

SUMMARY
Protective immunity against pathogens depends on the efficient generation of functionally diverse effector and memory T lymphocytes. However, whether plasticity during effector-to-memory CD8+ T cell differentiation affects memory lineage specification and functional versatility remains unclear. Using genetic fate mapping analysis of highly cytotoxic KLRG1+ effector CD8+ T cells, we demonstrated that KLRG1+ cells receiving intermediate amounts of activating and inflammatory signals, downregulated KLRG1 during the contraction phase in a Bach2-dependent manner, and differentiated into all memory T cell linages, including CX3CR1int peripheral memory cells and tissue-resident memory cells. ‘ExKLRG1’ memory cells retained high cytotoxic and proliferative capacity distinct from other populations, which contributed to effective anti-influenza and anti-tumor immunity. Our work demonstrates that developmental plasticity of KLRG1+ effector CD8+ T cells is important in promoting functionally versatile memory cells and long-term protective immunity.
IN BRIEF
Herndler-Brandstetter et al. demonstrate that KLRG1+ IL-7Rα+ effector CD8+ T cells downregulate KLRG1 in a Bach2-depenent manner and differentiate into long-lived circulating and tissue-resident ‘exKLRG1’ memory cells. Developmental plasticity of KLRG1+ effector cells therefore drives functional diversity within memory T cell lineages and promotes enhanced anti-influenza and anti-tumor immunity.
GRAPHICAL ABSTRACT
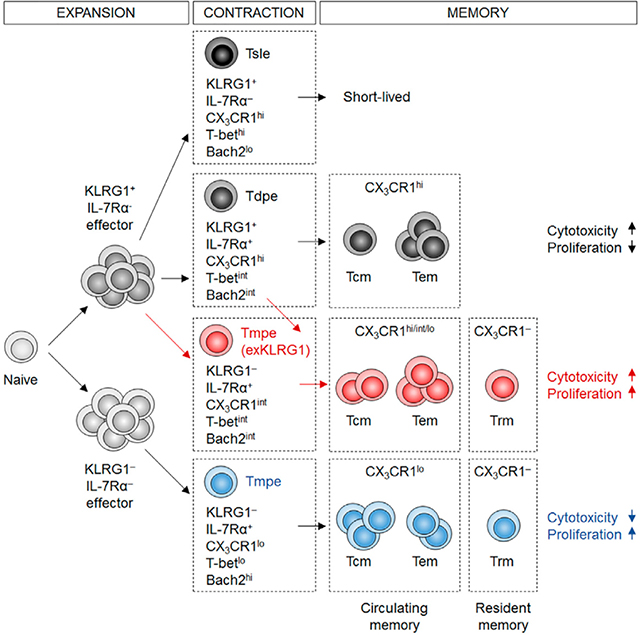
INTRODUCTION
CD8+ T cells are important in host protection from infectious and malignant diseases, and memory CD8+ T cell heterogeneity is a core feature of protective immune responses. Following a primary infection, naïve CD8+ T cells are activated by antigen-presenting cells, clonally expand and differentiate into short-lived effector and long-lived memory cell subsets (Jameson and Masopust, 2009; Mueller et al., 2013; Williams and Bevan, 2007). Two subsets of circulating memory CD8+ T cells with distinct migratory and effector properties have been described: central-memory T (Tcm) and effector-memory T (Tem) cells. Tcm cells express the lymph node (LN) homing receptors CCR7 and CD62L, have a high proliferative capacity but exhibit low cytotoxicity. In contrast, Tem cells lack CCR7 and CD62L, home to non-lymphoid tissues, have a lower proliferative capacity but display high cytotoxicity. In addition, tissue-resident memory T (Trm) cells constitute a recently identified memory cell lineage that does not recirculate but resides in barrier and non-barrier tissues (Mueller et al., 2013; Steinert et al., 2015). Trm cells are phenotypically distinct from recirculating Tcm and Tem cells, and represent the first line of defense upon reinfection at barrier sites, such as the skin and mucosal surfaces. However, considerable heterogeneity within each memory cell lineage has been reported (Kaech and Wherry, 2007; Mackay and Kallies, 2017). For example, a recently identified peripheral memory T (Tpm) cell population, which expresses intermediate levels of CX3CR1, shares features of both Tcm and Tem cells, and is chiefly responsible for the global surveillance of non-lymphoid tissues (Gerlach et al., 2016). So far, it remains unclear whether such heterogeneity originates from different effector cell precursors or differential states of activation.
Killer cell lectin-like receptor subfamily G, member 1 (KLRG1) is induced in highly cytotoxic and proliferative effector CD8+ T cells that received strong cumulative T cell receptor and inflammatory signals (Joshi et al., 2007; Sarkar et al., 2008; Xin et al., 2016). KLRG1, together with IL-7Rα, have been used as markers to identify effector CD8+ T cell subsets with distinct traits regarding effector function, migratory properties, long-term survival and multi-lineage memory potential (Buchholz et al., 2016; Chang et al., 2014; Joshi et al., 2007). KLRG1+ IL-7Rα− short-lived effector CD8+ T cells (Tsle) and KLRG1+ IL-7Rα+ double-positive effector CD8+ T cells (Tdpe) are thought to have a limited potential to become memory cells, and the KLRG1+ memory CD8+ T cells that develop, gradually decline over time (Joshi et al., 2007; Mackay et al., 2013; Obar and Lefrancois, 2010; Sheridan et al., 2014). In contrast, effector CD8+ T cells that do not express KLRG1 (KLRG1− IL-7Rα+) have been referred to as memory precursor effector cells (Tmpe), which display increased survival during the contraction phase and retain developmental plasticity, as they are able to differentiate into multiple memory cell lineages, including Tcm, Tem and Trm cells (Joshi et al., 2007; Mackay et al., 2013; Obar and Lefrancois, 2010).
The efficient generation of Tmpe cells has been used as an immune parameter critical for long-term protective immunity, in particular at barrier sites (Araki et al., 2009; Mackay et al., 2013; Pearce et al., 2009; Sheridan et al., 2014). However, the expression of proteins such as KLRG1 may not be stable but subject to spatiotemporal regulation by cell-intrinsic and -extrinsic signals (Gerlach et al., 2016; Joshi et al., 2007; Plumlee et al., 2015; Sarkar et al., 2008; Wherry et al., 2003), which may subsequently alter the fate of these effector cells. Whether KLRG1+ effector CD8+ T cell subsets display developmental plasticity, are able to efficiently differentiate into all memory cell lineages, and drive functional heterogeneity among distinct memory cell lineages remains unclear. So far, a fate mapping approach that tracks the fate of different subsets of effector CD8+ T cells in a dynamic and complex in vivo setting has not been reported.
In this study, we employed a Klrg1Cre reporter system, which enabled longitudinal tracking of KLRG1+ effector CD8+ T cells in vivo. By using this system, we demonstrated that KLRG1+ effector CD8+ T cells displayed developmental plasticity, as they were able to downregulate KLRG1 and to efficiently differentiate into all memory T cell lineages. We further showed that such plasticity was a driving force in generating phenotypic and functional diversity within Tcm, Tem and Trm cells. Finally, we showed that exKLRG1 memory cells mounted highly effective anti-influenza and anti-tumor responses, highlighting the functional significance of plasticity in effector-to-memory CD8+ T cell differentiation.
RESULTS
Generation and Validation of Klrg1Cre Reporter Mice
To investigate the role of plasticity in effector-to-memory CD8+ T cell differentiation and its impact on memory CD8+ T cell heterogeneity and recall responses, we generated a mouse strain that expressed an eGFP-Cre recombinase (Cre) fusion protein under the control of the Klrg1 gene (Klrg1Cre) (Figure S1A). We then crossed Klrg1Cre mice with Rosa26-flox-STOP-flox-eYFP (Rosa26eYFP) or Rosa26-flox-STOP-flox-tdTomato (Rosa26tdTomato) mice. Thus, the fluorescent reporter (eYFP or tdTomato) would permanently tag KLRG1-expressing cells. The analysis of Klrg1Cre/+Rosa26 tdTomato/+ mice revealed that naïve CD4+ and CD8+ T cells, Gr1+ cells, CD11c+ cells, and CD11b+ cells, which are known not to express KLRG1, did not express the fluorescent reporter (Figure S1B and data not shown). In contrast, cells that frequently express KLRG1, such as NK1.1+ cells, FoxP3+ regulatory T cells and CD8+ Tem cells, expressed the fluorescent reporter (Figures S1B and S1C).
To study the fate of KLRG1+ effector CD8+ T cells during infection in vivo, we employed a bacterial (ovalbumin (OVA)-expressing Listeria monocytogenes; LM) and a viral (OVA-expressing vesicular stomatitis virus; VSV) infection model (Figures S1D and S1E). Following infection with LM, transferred naïve CD44lo CD62Lhi Reporter− OVA-specific T cell receptor transgenic CD8+ T (OT-I) cells started to upregulate KLRG1 on day 4 p.i., and the vast majority of KLRG1-expressing cells were tagged with the fluorescent reporter (Reporter+ cells) by day 10 p.i. (Figures 1A and S1F). On day 5, the intensity of Klrg1 and iCre mRNA expression correlated with the efficiency of DNA recombination in the Rosa26 locus (Figure S1G). Cre expression, as determined by fluorescence of eGFP-Cre fusion protein, was restricted to KLRG1hi and KLRG1int effector cells and eGFP-Cre expression was hardly detectable in KLRG1lo effector cells (Figure S1H). The majority of the transferred KLRG1+ Reporter− effector OT-I cells were also faithfully tagged with the reporter 14 days post transfer (Figure S1I). In addition, both reporter strains (Klrg1Cre/CreRosa26eYFP/eYFP and Klrg1Cre/+Rosa26tdTomato/+) displayed similar KLRG1 fate mapping efficacy and development of exKLRG1 cells in the blood and spleen (Figures S1J and S1K). Together, these results indicate that KLRG1-expresssing cells are faithfully labeled by the fluorescent reporter once effector CD8+ T cells express sufficient levels of KLRG1.
Figure 1. Effector CD8+ T Cells Lose KLRG1 Expression and Differentiate into Long-Lived Memory Cells.

(A and B) Expression of KLRG1 and fate mapping in effector OT-I cells in the blood following LM infection.
(C) Frequency of KLRG1+, exKLRG1 and KLRG1− Reporter− cells among OT-I cells in the blood up to 120 days p.i. with LM.
(D) Percentage (top) and number (bottom) of OT-I cell subsets in the spleen and LN following LM infection. The numbers indicate fold difference in cell number between days 10 and 120.
(E) Frequency of KLRG1+, exKLRG1 and KLRG1− Reporter− cells among OT-I cells in the blood up to 100 days p.i. with VSV.
(F) Percentage of KLRG1+, exKLRG1 and KLRG1− Reporter− cells among OT-I cells in the spleen and LN 100 days p.i. with VSV.
Mean ± SEM are shown. Data are pooled from 2–4 independent experiments with 4–12 (C) or 3–12 mice per time point (D), or are representative of 2–3 independent experiments with 3–5 mice per time point (A, E, F). See also Figure S1 and S2.
KLRG1+ Effector CD8+ T Cells Lose KLRG1 Expression and Differentiate into Long-Lived ‘exKLRG1’ Memory Cells
As early as 6 days p.i. with LM, we identified effector OT-I cells in the blood that expressed KLRG1 previously (Reporter+) but lost KLRG1 expression thereafter (Figure 1A). We named this KLRG1− Reporter+ population ‘exKLRG1’ cells. Following infection with LM, exKLRG1 memory cells survived long-term and represented approximately 25% of the circulating OT-I memory cell pool (Figures 1B and 1C). We also detected long-lived exKLRG1 memory cells in the spleen and LN, where they represented 20% to 35% of the total memory OT-I cell population (Figure 1D). Remarkably, exKLRG1 and KLRG1− Reporter− cells had a comparable potential to become long-lived memory CD8+ T cells, as revealed by the fold decrease in cell number between days 10 and 120 p.i. with LM (Figure 1D). Infection with VSV also induced long-lived exKLRG1 memory cells in the blood, spleen and LN (Figures 1E and 1F). Although the intraclonal competition for antigenic and inflammatory signals has been shown to affect the generation of KLRG1+ effector CD8+ T cells, we could detect exKLRG1 memory cells irrespective of the number of naïve OT-I cell precursors transferred (Figures S2A–F). Furthermore, exKLRG1 memory cells also developed from endogenous CD8+ T cells in Klrg1Cre/+Rosa26tdTomato/+ mice and represented 20–40% of the long-lived OVA tetramer+ memory CD8+ T cell population following infection with LM (Figures S2G–J). Together, these results demonstrate that KLRG1+ effector CD8+ T cells are able to differentiate into exKLRG1 memory cells, which represent a significant proportion of the memory cell pool and are able to persist longer than KLRG1+ memory CD8+ T cells. Our results further indicate that a high proportion of cells previously defined as Tmpe cells are actually exKLRG1 cells.
KLRG1+ Effector CD8+ T Cells Downregulate KLRG1 and Differentiate into Trm Cells
Published studies suggest that KLRG1− Tmpe cells are the predominant effector cell subset able to differentiate into Tcm, Tem and Trm cell lineages, and thereby exhibit multipotency (Mackay et al., 2013; Obar and Lefrancois, 2010; Sheridan et al., 2014). Because our Klrg1Cre reporter model allowed us to follow the fate of KLRG1+ effector cells in vivo, we re-evaluated whether KLRG1+ effector CD8+ T cells were able to differentiate into Trm cells. Intravenous injection of anti-CD8α phycoerythrin antibody (Anderson et al., 2014) revealed that exKLRG1 memory OT-I cells were able to migrate to the lung parenchyma, while KLRG1+ memory OT-I cells could only be detected in the lung perivascular niche (Figure 2A–2C). In the liver, a preferred site of LM colonization, exKLRG1 and KLRG1− Reporter− memory OT-I cells expressed comparable levels of the Trm cell marker CD69 (Figure 2D). The number of exKLRG1 and KLRG1− Reporter− Trm cells in the lung and liver was comparable and both liver Trm cell subsets were equally dependent on the survival factor IL-15 (Figure 2C and 2E). Following infection with LM, about 50% of intraepithelial OT-I lymphocytes in the small intestine were exKLRG1 memory cells and KLRG1+ cells were largely excluded from this population (Figure 2F). Oral LM infection also generated exKLRG1 and KLRG1−Reporter− IELs that expressed the Trm cell markers CD69 and CD103 (Figure 2G), and exKLRG1 cells were enriched within the CD69+ CD103− cell population (Figure 2H).
Figure 2. KLRG1+ Effector CD8+ T Cells Differentiate into Long-Lived Trm Cells.

(A-C) Frequency and number of resident (i.v.−) and circulating (i.v.+) memory OT-I cell subsets in the lung 60 days p.i. with LM. Circulating cells were identified by i.v. injection of anti-CD8α antibody.
(D) Frequency of CD69+ memory OT-I cell subsets in the liver 62 days p.i. with LM.
(E) Frequency and number of CD69+ Trm cells in the liver 30 days p.i. with LM.
(F) Frequency and number of OT-I cell subsets in the intraepithelial lymphocyte fraction of the small intestine (SI-IEL) 10 and 120 days p.i. with LM. Numbers indicate fold difference in cell number between days 10 and 120.
(G) Expression of CD69 and CD103 in OT-I cell subsets in the SI-IEL of mice orally infected with LM (day 42 p.i.).
(H) Frequency of CD103− CD69+ or CD103+ CD69+ memory OT-I cell subsets in the SI-IEL.
Mean ± SEM are shown. ** P < 0.01, *** P < 0.001 and **** P < 0.0001 (unpaired two-tailed Student’s t-test). Data are representative of two independent experiments with 3 (A-C), 4–5 (D, E), or 8 mice (G, H), or are pooled from 2–3 independent experiments with 4–9 mice per time point (F). See also Movies S1 and S2.
Subcutaneous (s.c.) OVA injection of mice that were previously injected with naïve OT-I cells and infected with LM, revealed that up to 40% of OT-I memory cells recruited to the skin epidermis were descendants of KLRG1+ effector cells (Movie S1 and data not shown). Reporter+ Trm cells were also detected in the skin epidermis of uninfected mice following s.c. immunization with OVA plus poly (I:C) (Movie S2 and data not shown). Our results highlight a previously undefined potential of effector CD8+ T cells, which have previously expressed KLRG1, to differentiate into long-lived exKLRG1 Trm cells.
ExKLRG1 Effector CD8+ T Cells Express Key Molecules Associated with Effector Function, Survival, and Proliferation at an Intermediate Level
To better characterize the effector CD8+ T cell subsets that possess developmental plasticity, we first analyzed key molecules and transcription factors in effector cell subsets 8–11 days p.i. with LM. ExKLRG1 effector cells expressed intermediate levels of granzyme B (GzmB), T-bet, Ki-67 and Bcl-2, and similar levels of TCF-1 compared to KLRG1−Reporter− effector cells (Figure 3A). In addition, exKLRG1 effector cells expressed intermediate levels of Zeb2, Prdm1 and Bach2 (Figure 3B). The expression level of GzmB, T-bet, Ki-67 and Bcl-2 in exKLRG1 cells was closely associated with the expression levels observed in Tdpe cells (Figure S3A). Following infection with LM, effector CD8+ T cells rapidly up-regulated CX3CR1, which is used to identify 3 distinct effector CD8+ T cell subsets with different capacities to generate memory cells (Bottcher et al., 2015; Gerlach et al., 2016), but only KLRG1+ and exKLRG1 cells were able to maintain CX3CR1 expression during the early memory phase (30 – 60 days p.i.) (Figures 3C and 3D). IL-7Rα expression was downregulated in all effector cell subsets before the peak of expansion (day 5–6 p.i.) (Figure 3C), as reported previously (Joshi et al., 2007; Plumlee et al., 2015; Sarkar et al., 2008). Interestingly, the kinetics of IL-7Rα and CD62L re-acquisition was different among effector T cell subsets (Figures 3C and 3E): KLRG1−Reporter− effector cells exhibited the highest degree of IL-7Rα and CD62L re-acquisition, whereas exKLRG1 effector cells re-expressed intermediate levels of these molecules compared to KLRG1−Reporter− and KLRG1+Reporter+ cells (Figures 3C and 3E). Taken together, the development of exKLRG1 memory cells is linked to the degree of effector CD8+ T cell differentiation and proliferative history.
Figure 3. ExKLRG1 Effector CD8+ T cells Express Cytotoxicity, Survival, and Proliferation Molecules at an Intermediate Level.

(A) Expression of GzmB, T-bet, Ki-67, Bcl-2, and TCF-1 in splenic effector OT-I cell subsets 9–10 days p.i. with LM.
(B) Expression of effector and memory signature genes in splenic OT-I cell subsets 8–11 days p.i. with LM.
(C-E) Time-dependent expression of CX3CR1 and IL-7Rα in OT-I cell subsets in the blood following LM infection.
(F) Normalized ATAC-seq signal profiles across 7 gene loci in splenic naïve and effector OT-I cell subsets (8 days p.i. with LM). Peaks differentially expressed between OT-I cell subsets are highlighted in grey.
Mean ± SEM are shown. * P < 0.05, ** P < 0.01 and *** P < 0.001 (unpaired two-tailed Student’s t-test). Data are representative of 2–3 independent experiments with 4–8 mice (A, C), pooled from 2–3 independent experiments with 3–11 mice per time point (B, D, E), or 2 independent experiments with pooled cells from 2–3 mice (F). See also Figure S3.
To assess whether phenotypic and functional differences between memory CD8+ T cell subsets correlate with chromatin remodeling early during the effector phase, we analyzed the chromatin accessibility landscape in effector CD8+ T cell subsets using ATAC-seq. The analysis of 7 key loci revealed that exKLRG1 effector cells had an ATAC-seq signal profile that was different from KLRG1+Reporter+ and KLRG1−Reporter− effector cells (Figure 3F). ExKLRG1 effector cells displayed open chromatin states in both effector (Klrg1, Cx3cr1 and Gzma) and memory-related gene loci (Il7r, Il2, Tcf7 and Bach2). Chromatin remodeling in exKLRG1 effector cells may thus explain why exKLRG1 memory cells possess both a high cytotoxic and proliferative capacity.
Tdpe Cells, but not Tsle Cells, Efficiently Give Rise to ExKLRG1 Cells During the Contraction Phase
We next performed a series of effector cell transfer experiments to determine when and which KLRG1+ effector CD8+ T cell subsets differentiated into exKLRG1 memory cells. KLRG1+ Reporter+, KLRG1+ Reporter−, and early KLRG1−Reporter− cells (5–6 days p.i. with LM) were all able to differentiate into exKLRG1 cells by day 10–12 p.i. (Figure S4A and S4B) or day 28 p.i. (Figures 4A and 4B). The development of exKLRG1 cells from early KLRG1−Reporter− effector cells is explained by previous reports, in which early KLRG1− effector cells continue to give rise to KLRG1+ cells (Joshi et al., 2007; Plumlee et al., 2015; Sarkar et al., 2008). ExKLRG1 cells descending from early KLRG1−Reporter− effector cells expressed intermediate levels of CX3CR1, IL-7Rα and CD62L compared to KLRG1− Reporter− and KLRG1+ Reporter+ cells 12 and 28 days p.i. (Figures S4C and S4D). ExKLRG1 cells derived from KLRG1+ Reporter+ cells, however, displayed higher levels of IL-7Rα and CD62L, and lower levels of CX3CR1 compared to cells that did not lose KLRG1 12 and 28 days p.i. (Figures S4E and S4F). These results are consistent with our findings of higher IL-7Rα and CD62L, and lower CX3CR1 expression in exKLRG1 cells than KLRG1+ cells during memory CD8+ T cell differentiation (Figures 3C–3E).
Figure 4. Developmental Plasticity of Effector CD8+ T cell Subsets Following LM Infection.

(A) Expression of KLRG1, IL-7Rα and tdTomato in splenic effector OT-I cell subsets 6 days p.i. with LM (pre-transfer) and 22 days post-transfer (day 28 p.i.).
(B) Development of exKLRG1 memory cells (day 28 p.i.) from three different effector OT-I cell subsets 6 days p.i. with LM.
(C) Expression of KLRG1, IL-7Rα, tdTomato, CD62L and CX3CR1 in splenic effector OT-I cell subsets 9 days p.i. with LM (pre-transfer) and 26 days post-transfer (day 35 p.i.).
(D) Development of exKLRG1 memory cells (day 35 p.i.) from three different effector OT-I cell subsets 9 days p.i. with LM.
(E) Expression of CX3CR1 and CD62L in KLRG1+ and exKLRG1 memory cells 26 days post transfer of day 9 Tdpe cells. Host CD8+ T cells served as a control (gray line).
(F) Expression of CX3CR1 and CD62L in exKLRG1 and KLRG1− Reporter− memory cells 26 days post transfer of day 9 Tmpe cells. Host CD8+ T cells served as a control (gray line).
Mean ± SEM are shown. * P < 0.05 and ** P < 0.01 (unpaired two-tailed Student’s t-test). Data are representative of two independent experiments with 3–4 mice. See also Figure S4.
We then isolated KLRG1hi Reporter+ Tsle and Tdpe cells from the spleen of mice 9 days p.i. with LM, and transferred them into infection-matched WT mice (Figures 4C–4F). Thirty-five days p.i., about 15% of Tdpe and 3–5% of Tsle cells had differentiated into exKLRG1 memory cells (Figures 4C and 4D). We also isolated and transferred Reporter− Tmpe cells, and found that these cells at day 9–11 p.i. displayed a very low potential to develop into exKLRG1 cells (Figures 4C, 4D and S4G) compared to earlier KLRG1−Reporter− cells. Consistent with the results from day 5–6 early effector cell transfer experiments (Figures S4D and S4F), exKLRG1 cells derived from Tdpe or Tmpe cells expressed intermediate levels of CX3CR1 and CD62L compared to KLRG1+ and KLRG1−Reporter− cells (Figures 4E and 4F). A similar number of exKLRG1 and KLRG1− Reporter− cells was recovered, while the number of recovered KLRG1+ Reporter+ cells was 6.2 – 9.6 fold lower (Figure S4H). These results are in line with our findings in Figure 1D, and indicate that exKLRG1 and KLRG1− Reporter− cells have a similar high potential of survival, while only few KLRG1+ cells survive the contraction phase.
In the absence of antigen, transferred exKLRG1 and KLRG1− Reporter− memory cells (day 30 p.i. with LM) did not acquire KLRG1, and KLRG1+ Reporter+ memory cells did not differentiate into exKLRG1 cells (Figure S4I). Together, these results indicate that exKLRG1 cells develop during the late effector and contraction phase, and not during the memory phase, and Tdpe cells represent the preferential effector cell population differentiating into exKLRG1 memory cells.
Molecular Profiling of ExKLRG1 Memory CD8+ T Cells
To determine the lineage relationship of exKLRG1 memory cells, we performed genome-wide transcriptional profiling and compared the transcriptome of exKLRG1 memory OT-I cells with those of KLRG1− Reporter− and KLRG1+ memory OT-I cells. Principal component analysis (PCA) revealed that the transcriptome of exKLRG1 memory cells resembled that of KLRG1−Reporter− memory cells but not KLRG1+ memory cells (Figure S5A). Only 36 genes were differentially expressed (> 1.5-fold) between exKLRG1 and KLRG1− Reporter− memory cells, compared to 132 genes differentially expressed between exKLRG1 and KLRG1+ memory cells (Figures S5B and S5C). The analysis of signature genes involved in CD8+ T cell differentiation and function revealed that exKLRG1 memory cells had an intermediate expression level of Tem cell-associated molecules (Gzma, Zeb2, Prdm1, S1pr5, Cx3cr1, and T-bet) and Tcm cell-associated molecules (Id3, Socs3, Bach2, Myc, Bcl-2, Eomes, CD62L, CXCR3, CD43, and CCR7) (Figures S5D–S5G) (Best et al., 2013; Bottcher et al., 2015; Dominguez et al., 2015; Xin et al., 2016; Yang et al., 2011). These results indicate that exKLRG1 memory cells are a heterogeneous population consisting of Tcm and Tem cells, whereas KLRG1−Reporter− or KLRG1+ cells are enriched for Tcm or Tem cells, respectively.
Given the recent report about CX3CR1int Tpm cells (Gerlach et al., 2016), we analyzed to what extent the characteristics of exKLRG1 memory cells overlapped with those of Tpm cells. We found that exKLRG1 cells in the blood expressed intermediate levels of CX3CR1 7–60 days p.i. (Figure 3D), and expression of CX3CR1 was higher in circulating exKLRG1 Tem compared to exKLRG1 Tcm cells (Figure S5H). However, CX3CR1 expression on exKLRG1 memory cells was decreased by day 299 p.i (Figure 3D). Approximately 42% of CX3CR1int Tpm cells but only 22–27% of CX3CR1hi or CX3CR1lo memory cells were exKLRG1 cells, indicating that CX3CR1int Tpm cells were enriched in exKLRG1 cells (Figure S5I). Accordingly, about 70% of KLRG1− CX3CR1+ but only 27% of KLRG1− CX3CR1− memory cells expressed the Reporter (Figure S5J). However, neither exKLRG1 nor KLRG1− Reporter− Trm cells in the lung and small intestine expressed CX3CR1 (Figures S5K and S5L). As such, CX3CR1 may be used to distinguish circulating exKLRG1 from KLRG1− Reporter− early memory CD8+ T cells.
Functional analysis revealed that exKLRG1 memory cells in the spleen retained a higher cytotoxic capacity compared to KLRG1− Reporter− memory cells, as determined by GzmB production (Figure S5M). In contrast, the frequency of IL-2- and IFN-γ-producing cells upon restimulation with OVA peptide in vitro was similar between exKLRG1 and KLRG1− Reporter− memory CD8+ T cells (Figure S5N and data not shown). These results indicate that exKLRG1 memory cells acquire many features of KLRG1− Reporter− memory cells while retaining some characteristics reminiscent of their KLRG1+ effector cell origin.
ExKLRG1 Tcm and Tem Cells Retain High Responsiveness to IL-12
Bystander-mediated activation of memory CD8+ T cells is a key element in the early step of limiting pathogen invasion, and requires responsiveness to inflammatory cytokines but is independent of cognate antigen recognition (Chu et al., 2013; Soudja et al., 2012). We next investigated the responsiveness of exKLRG1 memory cells to inflammatory cytokines (IL-12 alone or in combination with IL-15 or type I interferons (IFN-α/β)) and found that KLRG1+ T cells exhibited the highest cytokine-driven cognate antigen-independent production of IFN-γ compared to KLRG1− T cell subsets (Figure 5A). Interestingly, a higher percentage of exKLRG1 Tcm and Tem cells produced IFN-γ compared to their corresponding KLRG1− Reporter− Tcm and Tem cell counterparts (Figure 5A). Although exKLRG1 cells were enriched in CX3CR1+ cells (Figures 3D and 3E), exKLRG1 cells produced higher amounts of IFN-γ than KLRG1− Reporter− cells within the CX3CR1+ or CX3CR1− population (Figure 5B). The increased cognate antigen-independent production of IFN-γ in exKLRG1 Tem cells was also confirmed in vivo by analyzing the recall response to wild-type Listeria monocytogenes, which did not express OVA antigen (Figures 5C). However, transferred KLRG1+ and exKLRG1 Tem cells had an equal capacity to expand upon secondary challenge (Figures S5O, right and S5P). Similarly, transferred exKLRG1 and KLRG1− Reporter− Tcm cells expanded equally well after re-challenge (Figures S5O, left and S5P). Our results indicate that the responsiveness of Tcm and Tem cells to the inflammatory cytokine IL-12 correlates with their distinct effector cell origin, and the high responsiveness of exKLRG1 memory cells to IL-12 is reminiscent of their KLRG1+ effector cell origin. Yet, the capacity of Tcm and Tem cells to proliferate upon secondary challenge is not affected by their effector cell origin.
Figure 5. ExKLRG1 Memory Cell Subsets Retain High Cytotoxic Capacity and Responsiveness to IL-12.

(A and B) Production of IFN-γ in splenic Tem (KLRG1+ CD62L− or KLRG1− CD62L−) and Tcm (KLRG1− CD62L+) OT-I cells (A) or Tem (KLRG1− CX3CR1+ CD62L−) and Tcm (KLRG1− CX3CR1+ CD62L+) OT-I cells (B) following stimulation with IL-12 (density plots), IL-12 + IFNα/β or IL-12 + IL-15 for 7 hours in vitro.
(C) Memory OT-I cells were generated as described in Figure S1E and mice were challenged 90 days later with Listeria monocytogenes, which did not express OVA. Production of IFN-γ in Tem (KLRG1+ CD62L− or KLRG1− CD62L−) and Tem (KLRG1− CD62L+) OT-I cells in spleen and liver (density plots) 12 hours after rechallenge.
(D) Expression of GzmB in circulating memory (KLRG1+CD69− or KLRG1−CD69−) and Trm (KLRG1−CD69+) cells within the endogenous OVA-tetramer+ CD44hi CD8+ T cell population in the liver 108 days p.i. with LM.
(E) Expression of GzmB in KLRG1− Reporter− Trm and exKLRG1 tetramer+ Trm cells in the liver 108 days p.i. with LM.
(F) Expression of GzmB in KLRG1− Reporter− Trm and exKLRG1 Trm cells within intraepithelial CD103+ CD69+ and CD103− CD69+ OT-I cell subsets 42 days after oral infection with LM (n=8).
Mean ± SEM are shown. * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001 (unpaired two-tailed Student’s t-test). The data are representative of two (C-F) or three (A, B) independent experiment with 3–7 mice. See also Figure S5.
ExKLRG1 Trm Cells Retain High Cytotoxic Capacity
We next investigated whether developmental plasticity of KLRG1+ effector CD8+ T cells also generated diversity among Trm cells. We found that the majority of CD69+ Trm cells in the liver expressed high levels of GzmB (Figure 5D). However, a higher frequency of CD69+ exKLRG1 Trm cells expressed GzmB compared to CD69+ KLRG1− Reporter− Trm cells (Figure 5E). We next analyzed the Trm cell subsets that were generated in the small intestine epithelium after oral infection with LM (Figure 2H). Both CD103+ and CD103− exKLRG1 Trm cells in the small intestine epithelium produced more GzmB than their KLRG1− Reporter− Trm cell counterparts (Figure 5F). These results indicate that similar to Tcm and Tem cells, Trm cells display functional diversity, which originates from two different effector cell populations, namely KLRG1+ and KLRG1− cells.
ExKLRG1 Memory CD8+ T Cells Mount Highly Effective Anti-Viral and Anti-Tumor Responses
We next determined the ability of exKLRG1 memory cells to mediate protective immunity. Although KLRG1− Reporter− cells expanded most efficiently at the site of viral infection (Figure 6A–6B), exKLRG1 and KLRG1+ memory cells, but not KLRG1− Reporter− memory cells, provided optimal anti-influenza immunity (Figure 6C). ExKLRG1 cells in the lung quickly re-expressed KLRG1 and upregulated the cytotoxicity marker CD107a to a level comparable to KLRG1+ cells (Figures 6D and 6E), indicating that cytolytic activity, rather than recall proliferation, correlated with enhanced protection, which is consistent with published results (Olson et al., 2013). On the other hand, we employed a mouse model of human melanoma, in which Tcm cells with high proliferative capacity drive potent anti-tumor immunity (Klebanoff et al., 2005). KLRG1− Reporter− cells and exKLRG1 cells proliferated and inhibited tumor growth more efficiently than KLRG1+ cells (Figures 6F and 6G). These results suggest that the heterogeneous and functionally versatile exKLRG1 memory CD8+ T cell population plays an important role in promoting highly effective immune responses in tissues and at barrier sites.
Figure 6. ExKLRG1 Memory CD8+ T cells Mount Potent Anti-Influenza and Anti-Tumor Responses.

(A) Schematic of the adoptive transfer and infection experiments.
(B) Mice receiving memory OT-I cell subsets were challenged i.n. with OVA-expressing influenza virus (FLU). Seven days later, the number of effector OT-I cells in the lung was determined.
(C) Viral RNA in the lung 7 days p.i. with FLU was determined by quantitative RT-PCR.
(D and E) Expression of KLRG1 and CD107a in effector OT-I cell subsets in the lung 7 days p.i. with FLU.
(F) Tumor size of mice s.c. injected with melanoma cells (B16-OVA) followed by adoptive transfer of memory OT-I cell subsets.
(G) Percentage of transferred OT-I cells within total CD8+ T cells in the LN, spleen and tumor at day 17 after tumor inoculation.
Data were analyzed by unpaired two-tailed Student’s t-test (B, C, E) or two-way ANOVA (F). Mean ± SEM are shown. * P < 0.05, ** P < 0.01 and *** P < 0.001. Data are pooled from 2–3 experiments with 10–11 mice (B and C), or representative of two (D, E) or three (F, G) independent experiments with 3–4 (D, E) and 5–6 (F, G) mice.
Bach2 Supports ExKLRG1 Memory Cell Development
We then analyzed which factor controlled the differentiation of KLRG1+ effector cells into exKLRG1 memory cells. Bach2 is a transcription repressor that restrains terminal differentiation and supports memory formation of lymphocytes including CD8+ T cells and B cells (Hu and Chen, 2013; Roychoudhuri et al., 2016; Shinnakasu et al., 2016). By using Bach2Flag reporter mice, we found that Bach2 expression was downregulated in Tsle cells, whereas Tdpe and Tmpe cells retained high expression of Bach2 (Figures S6A–F).
To determine whether Bach2 is required for the development of exKLRG1 memory cells, we transferred equal numbers of Klrg1Cre/+Rosa26tdTomato/+Bach2+/+ and Klrg1Cre/+Rosa26tdTomato/+Bach−/− OT-I cells into WT mice and infected them with LM (Figure S7A). As previously reported (Roychoudhuri et al., 2016), Bach2 deficiency in naïve OT-I cells led to a decrease in the total number of OT-I cells in the blood by day 10 p.i. with LM (Figure S7B). Accordingly, the number of KLRG1+, exKLRG1 and KLRG1− Reporter− cells in the spleen and LN, and the number of exKLRG1 Tcm and exKLRG1 Tem cells in the spleen was significantly reduced on day 27 p.i. in the absence of Bach2 (Figure 7A and Figure S7C). Importantly, Bach2 deficiency led to a decreased percentage of exKLRG1 memory cells within Reporter+ OT-I cells in the spleen (Figure 7B). Conversely, constitutive expression of Bach2 in naive OT-I cells by tamoxifen-mediated excision of the STOP cassette in the Rosa26 locus (Rosa26Bach2) (Kuwahara et al., 2016), resulted in considerably impaired expansion of effector CD8+ T cells in the spleen and liver on day 10 p.i. with LM (Figures 7C and 7D). Rosa26ERT2Cre/Bach2 effector OT-I cells exhibited lower Ki-67 expression and retained higher levels of Bcl-2 and CD62L compared with Rosa26ERT2Cre/+ effector OT-I cells, but failed to express KLRG1 (Figures 7E and S7F). As a result, Rosa26ERT2Cre/Bach2 memory OT-I cells accumulated in the LN on day 30 p.i. (Figure 7D), indicating that downregulation of Bach2 in naïve CD8+ T cells is essential for the complete differentiation of effector cells.
Figure 7. Bach2 Supports the Differentiation of KLRG1+ Effector Cells into ExKLRG1 Memory Cells.

(A) Naive Klrg1Cre/+Rosa26tdTomato/+Bach2+/+ and Klrg1Cre/+Rosa26tdTomato/+Bach2−/− OT-I cells from different CD45 congenic backgrounds were co-transferred into WT mice, followed by infection with LM one day later. The number of exKLRG1 Tcm and exKLRG1 Tem OT-I cells in the spleen 30 days p.i. with LM is shown.
(B) Percentage of exKLRG1 cells among Reporter+ donor OT-I cells in the spleen 10 and 30 days p.i. with LM.
(C) Schematic of the experimental protocol for (D-F). Tamoxifen was injected for four consecutive days to induce Cre-dependent Bach2 expression in OT-I Rosa26ERT2Cre/Bach2 mice. After 9 days, Bach2-expressed GFP+ naïve OT-I cells were sorted, mixed with Rosa26ERT2Cre/+ naïve OT-I cells at a 1:1 ratio, and transferred into C57BL/6 mice followed by infection with LM one day later.
(D) Relative frequency of Rosa26ERT2Cre/+ and Rosa26ERT2Cre/Bach OT-I cells 10 and 30 days p.i. with LM in the LN, spleen and liver.
(E and F) Expression of CD62L, KLRG1, Ki-67 and Bcl-2 in Rosa26ERT2Cre/+ and Rosa26ERT2Cre/Bach OT-I cells 10 days p.i. with LM.
(G) Schematic of the experimental protocol for (H).
(H) Expression of KLRG1 and Reporter in OT-I cells post-transfer of day 10 KLRG1+ effector OT-I cells into infection-matched WT mice (32 days p.i. with LM).
(I) Naive Klrg1Cre/+Rosa26tdTomato/+ and Klrg1Cre/+Rosa26tdTomato/Bach2 OT-I cells from different CD45 congenic backgrounds were co-transferred into WT mice, followed by infection with LM one day later. The number of KLRG1+, exKLRG1 and KLRG1−Reporter− OT-I cells in the spleen at days 10 and 70 p.i. with LM is shown.
(J) Percentage of exKLRG1 cells among Reporter+ OT-I cells in the spleen 70 days p.i. with LM.
(K) Number of exKLRG1 Tcm and exKLRG1 Tem OT-I cells in the spleen 70 days p.i. with LM.
Mean ± SEM are shown. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 (unpaired two-tailed Student’s t-test). Data are representative of two independent experiments with 5 (D-H) or 4–10 (A, B, I-K) mice. See also Figure S5, S6, and S7.
We next investigated the contribution of Bach2 for the development of exKLRG1 memory cells by crossing Klrg1Cre/+Rosa26tdTomato/+ mice with Bach2flox/− mice, in order to delete Bach2 only in effector cells that expressed KLRG1 (Figure S7D). However, we found that the deletion efficiency correlated with the intensity of KLRG1 expression: Bach2 was efficiently deleted in KLRG1hi cells, but only partially in KLRG1int and exKLRG1 cells (Figures S7E and S7I). Naïve OT-I cells and KLRG1− Reporter− memory OT-I cells had the same amount of the Bach2 floxed allele (Figures S7E and S7F), indicating that Klrg1Cre-mediated recombination did not occur in KLRG1− Reporter− memory cells. Therefore, we isolated KLRG1hi effector OT-I cells from mice engrafted with Klrg1Cre/+Rosa26tdTomato/+Bach2+/+ or Klrg1Cre/+Rosa26tdTomato/+Bach2flox/− OT-I cells and transferred them into infection-matched WT mice (Figure 7G). Whereas Klrg1Cre/+Rosa26tdTomato/+Bach2+/+ OT-I cells efficiently differentiated into exKLRG1 cells 22 days post transfer, Klrg1Cre/+Rosa26tdTomato/+Bach2flox/− OT-I cells were not able to differentiate into exKLRG1 cells (Figure 7H)._Conversely, Klrg1Cre-mediated specific induction of Bach2 in KLRG1+ effector OT-I cells promoted both Tcm and Tem exKLRG1 cell development 10 and 70 days p.i. with LM (Figures 7I–7K). The downregulation of Bach2 was regulated by an AKT-mTOR-Foxo1 pathway (Figures S7G–S7J), and inhibition of this pathway by Rapamycin (Araki et al., 2009) enhanced the conversion of KLRG1+ effector cells into exKLRG1 cells (Figures S7K–S7M). These findings suggest that Bach2 expressed in Tdpe and Tmpe cells plays an important role in the development of exKLRG1 memory cells.
DISCUSSION
By using a Klrg1Cre reporter system, we demonstrate that KLRG1+ IL-7Rα+ effector CD8+ T cells, which receive intermediate levels of activating and inflammatory signals, possess developmental plasticity, as they downregulate KLRG1 during the contraction phase, and enter the pool of long-lived KLRG1− IL-7Rα+ memory cells. The downregulation of KLRG1 in effector cells is not a singular, stand-alone event, but enables the development of exKLRG1 memory cells, which have a distinct molecular and functional profile, and promote long-lasting protective immunity in tissues and at barrier sites.
The phenotypic and functional diversity among effector cells is thought to shape memory CD8+ T cell heterogeneity and the graded activity of transcription factors in effector CD8+ T cells has been associated with distinct effector cell traits (Kaech and Cui, 2012). Here we demonstrate that exKLRG1 cells express T-bet, Bach2, Zeb2 and Bcl-2 at an intermediate level compared to KLRG1+ and KLRG1− Reporter− cells, indicating that T-betint Tdpe but not T-bethi Tsle cells possess developmental plasticity, as confirmed by our adoptive cell transfer experiments. We further show that the different properties of exKLRG1 and KLRG1− Reporter− memory cells may be due to differences in chromatin accessibility of key effector-(Klrg1, Cx3cr1 and Gzma) and memory-related gene loci (Il7r, Il2, Tcf7, and Bach2), as has been shown for naïve CD8+ T cells responding to viral infection (Scott-Browne et al., 2016), or for the specification of distinct CD8+ T cell fates (Gray et al., 2017; Yu et al., 2017).
ExKLRG1 memory cells express intermediate levels of CX3CR1 and CD62L reminiscent of recently identified CX3CR1int Tpm cells, which are the predominant memory CD8+ T cells surveying non-lymphoid tissues (Gerlach et al., 2016). We show that approximately 40% of CX3CR1int Tpm cells descend from exKLRG1 cells. However, exKLRG1 cells are also found within the CX3CR1− and CX3CR1hi cell populations, which exhibit characteristics of classical Tcm and Tem cells, respectively. These results indicate that exKLRG1 cells partly overlap with, but are not restricted to the CX3CR1int Tpm fraction. Consistent with the essential roles of IL-12 and T-bet in the induction of KLRG1 and CX3CR1 during effector CD8+ T cell differentiation, exKLRG1 Tcm and Tem cells retain higher responsiveness to IL-12, but similar proliferative capacity upon rechallenge, compared to KLRG1− Reporter− Tcm and Tem cells. This enhanced antigen-independent response of exKLRG1 memory cells may contribute to early anti-microbial protection meditated by bystander-activated memory CD8+ T cells.
CX3CR1 expression gradually declines in memory CD8+ T cells, in particular in exKLRG1 Tcm cells. Our results are consistent with previous findings that the frequency of CX3CR1hi cells is gradually decreased over time and CX3CR1int Tpm cells progressively convert into CX3CR1− cells (Gerlach et al., 2016). Together, these results suggest that CX3CR1 expression may only be suitable to distinguish early circulating exKLRG1 and KLRG1− Reporter− memory cells.
Our results demonstrate that the transcription repressor Bach2 does not only control TCR-driven transcriptional programs during the initial activation of CD8+ T cells to indirectly regulate memory CD8+ T cell development (Hu and Chen, 2013; Roychoudhuri et al., 2016), but also regulates developmental plasticity of KLRG1+ effector cells. Although high expression of Bach2 in our overexpression studies promotes both exKLRG1 Tcm and exKLRG1 Tem cell development, the enhancement of exKLRG1 Tcm cell development appears more pronounced. It is possible that the intermediate expression of Bach2 in exKLRG1 cells may equally promote Tcm and Tem cell differentiation. Alternatively, additional transcriptional regulators could be involved in controlling the differentiation of KLRG1+ effector cells into exKLRG1 Tem cells. In addition to the cell-intrinsic role of Bach2 in effector CD8+ T cell survival (Roychoudhuri et al., 2016), Bach2-mediated induction of CD62L may also enable exKLRG1 cells to access distinct survival niches, such as IL-7-producing cells in lymphoid organs (Hara et al., 2012; Jung et al., 2010).
ExKLRG1 cell development continuously occurred during the late effector and contraction phases, but not during the memory phase. It should be noted that although memory exKLRG1 cell development correlated with IL-7Rα re-acquisition, early exKLRG1 effector cells (day 5–7 p.i.) did not express IL-7Rα, indicating that downregulation of KLRG1 precedes the induction of IL-7Rα at this early time point. The different duration of KLRG1 expression may reflect different signal strength received by effector cells, which contributes to fate decision of KLRG1+ effector cells and may contribute to the functional heterogeneity within exKLRG1 cells. Noteworthy, the frequency of exKLRG1 memory cells reported in our study may be underestimated, because Cre-reporter strains may underreport Cre activity. We noticed that in about 5–10% of KLRG1+ effector cells, the expression of KLRG1 may have not been strong enough to drive Reporter expression. In fact, not all KLRG1+ effector cells expressed the eGFP-Cre fusion protein and the efficiency of Klrg1Cre-mediated deletion of Bach2 was dependent on the intensity of KLRG1 expression. Some KLRG1− Reporter− memory cells may share the features with exKLRG1 cells and therefore be descendants of effector cells that expressed low or intermediate levels of KLRG1.
As reported previously (Mackay et al., 2013; Sheridan et al., 2014), KLRG1+ cells are excluded from the Trm cell population, and adoptive transfer experiments of KLRG1− and KLRG1+ effector CD8+ T cells suggest that Trm cells derive from KLRG1− precursor cells (Mackay et al., 2013). However, temporal, spatial and proportional limitations of adoptive transfer experiments in previous studies may have led to underestimate KLRG1+ effector CD8+ T cell plasticity, and its impact on protective immunity at barrier sites. By using in vivo fate mapping, we demonstrate that effector CD8+ T cells that have previously expressed KLRG1 are able to migrate into tissue barrier sites, upregulate markers of tissue residency, and represent up to 50% of the Trm cell pool in the liver, lung, skin, and small intestine. Similar to circulating exKLRG1 memory CD8+ T cells, exKLRG1 Trm cells express higher levels of GzmB compared to KLRG1− Reporter− Trm cells. These results indicate that Trm cells with distinct functional characteristics originate from two different effector cell populations, namely from effector cells that have previously expressed KLRG1, and KLRG1− effector cells, although when and where KLRG1+ effector cells downregulate KLRG1 to become exKLRG1 Trm cells remains unclear. Following oral infection with LM, as much as 70% of CD103− CD69+ Trm cells in the small intestine epithelium are exKLRG1 cells, suggesting that exKLRG1 and KLRG1− Reporter− Trm cells. Because CD103− and CD103+ Trm cells may be differentially localized (Bergsbaken and Bevan, 2015), it is suggested that exKLRG1 and KLRG1− Reporter− Trm cells may, in part, seed distinct niches. In contrast to KLRG1+ memory cells, exKLRG1 memory cells express CXCR3, which has been shown to promote the formation of Trm cells (Fernandez-Ruiz et al., 2016; Shin and Iwasaki, 2012). This may allow exKLRG1 cells to enter the epithelium, gain close contact to IL-15-producing cells at barrier sites, and thereby receive the necessary signals for survival and tissue residency (Mortier et al., 2009).
In summary, we demonstrate that developmental plasticity of KLRG1+ effector CD8+ T cells is a driving force in promoting phenotypic and functional diversity among Tcm, Tem and Trm cells. KLRG1− IL-7Rα+ Tmpe cells with a multi-lineage memory potential are a heterogeneous population, containing cells with a history of KLRG1 expression. These findings redefine our understanding of effector-to-memory CD8+ T cell differentiation and the generation of functional heterogeneity, and could help facilitate the development of more effective T cell-based therapies and vaccines against malignancies and infectious diseases.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard A. Flavell (richard.flavell@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
OT-I (Jax 003831), UBC-GFP (Jax 004353), Rosa26eYFP (Jax 006148), Rosa26tdTomato (Jax 007908) mice were obtained from the Jackson Laboratory and Rag2−/− mice (RAGN12) were obtained from Taconic Biosciences. Bach2flox/flox and Rosa26Bach2 mice have been described previously (Kometani et al., 2013; Kuwahara et al., 2016). C57BL/6-Ly5.2/Cr (CD45.1) congenic mice were obtained from the National Cancer Institute. C57BL/6 mice were obtained from the National Cancer Institute, Charles River Laboratories and CLEA Japan. All animal studies were performed in accordance with the guidelines of the Office of Animal Research Support of Yale University and the Animal Experiment Committee of the RIKEN Yokohama Institute.
Generation of Klrg1Cre Mice
Klrg1Cre mice were generated with a standard cloning strategy. Homologous arms of the Klrg1 locus were isolated from genomic DNA by PCR. A 2.5 kb fragment containing exons 4 and 5 was used as the 5’ homology region and a 2.7 kb fragment containing 3’UTR was used as the 3’ homology region. The homologous arms were cloned into a reporter plasmid containing a Frt-flanked Neomycin resistance gene (Neo), a thymidine kinase, and a fragment encoding an IRES-eGFP-iCre fusion protein. The reporter cassette was introduced immediately after the stop codon to avoid disruption of cis-regulatory elements in the 3′ UTR and the polyadenylation signal. The targeting vector was electroporated into C57BL/6-derived JM8A ES cells. After the selection with G418, correctly targeted clones were identified by PCR. To obtain chimeric mice, correctly targeted ES clones were injected into C57BL/6 blastocysts, which were then implanted into CD1 pseudopregnant foster mothers. Male chimaeras were bred with C57BL/6 to screen for germline transmitted offspring. Germline transmitted mice were bred with FLPo (Jax 011065) mice to remove the Neo gene.
Generation of Bach2Flag Mice
To generate Bach2Flag mice, homologous arms of the Bach2 locus were cloned from genomic DNA by PCR. A 6.5 kb fragment was used as the 5’ homology region and a 2.5 kb fragment was used as the 3’ homology region. The homologous arms were cloned into a reporter plasmid containing a Frt-flanked Neo gene, a diphtheria toxin A (DTA), and a 3×Flag-tag which was inserted after the first methionine. The targeting vector was electroporated into C57BL/6-derived Bruce4 ES cells. After the selection with G418, correctly targeted clones were identified by PCR. The targeted ES clones were injected into blastocysts from BALB/c mice. The obtained chimeric mice were crossed with C57BL/6 mice to obtain germline transmitted animals. To remove the Neo cassette, the mice were further crossed to CAG-Flpe mice (Kanki et al., 2006).
Adoptive Cell Transfer, Infection and Tumor Challenge
Spleen and peripheral LN were isolated from Klrg1Cre/+Rosa26tdTomato/+OT-I or Klrg1Cre/CreRosa26eYFP/eYFPOT-I mice on a Rag2 sufficient or deficient background. Single cell suspension was generated by mechanical disruption and CD8+ T cells were enriched by using EasySep™ Mouse Naive CD8+ T cell Isolation Kit (STEMCELL Technologies) or a biotinylated antibody cocktail (anti-B220 (RA3–6B2), anti-CD4 (RM4–5), anti-CD11b (M1/70), anti-CD11c (N418), and anti-Gr1 (RB6–8C5) antibodies; BioLegend) and MojoSort™ Streptavidin Nanobeads (BioLegend). Naive CD44lo CD62Lhi CD8+ CD45.1+ Reporter− OT-I cells were further purified by a cell sorter (BD Biosciences). A total of 0.5 – 1×105 naïve OT-I cells were transferred into C57BL/6 mice (CD45.2), unless otherwise indicated. One day after adoptive transfer, mice were infected intravenously (i.v.) with 1 × 105 recombinant Listeria monocytogenes expressing OVA (LM) (Pope et al., 2001), or 1 × 106 plaque-forming units (PFU) recombinant vesicular stomatitis virus expressing OVA (VSV) (Kim et al., 1999). For oral LM infection, mice were orally infected with 1 × 108 CFU of LM. For tissue imaging, Klrg1Cre/+Rosa26tdTomato/+OT-I mice were crossed with UBC-GFP mice to detect transferred OT-I cells. For co-transfer experiments, naive control and Bach2 gene-manipulated OT-I cells, each with a different congenic marker, were mixed at a 1:1 ratio and transferred into C57BL/6 mice. For the in vivo induction of Bach2 in naïve CD8+ T cells, OT-I Rosa26ERT2Cre/Bach2 mice were administered orally with 2 mg of tamoxifen (Sigma-Aldrich) in sunflower seed oil (Sigma-Aldrich) once daily for four consecutive days, and Bach2-expressing GFP+ naïve OT-I cells were sorted 9 days after the initial treatment. For rapamycin treatment, OT-I-engrafted mice were administered i.p. with 300 μg kg−1 of rapamycin (Calbiochem) daily from day 8 to day 19 p.i. with LM.
CD45.1+ effector or memory OT-I cells were enriched from the spleen of LM-infected mice by using a biotin-anti-CD45.1 antibody and Streptavidin MicroBeads (Miltenyi Biotec), and further purified by a cell sorter (BD Biosciences). For effector cell transfer experiments, 2 – 5 × 105 effector OT-I cells were adoptively transferred into infection-matched WT mice. To determine the ability of exKLRG1 memory cells to mediate secondary responses, 1 × 104 memory OT-I cells were adoptively transferred into naïve WT mice that were infected i.n. with OVA-expressing influenza A/PR8 virus (H1N1; FLU) one day later. The lungs were collected 7–8 days p.i. and the amount of influenza virus RNA was determined by quantitative RT-PCR. For the in vivo tumor experiment, naïve WT mice were injected with B16 melanoma expressing OVA (1 × 105 cells) (Falo et al., 1995) intradermally into the flank. Five days later, memory OT-I cell subsets were transferred and the mice immediately immunized with 10 μg of OVA (Worthington Biochemical) and 2 μg of poly(I:C) (GE Healthcare) s.c. into the flank. Tumor growth was monitored by measurement of two perpendicular diameters of the skin tumor (mm2) by caliper every other day. To generate cognate antigen-independent recall responses, OT-I engrafted mice were reinfected i.v. with 1 × 106 wild-type Listeria (EGD) 90 days after primary LM infection.
Flow Cytometry
Fluorescent dye-labeled antibodies targeting Bcl2 (BCL/10C4), CCR7 (4B12), CD4 (RM4–5), CD8α (53–6.7), CD43 (1B11), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD62L (MEL-14), CD69 (H1.2F3), CD103 (2E7), CD107a (1D4B), CD127 (A7R34), CXCR3 (173), CX3CR1 (SA011F11), Eomes (Dan11mag), FLAG (DYKDDDDK) (L5), Foxp3 (FJK-16s), Granzyme B (GB11), IFN-γ (XMG1.2), IL-2 (JES6–5H4), Ki-67 (16A8), KLRG1 (2F1), NK1.1 (PK136), T-bet (4B10), and TCF1 (C63D9) were purchased from BioLegend, Thermo Fisher Scientific, BD Biosciences or Cell Signaling Technology. Rabbit anti-RFP (600–401-379 or 600–406-379) was purchased from Rockland. OVA-Kb tetramer was from MBL. For intracellular cytokine staining, splenocytes from infected mice were stimulated with SIINFEKL peptide in the presence of Protein Transport Inhibitor (BD Biosciences) or Brefeldin A (Biolegend) for 4–5 hours, followed by fixation and permeabilization according to the manufacturer’s instructions (BD Biosciences). T-bet, Eomes, Foxp3, and anti-tdTomato staining were performed by eBioscience™ Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific) according to the manufacturer’s instructions. The cells were analyzed on a LSRII or FACSCantoII (BD Biosciences) and data were analyzed using FlowJo software (Tree Star).
Intravascular CD8+ T Cell Staining
Mice were injected i.v. (retro-orbital) with 2 μg of anti-CD8α phycoerythrin antibody diluted in 100 μL of sterile PBS as previously described (Anderson et al., 2014). Mice were euthanized and tissues were collected five minutes after injection of the anti-CD8α antibody.
Isolation of Lymphocytes from Non-Lymphoid Organs
Peripheral blood lymphocytes (PBLs) were isolated from retro-orbital vein of infected mice. Liver and lung cell homogenates were digested for 1 hour at 37°C with digestion buffer (RPMI 1640 + 5% FBS + 100 U/ml DNase I (Sigma-Aldrich) + 0.2 mg/mL Collagenase IV (Sigma-Aldrich)). Liver homogenates were centrifuged at 300 rpm for 3 min to remove hepatocytes, and lung homogenates were run through a 70 μm cell strainer (BD Biosciences). Non-hepatic supernatant and lung lymphocytes were centrifuged at 1500 rpm for 10 min. The cell pellet was resuspended in 1 mL of RPMI 1640 + 5% FBS and mixed with 4 ml of 27.5% OptiPrep (Axis-Shield). To make a gradient, 1 mL of RPMI 1640 was layered on top of the cells, and centrifuged at 2000 rpm for 20 min. Lymphocytes were removed from the interface of the gradient. To isolate intraepithelial lymphocytes (IELs), small intestines were opened longitudinally and washed. Small intestines were then cut into short segments, which were transferred into 50 mL conical tubes, and incubated for 30 min at 37°C with gentle shaking in RPMI 1640 containing 2% FBS, 2 mM EDTA, and 1 mM dithioerythritol. Cell suspensions were passed through a 70 μm cell strainer. The cells were resuspended in 4 mL of 40% Percoll (GE Healthcare), and 4 mL of 80% Percoll was underlayed in a 15 mL conical tube. Percoll gradient separation was performed by centrifugation for 20 min at 600 × g at room temperature. IELs were collected at the interphase of the Percoll gradient. Tumor-infiltrating cell were isolated with mechanical disruption of the tumor through a 70 μm cell strainer and resuspended in 40% Percoll solution (GE Healthcare). Cells were collected from the interface of a 40:80 Percoll gradient after centrifugation at 2000 rpm for 20 min.
Intravital Two-Photon Microscopy
Transferred Klrg1Cre/+Rosa26tdTomato/+ UBC-GFP+ OT-I cells in the foot skin were imaged by an inverted TCS SP8 multiphoton microscope (Leica Microsystems) equipped with an HC FLUOTAR L 25×/0.95 W VISIR objective lens (Leica Microsystems) and four non-descanned Hybrid Detectors (Leica Microsystems). Multiphoton excitation was provided by a Chameleon Vision II Ti:Sapphire laser (Coherent) tuned to 910 nm and a Chameleon Compact OPO-converted Chameleon Ultra II Ti:Sapphire laser (Coherent) tuned to 1110 nm. For image acquisition, 466 μm × 466 μm x–y planes were scanned at a resolution of 0.9 μm per pixel and images of 21–29 x–y planes with 3 μm z spacing were formed after averaging eight video frames for each x–y plane. Three-dimensional stacks were captured every 30 s. Emission signals were separated by a dichroic mirror at 560 nm and then further separated by a dichroic mirror at 495 nm. A 460/50-nm emission filter was used for DAPI and second harmonic generation (SHG), a 525/50-nm emission filter for GFP, and a 585/40-nm emission filter for tdTomato.
Tissue Clearing
Tissue clearing was performed as reported previously (Tainaka et al., 2014). Briefly, small intestines and foot skins were fixed with 4% paraformaldehyde, washed with PBS, and then immersed in Reagent 1 containing 25% urea (Nacalai Tesque), 25% N,N,N’,N’-tetrakis(2-hydroxypropyl) ethylenediamine (Tokyo Chemical Industry), and 15% polyethylene glycol mono-p-isooctylphenyl ether/Triton X-100 (Nacalai Tesque). Samples were stored at 4°C in the dark until imaging.
RNA-Seq and Bioinformatics Analysis
Memory OT-I cell subsets were isolated from the spleen 104–110 days p.i. with LM and 70 days p.i. with VSV. Total RNA was extracted from at least 1 × 105 cells derived from 5–8 mice using TRIzol™ reagent (Thermo Fisher Scientific). For OT-I memory cell subsets isolated from LM-infected mice, the DNA library was constructed with a SureSelect Strand-Specific RNA Library Prep for Illumina Multiplexed Sequencing (Agilent) according to manufacturer’s instruction. The size range of the resulting DNA library was estimated on a 2100 Bioanalyzer (Agilent). After checking the molar concentration by qPCR using a KAPA Library Quantification Kit (Kapa Biosystems), the DNA library was subjected to sequencing on a HiSeq 1500 sequencer (Illumina) in a 49-bp single read mode. For OT-I memory cell subsets isolated from VSV-infected mice, the DNA library was subjected to sequencing on a HiSeq 2500 sequencer (Illumina) in a 75 bp paired-end read mode.
The raw data were processed with CASAVA 1.8.2 (Illumina) to generate fastq files. The sequence reads were aligned to the Mus musculus reference genome (GRCm38/mm10) using TopHat2 version 2.0.8 (Kim et al., 2013). Cufflinks version 2.1.1 (Trapnell et al., 2010) was used to calculate the fragments per kilobase of transcript per million fragments mapped (FPKM). Principal component analysis (PCA) was performed with R (http://www.r-project.org/), and visualization generated with ggplot2 package (Wickham, 2009). The Venn diagram was generated with VennDiagram package (Chen and Boutros, 2011). To identify differentially regulated genes, an absolute 1.5-fold-change difference between any two samples and FDR (Benjamini-Hochberg) ≤ 0.1 was used.
ATAC-Seq
To profile open chromatin, we used the ATAC-seq protocol developed by (Buenrostro et al., 2015). In brief, 5 × 104 naïve and effector (8 days p.i. with LM) OT-I cell subsets from the spleen were sorted, centrifuged in cold PBS buffer for 5 min at 500 g, resuspended in 100 μL cold lysis buffer, and centrifuged immediately for 10 min at 500 g. The nuclei pellets were resuspended in the transposition reaction mix (Nextera DNA Library Preparation Kit, Illumina) and incubated at 37°C for 30 min. Immediately following transposition, a MinElute PCR Purification Kit (Qiagen) was used to purify DNA. To amplify transposed DNA fragments, PCR with barcoded PCR primers (Buenrostro et al., 2013) and NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs) was performed. To reduce GC and size bias in PCR, the appropriate number of PCR cycles was determined using qPCR. The amplified library was purified using a MinElute PCR Purification Kit. ATAC-seq libraries were sequenced using Illumina HiSeq2500 (75 bp; paired-end), at an average sequencing depth per sample of 35 million reads.
Raw sequencing reads were first processed with cutadapt-1.8.3 (Martin, 2011) to trim adapters. Bowtie-2 (Langmead and Salzberg, 2012) was used then to align the trimmed reads to the mouse genome mm10 with default parameters. Duplicated reads were marked and removed from the analysis with Picard MarkDuplicate tool (http://broadinstitute.github.io/picard). Subsequently, BEDTools (Quinlan and Hall, 2010) was used to turn the alignment file to a bed file, which was the input of MACS2 (Zhang et al., 2008) to call peaks and produce BedGraph files. Visualizations of coverage on regions of interest were produced with IGV (Robinson et al., 2011).
Quantitative Reverse Transcription PCR (RT-PCR) Analysis
RNA was reverse transcribed into a single-strand cDNA using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific). Quantitative RT-PCR was performed on a 7500 Fast Real-Time PCR system (Applied Biosystems) or a CFX96 Real-Time System (Bio-Rad) using a SYBR FAST universal qPCR kit (KAPA Biosystems). Expression values were calculated using the comparative threshold cycle method and normalized to mouse Actb. Sequence-specific oligonucleotide primers were designed using Primer3Plus software, qPrimerDepot or were described elsewhere (Dominguez et al., 2015; Endrizzi and Jameson, 2003; Pillai et al., 2016). The oligonucleotide sequences are listed in the STAR methods section.
In Vitro Cell Cultures
Naïve CD8+ T cells were isolated from the spleen of C57BL/6J mice and were stimulated with Dynabeads Mouse T-Activator CD3/CD28 (Life Technologies) in the presence of 5 ng/mL of rIL-2 (R&D) for 2 days. Where indicated, 20 ng/mL of rapamycin or 1.0 μM of Foxo1 inhibitor (AS1842856; Calbiochem) was added to culture medium.
For retroviral transduction assay, naïve CD8+ T cells were stimulated as described above for 2 days and were spin-infected with retrovirus produced from pMCs-mock-IRES-GFP vector or pMCs-Foxo1-CA-IRES-GFP vector (Harada et al., 2010). 48 h after transduction, the GFP+ cells were sorted using a FACSAriaII for quantitative RT-PCR analysis.
For the in vitro IFN-γ production assay, splenocytes were stimulated with rIL-12 (Peprotech; 50 ng/mL) in combination with rIL-15 (Peprotech; 50 ng/mL) or rIFN-α/β (PBL Assay Science; 250 units/mL each) for 7 hours in the presence of Protein Transport Inhibitor (BD Biosciences).
Immunoblot Analysis
Cells were sorted and cell extracts were prepared using a Nuclear Extract Kit (Active Motif). The cell lysates were separated on 5–20 % SDS-PAGE and transferred to a PVDF membrane (Merck). The membrane was immunoblotted with anti-Bach2 (ab83364, Abcam), anti-FLAG (M2, Sigma-Aldrich), and anti-Actin (C-11, Santa Cruz Biotechnology). The blots were visualized with the SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific).
Statistical Analysis
All data were presented as mean ± standard error of the means (SEM). Comparisons between groups were analyzed by unpaired two-tailed Student’s t-test or two-way ANOVA. Statistical analysis was performed using Prism 6 (GraphPad). * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Data and Software Availability
Next-generation sequencing data have been deposited in Gene Expression Omnibus (GEO) under SuperSeries accession code GEO: GSE110707, which includes individual data sets GEO: GSE110629 (RNA-seq; LM), GEO: GSE110706 (RNA-seq; VSV) and GEO: GSE110876 (ATAC-seq).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Bcl2 (BCL/10C4) AF647 | BioLegend | Cat#633509, RRID:AB_2064149 |
| Anti-mouse B220 (RA3–6B2) BV510 | BioLegend | Cat#103247, RRID:AB_2561394 |
| Anti-mouse B220 (RA3–6B2) Biotin | BioLegend | Cat#103204, RRID:AB_312989 |
| Anti-mouse CCR7 (4B12) Biotin | BioLegend | Cat#120104, RRID:AB_389232 |
| Anti-mouse CD4 (RM4–5) FITC | BD Biosciences | Cat#553047, RRID:AB_394583 |
| Anti-mouse CD4 (RM4–5) Biotin | BioLegend | Cat#100508, RRID:AB_312711 |
| Anti-mouse CD4 (RM4–5) APC-Cy7 | BioLegend | Cat#100526, RRID:AB_312727 |
| Anti-mouse CD8α (53–6.7) PE | BioLegend | Cat#100708, RRID:AB_312747 |
| Anti-mouse CD8α (53–6.7) PE-Cy7 | BioLegend | Cat#100722, RRID:AB_312761 |
| Anti-mouse CD8α (53–6.7) BV510 | BioLegend | Cat#100752, RRID:AB_2563057 |
| Anti-mouse CD8α (53–6.7) PerCP | Biolegend | Cat#100732, RRID:AB_893423 |
| Anti-mouse CD8α (53–6.7) APC-Cy7 | BioLegend | Cat#100714, RRID:AB_312753 |
| Anti-mouse CD8α (53–6.7) Pacific Blue | BD Biosciences | Cat#558106, RRID:AB_397029 |
| Anti-mouse CD11b (M1/70) V500 | BD Biosciences | Cat#562127, RRID:AB_10893815 |
| Anti-mouse CD11b (M1/70) Biotin | BioLegend | Cat#101204, RRID:AB_312787 |
| Anti-mouse CD11c (N418) PE-Cy7 | Thermo Fisher Scientific | Cat#25–0114-82, RRID:AB_469590 |
| Anti-mouse CD11c (N418) Biotin | BioLegend | Cat#117304, RRID:AB_313773 |
| Anti-mouse CD43 (1B11) PE-Cy7 | BioLegend | Cat#121218, RRID:AB_528813 |
| Anti-mouse CD44 (IM7) V500 | BD Biosciences | Cat#560781, RRID:AB_1937328 |
| Anti-mouse CD44 (IM7) FITC | BioLegend | Cat#103022, RRID:AB_493685 |
| Anti-mouse CD44 (IM7) APC-Cy7 | BioLegend | Cat#103028, RRID:AB_830785 |
| Anti-mouse CD45.1 (A20) FITC | Thermo Fisher Scientific | Cat#11–0453-82, RRID:AB_465058 |
| Anti-mouse CD45.1 (A20) PE-Cy7 | BioLegend | Cat#110730, RRID:AB_1134168 |
| Anti-mouse CD45.1 (A20) Pacific Blue | BioLegend | Cat#110722, RRID:AB_492866 |
| Anti-mouse CD45.1 (A20) AF700 | BioLegend | Cat#110724, RRID:AB_493733 |
| Anti-mouse CD45.2 (104) FITC | BD Biosciences | Cat#553772, RRID:AB_395041 |
| Anti-mouse CD62L (MEL-14) APC-Cy7 | BioLegend | Cat#104428, RRID:AB_830799 |
| Anti-mouse CD62L (MEL-14) eFluor450 | Thermo Fisher Scientific | Cat#48–0621-82, RRID:AB_1963590 |
| Anti-mouse CD69 (H1.2F3) FITC | BioLegend | Cat#104506, RRID:AB_313109 |
| Anti-mouse CD69 (H1.2F3) PE-Cy7 | BD Biosciences | Cat#552879, RRID:AB_394508 |
| Anti-mouse CD69 (H1.2F3) Pacific Blue | BioLegend | Cat#104524, RRID:AB_2074979 |
| Anti-mouse CD103 (2E7) FITC | BioLegend | Cat#121420, RRID:AB_10714791 |
| Anti-mouse CD103 (2E7) Pacific Blue | BioLegend | Cat#121418, RRID:AB_2128619 |
| Anti-mouse CD107a (1D4B) AF488 | BioLegend | Cat#121608, RRID:AB_571983 |
| Anti-mouse CD127 (SB/199) APC | BD Biosciences | Cat#564175 |
| Anti-mouse CD127 (SB/199) BV711 | BD Biosciences | Cat#565490 |
| Anti-mouse CD127 (A7R34) eFluor450 | Thermo Fisher Scientific | Cat#48–1271-80, RRID:AB_2016629 |
| Anti-mouse CD127 (A7R34) Biotin | Thermo Fisher Scientific | Cat#13–1271-85, RRID:AB_466589 |
| Anti-mouse CXCR3 (173) PE-Cy7 | BioLegend | Cat#126516, RRID:AB_2245493 |
| Anti-mouse CX3CR1 (SA011F11) FITC | BioLegend | Cat#149020, RRID:AB_2565703 |
| Anti-mouse CX3CR1 (SA011F11) PE-Cy7 | BioLegend | Cat#149016, RRID:AB_2565700 |
| Anti-mouse CX3CR1 (SA011F11) Biotin | BioLegend | Cat#149018, RRID:AB_2565701 |
| Anti-mouse Eomes (Dan11mag) eFluor450 | Thermo Fisher Scientific | Cat#48–4875-80, RRID:AB_2574061 |
| Anti-mouse FLAG (DYKDDDDK) (L5) PE | BioLegend | Cat#637309, RRID:AB_2563147 |
| Anti-mouse Foxp3 (FJK-16s) APC | Thermo Fisher Scientific | Cat#17–5773-82, RRID:AB_469457 |
| Anti-mouse Gr1 (RB6–8C5) APC | BioLegend | Cat#108412, RRID:AB_313377 |
| Anti-mouse Gr1 (RB6–8C5) Biotin | BioLegend | Cat#108404, RRID:AB_313369 |
| Anti-mouse Granzyme B (GB11) AF647 | BioLegend | Cat#515405, RRID:AB_2294995 |
| Anti-mouse IFN-γ (XMG1.2) APC | BioLegend | Cat#505810, RRID:AB_315404 |
| Anti-mouse IL-2 (JES6–5H4) | BioLegend | Cat#503831, RRID:AB_2561749 |
| Anti-mouse Ki-67 (16A8) APC | BioLegend | Cat#652405, RRID:AB_2561929 |
| Anti-mouse Ki-67 (B56) AF647 | BD Biosciences | Cat#561126, RRID:AB_10611874 |
| Anti-mouse KLRG1 (2F1) FITC | BioLegend | Cat#138409, RRID:AB_10643998 |
| Anti-mouse KLRG1 (2F1) PE-Cy7 | BioLegend | Cat#138416, RRID:AB_2561736 |
| Anti-mouse KLRG1 (2F1) BV421 | BioLegend | Cat#138414, RRID:AB_2565613 |
| Anti-mouse KLRG1 (2F1) APC | BioLegend | Cat#138412, RRID:AB_10641560 |
| Anti-mouse KLRG1 (2F1) APC | Thermo Fisher Scientific | Cat#17–5893-82, RRID:AB_469469 |
| Anti-mouse NK1.1 (PK136) FITC | BioLegend | Cat#108705, RRID:AB_313392 |
| Anti-mouse T-bet (4B10) APC | BioLegend | Cat#644813, RRID:AB_10896913 |
| Anti-mouse TCF1 (C63D9) AF647 | Cell Signaling Technology | Cat#6709 |
| Anti-RFP | Rockland | Cat#600–401-379, RRID:AB_2209751 |
| Anti-RFP biotin | Rockland | Cat#600–406-379, RRID:AB_828390 |
| Anti-rabbit AF594 | Thermo Fisher Scientific | Cat#R37117, RRID:AB_2556545 |
| Anti-mouse FLAG (M2) | Sigma-Aldrich | Cat#F1804, RRID:AB_262044 |
| Anti-mouse Actin (C-11) | Santa Cruz Biotechnology | Cat#sc-1615, RRID:AB_630835 |
| Anti-mouse Bach2 | Abcam | Cat#ab83364, RRID:AB_1861444 |
| Bacterial and Virus Strains | ||
| Listeria monocytogenes (EGD) | Yoshikai Y (Kyushu University) | N/A |
| Listeria monocytogenes expressing OVA | Shen H (University of Pennsylvania) Pope C et al., 2001 | N/A |
| Vesicular stomatitis virus expressing OVA | Kim et al., 1999 | N/A |
| Influenza A/PR8 virus expressing OVA | Iwasaki A (Yale University), Garcia-Sastre A (Icahn School of Medicine at Mount Sinai) | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| T-Select H-2Kb OVA Tetramer-SIINFEKL-APC | MBL | Cat#TS-5001–2C |
| Rapamycin | Calbiochem | Cat#553210 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Sunflower seed oil | Sigma-Aldrich | Cat#S5007 |
| OptiPrep | Axis-Shield | Cat#1114542 |
| Percoll | GE Healthcare | Cat#17089101 |
| Collagenase IV | Sigma-Aldrich | Cat#C5138 |
| DNase I | Sigma-Aldrich | Cat#DN25 |
| TRIzol™ Reagent | Thermo Fisher Scientific | Cat#15596018 |
| Ovalbumin, Low Endo, Purified | Worthington Biochemical Corporation | Cat#LS003061 |
| Poly(I)・Poly(C) | GE Healthcare | Cat#27473201 |
| Foxo1 inhibitor | Calbiochem | Cat#AS1842856 |
| OVA (257–264) SIINFEKL | AnaSpec | Cat#AS-60193–1 |
| Protein Transport Inhibitor (containing Monensin) | BD Biosciences | Cat#554724 |
| Brefeldin A Solution | BioLegend | Cat#420601 |
| N,N,N’,N’-Tetrakis(2hydroxypropyl)ethylenediamine | Tokyo Chemical Industry | Cat#T0781 |
| Polyethylene glycol mono-p-isooctylphenyl ether (Triton X-100) | Nacalai Tesque | Cat#25987–85 |
| Urea | Nacalai Tesque | Cat#35904–45 |
| Dynabeads™ Mouse T-Activator CD3/CD28 | Thermo Fisher Scientific | Cat#11452D |
| Recombinant mouse IL-2 | R&D Systems | Cat#402-ML |
| Recombinant mouse IL-12 | PeproTech | Cat#210–12 |
| Recombinant mouse IL-15 | PeproTech | Cat#210–15 |
| Recombinant mouse IFN-α | PBL Assay Science | Cat#12100–1 |
| Recombinant mouse IFN-β | PBL Assay Science | Cat#12400–1 |
| PE Streptavidin | BioLegend | Cat#405204 |
| APC Streptavidin | BioLegend | Cat#405207 |
| Critical Commercial Assays | ||
| eBioscience™ FoxP3/transcription factor buffer set | Thermo Fisher Scientific | Cat#00–5523-00 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#554714 |
| Nuclear Extract Kit | Active Motif | Cat#40010 |
| Streptavidin MicroBeads | Miltenyi Biotec | Cat#130–048-102 |
| MojoSort™ Streptavidin Nanobeads | BioLegend | Cat#480016 |
| EasySep™ Mouse Naive CD8+ T cell Isolation Kit | STEMCELL Technologies | Cat#19858A |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| SureSelect Strand Specific RNA Library | Agilent | G9691A |
| Nextera DNA Library Preparation Kit | Illumina | Cat#FC-121–1030 |
| Deposited Data | ||
| Raw and processed data (RNA-seq and ATAC-seq) | This paper | GEO: GSE110707 |
| Experimental Models: Cell Lines | ||
| B16 melanoma expressing OVA | Fujii S (RIKEN) Falo DR et al., 1995 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 (B6) | National Cancer Institute | N/A |
| Mouse: C57BL/6 (B6) | Charles River Laboratories | N/A |
| Mouse: C57BL/6 (B6) | CLEA Japan | N/A |
| Ly5.2/Cr (CD45.1) | National Cancer Institute | N/A |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J | The Jackson Laboratory | JAX: 003831 |
| C57BL/6-Tg(UBC-GFP)30Scha/J | The Jackson Laboratory | JAX: 004353 |
| B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | The Jackson Laboratory | JAX: 006148 |
| B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX: 007914 |
| B6.Cg-Tg(Pgk1-flpo)10Sykr/J | The Jackson Laboratory | JAX: 011065 |
| C57BL/6-Tg(CAG-flpe)36Ito/ItoRbrc | RIKEN BioResource Center, Kanki et al., 2006 | RBRC01834 |
| B6.129S6-Rag2tm1Fwa N12 | Taconic Biosciences | RAGN12 |
| C57BL/6-Gt(ROSA)26Sortm9(Cre/ESR1)Arte | Taconic Biosciences | 10471 |
| Klrg1Cre | This paper | N/A |
| Bach2Flag | This paper | N/A |
| Bach2flox/flox | Kometani et al., 2013 | N/A |
| Rosa26Bach2 | Kuwahara et al., 2016 | N/A |
| Oligonucleotides | ||
| Primer for genotyping: Klrg1-Cre
Common: TTTGCCCAGATTTAGGCTTT Klrg1-Cre: CTGTGTCTGGTGTGGCTGAT Wild-type: ATTCACAGAAATGGCCTCCA |
This paper | N/A |
| Primer for genotyping: Bach2-Flag
Forward: GCTCATGGGGACCTGGGAGCTTTGG Reverse: CCGTTGGTCATTGAGGCCCAGGAGG |
This paper | N/A |
| Primer for quantitative PCR: Bach2 deleted
allele Forward flox: TGGCTCCCCCATGTATGTAT Reverse flox: ATCAGCGTCACGTCACAGAG Forward control: CCTAGGAAACTGGGATGACG Reverse control: GGGGTCTTCTTAAGGCCAAC |
This paper | N/A |
| Primer for quantitative RT-PCR: Actb
Forward: TTCTTTGCAGCTCCTTCGTT Reverse: ATGGAGGGGAATACAGCCC |
This paper | N/A |
| Primer for quantitative RT-PCR: Bach2
Forward: CACTGGTTGGACAGACGAAA Reverse: ACTGTAGCAGTGGCCCAAAG |
This paper | N/A |
| Primer for quantitative RT-PCR: Eomes
Forward: ATGTACGTTCACCCAGAATC Reverse: GTGCAGAGACTGCAACACTA |
Dominguez et al., 2015 | N/A |
| Primer for quantitative RT-PCR: Gzma
Forward: TCAGCTCCCTCTGAAACTCT Reverse: TCTCCACCAAAAGAGGTGAT |
Dominguez et al., 2015 | N/A |
| Primer for quantitative RT-PCR: iCre
Forward: CACCTGGAAGATGCTCCTGT Reverse: TCCCTCACATCCTCAGGTTC |
This paper | N/A |
| Primer for quantitative RT-PCR: Id3
Forward: ACTTACCCTGAACTCAACGC Reverse: CTCCAAGGAAACCAGAAGAA |
Dominguez et al., 2015 | N/A |
| Primer for quantitative RT-PCR: Influenza
A/PR8/M Forward: CGCTCAGACATGAGAACAGAATGG Reverse: TAACTAGCCTGACTAGCAACCTC |
Pillai et al., 2016 | N/A |
| Primer for quantitative RT-PCR: Klf2
Forward: AGCCTATCTTGCCGTCCTT Reverse: CCAACACGTTGTTTAGGTCCTC |
Endrizzi and Jameson, 2003 | N/A |
| Primer for quantitative RT-PCR: Prdm1
Forward: ACCAAGGAACCTGCTTTTCA Reverse: GGCATTCTTGGGAACTGTGT |
This paper | N/A |
| Primer for quantitative RT-PCR: S1pr5
Forward: GTACACCAAATGCCCAGCTT Reverse: CACTGGAGCACTGTGCAAAA |
This paper | N/A |
| Primer for quantitative RT-PCR: Zeb2
Forward: AGAAGCCACGATCCAGACC Reverse: GGCCATCTCTTTCCTCCAGT |
This paper | N/A |
| Recombinant DNA | ||
| pMCs-Foxo1-CA-IRES-GFP | Liu YC (La Jolla Institute) Harada et al., 2010 |
N/A |
| Software and Algorithms | ||
| BEDTools | Quinlan and Hall, 2010 | N/A |
| Bowtie 2 | Langmead and Salzberg, 2012 | http://bowtiebio.sourceforge.net/bowtie2/index.shtml |
| Casava 1.8.2 | Illumina | N/A |
| Cufflinks version 2.1.1 | Trapnell et al., 2010 | N/A |
| Cutadapt 1.8.3 | Martin, 2011 | N/A |
| FlowJo 9 and 10 | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
| GraphPad Prism 6.0 | GraphPad | https://www.graphpad.com |
| Imaris 8 | Bitplane | http://www.bitplane.com/download |
| MACS2 | Zhang et al., 2008 | N/A |
| Picard MarkDuplicate tool | N/A | http://broadinstitute.github.io/picard |
| R (for principal component analysis) | N/A | http://www.rproject.org/ |
| TopHat2 version 2.0.8 | Kim et al., 2013 | N/A |
| VennDiagram package | Chen and Boutros, 2011 | N/A |
HIGHLIGHTS.
KLRG1+ IL-7Rα+ effector cells lose KLRG1 and differentiate into exKLRG1 memory cells
ExKLRG1 memory cells comprise CX3CR1+ circulating and CX3CR1− tissue-resident cells
ExKLRG1 memory cells mount highly effective anti-viral and anti-tumor responses
Bach2 promotes exKLRG1 memory CD8+ T cell development
ACKNOWLEDGEMENTS
We thank J. Alderman, C. Lieber, E. Hughes-Picard and P. Musco for administrative assistance, J. Stein, L. Evangelisti and C. Hughes for generating Klrg1Cre mice, Y. Harada and L. Yun-Cai for providing Foxo1-CA cDNA, S. Fujii for providing B16-OVA, P. Ranney, G. Lyon, Z. Tobiasova and L. Borelli for technical assistance, and members of the Flavell and Okada lab for helpful discussions. This work was supported by an Erwin Schrödinger Fellowship (Austrian Science Fund; J3220-B19 to D.H.B.), the Ministry of Education, Culture, Sports, Science, and Technology (16K19166 to H.I., 22113006 to T.O., and 26221306, 21229007 to T.K.), an Austrian Marshall Plan Foundation Fellowship (V.P.), a University of Vienna KWA Scholarship (C.S.), NIH grant 1R01HG008383–01A1 (Y.K.), Japan Science and Technology Agency (CREST J098501018 to T.K., and PRESTO to T.O.), and the Howard Hughes Medical Institute (R.A.F.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, and Masopust D (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, and Ahmed R (2009). mTOR regulates memory CD8 T-cell differentiation. Nature 460, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, and Bevan MJ (2015). Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol 16, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW, and Immunological Genome Project C (2013). Transcriptional insights into the CD8+ T cell response to infection and memory T cell formation. Nat Immunol 14, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Hochst B, Eickhoff S, Rieckmann JC, Russo C, Bauer T, et al. (2015). Functional classification of memory CD8+ T cells by CX3CR1 expression. Nat Commun 6, 8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz VR, Schumacher TN, and Busch DH (2016). T Cell Fate at the Single-Cell Level. Annu Rev Immunol 34, 65–92. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Wherry EJ, and Goldrath AW (2014). Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, and Boutros PC (2011). VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T, Tyznik AJ, Roepke S, Berkley AM, Woodward-Davis A, Pattacini L, Bevan MJ, Zehn D, and Prlic M (2013). Bystander-activated memory CD8 T cells control early pathogen load in an innate-like, NKG2D-dependent manner. Cell Rep 3, 701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, and Kaech SM (2015). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med 212, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endrizzi BT, and Jameson SC (2003). Differential role for IL-7 in inducing lung Kruppel-like factor (Kruppel-like factor 2) expression by naive versus activated T cells. Int Immunol 15, 1341–1348. [DOI] [PubMed] [Google Scholar]
- Falo LD Jr., Kovacsovics-Bankowski M, Thompson K, and Rock KL (1995). Targeting antigen into the phagocytic pathway in vivo induces protective tumour immunity. Nat Med 1, 649–653. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, Lau LS, Mollard V, Cozijnsen A, Collins N, et al. (2016). Liver-Resident Memory CD8+ T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity 45, 889–902. [DOI] [PubMed] [Google Scholar]
- Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, de la Torre JC, and von Andrian UH (2016). The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 45, 1270–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SM, Amezquita RA, Guan T, Kleinstein SH, and Kaech SM (2017). Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8+ T Cell Terminal Differentiation and Loss of Multipotency. Immunity 46, 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Shitara S, Imai K, Miyachi H, Kitano S, Yao H, Tani-ichi S, and Ikuta K (2012). Identification of IL-7-producing cells in primary and secondary lymphoid organs using IL-7-GFP knock-in mice. J Immunol 189, 1577–1584. [DOI] [PubMed] [Google Scholar]
- Harada Y, Harada Y, Elly C, Ying G, Paik JH, DePinho RA, and Liu YC (2010). Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med 207, 1381–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G, and Chen J (2013). A genome-wide regulatory network identifies key transcription factors for memory CD8+ T-cell development. Nat Commun 4, 2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson SC, and Masopust D (2009). Diversity in T cell memory: an embarrassment of riches. Immunity 31, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YW, Rutishauser RL, Joshi NS, Haberman AM, and Kaech SM (2010). Differential localization of effector and memory CD8 T cell subsets in lymphoid organs during acute viral infection. J Immunol 185, 5315–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 12, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Wherry EJ (2007). Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki H, Suzuki H, and Itohara S (2006). High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp Anim 55, 137–141. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Schluns KS, and Lefrancois L (1999). Induction and visualization of mucosal memory CD8 T cells following systemic virus infection. J Immunol 163, 4125–4132. [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. (2005). Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 102, 9571–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kometani K, Nakagawa R, Shinnakasu R, Kaji T, Rybouchkin A, Moriyama S, Furukawa K, Koseki H, Takemori T, and Kurosaki T (2013). Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity 39, 136–147. [DOI] [PubMed] [Google Scholar]
- Kuwahara M, Ise W, Ochi M, Suzuki J, Kometani K, Maruyama S, Izumoto M, Matsumoto A, Takemori N, Takemori A, et al. (2016). Bach2-Batf interactions control Th2-type immune response by regulating the IL-4 amplification loop. Nat Commun 7, 12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, and Kallies A (2017). Transcriptional Regulation of Tissue-Resident Lymphocytes. Trends Immunol 38, 94–103. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat Immunol 14, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 171, pp-10. [Google Scholar]
- Mortier E, Advincula R, Kim L, Chmura S, Barrera J, Reizis B, Malynn BA, and Ma A (2009). Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity 31, 811–822. [DOI] [PubMed] [Google Scholar]
- Mueller SN, Gebhardt T, Carbone FR, and Heath WR (2013). Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 31, 137–161. [DOI] [PubMed] [Google Scholar]
- Obar JJ, and Lefrancois L (2010). Early signals during CD8 T cell priming regulate the generation of central memory cells. J Immunol 185, 263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JA, McDonald-Hyman C, Jameson SC, and Hamilton SE (2013). Effector-like CD8+ T cells in the memory population mediate potent protective immunity. Immunity 38, 1250–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, Jones RG, and Choi Y (2009). Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 460, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai PS, Molony RD, Martinod K, Dong H, Pang IK, Tal MC, Solis AG, Bielecki P, Mohanty S, Trentalange M, et al. (2016). Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 352, 463–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plumlee CR, Obar JJ, Colpitts SL, Jellison ER, Haining WN, Lefrancois L, and Khanna KM (2015). Early Effector CD8 T Cells Display Plasticity in Populating the Short-Lived Effector and Memory-Precursor Pools Following Bacterial or Viral Infection. Sci Rep 5, 12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope C, Kim SK, Marzo A, Masopust D, Williams K, Jiang J, Shen H, and Lefrancois L (2001). Organ-specific regulation of the CD8 T cell response to Listeria monocytogenes infection. J Immunol 166, 3402–3409. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nature biotechnology 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychoudhuri R, Clever D, Li P, Wakabayashi Y, Quinn KM, Klebanoff CA, Ji Y, Sukumar M, Eil RL, Yu Z, et al. (2016). BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers. Nat Immunol 17, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S, Kalia V, Haining WN, Konieczny BT, Subramaniam S, and Ahmed R (2008). Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. J Exp Med 205, 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Browne JP, Lopez-Moyado IF, Trifari S, Wong V, Chavez L, Rao A, and Pereira RM (2016). Dynamic Changes in Chromatin Accessibility Occur in CD8+ T Cells Responding to Viral Infection. Immunity 45, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, and Lefrancois L (2014). Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity 40, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, and Iwasaki A (2012). A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491, 463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, and Kurosaki T (2016). Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol 17, 861–869. [DOI] [PubMed] [Google Scholar]
- Soudja SM, Ruiz AL, Marie JC, and Lauvau G (2012). Inflammatory monocytes activate memory CD8+ T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 37, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, and Masopust D (2015). Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tainaka K, Kubota SI, Suyama TQ, Susaki EA, Perrin D, Ukai-Tadenuma M, Ukai H, and Ueda HR (2014). Whole-body imaging with single-cell resolution by tissue decolorization. Cell 159, 911–924. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, and Ahmed R (2003). Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 4, 225–234. [DOI] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York. [Google Scholar]
- Williams MA, and Bevan MJ (2007). Effector and memory CTL differentiation. Annu Rev Immunol 25, 171–192. [DOI] [PubMed] [Google Scholar]
- Xin A, Masson F, Liao Y, Preston S, Guan T, Gloury R, Olshansky M, Lin JX, Li P, Speed TP, et al. (2016). A molecular threshold for effector CD8+ T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat Immunol 17, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CY, Best JA, Knell J, Yang E, Sheridan AD, Jesionek AK, Li HS, Rivera RR, Lind KC, D’Cruz LM, et al. (2011). The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol 12, 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Zhang K, Milner JJ, Toma C, Chen R, Scott-Browne JP, Pereira RM, Crotty S, Chang JT, Pipkin ME, et al. (2017). Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat Immunol 18, 573–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Next-generation sequencing data have been deposited in Gene Expression Omnibus (GEO) under SuperSeries accession code GEO: GSE110707, which includes individual data sets GEO: GSE110629 (RNA-seq; LM), GEO: GSE110706 (RNA-seq; VSV) and GEO: GSE110876 (ATAC-seq).