

Abstract
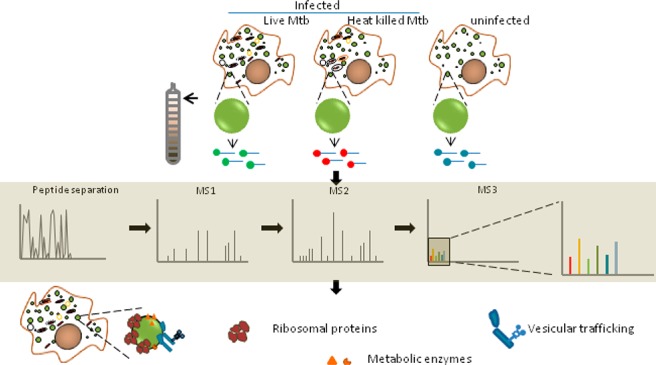
Growing evidence suggests the importance of lipid metabolism in pathogenesis of tuberculosis. Neutral lipids form the majority of lipids in a caseous granuloma, a pathology characteristic of tuberculosis. Cytosolic lipid droplets (LDs) of macrophages form the store house of these lipids and have been demonstrated to contribute to the inflammatory response to infection. The proteome of lipid droplets reflects the mechanisms of lipid metabolism active under a condition. However, infection induced changes in the proteome of these dynamic organelles remains elusive. Here, we employed quantitative proteomics to identify alterations induced upon infection with live Mycobacterium tuberculosis (Mtb) in comparison with heat killed bacilli or uninfected macrophages. We found increased abundance of proteins coupled with lipid metabolism, protein synthesis, and vesicular transport function in LDs upon infection with live Mtb. Using biochemical methods and microscopy, we validated ADP-ribosyltransferase (Arf)-like 8 (ARL8B) to be increased on the lipid droplet surface of live Mtb infected macrophages and that ARL8B is a bonafide LD protein. This study provides the first proteomic evidence that the dynamic responses to infection also encompass changes at the level of LDs. This information will be important in understanding how Mtb manipulates lipid metabolism and defense mechanisms of the host macrophage.
Keywords: tuberculosis, metabolism, lipid droplet, proteome
Mycobacterium tuberculosis (Mtb) is a successful human pathogen capable of growth within macrophages despite eliciting a robust innate and adaptive immune response. Part of its success is attributed to inhibition of phagolysosome maturation followed by egress to the cytosol.1 While the lysosome poses as a site of nutrient restriction, cytosolic escape ensures access to a wider range of nutrients. The acquisition of lipids is particularly challenging given the requirement of protein carriers or vesicular systems for lipid transfer.2 An emerging theme in TB pathogenesis is the manipulation of host lipid metabolism by Mtb whereby fatty acids and cholesterol are routed toward intracellular bacilli.3−8 Cytosolic lipid droplets (LDs) are an important source of cholesterol and fatty acids for intracellular mycobacteria,9 yet whether these dynamic organelles are actively modulated upon infection remains unaddressed.
LDs are phospholipid monolayer bound structures encasing neutral lipids such as triglycerides, cholesterol esters, ether lipids, diacylglycerides, and retinyl esters and are largely home to proteins that regulate lipid metabolism.10 The neutral lipid and protein composition of these organelles varies depending on cell type and metabolic state. For example, LDs of adipocytes undergoing lipolysis increase recruitment of ABHD5 and ATGL, enzymes that orchestrate triglyceride hydrolysis via regulated interactions with the coat protein Perilipin 1A.11 This regulates fatty acid supply from a major storage depot. Muscle cells are highly responsive to fatty acid availability. In the stimulated state, incoming fatty acids are metabolized via mitochondrial beta oxidation while in the basal state they are stored in the form of cytosolic LDs; the association of mitochondria with LDs is key to drive this process.12 The absence of Perilipin 1A in skeletal muscle cells indicates a different mechanism for regulation of lipid storage. While Perilipin 2 promotes LD stability, Perilipin 3 and Perilipin 5 enable mitochondrial oxidative metabolism of fatty acids in skeletal muscle cells.13−16 In macrophages, where Perilipin 2 serves as the major LD coat protein, regulation of fatty acid mobilization is not completely understood.17,18
Emerging evidence indicates cross talk between LDs and the ER membrane from which the droplets emerge, the mitochondria to which they provide fatty acids for oxidation, and the plasma membrane which regulates vectorial transfer of exogenous fatty acids to LDs. More recently, the physical interaction between various intracellular organelles has been quantitated using live cell microscopy, revealing interaction between LDs and ER, mitochondria, and lysosomes.19 Given the central role of LDs in nutrient balance within the cell and across organelles, the role of macrophage LDs during infection, wherein host and bacteria compete for nutrients, is an important area of investigation. Defined alterations in LD proteome have been previously reported during different physiological conditions, and differential localization of proteins partly mediated the metabolic changes.11 Therefore, determination of LD proteome is an important step in understanding infection induced LD protein changes and their role in host pathogen interaction.
In this study, we investigated the proteome level alterations in the LDs of human THP1 macrophages induced upon infection with live Mtb. Using subcellular fractionation to isolate LDs, followed by tandem mass tag (TMT) labeling of tryptic digests of proteins in the LD fraction, we identified a total of 418 proteins. Fifty seven proteins were found to increase, and 29 proteins were found to decrease in abundance in LD proteome from macrophages infected with live Mtb compared to heat killed Mtb. Proteins associated with protein synthesis, vesicular transport, and lysosomal function were specifically found to be more abundant when compared with cells infected with heat killed bacilli or uninfected cells. Our data identifies the first proteome level evidence that Mtb hijacks the macrophage LD during infection.
Results and Discussion
Qualitative Characterization of LDs Isolated from Mtb Infected Human Macrophages
THP1 monocyte derived macrophages contain lipid droplets in the basal state and exhibit Mtb infection induced alterations in lipid metabolism that guide the inflammatory response to infection.3,6 LDs of THP1 macrophages are mainly coated with Perilipin 2 (PLIN2) (Figure 1a) and to some extent with Perilipin 3 (PLIN3) but devoid of Perilipin 1 (PLIN1) (Figure S1). The presence of PLIN3 and PLIN1 detected with the same antibodies on cytosolic vesicles and LDs in 3T3L1 adipocytes confirmed PLIN2 to be the major LD coat protein of THP1 macrophages (Figure S1). The buoyant property of lipid droplets was utilized for their isolation using sucrose gradient ultracentrifugation (Figure 1b). The top 1 mL of the 11 mL gradient was treated as the LD fraction. This method led to purification of 80–100 μg of proteins in the LD fraction from 15 T175 flasks (4.5 × 108 THP1 macrophages). The relative purity of the LD fraction from contaminants of the endoplasmic reticulum, mitochondria, cytosol, and nuclei was assessed by immunoblotting for organelle specific markers. Relative enrichment of Perilipin 2 in fraction 1 and retention of all other organelle markers between fractions 5 and 11 revealed successful flotation based enrichment and purification of LDs in the topmost fraction (Figure 1c). Thin layer chromatography of the total cell lysates, post-nuclear supernatant, and the LD fraction revealed enrichment for neutral lipids and relative depletion of phospholipids in fraction 1 from the subcellular fractionation, confirming purity and enrichment of the LD fraction by our methodology (Figure 1d,e).
Figure 1.

Characterization of LDs of THP1 macrophages. (a) PLIN2 immunolocalization and BODIPY493/503 staining in THP1 macrophages. Individual channels in monochrome and the merged colored images are shown: PLIN2 (green), BODIPY493/503 (red), DAPI (blue). (b) Experimental schematic. (c) Immunoblotting for various subcellular markers on total cell lysate (T), nuclear pellet (N), post-nuclear supernatant (PNS), and 1 mL fractions isolated from top to bottom of the gradient after centrifugation. Blots include organelle markers PLIN2 (LD), Calnexin (ER, ab10286), GAPDH (cytoplasm), Lamin (nucleus), and VDAC2 (mitochondria). (d, e) Thin layer chromatogram of lipids isolated from indicated fractions. 0.2% of total cell lysate and PNS were loaded while 0.1% of the LD fraction was loaded. Standards for 1,3-diacylglycerol (1,3-DAG), cholesterol (C), cholesterol ester (CE), triacylglycerol (TG), phosphatidyl ethanolamine (PE), phosphatidyl choline (PC), and phosphatidyl serine (PS) were used as indicated.
The aim of this work was to understand how infection with the lipid metabolizing pathogen, Mtb, alters the LD proteome of human macrophages. Toward this goal, we compared macrophages infected with live and heat killed bacilli using TMT labeling based quantitative proteomics. The use of heat killed Mtb served as a control for particles with same size as that of live Mtb, with the retention of the lipidic surface that is recognized by the host macrophage as if it were live bacilli.20,21 Any active modulation of the host responses by proteins or newly synthesized metabolites would be missing in the case of infection with heat killed Mtb. This comparison would therefore allow us to look at actively induced changes rather than changes brought about by phagocytosis and surface recognition. The time point and multiplicity of infection was chosen such that infection induced cell death does not preclude equal isolation efficiency across conditions (Figure 2a,b). We determined that continuous infection for 14 h at multiplicity of infection (MOI) of 3 led to minimal necrotic cell death (Figure 2a) and equivalent abundance of total protein being isolated from macrophages infected with either live Mtb or heat killed Mtb (Figure 2b). Approximately 72% cells were found to be infected with at least one bacterium under these conditions of infection (Figure 2c).
Figure 2.

Selection of multiplicity of infection for lipid droplet isolation. (a) Cell death quantified by LDH release assay from supernatant of macrophages that were either uninfected or infected with the indicated multiplicity of infection of Mtb H37Rv. (b) Protein profile of lipid droplet fraction isolated from MOI3 and MOI5 of infection, obtained by silver staining of an SDS-PAGE gel. (c) Infectivity depicted from representative images upon infection at indicated multiplicity of infection, captured on a 43× objective using Floid microscope. Numbers indicate % infected cells as mean ± SD from ten representative fields (number of cells captured = 1572 for MOI1, 1161 for MOI3, and 1661 for MOI5). The top panel is a monochrome image of mCherry expressing strain of H37Rv, and the lower panel is a merge of the phase contrast image and the bacteria (red). Scale bar = 125 μm.
Alterations in the LD Proteome Induced upon Live Mtb Infection
On the basis of the above methodology, we isolated LDs from heat killed and live Mtb infected macrophages 14 h post-infection with MOI3 across two independent experiments and acetone precipitated the proteins, followed by trypsinization and TMT labeling of the peptides for quantitative mass spectrometry. Mass spectrometry analysis carried out in technical triplicates using high resolution mass spectrometry enabled the most comprehensive identification of proteins (Figure 3a). We identified a total of 418 proteins with high confidence (1% FDR) and completed an additional manual check of those identified with a single unique peptide for reliable spectra (Tables S1 and S2). Of these, 239 were identified with 2 or more unique peptides and 161 proteins were identified with a single unique peptide with 2 or more PSMs. Both PLIN2 and -3 were identified with 17 and 4 unique peptides, respectively, while no peptides were identified for PLIN1 as expected on the basis of immunostaining data.
Figure 3.

Summary of LD proteome in THP1 macrophages during Mtb infection. (a) Outline of the workflow: Lipid droplets were isolated from macrophages that were infected with either heat killed or live Mtb. The LD fraction was delipidated and trypsinized, and peptides thereof subjected to TMT labeling as indicated (in technical triplicates), followed by mass spectrometry. Experiments 1 and 2 are biological replicates (R1 and R2 indicate biological replicates for conditions, and T1, T2, and T3 indicate technical replicates). (b) Volcano plot representing significantly differentially abundant (FDR adjusted p-value <0.05) proteins; pink region reflects proteins increased by a ratio of >1.3 while blue region reflects protein decreased by a ratio <0.7 (live Mtb/heat killed Mtb). (c) Hierarchical clustering of differentially abundant proteins representing z scores of normalized abundance values from all technical and biological replicates. (d) Functional classification of 86 differentially abundant proteins listed in (c).
Prior to differential abundance analysis, we performed a surrogate variable analysis to regress out batch effects arising due to the specific mass spectrometry run (technical replicates) or day of performing lipid droplet isolation (biological replicates) (Table S3).22 This was performed for 258 proteins that were detected in all three technical replicates. Unsupervised hierarchical clustering revealed that indeed LD proteome abundance profiles of heat killed Mtb and live Mtb were reproducibly distinct (Figure S2). This analysis revealed 86 proteins to be statistically differentially abundant using a pairwise comparison of heat killed and live Mtb infection (adjusted p-value <0.05) (Figure 3b,c and Table S4). Of these, 57 were increased in abundance (fold change cutoff of >1.3-fold), and 29 were decreased in abundance (fold change cutoff of <0.7-fold) in the live versus heat killed group (Figure 3c and Table S4).
Gene Ontology based classification of these 86 differentially abundant proteins (Figure 3c) revealed enrichment for “catalytic activity” as the chief molecular function (Table S4). Further manual curation of the literature enabled us to classify the differentially abundant proteins better on the basis of the biological process (Figure 3d and Table S4). The majority of the differentially abundant proteins were found to be involved in “metabolism”, followed by “signal transduction” and “vesicular trafficking”. Within the category of metabolism, we found enzymes involved in lipid metabolism to be mainly increased: Hydroxysteroid 17-beta dehydrogenases (HSD17B7 and HSD17B4), lanosterol synthase (LSS), retinol dehydrogenase 10 (RDH10), long chain acyl-CoA synthases (ACSL3 and ACSL4), methyltransferase 7B (METTL7B), and zinc metallopeptidase STE24 (ZMPSTE24). In contrast, fatty acid binding protein 4 (FABP4) was significantly reduced upon live Mtb infection. Recent data on dual metabolomics and transcriptomics has revealed an increase in cholesterol biosynthetic flux in THP1 macrophages in response to cholesterol depletion imposed by intracellular Mtb.4 An increase in the abundance of proteins involved in cholesterol biosynthesis (HSD17B7, HSD17B4, and LSS) in the LD proteome may be an important aspect of regulating this process. Our previous work using pulse chase experiments has demonstrated increased lipolytic flux in Mtb infected macrophages with an increased rate of exogenous fatty acid flux toward intracellular bacteria rather than host lipid droplets.5 We therefore expected signatures of active lipolysis to be detected in the proteome analysis. While no lipase was detected, we found differential abundance of ACSL3, ACSL4, METTL7B, and FABP4. ACSL3 and METTL7B are known to localize to specialized domains of the endoplasmic reticulum where LD synthesis initiates23,24 while FABP4 is known to transfer fatty acids released from lipolysis for beta oxidation.25 A decrease in FABP4 and increase in ACSL3 and ACSL4 in live Mtb infected macrophage lipid droplet proteome suggest that free fatty acids liberated by lipolysis are likely not rapidly removed from the lipid droplets of live Mtb infected cells but may be utilized by ACSL3 and ACSL4 for re-esterification locally to other lipids. ZMPSTE24 is a lamin processing enzyme, deficiency or inhibition of which leads to lipodystrophy.26 The increased abundance of this protease in the lipid droplet proteome indicates a possible role in signaling that affects lipid metabolism during Mtb infection.
Proteins belonging to DNA binding function and immune response, like lymphocyte cytosolic protein (LCP1), S100 calcium binding protein 4 (S100A4), and S100A11, were only found to decrease in the LD proteome upon infection. LCP1, an actin modulator, is highly expressed in macrophages, monocytes, and neutrophils. In mice, it is known to be essential for an innate immune response to lung pathogens.27 S100A proteins are cytosolic calcium binding proteins that regulate many cellular functions. The levels of S100A8/A9 in murine TB lesions and serum of TB patients directly have been associated with the extent of inflammatory lung damage.28 In addition, S100A11 has been found to be part of a multigene blood based signature of active tuberculosis infection in humans.29 The regulated abundance of LCP1 and S100A proteins on lipid droplets might be important for the function of these proteins in inflammatory response to infection.
Proteins belonging to vesicular trafficking function were surprisingly only found to increase in the LD proteome upon infection. Fourteen proteins belong to this category which mainly includes small GTPases (ADP-ribosyltransferase (Arf)-like 8 (ARL8B), RAS related GTP binding proteins RAB1B, RAB6A, RAB7B, RAB8B, RAB10, and RAB21, and Rac family small GTPase 2 (RAC2)), followed by motor and cytoskeleton function associated with vesicle trafficking (myosin light chain components (MYL6 and MYL12A), myosin heavy chain 9 (MYH9), and tubulins (TUBB6, TUBA1A)).
ARL8B Localization to the LD Is Increased by Mtb
Enrichment of specific proteins associated with vesicular transport with LDs in live Mtb infected cells was intriguing. We biochemically verified one of the candidates, ARL8B, to be enriched in the LD fraction of live Mtb infected cells while the total cellular levels of ARL8B were comparable between heat killed and live Mtb infection (Figure 4a). We tested the possibility that ARL8B could have either increased in the LD proteome of live Mtb infected cells as an active phenomenon or decreased in the LD proteome in response to phagocytosis that perhaps live Mtb prevented. To test this, we compared the abundance of ARL8B in the LD fraction of live Mtb infected macrophages and uninfected macrophages. LD fraction from live Mtb infected macrophages exhibited increased abundance of ARL8B in comparison to uninfected macrophages, supporting an active modulation of the LD proteome (Figure 4b). No difference was found in the total levels of ARL8B between uninfected and live Mtb infected cell lysates. We sought to investigate the pattern of localization of endogenous ARL8B on macrophage LDs. Using commercially available antibodies specific to ARL8B, we observed punctate staining throughout the cytoplasm (data not shown), as reported previously.30 In order to minimize the cytosolic signal and ensure only membrane localized signal is detected, we permeabilized cells with 0.01% digitonin prior to fixing them. ARL8B positive punctae were clearly visible on the surface of LDs (Figure 4c). The density of the punctae was calculated per LD after normalizing for the LD surface area. The density of ARL8B fluorescence per LD was 6272–7784 (95% CI of median) in uninfected macrophages, 7652–9649 in macrophages infected with live Mtb, and 5748–7578 in macrophages infected with heat killed Mtb (Figure 4d).
Figure 4.

ARL8B is enriched in LDs of live Mtb infected macrophages. (a, b). Immunoblotting of total cell lysate (T) and LD fraction (LD) of macrophages infected with heat killed (HK) or live Mtb infected macrophages and uninfected macrophages with antibodies against ARL8B, PLIN2, and GAPDH. (c) Confocal images of endogenous ARL8B immunolocalization (green) relative to LDs stained with BODIPY665/676 (red) in THP1 macrophages. Nuclear staining with DAPI is shown in blue. Scale bar = 5 μm. (d) Quantification of ARL8B signal per LD in uninfected and infected (with live or heat killed (HK) Mtb) macrophages. ***p < 0.001 from the Kruskal–Wallis test. Data are from 311 to 361 LDs per group, representative of two independent experiments.
ARL8B is a small GTPase, which allows the movement of lysosomes toward the cell periphery, through its interaction with tubulin,31 and recruits protein machinery required for phagolysosome fusion.32,33 We identified several endosomal (RAB5 (early endosome), RAB7 (late endosome)) and lysosomal (lysosomal associated membrane protein 1 (LAMP1), late endosomal/lysosomal adaptor, MAPK and MTOR activator 1 (LAMTOR1), and scavenger receptor class B member 2 (SCARB2)) proteins in our LD proteome data and verified their presence using immunolocalization with PLIN2 (Figure S3a–o). These markers exhibited a range of colocalization rates with PLIN2, with the lowest for RAB5 and highest for RAB7 (Figure S3p). However, neither these nor other abundant lysosomal proteins such as sphignomyelinase were found to be differentially abundant like ARL8B, suggesting a specific role of ARL8B on lipid droplets. Consistent with published reports of ld-lysosome interaction,19 we observed dynamic “kiss-and-run” interactions (Figure S3q, Movies S1 and S2) or a more stable “hovering” type of interactions between lysosomes that moved along the surface of the LD (Figure S3q), consistent with recent spatiotemporal organelle interaction data from the Lippincott-Schwartz group.19 Together, these data suggested possible cross talk of lipid droplets with lysosome function but the enrichment of ARL8B to be independent of a generalized contamination of lysosomes to LDs of live Mtb infected cells.
To further test if ARL8B is a bonafide LD protein, we asked whether mCherry-tagged ARL8B can localize the fluorophore to LDs. We aimed to generate stable expression of this protein in THP1 monocytes and then differentiate them into macrophages. We consistently observed loss in fluorescence while trying to generate this line and therefore switched to generating a stable line of RAW264.7 macrophages expressing ARL8B-mCherry. As reported previously,31,34 ARL8B-mCherry localized to LAMP1+ lysosomes in RAW264.7 cells (Figure 5a). The colocalization rate of ARL8B-mCherry with LAMP1 was found to be approximately 60% (Figure 5b). To study LD localization, we treated these macrophages with oleic acid to induce LD biogenesis and imaged them using structured illumination microscopy. ARL8B-mCherry localized to BODIPY493/503 stained LDs in addition to defined ring-like structures and continuous tubular structures within the cytoplasm (Figure 5c). To confirm if ARL8B-mCherry can localize to purified LDs, we isolated LDs from ARL8B-mCherry and mCherry expressing cells treated with oleic acid to similar relative purity (Figure 5d). ARL8B-mCherry was found to be present in the LD fraction while mCherry was not (Figure 5e). Similar abundance of LAMP1 in the LD fraction of ARL8B-mCherry and mCherry overexpressing RAW cells verified that ARL8B localization in the LD fraction is independent of possible increased lysosomal contamination (Figure 5e). These data suggested that ARL8B is a bonafide LD protein in addition to its well-known lysosome associated function and likely affects lipid metabolism during Mtb infection.
Figure 5.

ARL8B is a bonafide LD associated protein. (a) ARL8B-mCherry colocalization with LAMP1+ organelles (lysosomes) in RAW264.7 cells. Scale bar = 10 μm. (b) Quantification of colocalization rate of ARL8B-mCherry and LAMP1 in RAW264.7 cells. (c) mCherry and ARL8B-mCherry (red) localization relative to lipid droplets stained with BODIPY665/676 (pseudocolored in green) in RAW264.7 cells loaded with oleic acid. The left panel shows the maximum intensity projection while the middle and right panel show indicated z stacks of a SIM image. Scale bar = 5 μm. (d) Thin layer chromatogram of lipids isolated from indicated fractions: total cell lysate (T) and LD fraction (LD). 0.2% of total cell lysate while 4% of the LD fraction was loaded. Standards for diacylglycerol (DAG), cholesterol (C), cholesterol ester (CE), and triacylglycerol (TG) are indicated. (e) Immunoblotting for mCherry, LAMP1, and PLIN2 in LDs isolated from RAW264.7 cells in (b) and (c). * represents possible degradation product of ARL8B-mCherry.
Active Manipulation of the LD Proteome by Mtb
Proteins could be observed to be higher in the live Mtb condition compared to the heat killed and also due to the possibility that response to heat killed Mtb might suppress the amount of protein localized to the lipid droplet while live Mtb may not be able to do so. Similarly, proteins that appeared to decrease by live Mtb infection could have been increased by macrophages in response to heat killed bacilli while live Mtb infection might not affect these proteins. In this study, we wanted to find proteins that were increased or decreased specifically by live Mtb infection, and the scale of the lipid droplet isolation experiment precluded us from performing a three-way comparison together. Therefore, we also performed TMT labeling of LD proteins of uninfected and live Mtb infected macrophages from an independent experiment (Table S5), and using the same criteria of >1.3- and <0.7-fold in all three technical replicates, we searched for differentially abundant proteins. Of 86 proteins previously identified to be differentially abundant between heat killed and live Mtb infection, 42 proteins were found to be differentially abundant in the same direction of change between uninfected and live Mtb infected macrophages (Figure S4a and Table S6). Proteins belonging to vesicular trafficking and protein synthesis were the most enriched class in this common set of proteins.
Ribosomal proteins were also part of this active manipulation of the LD proteome imposed by live Mtb. Ribosomal protein L11 was identified with the highest number of unique peptides (three) and peptide spectral matches (20 eight) from the group of ribosomal proteins found to be differentially abundant. We verified differential abundance of RPL11 in the LD fraction of live Mtb infected cells and uninfected cells (Figure S4b). In order to be able to detect low levels of possible ER contamination, we used a more sensitive antibody to calnexin. Using this antibody, we were able to identify contamination of calnexin in the LD fraction, consistent with its presence in our LD proteome data. Interestingly, while we saw an increase in RPL11 in the LD fraction of live Mtb infected cells, there was no change in the level of calnexin, consistent with our LD proteome data. Enrichment of neutral lipids as opposed to phospholipids further proved that the fraction was largely devoid of ER contamination (Figure S4c,d). This suggests that the increased abundance of ribosomal proteins in the LD fraction of live Mtb infected cells may be a selective enrichment of ribosome containing microdomains of the ER to be enriched20 on LDs rather than increased contamination of the endoplasmic reticulum in these samples. Interestingly, silencing of RpLP0, a ribosomal protein in Drosophila S2 cells, resulted in condensation of lipid droplets close to each other.35 Therefore, enrichment of ribosomal proteins may be significant to the mobilization of lipids from lipid droplets during TB infection.
Active modulation of the LD proteome was also evident for vesicular trafficking proteins such as ARL8B, RAC2, MYL6, MYL12A, MYH9, TUBB6, and TUBA1A. The alterations orchestrated by Mtb in the lipid droplet proteome could be a consequence of lipid changes that affect protein recruitment or a mechanism of lipid mobilization in infected cells. It is interesting to note that, even though we observe lipolysis in infected cells,5 we did not identify altered levels of lipases in the LD proteome of infected cells. This suggests that alternative mechanisms such as fragmentation of lipid droplets may be operative to bring about lipolysis. The molecular basis of this process is not well-defined, but the process is common to hormonally stimulated lipolysis wherein an increase in surface to volume ratio of the droplets ensures rapid degradation of the lipids.36 Previous studies have identified factors that regulate lipid droplet size and dispersion in other model systems. Silencing of the vesicular transport proteins Arf79F, garz (ARF-guanine nucleotide exchange factor), and COPI led to fewer and dispersed droplets. In addition, shifting from lipid rich media to starvation led to a loss of droplets from control cells while Arf79F silenced cells contained larger lipid droplets and exhibited poorer lipolysis. In a separate study, nonmuscle-myosin IIa (MYH-9) was found to play a role in dispersion of droplets, thereby affecting LD surface-to-volume ratio.37 Drawing parallels with our findings, we suspect vesicular transport pathways to drive metabolic changes rather than recruitment of lipases. We cannot rule out the role of mycobacterial secreted lipases in this process which may be difficult to identify despite a highly sensitive mass spectrometry approach owing to the dilution relative to the host proteome. Activity based proteomics has revealed a large number of putative lipases in Mtb proteome that are differentially regulated under normoxia and hypoxia.38,39 Recently, the mycobacterial protein LipY has been demonstrated to be important for lipid consumption from the host and assimilation into bacteria.8 On the basis of our findings, we speculate that host factors are made to synergize with bacterial cell surface/secreted lipases to divert host lipids toward bacteria. Mechanistically, we envisage secreted virulence factors of Mtb to play a role in this active manipulation of the proteome, and targeted deletions of these might be useful in the future to understand mechanisms of hijacking of the LD proteome by Mtb.
Conclusion
Mtb, a pathogen adept at utilizing host lipids, is understood as being capable of this feat by expression of a large number of genes involved in lipid metabolism. However, host macrophages utilize specific organelles for lipid storage and mobilization, and the bacteria must compete with the host machinery to make use of these lipids. Moreover, given the role of host triglyceride synthesis machinery in the inflammatory response to infection, the lipid droplet proteome may play a larger role in dictating host response to infection. This study was an attempt to address if the proteome of lipid droplets, organelles that store host lipids, is actively modulated by Mtb. Using quantitative proteomics, we found that Mtb could affect abundance of several proteins associated with lipid droplets. These proteins largely belonged to protein synthesis, vesicular trafficking, and lipid metabolism. Our data points toward possible mechanisms whereby Mtb might manipulate the host nutrient storage system to its advantage. Future studies will be aimed at understanding mechanisms of this manipulation by active processes of the bacilli.
Experimental Section
Cell Lines and Strains
THP1 monocytes (ECACC #88081201) were procured from Sigma. Mtb H37Rv was a kind gift from Dr. Vivek Rao. Monocytes were cultured in RPMI media containing 1 mM sodium pyruvate, 2 mM glutamax, and 10% fetal bovine serum (FBS). Monocyte to macrophage differentiation was induced by treatment with 100 nM phorbol myristate acetate (PMA) for 24 h followed by culturing in media without PMA for another 48 h. RAW264.7 macrophages (ATCC#TIB-71) were maintained in RPMI media containing 1 mM sodium pyruvate, 2 mM glutamax, and 10% fetal bovine serum (FBS). Mtb was cultured in 7H9 with 0.05% Tween 80 and ADC supplementation (Difco) under shaking conditions or in a roller bottle.
Infection
Mtb was grown to an OD600 nm of 0.4 to 0.6. Cultures were harvested by centrifugation at 3000 rpm in a swing out bucket rotor for 5 min, washed with phosphate buffered saline (PBS) with 0.05% Tween 80 twice, and then resuspended in PBS. Bacterial cell suspensions were centrifuged at 800 rpm in a swing out bucket rotor for 12 min. The supernatant was carefully transferred using a pipet into another tube and treated as the single cell suspension (SCS). Optical density of the SCS was measured at 600 nm, and bacterial density was calculated as per the conversion of 1 OD = 5 × 108 bacilli/mL. Macrophages were infected at a multiplicity of infection of 3 for 14 h in half the volume of media used for normally culturing the macrophages. For preparation of heat killed Mtb, the single cell suspension was heated at 95 °C for 15 min, cooled to RT, and then added in media for infection.
Infectivity Analysis
To quantify the percentage of infectivity of macrophages, PMA differentiated THP1 macrophages were infected with Mycobacterium tuberculosis (mCherry expressing) at MOIs of 1, 2, 3, and 5 for 16 hours. After 16 h of infection, cells were washed twice with PBS and fixed with 4% methanol free formaldehyde. These fixed cells were thereafter stained with cell mask deep red at 1 μg/mL concentration for half an hour at room temperature. These samples were washed thrice with PBS and imaged using an IN CELL 6000 high content image analyzer (GE health care) at 10× magnification. Percentage of infectivity of cells was estimated by an image analysis protocol generated using an IN CELL developer toolbox. Briefly, macrophages were counted using fluorescence in the cell mask channel, and cell mask signal colocalizing with mCherry (bacteria) fluorescence was used to identify infected cells.
LD Isolation
THP1 monocytes were seeded at a density of 3.0 × 107/T175 flask in 100 nM PMA for 24 h, followed by exchange with PMA free media and another 2 days in culture. At this point, they were infected as described above. LDs were isolated from 15 T175 flasks for each condition using previously described methods40 adapted to BSL3 conditions. Cells were gently scraped in media and then centrifuged in a swing out bucket rotor at 200g for 15 min. The cell pellet was washed twice in PBS and then resuspended in 2.5 mL of lysis buffer A’ (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 100 mM KCl buffer (pH 7.4)), followed by incubation on ice for 5 min. Cell lysis was completed using nitrogen cavitator by applying nitrogen gas at 750 psi for 5 min. The cell lysate was collected in 15 mL conical tubes to which 2.5 mL of Buffer B’ (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 100 mM KCl buffer, 1.08 M sucrose (pH 7.4)) was added and mixed. This was treated as the total cell lysate. The total cell lysate was centrifuged at 900g for 10 min. The supernatant so obtained was treated as the post-nuclear supernatant (PNS). The PNS was layered under a gradient (from top to bottom) of 1 mL of Buffer E (25 mM Tris HCl, 1 mM EDTA, 1 mM EGTA), 2 mL of Buffer D (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 100 mM KCl buffer, 0.13 M sucrose (pH 7.4)), and 2 mL of Buffer C (20 mM Tris, 1 mM EDTA, 1 mM EGTA, 100 mM KCl buffer, 0.27 M sucrose (pH 7.4)) in a quick seal bell top polyallomer tube (Beckman Coulter Cat #344622). A 1 mL cushion of Buffer B’ was layered under the post-nuclear supernatant. All the buffers used contained protease inhibitor cocktail (Roche). The tubes were sealed and centrifuged at 32 000 rpm for 4 h in a SW32.1 Ti rotor in a Beckman Coulter Optima XPN-100 ultracentrifuge. One milliliter fractions from the top were collected. For delipidation of proteins, the LD fraction (fraction 1 from the top) was mixed with 4 volumes of ice cold acetone and incubated overnight at −20 °C, followed by centrifugation at 13 000g for 30 min and acetone washing. The protein pellets were air-dried and then resuspended in 100 μL of 50 mM triethylammonium bicarbonate buffer (TEABC; pH 8.0) containing 0.1% SDS.
Quantitative Mass Spectrometry
Protein concentration of the LD fraction obtained from three independent experiments was estimated using Qbit. The extracted peptides were lyophilized, and an equal amount of protein from each condition was used for TMT labeling. Labels used for each condition, infected with heat killed Mtb (experiment 1), infected with live Mtb (experiment 1), infected with heat killed Mtb (experiment 2), infected with live Mtb (experiment 2), uninfected (experiment 3), infected with live Mtb (experiment 3), were TMT channels 126, 127, 128, 129, 130, and 131, respectively. Labeled peptides were purified, pooled, and analyzed in three technical replicates using an Orbitrap Fusion Tribrid mass spectrometer. Full details on sample preparation are provided in the Supporting Information.
Database Searches for Peptide and Protein Identification
Raw data files were processed to generate peak list files, and the data were searched against the human RefSeq protein database (version 75) (downloaded from NCBI, updated June, 2016) combined with common contaminants (total number of sequences: 26 976 proteins) appended using SequestHT and Mascot (version 2.5.1) search algorithms available through Thermo Proteome Discoverer 2.1 software suite (version 2.1.0.81/Thermo Fisher Scientific, Bremen Germany). The following search parameters were considered: maximum missed cleavage of 1, trypsin specified as proteolytic enzyme with minimum peptide length set to 7 amino acids. The precursor and fragment mass tolerance were set at 10 ppm and 0.05 Da, respectively. TMT modification at peptide N-termini and lysine residues and carbamidomethylation of cysteine were set as fixed modifications and oxidation of methionine, as variable modification. Reporter ion quantifier node was employed for peak integration and reporting intensities at MS3 level. A Sequest XCorr value of >2 or MASCOT Ion score of >30 was used for identification of peptides. Consensus workflow was employed to group PSMs and validate PSM and peptides as well as carry out protein grouping and report FDR at PSM, peptide, and protein level. For reporter ion quantification, both unique and razor peptides were considered. Reporter quantification was performed on the basis of reporter ion intensity. The percolator node available in the software suite was employed to calculate the false discovery rate (FDR) at the PSM level. The data were searched against both forward and a reverse decoy database, and the PSM validation was based on q-value. The peptide and protein validator nodes were employed in the consensus workflow to obtain peptides at 1% FDR. The FDR at PSM and peptide level was <1%, and at protein level, the FDR is <3%. For data normalization, the reporter ion intensities for each condition were normalized to the median of that sample. Normalized abundance values after regressing out technical and batch effects as described under “Statistical Rationale” (Supporting Information) was used for differential abundance analysis.
Microscopy
Cells were fixed in 4% methanol free formaldehyde and then permeabilized, blocked, and probed with primary antibodies overnight and Alexa tagged secondary antibodies for 1 h. The detailed protocol is provided in the Supporting Information. For detecting endogenous ARL8B, a similar procedure as above was followed except that cells were permeabilized with 0.01% digitonin (5 min) prior to fixing them with 4% methanol free formaldehyde (25 min). Nonspecific epitopes were blocked with 3% BSA in PHEM buffer containing 0.05% Tween 20. Subsequently, ARL8B antibody incubation (1:200) was performed at 4 °C for an overnight period. ARL8B immunoreactivity was detected using Alexa 488 conjugated goat antirabbit antibody, and LDs were stained using BODIPY 665/676. Coverslips were mounted in ProLong Diamond antifade reagent with or without DAPI (Life Technologies) and imaged using a Leica SP8 confocal microscope.
Cloning and Transfection
ARL8B-HA constructs were a kind gift from Dr. Mahek Sharma (IISER, Mohali). ARL8B was PCR amplified and cloned in frame with mCherry as a C terminal fusion tag under the CMV promoter in pcDNA3.1. The fragments were then subcloned using KpnI and NotI sites into pcDNA3.1 derivative in which the CMV promoter was replaced with the human EF1 promoter for stable expression in macrophages. For generating stable lines, RAW264.7 macrophages were transfected using lipofectamine LTX as per supplier’s instructions and 48 h later selected on 800 μg/mL G418 until only mCherry positive cells were selected. LDs from these and mCherry expressing stable RAW264.7 cells were isolated 48 h post-treatment with 150 μM oleic acid conjugated to BSA (Sigma O3008) with the same protocol as described for THP1 cells, using five T75 flasks for each group. For microscopy based experiments, cells were seeded at a density of 30 000 per well on a 24 well plate with a #1.5 coverslip. Six hours later, macrophages were loaded with oleic acid conjugated to BSA for 18 h and then fixed for microscopy.
Structured Illumination Microscopy
Samples were prepared as described for confocal microscopy. Structured illumination microscopy (SIM) images were collected with a N-SIM illumination system using a 100× oil objective lens with a numerical aperture of 1.4 at room temperature and equipped with an Andor iXon EMCCD Scientific Camera DU-897 and PCI-Controller DU-897 camera. Five phases and 3 rotation angles of the excitation grid were acquired for each z plane, with z spacing of 100 nm between planes in the 3D SIM mode. SIM processing was performed with the N-SIM module of the NIS Elements software (Nikon).
Acknowledgments
This work was supported by Wellcome Trust DBT India Alliance Intermediate Award to S.G. (IA/I/11/2500254). The authors acknowledge CSIR (STS0016 and BSC0403) for infrastructure support. K.S. acknowledges CSIR; D.M. and N.J. acknowledge DBT-India Alliance, and A.N. acknowledges UGC for fellowships. S.M.P. is a recipient of the INSPIRE Faculty award from Department of Science and Technology, Government of India.
Glossary
Abbreviations
- LD
lipid droplet
- Mtb
Mycobacterium tuberculosis
- TG
triacylglycerol
- ARL8B
ADP-ribosyltransferase (Arf)-like 8
- MOI
multiplicity of infection
- MS
mass spectrometry
- LC-MS
liquid chromatography-mass spectrometry
- HK
heat killed
- UI
uninfected
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.8b00301.
Supplementary methods; testing localization of PLIN1 and PIN3 in THP1 macrophages and 3T3L1 adipocytes; unsupervised hierarchical clustering of normalized protein abundance; peripheral association of endosomal and lysosomal proteins with LDs; comparison of uninfected and live Mtb infected cells; lists of proteins; normalized protein abundance and output of surrogate variable analysis; functional annotation of differentially abundant LD proteins in live and heat killed Mtb infected macrophages (PDF)
Movie S1. Confocal live cell movie of THP1 macrophages labeled with LipidTox (red) and lysotracker (green). This example shows kiss-and-run and hovering type interactions (AVI)
Movie S2. Confocal live cell movie of THP1 macrophages labeled with LipidTox (red) and lysotracker (green). This cell shows rare events of complete localization between lipid droplets and lysosomes as well as kiss-and-run and hovering type interactions (AVI)
Author Contributions
# D.M. and K.S. contributed equally. S.G. designed the research and wrote the manuscript. S.G. and T.S.K.P. designed the proteomic experiments. K.S. and D.M. performed LD isolation. D.M., A.N., N.J., S.G., and K.S. performed other experiments. S.M.P., T.S.K.P., and K.S. performed mass spectrometry and proteomic data analysis. S.M.P. and T.S.K.P. wrote the mass spectrometry analysis section of the manuscript. T.S.K.P. supervised proteomics and analysis. D.D. and R.K. performed data regression and statistical analysis and helped write relevant sections of the manuscript. D.M. performed validation experiments and helped write the manuscript. All the authors read and approved the manuscript.
The authors declare no competing financial interest.
Notes
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE41 partner repository with the data set identifier PXD008950. Post-analysis data are available within the paper and its Supporting Information files. All the codes for the statistical analysis can be accessed from this github repository (https://github.com/rintukutum/mtb-macrophage-LD).
Supplementary Material
References
- Simmons J. D.; Stein C. M.; Seshadri C.; Campo M.; Alter G.; Fortune S.; Schurr E.; Wallis R. S.; Churchyard G.; Mayanja-Kizza H.; Boom W. H.; Hawn T. R. (2018) Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat. Rev. Immunol. 18, 575–589. 10.1038/s41577-018-0025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C. L.; Walch L.; Verbavatz J. M. (2016) Lipids and Their Trafficking: An Integral Part of Cellular Organization. Dev. Cell 39, 139–153. 10.1016/j.devcel.2016.09.030. [DOI] [PubMed] [Google Scholar]
- Jaisinghani N.; Dawa S.; Singh K.; Nandy A.; Menon D.; Bhandari P. D.; Khare G.; Tyagi A.; Gandotra S. (2018) Necrosis Driven Triglyceride Synthesis Primes Macrophages for Inflammation During Mycobacterium tuberculosis Infection. Front. Immunol. 9, 1490. 10.3389/fimmu.2018.01490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M., Kogadeeva M., Gengenbacher M., McEwen G., Mollenkopf H. J., Zamboni N., Kaufmann S. H. E., and Sauer U. (2017) Integration of Metabolomics and Transcriptomics Reveals a Complex Diet of Mycobacterium tuberculosis during Early Macrophage Infection. mSystems 2, 10.1128/mSystems.00057-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaisinghani N.; Dawa S.; Singh K.; Nandy A.; Menon D.; Bhandari P.; Khare G.; Tyagi A.; Gandotra S. (2017) Necrosis triggered by Mycobacterium tuberculosis alters macrophage triglyceride metabolism and inflammatory response in a DGAT1-dependent manner. BioRxiv https://doi.org/10.1101/187104 10.3389/fimmu.2018.01490. [DOI] [Google Scholar]
- Ahsan F.; Maertzdorf J.; Guhlich-Bornhof U.; Kaufmann S. H. E.; Moura-Alves P. (2018) IL-36/LXR axis modulates cholesterol metabolism and immune defense to Mycobacterium tuberculosis. Sci. Rep. 8, 1520. 10.1038/s41598-018-19476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarova E. V.; Montague C. R.; La T.; Wilburn K. M.; Sukumar N.; Lee W.; Caldwell S.; Russell D. G.; VanderVen B. C. (2017) Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. eLife 6, e26969. 10.7554/eLife.26969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santucci P., Diomande S., Poncin I., Alibaud L., Viljoen A., Kremer L., de Chastellier C., and Canaan S. (2018) Delineating the Physiological Roles of the PE and Catalytic Domains of LipY in Lipid Consumption in Mycobacterium-Infected Foamy Macrophages. Infect. Immun. 86, 10.1128/IAI.00394-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barisch C.; Soldati T. (2017) Breaking fat! How mycobacteria and other intracellular pathogens manipulate host lipid droplets. Biochimie 141, 54–61. 10.1016/j.biochi.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Krahmer N.; Hilger M.; Kory N.; Wilfling F.; Stoehr G.; Mann M.; Farese R. V. Jr.; Walther T. C. (2013) Protein correlation profiles identify lipid droplet proteins with high confidence. Mol. Cell. Proteomics 12, 1115–1126. 10.1074/mcp.M112.020230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasaemle D. L.; Dolios G.; Shapiro L.; Wang R. (2004) Proteomic analysis of proteins associated with lipid droplets of basal and lipolytically stimulated 3T3-L1 adipocytes. J. Biol. Chem. 279, 46835–46842. 10.1074/jbc.M409340200. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Wang Y.; Li J.; Yu J.; Pu J.; Li L.; Zhang S.; Peng G.; Yang F.; Liu P.; et al. (2011) Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein a-I. J. Proteome Res. 10, 4757–4768. 10.1021/pr200553c. [DOI] [PubMed] [Google Scholar]
- Laurens C.; Bourlier V.; Mairal A.; Louche K.; Badin P. M.; Mouisel E.; Montagner A.; Marette A.; Tremblay A.; Weisnagel J. S.; Guillou H.; Langin D.; Joanisse D. R.; Moro C. (2016) Perilipin 5 fine-tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci. Rep. 6, 38310. 10.1038/srep38310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma M.; Hesselink M. K.; Sparks L. M.; Timmers S.; Ferraz M. J.; Mattijssen F.; van Beurden D.; Schaart G.; de Baets M. H.; Verheyen F. K.; Kersten S.; Schrauwen P. (2012) Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes 61, 2679–2690. 10.2337/db11-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington J. D.; Noland R. C.; Hebert R. C.; Masinter B. S.; Smith S. R.; Rustan A. C.; Ravussin E.; Bajpeyi S. (2015) Perilipin 3 Differentially Regulates Skeletal Muscle Lipid Oxidation in Active, Sedentary, and Type 2 Diabetic Males. J. Clin. Endocrinol. Metab. 100, 3683–3692. 10.1210/JC.2014-4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert M.; Parker B. L.; Chaudhuri R.; Fazakerley D. J.; Serup A.; Thomas K. C.; Krycer J. R.; Sylow L.; Fritzen A. M.; Hoffman N. J.; Jeppesen J.; Schjerling P.; Ruegg M. A.; Kiens B.; James D. E.; Richter E. A. (2016) mTORC2 and AMPK differentially regulate muscle triglyceride content via Perilipin 3. Mol. Metab. 5, 646–655. 10.1016/j.molmet.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S.; Yang W.; Kozusko K.; Saudek V.; Savage D. B. (2014) Perilipins 2 and 3 lack a carboxy-terminal domain present in perilipin 1 involved in sequestering ABHD5 and suppressing basal lipolysis. Proc. Natl. Acad. Sci. U. S. A. 111, 9163–9168. 10.1073/pnas.1318791111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S.; Cuervo A. M. (2015) Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 17, 759–770. 10.1038/ncb3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valm A. M.; Cohen S.; Legant W. R.; Melunis J.; Hershberg U.; Wait E.; Cohen A. R.; Davidson M. W.; Betzig E.; Lippincott-Schwartz J. (2017) Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. 10.1038/nature22369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod S.; Oschkinat H.; Enders J.; Schlegel B.; Brinkmann V.; Kaufmann S. H.; Haas A.; Schaible U. E. (2008) Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell. Microbiol. 10, 1530–1545. 10.1111/j.1462-5822.2008.01147.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa E.; Ishikawa T.; Morita Y. S.; Toyonaga K.; Yamada H.; Takeuchi O.; Kinoshita T.; Akira S.; Yoshikai Y.; Yamasaki S. (2009) Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin Mincle. J. Exp. Med. 206, 2879–2888. 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek J. T.; Johnson W. E.; Parker H. S.; Jaffe A. E.; Storey J. D. (2012) The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro S.; Ingelmo-Torres M.; Estanyol J. M.; Tebar F.; Fernandez M. A.; Albor C. V.; Gaus K.; Grewal T.; Enrich C.; Pol A. (2006) Identification and characterization of associated with lipid droplet protein 1: A novel membrane-associated protein that resides on hepatic lipid droplets. Traffic 7, 1254–1269. 10.1111/j.1600-0854.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- Kassan A.; Herms A.; Fernandez-Vidal A.; Bosch M.; Schieber N. L.; Reddy B. J.; Fajardo A.; Gelabert-Baldrich M.; Tebar F.; Enrich C.; Gross S. P.; Parton R. G.; Pol A. (2013) Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J. Cell Biol. 203, 985–1001. 10.1083/jcb.201305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W. J.; Sridhar K.; Bernlohr D. A.; Kraemer F. B. (1999) Interaction of rat hormone-sensitive lipase with adipocyte lipid-binding protein. Proc. Natl. Acad. Sci. U. S. A. 96, 5528–5532. 10.1073/pnas.96.10.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A. (2011) Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 96, 3313–3325. 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deady L. E.; Todd E. M.; Davis C. G.; Zhou J. Y.; Topcagic N.; Edelson B. T.; Ferkol T. W.; Cooper M. A.; Muenzer J. T.; Morley S. C. (2014) L-plastin is essential for alveolar macrophage production and control of pulmonary pneumococcal infection. Infect. Immun. 82, 1982–1993. 10.1128/IAI.01199-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal R.; Monin L.; Torres D.; Slight S.; Mehra S.; McKenna K. C.; Fallert Junecko B. A.; Reinhart T. A.; Kolls J.; Baez-Saldana R.; Cruz-Lagunas A.; Rodriguez-Reyna T. S.; Kumar N. P.; Tessier P.; Roth J.; Selman M.; Becerril-Villanueva E.; Baquera-Heredia J.; Cumming B.; Kasprowicz V. O.; Steyn A. J.; Babu S.; Kaushal D.; Zuniga J.; Vogl T.; Rangel-Moreno J.; Khader S. A. (2013) S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am. J. Respir. Crit. Care Med. 188, 1137–1146. 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M. P.; Graham C. M.; McNab F. W.; Xu Z.; Bloch S. A.; Oni T.; Wilkinson K. A.; Banchereau R.; Skinner J.; Wilkinson R. J.; Quinn C.; Blankenship D.; Dhawan R.; Cush J. J.; Mejias A.; Ramilo O.; Kon O. M.; Pascual V.; Banchereau J.; Chaussabel D.; O’Garra A. (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977. 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuli A.; Thiery J.; James A. M.; Michelet X.; Sharma M.; Garg S.; Sanborn K. B.; Orange J. S.; Lieberman J.; Brenner M. B. (2013) Arf-like GTPase Arl8b regulates lytic granule polarization and natural killer cell-mediated cytotoxicity. Mol. Biol. Cell 24, 3721–3735. 10.1091/mbc.e13-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann I.; Munro S. (2006) An N-terminally acetylated Arf-like GTPase is localised to lysosomes and affects their motility. J. Cell Sci. 119, 1494–1503. 10.1242/jcs.02958. [DOI] [PubMed] [Google Scholar]
- Khatter D.; Raina V. B.; Dwivedi D.; Sindhwani A.; Bahl S.; Sharma M. (2015) The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci. 128, 1746–1761. 10.1242/jcs.162651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S.; Sharma M.; Ung C.; Tuli A.; Barral D. C.; Hava D. L.; Veerapen N.; Besra G. S.; Hacohen N.; Brenner M. B. (2011) Lysosomal trafficking, antigen presentation, and microbial killing are controlled by the Arf-like GTPase Arl8b. Immunity 35, 182–193. 10.1016/j.immuni.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw R. D.; Callahan J. W.; Mahuran D. J. (2006) The Arf-family protein, Arl8b, is involved in the spatial distribution of lysosomes. Biochem. Biophys. Res. Commun. 344, 1186–1191. 10.1016/j.bbrc.2006.03.221. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Walther T. C.; Rao M.; Stuurman N.; Goshima G.; Terayama K.; Wong J. S.; Vale R. D.; Walter P.; Farese R. V. (2008) Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453, 657–661. 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paar M.; Jungst C.; Steiner N. A.; Magnes C.; Sinner F.; Kolb D.; Lass A.; Zimmermann R.; Zumbusch A.; Kohlwein S. D.; Wolinski H. (2012) Remodeling of lipid droplets during lipolysis and growth in adipocytes. J. Biol. Chem. 287, 11164–11173. 10.1074/jbc.M111.316794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfisterer S. G.; Gateva G.; Horvath P.; Pirhonen J.; Salo V. T.; Karhinen L.; Varjosalo M.; Ryhanen S. J.; Lappalainen P.; Ikonen E. (2017) Role for formin-like 1-dependent acto-myosin assembly in lipid droplet dynamics and lipid storage. Nat. Commun. 8, 14858. 10.1038/ncomms14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallman K. R.; Levine S. R.; Beatty K. E. (2016) Small-Molecule Probes Reveal Esterases with Persistent Activity in Dormant and Reactivating Mycobacterium tuberculosis. ACS Infect. Dis. 2, 936–944. 10.1021/acsinfecdis.6b00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega C.; Anderson L. N.; Frando A.; Sadler N. C.; Brown R. W.; Smith R. D.; Wright A. T.; Grundner C. (2016) Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence. Cell Chem. Biol. 23, 290–298. 10.1016/j.chembiol.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samsa M. M.; Mondotte J. A.; Iglesias N. G.; Assuncao-Miranda I.; Barbosa-Lima G.; Da Poian A. T.; Bozza P. T.; Gamarnik A. V. (2009) Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5, e1000632 10.1371/journal.ppat.1000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino J. A.; Csordas A.; Del-Toro N.; Dianes J. A.; Griss J.; Lavidas I.; Mayer G.; Perez-Riverol Y.; Reisinger F.; Ternent T.; Xu Q. W.; Wang R.; Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.