

Abstract
Although shotgun metagenomic sequencing of microbiome samples enables partial reconstruction of the strain-level community structure, it remains difficult to obtain high-quality microbial genome drafts without isolation and culture. Here we present a novel application of read clouds, short read sequences tagged with long-range information, to microbiome samples. We present Athena, a de novo assembler that uses read clouds to improve metagenomic assemblies. We apply this approach to sequence stool samples from two healthy individuals, and compare it to existing short-read and synthetic long-read metagenomic sequencing techniques. Read cloud metagenomic sequencing and Athena assembly produce the most complete individual genome drafts with high contiguity (>200 kbp N50, <10 contigs), even for bacteria that have relatively low (20x) raw short-read sequence coverage. We also sequence a complex marine sediment sample and generate 24 intermediate-quality genome drafts (>70% complete, <10% contaminated), nine of which are complete (>90% complete, <5% contaminated). Thus, our approach allows culture-free generation of high-quality microbial genome drafts using a single shotgun experiment.
Introduction
Short-read sequencing and assembly have played an instrumental role in advancing the study of microbial genomes beyond the minority of organisms that have been isolated and cultured1. This has greatly expanded our understanding of the genomic structure and dynamics of complex microbial communities that range from the human microbiome2–4 to environmental communities in the ocean, soil, and beyond5–8. However, the precise gene coding potential and consequent functional capabilities of organisms within these complex systems remains poorly understood.
Despite large-scale sequencing efforts of cultured isolates, analysis of sequences from diverse environmental samples has revealed that major novel taxonomic lineages are entirely unrepresented in current reference collections9,10, such as Refseq11. For example, even prevalent clades within heavily sequenced niches, such as Clostridiales and Bacteroides within the human gut, do not currently have a collection of isolate reference genomes that represent organisms observed in metagenomic shotgun sequencing4. Thus methods that accelerate the generation of high-quality genome drafts from shotgun sequencing of microbiome samples are needed.
Metagenomic shotgun sequencing, with the aid of specialized computational techniques, has also been used to generate draft genomes for individual taxa without the use of culture. The computational techniques developed include dedicated metagenomic assemblers12–14, and metagenome draft binning based on sequence similarity15,16 and coverage depth covariance17–19. Binning techniques can group assembled sequences into more comprehensive drafts, but these techniques often fail to properly assign sequences that are shared between multiple bacterial strains. Furthermore, sequencing reads produced by existing high throughput methods (typically 100–250 base pairs) are too short to span many types of shared or duplicated sequences, and as a result, regions containing these types of sequences remain unassembled.
In principle, long-read sequencing approaches can be used to address these issues. Long-read platforms such as Pacific Biosciences’ Single Molecule Real Time sequencing approach have been successfully applied to close genomes of cultured isolates20–22 and dominant organisms within more complex mixtures23. However, these single molecule platforms have lower throughput and a higher error rate in comparison to short reads. These single molecule platforms also typically require higher input DNA mass (~100ng), which prevents their application to biological samples containing insufficient high-molecular-weight DNA.
Synthetic long read (SLR) approaches, such as Illumina Truseq Synthetic Long Reads24, use long fragment partitioning and short-read barcoding to obtain virtual long read sequences, which can in theory be used to improve metagenomic assembly. Deep sequencing applied to a healthy human stool sample using this SLR approach has allowed assembly of more contiguous genome sequences from a subset of constituent bacteria25. However, SLR sequencing applied to more complex environmental samples, such as soil, has not yet resulted in improved genome assemblies26,27. This is most likely due to both the higher species richness of these samples and the limited overall throughput of the SLR approach.
A recent method, introduced by 10x Genomics, streamlines the short-read barcoding process by using more than a million droplet partitions to yield uniquely barcoded short-read fragments from one or a few long molecules trapped in each droplet partition28. Sequencing of libraries generated by this method yields shallow-coverage groups of barcode-sharing reads, which we will refer to as read clouds29 (also referred to as linked-reads28). Though both read cloud and SLR approaches use long fragment partitioning, read clouds trade off shallower short-read coverage of each individual long fragment for a larger total number of long fragments sequenced (Supplementary Note 1). This method and similar ones predating it have demonstrated utility for this approach in reference-based human haplotype phasing28,30–33, and also in resolving complex structural variations in human genomes34. To date, their potential for de novo metagenomic sequence assembly has yet to be explored.
Here we apply read clouds, generated by the 10x Genomics Chromium method, to sequence human and marine microbiome samples. We also introduce an assembler, Athena, that uses the barcode information from read clouds to produce high-quality genome drafts from a single shotgun sequencing experiment.
Results
Read cloud sequencing and Athena Assembly
We developed the Athena assembler to use long-range information encoded within barcoded short-read sequences. In our approach, we extract long DNA fragments and use the 10x Genomics Chromium platform to obtain barcoded short reads for our samples (Figure 1a). The resulting short reads are first stripped of their barcodes and jointly assembled using a standard short-read assembler (Online Methods) to obtain an initial assembly of the metagenome in the form of sequence contigs. These seed contigs are then provided to the Athena assembler for further metagenome sequence assembly (Figure 1b). The same barcoded short reads are mapped back to the seed contigs and read pairs that span contigs are used to form edges in a scaffold graph. Branches in this scaffold graph correspond to ambiguities encountered by the short-read assembler. At each edge, Athena examines the short-read mappings together with the attached barcodes to propose a simpler subassembly problem of a pooled subset of barcoded reads that can potentially assemble through branches in the scaffold graph (Supplementary Note 2). The selection of this read subset removes the majority of reads considered during the initial assembly while retaining reads that cover the local target sequence, isolating the local subassembly problem from the broader metagenome. The much smaller and independent subassembly problems are performed separately for every edge in the scaffold graph to yield longer, overlapping subassembled contigs that resolve branches in the scaffold graph. The initial seed contigs and intermediary subassembled contigs are then passed as reads to the long read De Bruijn graph-based assembler, Flye35,36, which determines how to assemble the target genome from these much longer contigs. The resulting metagenome assembly consists of more complete sequence contigs resolving repeats that are too difficult to assemble with short-read techniques alone. Athena is free open-source software (https://github.com/abishara/athena_meta).
Figure 1. Overview of the read cloud shotgun sequencing and assembly approach.
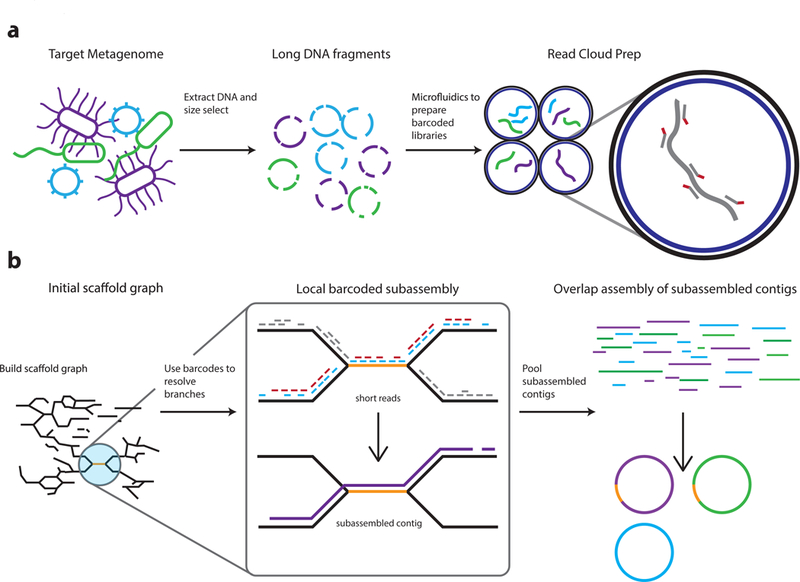
a) DNA is first extracted from microbiome samples and is size selected to enrich for long DNA fragments. The long fragments are then diluted and undergo sparse partitioning across more than a million droplet partitions (using, for example, the 10X Genomics Chromium library preparation platform). Degenerate amplification of these long fragments is then performed within these partitions to obtain barcoded traditional libraries -- each with a barcode unique to its partition. These libraries are then pooled and sequenced with an Illumina instrument.
b) The Athena assembler uses read clouds to yield more complete drafts in which genomic repeats are also accurately placed. An example repeat that is resolved and placed by Athena is shown in orange. 1) Read clouds are first assembled with standard short-read techniques to obtain seed contigs, input reads are mapped back to these seed contigs, and read pairs that span two seed contigs are used to build a scaffold graph containing unresolvable branches. 2) At each edge, Athena proposes a much simpler subassembly problem on a pooled subset of barcoded reads informed by the scaffold graph mappings. Example short reads with red and blue barcodes are passed to a short-read assembler to perform subassembly, which yields a longer subassembled contig that disambiguates branches in the scaffold graph. 3) The resulting subassembled contigs, together with the initial seed contigs, are then passed as reads to the long read De Bruijn graph based assembler Flye for final assembly. The resulting draft assembly metagenome produces more complete and more contiguous drafts in which repeats are also assembled and correctly placed.
Assembly of a mock metagenome community
As a first validation of our approach, we applied Athena to assemble a read cloud library of a staggered mixture of genomic DNA from 20 bacterial strains (ATCC MSA−1003, Online Methods). Groups of bacterial strains within the genomic DNA mixture were present in staggered abundances as high as 18% and as low as 0.02% (Supplementary Table 1). The read cloud library was prepared directly from genomic DNA supplied by ATCC and sequenced on one full lane of an Illumina HiSeq 4000 sequencer, which yielded roughly 74Gbp of raw short-read sequences.
We assembled the read cloud library of the 20 strain mixture using Athena and evaluated the overall draft quality against the available closed reference genomes. To compare against conventional short-read assembly, which does not leverage the read cloud barcode information, we also assembled the raw barcode-stripped read cloud sequencing data using a standard short-read assembler (Online Methods). The assembled metagenome drafts of each approach were evaluated using MetaQUAST37 to assess contiguity, base-error rates, and mis-assemblies (Supplementary Table 2). Athena-assembled drafts were significantly more contiguous than short-read assembled drafts with a median contig N50 increase of 7.6-fold for organisms with a minimum of 20x raw short read coverage (0.18% reported DNA fraction; Supplementary Figure 1). This contiguity was achieved without sacrificing overall accuracy when compared against conventional short-read assembly. We found Athena assembly to be comparable to short-read assembly on two important metrics: base-error rates (8.97 vs. 10.45 mismatches per 100kbp, respectively) and also the total number of mis-assemblies (67 vs. 61, respectively).
We then identified 16S/23S rRNA operons within drafts from both approaches and compared the placement of these repeats (5–7kbp in size) against the available closed reference genomes to ensure correct placement. Conventional short-read assembly was unable to correctly assemble and place a single rRNA operon. By contrast, Athena read cloud assembly produced 41 copies of the complete rRNA operon across multiple species (Supplementary Table 1). All 41 assembled rRNA operons were correctly assigned to their respective genome and only three were determined to be mis-assembled (Supplementary Note 3).
Sequencing and assembly of the human intestinal microbiome
To test the generalizability of this approach to natural biological samples, we next applied read cloud sequencing and Athena assembly to stool samples from two healthy human participants, P1 and P2. We used the Puregene DNA extraction kit following enzymatic cell lysis to extract DNA from sample P1 and the Qiagen DNA extraction kit following mechanical cell lysis to extract DNA from sample P2. To evaluate performance against alternative metagenomic sequence assembly approaches, we also prepared standard Illumina Truseq short read and Illumina Truseq SLR sequencing libraries from extracted DNA. Read cloud and SLR library preparations both require long DNA fragments whereas Truseq library preparation does not. Thus, extracted DNA to be used in read cloud and SLR libraries was first subjected to size selection (Online Methods, Supplementary Table 3). For each stool sample, prepared short read Truseq and read cloud libraries were multiplexed together and sequenced using an Illumina HiSeq 4000 sequencer yielding roughly 40Gbp of raw short-read sequences per library. SLR libraries cannot be multiplexed, so each of the two SLR libraries was given its own full lane of sequencing on a HiSeq 4000, yielding roughly 102Gbp of raw short-read sequences for each library (Supplementary Table 4).
Genus-level community compositions for each of the three sequencing approaches were first assessed using k-mer based short-read classifications (Figure 2a,b). Though some less abundant genera differed in their abundance rank, the community composition was largely concordant between all approaches tested (Supplementary Note 4).
Figure 2. Composition of stool microbiome communities from two healthy human participants.
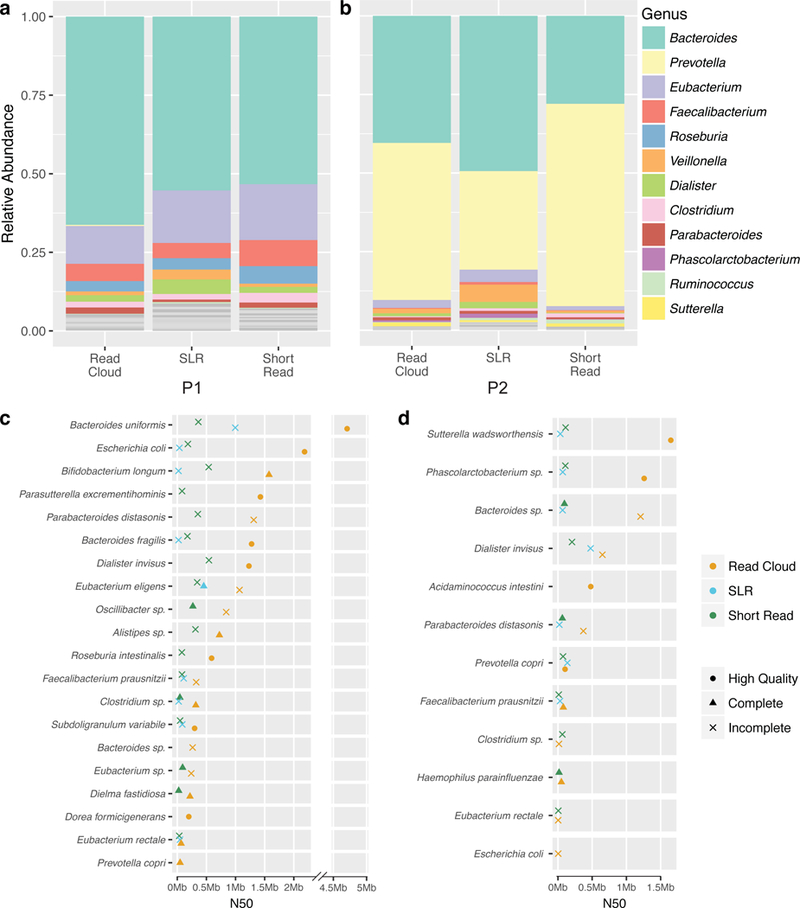
a, b) Relative abundances of genera as determined by short-read classification for each of the three libraries from samples P1 and P2. The relative representation of genera appears fairly concordant between the three different library preparation methods (read cloud, SLR, short read) for each sample. Sample P1 is more diverse than sample P2 at the genus-level.c, d) Comparisons of genome draft contiguity, as measured by N50, for taxa that were present in samples P1 and P2. The read cloud approach results in a larger number of more contiguous genome drafts than the short read or SLR approaches. Results are only displayed for the largest bin of each taxon determined to be present. The completeness and contamination of genome drafts for these taxa was determined by assessing the presence of lineage-specific single copy core genes as predicted by checkM. Genome drafts were designated as incomplete (‘x’, <90% completeness), complete (circle, >90% completeness and <5% contamination), high quality (triangle, complete and with at least 18 tRNAs, as well as at least one of each of the 5S, 16S, and 23S rRNA genes). Read cloud sequencing and assembly produces many high-quality and complete drafts. The read cloud drafts are much more contiguous as compared to those obtained from SLR and short read sequencing.
To compare performance of the three sequencing approaches, the appropriate assembly approach was applied to each sequenced library to obtain initial metagenomic drafts. Short read, read cloud, and SLR libraries were assembled using a conventional short-read assembler, Athena, and a two-stage assembly process25, respectively (Online Methods). Despite high raw short-read sequence for the SLR libraries (~102Gbp per sample for both P1 and P2), the total sequence in the form of virtual long reads was low (0.64Gbp for P1 and 0.55Gbp for P2, Supplementary Table 4).
Read cloud sequencing and assembly resulted in much longer microbial sequence contigs compared to both SLR and short-read sequencing and assembly. Nearly 144Mbp of sequence from P1 and 40Mbp of sequence from P2 were assembled using read clouds into contigs with a minimum size of 100kbp, compared to just 68Mbp and 22Mbp using short reads, and 26Mbp and 14Mbp using SLRs (Supplementary Figure 2). The overall size of the read cloud metagenome drafts was also larger compared to the SLR metagenome drafts (345Mbp vs 55Mbp in P1 and 229Mbp vs 31Mbp in P2), highlighting the benefit of increased throughput of our approach that allows assembly of lower-abundance organisms.
Read clouds produce high-quality genomes for individual bacterial species
To assess the ability of each approach to produce genome drafts for constituent bacteria, we binned metagenome draft contigs and used annotations of contigs to obtain genus-level and/or species-level assignments for each resulting bin (Online Methods, Supplementary Figure 3, Supplementary Tables 5 and 6). The resulting bins were evaluated as genome drafts by the presence of lineage-specific single copy core genes to determine completeness and contamination. Using previously described criteria, we refer to a genome bin as a complete genome draft if it is >90% complete and <5% contaminated as assessed by checkM38. We refer to the subset of these complete genome drafts as high quality, adopting a previously defined standard39, if the draft also contains at least 18 tRNA loci and at least one copy each of 5S, 16S and 23S. We also designate less complete genome bins that were >70% complete and <10% contaminated as intermediate-quality genome drafts.
Read cloud sequencing yielded complete and high-quality genome drafts for bacteria from both samples P1 and P2 (Figure 2c,d, Supplementary Note 5). Our most contiguous, high-quality read cloud draft was for Bacteroides uniformis in sample P1, which was contained completely in three contigs of sizes 4.7Mbp, 369kbp, and 25kbp. Several other bacteria from P1 were also well-assembled including Bifidobacterium longum, Escherichia coli, and Bacteroides fragilis. Alignments of input short reads to the assembled genome drafts from each sequenced library of samples P1 and P2 allowed estimation of short-read coverage of individual organisms within these libraries (Supplementary Table 6). Read cloud and short read libraries showed overall concordance with each other, and also discordance with the SLR libraries, in terms of raw short-read coverage of individual taxa in both samples. All three approaches yielded fewer complete and high-quality genome drafts from sample P2 as compared to sample P1. Examination of per-taxon coverage in sample P2 libraries revealed this sample to be largely dominated by a small number of highly abundant taxa, and as a result, libraries of sample P2 contained far fewer well-covered taxa than libraries of sample P1.
Though read cloud assembly and binning yielded a single high-quality genome draft that was annotated as Prevotella copri in sample P2, the N50 of 103kbp for this read cloud draft was unexpectedly low given its 2,836x short read coverage. Analysis of short reads originating from this genome bin in the read cloud library illuminated the unusual presence of five high-copy (>10 copies) genomic elements that likely impeded improvements in assembly by our approach (Supplementary Note 6).
The read cloud approach was superior to both the short read and SLR approaches in its ability to generate genome drafts for individual bacterial species (Figure 3, Supplementary Figure 4). The combined results from read cloud sequencing of samples P1 and P2 yielded a total of 51 intermediate-quality drafts, of which 27 were complete. The short read approach yielded fewer with 43 intermediate-quality drafts, of which only 18 were complete. SLR sequencing produced a total of only two intermediate-quality drafts, of which one was complete, despite receiving twice the amount of raw short read sequencing for each sample (due to the inability to multiplex SLR libraries). Read clouds produced the most complete drafts that were also highly contiguous (N50 > 200kbp) with a total of 16, compared to just one each from short read and SLR approaches. Read clouds were able to produce complete genome drafts, a large fraction of which were also highly contiguous, with as little as 20x short read coverage for some bacteria (Figure 3b, c). The short read approach also produced multiple complete drafts at low coverage. However, the resulting drafts from short reads were fragmentary compared to the read cloud drafts, even for bacteria with high short read coverage. Of all three tested approaches, read clouds were the only approach capable of producing high-quality drafts (Figure 3d,3e,3f).
Figure 3. Combined genome draft results of read cloud, SLR, and short read approaches applied to healthy human stool samples.
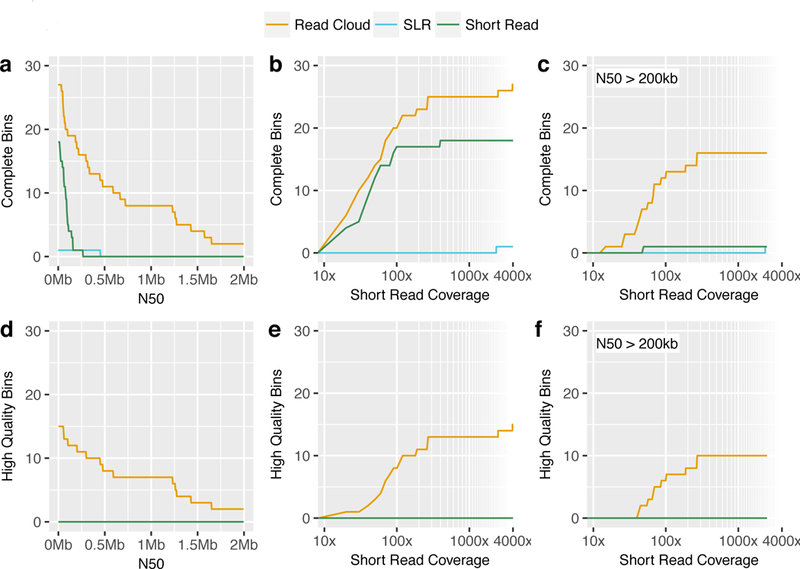
Under various performance metrics, read clouds (gold) consistently display superior performance in their ability to produce many complete and high-quality genome drafts as compared to either SLRs (blue) or short reads (green) approaches. Performance was also superior even in low short read coverage regimes (defined as <50x coverage). Counts include all complete/high-quality genome bins for all taxa in each approach.
a) Number of complete genome bins (>90% completeness, <5% contamination) with a minimum N50.
b) Number of complete genome bins with a minimum short read coverage depth. Genome bins with lower short read coverage correspond to less abundant organisms.
c) Number of complete genome bins with an N50 of >200kb and a minimum short read coverage depth.
d) Number of high-quality genome bins (complete and with at least 18 tRNAs, as well as at least one instance each of the 5S, 16S, and 23S rRNA genes) with a minimum N50.
e) Number of high-quality genome bins with a minimum short read coverage depth.
f) Number of high-quality genome bins with an N50 of >200kb and a minimum short read coverage depth.
We next assessed differences between the three approaches in their ability to produce complete drafts for particular taxa (Figure 4). Read clouds produced by far the most complete and high-quality genome drafts in which all contigs were clustered into a single bin. In contrast, short read genomes were most frequently split across two or more bins. For the majority of taxa discovered in samples P1 and P2, read clouds also successfully assembled and binned more genes together than either short reads or SLRs.
Figure 4. Completeness of genome bins produced by read cloud, SLR, and short read sequencing for various taxa present in healthy human stool samples.
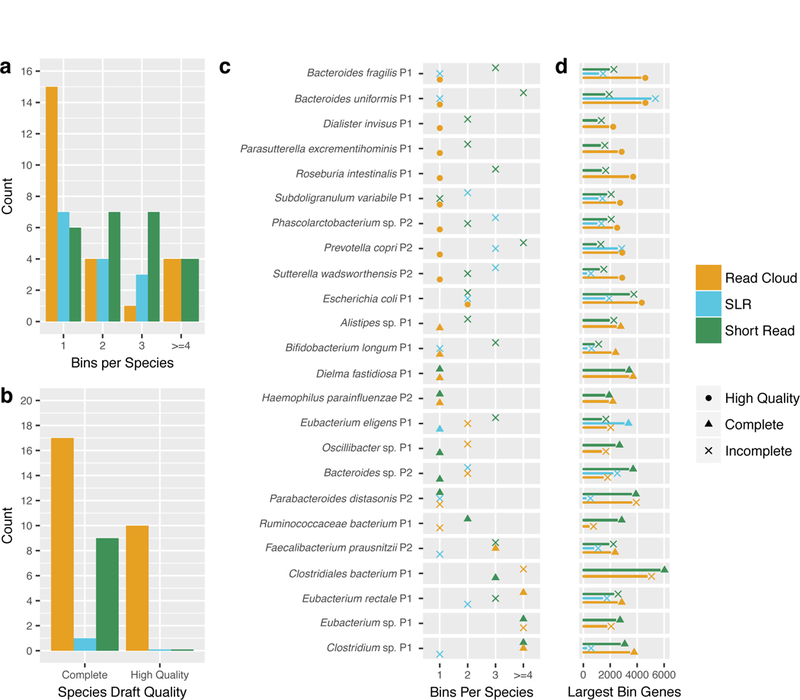
Read clouds (gold) consistently yield more complete and high-quality genome drafts for taxa within singleton bins, as compared to SLR (blue) and short read sequencing (green), both of which split sequence contigs from single genomes into two or more genome bins. Taxa are only shown if represented in at least two approaches and at least one approach produced a complete bin.
a) Counts of the number of bins containing sequence for each taxon for each of the three approaches. Read clouds produced the most singleton bins for the taxa considered.
b) Counts of complete and high-quality drafts for each approach. Read clouds produced the most complete genome drafts in singleton bins with 14. Ten of the 14 singleton bin complete genome drafts were designated as high quality.
c) For each approach, the total number of genome bins annotated as belonging to a particular taxon. The largest bin produced by an approach for a particular taxon is designated as a incomplete (‘x’), complete (circle), or high-quality (triangle) genome draft. For nearly all taxa that received a complete or high-quality genome draft from a particular approach, only a single genome bin was annotated as belonging to these taxa. However, for some taxa, such as Escherichia coli and Clostridiales bacterium, these complete or high-quality genome drafts were accompanied by a few much smaller incomplete bins that were also annotated as belonging to these taxa.
d) Counts of the number of genes present in the largest bin for a particular taxon and approach. The read cloud approach yields the bins containing the largest number of genes for the majority of taxa. The SLR bin annotated as Bacteroides uniformis in sample P1 contains more genes, but was determined to be 15% contaminated. This suggests that such some of these genes assigned to the SLR bin for Bacteroides uniformis are likely from other organisms.
To assess whether performance gains of read clouds over short reads are retained if overall sequencing depth is reduced, we also evaluated performance on in silico downsampled datasets of the sequenced mock community sample and a human stool sample. Comparisons of assembly results between the full sequenced datasets and downsampled datasets (8Gbp overall sequencing) revealed the read cloud performance gains over short reads to be depth-dependent, and that these gains diminish with lower overall sequencing depths (Supplementary Note 7).
Alignments of read cloud genomes against closed reference genomes
Comparisons of our high-quality drafts against available closed reference genomes show both cases where genome structure is largely maintained, and also cases where large structural rearrangements are apparent (Figure 5). Both Dialister invisus and Eubacterium eligens were present and assembled into high-quality genome drafts in both samples P1 and P2. Alignments of both D. invisus drafts from samples P1 and P2 illustrated large scale rearrangement with respect to the available reference genome. Inspection of these reference alignments indicates that the D. invisus strains generated by the read clouds in each sample are largely structurally divergent from each other as well. Interestingly, the draft recovered for E. eligens from sample P2 was structurally similar to the reference genome, whereas the draft recovered from sample P1 displayed two large scale inversions. Despite structural concordance in most our assembled drafts to the available reference genomes, all of them deviated substantially from the available references in sequence identity for alignable bases and also the total number of bases that were unalignable (Supplementary Table 7). The median nucleotide sequence identity was 98.5% and the median fraction of reference-unaligned bases in each draft was 15.7%.
Figure 5. Comparisons of representative read cloud genome drafts to reference genomes, and corresponding short read and SLR drafts.
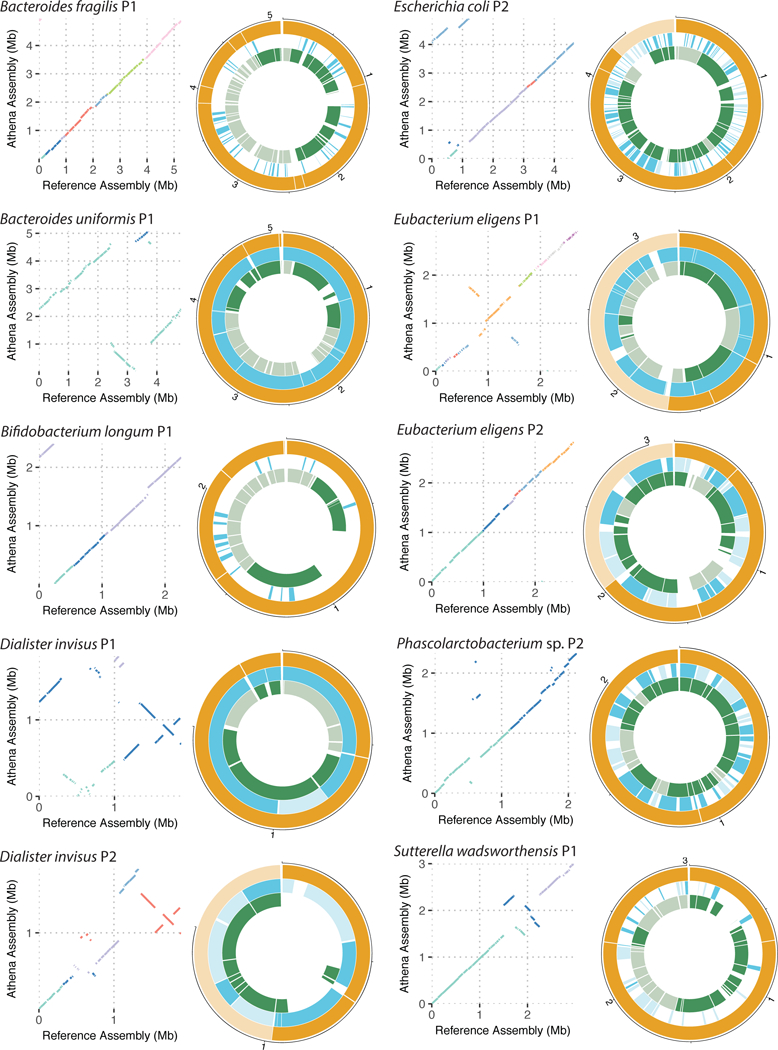
Dot-plot alignments between read cloud drafts (y-axis) and the closest available reference genome (x-axis) are shown. For each dot-plot, a given color corresponds to the alignment of a single contig in the read cloud draft against the available reference. Large-scale structural concordance and also differences including inversions are visually apparent. Alignments of SLR and short read drafts to the read cloud drafts for each taxon are also shown. In all cases, read cloud drafts were the most contiguous. For each approach, contigs belonging to the largest genome bin for a particular taxa are given a darker color, and the rest of the contigs in other bins are represented with a lighter color.
For the organisms assembled into high-quality drafts using read clouds, alignments of the corresponding SLR and short read drafts illustrate the fragmentary nature of the drafts recovered by these two approaches. Organisms that were not present at high enough abundances within each of the samples received only sparse virtual long read coverage in the SLR libraries, such that further sequence assembly of these virtual long reads into sequence contigs was generally not possible. Although the short read approach did not suffer from the same throughput limitation, it was nonetheless only capable of producing fragmentary genome drafts. The read cloud approach was the only one capable of producing high-quality and highly contiguous genome drafts de novo from the studied human stool samples.
Assembly of a marine sediment microbial community
To test the ability of read clouds to generate genome drafts from samples that are generally regarded as more complex than human stool microbiomes, we applied read cloud sequencing and Athena to deep-sea marine sediment obtained approximately 115 kilometers off the coast near San Francisco, California. DNA was extracted from this sample using a combination of mechanical bead-beating based and chemical lysis, and subjected to a size selection to enrich for long DNA fragments (Online Methods). A read cloud library was prepared and sequenced on one full lane and a quarter lane on an Illumina HiSeq 4000 flow cell, yielding roughly 72Gbp of raw short-read sequences (Supplementary Table 4). To successfully assemble this sample, which is significantly more complex than our human stool samples, we applied a specialized short-read assembler designed for use with large and complex metagenomes (Online Methods). Modifications were also made to Athena to successfully assemble the sequencing data using the read cloud barcode information (Online Methods).
The short-read assembled metagenome was 5.3Gbp, as compared to just 574Mbp from the combined metagenomes of the human stool samples, suggesting a much higher species-richness in our marine sediment sample (Supplementary Note 8). Athena read cloud assembly produced more large sequence contigs (351Mbp vs. 135Mbp in contigs >10kbp; Supplementary Figure 5) and 16S rRNA sequences (130 vs 23) than short-read assembly alone.
We next assessed the ability of each assembly approach to produce genome drafts from the marine microbiome (Online Methods, Supplementary Table 8). Read cloud sequencing and Athena assembly consistently produced more genome drafts than short-read assembly alone (Figure 6). Athena assembly produced nine complete genome drafts, of which eight were also high quality. Short read assembly was unable to produce a single complete or high-quality draft. Athena produced 49 intermediate-quality genome drafts, of which 24 also contained assembled 16S rRNA sequences. Short-read assembly produced 28 intermediate-quality genome drafts, of which only four contained 16S rRNA sequences. Alignments of input short reads to the assembled genome drafts from the read cloud library of the marine sediment sample allowed estimation of short-read coverage of individual organisms within this sample (Supplementary Table 9). Higher quality drafts tended to be more well-covered within our sequenced sample, with high-quality genome bins and intermediate-quality genome bins having median coverages of 27x and 13x respectively.
Figure 6. Comparison of marine sediment genome drafts generated by read cloud sequencing with standard short-read vs. Athena assembly.
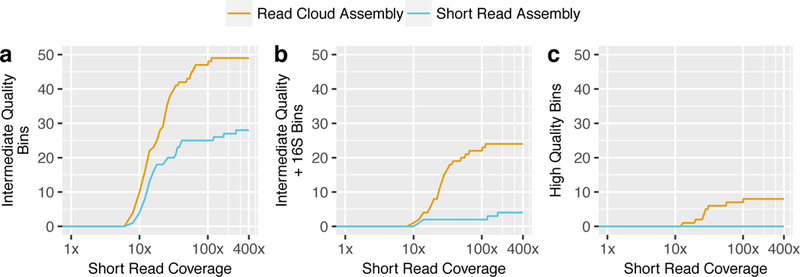
Athena read cloud assembly (gold) consistently produced more genome drafts than standard short-read assembly (blue) with genome bins assessed as genome drafts under various quality criteria. Athena read cloud assembly allowed significantly more 16S rRNA (16S) taxonomic sequences to be assigned to genome drafts than short-read assembly. The number of a) intermediate-quality (>70% completeness and <10% contamination) genome drafts b) intermediate-quality genome drafts with assembled 16S rRNA sequences, and c) high-quality genome drafts with assembled 16S rRNA sequences with a minimum short read coverage depth are shown.
Discussion
We present a novel approach using read clouds to generate de novo genome drafts from microbiome samples with the use of a single shotgun sequencing experiment. Application of our approach across diverse samples will provide high-quality genome drafts across the microbial tree of life, increasing the comprehensiveness of reference collections without the need for laborious isolation and culture. Our work is an important step towards enabling fine-grained comparative genomics for microorganisms within complex communities.
We anticipate that our read cloud sequencing approach will benefit from future improvements in both DNA extraction techniques and long fragment barcoding approaches. Our approach currently requires relatively high input DNA mass, as the application of a size selection following existing mechanical lysis techniques incurs significant loss. Improvements to DNA extraction that better preserve high molecular weight DNA across all constituent bacteria will enhance the usability of this and other approaches. Although our approach produced highly contiguous drafts for many taxa present in our human microbiome samples, the genome draft for a highly abundant Prevotella copri strain was notably fragmented. We found this strain to contain several high-copy genomic repeat elements that likely complicated correct resolution of local genomic structure during subassembly in Athena. The current 10x Genomics Chromium method currently groups several (~10) long fragments per barcode. Improvements that allow only a single long fragment per partition would greatly reduce the complexity of each subassembly task within Athena, and potentially allow read clouds to better assemble organisms with these high-copy repeats.
Further development of binning methods that take advantage of the read cloud barcode information will allow recovery of even more individual microbial genome drafts from the communities presented. Our current approach to produce individual genome drafts leveraged both our Athena assembler to improve metagenomic contig assembly, as well as existing binning tools that were designed for use with conventional short read assembly techniques. These binning tools cluster contigs into groups with similar nucleotide composition (e.g. tetramer frequencies) and coverage depth. Although application of these tools worked well when applied to our improved metagenome draft contigs, they were unable to properly deconvolve a few members of some genera in our stool microbiome samples, such as Bacteroides and Faecalibacterium, and likely members of many less characterized genera within our marine sediment samples. Multiple species belonging to each of these genera are likely present in similar abundances and have similar nucleotide compositions, such that the current metrics do not allow contigs from these taxa to be correctly separated into individual draft genomes. Read clouds have the potential to solve this issue. Pairs of sequences sharing many barcodes are indicative of sequences originating from the same input DNA fragments, which should then be binned together. Binning approaches that aim to incorporate this linkage information will likely provide a stronger signal that can further disentangle closely related taxa within complex metagenomic samples.
Of the methods evaluated, our read cloud approach was the only one capable of generating complete and high-quality genome drafts for the marine sediment sample. Read clouds also generated more intermediate quality genome drafts, with nearly half of these including the 16S rRNA gene. The added ability to link genomic sequences with 16S rRNA sequence provides an opportunity to improve functional characterization of the vast number of environmental samples for which taxonomic composition (i.e. 16S rRNA datasets), but not functional characterization (i.e., metagenomic data), is readily available. Extensions of binning approaches to use the linkage information present in read clouds will likely allow the generation of far more complete bins from these complex samples. Further applications of our read cloud approach to diverse environmental samples, especially those in which isolation and culture have been limited, will help illuminate the vast microbial life that is currently unknown.
Methods
Healthy subject recruitment
Two healthy adult volunteers were recruited at Stanford University and consented to provide stool biospecimens under the auspices of a protocol approved by the Stanford University Institutional Review Board (PI: Dr. Ami Bhatt). Informed consent was obtained and we complied with all relevant ethical regulations. The subjects had no gastrointestinal disease or antibiotic use in the 6 months prior to sample collection.
Sample Collection
Healthy volunteer stool samples:
A single stool sample was obtained from each of the two healthy volunteers. Stool samples were placed at 4˚C immediately upon collection, and processed for storage at −80˚C the same day. Stool samples were aliquoted into 2mL cryovial tubes with no preservative. Samples were stored at −80˚C until extraction.
Marine sediment sample:
A deep-sea sediment core was collected using an MC-800 multicorer aboard the R/V Oceanus (expedition #1703A) 115 km off the coast near San Francisco, CA, USA in March of 2017 (36.61°N, 123.38°W; water depth 3535 m). The core was stored at 4C until extruded and sectioned within 24 hours of collection. Approximately 2g of sediment was sampled from the top 2.5 cm of sampled core using a cut-off syringe, flash frozen in liquid nitrogen, and stored at −80C until extraction.
DNA preparation
ATCC 20 mock metagenome sample:
DNA from ATCC 20 Strain Staggered Mix Genomic Material was used directly without size selection for the mock metagenome. A single read cloud library was prepared for sequencing with the 10x Genomics Chromium (10x Genomics, Pleasanton, CA) according to the manufacturer’s standard protocol.
Healthy volunteer stool samples:
DNA was extracted from Participant 1 (P1) stool with the Qiagen Gentra Puregene Yeast/Bacteria kit according to the manufacturer’s standard protocol with two modifications: a chilling step at −80˚C for five minutes prior to DNA precipitation, and DNA precipitation with 14,000g, 20 minute centrifugation at 4˚C. DNA was extracted from Participant 2 (P2) stool with the Qiagen QIamp Stool Mini Kit according to the manufacturer’s standard protocol, modified with an additional step after addition of buffer ASL. The additional step was 7 cycles of alternating 30 second periods of beating with zirconia beads in a Minibeadbeater (Biospec Products, Bartlesville, OK) and chilling on ice. DNA concentration was measured using Qubit fluorometric quantitation (see Supplementary Table 3 for measured concentrations).
DNA that was to be taken forward for to 10x Chromium preparation was size-selected with the BluePippin instrument targeting the 10kb-50kb size range, the maximum yielding measurable output. DNA for the SLR library preparation was size-selected with the BluePippin instrument targeting the 8–12kb size range as per the manufacturer’s recommended protocol. DNA for Truseq conventional short read library preparation was not size-selected. Libraries were prepared for sequencing with the 10x Genomics Chromium (10x Genomics, Pleasanton, CA), the Illumina Truseq SLR kit, or Illumina Truseq Nano kit according to the respective manufacturer’s standard protocol. Library fragment size was quantified with the Agilent 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA) using the High Sensitivity DNA kit.
Marine sediment sample:
DNA was extracted using the RNeasy PowerSoil DNA elution kit (Qiagen, Hilden, Germany; cat. no. 12867–25) in combination with the RNeasy PowerSoil Total RNA kit (Qiagen, Hilden, Germany; cat. no. 12866–25). The protocol was modified from the manufacturer’s instructions to include a bead-beating step of 5.5m/s for 2X 45s using a FastPrep-24 (MP Biomedicals, Santa Ana, CA, USA; cat. no. 116005500). DNA was eluted in 100ul DNase, RNase-free water and stored at −80C until further processing. DNA was then size-selected with the BluePippin instrument targeting the 10kb-50kb size range (the maximum yielding measurable output), and a library was prepared for sequencing with the 10x Genomics Chromium (10x Genomics, Pleasanton, CA), according to the manufacturer’s standard protocol.
Sequencing
Chromium libraries
Chromium libraries from the mock metagenome, healthy stool samples, and ocean sediment were sequenced with 2×151bp sequencing on an Illumina HiSeq 4000. The healthy stool samples were allocated a half lane each. The marine sediment was allocated a quarter lane and a full lane. The mock metagenome was allocated one lane. (See Supplementary Table 4 for total Gbp coverage). Resulting sequences were demultiplexed and barcoded with the 10x Longranger v2.1.3 mkfastq tool to generate raw reads, then subjected to quality control.
Truseq libraries
DNA from the healthy stool samples was prepared for sequencing with the Illumina Truseq library prep kit according to the manufacturer’s standard protocol and subjected to 2×101bp sequencing on an Illumina HiSeq 4000. Each library was allocated a half lane of sequence coverage (see Supplementary Table 4 for total Gbp coverage). Raw reads were then subjected to quality control (see below).
Synthetic long read libraries
DNA from the healthy stool samples was prepared for sequencing with the Illumina Truseq Synthetic Long Read library prep kit according to the manufacturer’s standard protocol. These libraries use the sample barcode to identify the 384 molecular partitions, so samples cannot be multiplexed. Thus, each library was necessarily allocated one full lane of 2×151bp coverage on an Illumina HiSeq 4000 (see Supplementary Table 4 for total Gbp coverage). Raw reads were then subjected to quality control (see below).
Quality control
Following sequencing, all libraries were trimmed using cutadapt40 v1.8.1 using a minimum length of 60bp and minimum terminal base score of 20 (with the exception of the ATCC mock metagenome reads, which were trimmed with a minimum trimmed read length of 80bp and minimum terminal base score of 35, as well as 8bp removed from the 5’ end and 15bp removed from the 3’ end due to low read quality). Reads were synced and orphans (reads whose pair mates were filtered out) were placed in a separate single-ended fastq file with an in-house script.
Assembly of mock metagenome and human stool samples
Data from read cloud 10x Genomics Chromium and short read Truseq libraries were assembled using MetaSPAdes v3.11.1 41 with default parameters. For read cloud libraries, MetaSPAdes assembled seed contigs were then assembled with Athena (Supplementary Note 2).
Synthetic long reads were assembled with a two stage process: (1) synthetic long reads were assembled from trimmed sequencing reads with TruSPAdes42 v3.11.1 with default parameters, (2) these assembled synthetic long reads were then further assembled into contigs with CANU v1.5 43 with the following parameters: errorRate=0.06, genomeSize=45.00m, contigFilter=“2 2000 1.0 1.0 2”, stopOnReadQuality=false.
Assembly of marine sediment sample
Data from the marine sediment read cloud library was assembled using MEGAHIT v1.1.2 44 with default parameters. MEGAHIT short-read assembled contigs were then used as seed contigs and assembled with Athena (Supplementary Note 2).
To make Athena assembly tractable on complex metagenomes, Athena was modified to only perform subassembly for well-covered seed contigs with a minimum short read sequence coverage of 20x. MEGAHIT contigs excluded from Athena assembly were then mapped back to the initial Athena draft, and each of these contigs was included in the final output if more than 2000 bases did not align to the initial draft.
Assembly classification, genome draft binning, and gene identification
For each approach, raw short reads were aligned to assembled contigs with BWA v0.7.10 45 to generate contig coverage profiles. Contigs were then binned with Metabat v2.12.1 16 to form genome drafts. Bins were evaluated with Metaquast v4.6.0 46 for assembly size and contiguity, CheckM v1.0.7 47 for completeness and contamination as genome drafts, Prokka v1.12 48 for gene content, Aragorn v1.2.36 49 to count tRNA sequences, and Barrnap v0.7 50 to count 5S, 16S and 23S ribosomal RNA loci. We define an “intermediate quality” genome as one with >70% completeness and <10% contamination. We adopt previously described standard defining a “high quality” genome as one containing at least 18 tRNA loci, at least one copy each of 5S, 16S and 23S, >90% completeness and <5% contamination39.
Individual contigs from all assemblies were assigned taxonomic classifications using Kraken v0.10.6 51 with a custom database constructed from the Refseq and Genbank52,53 bacterial genome collections. Each genome draft was assigned a species-level label if >60% of total bases within the draft shared a species-level classification. Otherwise, drafts were assigned the majority genus-level label.
Code availability
The Athena assembler together with a demonstration dataset can be found at https://github.com/abishara/athena_meta. This example contains a subset of the read clouds from the ATCC 20 mock metagenome, for which assembly with Athena yields the full Lactobacillus gasseri genome in two sequence contigs. The binning, annotation, and evaluation workflow can be found at https://github.com/elimoss/metagenomics_workflows.
Data availability
The datasets generated during the current study are available in the NCBI Sequence Read Archive under Bioproject accession PRJNA380276. 10x read barcodes have been encoded as sample barcodes, and must be reformatted as molecular barcodes for use with Athena.
Supplementary Material
Acknowledgements
The authors would like to thank Ekaterina Tkachenko for assistance preparing Truseq libraries, and Michael Snyder and members of the Bhatt lab for helpful feedback. The authors would also like to thank Hongxia Xu at Illumina for sharing read cloud sequencing data of ATCC 20 for the mock metagenome. This work was supported by NCI K08 CA184420, the Amy Strelzer Manasevit Award from the National Marrow Donor Program, and a Damon Runyon Clinical Investigator Award to A.S.B.. E.L.M. was supported by National Science Foundation Graduate Research Fellowship DGE-114747. A.B. was supported by the Stanford Genome Training Program (SGTP; NIH/NHGRI) and the Training Grant of the Joint Initiative for Metrology in Biology (JIMB; NIST). A.E.D. and the marine sample collection and extraction was supported by National Science Foundation OCE-1634297. A.E.P. was supported by the Center for Dark Energy Biosphere Investigations Postdoctoral Fellowship. Access to shared compute resources was supported in part by NIH P30 CA124435 using the Stanford Cancer Institute Shared Resource Genetics Bioinformatics Service Center.
Footnotes
Competing financial interests
S.B. is an employee and owns stock in Illumina. Shotgun sequencing products developed, marketed and/or sold by Illumina were used in this manuscript.
References
- 1.Schloss PD & Handelsman J Metagenomics for studying unculturable microorganisms: cutting the Gordian knot. Genome Biol. 6, 229 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd-Price J et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature (2017). doi: 10.1038/nature23889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kashtan N et al. Single-cell genomics reveals hundreds of coexisting subpopulations in wild Prochlorococcus. Science 344, 416–420 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Baker BJ, Lazar CS, Teske AP & Dick GJ Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3, 14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eyice Ö et al. SIP metagenomics identifies uncultivated Methylophilaceae as dimethylsulphide degrading bacteria in soil and lake sediment. ISME J. 9, 2336–2348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He Y et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat Microbiol 1, 16035 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Brown CT et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Hug LA et al. A new view of the tree of life. Nat Microbiol 1, 16048 (2016). [DOI] [PubMed] [Google Scholar]
- 11.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–45 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng Y, Leung HCM, Yiu SM & Chin FYL IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Namiki T, Hachiya T, Tanaka H & Sakakibara Y MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 40, e155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cleary B et al. Detection of low-abundance bacterial strains in metagenomic datasets by eigengenome partitioning. Nat. Biotechnol. 33, 1053–1060 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y-W, Tang Y-H, Tringe SG, Simmons BA & Singer SW MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2, 26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang DD, Froula J, Egan R & Wang Z MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen HB et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Alneberg J et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Popic V, Kuleshov V, Snyder M & Batzoglou S GATTACA: Lightweight Metagenomic Binning With Compact Indexing Of Kmer Counts And MinHash-based Panel Selection. bioRxiv 130997 (2017). doi: 10.1101/130997 [DOI] [Google Scholar]
- 20.Koren S et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 30, 693–700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chin C-S et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Loman NJ, Quick J & Simpson JT A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 12, 733–735 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Leonard MT et al. The methylome of the gut microbiome: disparate Dam methylation patterns in intestinal Bacteroides dorei. Front. Microbiol. 5, 361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voskoboynik A et al. The genome sequence of the colonial chordate, Botryllus schlosseri. Elife 2, e00569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuleshov V et al. Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome. Nat. Biotechnol. 34, 64–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharon I et al. Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res. 25, 534–543 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White RA 3rd et al. Moleculo Long-Read Sequencing Facilitates Assembly and Genomic Binning from Complex Soil Metagenomes. mSystems 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng GXY et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 34, 303–311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bishara A et al. Read clouds uncover variation in complex regions of the human genome. Genome Res. 25, 1570–1580 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuleshov V et al. Synthetic long-read sequencing reveals intraspecies diversity in the human microbiome. Nat. Biotechnol. advance online publication, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peters BA et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 487, 190–195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kitzman JO et al. Haplotype-resolved genome sequencing of a Gujarati Indian individual. Nat. Biotechnol. 29, 59–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amini S et al. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet. 46, 1343–1349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spies N et al. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 14, 915–920 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin Y et al. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. U. S. A. 113, E8396–E8405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kolmogorov M, Yuan J, Lin Y & Pevzner P Assembly of Long Error-Prone Reads Using Repeat Graphs. bioRxiv 247148 (2018). doi: 10.1101/247148 [DOI] [PubMed] [Google Scholar]
- 37.Mikheenko A, Saveliev V & Gurevich A MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32, 1088–1090 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowers RM et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 41.Bankevich A et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 19, 455–477 (2012–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bankevich A & Pevzner PA TruSPAdes: barcode assembly of TruSeq synthetic long reads. Nat. Methods 13, 248–250 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Koren S et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. (2017). doi: 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li D, Liu C-M, Luo R, Sadakane K & Lam T-W MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mikheenko A, Saveliev V & Gurevich A MetaQUAST: evaluation of metagenome assemblies. Bioinformatics btv697 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seemann T Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).24642063 [Google Scholar]
- 49.Laslett D & Canback B ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seemann T barrnap. (Github). [Google Scholar]
- 51.Wood D & Salzberg S Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tatusova T, Ciufo S, Fedorov B, O’Neill K & Tolstoy I RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42, D553–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benson DA et al. GenBank. Nucleic Acids Res. 41, D36–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available in the NCBI Sequence Read Archive under Bioproject accession PRJNA380276. 10x read barcodes have been encoded as sample barcodes, and must be reformatted as molecular barcodes for use with Athena.