rather than a metal chelator.17,58–60 As such, we paired a GalNAc targeting moiety with the bisthiosemicarbazone complex Cu(gtsm) (Figure 1D), which is a well-established copper ionophore that has been explored as a potential therapeutic for neurodegenerative disorders31,39,61,62 and cancer36,63. The metal binding and Cu release properties of Cu(gtsm) complex are well-studied and characterized. The gtsm ligand binds Cu2+ (Kd ~ 10−18 M−1) much more tightly than Cu+ (Kd ~ 10−13 M−1),64 and releases Cu+ upon reduction in the intracellular medium where it can metalate copper-binding small molecules and proteins.31,36,65 We reasoned that the significant hydrophilicity conferred by the GalNAc subunit may preclude the ionophore from passive diffusion and encourage receptor-mediated uptake. We chose to link the ionophore and targeting GalNAc ligand with a triethylene glycol chain for enhanced hydrophilicity owing to literature precedent.50
Cu(gtsm) was synthesized using literature methods.66 For Gal-Cu(gtsm), we employed a convergent synthetic approach where we synthesized the ionophore and targeting group fragments independently, then linked them together through a transamination reaction in the penultimate step (Scheme 1).67,68 Briefly, we installed the glyoxal unit with an acetal protecting group onto thiosemicarbazide 1, then unmasked acetal 2 with lithium tetrafluoroborate to give iminoacetaldehyde 3.69 Condensation with 4,4-dimethyl-3-thiosemicarbazide gave the asymmetrically substituted gtsm analogue 4. We reacted oxazoline 570 with azidotriethylene glycol in the presence of trimethylsilyl trifluoromethanesulfonate to access the azide-protected peracetylated GalNAc 6. The azide and O-acetyl groups were deprotected in a single palladium-catalyzed hydrogenation reaction to give GalNAc amine 7. Transamination between 4 and 7 gave apo-Gal-H2gtsm, which we then metalated with copper diacetate to give Gal-Cu(gtsm).
Scheme 1. Synthetic route for accessing Gal-Cu(gtsm).
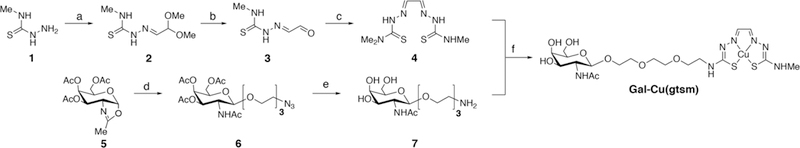
a) 2,2-dimethoxyacetaldehyde, MeOH, r.t., quantitative yield. b) LiBF4, MeCN, H2O, r.t., 81% yield. c) 4,4-dimethyl-3-thiosemicarbazide, DMF, 4Å MS, 60˚C, 38% yield. d) HO-TEG-N3, TMSOTf, DCE, r.t. 81% yield. e) Pd/C, H2, MeOH, 92% yield. f) i. MeCN, 80˚C, 32% yield. ii. Cu(OAc)2, DMF, r.t. 73% yield.
Log P measurements with a shake-flask octanol-water paritition experiment confirmed that Gal-Cu(gtsm) (log P = –1.03 ± 0.10) is more hydrophilic than Cu(gtsm) (log P =1.39 ± 0.14).66,71 Interestingly, we also found that Gal-Cu(gtsm) forms colloids in aqueous buffer with a number-weighted hydrodynamic diameter of 4.60 ± 0.69 nm whereas Cu(gtsm) does not. Cu(gtsm) and Gal-Cu(gtsm) demonstrate similar Cu-ligand charge transfer absorption band characteristics (Figure S1). We performed further in vitro spectroscopic studies to evaluate the stability of Gal-Cu(gtsm) under physiologically relevant conditions. We found that the Gal-Cu(gtsm) complex is stable in aqueous buffers ranging from pH = 3 to 8 (Figure S2). The complex also retains copper selectively in the presence of 1000-fold excess of Zn2+ and Fe2+ (Figure S3), which are two other abundant intracellular transition metal ions.72
ASGPR-Dependent Copper Delivery in Cell Culture.
With the two ionophores in hand, we characterized the behavior of Cu(gtsm) and Gal-Cu(gtsm) in cell lines with high ASGPR expression (HepG2) or no ASGPR expression (HEK 293T). We evaluated ionophore toxicity in these cell lines using propidium iodide staining as a proxy for cell viability, finding that that Cu(gtsm) treatment is toxic at an 8 µM dose in HEK 293T cells, whereas Gal-Cu(gtsm) is non-toxic at a 500 µM dose in both cell lines (Figure S4). We then evaluated the intracellular Cu delivery abilities of the two ionophores using inductively coupled plasma-mass spectrometry (ICP-MS) to analyze total copper levels. We find that Cu(gtsm) elicits dose-dependent increases in cellular copper content for both cell types, whereas Gal-Cu(gtsm) only demonstrates a robust dose-dependent copper delivery response in the ASGPR-expressing HepG2 cells (Figure S5).
Figures 2A-B highlights these differences at an equimolar 4 µM copper dose. We treated HepG2 and HEK 293T cells with either vehicle control, 4 µM Cu(gtsm), or 4 µM Gal-Cu(gtsm) for three hours. Figure 2 displays 63Cu/31P ratios from ICP-MS data as a metric for characterizing copper delivery for each treatment condition, with the 31P signal normalizing for number of cells based on cellular phosphate content. Figure 2A shows that Gal-Cu(gtsm) leads to a 4-fold Cu increase relative to the basal levels in HepG2 cells, while Cu(gtsm) treatment leads to a 45-fold increase in Cu content. In HEK 293T cells (Figure 2B) that do not express ASGPR, Gal-Cu(gtsm) treatment does not increase cellular copper beyond basal levels, while Cu(gtsm) treatment leads to a 100-fold increase in Cu levels. The data show that Cu(gtsm) delivers copper indiscriminately to both cell types, likely via passive diffusion since Cu(gtsm) is small, hydrophobic, and cell permeable. Conversely, the hydrophilic Gal-Cu(gtsm) experiences limited passive diffusion and does not deliver copper to HEK 293T cells lacking ASGPRs, whereas copper delivery only occurs in the ASGPR-expressing HepG2 cells upon Gal-Cu(gtsm)/ASGPR recognition and internalization.
Figure 2.
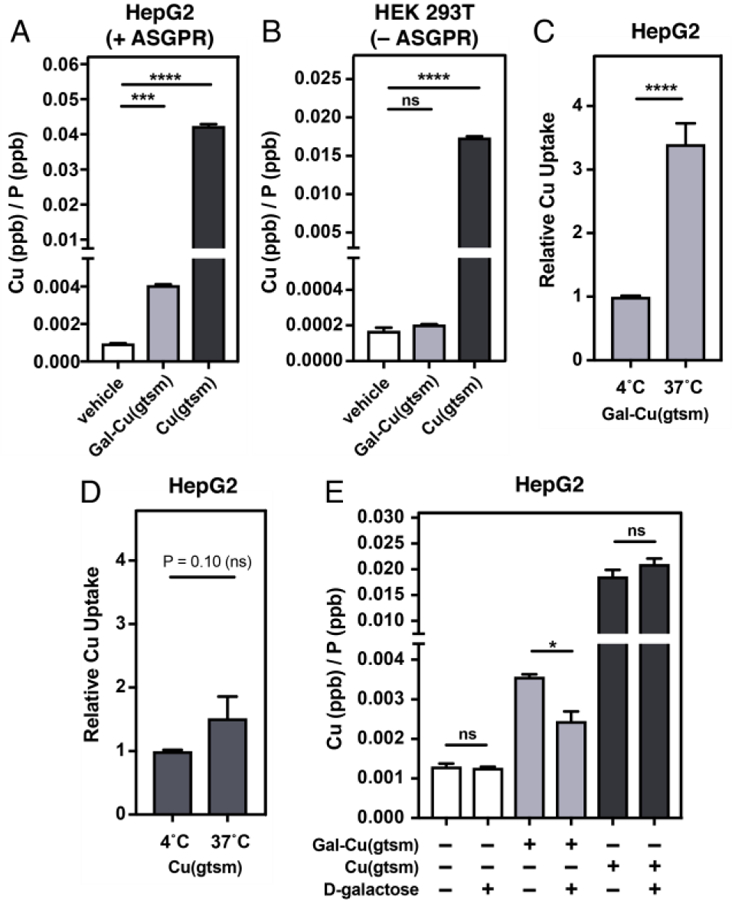
ICP-MS measurements reveal that Cu supplementation via Gal-Cu(gtsm) is ASGPR-dependent. (A) ICP-MS studies comparing Cu levels upon 4 µM Cu-ionophore treatment (0.2% DMSO in serum-free DMEM) over a three-hour time course in HepG2 (ASGPR-expressing) cells. (B) ICP-MS studies upon 4 µM Cu-ionophore treatment (0.2% DMSO in serum-free DMEM) over a three-hour time course in HEK 293T (no ASGPR expression) cells. (C-D) HepG2 cells were treated with Gal-Cu(gtsm) (20 µM, C) or Cu(gtsm) (4 µM, D) over a one-hour period at either 4˚C or 37˚C. Data plotted relative to Cu/P ratio for each ionophore at 4˚C. (E) HepG2 cells were treated with or without D-galactose (1 M in serum-free DMEM) as a competitive ASGPR ligand fifteen minutes prior to treatment with vehicle, Cu(gtsm) (4 µM), or Gal-Cu(gtsm) (20 µM) over a one-hour period. Error bars = SEM (n = 6). Statistical analyses were performed using a one-way ANOVA with Bonferroni’s multiple comparisons test (A-B) or a two-tailed Student’s t test (C) where *P ≤ 0.05, ***P ≤ 0.001, and ****P ≤ 0.0001.
In support of this mechanistic proposal, we performed temperature dependent uptake studies at 4˚C and 37˚C. Gal-Cu(gtsm) demonstrates much higher uptake at 37˚C than at 4˚C, which suggests that Gal-Cu(gtsm)-mediated Cu delivery is an active uptake process (Figure 2C). In contrast, we did not observe any significant differences in Cu delivery ability for Cu(gtsm) between 4˚C and 37˚C, which suggests that Cu delivery via Cu(gtsm) occurs primarily through a passive diffusion mechanism (Figure 2D). As further support for an ASGPR-mediated Cu delivery mechanism for Gal-Cu(gtsm), we performed a competition experiment where we treated HepG2 cells with vehicle control or ionophore in the presence of D-galactose (1 M) for one hour. The presence or absence of D-galactose does not alter Cu content upon vehicle or Cu(gtsm) treatment in HepG2 cells; however, Cu delivery via Gal-Cu(gtsm) is significantly attenuated in the presence of D-galactose (Figure 2E). We also performed confocal microscopy imaging of HepG2 cells treated with Cu(gtsm) and Gal-Cu(gtsm) using the copper sensing CF4 fluorescent probe73 and observed increased fluorescence upon ionophore treatment (Figure S6).
Though the untargeted Cu(gtsm) ionophore delivered more total copper than Gal-Cu(gtsm) under equimolar doses to both HEK 293T and HepG2 cells, the salient finding for expanding the TIMS platform to animal studies was that copper delivery via Gal-Cu(gtsm) occurred exclusively in the ASGPR-expressing HepG2 cells. With these promising cell culture results in hand, we sought to identify whether Gal-Cu(gtsm) could selectively provide Cu supplementation to the liver with minimal off-target delivery in the complex biological milieu of live mice.
Evaluating Liver Target Specificity of Copper Supplementation by Gal-Cu(gtsm) in Living Mice.
We chose to characterize liver-targeted copper supplementation in mice because they are common model organisms for elucidating the pathology of metabolic disorders such as NAFLD. Our laboratory recently developed a Copper Caged Luciferin-1 (CCL-1) probe that reports on dynamic changes to the loosely bound, labile Cu+ pool in transgenic bioluminescent mice (Figure 3A).27 Here we utilize CCL-1 to characterize liver-directed copper supplementation in the firefly luciferase-expressing FVB-luc+ mouse strain, where luciferase expression enables detection of changes in copper pools throughout the entire mouse. We also imaged ionophore-treated mice with D-luciferin (D-Luc) to normalize the CCL-1 signal and account for CCL-1 cleavage-independent bioluminescent effects. Male FVB-luc+ mice (11–14 weeks old) received intraperitoneal (i.p.) injections of vehicle control, Gal-Cu(gtsm), or Cu(gtsm) at equivalent copper concentrations (0.75 mg Cu/kg mouse). We selected this copper dose based on similar i.p. doses utilized by others to study the therapeutic effects of Cu(gtsm) and its derivatives.63,74 After six hours, the same mice were anesthetized with isoflurane and given scapular subcutaneous (s.c.) injections of either CCL-1 or D-Luc (0.1 µmol) and imaged with an In Vivo Imaging System (IVIS) every 5 minutes over a 40-minute period. Representative images of CCL-1 injected mice six hours following vehicle or Gal-Cu(gtsm) treatment are provided in Figure 3B. A full panel of images for CCL-1 and D-Luc injected mice both 6 and 24 hours after ionophore treatment is provided in Figures S7, S8.
Figure 3.
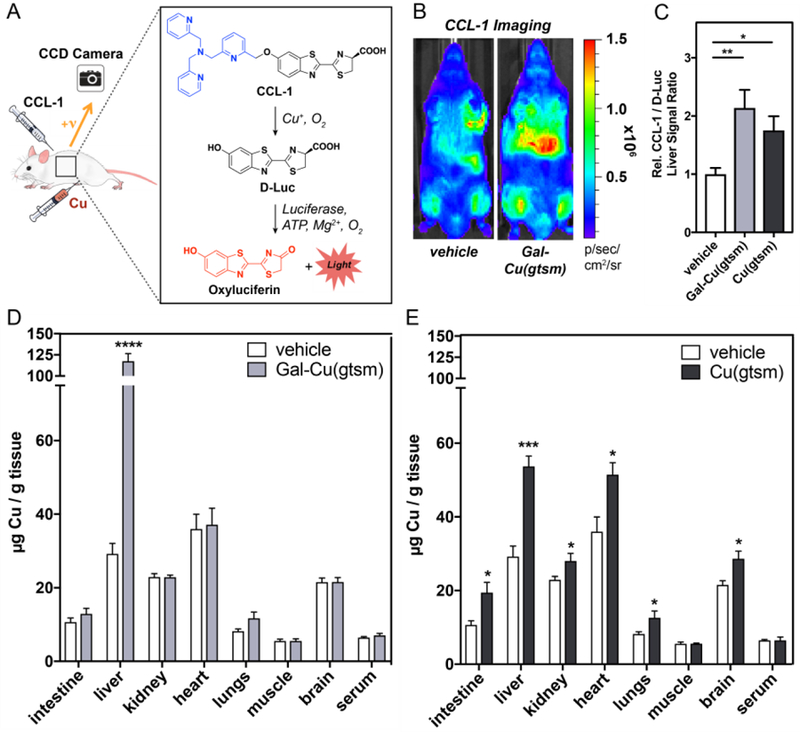
In vivo bioluminescence imaging and ex vivo tissue ICP-MS analysis reveal a tissue-selective increase in hepatic copper stores after Gal-Cu(gtsm) supplementation. (A) Schematic describing Cu+-dependent cleavage of CCL-1 to give bioluminescent signal. Free D-luciferin is released upon binding between Cu and CCL-1, which then interacts with luciferase and emits a photon. (B) Representative images of mice injected s.c. with CCL-1 6 hours after vehicle or Gal-Cu(gtsm) i.p. administration. Images for Cu(gtsm)-treated mice are given in Figure S7. (C) Data plotted represents CCL-1 total integrated photon flux as a ratio over D-Luc total integrated photon flux. Total integrated photon flux was collected 5–45 minutes post injection in a region of interest drawn over the liver. Liver signal ratio is normalized to vehicle control. Error bars = SEM (n = 5–7). Statistical analysis performed with a one-way ANOVA with Bonferroni’s multiple comparisons test where *P ≤ 0.05. (D-E) Mice were injected i.p. with 0.75 mg equivalent Cu/kg mouse of Gal-Cu(gtsm) or Cu(gtsm) six hours prior to blood and tissue collection. Tissue copper levels relative to tissue wet weight was determined using ICP-MS assays. Error bars = SEM (n = 5). Statistical analyses were performed with a two-tailed Student’s t test where *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001.
Figure 3C plots the ratio of CCL-1/D-Luc integrated photon fluxes in the liver region, six hours after ionophore supplementation. We find high increases in liver CCL-1/D-Luc signal ratio responses, suggesting increases in labile copper levels for both the targeted Gal-Cu(gtsm) and Cu(gtsm) compounds. There are two potential factors that may contribute to the observed differences between cell culture and live animal TIMS. First, there is a much higher expression of ASGPR in primary hepatocytes (ca. 500,000 ASGPR/cell) than in HepG2 cells (ca. 76,000 ASGPR/cell).47,75 Second, Cu(gtsm) delivery occurs through non-specific passive diffusion. In cell culture, this property results in high levels of copper delivery. However, in animals, this property leads to non-specific copper delivery to off-target organs.
The greater propensity for off-target copper accumulation with Cu(gtsm) is supported by ex vivo tissue metal analysis via ICP-MS to evaluate total copper levels across organs after Cu(gtsm) and Gal-Cu(gtsm) supplementation at a 0.75 mg Cu/kg dose. We first performed ICP-MS analysis on the stock solutions of Cu(gtsm) and Gal-Cu(gtsm) to confirm that we are indeed administering equivalent copper doses between the ionophores (Figure S9). We collected blood and harvested organs at two different timepoints (6 hours, 24 hours) to study the time-dependence of copper content and distribution across organs, then processed tissues for ICP-MS analysis. Gal-Cu(gtsm) supplementation results in a 300% increase in liver copper levels after 6 hours, with no significant differences in Cu levels in the other extracted organs compared to basal conditions (Figure 3D). Half of this copper is excreted 24 hours following Gal-Cu(gtsm) supplementation, with a 150% increase in liver copper relative to basal levels. Interestingly, copper levels in serum and other organs remains at basal levels at 24 hours (Figure S10), implying that the supplemented hepatic copper pool is not re-distributed from the liver to other organs over time. We find that hepatic copper stores are cleared to basal levels after 72 hours (Figure S11).
In contrast, untargeted Cu(gtsm) ionophore treatment leads to a 90% increase in liver copper content at the 6-hour timepoint (Figure 3E), with substantial off-target copper delivery to organs including the intestines, kidney, heart, lungs, and brain. At 24 hours, there remains a statistically significant increase in liver, heart, and lung copper relative to basal levels (Figure S10). Though total hepatic copper levels remain substantially elevated at 24 hours for both Gal-Cu(gtsm) and Cu(gtsm), interestingly, we find that the bioluminescent CCL-1/D-luc liver signal returns to basal levels after 24 hours (Figure S8). These observations suggest a potential difference in labile copper status at 6 hours compared to 24 hours, where the delivered copper becomes more tightly sequestered by metal storage proteins at longer timepoints. Furthermore, we find that the administration of Gal-Cu(gtsm) or Cu(gtsm) offers minimal perturbation to iron and zinc levels across tissues relative to basal conditions (Figures S12, S13). These data indicate that appending a N-acetylgalactosamine moiety to the Cu(gtsm) framework can enable copper supplementation selectively to the liver in whole animal settings, likely through an ASGPR-mediated pathway.
Evaluating Toxicity Differences Between Targeted Gal-Cu(gtsm) and Untargeted Cu(gtsm) Supplements.
Despite the fact that Gal-Cu(gtsm) delivers ca. 4 times more hepatic copper than Cu(gtsm) at an equivalent dose, we find that Gal-Cu(gtsm) treatment is non-toxic while Cu(gtsm) treatment incurs significant hepatocellular injury as indicated by liver histology and toxicity assays (Figure 4). Indeed, hematoxylin & eosin (H&E) staining on liver tissue slices harvested from mice 6 hours after ionophore treatment at 0.75 mg Cu/kg mouse (Figure 4A) show that Gal-Cu(gtsm)-treated liver slices largely resemble those of the vehicle-treated control mice, whereas H&E histology reveals significant hepatocellular damage upon Cu(gtsm) supplementation, with features characteristic of hydropic degeneration that occurs upon acute liver injury (Figure 4A).76
Figure 4.
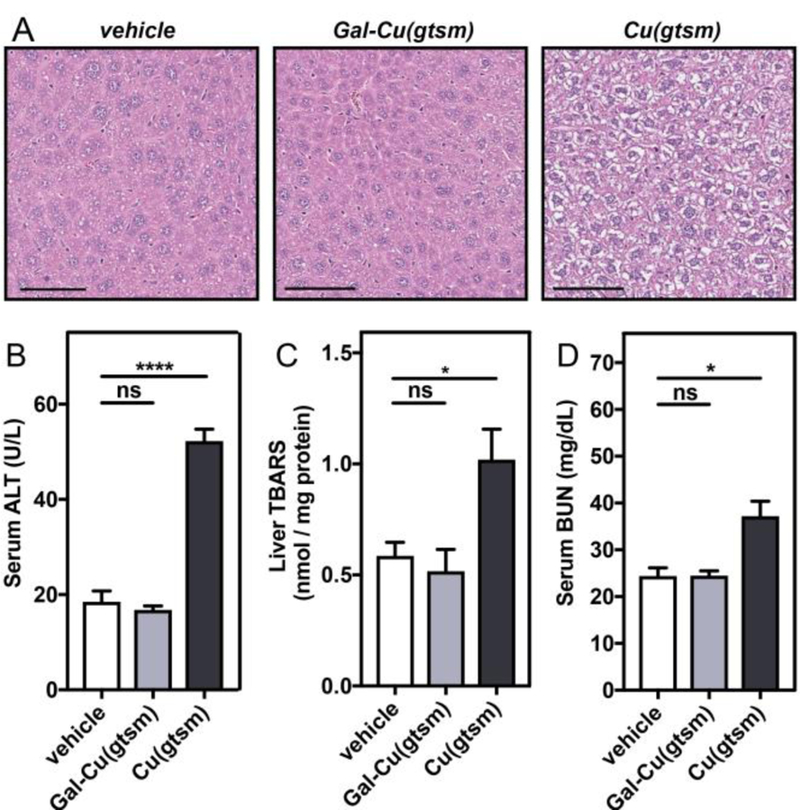
Gal-Cu(gtsm) treatment is non-toxic despite delivering more copper than Cu(gtsm). (A) Representative liver tissue slices from H&E staining show significant hydropic degeneration (wispy/white cytosolic areas surrounding nuclei) upon Cu(gtsm) treatment. Liver sections were isolated six hours following vehicle or 0.75 mg Cu/kg mouse ionophore i.p. injections. Scale bar = 100 µM. (B-D) Toxicity assays were performed on serum (B,D) or liver lysate (C) collected from mice treated with Cu(gtsm) or Gal-Cu(gtsm) at 0.75 mg Cu/kg mouse after 6 hours to evaluate liver (B,C) and kidney (D) toxicity. Error bars = SEM (n = 5). Statistical analyses were performed with a one-way ANOVA with Bonferroni’s multiple comparisons test where *P ≤ 0.05, ****P ≤ 0.0001.
To further assess potential ionophore toxicity, we also performed alanine transaminase (ALT) activity and liver thiobarbituric acid reactive substances (TBARS) assays on serum and liver lysate collected from mice treated with Cu(gtsm) or Gal-Cu(gtsm) at the same Cu dose after 6 hours (Figure 4B-C). Serum ALT activity assays are commonly used to evaluate hepatocellular injury and evaluate liver health, with increased serum ALT activity indicating potential liver damage. We find virtually the same serum ALT levels for Gal-Cu(gtsm) compared to the vehicle control, but much higher serum ALT levels after Cu(gtsm) treatment (Figure 4B). Likewise, the TBARS assay, which quantifies byproducts like malondialdehyde generated from lipid peroxidation during oxidative tissue damage, indicates there is significant oxidative stress-induced liver damage in Cu(gtsm)-treated mice compared to Gal-Cu(gtsm) or vehicle-treated mice (Figure 4C). In both assays, liver health recovers to some extent after 24 hours, but there are still significant elevations in ALT activity and TBARS levels relative to basal conditions (Figure S14).
Finally, we investigated off-target toxicity using blood urea nitrogen (BUN) assays, which are commonly elevated under conditions of kidney damage. Figure 4D shows elevated BUN levels for Cu(gtsm), but not for Gal-Cu(gtsm), treatment, which further supports the proposal that targeted copper delivery via Gal-Cu(gtsm) does not promote damage due to off-target copper accumulation. Indeed, previous studies by Cater et al. found significant kidney necrosis occurring upon long-term administration of Cu(gtsm).36 These studies indicate that targeted copper supplementation via Gal-Cu(gtsm) is non-toxic, whereas untargeted supplementation via Cu(gtsm) incurs hepatic and renal damage.
Selective Upregulation of Copper Trafficking and Storage Proteins as a Mechanism for Minimizing Copper-Induced Toxicity.
We speculate that the lack of toxicity resulting from copper supplementation using Gal-Cu(gtsm) compared to its generic non-targeted Cu(gtsm) counterpart originates from their disparate mechanisms for delivery, as hypothesized in Figure 1. In the case of Cu(gtsm) and other conventional copper ionophores, the metal complexes passively diffuse through the cell membrane and release copper upon encountering the reducing cytosolic medium.65,77 This process is unregulated, and the inability of the cell to buffer such rapid excesses of cytosolic copper likely promotes oxidative stress and damage via Fenton-like chemistry and other redox-mediated pathways. Meanwhile, it is well-established that ASGPR-mediated delivery occurs through clathrin-mediated endocytosis, where ASGPR-ligand recognition is followed by endolysosomal processing.47 In this case, copper release from Gal-Cu(gtsm) is then compartmentalized and localized to endosomes and lysosomes, in contrast to unregulated Cu+ release in the cytosol. Importantly, there is mounting evidence that the lysosome is a central organelle for copper storage and homeostasis from algae to mammals.44,45 Copper delivery via Gal-Cu(gtsm) may then conceivably occur with far greater control, where the lysosome can recruit copper trafficking proteins in a regulated manner based on homeostatic cues within the cell.44–46,78
To test this prediction, we performed Western blot analysis on mouse liver lysates to investigate how these different routes of copper supplementation may affect key players in copper storage and trafficking. Figure 5 shows SDS/PAGE analysis of liver lysates from mice treated with vehicle, Cu(gtsm), and Gal-Cu(gtsm) after 6 or 24 hours of treatment at equal protein loading. Expanded blots for all proteins are provided in Figure S15. We first blotted for ATP7B, the major copper export protein in the liver, and found increased ATP7B expression at both timepoints for Cu(gtsm) and Gal-Cu(gtsm). This observation is consistent with the known role of ATP7B to facilitate the removal of excess hepatic copper via biliary excretion.46,79 Next, we investigated the expression patterns of CTR1, a copper trafficking protein that imports copper into the cell from the extracellular matrix, and transports copper from the lysosome to the cytosol in concert with CTR2.45,78 We find increased expression of both the monomer and truncated forms of CTR1 at both 6 and 24 hour timepoints for Gal-Cu(gtsm) supplementation alone. Thiele and coworkers have previously suggested that the truncated CTR1 form may play an important role in copper transport from the lysosome to cytosol.78 Because we observe increased CTR1 expression only under conditions of endolysosomal Cu delivery, we hypothesize that CTR1 plays a role in copper transport from the lysosome to cytosol. Finally, metallothioneins (MTs) are key metal storage proteins in the cytosol. MT-I/MT-II expression increases dramatically upon Cu(gtsm) and Gal-Cu(gtsm) treatment after 6 hours and 24 hours. Blots for the copper chaperone for superoxide dismutase (CCS) did not demonstrate any clear differences between treatment conditions (Figure S15). Further studies are necessary to more conclusively disentangle the relationship between Gal-Cu(gtsm) uptake and lysosomal homeostatic copper regulation and will be the subject of future investigation.
Figure 5.
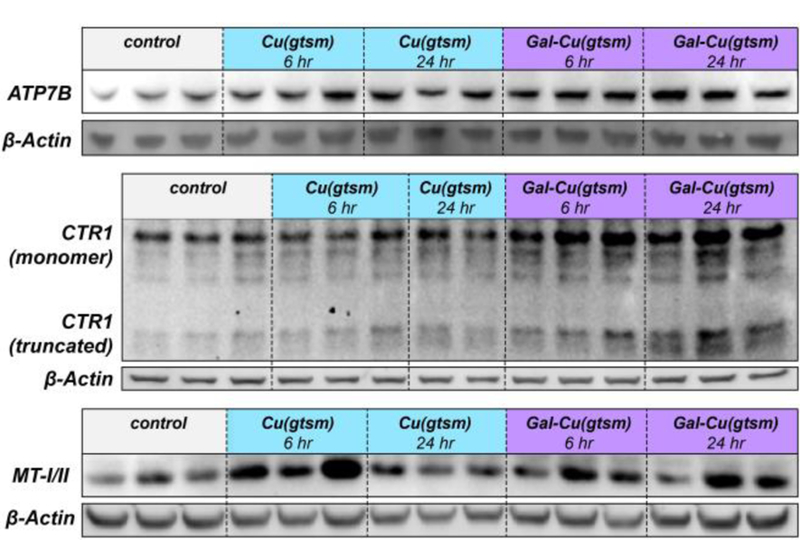
Western blots demonstrate how differences in copper delivery route affect copper storage and trafficking protein expression. SDS/PAGE analysis of liver extracts from mice (n = 2–3) sacrificed either 6 or 24 hours after treatment with vehicle, Cu(gtsm), or Gal-Cu(gtsm) (0.75 mg Cu/kg mouse). Tissues were probed for ATP7B, CTR1, and MT-I/II. For CTR1, the monomer shown is for the unglycosylated region (~25 kDa).
CONCLUDING REMARKS
In this report, we have introduced the concept of Targeted Ionophore-based Metal Supplementation (TIMS), a general strategy to deliver metals in a site-specific manner within living organisms. As an initial proof of concept demonstration of this approach, we synthesized a hepatic copper delivery agent and validated liver-directed copper supplementation in cell culture and mice through in vivo bioluminescence imaging and ex vivo tissue metal analysis via ICP-MS. We showed that the targeted Gal-Cu(gtsm) ionophore brings far more copper to the liver and minimal Cu to other organs compared to the generic untargeted Cu(gtsm) ionophore. Moreover, receptor-mediated metal supplementation proceeds in a non-toxic manner at the investigated copper dose relative to Cu(gtsm). This work establishes that the TIMS approach is a viable strategy for delivering metal nutrients in a site-specific manner with minimal off-target metal accumulation.
The modular nature of the targeted ionophore building blocks should enable the delivery of other metals and metal-based probes and therapeutics to other organs and sites of interests. We envisage that the TIMS strategy will facilitate both fundamental and therapeutic applications. The site-specific nature of metal delivery may enable us to uncover and elaborate upon the roles that metals play in cellular signaling and function in specific organs of interest at the animal level. As one possible example, Gal-Cu(gtsm) could be used as a tool for studying the complex relationships between copper and lipid signaling in the liver.25 Likewise, pairing copper ionophores with other tissue-specific targeting groups80 should further our understanding of the diverse roles that copper plays in different tissue and organ systems.
From a therapeutic perspective, TIMS offers a potential starting point to addressing metal deficiencies in a broad range of diseases with minimal off-target effects. Studies are currently underway in our laboratories to utilize Gal-Cu(gtsm) to study the potential roles and impact of liver-targeted copper supplementation in NAFLD and other metabolic disorders. Gal-Cu(gtsm) may also find potential use in treating hepatocellular carcinomas with minimal off-target effects, as cancer cells demonstrate heightened sensitivity to copper-induced toxicosis.77,81,82 Beyond the liver, one could use TIMS to address diseases in organs such as adipose tissue, heart, and brain. Indeed, our laboratory recently discovered that dynamic copper binding to PDE3B activates lipolysis in adipose tissue.12 As such, this TIMS strategy offers a general approach to open new areas for metals in medicine.
EXPERIMENTAL DETAILS
General Synthetic and Characterization Methods.
Reactions using air- or moisture-sensitive reagents were conducted in flame-dried glassware under an inert atmosphere of N2. When dry solvent was required, solvent was passed over activated alumina prior to use. All commercially purchased chemicals were used as received without further purification. 4,4-dimethyl-3-thiosemicarbazide was purchased from TCI America. All other chemicals and solvents purchased from Sigma Aldrich. Cu(gtsm)66, CCL-127, HO-TEG-N383, oxazoline 5,70 were synthesized according to previously reported procedures. Silica gel P60 (SiliCycle) was used for column chromatography and SiliCycle 60 F254 silica gel (pre-coated sheets, 0.25 mm thick) were used for analytical thin layer chromatography. 1H and 13C NMR spectra were collected at 298 K in deuterated solvents from Cambridge Isotope Laboratories (Cambridge, MA) at 25 °C on Bruker AV-300, AVQ-400, AVB-400, AV-500, DRX-500, or AV-600 instruments at the College of Chemistry NMR Facility at the University of California, Berkeley. Chemical shifts for protons are reported in parts per million downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CHCl3: δ 7.26; CH3OH δ 3.31, DMSO: δ 2.50). Chemical shifts for carbon are reported in parts per million downfield from tetramethylsilane and are referenced to the carbon resonances of the solvent (CDCl3 δ 77.16; DMSO δ 39.52). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd= doublet of doublets, t = triplet, m = multiplet), coupling constants in Hertz, and integration. Low-resolution electrospray mass spectral analyses were performed using a LC−MS (Advion Expression-L Compact MS, ESI source). High-resolution mass spectral analyses (ESI-MS) were carried out at the College of Chemistry Mass Spectrometry Facility at the University of California, Berkeley.
(E)-2-(2,2-Dimethoxyethylidene)-N-methylhydrazine-1-carbothio-amide (2).
Synthesis of 2 was adapted from previously reported procedures.69 4-Methyl-3-thiosemicarbazide (1.00 equiv, 10.35 g, 98.4 mmol) was added to a 1 L round bottom flask equipped with a stir bar followed by methanol (500 mL). 2,2-Dimethoxyacetaldehyde (60% wt. solution in water) (1.01 equiv, 15.0 mL, 99.43 mmol) was added via syringe, and the resulting solution was stirred overnight, after which a significant white precipitate developed. Volatiles were removed in vacuo to yield 2 as a white solid (18.9 g, quantitative yield). 1H NMR (400 MHz, CDCl3) δ 9.20 (s, 1H), 7.37 (s, 1H), 7.04 (d, J = 4.9 Hz, 1H), 4.80 (d, J = 4.9 Hz, 1H), 3.39 (s, 6H), 3.20 (d, J = 4.9 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 178.28, 140.78, 102.08, 53.44, 30.88. ESI-MS(+) calcd. for C6H13N3O2SNa [M+Na]+ m/z: 214.1; found 214.6.
(E)-N-Methyl-2-(2-oxoethylidene)hydrazine-1-carbothioamide (3).
Acetal 2 (1.00 equiv, 5.63 g, 29.4 mmol) and lithium tetrafluoroborate (2.00 equiv, 5.52 g, 58.8 mmol) was added to a 2 L round bottom flask equipped with a stir bar. Acetonitrile (750 mL) was added to the flask, followed by water (15 mL). The reaction mixture was stirred at room temperature for 6 hours, during which the reaction mixture transformed from a neon yellow to orange color. The reaction mixture was concentrated, then diluted with ethyl acetate (300 mL). The organic layer was washed with saturated NaHCO3 (50 mL), water (50 mL), and brine (50 mL), dried over Na2SO4, filtered, then concentrated in vacuo. The resulting brown solid was sonicated and extracted with hexane/ethyl acetate 1/1, v/v (400 mL) then filtered. The filtrate was concentrated in vacuo to obtain 3 as an orange powder (3.41 g, 81% yield). 1H NMR (600 MHz, DMSO-d6) δ 12.24 (s, 1H), 9.46 (d, J = 7.8 Hz, 1H), 9.00 (d, J = 5.7 Hz, 1H), 7.44 (dd, J = 7.8, 1.1 Hz, 1H), 3.01 (d, J = 4.6 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 191.30, 178.33, 138.58, 39.52, 31.17. ESI-MS could not be obtained.
(E)-N,N-Dimethyl-2-((E)-2-(2-(methylcarbamothioyl)hydrazineey-lidene)ethylidene)hydrazine-1 carbothioamide (4).
Compound 3 (1.00 equiv, 1.70 g, 11.71 mmol), 4,4-dimethyl-3-thiosemicarbazide (1.05 equiv, 1.47 g, 12.30 mmol), activated 4 Å MS, and DMF (50 mL) were added to a 100 mL round bottom flask equipped with a stir bar and condenser. The reaction mixture was stirred at 60˚C overnight under nitrogen to yield a dark brown solution. The reaction mixture was filtered to remove sieve dust and other particulates, then distilled to remove DMF. The resulting crude brown solid was sonicated and washed with ethanol (3 × 20 mL) and methanol (1 × 3 mL) to yield 4 as a beige powder (1.08 g, 38% yield). Subsequent filtrate precipitation at −20˚C yielded additional material albeit of reduced purity. 1H NMR (400 MHz, DMSO-d6) δ 11.67 (s, 1H), 11.18 (s, 1H), 8.42 (d, J = 4.8 Hz, 1H), 7.88 (d, J = 8.5 Hz, 1H), 7.74 (d, J = 8.5 Hz, 1H), 3.25 (s, 6H), 2.97 (d, J = 4.5 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 180.11, 177.54, 142.26, 140.66, 41.94, 39.52, 30.94. ESI-MS(–) calcd. for C7H13N6S2 [M-H]− m/z: 245.1; found 245.2.
(2R,3R,4R,5R,6R)-5-Acetamido-2-(acetoxymethyl)-6-(2-(2-(2-azid-oethoxy)ethoxy)ethoxy)tetrahydro-2H-pyran-3,4-diyldiacetate (6).
Compound 5 (1.00 equiv, 3.3 g, 10.02 mmol), HO-TEG-N3 (1.10 equiv, 1.93 g, 11.00 mmol), and DCE (60 mL) were added to a 200 mL round bottom flask followed by a scoop of activated 4 Å molecular sieves. After stirring for 5 minutes, trimethylsilyl trifluoromethanesulfonate (0.50 equiv, 0.91 mL, 5.01 mmol) was added via syringe to the reaction mixture and stirred overnight. The reaction mixture was filtered, and the filtrate was washed with a saturated sodium bicarbonate solution. The organic layer was dried with Na2SO4, filtered, concentrated in vacuo, then purified with silica gel chromatography (ethyl acetate to 9/1 ethyl acetate/methanol) to yield 6 as a pale orange oil (3.65 g, 72% yield). 1H NMR (600 MHz, CDCl3) δ 6.14 (d, J = 9.3 Hz, 1H), 5.32 (dd, J = 3.4, 1.1 Hz, 1H), 5.05 (dd, J = 11.2, 3.4 Hz, 1H), 4.78 (d, J = 8.6 Hz, 1H), 4.22 (dt, J = 11.2, 8.9 Hz, 1H), 4.19 – 4.10 (m, 2H), 3.93 – 3.83 (m, 3H), 3.76 – 3.65 (m, 4H), 3.63 (dd, J = 7.1, 3.2 Hz, 4H), 3.51 – 3.39 (m, 2H), 2.15 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H), 1.98 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 170.54, 170.42, 170.39, 170.29, 102.15, 71.56, 70.82, 70.64, 70.56, 70.32, 69.72, 68.51, 66.65, 61.50, 50.73, 50.47, 23.12, 20.68, 20.62, 20.61. ESI-MS(+) calcd. for C20H32N4NaO11 [M+Na]+ m/z: 527.2, found: 527.7.
N-((2R,3R,4R,5R,6R)-2-(2-(2-(2-Aminoethoxy)ethoxy)ethoxy)-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-3-yl)acetami-de (7).
Compound 6 (1.00 equiv, 1.36 g, 2.70 mmol) was added to a 100 mL round bottom flask equipped with a stir bar and dissolved in methanol (15 mL). Palladium (10%) on activated carbon (~50 mg) was slowly added to the flask, which was subsequently placed under vacuum and backfilled with hydrogen gas three times. The reaction mixture was stirred under a hydrogen gas balloon for 20 hours. The reaction mixture was filtered over Celite, which was then rinsed with methanol (40 mL). The filtrate was concentrated to yield 7 as a pale yellow semisolid (0.88 g, 92% yield), which was then carried forward to the next step. 1H NMR (500 MHz, CD3OD) δ 4.39 (d, J = 8.4 Hz, 1H), 4.01 – 3.87 (m, 2H), 3.83 (d, J = 3.4 Hz, 1H), 3.80 – 3.54 (m, 12H), 3.49 (t, J = 6.2 Hz, 1H), 2.91 (t, J = 5.3 Hz, 1H), 1.98 (s, 3H). 13C NMR (151 MHz, MeOD) δ 170.38, 103.55, 76.62, 73.00, 71.32, 71.21, 71.00, 70.81, 69.92, 69.64, 62.52, 53.94, 49.58, 23.16. ESI-MS(+) calcd. for C14H29N2O8 [M+H]+ m/z: 353.2, found: 353.4.
apo-Gal-H2gtsm.
Compound 4 (1.00 equiv, 0.56 g, 2.27 mmol), compound 7 (1.10 equiv, 0.88 g, 2.50 mmol), and acetonitrile (100 mL) were added to a 250 mL round bottom flask equipped with a reflux condenser and stir bar. The reaction mixture was refluxed at 82˚C overnight, in which the reaction mixture turned from a light orange to brown color. The reaction mixture was cooled to room temperature and Celite (10 g) was added to the flask, then concentrated in vacuo. The crude material loaded onto Celite was subjected to flash silica gel chromatography (100% DCM to 9/1 DCM/MeOH to 4/1 DCM/MeOH gradient) to afford apo-Gal-H2gtsm as a beige solid (436 mg, 32% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.80 (d, J = 18.1 Hz, 2H), 8.51 (d, J = 4.7 Hz, 1H), 8.43 (t, J = 5.8 Hz, 1H), 7.73 (s, 2H), 7.61 (d, J = 9.0 Hz, 1H), 4.62 – 4.55 (m, 3H), 4.50 (d, J = 4.3 Hz, 1H), 4.28 (d, J = 8.4 Hz, 1H), 3.78 (dt, J = 10.1, 4.2 Hz, 1H), 3.74 – 3.62 (m, 5H), 3.61 – 3.38 (m, 15H), 3.32 – 3.23 (m, 1H), 2.96 (d, J = 4.5 Hz, 3H), 1.80 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 177.57, 177.05, 169.63, 140.51, 140.00, 101.40, 75.36, 71.66, 69.81, 69.66, 68.30, 67.67, 67.60, 60.55, 52.03, 43.15, 39.52, 30.99, 23.17. ESI-MS(–) calcd. for C19H34N7O8S2 [M-H]− m/z: 552.2; found 552.4.
Gal-Cu(gtsm).
Compound 8 (1.00 equiv, 0.24 g, 0.43 mmol) and copper acetate (1.02 equiv, 87 mg, 0.44 mmol) were added to a 25 mL round bottom flask equipped with a stir bar. Anhydrous DMF (4.7 mL) was added, and the reaction mixture immediately adopted a crimson red color. The resulting solution was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo, and ethanol (2.5 mL) was added to precipitate a dark red solid, which was then frit filtered and rinsed with ethanol (2.5 mL) and diethyl ether (2X5 mL), giving Gal-Cu(gtsm) as a dark red powder (192 mg, 73% yield). High-resolution ESI-MS(+) calcd. for C19H34CuN7O8S2 [M+H]+ m/z: 615.1201, found: 615.1202.
Cell Culture Procedures.
Cells were maintained by the UC Berkeley Tissue Culture Facility. HEK 293T and HepG2 cells were maintained as a monolayer in exponential growth at 37°C in a 5% CO2 atmosphere. HEK 293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Hyclone), and glutamax (Gibco). HepG2 cells were maintained in low glucose DMEM with L-glutamine and sodium pyruvate (Gibco). One day before ionophore treatment, cells were passaged and plated in DMEM with glutamax supplemented with 10% FBS on either poly D-lysine-coated (HEK 293T) or gelatin-coated (HepG2) sterile 6-well Corning polystyrene plates. Cells were grown to 60 to 80% confluency prior to ionophore treatment.
Cellular Ionophore Treatment and ICP-MS Assays.
Cells were washed twice with serum-free DMEM. 2 mM stock solutions of Cu(gtsm) or Gal-Cu(gtsm) were diluted in serum-free DMEM to a final ionophore concentration of 4 µM. 2 mL of vehicle (0.2% DMSO in DMEM), 4 µM Cu(gtsm), or 4 µM Gal-Cu(gtsm) were then added to the 6-well plates and incubated for three hours. For the galactose competition experiment, cells were washed twice with serum-free DMEM, then incubated with 2 mL serum-free DMEM either with or without 1 M D-galactose (Sigma Aldrich) for 15 minutes. 4 µL of DMSO, 2 mM Cu(gtsm), or 10 mM Gal-Cu(gtsm) was diluted with 300 µL of cell media, which was then mixed back into each well. Cells were then incubated for one hour. Cells were then rinsed twice with ice-cold EDTA (1 mM in 50 mM HEPES buffer, pH = 7.4) to remove cell surface-bound copper and rinsed twice with ice-cold 50 mM HEPES buffer (pH = 7.4), followed by the addition of 215 µL concentrated nitric acid (BDH Aristar Ultra). The plates were sealed with Parafilm and incubated on a shaker overnight. Samples (150 µL) were further diluted in 2 mL 2% nitric acid (made freshly from concentrated nitric acid and Milli-Q water) in 15 mL tubes (Sarstedt) and analyzed on a Thermo Fisher iCAP Qc ICP mass spectrometer in kinetic energy discrimination (KED) mode against a standard curve of known copper and phosphorus concentrations (CMS-5, Inorganic Ventures), with Ga (20 µg/L, Inorganic Ventures) as an internal standard. Each experiment was carried out twice and each condition was repeated in at least triplicate.
Animals.
FVB-luc+ (FVB-Tg (CAG-luc,-GFP)L2G85Chco/J) mice were obtained from our in-house breeding colony. Mice were group housed on a 12:12 hour light-dark cycle at 22 °C with free access to food and water. All animal studies were approved by and performed according to the guidelines of the Animal Care and Use Committee of the University of California, Berkeley.
General Animal Imaging Methods and Data Analysis.
A Xenogen IVIS Spectrum instrument (Caliper Life Sciences) was used for bioluminescence imaging in all animal experiments, and image analysis was performed using the Living Image software. The total photon flux for each animal was determined by drawing a region of interest around the liver and integrating photon flux over the total imaging period (area under the curve). We selected our liver region of interest based on comparing the bioluminescence data in vivo and ex vivo, as described in our previous studies with the CCL-1 probe.27 The same region of interest around the liver was applied to analyzing both the CCL-1 and D-Luc data. The data plotted in Figure 3A represents the ratio of CCL-1 integrated photon flux to basal D-luc integrated photon flux. Mice were anesthetized prior to injection and during imaging via inhalation of isoflurane. Isoflurane was purchased from Phoenix Pharmaceuticals, Inc. DMSO was purchased from Sigma-Aldrich, Dulbecco’s phosphate buffered saline was purchased from Gibco, and medical-grade oxygen was purchased from Praxair.
In Vivo Imaging with CCL-1.
FVB-luc+ mice were given intraperitoneal (i.p.) injections of vehicle (50 µL 1:1 DMSO:DPBS), 3.46 mg/kg Cu(gtsm), or 7.26 mg/kg Gal-Cu(gtsm) under anesthesia with isoflurane. The ionophore concentrations were chosen to give an equivalent copper dose of 0.75 mg Cu/kg mouse at 0.6 mg Cu/mL vehicle. Six hours later, the same mice were anesthetized and subjected to scapular subcutaneous (s.c.) injection of CCL-1 (0.1 µmol in 50 µL DMSO/150 µL DPBS) or D-luciferin (0.1 µmol in 50 µL DMSO/150 µL DPBS). Five minutes after s.c. injection, mice were transferred to a Xenogen IVIS Spectrum and imaged for 40 min under 2% isoflurane anesthesia to characterize ionophore-treated liver signal response.
Tissue Harvesting and Serum Isolation.
FVB-luc+ mice were heavily anesthetized and blood was collected via cardiac puncture. Mice were immediately euthanized by cervical dislocation. Tissues were harvested, rinsed twice with DPBS, snap-frozen under liquid nitrogen, and placed on dry ice in cryotubes and stored at −80ºC until analysis. Serum was isolated by allowing blood samples to coagulate for 1 hour at room temperature, centrifuging at 1,500 g for 15 minutes at 4˚C, then collecting the serum supernatant. Samples were aliquoted, snap-frozen, and stored at −80˚C until analysis.
Tissue Copper Analysis with ICP-MS.
20–100 mg portions of the harvested tissues were digested in concentrated nitric acid (100 mg tissue/mL HNO3, BDH Aristar Ultra) at 95 ºC for 2 h in 1.5 mL tubes (Sarstedt) with small holes poked in the caps with an 18G needle. After overnight incubation at room temperature, samples were diluted into freshly prepared 2% nitric acid and doped with a gallium internal standard (Inorganic Ventures, diluted from 1 ppm in 2% nitric acid to a 20 ppb final concentration). The copper content was determined by measuring 63Cu using a Thermo Fisher iCAP-Qc ICP-MS in Kinetic Energy Discrimination (KED) mode. Measurements were normalized to a standard curve of known copper concentrations doped with 20 ppb Ga. The standard curve was diluted from CMS-5 (Inorganic Ventures).
Liver Tissue Histology.
FVB-luc+ mice were injected i.p. with vehicle (50 µL 1:1 DMSO:DPBS), Cu(gtsm), or Gal-Cu(gtsm) at a 0.75 mg Cu/kg mouse dose. After six hours, mice were euthanized with CO2 asphyxiation followed by cervical dislocation and liver tissue was extracted. Liver sections were fixed in a 10% formalin in PBS solution and sent to Histowiz Inc. (Brooklyn, NY) for further processing and hematoxylin & eosin staining.
Liver Tissue Lysis.
Frozen mouse livers were minced into 50 mg samples on dry ice and homogenized in ice-cold RIPA buffer (25 mM Tris•HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitor cocktail with EDTA (Roche) at 100 mg/mL using a hand-held mechanical homogenizer. Homogenates were incubated on ice for 30 min and centrifuged at 12,000 × g for 20 min at 4 ºC. The soluble protein lysates were collected from underneath the upper lipid layer with a pipette and transferred to new tubes. Protein concentration was determined using a detergent-compatible Bradford Assay (Pierce).
Toxicity Assays of Liver Lysate and Serum.
Liver lysate (TBARS Assay via TCA Method, Cayman Chemical) and serum (BUN Assay, Invitrogen; ALT Assay, Cayman Chemical) samples were processed according to manufacturer instructions; sample concentration was selected based on initial dilution screening to fall within linear range of the standard curve. All samples were analyzed in triplicate.
Western Blot Analysis.
Protein lysates were denatured in NuPAGE lithium dodecyl sulfate sample buffer (Invitrogen) containing 10% v/v ß-mercaptoethanol as a reducing agent. The samples (20 µg for CCS, CTR1, ATP7B; 50 µg for MT) were resolved by SDS-PAGE using 15-well or 17-well NuPAGE 4–12% Bis-Tris gels (Invitrogen) with MES SDS running buffer (Invitrogen) with 10 µL of loading sample. Proteins were transferred to a polyvinylidene difluoride membrane (BioRad, Munich, Germany) with the use of the Trans-Blot Turbo transfer system (BioRad, Munich, Germany)). The membranes were blocked in 5% non-fat dry milk in TBST buffer (10 mM Tris, pH 7.5, 100 mM NaCl, 0.1% Tween-20) for 1 hour at room temperature. After blocking, the membranes were incubated at 4 ºC overnight with primary antibodies diluted with TBST buffer containing 5% bovine serum albumin (BSA). The anti-ATP7B (NB100–360, Novus Biologics) and anti-metallothionein (sc-11377, Santa Cruz Biotechnology) were used at 1:250 dilution. The anti-CTR1 antibody (13086, Cell Signaling Technology) was used at 1:1000 dilution. The anti-CCS (sc-20141, Santa Cruz Biotechnology) was used at 1:500 dilution. The membranes were washed 3 times for 5 minutes in TBST and incubated for 1 hour at room temperature with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG secondary antibody (sc-2004, Santa Cruz Biotechnology) at a 1:2000 dilution in TBST containing 5% BSA. The membranes were washed 5 times for 5 minutes in TBST, then visualized using enhanced chemiluminescence (Western Lighting Plus for visualizing CCS and ATP7B, Perkin Elmer; Western Clarity Max for visualizing CTR1 and MT, Bio-Rad) recorded on a BioRad GelDoc imaging station. ß-actin was probed to determine equal loading using anti-ß-actin (sc-69879, Santa Cruz Biotechnology) and AlexaFluor 647-conjugated anti-mouse IgG (A31571, Molecular Probes) antibodies at 1:5000 and 1:2500 dilutions, respectively, with visualization using fluorescence recorded on a BioRad GelDoc imaging station.
Supplementary Material
ACKNOWLEDGMENT
We thank Alison Killilea and Carissa Tasto at UC Berkeley’s Cell Culture Facility for expert technical assistance. We thank National Institutes of Health (NIH) (Grants GM79465 ES4705 to C.J.C. and Grant R01DK101293 to A.S.) for support. T.A.S. thanks the NIH Ruth L. Kirschstein National Research Service Award (Grant F32 GM122248) for support and Dr. Lakshmi Krishnamoorthy for helpful discussions. W.C. was supported by the UC Berkeley Amgen Scholars Program. M.C.H. was supported by the University of California President’s Postdoctoral Program. T.D. was partially supported by NIH Chemical Biology Training Grant T32 GM066698. C.J.C. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional experimental details for log P determination, spectroscopy, dynamic light scattering, and confocal microscopy imaging experiments. Additional figures in-cluding representative bioluminescence images of iono-phore-treated mice imaged with CCL-1 or D-luc, ICP-MS analysis of ionophore stock solution, ICP-MS analysis of tissues from ionophore-treated mice 24 hours after ionophore injection, ICP-MS study of hepatic copper clearance, ICP-MS assays of iron and zinc content across tissues upon copper ionophore supplementation, toxicity assays at 24 hour timepoint, expanded Western blots, NMR and LC-MS spectra (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).Lippard SJ; Berg JM Principles of Bioinorganic Chemistry; Principles of Bioinorganic Chemistry; University Science Books: Mill Valley, CA, 1994. [Google Scholar]
- (2).Ferguson-Miller S; Babcock GT Chem. Rev 1996, 96, 2889. [DOI] [PubMed] [Google Scholar]
- (3).McCord JM; Fridovich IJ Biol. Chem 1969, 244, 6049. [PubMed] [Google Scholar]
- (4).Hart PJ; Balbirnie MM; Ogihara NL; Nersissian AM; Weiss MS; Valentine JS; Eisenberg D Biochemistry 1999, 38, 2167. [DOI] [PubMed] [Google Scholar]
- (5).Prigge ST; Mains RE; Eipper BA; Amzel LM Cell. Mol. Life Sci. C 2000, 57, 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Friedman S; Kaufman SJ Biol. Chem 1966, 241, 2256. [PubMed] [Google Scholar]
- (7).Solomons NW J. Am. Coll. Nutr 1985, 4, 83. [DOI] [PubMed] [Google Scholar]
- (8).Bost M; Houdart S; Oberli M; Kalonji E; Huneau JF; Margaritis IJ Trace Elem. Med. Biol 2016, 35, 107. [DOI] [PubMed] [Google Scholar]
- (9).Brady DC; Crowe MS; Turski ML; Hobbs GA; Yao X; Chaikuad A; Knapp S; Xiao K; Campbell SL; Thiele DJ; Counter CM Nature 2014, 509, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Turski ML; Brady DC; Kim HJ; Kim B-E; Nose Y; Counter CM; Winge DR; Thiele DJ Mol. Cell. Biol 2012, 32, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dodani SC; Firl A; Chan J; Nam CI; Aron AT; Onak CS; Ramos-Torres KM; Paek J; Webster CM; Feller MB; Chang CJ Proc. Natl. Acad. Sci 2014, 111, 16280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Krishnamoorthy L; Cotruvo JA; Chan J; Kaluarachchi H; Muchenditsi A; Pendyala VS; Jia S; Aron AT; Ackerman CM; Wal MN Vander; Guan T; Smaga LP; Farhi SL; New EJ; Lutsenko S; Chang CJ Nat. Chem. Biol 2016, 12, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vulpe C; Levinson B; Whitney S; Packman S; Gitschier J Nat. Genet 1993, 3, 7. [DOI] [PubMed] [Google Scholar]
- (14).Roberts BR; Tainer JA; Getzoff ED; Malencik DA; Anderson SR; Bomben VC; Meyers KR; Karplus PA; Beckman JS J. Mol. Biol 2007, 373, 877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).McAllum EJMC; Lim NK; Hickey JL; Paterson BM; Donnelly PS; Li Q; Liddell JR; Barnham KJ; White AR; Crouch PJ Amyotroph. Lateral Scler. Front. Degener 2013, 14, 586. [DOI] [PubMed] [Google Scholar]
- (16).Bush AI J. Alzheimer’s Dis 2008, 15, 223. [DOI] [PubMed] [Google Scholar]
- (17).Hayne DJ; Lim S; Donnelly PS Chem. Soc. Rev 2014, 43, 6701. [DOI] [PubMed] [Google Scholar]
- (18).Perez LR; Franz KJ Dalton Trans 2010, 39, 2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Elsherif L; Ortines RV; Saari JT; Kang Y J. Exp. Biol. Med 2003, 228, 811. [DOI] [PubMed] [Google Scholar]
- (20).Klevay LM Biol. Trace Elem. Res 1983, 5, 245. [DOI] [PubMed] [Google Scholar]
- (21).Stättermayer AF; Traussnigg S; Aigner E; Kienbacher C; Huber-Schönauer U; Steindl-Munda P; Stadlmayr A; Wrba F; Trauner M; Datz C; Ferenci PJ Trace Elem. Med. Biol 2017, 39, 100. [DOI] [PubMed] [Google Scholar]
- (22).Aigner E; Strasser M; Haufe H; Sonnweber T; Hohla F; Stadlmayr A; Solioz M; Tilg H; Patsch W; Weiss G; Stickel F; Datz C Am. J. Gastroenterol 2010, 105, 1978. [DOI] [PubMed] [Google Scholar]
- (23).Aigner E; Weiss G; Datz C World J. Hepatol 2014, 7, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huster D; Purnat TD; Burkhead JL; Ralle M; Fiehn O; Stuckert F; Olson NE; Teupser D; Lutsenko SJ Biol. Chem 2007, 282, 8343. [DOI] [PubMed] [Google Scholar]
- (25).Burkhead JL; Lutsenko S Lipid Metab 2013, 39.
- (26).Morrell A; Tallino S; Yu L; Burkhead JL IUBMB Life 2017, 69, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Heffern MC; Park HM; Au-Yeung HY; Van de Bittner GC; Ackerman CM; Stahl A; Chang CJ Proc. Natl. Acad. Sci 2016, 113, 14219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Weekley CM; He C Curr. Opin. Chem. Biol 2017, 37, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Helsel ME; Franz KJ Dalt. Trans 2015, 44, 8760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sarkar B; Lingertat-Walsh K; Clarke JT R. J. Pediatr 1993, 123, 828. [DOI] [PubMed] [Google Scholar]
- (31).Crouch PJ; Hung LW; Adlard P. a; Cortes M; Lal V; Filiz G; Perez KA; Nurjono M; Caragounis A; Du T; Laughton K; Volitakis I; Bush AI; Li Q-X; Masters CL; Cappai R; Cherny RA; Donnelly PS; White AR; Barnham KJ Proc. Natl. Acad. Sci 2009, 106, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Donnelly PS; Liddell JR; Lim S; Paterson BM; Cater MA; Savva MS; Mot AI; James JL; Trounce IA; White AR; Crouch PJ Proc. Natl. Acad. Sci. U. S. A 2012, 109, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Paterson BM; Donnelly PS Chem. Soc. Rev 2011, 40, 3005. [DOI] [PubMed] [Google Scholar]
- (34).Cherny RA; Atwood CS; Xilinas ME; Gray DN; Jones WD; McLean CA; Barnham KJ; Volitakis I; Fraser FW; Kim YS; Huang X; Goldstein LE; Moir RD; Lim JT; Beyreuther K; Zheng H; Tanzi RE; Masters CL; Bush AI Neuron 2001, 30, 665. [DOI] [PubMed] [Google Scholar]
- (35).Halliwell B; Gutteridge JM Biochem. J 1984, 219, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cater MA; Pearson HB; Wolyniec K; Klaver P; Bilandzic M; Paterson BM; Bush AI; Humbert PO; La Fontaine S; Donnelly PS; Haupt Y ACS Chem. Biol 2013, 8, 1621. [DOI] [PubMed] [Google Scholar]
- (37).Shimada K; Reznik E; Stokes ME; Krishnamoorthy L; Bos PH; Song Y; Quartararo CE; Pagano NC; Carpizo DR; deCarvalho AC; Lo DC; Stockwell BR Cell Chem. Biol 2018, 25, 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).John EK; Green MA J. Med. Chem 1990, 33, 1764. [DOI] [PubMed] [Google Scholar]
- (39).Torres JB; Andreozzi EM; Dunn JT; Siddique M; Szanda I; Howlett DR; Sunassee K; Blower PJ J. Nucl. Med 2016, 57, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Younossi Z; Anstee QM; Marietti M; Hardy T; Henry L; Eslam M; George J; Bugianesi E Nat. Rev. Gastroenterol. Hepatol 2018, 15, 11. [DOI] [PubMed] [Google Scholar]
- (41).Byrne CD; Targher GJ Hepatol 2015, 62, S47. [DOI] [PubMed] [Google Scholar]
- (42).Leung TM; Nieto NJ Hepatol 2013, 58, 395. [DOI] [PubMed] [Google Scholar]
- (43).Wu X; Zhang L; Gurley E; Studer E; Shang J; Wang T; Wang C; Yan M; Jiang Z; Hylemon PB; Sanyal AJ; Pandak WM; Zhou H Hepatology 2008, 47, 1905. [DOI] [PubMed] [Google Scholar]
- (44).Blaby-Haas CE; Merchant SS J. Biol. Chem 2014, 289, 28129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Polishchuk EV; Polishchuk RS Metallomics 2016, 8, 853. [DOI] [PubMed] [Google Scholar]
- (46).Polishchuk EV; Concilli M; Iacobacci S; Chesi G; Pastore N; Piccolo P; Paladino S; Baldantoni D; Van Ijzendoorn SCD; Chan J; Chang CJ; Amoresano A; Pane F; Pucci P; Tarallo A; Parenti G; Brunetti-Pierri N; Settembre C; Ballabio A; Polishchuk RS Dev. Cell 2014, 29, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).D’Souza AA; Devarajan PV J. Control. Release 2015, 203, 126. [DOI] [PubMed] [Google Scholar]
- (48).Mamidyala SK; Dutta S; Chrunyk BA; Préville C; Wang H; Withka JM; McColl A; Subashi TA; Hawrylik SJ; Griffor MC; Kim S; Pfefferkorn JA; Price DA; Menhaji-Klotz E; Mascitti V; Finn MG J. Am. Chem. Soc 2012, 134, 1978. [DOI] [PubMed] [Google Scholar]
- (49).Julyan PJ; Seymour LW; Ferry DR; Daryani S; Boivin CM; Doran J; David M; Anderson D; Christodoulou C; Young AM; Hesslewood S; Kerr DJ J. Control. Release 1999, 57, 281. [DOI] [PubMed] [Google Scholar]
- (50).Rensen PCN; Van Leeuwen SH; Sliedregt LAJM; Van Berkel TJC; Biessen EA L. J. Med. Chem 2004, 47, 5798. [DOI] [PubMed] [Google Scholar]
- (51).Lee K; Rafi M; Wang X; Aran K; Feng X; Lo Sterzo C; Tang R; Lingampalli N; Kim HJ; Murthy N Nat. Mater 2015, 14, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Nair JK; Willoughby JLS; Chan A; Charisse K; Alam MR; Wang Q; Hoekstra M; Kandasamy P; Kel’in AV; Milstein S; Taneja N; O’Shea J; Shaikh S; Zhang L; van der Sluis RJ; Jung ME; Akinc A; Hutabarat R; Kuchimanchi S; Fitzgerald K; Zimmermann T; van Berkel TJC; Maier MA; Rajeev KG; Manoharan MJ Am. Chem. Soc 2014, 136, 16958. [DOI] [PubMed] [Google Scholar]
- (53).Rozema DB; Lewis DL; Wakefield DH; Wong SC; Klein JJ; Roesch PL; Bertin SL; Reppen TW; Chu Q; Blokhin AV; Hagstrom JE; Wolff JA Proc. Natl. Acad. Sci 2007, 104, 12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rouet R; Thuma BA; Roy MD; Lintner NG; Rubitski DM; Finley JE; Wisniewska HM; Mendonsa R; Hirsh A; de Oñate L; Compte Barrón J; McLellan TJ; Bellenger J; Feng X; Varghese A; Chrunyk BA; Borzilleri K; Hesp KD; Zhou K; Ma N; Tu M; Dullea R; McClure KF; Wilson RC; Liras S; Mascitti V; Doudna JA J. Am. Chem. Soc 2018, 140, 6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Pujol AM; Cuillel M; Renaudet O; Lebrun C; Charbonnier P; Cassio D; Gateau C; Dumy P; Mintz E; Delangle PJ Am. Chem. Soc 2011, 133, 286. [DOI] [PubMed] [Google Scholar]
- (56).Pujol AM; Cuillel M; Jullien AS; Lebrun C; Cassio D; Mintz E; Gateau C; Delangle P Angew. Chemie - Int. Ed 2012, 51, 7445. [DOI] [PubMed] [Google Scholar]
- (57).Stokmaier D; Khorev O; Cutting B; Born R; Ricklin D; Ernst TOG; Böni F; Schwingruber K; Gentner M; Wittwer M; Spreafico M; Vedani A; Rabbani S; Schwardt O; Ernst B Bioorganic Med. Chem 2009, 17, 7254. [DOI] [PubMed] [Google Scholar]
- (58).Choi JS; Braymer JJ; Nanga RPR; Ramamoorthy A; Lim MH Proc. Natl. Acad. Sci 2010, 107, 21990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Sharma AK; Pavlova ST; Kim J; Finkelstein D; Hawco NJ; Rath NP; Kim J; Mirica LM J. Am. Chem. Soc 2012, 134, 6625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Haas KL; Franz KJ Chem. Rev 2009, 109, 4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Bica L; Liddell JR; Donnelly PS; Duncan C; Caragounis A; Volitakis I; Paterson BM; Cappai R; Grubman A; Camakaris J; Crouch PJ; White AR PLoS One 2014, 9. [DOI] [PMC free article] [PubMed]
- (62).Crouch PJ; Barnham KJ Acc. Chem. Res 2012, 45, 1604. [DOI] [PubMed] [Google Scholar]
- (63).Palanimuthu D; Shinde SV; Somasundaram K; Samuelson AG J. Med. Chem 2013, 56, 722. [DOI] [PubMed] [Google Scholar]
- (64).Xiao Z; Donnelly PS; Zimmermann M; Wedd AG Inorg. Chem 2008, 47, 4338. [DOI] [PubMed] [Google Scholar]
- (65).Price KA; Crouch PJ; Volitakis I; Paterson BM; Lim S; Donnelly PS; White AR Inorg. Chem 2011, 50, 9594. [DOI] [PubMed] [Google Scholar]
- (66).Dearling JLJ; Lewis JS; Mullen GED; Welch MJ; Blower PJ J. Biol. Inorg. Chem 2002, 7, 249. [DOI] [PubMed] [Google Scholar]
- (67).Klayman DL; Lin AJ Org. Prep. Proced. Int 1984, 16, 79. [Google Scholar]
- (68).Brett MP; John AK; Denis BS; Jonathan MW; Donnelly PS Inorg. Chem 2010, 49, 1884.20055473 [Google Scholar]
- (69).Barnham KJ; Donnelly PS; White AR Metal delivery agents and therapeutic uses of the same PCT/AU2007/001792.
- (70).Wang W; Jin C; Guo L; Liu Y; Wan Y; Wang X; Li L; Zhao W; Wang PG Chem. Commun 2011, 47, 11240. [DOI] [PubMed] [Google Scholar]
- (71).Andrés A; Rosés M; Ràfols C; Bosch E; Espinosa S; Segarra V; Huerta JM Eur. J. Pharm. Sci 2015, 76, 181. [DOI] [PubMed] [Google Scholar]
- (72).Carter KP; Young AM; Palmer AE Chem. Rev 2014, 114, 4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Xiao T; Ackerman CM; Carroll EC; Jia S; Hoagland A; Chan J; Thai B; Liu CS; Isacoff EY; Chang CJ Nat. Chem. Biol 2018, 14, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Bremer PT; Pellett S; Carolan JP; Tepp WH; Eubanks LM; Allen KN; Johnson EA; Janda KD J. Am. Chem. Soc 2017, 139, 7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Li Y; Huang G; Diakur J; Wiebe LI Curr. Drug Deliv 2008, 5, 299. [DOI] [PubMed] [Google Scholar]
- (76).Zachary JF Pathologic Basis of Veterinary Disease; 6th ed.; Elsevier: St Louis, MO., 2017. [Google Scholar]
- (77).Tardito S; Bassanetti I; Bignardi C; Elviri L; Tegoni M; Mucchino C; Bussolati O; Franchi-Gazzola R; Marchiò L J. Am. Chem. Soc 2011, 133, 6235. [DOI] [PubMed] [Google Scholar]
- (78).Ohrvik H; Nose Y; Wood LK; Kim B-E; Gleber S-C; Ralle M; Thiele DJ Proc. Natl. Acad. Sci 2013, 110, E4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Harada M; Sakisaka S; Terada K; Kimura R; Kawaguchi T; Koga H; Taniguchi E; Sasatomi K; Miura N; Suganuma T; Fujita H; Furuta K; Tanikawa K; Sugiyama T; Sata M Gastroenterology 2000, 118, 921. [DOI] [PubMed] [Google Scholar]
- (80).Alsaggar M; Liu DJ Drug Target 2018, 26, 385. [DOI] [PubMed] [Google Scholar]
- (81).Denoyer D; Pearson HB; Clatworthy SAS; Smith ZM; Francis PS; Llanos RM; Volitakis I; Phillips WA; Meggyesy PM; Masaldan S; Cater MA Oncotarget 2016, 7, 37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Skrott Z; Mistrik M; Andersen KK; Friis S; Majera D; Gursky J; Ozdian T; Bartkova J; Turi Z; Moudry P; Kraus M; Michalova M; Vaclavkova J; Dzubak P; Vrobel I; Pouckova P; Sedlacek J; Miklovicova A; Kutt A; Li J; Mattova J; Driessen C; Dou QP; Olsen J; Hajduch M; Cvek B; Deshaies RJ; Bartek J Nature 2017, 552, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Brun MA; Tan KT; Griss R; Kielkowska A; Reymond L; Johnsson KJ Am. Chem. Soc 2012, 134, 7676. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.