

Abstract
Side chains cover protein surfaces and are fundamental to processes as diverse as substrate recognition, protein folding and enzyme catalysis. However, characterisation of side-chain motions has so far been restricted to small proteins and methyl-bearing side chains. Here we present a class of methods, based on 13C-detected NMR spectroscopy, to more generally quantify motions and interactions of side chains in medium-to-large proteins. A single, uniformly isotopically labelled sample is sufficient to characterise the side chains of six different amino acid types. Side-chain conformational dynamics on the millisecond time-scale can be quantified by incorporating chemical exchange saturation transfer (CEST) into the presented methods, whilst long-range 13C-13C scalar couplings reporting on nanosecond to millisecond motions can be quantified in proteins as large as 80 kDa. The presented class of methods promises characterisation of side-chain behaviour at a level that has so far been reserved for the protein backbone.
Analysis of side-chain motions by NMR has so far been restricted to small proteins and methyl-bearing side chains. Here, the authors present NMR methods based on 13C direct detection of highly deuterated protein samples that yield sharp and well-resolved signals and allow the characterisation of side-chain conformational dynamics of six different amino acid types in medium-to-large proteins.
Introduction
Proteins are dynamic entities whose molecular function is intrinsically related to their structure and dynamic sampling, both in the immediate vicinity of active sites and in regulatory sites1,2. Proteins are often viewed as representations of their backbone and most experimental studies of protein dynamics to date have primarily focussed on the protein backbone, with much less attention paid to side chains3–9. Whilst knowledge of the behaviour of the protein backbone is essential in order to understand many aspects of protein function, bringing side chains into focus is crucial. It is the side chains that give the amino acids in proteins their unique chemical diversity, for example, side chains form critical parts of many active sites of enzymes and the side chains presented on the protein surface are key to substrate recognition and binding events. In many cases, the dynamics and interactions of side chains are as, if not more, important to the biological function than the overall backbone conformation. In addition, the motions of side chains are often decorrelated from the backbone10–12. In order to understand the mechanisms of enzymes and characterise macromolecular interactions and regulation, it is imperative to be able to specifically characterise the side-chain structure and movements.
Nuclear magnetic resonance (NMR) spectroscopy is uniquely situated to generally characterise the interactions and conformational sampling of side chains in proteins. However, most experimental studies of protein motions using NMR spectroscopy to date have focussed on the backbone or methyl groups, and detailed information regarding side-chain behaviour has been restricted to small proteins13,14 and methyl-bearing side chains15–17. The limitations of currently available approaches are mainly due to line broadening and signal loss as a result of efficient relaxation pathways, as well as insufficient resolution and signal overlap in the NMR correlation maps used to extract parameters reporting on dynamics and interactions.
Here, we present a class of NMR methods that allows a more general characterisation of side chains in medium-to-large proteins. The presented class of methods is anchored in 13C-direct detection18 NMR spectroscopy of per-deuterated proteins. The slow relaxation of the aliphatic 13C nuclei in deuterated proteins results in sharp NMR signals, whilst the correlation of two 13C chemical shifts results in high-resolution and well-resolved two-dimensional NMR spectra. It is shown that the slow transverse 13C relaxation rates enable a large range of NMR experiments to be performed to characterise the structure, interactions and dynamics of side chains in medium-to-large (< 82 kDa) proteins. The presented experiments include a quantification of long-range 13C–13C scalar couplings13 reporting on the sampling of side-chain dihedral angles and chemical exchange saturation transfer (CEST) experiments19 to characterise low-populated states.
Results and discussion
Side-chain 13C–13C correlation spectra
The BMRB20 databank provides a large database of assigned NMR chemical shifts, including the side-chain 13C chemical shift. Analysis of these data reveals six side chains (Fig. 1; Supplementary Fig. 1) with a ‘terminal’ 13C (13Ct), which is directly bonded to just one other 13C and which has a chemical shift distribution that is isolated from its directly bonded penultimate 13C (13Cp). This distinct chemical shift profile means that the terminal 13Ct can be selectively excited in an NMR experiment using frequency-selective pulses. Selective excitation enables spectral filtering and observation of specific residue types in multidimensional NMR spectra, even when using uniformly isotopically labelled proteins.
Fig. 1.
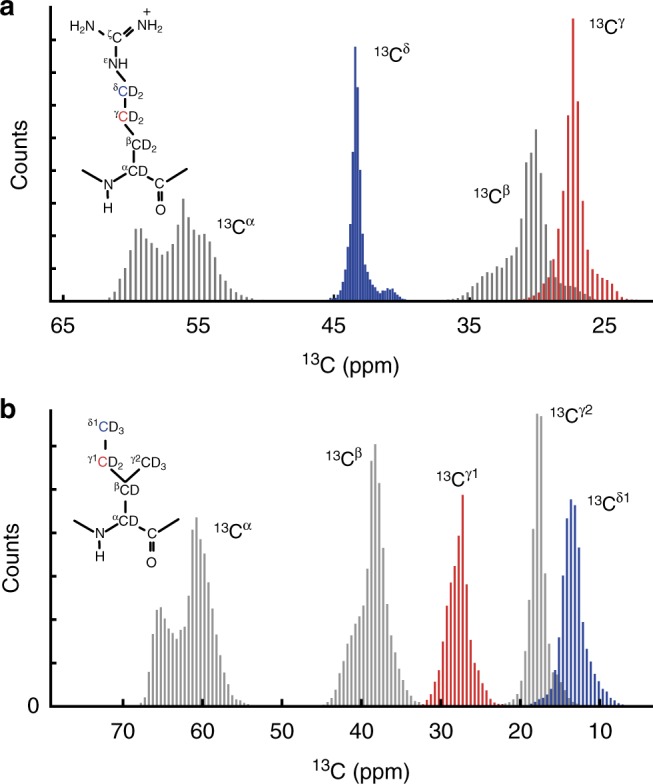
Examples of aliphatic 13C chemical shift distributions. Distribution of the assigned 13C chemical shifts of (a) arginine and isoleucine (b) residues extracted from the BMRB20 databank. Terminal 13Ct, directly coupled to just one other 13C are coloured blue and the bonded, penultimate 13Cp, are coloured red. For clarity, only one of the two 13Ct–13Cp pairs have been highlighted in isoleucine (See Supplementary Fig. 1 for other sites)
The core element of the presented side-chain-specific NMR experiments, Fig. 2a, correlates the terminal carbon 13Ct with its directly bonded penultimate 13Cp within a protein side chain. The terminal 13Ct includes 13Cδ in the arginine side chains, 13Cδ1 and 13Cγ2 in isoleucine, 13Cε in lysine, 13Cδ in proline, 13Cγ2 in threonine and 13Cγ in valine residues. The fact that the magnetisation of interest starts and is detected on the terminal 13Ct has some key advantages. The homonuclear coupling pattern for 13Ct is a simple doublet, which allows for the magnetisation of interest to be transferred completely between 13Ct and 13Cp using simple INEPTs and also facilitates virtual decoupling in the 13Ct detected dimension.
Fig. 2.
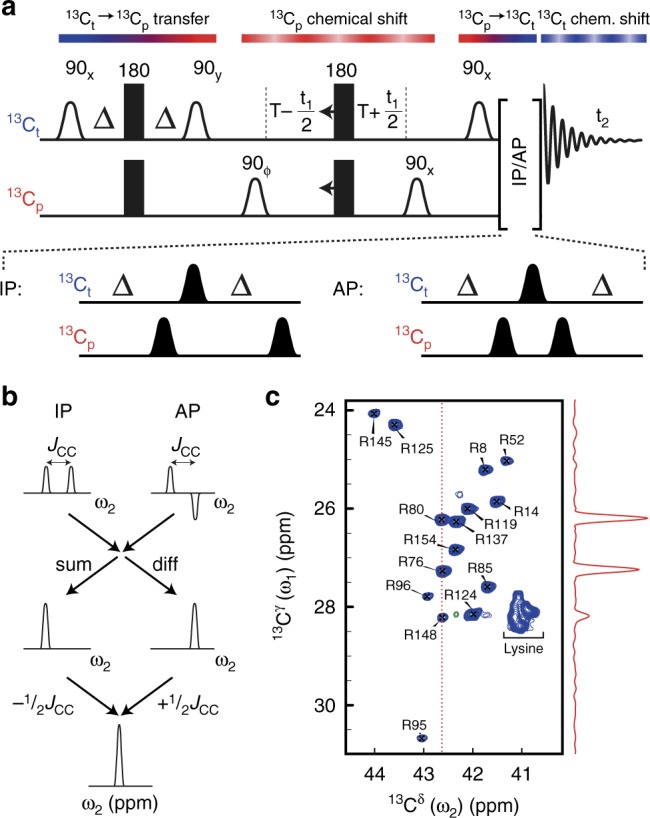
13C–13C side-chain correlation spectra of per-deuterated proteins. a Schematic representation of the NMR pulse sequence used to obtain 13C–13C side-chain correlation spectra. The flow of the magnetisation between 13Ct (blue) and 13Cp (red) is shown above the sequence with colour gradients. The following delays are used: Δ = 1/(4JCC) ≈ 7.1 ms, T = 1/(2JCC) ≈ 14.1 ms, where JCC is the one-bond 13C–13C scalar coupling constant. Rectangular pulses are high-power and not selective, bell-shaped pulses are frequency selective (90°: white outlined, 180°: black). Deuterium, 2H, is decoupled throughout the sequence and frequency discrimination is obtained by states–TPPI of phase ϕ21. b Schematic representation of post-processing to obtain the decoupled spectrum. c Arginine 13Cδ–13Cγ correlation of the 18-kDa protein T4L L99A, obtained on a 1.4 mM sample at a static field of 14.1 T at 278 K in 37 min
In the core experiment, Fig. 2a and Supplementary Fig. 2, magnetisation is initially transferred from 13Ct to 13Cp, then labelled with the chemical shift of 13Cp during t1 and finally transferred back to 13Ct for detection. The doublet peak splitting observed for 13Ct because of the evolution of the single one-bond 13Ct–13Cp scalar coupling during acquisition (t2) can conveniently be resolved by recording two sub-spectra, in-phase (IP) and anti-phase (AP)22. Figure 2b shows how subsequent post-processing is used to virtually ‘decouple’ the spectra in the direct 13Ct dimension, so that single peaks are observed in multidimensional correlation spectra. One-bond 13C–13C couplings in indirect dimensions (t1) are removed using constant-time evolutions23, and evolutions of the 2H–13C couplings are efficiently suppressed using 2H decoupling (see Supplementary Fig. 2).
Application of the method to observe 13C–13C correlations in the 18-kDa L99A mutant of T4 lysozyme (T4L L99A) is shown for arginine 13Cδ–13Cγ in Fig. 2c, and for other side-chain correlations in Supplementary Figs. 3 and 4. Of importance is that (1) the NMR correlation spectrum in Fig. 2c is obtained in 37 min at 278 K, where the rotational correlation time of the 18-kDa T4L L99A mimics that of an ~30-kDa protein at room temperature, (2) six different 13C–13C correlation maps can be obtained in less than 12 min (see Supplementary Figs. 3, 4) and (3) a single uniformly [2H,13C] isotopically labelled sample could be used to characterise six side chains. The lack of directly bonded protons and absence of efficient relaxation pathways for aliphatic 13C in highly deuterated proteins dramatically reduces the 13C relaxation rates. For T4L L99A, the isoleucine 13Cδ1 non-selective longitudinal relaxation rates, R1, range between 0.12 and 0.24 s−1 at 278 K and a field of 14.1 T (see Supplementary Table 1 and Supplementary Fig. 5). This necessitates longer recycling delays and fewer scans per unit time, and the lower gyromagnetic ratio of 13C compared with 1H leads to an intrinsic lower signal-to-noise. However, reduced relaxation rates also lead to small transverse relaxation rates, between 2.7 and 8.8 s−1 for isoleucine 13Cδ1 in T4L L99A at 278 K and 14.1 T (see Supplementary Table 2 and Supplementary Fig. 7), which in turn lead to very sharp signals. For example, with the 13C-detected method, it became possible to observe sites in arginine side chains of T4L L99A that were not detectable in a 1H-detected equivalent experiment, even when using a highly optimised isotope-labelling scheme (Supplementary Fig. 6). Another striking advantage of 13C–13C correlation spectra compared with 1H–13C spectra is the substantially better chemical shift dispersion in the directly detected dimension, resulting in significantly better peak separation (see Supplementary Fig. 6 for a comparison). Moreover, as compared with 1H–13C spectra, the 13C–13C correlation maps directly provide the chemical shift of two aliphatic 13C that are both known to report on the structure and sampling of side chains24,25.
Direct-detected 13C spectra of a 82-kDa protein
The excellent spectra obtained on the medium-sized T4L L99A protein at low temperature and the favourable 13C transverse relaxation rates show that 13C-direct detection is ideally suited for characterising side chains in large proteins. Side-chain 13C–13C correlation maps were recorded for the significantly larger 82-kDa malate synthase G (MSG) protein26. Figure 3a and c shows the isoleucine 13Cδ1–13Cγ1 and valine 13Cγ–13Cβ correlation maps of MSG, respectively, where excellent chemical shift dispersion and resolution result in nearly no overlap of peaks even in this large and uniformly [2H,13C] isotopically labelled system. The slow transverse relaxation rate of aliphatic 13C in per-deuterated proteins means that the experiment shown in Fig. 2a easily can be extended to obtain numerous other NMR parameters reporting on motions, conformations and interactions. TOtal Correlated SpectroscopY (TOCSY) NMR experiments are typically used to aid side-chain chemical shift assignment28 of proteins and a couple of representative examples are shown in Fig. 3b for MSG, with details of the sequence given in Supplementary Fig. 8.
Fig. 3.
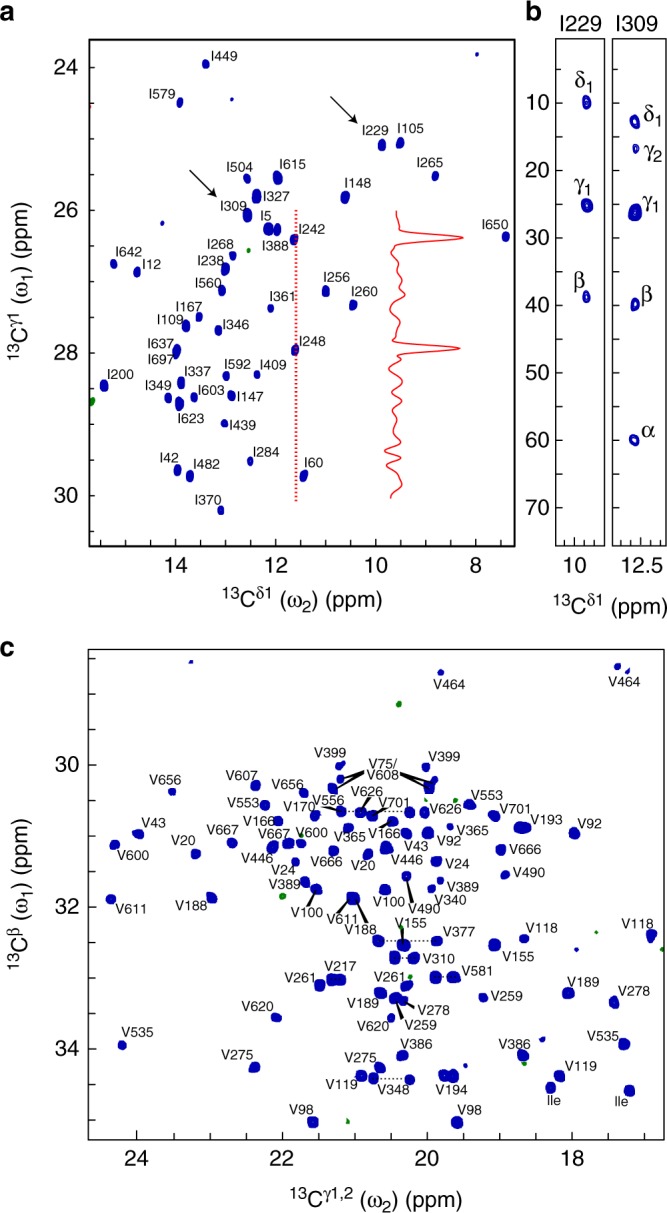
13C–13C side-chain correlation spectra of a large protein. a Isoleucine 13Cδ1–13Cγ1 and c valine 13Cγ–13Cβ correlation spectra of a 0.5 mM sample of the 82-kDa protein MSG. Each of the two spectra in a and c were recorded in 7.5 h at 18.8 T using a standard helium-cooled Bruker TCI radio-frequency probe. b Correlating the 13Cδ1–13Cγ1 of isoleucine with the rest of the side chain 13C for chemical shift assignment. Representative TOCSY strips from a three-dimensional 13C-detected CC-TOCSY experiment (see Supplementary Fig. 8) for I229 and I309 (marked with black arrows in a. Chemical shift assignments in a and c were obtained from (refs. 26,27) combined with the 3D-CC-TOCSY. Peaks labelled Ile in c are isoleucine 13Cγ2–13Cβ correlations. All experimental parameters are given in Supplementary Materials and Methods
Characterisation of millisecond dynamics
A full characterisation of the role of side chains requires an appreciation of their motions within the protein. Of substantial importance is that the method detailed above can be extended to allow quantification of side-chain motions across a wide range of timescales. Although many experiments will be possible using the new scheme, two examples, which together report on side-chain motions on timescales from nanoseconds to milliseconds, were chosen here to highlight the versatile applicability.
Protein dynamics and conformational exchange on the millisecond timescale have been shown to be important for the function of many proteins29 and the chemical exchange saturation transfers (CEST)19,30 NMR experiment, amongst others, allows a quantification of exchange events on this timescale. CEST experiments have recently been adapted for side chains in small proteins14, in a 1H-detected manner. Integrating CEST with the 13C-detected method described above (see Supplementary Fig. 9) allows quantification of side-chain conformational exchange in medium-to-large proteins between the ground state (G) and an excited state (E). Examples of CEST profiles are shown in Fig. 4 and Supplementary Fig. 9, where the conformational exchange of T4L L99A at 278 K is quantified by a 13C-detected CEST experiment. The calculated overall exchange rate, kex = 128 ± 18 s−1, and the population of the excited state, pE = 1.15 ± 0.11% (see Supplementary Fig. 10), agree well with previous studies11 showing that reliable parameters are derived from 13C-detected CEST experiments. The chemical shifts of aliphatic 13C within the protein side chains report on the rotameric sampling of the side chain24, and the results of the CEST experiments in Fig. 4 also report on the rotameric sampling in the excited state. Specifically, from the chemical shifts obtained for V103 in the ground and excited states, ϖG and ϖE, respectively, it can be calculated24 that the χ1 angle of V103 changes from a predominantly trans conformation (91% trans, 9% gauche-plus) in the ground state to a mainly gauche-minus conformation in the excited state (26% gauche-plus, 16% trans, 58% gauche-minus), which is in agreement with the existing structures of T4L L99A in the ground and the excited state11. Access to the 13C chemical shifts in the excited state via the 13C-detected CEST experiment also reveals that the V103 side chain is more dynamic in the excited state with a broader distribution of side-chain rotamers.
Fig. 4.
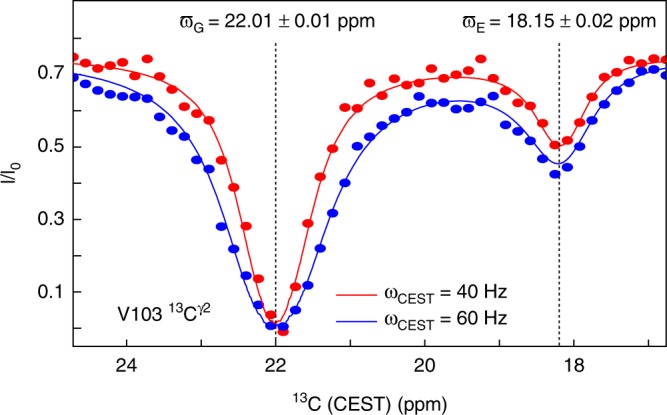
Quantifying side-chain motions on the millisecond timescale. CEST profiles to quantify millisecond chemical exchange of V103 13Cγ2 in T4L L99A at 278 K are shown. The circles represent experimentally obtained normalised intensities, and the lines are the result of a least-squares fit. Uncertainties in the reported ϖG and ϖE are obtained using the covariance method in the least-squares fit (see Materials and Methods for full details)
Measuring long-range 13C–13C scalar couplings
The second application involves the measurement of three-bond 13Cδ–13Cα scalar couplings13, 3JCαCδ, reporting on the conformational sampling of the side-chain χ2 dihedral angle on a timescale from nanoseconds to milliseconds. The three-bond scalar coupling 3JCαCδ relates to the side-chain χ2 dihedral angle via a Karplus relationship, where large coupling constants are observed when the χ2 angle is in a trans conformation, and small couplings are observed when χ2 is in a gauche conformation. Intermediate values of the coupling constant are observed when the side chain is dynamic about the χ2 dihedral angle. Extending the 13C–13C correlation experiment to measure these couplings (see Supplementary Fig. 10) showed that the majority of the arginine side chains in T4L L99A are predominantly in a trans conformation around the χ2 angle (see Supplementary Table 3) in agreement with crystallographic data. For the two arginine side chains of T4L L99A shown in Fig. 5a, the intermediate value of the coupling constant for R14 shows that this side chain is flexible and dynamic around the χ2 angle, while the high value observed for the R148 side chain shows a rigid trans conformation. This is in good agreement with previous characterisations of the dynamics of the arginine side chains of T4L L99A6,31,32, as well as with the structure of T4L L99A, where R14 is on the surface and R148 is engaged in a salt bridge with D10. Long-range 13Cδ–13Cα scalar couplings were also measured for isoleucine residues in the 82 -kDa MSG protein (Fig. 5b; Supplementary Table 4). Of the 44 isoleucine residues in MSG, 13Cδ–13Cα scalar couplings were obtained for 32, allowing a quantification of the dynamics of these side chains. Access to both the 13Cδ1 chemical shift and the 13Cδ1–13Cα scalar coupling allows a full characterisation of the rotameric sampling about the χ2 angle. For example, a large 13Cδ1 chemical shift and a small 13Cδ1–13Cα coupling for I200 in MSG show that this residue is restrained in a rare gauche-plus state (see Supplementary Table 4)25, in agreement with the crystal structure of MSG.
Fig. 5.
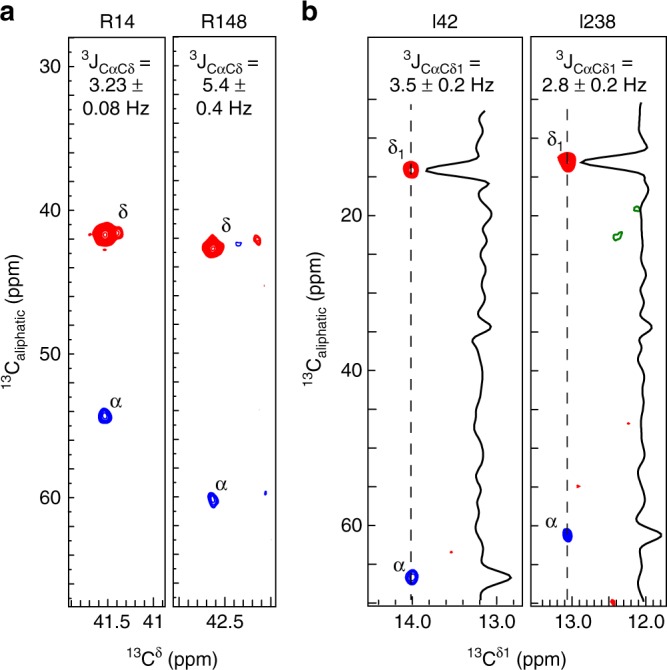
Quantifying long-range scalar 13C–13C couplings. a Two examples of strips used to quantify long-range 13Cδ–13Cα scalar couplings in the arginine side chains of T4L L99A at 18.8 T and 278 K. The R14 side chain is flexible, while the R148 is rigid in agreement with relaxation measurements6. b Strips from a three-dimensional experiment to quantify long-range 13Cδ1–13Cα scalar couplings in isoleucine side chains of the 82-kDa MSG at 14.1 T and 310 K. The I238 side chain is dynamic around the χ2 angle, as seen by the intermediate value of the 3JCαCδ1 coupling constant, whereas I42 is more rigid in a trans conformation. Positive contours are depicted as red, while negative contours are in blue. Values for 3JCαCδ1 in a and b are mean ± s.d.
A strategy to characterise side chains in proteins using 13C-direct-detected NMR has been developed. Applications of 13C-detection to macromolecules has so far mainly been employed to investigate intrinsically disordered regions18,22, where many experiments are now available to exploit the improved resolution of 13C compared with 1H. Existing methods to characterise side chains in medium-to-large proteins have been limited to methyl-bearing residues and require specific labelling. The presented method employs 13C-detection and capitalises on the distinct chemical shift profiles of side chains to specifically observe different residues in a single uniformly labelled sample. As such, it allows a more general characterisation of side chains for investigations in large proteins (~82 kDa) and for a quantification of the dynamics of these side chains. The slow transverse 13C relaxations rates in fully deuterated proteins enables a large range of NMR experiments to characterise side-chain structure, interactions and dynamics. A few examples of experiments integrated with the side-chain 13C–13C-direct-detected method are detailed above. Combined with recent developments of hardware aimed at 13C-direct-detected NMR spectroscopy, we envisage that the new method will be particularly useful for experimentally quantifying functional side chains, at atomic resolution, in medium-to-large proteins. Forthcoming applications of this suite of methods promise a burgeoning appreciation of the role side chains play in orchestrating protein function.
Methods
Sample preparations
Isotopically labelled T4 lysozyme mutant C54T/C97A/L99A (T4L L99A) was expressed and purified as described previously33. Briefly, a codon-optimised form of the gene containing the mutations L99A, C54A and C97A in a kanamycin-resistant, pET-29b vector was transformed into BL21 (DE3) chemically competent E. coli cells. A single colony was used to inoculate 5-mL culture, which was grown overnight at 37 °C. This was used to inoculate 50 mL of minimal M9 media made with 2H2O and supplemented with 1 g L−1 [1H,15N]-ammonium chloride as the sole nitrogen source. For the uniformly labelled [2H,13C,15N] sample, 2 g L−1 [2H, 13C]-glucose was added as the sole carbon source. For the [1H13C-Lys,Arg; 2H12C] isotopically labelled sample, 2 g L−1 [2H, 12C]-glucose was added to the media, and 0.15 g L−1 [1H13C]-L-lysine, and 0.15 g L−1 [1H,13C]-L-arginine was later added 1 h prior to induction. This culture was grown overnight at 37 °C and used to inoculate a final 2-L culture. The final culture was grown to OD600 ≈ 0.6 before a 16-h induction with 1 mM IPTG at 18 °C. The T4L L99A protein was initially purified by ion-exchange chromatography. After lysis by sonication in (50 mM NaPO4 (pH 6.5), 2 mM EDTA, 5 mg of DNAse1 (Sigma) and 1 cOmpleteTM Mini Protease Inhibitor Cocktail tablet (Sigma) per 50-mL lysate), the soluble fraction was loaded onto a 5-mL HiTrap SP Sepharose FF column (GE Healthcare) (50 mM NaPO4 (pH 6.5), 2 mM EDTA). Protein was eluted from the column using a gradient of 1 M NaCl and T4L99A eluted at ~300 mM NaCl. Pooled fractions were further purified by size-exclusion chromatography using a Superdex S75 gel filtration column (GE Healthcare) (50 mM NaPO4 (pH 5.5), 2 mM EDTA and 25 mM NaCl). Pooled fractions were buffer exchanged into the final NMR buffer (50 mM NaPO4 (pH 5.5), 2 mM EDTA, 25 mM NaCl and 2 mM NaN3). The NMR sample contained 1.4 mM protein in 95%/5% 1H2O/2H2O for the uniformly [2H13C15N] labelled sample and 1.5 mM protein in 100% 2H2O for the [1H13C-Lys,Arg; 2H12C] labelled sample.
Isotopically labelled MSG was produced as described previously26. The gene with a C-terminal hexahistidine tag in an kanamycin-resistant pET-28a vector was transformed into BL21 (DE3) chemically competent E. coli cells. A single colony was used to inoculate 5-mL culture, which was grown overnight at 37 °C. This was used to inoculate 50 mL of M9 minimal media made with 2H2O and supplemented with 1 g L−1 [1H,15N]-ammonium chloride and 2 g L−1 of [2H,13C]-glucose as the sole nitrogen and carbon sources, respectively. The pre-culture was used to inoculate 1 L of M9 media, which was grown at 37 °C to OD600≈0.45 before a 1-in-2 dilution to make the final 2-L culture volume. This final culture was grown to OD600 ≈ 0.45 before a > 16-h induction with 1 mM IPTG at 16 °C. The MSG protein was initially purified by affinity chromatography. After lysis by sonication (20 mM Tris, 300 mM NaCl, 20 mM 2-mercaptoethanol, 10 mg of DNase1 (Sigma), 10 mg of hen egg lysozyme (Sigma) and 1 cOmpleteTM Mini Protease Inhibitor Cocktail tablet (Sigma) per 50-mL lysate), the soluble fraction was loaded onto a HisTrap 5-mL HP column (GE Healthcare) (20 mM Tris, 300 mM NaCl and 20 mM 2-mercaptoethanol, pH 7.8). Protein was eluted from the column using a gradient of 250 mM imidazole in the same buffer and the MSG protein eluted at ~90 mM imidazole. Protein in pooled fractions was unfolded (20 mM Tris, 100 mM NaCl, 10 mM 2-mercaptoethanol and 6 M guanidium chloride, pH 7.8) for 1 h at room temperature to allow full exchange of amide protons. Protein was refolded by rapid dilution (~1:50) into ice-cold refolding buffer (20 mM Tris, 100 mM NaCl, 10 mM 2-mercaptoethanol, 5 mM MgSO4, 10% sucrose and 6 cOmpleteTM Mini Protease Inhibitor Cocktail tablets (Sigma) per litre buffer, pH 7.8). The protein was incubated, stirring at room temperature for 2 h before purification by affinity chromatography as described above. The pooled fractions were further purified by size-exclusion chromatography using a Superdex 200 5/150 gel filtration column (GE Healthcare) (20 mM NaPO4, 5 mM dithiothreitol) and the MSG eluted at ~75 mL. Overall, 20 mM MgCl2, 0.05% NaN3 and 10% 2H2O was added to the samples to make up the final NMR buffer.
Database extractions
The 13C chemical shift distributions for arginine, isoleucine, valine, threonine, lysine and proline shown in Supplementary Fig. 1 were extracted from the BMRB database20 using the PACSY database34 and the provided API (https://github.com/uwbmrb/BMRB-API), which was embedded in an in-house written python script.
NMR spectroscopy
All 13C-detected experiments were performed on a Bruker Avance II 600-MHz spectrometer using a 13C-optimised TXO coldprobe or a Bruker Avance HD 800-MHz spectrometer using an HCN inverse TCI coldprobe equipped with cooled 1H and 13C preamplifiers.
NMR experiments on T4L L99A
The two-dimensional 13C–13C correlation spectra in Fig. 2c, Supplementary Figs. 3 and 4 were all recorded at a static magnetic field of 14.1 T and a temperature of 278 K. The pulse sequence used was the one shown in Fig. 2a and Supplementary Fig. 2. Deuterium 2H decoupling was achieved using a constant-wave (CW) field applied at 1.1 kHz, which in our hands gave significantly better decoupling than using composite decoupling schemes, e.g. WALTZ1635. During the INEPT transfers, the 2H decoupling field was centred on the frequency of the 2H bound to 13Ct, while for indirect chemical shift evolution, the 2H decoupling field was centred on the frequency of the 2H bound to 13Cp. For example, for arginine 13Cδ–13Cγ, the 2H decoupling field was centred at 1.55 ppm (2Hγ) during the indirect chemical shift evolution and at 3.10 ppm elsewhere. Frequency-selective 90° (180°) pulses were applied with RE-BURP (E-BURP)36 shapes. Spectra shown to the left (right) in Supplementary Figs. 2 and 3 were recorded with 10 (33) complex points in the indirect dimension, 4 scans per transient and a recovery delay of 4 s, leading to a total acquisition time of 12 min (38 min) per spectrum.
The 13C-detected CEST experiments were recorded at 14.1 T using the pulse sequence shown in Supplementary Fig. 9. CEST experiments to characterise valine residues were recorded with 22 complex points in the 13Cβ dimension (sweep width of 800 Hz) and 50 CEST saturation points between −600 and 600 Hz. Eight scans were recorded per transient and a recovery delay of 4 s was used, leading to a total acquisition time of 40 h. CEST experiments to characterise arginine and threonine residues were recorded with 30 complex points in the 13Cγ dimension (sweep width of 1136 Hz) and 42 CEST saturation points between −300 and 300 Hz. Sixteen scans were recorded per transient and a recovery delay of 4.4 s was used, leading to a total acquisition time of 98 h. The CEST field, ωCEST, and its inhomogeneity was obtained, as described by Guenneugues et al.37, using a 20 mM sample of [U-2H,13C]-isoleucine. Uncertainties in I/I0 were estimated from duplicate measurements.
The 3D long-range 13C–13C scalar coupling experiment shown in Fig. 5a was recorded at 18.8 T and obtained using the pulse sequence shown in Supplementary Fig. 11. The spectrum was recorded with 42 complex points in the indirect 13Cγ1 constant-time period (t2), 24 complex points in the indirect t1 period, 8 scans per transient and a recovery delay of 4 s, leading to a total acquisition time of 75 h. Further experimental details are given in the legend of Supplementary Fig. 11.
NMR experiments on MSG
The two-dimensional 13C–13C correlation spectra in Fig. 1e and S8 were recorded at a static magnetic field of 18.8 T and a temperature of 310 K. The pulse sequence used was the one shown in Fig. 2a and Supplementary Fig. 2. The two spectra in Fig. 3 were recorded with 43 complex points in the indirect constant-time dimension, 16 scans per transient and a recovery delay of 10 s, leading to a total acquisition time of 7.5 h per spectrum.
The 3D 13C–13C-TOCSY spectrum shown in Fig. 3b was recorded using the pulse sequence described in Supplementary Fig. 8. The spectrum was recorded with 54 complex points in the indirect 13Cγ1 constant-time period (t2), 24 complex points in the indirect t1 period, 4 scans per transient and a recovery delay of 10 s, leading to a total acquisition time of 120 h. Further experimental details are given in the legend to Supplementary Fig. 8.
The 3D long-range 13C–13C scalar coupling experiment shown in Fig. 5b was obtained using the pulse sequence shown in Supplementary Fig. 11. The spectrum was recorded with 42 complex points in the indirect 13Cγ1 constant-time period (t2), 27 complex points in the indirect t1 period, 8 scans per transient and a recovery delay of 7.8 s, leading to a total acquisition time of 160 h. Further experimental details are given in the legend to Supplementary Fig. 11.
Chemical shift assignments
The side-chain chemical shift assignment of T4L L99A was obtained based on the assignments published previously6,11 combined with 1H and 13C-detected CC-TOCSY experiments and the 1H and 13C-detected long-range 3JCαCδ experiments. The stereospecific assignment of the valine 13Cγ was taken from ref. 38.
The chemical shift assignment of the MSG valine 13Cγ–13Cβ correlation spectrum was transferred from a previously published assignment26,27, that in turn was based on 1H-detected experiments. The chemical shift assignment of the isoleucine 13Cδ1–13Cγ1 spectrum was obtained by a combination of a previous assignment26,27 and the 13C-detected TOCSY experiment (Fig. 3b; Supplementary Fig. 8) and the 13C-detected long-range 3JCαCδ experiment (Fig. 5b; Supplementary Fig. 11).
Data analysis
All NMR spectra were processed using nmrPipe39 and initially analysed using NMRFAM-Sparky40 or CCPN41. Peak intensities in experiments reporting on relaxation rates and CEST, Fig. 4 and Supplementary Figs. 5, 7, 9 and 10, were obtained using FuDA42,43.
Long-range scalar couplings were derived by first obtaining the peak heights of the peaks corresponding to the diagonal peak, Id = I(13Cδ–13Cγ–13Cδ) and the cross-peak Ic = I(13Cα–13Cγ–13Cδ) using the inbuilt tools of NMRFAM-Sparky and CCPN. The long-range scalar coupling was then calculated according to4 Ic/Id = tan2(2πJCCTJ), where JCC is the three-bond scalar coupling and TJ is the coupling evolution delay in the pulse sequence in Supplementary Fig. 11. As described previously13, passive couplings cancel by taking the ratio of Ic and Id, such that this ratio only reports on the long-range coupling. For the 1H-detected experiments, the error was determined as the root-mean-square deviation of two experiments. For the other long-range scalar coupling experiments, errors in the obtained Ic and Id were estimated from RMSD of the spectral region, where no peaks were observed. The uncertainty of the calculated 3JCC was estimated using a Monte-Carlo procedure to propagate the errors from the intensities.
Carbon 13C-detected CEST experiments were analysed using in-house written software in python using the LMFIT44 module for least-squares minimisation of the target function
| 1 |
where and σ are the experimentally obtained normalised intensities and their uncertainty, respectively. The sum is over the different CEST saturation points and CEST field strengths, ωCEST. are the calculated normalised intentities calculated as a function of the model parameters p. Briefly, calculated intensities were obtained by evolving the spin system according to the Liouvillian described previously14,19,45,46, also taking into account the one-bond 13C–13C scalar coupling, which generally leads to a simple line broadening of the CEST profiles. An inhomogeneity of the saturation field of 5% was used. The model parameters consist of the chemical shifts of the ground and excited states, ϖG and ϖE, respectively, the longitudinal relaxation rate that was assumed to be identical in the ground and excited state, R1G = R1E = R1 and the transverse relaxation rate in the ground and excited states, R1G, R2E. For the analysis of R125, it was imposed that R2G = R2E, since the data did not contain enough information to determine R2E; for other fits, both R2G and R2E were determined.
The overall exchange range, kex, and the population of the excited state, pE, were treated as global parameters. Uncertainties of the obtained residue-specific parameters were estimated using the covariance method47, as implemented in LMFIT. The uncertainties of kex and pE were estimated by performing a grid search (see Supplementary Fig. 10).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Dr Vitali Tugarinov and Prof. Lewis E. Kay for kindly providing expression constructs of MSG and Dr Angelo Figueiredo and Dr Christopher Waudby for helpful discussions. Mr Harold W. Mackenzie is acknowledged for assisting with some of the pulse sequences. The Wellcome Trust (101569/z/13/z) and the Biotechnology and Biological Science Research Council (BBSRC; BB/R000255/1) are acknowledged for supporting the ISMB NMR facility at University College London. The Medical Research Council/Francis Crick Biomedical NMR centre is acknowledged for access to high-field NMR spectrometers. R.B.D. acknowledges the Wellcome Trust (102404/13/Z/13) for a PhD studentship. This research is supported by the Leverhulme Trust (RPG-2016-268).
Author contributions
R.B.P. produced isotope-labelled samples. R.B.P. and D.F.H. designed, performed and analysed all experiments. R.B.P. and D.F.H. wrote the paper.
Data availability
The data that support the findings of this study and the pulse programmes (Bruker) are available from the corresponding author upon reasonable request. The source data underlying Figs. 1, 4 and Supplementary Figs. 1, 5, 7 and 9 are provided as a Source Data file.
Code availability
The pulse programmes (sequences) are available for Bruker spectrometers from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Journal Peer Review Information: Nature Communications thanks Haribabu Arthanari, Ad Bax, and other anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-019-09743-4.
References
- 1.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang H, et al. Protein conformational dynamics probed by single-molecule electron transfer. Science. 2003;302:262–266. doi: 10.1126/science.1086911. [DOI] [PubMed] [Google Scholar]
- 3.Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheppard D, Sprangers R, Tugarinov V. Experimental approaches for NMR studies of side-chain dynamics in high-molecular-weight proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2010;56:1–45. doi: 10.1016/j.pnmrs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 6.Werbeck ND, Kirkpatrick J, Hansen DF. Probing arginine side-chains and their dynamics with carbon-detected NMR spectroscopy: application to the 42 kDa human histone deacetylase 8 at high pH. Angew. Chem. Int. Ed. Engl. 2013;52:3145–3147. doi: 10.1002/anie.201209385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trbovic N, et al. Protein side-chain dynamics and residual conformational entropy. J. Am. Chem. Soc. 2009;131:615–622. doi: 10.1021/ja806475k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esadze A, Li DW, Wang T, Brüschweiler R, Iwahara J. Dynamics of lysine side-chain amino groups in a protein studied by heteronuclear 1H−15N NMR spectroscopy. J. Am. Chem. Soc. 2011;133:909–919. doi: 10.1021/ja107847d. [DOI] [PubMed] [Google Scholar]
- 9.Stafford KA, Ferrage F, Cho JH, Palmer AG. Side chain dynamics of carboxyl and carbonyl groups in the catalytic function of Escherichia coli Ribonuclease H. J. Am. Chem. Soc. 2013;135:18024–18027. doi: 10.1021/ja409479y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson KM, et al. Direct observation of the ion-pair dynamics at a protein–DNA interface by NMR spectroscopy. J. Am. Chem. Soc. 2013;135:3613–3619. doi: 10.1021/ja312314b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouvignies G, et al. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature. 2011;477:111–117. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeymer C, Werbeck ND, Zimmermann S, Reinstein J, Hansen DF. Characterizing active site conformational heterogeneity along the trajectory of an enzymatic phosphoryl transfer reaction. Angew. Chemie Int. Ed. 2016;55:11533–11537. doi: 10.1002/anie.201606238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bax A, Max D, Zax D. Measurement of long-range 13C-13C J couplings in a 20 kDa protein-peptide complex. J. Am. Chem. Soc. 1992;114:6923–6925. doi: 10.1021/ja00043a052. [DOI] [Google Scholar]
- 14.Bouvignies G, Vallurupalli P, Kay LE. Visualizing side chains of invisible protein conformers by solution NMR. J. Mol. Biol. 2014;426:763–774. doi: 10.1016/j.jmb.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 15.Kitevski-LeBlanc JL, et al. Investigating the dynamics of destabilized nucleosomes using methyl-TROSY NMR. J. Am. Chem. Soc. 2018;140:4774–4777. doi: 10.1021/jacs.8b00931. [DOI] [PubMed] [Google Scholar]
- 16.Rosenzweig R, Kay LE. Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu. Rev. Biochem. 2014;83:291–315. doi: 10.1146/annurev-biochem-060713-035829. [DOI] [PubMed] [Google Scholar]
- 17.Huang C, Kalodimos CG. Structures of large protein complexes determined by nuclear magnetic resonance spectroscopy. Annu. Rev. Biophys. 2017;46:317–336. doi: 10.1146/annurev-biophys-070816-033701. [DOI] [PubMed] [Google Scholar]
- 18.Felli IC, Pierattelli R. Novel methods based on (13)C detection to study intrinsically disordered proteins. J. Magn. Reson. 2014;241:115–125. doi: 10.1016/j.jmr.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Vallurupalli P, Bouvignies G, Kay LE. Studying ‘invisible’ excited protein states in slow exchange with a major state conformation. J. Am. Chem. Soc. 2012;134:8148–8161. doi: 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- 20.Markley JL, et al. BioMagResBank (BMRB) as a partner in the Worldwide Protein Data Bank (wwPDB): new policies affecting biomolecular NMR depositions. J. Biomol. NMR. 2008;40:153–155. doi: 10.1007/s10858-008-9221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marion D, Ikura M, Tschudin R, Bax A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J. Magn. Reson. 1989;85:393–399. [Google Scholar]
- 22.Bermel W, Bertini I, Felli IC, Kümmerle R, Pierattelli R. Novel 13C direct detection experiments, including extension to the third dimension, to perform the complete assignment of proteins. J. Magn. Reson. 2006;178:56–64. doi: 10.1016/j.jmr.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Santoro J, King GC. A constant-time 2D overbodenhausen experiment for inverse correlation of isotopically enriched species. J. Magn. Reson. 1992;97:202–207. [Google Scholar]
- 24.Hansen DF, Kay LE. Determining valine side-chain rotamer conformations in proteins from methyl 13C chemical shifts: application to the 360 kDa half-proteasome. J. Am. Chem. Soc. 2011;133:8272–8281. doi: 10.1021/ja2014532. [DOI] [PubMed] [Google Scholar]
- 25.Hansen DF, Neudecker P, Kay LE. Determination of isoleucine side-chain conformations in ground and excited states of proteins from chemical shifts. J. Am. Chem. Soc. 2010;132:7589–7591. doi: 10.1021/ja102090z. [DOI] [PubMed] [Google Scholar]
- 26.Tugarinov V, Muhandiram R, Ayed A, Kay LE, Four-dimensional NMR. spectroscopy of a 723-residue protein: chemical shift assignments and secondary structure of malate synthase G. J. Am. Chem. Soc. 2002;124:10025–10035. doi: 10.1021/ja0205636. [DOI] [PubMed] [Google Scholar]
- 27.Tugarinov V, Kay LE. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]
- 28.Kerfah R, Hamelin O, Boisbouvier J, Marion D. CH3-specific NMR assignment of alanine, isoleucine, leucine and valine methyl groups in high molecular weight proteins using a single sample. J. Biomol. NMR. 2015;63:389–402. doi: 10.1007/s10858-015-9998-4. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- 30.Fawzi NL, Ying J, Ghirlando R, Torchia DA, Clore GM. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature. 2011;480:268–272. doi: 10.1038/nature10577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerecht K, Figueiredo AM, Hansen DF. Determining rotational dynamics of the guanidino group of arginine side chains in proteins by carbon-detected NMR. Chem. Commun. 2017;53:10062–10065. doi: 10.1039/C7CC04821A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackenzie HW, Hansen DF. Arginine side-chain hydrogen exchange: quantifying arginine side-chain interactions in solution. ChemPhysChem. 2019;20:252–259. doi: 10.1002/cphc.201800598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vallurupalli P, Hansen DF, Lundström P, Kay LE. CPMG relaxation dispersion NMR experiments measuring glycine 1H alpha and 13C alpha chemical shifts in the ‘invisible’ excited states of proteins. J. Biomol. NMR. 2009;45:45–55. doi: 10.1007/s10858-009-9310-6. [DOI] [PubMed] [Google Scholar]
- 34.Lee W, et al. PACSY, a relational database management system for protein structure and chemical shift analysis. J. Biomol. NMR. 2012;54:169–179. doi: 10.1007/s10858-012-9660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaka AJ, Keeler J, Frenkiel T, Freeman R. An improved sequence for broadband decoupling: WALTZ-16. J. Magn. Reson. 1983;52:335–338. [Google Scholar]
- 36.Geen H, Freeman R. Band-selective radiofrequency pulses. J. Magn. Reson. 1991;93:93–141. [Google Scholar]
- 37.Guenneugues M, Berthault P, Desvaux H. A method for determining B1 field inhomogeneity. Are the biases assumed in heteronuclear relaxation experiments usually underestimated? J. Magn. Reson. 1999;136:118–126. doi: 10.1006/jmre.1998.1590. [DOI] [PubMed] [Google Scholar]
- 38.Mulder FA, Hon B, Mittermaier A, Dahlquist FW, Kay LE. Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 2002;124:1443–1451. doi: 10.1021/ja0119806. [DOI] [PubMed] [Google Scholar]
- 39.Delaglio F, et al. Nmrpipe - a multidimensional spectral processing system based on Unix Pipes. J. Biomol. Nmr. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 40.Lee W, Tonelli M, Markley JL. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015;31:1325–1327. doi: 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vranken WF, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 42.Kristensen, S. M. & Hansen, D. F. FuDA: a function and data fitting and analysis package. (www.ucl.ac.uk/hansen-lab/) (2006).
- 43.Hansen DF, et al. Exchange-free measure of 15N transverse relaxation: NMR eval folding Int with pervasive chemical exchange. J. Am. Chem. Soc. 2007;129:11468–11479. doi: 10.1021/ja072717t. [DOI] [PubMed] [Google Scholar]
- 44.Newville, M., Stensitzki, T., Allen, D. B., & Ingargiola, A. LMFIT: non-linear least-square minimization and curve-fitting for python. https://lmfit.github.io/lmfit-py10.5281/zenodo.11813 (2014).
- 45.Vallurupalli P, Hansen DF, Stollar E, Meirovitch E, Kay LE. Measurement of bond vector orientations in invisible excited states of proteins. Proc Natl Acad Sci USA. 2007;104:18473–18477. doi: 10.1073/pnas.0708296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen DF, Vallurupalli P, Lundstrom P, Neudecker P, Kay LE. Probing chemical shifts of invisible states of proteins with relaxation dispersion NMR spectroscopy: how well can we do? J Am Chem Soc. 2008;130:2667–2675. doi: 10.1021/ja078337p. [DOI] [PubMed] [Google Scholar]
- 47.Press, W. H., Teukolsky, S. A., Vetterling, W. T. & Flannery, B. P. Numerical Recipes in C. Technometrics29, 682–687 (1992).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study and the pulse programmes (Bruker) are available from the corresponding author upon reasonable request. The source data underlying Figs. 1, 4 and Supplementary Figs. 1, 5, 7 and 9 are provided as a Source Data file.
The pulse programmes (sequences) are available for Bruker spectrometers from the corresponding author upon request.