

Abstract
Background
Follistatin‐like 1 (Fstl1) is a glycoprotein expressed throughout embryonic development. Homozygous loss of Fstl1 in mice results in skeletal and respiratory defects, leading to neonatal death due to a collapse of the trachea. Furthermore, Fstl1 conditional deletion from the endocardial/endothelial lineage results in postnatal death due to heart failure and profound atrioventricular valve defects. Here, we investigated patients with phenotypes similar to the phenotypes observed in the transgenic mice, for variants in FSTL1.
Methods
In total, 69 genetically unresolved patients were selected with the following phenotypes: campomelic dysplasia (12), small patella syndrome (2), BILU (1), and congenital heart disease patients (54), of which 16 also had kyphoscoliosis, and 38 had valve abnormalities as their main diagnosis. Using qPCR, none of 69 patients showed copy number variations in FSTL1. The entire gene body, including microRNA‐198 and three validated microRNA‐binding sites, were analyzed using Sanger sequencing.
Results
No variants were found in the coding region. However, 8 intronic variants were identified that differed significantly in their minor allele frequency compared to controls. Variant rs2272515 was found to significantly correlate (p < 0.05) with kyphoscoliosis.
Conclusion
We conclude that pathogenic variants in FSTL1 are unlikely to be responsible for skeletal or atrioventricular valve anomalies in humans.
Keywords: Fstl1, gene expression, heart development, skeletal abnormalities
1. INTRODUCTION
Approximately 3%–4% of all life‐born babies have a congenital malformation. In a minority of these cases, the underlying cause is identified as a chromosomal abnormality, a familial or de novo pathogenic variations, or as an environmental cause (e.g., pollution, alcohol abuse), or a combination of these conditions (Kuciene & Dulskiene, 2008; Liu et al., 2009). However, in the vast majority of congenital abnormalities, the cause is unknown.
We have recently prepared a global and conditional knockout of Follistatin‐like 1 (Fstl1) (Prakash et al., 2017; Sylva et al., 2011) which both show congenital abnormalities (see below). FSTL1 is a secreted glycoprotein (OMIM 605547), belonging to SPARC family of proteins (Sylva, Moorman, & Hoff, 2013), and is a potential prognostic marker for cardiovascular disease (Widera et al., 2012, 2009). FSTL1 expression is induced by TGFβ and thought to be an extracellular regulator of BMP signaling (for review, see (Mattiotti, Prakash, Barnett, & Hoff, 2018; Sylva et al., 2013)). The human FSTL1 gene is located on chromosome 3q13.33 and comprises 11 exons. Exons 2 to 11 encode the 308 amino acid long protein. The last exon also contains the coding sequence for microRNA‐198 (MIR198). As a consequence, the FSTL1 primary transcript serves as mRNA or pre‐miRNA in a mutually exclusive way (Hinske, Galante, Kuo, & Ohno‐Machado, 2010; Sundaram et al., 2013, 2017). Moreover, multiple microRNA‐binding sites are present in the 3’UTR of FSTL1, of which three (miR‐206, miR‐32–5p, and miR‐27a) have been shown to be functional and suppress FSTL1 expression (for review, see: (Mattiotti et al., 2018)). Thus far, no human congenital disease has been associated with alterations in FSTL1. In the analysis of the exomes of over 120,000 individuals, no homozygous loss‐of‐function variations were observed, and heterozygous ones have only been described in twenty‐five individuals (http://gnomad.broadinstitute.org/gene/ENSG00000163430).
Homozygous knockout (Fstl1 KO) mice are born in normal Mendelian ratios but suffocate within hours due to the inability to breathe as a consequence of the absence and/or hypoplasia of tracheal cartilage rings. These KO mice show extensive skeletal defects, including abnormal spine curvature, bending of long bones, displacement of the atlas, the absence of the patella, dislocation of the hip and digit defects as well as cardiac, lung and ureter abnormalities (Geng et al., 2011; Sylva et al., 2011; Xu et al., 2012). The phenotype of Fstl1 KO mice is comparable to a rare genetic disorder known as Campomelic Dysplasia (CD; OMIM 114290) (Mansour, Hall, Pembrey, & Young, 1995). In the majority of CD cases, the underlying cause is a loss of function of one SOX9 allele (OMIM 608160) (Corbani et al., 2011). However, in approximately 5% of CD cases, no underlying genetic cause can be identified (Unger, Scherer, & Superti‐Furga, 1993). Other rare genetic syndromes that show (partial) overlap with the phenotype of Fstl1 KO mice are small patella syndrome (SPS; OMIM 147891) (see Table 3) (Offiah et al., 2002), caused by heterozygous loss‐of‐function pathogenic variants in TBX4 (OMIM 601719) (Bongers et al., 2004), and BILU (OMIM 609296) (B‐cell Immunodeficiency, Limb anomalies, and Urogenital malformations) (see Table 3) (Edery et al., 2001; Hugle et al., 2011). The finding that Fstl1 KO mice display both abnormal spine curvature and respiratory distress at birth can also point toward kyphoscoliosis, a musculoskeletal disorder characterized by an abnormal curve of the spine that causes extrapulmonary restriction of the lungs that further results in impairment of pulmonary functions (Pajdzinski et al., 2017). In addition, these patients often have psychological problems (for example, anxiety, difficulty in performing daily activities) which are also observed in mice, in which Fstl1 is genetic deleted from the small dorsal root ganglion neurons (Li et al., 2011).
Table 3.
Summary of clinical and phenotypical data
| Malformation | Fstl1 KO | Fstl1 endoKO | # Skeletal Dysplasia patients (n = 15) | # Congenital heart defect patients (n = 54) |
|---|---|---|---|---|
| Skeletal defects | ||||
| Kyphoscoliosis | + | ‐ | 12 | 16 |
| Bending of long bones | + | ‐ | 12 | 12 |
| Digit malformation | + | ‐ | 13 | ‐ |
| Small/Absent patella | + | ‐ | 2 | ‐ |
| Valves defects | ||||
| Mitral valve | ‐ | + | ‐ | 39 |
| Tricuspid valve | ‐ | + | ‐ | 19 |
| Aortic valve | ‐ | ‐ | ‐ | 14 |
| Pulmonary valve | ‐ | ‐ | ‐ | 12 |
| Other cardiac defects | ||||
| Enlarged heart | + | + | ‐ | 3 |
| Atrioventricular septum | ‐ | ‐ | ‐ | 1 |
| Atrial septum | ‐ | ‐ | ‐ | 7 |
| Ventricular septum | ‐ | ‐ | ‐ | 8 |
| Outflow tract | ‐ | ‐ | ‐ | 15 |
| Conduction anomalies | ns | + | ‐ | 23 |
| Other defects | ||||
| Lung | + | + | 12 | 12 |
| Renal | + | ns | 13 | 2 |
ns, not studied. Summary of the congenital anomalies observed in Fst1 KO mice, Fstl1 endoKO mice and the 69 selected patients.
Besides the full Fstl1 knockout, a set of conditional knockouts has also been described. Cardiomyocyte‐specific deletion of Fstl1 using αMHC‐Cre (Shimano et al., 2011) or fibroblasts specific deletion using S100A4‐Cre (Tanaka et al., 2016) resulted in viable offspring with no reported developmental defects. However, conditional ablation of Fstl1 from the endocardial/endothelial lineage using Tie2‐Cre (Fstl1 endoKO) results in neonatal lethality between 2 and 4 weeks after birth (Prakash et al., 2017). These Fstl1 endoKO mice showed profound cardiac and valve defects, including an enlarged stressed heart and long and thick atrioventricular valves, displaying a myxomatous phenotype and being dysfunctional (Prakash et al., 2017). The Fstl1 endoKO mice also have cardiac conduction abnormalities including fragmented QRS and increased QT intervals. Besides the cardiac phenotype, the lungs are also affected showing impaired remodeling of the small pulmonary vasculature (Tania et al., 2017).
This study aimed at identifying whether variations in FSTL1 underlie human congenital abnormalities that show phenotypic similarity with those that are observed in the Fstl1 KO and/or Fstl1 endoKO mice. To this end, we selected 69 patients with genetically unresolved campomelic dysplasia, small patella syndrome, BILU syndrome, kyphoscoliosis and/or defects of the mitral or tricuspid valves, and Sanger sequenced the FSTL1 gene body and determined the copy number.
2. MATERIAL AND METHODS
2.1. Editorial policies and ethical considerations
This study was approved by the Medical Ethical Committee at the Academic Medical Center in Amsterdam. Written informed consent was obtained from all participants.
2.2. Patient recruitment
Genomic DNA was obtained of 12 patients suffering from unexplained campomelic dysplasia, that is, no pathogenic variants for SOX9, two patients suffering from unexplained small patella syndrome, that is, no pathogenic variants for TBX4, and one patient suffering from BILU syndrome. Furthermore, from the CONCOR repository (van der Velde et al., 2005), which comprises DNA of more than 16,000 adult patients with a congenital abnormality, 54 patients were selected with an unexplained congenital abnormality, comprising kyphoscoliosis and/or cardiac valve defects. Anonymized samples were analyzed for variations in the coding sequence of FSTL1 (NM_007085.5) by using Sanger sequencing and for copy number variation using genomic qPCR.
2.3. Sequence analysis of FSTL1
Using the primers listed in Table 1, we PCR amplified the FSTL1 coding region of the exons and surrounding regions which are considered the splice donor and acceptor sites. We included, furthermore, the parts of the 3’UTR that code the validated miRNA‐binding sites (MIR206/32–5p/MIR‐27a) and miRNA (MIR198). After amplifications, the PCR product was diluted and sequenced using Big Dye Terminator (BDT). The analysis for variances was done manually.
Table 1.
Primer sequences for amplification
| Amplicon | Forward primer | Reverse primer |
|---|---|---|
| Exon1 & 2 | CGCTCTCCCTCCATGTTAGC | TTCCCCCTACCCCTTTGTGG |
| Exon3 | GAAATCATTGGCACTGCCTG | AGCTGTTTAAGACCATGAGCC |
| Exon4 | AGCTTTGAGAGGCTGTGGTC | TCTCTGGTGGTGGAGAGTCC |
| Exon5 | ATCTTATGAGCTTGGCAGGG | GAAAAGGTTACAAACATTCCCC |
| Exon6 | GTCTGGGAAGGTGTGATCGG | TGCCAGATATCACTGGGGTC |
| Exon7 | GGGAAAGGGTTTAAGTCCCC | TGGAGTAATGATGGAACAGGG |
| Exon8 | CCACCAATCCTCTCTATCTTGG | GCTTACTTCCTTTCTTCTGAGCG |
| Exon9 | CACCCCAACTTCTCTCATGC | TAGCAAAGATGCCCTGTTCC |
| Exon10 | CACTGGGCAGTGTTTGAATG | AGGTAGATCTTGGCGGATTC |
| Exon11 & MIR32–5pa bs | TTCTCTCTGGCATCGTGTGC | ACGCCTATTTTCTCTTGCATCT |
| MIR198 & MIR27a bs | GAAAGACACAGGACTAACTGT | CCACTATCTGCCAGAGGAGCA |
| MIR206 bs | CACTGGATCTTAACAGATGC | CTTCAAACTCCACTTGTTCAGT |
Sequence of the forward and reverse primers used to analyze the coding region of FSTL1 locus (NM_007085.5); bs: binding site.
2.4. Copy number variation analysis
Using the primer sequences listed in Table 2, copy number variation (CNV) analysis was performed. Three regions of the FSTL1 locus were amplified with triplicate measurements (Figure 1). As a reference, a genomic DNA region (Chr3q26.2) in which no known copy number variations occur in healthy controls was amplified (http://exac.broadinstitute.org/gene/ENSG00000206120). Quantitative PCR (qPCR) was performed using the LightCycler 480 (Roche) and the LightCycler 480 SYBR Green I Master (Roche). The qPCR data were analyzed using the LinRegPCR program (Ruijter et al., 2013).
Table 2.
Primer sequences for copy number variation (CNV)
| FSTL1 CNV | Forward primer | Reverse primer |
|---|---|---|
| Start of Intron 2 | GCGATGCTAAGGGTGTGGTT | GGGAGAAGAAATGGAATGCACAT |
| End of Intron 2 | CCCATGTCTGCCACTGTCTT | CACACGTCTGAACAGGGGAA |
| 3´‐UTR | GCGTGGATGCTGGAGGTCTG | AACTTAGCAGGGTGTCCCCGGAG |
| Chr3q26.2 | AAGACCTCTTTCTACCACATGGT | AAATCTGAAGAAGCCACGCTT |
Sequences of the forward and reverse primers used to determine the CNV in the reference region and in three different regions of FSTL1.
Figure 1.
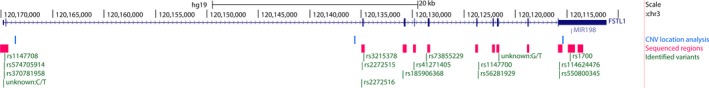
Representation of Fstl1 gene. The Fstl1 locus according to human genome GRCh37/hg19. In graph is in order indicated: scale and the genome location in chr3 (black), the FSTL1 gene (dark blue), the miRNA198 (red), the 3 regions used for copy number variation qPCR (light blue), the regions sequenced for the Sanger analysis (pink) and the identified SNP (green)
2.5. Statistical analysis
To compare minor allele frequency between the control population and the selected patient groups, a z‐test analysis was performed. Within patients with cardiac defects, association of a specific genotype with a particular diagnosis was performed using Fisher´s exact test. Differences with p < 0.05 were considered statistically significant.
3. RESULTS
3.1. Clinical data
Based on the phenotype of the Fstl1 KO mice, we selected 15 patients with unexplained skeletal dysplasia, of which 12 have campomelic dysplasia, 2 have small patella syndrome, and 1 with BILU. Moreover, when analyzing conditional KO mice, cardiac abnormalities have become apparent due to deletion of Fstl1 from the endocardial/endothelial lineage. Based on this observation, we selected an additional group of 54 patients from the CONCOR biobank having abnormalities similar to the phenotype of Fstl1 KO and Fstl1 endoKO mice, but with an unknown genetic cause. Of these patients, 39 showed congenital abnormalities related to either the mitral or the tricuspid valve and 16 showed, besides congenital cardiac defects also kyphoscoliosis, skeletal and lung defects. A summary of the mouse phenotypes, congenital malformations, and the number of patients in each category are shown in Table 3.
3.2. FSTL1 copy number variation
To investigate whether copy number variations, either duplication or deletions, were present in the FSTL1 locus in our patient groups, we used qPCR with three different primer sets for three loci, effectively covering the entire genomic locus of FSTL1 in combination with the reference region Chr3q26.2 (Table 2 and Figure 1). Control DNA of 6 randomly selected unaffected healthy individuals was taken as reference. One out of the 15 unexplained skeletal dysplasia patients displayed an abnormal copy number variation. To follow‐up and confirm these findings, we attempted to perform an array CGH. However, an initial quality check showed that the DNA was too degraded to undergo array CGH, likely explaining the spurious qPCR findings in FSTL1. In the 54 patients with congenital heart disease, we did not find any CNV in FSTL1.
3.3. Identification of two novel intronic variants
FSTL1 encodes a 308‐amino acid protein with an N‐terminal signal peptide of 20 amino acids. FSTL1 is composed of 11 exons, the first exon encodes the noncoding 5’UTR, and exon 11 encodes a small part of coding sequence and a long 3’UTR. In this 3’UTR, MIR198 is encoded and 3 functionally validated binding sites for miRNAs are present (Figure 1). Using PCR, all the FSTL1 coding exons and their immediately flanking regions, the MIR198 and the miR‐binding sites, were amplified and sequenced. We found 16 nucleotide variations in our patients compared to the human reference genome (Hg19). We did not identify any protein‐altering variants in any of our patient groups. Only variations in the noncoding intronic regions of FSTL1 were found (Table 4 and shown in Figure 1). Out of the 16 identified variations, 14 are known polymorphisms and 2 are novel variants not present in over 120 k exomes (http://gnomad.broadinstitute.org/). The first novel variant is located in intron 1 (chr3:120,169,715); it is a C > T nucleotide change in a patient with atrioventricular and outflow tract valves defects. This portion of the FSTL1 gene is an epigenetic dense region and likely to be a part of the promoter of Fstl1, based on the integration of multiple epigenetic marks (van Duijvenboden, Boer, Capon, Ruijter, & Christoffels, 2016). The second novel variant is positioned at chr3:120,121,556 and is a G > T nucleotide change located in intron 9, in a patient with campomelic dysplasia. In this region, there is no evidence of evolutionary conservation or regulatory regions. Both SNPs are outside the splicing donor/acceptor sites.
Table 4.
Identified variants
| RS number | Nucleotide change | Position Hg19 | Location |
|---|---|---|---|
| Unknown | C/T | chr3:120169714 | Intronic (30 bp downstream exon 1) |
| rs370781958 | C/A | chr3:120169697 | Intronic (47 bp downstream exon 1) |
| rs574705914 | C/T | chr3:120169668 | Intronic (76 bp downstream exon 1) |
| rs1147708 | C/T | chr3:120169641 | 5’UTR |
| rs2272516 | C/A | chr3:120134901 | Intronic (27 bp upstream exon 3) |
| rs2272515 | T/C | chr3:120134883 | Intronic (9 bp upstream exon 3) |
| rs3215378 | ‐/G | chr3:120134720 | Intronic (49 bp downstream exon 3) |
| rs185906368 | C/T | chr3:120130874 | Intronic (44 bp upstream exon 4) |
| rs41271405 | T/C | chr3:120129860 | Intronic (29 bp upstream exon 5) |
| rs73855229 | C/T | chr3:120128618 | Intronic (109 bp upstream exon 6) |
| rs56281929 | T/G | chr3:120123572 | Intronic (128 bp downstream exon 7) |
| rs1147700 | A/C | chr3:120123556 | Intronic (140 bp downstream exon 7) |
| Unknown | G/T | chr3:120121556 | Intronic (99 bp downstream exon 9) |
| rs550800345 | C/T | chr3:120115713 | 3’UTR |
| rs114624476 | T/C | chr3:120115683 | 3’UTR |
| rs1700 | C/T | chr3:120114637 | 3’UTR |
Summary of the different nucleotide variations found in 69 selected patients.
3.4. Differences in frequency of intronic variants
Comparison of the minor allele frequency (MAF) of the intronic variants identified in our patients with those of 1,000 genomes project (www.ensembl.org) revealed that 3/11 variants identified in the skeletal dysplasia group, and 5/10 in the congenital heart defect patients, differed significantly (Table 5). However, none of these variants have been associated with a pathological function. Due to the variability in the phenotype observed in these patients, we tried to correlate the clinical defects reported in our patients, with the identified variants. We observed an association between variation rs2272515 and kyphoscoliosis (p = 0.0487), and a trend (p = 0.0508) with pulmonary valve defects.
Table 5.
Variant frequency
| RS number |
MAF control 1000G (2504) |
MAF skeletal dysplasia (15) | p value | MAF congenital heart defect (54) | p value |
|---|---|---|---|---|---|
| Unknown | ‐ | ‐ | ‐ | 0.01 (1) | 2.7E−05* |
| rs370781958 | 0.01 (43) | ‐ | ‐ | 0.01 (1) | 0.94 |
| rs574705914 | <0.01 (17) | ‐ | ‐ | 0.01 (1) | 0.31 |
| rs1147708 | 0.16 (532/87) | 0.07 (1) | 0.09 | 0.06 (6) | 0.01* |
| rs2272516 | 0.09 (142/19) | 0.27 (4) | 4.5E−3* | 0.01 (1) | 0.14 |
| rs2272515 | 0.40 (917/251) | 0.40 (6) | 0.31 | 0.61 (21/10) | 3.1E−07* |
| rs3215378 | 0.28 (914/251) | 0.40 (6) | 0.32 | ‐ | ‐ |
| rs185906368 | 0.05 (209/17) | 0.07 (1) | 0.70 | 0.07 (7) | 0.44 |
| rs41271405 | 0.04 (193/6) | 0.07 (1) | 0.83 | ‐ | ‐ |
| rs73855229 | 0.02 (88/3) | 0.07 (1) | 0.56 | ‐ | ‐ |
| rs56281929 | 0.04 (197/6) | 0.07 (1) | 0.82 | ‐ | ‐ |
| rs1147700 | 0.3 (931/323) | 0.27 (4) | 0.03* | ‐ | ‐ |
| Unknown | ‐ | 0.07 (1) | 1.2E−4* | ‐ | ‐ |
| rs550800345 | <0.01 (1) | ‐ | ‐ | 0.01 (1) | 2.4E−06* |
| rs114624476 | <0.01 (15) | ‐ | ‐ | 0.02 (2) | 5.5E−3* |
| rs1700 | 0.08 (381/22) | 0.13 (2) | 0.89 | 0.08 (9) | 0.95 |
Data on the frequency and statistics of the identified nucleotide variations. If available, the reference of the single nucleotide polymorphism identification number (RS) is given in the first column. The minor allele frequency (MAF) of controls as identified in the 1000 genome project, as well as in the two patient groups, is shown. In brackets, the number of heterozygous and, when applicable, homozygous individual is indicated.
p‐values less than 0.05 were considered significant.
4. DISCUSSION
Deletion of Fstl1 in mice results in skeletal and respiratory abnormalities (Sylva et al., 2011) and conditional deletion from endocardial/endothelial lineage shows profound AV valve defects (Prakash et al., 2017). To the best of our knowledge, homozygous loss‐of‐function pathogenic variants in FSTL1 have never been reported, and heterozygous ones have only been described in 35 individuals. In line with these findings, the estimated probability of loss‐of‐function intolerance of FSTL1 is 0.96 (http://exac.broadinstitute.org/gene/ENSG00000163430), comparing the exomes of over 120,000 humans. This suggests that pathogenic variations are not tolerated in the coding region of FSTL1 gene. This finding is not surprising considering that complete disruption of Fstl1 (Fstl1 KO) in mice results in death of the newborns due to their inability to breathe. Furthermore, these Fstl1 KO mice display major skeletal defects, including bending of the spine and long bones, and an enlarged heart (Sylva et al., 2011). Conditional removal of Fstl1 from the endocardial/endothelial cell lineage (Fstl1 endoKO) results in mice that die between 2 and 4 weeks after birth with heart failure with preserved ejection fraction and dysfunctional valves (Prakash et al., 2017). The mitral valve displays a myxomatous phenotype with mitral valve prolapse and regurgitation. It should be noted, that in different genomewide association studies (GWAS), FSTL1 has never been associated with mitral valve prolapse (Dina et al., 2015; Freed et al., 2003; Nesta et al., 2005). Conditional removal of Fstl1 from the cardiomyocytes (Shimano et al., 2011) or from the cardiac fibroblasts (S100A or Col1a1) (Maruyama et al., 2016) did not result in any gross abnormalities in mice.
To identify potential carriers of variations in the FSTL1 gene, we compared the phenotypes of the Fstl1 KO and endoKO mice to rare congenital malformations reported in humans. We identified a total of 69 patients comprising the following groups: (a) skeletal defects, such as campomelic dysplasia, small patella syndrome and BILU syndrome, and (b) patients with cardiac anomalies, valve defects in the majority, in combination with or without kyphoscoliosis. Importantly, in none of these selected patients, a known genetic defect was reported. Analysis of the coding sequence with respect to the copy number of the FSTL1 gene revealed no genetic abnormalities in either of these 69 patients. In 52 patients, a total of 16 variations were found. These variations were only found in the noncoding region of the FSTL1 gene. Eleven were found in patients with skeletal dysplasia, and three of these SNP showed significant differences in minor allele frequency (MAF) compared to control populations. Ten of these SNPs were found in patients from the cardiac anomalies group, and five of these SNP showed significant differences in MAF compared to control populations. The intronic variant rs2272515 significantly correlates with kyphoscoliosis in our population. This variant is located 9 bp upstream exon 3 in the splice acceptor sequence and could, provided it is biologically functional, affect mRNA maturation causing a truncated form of the protein. In the congenital heart disease patients, 3 SNPs are located in the 3’UTR of FSTL1. The location of these SNPs was not in the encoded miRNA or in either of the validated miRNA‐binding site. To evaluate whether these SNPs might affect an unknown microRNA‐binding site, www.microRNA.org and www.targetscan.org were used to predict miRNA‐binding sites in the region of the identified SNP. This analysis revealed that SNP rs1700 overlapped with the predicted binding site for MIR154, which is known to promote myocardial fibrosis (Dong, Liu, & Wang, 2018) and regulating the metastatic behavior of nonsmall lung cancer cells (Liu et al., 2018). As both biological effects of MIR154 are among the biological functions of FSTL1 (for review see (Mattiotti et al., 2018)), this might be an inroad for further analysis. However, due to the relative small patient group, the frequency of this variant is not significantly different between the patients and control groups.
In conclusion, we did not find any pathogenic variations in the coding region of the FSTL1 gene in 69 patients suffering from rare, and genetically unresolved, skeletal or cardiac disorders, that resembled the phenotypes observed in complete or conditional Fstl1 KO mice. However, we identified 8 intronic FSTL1 variants that significantly differed from control populations in minor allele frequency, and found two novel/unique intronic variants. Taken together, we conclude that pathogenic variations in FSTL1 are likely not responsible for skeletal or atrioventricular valve anomalies in humans.
CONFLICT OF INTEREST
The authors state that there are no conflicts of interest to disclose.
ACKNOWLEDGMENTS
The authors thank Prof Dr Gerd Scherer to kindly provide genomic DNA from campomelic dysplasia patients, Dr David Webster for unexplained small patella syndrome patients and Dr Sonja de Munnik for BILU patients. This work is funded by the Dutch Heart Foundation (CVON CONCOR‐genes project), and European Commission Seventh Framework Programme FP7 People: Marie‐Curie Actions, CardioNet ITN 2011 GA289600.
Prakash S, Mattiotti A, Sylva M, Mulder BJM, Postma AV, van den Hoff MJB. Identifying pathogenic variants in the Follistatin‐like 1 gene (FSTL1) in patients with skeletal and atrioventricular valve disorders. Mol Genet Genomic Med. 2019;7:e567 10.1002/mgg3.567
Stuti Prakash and Andrea Mattiotti contributed equally.
REFERENCES
- Bongers, E. M. , Duijf, P. H. , van Beersum, S. E. , Schoots, J. , Van Kampen, A. , Burckhardt, A. , … van Bokhoven, H. (2004). Mutations in the human TBX4 gene cause small patella syndrome. The American Journal of Human Genetics, 74(6), 1239–1248. 10.1086/421331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbani, S. , Chouery, E. , Eid, B. , Jalkh, N. , Ghoch, J. A. , & Megarbane, A. (2011). Mild Campomelic Dysplasia: Report on a Case and Review. Molecular Syndromology, 1(4), 163–168. 10.1159/000322861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina, C. , Bouatia‐Naji, N. , Tucker, N. , Delling, F. N. , Toomer, K. , Durst, R. , … Jeunemaitre, X. . (2015). Genetic association analyses highlight biological pathways underlying mitral valve prolapse. Nature Genetics, 47(10), 1206–1211. 10.1038/ng.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, P. , Liu, W. J. , & Wang, Z. H. (2018). MiR‐154 promotes myocardial fibrosis through beta‐catenin signaling pathway. European Review for Medical and Pharmacological Sciences, 22(7), 2052–2060. [DOI] [PubMed] [Google Scholar]
- Edery, P. , Le, D. F. , Briard, M. L. , Debre, M. , Munnich, A. , Griscelli, C. , … Lyonnet, S. (2001). B cell immunodeficiency, distal limb abnormalities, and urogenital malformations in a three generation family: A novel autosomal dominant syndrome? Journal of Medical Genetics, 38(7), 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed, L. A. , Acierno, J. S. Jr , Dai, D. , Leyne, M. , Marshall, J. E. , Nesta, F. , … Slaugenhaupt, S. A. (2003). A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. American Journal of Human Genetics, 72(6), 1551–1559. 10.1086/375452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng, Y. , Dong, Y. , Yu, M. , Zhang, L. , Yan, X. , Sun, J. , … Ning, W. (2011). Follistatin‐like 1 (Fstl1) is a bone morphogenetic protein (BMP) 4 signaling antagonist in controlling mouse lung development. Proceedings of the National Academy of Sciences USA, 108(17), 7058–7063. 10.1073/pnas.1007293108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinske, L. C. , Galante, P. A. , Kuo, W. P. , & Ohno‐Machado, L. (2010). A potential role for intragenic miRNAs on their hosts' interactome. BMC Genomics, 11, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugle, B. , Hoffman, H. , Bird, L. M. , Gebauer, C. , Suchowerskyj, P. , Sack, U. , … Schuster, V. (2011). Hoffman syndrome: New patients, new insights. American Journal of Medical Genetics, 155A(1), 149–153. [DOI] [PubMed] [Google Scholar]
- Kuciene, R. , & Dulskiene, V. (2008). Selected environmental risk factors and congenital heart defects. Medicina (Kaunas), 44(11), 827–832. 10.3390/medicina44110104 [DOI] [PubMed] [Google Scholar]
- Li, K. C. , Zhang, F. X. , Li, C. L. , Wang, F. , Yu, M. Y. , Zhong, Y. Q. , … Zhang, X. (2011). Follistatin‐like 1 suppresses sensory afferent transmission by activating Na+, K+‐ATPase. Neuron, 69(5), 974–987. 10.1016/j.neuron.2011.01.022 [DOI] [PubMed] [Google Scholar]
- Liu, S. , Liu, J. , Tang, J. , Ji, J. , Chen, J. , & Liu, C. (2009). Environmental risk factors for congenital heart disease in the Shandong Peninsula, China: A hospital‐based case‐control study. Journal of Epidemiology, 19(3), 122–130. 10.2188/jea.JE20080039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Tan, X. , Liu, W. , Chen, X. , Hou, X. , Shen, D. , … Cao, G. (2018). Follistatin‐like protein 1 plays a tumor suppressor role in clear‐cell renal cell carcinoma. Chinese Journal of Cancer, 37(1), 2 10.1186/s40880-018-0267-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour, S. , Hall, C. M. , Pembrey, M. E. , & Young, I. D. (1995). A clinical and genetic study of campomelic dysplasia. Journal of Medical Genetics, 32(6), 415–420. 10.1136/jmg.32.6.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama, S. , Nakamura, K. , Papanicolaou, K. N. , Sano, S. , Shimizu, I. , Asaumi, Y. , … Walsh, K. (2016). Follistatin‐like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Molecular Medicine, 8(8), 949–966. 10.15252/emmm.201506151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiotti, A. , Prakash, S. , Barnett, P. , & van den Hoff, M. J. B. (2018). Follistatin‐like 1 in development and human diseases. Cellular and Molecular Life Sciences, 75(13), 2339–2354. 10.1007/s00018-018-2805-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesta, F. , Leyne, M. , Yosefy, C. , Simpson, C. , Dai, D. , Marshall, J. E. , … Levine, R. A. (2005). New locus for autosomal dominant mitral valve prolapse on chromosome 13: Clinical insights from genetic studies. Circulation, 112(13), 2022–2030. 10.1161/CIRCULATIONAHA.104.516930 [DOI] [PubMed] [Google Scholar]
- Offiah, A. C. , Mansour, S. , McDowall, S. , Tolmie, J. , Sim, P. , & Hall, C. M. (2002). Surviving campomelic dysplasia has the radiological features of the previously reported ischio‐pubic‐patella syndrome. Journal of Medical Genetics, 39(9), e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajdzinski, M. , Mlynarczyk, P. , Milkowska‐Dymanowska, J. , Bialas, A. J. , Afzal, M. A. M. , Piotrowski, W. J. , & Gorski, P. (2017). Kyphoscoliosis ‐ what can we do for respiration besides NIV? Advances in Respiratory Medicine, 85(6), 352–358. [DOI] [PubMed] [Google Scholar]
- Prakash, S. , Borreguero, L. J. J. , Sylva, M. , Flores Ruiz, L. , Rezai, F. , Gunst, Q. D. , … van den Hoff, M. J. B. (2017). Deletion of Fstl1 (Follistatin‐Like 1) from the endocardial/endothelial lineage causes mitral valve disease. Arteriosclerosis, Thrombosis, and Vascular Biology, 37:116–130. [DOI] [PubMed] [Google Scholar]
- Ruijter, J. M. , Pfaffl, M. W. , Zhao, S. , Spiess, A. N. , Boggy, G. , Blom, J. , … Vandesompele, J. (2013). Evaluation of qPCR curve analysis methods for reliable biomarker discovery: Bias, resolution, precision, and implications. Methods, 59, 32–46. 10.1016/j.ymeth.2012.08.011 [DOI] [PubMed] [Google Scholar]
- Shimano, M. , Ouchi, N. , Nakamura, K. , van Wijk, B. , Ohashi, K. , Asaumi, Y. , … Walsh, K. (2011). Cardiac myocyte follistatin‐like 1 functions to attenuate hypertrophy following pressure overload. Proceedings of the National Academy of Sciences of the United States of America, 108(43), E899–E906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram, G. M. , Common, J. E. , Gopal, F. E. , Srikanta, S. , Lakshman, K. , Lunny, D. P. , … Sampath, P. (2013). 'See‐saw' expression of microRNA‐198 and FSTL1 from a single transcript in wound healing. Nature, 495(7439), 103–106. 10.1038/nature11890 [DOI] [PubMed] [Google Scholar]
- Sundaram, G. M. , Ismail, H. M. , Bashir, M. , Muhuri, M. , Vaz, C. , Nama, S. , … Sampath, P. (2017). EGF hijacks miR‐198/FSTL1 wound‐healing switch and steers a two‐pronged pathway toward metastasis. Journal of Experimental Medicine, 214(10), 2889–2900. 10.1084/jem.20170354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylva, M. , Moorman, A. F. , & van den Hoff, M. J. (2013). Follistatin‐like 1 in vertebrate development. Birth Defects ResCEmbryoToday, 99(1), 61–69. 10.1002/bdrc.21030 [DOI] [PubMed] [Google Scholar]
- Sylva, M. , Li, V. S. , Buffing, A. A. , van Es, J. H. , van den Born, M. , van der Velden, S. , … van den Hoff, M. J. (2011). The BMP antagonist follistatin‐like 1 is required for skeletal and lung organogenesis. PLoSONE, 6(8), e22616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, K. , Valero‐Munoz, M. , Wilson, R. M. , Essick, E. E. , Fowler, C. T. , Nakamura, K. , … Sam, F. (2016). Follistatin like 1 Regulates Hypertrophy in Heart Failure with Preserved Ejection Fraction. JACC Basic Translational Science, 1(4), 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tania, N. P. , Maarsingh, H. , Bos, I. S. T. , Mattiotti, A. , Prakash, S. , Timens, W. , … Gosens, R. (2017). Endothelial follistatin‐like‐1 regulates the postnatal development of the pulmonary vasculature by modulating BMP/Smad signaling. Pulmonary Circulation, 7(1), 219–231. 10.1177/2045893217702340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger, S. , Scherer, G. , & Superti‐Furga, A. (1993). Campomelic Dysplasia. PMID: 20301724 [PubMed]
- van der Velde, E. T. , Vriend, J. W. , Mannens, M. M. , Uiterwaal, C. S. , Brand, R. , & Mulder, B. J. (2005). CONCOR, an initiative towards a national registry and DNA‐bank of patients with congenital heart disease in the Netherlands: Rationale, design, and first results. European Journal of Epidemiology, 20(6), 549–557. 10.1007/s10654-005-4264-9 [DOI] [PubMed] [Google Scholar]
- van Duijvenboden, K. , de Boer, B. A. , Capon, N. , Ruijter, J. M. , & Christoffels, V. M. (2016). EMERGE: A flexible modelling framework to predict genomic regulatory elements from genomic signatures. Nucleic Acids Research, 44(5), e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widera, C. , Giannitsis, E. , Kempf, T. , Korf‐Klingebiel, M. , Fiedler, B. , Sharma, S. , … Wollert, K. C. (2012). Identification of follistatin‐like 1 by expression cloning as an activator of the growth differentiation factor 15 gene and a prognostic biomarker in acute coronary syndrome. Clinical Chemistry, 58(8), 1233–1241. 10.1373/clinchem.2012.182816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widera, C. , Horn‐Wichmann, R. , Kempf, T. , Bethmann, K. , Fiedler, B. , Sharma, S. , … Wollert, K. C. (2009). Circulating concentrations of follistatin‐like 1 in healthy individuals and patients with acute coronary syndrome as assessed by an immunoluminometric sandwich assay. Clinical Chemistry, 55(10), 1794–1800. 10.1373/clinchem.2009.129411 [DOI] [PubMed] [Google Scholar]
- Xu, J. , Qi, X. , Gong, J. , Yu, M. , Zhang, F. , Sha, H. , & Gao, X. (2012). Fstl1 antagonizes BMP signaling and regulates ureter development. PLoSONE, 7(4), e32554. [DOI] [PMC free article] [PubMed] [Google Scholar]