

Abstract
Background
Mutations in mediator of RNA polymerase II transcription subunit 12 homolog (MED12, OMIM 300188) cause X‐linked intellectual disability (XLID) disorders including FG, Lujan, and Ohdo syndromes. The Gli3‐dependent Sonic Hedgehog (SHH) signaling pathway has been implicated in the original FG syndrome and Lujan syndrome. How are SHH‐signaling defects related to the complex clinical phenotype of MED12‐associated XLID syndromes are not fully understood.
Methods
Quantitative RT‐PCR was used to study expression levels of three SHH‐signaling genes in lymophoblast cell lines carrying four MED12 mutations from four unrelated XLID families. Genotype and phenotype correlation studies were performed on these mutations.
Results
Three newly identified and one novel MED12 mutations in six affected males from four unrelated XLID families were studied. Three mutations (c.2692A>G; p.N898D, c.3640C>T; p.R1214C, and c.3884G>A; p.R1295H) are located in the LS domain and one (c.617G>A; p.R206Q) is in the L domain of MED12. These mutations involve highly conserved amino acid residues and segregate with ID and related congenital malformations in respective probands families. Patients with the LS‐domain mutations share many features of FG syndrome and some features of Lujan syndrome. The patient with the L‐domain mutation presented with ID and predominant neuropsychiatric features but little dysmorphic features of either FG or Lujan syndrome. Transcript levels of three Gli3‐dependent SHH‐signaling genes, CREB5, BMP4, and NEUROG2, were determined by quantitative RT‐PCR and found to be significantly elevated in lymphoblasts from patients with three mutations in the MED12‐LS domain.
Conclusions
These results support a critical role of MED12 in regulating Gli3‐dependent SHH signaling and in developing ID and related congenital malformations in XLID syndromes. Differences in the expression profile of SHH‐signaling genes potentially contribute to variability in clinical phenotypes in patients with MED12‐related XLID disorders.
Keywords: MED12, Mutation, qRT‐PCR, SHH Signaling, XLID
1. INTRODUCTION
Mediator of RNA polymerase II transcription subunit 12 homolog (MED12) encodes a core component of a multi‐subunit mediator complex, which includes MED12, MED13, cyclin C, and CDK8 (Yin & Wang, 2014). Mediator is a large, evolutionarily conserved protein complex that plays an important role in gene regulation (Yin & Wang, 2014). The mediator complex consists of approximately 30 subunits that are assembled into four functional modules interacting with signaling proteins in the vertebral endoderm (Shin et al., 2008) and neuronal development (Wang, Yang, Uno, Roeder, & Guo, 2006) and the canonical Wnt signaling pathway (Rocha, Scholze, Bleiss, & Schrewe, 2010). GLI family zinc family protein 3 (GLI3) binds directly to the PQL domain of MED12, which suppresses the enhanced GLI3‐dependent transactivation induced by Sonic hedgehog (SHH) signaling (Zhou, Kim, Ishii, & Boyer, 2006). SHH signaling has critical functions in the development of the central nervous system including neural tube patterning, neuronal cell differentiation and survival (Ho & Scott, 2002).
MED12 mutations have been associated with a wide phenotypic spectrum of MED12‐related disorders (Charzewska et al., 2018). Somatic mutations in MED12 have been reported in leiomyosarcoma (Ravegnini et al., 2013) and prostate cancer (Kämpjärvi et al., 2015). Germline MED12 mutations were originally found in patients with FG and Lujan syndromes (Risheg et al., 2007; Schwartz et al., 2007) and later in patients with X‐linked Ohdo syndrome and isolated XLID disorders (Bouazzi, Lesca, Trujillo, Alwasiyah, & Munnich, 2015; Callier et al., 2013; Isidor et al., 2013; Langley et al., 2015; Lesca et al., 2013; Tzschach et al., 2015; Vulto‐van Silfhout et al., 2013; Yamamoto & Shimojima, 2015). Mutations that cause FG syndrome (p.R961W) and Lujan syndrome (p.N1007S) were found to compromise the mediator‐imposed constraint on GLI3‐dependent SHH signaling and result in increased transcript levels for multiple GLI3 target genes in lymphoblasts from patients (Zhou et al., 2012). How GLI3–SHH‐signaling defects relate to the complex clinical phenotypes of MED12‐associated XLID syndromes are not known.
We report genetic and functional characterizations of four MED12 mutations in patients with XLID disorders. Patients with LS‐domain mutations show ID and congenital malformations overlapping with FG and Lujan syndromes. Transcript levels of three SHH/GLI3‐signaling genes, BMP4(OMIM112262), CREB5, NEUROG2 (OMIM606624), were found to be significantly elevated in lymphoblasts from patients with these mutations. The patient with a change in the L domain only had elevated expression of BMP4 and presented with ID and predominant psychiatric phenotypes. These results expand genotype and phenotype spectrums of MED12‐mediated XLID syndromes and support a critical role of MED12‐regulated Gli3‐dependent SHH signaling in the expression of disease phenotypes.
2. MATERIALS AND METHODS
2.1. Study patients and controls
Patients with XLID and control males with normal cognitive function were recruited by the Greenwood Genetic Center (Greenwood, SC) and the Johns Hopkins University (Baltimore, MD). Human subject research protocols for these studies were approved by Institutional Review Boards (IRBs) at the respective institutions. An informed consent was obtained from each study patient and/or their parents or legal guardians. These patients were all evaluated by clinical geneticists and underwent comprehensive laboratory studies for ID. All patients were found to have a normal karyotype, negative molecular testing for fragile X syndrome, and a negative screen for common inborn errors of metabolism. For each individual, 5–10 ml of blood was collected to establish EB‐transformed lymphoblast cell lines. Genomic DNA samples from affected probands males with XLID were used for sequencing and mutation screening. A cohort of >800 males with normal cognitive function from the Greenwood Genetic Center and Johns Hopkins University were used as controls. Additional reference data include the male portion of samples (n = 525) from the 1,000 Genomes project (Integrated Phase 1, version 3:20101123).
2.2. X chromosome exome sequencing and exon‐based resequencing
Sequence libraries were prepared using a TruSeqTM Genomic DNA Library Preparation kit (Illumina), enriched for the human X chromosome Exome using a SureSelect Target Enrichment kit (Agilent), and sequenced using the 75 bp pair‐end sequence module on the HiSeq2000 (Illumina). Alignment of the fastq reads, base recalibration, and variant calling were completed using Bowtie2 and Unified Genotyper (GATK). Disease‐causing mutations were enriched using the nonclinical portion of the dbSNP, the male‐restricted portion of the 1,000 Genomes, Exome Variant Server datasets, and an affected sib‐pair/cross‐cohort‐based algorithm (Niranjan et al., 2015). Evolutionary conservation of the amino acid residues involved in the identified mutations was evaluated by multiple sequence alignment of HomoloGene (http://www.ncbi.nlm.nih.gov/homologene/). Standard bioinformatics algorithms including SIFT (http://sift.jcvi.org) and PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) were used to predict the functional impact of the identified mutations (Niranjan et al., 2015). Additional exon‐based resequencing was conducted to identify novel MED12 mutations in patients with intellectual disability and a pedigree consistent with X‐linked inheritance (Raymond et al., 2007). Sanger sequencing was used for validation, segregation analysis, and polymorphism studies of each mutation using the BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI3100 automatic DNA analyzer (Applied Biosystems) following manufacturer's instruction. Variant analysis of MED12 (NM_005120.2) was completed using standard sequence alignment software (CodonCode and MacVector) followed by manual investigations of the chromatograms.
2.3. Real‐time quantitative PCR analysis for transcript levels of SHH‐signaling genes
EBV‐transformed lymphoblastoid cell lines from patients and controls were cultured in RPMI 1640 medium with 15% FBS (Sigma) and 1% penicillin–streptomycin (100 U penicillin; 0.1 mg/ml streptomycin) at 37°C, 10% CO2. Cells were harvested at log phase. Total RNA was prepared from cultured lymphoblasts from individual patients and normal controls using a Qiagen RNA preparation kit. cDNA from these samples was synthesized using a M‐MLV RT kit (Promega). Real‐time qPCR was conducted using an absolute SYBR Green mix (Applied Biosystems) in an iCycler (BioRad). After initial denaturation at 95.0ºC for 3 min, the reaction was cycled for 35 times at 95.0ºC for 30 s and 60.0ºC for 45 s. Input samples were normalized using a simultaneous quantification of β‐actin. qPCR Primers used in this study are given in Table S1. Lymphoblast cell lines from three unrelated normal males and from the original Lujan family with a p.N1007S mutation were used as controls. Each data set for the transcript levels was generated from triplicate studies. Statistical analyses of qPCR data were performed using two‐tailed t test for comparison of the means of two independent samples. Data were presented as mean ± SEM; p < 0.05 was considered statistically significant.
3. RESULTS
3.1. Identification and pedigree analysis of MED12 mutations in XLID families
Exon resequencing of 718 X‐linked genes (Tarpey et al., 2009) found a c.3884G>A; p.R1295H mutation in two affected brothers (II‐3 and II‐6) and their mother who is heterozygous (K9338) (Figure 1a). Sequencing of MED12 in a cohort of XLID families identified c.617G>A; p.R206Q mutation in an affected male and his unaffected mother (K8935) (Figure 1b) and a c.2692A>G; p.N898D mutation in an affected male and his unaffected mother in a family suspected to have FG syndrome (K9467) (Figure 1c). X chromosome exome sequencing identified a c.3640C>T; p.R1214C in two affected brothers and their unaffected mother in an XLID family (L08–2677) (Niranjan et al., 2015) (Figure 1d). The p.N898D, p.R1214C, and p.R1295H mutations are located in the LS domain while the p.R206Q mutation is located in the L domain of MED12 (Figure 2a). All four mutations involve highly conserved amino acid residues during evolution, and none was found in >1,325 normal X chromosomes (Figure 2b). The Combined Annotation Dependent Depletion (CADD) scores for all four mutations are in the range of 23.2 to 31 suggesting a high likelihood of deleterious effects (Table 1) (GRCh37‐v1.4, https://cadd.gs.washington.edu).
Figure 1.
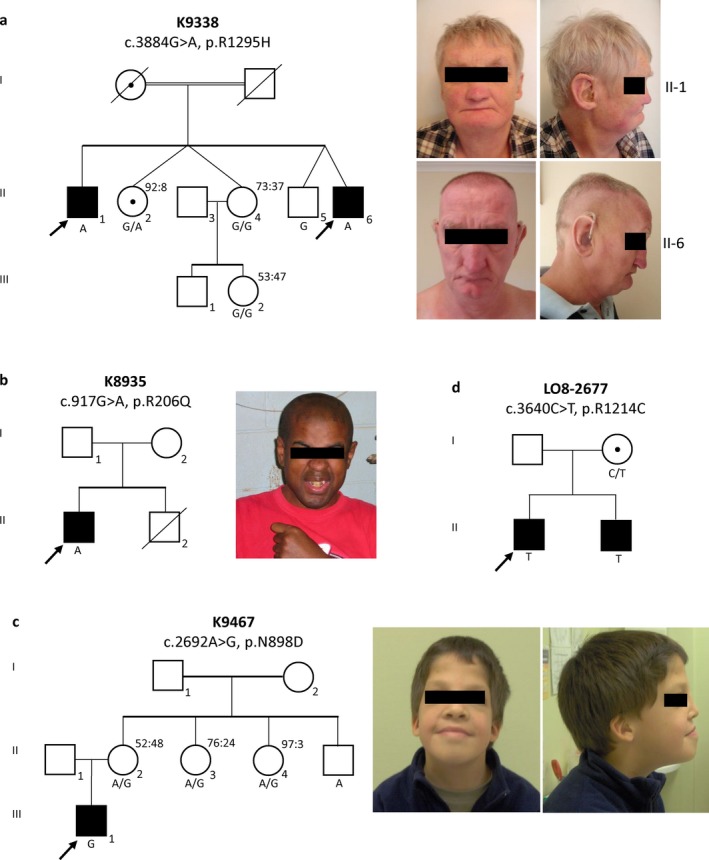
Pedigree Analysis and Clinical Features of XLID Probands with MED12 Mutations. Panel a: Segregation of c.3884G>A; p.R1295H in pedigree of K9338; confirmed genotypes (WT, G; mutation, A) are indicated below individual symbols; skewed X chromosome inactivation data in female carriers are provided for II2, II4, III2. Facial features of one affected male are shown. Panel b: Segregation of c.617G>A; p.R206Q in pedigree K8935. Confirmed genotypes (WT, G; mutation, A) were shown below the affected male. Facial features of the affected male are shown. Panel c: Segregation of c.2692A>G; p.N898D in pedigree K9467. Confirmed genotypes (WT, A; mutation, G) are indicated below individual symbols; data for skewed X chromosome inactivation are provided for II2, II3, and II4. Facial features of the affected male are shown. Panel d: Segregation of c.3640C>T; p.R1214C in pedigree of LO8–2677. Confirmed genotypes (WT, C; mutation, T) are indicated below individual symbols. For all pedigrees: square symbol, male; circle symbol, female; open symbol, unaffected; filled symbol, affected; circle with a center dot, confirmed female carrier
Figure 2.
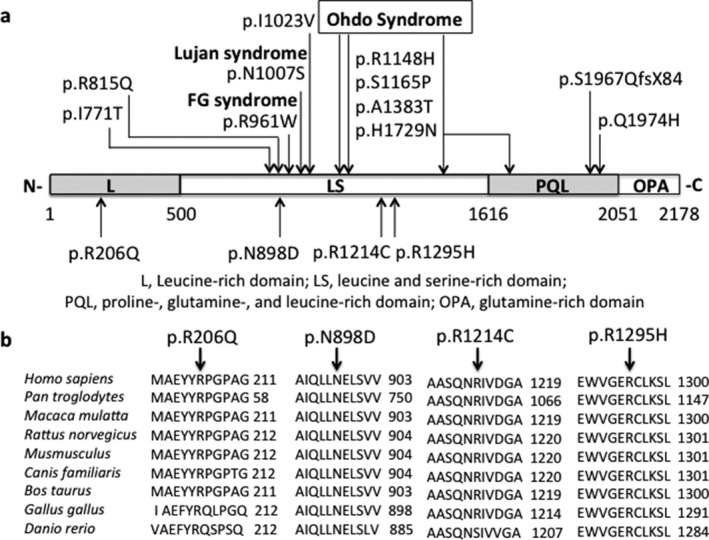
Disease‐causing MED12 Mutations Involve Highly Conserved Amino Acid Residues and are Clustered around the Established Functional Domains. Panel a: Distribution of ID‐associated mutations over functional domains of MED12. Known ID‐associated mutations(Graham & Schwartz, 2013) are presented above and the four mutations of MED12 (NM_005120.2) in this study are below the symbols of protein domains. Note that 3 mutations p.N898D, p.R1214C, and p.R1295H are located within the LS domain where the recurrent mutations for FG and Lujan syndromes occur and one, p.R206Q, is located within the L domain. L, leucine‐rich domain; LS, leucine serine‐rich domain; PQL, proline‐, glutamine‐, and leucine‐rich domain; OPA, glutamine‐rich domain. Panel b. MED12mutations involve highly evolutionarily conserved amino acid residues as shown in a multispecies sequence alignment. Mutations responsible for FG, Lujan, and Ohdo syndrome are based on previously published studies(Graham & Schwartz, 2013)
Table 1.
Clinical features of MED12 mutations responsible for XLID syndromes
| Pedigree ID_Syndrome | FG syndrome | Lujan syndrome | XLID_K8935_II‐1 | XLID_K9467_III‐1 | XLID_L08‐2677_II‐1 | XLID_L08‐2677_II‐2 | XLID_K9338_II‐1 | XLID_K9338_II‐6 |
|---|---|---|---|---|---|---|---|---|
| GRCh37‐v1.4 | ChrX:70340884 | ChrX:70346825 | ChrX:70349228 | ChrX:70349228 | ChrX:70349901 | ChrX:70349901 | ||
| Mutation | c.617G>A; p.R206Q | c.2692A>G; p.N898D | c.3640C>T; p.R1214C | c.3640C>T; p.R1214C | c.3884G>A; p.R1295H | c.3884G>A; p.R1295H | ||
| CADDa | 23.2 | 26.2 | 31 | 31 | 23.4 | 23.4 | ||
| Evaluation Age (year) | 15 | 17 | 14 | 11 | ||||
| Weight (kg) | 5th centile | 5th% | failure to thrive | failure to thrive | ||||
| Height (cm) | 10 th centile | 5th% | 25th centile (at 54 years) | 60th centile (at 49 years) | ||||
| OFC (cm) | Relative macrocephaly | Macrocephaly | Macrocephaly | >97th centile | 50–75th centile | 10–25th centile | Macrocephalic | 75th centile (at 49 years) |
| Appearance | Marfanoid habitus | Height and weight >97%tile | Asthenic build | Asthenic build | Asthenic build | Poor muscle bulk | ||
| Motor Development | Delayed walking | Delayed walking | Delayed walking | Delayed; clumsy walking | Delayed; walking after 2 years | |||
| Language Development | Delayed; first word at 3 years | Delayed speech | Delayed speech | Speech unintellligible until 5 years | Delayed speech | |||
| Intellectual Disability | Moderate to severe | Mild to moderate | Mild to moderate (IQ 40–85) | Mild to moderate (IQ 58) | Mild to moderate | Mild to moderate | Mild to moderate (IQ 58) | Moderate |
| Behaviors |
Friendly personality Short attention span Temper tantum |
Hyperactivity Emotional lability Aggressiveness |
Aggression Panic disorders Agoraphobia |
Psychological lability Short attention span Easily frustrated |
Friendly personality Excessive talkativeness |
Friendly personality Excessive talkativeness |
Impulsiveness Restlessness Temper tantrums |
|
| Craniofacial | Tall forehead Frontal hair upsweep | Tall forehead | Tall and broad forehead Frontal upsweep | Tall forehead | Tall forehead | |||
|
Long and narrow face Maxillary hypoplasia |
Long and narrow face Maxillary hypoplasia |
Maxillary hypoplasia |
Long and narrow face Maxillary hypoplasia |
Long and narrow face | Long face; prominent supraorbital ridge | Long face with maxillary hypoplasia | ||
| Hypertelorism; downslanting palpebral fissures | Downslanting palpebral fissures | Downslanting palpebral fissures |
Downslanting palpebral fissures Hypertelorism Astigmatism, hyeropia |
Telecanthus | Telecanthus | Epicanthus; downslanting palpebral fissures; cataract | Strabismus | |
| Small prominent ears with simplified helical pattern | Hypernasal speech | Astigmatism, exotropia, and mild hyperopia | Small posteriorly rotated ears | Normal ears | Normal ears | Normal ears | ||
| Dental crowding Micro/retrognathia | Dental crowding; micro/retrognathia | Dental crowding and prognathism | High‐arched palate | High‐arched palate | ||||
| Musculoskeletal |
Pectus extracavatum Scoliosis Joint contracture |
Mild pectus carinatum |
Severe pectus carinatum Moderate scoliosis Mild contracture of elbows |
Thoracic kyphosis; | Marked thoracic kyphosis | |||
| Broad thumb; syndactyly; persistent fetal finger pads | Long extensible digits; broad thumbs | Long hands (length >97th%) | Angulation of distal phalanges; broad big toes |
Hand length: 25–50th centile Middle finger length: 50th centile Arm span‐to‐height ratio: 1.03 Upper‐to‐lower segment ratio: 0.89 |
Hand length: 25–50th centile Middle finger length: 25–50th centile Arm span‐to‐height ratio: 1.05 Upper‐to‐lower segment ratio: 0.71 Slender fingers; hyperextensibility; fifth finger clinodactyly; pes planus |
Pes cavus | Finger contractures at PIP joints; Hammer toes; wide gap between first and second toes | |
| Gastrointestinal | Anal anomaly; constipation | Imperforate anus Constipation | Severe constipation; megacolon | Anal stricture and stenosis; umblical and inguinal hernia | Constipation | |||
| Genitourinary | Genitourinary anomaly | Hydroceles | Cryptorchidism | Posteriour urethral valves | Undescended testicles | Undescended testicles | ||
| Neurological | Hypotonia | Hypotonia | Hypotonia | Seizure disorders; wide spaced gait | Seizure disorders | Uncoordinated gait; hearing loss | Hypotonia; hearing loss | |
| Agenesis of corpus callosum | Agenesis of corpus callosum | Normal head CT | Normal head CT | Agenesis of corpus callosum; enlarged ventricules on MRI |
Combined Annotation Dependent Depletion (CADD); https://cadd.gs.washington.edu
3.2. Increased expression of SHH‐signaling genes in lymphoblasts with LS‐domain mutations
Increased transcript levels for SHH‐signaling genes have been reported in cell lines from patients with FG and Lujan syndrome (Zhou et al., 2012). We thus performed a real‐time qRT‐PCR quantification of transcript levels of three genes (CREB5, BMP4, and NEUROG2)in the SHH‐signaling pathway in lymphoblasts from patients with four MED12 mutations (Figure 3). Transcript levels for these genes are significantly increased for the three LS‐domain mutations. The L‐domain mutation, p.R206Q, shows only modest increase in the transcript of BMP4 (Figure 3). Expression of HPRT1, a house‐keeping gene not known to be regulated by SHH/GLI3 signaling, showed no significant difference in lymphoblast cell lines between normal controls and patients with these MED12 mutations (Figure S1). These results support that the MED12‐LS domain plays a major role in the negative regulation of Gli3‐dependent SHH signaling (Zhou et al., 2012), and these mutations found in patients with ID and congenital malformations show deleterious effects on MED12 function.
Figure 3.
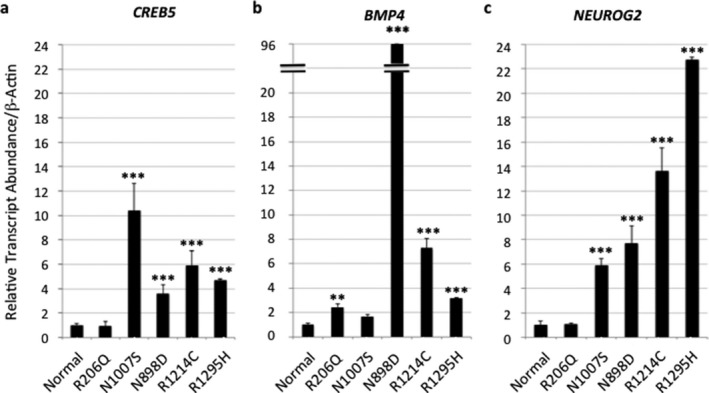
Real‐time Quantitative PCR Analysis of Transcript Levels of Three SHH‐Signaling Genes in Lymphoblasts from Patients with XLID. Note that compared to normal control lymphoblasts (n = 3), the transcript levels for all three genes, CREB5, BMP4, NEUROG2 in the SHH‐signaling pathway show a significant increase in lymphoblasts carrying three MED12 mutations (p.N898D; p.R1214C; and p.R1295H) within the LS domain but minimal changes for the mutation, p.R206Q, within the L domain. N1007S is the lymphoblast cell line from the probands of the original Lujan syndrome family(Schwartz et al., 2007). Means ± SEM from the triplicate studies of each lymphoblast cell line were shown. **, p < 0.01; ***, p < 0.001
3.3. Expanding phenotypic spectrum of MED12‐related XLID disorders
MED12‐related XLID disorders exhibit a wide phenotypic spectrum in impairment of cognition, behavioral defects, and multiple congenital anomalies (Graham & Schwartz, 2013). “The patient with p.N898D mutation presented with macrocephaly, tall and broad forehead, downslanting palpebral fissures, small and posteriorly rotated ears, broad big toes, imperforate anus and constipation, and behavioral profile that mostly resemble patients with FG syndrome. Patients with p.R1214C and p.R1295H mutations presented with features that resemble both Lujan and FG syndromes. These features include craniofacial features, behavioral profile, anal anomalies, and severe constipation that are common in patients with FG syndrome as well as long and thin face, high‐arch palate, asthenic body build, reduced muscle mass, long and slender fingers, joint hyperextensibility that are seen in patients with Lujan syndrome. The patient with p.R206Q mutation presented with ID and predominant neuropsychiatric phenotype but without significant dysmorphic features or congenital malformations of either FG or Lujan syndromes (Table 1).”
4. DISCUSSION
Classical features of FG syndrome include ID, craniofacial features such as macrocephaly, prominent forehead, hypertelorism, downslanting palpebral fissures, small ears, musculoskeletal anomalies such as pectus deformities, broad thumbs, gastrointestinal defects such as anal anomaly with severe constipation, and a characteristic behavioral profile with friendly personality and short attention span (Clark et al., 2009; Opitz & Kaveggia, 1974). A recurrent mutation, p.R961W, in the MED12‐LS domain was identified in the original and five additional families with clinical features suggestive of FG syndrome (Risheg et al., 2007). Patients from the original Lujan syndrome family presented with X‐linked intellectual disability, hypotonia, macrocephaly, marfanoid habitus with long fingers with extensible joints, craniofacial features such as long and narrow face, high‐arched palate with hypernasal voice, and structural brain anomalies such as agenesis of corpus callosum (Lujan, Carlin, & Lubs, 1984). A single missense mutation in MED12‐LS domain, N1007S, has been identified in the original Lujan syndrome family (Schwartz et al., 2007). The patient with p.N898D mutation presented with features that mostly resemble FG syndrome; patients with p.R1214C and p.R1295H mutations presented with features that overlap with FG and Lujan syndromes; the patient with p.R206Q mutation showed severe psychiatric phenotype but without characteristic dysmorphic features or malformations of either FG syndrome or Lujan syndromes (Table 1). Taken together, our current studies on these four MED12 mutations expand the genotype and phenotype spectrums of the MED12‐related X‐linked ID disorders.
MED12 consists of several structurally and functionally defined domains including a N‐terminal leucine‐rich domain (L), a leucine‐ and serine‐rich domain (LS), a proline‐, glutamine‐, and leucine‐rich domain (PQL), and a C‐terminal opposite paired domain (OPA). The PQL domain mediates protein–protein interaction while function for other domains remain largely unknown (Zhou et al., 2006). Classical FG and Lujan syndromes are caused by mutations in the LS domain, which is highly conserved (Figure 2a). Previous studies have shown that MED12 mutations in FG and Lujan syndromes disrupt a mediator‐imposed constraint on GLI3‐dependent SHH signaling (Zhou et al., 2012). Expression levels of multiple SHH/GLI3 target genes including GLI3, ASCL1, BMP4, CREB5, and NEUROG2were significantly elevated in patients’ lymphoblast cell lines (Zhou et al., 2012). Increased expression of these genes was inhibited by a SHH antagonist, cyclopamine, suggesting that their induction was SHH/GLI3 dependent (Zhou et al., 2012).
By examining transcript levels of three Gli3‐dependent SHH‐signaling genes, CREB5, BMP4, and NEUROG2,in lymphoblasts, we found that p.N898D, p.R1214C, and p.R1295H mutations are associated with increased expression of all three genes. Interestingly, p.N898D mutation was found to associate with a significant increase in expression of BMP4 but moderate increase in expression of NEUROG22.In contrast, p.R1214C and p.R1295H mutations were found to associate with significant increase in NEUROG2 but moderate increase in BMP4. p.R206Q was found to associate with a mild increase in the expression of BMP4 only. GLI3‐dependent SHH‐signaling pathway plays a critical role in development of multiple organs including brain, neural tube, developing limbs, and the gut (Villavicencio, Walterhouse, & Lannaccone, 2000). We speculate that severe disturbance in SHH‐signaling genes likely play an important role in craniofacial anomalies and multiple organ malformations in these patients. We further speculate that differences in the expression profile of SHH‐signaling genes potentially contribute to variable expression of phenotypes in patients with MED12‐related XLID syndromes.
The Ohdo syndrome is defined by several mutations in LS and PQL domains (p.R1148H, p.S1165P, p.A1383T, p.H1729N). Core clinical features of patients include moderate to severe intellectual disability, autistic behaviors, craniofacial anomalies such as microcephaly, blepharophimosis, prominent and bulbous nose, ear anomalies, and moderate to severe hypotonia. Skeletal, gastrointestinal, and genital urinary anomalies that are common in the FG‐Lujan syndrome spectrum are relatively mild or absent in patients with Ohdo syndrome. The molecular mechanisms that underlie Ohdo syndrome have not been fully characterized. MED12 has also been found to play key roles in the regulation of REST‐dependent epigenetic silencing of neuronal gene expression (Ding et al., 2008) and immediate early gene expression (Donnio et al., 2017). Systematic functional characterizations of these mutations are warranted.
CONFLICT OF INTEREST
All authors declare no conflict of interest in the study.
Supporting information
ACKNOWLEDGMENTS
This study was supported in part by grants from NIH (HD26202) to C. E. S; (NS73854) to C.E.S. and T.W; NHMRC (1155224 and 1091593) to J.G; Department of Disabilities and Special Needs of South Carolina; and the Greenwood Genetic Center Foundation. We thank Ellen Boyd from Fullerton Genetics, NC for patient data. The cooperation of patients and their families is gratefully acknowledged.
Srivastava S, Niranjan T, May MM, et al. Dysregulations of sonic hedgehog signaling in MED12‐related X‐linked intellectual disability disorders. Mol Genet Genomic Med. 2019;7:e569 10.1002/mgg3.569
Contributor Information
Charles E. Schwartz, Email: ceschwartz@ggc.org.
Tao Wang, Email: twang9@jhmi.edu.
REFERENCES
- Bouazzi, H. , Lesca, G. , Trujillo, C. , Alwasiyah, M. , & Munnich, A. (2015). Nonsyndromic X‐linked intellectual deficiency in three brothers with a novel MED12 missense mutation [c.5922G>T (p.Glu1974His)]. Clinical Case Reports, 3, 604–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callier, P. , Aral, B. , Hanna, N. , Lambert, S. , Dindy, H. , Ragon, C. … Faivre, L. . (2013). Systematic molecular and cytogenetic screening of 100 patients with marfanoid syndromes and intellectual disability. Clinical Genetics, 84, 507–521. 10.1111/cge.12094 [DOI] [PubMed] [Google Scholar]
- Charzewska, A. , Maiwald, R. , Kahrizi, K. , Oehl‐Jaschkowitz, B. , Dufke, A. , Lemke, J. , … Kalscheuer, V. (2018). The power of the Mediator complex‐Expanding the genetic architecture and phenotypic spectrum of MED12‐related disorders. Clinical Genetics, 94, 450–456. [DOI] [PubMed] [Google Scholar]
- Clark, R. , Graham, J. J. , Friez, M. , Hoo, J. , Jones, K. , McKeown, C. , … Stevenson, R. (2009). FG syndrome, an X‐linked multiple congenital anomaly syndrome: The clinical phenotype and an algorithm for diagnostic testing. Genetics in Medicine, 11, 769–775. 10.1097/GIM.0b013e3181bd3d90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, N. , Zhou, H. , Esteve, P.‐O. , Chin, H. , Kim, S. , Xu, X. , … Boyer, T. (2008). Mediator Links Epigenetic Silencing of Neuronal Gene Expression with X‐Linked Mental Retardation. Molecular Cell, 31, 347–359. 10.1016/j.molcel.2008.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnio, L. , Bidon, B. , Hashimoto, S. , May, M. , Epantchintsev, A. , Ryan, C. , … Egly, J. (2017). MED12‐related XLID disorders are dose‐dependent of immediate early genes (IEGs) expression. Human Molecular Genetics, 26(11), 2062–2075. 10.1093/hmg/ddx099 [DOI] [PubMed] [Google Scholar]
- Graham, J. , & Schwartz, C. (2013). MED12 related disorders. American Journal of Medical Genetics, 161A, 2734–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho, K. , & Scott, M. (2002). Sonic hedgehog in the nervous system: Functions, modifications and mechanisms. Current Opinion in Neurobiology, 12, 57–63. 10.1016/S0959-4388(02)00290-8 [DOI] [PubMed] [Google Scholar]
- Isidor, B. , Lefebvre, T. , Le Vaillant, C. , Caillaud, G. , Faivre, L. , Jossic, F. , … David, A. (2013). Blepharophimosis, Short Humeri, Developmental Delay and Hirschsprung Disease: Expanding the Phenotypic Spectrum of MED12 Mutations. American Journal of Medical Genetics, 164A, 1821–1825. [DOI] [PubMed] [Google Scholar]
- Kämpjärvi, K. , Kim, N. , Keskitalo, S. , Clark, A. , vonNandelstadh, P. , Turunen, M. , … Vahteristo, P. (2015) Somatic MED12 mutations in prostate cancer and uterine leiomyomas promote tumorigenesis through distinct mechanisms. Prostate, 76(1):22–31. [DOI] [PubMed] [Google Scholar]
- Langley, K. , Brown, J. , Gerber, R. , Fox, J. , Friez, M. , Lyons, M. , & Schrier Vergano, S. (2015). Beyond Ohdo Syndrome: A Familial Missense Mutation Broadens the MED12 Spectrum. American Journal of Medical Genetics, 167A, 3180–3185. [DOI] [PubMed] [Google Scholar]
- Lesca, G. , Moizard, M.‐P. , Bussy, G. , Boggio, D. , Hu, H. , Haas, S. , … Lespinasse, J. (2013). Clinical and Neurocognitive Characterization of a Family With a Novel MED12 Gene Frameshift Mutation. American Journal of Medical Genetics, 161A, 3063–3071. [DOI] [PubMed] [Google Scholar]
- Lujan, J. , Carlin, M. , & Lubs, H. (1984). A form of X‐linked mental retardation with marfanoid habitus. American Journal of Medical Genetics, 17, 311–322. 10.1002/ajmg.1320170124 [DOI] [PubMed] [Google Scholar]
- Niranjan, T. , Skinner, C. , May, M. , Turner, T. , Rose, R. , Stevenson, R. , … Wang, T. (2015). Affected Kindred Analysis of Human X Chromosome Exomes to Identify Novel X‐Linked Intellectual Disability Genes. PLoS ONE, 10, e0116454 10.1371/journal.pone.0116454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz, J. , & Kaveggia, E. (1974). Studies of malformation syndromes of man 33: The FG syndrome. An X‐linked recessive syndrome of multiple congenital anomalies and mental retardation. Zeitschrift Für Kinderheilkunde, 117, 1–18. [DOI] [PubMed] [Google Scholar]
- Ravegnini, G. , Mariño‐Enriquez, A. , Slater, J. , Eilers, G. , Wang, Y. , Zhu, M. , … Fletcher, J. (2013). MED12 mutations in leiomyosarcoma and extrauterine leiomyoma. Modern Pathol, 26, 743–749. 10.1038/modpathol.2012.203 [DOI] [PubMed] [Google Scholar]
- Raymond, F. , Tarpey, P. , Edkins, S. , Tofts, C. , O'Meara, S. , Teague, J. , … Barthorpe, s., Buck, G. and al, e., (2007). Mutations in ZDHHC9, which encodes a palmitoyltransferase of NRAS and HRAS, cause X‐linked mental retardation associated with a marfanoid habitus. American Journal of Human Genetics, 80, 982–987. 10.1086/513609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risheg, H. , Graham, J. J. , Clark, R. , Rogers, R. , Opitz, J. , Moeschler, J. , … Friez, M. (2007). A recurrent mutation in MED12 leading to R961W causes Opitz‐Kaveggia syndrome. Nature Genetics, 39, 451–453. 10.1038/ng1992 [DOI] [PubMed] [Google Scholar]
- Rocha, P. , Scholze, M. , Bleiss, W. , & Schrewe, H. (2010). Med12 is essential for early mouse development and for canonical Wnt and Wnt/PCP signaling. Development, 137, 2723–2731. 10.1242/dev.053660 [DOI] [PubMed] [Google Scholar]
- Schwartz, C. , Tarpey, P. , Lubs, H. , Verloes, A. , May, M. , Risheg, H. … Stevenson, R. (2007). The original Lujan syndrome family has a novel missense mutation (p. N1007S) in the MED12 gene. Journal of Medical Genetics, 44, 472–477. 10.1136/jmg.2006.048637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, C. , Chung, W. , Hong, S. , Ober, E. , Verkade, H. , Field, H. , … Stainier, D. (2008). Multiple roles for Med12 in vertebrate endoderm development. Developmental Biology, 317, 467–479. 10.1016/j.ydbio.2008.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey, P. , Smith, R. , Pleasance, E. , Whibley, A. , Edkins, S. , Hardy, C. , … Stratton, M. R. . (2009). A systematic, large scale resequencing screen of X‐chromosome coding exons in mental retardation. Nature Genetics, 41, 535–543. 10.1038/ng.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschach, A. , Grasshoff, U. , Beck‐Woedl, S. , Dufke, C. , Bauer, C. , Kehrer, M. , … Bauer, P.. (2015). Next‐generation sequencing in X‐linked intellectual disability. European Journal of Human Genetics, 23, 1513–1518. 10.1038/ejhg.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villavicencio, E. , Walterhouse, D. , & Lannaccone, P. (2000). The Sonic Hedgehog–Patched–Gli Pathway in Human Development and Disease. American Journal of Human Genetics, 67, 1047–1054. 10.1016/S0002-9297(07)62934-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulto‐van Silfhout, A. , de Vries, B. , van Bon, B. , Hoischen, A. , Ruiterkamp‐Versteeg, M. , Gilissen, C. , … de Brouwer, A. (2013). Mutations in MED12 Cause X‐Linked Ohdo Syndrome. American Journal of Human Genetics, 92, 401–406. 10.1016/j.ajhg.2013.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Yang, N. , Uno, E. , Roeder, R. , & Guo, S. (2006). A subunit of the mediator complex regulates vertebrate neuronal development. Proceedings of the National Academy of Sciences, 103, 17284–17289. 10.1073/pnas.0605414103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, T. , & Shimojima, S. (2015). A novel MED12 mutation associated with non‐specific X‐linked intellectual disability. Human Genome Variation, 2, 15018 10.1038/hgv.2015.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, J. , & Wang, G. (2014). The Mediator complex: A master coordinator of transcription and cell lineage development. Development, 141, 977–987. 10.1242/dev.098392 [DOI] [PubMed] [Google Scholar]
- Zhou, H. , Kim, S. , Ishii, S. , & Boyer, T. (2006). Mediator modulates Gli3‐dependent Sonic hedgehog signaling. Molecular and Cellular Biology, 26, 8667–8682. 10.1128/MCB.00443-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Spaeth, J. , Kimb, N. , Xu, X. , Friezc, M. , Schwartz, C. , & Boyerb, T. (2012). MED12 mutations link intellectual disability syndromes with dysregulated GLI3‐dependent Sonic Hedgehog signaling. Proceedings of the National Academy of Sciences, 109, 19763–19768. 10.1073/pnas.1121120109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials