

Summary/Abstract
The PARP family of ADP-ribosyl transferases contains 17 members in human cells, most of which catalyze the transfer of the ADP-ribose moiety of NAD+ onto their target proteins. This post-translational modification plays important roles in cellular signaling, especially during cellular stresses, such as heat shock, inflammation, unfolded protein responses, and DNA damage. Knowing the specific proteins that are substrates for individual PARPs, as well as the specific amino acid residues in a give target protein that are ADP-ribosylated, is a key step in understanding the biology of individual PARPs. Recently, we developed a robust NAD+ analog-sensitive approach for PARPs, which allows PARP-specific ADP-ribosylation of substrates that is suitable for subsequent copper-catalyzed azide-alkyne cycloaddition (‘click chemistry’) reactions. When coupled with proteomics and mass spectrometry, the analog-sensitive PARP approach can be used to identify the specific amino acids that are ADP-ribosylated by individual PARP proteins. In this chapter, we describe the key facets of the experimental design and application of the analog-sensitive PARP methodology to identify site-specific modification of PARP target proteins.
Keywords: ADP-ribosylation, Analog-sensitivity, Automodification, Click chemistry, Mono(ADP-ribosyl)ation, Mutation, NAD+ analog, PARP, Poly(ADP-ribose) polymerase, Poly(ADP-ribosyl)ation, Post-translational modification
1. Introduction
ADP-ribosylation of proteins by PARP family members is a posttranslational modification that plays important roles in a variety of cellular processes, from the regulation of chromatin and transcription to protein translation and stability [3, 11, 17]. A number of studies over the past five years have led to development of methods for detecting site-specific ADP-ribosylation of proteins [7]. Nonetheless, the study of how PARPs function in cells has been hindered by the lack of a robust and definitive method for identifying the substrate proteins of individual PARP enzymes. This is due to a number of technical and biological issues. First, all PARPs use the same cofactor, NAD+, as a donor of ADP-ribose groups to catalyze ADP-ribosylation, providing no selectivity at the level of enzymology [3, 11, 17]. Second, PARP inhibitors are insufficiently selective at inhibiting a single PARP protein’s catalytic activity without off-target effects within the PARP family [10, 25]. Finally, a number of studies have shown that PARP proteins cooperate and exhibit considerable functional interplay in vivo (see for example: [1, 13, 15, 16, 18, 21, 22, 24]), suggesting that attributing ADP-ribosylation events to specific PARP enzymes using experimental approaches based knockout, knockdown, or chemical inhibition may be misleading.
Similar challenges exist for the identification of phosphorylation events catalyzed by individual kinases. The experimental solutions found for kinases have been instructive for other protein modifying enzymes. In this regard, Shokat and colleagues developed and applied an analog-sensitive (“bump and hole”) approach in an effort to understand how individual kinases contribute to complex and interdependent phosphorylation-driven signal transduction events [2]. Recently, we and others have adopted this approach in an effort to make a similar advance in understanding PARP-specific ADP-ribosylation [4, 5, 12]. In the analog-sensitive approach, a “gatekeeper” mutation is engineered in the active site of an enzyme to create a “hole” that can be complemented by the addition of a chemical moiety (a “bump”) on the small molecule substrate (e.g., ATP for kinases, NAD+ for PARPs) [2]. This approach creates an enzyme capable of using an unnatural substrate that is unusable by wild-type enzymes. Critical considerations for this approach are (1) the extent to which the modified small molecule substrate is unusable by the wild-type enzyme, (2) the amount of catalytic activity for the analog-sensitive enzyme with the chemically-modified small molecule substrate, and (3) the location of the gatekeeper mutation within the substrate binding pocket. With respect to the latter, burying the mutation within the substrate binding pocket is essential, since it allows for the preservation of contacts between the enzyme and its target protein, allowing for high fidelity modification [4].
We have recently reported an NAD+ analog-sensitive PARP approach using a single bi-functional and “clickable” NAD+ analog [12] (Figure 1, A and B). In a focused mutagenic screen of PARP-1 with a set of 11 NAD+ analogs, we identified two gatekeeper residues, leucine 877 (L877) and isoleucine 895 (I895), that confer analog-sensitivity with a subset of NAD+ analog when mutated to alanine. Both of these residues are highly conserved across the PARP family (Figure 1C). Mutation of residues homologous to L877 in PARP-2 and PARP-3 (i.e., L443 and L394, respectively) confers analog-sensitivity to those enzymes as well. We then functionalized the R group of the most active NAD+ analog, 8-Butylthio-NAD+, with an alkyne moiety to generate 8-Bu(3-yne)T-NAD+ (Figure 1B). Catalytic activity of PARP-1(L877A), PARP-2(L443), and PARP-3(L394A) with 8-Bu(3-yne)T-NAD+ allows for PARP-specific modification of protein targets with an unnatural alkyne-containing 8-Bu(3-yne)T-ADP-ribose, which is suitable for subsequent copper-catalyzed azide-alkyne cycloaddition (‘click chemistry’) reactions (Figure 2). The protein-linked 8-Bu(3-yne)T-ADP-ribose can be conjugated (“clicked”) to a fluor, biotin, or solid supports (e.g., agarose) (Figure 2). Combining this analog-sensitive PARP approach with previously described proteomic and mass spectrometry-based methods for identification of site-specific ADP-ribosylation (e.g., hydroxylamine-dependent identification glutamate and aspartate ADP-ribosylation [27]) allows for systematic and robust identification of PARP-specific modification events (Figure 2). The biological functions of these sites can then be explored in a variety of PARP-specific assays with ADP-ribosylation site point mutants. Identifying the target proteins of specific PARP family members will aid in developing a more thorough understanding of the biological functions of individual PARP proteins, and potentially guide a more effective and targeted use of PARP inhibitors for the treatment of human diseases.
Figure 1. The asPARP approach and a multiple sequence alignment of the human PARP family of proteins focusing on the D-loop.
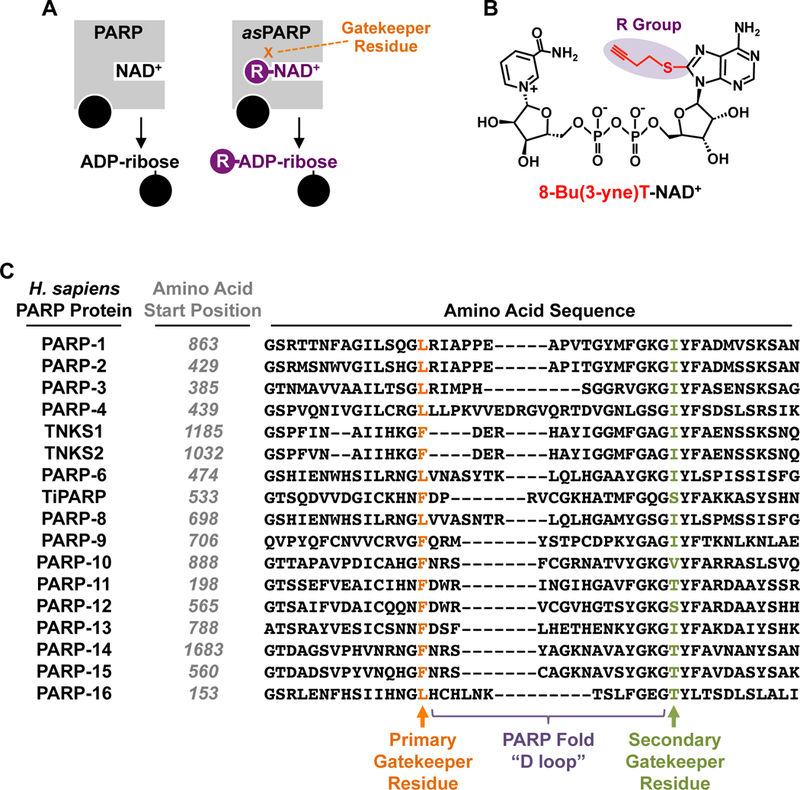
(A) Schematic representation of the asPARP approach using a single bi-functional and “clickable” NAD+ analog.
(B) Chemical structure of the clickable NAD+ analog 8-Bu(3-yne)T-NAD+.
(C) Multiple sequence alignment of a portion of the PARP catalytic domain, focusing on the D-loop, for all proteins in the human PARP family. The gatekeeper residues that confer NAD+ analog sensitivity are highlighted. For each PARP protein, the number of the first amino acid shown in the linear sequence of that PARP protein is indicated. “Primary Gatekeeper” refers to the amino acid in the PARP-1 catalytic domain where the most robust analog sensitivity was observed upon mutation to alanine using an automodification-based screen with a library of NAD+ analogs [12]. “Secondary Gatekeeper” refers to a second amino acid whose mutation also conferred analog sensitivity, but to a lesser extent and within a more constrained chemical space of NAD+ analogs [12]. These two sites are separated by the PARP “D loop,” but are located in proximity in three-dimensional space [12]. Although primary gatekeeper residue has been most useful for the asPARP approach to date, other NAD+ analogs screened in the future may show preferential activity with the secondary gatekeeper residue.
Figure 2. PARP-specific identification of target proteins and ADP-ribosylation sites using asPARPs and a clickable NAD+ analog.
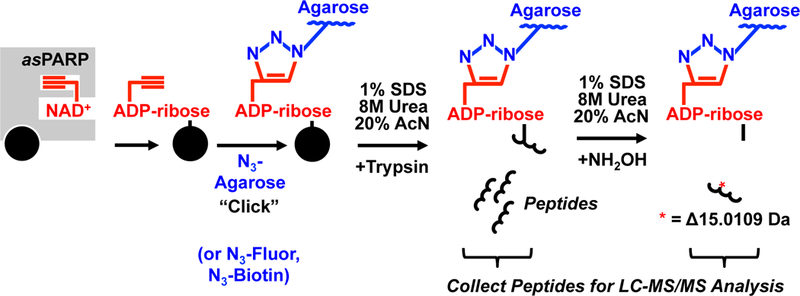
Schematic representation of the asPARP methodology used to determine aspartate and glutamate amino acids ADP-ribosylated by an specific PARP family member. In this approach, an asPARP protein (grey square) uses the alkyne group-containing NAD+ analog 8-Bu(3-yne)T-NAD+ (shown in red) to catalyze transfer of 8-Bu(3-yne)T-ADP-ribose (also shown in red) onto a target protein (black circle). The alkyne group of 8-Bu(3-yne)T-ADP-ribose is used to covalently conjugate the PARP target protein to azido-agarose resin (N3-Agarose) (shown in blue) through the use of an orthogonal copper-catalyzed cycloaddition reaction, also known as “click” chemistry. Following conjugation, the resin is washed thoroughly with different strong denaturants and subjected to trypsinization to release unconjugated peptides of PARP target proteins, which can be collected and analyzed by LC-MS/MS analysis. After trypsinization, the resin is washed thoroughly again and treated with hydroxylamine (NH2OH) to release the conjugated peptides. The released conjugated peptides will exhibit a molecular weight change of 15.0109 Daltons and a signature m/z shift, which can be used to determine the specific site of ADP-ribosylation by mass spectrometry.
2. Materials
All reagents should be the highest quality molecular biology and biochemistry grade.
2.1. asPARP-dependent labeling of PARP target proteins
50 mL conical tubes.
9x PARP Reaction Buffer: 270 mM HEPES (pH 8.0), 45 mM MgCl2, 45 mM CaCl2, 0.09% NP-40, 9 mM DTT, 900 µg/mL sonicated salmon sperm DNA (sssDNA), 900 µg/mL bovine serum albumin. The sssDNA is only required for DNA-dependent PARPs, such as PARPs 1, 2, and 3.
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
Purified recombinant analog-sensitive PARP (asPARP) protein: N-terminal FLAG epitope-tagged PARP-1(L877A), PARP-2(L443A), or PARP-3(L394A) expressed in insect cells using a baculovirus expression vector and purified using anti-FLAG immunoaffinity chromatography, as described previously [12]. Other purified recombinant asPARP of interested can be substituted for the asPARP-1, 2, and 3 proteins.
Nuclear extract: Nuclear extract from HeLa S3 cells, or a cell type of interest, prepared as previously described [9].
1.7 mL microcentrifuge tubes.
8-Bu(3-yne)T-NAD+ (Biolog Life Science Institute).
2.2. Methanol:chloroform precipitation of the asPARP labeling reaction
100% Methanol.
50 mL conical tube.
100% Chloroform.
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
Urea Solubilization Buffer: 200 mM HEPES (pH 8.0), 8 M urea, 1 M NaCl, 4% CHAPS, 2x Protease Inhibitor Cocktail (e.g., Roche Complete Protease Inhibitor) (See Note 1).
2.3. Visualization of 8-Bu(3-yne)T-ADP-ribosylated proteins by in-gel fluorescence (Optional steps)
2.3.1. Fluorescent labeling of 8-Bu(3-yne)T-ADP-ribosylated proteins using click chemistry
1.7 mL microcentrifuge tubes.
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
100 mM Copper sulfate (CuSO4) dissolved in MilliQ H2O (See Note 1).
500 mM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) dissolved in MilliQ H2O (See Note 2).
Azido-tetramethylrhodamine (N3-TAMRA), a clickable form of the fluorescent dye rhodamine.
Aminoguanidine hydrochloride (See Note 1).
Sodium ascorbate (See Note 1).
2.3.2. Methanol:chloroform precipitation of fluorescently labeled PARP target proteins
100% Methanol.
100% Chloroform.
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
1x SDS Loading Buffer: 60 mM Tris•HCl (pH 6.8), 10% Glycerol, 0.01% bromophenol blue, 2% SDS, 5% % mercaptoethanol.
2.3.3. In-gel fluorescence visualization of 8-Bu(3-yne)T-ADP-ribosylated proteins
10% acrylamide-SDS gel.
100% Methanol.
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
2.4. Preparation of azido-agarose resin
Azido-agarose Beads (e.g., Click Chemistry Tools; Catalog no. 1038–2).
1.7 mL microcentrifuge tubes.
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
2.5. Bio-orthogonal conjugation of labeled proteins to agarose
100 mM Copper sulfate (CuSO4) dissolved in MilliQ H2O (See Note 1).
500 mM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA) dissolved in MilliQ H2O (See Note 2).
2 mL microcentrifuge tubes.
Azido-agarose Beads (e.g., Click Chemistry Tools; Catalog no. 1038–2).
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
Aminoguanidine hydrochloride (See Note 1).
Sodium ascorbate (See Note 1).
Parafilm.
Aluminum foil.
2.6. Cysteine alkylation
2 mL microcentrifuge tubes.
Azido-agarose Beads (e.g., Click Chemistry Tools; Catalog no. 1038–2).
MilliQ H2O or otherwise >18 MΩ double distilled H2O.
SDS Wash Buffer: 100 mM Tris•HCl (pH 8.0), 1% SDS, 250 mM NaCl, 5 mM EDTA.
Dithiothreitol (DTT).
Iodoacetamide.
2.7. Washing and proteolysis of 8-Bu(3-yne)T-ADP-ribosylated proteins conjugated to agarose resin
Razor Blade
2 mL bed volume single use chromatography column (e.g., Pierce; Catalog no. 89896).
Azido-agarose Beads (e.g., Click Chemistry Tools; Catalog no. 1038–2).
SDS Wash Buffer: 100 mM Tris•HCl (pH 8.0), 1% SDS, 250 mM NaCl, 5 mM EDTA.
Urea Wash Buffer: 100 mM Tris•HCl (pH 8.0), 8 M urea (See Note 1).
20% acetonitrile.
Trypsin Digestion Buffer: 100 mM Tris•HCl (pH 8.0), 2 mM CaCl2, 10% acetonitrile.
1.7 mL microcentrifuge tubes.
Mass spectrometry grade trypsin (e.g., Promega Trypsin Gold; Catalog no. V5280).
C18 StageTips (Thermo).
2.8. Washing and NH2OH elution of 8-Bu(3-yne)T-ADP-ribosylated peptides conjugated to agarose resin
Razor Blade
2 mL bed volume single use chromatography column (e.g., Pierce; Catalog no. 89896).
Azido-agarose Beads (e.g., Click Chemistry Tools; Catalog no. 1038–2).
SDS Wash Buffer: 100 mM Tris•HCl (pH 8.0), 1% SDS, 250 mM NaCl, 5 mM EDTA.
Urea Wash Buffer: 100 mM Tris•HCl (pH 8.0), 8M urea (See Note 1).
20% acetonitrile.
Peptide Elution Buffer: 100 mM HEPES (pH 8.5).
1.7 mL microcentrifuge tubes.
Hydroxylamine (NH2OH; Sigma).
Mass spectrometry grade trypsin (e.g., Promega Trypsin Gold; Catalog no. V5280).
C18 StageTips (Thermo).
3. Methods
3.1. asPARP-dependent labeling of PARP target proteins.
In 50 mL conical tube, combine 111 µL of 9x PARP Reaction Buffer, 584 µL MilliQ H2O, and 30 µL of asPARP protein (0.667 mg/mL; 20 µg total) and incubate for 5 minutes at room temperature. The 5 minute incubation of the asPARP protein with sssDNA is only required for DNA-dependent PARPs, such as PARPs 1, 2, and 3.
Thaw 250 µL of Nuclear Extract (4 mg/mL; 1 mg total) and bring it to room temperature.
Add 250 µL of room temperature Nuclear Extract to the reaction described in Step 1 above in the 50 mL conical tube and mix by gentle pipetting or gentle swirling, followed by a 5 minute incubation at room temperature. During this time, the asPARP protein will interact with proteins in the nuclear extract.
Thaw and bring 25 µL of 8-Bu(3-yne)T-NAD+ to room temperature in a 1.7 mL microcentrifuge tube.
Add 25 µL of 8-Bu(3-yne)T-NAD+ (10 mM; 250 μM final concentration) to the reaction described in Step 3 in the 50 mL conical tube for a final reaction volume of 1 mL. Mix by gentle pipetting or gentle swirling, followed by a 15 minute incubation at room temperature.
3.2. Methanol:chloroform precipitation of the asPARP labeling reaction
Stop the labeling reaction by from 3.1, Step 5 by the addition of 4 mL of 100% methanol in the 50 mL conical tube, followed by thorough mixing.
Add 1 mL of 100% chloroform to the 50 mL conical tube and mix thoroughly.
Add 3 mL of MilliQ H2O to the 50 mL conical tube and vortex until a cloudy white protein precipitate forms and the solution is mixed to homogeneity.
Centrifuge the 50 mL conical tube at 1000 RCF for 10 minutes to separate the aqueous and hydrophobic layers.
Remove as much of the top layer (containing a methanol and water solution) as possible and discard it, taking care not to disrupt the interface.
Add 4 mL of 100% methanol to the 50 mL conical and swirl the tube gently to wash the precipitated proteins at the interface until the solution is fully mixed.
Centrifuge the 50 mL conical tube at 1000 RCF for 10 minutes to collect the precipitated proteins in a pellet.
Decant the supernatant, taking care not to disturb the protein pellet. Allow the pellet to air dry briefly, but do not over-dry it (it should be damp, not completely dehydrated).
Dissolve the protein pellet in 1 mL of freshly prepared Urea Solubilization Buffer in the 50 mL conical tube by gentle swirling. Transfer the dissolved proteins to a new 1.5 mL microcentrifuge tube (See Note 3).
Centrifuge the solution for 10 minutes in a microcentrifuge tube at maximum speed at room temperature to clarify.
Carefully move the supernatant containing the labeled and solubilized proteins in Urea Solubilization Buffer into a fresh 2 mL microcentrifuge tube and store on ice.
3.3. Visualization of 8-Bu(3-yne)T-ADP-ribosylated proteins by in-gel fluorescence (Optional steps)
3.3.1. Fluorescent labeling of 8-Bu(3-yne)T-ADP-ribosylated proteins using click chemistry
Transfer 50 µL from the 1 mL total of labeled and solubilized proteins from 3.2. Step 11 into a fresh 1.7 mL microcentrifuge tube.
Add 45 µL of MilliQ H2O and mix gently. Store on ice.
Prepare the copper catalyst by combining 10 µL of 100 mM CuSO4 and 10 µL of 500 mM THPTA (1:1 ratio) in a 1.5 mL microcentrifuge tube by pipetting up and down . Store at room temperature until use (See Note 4).
- Add the following in the order listed to the 95 µL of labeled, solubilized, and diluted proteins from Step 2, with each addition followed by gentle mixing to homogeneity by flicking of the tube:
- 1 µL of N3-TAMRA.
- 2 µL of 50:250 mM CuSO4:THPTA pre-formed catalytic complex.
- 1 µL of 500 mM Aminoguanidine hydrochloride.
- 1 µL of 500 mM Sodium ascorbate.
Incubate the reaction in the dark for 2 hours to allow bio-orthogonal conjugation.
3.3.2. Methanol:chloroform precipitation of fluorescently labeled PARP target proteins
Add 400 µL of 100% methanol to the bio-orthogonal conjugation reaction from 3.3.1 Step 5, followed by thorough mixing.
Add 100 µL of chloroform and mix thoroughly.
Add 300 µL of MilliQ H2O and vortex the solution until a protein precipitate forms and the solution is mixed to homogeneity.
Centrifuge the mixture in a microcentrifuge at 1000 RCF for 10 minutes to separate the aqueous and hydrophobic layers.
Remove as much of the top layer (containing a methanol and water solution) as possible and discard it, taking care not to disrupt the interface.
Add 400 µL of 100% methanol to the tube and flick it gently to wash the precipitated proteins at the interface until the solution is fully mixed.
Centrifuge the mixture in a microcentrifuge at 1000 RCF for 10 minutes to collect the precipitated proteins in a pellet.
Decant the supernatant, taking care not to disturb the protein pellet. Allow the pellet to air dry briefly, but do over-dry it (it should be damp, not completely dehydrated). The protein pellets should look similar to those shown in Figure 3A (See Note 5).
Add 25 µL of 1x SDS-PAGE Loading Buffer to each pellet and incubate at 65°C with occasional flicking until protein pellet is dissolved and is no longer visible (~15 to 20 minutes) (See Note 6).
Denature the proteins fully by incubation at 100°C for 5 minutes.
Remove any insoluble proteins by centrifugation for 10 minutes at maximum speed in a microcentrifuge.
Transfer 25 µL of the soluble proteins in 1x SDS-PAGE Loading Buffer into a fresh 1.7 mL microcentrifuge tube.
Figure 3. Examples of asPARP-1-dependent labeling of proteins and in-gel fluorescence analyses.
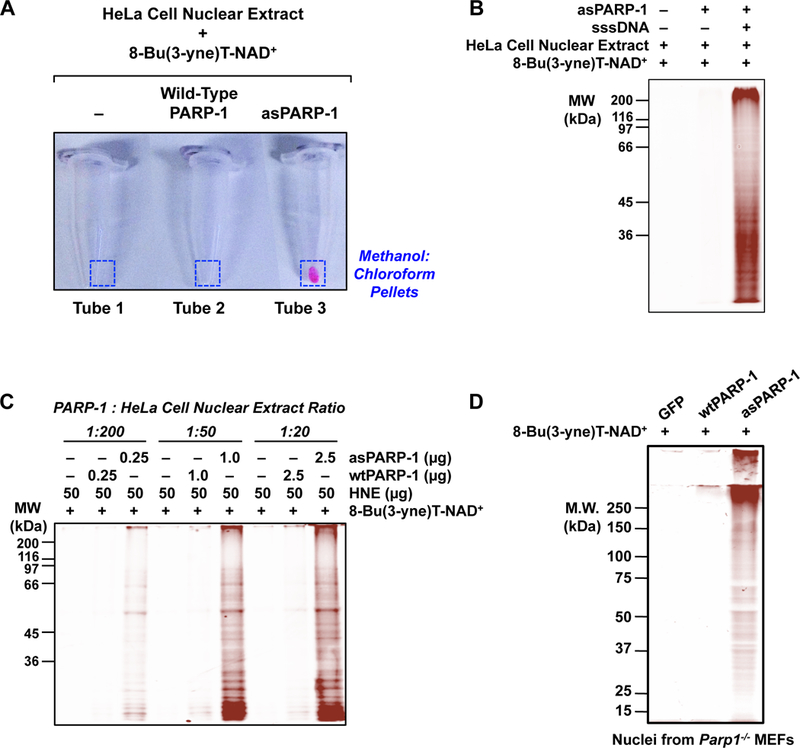
(A) An example of fluor incorporation into methanol:chloroform extracted protein pellets following bio-orthogonal conjugation of 8-Bu(3-yne)T-ADP-ribosylated proteins with azido-TAMRA dye. Only the combination of asPARP-1, 8-Bu(3-yne)T-NAD+, and HeLa cell nuclear extract proteins yields a detectable fluor-labeled product.
(B) An example of DNA- and asPARP-1-dependent 8-Bu(3-yne)T-ADP-ribosylation of HeLa cell nuclear extract proteins, with subsequent visualization of the PARP-1 target proteins by ingel fluorescence. The 8-Bu(3-yne)T-ADP-ribosylated proteins were visualized by bio-orthogonal conjugation with azido-TAMRA dye following separation by SDS-PAGE. Sheared salmon sperm DNA (sssDNA) was added to activate the asPARP-1 catalytic activity. Molecular weight markers (in kiloDaltons) are shown.
(C) The assay shown is similar to the one shown in (B), except that different mass ratios of PARP-1 protein to HeLa cell nuclear extract protein were used: 1:200, 1:50, or 1:20. Wild-type PARP-1 (wtPARP-1) or analog-sensitive PARP-1 (asPARP-1) proteins were mixed with HeLa cell nuclear extract protein in the ratios specified and incubated with 8-Bu(3-yne)T-NAD+. The 8-Bu(3-yne)T-ADP-ribosylated proteins were visualized by bio-orthogonal conjugation with azido-TAMRA dye following separation by SDS-PAGE. Sheared salmon sperm DNA (sssDNA) was added to each reaction to activate the asPARP-1 catalytic activity. Molecular weight markers (in kiloDaltons) are shown.
(D) Intact nuclei isolated from PARP-1 knockout mouse embryonic fibroblasts (Parp1−/− MEFs) with ectopic expression of GFP (as a control), wild-type PARP-1 (wtPARP-1), or analog-sensitive PARP-1 (asPARP-1), were incubated with 250 µM 8-Bu(3-yne)T-NAD+ to stimulate asPARP-1-mediated ADP-ribosylation. The 8-Bu(3-yne)T-ADP-ribose-labeled nuclear proteins were then clicked to azido-rhodamine, extracted with methanol:chloroform, and separated by SDS-PAGE. The gel was analyzed on a fluorescence imager.
3.3.3. In-gel fluorescence visualization of 8-Bu(3-yne)T-ADP-ribosylated proteins
Load 10 to 20 µL of the solubilized and labeled protein solution on a 10% acrylamide-SDS gel and run at 20 mA per gel until the non-clicked TAMRA at the dye front runs out of gel (See Note 7).
Remove the resolving portion of the SDS-PAGE gel with a clean razor blade and wash the gel in a clean vessel twice for 10 minutes each in a solution of 10% methanol in MilliQ H2O.
Wash the gel twice for 10 minutes each in MilliQ H2O.
Visualize the fluor-labeled 8-Bu(3-yne)T-ADP-ribosylation in the gel using a fluorescence-capable imaging system (e.g., Bio-Rad Pharos FX plus, Bio-Rad Chemidoc, or equivalent) (Figure 3B).
3.4. Preparation of azido-agarose resin
Transfer 200 µL of a 50% slurry of azido-agarose beads into a 1.7 mL microcentrifuge tube.
Add 1 mL of MilliQ H2O to the azido-agarose resin and mix well by gentle inversion of the tube.
Collect the azido-agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Remove and discard the supernatant, taking care not to disturb the resin pellet.
Repeat Steps 2 through 4 twice for a total of 3 washes of the azido-agarose resin. Resuspend the final washed azido-agarose pellet, which should have ~100 µL of bed volume, in 820 µL of MilliQ H2O.
3.5. Bio-orthogonal conjugation of labeled proteins to Agarose
Prepare the copper catalyst by combining 30 µL of 100 mM CuSO4 made in H2O and 30 µL of 500 mM THPTA (1:1 ratio) by pipetting up, and down and store at room temperature until use (See Note 4).
- Move the 2 mL tube from 3.3 Step 11 containing 8-Bu(3-yne)T-ADP-ribosylated Proteins in Urea Solubilization Buffer from ice to room temperature (See Note 8) and add the following components one at a time, in order, with each step followed by gentle flicking of the tube until the added components are mixed to homogeneity:
- 100 µL bed volume equivalent of washed azido-agarose beads resuspended in 820 µL of MilliQ H2O, as described in 3.4 Step 5.
- 40 µL of 50:250 mM CuSO4:THPTA in a pre-formed catalytic complex, as described in 3.5 Step 1.
- 20 µL of 500 mM Aminoguanidine hydrochloride.
- 20 µL of 500 mM Sodium ascorbate.
Wrap parafilm around the closed lid of the 2 mL microcentrifuge tube following addition of all of the copper-catalyzed click chemistry components (See Note 9), cover the tube with aluminum foil (See Note 10), and incubate on a rotisserie-style mixer at a low speed (~10– 15 RPM) for 18 hours at room temperature.
3.6. Cysteine alkylation
Collect the “clicked” agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF in a microcentrifuge.
Aspirate the supernatant and discard, taking care not to disturb the pellet.
Resuspend the agarose resin in 1.8 mL MilliQ H2O and wash resin by gentle inversion of the microcentrifuge tube until the agarose resin is thoroughly mixed.
Collect the agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Aspirate the supernatant and discard, taking care not to disturb the pellet.
Resuspend the agarose resin in 1 mL of SDS Wash Buffer (100 mM Tris•HCl, pH 8.0, 1% SDS, 250 mM NaCl, 5 mM EDTA) supplemented with 1 mM DTT (freshly made) and incubate the resin in SDS Wash Buffer at 70°C for 15 minutes.
Move the 2 mL tube from 70°C to room temperature and allow the tube to cool to room temperature,
Collect the agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Aspirate the supernatant and discard, taking care not to disturb the pellet.
Resuspend the agarose resin in 1 mL of SDS Wash Buffer containing 40 mM iodoacetamide (freshly made) and incubate at room temperature for 30 minutes in the dark in order to alkylate all DTT-reduced cysteine residues.
3.7. Washing and proteolysis of 8-Bu(3-yne)T-ADP-ribosylated proteins conjugated to agarose resin
Using a clean razor blade, cut the very end off of a 1 mL pipette tip and transfer the agarose resin in SDS Wash Buffer with iodoacetamide to a single use column with the capacity for 2 mL of bed volume. Allow the resin to pack by gravity flow.
Wash the resin in the single use column by application of 10 washes of 2 mL each with SDS Wash Buffer, allowing the buffer to flow through the column by gravity.
Wash the resin by application of 10 washes of 2 mL each with Urea Wash Buffer, allowing the buffer to flow through the column by gravity.
Wash the resin by application of 10 washes of 2 mL each with 20% acetonitrile, allowing the buffer to flow through the column by gravity
Wash the resin by application of 5 washes of 2 mL each with Trypsin Digestion Buffer, allowing the buffer to flow through the column by gravity.
Using a clean razor blade, cut the very end off of a 1 mL pipette tip and resuspend the resin in the single use column in 500 µL of Trypsin Digestion Buffer by pipetting up and down. Move the resuspended resin to a new 1.7 mL tube.
Put a clean stopper onto the bottom of the single use column in order to stop the flow of buffer.
Using a clean razor blade, cut the very end off of a 1 mL pipette tip and wash the capped 2 mL single use column with an additional 500 µL of Trypsin Digestion Buffer to collect any remaining resin. Add it to the 1.7 mL tube containing the bulk of resin from Step 6.
Proteolyze the bead-conjugated 8-Bu(3-yne)T-ADP-ribosylated nuclear extract proteins in Trypsin Digestion Buffer by the addition of 1 µg of Trypsin (MS grade), followed by incubation overnight at room temperature with slow mixing (~10–15 RPM) on a rotisserie-style rotating mixer.
Collect the agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Move all 1 mL of the supernatant, which contains the trypsin-digested peptides, to new 1.7 mL tube, taking care not to disturb the pellet.
Add 500 µL of Trypsin Digestion Buffer to the pelleted resin and rinse the resin by gentle inversion of the tube.
Pellet the agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Move all 500 µL of the supernatant into the 1.7 mL tube containing the rest of the tryptic digest from Step 11, taking care not to disturb the pellet.
Desalt the peptides from the tryptic digestion using a C18 StageTip, as described in the manufacturer’s instructions.
Lyophilize the desalted peptides and store them at −20°C until they are preparation for LC-MS/MS analysis, as previously described [27].
3.8. Washing and NH2OH elution of 8-Bu(3-yne)T-ADP-ribosylated peptides conjugated to agarose resin
Using a clean razor blade, cut the very end off of a 1 mL pipette tip and resuspend the agarose resin from 3.7 Step 13 (See Note 11) in 1 mL of SDS Wash Buffer by gentle pipetting.
Transfer the agarose resin resuspended in SDS Wash Buffer to a single use column with the capacity for 2 mL of bed volume and allow the resin to pack by gravity flow.
Wash the resin in the single use column by application of 10 washes of 2 mL each with SDS Wash Buffer, allowing the buffer to flow through the column by gravity.
Wash the resin by application of 10 washes of 2 mL each with Urea Wash Buffer, allowing the buffer to flow through the column by gravity.
Wash the resin by application of 10 washes of 2 mL each with 20% acetonitrile, allowing the buffer to flow through the column by gravity.
Wash the resin by application of 5 washes of 2 mL each with Peptide Elution Buffer, allowing the buffer to flow through the column by gravity.
Put a clean stopper onto the bottom of the single use column in order to stop the flow of buffer.
Using a clean razor blade, cut the very end off of a 1 mL pipette tip and resuspend the resin in the single use column in 500 µL of Peptide Elution Buffer by pipetting up and down. Move the resuspended resin to a new 1.7 mL tube.
Wash the capped 2 mL single use column with an additional 500 µL of Peptide Elution Buffer to collect any remaining resin, adding it to the 1.7 mL tube containing the bulk of the agarose resin from Step 8.
Add hydroxylamine (NH2OH) at a final concentration of 0.5 M to the washed agarose resin, which has been resuspended in 1 mL of Peptide Elution Buffer.
Incubate the agarose resin in Peptide Elution Buffer containing 0.5 M NH2OH overnight at room temperature with slow mixing (~10–15 RPM) on a rotisserie-style rotating mixer to elute the glutamate- and aspartate-modified 8-Bu(3-yne)T-ADP-ribosylated peptides from the resin, as previously described [27].
Collect the agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Move all 1 mL of the supernatant, which contains the hydroxylamine eluted peptides, to a new 1.7 mL tube, taking care not to disturb the pellet.
Add 500 µL of Peptide Elution Buffer to the pelleted resin and rinse the resin by gentle inversion of the tube.
Collect the agarose resin by centrifugation at room temperature for 1 minute at 1000 RCF.
Move all 500 µL of the supernatant above the agarose resin into the 1.7 mL tube containing the rest of the hydroxylamine elution from Step 13, taking care not to disturb the pellet.
Desalt the peptides from the hydroxylamine elution using a C18 StageTip, as described in the manufacturer’s instructions.
Lyophilize the desalted peptides and store them at −20°C until they are prepared for LC-MS/MS analysis as previously described [27].
3.9. Expected Results, Problems and Pitfalls, Alternate Approaches, and Follow-up Experiments
The experiment as described herein should reveal the sites of aspartate- and glutamate-mediated ADP-ribosylation on the target proteins of specific PARPs. We have previously used this protocol to identify hundreds of target proteins and the specific sites of modification for PARP-1, PARP-2, and PARP-3 [12]. For each of these PARPs, more sites than targets were identified, indicating multiple modification events for each substrate protein. We expect similar outcomes with other PARP family members as well (See Notes 12 and 13).
The asPARP approach is a useful, but technically challenging experimental approach, with a variety of potential pitfalls that range from seemingly mundane technical details to the overall experimental design (See Notes 14 – 16).
A variety of alternate approaches may help those who undertake this protocol achieve the best possible results (See Notes 17 and 18).
One outcome of the asPARP approach described herein is the identification of possibly hundreds of sites of ADP-ribosylation on PARP target proteins. With respect to understanding PARP biology, however, this is the first step. Follow-up experiments are required to achieve maximum utility of this approach (See Notes 19 – 22).
Figure 4. Using asPARP methodologies for the discovery of PARP target proteins.
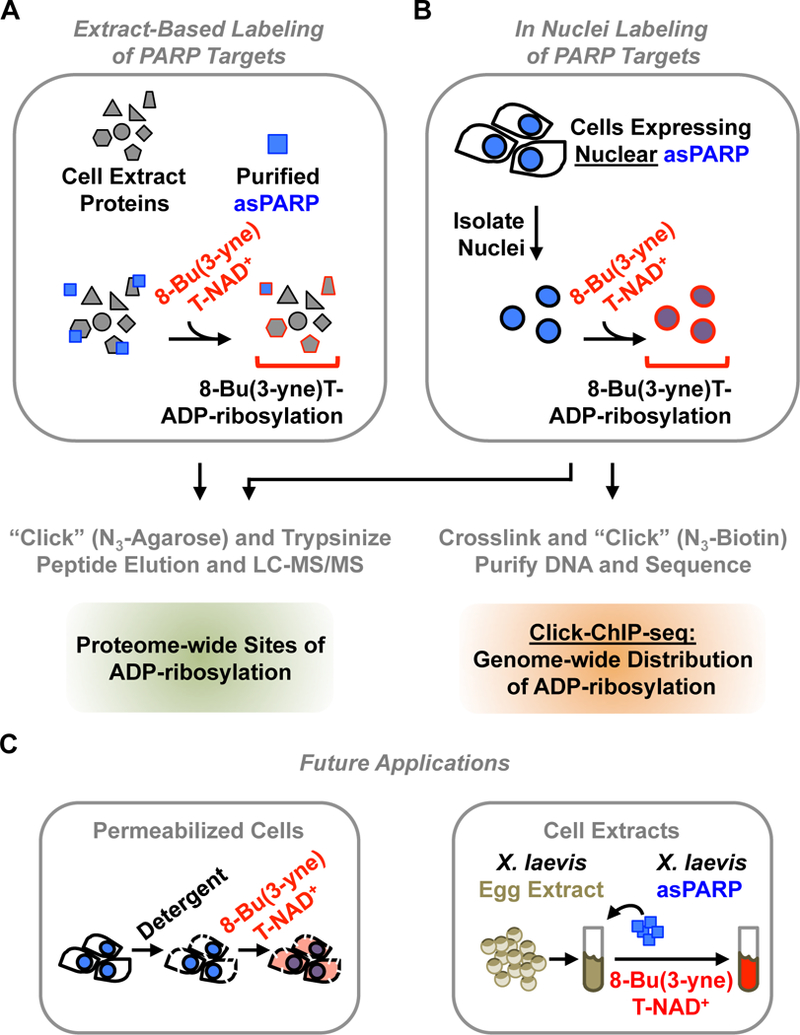
A schematic representation of different proven, as well as promising, applications of asPARP-dependent target labeling methodologies.
(A) In extract-based methods, a purified asPARP protein is used with a cellular extract and 8-Bu(3-yne)T-NAD+, as described in Figure 2. Sites of ADP-ribosylation on PARP target proteins can be identified by LC-MS/MS.
(B) For PARP proteins, which are expressed in the nucleus, in nuclei labeling is possible. Nuclei from nuclear asPARP-expressing cells are isolated and incubated with 8-Bu(3-yne)T-NAD+, stimulating spontaneous resumption of asPARP-driven 8-Bu(3-yne)T-ADP-ribosylation of nuclear PARP target proteins, as described in Figure 6. Sites of ADP-ribosylation on PARP target proteins can be identified by LC-MS/MS. Alternatively, sites of ADP-ribosylation across the genome can be determined by Click-ChIP-seq [12]. In click ChIP-seq, labeled nuclei are crosslinked with formaldehyde, clicked to azido-biotin (N3-biotin), and sonicated to shear the genomic DNA. This is followed by streptavidin-based purification of ADP-ribosylated chromatin and subsequent high throughput sequencing of the purified DNA.
(C) The asPARP technology may be developed further in the future to include more relevant or faithful biological contexts that recapitulate those that are found in vivo. (Left) Advances may include opportunities for delivering the clickable NAD+ analog directly into cells. This may entail careful permeabilization of cells using detergents, or perhaps even the development of new cell-permeable NAD+ analogs. (Right) Advances may also include opportunities to take advantage of complex biochemical extract preparations, such as the X. laevis egg extract system, where concerns such as dilution and unnatural buffer composition are less of an issue than with traditional extract preparation methods. Nearly all of the human PARP proteins have a X. laevis homolog with an identical primary gatekeeper residue. Furthermore, X. laevis egg extract preparations have been used with unparalleled success to reconstruct many dynamic biological processes [8], suggesting that the development of this system may offer an opportunity to uncover important PARP targets and molecular mechanisms that have remained conserved through evolution.
Figure 5. Flowchart for the selection and PARP target proteins and the functional analysis of ADP-ribosylation.

As described in the text.
Figure 6. Site-specific mutagenesis strategies to study ADP-ribosylation.
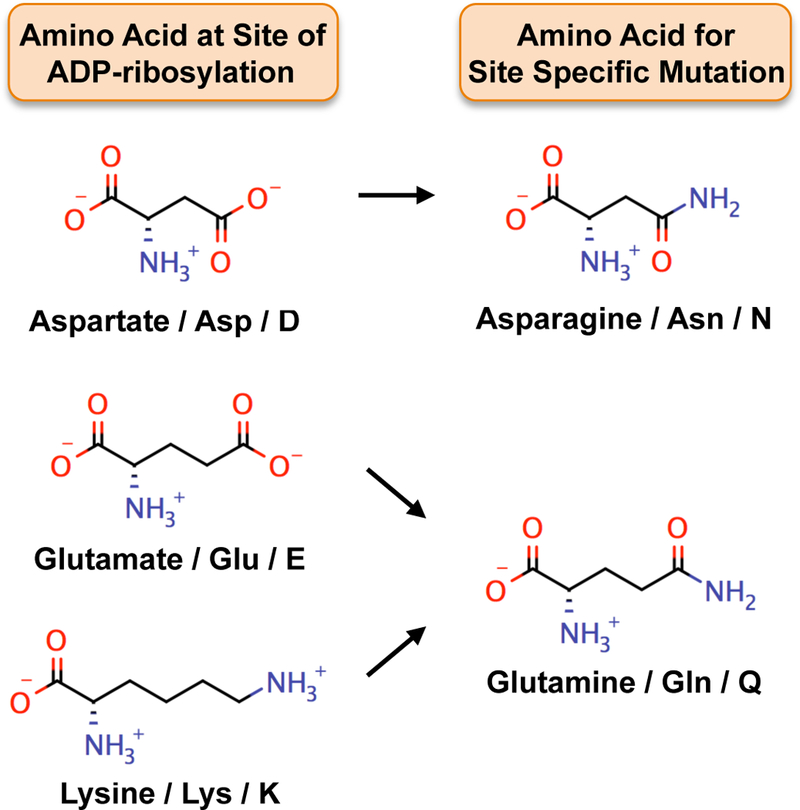
Mutating amino acids that are sites of ADP-ribosylation, such as aspartate, glutamate, and lysine, to structurally similar amino acids, such as asparagine and glutamine, can be an effective way to mimic the un-modified state. Mutation to asparagine (similar to aspartate) and glutamine (similar to glutamate and lysine), which are refractory to ADP-ribosylation, can facilitate understanding of how ADP-ribosylation of specific amino acids modulates the function of PARP target proteins.
Footnotes
Notes
Make this buffer fresh for every use.
Make THPTA in MilliQ H2O and store in single use aliquots at −20°C.
Take care to assure that the protein pellet does not overdry and remains covered in resolubilization buffer. Gently swirl the precipitated protein pellet in Urea Solubilization Buffer to avoid it adhering nonspecifically to the plastic surface of the side of tube.
This solution should turn bright blue as THPTA chelates the divalent copper cation.
At this point, the vast majority of free N3-TAMRA should have been washed away during the second methanol wash, so retention of the TAMRA dye in the protein pellet is a good indicator of successful 8-Bu(3-yne)T-ADP-ribosylation of PARP target proteins (Figure 3A).
Do not vortex. Take care to ensure that the protein pellet remains in SDS Loading Buffer following drying and does not encounter the bare plastic of the side of the tube, since the pellet can adhere nonspecifically and will can be difficult to dissolve. Gently flick the tube during solubilization of the protein pellet.
The loading volume that is used for the gel will depend on the size of the wells in the gel that is being used. The goal is to get as much labeled protein as possible into the gel in order to get as much fluorescence signal as possible during imaging. As long as the amount of protein running in the lane does not prevent resolution of distinct protein bands, aim to add ~75% of the well volume for your particular gel format.
If you removed 50 µL of buffer from this tube to perform an in-gel fluorescence assay, add 50 µL of Urea Solubilization Buffer to replace that volume at this time.
Oxygen is detrimental to the copper-catalyzed bio-orthogonal conjugation reaction.
Azide groups are light sensitive. Therefore, the reaction should be kept in the dark as much as is possible in order to protect the azido-agarose resin.
At this point, the agarose resin should contain only those peptides that are still covalently linked to the agarose resin through their azide-clicked 8-Bu(3-yne)T-ADP-ribosylation site.
Exactly how many targets one should expect may vary from one PARP to another, but approximately 100 target proteins with two to four ADP-ribosylation sites per target is a reasonable expected outcome for each PARP protein. The amount of labeling in the assays, as visualized by the in-gel fluorescence assay and detected by mass spectrometry, may also vary depending on the nature and activity of the PARP protein studied, as well as the experimental conditions. Poly(ADP-ribosyl) transferases, such as PARP-1, may add more 8-Bu(3-yne)T-ADP-ribose per reaction than mono(ADP-ribosyl) transferases, such as PARP-3, but this labeling may represent fewer sites or targets [12]. Furthermore, the nature of the gatekeeper residue may also affect the extent of labeling. A little over half of the PARP family encodes a phenylalanine instead of leucine at the PARP-1 L877 paralogous site (Figure 1C). Although our unpublished data indicate that the asPARP approach works for PARPs with phenylalanine at this site, this has not yet been explored in detail. Finally, asPARPs that retain catalytic activity with natural NAD+ used in conjunction with extracts that have high levels of endogenous NAD+ may exhibit lower labeling efficiency due to competition between 8-Bu(3-yne)T-NAD+ and NAD+ in the extract. Given these considerations, the extent of labeling cannot be predicted in advance.
Certain measurable outcomes can serve as good indicators if the asPARP approach is working appropriately. Perhaps the best indicator is a substantial difference in labeling between wild-type PARP protein and the asPARP protein (Figure 3B). Differences in the extent of labeling can be observed over a range of extract protein concentrations in the reaction (see below) (Figure 3C). As such, extract titration experiments can be a useful way of determining the most effective ratio of extract protein:PARP protein to use. Addition of the asPARP mutant to a cell extract, or expression in cells prior to performing the assay (see below), should result in substantially increased labeling by in-gel fluorescence and ADP-ribosylation site detection by mass spectrometry compared to either wild-type PARP or a control protein, such as GFP. Moreover, in the in-gel fluorescence assays, visualization of distinct bands at molecular weights that differ from the PARP being studied are a good indicator of effective labeling of target proteins (Figure 3, B and C).
One particularly challenging technical aspect of the experiment is the methanol:chloroform precipitation of the extract proteins after labeling. This is an important step in the protocol and it must be mastered to achieve good results with the asPARP approach. As described in Note 4 above, preventing the pellet from the methanol:chloroform precipitation from overdrying is very important; an overdry pellet can be difficult (perhaps even impossible) to resolubilize. This technical consideration is especially important for the in-gel fluorescence assay, where insoluble proteins will run poorly in the gel and may autofluoresce, producing results that are difficult to interpret. We recommend that you practice the methanol:chloroform extractions a few times prior to running these experiments, judging success when the clarity of protein banding in a Coomassie-stained gel is equivalent for the resolubilized methanol:chloroform precipitated proteins and unprecipitated proteins from the cognate protein extract.
As might be expected, another important aspect of the success with this experimental approach is the concentration of the asPARP protein - either added as a purified recombinant protein to a cell extract or expressed ectopically in cells - since the presence and activity of the asPARP protein is necessary for efficient 8-Bu(3-yne)T-ADP-ribosylation of target proteins. For nuclear extract-based approaches with the DNA-dependent PARPs, we have found that a 1:50 mass ratio of asPARP protein and nuclear extract proteins produces robust labeling of extract proteins (Figure 3C). Although this ratio may not be a universally appropriate for all types of extracts (e.g., cytoplasmic versus nuclear), all cell types, or all PARP family members, it should be a good starting point for labeling reactions when testing other conditions or other PARPs. For ectopic expression in cells, we have observed strong labeling of target proteins using 8-Bu(3-yne)T-NAD+ when (1) expression of the endogenous PARP protein is readily detectable by Western blotting in the cell type of interest, indicating a robust biological system for that PARP protein, (2) the asPARP protein is ectopically expressed from a transgene to levels similar to the endogenous PARP protein, and (3) expression of the endogenous PARP protein is reduced by RNAi-mediated knockdown or genetic knockout [12] (Figure 3D). Without methods for robust expression or purification of the asPARP protein, this method will be limited. Alternate approaches for asPARP protein production or different cell lines for asPARP expression may provide a way around these issues.
A significant challenge with the asPARP approach is that the clickable NAD+ analog, 8-Bu(3-yne)T-NAD+, which works in conjunction with the asPARP protein, is not cell permeable. In the experiment described above, the use of cellular extracts to perform a simple reconstitution assay with the DNA dependent PARPs overcomes this issue (Figure 4A). However, the extract-based approach is not ideal; the extract proteins are diluted, are in a non-native solvent, and exist without the organizational context of an intact cell. Nonetheless, the extract-based approach remains a reasonable first step for addressing fundamental questions in the PARP field in spite of this particular drawback. Note, for example, that we have performed a simple reconstitution of the ADP-ribosylation events catalyzed during DNA damage using nuclear extracts, DNA-dependent PARPs, and sonicated DNA as an activator of catalytic activity [12]. An extension of this experiment could use DNA activators that differ in the type of DNA lesion or the base sequence, or perhaps chromatin in the form of trinucleosomes without free DNA ends [6, 20] as a substrate to simulate undamaged DNA in chromatin. Such experiments could reveal if or how different DNA structures might affect PARP target selection. In this way, by pairing asPARP proteins with cell compartment-specific catalytic activators in extracts in vitro, one can overcome the limitations of the extract-based approach.
As noted above, the lack of cell permeability of the NAD+ analog is a potential issue in the design of the asPARP experiments. One way that we have worked around this for the nuclear PARPs is to perform the labeling reaction in intact nuclei, where ADP-ribosylation spontaneously resumes upon incubation of the nuclei with NAD+ or NAD+ analog (Figure 3D and 4B) [12]. In this regard, we have expressed asPARP-1 in Parp1−/− MEFs isolated from PARP-1 knockout mice and then perform the asPARP-1 reaction in isolated nuclei by the addition of 8-Bu(3-yne)T-NAD+. Using this approach, we identified PARP-1 protein targets, as well as genomic loci ADP-ribosylated by PARP-1, in a technique we call click-ChIP-seq (Figure 4B) [12]. While this particular elaboration of the asPARP approach, the nuclear ADP-ribosylation assay, is appropriate for studying the role of PARPs in nuclear biology, it will have limited utility for non-nuclear PARPs. Other ways to augment the basic approach outlined in this protocol might be to use chemically permeabilized cells [3], transfection of the NAD+ analog [17], or possibly X. laevis egg extract-based systems (Figure 4C). With respect to the latter, most mammalian PARP family members contain a conserved homolog in X. laevis and extracts from X. laevis have proven to be very useful for reconstituting highly complex biological processes in vitro [8].
Identification of sites of ADP-ribosylation using mass spectrometry has historically been a difficult problem due to the fact that ADP-ribosylation modifications can be exceptionally heterogeneous [e.g., branched chains of poly(ADP-ribose) of variable length] and are labile. The approach outlined in this protocol solves these problems by using a previously published method of ADP-ribosylation site identification. This approach uses hydroxylamine to elute ADP-ribosylated peptides covalently linked to a solid support through 8-Bu(3-yne)T-ADP-ribose attached at glutamate or aspartate residues. The hydroxylamine cleaves the 8-Bu(3-yne)T-ADP-ribose, leaving a residual chemical fragment at the site of ADP-ribosylation, which results in a predictable and readily detectable m/z shift when analyzed by mass spectrometry. This approach, which can be used in place of direct identification ADP-ribose, is limited by the fact that the hydroxylamine elution and LC-MS/MS analysis only allows for identification of ADP-ribosylated glutamates and aspartates. While aspartates and glutamates may account for at least half of the population of amino acids ADP-ribosylated by PARPs in cells [7], the modification of other amino acids by PARPs, such as lysine, may be equally important. Efforts continue to develop approaches for the identification of all ADP-ribosylated residues, relying largely on enzymatic cleavage to reduce poly(ADP-ribose) to mono(ADP-ribose) as a means to reduce heterogeneity [19] or to cleave mono(ADP-ribose) to smaller moieties that are more stable during mass spectrometry [23]. These methods, some of which may require additional mass spectrometry approaches (e.g., electron transfer dissociation) [19, 26], can also be applied to the asPARP technique. Enzymatic cleavage of ADP-ribosylation should not be hindered with analog ADP-ribose derived from 8-Bu(3-yne)T-ADP-ribose. As such, the asPARP approach described herein should continue to be useful for the identification of PARP-specific ADP-ribosylation sites regardless of how mass spectrometry methods advance in the field.
A variety of follow-up experiments are needed to determine the biological relevance of individual ADP-ribosylation sites from the omics-level data set. In our experience, the process should include the following steps: (1) determining a relevant PARP target protein to study in detail, (2) confirmation that the target protein is ADP-ribosylated in a relevant cellular and biological context, (3) demonstration of ADP-ribosylation-dependent functional interplay between the PARP protein and its target, and (4) demonstration that specific sites of ADP-ribosylation on the target protein mediate the PARP-dependent biological outcomes through mutagenesis of the sites (Figure 5).
When narrowing down the list of potential PARP targets to explore in detail, we have found it helpful to incorporate independent experimental data to determine which hits may be fruitful to follow up on. This may include independent observations about the broader biology or physiology, and how they may relate to the underlying molecular and biochemical mechanisms driven by a particular target protein or group of target proteins. For example, a PARP-dependent cellular outcome that requires RNA splicing may point the way to an investigation of ADP-ribosylation of a splicing factor. Alternatively, approaches that link ADP-ribosylation of specific target proteins with genomic ADP-ribosylation events at specific loci (e.g., through Click-ChIP-seq) may support hypotheses about specific PARP target proteins [12]. Finally, cases where genetic or chemical ablation of the PARP being studied phenocopies modulation of the prospective target protein, either positively or negatively, can help point the way to relevant target proteins to study in more detail.
Once a prospective target is identified, confirmation of ADP-ribosylation by the PARP of interest is essential. This can be accomplished by immunoprecipitation of the target protein from cells (either endogenous or ectopically expressed), followed by Western blotting with an anti-ADP-ribose antibody or detection reagent. By coupling this experiment with knockdown or knockout of the PARP of interest, initial links between the PARP and its target protein can be established. Alternatively, confirmation of ADP-ribosylation by the PARP of interest can be accomplished using purified proteins in a biochemical assay. These cell-based and biochemical experiments lead directly into functional testing of the consequences of ADP-ribosylation. The functional assays will depend on the activities or cellular functions of the target protein being studied. Does it have an enzymatic activity? DNA- or RNA-binding activity? Specific protein interaction partners? A distinct subcellular localization? For example, we recently studied PARP-1-mediated ADP-ribosylation of the E subunit of the negative elongation factor (NELF) complex, which is an RNA binding protein that binds at the promoters of RNA polymerase II transcribed genes [12]. We determined the effect of PARP-1-mediated ADP-ribosylation of NELF-E on (1) its RNA binding activity using an electrophoretic mobility shift assay and (2) its promoter localization using ChIP-seq [12]. When designing an assay to test the functional consequences of ADP-ribosylation, one should consider that ADP-ribosylation typically inhibits the normal functions of the protein being modified. Although this generality is not always true (see for example: [14, 15]), the large and negatively charged nature of poly(ADP-ribose) should be considered when generating hypotheses regarding how it might modulate the molecular functions of a PARP target protein [11].
After establishing the initial functional links between the PARP of interest and a prospective target protein, the most definitive way to study the effects of ADP-ribosylation is through mutagenesis of the modification sites. The function of the mutant target protein can then be compared to the wild-type protein in biochemical or cell-based assays. For mutational analyses, target amino acids are typically mutated to alanine, but for ADP-ribosylation, asparagine may be a more appropriate choice for aspartate and glutamine may be a more appropriate choice glutamate and lysine. Asparagine and glutamine are structurally similar to the cognate amino acids they would replace, but they are refractory to ADP-ribosylation (Figure 6). Finally, in more detailed analyses, different versions of the asPARP protein or the target protein with functional domains mutated or deleted can be used to understand structure-function relationships leading to site-specific ADP-ribosylation.
References
- 1.Bai P, Canto C (2012) The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell metabolism 16:290–295 [DOI] [PubMed] [Google Scholar]
- 2.Bishop A, Buzko O, Heyeck-Dumas S et al. (2000) Unnatural ligands for engineered proteins: new tools for chemical genetics. Annual review of biophysics and biomolecular structure 29:577–606 [DOI] [PubMed] [Google Scholar]
- 3.Cambronne XA, Stewart ML, Kim D et al. (2016) Biosensor reveals multiple sources for mitochondrial NAD(+). Science 352:1474–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter-O’connell I, Jin H, Morgan RK et al. (2014) Engineering the substrate specificity of ADP-ribosyltransferases for identifying direct protein targets. Journal of the American Chemical Society 136:5201–5204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter-O’connell I, Jin H, Morgan RK et al. (2016) Identifying family-member-specific targets of mono-ARTDs by using a chemical genetics approach. Cell reports 14:621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark NJ, Kramer M, Muthurajan UM et al. (2012) Alternative modes of binding of poly(ADP-ribose) polymerase 1 to free DNA and nucleosomes. J Biol Chem 287:32430–32439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniels CM, Ong SE, Leung AK (2015) The promise of proteomics for the study of ADP-ribosylation. Molecular cell 58:911–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Desai A, Murray A, Mitchison TJ et al. (1999) The use of Xenopus egg extracts to study mitotic spindle assembly and function in vitro. Methods in cell biology 61:385–412 [DOI] [PubMed] [Google Scholar]
- 9.Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic acids research 11:1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekblad T, Camaioni E, Schuler H et al. (2013) PARP inhibitors: polypharmacology versus selective inhibition. The FEBS journal 280:3563–3575 [DOI] [PubMed] [Google Scholar]
- 11.Gibson BA, Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature reviews. Molecular cell biology 13:411–424 [DOI] [PubMed] [Google Scholar]
- 12.Gibson BA, Zhang Y, Jiang H et al. (2016) Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science [DOI] [PMC free article] [PubMed]
- 13.Huber A, Bai P, De Murcia JM et al. (2004) PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA repair 3:1103–1108 [DOI] [PubMed] [Google Scholar]
- 14.Jwa M, Chang P (2012) PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nature cell biology 14:1223–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leung AK, Vyas S, Rood JE et al. (2011) Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular cell 42:489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loseva O, Jemth AS, Bryant HE et al. (2010) PARP-3 is a mono-ADP-ribosylase that activates PARP-1 in the absence of DNA. J Biol Chem 285:8054–8060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mao Z, Hine C, Tian X et al. (2011) SIRT6 promotes DNA repair under stress by activating PARP1. Science 332:1443–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menissier De Murcia J, Ricoul M, Tartier L et al. (2003) Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. The EMBO journal 22:2255–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Messner S, Altmeyer M, Zhao H et al. (2010) PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic acids research 38:6350–6362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muthurajan UM, Hepler MR, Hieb AR et al. (2014) Automodification switches PARP-1 function from chromatin architectural protein to histone chaperone. Proceedings of the National Academy of Sciences of the United States of America 111:12752–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quenet D, Gasser V, Fouillen L et al. (2008) The histone subcode: poly(ADP-ribose) polymerase-1 (Parp-1) and Parp-2 control cell differentiation by regulating the transcriptional intermediary factor TIF1beta and the heterochromatin protein HP1alpha. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 22:3853–3865 [DOI] [PubMed] [Google Scholar]
- 22.Schreiber V, Ame JC, Dolle P et al. (2002) Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem 277:23028–23036 [DOI] [PubMed] [Google Scholar]
- 23.Vivelo CA, Leung AK (2015) Proteomics approaches to identify mono-(ADP-ribosyl)ated and poly(ADP-ribosyl)ated proteins. Proteomics 15:203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vyas S, Matic I, Uchima L et al. (2014) Family-wide analysis of poly(ADP-ribose) polymerase activity. Nature communications 5:4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wahlberg E, Karlberg T, Kouznetsova E et al. (2012) Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nature biotechnology 30:283–288 [DOI] [PubMed] [Google Scholar]
- 26.Zee BM, Garcia BA (2010) Electron transfer dissociation facilitates sequencing of adenosine diphosphate-ribosylated peptides. Analytical chemistry 82:28–31 [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Wang J, Ding M et al. (2013) Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nature methods 10:981–984 [DOI] [PubMed] [Google Scholar]