

Abstract
Background
This study summarizes the results of prenatal diagnosis due to a history of de novo mutation in a previous pregnancy, in a tertiary center in Israel, over a 10‐year period.
Methods
We sorted all cases of de novo mutations from a pool of 2,260 pregnancies for which prenatal molecular diagnosis was applied, between the years 2008 and 2017. We identified 122 molecular prenatal diagnosis performed for de novo mutations, in 90 women.
Results
While the total number of yearly prenatal diagnoses stayed stable, a linear increase was detected in the number of cases for which the procedure was done due to a previous de novo mutation: from 3 cases in 2008 to 24 cases in 2017. The most common diseases were Rett syndrome (19), neurofibromatosis Type‐1 (12) and Tuberous sclerosis (5). Recurrence occurred in 3 of the 90 women (3.3%) and hotspot mutations were identified in two genes accounting for 11 cases. We did not find a difference in paternal age at first occurrence of the de novo mutation between the study group and the control group.
Conclusion
The large increase in the annual number of prenatal diagnoses performed due to a previous pregnancy with a de novo mutation reflects the growing understanding regarding the role of these mutations in the pathogenesis of genetic diseases.
Keywords: de novo, hot spots, mutations, neurofibromatosis Type‐1, prenatal diagnosis, Rett syndrome
1. INTRODUCTION
Prenatal diagnosis for monogenic diseases is on the rise. Two main factors contribute to this trend. The first is pre‐conception screening programs that intend to identify carriers for autosomal recessive and X‐linked diseases. If both parents are found to be carriers for a recessive disorder or if the female is found to be a carrier for an X‐linked disorder, prenatal diagnosis is offered to the parents. The chance of having an affected baby in these circumstances is 25%. The second is families in which a genetic disease is identified and verified by a molecular analysis. Recurrence rates are usually between 25%–50% depending on the inheritance mode; autosomal recessive, autosomal dominant or X‐linked. A third and less common cause for prenatal diagnosis is a previous diagnosis of a de novo mutation. A human zygote inherits half of its genome from the female and the other half from the male. With the genetic information passed on from generation to generation, a small number of novel genetic changes, de novo mutations, are formed in each individual. Such changes can occur during the formation of the gametes or postzygotically (Lynch, 2010; Roach et al., 2010). Genome‐wide next generation sequencing studies have estimated the rate of de novo single nucleotide variants (SNVs) in humans at 1.0–1.8 × 10–8 per nucleotide per generation (Francioli et al., 2015; Gilissen et al., 2014; Goldmann et al., 2016; Michaelson et al., 2012; Rahbari et al., 2016; Roach et al., 2010). This number translates into an average of 60 de novo SNVs in the genome of a newly formed embryo, however only one to two of these affect the coding sequence (Francioli et al., 2015; Gilissen et al., 2014; Goldmann et al., 2016; Kong et al., 2012; Michaelson et al., 2012), and only a fraction of those causes genetic diseases. Novel mutations continue to arise throughout postnatal and adult life in both somatic and germ cells. Mutations present in the germ cells can be transmitted to the next generation (Campbell, Shaw, Stankiewicz, & Lupski, 2015). Accordingly, the recurrence rate for a couple that have a child with a genetic disease caused by a de novo mutation is higher than that of the general population and is estimated at 1%–4% (Campbell et al., 2014). The single most important known risk factor contributing for de novo mutations is advanced paternal age at conception (Kong et al., 2012). Interestingly de novo mutations are not equally distributed throughout the human genome and occur more often in genes belonging to RAS–MAPK pathway (Goriely & Wilkie, 2010; Yoon et al., 2013).
In order to prevent recurrence in such families we offer them prenatal diagnosis by amniocentesis between weeks 17–22 of the pregnancy. Here we summarize the results of 90 women who performed prenatal diagnosis due to a history of de novo mutation in a previous pregnancy, over a 10‐year period.
2. METHODS
2.1. Ethical compliance
The study approved by the ethics committee was performed at the Institute of Human Genetics at the Sheba Medical Center, Israel.
2.2. Data collection
Information was collected from a pool of medical files from 2,260 women who performed amniocentesis due to an increased risk for a genetic disease in the previous child or fetus. Information retrieved included: genetic diagnosis, gene, mutation, paternal age at the time of conception with the de novo mutation pregnancy, outcome of the prenatal diagnosis in following pregnancies focusing on whether the de novo mutation had reoccurred. Some families repeated the prenatal diagnoses in more than one pregnancy but were included only once. A control group was obtained from the Israeli Ministry of Health and included the paternal age at the time of conception from 1,248,955 live births, between the years 2008–2015 (data on the years 2016–2017 are not yet available).
2.3. Statistical analysis
The statistical analysis was carried out with a one sample T‐test.
Sequence references of MECP2 (NG_007107.2), FGFR3 (NG_012632.1), TSC1 (NG_012386.1) and COL1A2 (NG_007405.1) were obtained from GenBank (https://www.ncbi.nlm.nih.gov/genbank/).
3. RESULTS
Between 2008 and 2017, 122 prenatal diagnosis procedures were performed in 90 pregnancies for a previously identified de novo mutation. Thirty‐one women performed the procedure more than once. During this period, we have witnessed an almost 10‐fold increase in the rate of procedures for de novo mutations: from 3 in 2007 to 24 in 2017. Interestingly the overall rate of molecular prenatal diagnosis procedures did not increase during this period with an average of 222 procedures per year (Figure 1).
Figure 1.
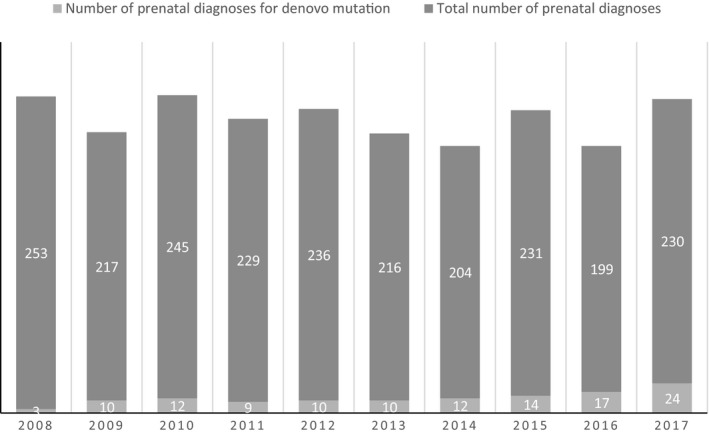
Number of prenatal diagnoses for de novo mutations between 2008–2017
The two most common de novo conditions for prenatal diagnosis were Rett syndrome and neurofibromatosis Type‐1, accounting for more than 30% of the cases. Other de novo diseases that appeared multiple times are shown in Figure 2. Two mutations were found to occur more than once in our families, representing mutational hotspots: The mutation R168X (c.502 C>T) in MECP2 (Rett syndrome; OMIM: #300005) repeated itself in eight different families and G380R (c.1138 C>A) in FGFR3 (Achondroplasia; OMIM: #134934) was found in three different families.
Figure 2.
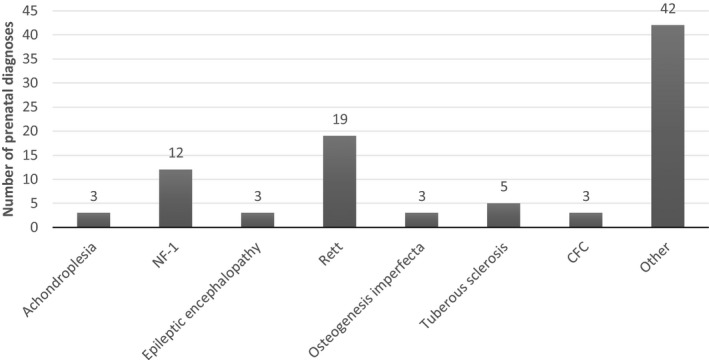
Common de novo syndromes
In three families (3.33%), we observed recurrence of the de novo mutation in a following pregnancy: a family with osteogenesis imperfecta and a mutation in COL1A2 (OMIM: #120160), a family with Rett syndrome and a mutation in the MECP2 and a family with tuberous sclerosis and a mutation in the TSC1 (OMIM: #605284; Table 1). These cases represent germinal mosaicism in one of the parents and the overall recurrence rate (3.33%) is similar to what has previously been described in the literature (Campbell et al., 2014). Sanger sequencing did not detect low‐level mosaicism of the de novo mutations in any of the parents.
Table 1.
Mutations that reoccurred in the same family
| Syndrome | Mutation | No. of recurrences |
|---|---|---|
| Osteogenesis imperfecta | COL1A2‐G994D (c.2981 G>A) | 1 recurrence |
| Rett | MECP‐R294X (c.880 C>T) | 1 recurrence |
| Tuberous sclerosis | TSC1‐R786X (c.2356 C>T) | 1 recurrence |
GenBank reference no: COL1A2 (NG_007405.1), MECP2 (NG_007107.2), TSC1 (NG_012386.1).
Surprisingly, we did not find a difference in paternal age at the time of conception of the pregnancy with the de novo mutation (32.37 ± 0.3251) and a control group composed of the average paternal age in Israel between the years 2008–2015 in over 1,000,000 fathers (32.82; p = 0.629) (Table 2).
Table 2.
Paternal age in the study group and in the general population
| Average paternal age | Number of cases | SE | |
|---|---|---|---|
| Study group | 32.37 | 80 | 0.3251 |
| General population | 32.823 | 1,248,955 |
p = 0.629.
4. DISCUSSION
We have found a steady rise that resulted in an eightfold increase in prenatal diagnosis due to de novo mutations, over a 10‐year period. Three explanations could account for this sharp increase. The first is the explosive use of whole exome sequencing (WES) for the identification of genetic diseases that has resulted in a substantial rise in the identification of de novo mutations. However as shown in Figure 2, most of the diseases for which prenatal diagnosis were performed were diagnosed on a clinical basis without the need for WES and therefore this explanation by itself cannot be responsible for the increase. The second explanation is growing awareness to the increased recurrence rates due to gonadal mosaicism among medical staff. Improvement in medical services, better education of physicians and nurses, the growing availability and utilization of genetic counseling and prenatal services may explain the large increase in pregnant women with previous offspring affected by de novo mutations sent for prenatal diagnosis. The last explanation is a change in referrals characteristics to our center. Over the last decade, new genetic centers have opened, the mixture of cases in our center has changed and the more complex ones were sent to us. Paternal age has been on the rise in the last decades, however in our cohort this explanation cannot account for the increase (Table 2). Taken together the growing number of prenatal diagnoses for de novo mutations reflects the growing understanding that such mutations contribute a significant portion of morbidity due to genetic diseases in the general population.
The main reasons for the creation of de novo mutations are incorrect incorporation of nucleotides by DNA polymerases ε and δ (Korona, LeCompte, & Pursell, 2010; Schmitt, Matsumoto, & Loeb, 2009; Ségurel, Wyman, & Przeworski, 2014) and by failure of the proofreading subunit present in both polymerases to correct these errors (Ségurel et al., 2014). Their occurrence across the genome is not completely random and factors that play a role in the genome mutability include the local base‐pair context, recombination rate and the timing of replication (Goldmann et al., 2016; Michaelson et al., 2012; Stamatoyannopoulos et al., 2009). Timing of replication refers to the order in which different regions of the genome are replicated during the S‐phase of the cell cycle. On the average those that are replicated late display more newly acquired changes than parts that are replicated early, possibly due to depletion of dNTPs at the end of replication, to alterations in polymerase activity or decreased repair activity (Chen et al., 2010; Koren et al., 2012; Ségurel et al., 2014). Occasionally, multiple de novo mutations may occur very close to the other in a given individual, thus creating “mutational clusters.” In addition, de novo mutations may appear at the same location in several unrelated individuals thus pointing to the existence of mutational hotspots (Chan & Gordenin, 2015). Mutational hotspots are the probable explanation for the recurrence of the same mutations in our cohort (Table 1).
About 80% of the de novo germline point mutations form on the paternal allele, findings that can be explained by the constant division of the spermatogonial cells throughout life resulting in the accumulation of de novo mutations. Amazingly, a large increase with paternal age has been observed for a small subset of de novo mutations that are highly recurrent and localize to specific nucleotides in the genome. Some investigators have hypothesized that spermatogonial stem cells with mutations in genes in the RAS–MAPK pathway exert a growth advantage that may lead to their clonal expansion in the testis (Goriely & Wilkie, 2010; Yoon et al., 2013). Such positively selected mutations offer sperm cells a selective advantage in the testis despite being detrimental at the organism level and have been described in a host of genetic diseases including Apert, Crouzon, and Pfeiffer syndromes (Goriely, McVean, Röjmyr, Ingemarsson, & Wilkie, 2003; Maher et al., 2016), Noonan, and Costello syndromes (Goriely et al., 2009; Maher et al., 2016; Yoon et al., 2013), Muenke syndrome, achondroplasia and thanatophoric dysplasia (Goriely et al., 2009; Maher et al., 2016), and multiple endocrine neoplasia (Choi, Yoon, Calabrese, & Arnheim, 2012). Indeed 22% of the de novo mutations that we have found are in genes that belong to the RAS–MAPK (Table 3).
Table 3.
RAS‐MAP kinase associated diseases
| Genetic disease | Number of cases |
|---|---|
| Achondroplasia | 3 |
| Cardio‐facio‐cutaneous | 3 |
| Neurofibromatosis Type‐1 | 12 |
| PTEN | 1 |
| Thanatophoric dysplasia | 1 |
In 3 of the 90 pregnancies (3%), we have detected recurrence of the de novo mutation, indicating germline mosaics in one of the parents (Table 1). These figures are compatible with previous reports in the medical literature (Campbell et al., 2014).
de novo mutations can be further delineated to include two additional subgroups: approximately 4% originate from parental mosaicism detectable in blood samples of one of the parents (Rahbari et al., 2016), for which recurrence risk is higher and has been estimated at above 5% (Campbell et al., 2014). In contrast, postzygotic events in the embryo account for 7% of what may initially appear as de novo mutations (Acuna‐Hidalgo et al., 2015; Besenbacher et al., 2015; Dal et al., 2014) and for these recurrence risks in following pregnancies are similar to the general population (Biesecker & Spinner, 2013). Differentiating these two subgroups by targeted deep sequencing of blood samples from the parents and affected offspring may provide a personalized and more accurate estimate of the recurrence risk (Acuna‐Hidalgo, Veltman, & Hoischen, 2016).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTION
Conceptualization: E.P. H.Y.; Methodology: O.E., M.B., H.R.W., H.P., E.P.; Investigation: O.E., M.B., H.R.W., L.G., H.P., T.Z.B., E.P.; Writing: O.E., L.G., T.Z.B., E.P.; Supervision H.Y., E.P.; Manuscript editing and revision for intellectual content: all authors.
Eyal O, Berkenstadt M, Reznik‐Wolf H, et al. Prenatal diagnosis for de novo mutations: Experience from a tertiary center over a 10‐year period. Mol Genet Genomic Med. 2019;7:e573 10.1002/mgg3.573
REFERENCES
- Acuna‐Hidalgo, R. , Bo, T. , Kwint, M. P. , Van De Vorst, M. , Pinelli, M. , Veltman, J. A. , … Gilissen, C. (2015). Post‐zygotic point mutations are an underrecognized source of de novo genomic variation. The American Journal of Human Genetics, 97(1), 67–74. 10.1016/j.ajhg.2015.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acuna‐Hidalgo, R. , Veltman, J. A. , & Hoischen, A. (2016). New insights into the generation and role of de novo mutations in health and disease. Genome Biology, 17, 241 10.1186/s13059-016-1110-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besenbacher, S. , Liu, S. , Izarzugaza, J. M. , Grove, J. , Belling, K. , Bork‐Jensen, J. , … Rubio‐García, A. (2015). Novel variation and de novo mutation rates in population‐wide de novo assembled Danish trios. Nature Communications, 6, 5969 10.1038/ncomms6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker, L. G. , & Spinner, N. B. (2013). A genomic view of mosaicism and human disease. Nature Reviews Genetics, 14, 307 10.1038/nrg3424 [DOI] [PubMed] [Google Scholar]
- Campbell, I. M. , Shaw, C. A. , Stankiewicz, P. , & Lupski, J. R. (2015). Somatic mosaicism: Implications for disease and transmission genetics. Trends in Genetics, 31, 382–392. 10.1016/j.tig.2015.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, I. M. , Stewart, J. R. , James, R. A. , Lupski, J. R. , Stankiewicz, P. , Olofsson, P. , & Shaw, C. A. (2014). Parent of origin, mosaicism, and recurrence risk: Probabilistic modeling explains the broken symmetry of transmission genetics. The American Journal of Human Genetics, 95, 345–359. 10.1016/j.ajhg.2014.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, K. , & Gordenin, D. A. (2015). Clusters of multiple mutations: Incidence and molecular mechanisms. Annual Review of Genetics, 49, 243–267. 10.1146/annurev-genet-112414-054714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. L. , Rappailles, A. , Duquenne, L. , Huvet, M. , Guilbaud, G. , Farinelli, L. , … Thermes, C. (2010). Impact of replication timing on non‐CpG and CpG substitution rates in mammalian genomes. Genome Research, 20, 447–457. 10.1101/gr.098947.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. K. , Yoon, S. R. , Calabrese, P. , & Arnheim, N. (2012). Positive selection for new disease mutations in the human germline: Evidence from the heritable cancer syndrome multiple endocrine neoplasia type 2B. PLoS Genetics, 8, e1002420 10.1371/journal.pgen.1002420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal, G. M. , Ergüner, B. , Sağıroğlu, M. S. , Yüksel, B. , Onat, O. E. , Alkan, C. , & Özçelik, T. (2014). Early postzygotic mutations contribute to de novo variation in a healthy monozygotic twin pair. Journal of Medical Genetics, 51, 455–459. [DOI] [PubMed] [Google Scholar]
- Francioli, L. C. , Polak, P. P. , Koren, A. , Menelaou, A. , Chun, S. , Renkens, I. , … Slagboom, P. E. (2015). Genome‐wide patterns and properties of de novo mutations in humans. Nature Genetics, 47, 822 10.1038/ng.3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilissen, C. , Hehir‐Kwa, J. Y. , Thung, D. T. , van de Vorst, M. , van Bon, B. W. , Willemsen, M. H. , … Leach, R. (2014). Genome sequencing identifies major causes of severe intellectual disability. Nature, 511, 344 10.1038/nature13394 [DOI] [PubMed] [Google Scholar]
- Goldmann, J. M. , Wong, W. S. , Pinelli, M. , Farrah, T. , Bodian, D. , Stittrich, A. B. , … Vockley, J. G. (2016). Parent‐of‐origin‐specific signatures of de novo mutations. Nature Genetics, 48, 935 10.1038/ng.3597 [DOI] [PubMed] [Google Scholar]
- Goriely, A. , Hansen, R. M. , Taylor, I. B. , Olesen, I. A. , Jacobsen, G. K. , McGowan, S. J. , … Wilkie, A. O. (2009). Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nature Genetics, 41, 1247 10.1038/ng.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriely, A. , McVean, G. A. , Röjmyr, M. , Ingemarsson, B. , & Wilkie, A. O. (2003). Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science, 301, 643–646. 10.1126/science.1085710 [DOI] [PubMed] [Google Scholar]
- Goriely, A. , & Wilkie, A. O. (2010). Missing heritability: Paternal age effect mutations and selfish spermatogonia. Nature Reviews Genetics, 11, 589 10.1038/nrg2809-c1 [DOI] [PubMed] [Google Scholar]
- Kong, A. , Frigge, M. L. , Masson, G. , Besenbacher, S. , Sulem, P. , Magnusson, G. , … Wong, W. S. (2012). Rate of de novo mutations and the importance of father's age to disease risk. Nature, 488, 471 10.1038/nature11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren, A. , Polak, P. , Nemesh, J. , Michaelson, J. J. , Sebat, J. , Sunyaev, S. R. , & McCarroll, S. A. (2012). Differential relationship of DNA replication timing to different forms of human mutation and variation. The American Journal of Human Genetics, 91, 1033–1040. 10.1016/j.ajhg.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korona, D. A. , LeCompte, K. G. , & Pursell, Z. F. (2010). The high fidelity and unique error signature of human DNA polymerase ε. Nucleic Acids Research, 39, 1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. (2010). Rate, molecular spectrum, and consequences of human mutation. Proceedings of the National Academy of Sciences of the United States of America, 107, 961–968. 10.1073/pnas.0912629107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, G. J. , McGowan, S. J. , Giannoulatou, E. , Verrill, C. , Goriely, A. , & Wilkie, A. O. (2016). Visualizing the origins of selfish de novo mutations in individual seminiferous tubules of human testes. Proceedings of the National Academy of Sciences of the United States of America, 113, 2454–2459. 10.1073/pnas.1521325113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelson, J. J. , Shi, Y. , Gujral, M. , Zheng, H. , Malhotra, D. , Jin, X. , … Wu, W. (2012). Whole‐genome sequencing in autism identifies hot spots for de novo germline mutation. Cell, 151, 1431–1442. 10.1016/j.cell.2012.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari, R. , Wuster, A. , Lindsay, S. J. , Hardwick, R. J. , Alexandrov, L. B. , Al Turki, S. , … Stratton, M. R. (2016). Timing, rates and spectra of human germline mutation. Nature Genetics, 48, 126 10.1038/ng.3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach, J. C. , Glusman, G. , Smit, A. F. , Huff, C. D. , Hubley, R. , Shannon, P. T. , … Shendure, J. (2010). Analysis of genetic inheritance in a family quartet by whole‐genome sequencing. Science, 328, 636–639. 10.1126/science.1186802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt, M. W. , Matsumoto, Y. , & Loeb, L. A. (2009). High fidelity and lesion bypass capability of human DNA polymerase δ. Biochimie, 91, 1163–1172. 10.1016/j.biochi.2009.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségurel, L. , Wyman, M. J. , & Przeworski, M. (2014). Determinants of mutation rate variation in the human germline. Annual Review of Genomics and Human Genetics, 15, 47–70. 10.1146/annurev-genom-031714-125740 [DOI] [PubMed] [Google Scholar]
- Stamatoyannopoulos, J. A. , Adzhubei, I. , Thurman, R. E. , Kryukov, G. V. , Mirkin, S. M. , & Sunyaev, S. R. (2009). Human mutation rate associated with DNA replication timing. Nature Genetics, 41, 393 10.1038/ng.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, S. R. , Choi, S. K. , Eboreime, J. , Gelb, B. D. , Calabrese, P. , & Arnheim, N. (2013). Age‐dependent germline mosaicism of the most common noonan syndrome mutation shows the signature of germline selection. The American Journal of Human Genetics, 92, 917–926. [DOI] [PMC free article] [PubMed] [Google Scholar]