

Abstract
Comprehensive study of novel microbial organisms capable of degrading fluorene is crucial to develop essential strategies for further application on enhanced bioremediation technologies. Many fluorene-degrading bacteria have been studied; however, little information about the genome sequences of these organisms, which would facilitate investigation of the molecular mechanisms of fluorene degradation, is available. In this study, a bacterial strain designated SMT-1, which uses fluorene as its sole carbon source, was isolated from Laogang landfill in Shanghai, People’s Republic of China, and identified as a Pseudomonas sp., based on 16S rRNA gene sequence analysis. Maximum growth and degrading activity of strain SMT-1 were observed at 30°C, pH 7.0 and 200 r/min in mineral salt medium containing 0.4 mm fluorene. We obtained a draft genome sequence of strain SMT-1 to gain insight into the genetic mechanisms for the degradation of aromatic compounds. Sequences greater than 1 kb in length were obtained by Illumina sequencing; strain SMT-1 was found to contain 5542 predicted genes. This working draft genome comprises 68 contigs and DNA scaffolds and has a total size of 6 108 237 bp and a calculated G + C content of 61.59%. Amino acid metabolism clusters were enriched in SMT-1 genes annotation, with the highest abundant observed for the “ABC transporters” subcategories, followed by transcription, energy production and conversion, and inorganic ion transport and metabolism. The genomic information for SMT-1 provides a useful resource for elucidating the molecular mechanism of fluorene catabolism.
Keywords: Pseudomonas sp. strain SMT-1, fluorene, genome, illumina sequencing, contigs
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are of concern to human health and the environment due to their toxicity and potential to move through various trophic levels in an ecosystem. Some low-molecular-weight PAHs are acutely toxic1 and most high-molecular-weight compounds have mutagenic and potential carcinogenic effects.2 Due to their hydrophobic nature, most PAHs bind to particulates in soil and sediments, rendering them less available for biological uptake.3 Fluorene is among the most prevalent and persistent PAHs found in the environment, and it is listed as a priority pollutant by the US Environmental Protection Agency due to its toxicity and persistence in terrestrial and aquatic environments.4 Some microorganisms have an innate capacity to degrade chemicals that pollute the environment. Fluorene biodegradation at a hazardous-waste site by the indigenous microflora has been reported.5 In a previous study, a strain of Pseudomonas vesicularis that used fluorene as the sole source of carbon and energy was isolated, but no metabolites from the bacterial degradation of fluorene have yet been described.6 Many fluorene-degrading bacteria, including Arthrobacter, Brevibacterium, Burkholderia, Mycobacterium, Pseudomonas, and Sphingomonas7–9 have been used in studies on biodegradation of fluorene. To date, none of the fluorene-degrading bacteria genomes except Sphingobium sp. strain LB12610 has been sequenced to investigate the molecular mechanism. Characterizing additional fluorene-degrading strains may increase our understanding of the molecular mechanism of fluorene degradation. Here, we report the isolation, characterization, and draft genome sequence of Pseudomonas sp. strain SMT-1, an efficient degrader of fluorene. This strain could also degrade fluoranthene, phenanthrene, and dibenzofuran (DBF) as the primary substrate. The genome of strain SMT-1 was sequenced using a whole-genome shotgun strategy on the Illumina MiSeq platform (2 × 250 bp paired end) at a Shanghai Personal Biotechnology Co. Ltd. The draft genome sequence of strain SMT-1 enabled us to identify genes related to fluorene degradation; further analysis of these genes will provide valuable information for elucidating the full mechanism of fluorene degradation and metabolic pathways.
Materials and Methods
Growth conditions and main features of the organism
The initial enrichment culture was started by adding 1 g soil sample into 50 mL sterile mineral salt medium (MSM) containing 0.1% fluorene. This inoculum was used to select a strain capable of degrading fluorene. Maximum growth of the new isolate, strain SMT-1, was observed under aerobic conditions at 30°C, pH 7.0, and shaker speed of 200 rev/min, in MSM containing 0.4 mm fluorene as the carbon source. The optimal conditions for strain storage are 50% glycerol at −80°C. Evolutionary similarity analysis was performed by comparing16S ribosomal RNA (rRNA) gene sequences using Basic Local Alignment Search Tool (BLAST; http://blast.ncbi.nlm.nih.gov/Blast.cgi). This analysis revealed that the isolate might be Pseudomonas species. Strain SMT-1 has the following morphological characteristics: rod-shaped, wrinkled appearance, reddish brown in color, and Gram negative (Figure 1). Physiological and biochemical characteristics, such as utilization of different carbon sources and enzymatic properties, were determined by China Center for Type Culture Collection (CCTCC). Compared with other reported Pseudomonas species, strain SMT-1 has a wide substrate range (Table 5). This strain has been deposited at the CCTCC, under reference number AB2018208.
Figure 1.
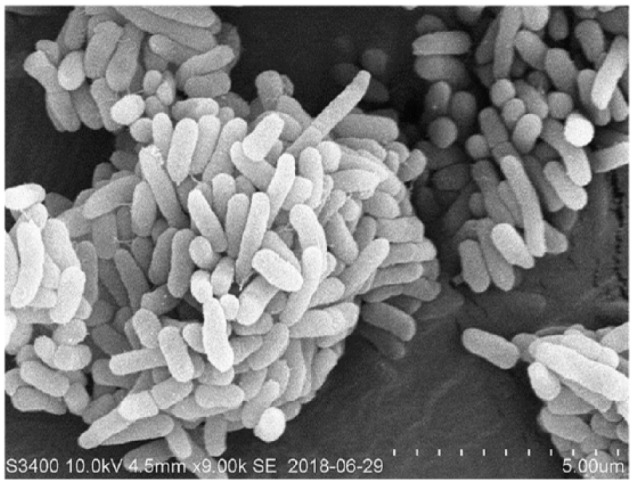
Scanning electron microscope (SEM) image showing the rod-shaped bacteria isolate SMT-1. SMT-1 bacteria wrinkled in appearance, are reddish brown in color, and are Gram negative.
Table 5.
Biochemical and physiological characteristics of strain SMT-1 and comparisons with other reported Pseudomonas species.
| Test | Substrate | Biochemical reactions | Results | |
|---|---|---|---|---|
| SMT-1 | Pseudomonas geniculata N1 | |||
| ONPG | O-nitrophenyl-galactoside | Beta-galactosidase | + | + |
| ADH | Arginine | Arginine dihydrolase | + | + |
| LDC | Lysine | Lysine decarboxylase | − | + |
| ODC | Ornithine | Aminase decarboxylation | − | − |
| CIT | Sodium citrate | Citric acid utilization | + | + |
| H2S | Sodium thiosulfate | H2S generation | + | − |
| URE | Urea | Urease | − | + |
| TDA | Tryptophan | Tryptophan deaminase | − | − |
| IND | Tryptophan | Indole production | − | − |
| VP | Pyruvate | 3-Hydroxybutanone produces acetyl methyl carbinol | + | |
| GEL | Kohn gelatin | Gelatinase | − | + |
| GLU | glucose | Fermentation/oxidation (4) | + | − |
| MAN | Mannitol | Fermentation/oxidation (4) | + | − |
| INO | Inositol | Fermentation/oxidation (4) | − | − |
| SOR | Sorbitol | Fermentation/oxidation (4) | − | − |
| RHA | Rhamnose | Fermentation/oxidation (4) | − | − |
| SAC | Sucrose | Fermentation/oxidation (4) | − | − |
| MEL | Midiose | Fermentation/oxidation (4) | + | − |
| AMY | Amygdalin | Fermentation/oxidation (4) | − | − |
| ARA | Arabic candy | Fermentation/oxidation (4) | + | − |
: positive reaction; −: negative reaction.
Nucleotide sequence accession numbers. The whole-genome shotgun sequencing data have been deposited at DDBJ/ENA/GenBank under accession number QJOV00000000. The version described in this article is the first version QJOV01000000.
Genome sequencing and assembly
The draft genome sequence was generated by whole-genome shotgun strategy using Illumina technology. The total amount of sequence data comprises 1 224 164 919 bases. A next-generation sequencing (NGS) DNA library was constructed and sequenced using the Illumina MiSeq platform (2 × 250 bp paired end), which generated 4 900 736 reads (Table S1). The raw read sequences were cleaned up using adapter removal (version 2.1.7)11 to remove the low-quality ends. The quality-controlled reads were assembled using SOAPec (v2.0),12 with a default set of k-mer sizes and options; hence, some contigs and fold coverage were eliminated. After filtering, a total of 4 769 110 (97.31%) high-quality paired end reads were obtained and assembled into 68 high-quality contigs providing a 183× coverage of the genome. The basic situation of data filtering and quality control information of the Illumina sequencing is shown in supplementary Table S2. A summary of the sequencing project details is shown in Table 1.
Table 1.
Sequencing project details.
| No. | Description | Term |
|---|---|---|
| 1 | Sequencing platforms | Illumina MiSeq |
| 2 | Fold coverage | 183× |
| 3 | DDBJ/ENA/GenBank ID | QJOV00000000 |
| 4 | GenBank Date of Release | 10 June 2018 |
| 5 | Bio project | PRJNA473277 |
| 6 | Gene annotation method | NCBI Proka version 4.32 April 201513 software |
| 7 | Assemblers | A5-miseq v2015052214 |
| 8 | Finishing quality | High-quality draft |
| 9 | Libraries used | Paired-end next-generation sequencing (NGS) |
| 10 | Bio sample | SAMN09269772 |
| 11 | SUBID | SUB4089701 |
| 12 | Organism | Pseudomonas sp. SMT-1 |
Coassembly of the sequence was performed using A5-miseq tools v20150522,14 which allows evaluation of assembly quality, automation of the process of adapter trimming, quality filtering, error correction, contig and scaffold generation, and detection of misassembles.
A summary of genomic assembly statistics is shown in Table 2.
Table 2.
Genomic assembly statistics.
| Sample | Property | Contig | Scaffold |
|---|---|---|---|
| SMT-1 | Shortest (bp) | 1326 | 1326 |
| Longest (bp) | 550 016 | 550 016 | |
| Total sequence number | 68 | 68 | |
| N20 (bp) | 351 711 | 351 711 | |
| N50 (bp) | 198 327 | 198 327 | |
| N90 (bp) | 58 153 | 58 153 | |
| N number | 0 | 0 | |
| N rate | 0 | 0 | |
| Total sequence length | 6 108 237 | 6 108 237 | |
| GC content | 61.59 | 61.59 | |
| Sequences greater than 1 kb | 68 | 68 |
Abbreviations: GC, guanine-cytosine; N number, uncertain number of bases; N rate, the ratio of the number of bases to the length of the splicing sequence.
Genome properties
The draft genome of Pseudomonas sp. strain SMT-1 is 6 108 237 bp in length with a calculated G + C content of about 61.59% (Table 3). The protein coding density is 0.919 genes per kb with an average intergenic length of 951.4 bp (Table S3). Of 5828 genes, 5542 were predicted protein-coding genes, accounting for 87.50% of the total sequences. There were 92 RNA genes identified in the genome, which were further partitioned into 71 transfer RNA (tRNA) genes, 5S rRNA genes, 6 16S rRNA genes, 5 23S rRNA genes, and 4 non-coding RNA (ncRNA) genes.
Table 3.
Genome composition.
| Characteristics | Value | Total (%) |
|---|---|---|
| Genome size (bp) | 6 108 237 | 100.00 |
| Genes (total) | 5828 | 100.00 |
| CDS (total) | 5736 | 100.00 |
| Genes (coding) | 5542 | 95.1 |
| CDS (coding) | 5542 | 96.6 |
| Genes (RNA) | 92 | 1.6 |
| Pseudo genes (total) | 194 | 3.3 |
| G + C (bp) | 68rc | 61.59% |
| DNA scaffolds | 68 | 100% |
| Gene average length (bp) | 951.4 bp | |
| Genes assigned to COGs | 4882 | 86.88% |
| CRISPR repeats | 3 | 0.0086% |
Most of the predicted genes (64.9%) could be assigned to one of 25 functional clusters of orthologous groups (COGs), and 23.2% were classified as proteins of unknown function, while the remaining genes were annotated as hypothetical proteins. The distribution of genes into COGs’ functional categories is presented in Table 4.
Table 4.
Number of genes associated with general COG functional categories.
| Category | Number of genes | Percentage | Description |
|---|---|---|---|
| A | 1 | 0.2 | RNA processing and modification |
| B | 0 | 0.00 | Chromatin structure and dynamics |
| C | 326 | 5.9 | Energy production and conversion |
| D | 34 | 0.61 | Cell cycle control, cell division, chromosome partitioning |
| E | 432 | 7.8 | Amino acid transport and metabolism |
| F | 97 | 1.8 | Nucleotide transport and metabolism |
| G | 210 | 3.8 | Carbohydrate transport and metabolism |
| H | 159 | 2.9 | Coenzyme transport and metabolism |
| I | 136 | 2.5 | Lipid transport and metabolism |
| J | 190 | 3.4 | Translation, ribosomal structure, and biogenesis |
| K | 413 | 7.5 | Transcription |
| L | 227 | 4.1 | Replication, recombination, and repair |
| M | 298 | 5.4 | Cell wall/membrane/envelope biogenesis |
| N | 41 | 0.7 | Cell motility |
| O | 173 | 3.1 | Posttranslational modification, protein turnover, chaperones |
| P | 319 | 5.8 | Inorganic ion transport and metabolism |
| Q | 113 | 2.0 | Secondary metabolites biosynthesis, transport, and catabolism |
| S | 0 | 0.00 | General function prediction only |
| T | 1287 | 23.2 | Function unknown |
| U | 274 | 4.9 | Signal transduction mechanisms |
| V | 83 | 1.5 | Intracellular trafficking, secretion, and vesicular transport |
| W | 68 | 1.2 | Defense mechanisms |
| X | 1 | 0.01 | Extracellular structures |
| Y | 0 | 0.00 | Nuclear structure |
| Z | 0 | 0.00 | Cytoskeleton |
| – | 737 | 13.1162 | Not in EggNOG |
The total is based on the total number of protein-coding genes in the genome.
Genome annotation
Scaffolds were submitted to GenBank for gene annotation, using the National Center for Biotechnology Information (NCBI) Prokaryotic Genome Annotation Pipeline version 4.32, April 2015,13 and GeneMark (http://exon.gatech.edu). The 16S rRNA gene sequence of SMT-1 shared 99% similarity with Pseudomonas stutzeri B15 and several Pseudomonas species. The Average Nucleotide Identity (ANI) of the SMT-1 genome to near-neighbor P stutzeri B15 was calculated using both best hits (1-way ANI) and reciprocal best hits (2-way ANI) between 2 genomic datasets, as calculated in Goris et al.15 Typically, the ANI values between genomes of the same species are above 95%. Values below 75% are not to be trusted. Strain SMT-1 and the closest related P stutzeri B15 share 97.67% nucleotide identity distributions (Figure 2). In the EggNOG annotations database, most genes were assigned to “function unknown,” “amino acid transport and metabolism,” “transcription,” and “inorganic ion transport and metabolism” (Table 4). Seventy-four genes in strain SMT-1 were annotated as being involved in the metabolism of xenobiotic and aromatic compounds and the metabolism of aromatic compounds, such as catechol 1,2-dioxygenase and protocatechuate. Two of these genes encode the alpha and beta subunit of 3,4 dioxygenase, which is an aromatic ring-hydroxylating enzyme and dehydrogenase involved in the degradation of polycyclic aromatic compounds.16 In addition, several putative monooxygenases and dioxygenases were identified in the genome, and 619 protein-coding sequences were annotated as membrane transport proteins, which would further explain SMT-1 ability to degrade various aromatic compounds. The genome also contains other enzyme-encoding genes, such as ferredoxin reductase, which functions as an electron transporter during fluorene degradation. Genes associated with benzoate degradation, along with some membrane-transport proteins and transcriptional regulators, were also annotated in the genome of strain SMT-1.17 CRISPR finder (http://crispr.i2bc.paris-saclay.fr/Server/) was used to predict direct repeats in the whole genome (forward repeats) and spacers.18 Three CRISPR with lengths 120 bp (0.0020%), 328 bp (0.0054%), and 73 bp (0.0012%) were identified in strain SMT-1 (Table S4).
Figure 2.

Average Nucleotide Identity (ANI) distribution. ANI was calculated using both best hits (1-way ANI) and reciprocal best hits (2-way ANI) between 2 genomic datasets of strain SMT-1 and Pseudomonas stutzeri B15, hence found to be share 97.67% nucleotide identity distributions.
Cell growth and fluorene degradation
Determination of optimal cell growth and fluorene degradation analysis was performed up to 10 days under several physiological conditions including different ranges of pH. Slight growth of strain SMT-1 was observed in the pH range 7.0 to 9.0 (Figure 3A), while an optimum growth and degradation of fluorene was substantial at 30°C, pH 7.0, and 200 r/min in MSM containing 0.4 mm fluorene. Strain SMT-1, which efficiently degrades fluorene, was isolated from a landfill in Shanghai industrial zone. This strain could use fluorene as a carbon source and degrade approximately 0.4 mM in 8 days under neutral conditions at 30°C and 200 r/min. The degradation of fluorene by strain SMT-1 under different pH values was shown in Figure 3B. Pseudomonas sp. strain SMT-1 can also grow on and degrade fluoranthene, phenanthrene, and DBF as the primary substrate. The degradation comparison of these different PAHs by Pseudomonas sp. strain SMT-1 in addition to fluorene was conducted under similar conditions at 30°C, pH 7.0, and 200 r/min. The degradation results within 6 consecutive days were fluorene (85%), phenanthrene (48.4%), DBF (47.5%), and fluoranthene (29.1%; Figure 4). To detect fluorene degradation, the fluorene present in the culture medium was quantified by high-performance liquid chromatography (HPLC; Agilent 1200 series) equipped with an Eclipse XDB-C18 column (column size, 250 × 64.6 mm; particle size, 5 mm; Agilent), ultraviolet (UV) visible detector set at 254 nm. A mixture of MeOH and ddH2O (80:20 v/v) was used as the mobile phase, at a flow rate of 0.8 mL/min and 5 µL injection volume. An important gene, such as α and β subunits of a dioxygenase complex (FlnA1A2), showing 36% and 35% sequence identity, respectively, with the subunits of an angular dioxygenase from Terrabacter sp. DBF63, fluorene mono-oxygenase, 9-hydroxyfluorene dehydrogenase, and large and small subunits of benzoate 1,2-dioxygenase, involved in the upper fluorene biodegradation pathway, were identified in the fluorene degrader Pseudomonas sp. SMT-1. The presence of these enzymes explains the fluorene degradation capacities of Pseudomonas sp. strain SMT-1. The genes for protocatechuate 3,4-dioxygenase and catechol 1,2-dioxygenase of the central protocatechuate catabolic pathway for aromatic degradation were also present in strain SMT-1 genome. The possible metabolic pathway used by strain SMT-1 fluorene biodegradation is predicted to be through mono-oxygenation of fluorene at the C-9 position to give 9-fluorenol, which is then dehydrogenated to 9-fluorenone to form phthalic acid and protocatechuic acid as intermediate products.8 A series of experiments were conducted to identify the morphological, physiological, and biochemical characteristics of the strain SMT-1. The morphology was studied using scanning electron microscope (SEM). The physiological and biochemical characteristics, such as the utilization of different carbon sources and enzymatic properties (Table 5), were determined by CCTCC.
Figure 3.

(A) Growth of strain SMT-1 at different pH values and (B) fluorene degradation by strain SMT-1 under different pH values.
Figure 4.

Degradation comparison of different polycyclic aromatic hydrocarbons (PAHs) by Pseudomonas sp. strain SMT-1. (A) Growth of strain SMT-1 on different polycyclic aromatic hydrocarbons. (B) Degradation results within 6 consecutive days: fluorene (85%), phenanthrene (48.4%), dibenzofuran (47.5%), and fluoranthene (29.1%).
Results
In this study, strain SMT-1, which is capable of degrading fluorene and other aromatic compounds, was isolated, characterized, and sequenced. Sequencing of an NGS DNA library generated about 4 769 110 (97.31%) of clean reads providing 183× coverage of the genome. After sequence assembly, the draft genome of strain SMT-1 was obtained, containing 68 scaffolds and consisting of 6 108 237 bp, with a G + C content of 61.59% (Table 3). The genome contains 5542 predicted genes, including 92 rRNA genes, with an average length of 951.4 bp. A total of 68 rc genes were annotated in the NCBI-nr database. The genome annotation has revealed that more genes were enriched in amino acid metabolism pathways, more specifically, ATP-binding cassette (ABC) transporter subcategories, followed by transcription, energy production and conversion, and inorganic ion transport and metabolism. The degradation comparison results between different PAHs by strain SMT-1 within 6 consecutive days were 85% for fluorene, 48.4% for phenanthrene, 47.5% for DBF, and 29.1% for fluoranthene (Figure 4); these degradation characteristics revealed that strain SMT-1 is highly effective in fluorene degradation and adaptive to a substrate range. Three CRISPR with lengths 120 bp, 328 bp, and 73 bp were identified in SMT-1. The genome analyses of strain SMT-1 revealed the presence of important genes potentially involved in the metabolism of aromatic compounds.
Discussion
Polycyclic aromatic hydrocarbons are widespread environmental contaminants that can be released during the burning or disposal of organic matter and petroleum products. These compounds are of great concern due to their negative effects on the environment and human health; therefore, it is crucial to remove them from the environment. Various techniques have been used to remove PAHs, including biodegradation19 and photochemical degradation. However, biodegradation is a major mechanism and a viable bioremediation technology for organic pollutants.20 Nevertheless, many factors influence microorganisms to metabolize pollutants as substrates. Intensive study of novel microorganisms and their physiological and molecular mechanisms and catabolic pathways is an effective approach to identify significant factors for efficient elimination of pollutants. This study reports the isolation, characterization, and genome sequence analysis of Pseudomonas sp. strain SMT-1, which is capable of degrading fluorene. Pseudomonas sp. strain SMT-1 was selected from other screened species, and its genome was sequenced, based on its ability to grow on and degrade fluorene, its metabolic versatility, and relevant biotechnological diversity of applications. Optimal cell growth and fluorene degradation analysis was performed for up to 10 days under several conditions. An optimum growth and degradation of fluorene by strain SMT-1 was observed at 30°C, pH 7.0, and 200 r/min in MSM containing 0.4 mm fluorene. Strain SMT-1 contains important genes involved in the upper fluorene biodegradation pathway. The presence of these enzymes indicates that Pseudomonas sp. strain SMT-1 is an important candidate for fluorene catabolism. A possible metabolic pathway used by strain SMT-1 in the biodegradation of fluorene was predicted to occur through mono-oxygenation of fluorene at the C-9 position to yield 9-fluorenol, which is then dehydrogenated to 9-fluorenone to form phthalic acid and protocatechuic acid as intermediate products.8 Analysis of strain SMT-1 draft genome provided basic knowledge of the mechanisms used to metabolize fluorene in this bacterium.
In conclusion, a novel Pseudomonas sp. strain SMT-1, capable of degrading fluorene, was isolated from landfill soil in Shanghai industrial zone, characterized, and its genome sequenced. Fluorene is a highly persistent and toxic organic compound and presents a serious problem for wild life and human health. Biodegradation of fluorene is a mechanism used to clean up polluted environments; hence, sequencing the genomes of novel environmental microbes to unveil the metabolic processes of fluorene degradation will support in the construction of effective biodegradation processes. By sequencing Pseudomonas sp. strain SMT-1 genome, we identified genes associated with fluorene degradation. Further analysis of these genes will provide useful information for elucidating the complete mechanism of fluorene catabolism.
Supplemental Material
Supplemental material, Supporting_information for Isolation, Characterization, and Genomic Analysis of Pseudomonas sp. Strain SMT-1, an Efficient Fluorene-Degrading Bacterium by Mulugeta Desta, Weiwei Wang, Lige Zhang, Ping Xu and Hongzhi Tang in Evolutionary Bioinformatics
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Chinese National Science Foundation for Excellent Young Scholars (31422004), Science and Technology Commission of Shanghai Municipality (17JC1403300), and by the “Shuguang Program” (17SG09) supported by the Shanghai Education Development Foundation and the Shanghai Municipal Education Commission.
Declaration of conflicting interest:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: HZT outset and designed experiments. MD, WW, and ZLG performed experiments. HZT and PX contributed reagents and materials. MD performed all the bioinformatics analyses and wrote the manuscript. All authors discussed and revised the manuscript. All authors commented on the manuscript before submission. All authors read and approved the final manuscript.
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Darville R, Wilhm J. The effect of naphthalene on oxygen consumption and haemoglobin concentration of Chironomus attenuatus and oxygen consumption and the cycle of Tanytarsus dissimilis. Environ Toxicol Chem. 1984;3:135–141. [Google Scholar]
- 2. Mortelmans K, Wawarth T, Lawlor W, Speck B, Tainer Zeiger E. Salmonella mutagenicity tests II. Results from the testing of 270 chemicals. Environ Mutagen. 1986;8:1–119. [PubMed] [Google Scholar]
- 3. Peng R, Ai-Sheng X, Yong X, et al. Microbial biodegradation of polyaromatic hydrocarbons. FEMS Microbol Rev. 2008;32:927–955. [DOI] [PubMed] [Google Scholar]
- 4. Keith LH, Telliard WA. Priority pollutants: A perspective view. Environ Sci Technol. 1979;13:416–423. [Google Scholar]
- 5. Lee M, Wilson T, Ward H. Microbial degradation of selected aromatics in hazardous waste site. Dev Ind Microbiol. 1984;50:557–565. [Google Scholar]
- 6. Weissenfels W, Beyer M, Klein J. Degradation of fluorene and fluoranthene by pure bacterial cultures. Appl Microbiol Biotechnol 1990;32:479–484. [DOI] [PubMed] [Google Scholar]
- 7. Baboshin M, Akimov V, Baskunov B, Born T, Khan S, Golovleva L. Conversion of polycyclic aromatic hydrocarbons by Sphingomonas sp. VKM B-2434. Biodegradation. 2008;19:567–576. [DOI] [PubMed] [Google Scholar]
- 8. Boldrin B, Tiehm A, Fritzsche C. Degradation of phenanthrene, fluorene, fluoranthene, and pyrene by a Mycobacterium sp. Appl Environ Microbiol. 1993;59:1927–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wattiau P, Bastiaens L, van Herwijnen R, et al. Fluorene degradation by Sphingomonas sp. LB126 proceeds through protocatechuic acid: a genetic analysis. Res Microbiol. 2011;152:861–872. [DOI] [PubMed] [Google Scholar]
- 10. Augelletti F, Tremblay J, Agathos SN, Stenuit B. Draft whole-genome sequence of the fluorene-degrading Sphingobium sp. strain LB126, isolated from polycyclic aromatic hydrocarbon-contaminated soil. Genome Announc. 2018;6:e0024918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schubert M, Lindgreen S, Orlando L. Adapter removal v2: rapid adapter trimming, identification, and read merging. BMC Res Notes. 2016;9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. John B, Alexandre L, Mark B. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coil D, Guillaume J, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinf. 2014;31:587–589. [DOI] [PubMed] [Google Scholar]
- 15. Goris J, Konstantinidis T, Klappenbach JA, Coenye P, Vandamme P, Tiedje M. DNA-DNA hybridization values and their relation to whole genome sequence. Int J Syst Evol Microbiol. 2007;57:81–91. [DOI] [PubMed] [Google Scholar]
- 16. Sampedro I, Parales RE, Krell T, Hill JE. Pseudomonas chemotaxis. FEMS Microbiol Rev. 2015;39:17–46. [DOI] [PubMed] [Google Scholar]
- 17. Dos Santos V, Heim E, Moore R, Stratz M, Timmis K. Insights into the genomic basis of niche specificity of Pseudomonas putida KT2440. Environ Microbiol. 2004;6:1264–1286. [DOI] [PubMed] [Google Scholar]
- 18. Bland C, Ramsey TL, Sabree F, et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinf. 2007;8:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abdel-Shafy HI, Mansour MSM. A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation [published online ahead of print December 18, 2015]. Egypt J Pet. doi:10.1016/j.ejpe.2015.03.011. [Google Scholar]
- 20. Ghosal D, Ghosh S, Dutta T, Ahn Y. Current state of knowledge in microbial degradation of polycyclic aromatic hydrocarbons (PAHs): a review. Front Microbiol. 2016;7:1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supporting_information for Isolation, Characterization, and Genomic Analysis of Pseudomonas sp. Strain SMT-1, an Efficient Fluorene-Degrading Bacterium by Mulugeta Desta, Weiwei Wang, Lige Zhang, Ping Xu and Hongzhi Tang in Evolutionary Bioinformatics