

Abstract
In the last decade, cancer therapies have increasingly taken the form of combination treatments in which biologic agents play a crucial role. In breast cancer, the treatment strategy is adjusted to intrinsic subtypes such as human epidermal growth factor receptor-2(HER2)-positive. With the introduction of trastuzumab, a monoclonal antibody against HER2, survival has significantly improved in early and metastatic breast cancer. Trastuzumab's patent has now expired and biosimilars are moving into the market. Several clinical trials have led to the approval of 5 different biosimilar trastuzumabs. Results proved similarity between the proposed biosimilar and the reference product without significant differences in efficacy and safety, although follow-up has been short. However, the shorter drug development process with its goal of showing similarity rather than patient benefit uses surrogate endpoints such as pathologic complete response and overall response rate, not survival endpoints in terms of the ‘gold standarD' in evaluating cancer therapies. The aim of this article is to give insight into how to plan and perform a clinical trial to prove equivalence between a biosimilar trastuzumab and its reference product and to elucidate the setup and outcome of published clinical trials.
Key Words: Biosimilars, Trastuzumab, Herceptin, HER2-directed treatment
Introduction
The goal of the development process of biosimilars is to demonstrate similarity to the reference product (RP) rather than patient benefit per se, as this was established for the RP. The development process is a stepwise approach that begins with a comparative analytical characterization and a comprehensive functional and preclinical assessment, including the mode of action (MOA). The next step includes phase I studies to prove similarity with regard to clinical pharmacokinetics (PK) and pharmacodynamics (PD) in comparison with the RP. These studies are recommended to be conducted in healthy subjects. The extensive testing for PK and PD similarity leads to a lack of phase II studies. Instead of additional phase II studies, at least 1 comparative clinical phase III study has to be conducted to confirm equivalence for efficacy, safety, and risk of immunogenicity and to demonstrate no clinically meaningful differences. The clinical phase III study is recommended to be done in the most homogenous and sensitive patient population to sufficiently detect potential clinical differences between the biosimilar and the RP. If high similarity is shown in all steps (totality of evidence), the biosimilar product can be submitted for approval. In Europe, the European Medicines Agency (EMA) and in the U.S. the Federal Drug Administration (FDA) are responsible for approval [1,2,3,4].
Clinical Development
Study Design
All PK/PD/MOA studies are recommended to be conducted in healthy subjects to ensure a homogenous and immunocompetent population and enable a sensitive comparison between the biosimilar and the RP [5]. This is highly important as the study results provide the scientific basis and justify extrapolation from the tested indication to other indications that were not evaluated in a prospective clinical trial (Thill M, Thatcher N, Hanes V, Lyman G: Biosimilars. What the oncologist should know. Future Oncol 2018, accepted). Therefore, the phase III study has to be carefully planned in patients that are sensitive enough to discover clinically meaningful differences between both products [1,4,6].
The goal of the prospective head-to-head comparison is to show similar efficacy and a similar safety profile, not clinical efficacy per se. For this reason, the equivalence study design is the most recommended (fig. 1). This design includes prespecified equivalence margins, an upper and a lower margin, to demonstrate that the efficacy of the biosimilar lies within these margins [1,4]. The equivalence margins should be adequate and scientifically justified. Generally, 90% confidence intervals (CIs) for equivalence studies are recommended [7,8]. The efficacy endpoint could be that of clinical benefit or, alternatively, a meaningful surrogate for efficacy. Ideally, the safety, including immunogenicity, of the biosimilar should be assessed in the same study as its efficacy.
Fig. 1.
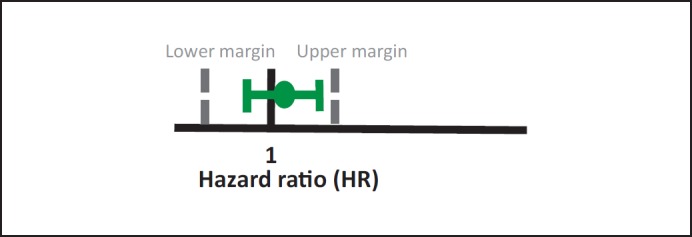
Equivalence design.
Study Population
The regulatory guidelines recommend, as mentioned above, the most sensitive patient population to detect potential and clinically meaningful differences [1]. In the case of trastuzumab, 2 different populations are possible: early breast cancer (eBC) or metastatic breast cancer (mBC). The clinical studies that have so far evaluated trastuzumab biosimilars had been conducted in these 2 settings, as neoadjuvant or metastatic treatment. Both settings have advantages and disadvantages, for mBC with longer treatment duration but less data on long-term efficacy and for eBC with less safety data [9]. However, a metastatic and pretreated population with pretreatment-associated changes in the cancer cell clone may be not as sensitive as the treatment-naive neoadjuvant population. It might play a role to trust in extrapolation, whether data from mBC are extrapolated to eBC or, better still, vise versa.
Efficacy Endpoints
The primary endpoints of a biosimilar clinical prospective trial are chosen to detect clinically meaningful differences between the biosimilar and its RP. However, the endpoints that are recommended in the guidelines [1,3,10] are different from those used for the approval of the RP [1]. Overall survival (OS) has been considered to be the ‘gold standarD' when cancer therapies are evaluated; however, as mentioned above, the key message in the development process of a biosimilar is to show similarity and not patient benefit. Therefore, no survival endpoints such as event-free survival (EFS), disease-free survival, progression-free survival (PFS), and OS, the preferred endpoints to prove efficacy in cancer, are recommended to be used (table 1). These endpoints may not be feasible due to the long follow-up needed and not sensitive enough to show differences between the biosimilar and the RP. Instead, surrogate endpoints are preferable, such as pathologic complete response (pCR) and overall response rate (ORR). Since its value was proven in various meta-analyses [11,12] in human epidermal growth factor receptor-2 (HER2)-positive breast cancer, pCR can be recommended as a reliable endpoint, as can ORR in first-line mBC.
Table 1.
Differences in clinical trials for biosimilars and reference product
| Biosimilar | Reference product | |
|---|---|---|
| Patient population Clinical design Study endpoints |
sensitive and homogeneous comparative vs. innovator (equivalence studies) sensitive clinically validated pharmacodynamic markers; overall response rate, pathologic complete response |
any superiority vs. standard of care clinical outcomes data (overall/progression-free survival) or accepted/established surrogates |
| Safety Immunogenicity (tested in most sensitive population) |
similar safety profile to innovator similar immunogenicity profile to innovator |
acceptable risk/benefit profile vs. standard of care acceptable risk/benefit profile vs. standard of care |
| Extrapolation | possible if justified | not allowed |
Equivalence Margins
Jackisch et al. [13] presented in an evaluation of the sensitivity of endpoints for eBC and mBC in similarity studies of trastuzumab RP and biosimilars that the shorter-term endpoint of ORR for the measurement of equivalence led to a substantial difference in long-term PFS despite prior findings suggesting ORR being a surrogate for PFS in mBC. They found that at an equivalence margin of 10% for ORR was associated with a difference in PFS of 3.2 months. For an equivalence margin of 15%, a difference of 4.4 months could be ascertained. For a stricter margin of 5%, the number of patients had to be increased from 924 (10% margin) to 3,742. Regarding total pCR (tpCR; no residual tumor in breast and lymph node, residual in situ carcinoma allowed) as a surrogate endpoint for EFS, they calculated for a 10% margin a difference of 3.8% and for a 15% margin a difference of 6.8% in 3-year EFS. To summarize, the lower the lower margin, the higher the potential loss in long-term efficacy. The authors concluded that tpCR is a more sensitive endpoint in eBC than ORR in mBC in HER2-positive patients in order to establish similarity between a trastuzumab biosimilar and its RP. The use of tpCR was associated with a lower potential loss in long-term efficacy than ORR in HER2-positive mBC patients. Therefore, the neoadjuvant setting is the preferred setting for the evaluation of clinical similarity between a proposed trastuzumab biosimilar and the trastuzumab RP [13].
Clinical Trials for Biosimilar Trastuzumab
Efficacy Data
Several clinical trials have been already conducted and published to demonstrate similarity between a biosimilar trastuzumab and the RP. The results led to the approval of all of them in the EU or in the US. Table 2 summarizes the phase III trials of the already approved biosimilar trastuzumabs, including their results and their current drug status.
Table 2.
Phase III trials evaluating trastuzumab biosimilars
| Agent | Company | Phase | n | Indication | Endpoint | Study design | Trial status | Results | Drug status |
|---|---|---|---|---|---|---|---|---|---|
| CT-P6 (Herzuma®) | Celltrion Healthcare(Incheon, ROK)/ TevaPharmaceuticals(Petach Tikwa, Israel) | pooledI/IIB–III | 475 | first line mBC |
ORR | equivalence | completed | CT-P6 vs. trastuzumab RPa: ORR 57 vs. 62%; mTTP 11.07 vs. 12.52 months [14] | marketed in South Korea following approval in January 2014; submitted to FDA in July 2017 and rejected in April 2018; resubmitted to FDA in May 2018 |
| III | 549 | eBC (NAT) |
pCR | equivalence | completed | non-inferior pCR (ypt0/is ypN0); 46.8% (95% CI 40.4–53.2) vs. 50.4% (95% CI 44.1–56.7) (CT-P6 vs. trastuzumab RP); 95% CI of the estimated treatment outcome difference (−0.04 (95% CI −0.12 to 0.05)) was within the equivalence margin, RR 0.92 [15] | |||
| PF-05280014 (Trazimera®) | Pfizer (New York, NY, USA)/Hospira (Lake Forest, IL, USA) | III | 707 | mBC | ORR | equivalence | completed | RR 0.940 (95% CI 0.842–1.049) over trastuzumab; 95% CI within the prespecified equivalence margin of 0.80–1.25; ORR 62.5% (95% CI 57.2–67.6%) for PF-05280014 vs. 66.5% (95% CI 61.3–71.4%) for trastuzumab RP; | phase I study completed; phase III study ongoing, expected to be completed March 2018; positive phase III results reported in November 2016 and September 2017; submitted to EMA and FDA for approval in September 2017; rejected by FDA in April 2018; approved by EMA in June 2018 |
| PFS (median 12.16 months for PF-05280014 vs. 12.06 months for trastuzumab; 1-year rate 54 vs. 51%) or OS (median: not reached in either group; 1-year rate 89.31 vs. 87.36%) [16] | |||||||||
| III | 226 | eBC (NAT) |
PK endpoints |
non-inferiority | completed | PF-05280014 vs. trastuzumab RP: non-inferior pCR 47 vs. 50%; lower limit of 95% CI (−8.02, 6.49%) for the stratified difference between groups was above the non-inferiority margin (−12.5%) (NCT02187744) [17] | |||
| ABP980 (Kanjinti®) | Amgen (Thousand Oaks, CA, USA) | III | 827 | eBC (NAT) |
pCR | equivalence | completed | non-inferior pCR 48% (95% CI 43–53) for ABP 980 41%, (95% CI 35–46) for trastuzumab RP (RD 7.3, 90% CI 1.2–13.4; RR 1.188, 90% CI 1.033–1.366); central assessed pCR 48 vs. 42% (RD 5.8, 90% CI −0.5 to 12.0 and RR 1.142, 90% CI 0.993–1.312) [18] | approved by EMA in March 2018; submitted to FDA for approval in July 2017; rejected by FDA in June 2018 |
| SB3 (Ontruzant®, EU; Samfenet®, R.O.K.) | Samsung Bioepis (Biogen/Samsung) (Incheon, ROK)/Daewoong Pharmaceuticals (Seoul, ROK)/Merck (MSD)(Kenilworth, NJ, USA) | III | 875 | eBC | pCR | equivalence | completed | SB3 vs. trastuzumab RP: equivalent bpCR 51.7 and 42% with SB3 and trastuzumab RP; adjusted ratio of bpCR: 1.259 (95% CI 1.085–1.460), within the predefined equivalence margins; adjusted difference: 10.70% (95% CI 4.13–17.26%), with the lower limit contained within and the upper limit outside the equivalence margin; tpCR: 45.8 and 35.8% | approved by EMA in November 2017; approved by Korea's MFDS in November 2017; submitted to FDA for approval in December 2017 |
| ORR 96.3% for SB3 vs. 91.2% for trastuzumab RP, adjusted ratio 1.259 (95% CI 1.085–1.460) [19]; | |||||||||
| EFS: comparable between groups, HR (SB3/trastuzumab RP) 0.94 (95% CI 0.59–1.51); 12-month EFS 93.7% for SB3 vs. 93.4% for trastuzumab RP; final analysis after 438 days: EFS rate for SB3 (92.2%) vs. trastuzumab RP (91.6%) [20] | |||||||||
| MYL-1410 (Ogivri®) | Mylan (Canonsburg, PA, USA)/Biocon (Bangalore, India) | III | 458 | mBC | ORR | equivalence | completed | MYL-1410 vs. trastuzumab RP: non-inferior ORR: 69.6 vs. 64% (HR 1.09; 95% CI 0.95–1.24); ORR difference (5.53; 95% CI −3.08 to 14.04) 48-week PFS (44.3 vs 44.7%; −0.4%; 95% CI −9.4 to 8.7%), OS (89.1 vs. 85.1%; 4.0%; 95% CI −2.1 to 10.3%; p = 0.13) [21] | received approval from FDA December 2017; submitted to EMA for approval in November 2016; withdrawn from EMA August 2017; resubmitted to EMA for approval in December 2017; |
| BCD-022 (HERtiCAD®) | Biocad (Saint Petersburg, Russia) | III | 126 | mBC | ORR | non-inferiority | completed | BCD-022 vs. trastuzumab RP: ORR 53.6 vs. 53.7%; progression rate not different: 21.45 vs. 20.4%) | received approval from Russian regulatory body January 2016 |
aGenentech, San Francisco, CA, USA.
RP, reference product; PK, pharmacokinetics; RD, risk difference; RR, risk ratio; pCR, pathologic complete response; bpCR, breast pathologic complete response; tpCR, total pathologic complete response; eBC, early breast cancer; mBC, metastatic breast cancer; EFS, event-free survival; HR, hazard ratio; CI, confidence interval; ORR, overall response rate; OS, overall survival; mTTP = median time to progression; ROK, Republic of Korea; EU, European Union; FDA, Federal Drug Administration; EMA, European Medicines Agency; MFDS, Ministry of Food and Drug Safety; NAT, neoadjuvant treatment; PFS, progression-free-survival.
CT-P6 (Herzuma®, Celltrion Healthcare, Incheon, ROK/Teva Pharmaceuticals, Petach Tikwa, Israel) and PF-05280014 (Trazimera®, Pfizer, New York, NY, USA/Hospira, Lake Forest, IL, USA) were evaluated in both mBC and eBC [14,15,16,17], ABP 980 (Kanjinti®, Amgen, Thousand Oaks, CA, USA) [18] and SB3 (Ontruzant®, Samsung Bioepis, Incheon, ROK/Daewoong Pharmaceuticals, Seoul, ROK/Merck (MSD), Kenilworth, NJ, USA) [19,20] only in eBC, and MYL-1410 (Ogivri®, Biocon, Bangalore, India/ Mylan, Canonsburg, PA, USA) [21] and BCD-022 (HERtiCAD®, Biocad, Saint Petersburg, Russia) only in mBC (NCT01764022). All biosimilars were evaluated with the recommended equivalence trial design, except for BCD-022 which was evaluated in a non-inferiority trial. The primary endpoint was pCR for eBC and ORR for mBC.
In addition to the proposed trastuzumab biosimilar or the RP, all trials used anthracycline/taxane-based chemotherapy in the neoadjuvant setting and a taxane-based chemotherapy in the metastatic setting and demonstrated no significant differences between the biosimilar trastuzumab and the trastuzumab RP.
In the Lilac trial (ABP 980 vs. trastuzumab RP) which enrolled 827 patients [18], 725 patients were randomized to receive ABP 980 or the trastuzumab RP. The primary endpoint was pCR which was achieved in 172 patients. 2 sensitivity analyses for pCR were done, 1 locally assessed, 1 centrally. In the local analysis, the lower margin of the 90% CIs for risk ratio and risk difference showed non-inferiority, but the upper margin exceeded the predefined equivalence margins, meaning that non-superiority was inconclusive. In the central laboratory evaluation of tumor samples, ABP 980 and the trastuzumab RP were within the predefined equivalence margins, indicating similar efficacy (table 2) [18].
SB3 was also evaluated in the neoadjuvant setting. The primary objective was to compare the breast pCR (bpCR) rate between SB3 and the trastuzumab RP. Equivalence was declared if the 95% CI of the ratio was within 0.785-1.546 or the 95% CI of the difference was within the lower and upper equivalence margin of ±13%. 875 patients were included in the trial. 764 patients (SB3, n = 380; trastuzumab RP, n = 384) completed the study. bpCR was similar in both arms, and tpCR was also equivalent with 45.8% in the SB3 group and 35.8% for the trastuzumab RP [19]. In the final efficacy analysis (after 438 days), both the EFS and the OS rate were similar between the 2 groups [20].
The Heritage study enrolled 458 metastatic patients. The primary endpoint, ORR, was equivalent between both arms. The ORR difference was within the prespecified equivalence margins of ±15%. The survival analysis after 48 months showed again equivalent results for PFS. OS was not statistically different [21].
PF-05280014 was evaluated in 707 metastatic patients and showed results in line with the results of the Heritage study. Regarding ORR, equivalent results could be observed in the biosimilar group versus the trastuzumab RP group with no differences in PFS or OS [16].
CT-P6 was evaluated in both the metastatic and the neoadjuvant setting. Similar results could be detected in the metastatic setting with 475 enrolled patients [14,15]. In the study conducted in the neoadjuvant setting, 549 patients were randomized between CT-P6 and the trastuzumab RP. No significant differences were shown regarding the primary endpoint pCR (ypt0/is ypN0) for CT-P6 versus the trastuzumab RP [15].
BCD-022 was only evaluated in a non-inferiority design and had a prespecified lower margin of −20% (NCT01764022). Moreover, the study is still not available as a full paper, therefore data should be viewed with caution. Concerning the other biosimilar trastuzumabs, different equivalence margins were used in the studies.
MYL-1410 and CT-P6 were evaluated with equivalence margins of ±15% [15,21], whereas ABP-980 and SB3 were tested with margins of ±13% [18,20]. Referring to the work of Jackisch et al. [13], it is theoretically possible, albeit remaining unclear, that a 2% difference in prespecification of the equivalence margins will lead to a potential loss of long-term efficacy in terms of EFS or PFS.
Safety Data
In all studies listed in table 2, the safety analyses demonstrated no significant differences between the biosimilar product and the RP. In the LILAC trial [18], 15% (n = 54) in the Kanjinti (ABP 980) group and 14% (n = 51) in the trastuzumab RP group demonstrated adverse events (AE) ≥ grade 3 during the neoadjuvant phase. Of the ≥ grade 3 AE, neutropenia was most frequently observed at 6% (n = 21) in both groups. In the adjuvant phase, AE ≥ grade 3 occurred in 9% (n = 30) of patients continuing ABP 980 and in 6% (n = 11) continuing trastuzumab. The LILAC trial is the only trial including a switch from the trastuzumab RP to ABP-980. AE ≥ grade 3 occurred in 8% (n = 13) of the switch population. The most frequent AE ≥ grade 3 were infections and infestations, neutropenia, and infusion reactions with 1% each. Regarding cardiac safety, no significant differences were reported [18].
The safety analysis of the neoadjuvant phase evaluating the biosimilar trastuzumab Ontruzant (SB3) versus trastuzumab RP showed an overall AE rate (≥1 AE) of 96.6 versus 95.2%; 10.5 and 10.7% had a serious adverse event (SAE) [19]. In the final safety and immunogenicity analyses, the incidence of treatment-emergent adverse events (TEAE) was comparable between the 2 groups (SB3, 97.5% and trastuzumab RP, 96.1%) during the whole study period. TEAE ≥ grade 3 occurred in 27 of 437 (6.2%) patients in the SB3 arm and in 36 (8.2%) of the trastuzumab RP arm. Regarding immunogenicity, the overall incidence of antidrug antibodies (ADAs) and neutralizing antibodies (NAbs) was low in both treatment groups. NAbs occurred in 0.5% (n = 2) and ADAs 0.7% (n = 3). The incidence of a decrease in left ventricular ejection fraction (LVEF) of <50% was comparable (SB3: 3.7%, n = 16 and trastuzumab RP: 2.8%, n = 12). However, the analysis was done after a short follow-up of 437 days in the SB3 group and 438 days in the trastuzumab RP arm [20].
In the HERITAGE study, the overall TEAE incidence (at least 1 TEAE) was 96.8% (n = 239) in the biosimilar group and 94.7% (n = 233) in the trastuzumab RP group [21]. The majority of events were mild or moderate in both groups. 7 (2.8%) patients in each group reported an AE leading to discontinuation of treatment. Neutropenia, leukopenia, and anemia had an incidence of 57.5% (n = 142), 17% (n = 42), and 16.2% (n = 40), respectively, in the biosimilar group and 53.3% (n = 131), 20.7% (n = 51), and 16.3% (n = 40), respectively, in the trastuzumab RP group. AE ≥ grade 3 were reported for 312 (63.3%) of all participants, with neutropenia in 44.8% (n = 221) and leukopenia in 14% (n = 69) most frequently reported. The most frequent non-hematologic TEAE included peripheral neuropathy for the biosimilar versus the trastuzumab RP group in 23.1% (n = 57) and 24.8% (n = 61), diarrhea in 20.6% (n = 51) and 20.7% (n = 51), asthenia in 21.9% (n = 54) and 16.3% (n = 40), and nausea in 19.8% (n = 49) and 13.8% (n = 34) [21].
Regarding SAE, no significant difference was observed between the biosimilar and the RP trastuzumab group (38.1%, n = 94 vs. 36.2%, n = 89). Regarding LVEF in the biosimilar group (median 64%; range 51-82%) and in the trastuzumab RP group (median 63%; range 51-84%), no appreciably change was noted at week 24 (proposed biosimilar group: median −1%; range −13 to 21%; trastuzumab RP group: median −1%; range −19 to 13%). Finally, 5.9% (n = 14) in the biosimilar group and 8.9% (n = 21) in the trastuzumab RP group had ADAs prior to study treatment [21].
In the safety analysis of Herzuma (CT-P6) versus the trastuzumab RP, serious TEAE occurred in 7% (n = 19) versus 8% (n = 22), respectively. Frequent SAE were febrile neutropenia in 1% in each arm. TEAE ≥ grade 3 occurred in 6% (n = 17) in the CT-P6 group versus 8% (n = 23) in the trastuzumab RP group; the most frequently reported AE was neutropenia in 4% (n = 10) versus 5% (n = 14) [15].
Finally, the safety and immunogenicity analysis between Trazimera (PF-05280014) and the trastuzumab RP yielded similar results between the treatment groups; however, follow-up was quite short with 378 days post-randomization [16].
Conclusion
All trials that have led to an approval of a biosimilar trastuzumab by the EMA or FDA observed no significant differences compared to the trastuzumab RP regarding hematologic and non-hematologic safety, cardiotoxicity, and immunogenicity. The use of these biosimilar trastuzumabs is as safe as the use of the trastuzumab RP; however, the follow-up of all studies has been quite short. Due to the use of the surrogate endpoints pCR for eBC and ORR for mBC, further follow-up is warranted. A biosimilar trastuzumab registry would be the best way of collecting follow-up data on efficacy and safety; however, no such registry exists at the moment. A registry would be even more important with regard to an optimal HER2-directed treatment that consists of the combination of trastuzumab and pertuzumab. Certain trials are currently ongoing such as the neoadjuvant GeparX trial by the German Breast Group, but results are pending [22].
The use of biosimilar trastuzumab is rapidly increasing and will play an important role in breast cancer treatment. Clinicians have to observe correct pharmacovigilance and precise documentation and should avoid extensive multiple switching between the different biosmilar trastuzumabs in order to obtain clear and reproducible safety results.
Disclosure Statement
M.T. received consulting or advisory honoraria from: Amgen, AstraZeneca, Celgene, Daiichi Sankyo, Eisai, Genomic Health, Hexal, Lilly, MSD, Myriad, Neodynamics, Norgine, Novartis, Pfizer, pfm Medical, Roche, RTI Surgical, SurgicEye, Teva; speaker honoraria from: Amgen, AstraZeneca, Celgene, Eisai, Genomic Health, Hexal, Lilly, Myriad, Neodynamics, Novartis, Pfizer, pfm Medical, Roche, RTI Surgical, SurgicEye, Teva; manuscript support from: Amgen, Roche, Celgene; and trial funding from: Genomic Health
References
- 1.European Medicines Agency Guideline on similar biological medicinal products containing monoclonal antibodies - non-clinical and clinical issues (EMA/CHMP/BMWP/ 403543/2010; adoption by CHMP 30 May 2012) www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. [Google Scholar]
- 2.European Medicines Agency Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substances: non-clinical and clinical issues (EMEA/CHMP/BMWP/42832/2005). Effective date 1 June 2006 (revised 3 June 2013) www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003920.pdf. [Google Scholar]
- 3.Food and., Drug Administration Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for industry. FDA2016. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf; accessed December 2018. [Google Scholar]
- 4.Food and., Drug Administration Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. FDA2015. www.fda.gov/downloads/drugs/guidances/ucm291128.pdf; accessed December 2018. [Google Scholar]
- 5.European Medicines Agency Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues (EMEA/CHMP/BMWP/42832/2005 Rev 1), 18 December 2014. www.ema.europa.eu/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-biotechnology-derived-proteins-active_en-2.pdf; accessed December 2018. [Google Scholar]
- 6.Jacobs I, Ewesuedo R, Lula S, Zacharchuk C. Biosimilars for the treatment of cancer: a systematic review of published evidence. BioDrugs. 2017;31:1–36. doi: 10.1007/s40259-016-0207-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ICH Harmonised Tripartite Guideline Statistical principles for clinical trials E9. 5 February 1997. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf; accessed December 2018. [PubMed] [Google Scholar]
- 8.ICH Harmonised Tripartite Guideline Choice of control group and related issues in clinical trials E10. 20 July 2000. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E10/Step4/E10_Guideline.pdf; accessed December 2018. [Google Scholar]
- 9.Nixon NA, Hannouf MB, Verma S. The evolution of biosimilars in oncologywith a focus on trastuzumab. Curr Oncol. 2018;25((suppl 1)):S171–S179. doi: 10.3747/co.25.3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thill M. New frontiers in oncology: biosimilar monoclonal antibodies for the treatment of breast cancer. Expert Rev Anticancer Ther. 2015;15:331–338. doi: 10.1586/14737140.2015.993318. [DOI] [PubMed] [Google Scholar]
- 11.Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384:164–172. doi: 10.1016/S0140-6736(13)62422-8. [DOI] [PubMed] [Google Scholar]
- 12.Spring LM, Fell G, Arfe A, et al. Pathological complete response after neoadjuvant chemotherapy and impact on breast cancer recurrence and mortalitystratified by breast cancer subtypes and adjuvant chemotherapy usage: individual patient-level meta-analyses of over 27,000 patients. SABCS. 2018:GS2–G03. [Google Scholar]
- 13.Jackisch C, Scappaticci FA, Heinzmann D, et al. Neoadjuvant breast cancer treatment as a sensitive setting for trastuzumab biosimilar development and extrapolation. Future Oncol. 2015;11:61–71. doi: 10.2217/fon.14.187. [DOI] [PubMed] [Google Scholar]
- 14.Im YH, Odarchenko P, Grecea D, et al. Double-blind, randomizedparallel groupphase III study to demonstrate equivalent efficacy and comparable safety of CT-P6 and trastuzumab both in combination with paclitaxel in patients with metastatic breast cancer (mBC) as first-line treatment. J Clin Oncol. 2013;31:629. [Google Scholar]
- 15.Stebbing J, Baranau Y, Baryash V, et al. CT-P6 compared with reference trastuzumab for HER2-positive breast cancer: a randomiseddouble-blind, active-controlled, phase 3 equivalence trial. Lancet Oncol. 2017;18:917–928. doi: 10.1016/S1470-2045(17)30434-5. [DOI] [PubMed] [Google Scholar]
- 16.Pegram MD, Bondarenko I, Zorzetto MMC, et al. PF-05280014 (a trastuzumab biosimilar) plus paclitaxel compared with reference trastuzumab plus paclitaxel for HER2-positive metastatic breast cancer: a randomiseddouble-blind study. Br J Cancer. 2018 doi: 10.1038/s41416-018-0340-2. DOI: 10.1038/s41416-018-0340-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lammers PE, Dank M, Masetti R, et al. Neoadjuvant PF-05280014 (a potential trastuzumab biosimilar) versus trastuzumab for operable HER2+ breast cancer. Br J Cancer. 2018;119:266–273. doi: 10.1038/s41416-018-0147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Von Minckwitz G, Colleoni M, Kolberg HC, et al. Efficacy and safety of ABP 980 compared with reference trastuzumab in women with HER2-positive early breast cancer (LILAC study): a randomiseddouble-blind, phase 3 trial. Lancet Oncol. 2018;19:987–998. doi: 10.1016/S1470-2045(18)30241-9. [DOI] [PubMed] [Google Scholar]
- 19.Pivot X, Bondarenko I, Nowecki Z, et al. Phase IIIrandomized, double-blind study comparing the efficacysafety, and immunogenicity of SB3 (trastuzumab biosimilar) and reference trastuzumab in patients treated with neoadjuvant therapy for human epidermal growth factor receptor 2-positive early breast cancer. J Clin Oncol. 2018;36:968–974. doi: 10.1200/JCO.2017.74.0126. [DOI] [PubMed] [Google Scholar]
- 20.Pivot X, Bondarenko I, Nowecki Z, et al. A phase III study comparing SB3 (a proposed trastuzumab biosimilar) and trastuzumab reference product in HER2-positive early breast cancer treated with neoadjuvant-adjuvant treatment: final safetyimmunogenicity and survival results. Eur J Cancer. 2018;93:19–27. doi: 10.1016/j.ejca.2018.01.072. [DOI] [PubMed] [Google Scholar]
- 21.Rugo HS, Barve A, Waller CF, et al.;, Heritage Study Investigators Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)-positive metastatic breast cancer: a randomized clinical trial. JAMA. 2017;317:37–47. doi: 10.1001/jama.2016.18305. [DOI] [PubMed] [Google Scholar]
- 22.Kummel S, Wimberger P, von Minckwitz G, Nekljudova V, Denkert C, Just M. Investigating denosumab as an add-on neoadjuvant treatment for RANK/L-positive or RANK/L-negative primary breast cancer and two different nab-paclitaxel schedules: 2 × 2 factorial design (GeparX) - an interim safety analysis. J Clin Oncol. 2018;36 abstr 569. [Google Scholar]