

Abstract

B(C6F5)3 enables the metal-free unprecedented substrate-controlled direct α-stereoselective synthesis of deoxyglycosides from glycals. 2,3-Unsaturated α-O-glycoside products are obtained with deactivated glycals at 75 °C in the presence of the catalyst, while 2-deoxyglycosides are formed using activated glycals that bear no leaving group at C-3 at lower temperatures. The reaction proceeds in good to excellent yields via concomitant borane activation of glycal donor and nucleophile acceptor. The method is exemplified with the synthesis of a series of rare and biologically relevant glycoside analogues.
Introduction
Deoxyglycosides are an important class of carbohydrates commonly found in nature as part of biologically active glycoconjugates.1 These compounds are characterized by the lack of substitution at one or several positions around the carbohydrate ring, often at C-2, which makes them more challenging to synthesized than their fully oxygenated counterparts. The absence of directing groups adjacent to the anomeric center to bias the nucleophile approach during the glycosylation reaction, often leads to mixtures of anomers and/or Ferrier-type products.2 Owing to their relevance in drug discovery, research efforts have focused in achieving their stereoselective synthesis.3
Coupling reactions involving glycals are one of the most atom-efficient routes to access deoxyglycosides.4 Traditional methods to yield the corresponding 2-deoxyglycoside rely on acid catalyzed direct addition of an alcohol to the glycal, which often leads to either moderate to low yields and/or variable selectivities which are dependent on the nature of the OH nucleophile (e.g., primary vs secondary, axial vs equatorial) with 2,3-unsaturated glycosides and hydrolyzed starting material as common side products.2a,3a,3q However, most acid-catalyzed processes to access 2,3-unsaturated glycosides directly, which are also versatile synthons in organic chemistry,5 tend to use harsh promoters, required specific protected building blocks, and often lead to moderate overall yields and diastereocontrol, which has limited their utility.3a Given the range of glycoside donor and OH nucleophile reactivity profiles, there is currently no universal Lewis acid glycosylation promoter that can be used for the activation of glycals.3 Thus, there is a need to find improved and more general catalysts to access these high value glycosides.
Our group has been interested over the past few years in the development of practical, selective and catalytic methods for the direct activation of glycals using thiourea-based organocatalysts,4b−4d as well as palladium and gold catalytic activators.9 Encouraged by our previous work, we undertook synthetic studies toward the development of a metal-free and improved organocatalytic method for the activation of glycals.
Trivalent boron reagents are often employed as Lewis acids because of their ubiquitous electrophilic nature and ability to reversibly form bonds with oxygen and thus are attractive catalysts in glycosylation chemistry including examples in regioselective glycosylations.3a,3p,6 Among the boron-based Lewis acids available, B(C6F5)3 (BCF) has demonstrated extensive versatility in a wide variety of reactions including borylation, hydrogenation, hydrosilylation, frustrated Lewis pair (FLP) chemistry, and Lewis acid catalysis.7 In the context of glycosylation chemistry, the utility of BCF has only been shown in the activation of fully substituted trichloroacetimidate glycosyl donors in good to excellent yields and moderate to good diastereoselectivity,8 with no examples reported in the synthesis of deoxyglycosides.
Herein we describe the metal-free, atom-economic, and versatile substrate-controlled borane-catalyzed highly α-stereoselective synthesis of deoxyglycosides directly from glycals (Scheme 1).
Scheme 1. BCF-Catalysed Synthesis of 2,3-Unsaturated Glycosides from “Disarmed” Glycals (Pathway A) and 2-α-Deoxyglycosides from “armed” Glycals (Pathway B).

Results and Discussion
Initial studies began by screening BCF for its ability to promote the stereoselective glycosylation of peracetylated galactal 1a with glucoside acceptor 2a in the presence of different catalyst loadings, solvents, and temperatures. It was found that 5 mol % BCF in toluene at 75 °C was the optimum condition to yield the corresponding 2,3-unsaturated glycoside 3a after 2 h (88%, α:β 30:1, entry 1 in Table 1). Reactions were less efficient at lower catalyst loadings or at lower temperatures; changing the solvent to CH2Cl2 or CH3CN was also detrimental to the reaction (see Table S1 in the Supporting Information (SI) for solvent and temperature screen details). Having established the optimal reaction conditions, our attention then turned to exploring the substrate scope of the reaction between 1a and a range of OH nucleophiles 2b–2k (Table 1). In all cases, reactions proceeded smoothly within 1.5–4 h and in good to excellent yields and a clear preference for the α-products, demonstrating the reaction is tolerant of primary, secondary, and phenolic OH nucleophiles, as well as common alcohol protecting groups (e.g., acetals, ethers, and esters).
Table 1. Glycosylation Reactions with Galactal 1ac.


Isolated yield.
Determined by 1H-NMR.
Reaction did not proceed in the absence of catalyst.
Activation with BF3.OEt2 afforded 3a in 19% as a 6:1 mixture of anomers.
Glycosylations with primary alcohols 2b–2e afforded the corresponding 2,3-unsaturated glycosides in 72–86% yield within 2 h and with a 30:1 α:β ratio (entries 2–5). Reactions with secondary alcohols such as 4-methoxyphenol 2i, cholesterol 2j, or N-hydroxysuccinimide 2k (entries 9–11) proceeded smoothly giving the desired products in similar high α-selectivity (>30:1 to >20:1, α/β ratio) and yields of 77–82%. Reactions with glycosides 2g, 2h, and propargyl alcohol 2f (entries 6–8) prove to be more challenging and afforded the products in lower yields and stereoselectivity, albeit in favor of the α-products (67–86%, 6:1–7:1 α:β ratio). The ability to effectively activate galactals is noteworthy, as generally glycal substrates that favor a bigger shift toward 5H4 conformations (e.g., glucals) undergo rearrangement more readily than their C-4 epimer galactals where the equilibria is shifted toward the 4H5 form and as a result galactals often give mixtures of products, as well as lower overall yields.10
The scope of the reaction was further investigated with regards to glycal donor. To this end, a series of peracetylated glycals: d-glucal 1b, l-fucal 1c, d-xylal 1d and l-rhamnal 1e were subjected to the reaction conditions with 2e as the model OH nucleophile (Scheme 2). In general, moderate to good yields (65–86%) and α-selectivities were obtained in all cases leading to the formation of 2-deoxy and 2,6-dideoxy Ferrier-type products. Best diastereoselectivities were observed for 2,6-dideoxyglycals (15:1–30:1 α:β, 1c-1e), while glucal 1b yielded 4b in 86% yield and a 3:1 α:β ratio.
Scheme 2. Reaction of Glycals 1a–1e, with Glycosyide Acceptors 2e.

The synthetic utility of our strategy was further exemplified on the preparation of rare glycoside analogues 6–8, which are often difficult to access by traditional methods11 bearing a Boc-protected amino propyl linker that could be used for array conjugation (Scheme 3). Glycosylation of 3-(Boc-amino)-1-propanol with 1c, followed by ester deprotection gave 2,3-unsaturated fucoside 5 in 83% yield (2 steps) and a 10:1 α/β ratio. Alkene reduction of 5 with Rh–Al2O3 afforded α-l-Rhodinose 6 in 76% yield. Alternatively, subjecting 5 to reduction followed by treatment with Dess-Martin periodinane gave α-l-cinerulose 7 (55%), while direct alcohol oxidation of 5 yields α-l-aculose 8 (79%).
Scheme 3. Synthesis of Rare Glycosides 6–8.

Next, we explored whether “armed” glycosides lacking a leaving group at C-3 could undergo BCF-activation and give substitution products selectively. To probe this, reactions between perbenzylated galactal 9a, acceptor 2a and BCF were screened at different catalyst loadings, solvents and temperatures as before. Best results were found when 5 mol % B(C6F5)3 was used in toluene at 50 °C to give 2-deoxyglycoside 10a after 2 h (88%, α/β > 30:1, entry 1, Table 2). As before, reactions were less efficient at lower catalyst loadings or temperatures below 50 °C and changing the solvent to CH2Cl2, CH3CN or CF3Ph was also detrimental (SI Table S2 for full details).
Table 2. Reaction of Glycals 9a–d and 11a–c with Model Glycosyide Acceptors 2a, 2e, or 2g.

| entry | R1 | R2 | R3 | product | time (h) | yield (%)a | α/βb | |
|---|---|---|---|---|---|---|---|---|
| 1 | 9a | Bn | Bn | Bn | 10a | 1 | 88d,e | 21:1 |
| 2 | 9a | Bn | Bn | Bn | 10c | 1 | 94 | 26:1 |
| 3 | 9a | Bn | Bn | Bn | 10e | 2 | 84 | 30:1 |
| 4 | 9a | Bn | Bn | Bn | 10g | 7 | 62 | 25:1 |
| 5 | 9a | Bn | Bn | Bn | 10h | 7 | 68 | 20:1 |
| 6 | 9b | TBS | TBS | TBS | 11b | 1 | 92 | 30:1 |
| 7 | 9c | MOM | MOM | MOM | 11c | 1 | 71 | 20:1 |
| 8 | 9d | Si(tBu)2 | MOM | 11d | 1 | 82 | 20:1 | |
| 9 | 11a | Bn | Bn | Bn | 12ac | 17 | 54 | 30:1 |
| 10 | 11b | O[Si(i-Pr)2]2 | Bn | 12b | 17 | 78 | 30:1 | |
| 11 | 11c | O[Si(i-Pr)2]2 | TIPS | 12c | 17 | 75 | 30:1 | |
| 12 | 11c | O[Si(i-Pr)2]2 | TIPS | 13 | 17 | 86 | 30:1 | |
| 13 | 11c | O[Si(i-Pr)2]2 | TIPS | 14 | 17 | 64 | 20:1 | |
Isolated yield.
Determined by 1H NMR.
10% Ferrier product 12a′ also isolated.
Reaction did not proceed in the absence of catalyst.
Reaction with BF3.OEt2 afforded 10a (<35%) and a mixture of products and starting material.
To explore the substrate scope of the glycosylation, galactals 9b–d and glucals 11a–c were reacted with a range of primary and secondary OH nucleophiles 2a, 2e, or 2g under the optimized reaction conditions. In all cases, reactions proceeded smoothly in yields of 62–94% and high α-selectivity (20:1–30:1 α:β), with secondary OHs requiring longer reaction times (entries 4 and 5 vs 1–3). Subsequently, a series of differentially protected galactals 9b–d and glucals 11a–c bearing benzyl, methoxymethyl acetal, silyl ethers and acetals, and siloxane protecting groups were prepared and subjected to the reaction conditions to investigate the effect of glycal donor on the reaction. Pleasingly, reactions involving all galactals were complete within 1–7 h, in good yields (71–82%) and high α-selectivities (20:1 to 30:1 α/β) (entries 6–8). The reaction was also amenable to glycosylations with glucal substrates, albeit required longer reaction times (17 h) and afforded the glycoside products in moderate to good yields (54–86%) with similarly high α-stereocontrol. Siloxane protected donors 11b and 11c gave better yields that perbenzylated glucal 11a (entries 9–13) as expected.4c These results further highlight that the catalytic system works well across a range of reactivity profiles in both the glycal moiety and nucleophile acceptor.
To probe the mechanism of this versatile reaction, deuterated perbenzylated galactal 15 was reacted with 2a to yield α-glycoside 16a and 16b (90% yield) as a 2:1 mixture of cis/trans products, with a preference for syn addition of both H and O-nucleophile across the double bond (Scheme 4A). Addition of K2CO3 to the reaction between either 1a or 9a with 2a inhibited the reaction, which supports an acid catalyzed process. Monitoring the reaction between 1a or 9a and hexafluoroisopropanol 17 (Scheme 4B) by 1H NMR over 60 min at 45 °C or 90 min at RT, respectively, only showed anomeric signals corresponding to the starting material and product, without any observable changes in the anomeric ratio of the product throughout the time scales of the reaction (SI Figures S1 and S3).13 Moreover, subjecting a 4:1 α/β-anomeric mixture of 10a to the reaction conditions in the presence of acceptor 2a gave no change in the anomeric ratio (see SI for details). These results suggest the reaction proceeds via short-lived intermediates and that the high α-selectivity is not likely the result of anomerization. 19F-NMR of the reactions (SI Figures S2 and S4) showed the appearance of fluorinated signals assigned to products 18 and 19, respectively, and also shifts associated with the formation of other BCF species, suggesting the presence of BCF-adducts. Moreover, 1H NMR spectroscopy studies in Toluene-d8 of a 1:1 mixture of BCF with galactal donor 1a or 9a, also showed H-shifts associated with the enol ether alkene protons, in each case (SI Figures S5 and S7). 19F-NMR of the same mixtures showed additional signals associated with several distinct BCF-species (SI Figures S6 and S8), suggesting activation of the glycal enol ethers by BCF can take place and formation of adducts. Interestingly, 1H NMR equimolar mixtures of B(C6F5)3 and OH nucleophile 2a at room temperature showed proton shifts associated with 2a (Figure S9). This effect was more evident in the 19F-NMR spectra of the same mixtures (SI Figure S10) which showed the shift of the fluorine signals from the catalysts and appearance of different fluorinated species, further supporting the formation of an adduct between the catalyst and the OH nucleophile. This is in agreement to previous reports of glycosyl acceptor activation with boron-based catalysts such as BCF and PhBF2 in the acid–base activation of trichloroacetimidate glycosyl donors.8b,12
Scheme 4. Model Glycosylations of 2a or 17 with Glycal Donors 15, 1a, or 9a.

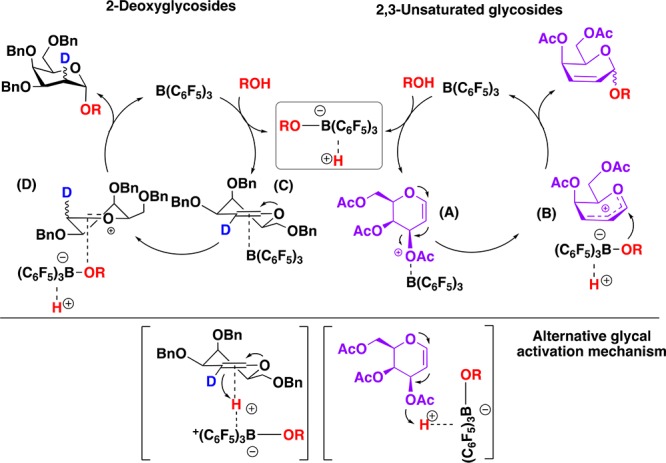
As our preliminary findings suggest, BCF could act as a Lewis acid to promote the effective allylic rearrangement13 (A) of deactivated glycals such as 1a to form transient oxocarbenium ion (B) that can undergo nucleophilic substitution by the BCF-activated nucleophile adduct (H—BCF—OR) in a stereoselective manner to give 2,3-unsaturated glycosides. In the presence of more reactive glycals, which lack a leaving group at C-3 (e.g., 9a), enol ether direct activation to form oxacarbenium ion (D) might take place, which after nucleophilic substitution by the BCF-activated nucleophile and concomitant protonolysis leads to deoxyglycoside products. In both instances, there is a clear preference for an α-face nucleophilic approach, likely due to sterics and a favorable anomeric effect14 (Scheme 5, top). However, in the presence of Lewis basic oxygen atoms, BCF coordination to the pyran oxygen in the donor is also possible. Therefore, an acid–base catalyzed mechanism whereby the boron ate adduct promotes both oxocarbenium ion formation and nucleophile activation can not be discarded and it is likely to occur in parallel (Scheme 5, bottom). Further investigations are ongoing to better understand the mechanism of this reaction.
Scheme 5. Proposed Mechanism.
Conclusions
In summary, we have described the unprecedented BCF-catalyzed substrate-controlled stereoselective synthesis of α-deoxyglycosides directly from glycals. We show that 2,3-unsaturated α-O-glycoside products are obtained with deactivated glycals at 75 °C, while 2-α-deoxyglycosides are formed with activated glycals lacking a leaving group at C-3 at slightly lower temperatures. This metal-free and versatile reaction is applicable to a range of glycal donors and nucleophile acceptors, and is tolerant of most common protecting groups. The reaction proceeds with good to excellent yields and high selectivity for the α-anomer. We exemplify the robustness and utility of the approach in the stereoselective synthesis of a series of oligosaccharides, glycosyl-amino acids, and other glyco-conjugates including rare glycosides analogues of α-l-Rhodinose α-l-cinerulose and α-l-aculose. Work from our lab is currently underway to exploit this chemistry for the stereoselective synthesis of other important glycosides.
Experimental Section
General Experimental Procedures
Chemicals were purchased and used without further purification. Glycal donors 1a–1e, 9a, and 11a were purchased from Carbosynth and OH acceptors 2b, 2e, 2f, 2h, 2i, 2j, and 2k were obtained from Sigma-Aldrich. Galactal donors 9b and 9c and glycosyl acceptors 2a, 2c, and 2d were prepared following literature procedures,4d while glucal 11b and 11c and glycosyl acceptor 2g were synthesized by Balmond et al. reported methods.4c Dry solvents were obtained by distillation using standard procedures or by passage through a column of anhydrous alumina using equipment from Anhydrous Engineering (University of Bristol) based on the Grubbs’ design. Reactions requiring anhydrous conditions were performed under nitrogen; glassware and needles were either flame-dried immediately prior to use or placed in an oven (150 °C) for at least 2 h and allowed to cool either in a desiccator or under reduced pressure; liquid reagents, solutions, or solvents were added via syringe through rubber septa; solid reagents were added via Schlenk type adapters. Teflon rings were used between the joints of the condensers and round-bottom flasks. Reactions were monitored by TLC on Kieselgel 60 F254 (Merck). Detection was by examination under UV light (254 nm) and by charring with 10% sulfuric acid in ethanol. Flash column chromatography was performed using silica gel [Merck, 230–400 mesh (40–63 μm)]. Extracts were concentrated in vacuo using both a Büchi rotary evaporator (bath temperatures up to 40 °C) at a pressure of either 15 mmHg (diaphragm pump) or 0.1 mmHg (oil pump), as appropriate, and a high vacuum line at room temperature. 1H NMR and 13C NMR spectra were measured in the solvent stated at 400 or 500 MHz. Chemical shifts are quoted in parts per million from residual solvent peak (CDCl3: 1H–7.26 ppm and 13C–77.16 ppm) and coupling constants (J) given in Hertz. Multiplicities are abbreviated as b (broad), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), or combinations thereof. Mass spectra were determined by the University of Bristol mass spectrometry service by electrospray ionization (SI) modes. The units of the specific rotation, (deg·mL)/(g·dm), are implicit and are not included with the reported value. Concentration c is given in g/100 mL.
General Glycosylation Procedure
Glycal donor (1.0 equiv), OH nucleophile acceptor (0.75 equiv), and B(C6F5)3 (5 mol %) were weighed into an oven-dried microwave vial, sealed and placed under vacuum for 1 h. Then the vial was filled with N2 followed by the addition of ∼1.0 mL of anhydrous toluene. The solutions were stirred and heated at 75 °C for Ferrier glycosylation and 50 °C for 2-deoxy glycosylation until the reaction was determined to be complete by either TLC or NMR analysis of the crude material (times are given in Tables S1 and S2 and Tables 1 and 2 of the main manuscript). The reaction mixture was concentrated in vacuo and the dried residue was purified by silica gel column chromatography.
Methyl-6-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside 3a)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol), and acceptor 2a (64 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 6:1 to 3:1) 3a as a colorless oil (82 mg, 88%). Spectroscopic data in agreement with literature.15
Benzyl-4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranoside (3b)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2b (15 mg, 0.14 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 12:1 to 8:1) 3b as a colorless oil (37 mg, 84%). Spectroscopic data in agreement with literature.15
Methyl-6-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranosyl)-2,3,4-tri-O-benzoyl-α-d-glucopyranoside (3c)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2c (70 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane: EtOAc, 5:1 to 2:1) 3c as a colorless oil (72 mg, 72%). 1H NMR (500 MHz, CDCl3) δ 7 δ 7.98 (dd, J = 18.7, 7.7 Hz, 5H, Ar–H), 7.87 (d, J = 7.8 Hz, 2H, Ar–H), 7.55–7.52 (m, 2H, Ar–H), 7.39 (t, J = 7.7 Hz, 4H, Ar–H), 7.32–7.28 (m, 2H, Ar–H), 6.20–6.09 (m, 2H, H-2, H-2’), 6.04 (dd, J = 10.0, 3.0 Hz, 1H, H-3′), 5.72 (t, J = 9.8 Hz, 1H, H-4), 5.30–5.26 (m, 2H, H-1, H-3), 5.13 (d, J = 2.9 Hz, 1H, H-1’), 5.04 (dd, J = 5.5, 2.5 Hz, 1H, H-4’), 4.39 (ddd, J = 7.8, 5.4, 2.4 Hz, 1H, H-5′), 4.26 (dt, J = 10.4, 3.7 Hz, 1H, H-5), 4.16–3.99 (m, 3H, H-6a, H-6a’, H-6b’), 3.74 (dd, J = 11.1, 3.0 Hz, 1H, H-6b), 3.50 (s, 3H, OCH3), 2.07 (s, 3H, COCH3), 1.86 (s, 3H, COCH3).13C NMR (126 MHz, CDCl3) δ 170.5 (COCH3), 170.3 (COCH3), 165.8 (COPh), 165.8 (COPh), 165.2 (COPh), 133.5 (Ar–C), 133.4 (Ar–C), 133.1 (Ar–C) 130.1 (Ar–C), 129.9 (Ar–C), 129.8 (Ar–C), 129.2 (C-2’), 129.1 (Ar–C), 128.5 (Ar–C), 128.4 (Ar–C), 128.3 (Ar–C), 125.3, (C-3′) 97.1 (C-1), 93.9 (C-1′), 72.1(C-3), 70.6 (C-2), 69.4 (C-4), 68.3 (C-5), 66.6 (C-5′), 66.1 (C-6), 62.6 (2C- 4′, 6′), 55.7 (OCH3), 20.8 (COCH3), 20.5 (COCH3); ESI-HRMS for C38H38O14Na+ (MNa+) calculated: 741.2159; found: 741.2161
Thiophenyl-6-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranosyl)-2,3,4-tri-O-benzoyl-α-d-glucopyranoside (3d)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2d (80 mg, 0.14 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 5:1 to 1:1) 3d as a colorless oil (85 mg, 72%). 1H NMR (500 MHz, CDCl3) δ 8.00–7.96 (m, 2H, Ar–H), 7.96–7.91 (m, 2H, Ar–H), 7.82–7.78 (m, 2H, Ar–H), 7.57–7.50 (m, 4H, Ar–H), 7.41 (m, 5H, Ar–H), 7.35–7.25 (m, 5H, Ar–H), 6.10 (ddd, J = 10.1, 5.5, 1.0 Hz, 1H, H-2’), 5.96 (dd, J = 10.0, 3.1 Hz, 1H, H-3′), 5.91 (t, J = 9.5 Hz, 1H, H-3), 5.63 (t, J = 9.7 Hz, 1H, H-4), 5.49 (t, J = 9.7 Hz, 1H, H-2), 5.12 (d, J = 3.1 Hz, 1H, H-1’), 5.08 (d, J = 10.0 Hz, 1H, H-1), 4.97 (dd, J = 5.5, 2.5 Hz, 1H, H-4’), 4.32 (ddd, J = 7.8, 5.4, 2.5 Hz, 1H, H-5′), 4.16–4.08 (m, 2H, H-6a′. H-6b′), 4.07–4.00 (m, 2H, H-5, H-6a), 3.83–3.77 (m, 1H, H-6b), 2.08 (s, 3H, COCH3), 1.93 (s, 3H, COCH3). 13C NMR (126 MHz, CDCl3) δ 170.5 (COCH3), 170.3 (COCH3), 165.8 (COPh), 165.1 (COPh), 165.0 (COPh), 133.5 (Ar–C), 133.2 (Ar–C), 132.6 (Ar–C), 132.1 (Ar–C), 130.1 (C-3′), 129.9 (Ar–C), 129.8 (Ar–C), 129.7 (Ar–C), 129.2 (Ar–C), 128.9 (Ar–C), 128.9 (Ar–C), 128.8 (Ar–C), 128.5 (Ar–C), 128.4 (Ar–C), 128.3 (Ar–C), 128.2 (Ar–C), 125.3 (C-2’), 93.8 (C-1′), 85.9 (C-1), 77.2 (C-5), 74.3 (C-3), 70.5 (C-2), 69.5 (C-4) 66.7 (C-5′) 66.5 (C-6), 62.7 (C-6′), 62.6 (C-4′), 20.8 (COCH3), 20.6 (COCH3). ESI-HRMS for C43H40O13SNa+ (MNa+) calculated: 819.2087; found: 819.2103.
6-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranosyl)-1,2:4,5-di-O-isopropylidene-α-d-galacopyranoside (3e)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol), and acceptor 2e (36 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 7:1 to 4:1) 3e as a colorless oil (49 mg, 75%). Spectroscopic data in agreement with literature.15
2′-Propyn-1′-yl-4,6-di-O-acetyl-2,3-dideoxy-α/β-d-threo-hex-2-enopyranoside (3f)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2f (7.73 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 8:1 to 4:1) 3f as a colorless oil (32 mg, 86%). Spectroscopic data in agreement with literature.15
Methyl 3-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranosyl)-4,6-O-benzylidene 2-O-benzyl-α-d-glucopyranoside (3g)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2g (51 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 5:1 to 3:1) 3g as a colorless oil (55 mg, 68%)1H NMR (500 MHz, CDCl3) δ 7.42 (dd, J = 6.7, 2.8 Hz, 2H, Ar–H), 7.37–7.32 (m, 7H, Ar–H), 7.32–7.29 (m, 1H, Ar–H), 6.07 (dd, J = 3.2, 1.8 Hz, 2H, H-2′, H-3′), 5.54 (d, J = 2.0 Hz, 1H, H-1), 5.51 (s, 1H, H-PhCH), 5.00 (dd, J = 4.3, 2.7 Hz, 1H, H-4’), 4.81–4.69 (m, 1H, PhCHH), 4.57 (d, J = 12.2 Hz, 1H, PhCHH), 4.54 (d, J = 3.7 Hz, 1H, H-1), 4.51–4.46 (m, 1H, H-5′), 4.41 (t, J = 9.4 Hz, 1H, H-3), 4.25 (ddt, J = 13.2, 7.3, 3.3 Hz, 2H, H-6a′, H-6a), 4.05 (dd, J = 11.2, 7.2 Hz, 1H, H-6b’), 3.82 (td, J = 9.9, 4.8 Hz, 1H, H-5), 3.69 (t, J = 10.3 Hz, 1H, H-6b), 3.56 (t, J = 9.5 Hz, 1H, H-4), 3.46 (ddd, J = 13.1, 9.4, 3.6 Hz, 1H, H-2), 3.35 (s, 3H, OCH3), 2.06 (d, J = 6.1 Hz, 6H, 2 COCH3). 13C NMR (126 MHz, CDCl3) δ 170.8 (COCH3), 170.4 (COCH3), 138.2 (Ar–C), 137.2 (Ar–C), 130.8 (C-3′), 129.0 (Ar–C), 128.4 (Ar–C), 128.3 (Ar–C), 128.1 (Ar–C), 127.9 (Ar–C), 126.0 (Ar–C), 124.9 (C-2′), 101.4 (C-PhCH), 99.1 (C-1), 93.6 (C-1′), 82.8 (C-4), 78.1 (C-2), 73.4 (C-PhCH2), 73.1 (C-3), 69.1 (C-6), 66.4 (C-5′), 62.7 (C-4′), 62.2 (C-6′), 62.0 (C-5), 55.2 (OCH3), 20.8 (COCH3), 20.70 (COCH3). ESI-HRMS for C31H36O11Na+ (MNa+) calculated: 607.2155; found: 607.2155.
3-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2- enopyranosyl)-l,2:5,6-di-O-isopropylidene-α-d-glucofuranose (3h)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol), and acceptor 2h (36 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 7:1 to 3:1) 3h as a colorless oil (44 mg, 67%). 1H NMR (500 MHz, CDCl3) δ 6.16 (ddd, J = 10.0, 5.5, 1.1 Hz, 1H, H-2’), 6.09–5.99 (m, 1H, H-3′), 5.90 (d, J = 3.6 Hz, 1H, H-1), 5.39–5.34 (m, 1H, H-1′), 5.04 (dt, J = 5.5, 2.8 Hz, 1H, H-4′), 4.63 (d, J = 3.6 Hz, 1H, H-2), 4.41–4.28 (m, 3H, H-5′, H-3, H-6a′), 4.25–4.18 (m, 2H, H-4, H-6b′), 4.16–4.08 (m, 2H, H-5, H-6a), 3.99 (dd, J = 8.6, 5.1 Hz, 1H, H-6b), 2.13 (s, 3H), 2.10 (s, 3H), 1.52 (s, 3H), 1.42 (s, 3H), 1.33 (d, J = 4.1 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 170.8 (COCH3), 170.3 (COCH3), 130.0 (C-3′), 125.4 (C-2′), 112.0 (4°C), 109.17(4°C), 105.4 (C-1), 94.9 (C-1′), 84.3 (C-2), 81.3 (C-4), 80.77 (C-3), 72.7 (C-5), 67.8 (C-6), 67.2 (C-5′), 63.1 (C-6’), 62.8 (C-4′), 27.0 (CCH3), 26.9 (CCH3), 26.5 (CCH3), 25.4 (CCH3), 20.8 (COCH3), 20.7 (COCH3). ESI-HRMS for C22H32O11Na+ (MNa+) calculated: 495.1842; found: 495.1832.
4-Methoxyphenyl,4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranoside (3i)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2i (17 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 9:1 to 3:1) 3i as a colorless oil (36 mg, 82%)1H NMR (500 MHz, CDCl3) δ 7.10–7.05 (m, 4H, Ar–H), 6.88–6.83 (m, 2H, Ar–H), 6.27 (ddd, J = 9.9, 5.4, 1.0 Hz, 1H, H-2), 6.21 (dd, J = 10.0, 3.0 Hz, 1H, H-3), 5.64 (dt, J = 3.0, 0.6 Hz, 1H, H-1), 5.13 (dd, J = 5.4, 2.5 Hz, 1H, H-4), 4.54 (ddd, J = 7.7, 5.3, 2.5 Hz, 1H, H-5), 4.29–4.23 (m, 2H, H-6a, H-6b), 3.80 (s, 3H, OCH3), 2.12 (s, COCH3), 1.98 (s, 3H, COCH3). 13C NMR (126 MHz, CDCl3) δ 170.6 (COCH3), 170.4 (COCH3), 155.2 (Ar–C), 151.0 (Ar–C), 129.9 (C-3), 125.9 (C-2), 118.8 (Ar–C), 114.5 (Ar–C), 93.7 (C-1), 67.5 (C-5), 62.6 (C-4), 62.5 (C-6), 55.7 (OCH3), 20.8 (COCH3), 20.7 (COCH3). ESI-HRMS for C17H20O7Na+ (MNa+) calculated: 359.1107; found: 359.1117.
Cholesteryl-4,6-diacetyl-2,3-dideoxy-α-d-threo-2-enopyranoside (3j)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2j (53 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 4:1 to 2:1) 3j as a colorless oil (65 mg, 79%) 1H NMR (500 MHz, Chloroform-d) δ 6.12 (dd, J = 10.0, 5.3 Hz, 1H, H-3), 6.03 (dd, J = 10.0, 3.0 Hz, 1H, H-2), 5.38 (dd, J = 4.9, 2.4 Hz, 1H, C = CH), 5.23 (d, J = 3.1 Hz, 1H, H-1), 5.04 (dd, J = 5.4, 2.5 Hz, 1H, H-4), 4.43 (ddd, J = 7.7, 5.6, 2.5 Hz, 1H, H-5), 4.27–4.20 (m, 2H, H-6a, H-6b), 3.59 (m, 1H), 2.44 (ddd, J = 13.4, 5.2, 2.1 Hz, 1H), 2.39–2.24 (m, 1H), 2.09 (d, J = 5.2 Hz, 6H, 2 COCH3), 2.05–1.95 (m, 3H), 1.93–1.79 (m, 3H), 1.63–1.05 (m, 16H), 1.02 (s, 5H), 0.93 (d, J = 6.5 Hz, 4H), 0.88 (dd, J = 6.6, 2.2 Hz, 8H), 0.69 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 170.7 (COCH3), 170.4 (COCH3), 140.8 (C = CH), 131.2 (C-2), 125.0 (C-3), 121.8 (C = CH), 92.4 (C-1), 78.0, 66.7 (C-5), 63.0 (2C-6, 4)), 56.8 (CH), 56.2 (CH), 50.2 (CH), 42.3 (4 °C), 40.4 (CH2), 39.8 (CH2), 39.5 (CH2), 37.2 (4 °C), 36.7 (CH2), 36.2 (CH), 35.8, 31.9, 31.9, 28.2, 28.0, 24.3, 23.8, 22.8, 22.6, 21.1 (COCH3), 20.8 (COCH3), 19.3 (CH3), 18.7 (CH3), 11.9 (CH3). ESI-HRMS for C37H58O6Na+ (MNa+) calculated: 621.4131; found: 621.4126.
N-Succinimido-4,6-diacetyl-2,3-dideoxy-α-d-threo-2-enopyranoside (3k)
Following the general glycosylation procedure, donor 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2k (16 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 3:1 to 1:1) 3k as a colorless oil (41 mg, 91%). 1H NMR (500 MHz, CDCl3) δ 6.36 (ddd, J = 10.0, 5.6, 1.2 Hz, 1H, H-3), 6.20 (ddd, J = 10.0, 3.1, 0.6 Hz, 1H, H-2), 5.64 (ddd, J = 3.2, 1.2, 0.6 Hz, 1H, H-1), 5.15 (ddd, J = 5.6, 2.7, 0.6 Hz, 1H, H-4), 4.81 (td, J = 6.3, 2.7 Hz, 1H, H-5), 4.33 (dd, J = 11.3, 6.2 Hz, 1H, H-6a), 4.07 (dd, J = 11.3, 6.5 Hz, 1H, H-6b), 2.75 (s, 4H, 2CH2), 2.08 (s, 3H, COCH3), 2.07 (s, 3H, COCH3). 13C NMR (126 MHz, CDCl3) δ 171.0 (2 NHS CO), 170.6 (COCH3), 170.1 (COCH3), 128.8 (C-3), 125.9 (C-2), 97.5 (C-1), 68.3 (C-5), 61.9 (C-4), 61.8 (C-6), 25.5 (2 CH2), 20.8 (COCH3), 20.7 (COCH3). ESI-HRMS for C14H17NO8Na+ (MNa+) calculated: 350.0852; found: 350.0862.
6-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-erythro-hex-2-enopyranosyl)-1,2:4,5-di-O-isopropylidene-α-d-galacopyranoside (4b)
Following the general glycosylation procedure, donor 1b (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.01 mmol) and acceptor 2e (36 mg, 0.14 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 8:1 to 4:1) 4b as a yellow oil (56 mg, 86%). Spectroscopic data in agreement with literature.9b
4-O-(acetyl)-2,3,6-trideoxy-α-l-hex-2-enopyranosyl-(1 → 6)-1,2;3,4-di-O-isopropylidene-α-d-galactopyranoside (4c)
Following the general glycosylation procedure, donor 1c (50 mg, 0.12 mmol), B(C6F5)3 (6 mg 0.011 mmol), and acceptor 2e (46 mg, 0.10 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 8:1 to 5:1) 4c as a yellow oil (52 mg, 71%). 1H NMR (500 MHz, CDCl3) δ 6.14–5.95 (m, 2H, H-2′, H-3′), 5.53 (d, J = 5.0 Hz, 1H, H-1), 5.10 (d, J = 1.8 Hz, 1H, H–I′), 4.92 (dd, J = 4.4, 2.5 Hz, 1H, H-4’), 4.60 (dd, J = 7.9, 2.4 Hz, 1H, H-3), 4.31 (dd, J = 5.1, 2.4 Hz, 1H, H-2), 4.28–4.21 (m, 2H, H-5′, H-4), 3.99–3.91 (m, 2H, H-5, H-6a), 3.72–3.64 (m, 1H, H-6b), 2.10 (s, 3H, COCH3), 1.53 (s, 3H, CCH3), 1.45 (s, 3H, CCH3), 1.33 (s, 5H, CCH3), 1.22 (d, J = 6.6 Hz, 3H, CCH3). 13C NMR (126 MHz, CDCl3) δ 170.7 (COCH3), 130.5 (C-3′), 125.8 (C-2′), 109.2 (CCH3), 108.5 (CCH3), 96.3 (C-1), 94.0 (C-1′), 71.1 (C-5′), 70.6 (C-3), 70.5 (C-2), 67.0 (C-5), 66.1 (C-6), 65.1 (C4′), 64.6 (C-4), 26.1 (CCH3), 26.0 (CCH3), 24.9 (CCH3), 24.5 (CCH3), 20.9 (COCH3), 15.9 (CCH3). ESI-HRMS for C20H30O9Na+ (MNa+) calculated: 437.1788; found: 437.1788.
6-O-(R-2,3-dihydro-2H-pyran-4-yl acetate)-1,2:4,5-di-O-isopropylidene-α-d-galacopyranoside (4d)
Following the general glycosylation procedure, donor 1d (50 mg, 0.18 mmol), B(C6F5)3 (6 mg 0.011 mmol) and acceptor 2e (36 mg, 0.14 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 10:1 to 6:1) 4d as a colorless oil (56 mg, 75%). 1H NMR (400 MHz, CDCl3) δ 6.09–5.99 (m, 2H, H-2′, H-3′), 5.54 (d, J = 5.1 Hz, 1H, H-1), 5.09 (d, J = 2.3 Hz, 1H, H-1′), 4.95–4.91 (m, 1H, H-4′)), 4.59 (dd, J = 7.9, 2.4 Hz, 1H, H-3), 4.30 (dd, J = 5.1, 2.4 Hz, 1H, H-2), 4.23–4.15 (m, 2H, H-4, H-6a), 3.99 (ddd, J = 7.1, 4.8, 1.9 Hz, 1H, H-5), 3.90–3.72 (m, 3H, H-6b, H-5a′, H-5b′), 2.08 (s, 3H, COCH3), 1.52 (s, 3H, CCH3), 1.43 (s, 3H CCH3), 1.32 (s, 6H, 2 CCH3). 13C NMR (101 MHz, CDCl3) δ 170.6 (COCH3), 130.9 (C-3′), 124.7 C-2′), 109.3 (CCH3), 108.5 (CCH3), 96.3 (C-1), 93.5 (C-1′), 71.2 (C-4′), 70.7 (C-3), 70.4 (C-2), 67.4 (C-5), 67.3 (C-5′), 63.4 (C-4), 61.4 (C-6), 26.0 (CCH3), 26.0 (CCH3), 24.9 (CCH3), 24.5 (CCH3), 21.1 (COCH3).). ESI-HRMS for C19H28O9Na+ (MNa+) calculated: 423.1631; found: 423.1623.
4-O-(acetyl)-2,3,6-trideoxy-α-l-hex-2-enopyranosyl-(1 → 6)-1,2;3,4-di-O-isopropylidene-α-d-galactopyranoside (4e)
Following the general glycosylation procedure, donor 1e (50 mg, 0.12 mmol), B(C6F5)3 (6 mg 0.011 mmol) and acceptor 2e (46 mg, 0.10 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 15:1 to 8:1) 4e as a yellow oil (48 mg, 65%). Spectroscopic data in agreement with literature.9b
3-N-Boc-propyl acetyl-2,3,6-trideoxy-α-l-hex-2-enopyranosyl (4f)
Following the general glycosylation procedure, donor 1e (50 mg, 0.12 mmol), B(C6F5)3 (6 mg 0.011 mmol), and acceptor 2l (31 mg, 0.10 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 5:1 to 3:1) 4f as a yellow oil (50 mg, 85%). 1H NMR (500 MHz, CDCl3) δ 1.24 (d, J = 6.6 Hz, 3H, CCH3), 1.45 (s, 9H, OC(CH3)3), 1.80 (dt, J = 11.6, 5.9 Hz, 2H, CH2), 2.12 (s, 3H, COCH3), 3.18–3.29 (m, 2H, NHCH2), 3.56 (dt, J = 9.9, 6.0 Hz, 1H, OCHH), 3.82–3.87 (m, 1H. OCHH), 4.23 (qd, J = 6.7, 2.3 Hz, 1H, H-5), 4.70 (s, 1H, NH), 4.93 (dd, J = 5.4, 2.5 Hz, 1H, H-4), 5.02 (d, J = 2.9 Hz, 1H, H-1), 6.02 (ddt, J = 9.9, 3.1, 0.7 Hz, 1H, H-2), 6.09 (ddd, J = 10.0, 5.4, 1.0 Hz, 1H, H-3). 13C NMR (126 MHz, CDCl3) δ 170.7 (COCH3), 155.9 (NHCO), 130.3 (C-2), 125.9 (C-3), 94.3 (C-1), 69.5 (OC(CH3)3) 66.3 (OCH2), 65.0 (C-4), 64.7 (C-5), 38.2 (NHCH2), 29.9 (CH2), 28.4 (OC(CH3)3), 20.9(COCH3), 16.1. ESI-HRMS for C16H27NO6Na+ (MNa+) calculated: 352.1736; found: 352.1748
(3-N-Boc-propyl)-acetyl-2,3,6-trideoxy-α-l-hex-2-enopyranosyl (5)
To a stirring solution of glycoside 4f (200 mg, 0.18 mmol) in 5 mL of methanol, 20 mol % K2CO3 (36 mg, 0.14 mmol) was added. The solution was stirred at RT until the reaction was determined to be complete by TLC. The reaction mixture was concentrated in vacuo and the dried residue diluted in CHCl3 (20 mL) and washed with water (20 mL), brine (20 mL), and dried over anhydrous MgSO4. The organic phase was concentrated in vacuo and purified by silica gel column chromatography (Hexane:EtOAc, 3:1 to 1:1) to afford 5 as a colorless oil (170 mg, 75%). 1H NMR (500 MHz, CDCl3) δ 1.30 (dd, J = 6.7, 2.3 Hz, 3H, CCH3), 1.44 (s, 9H, OC(CH3)3), 1.79 (p, J = 6.9 Hz, 2H, CH2), 3.24 (s, 2H, NHCH2), 3.51–3.63 (m, 2H, OCHH, H-4), 3.84 (dddd, J = 10.4, 9.3, 5.6, 2.8 Hz, 1H, OCHH), 4.11 (qd, J = 6.7, 3.2 Hz, 1H, H-5), 4.72 (s, 1H, NH), 4.95 (s, 1H, H-1), 5.84–5.92 (m, 1H, H-2), 6.15–6.22 (m, 1H, H-3). 13C NMR (126 MHz, CDCl3) δ 155.9 (NHCO), 130.3 (C-3), 128.1 (C-2), 94.6 (C-1), 79.1 (OC(CH3)3), 66.4 (C-5), 66.2 (OCH2), 63.9 (C-4), 38.2 (NHCH2), 29.9 (CH2), 28.4 (OC(CH3)3), 16.1 (CCH3). ESI-HRMS for C14H25NO5Na+ (MNa+) calculated: 310.1630; found: 310.1629.
(3-N-Boc-propyl)-α-l-Rhodinoside (6)
To a stirring solution of glycoside 5 (100 mg 0.348 mmol) in 2 mL of a 1:6 mixture of ethyl acetate;toluene at RT, 5 mol % of Rh–Al2O3 (25 mg) was added. The reaction mixture was placed under a H2 atmosphere (balloon) and was stirred at RT for 5h. The reaction mixture was filtered through diatomaceous earth and the filtrate was concentrated under vacuum. The dry residue was purified by silica gel column chromatography (Hexane:EtOAc, 5:1 to 2:1) 6 as a colorless oil (77 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 4.87 (s, 1H, NH), 4.73–4.77 (m, 1H, H-1), 3.90 (q, J = 6.7, 6.0 Hz, 1H, H-5), 3.70 (ddd, J = 9.9, 7.2, 5.1 Hz, 1H, OCHH), 3.55 (s, 1H, H-4), 3.39–3.48 (m, 1H, OCHH), 3.19 (dd, J = 26.9, 5.8 Hz, 2H, NHCH2), 2.08 (s, 1H, OH), 1.89–2.02 (m, 2H, H-2a, H-3a), 1.67–1.81 (m, 3H, H-3b, CH2), 1.48–1.55 (m, 1H, H-2b), 1.41 (s, 9H, OC(CH3)3), 1.15 (d, J = 6.6 Hz, 3H, CCH3),13C NMR (126 MHz, CDCl3) δ 155.9 (NHCO), 97.2 (C-1), 73.9 (OC(CH3)3), 67.2 (C-5), 66.2 (C-4), 65.4 (OCH2), 38.7 (NHCH2), 29.5 (CH2), 28.4 (OC(CH3)3), 25.7 (C-3), 23.4 (C-2), 17.1 (CCH3). ESI-HRMS for C14H27NO5Na+ (MNa+) calculated: 312.1787; found: 312.1781.
(3-N-Boc-propyl)-α-l-Cineruloside (7)
To a stirring solution of 5 (100 mg 0.348 mmol) in 2 mL of a 1:6 mixture of ethyl acetate;toluene at RT, 5% Rh–Al2O3 (25 mg) was added. The reaction mixture was placed under a H2 atmosphere (balloon) and was stirred at room temperature for 5 h. The reaction mixture was filtered through diatomaceous earth, and the filtrate was concentrated under vacuum. To the dried residue, Dess-Martin periodinane (221 mg 0.52 mmol) in 5 mL CH2Cl2 solvent were added and the reaction left to stir for another 5h at RT. The reaction mixture was filtered through diatomaceous earth, and the filtrate was concentrated under vacuum. The dry residue was purified by silica gel column chromatography (Hexane:EtOAc, 4:1 to 2:1) 7 as a colorless oil (77 mg, 76%). 1H NMR (500 MHz, CDCl3) δ 4.97 (t, J = 4.7 Hz, 1H, H-1), 4.73 (s, 1H, NH), 4.24 (q, J = 6.7 Hz, 1H, H-5), 3.83 (ddd, J = 10.0, 6.8, 5.5 Hz, 1H, OCHH), 3.55 (ddd, J = 10.0, 6.5, 5.4 Hz, 1H, OCHH), 3.25 (q, J = 6.0 Hz, 2H, NHCH2), 2.53 (ddd, J = 16.1, 8.0, 5.7 Hz, 1H, H-3b), 2.44 (ddd, J = 16.1, 8.7, 5.8 Hz, 1H, H-3a), 2.25–2.32 (m, 1H, H-2a), 2.01 (dddd, J = 14.1, 8.3, 5.7, 4.3 Hz, 1H, H-2b), 1.75–1.85 (m, 2H, CH2), 1.44 (s, 9H, OC(CH3)3), 1.29 (d, J = 6.7 Hz, 3H, CCH3).13C NMR (126 MHz, CDCl3) δ 210.3 (C-4, C2CO), 155.9 (NHCO), 96.8 (C-1), 79.2 (OC(CH3)3), 71.0 (C-5), 65.8 (OCH2), 38.3 (NHCH2), 33.6 (C-3), 29.8 (CH2), 29.0 (CH2), 28.4 (OC(CH3)3), 14.9 (CCH3). ESI-HRMS for C14H25NO5Na+ (MNa+) calculated: 310.1630; found: 310.1630.
(3-N-Boc-propyl)-α-l-Aculoside (8)
To a flask loaded with 5 (100 mg 0.18 mmol), Dess-Martin periodinane (221 mg 0.52 mmol) in 5 mL CH2Cl2 solvent was added and the reaction left to stir for 6 h at RT. The reaction mixture was then filtered through diatomaceous earth, and the filtrate was concentrated under vacuum. The dry residue was purified by silica gel column chromatography (Hexane:EtOAc, 5:1 to 3:1) 8 as a colorless oil (84 mg 86% yield). 1H NMR (500 MHz, CDCl3) δ 6.84 (dd, J = 10.2, 3.5 Hz, 1H, H-2), 6.09 (d, J = 10.4 Hz, 1H, H-3), 5.18 (d, J = 3.5 Hz, 1H, H-1), 4.67 (s, 1H, NH), 4.55 (q, J = 6.8 Hz, 1H, H-5), 3.92 (dt, J = 9.9, 6.0 Hz, 1H, OCHH), 3.64 (dt, J = 9.9, 6.0 Hz, 1H, OCHH), 3.21–3.31 (m, 2H, NHCH2), 1.83 (p, J = 6.6 Hz, 2H, CH2), 1.45 (s, 9H, OC(CH3)3), 1.40 (d, J = 6.8 Hz, 3H, CCH3),13C NMR (126 MHz, CDCl3) δ 196.9 (C-4 CO), 155.9 (NHCO), 143.3 (C-2), 127.3 (C-3), 93.3 (C-1), 70.4 (OC(CH3)3), 67.2 (C-5), 38.0 (CH2), 30.0 (CH2), 29.7 (CH2), 28.0, (OC(CH3)3), 15.3 (CCH3). ESI-HRMS for C14H23NO5Na+ (MNa+) calculated: 308.1474; found: 308.1461.
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4,6-tri-O-benzyl-α-d-lyxo-hexapyranosyl)-α-d-glucopyranoside (10a)
Following the general glycosylation procedure. Donor 9a (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol), and acceptor 2a (42 mg, 0.09 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 6:1 to 3:1) 10a as a colorless oil (82 mg, 88%). Spectroscopic data in agreement with literature.9a
Benzyl 2-deoxy-3,4,6-tri-O-benzyl-α-d-lyxo-hexapyranoside (10b)
Following the general glycosylation procedure. Donor 9a (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol) and acceptor 2b (10 mg, 0.09 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 20:1 to 10:1) 10b as a colorless oil (46 mg, 94%). Spectroscopic data in agreement with literature.9a
6-O-(3,4,6-tri-O-benzyl-α-d-lyxo-hexapyranosyl)-1,2,3,4-di-O-isopropylidene-α-d-galactopyranoside (10e)
Following the general glycosylation procedure. Donor 9a (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol), and acceptor 2e (23 mg, 0.09 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 6:1 to 4:1) 10e as a colorless oil (50 mg, 84%). Spectroscopic data in agreement with literature.4
Methyl 3-O-benzyl-2-O-(2-deoxy-3,4,6-tri-O-benzyl-α-d-lyxo-hexapyranosyl)-4,6-O-benzylidene-α-d-glucopyranoside (10g)
Following the general glycosylation procedure. Donor 9a (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol), and acceptor 2e (34 mg, 0.09 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 6:1 to 4:1) 10g as a colorless oil (45 mg, 62%). Spectroscopic data in agreement with literature.9a
3-O-(2-deoxy-3,4,6-Tri-O-benzyl-α-d-lyxo-hexapyranoside)-1,2:5,6-di-O-isopropylidene-α-d-glucofuranoside (10h)
Following the general glycosylation procedure. Donor 9a (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol) and acceptor 2h (23 mg, 0.09 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 6:1 to 3:1) 10h as a colorless oil (41 mg, 68%). Spectroscopic data in agreement with literature.9d
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4,6-Tri-O-tert-butyldimethylsilyl-α-d-lyxo-hexapyranosyl)-α-d-glucopyranoside (11b)
Following the general glycosylation procedure. Donor 9b (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol) and acceptor 2a (36 mg, 0.08 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 10:1 to 8:1) 11b as a colorless oil (68 mg, 92%). Spectroscopic data in agreement with literature.9a
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4,6-tri-O-methoxymethyl ether-α-d-lyxo-hexapyranosyl)-α-d-glucopyranoside (11c)
Following the general glycosylation procedure, donor 9c (50 mg, 0.10 mmol), B(C6F5)3 (5 mg 0.009 mmol) and acceptor 2a (63 mg, 0.11 mmol) to afford following purification by silkica gel column chromatography (Hexane:EtOAc, 7:1 to 5:1) 11c as a colorless oil (71 mg, 71%). Spectroscopic data in agreement with literature.9a
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-4,6-O-[Bis(tert-butyl)silylene]-3-O-methoxymethyl ether-α-d-lyxo-hexapyranosyl)-α-d-glucopyranoside (11d)
Following the general glycosylation procedure, donor 9d (50 mg, 0.10 mmol), B(C6F5)3 (4 mg 0.008 mmol) and acceptor 2a (53 mg, 0.11 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 10:1 to 7:1) 11d as a colorless oil (74 mg, 82%). 1H NMR (400 MHz, CDCl3) δ 7.31 (dddd, J = 20.0, 16.0, 8.5, 5.8 Hz, 15H, Ar–H), 5.02–4.93 (m, 3H, H-1′, PhCHH), 4.83–4.77 (m, 3H, PhCHH, OCH-H), 4.74–4.65 (m, 2H, PhCHH, OCH-H), 4.61 (d, J = 3.5 Hz, 1H, H-1), 4.55 (d, J = 11.4 Hz, 1H, PhCHH), 4.38 (d, J = 2.0 Hz, 1H, H-4′), 4.05–3.96 (m, 3H, H-3, H-6a′, H-6b′), 3.88 (ddd, J = 12.2, 4.4, 2.7 Hz, 1H, H-3′), 3.78 (m, 2H, H-5, H-6a), 3.63 (d, J = 9.6 Hz, 1H, H-6b), 3.54 (dd, J = 9.7, 3.6 Hz, 1H, H-2), 3.51–3.45 (m, 1H, H-4), 3.42 (d, J = 3.1 Hz, 4H, H-5′, CH2OCH3), 3.38 (s, 3H, OCH3), 2.18 (td, J = 12.4, 3.6 Hz, 1H, H-2a′), 1.82 (dd, J = 12.5, 4.7 Hz, 1H, H-2b′), 1.04 (s, 18H, 2C(CH3)3). 13C NMR (101 MHz, CDCl3) δ 138.6 (Ar–C), 138.3 (Ar–C), 138.1 (Ar–C), 128.5 (Ar–C), 128.4 (Ar–C), 127.9 (Ar–C), 127.7 (Ar–C), 127.2 (Ar–C), 98.2 (C-1′), 97.9 (C-1), 95.0 (OCH2–CH3), 82.2 (C-3), 80.1 (C-2), 78.1 (C-4), 75.8 (PhCH2), 74.8 (PhCH2), 73.3 (PhCH2), 72.1 (C-3′), 70.4 (C-4′), 69.7 (C-5), 67.56 (2C, C-6′, C-5′), 65.9 (C-6), 55.7 (OCH2CH3), 55.1 (OCH3), 30.1 (C-2′), 27.6 (C(CH3)3), 27.4 (C(CH3)3), 23.4 (C(CH3)3), 20.8 (C(CH3)3). ESI-HRMS for C44H62O11SiNa+ (MNa+) calculated: 817.3959; found: 817.3965.
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4,6-tri-O-benzyl-α-d-lyxo-hexapyranosyl)-α-d-glucopyranoside (12a) and Methyl-6-O-(4,6-di-O-acetyl-2,3-dideoxy-α-d-erythreo-hex-2-enopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside(12a′)
Following the general glycosylation procedure. Donor 11a (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol) and acceptor 2a (42 mg, 0.09 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 6:1 to 3:1) 12a as a colorless oil (44 mg, 54%) and 12a′ as a colorless oil (7 mg, 10%). Spectroscopic data in agreement with literature.4c
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-6-O-benzyl-α-d-erythro-hexapyranosyl)-α-d-glucopyranoside (12b)
Following the general glycosylation procedure, donor 11b (50 mg, 0.10 mmol), B(C6F5)3 (3 mg 0.006 mmol), and acceptor 2a (36 mg, 0.08 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 12:1 to 8:1) 12b as a colorless oil (56 mg, 78%). Spectroscopic data in agreement with literature.9a
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-6-O-triisopropylsilyl-α-d-erythro-hexapyranosyl)-α-d-glucopyranoside (12c)
Following the general glycosylation procedure, donor 11c (50 mg, 0.10 mmol), B(C6F5)3 (2 mg 0.005 mmol) and acceptor 2a (32 mg, 0.07 mmol) to afford following purification by silica gel column chromatography (Hexane:EtOAc, 12:1 to 8:1) 12c as a colorless oil (51 mg, 73%). Spectroscopic data in agreement with literature.9a
6-O-(2-deoxy-3,4-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-6-O-triisopropylsilyl-α-d-erythro-hexapyranosyl)-1,2,3,4-di-O-isopropylidene-α-d-galactopyranoside (13)
Following the general glycosylation procedure, donor 11c (50 mg, 0.10 mmol), B(C6F5)3 (2 mg 0.005 mmol), and acceptor 2e (18 mg, 0.07 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 16:1 to 10:1) 13 as a colorless oil (48 mg, 86%). 1H NMR (400 MHz, CDCl3) δ 0.83–1.18 (m, 49H, 7× SiCH(CH3)2), 1.32 (s, 6H, 2 O2CCH3), 1.43 (s, 3H, O2CCH3), 1.51 (s, 3H, O2CCH3), 1.66 (ddd, J = 13.3, 11.4, 3.7 Hz, 1H, H-2a′), 2.10 (dd, J = 12.9, 5.3 Hz, 1H, H-2b′), 3.49 (dd, J = 9.4, 8.3 Hz, 1H, H-4′), 3.54–3.61 (m, 1H, H-5′), 3.65 (dd, J = 10.7, 6.2 Hz, 1H, H-6b′), 3.79 (ddd, J = 17.8, 10.7, 6.4 Hz, 2H, H-6b, H-6a′), 3.97 (td, J = 6.5, 1.6 Hz, 1H, H-5), 4.00–4.08 (m, 2H, H-6a, H-3′), 4.20 (dd, J = 7.9, 1.8 Hz, 1H, H-4), 4.30 (dd, J = 5.0, 2.4 Hz, 1H, H-2), 4.60 (dd, J = 7.9, 2.3 Hz, 1H, H-3), 4.95 (d, J = 3.1 Hz, 1H, H-1′), 5.51 (d, J = 5.0 Hz, 1H, H-1). 13C NMR (101 MHz, CDCl3) δ 109.2 (O2CCH3), 108.4 (O2CCH3), 96.3 (C-1), 96.0 (C-1′), 74.6 (C-4′), 73.4 (C-5′), 71.6 (C-3′), 71.1 (C-4), 70.7 (2C, C-2, C-3), 65.5 (C-5), 64.5 (C-6′), 63.3 (C-6), 38.0 (C-2′), 26.0 (O2CCH3), 25.9 (O2CCH3), 24.9 (O2CCH3), 24.4 (O2CCH3), 18.0, 17.9, 17.6, 17.4, 17.37, 17.3, 17.2 (Si(CH(CH3)2)), 13.0, 12.8, 12.4, 12.3, 12.0 (Si(CH(CH3)2)). ESI-HRMS for C39H76O11Si3Na+ (MNa+) calculated: 827.4593; found: 827.4586
Methyl-3-O-benzyl-2-O-(2-deoxy-3,4-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-6-O-triisopropylsilyl-α-d-erythro-hexapyranosyl)-4,6-O-benzylidene-α-d-glucopyranoside (14)
Following the general glycosylation procedure, donor 11c (50 mg, 0.10 mmol), B(C6F5)3 (2 mg 0.005 mmol), and acceptor 2g (26 mg, 0.07 mmol) to afford following purification by column chromatography (Hexane:EtOAc, 10:1 to 5:1) 14 as a colorless oil (41 mg, 64%). Spectroscopic data in agreement with literature.9a
Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4,6-tri-O-benzyl-(axial/equatorial)2-2H-α-d-lyxo-hexapyranosyl)-α-d-glucopyranoside (16a/16b)
Following the General Glycosylation Procedure, galactal 9 (50 mg, 0.120 mmol), B(C6F5)3 (3 mg 0.006 mmol), and acceptor 2a (42 mg, 0.090 mmol). Following purification by silica gel column chromatography (8:1 to 4:1, Hexane:EtOAc) to afford glycoside 16a/16b as an oil (70 mg, 88%).9a
Hexafluoroisopropayl-4,6-di-O-acetyl-2,3-dideoxy-α-d-threo-hex-2-enopyranoside (18)
Following the General Glycosylation Procedure, galactal 1a (50 mg, 0.18 mmol), B(C6F5)3 (5 mg 0.009 mmol) acceptor 17 (42 mg, 0.090 mmol) at 45°C. Following purification by silica gel column chromatography (6:1 to 4:1, Hexane:EtOAc) product 18 was obtained as an oil (45 mg, 87%). 1H NMR (500 MHz, CDCl3) δ 6.29 (ddd, J = 10.0, 5.7, 1.2 Hz, 1H, H-3), 6.09 (dd, J = 10.0, 3.0 Hz, 1H, H-2), 5.34 (d, J = 2.6 Hz, 1H, H-1), 5.08 (dd, J = 5.7, 2.5 Hz, 1H, H-4), 4.63 (hept, J = 5.4 Hz, 1H, HC(CF3)2), 4.35 (ddd, J = 7.3, 4.6, 2.4 Hz, 1H, H-5), 4.30 (dd, J = 11.6, 4.6 Hz, 1H, H-6a), 4.18 (dd, J = 11.6, 7.5 Hz, 1H, H-6b), 2.09 (s, 3H, COCH3), 2.07 (s, 3H, COCH3). 13C NMR (126 MHz, CDCl3) δ 170.5 (COCH3), 170.1 (COCH3), 127.9(C-2), 126.9 (C-3), 122.9 (CF3), 122.2 (CF3), 94.8 (C-1), 71.6–71.1 (C(CF3)2), 68.1 (C-5), 62.4 (C-6), 62.1 (C-4), 20.7 (COCH3), 20.5 (COCH3). 19F NMR (470 MHz, CDCl3) δ −73.52 (m, CF3), −73.61 (m, CF3). ESI-HRMS for C13H14F6O6Na+ (MNa+) calculated: 403.0592; found: 403.0600.
Hexafluoroisopropayl-2-deoxy-3,4,6-tri-O-benzyl-α-D-lyxo-hexapyranoside (19)
Following the General Glycosylation Procedure, galactal 9a (50 mg, 0.120 mmol), B(C6F5)3 (3 mg 0.006 mmol) acceptor 17 (42 mg, 0.090 mmol) at RT. Following purification by silica gel column chromatography (12:1 to 8:1, Hexane:EtOAc) product 19 was obtained as an oil (48 mg, 92%). 1H NMR (500 MHz, CDCl3) δ 7.46–7.23 (m, 15H, Ar–H), 5.28 (d, J = 3.6 Hz, 1H, H-1), 4.97 (d, J = 11.5 Hz, 1H, PhCHH), 4.67–4.61 (m, 3H, PhCHH. PhCHH), 4.50 (q, J = 11.9 Hz, 3H, PhCHH, HC(CF3)2), 4.06–3.99 (m, 2H, H-4, H-5), 3.95 (ddd, J = 12.1, 4.6, 2.3 Hz, 1H, H-3), 3.63 (dd, J = 9.3, 7.0 Hz, 1H, H-6a), 3.58 (dd, J = 9.3, 5.9 Hz, 1H, H-6b), 2.37 (td, J = 12.7, 3.9 Hz, 1H, H-2a), 2.19 (dd, J = 13.2, 4.6 Hz, 1H, H-2b). 13C NMR (126 MHz, CDCl3) δ 138.6 (Ar–C), 138.1 (Ar–C), 138.0 (Ar–C), 128.5 (Ar–C), 128.4 (Ar–C), 128.3 (Ar–C), 128.2 (Ar–C), 127.7 (Ar–C), 127.7 (Ar–C), 127.6 (Ar–C), 127.6 (Ar–C), 127.4 (Ar–C), 122.4 (CF3). 120.2 (CF3), 100.0 (C-1), 74.4 (PhCH2), 73.8 (C-3), 73.4 (PhCH2), 72.5 (C-4), 71.5 (C(CF3)2) 71.4 (C-5), 70.7 (PhCH2), 68.8 (C-6), 30.2 (C-2). 19F NMR (470 MHz, CDCl3) −73.24 (m, CF3), −73.40 (m, CF3). ESI-HRMS for C30H30F6O5Na+ (MNa+) calculated: 607.1895; found:607.1903.
Synthesis of Methyl-2,3,4-tri-O-benzyl-6-O-(2-deoxy-3,4,6-tri-O-benzyl-α/b-d-lyxohexapyranosyl)-α-d-glucopyranoside (10a)
The glycosyl donor 9a (1 equiv) and acceptor 2a (0.83 equiv) were weighed into a microwave vial and placed under vacuum for 1 h, after which time the microwave vial was filled with N2. A solution mixture containing (R)-3,3′-Bis[3,5-bis(trifluoromethyl)phenyl]-1,1′-binaphthyl-2,2′-diyl hydrogenphosphate (0.1 equiv) and thiourea (0.1 equiv) in anhydrous CH3CN (1 mL) was stirred for 30 min, before adding it to the microwave vial containing 1a and 2a.The reaction mixture was stirred at RT for 4 h and then was purified by silica gel column chromatography (Hexane:EtOAc, 7:1 to 4:1) affording disaccharide 3a as a colorless oil (66 mg 70%, 4:1 α:β). The spectroscopic data was in agreement with previously reported data.15
In Situ Anomerization Test of 10a in the Presence of 2a
Disaccharide 10a (4:1 α:β, 1 equiv), acceptor monosaccharide 2a (1 equiv), and B(C6F5)3 (5 mol %) were weighed into an oven-dried microwave vial, sealed and placed under vacuum for 1 h. Then the vial was filled with N2 followed by the addition of ∼1.0 mL of anhydrous toluene. The solutions were stirred and heated at 50 °C for 2 h without observing any change in the anomeric ratio (4:1 α/β) as monitored by NMR of the crude mixture.
Acknowledgments
This research was supported by ERC–COG: 648239 (M.C.G. and A.S.) and RS Newton International fellowship NF150783 (C.P.-N).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b02613.
Reaction optimization tables and NMR reaction studies and NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a He X. M.; Liu H. W. Mechanisms of enzymatic C-O bond cleavages in deoxyhexose biosynthesis. Curr. Opin. Chem. Biol. 2002, 6, 590–597. 10.1016/S1367-5931(02)00367-8. [DOI] [PubMed] [Google Scholar]; b Lindhorst T. K.Glycoscience: Chemistry and Chemical Biology;Springer: Berlin, 2001; pp 2393–2439. [Google Scholar]
- a Marzabadi C. H.; Franck R. W. The synthesis of 2-deoxyglycosides: 1988–1999. Tetrahedron 2000, 56, 8385–8417. 10.1016/S0040-4020(00)00691-8. [DOI] [Google Scholar]; b Williams R.; Galan M. C. Recent Advances in Organocatalytic Glycosylations. Eur. J. Org. Chem. 2017, 2017, 6247–6264. 10.1002/ejoc.201700785. [DOI] [Google Scholar]; c Medina S.; Galan M. C. Recent developments in the stereoselective synthesis of deoxy glycosides. Carbohydr. Chem. 2015, 41, 59–89. 10.1039/9781782620600-00059. [DOI] [Google Scholar]
- a Benito-Alifonso D.; Galan M. C.. Bronsted and Lewis Acid Catalyzed Glycosylation. In Selective Glycosylations: Synthetic Methods and Catalysis; Bennett C. E., Ed.; 2017; pp 155–171. [Google Scholar]; b McKay M. J.; Nguyen H. M. Recent Advances in Transition Metal-Catalyzed Glycosylation. ACS Catal. 2012, 2, 1563–1595. 10.1021/cs3002513. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Balmond E. I.; Galan M. C.; McGarrigle E. M. Recent Developments in the Application of Organocatalysis to Glycosylations. Synlett 2013, 24, 2335–2339. 10.1055/s-0033-1338970. [DOI] [Google Scholar]; d Li X. H.; Zhu J. L. Glycosylation via Transition-Metal Catalysis: Challenges and Opportunities. Eur. J. Org. Chem. 2016, 2016, 4724–4767. and references therein 10.1002/ejoc.201600484. [DOI] [Google Scholar]; e Pradhan T. K.; Lin C. C.; Mong K. K. T. Preparation of a Protected 3-Deoxy-D-manno-oct-2-ulosonate Glycal Donor for the Synthesis of beta-KDO-Containing Oligosaccharides. Org. Lett. 2014, 16, 1474–1477. 10.1021/ol500275j. [DOI] [PubMed] [Google Scholar]; h Wang H.; Tao J. Y.; Cai X. P.; Chen W.; Zhao Y. Q.; Xu Y.; Yao W.; Zeng J.; Wan Q. Stereoselective Synthesis of alpha-Linked 2-Deoxy Glycosides Enabled by Visible-Light-Mediated Reductive Deiodination. Chem. - Eur. J. 2014, 20, 17319–17323. 10.1002/chem.201405516. [DOI] [PubMed] [Google Scholar]; i Song W. Z.; Zhao Y.; Lynch J. C.; Kim H.; Tang W. P. Divergent de novo synthesis of all eight stereoisomers of 2,3,6-trideoxyhexopyranosides and their oligomers. Chem. Commun. 2015, 51, 17475–17478. 10.1039/C5CC07787G. [DOI] [PubMed] [Google Scholar]; j Thombal R. S.; Jadhav V. H. Facile O-glycosylation of glycals using Glu-Fe3O4-SO3H, a magnetic solid acid catalyst. RSC Adv. 2016, 6, 30846–30851. 10.1039/C6RA03305A. [DOI] [Google Scholar]; k Tanaka H.; Yoshizawa A.; Takahashi T. Direct and stereoselective synthesis of beta-linked 2,6-deoxyoligosaccharides. Angew. Chem., Int. Ed. 2007, 46, 2505–2507. 10.1002/anie.200604031. [DOI] [PubMed] [Google Scholar]; l Verma V. P.; Wang C. C. Highly Stereoselective Glycosyl-Chloride-Mediated Synthesis of 2-Deoxyglucosides. Chem. - Eur. J. 2013, 19, 846–851. 10.1002/chem.201203418. [DOI] [PubMed] [Google Scholar]; m Zhu D. Y.; Baryal K. N.; Adhikari S.; Zhu J. L. Direct Synthesis of 2-Deoxy-beta-Glycosides via Anomeric O-Alkylation with Secondary Electrophiles. J. Am. Chem. Soc. 2014, 136, 3172–3175. 10.1021/ja4116956. [DOI] [PubMed] [Google Scholar]; n Beale T. M.; Moon P. J.; Taylor M. S. Organoboron-Catalyzed Regio- and Stereoselective Formation of beta-2-Deoxyglycosidic Linkages. Org. Lett. 2014, 16, 3604–3607. 10.1021/ol501711v. [DOI] [PubMed] [Google Scholar]; o Soliman S. E.; Bennett C. S. Reagent-Controlled Synthesis of the Branched Trisaccharide Fragment of the Antibiotic Saccharomicin B. Org. Lett. 2018, 20, 3413–3416. 10.1021/acs.orglett.8b01355. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Lloyd D.; Bennett C. S. An Improved Approach to the Direct Construction of 2-Deoxy-beta-Linked Sugars: Applications to Oligosaccharide Synthesis. Chem. - Eur. J. 2018, 24, 7610–7614. 10.1002/chem.201800736. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Bennett C. S.; Galan M. C. Methods for 2-Deoxyglycoside Synthesis. Chem. Rev. 2018, 118, 7931–7985 . and references therein 10.1021/acs.chemrev.7b00731. [DOI] [PMC free article] [PubMed] [Google Scholar]; r Bradshaw G. A.; Colgan A. C.; Allen N. P.; Pongener I.; Boland M. B.; Ortin Y.; McGarrigle E. M. Stereoselective organocatalyzed glycosylations - thiouracil, thioureas and monothiophthalimide act as Bronsted acid catalysts at low loadings. Chem. Sci. 2019, 10, 508–514. 10.1039/C8SC02788A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhao G. Y.; Wang T. Stereoselective Synthesis of 2-Deoxyglycosides from Glycals by Visible-Light-Induced Photoacid Catalysis. Angew. Chem., Int. Ed. 2018, 57, 6120–6124. 10.1002/anie.201800909. [DOI] [PubMed] [Google Scholar]; b Medina S.; Harper M. J.; Balmond E. I.; Miranda S.; Crisenza G. E. M.; Coe D. M.; McGarrigle E. M.; Galan M. C. Stereoselective Glycosylation of 2-Nitrogalactals Catalyzed by a Bifunctional Organocatalyst. Org. Lett. 2016, 18, 4222–4225. 10.1021/acs.orglett.6b01962. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Balmond E. I.; Benito-Alifonso D.; Coe D. M.; Alder R. W.; McGarrigle E. M.; Galan M. C. A 3,4-trans-Fused Cyclic Protecting Group Facilitates alpha-Selective Catalytic Synthesis of 2-Deoxyglycosides. Angew. Chem., Int. Ed. 2014, 53, 8190–8194. 10.1002/anie.201403543. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Balmond E. I.; Coe D. M.; Galan M. C.; McGarrigle E. M. Alpha-Selective Organocatalytic Synthesis of 2-Deoxygalactosides. Angew. Chem., Int. Ed. 2012, 51, 9152–9155. 10.1002/anie.201204505. [DOI] [PubMed] [Google Scholar]; e Sherry B. D.; Loy R. N.; Toste F. D. Rhenium(V)-catalyzed synthesis of 2-deoxy-alpha-glycosides. J. Am. Chem. Soc. 2004, 126, 4510–4511. 10.1021/ja031895t. [DOI] [PubMed] [Google Scholar]; f Das S.; Pekel D.; Neudorfl J. M.; Berkessel A. Organocatalytic Glycosylation by Using Electron- Deficient Pyridinium Salts. Angew. Chem., Int. Ed. 2015, 54, 12479–12483. 10.1002/anie.201503156. [DOI] [PubMed] [Google Scholar]; g Liu J. L.; Zhang Y. T.; Liu H. F.; Zhou L.; Chen J. N-Heterocyclic Carbene Catalyzed Stereoselective Glycosylation of 2-Nitrogalactals. Org. Lett. 2017, 19, 5272–5275. 10.1021/acs.orglett.7b02543. [DOI] [PubMed] [Google Scholar]
- a Schmidt R. R.; Angerbauer R. Convenient Preparation of 2,3-Unsaturated N-Galactosyl Derivatives. Carbohydr. Res. 1979, 72, 272–275. 10.1016/S0008-6215(00)83948-8. [DOI] [Google Scholar]; b Fraser-Reid B. Some progeny of 2,3-unsaturated sugars - They little resemble grandfather glucose: Twenty years later. Acc. Chem. Res. 1996, 29, 57–66. 10.1021/ar950104s. [DOI] [Google Scholar]; c Csuk R.; Schaade M.; Krieger C. Synthesis of C-glycosides from glycals or vinylogous lactones and trimethylsilyl ketene acetals. Tetrahedron 1996, 52, 6397–6408. 10.1016/0040-4020(96)00275-X. [DOI] [Google Scholar]; d Borrachero-Moya P.; Cabrera-Escribano F.; Gomez-Guillen M.; Paredes-Leon M. d. R. Synthesis of 4-(4,6-di-0-benzyl-2,3-dideoxy-beta-D-erythro-hex-2-enopyranosyl)pyrazoles from 3,4,6-tri-O-acetyl-D-glucal. Carbohydr. Res. 1998, 308, 181–190. 10.1016/S0008-6215(98)00059-7. [DOI] [Google Scholar]; e Gomez A. M.; Lobo F.; Uriel C.; Lopez J. C. Recent Developments in the Ferrier Rearrangement. Eur. J. Org. Chem. 2013, 2013, 7221–7262. 10.1002/ejoc.201300798. [DOI] [Google Scholar]; f Ferrier R. J. Unsaturated Carbohydrates. Part II. Three Reactions Leading to Unsaturated Glycopyranosides. J. Chem. Soc. 1964, 5443–5449. 10.1039/jr9640005443. [DOI] [Google Scholar]; g Ferrier R. J.; Hoberg J. O. Synthesis and reactions of unsaturated sugars. Adv. Carbohydr. Chem. Biochem. 2003, 58, 55–119. 10.1016/S0065-2318(03)58003-9. [DOI] [PubMed] [Google Scholar]
- a Lee D.; Taylor M. S. Borinic Acid-Catalyzed Regioselective Acylation of Carbohydrate Derivatives. J. Am. Chem. Soc. 2011, 133, 3724–3727. 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]; b Beale T. M.; Moon P. J.; Taylor M. S. Organoboron-Catalyzed Regio- and Stereoselective Formation of beta-2-Deoxyglycosidic Linkages. Org. Lett. 2014, 16, 3604–3607. 10.1021/ol501711v. [DOI] [PubMed] [Google Scholar]
- Lawson J. R.; Melen R. L. ris(pentafluorophenyl)borane and Beyond: Modern Advances in Borylation Chemistry. Inorg. Chem. 2017, 56, 8627–8643. 10.1021/acs.inorgchem.6b02911. [DOI] [PubMed] [Google Scholar]
- a Karimov R. R.; Tan D. S.; Gin D. Y. Rapid assembly of the doubly-branched pentasaccharide domain of the immunoadjuvant jujuboside A via convergent B(C6F5)3-catalyzed glycosylation of sterically-hindered precursors. Chem. Commun. 2017, 53, 5838–5841. 10.1039/C7CC01783A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mishra K. B.; Singh A. K.; Kandasamy J. Tris(pentafluorophenyl)borane-Promoted Stereoselective Glycosylation with Glycosyl Trichloroacetimidates under Mild Conditions. J. Org. Chem. 2018, 83, 4204–4212. 10.1021/acs.joc.8b00215. [DOI] [PubMed] [Google Scholar]
- a Sau A.; Williams R.; Palo-Nieto C.; Franconetti A.; Medina S.; Galan M. C. Palladium-Catalyzed Direct Stereoselective Synthesis of Deoxyglycosides from Glycals. Angew. Chem., Int. Ed. 2017, 56, 3640–3644. 10.1002/anie.201612071. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sau A.; Galan M. C. Palladium-Catalyzed alpha-Stereoselective O-Glycosylation of O(3)-Acylated Glycals. Org. Lett. 2017, 19 (11), 2857–2860. 10.1021/acs.orglett.7b01092. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Palo-Nieto C.; Sau A.; Williams R.; Galan M. C. Cooperative Bronsted Acid-Type Organocatalysis for the Stereoselective Synthesis of Deoxyglycosides. J. Org. Chem. 2017, 82, 407–414. 10.1021/acs.joc.6b02498. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Palo-Nieto C.; Sau A.; Galan M. C. Gold(I)-Catalyzed Direct Stereoselective Synthesis of Deoxyglycosides from Glycals. J. Am. Chem. Soc. 2017, 139, 14041–14044. 10.1021/jacs.7b08898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Thiem J.; Ossowski P. Studies of Hexuronic Acid Ester Glycals and the Synthesis of 2-Deoxy-Beta-Glycoside Precursors. J. Carbohydr. Chem. 1984, 3, 287–313. 10.1080/07328308408058822. [DOI] [Google Scholar]; b Rico M.; Santoro J. Complete Analysis of H-1 Nmr-Spectra of Acetylated Glycals - Conformational Study. Org. Magn. Reson. 1976, 8, 49–55. 10.1002/mrc.1270080112. [DOI] [Google Scholar]
- For other elegant examples on de novo synthesis of d- and l-rare deoxyglycosides, see:; a Shan M.; O’Doherty G. A. De novo asymmetric syntheses of SL0101 and its analogues via a palladium-catalyzed glycosylation. Org. Lett. 2006, 8 (22), 5149–52. 10.1021/ol062076r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Song W. Z.; Wang S. J.; Tang W. P. De Novo Synthesis of Mono- and Oligosaccharides via Dihydropyran Intermediates. Chem. - Asian J. 2017, 12 (10), 1027–1042. 10.1002/asia.201700212. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Kumar V.; Dere R. T.; Schmidt R. R. Glycoside Bond Formation via Acid-Base Catalysis. Org. Lett. 2011, 13, 3612–3615. 10.1021/ol201231v. [DOI] [PubMed] [Google Scholar]
- Deraadt A.; Ferrier R. J. Unsaturated Carbohydrates 0.30. Syntheses and Reactions of Saturated and 2,3-Unsaturated Vinyl and 1’-Substituted-Vinyl Glycosides. Carbohydr. Res. 1992, 216, 93–107. 10.1016/0008-6215(92)84153-J. [DOI] [Google Scholar]
- a Ayala L.; Lucero C. G.; Romero J. A. C.; Tabacco S. A.; Woerpel K. A. Stereochemistry of nucleophilic substitution reactions depending upon substituent: Evidence for electrostatic stabilization of pseudoaxial conformers of oxocarbenium ions by heteroatom substituents. J. Am. Chem. Soc. 2003, 125, 15521–15528. 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]; b Cumpstey I. On a so-called “kinetic anomeric effect” in chemical glycosylation. Org. Biomol. Chem. 2012, 10, 2503–2508. 10.1039/c2ob06696c. [DOI] [PubMed] [Google Scholar]; c Filloux C. M. The Problem of Origins and Origins of the Problem: Influence of Language on Studies Concerning the Anomeric Effect. Angew. Chem., Int. Ed. 2015, 54, 8880–8894. 10.1002/anie.201411185. [DOI] [PubMed] [Google Scholar]
- Balamurugan R.; Koppolu S. R. Scope of AuCl3 in the activation of per-O-acetylglycals. Tetrahedron 2009, 65, 8139–8142. 10.1016/j.tet.2009.07.087. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.