

Abstract
Bronchopulmonary dysplasia (BPD), characterized by alveoli simplification and dysmorphic pulmonary microvasculature, is a chronic lung disease affecting prematurely born infants. Pulmonary hypertension (PH) is an important BPD feature associated with morbidity and mortality. In human BPD, inflammation leads to decreased fibroblast growth factor 10 (FGF10) expression but the impact on the vasculature is so far unknown. We used lungs from Fgf10+/− versus Fgf10+/+ pups to investigate the effect of Fgf10 deficiency on vascular development in normoxia (NOX) and hyperoxia (HOX, BPD mouse model). To assess the role of fibroblast growth factor receptor 2b (Fgfr2b) ligands independently of early developmentaldefects, we used an inducible double transgenic system in mice allowing inhibition of Fgfr2b ligands activity. Using vascular morphometry, we quantified the pathological changes. Finally, we evaluated changes in FGF10, surfactant protein C (SFTPC), platelet endothelial cell adhesion molecule (PECAM) and alpha-smooth muscle actin 2 (α-SMA) expression in human lung samples from patients suffering from BPD. In NOX, no major difference in the lung vasculature between Fgf10+/− and control pups was detected. In HOX, a greater loss of blood vessels in Fgf10+/− lungs is associated with an increase of poorly muscularized vessels. Fgfr2b ligands inhibition postnatally in HOX is sufficient to decrease the number of blood vessels while increasing the level of muscularization, suggesting a PH phenotype. BPD lungs exhibited decreased FGF10, SFTPC and PECAM but increased α-SMA. Fgf10 deficiency-associated vascular defects are enhanced in HOX and could represent an additional cause of morbidity in human patients with BPD.
Introduction
Fibroblast growth factor 10 (FGF10) protein deficiency has been reported in patients with bronchopulmonary dysplasia (BPD) (1). BPD is a chronic lung disease of prematurely born infants and remains a leading cause of morbidity and mortality. In humans, inflammation is known to increase risk for BPD (2,3). Supporting a functional link between inflammation and FGF10-messenger ribonucleic acid (mRNA) expression, inflammatory mediators activated by bacterial-derived lipopolysaccharides (LPS) (4) such as NF-κB, Sp1 and Sp3 were found to inhibit Fgf10 transcription in mouse lung explants (5,6). Indeed, the inhibition of Fgf10 expression is mediated by LPS receptors (toll-like receptors 2 and 4) activation (1). In addition to inflammation-induced FGF10 deficiency, congenital FGF10 expression deficiency [as well as deficiency in the FGF10 receptor, fibroblast growth factor receptor 2b (FGFR2b)] has been reported in humans suffering from aplasia of lacrimal and salivary glands (ALSG) and lacrimo-auriculo-dento-digital (LADD) syndromes (7–10). Patients with heterozygous FGF10 loss-of-function exhibit significant decrease in inspiratory vital capacity (IVC), forced expiratory volume 1 (FEV1) and FEV1/IVC ratio compared to non-carrier siblings and to predicted reference values (11). These data are consistent with chronic obstructive pulmonary disease. As both patients with BPD and ALSG or LADD syndrome are characterized by developmental abnormalities, we hypothesized that dysregulation of FGF10 expression contributes to the associated lung pathogenesis in these patients.
Fgf10 has been shown using animal models to be a key developmental regulator during lung development. This is illustrated by the fact that Fgf10 null pups die at birth due to lung agenesis (12). Fgf10 is localized in the mesenchyme of the developing lung from the onset of organogenesis and encodes a secreted protein, which acts in a paracrine fashion mainly through the receptor Fgfr2b, expressed in the lung epithelium (13–15). During the initial phases of the pseudoglandular stage of lung development [embryonic day (E) 9.5 through 16.5 in mice], Fgf10 expression adjacent to the nascent epithelial buds as well as its activity in vitro on isolated lung epithelium grown in matrigel suggested that Fgf10 plays a crucial role in controlling branching morphogenesis (16). We reported that congenital Fgf10 deficiency (mice heterozygous for Fgf10, Fgf10+/−) disrupts the formation of the alveolar lineage in a quantitative and qualitative manner leading to increased alveolar epithelial cell type I and decreased alveolar epithelial cell type II (AT2). Furthermore, surfactant protein B and C (Sftpb and Sftpc) levels were reduced in the mutants and such decreases are associated with the observed fatal hyperoxia (HOX)-induced lung injury in a mouse model of BPD (17).
During the pseudoglandular stage, Fgf10 also indirectly orchestrates the development of the adjacent mesenchyme, which contains the progenitors of multiple important lung cell types, including the endothelium, the vascular and airway smooth muscle cells, the alveolar myofibroblasts, the lipofibroblasts, the interstitial fibroblasts and the nerve cells (for a review, see (18,19)). Its impact on the mesenchyme is not surprising as Fgf10 signalling is embedded in an interactive signalling network comprising major pathways such as retinoic acid, sonic hedgehog, bone morphogenetic protein 4, transforming growth factor beta-1, Wnt, platelet-derived growth factor and vascular endothelial growth factor. In addition to a developmental role, Fgf10 is a major player in regeneration of the lung after injury. In bleomycin-induced lung fibrosis, Fgf10 overexpression is both protective and therapeutic. Fgf10 is also involved in the regeneration of the bronchial lung epithelium after naphthalene injury (20,21).
The evidence confirming the key role of Fgf10 in embryonic lung development is strong. However, less is known about the impact of constitutive Fgf10 deficiency on the formation and maintenance of the pulmonary vasculature and in the context of normoxia (NOX) and HOX-induced lung injury.
To demonstrate the effect of Fgf10 deficiency on postnatal pulmonary vasculature in normoxic condition, we used a constitutive heterozygous Fgf10+/− mouse line. Since oxygen toxicity is one of the major risk factors contributing to BPD, we used the HOX-induced BPD mouse model (85% oxygen from P0–P8) to investigate the impact of Fgf10 deficiency in hyperoxic condition. As 100% of the Fgf10+/− mutant pups are dead at P8 during HOX exposure (17), we chose P3 as a time point for further analysis of the lung vasculature. The analysis of lung vascular morphometry to identify changes in lung vascular structure consists of quantification of vessel count and extent of vessel muscularization. We also used gene arrays on Fgf10+/− versus Fgf10+/+ control littermate whole lungs exposed to NOX or HOX to identify transcriptomic changes potentially linked to vascular formation. We also performed immunofluorescence (IF) and reverse transcriptase-quantitative polymerase chain reaction on experimental and control lungs at different stages of development.
Finally, in human lung samples of patients suffering from BPD, we used immunohistochemistry (IHC) and IF to evaluate the changes in FGF10, SFTPC, platelet endothelial cell adhesion molecule (PECAM) and ACTA2 expressions. For the first time, we report the consequence of Fgf10 deficiency in mice on postnatal lung vasculature and during HOX-induced lung injury and suggest that defective FGF10 signalling in human patients with BPD is causative for some of the vascular defects.
Results
FGF10 and its receptor FGFR2 are expressed in lungs from patients with BPD
Using IHC, we found that FGF10 and its receptor FGFR2 are expressed in the lung parenchyma of patients with BPD. In particular, FGF10 and FGFR2 positive immunoreactivity are localized in relevant lung histopathological features of the disease, such as remodelled pulmonary vasculature (Fig. 1A) as well as in alveolar septa and interstitial fibrotic regions (Supplementary Material, Fig. S1A and C) and in distorted conducting airways (Supplementary Material, Fig. S1B and D).
Figure 1.
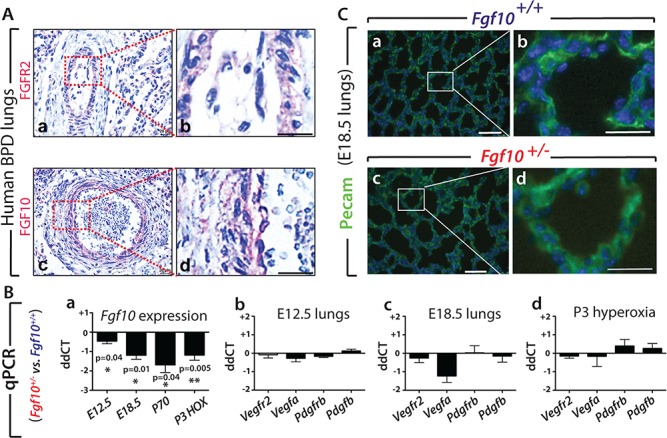
Expression of FGF10 and associated targets in human and mouse lungs. (A) FGFR2 receptor and its ligand FGF10 are expressed in lungs from patients with BPD (n = 4). (Aa) FGFR2 and (Ac) FGF10 expression in remodelled pulmonary vasculature. (Ab) and (Ad) respective high magnification. Scale bar: 20 μm. (B–C) Analysis of vascular smooth muscle cell and endothelial cell markers did not show changes in Fgf10+/− versus Fgf10+/+ control lungs at E12.5, E18.5 and P3 (HOX). (Ba) qPCR analysis showed significant decrease of Fgf10 expression in Fgf10+/− versus Fgf10+/+ lungs at E12.5, E18.5, P70 and P3 HOX, n = 4 each. (Bb–Bd) Comparative expression by qPCR analysis in Fgf10+/− versus Fgf10+/+ control lungs at (b) E12.5, (c) E18.5 and (d) P3 HOX of Vegfr2, Vegfa, Pdgfrb and Pdgfb. No significant difference is observed even though a trend towards a decrease in Vegfa is observed at E18.5. (C) IF for PECAM staining on E18.5 (a,b) Fgf10+/+ and (c,d) Fgf10+/− lungs (n = 4 each) showed slightly lower immunoreactivity in Fgf10+/− lungs. Scale bar: a, c, 100 μm; b, d, 30 μm.
Expression levels of vascular smooth muscle cell and endothelial cell markers are indistinguishable between Fgf10+/- versus control Fgf10+/+ lungs in NOX and HOX
The formation of the vascular system during development was first investigated by examining the expression levels of key vascular genes, previously reported to be regulated in the lungs of Fgf10 hypomorphic (Fgf10LacZ/−) embryos displaying one copy of the Fgf10LacZ hypomorphic allele and one copy of the deleted (Fgf10−) allele (22). We compared the expression of these genes in Fgf10+/− versus control lungs. The decrease in Fgf10 expression in experimental lungs was validated by qPCR at different time points (Fig. 1Ba). At all the stages considered [embryonic day (E) 12.5, E18.5, P70], Fgf10 was reduced but at different levels indicating potential compensatory mechanisms at work allowing to regulate Fgf10 transcription from the remaining Fgf10 wild-type allele. Decrease in Fgf10 expression in the P3 lungs between the two genotypes was also detected in the context of HOX exposure (Fig. 1Ba).
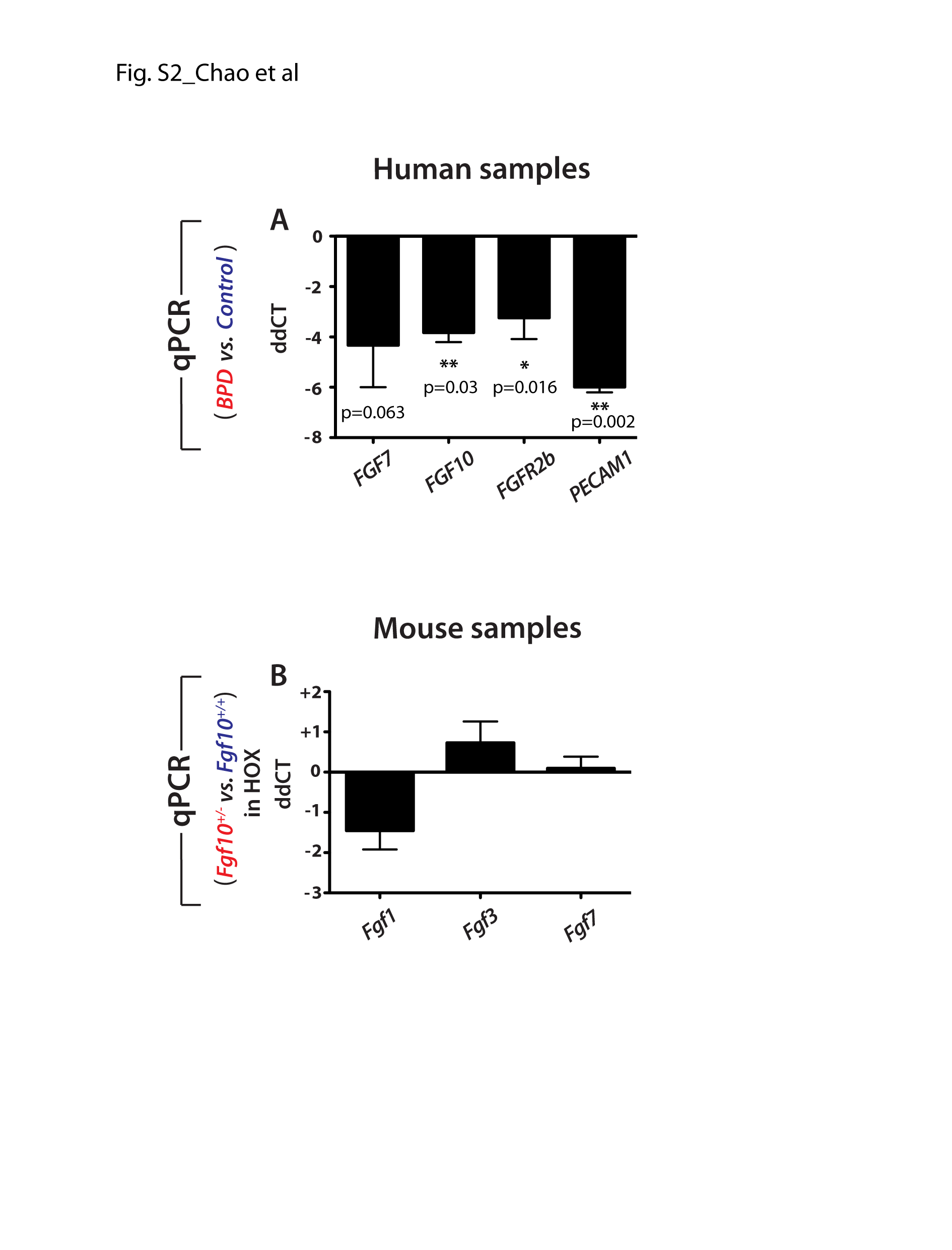
Next, we determined by qPCR in Fgf10+/− versus control lungs at the time points mentioned before (Fig. 1Bb–d) the expressions of Vegfr2, its associated ligand Vegfa, as well as the vascular smooth muscle receptor platelet derived growth factor receptor beta (Pdgfrb) and its associated ligand Pdgfb. For all these time points and conditions, we found that the expression of these genes was not severely affected by loss of one copy of the Fgf10 allele, even though a decrease was observed at E18.5 for Vegfa. IF in lungs at E18.5 for Pecam to label the endothelial cells indicated a strong and very defined signal in control lungs (Fig. 1Ca, b; n = 4) and an equally strong but more diffuse signal in mutant lung (Fig. 1Cc, d; n = 4). As the samples were processed under the same conditions, this difference could reflect potential endothelial defects in Fgf10+/− lungs. Interestingly, a significant decrease in PECAM1 mRNA expression was detected in BPD versus control lungs (Supplementary Material, Fig. S2A).
Morphometry analysis of mutant versus control lungs in NOX suggests only minor defects in the status of the vasculature upon reduced Fgf10 expression
To further examine the status of the vasculature in mutant versus control lungs in NOX, we quantified at P3 the number of vessels and extent of muscularization of these vessels. The analysis was based on detection of muscle tissue (α-SMA staining) surrounding the vessel endothelium (von Willebrand staining, vWF) (Fig. 2A). Vessels were subdivided into four groups based on their diameters (10–20 μm, 20–70 μm, 70–150 μm and >150 μm). The number of blood vessels in Fgf10+/− versus control lungs in NOX indicated no difference in total vessel count. However, a significant reduction was observed in the number of 70–150 μm vessels (P = 0.049) (Fig. 2B). No difference in muscularization was noted (Fig. 2Ca–d).
Figure 2.
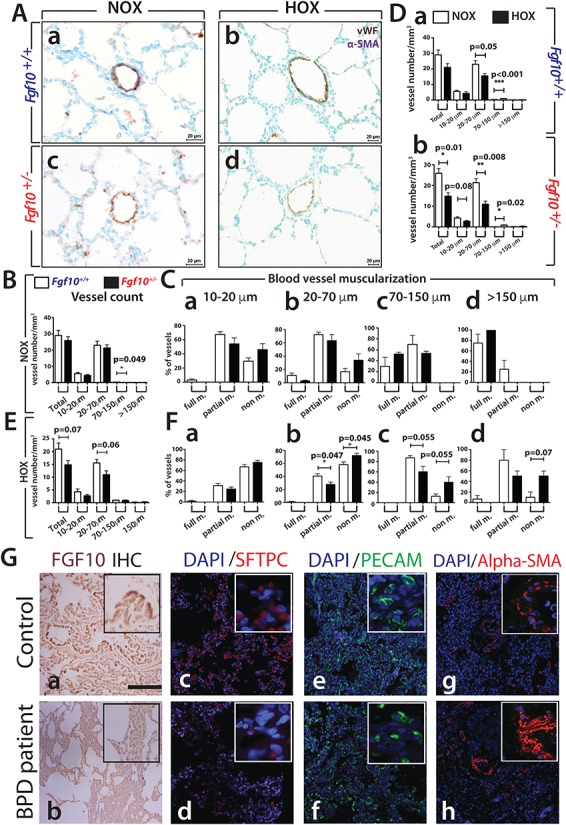
Analysis of the vascular defects in mouse and human lungs. (A–F) Analysis of the vascular defects in P3 Fgf10+/− versus Fgf10+/+ control lungs in HOX. (A) IHC for vWF and α-SMA of (a, b) Fgf10+/+ and (c, d) Fgf10+/− lungs between NOX (Fgf10+/+ n = 6, Fgf10+/− n = 3) and HOX (n = 4 each). (B) Total vessel count in NOX between P3 Fgf10+/+ and Fgf10+/− lungs. (C) Blood vessel muscularization in NOX between Fgf10+/+ and Fgf10+/− lungs for blood vessels at (a) 10–20 μm, (b) 20–70 μm, (c) 70–150 μm and (d) >150 μm. (D) Total vessel count between NOX and HOX in (a) Fgf10+/+ and (b) Fgf10+/− lungs. (E) Total vessel count in HOX between P3 Fgf10+/+ and Fgf10+/− lungs. (F) Blood vessel muscularization in HOX between Fgf10+/+ and Fgf10+/− control lungs for blood vessels at (a) 10–20 μm, (b) 20–70 μm, (c) 70–150 μm and (d) >150 μm. Scale: Aa–d, 20 μm. (G) Immunofluorescence for FGF10, SFTPC, PECAM and ACTA2 in lungs of BPD (n = 4) versus control non-BPD (n = 2) patients. (a, b) FGF10 expression, (c, d) mature-SFTPC expression, (e, f) PECAM expression and (g, h) ACTA2 expression. Scale bars A–F: 100 μm, insets in (A–F): 25 μm.
Decreased number of blood vessels and decreased muscularization is observed in Fgf10+/− versus control lungs exposed to HOX
Next, we compared the vascular response of mutant and control lungs to HOX. In both genotypes, we observed a drop in total blood vessel count upon HOX injury, the 20–70 μm blood vessels being the most affected (Fig. 2Da and b). Interestingly, in control lungs, HOX significantly increased the number of 70–150 μm blood vessels (Fig. 2Da, P < 0.001). In Fgf10+/– lungs, HOX significantly reduced the total blood vessel count (P = 0.01) (Fig. 2Db). As for the control lungs, we observed a decrease in the number of 20–70 μm blood vessels (P = 0.008) and an increase in the number of 70–150 μm blood vessels (P = 0.02).
The direct comparison of the Fgf10+/− versus control lungs in HOX (Fig. 2E) indicated decreased vessel number in mutant lungs (P = 0.07), which is mostly affecting the 20–70 μm blood vessels (P = 0.06). A significant loss of muscularization (decrease in partial muscularized but increase in non-muscularized vessels) was seen for the 20–70 μm blood vessel group (Fig. 2Fb) and reduced muscularization was observed for the 70–150 μm blood vessels (Fig. 2Fc). Interestingly, despite these defects, we failed to see any evidence of lung hemorrhage in Fgf10+/− lungs in HOX at P3 (Fig. 2A), suggesting that in our conditions, the contribution of the vascular system to the previously observed lethal phenotype is likely minor (17).
FGF10 expression is reduced in patients with BPD and is associated with epithelial and vascular defects
We further validated in human lungs the observations made in mice. We obtained samples from either patients with BPD (n = 4) or neonates who died from causes unrelated to lung disease (n = 2). In non-BPD control lungs, FGF10 protein was detected around the conducting airways and within the lung interstitial tissue. However, lungs from patients with BPD displayed lower FGF10 protein expression (Fig. 2Ga and b), confirming the previously reported decrease in FGF10 in BPD lungs (1). qRT-PCR analysis was carried out for FGF10 and associated ligands as well as FGFR2b in human BPD (n = 3) versus control (n = 4) lungs (Supplementary Material, Fig. S2A). We confirmed the decrease in FGF10 expression as well as FGF7 and FGFR2b. No changes in the expression of FGF1 and FGF3 were detected. In mouse, the expression of Fgf1, 3 and 7 is not changed between Fgf10+/− and wild type (WT) lungs (n = 4 each group) (Supplementary Material, Fig. S2B).
Mature SFTPC, a secreted protein expressed by AT2 cells, which was strongly detected in control lungs, was much reduced in BPD samples (Fig. 2Gc and d). PECAM, an endothelial marker, was also reduced in BPD versus control lungs (Fig. 2Ge and f). In line with the known pulmonary hypertension (PH) phenotype, alpha-smooth muscle actin (α-SMA) expression was increased in patients with BPD (Fig. 2Gg and h).
Transcriptomic analysis of the Fgf10+/− and control lungs at P3 in NOX or HOX
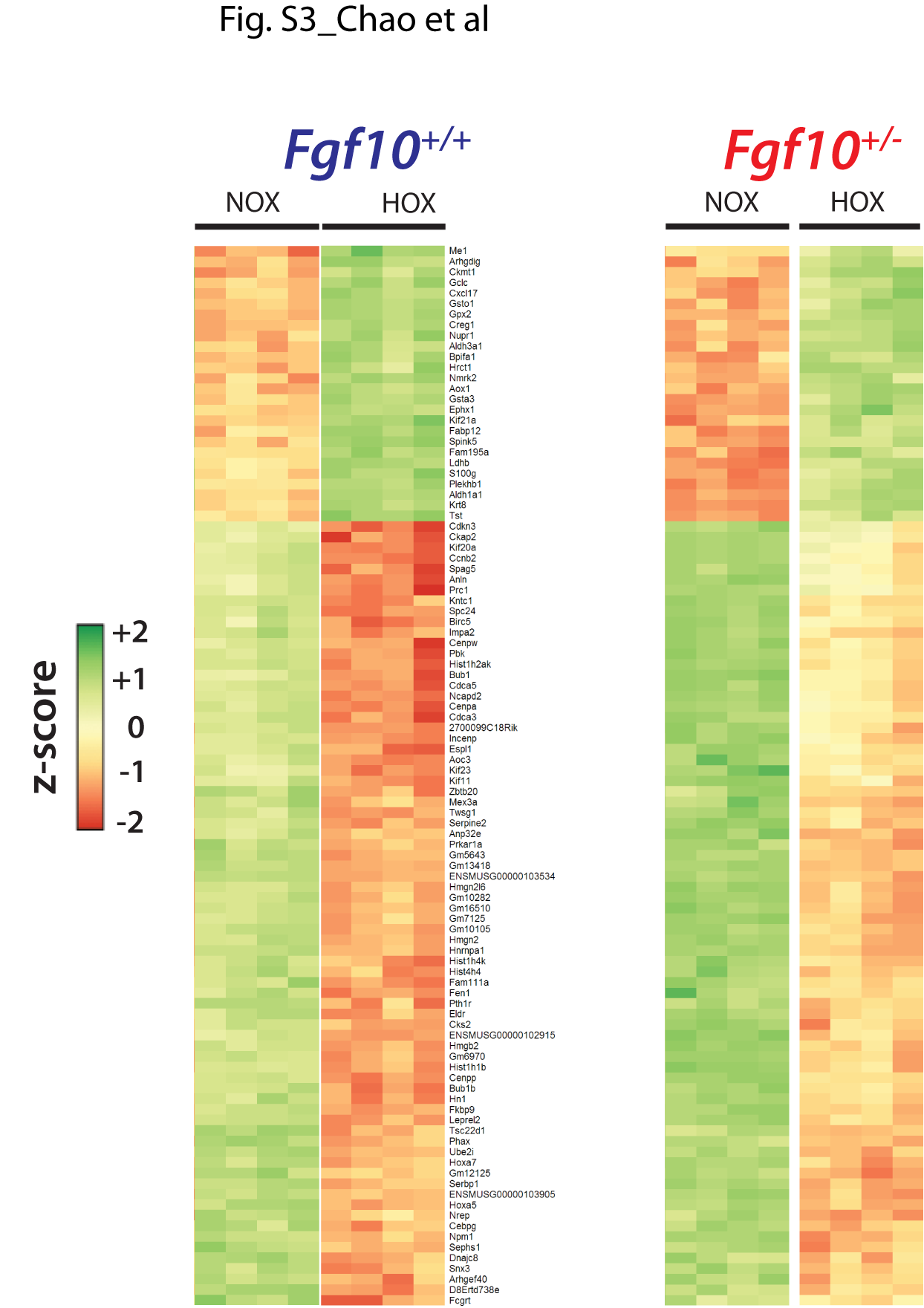
Next, we compared the gene arrays from HOX- versus NOX-exposed Fgf10+/− and control P3 lungs (Fig. 3A). Volcano plots revealed that HOX treatment was indeed effective in triggering changes in gene expression profiles in both control and experimental lungs (Fig. 3B and C, respectively). However, changes between the two genotypes, either in NOX or HOX, were more modest (Fig. 3D and E, respectively). The expression changes in response to HOX correlated between the two genotypes (Fig. 3F). The light blue points within the central circle represent genes that are not considerably regulated by HOX in either genotype. The dark blue points represent genes that are similarly regulated by HOX in both genotypes. The red points represent genes that show a more than 2-fold differential regulation between the genotypes. To support this result, we identified sets of genes differentially expressed in HOX versus NOX (selected according to their P-value) in control P3 lungs (Fig. 3G). As expected from the correlation analysis, these genes were similarly differentially regulated in HOX versus NOX in Fgf10+/− P3 lungs (Fig. 3G). (See Supplementary Material, Fig. S3 for higher magnification of the heatmaps shown in Fig. 3G)
Figure 3.
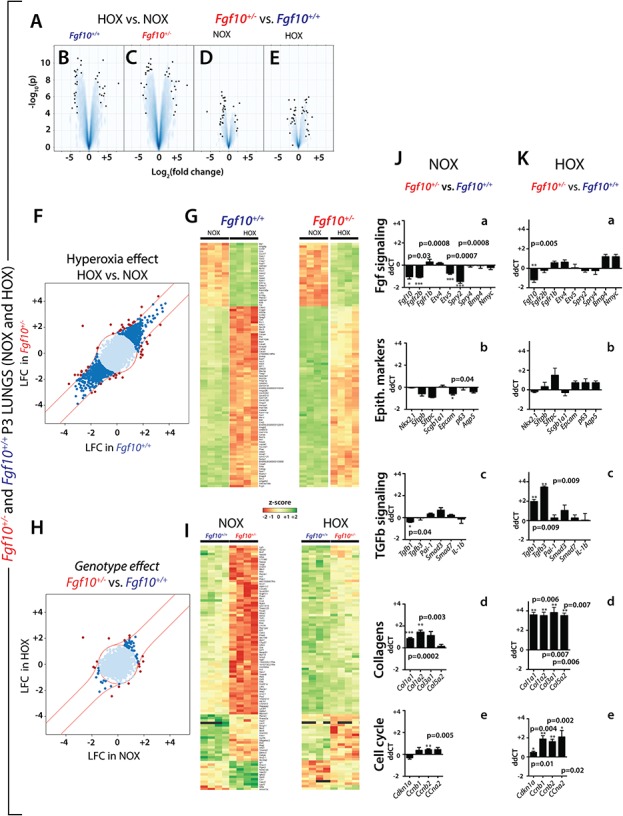
Identification of sets of genes differentially expressed in HOX versus NOX (selected according to their P-value) in whole lungs from WT or Fgf10+/− (A) volcano plots. (B) Volcano plot for HOX versus NOX in Fgf10+/+ (control) lungs (n = 4 each). (C) Volcano plot for HOX versus NOX in Fgf10+/− lungs (n = 4 each). (D) Volcano plot from Fgf10+/− versus control lungs in NOX. (E) Volcano plot in Fgf10+/− versus control lungs in HOX. (F) HOX effect HOX versus NOX. (G) Left heat map: top 100 regulated genes selected based on their P-value between NOX and HOX in control lungs (n = 4 each), right heat map: analysis of the same top 100 genes between NOX and HOX in Fgf10+/− lungs (n = 4 each). (H) Genotype effect Fgf10+/− versus control lungs. (I) Left heat map: top 100 regulated genes between Fgf10+/− versus control lungs in NOX (n = 4 each), right heat map: analysis of the same top 100 genes between Fgf10+/− versus control lungs in HOX (n = 4 each). (J–K) qPCR validation of gene expression in Fgf10+/− versus control lungs in NOX (n = 4 each) (J) and HOX (n = 4 each) (K) for (a) Fgf signalling, (b) Epithelial markers, (c) Tgfb signalling, (d) collagens and (e) cell cycle.
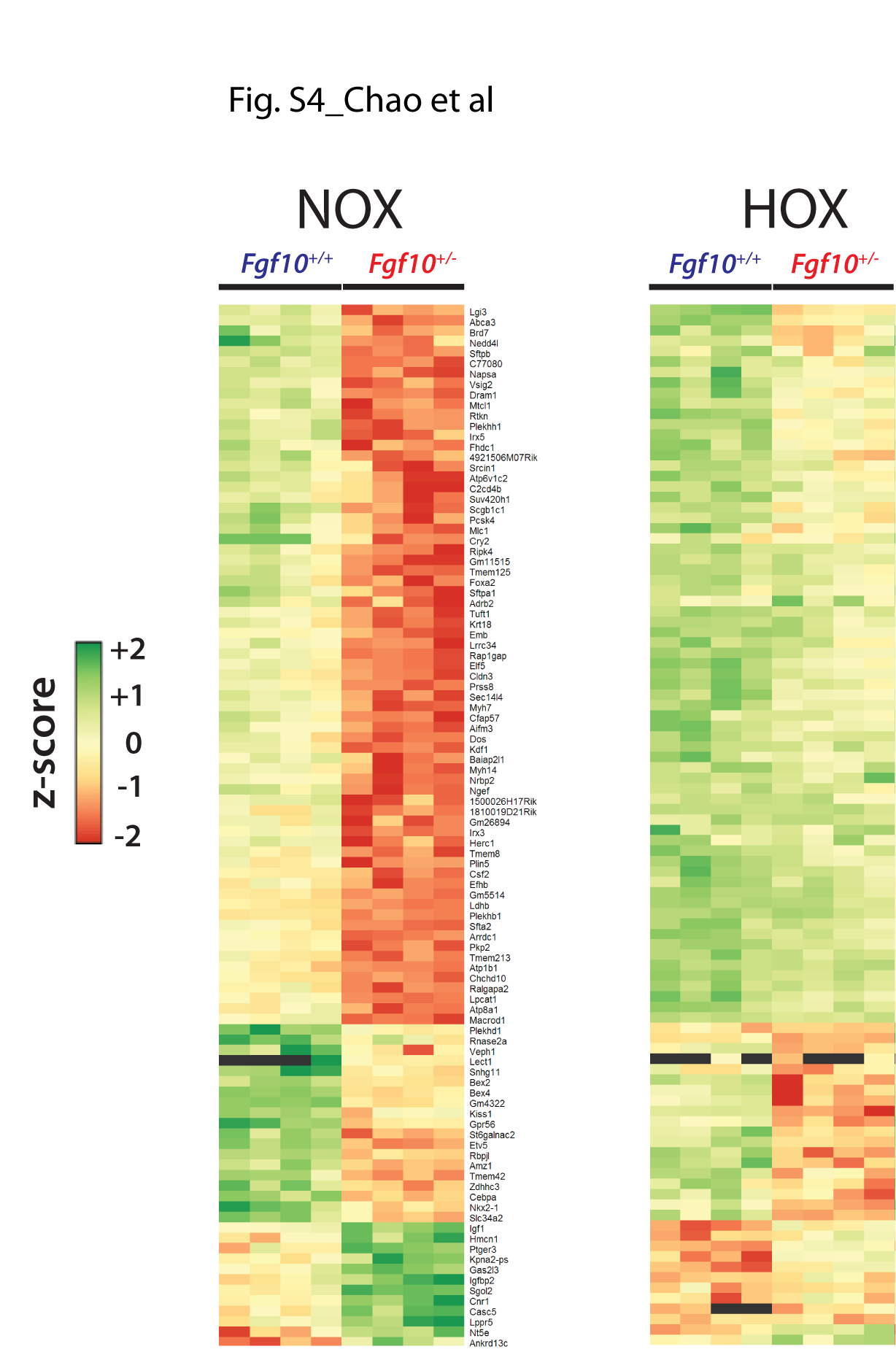
In contrast, the differential expression profiles due to the genotype did not correlate between the hyperoxic and normoxic conditions (Fig. 3H). To further define the genes affected by the decrease in Fgf10 expression in NOX, we identified the top 100 genes regulated between Fgf10+/− versus control in NOX (Fig. 3I). Interestingly, these genes respond to HOX by being globally up-regulated (top part of the heatmap) or down-regulated (low part of the heatmap) (Fig. 3I, right heatmap). (See Supplementary Material, Fig. S4 for higher magnification of the heatmaps shown in Fig. 3I.)
Next, we compared by qPCR the changes in Fgf signalling and the associated epithelial markers between Fgf10+/− and control lungs in NOX (Fig. 3Ja and b). As expected, the decrease in Fgf10 expression is associated with decreased Fgfr2b, Etv5 and Spry2, which support impaired Fgf10 signalling in NOX. The decrease in epithelial markers, particularly significant for Epcam, was also observed (Fig. 3Jb). Fgf10+/− lungs at P3 do not exhibit significantly increased Tgfβ signalling (Fig. 3Jc). Nonetheless, we observe a modest increase in collagen expression (Fig. 3Jd) and except for Cncb2, no significant change in cell cycle gene expression was detected (Fig. 3Je).
The same analysis in HOX showed significant decreased expression of Fgf10 and an increase of epithelial markers (Fig. 3Ka and b). Further, we found significant increase in Tgfβ1 and Tgfβ3 and drastic increase in all the collagen genes (Fig. 3Kc and d).
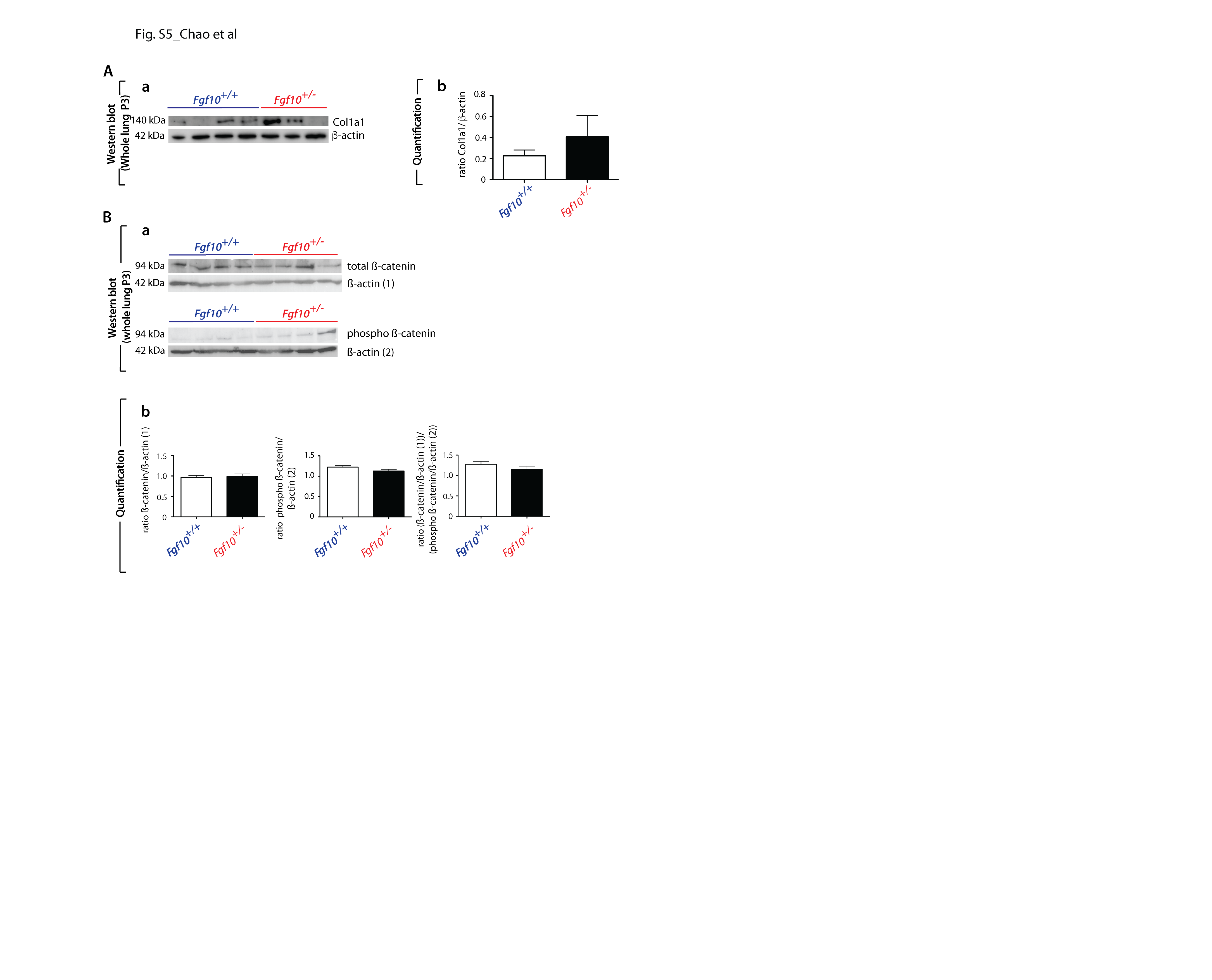
These results are supported by our previous report (17). Increased phospho-SMAD3 indicating activated Tgf-β signalling was detected in Fgf10+/− versus control mice in HOX P3 lung. In addition, our western blot results show a trend towards increase collagen levels in Fgf10+/− versus control mice (Supplementary Material, Fig. S5A).
Finally, cell cycle genes observed in the gene array, which regulate cell cycle either negatively (cyclin-dependent kinase inhibitor 1A, Cdkn1a) or positively (Ccnb1, Ccnb2, Ccna2) are significantly up-regulated (Fig. 3Ke).
In order to identify the significant genes/pathways differentially affected by HOX versus NOX between Fgf10+/− versus control, we carried out an analysis of the interaction HOX × genotype (Fig. 4). This analysis relies on the comparison of the genes differentially expressed in (HOX versus NOX in Fgf10+/− lungs) with those differentially expressed in (HOX versus NOX in control lungs). The volcano plot identified a set of genes, which are up- and down-regulated (Fig. 4A). The top 100 regulated genes according to their P-values are represented in the corresponding heat maps (Fig. 4B and C). As expected from the interaction analysis, the differential regulation observed in HOX (Fig. 4B) is no longer maintained in NOX (Fig. 4C). (Please see higher magnification of the heatmaps in the Supplementary Material, Fig. S6). For example, genes, which expression was not drastically changed in HOX between the two genotypes (located in the top part of the heatmap) are differentially regulated in NOX conditions. Furthermore, genes that were down-regulated in HOX conditions are up-regulated in NOX conditions. These sets of genes, which do not show a parallel increase or decrease in HOX versus NOX, are therefore likely to contribute to the lethal phenotype observed in the Fgf10+/− mice in HOX. KEGG analysis reveals that the main processes altered differentially are spliceosomes, protein processing in endoplasmic reticulum, metabolism of xenobiotics by cytochrome P450, antigen processing and presentation (Fig. 4D).
Figure 4.
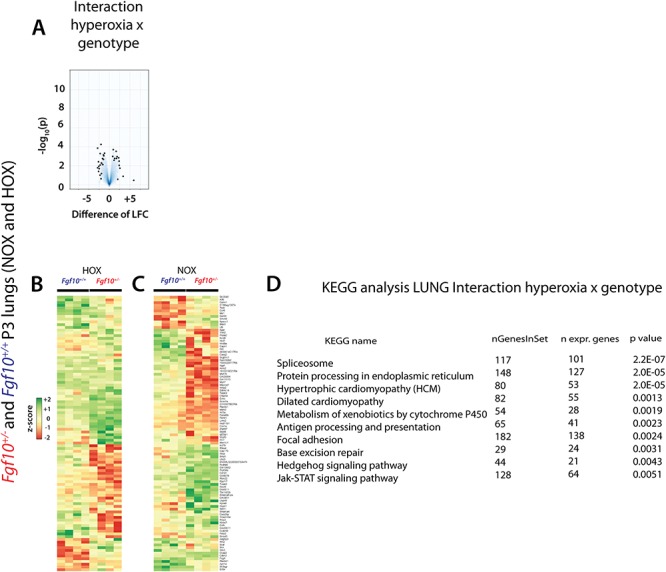
Interaction HOX × genotype analysis to identify the significant genes/pathways differentially affected by HOX in Fgf10+/− versus control P3 lungs. (A) Volcano plot. (B) Top 100 regulated genes in HOX in Fgf10+/− versus control lungs (n = 4 each). (C) Analysis of the same top 100 genes in NOX between Fgf10+/− and control lungs (n = 4 each). (D) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis between Fgf10+/− versus control lungs in HOX versus NOX.
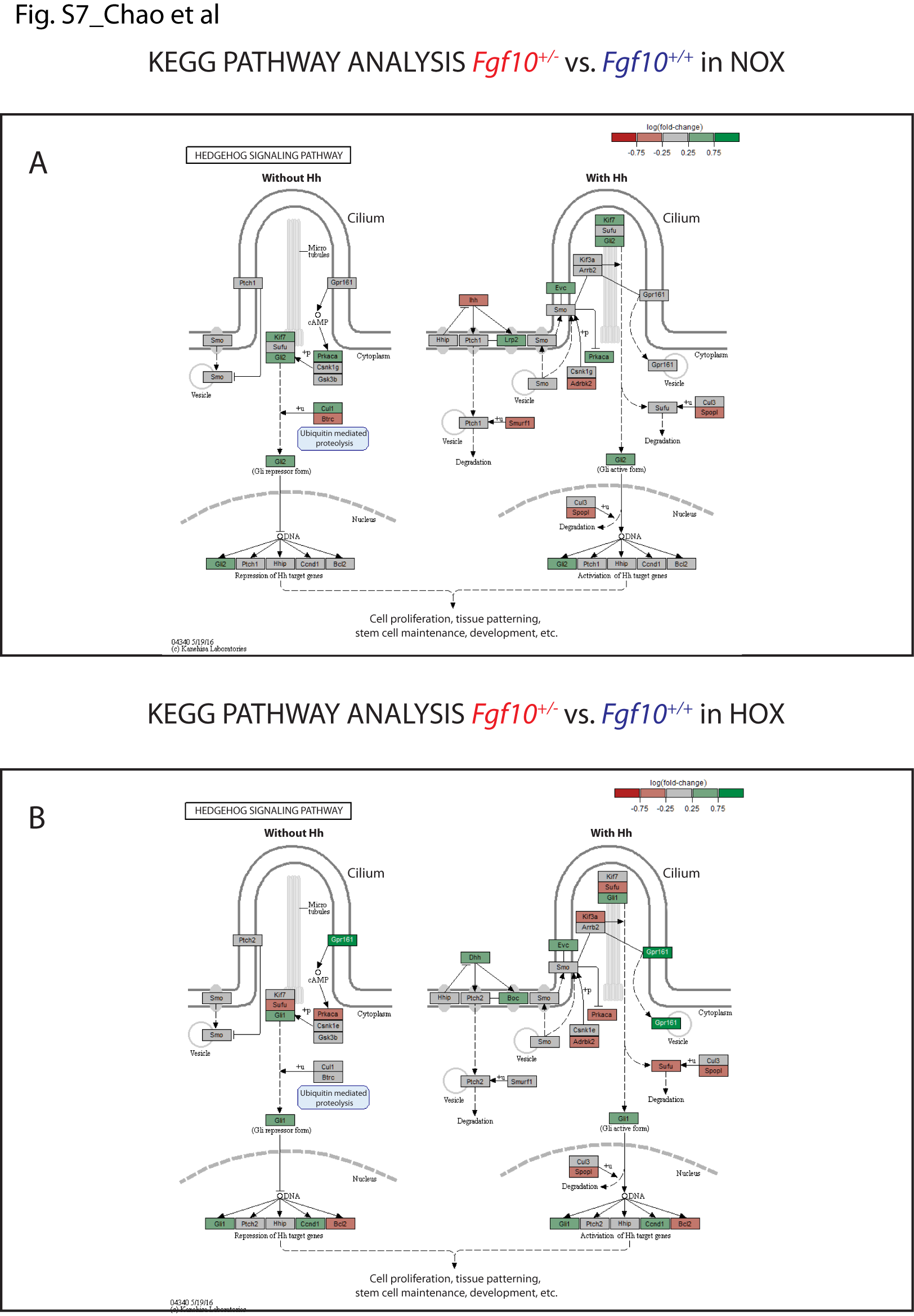
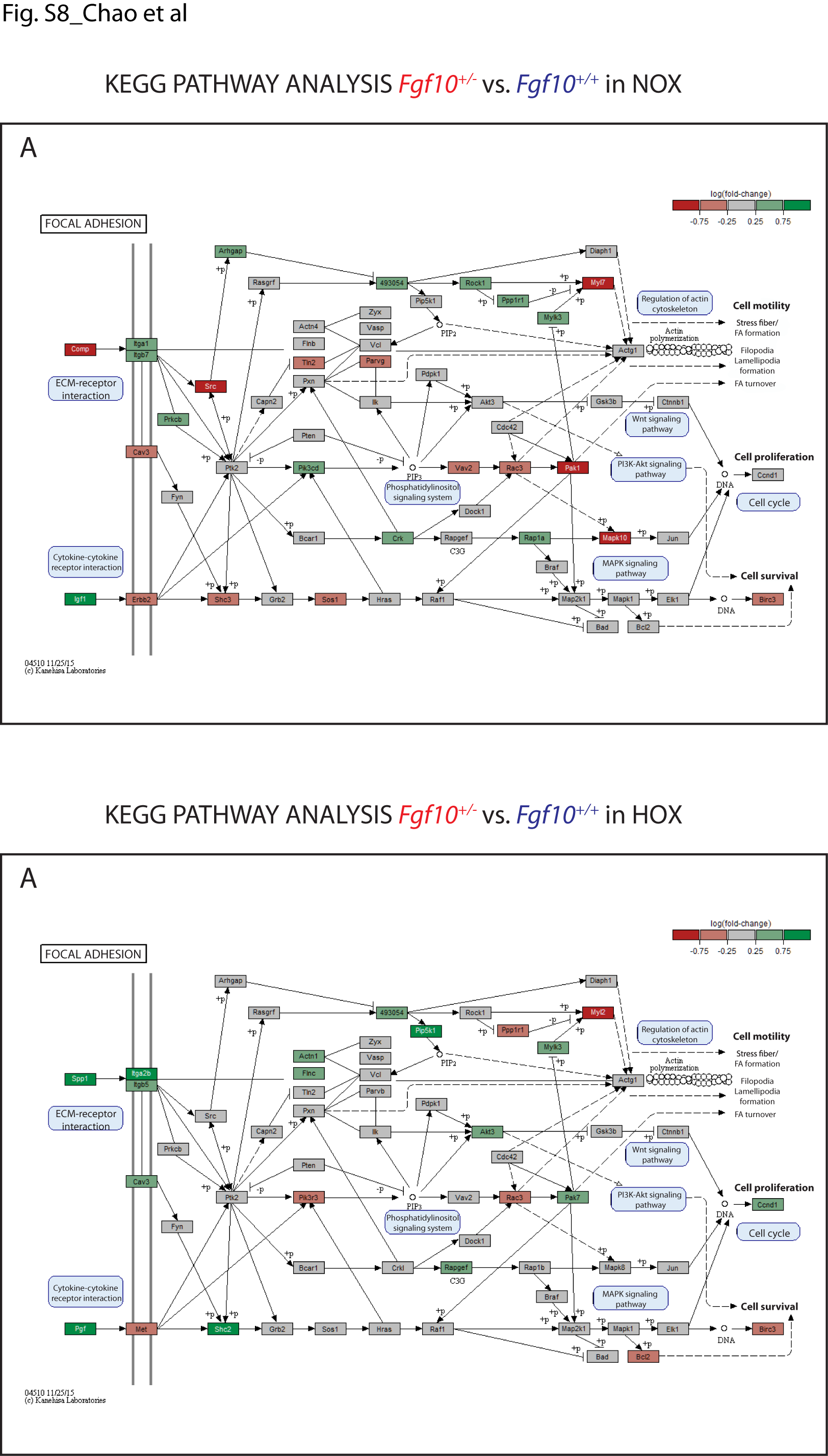
Regarding the changes in the Hedgehog (Hh) pathway (Supplementary Material, Fig. S7), our data show an increase of expression in the Fgf10+/− versus control mice in some elements of the Hh pathway. As reported previously (23,24), we have indications that Hh signalling to the lung mesenchyme regulates capillary network formation of the developing distal lung and through the regulation of expression of Vegfa in sub-mesothelial lung mesenchyme. The main difference observed in NOX and HOX is an increase in the expression of the transcriptional factor Gli2 and 1, respectively. Interestingly, we previously reported that Sonic Hedgehog (Shh) down-regulates Fgf10 expression in the mesenchyme (16) and it was recently shown that Fgf10 induces Shh expression in the epithelium (25). From the developmental studies, it is therefore not surprising that Fgf10 could impact Shh expression and a down-regulation of Shh expression and activity should be expected in the Fgf10+/− versus control lungs. However, our data are not sufficient to prove a substantial repression of the pathway, further analysis of the protein localization or post translational modification would be needed. The changes in the focal adhesion (Supplementary Material, Fig. S8) and Janus kinases (Jak)-Signal Transducer and Activator of Transcription proteins (STAT) (Supplementary Material, Fig. S9) signalling pathways appear more marked in the Fgf10+/− versus control mice, but once again we cannot determine with absolute certainty the exact contribution of each of these pathways to the observed phenotype, especially when the main change in downstream elements of the pathway involves cyclin D1, which is a common target for multiple effector pathways. We also investigated the expression of different elements of the Wnt pathway using KEGG pathway analyses in NOX and HOX, finding a significant alteration of expression in many of them (Supplementary Material, Fig. S10A and B). In particular, the Wnt canonical pathway in HOX shows an up-regulation through the increased expression of Wnt10b, a gene encoding an activator of the pathway, and the down-regulation of adenomatous polyposis coli (Apc), a gene encoding a negative regulator of β-catenin activity. These findings suggest an activation of the β-catenin nuclear activity and are supported by an increase expression of the T-cell factor/lymphoid enhancer factor (TCF/LEF) downstream targets in the analyzed sample, in particular, Fosl1, Wisp1 and Cyclin D1. However, western blot analysis using whole lung protein extract shows no major difference in phospho-ß-catenin between Fgf10+/− versus WT lungs (Supplementary Material, Fig. S5B).
Transcriptomic analysis of Fgf10+/− versus control lungs supports the correlation between decrease Fgf10 expression and vascular defects
Next, a KEGG pathway analysis for the genes involved in vascular smooth muscle cell contraction and Vegf signalling between the two genotypes in NOX (Fig. 5) and HOX (Supplementary Material, Fig. S11) was carried out. Figure 5A indicates that 60.9% of the genes controlling vascular smooth muscle cell contraction were differentially expressed between the two genotypes in NOX (41.3% up-regulated, 19.6% down-regulated). Among the down-regulated genes, we found genes encoding for transmembrane proteins [e.g. Ramp1 (receptor activity modifying protein 1), Kcnmb2, Adora2] and cytosolic proteins [e.g. Itpr3 (inositol 1,4,5-trisphosphate receptor type 3), Pla2g1 (phospholipase A2 group I), Arhgef, Adcy8]. In particular, Ramp1 is a receptor compound for intermedin/adrenomedullin leading to stabilization of endothelial barrier function and attenuation of ventilator-induced lung injury in mice (26). Furthermore, adrenomedullin acts through Ramp1 and is proposed to have an antifibrotic effect in human lung fibroblast (27). Ramp1, as part of the CGRP1 receptor, regulates the pulmonary circulation (28). In addition, Caveolin-1 interacts with Itpr3 to regulate Ca2+ entry in endothelial cells (29). Interestingly, Pla2g1 inactivation reduces atherosclerosis in mice (30).
Figure 5.
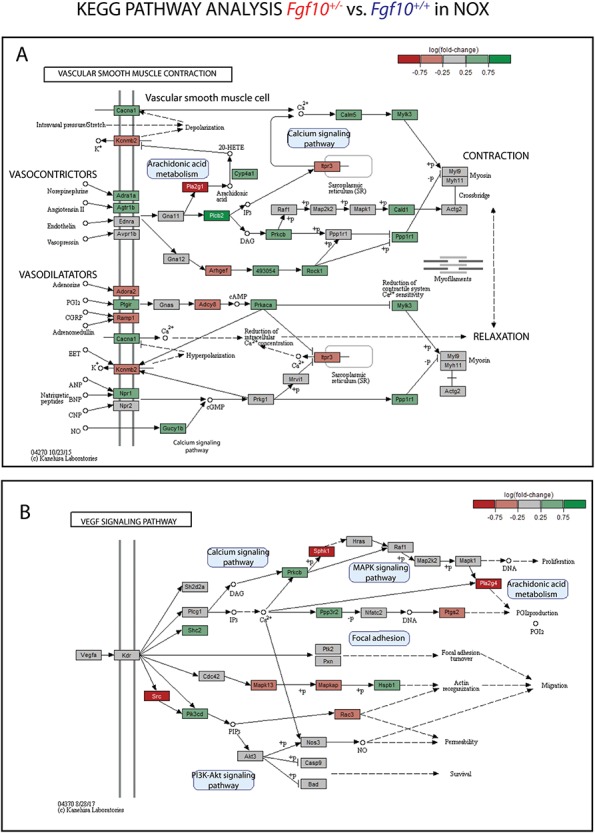
Transcriptomic analysis of Fgf10+/− versus control P3 lungs in NOX suggests that reduced Fgf10 expression could lead to vascular defects. KEGG pathway analysis of the expression of genes involved in (A) vascular smooth muscle cell contraction and (B) Vegf signalling pathway in Fgf10+/- versus control P3 lungs in NOX (n = 4 each). Note that 60.9% of the genes are affected for the (A) vascular smooth muscle cell contraction and 42.9% of the genes are affected for the (B) Vegf signalling pathway.
Among the genes up-regulated, we found genes encoding for transmembrane proteins [e.g. Ptgir (prostaglandin I2 receptor), Cacna1, Adra1a, Agtr1b, Npr1] and cytosolic proteins (e.g. Rock1, Plcb2, Prkaca). In particular, Ptgir, a major regulator in the prostacyclin pathway, has been shown to be dysregulated in congenital diaphragmatic hernia leading to PH (31). Rho A/rho kinase (Rock) pathway plays an important role in the pathogenesis of human PH (32). Furthermore, Rock1 contributes to the profibrotic response of endothelial cells in experimental pulmonary fibrosis (33). Figure 5B indicates that 42.9% of the genes involved in Vegf signalling were differentially expressed (17.9% up-regulated, 25% down-regulated). Among the down-regulated genes, we found genes encoding for cytosolic proteins [e.g. Src, Sphk1 (sphingosine kinase 1), Pla2g4, Rac3]. Src-family kinases, a family of non-receptor tyrosine kinases, are major regulators of vascular smooth muscle function in health and disease (34). Sphk1 is involved in endothelial cell motility and angiogenesis (35). Sphk1 deficiency increased pulmonary vascular hyperresponsiveness, which is one aspect in pulmonary arterial hypertension pathogenesis (36). Pla2 (phospholipase A2) activity plays a substantial role in protecting pulmonary microvascular endothelial cells against oxidative stress (37). Among the genes up-regulated, we found genes encoding for cytosolic proteins [e.g. Pik3cd (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta), Shc2, Prkcb, Hspb1]. In particular, inhibition of Pik3cd has been shown to attenuate antigen-induced airway inflammation and hyperresponsiveness by preventing vascular leakage in mice (38). Interestingly, while the percentage of genes being differentially expressed in vascular smooth muscle cell contraction pathway are very similar in HOX (Supplementary Material, Fig. S11A) and NOX (Fig. 5A), the total number of genes being affected in the Vegf signalling pathway is much lower in HOX (Supplementary Material, Fig. S11B) compared to NOX (Fig. 5B). Especially, the number of down-regulated genes decreases from 25% to 11%. One reason is the up-regulation of genes upon HOX exposure, which are previously down-regulated in NOX, resulting in no more differential expression when comparing to the control group. For example, Pla2g4 is down-regulated in NOX but up-regulated in HOX supporting its protective role in oxidative stress. Taken together, the transcriptomic analysis of Fgf10+/− versus control lungs in NOX suggests that reduced Fgf10 expression could lead to vascular defects by dysregulating genes that are playing a crucial role in endothelial barrier function, atherosclerosis, pulmonary circulation/ hypertension, vascular smooth muscle function, airway inflammation and endothelial oxidative stress.
Increased myosin light chain phosphorylation is observed in BPD versus control lungs
Given the transcriptomic data regarding smooth muscle cell contraction in NOX (Fig. 5) and HOX (Supplementary Material, Fig. S11) between Fgf10+/− and WT lungs, we examined by IHC the extend of phosphorylation of two key myosin light chain proteins identified in our gene array, myosin light chain 9 (MYL9) and MYL6b in BPD (n = 3) versus control (n = 4) lungs. Both MYL9 and MYL6b are detected in pulmonary vessels. MYL9 expression appears to be decreased in BPD versus control (Supplementary Material, Fig. S12a and b) while the expression of MYL6b is unchanged (Supplementary Material, Fig. S12e and f). Analysis of the expression of the corresponding phosphorylated forms indicates a clear increase in the context of BPD (Supplementary Material, Fig. S12c, d, g and h), thereby supporting our transcriptomic data. A similar analysis was carried out in mice (Supplementary Material, Fig. S13). As the KEGG analysis showed that MYL9 in NOX and MYL6b in HOX could be potentially regulated, we focused on their expression by IHC in these two conditions. In NOX, MYL9 expression (as well as its corresponding phosphorylated form) does not display any difference between Fgf10+/− and control lungs (Supplementary Material, Fig. S13Aa–d). However, in HOX, MYL6b and its phosphorylated form exhibited the same pattern as in human BPD (Supplementary Material, Fig. S13Ba–d).
Blockade of Fgfr2b ligands activity postnatally leads to decreased blood vessel number and increased muscularization
Fgf10+/− lungs display ‘build in’ developmental defects (17), which could cause the vascular defects observed. While the Fgf10+/− mice are useful to understand human pathologies associated with reduced FGF10 expression such as patients with ALSG and LADD syndromes (7,10), it is very unlikely that this model is representative of the majority of cases with BPD, where due to inflammation, FGF10 expression is reduced during the saccular/alveolar stages of lung development. Supporting this conclusion, mutation in the FGF10 gene has not been reported so far in the context of BPD. In order to investigate the impact of Fgfr2b ligands inhibition postnatally, we developed a transgenic mouse model [Rosa26rtTA/rtTA, Tg(Tet(O)sFgfr2b)/+] allowing the expression of a soluble form of the Fgf10 receptor Fgfr2b upon exposure of the mice to doxycycline. This system is well established in our laboratory and has been validated in many organs (21,39–42). We have therefore exposed the nursing females to dox food immediately after the birth of double transgenic [DTG, Rosa26rtTA/rtTA, Tg(Tet(O)sFgfr2b)/+] and control single transgenic (STG, Rosa26rtTA/rtTA, +/+) pups and analyzed the lungs for vascular defects at P3 in HOX using the vWF and α-SMA staining (Fig. 6Aa and b) as previously described. The quantification of the vessel number indicates a significant decrease of the 20–70 μm blood vessels in the DTG versus control lungs (P = 0.04, n = 5) as well as decreased total blood vessels (Fig. 6B). Further analysis of the muscularization status indicates increased muscularization of both 10–20 μm and 20–70 μm vessels (Fig. 6Ca and b). This is a feature seen in the context of PH and indicates that the blockade of Fgfr2b ligands postnatally is sufficient to elicit the PH phenotype, a characteristic defect seen in BPD.
Figure 6.
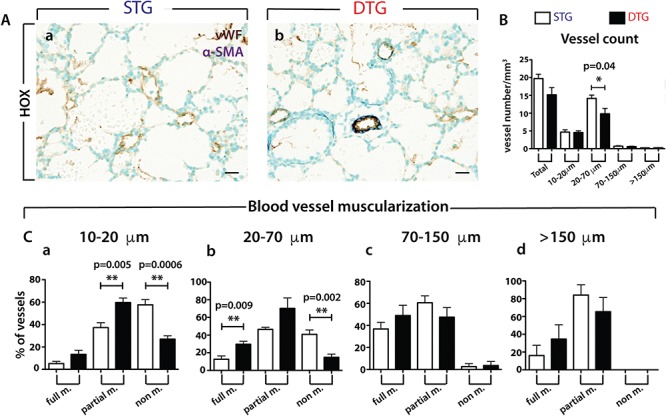
Analysis of the vascular defects in P3 double transgenic (DTG: Rosa26rtTA/rtTA, Tg(Tet(O)sFgfr2b)/+) and control single transgenic (STG: Rosa26rtTA/rtTA, +/+) lungs in HOX. (A) IHC for vWF and α-SMA of (a) STG and (b) DTG lungs (n = 5 each). (B) Total vessel count in STG and DTG lungs. (C) Blood vessel muscularization between STG and DTG lungs for blood vessels at (a) 10–20 μm, (b) 20–70 μm, (c) 70–150 μm and (d) >150 μm. Scale: Aa, b, 20 μm.
Discussion
We have previously reported a clear link between Fgf10 deficiency and vascular defects in Fgf10LacZ/− hypomorphic lungs (these lungs express only 20% of the normal level of Fgf10) (22). We showed that Vegfa is a target of Fgf10 in the developing lung epithelium and that decreased Fgf10 levels eventually translate into decreased Vegfa and associated vascular defects. Vegfa is not significantly reduced in E18.5 Fgf10+/− versus WT lungs. Only a trend towards reduction is visible in the present study (Fig. 1Ab). In the Fgf10LacZ/− hypomorphic lungs, the vascular tree is severely underdeveloped (22). Our quantification of the number and size of blood vessels indicates that significant vascular defects are not observed in P3 Fgf10+/− lungs in NOX. However, such defects emerge after HOX exposure and are characterized by a significant drop in the number of blood vessels and by an increase of the more immature (smooth muscle actin-negative) blood vessels. However, these defects did not appear to lead to lung hemorrhages, either at P3 or at later time points analyzed, suggesting that the observed lethality is not due to a failing vascular system (17). Interestingly, vascular defects, primarily PH, have been proposed as the main causes of lethality in infants with BPD (43). The PH phenotype is not observed in Fgf10+/− lungs but is present upon Fgfr2b ligands inhibition postnatally. This suggests that developmental defects upon reduction of Fgf10 expression could be protective postnatally against PH. This could be a possibility, for example, if the smooth muscle progenitor cells are impaired and do not respond to proliferative stimuli following HOX exposure. Interestingly, we reported defective smooth muscle cells formation in the Fgf10 hypomorphic lung (44). Along these lines, a decrease in Fgf10 expression starting during the saccular phase that would allow normal smooth muscle cell formation could then be causative for the PH phenotype. Alternatively, Fgf10 decrease during the saccular phase is not sufficient to trigger a PH phenotype as additional Fgfr2b ligands, such as Fgf1 and Fgf7 could act redundantly with Fgf10 and will have to be inhibited also for the vascular phenotype to appear. Our results allow for the first time to propose vascular defects caused by inhibition of Fgfr2b ligands activity postnatally. It is still unclear if these defects are the consequences of direct or indirect effect of Fgfr2b ligands on the endothelium and/or vascular smooth muscle cells. Interestingly, the simultaneous deletion of Fgfr1 and Fgfr2 in endothelial cells while allowing normal vascular homeostasis leads to impaired neovascularization of the skin and retina after injury and is associated with delayed wound healing (45). In addition, Fgfr1 and Fgfr2 in endothelial cells play a critical role for cardiac functional recovery and vascular remodelling following cardiac ischemia-reperfusion injury, without affecting the cardiac hypertrophic response suggesting that from the activation of the Fgf pathway in endothelial cells after ischemia injury could be beneficial (46).
The presence of more immature (smooth muscle actin-negative) blood vessels in our experimental mice in HOX raises the possibility that Fgf10 signalling contributes to the formation of vascular smooth muscle cells during organogenesis via its action on Pdgfb (22) or via a more direct action on smooth muscle cell progenitors (47).
Interestingly, recombinant Fgf10 has been shown to act directly on mesenchymal-derived cells such as cardiomyocytes (48), adipocyte stem cells (49) and lipofibroblasts (50), strengthening the concept that in the lung, Fgf10 does not only act on the epithelium. Fgf10 has also been shown to directly support the proliferation of endothelial cells (51).
It is therefore tempting to subdivide the effects of decreased Fgf10 expression on the vasculature into two classes: one class linked to developmental defects where the blood vessels are more sensitive and immature and one class linked to the role of Fgf10 in repair as a proliferative factor for the endothelium, thereby limiting the vascular damages triggered by HOX.
In conclusion, constitutive decrease in Fgf10 mRNA levels in mice leads to lung congenital defects, which are compatible with postnatal survival, but which compromise the ability of the lungs to cope with sub-lethal hyperoxic injury. In particular, we provide evidence that Fgf10 heterozygous mice display pulmonary vasculature defects during normal lung development and during injury. This knowledge may be critical in designing therapies to prevent lung injury in neonates at risk for BPD and in adult lung disorders characterized by FGF10 deficiency.
Materials and Methods
Ethical approvals
Animal studies were performed in accordance with the National Institutes of Health Guidelines for the Use of Laboratory Animals and approved by the Federal Authorities for Animal Research of the Regierungspraesidium Giessen, Hessen, Germany protocols 105/2011. For the human studies written informed consent was received from participants prior to inclusion in the study at Columbia University (New York, USA). Participants were identified by number, not by name. In addition, human lung tissues were also obtained from Erasmus MC Sophia Children's Hospital (Rotterdam, The Netherlands). Human lung tissue donation was approved by the ethics committee of the Erasmus MC Sophia Children's Hospital (Rotterdam, The Netherlands).
Mice
Fgf10+/− mice were generated by crossing Fgf10flox/flox mice (Fgf10tm1.2Sms/J, Jackson laboratory stock 023729) with Cytomegalovirus (CMV)-Cre mice [B6.C-Tg(CMV-cre)1Cgn/J, Jackson laboratory stock 006054]. The resulting Fgf10+/− mice (Fgf10tm1.1Sms/J) were crossed for at least five generations with C57BL/6 mice. Fgf10+/− and Fgf10+/+ embryonic and newborn littermates were used.
Human samples
Lung sections from four patients with BPD were obtained. Two males (25 and 34 weeks of gestational age) and two females (23 and 24 weeks of gestational age) underwent invasive ventilation for either 1 or 4 months and died between 1 to 6 months after birth. Diagnosis of BPD was confirmed post-mortem by resident pathologists in all patients. Lung sections from two females (37 weeks and full term) who died (7 days and 13 months, respectively) from causes other than lung disease were obtained. Further samples were obtained for MYL6b and MYL9 immunostaining. Lung sections from three infants diagnosed with BPD (25, 28, 31 weeks of gestational age) were used. Lung sections from three infants (24, 26, 33 weeks of gestational age) who died from causes other than BPD were used as controls. RNA from three patients (two with 26 weeks and one with 27 weeks of gestational age) with BPD and from four controls (27, 33, 38 and 39 weeks of gestational age) were used for qRT-PCR.
Left lobe perfusion, isolation and tissue processing
The left lobe was perfused through the trachea with a pressure of 20 cm H2O with 5 ml phosphate buffered saline (PBS) followed by 5 ml, 4% paraformaldehyde (PFA). The trachea was tied off with a string and the lung was removed and placed in 4% PFA for max. 24 h at 4°C. Lungs were then progressively dehydrated (30%, 50%, 70%, 99.6% ethanol, each 3 h) and embedded with a Leica embedding machine (EG 1150C). Paraffin blocks were kept cold and 5 μm sections were cut.
Immunostaining
Immunofluorescence staining for SFTPC, PECAM and α-SMA
Paraffin sections were deparaffinized, blocked with 3% bovine serum albumin (BSA) and 0.4% Triton X-100 [in tris-buffered saline (TBS)] at room temperature (RT) for 1 h and then incubated with primary antibodies against mature-SFTPC (Seven Hills, 1:1000), PECAM (Abcam, 1:100) and α-SMA (Sigma-Aldrich, 1:400) at RT for 1 h or at 4°C overnight. After incubation with primary antibodies, slides were washed three times in TBST (TBS buffer + 0.1% tween 20) for 5 min, incubated with secondary antibodies at RT for 1 h and then washed three times in TBST before being mounted with Prolong Gold Anti-fade Reagent with DAPI (4’,6-diamidino-2-phenylindole; Invitrogen) (Table 1).
Table 1.
Antibodies for immunohistochemistry
| Antibody | Company (Catalog number) | Dilution |
|---|---|---|
| FGF10 | Antibodies-online (ABIN360398) | 1:30 (human) |
| FGFR2 (BEK) | Santa Cruz (sc-122) | 1:300 (human) |
| SFTPC | Seven Hills (WRAB-9337) | 1:100 (human) |
| PECAM/CD31 | BD Pharmingen (550274) | 1:100 (mouse and human) |
| α-SMA | Sigma-Aldrich (A2547) | 1:600 (mouse) |
| 1:400 (human) | ||
| von-Willebrand-factor (VWF) | Dako (A0082) | 1:1200(mouse) |
| MYL6b | Merck (AB3381) | 1:100 (mouse) |
| 1:200 (human) | ||
| Phospho MYL6b | BosterBio (A14059) | 1:600 (mouse) |
| 1:200 (human) | ||
| MYL9 | Abcam (ab64161) | 1:200 (mouse and human) |
| Phospho MYL9 | Abcam (ab2480) | 1:100 (mouse and human) |
FGFR2 and FGF10 staining
Immunohistochemistry for FGFR2 and FGF10 was performed on lung tissue sections from patients with BPD. First, paraffin sections were deparaffinized. For FGFR2 staining, antigen unmasking were done by incubation of the slides in boiling buffer solution for 15 min, then blocked with 3% BSA for 60 min in RT and incubated with the primary antibody (FGFR2, 1:300) over night at 4°C. After washing steps, incubation with secondary antibody was performed before covering the slides with DAPI solution. For FGF10 staining, antigen unmasking were done by incubation of the slides in boiling buffer solution for 25 min, then treated with proteinase K for 15 min at RT. Afterwards slides were blocked with 10% BSA for 60 min in RT followed by incubation of the primary antibody (FGF10, 1:30) at 4°C overnight. After washing steps, incubation with secondary antibody was performed before covering the slides with DAPI solution (Table 1).
α- SMA/vWF double staining
The 3 μm sections were deparaffinized, endogenous peroxidase was removed by incubation with 3% H2O2-methanol solution for 20 min, then washed with Aqua Dest and PBS each 2 × 5 min. For antigen retrieval, sections were trypsinized for 10 min at 37°C, then washed with PBS (4 × 5 min) and immersed in blocking buffer (10% BSA) for a further 20 min. After washing with PBS (4 × 5 min), sections were blocked with Rodent Block M (Biocare Medical MM horseradish peroxidase (HRP)-Polymer Kit, Cat. No. 1-800-799-9499) for 30 min, washed with PBS, incubated with primary antibody (alpha-actin 1:800 diluted, 30 min) and HRP polymer (20 min) before immersed in substrate solutions (Vip substrat Kit, vector, 30 s–4 min). Slides were monitored under the microscope for staining progression and washed with tap water for 5 min. Before staining for vWF, sections were immersed in blocking buffer (10% BSA) for 15 min, washed with PBS, blocked with serum (ImmPRESS Kit Anti-rabbit Ig), then sections were incubated with vWF antibody (1:1200 diluted) at 37°C for 30 min and washed again. After incubation with secondary antibody (Anti-rabbit Ig peroxidase, ImmPRESS reagent), sections were stained with diaminobenzidine (DAB) substrate Kit (SK-4100, vector) and staining progression was observed under the microscope. After washing with tap water, sections were counterstained with methyl green for nuclei on a heating plate (60°C), progressively dehydrated and coverslipped (Table 1).
Immunostaining for MYL6b, phospho-MYL6b, MYL9 and phospho-MYL9 in mouse and human lungs
The 3 μm sections were deparaffinized. For antigen retrieval, sections were heated in a microwave for 25 min in Rodent Decloaker (Biocare/Zytomed Systems), then washed with PBS (4 × 5 min) and immersed in blocking buffer (10% BSA) for a further 10 min. After washing with PBS (4 × 5 min), sections were blocked with Rodent Block M (Biocare/Zytomed Systems) for 30 min. For human samples Protein-Block from ZytoChem-Plus AP Polymer-Kit was used instead of Rodent Block M for 5 min. Slides were washed with PBS and incubated with primary antibody overnight. After washing, slides with human samples were incubated with PostBlock solution for 20 min before incubation with AP-Polymer Reagent for 30 min. Slides with mouse samples were not incubated with PostBlock solution. Next, slides were washed and immersed in substrate solutions (RedChromogen, 2–10 min). Slides were monitored under the microscope for staining progression and washed with tap water for 5 min. After washing, sections were counterstained with hematoxylin for nuclei on a heating plate (60°C), progressively dehydrated and coverslipped (Table 1).
Image acquisition
Fluorescent images were acquired using Leica DM5500 B fluorescence microscope connected to Leica DFC360 FX camera. Lung tissue sections without immunofluorescence were analyzed under light microscope and representative photomicrographs were taken by usage of computer software for image analysis (QWin, Leica, Wetzlar, Germany).
RNA extraction
After lung function measurements were taken, the right bronchus was clamped and either cranial and accessory or caudal and medial lobes were removed, placed in TRIZOL, homogenized in GentleMACs and frozen in liquid nitrogen for RNA extraction. RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to manufacturer's instructions.
Quantitative real-time RT-PCR
RNA was reverse-transcribed (QuantiTect Reverse Transcription Kit, Cat. No. 205313, Qiagen GmbH, Deutschland, Hilden). cDNA was diluted to a concentration of 5 ng/μl. Primers were designed using Roche Applied Sciences online Assay Design Tool. All primers were designed to span introns and blasted using NCBI software for specificity. Sybr Green Master Mix (Invitrogen, Cat. No.11733-038) was used for RT-PCR with a Roche LightCycler 480 machine. Samples were run in triplicates using Hprt as a reference gene. The dCT and ddCT values were calculated as follows: dCT (WT) = CT (WT, reference gene) – CT (WT, gene of interest), dCT (Mutant) = CT (Mutant, reference gene) – CT (Mutant, gene of interest), ddCT = dCT (Mutant) – dCT (WT). Mouse primers are listed below (Table 2).
Table 2.
Human and mouse primers forward/reverse
| Human gene | Forward sequence (5'-3') | Reverse sequence (5'-3') |
|---|---|---|
| FGF7 | AAGGGACCCAAGAGATGAAGA | CCTTTGATTGCCACAATTCC |
| FGF10 | GAAGGAGAACTGCCCGTACA | GGCAACAACTCCGATTTCTACT |
| FGFR2b | GATAAATAGTTCCAAT | TGCCCTATATAATTG |
| GCAGAAGTGCT | GAGACCTTACA | |
| PECAM1 | CTGTCCCTGATGCCGTG | AGCAGGGCAGGTTCATAAAT |
| PGBD | TGTCTGGTAACGGCAATGCG | CCCACGCGAATCACTCTCAT |
| Mouse gene | Forward sequence (5'-3') | Reverse sequence (5'-3') |
| Fgf1 | GTAGTTTCCTAGAGGCAGGTTG | TGATAAAGTGGAGTGAAGAGAGC |
| Fgf3 | GATTACTGCGGTGGAAGTGG | CCGTTCCACAAACTCACACTC |
| Fgf7 | ACTATCTGCTTATAAAATGGCTGCT | GTGGGGCTTGATCATCTGAC |
| Vegfr2 | CAGTGGTACTGGCAGCTAGAAG | ACAAGCATACGGGCTTGTTT |
| Vegfa | AAAAACGAAAGCGCAAGAAA | TTTCTCCGCTCTGAACAAGG |
| Pdgfb | GAGTCGGCATGAATCGCT | CAGCCCCATCTTCATCTACG |
| Pdgfrb | GGAGAACAGAGAGGAAACAGT | GCGGAAAACCCTGAGAGAAT |
| Hprt | CCACAGGACTAGAACACCTGCTAA | CCTAAGATGAGCGCAAGTTGAA |
Western blot
Loading buffer was added to protein samples from the lungs (5% SDS in bromophenol blue and β-mercaptoethanol), denatured for 8 min at 98°C and cooled on ice. 15 μg of protein sample was loaded on a 8–10% polyacrylamide gel and run at 35 mA per gel for ~1 h. Samples were then transferred electrically to a polyvinylidene fluoride membrane (Amersham, Germany) by semi-dry electro blotting (70 mA per gel, gel size, 7 × 9 cm) for 90 min. The membrane was blocked with 5% milk in TBS-T blocking buffer at RT on a shaker for 1 h followed by incubation with primary antibody overnight (Supplementary Material, Table S2) at 4°C. After washing with 1 × TBS-T four times for 15 min each, the membrane was incubated with the respective HRP-labelled secondary antibody (dilution 1:2000) at RT for 1 h followed by four times washing with 1 × TBS-T buffer for 15 min each. The protein bands were detected by Enhanced Chemiluminescence (Amersham, Germany) treatment and emitted signals were detected with a chemiluminescence imager. Quantification of bands was done by using ImageJ software Table 3.
Table 3.
Antibodies used for western blotting
| Primary antibody (rabbit) | Company (Catalog number) | Dilution | Secondary antibody | Company (Catalog number) | Dilution |
|---|---|---|---|---|---|
| COL1A1 | Meridian Bioscience (T40777R) | 1:1000 | Swine anti rabbit HRP | Dako (P0217) | 1:2000 |
| β-ACTIN | Abcam (ab8227) | 1:30.000 | Swine anti rabbit HRP | Dako (P0217) | 1:2000 |
| Phospho-ß-CATENIN | Cell Signaling (9566) | 1:1000 | Anti-Rabbit IgG (H+L), HRP Conjugate | Promega W401B | 1:5000 |
| Total β-CATENIN | Cell Signaling (9582) | 1:1000 | Anti-Rabbit IgG (H+L), HRP Conjugate | Promega W401B | 1:5000 |
| β-ACTIN | BioLegend (664802) | 1:2500 | HRP Goat anti-rat IgG | BioLegend 405405 | 1:5000 |
HOX injury (BPD mouse model)
Newborn pups were subjected to HOX (85% O2) injury from P0–P8 in a chamber (Proox Model 110, Biospherix). To minimize oxygen toxicity and bias, nursing dams were rotated every 24 h between NOX and HOX. Pups and dams received food and water ad libitum.
Vascular morphometry
The degree of muscularization was determined from 3 μm mouse lung sections. Morphological assessment of lung vessel muscularization (outer diameter: 10–20 μm, 20–70 μm, 70–150 μm and >150 μm) was performed via computer-assisted analysis (Leica Q Win Standard analyzing software) at 40× or 63× magnification under a microscope. The analytical software detected the vessels’ endothelium that appeared in a brown color (vWF staining) and the muscle tissue surrounding the vessels that appeared in a violet color (α-SMA staining). The software distinguished between non-muscularized vessels (no smooth muscle cells detectable with α-SMA staining), partially muscularized (minimum one smooth muscle cell and maximum 75% of the vessel circumference with α-SMA staining) and fully muscularized (>75% of the vessel circumference with α-SMA staining).
Gene arrays
The data from the microarray experiment have been deposited in NCBI’s Gene Expression Omnibus (GEO) and is accessible through GEO Series accession number GSE113104.
Statistical analyses
Significance was determined by two-tailed Student's t-test using GraphPad PRISM statistical analysis software. All data are presented as mean ± SEM. Values of P < 0.05 were considered significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We would like to thank Kerstin Goth and Jana Rostkovius for managing and genotyping the mice used for experiments. We want to thank Ewa Bieniek for assistance in immunostaining. We also thank Caterina Tiozzo for giving us the human BPD samples.
Conflict of Interest statement. None declared.
Funding
National Heart, Lung, and Blood Institute (NHLBI) support [HL107307 to S.B.]; Deutsche Forschungsgemeinschaft [DFG; BE4443/1-1, BE4443/4-1, BE4443/6-1, KFO309 P7 and SFB1213-projects A02 and A04 to S.B.]; Landes-Offensive zur Entwicklung Wissenschaftlich-Ökonomischer Exzellenz (LOEWE); Universitätsklinikum Giessen & Marburg (UKGM); Universities of Giessen and Marburg Lung Center (UGMLC); DZL and COST [BM1201]; Whenzhou Medical University and the National Natural Science Foundation of China [grant number 81472601 to J.S.Z.]; Forschungsförderung of the UKGM and the Excellence Cluster Cardio-Pulmonary System (ECCPS) [to C.M.C.]; The ECCPS [to E.E.A.]; Program of Competitive Growth and State Assignment [20.5175.2017/6.7 to A.A.R.]. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions
Concept and design: CMC, SB and JSZ. Acquisition of data: CMC, AM, DK, SR, JW, MK and FY. Analysis and interpretation: CMC, AM, JW, DK, EEA, FY and SB. Drafting and editing of the manuscript: CMC, AM, EEA, HE, KPZ, GB, AR, RTS, IR, REM, RR, SB and JSZ. All authors read and approved the final manuscript.
References
- 1. Benjamin J.T., Smith R.J., Halloran B.A., Day T.J., Kelly D.R. and Prince L.S. (2007) FGF-10 is decreased in bronchopulmonary dysplasia and suppressed by toll-like receptor activation. Am. J. Physiol. Lung Cell. Mol. Physiol., 292, L550–L558. [DOI] [PubMed] [Google Scholar]
- 2. Watterberg K.L., Demers L.M., Scott S.M. and Murphy S. (1996) Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops.Pediatrics, 97, 210–215. [PubMed] [Google Scholar]
- 3. Klinger G., Levy I., Sirota L. Boyko V., Lerner-Geva L. and Reichman B. (2010) Outcome of early-onset sepsis in a national cohort of very low birth weight infants. Pediatrics, 125, e736–e740. [DOI] [PubMed] [Google Scholar]
- 4. Stoll B.J., Hansen N.I., Bell E.F. Shankaran S., Laptook A.R. Walsh M.C., Hale E.C., Newman N.S., Schibler K., Carlo W.A. et al. (2010) Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics, 126, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benjamin J.T., Carver B.J., Plosa E.J., Yamamoto Y., Miller J.D. Liu J.H., van der Meer R., Blackwell T.S. and Prince L.S. (2010) NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression. J. Immunol., 185, 4896–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carver B.J., Plosa E.J., Stinnett A.M. Blackwell T.S. and Prince L.S. (2013) Interactions between NF-kappaB and SP3 connect inflammatory signaling with reduced FGF-10 expression. J. Biol. Chem., 288, 15318–15325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Entesarian M., Dahlqvist J., Shashi V. Stanley C.S., Falahat B. Reardon W. and Dahl N. (2007) FGF10 missense mutations in aplasia of lacrimal and salivary glands (ALSG). Eur. J. Hum. Genet., 15, 379–382. [DOI] [PubMed] [Google Scholar]
- 8. Entesarian M., Matsson H., Klar J., Bergendal B. Olson L., Arakaki R., Hayashi Y., Ohuchi H., Falahat B., Bolstad A.I. et al. (2005) Mutations in the gene encoding fibroblast growth factor 10 are associated with aplasia of lacrimal and salivary glands. Nat. Genet., 37, 125–127. [DOI] [PubMed] [Google Scholar]
- 9. Milunsky J.M., Zhao G., Maher T.A., Colby R. and Everman D.B. et al. (2006) LADD syndrome is caused by FGF10 mutations. Clin. Genet., 69, 349–354. [DOI] [PubMed] [Google Scholar]
- 10. Rohmann E., Brunner H.G., Kayserili H., Uyguner O.,Nurnberg G., Lew E.D., Dobbie A., Eswarakumar V.P., Uzumcu A., Ulubil-Emeroglu M. et al. (2006) Mutations in different components of FGF signaling in LADD syndrome. Nat. Genet., 38, 414–417. [DOI] [PubMed] [Google Scholar]
- 11. Klar J., Blomstrand P., Brunmark C., Badhai J., Hakansson H.F., Brange C.S., Bergnedal B. and Dahl N. (2011) Fibroblast growth factor 10 haploinsufficiency causes chronic obstructive pulmonary disease. J. Med. Genet., 48, 705–709. [DOI] [PubMed] [Google Scholar]
- 12. Sekine K., Ohuchi H., Fujiwara M., Yamasaki M., Yoshizawa T., Sato T., Yagishita N., Matsui D., Koga Y. Itoh N. et al. (1999) Fgf10 is essential for limb and lung formation. Nat. Genet., 21, 138–141. [DOI] [PubMed] [Google Scholar]
- 13. Arman E., Haffner-Krausz R., Gorivodsky M. and Lonai P. (1999) Fgfr2 is required for limb outgrowth and lung-branching morphogenesis. Proc. Natl. Acad. Sci. U. S. A., 96, 11895–11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Moerlooze L., Spencer-Dene B., Revest J.M., Hajihosseini M., Rosewell I. and Dickson C. (2000) An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal–epithelial signalling during mouse organogenesis. Development, 127, 483–492. [DOI] [PubMed] [Google Scholar]
- 15. Ohuchi H., Hori Y., Yamasaki M., Harada H., Sekine K., Kato S. and Itoh N. (2000) FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem. Biophys. Res. Commun., 277, 643–649. [DOI] [PubMed] [Google Scholar]
- 16. Bellusci S., Grindley J., Emoto H., Itoh N. and Hogan B.L. (1997) Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development, 124, 4867–4878. [DOI] [PubMed] [Google Scholar]
- 17. Chao C.M., Yahya F., Moiseenko A., Tiozzo C., Shrestha A., Ahmadvand N., El Agha E., Quantius J., Dilai S. Kheirollahi V. et al. (2017) Fgf10 deficiency is causative for lethality in a mouse model of bronchopulmonary dysplasia. J. Pathol., 241, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El Agha E. and Bellusci S. (2014) Walking along the fibroblast growth factor 10 route: a key pathway to understand the control and regulation of epithelial and mesenchymal cell-lineage formation during lung development and repair after injury. Scientifica (Cairo), 2014, 538379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chao C.M., El Agha E., Tiozzo C. Minoo P. and Bellusci S. (2015) A breath of fresh air on the mesenchyme: impact of impaired mesenchymal development on the pathogenesis of bronchopulmonary dysplasia. Front. Med. (Lausanne), 2, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gupte V.V., Ramasamy S.K., Reddy R., Lee J., Weinreb P.H., Violette S.M., Guenther A., Warburton D., Driscoll B., Minoo P. et al. (2009) Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med., 180, 424–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volckaert T., Dill E., Campbell A., Tiozzo C., Majika S., Bellusci S. and De Langhe S.P. (2011) Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Invest., 121, 4409–4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ramasamy S.K., Mailleux A.A., Gupte V.V., Mata F., Sala F.G., Veltmaat J.M., Del Moral P.M., De Langhe S., Parsa S. and Kelly L.K. (2007) Fgf10 dosage is critical for the amplification of epithelial cell progenitors and for the formation of multiple mesenchymal lineages during lung development. Dev. Biol., 307, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yao Q., Renault M.A., Chapouly C., Vandierdonck S., Belloc I., Jaspard-Vinassa B., Daniel-Lamazière J.M., Laffargue M., Merched A., Desgranges C. and Gadeau A.P. (2014) Sonic hedgehog mediates a novel pathway of PDGF-BB-dependent vessel maturation. Blood ,123(15), 2429–2437. [DOI] [PubMed] [Google Scholar]
- 24. White A.C., Lavine K.J. and Ornitz D.M. (2007) FGF9 and SHH regulate mesenchymal Vegfa expression and development of the pulmonarycapillary network. Development, 134(20), 3743–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herriges J.C., Verheyden J.M., Zhang Z., Sui P., Zhang Y., Anderson M.J., Swing D.A., Zhang Y., Lewandoski M. and Sun X.(2015) FGF-regulated ETV transcription factors control FGF-SHH feedback loop in lung branching. Dev. Cell, 35(3), 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Muller-Redetzky H.C., Kummer W., Pfeil U., Hellwig K., Will D., Paddenberg R., Tabeling C., Hippenstiel S., Suttorp N. and Witzenrath M. (2012) Intermedin stabilized endothelial barrier function and attenuated ventilator-induced lung injury in mice. PLoS One, 7, e35832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hao S.L., Yu Z.H., Qi B.S., Luo J.Z. and Wang W.P. (2011) The antifibrosis effect of adrenomedullin in human lung fibroblasts. Exp. Lung Res., 37, 615–626. [DOI] [PubMed] [Google Scholar]
- 28. Qing X., Svaren J. and Keith I.M. (2001) mRNA expression of novel CGRP1 receptors and their activity-modifying proteins in hypoxic rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol., 280, L547–L554. [DOI] [PubMed] [Google Scholar]
- 29. Sundivakkam P.C., Kwiatek A.M., Sharma T.T., Minshall R.D. and Tiruppathi C. (2009) Caveolin-1 scaffold domain interacts with TRPC1 and IP3R3 to regulate Ca2+ store release-induced Ca2+ entry in endothelial cells. Am. J. Physiol. Cell Physiol., 296, C403–C413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hollie N.I., Konaniah E.S., Goodin C. and Hui D.Y. (2014) Group 1B phospholipase A(2) inactivation suppresses atherosclerosis and metabolic diseases in LDL receptor-deficient mice. Atherosclerosis, 234, 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mous D.S., Buscop-van Kempen M.J., Wijnen R.M.H., Tiobboel D. and Rottier R.J. (2017) Changes in vasoactive pathways in congenital diaphragmatic hernia associated pulmonary hypertension explain unresponsiveness to pharmacotherapy. Respir. Res., 18, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishikura K., Yamada N., Ito M., Ota S., Nakamura M., Isaka N. and Nakano T. (2006) Beneficial acute effects of rho-kinase inhibitor in patients with pulmonary arterial hypertension. Circ. J., 70, 174–178. [DOI] [PubMed] [Google Scholar]
- 33. Knipe R.S., Probst C.K., Lagares D., Franklin A., Spinney J.J., Brazee P.L., Grasberger P., Zhang L., Black K.E., Sakai N. et al. (2018) The rho kinase isoforms ROCK1 and ROCK2 each contribute to the development of experimental pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol., 58, 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. MacKay C.E. and Knock G.A. (2015) Control of vascular smooth muscle function by Src-family kinases and reactive oxygen species in health and disease. J. Physiol., 593, 3815–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fu P., Ebenezer D.L., Berdyshev E.V., Bronova I.A., Shaaya M., Harijith A. and Natarajan V. (2016) Role of sphingosine kinase 1 and S1P transporter Spns2 in HGF-mediated lamellipodia formation in lung endothelium. J. Biol. Chem., 291, 27187–27203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Haberberger R.V., Tabeling C., Runciman S., Gutbier B., Konig P., Andratsch M., Schutte H., Suttorp N., Gibbins I. and Witzenrath M. (2009) Role of sphingosine kinase 1 in allergen-induced pulmonary vascular remodeling and hyperresponsiveness. J. Allergy Clin. Immunol., 124, 933–941e931-939. [DOI] [PubMed] [Google Scholar]
- 37. Lien Y.C., Feinstein S.I., Dodia C. and Fisher A.B. (2012) The roles of peroxidase and phospholipase A2 activities of peroxiredoxin 6 in protecting pulmonary microvascular endothelial cells against peroxidative stress. Antioxid. Redox Signal., 16, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee K.S., Kim S.R., Park S.J., Min K.H., Lee K.Y., Choe Y.H., Park S.Y., Chai O.H., Zhang X., Song C.H. et al. (2008) Mast cells can mediate vascular permeability through regulation of the PI3K-HIF-1alpha-VEGF axis. Am. J. Respir. Crit. Care Med., 178, 787–797. [DOI] [PubMed] [Google Scholar]
- 39. Parsa S., Kuremoto K., Seidel K., Tabatabai R., Mackenzie B., Yamaza T., Akiyama K., Branch J., Koh C.J., Al Alam D. et al. (2010) Signaling by FGFR2b controls the regenerative capacity of adult mouse incisors. Development, 137, 3743–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parsa S., Ramasamy S.K., De Langhe S., Gupte V.V., Haigh J.J., Medina D. and Bellusci S. (2008) Terminal end bud maintenance in mammary gland is dependent upon FGFR2b signaling. Dev. Biol., 317, 121–131. [DOI] [PubMed] [Google Scholar]
- 41. Volckaert T., Campbell A., Dill E., Li C., Minoo P. and De Langhe S. (2013) Localized Fgf10 expression is not required for lung branching morphogenesis but prevents differentiation of epithelial progenitors. Development, 140, 3731–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. MacKenzie B., Henneke I., Hezel S., Al Alam D., El Agha E., Chao C.M., Quantius J., Wilhelm J., Jones M., Goth K. et al. (2015) Attenuating endogenous Fgfr2b ligands during bleomycin-induced lung fibrosis does not compromise murine lung repair. Am. J. Physiol. Lung Cell. Mol. Physiol., 308, L1014–L1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kool H., Mous D., Tibboel D., de Klein A. and Rottier R.J. (2014) Pulmonary vascular development goes awry in congenital lung abnormalities. Birth Defects Res. C Embryo Today, 102, 343–358. [DOI] [PubMed] [Google Scholar]
- 44. Mailleux A.A., Kelly R., Veltmaat J.M., De Langhe S.P., Zaffran S., Thiery J.P. and Bellusci S. (2005) Fgf10 expression identifies parabronchial smooth muscle cell progenitors and is required for their entry into the smooth muscle cell lineage. Development, 132, 2157–2166. [DOI] [PubMed] [Google Scholar]
- 45. Oladipupo S.S., Smith C., Santeford A., Park C., Sene A., Wiley L.A., Osei-Owusu P., Hsu J., Zapata N., Liu F. et al. (2014) Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc. Natl. Acad. Sci. U. S. A., 111, 13379–13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. House SL, Castro A.M., Lupu T.S., Weinheimer C., Smith C., Kovacs A. and Ornitz D.M. (2016) Endothelial fibroblast growth factor receptor signaling is required for vascular remodeling following cardiac ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol., 310, H559–H571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moiseenko A., Kheirollahi V., Chao C.M., Ahmadvand N., Quantius J., Wilhelm J., Herold S., Ahlbrecht K., Morty R.E., Rizvanov A.A. et al. (2017) Origin and characterization of alpha smooth muscle actin-positive cells during murine lung development. Stem Cells, 35, 1566–1578. [DOI] [PubMed] [Google Scholar]
- 48. Rochais F., Sturny R., Chao C.M., Mesbah K., Bennett M., Mohun T.J., Bellusci S. and Kelly R.G. (2014) FGF10 promotes regional foetal cardiomyocyte proliferation and adult cardiomyocyte cell-cycle re-entry. Cardiovasc. Res., 104, 432–442. [DOI] [PubMed] [Google Scholar]
- 49. Sakaue H., Konishi M., Ogawa W., Asaki T., Mori T., Yamasaki M., Takata M., Ueno H., Kato S., Kasuga M. et al. (2002) Requirement of fibroblast growth factor 10 in development of white adipose tissue. Genes Dev., 16, 908–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Al Alam D., El Agha E., Sakurai R., Kheirollahi V.,Moiseenko A., Danapoulos S., Shrestha A., Schmoldt C., Quantius J., Herold S. et al. (2015) Evidence for the involvement of fibroblast growth factor 10 in lipofibroblast formation during embryonic lung development. Development, 142, 4139–4150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sugimoto K., Yoshida S., Mashio Y., Toyota N., Xing Y., Xu H., Fujita Y., Huang Z., Touma M. and Wu Q. (2014) Role of FGF10 on tumorigenesis by MS-K. Genes Cells, 19, 112–125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.