

Abstract
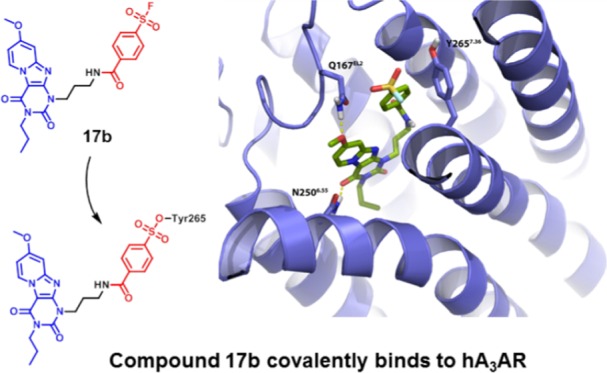
The development of covalent ligands for G protein-coupled receptors (GPCRs) is not a trivial process. Here, we report a streamlined workflow thereto from synthesis to validation, exemplified by the discovery of a covalent antagonist for the human adenosine A3 receptor (hA3AR). Based on the 1H,3H-pyrido[2,1-f]purine-2,4-dione scaffold, a series of ligands bearing a fluorosulfonyl warhead and a varying linker was synthesized. This series was subjected to an affinity screen, revealing compound 17b as the most potent antagonist. In addition, a nonreactive methylsulfonyl derivative 19 was developed as a reversible control compound. A series of assays, comprising time-dependent affinity determination, washout experiments, and [35S]GTPγS binding assays, then validated 17b as the covalent antagonist. A combined in silico hA3AR-homology model and site-directed mutagenesis study was performed to demonstrate that amino acid residue Y2657.36 was the unique anchor point of the covalent interaction. This workflow might be applied to other GPCRs to guide the discovery of covalent ligands.
Introduction
The adenosine A3 receptor (A3AR) is one of four G protein-coupled receptor subtypes stimulated by adenosine.1 Different from the other subtypes (A1, A2A, and A2B) A3AR was identified by molecular biology studies prior to its pharmacological characterization.2 The initial studies indicated its important role in both physiological and pathophysiological conditions, such as cell proliferation, cell differentiation, neuroprotection, cardioprotection, and apoptosis.3 Nevertheless, the medical relevance of the human adenosine A3 receptor (hA3AR) is enigmatic due to its dichotomy in different therapeutic applications.3 In this regard, there is a continuing interest in the development of selective ligands of the hA3AR to investigate its pharmacological effects. For instance, selective A3AR antagonists have been applied for the treatment of glaucoma4 and respiratory tract inflammation such as asthma.5 In particular, a tricyclic xanthine derivative, 1-benzyl-8-methoxy-3-propyl-1H,3H-pyrido[2,1-f]purine-2,4-dione (compound 1, Figure 1A), has been reported to exert high affinity for the hA3AR.6−8
Figure 1.

(A) Reference antagonist (1) for hA3AR. (B) DU172 (2), a covalent antagonist for hA1AR.
Initial efforts to study the structural biology of GPCRs suffered from numerous limitations, such as low expression, dynamic conformational states, and inherent instability. Covalent ligands, i.e., compounds that irreversibly bind to the receptor and possess a reactive moiety to target specific amino acid residues, helped to solve some of these obstacles.9 This is also the case for adenosine receptors. For example, the structure of the human adenosine A1 receptor, having the highest similarity to the hA3AR among all adenosine receptor subtypes (61% of sequence homology),10 has been elucidated by X-ray crystallography with a covalent antagonist DU172 (2) (Figure 1B).11 However, the application of covalent ligands in hA3AR studies has been limited to the characterization of the receptor type,12−14 far from providing a comprehensive study of receptor structure elucidation, pharmacological characteristics, and ligand–receptor binding description.
To this end, we devoted our efforts to the discovery of a well-defined covalent antagonist based on xanthine analogue 1 mentioned above. Inspired by the resemblance in the chemical structure between the potent hA3AR antagonist 1 and irreversible adenosine A1 receptor antagonist 2, we incorporated the reactive moiety, a fluorosulfonyl benzoyl group, connected to a spacer, at the N1 position of the scaffold. Using a structured approach to bring the reactive fluorosulfonyl group in close proximity to a nucleophilic amino acid residue, we diversified the type of linker, linker length, and position of the fluorosulfonyl substituent on the phenyl group, resulting in a series of analogues with a wide range of affinities. Our efforts led to the discovery of a best-in-class antagonist, 17b, which is bound to the hA3AR with an apparent affinity in the nanomolar range. To retain the chemical structure similarity, we replaced the warhead with a methylsulfonyl moiety to obtain a nonreactive derivative 19 as a reversible control compound. 17b was then validated to covalently bind and inactivate the hA3AR in an insurmountable manner. Molecular modeling suggested the fluorosulfonyl functionality of 17b in close proximity to Y2657.36, which was identified as the unique anchor point of the covalent interaction in a subsequent mutagenesis study. The confirmed binding mode between this novel covalent antagonist and hA3AR opens the door for exploring other ligand binding motifs and will benefit receptor stabilization and further structure elucidation of the hA3AR.
Results and Discussion
Design of Covalent hA3AR Antagonists
In previous studies, our research group disclosed several series of hA3AR antagonists based on the pyrido[2,1-f]purine-2,4-dione scaffold.6−8 Using compound 1, a nanomolar probe from the previous series, as the starting point, we further designed and synthesized compounds based on a previously suggested binding mode of the pyrido[2,1-f]purine-2,4-dione scaffold.7 When examining the suggested binding mode of this scaffold, we noted that this scaffold inserted into the binding pocket with a receptor interaction between TM3, TM6, and EL2. Two key H-bonds include the carbonyl-oxygen at the C4-position with residue N2506.55 and the methoxy substituent at the C8-position bonding to Q167EL2. Taking this into account, we reasoned that the only available space to incorporate the reactive warhead is limited to N1-position substituents.
To explore the chemical space required to optimally position the warhead in close proximity to a nucleophilic amino acid residue, we examined various linker systems, connecting the warhead and the pyrido[2,1-f]purine-2,4-dione scaffold. First, variation in the length of the spacer, between two and four carbon atoms, may offer more steric freedom allowing the fluorosulfonyl group to orient toward an adjacent nucleophilic residue in the receptor binding site.15,16 Additionally, the type of chemical bond connecting the warhead to the spacer was varied between the slightly differently oriented ester or amide bond. Finally, since the exact position of an appropriate nucleophilic residue is unknown, the sulfonyl fluoride moiety was positioned at either the 3- or 4-position of the phenyl ring. To this end, four series of compounds 13a–c, 14a–c, 17a–c, and 18a–c, bearing three different spacer lengths, ester or amide linkage, and 3- or 4-fluorosulfonylphenyl warhead were targeted for synthesis.
Synthesis
Scaffold
The scaffold, 8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2, 4-dione (1), was synthesized according to the previously published procedure.6−8 Starting from the commercially available benzylurea (3), the fused tricyclic intermediate (6) was generated by excess N-bromosuccinimide (NBS) bromination and 4-methoxypyridine cyclization (Scheme 1). Then, alkylation at the N3-nitrogen by 1-bromopropane in dry dimethylformamide (DMF), using dry potassium carbonate as a weak base, afforded the reference compound (1) in 73% yield. Removing the benzyl protecting group by palladium hydroxide afforded the fused xanthine core (7).
Scheme 1. Synthetic Route toward Scaffold 7.

Reagents and conditions: (a) (i) Ac2O, 80 °C, 2 h; (ii) Et2O, room temperature (rt), 1 h; (iii) 3 M NaOH, 85 °C, 1 h; (iv) HCl (37%), 25%; (b) (i) NBS, MeCN, 80 oC; (ii) 4-methoxypyridine, 80 °C, 64%; (c) 1-bromopropane, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), MeCN, 70 °C, 73%; (d) Pd(OH)2/C, HCOONH4, EtOH, reflux, 40%.
Ester Linker
The fluorosulfonyl warhead is notorious for its reactivity, resulting in undesired side reactions or hydrolysis under several harsh reactions.17 So, we adopted a convergent synthetic strategy in which the fluorosulfonylphenyl linker unit was prepared separately and attached directly to scaffold 7 at the N3 position. This approach offers flexibility to accommodate a variety of different linker lengths. The warhead was synthesized from commercially available chlorosulfonylbenzoic acids (8a and 8b) (Scheme 2), followed by a 2 M solution of potassium bifluoride treatment to afford fluorosulfonylbenzoic acids (9a and 9b) in good yields.18 The next step converted the carboxylic acids to acid chlorides (10a and 10b) by excess thionyl chloride treatment. These acyl chlorides are susceptible to hydrolysis and were thus used in the next step reaction without further purification. To incorporate the acyl chlorides with the corresponding bromoalkylalcohols, compounds 10a and 10b were heated to 100 °C with the addition of bromoalkylalcohols to afford the desired bromoalkyl fluorosulfonylbenzoates (11a–c and 12a–c) in decent yields. The final step was to couple the core to the corresponding bromoalkyl fluorosulfonylbenzoates. To preserve the functional fluorosulfonyl group, the reactions were carried out under mild conditions at low temperatures. Additionally, excess DMF was removed by multiple washing steps, instead of vacuum removal at high temperatures. Six final products (13a–c and 14a–c) were obtained in acceptable yields.
Scheme 2. Synthetic Route toward the Bromoalkyl Fluorosulfonylbenzoates 13a–c and 14a–c.

Reagents and conditions: (a) 2 M KHF2 solution, dioxane, rt, 1 h, 87–90%; (b) SOCl2 reflux; (c) corresponding bromoalkylalcohol, anhydrous dioxane, 100 °C, 18h, 55–83%, (d) 11a–c or 12a–c, K2CO3, anhydrous DMF, 50 °C, 5–57%.
Amide Linker
A similar synthetic approach was initially pursued to prepare analogues with an amide linker. However, the basicity and instability of bromoalkylamine caused complex side reactions with itself and with the warhead, ending up with an unacceptably low yield of amide-linked building blocks. An alternative synthetic route was devised, where 1-phthalimidopropyl bromide was attached directly to the N3 position of scaffold 6, to afford the substituted intermediates 15a–c (Scheme 3). Liberation of the amine took place by treatment with hydrazine monohydrate in methanol to obtain compound 16a–c in moderate yield. Then 16b and 16c were acylated with acyl chlorides 10a and 10b, respectively, to obtain 17c and 18b. However, impurities brought by the acylation reaction were not easily removed by column chromatography or preparative thin-layer chromatography (TLC). To overcome this, we used peptide coupling conditions with the corresponding benzoic acids (9a and 9b) to convert the free amine to the target compounds (17a,b, 18a, and 18c) in good yields (Scheme 3). A similar synthetic strategy was adapted to obtain reversible ligand 19 as a control compound.
Scheme 3. Synthetic Route toward the Amide-Linker Antagonists 17a–c, 18a–c, and 19.

Reagents and conditions: (a) N-(bromoalkyl)phthalimide, K2CO3, DMF, 100 °C, 5–96%; (b) N2H4·H2O, MeOH reflux, 86–90%; (c) 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), corresponding acid (9a,b), CHCl3 or CH2Cl2, rt; and (d) SOCl2, K2CO3, dry DMF, 40 °C, 3–78%
Pharmacological Evaluation
Determination of the Apparent Affinity (Ki) of Synthetized Ligands
To determine the binding affinity for the hA3AR, all compounds were tested in a radioligand displacement binding assay in the presence of 10 nM [3H]PSB-11 at 25 °C according to previously reported procedures.7,19 All compounds were able to concentration-dependently inhibit specific [3H]PSB-11 binding to the hA3AR. As detailed in Table 1, all putative covalent compounds, except the two carbon linker compounds (13a, 14a, 17a, and 18a), displayed high affinities for the hA3AR (Ki < 100 nM). It should be mentioned that the putative covalent nature of the interaction between the hA3AR and ligands precludes the determination of equilibrium binding parameters. Therefore, we expressed the ligands’ affinity for the hA3AR as “apparent Ki”. Of note, 17b, bearing three carbon atoms with amide linkage and positioning the sulfonyl fluoride at the 4-position of the phenyl ring, interacted with the hA3AR with comparable affinity (10 nM) as the parent compound 1. High affinity is desirable for covalent ligand design, as it allows sufficient receptor occupancy with the electrophilic warhead in proximity to a nucleophilic residue in the binding site over time, concomitant with putatively negligible or less interaction with off-targets. Thus, we chose compound 17b for further studies. However, featuring an electrophilic fluorosulfonyl functionality, 17b was no longer a close analogue of compound 1, whereas a nonreactive control compound, chemically similar to the designed covalent ligand, is needed for the further pharmacological characterization.
Table 1. Apparent Affinities of Pyrido[2,1-f]purine-2,4-dione Derivatives 13–19.
| compound | n | X | R1 | pKi ± SEMa or disp. at 10 μm (%) |
|---|---|---|---|---|
| 13a | 1 | O | 4-SO2F | 6.7 ± 0.1 |
| 13b | 2 | O | 4-SO2F | 7.7 ± 0.1 |
| 13c | 3 | O | 4-SO2F | 7.5 ± 0.1 |
| 14a | 1 | O | 3-SO2F | 6.4 ± 0.1 |
| 14b | 2 | O | 3-SO2F | 7.0 ± 0.05 |
| 14c | 3 | O | 3-SO2F | 7.1 ± 0.05 |
| 17a | 1 | NH | 4-SO2F | 27% |
| 17b (LUF7602) | 2 | NH | 4-SO2F | 8.0 ± 0.05 |
| 17c | 3 | NH | 4-SO2F | 7.5 ± 0.05 |
| 18a | 1 | NH | 3-SO2F | 18% |
| 18b | 2 | NH | 3-SO2F | 7.5 ± 0.01 |
| 18c | 3 | NH | 3-SO2F | 6.8 ± 0.1 |
| 19 (LUF7714) | 2 | NH | 4-SO2Me | 6.3 ± 0.03 |
Data are expressed as means ± standard error of the mean (SEM) of three separate experiments each performed in duplicate. Apparent affinity determined from the displacement of specific [3H]PSB-11 binding from the hA3AR stably expressed on Chinese hamster ovary (CHO) cell membranes at 25 °C during 2 h of incubation.
A nonsubstituted phenyl to replace the warhead might impose different steric and electronic characteristics of the ligand. To avoid this, we performed a conservative structural modification to replace the reactive warhead with an electron-withdrawing methylsulfonyl group, yielding derivative 19 as a nonreactive control compound.
To better understand the time-dependent binding characteristics of these compounds, we carried out radioligand displacement assays under two different protocols. In detail, the CHO cell membranes overexpressing the hA3AR were either preincubated with the indicated compound for 4 h, followed by a 0.5 h co-incubation or only co-incubated for 0.5 h with the radioligand [3H]PSB-11. As detailed in Table 2, both compounds had comparable binding affinity in the low micromolar range (pKi = 6.9 ± 0.06 for 17b and pKi = 6.2 ± 0.03 for 19) at 0.5 h incubation time. However, compound 17b showed a significantly increased affinity (pKi = 8.0 ± 0.01) when it was preincubated with the hA3AR, whereas the affinity of compound 19 did not change (pKi = 6.1 ± 0.06). The effect of preincubation on the affinity of 17b and 19 is illustrated in Figure 2, i.e., the [3H]PSB-11 displacement curve was shifted to the left with an increased incubation time for compound 17b (Figure 2A), whereas no difference was observed for compound 19 (Figure 2B).
Table 2. (Apparent) Affinities of 17b and 19 for All Adenosine Receptor Subtypes, hA3AR-WT, and hA3AR-Y265F7.36 a.
| hA1ARb | hA2AARc | hA2BARd | hA3AR | hA3AR-WTg | hA3AR-Y265F7.36 h | ||
|---|---|---|---|---|---|---|---|
| cpd | pKi ± SEM | displ. (%) at 1 μm | pKie (pre-0 h) | pKif (pre-4 h) | pIC50 ± SEMf | ||
| 17bi | 6.1 ± 0.03 | 5.9 ± 0.09 | 0% (7, −7) | 6.9 ± 0.06 | 8.0 ± 0.01** | 7.8 ± 0.05 | 6.0 ± 0.3* |
| 19 | 4.8 ± 0.20 | 5.2 ± 0.20 | 0% (−10, −13) | 6.2 ± 0.03 | 6.1 ± 0.06NS | 5.9 ± 0.02 | 6.1 ± 0.1NS |
Values represent mean ± SEM of three separate experiments, each performed in duplicate, or percentage displacement at 1 μm of two separate experiments, each performed in duplicate.
Affinity determined from the displacement of specific [3H]DPCPX binding on CHO cell membranes stably expressing human adenosine A1 receptors at 25 °C during 2 h of incubation.
Affinity determined from the displacement of specific [3H]ZM241385 binding on HEK293 cell membranes stably expressing human adenosine A2A receptors at 25 °C during 2 h of incubation.
% displacement at 1 μm concentration of specific [3H]PSB-603 binding on CHO cell membranes stably expressing human adenosine A2B receptors at 25 °C during 2 h of incubation.
Displacement of specific [3H]PSB-11 binding on CHO cell membranes stably expressing the hA3AR at 25 °C during 0.5 h of incubation.
Displacement of specific [3H]PSB-11 binding from CHO cell membranes stably expressing the hA3AR preincubated with an antagonist for 4 h at 25 °C, followed by a 0.5 h of co-incubation with [3H]PSB-11. P < 0.01** compared with the pKi values in displacement experiments during 0.5 h of incubation time; NS: no significant difference compared with the pKi values in displacement experiments during 0.5 h of incubation time; Student’s test.
Displacement of specific [3H]PSB-11 binding from CHO-K1 cell membranes transiently transfected with hA3AR-WT at 25 °C during 2 h of incubation.
Displacement of specific [3H]PSB-11 binding from CHO-K1 cell membranes transiently transfected with hA3AR-Y265F7.36 at 25 °C during 2 h of incubation. P < 0.01* compared with the pIC50 values in displacement experiments on hA3AR-WT. NS: no significant difference compared with the pIC50 values in displacement experiments on hA3AR-WT membranes; Student’s test.
For 17b, pKi values are apparent affinity values as no dynamic equilibrium can be obtained.
Figure 2.

(A) Displacement of [3H]PSB-11 binding from the hA3AR at 25 °C by 17b with and without preincubation of 4 h. (B) Displacement of [3H]PSB-11 binding from the hA3AR at 25 °C by 19 with and without preincubation of 4 h. Data represent the mean ± SEM of three individual experiments performed in duplicate.
Presumably, this time-dependent binding affinity of compound 17b (i.e., resulting from an increased receptor occupancy over time) is a result of an increasing level of covalent binding. Similar results on other GPCRs, such as β2 adrenergic receptor20 and A2A adenosine receptor,21 showed that covalent bond formation generates an increased affinity over time. Meanwhile, control compound 19 showed no substantial pKi shift in affinity at the two incubation times, indicating that a dynamic equilibrium was achieved at both incubation times. We can thus speculate that the possible covalent interaction between compound 17b and the receptor may be attributed to the presence of a reactive warhead.
Finally, we tested 17b and 19 for their affinity on the other adenosine receptor subtypes and learned that the two compounds were at least modestly selective for the hA3AR (Table 2).
Kinetic Characterization of the Covalent Ligand
Subsequently, the significant shift in apparent Ki drove us to explore the binding kinetic profile of 17b at the hA3AR, specifically its dissociation rate and residence time (RT). Previously, the kon (k1 = 0.281 ± 0.04 × 108 M–1 min–1) and koff (k2 = 0.3992 ± 0.02 min–1) values of [3H]PSB-11 at 25 °C had been determined in our laboratory by traditional association and dissociation assays. Here, we performed a competition association assay to characterize the binding kinetics of 17b and 19 following previously reported procedures from our research group.7 Using the on- and off-rate constants from [3H]PSB, the kon (k3) and koff (k4) values for 17b were determined using the equations from the (equilibrium) Motulsky and Mahan model.2217b had a much slower association rate (kon = 3.48 ± 0.22 × 105 M–1 min–1) than the radioligand and a negligible dissociation rate (koff = 1.38 ± 0.22 × 10–12 min–1), yielding an almost infinite residence time (RT = 7.63 ± 1.19 × 1011 min), indicative of irreversible receptor binding by 17b. The inadequacy of the Motulsky–Mahan equations to fit this data is further evidence for the nonequilibrium features of the binding of 17b to the receptor. Compound 19 showed fast association and dissociation rate constants (Figure 3). Unfortunately, the data did not converge in the fitting procedure, possibly due to the low binding affinity of compound 19 (Ki = 525 nM).
Figure 3.
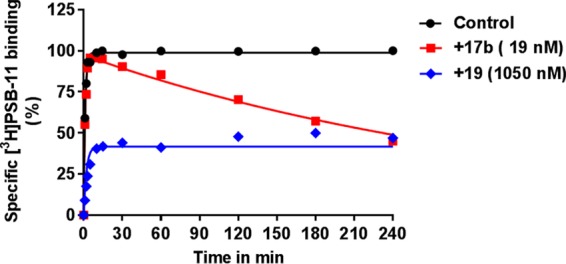
Competition association assay of [3H]PSB-11 in the absence (control) or presence of 17b and 19 at the indicated concentration. Association and dissociation rate constants for the unlabeled ligands were calculated by fitting the data to the equations described in the Experimental Section (“data analysis”). Representative graphs are from one experiment performed in duplicate.
As detailed in Figure 3, the control curve represented the association curve of radioligand [3H]PSB-11 alone, approaching equilibrium over time. Compound 19 equally associated with and dissociated from the receptor and reached equilibrium within 30 min, evidenced by the same curve shape as the control curve. Of note, 17b’s behavior caused an initial “overshoot” of the competition association curve, followed by a linear decline over time indicating that no equilibrium was reached. The shape of 17b’s kinetic curve is a quintessential example for the irreversible interaction, similar to the reported covalent ligands’ behavior for the adenosine A2A receptor21 and mGlu2 receptor.23
Wash-Resistant Interaction between 17b and hA3AR
Inspired by the negligible dissociation of compound 17b from the hA3AR, we performed a “washout” experiment to ascertain the irreversible binding between the ligand and the receptor. A protocol previously reported by our laboratory21 was adapted. We first exposed hA3AR cell membranes to 17b or 19 both at 10-fold Ki for 2 h, and without washing the samples were supplemented with [3H]PSB-11 to assess the competitive binding capacity of the receptor (“control group” in Figure 4). For washed samples, hA3AR cell membranes were subjected to four-cycle washing steps to remove unbound ligand following the preincubation (“4× wash group” in Figure 4), after which the membranes were exposed to [3H]PSB-11 to determine the remaining binding capacity. In the absence of the ligand (labeled “+ vehicle” in Figure 4), we normalized membranes’ binding ability to 100%. Following preincubation with 17b, membranes containing the hA3AR lost most of the ability to bind to the radioligand (11.3 ± 1.2% binding remaining). Furthermore, after the preincubation, membranes were washed by cycles of centrifugation in an attempt to regenerate binding capacity. However, washing steps failed to restore hA3AR binding of [3H]PSB-11 (8.7 ± 3.8%). This was in contrast to preincubation of the hA3AR-expressing membranes with ligand 19, in which binding function was completely restored from 19.8 ± 4.7 to 97.6 ± 4.5% following four washing steps. This result indicates that 19 is a reversible ligand which can be rapidly washed off the membranes, whereas 17b forms a wash-resistant bond between the ligand and the receptor. Similar experiments on other GPCRs, such as adenosine A124,25 and A2A21 receptors and the metabotropic glutamate receptor 2 (mGluR2),23 demonstrated that the covalent interaction between the ligand and the receptor resulted in a wash-resistant bond formation.
Figure 4.
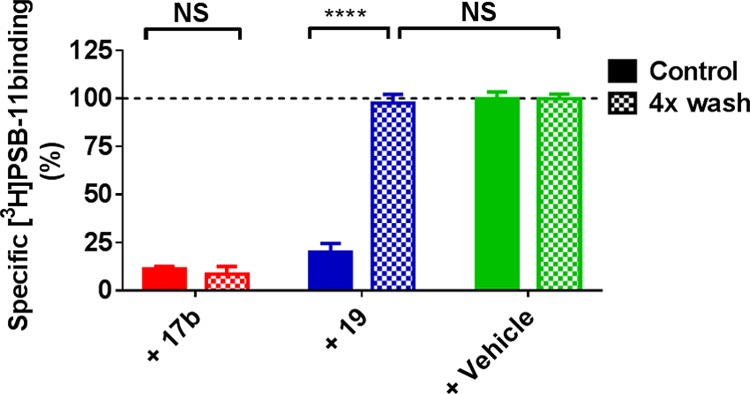
hA3AR membranes preincubated with buffer (vehicle) or a 10 × Ki concentration of indicated ligand, followed by no washing (control) or four-cycle washing treatment (4× wash) before being exposed to [3H]PSB-11. Data represent the mean ± SEM of three individual experiments performed in duplicate, normalized to the vehicle (set at 100%). Statistics were determined using unpaired Student’s t-test. NS: no significant difference, ****P < 0.0001, significant difference between indicated groups.
Insurmountable Antagonism Caused by Covalent Interaction
To further evaluate the effect of irreversible inhibition by covalent ligand 17b on receptor function, we performed a membrane functional assay using [35S]GTPγS, which is a typical readout for the activation of receptor-coupled Gi/o proteins.26 Pretreatment of the hA3AR with increasing concentrations of ligand 17b, prior to the stimulation with hA3AR agonist 1-[2-chloro-6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-1-deoxy-N-methyl-β-d-ribofuranuronamide (2-Cl-IB-MECA), produced rightward shifts of agonist concentration–response curves with a concomitant decline in maximal stimulation (Figure 5A). Therefore, the covalent ligand 17b generated insurmountable antagonism in the preincubation experiment. In contrast, pretreatment of the hA3AR with 19, followed by 2-Cl-IB-MECA agonist exposure resulted in surmountable antagonism (Figure 5B), i.e., shifting dose–response curves to the right with no alteration of its maximum effect. The extent of the shifts was used to construct a Schild plot as previously described,7 which would have a slope of unity if the interaction is competitive and the pA2-value corresponds to the pKi value of the antagonist. The slope for 19 was found to be 1.1 ± 0.1 and the compound’s pA2 value was 5.9 ± 0.1, comparable with its pKi value (6.3 ± 0.03), suggesting that 19 competed with 2-Cl-IB-MECA for the same receptor binding site.
Figure 5.

Effects of 17b and 19 on hA3AR activation as measured by [35S]GTPγS binding. (A, B) Compound 17b (A) or 19 (B) was preincubated with the hA3AR stably expressed on CHO cell membranes (25 °C) for 60 min prior to the addition of 2-Cl-IB-MECA at a concentration ranging from 0.1 nM to 10 μm for 30 min. (C, D) Compound 17b (C) or 19 (D) were co-incubated with 2-Cl-IB-MECA, at a concentration ranging from 0.1 nM to 10 μm, for 30 min. The agonist curves were generated in the presence of increasing concentrations of antagonists, such as 0.3-, 1-, 3-, and 10-fold Ki values, respectively. Data are from three independent experiments performed in duplicate, normalized according to the maximal response (100%) produced by 10 μm 2-Cl-IB-MECA alone. The shift in agonist EC50 values was determined to perform Schild analyses.
To unravel the molecular mechanism responsible for the insurmountable antagonism of 17b, we also co-incubated either 17b or 19 with the hA3AR in the presence of 2-Cl-IB-MECA. Both ligands produced a rightward shift of the agonist’s concentration–response curve (Figure 5C,D) with no suppression of maximal response, indicative of surmountable antagonism. The Schild plot showed that both antagonists inhibited receptor activation in a competitive manner, with their Schild-slopes close to unity (1.1 ± 0.1 for 17b, 1.0 ± 0.1 for 19, Table 3). In addition, 19’s pA2 value was in agreement with that from the preincubation experiments (6.2 ± 0.1, Table 3), and the pA2 value of 17b was also comparable with its pKi value (7.4 ± 0.1 vs 8.0 ± 0.05). Taken together, both ligands fully competed with 2-Cl-IB-MECA bound to the hA3AR. Notably, it is likely that the insurmountable behavior relates to the covalent binding of 17b due to an irreversible blockade that reduces the total receptor population available.
Table 3. Functional Analysis of hA3AR Antagonism from [35S]GTPγS Binding Assaysa.
| preincubation |
co-incubation |
||||
|---|---|---|---|---|---|
| compound | pA2 | Schild slope | pA2 | Schild slope | mode of antagonism |
| 17b | NA | NA | 7.4 ± 0.1 | 1.1 ± 0.1 | competitive insurmountable |
| 19 | 5.9 ± 0.1 | 1.1 ± 0.1 | 6.2 ± 0.1 | 1.0 ± 0.1 | competitive surmountable |
Values represent mean ± SEM of three separate experiments each performed in duplicate.
Binding Model for 17b in the hA3AR Receptor-Binding Pocket
To examine the interaction between receptor residues possibly involved in covalent binding, we docked 17b into a ligand optimized homology model on the basis of the A2A receptor crystal structure (PDB: 4EIY(27)), as described previously.7 As detailed in Figure 6, the core structure of compound 17b interacted with the TM3, TM6, and EL2 regions. Additionally, the carbonyl-oxygen at the C4-position participated in H-bond formation with residue N2506.55 and the methoxyl moiety at the C8-position functioned as H-bond acceptor with Q167EL2. Interestingly, the latter is a unique residue in the hA3AR, as it is not conserved in other subtypes of adenosine receptors. Due to the flexibility of the three carbon linkers, the tyrosine residue Y2657.36 is in close proximity of the ligand, and could therefore interact with the 4-fluorosulfonylbenzoic warhead to form a covalent sulfonyl amide. Similarly, the same residue Y2717.36 located within the human adenosine A1 receptor has also been reported to covalently interact with the fluorosulfonyl warhead of compound 2.11 Comparison of the binding modes of compound 2 and ligand 17b in an A1/A3 receptor overlay showed that key interactions between ligands and binding sites are preserved, such as a hydrogen bond with N6.55 (Figure S1).
Figure 6.

Proposed binding mode of compound 17b (green carbon sticks) in a homology model (violet ribbons) of the hA3AR. The hA3AR homology model was based on the high-resolution antagonist-bound crystal structure of the adenosine A2A receptor (PDB: 4EIY(27)). Atom color code: red = oxygen, blue = nitrogen, white = hydrogen, yellow = sulfur, cyan = fluorine. Hydrogen bonds between the ligand and receptor are indicated by yellow dashed lines. Residue Y2657.36 is in the proximity of the fluorosulfonyl warhead.
Y2657.36 as an Anchor Point for the Covalent Bond
Based on the docking study, we postulated that Y2657.36 is the anchor point for covalent bond formation. To investigate our hypothesis this tyrosine was mutated to phenylalanine (hA3AR-Y265F7.36), to remove the nucleophilic reactivity of the phenolic hydroxyl group. First, we performed standard [3H]PSB-11 displacement assays to investigate the binding affinity of 17b and 19 using CHO-K1 cell membranes transiently transfected with either wild type (hA3AR-WT) or mutant receptors (hA3AR-Y265F7.36). As shown in Table 2 and Figure 7, the affinity of control compound 19 on hA3AR-Y265F7.36 (pIC50 = 6.09 ± 0.11) was similar to the affinity to hA3AR-WT (pIC50 = 5.95 ± 0.03), indicating that the mutation has no impact on the binding affinity of the reversible ligand. In marked contrast, 17b’s affinity was decreased nearly 43-fold relative to the WT, from an IC50 value of 27 to 1072 nM, indicative of the loss of irreversible interaction. Moreover, there were no marked affinity differences on hA3AR-Y265F7.36 between 17b and 19. This suggests that the chemically dissimilar ligands 17b (reactive) and 19 (nonreactive) exhibit a similar binding interaction with hA3AR-Y265F7.36. We thus speculate that the amino acid in position 7.36 plays a prominent role in the covalent bond formation between the fluorosulfonyl warhead and the receptor. To support this idea, we repeated the washout assay on hA3AR-Y265F7.36. Membranes treated with 17b at 10-fold IC50 inhibited the specific [3H]PSB-11 binding to 7.2 ± 0.6%. After extensive washing, hA3AR-Y265F7.36 showed a complete recovery of [3H]PSB-11 binding to 91 ± 2% (Figure 7C). This full recovery for mutant hA3AR-Y265F7.36 is in sharp contrast to the findings in the wild-type washout assay (Figure 4), indicating that Y265F7.36 completely prevented the wash-resistant bond formation. In other words, Y2657.36 is the unique amino acid residue involved in the covalent attachment of 17b’s fluorosulfonyl group within the hA3AR binding pocket. A similar approach was also adopted to pinpoint the anchor point between covalent probes and other subtypes of GPCRs, such as the adenosine A2A receptor,21 mGlu2 receptor,23 and cannabinoid CB1 receptor.28
Figure 7.

(A, B) Displacement of specific [3H]PSB-11 binding from transiently transfected hA3AR-WT and hA3AR-Y265F7.36 at 25 °C by compound 17b (A) and 19 (B) during incubation of 2 h. (C) hA3AR-Y265F7.36 cell membranes were pretreated with buffer (vehicle) or 10 × IC50 of compound 17b for 2 h followed by no washing (control) or four-cycle washing treatment (4× wash) before being exposed to [3H]PSB-11. Data represent the mean ± SEM of three individual experiments performed in duplicate, normalized to the vehicle (set at 100%). NS: no significant difference between groups; ***Significant difference between groups (P < 0.001); Student’s t-test.
17b can be a useful structural biology tool as it would be expected to stabilize the 7TM domain in its inactive state, thereby potentially facilitating crystallization of the receptor material. This could be highly valuable for the structure elucidation of the hA3AR, which up to now remains unreported. Furthermore, understanding the precise molecular interactions between the ligand and the receptor may stimulate the more rational design of novel ligands. Such ligands may have improved receptor subtype selectivity, fewer undesirable side effects, and enhanced potency and efficacy, leading to potentially attractive therapeutic agents that produce their effects by modulating the functionality of the adenosine system. Given that GPCR-targeted covalent drugs went through clinical success across various indications,29 our covalent compound 17b may serve as a probe to explore the problematic translation of hA3AR ligands into the clinical utility in certain disease states such as eye disorder glaucoma, in which an increased A3 adenosine receptor mRNA and protein levels have been detected.
Conclusions
By introducing a reactive sulfonyl fluoride warhead onto the 1-benzyl-3-propyl-1H,3H-pyrido [2,1-f]purine-2,4-dione scaffold, we designed and synthesized a series of novel covalent hA3AR antagonists. Compound 17b acted as the most potent antagonist, with a time-dependent apparent affinity in the low nanomolar range. Meanwhile, we removed the warhead and inserted a methylsulfonyl moiety into the scaffold, to obtain ligand 19 as a reversible control compound. Ligand 17b was then validated as a covalent antagonist through its wash-resistant nature and insurmountable antagonism in [35S]GTPγS binding assays. In silico homology-docking suggested that Y2657.36 is responsible for the covalent interaction. Site-directed mutagenesis showed that removal of the nucleophilic tyrosine phenolic hydroxyl group resulted in the complete loss of covalent binding, validating that Y2657.36 is the only anchor point of reactive covalent ligand 17b. The results contribute to a better understanding of pharmacological behaviors caused by covalent interaction with GPCRs. In the end, we developed a structured approach to quickly obtain a well-defined covalent ligand. Besides, we envisioned that a methylsulfonyl replacement would be suitable for providing a nonreactive sulfonyl-bearing control compound. The rational design of covalent probes may have further value in receptor structure elucidation or in new technologies such as affinity-based protein profiling15,30 with the perspective of imaging or structurally probing GPCRs.
Experimental Section
Chemistry
All solvents and reagents were purchased from commercial sources and were of analytical grade. Demineralized water is simply referred to as H2O, and was used in all cases unless stated otherwise (i.e., brine). 1H were recorded on a Bruker AV 400 liquid spectrometer (1H NMR, 400 MHz) at ambient temperature and 13C NMR spectra were recorded on a Bruker AV 600 liquid spectrometer (13C NMR, 125 MHz) at indicated temperature. Chemical shifts are reported in parts per million (ppm), using residual solvent as the internal reference in all cases. The values are given in δ scale. Coupling-constants are reported in Hz and are designated as J. Analytical purity of the final compounds was determined by high-performance liquid chromatography (HPLC) with a Phenomenex Gemini 3 μm C18 110 Å column (50 × 4.6 mm, 3 μm), measuring UV absorbance at 254 nm. Sample preparation and the HPLC method were as follows: 0.3–1.0 mg of compound was dissolved in 1 mL of a 1:1:1 mixture of MeCN/H2O/tBuOH and eluted from the column within 15 min at a flow rate of 1.3 mL min–1 with a three-component system of H2O/MeCN/1% trifluoroacetyl (TFA) in H2O. The elution method was set up as follows: 1–4 min isocratic system of H2O/MeCN/1% TFA in H2O, 80:10:10, from the 4th min, a gradient was applied from 80:10:10 to 0:90:10 within 9 min, followed by 1 min of equilibration at 0:90:10 and 1 min at 80:10:10. All final compounds showed a single peak at the designated retention time and are at least 95% pure. Liquid chromatography–mass spectrometry (LC–MS) analyses were performed using a Thermo Finnigan Surveyor–LCQ Advantage Max LC–MS system and a Gemini C18 Phenomenex column (50 × 4.6 mm2, 3 μm). High-resolution mass spectrometry (HRMS) analyses were performed using a Thermo Scientific LTQ Orbitrap XL Hybrid Ion Trap-Orbitrap Mass Spectrometer. The sample preparation was the same as for HPLC and HRMS analyses. The compounds were eluted from the column within 15 min after injection, with a three-component system of H2O/MeCN/0.2% TFA in H2O, decreasing polarity of the solvent mixture in time from 80:10:10 to 0:90:10. Thin-layer chromatography (TLC) was routinely performed to monitor the progress of reactions, using aluminum-coated Merck silica gel F254 plates. Purification by column chromatography was achieved using the Grace Davison Davisil silica column material (LC60A 30–200 μm). Solutions were concentrated using a Heidolph Laborota W8 2000 efficient rotary evaporation apparatus. All reactions in the synthetic routes were performed under a nitrogen atmosphere unless stated otherwise. The procedure for a series of similar compounds is given as a general procedure for all within that series, annotated by the numbers of the compounds.
1-Benzyl-8-methoxy-3-propyl-1H,3H-pyrido[2,1-f]purine-2,4-dione (1)7,8
To a stirred suspension of 6 (6.0 g, 19 mmol, 1.0 equiv) in MeCN (120 mL) were added 1-bromopropane (5.6 mL, 57 mmol, 3.0 equiv) and DBU (50 mL, 57 mmol, 3.0 equiv). This mixture was stirred at 70 °C overnight. The conversion of the starting material was confirmed by TLC (2% MeOH in CH2Cl2) and the solvent was removed under vacuum. The residue was suspended in CH2Cl2 (200 mL) and the organic phase was washed with 1 M HCl (200 mL), H2O (200 mL), and brine (200 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude was purified by column chromatography (0.5% MeOH in CH2Cl2) to obtain 1 as a white solid (5.0 g, 14 mmol, 73%). 1H NMR (400 MHz, CDCl3): δ 8.82 (d, J = 7.6 Hz, 1H), 7.58–7.51 (m, 2H), 7.34–7.22 (m, 3H), 6.98 (d, J = 2.0 Hz, 1H), 6.74 (dd, J = 7.4, 2.2 Hz, 1H), 5.36 (s, 2H), 4.04–3.97 (m, 2H), 3.92 (s, 3H), 1.76–1.65 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H)
6-Amino-1-benzyl-1,3-dihydropyrimidine-2,4-dione (5)7,8
The synthesis of the compounds was performed as adapted from the procedure reported before.7,8 Benzylurea (3) (25 g, 167 mmol, 1.0 equiv) and 4 (16 g, 191 mmol, 1.1 equiv) were dissolved in acetic anhydride (100 mL). This mixture was stirred at 80 °C for 2 h. After the mixture was cooled to room temperature, diethyl ether (150 mL) was added followed by 1 h of stirring at room temperature. The precipitate was filtered off and suspended in a mixture of EtOH (75 mL) and H2O (150 mL). This mixture was heated to 85 °C and 3 M NaOH (aq.) (50 mL) was added dropwise. After 1 h, the mixture was concentrated and neutralized by the dropwise addition of HCl (37%). The precipitate was filtered off and washed with acetone, obtaining 5 as a white solid (9.0 g, 42 mmol, 25%). 1H NMR (400 MHz, DMSO-d6): δ 10.42 (brs, 1H), 7.48–7.08 (m, 5H), 6.85 (brs, 2H), 5.03 (s, 2H), 4.60 (s, 1H)
1-Benzyl-8-methoxy-1H,3H-pyrido[2,1-f]purine-2,4-dione (6)7,8
To the intermediate (5) (9.0 g, 42 mmol, 1.0 equiv) and NBS (15 g, 83 mmol, 2.0 equiv) was added MeCN (100 mL). This mixture was stirred at 80 °C. After 1.5 h, the conversion of the starting material was confirmed by TLC (10% MeOH in CH2Cl2), 4-methoxypyridine (13 g, 125 mmol, 3.0 equiv) was added and the reaction mixture was stirred at 80 °C for 4.5 h. After cooling to room temperature, the precipitate was filtered off and washed with diethyl ether and MeOH, yielding product 6 as a white solid (8.5 g, 26 mmol, 64%). 1H NMR (400 MHz, DMSO-d6): δ 11.31 (br s, 1H), 8.70 (d, J = 7.2 Hz, 1H), 7.38–7.16 (m, 6H), 6.90 (dd, J = 7.4, 2.2 Hz, 1H), 5.18 (s, 2H), 3.89 (s, 3H)
8-Methoxy-3-propyl-1H,3H-pyrido[2,1-f]purine-2,4-dione (7)7,8
To a mixture of intermediate 1 (1.1 g, 3.0 mmol, 1.0 equiv), Pd(OH)2/C (2.0 g, 14 mmol, 1.0 equiv), and ammonium formate (0.20 g, 3.0 mmol, 1.0 equiv) was added EtOH (250 mL). During the reaction, five portions of ammonium formate (0.20 g, 3.0 mmol, 1.0 equiv) was added, after which completion of the reaction was observed by TLC (5% MeOH in CH2Cl2). The reaction was filtered over Celite and the residue was extracted with hot DMF. Purification of the crude product was performed by column chromatography using 2–10% MeOH in CH2Cl2 to obtain 5 as a white solid (0.30 g, 1.2 mmol, 40%). 1H NMR (400 MHz, DMSO-d6): δ 12.05 (s, 1H), 8.73 (d, J = 7.2 Hz, 1H), 7.12 (d, J = 2.0 Hz, 1H), 6.89 (dd, J = 7.4, 2.6 Hz, 1H), 3.90 (s, 3H), 3.85–3.78 (m, 2H), 1.64–1.52 (m, 2H), 0.88 (t, J = 7.4 Hz, 3H)
General Procedure for the Synthesis of Fluorosulfonylbenzoic Acids (9a,b)
To a solution of chlorosulfonylbenzoic acid (8a,b) (2.2 g, 10 mmol, 1.0 equiv) in dioxane (25 mL) was added a solution of HF/KF (15 mL, 2.0 M, 3.0 equiv). The mixture was stirred at room temperature. After 1 h, the reaction mixture was diluted with EtOAc (80 mL). The organic phase was washed with H2O (50 mL), dried over MgSO4, filtered, and concentrated in vacuo.
3-(Fluorosulfonyl)benzoic Acid (9a)
White solid (1.9 g, 8.7 mmol, 87%). 1H NMR (400 MHz, DMSO-d6): δ 8.47–8.44 (m, 2H), 8.4 (d, J = 8.0 Hz, 1H), 7.94 (t, J = 7.6 Hz, 1H).
4-(Fluorosulfonyl)benzoic Acid (9b)
White solid (2.0 g, 9.0 mmol, 90%). 1H NMR (400 MHz, DMSO-d6): δ 13.86 (s, 1H), 8.28 (s, 4H)
General Procedure for the Synthesis of Bromoalkyl (fluorosulfonyl)benzoates (11a–c and 12a–c)
A mixture of thionyl chloride (8 mL) and fluorosulfonylbenzoic acid (9a,b) (1 equiv) was refluxed at 75 °C for 3 h. The solvent was removed under vacuum and the product was used in the next step without further analysis. Dry dioxane (6 mL) was added to the (fluorosulfonyl)benzoyl chloride (10a,b). To this solution, the corresponding bromoalkylalcohol (0.85 equiv) was added and the mixture was refluxed overnight. After the completion of the reaction was observed by TLC (CH2Cl2), the volatiles were removed in vacuo and the crude product was purified by column chromatography using CH2Cl2 as an eluent to afford the products.
2-Bromoethyl-4-(fluorosulfonyl)benzoate (11a)
Colorless oil (0.088 g, 0.28 mmol, 23%) 1H NMR (400 MHz, DMSO-d6): δ 8.31 (d, J = 8.2 Hz, 2H), 8.11 (d, J = 8.5 Hz, 2H), 4.69 (t, J = 5.9 Hz, 2H), 3.67 (t, J = 5.9 Hz, 2H).
3-Bromopropyl-4-(fluorosulfonyl)benzoate (11b)
White solid (2.0 g, 6.2 mmol, 50%) 1H NMR (400 MHz, CDCl3): δ 8.27 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 8.4 Hz, 2H), 4.54 (t, J = 6.0 Hz, 2H), 3.54 (d, J = 6.4 Hz, 2H), 2.35 (m, 2H).
4-Bromobutyl-4-(fluorosulfonyl)benzoate (11c)
White solid (0.30 g, 0.89 mmol, 45%) compound was used without further purification.
2-Bromoethyl-3-(fluorosulfonyl)benzoate (12a)
Colorless oil (0.51 g, 1.7 mmol, 55%) 1H NMR (400 MHz, CDCl3): δ 8.69 (s, 1H), 8.47 (d, J = 7.6 Hz, 1H), 8.25–8.20 (m, 1H), 7.78 (t, J = 8.0 Hz, 1H), 4.71 (t, J = 6.0 Hz, 2H), 3.68 (t, J = 6.0 Hz, 2H).
3-Bromopropyl-3-(fluorosulfonyl)benzoate (12b)
Colorless oil (0.12 g, 0.38 mmol, 23%) 1H NMR (400 MHz, CDCl3) δ 8.65 (t, J = 1.6 Hz, 1H), 8.44 (d, J = 7.8 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 7.76 (t, J = 7.9 Hz, 1H), 4.55 (t, J = 6.1 Hz, 1H), 3.55 (t, J = 6.4 Hz, 1H), 2.37 (p, J = 6.3 Hz, 1H).
4-Bromobutyl-3-(fluorosulfonyl)benzoate (12c)
Colorless Oil (0.84 g, 2.5 mmol, 83%,) 1H NMR (400 MHz, CDCl3): δ 8.65 (s, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 7.78 (t, J = 7.6 Hz, 1H), 4.44 (t, J = 6.0 Hz, 2H), 3.50 (t, J = 6.4 Hz, 2H), 2.11–1.85 (m, 4H).
General Procedure for the Synthesis of 13a–c and 14a–c
The synthesis of these compounds was adapted from the conditions previously described by Priego et al.6 The scaffolds 8-methoxy-3-propyl-1H,3H-pyrido[2,1-f]purine-2,4-dione 7 (1.0 equiv) and K2CO3 (1.6 equiv) were suspended in anhydrous DMF. The mixture was added dropwise to a stirred solution of the corresponding bromoalkyl (fluorosulfonyl)benzoate (11a–c or 12a–c) (1.0 equiv) in anhydrous DMF (4 mL). The reaction was stirred at 50 °C overnight. After the conversion was observed by TLC, an excess amount of CH2Cl2 was added. Then the mixture was washed with 1 M HCl (aq.), water, and brine. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography, followed by prep TLC to further purify the compound if necessary.
2-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purine-1(2H)-yl)ethyl 4-(fluorosulfonyl)benzoate (13a)
Prepared from 11a and purified by column chromatography (1% CH3OH in CH2Cl2) to give the desired product as a white solid (0.038 g, 0.07 mmol, 52%). 1H NMR (400 MHz, CDCl3): δ 8.80 (d, J = 8.0 Hz, 1H), 8.17 (d, J = 8.0 Hz, 2H), 7.98 (d, J = 8.4 Hz, 2H), 6.76–6.73 (m, 2H), 4.78 (t, J = 4.8 Hz, 2H), 4.64 (t, J = 5.2 Hz, 2H), 4.00 (t, J = 7.6 Hz, 2H), 3.89 (s, 3H), 1.73–1.62 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H). MS: [ESI + H]+: 505.1. HPLC: 9.99 min
3-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purine-1(2H)-yl)propyl 4-(fluorosulfonyl)benzoate (13b)
Prepared from 11b and purified by column chromatography (1% CH3OH in CH2Cl2) to give the desired product as a white solid (0.096 g, 0.19 mmol, 76%). 1H NMR (400 MHz, CDCl3): δ 8.76 (d, J = 7.2 Hz, 1H), 8.23 (d, J = 8.0 Hz, 2H), 8.06 (d, J = 8.4 Hz, 2H), 6.77 (d, J = 2.4 Hz, 1H), 6.73 (dd, J = 7.2, 2.4 Hz, 1H), 4.50 (t, J = 6.0 Hz, 2H), 4.41 (t, J = 6.8 Hz, 2H), 4.00 (t, J = 7.2 Hz, 2H), 3.90 (s, 3H), 2.38 (pentet, J = 6.0 Hz, 2H), 1.71 (sextet, J = 7.2 Hz, 2H), 0.99 (t, J = 7.6 Hz, 3H). MS: [ESI + H]+: 519.1. HPLC: 10.18 min
4-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purine-1(2H)-yl)butyl 4-(fluorosulfonyl)benzoate (13c)
Prepared from 11c and purified by column chromatography (2% CH3OH in CH2Cl2) to give the desired product as a white solid (0.010 g, 0.019 mmol, 5.2%) 1H NMR (400 MHz, CDCl3): δ 8.83 (dd, J = 7.6, 0.8 Hz, 1H), 8.27 (d, J = 8.0 Hz, 2H), 8.07 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 2.4 Hz, 1H), 6.76 (dd, J = 7.6, 2.4 Hz, 1H), 4.46 (t, J = 6.4 Hz, 2H), 4.28 (t, J = 6.8 Hz, 2H), 4.02 (t, J = 7.2 Hz, 2H), 3.93 (s, 3H), 2.05–1.90 (m, 4H), 1.77–1.68 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H). MS: [ESI + H]+: 533.1. HPLC: 9.40 min
2-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purine-1(2H)-yl)ethyl 3-(fluorosulfonyl)benzoate (14a)
Prepared from 12a and without purification to give the desired product as a white solid (0.19 g, 0.36 mmol, 57%). 1H NMR (400 MHz, CDCl3): δ 8.80 (d, J = 7.2 Hz, 1H), 8.51 (s, 1H), 8.36 (d, J = 7.6 Hz, 1H), 8.14–8.09 (m, 1H), 7.66 (t, J = 7.8 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.74 (dd, J = 7.6, 2.6 Hz, 1H), 4.78 (t, J = 4.8 Hz, 2H), 4.65 (t, J = 4.8 Hz, 2H), 4.04–3.97 (m, 2H), 3.90 (s, 3H), 1.68 (sextet, J = 7.6 Hz, 2H), 0.96 (t, J = 7.4 Hz, 3H). MS: [ESI + H]+: 505.1. HPLC: 8.47 min
3-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purine-1(2H)-yl)propyl 3-(fluorosulfonyl)benzoate (14b)
Prepared from 12b and purified by column chromatography (1% CH3OH in CH2Cl2) to give the desired product as a white solid (0.035 g, 0.068 mmol, 34%). 1H NMR (400 MHz, CDCl3): δ 8.74 (d, J = 7.6 Hz, 1H), 8.65 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.72 (dd, J = 7.2, 2.4 Hz, 1H), 4.51 (t, J = 6.0 Hz, 2H), 4.41 (t, J = 6.0 Hz, 2H), 3.99 (t, J = 7.6 Hz, 2H), 3.91 (s, 3H), 2.39 (pentet, J = 6.0 Hz, 2H), 1.70 (sextet, J = 7.6 Hz, 2H), 0.98 (t, J = 7.6 Hz, 3H). MS: [ESI + H]+: 519.1. HPLC: 8.84 min
4-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purine-1(2H)-yl)butyl 3-(fluorosulfonyl)benzoate (14c)
Prepared from 12c and purified by column chromatography (first 30% DCM in EtOAc). Further purification by another column (4:1 = methyl tert-butyl ether/petroleum ether) gives the desired product as a white solid (0.20 g, 0.37 mmol, 38%). 1H NMR (400 MHz, CDCl3): δ 8.85 (d, J = 7.2 Hz, 1H), 8.67 (s, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.21 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 8.0 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.77 (dd, J = 7.2, 2.4 Hz, 1H), 4.49 (t, J = 6.4 Hz, 2H), 4.30 (t, J = 7.2 Hz, 2H), 4.08–4.01 (m, 2H), 3.95 (s, 3H), 2.10–2.00 (m, 2H), 2.00–1.89 (m, 2H), 1.81–1.69 (m, 2H), 1.01 (t, J = 7.2 Hz, 3H). MS: [ESI + H]+: 533.1. HPLC: 9.14 min
General Procedure for the Synthesis of 1-(2-(1,3-Dioxoisoindolin-2-yl)alkyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (15a–c)
To a mixture of the core (7) (0.8 mmol, 1 equiv), N-(bromoalkyl)phthalimide (1.2 mmol, 1.5 equiv), and K2CO3 (1.2 mmol, 1.5 equiv) was added anhydrous DMF (8 mL). The mixture was refluxed at 100 °C. After completion of the reaction, monitored by TLC (1% MeOH in CH2Cl2), the mixture was concentrated in vacuo and diluted with EtOAc (30 mL). The organic layer was washed with H2O (3 × 30 mL) and brine (15 mL), and dried over MgSO4. The solvent was evaporated under reduced pressure and the residue was purified by column chromatography using 1% MeOH as an eluent to give 15a–c as solids.
1-(2-(1,3-Dioxoisoindolin-2-yl)ethyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (15a)
Prepared from N-(2-bromoethyl)phthalimide and purified by column chromatography to give the desired product as a white solid (0.20 g, 0.44 mmol, 5%). 1H NMR (CDCl3): δ 8.77 (d, J = 6.8 Hz, 1H), 7.73 (s, 2H), 7.64 (s, 2H), 6.69 (d, J = 14.0 Hz, 2H), 4.53 (s, 2H), 4.17 (s, 2H), 3.89 (d, J = 6.2 Hz, 2H), 3.85 (s, 3H), 1.58–1.45 (m, 3H), 0.86 (t, J = 7.2 Hz, 3H).
1-(2-(1,3-Dioxoisoindolin-2-yl)propyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (15b)
Prepared from N-(3-bromoethyl)phthalimide and purified by column chromatography to give the desired product as a yellow solid (0.31 g, 0.66 mmol, 66%). 1H NMR (400 MHz, CDCl3): δ 8.81–8.75 (m, 1H), 7.86–7.76 (m, 2H), 7.73–7.61 (m, 2H), 6.80 (s, 1H), 6.72 (dd, J = 7.2, 2.4 Hz, 1H), 4.29 (t, J = 6.8 Hz, 2H), 4.04–3.93 (m, 2H), 3.90 (s, 3H), 3.86–3.78 (m, 2H), 2.35–2.20 (m, 2H), 1.78–1.60 (m, 2H), 1.06–0.87 (m, 3H).
1-(2-(1,3-Dioxoisoindolin-2-yl)butyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (15c)
Prepared from N-(4-bromoethyl)phthalimide and purified by column chromatography to give the desired product as a white solid (0.37 g, 0.76 mmol, 96%). 1H NMR (400 MHz, CDCl3): δ 8.82 (d, J = 7.2 Hz, 1H), 7.82 (dd, J = 5.2, 2.8 Hz, 2H), 7.70 (dd, J = 5.2, 2.8 Hz, 2H), 6.93 (d, J = 2.4 Hz, 1H), 6.74 (dd, J = 7.2, 2.4 Hz, 1H), 4.22 (d, J = 7.2 Hz, 2H), 4.04–3.96 (m, 2H), 3.92 (s, 3H), 3.75 (d, J = 7.2 Hz, 2H), 1.95–1.85 (m, 2H), 1.85–1.77 (m, 2H), 1.74–1.65 (m, 3H), 0.97 (d, J = 7.6 Hz, 3H).
General Procedure for the Synthesis of 1-(2-Aminoalkyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (16a–c)
To a stirred suspension of 15a–c (0.66 mmol, 1 equiv) in MeOH (8 mL) was added excess hydrazine monohydrate (4.8 mL, 99 mmol). The mixture was stirred for 2–4 h at reflux. After conversion of the starting material, the mixture was cooled to room temperature. The solvents were removed under vacuum and the residue was dissolved in 2 M NaOH (aq.) (25 mL). This aqueous phase was extracted three times with CH2Cl2 (25 mL). The organic layers were combined, dried over MgSO4, and concentrated in vacuo to obtain 16a–c.
1-(2-Aminoethyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (16a)
Prepared from 15a and purified by column chromatography to give the desired product as a white solid (0.13 g, 0.39 mmol, 90%). 1H NMR (400 MHz, CDCl3): δ 8.84 (d, J = 7.6 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 7.2, 2.4 Hz, 1H), 4.27 (t, J = 6.4 Hz, 2H), 4.05–3.99 (m, 2H), 3.93 (s, 3H), 3.15 (t, J = 6.4 Hz, 2H), 1.78–1.67 (m, 2H), 0.99 (d, J = 7.6 Hz, 3H).
1-(3-Aminopropyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (16b)
Prepared from 15b and purified by column chromatography to give the desired product as a white solid (0.25 g, 0.75 mmol, 97%). 1H NMR (400 MHz, CDCl3): δ 8.82 (d, J = 7.2 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 7.6, 2.4 Hz, 1H), 4.29 (t, J = 6.8 Hz, 2H), 4.07–3.98 (m, 2H), 3.93 (s, 3H), 2.75 (t, J = 6.6 Hz, 2H), 1.98 (p, J = 6.6 Hz, 2H), 1.78–1.65 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H).
1-(4-Aminobutyl)-8-methoxy-3-propyl-1H,3H-pyrido-[2,1-f]purine-2,4-dione (16c)
Prepared from 15c and purified by column chromatography to give the desired product as a white solid (0.23 g, 0.66 mmol, 86%). 1H NMR (400 MHz, CDCl3): δ 8.84 (d, J = 7.6 Hz, 1H), 6.97 (d, J = 2.4 Hz, 1H), 6.75 (dd, J = 7.2, 2.4 Hz, 1H), 4.20 (t, J = 7.2 Hz, 2H), 4.06–3.98 (m, 2H), 3.92 (s, 3H), 2.77 (d, J = 6.8 Hz, 2H), 1.92–1.82 (m, 2H), 1.78–1.66 (m, 2H), 1.63–1.53 (m, 2H), 0.99 (t, J = 7.2 Hz, 3H).
4-((2-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)-yl)ethyl)carbamoyl)benzenesulfonyl Fluoride (17a)
EDC (0.12 g, 0.60 mmol, 1.2 equiv) was dissolved in CHCl3 (4 mL). To this stirring solution was added the acid (9a) (0.11 g, 0.55 mmol, 1.1 equiv). The amine (16a) (0.16 g, 0.50 mmol, 1.0 equiv) was suspended in CHCl3 (6 mL) and then was added dropwise via an automatic syringe at a rate of 0.2 mL min–1. The reaction was stirred for 1.5 h at room temperature and monitored by TLC (CH2Cl2/acetone = 3:2). After completion, the solvent was removed under vacuum and the residue was redissolved in CHCl3 (40 mL). The organic layer was washed with 1 M HCl (40 mL) and H2O (2 × 40 mL), dried over MgSO4, and concentrated in vacuo to obtain 17a as a white solid (0.20 g, 0.39 mmol, 78%). 1H NMR (400 MHz, CDCl3): δ 8.83 (d, J = 7.2 Hz, 1H), 8.07–8.00 (m, 5H), 6.91 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 7.2, 2.0 Hz, 1H), 4.62–4.55 (m, 2H), 4.03 (t, J = 7.6 Hz, 2H), 3.95 (s, 3H), 3.94–3.89 (m, 2H), 1.68 (sextet, J = 7.6 Hz, 2H), 0.97 (t, J = 7.2 Hz, 3H). MS: [ESI + H]+: 504.1. HPLC: 7.93 min.
4-((3(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)yl)propyl)carbamoyl)benzenesulfonyl Fluoride (17b)
A suspension of EDC (0.22 g, 0.80 mmol, 1.5 equiv) and 9a (0.16 g, 0.80 mmol 1.05 equiv) was dissolved in CH2Cl2 (4 mL). To this stirring solution amine was added (16b) (0.25 g, 0.76 mmol, 1.0 equiv) at room temperature. The reaction was stirred for 2 h and monitored by TLC (3% MeOH in CH2Cl2). After completion, the solvent was removed in vacuo and the residue was dissolved in CHCl3 (40 mL). The organic layer was washed with 1 M HCl (40 mL) and twice with H2O (2 × 40 mL), dried over MgSO4, and concentrated in vacuo. The product was purified by column chromatography using 2% MeOH in CH2Cl2 to afford the title compound as a white solid (0.26 g, 0.50 mmol, 66%). 1H NMR (400 MHz, CDCl3) δ: 8.86 (d, J = 7.2 Hz, 1H), 8.38 (t, J = 5.6 Hz, 1H), 8.25 (d, J = 8.4 Hz, 2H), 8.16 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 2.4 Hz, 1H), 6.81 (dd, J = 7.2, 2.4 Hz, 1H), 4.35 (t, J = 6.0 Hz, 2H), 4.05 (t, J = 7.6 Hz, 2H), 3.92 (s, 3H), 3.47 (q, J = 6.4 Hz, 2H), 2.19–2.13 (m, 2H), 1.73 (sextet, J = 7.6 Hz, 2H), 1.00 (t, J = 7.6 Hz, 3H). 13C NMR (600 MHz, DMSO-d6, 348 K) δ 164.0, 160.9, 153.4, 150.6, 150.5, 149.1, 141.3, 133.3 (d, J = 96 Hz), 128.5, 127.9, 127.3, 107.0, 99.8, 95.4, 55.7, 41.5, 40.6, 36.8, 27.1, 20.5, 10.6. MS: [ESI + H]+: 518.1. HRMS-ESI+: [M + H]+ calcd: 518.1510 found: 518.1540, C23H25O6N5FS. HPLC: 8.27 min.
4-((4-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)-yl)butyl)carbamoyl)benzenesulfonyl Fluoride (17c)
Acid 9a (0.11 g, 0.53 mmol, 1.5 equiv) was dissolved in an excess of thionyl chloride (20 mL) at 75 °C under nitrogen for 3 h. After removal of solvent and other volatiles under vacuum, 10a was obtained as a colorless oil. Subsequently, amine 16c (0.12 g, 0.35 mmol, 1.0 equiv), K2CO3 (0.073 g, 0.53 mmol, 1.5 equiv), and dry DMF were added and the reaction as stirred at 40 °C overnight. After completion of the reaction, 1 M HCl (200 mL) was added and extracted with CH2Cl2 (150 mL). The organic layer was washed with water (100 mL) and brine (100 mL). The organic layer was dried, filtered, and concentrated in vacuo. The residue was purified by column chromatography using CH2Cl2 with 1% methanol as the eluent to give 17c as a white solid (5.0 mg, 0.0094 mmol, 4%). 1H NMR (400 MHz, CDCl3): δ 8.87 (d, J = 7.6 Hz, 1H), 8.17 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 8.4 Hz, 2H), 7.54 (brs, 1H), 6.85 (s, 1H), 6.80 (dd, J = 7.2, 2.4 Hz, 1H), 4.29 (t, J = 7.6 Hz, 2H), 4.06 (t, J = 7.6 Hz, 2H), 3.92 (s, 3H), 3.68 (q, J = 6.0 Hz, 2H), 2.01 (pent, J = 6.8 Hz, 2H), 1.84–1.70 (m, 4H), 1.02 (t, J = 7.6 Hz, 3H). MS: [ESI + H]+: 532.3. HPLC: 8.28 min.
3-((2-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)-yl)ethyl)carbamoyl)benzenesulfonyl Fluoride (18a)
EDC (0.12 g, 0.60 mmol, 1.2 equiv) was dissolved in CHCl3 (4 mL). To this stirring solution was added acid 9b (0.11 g, 0.55 mmol, 1.1 equiv). Amine 16a (0.16 g, 0.50 mmol, 1.0 equiv) was suspended in CHCl3 (6 mL) and then was added dropwise via an automatic syringe at a rate of 0.2 mL min–1. The reaction was stirred for 3 h at room temperature and monitored by TLC (CH2Cl2/acetone = 3:2). After completion, the solvent was removed in vacuo and the residue was resolubilized in CHCl3 (40 mL). The organic layer was washed with 1 M HCl (40 mL) and twice with H2O (2 × 40 mL), dried over MgSO4, and concentrated in vacuo to give 18a as a white solid (0.17 g, 0.35 mmol, 70%). 1H NMR (400 MHz, CDCl3): δ 8.81 (d, J = 7.6 Hz, 1H), 8.37 (s, 1H), 8.29 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 7.6 Hz, 1H), 8.04 (br s, 1H), 7.71 (t, J = 8.0 Hz, 1H), 7.02 (d, J = 2.4 Hz, 1H), 6.77 (dd, J = 7.6, 2.4 Hz, 1H), 4.61–4.54 (m, 2H), 4.03 (t, J = 7.6 Hz, 2H), 3.96 (s, 3H), 3.94–3.89 (m, 2H), 1.76–1.63 (m, 2H), 0.97 (t, J = 7.6 Hz, 3H). MS: [ESI + H]+: 504.1. HPLC: 7.67 min.
3-((3-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)-yl)propyl)carbamoyl) benzenesulfonyl Fluoride (18b)
Acid 9b (0.42 g, 2.0 mmol, 3.0 equiv) was dissolved in thionyl chloride (20 mL) and stirred for 3 h at 75 °C. The thionyl chloride was evaporated and the residue was co-evaporated twice with toluene. Then, amine 14b (0.23 mg, 0.7 mmol, 1.00 equiv), K2CO3 (0.073 g, 0.53 mmol, 1.5 equiv), and dry DMF were added and the reaction was stirred at 40 °C overnight. 1 M HCl (200 mL) was added and extracted with CH2Cl2 (150 mL). The organic layer was washed with water (100 mL) and brine (100 mL). The organic layer was dried, filtered, and concentrated in vacuo. The residue was purified by column chromatography using CH2Cl2 with 1% methanol as the eluent to give 18b as a white solid (0.0050 g, 0.01 mmol, 2.7%). 1H NMR (400 MHz, CDCl3) δ: 8.88 (d, J = 7.2 Hz, 1H), 8.68 (s, 1H), 8.55–8.50 (m, 2H), 8.20 (d, J = 8.0 Hz, 1H), 7.82 (t, J = 8 Hz, 2H), 7.00 (d, J = 2.4 Hz, 1H), 6.80 (dd, J = 7.2, 2.4 Hz, 1H), 4.33 (t, J = 6.0 Hz, 2H), 4.05 (t, J = 7.6 Hz, 2H), 3.94 (s, 3H), 3.47 (q, J = 6.0 Hz, 2H), 2.17–2.12 (m, 2H), 1.74 (sextet, J = 7.6 Hz, 2H), 1.00 (t, J = 7.6 Hz, 3H). MS: ESI [M + H]+: 518.1 HPLC: 8.28 min.
3-((4-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)-yl)butyl)carbamoyl)benzenesulfonyl Fluoride (18c)
EDC (0.13 g, 0.69 mmol, 1.2 equiv) was dissolved in CH2Cl2 (3 mL). Acid 9b (0.13 g, 0.63 mmol, 1.1 equiv) was added to this solution and the mixture was stirred. Amine 16c (0.20 g, 0.57 mmol, 1 equiv) was dissolved in CHCl3 (8 mL) and added dropwise via an automatic syringe at a rate of 0.2 mL min–1 to the stirring solution. After 3 h at room temperature, the reaction was completed and the mixture was concentrated in vacuo. The residue was dissolved in CH2Cl2 (40 mL) and washed with 1 M HCl (40 mL) and twice with H2O (2 × 40 mL). The organic layer was dried over MgSO4 and concentrated in vacuo. Purification by column chromatography (CH2Cl2/acetone = 3:2) gave 18c as a white solid (0.14 g, 0.26 mmol, 47%). 1H NMR (400 MHz, CDCl3): δ 8.84 (d, J = 7.2 Hz, 1H), 8.54 (s, 1H), 8.36 (d, J = 7.6 Hz, 1H), 8.12 (d, J = 7.6 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.62 (br s, 1H), 6.84 (s, 1H), 6.80–6.70 (m, 1H), 4.27 (t, J = 7.2 Hz, 2H), 4.04 (t, J = 8.0 Hz, 2H), 3.89 (s, 3H), 3.74–3.60 (m, 2H), 2.07–1.92 (m, 2H), 1.85–1.64 (m, 4H), 0.98 (t, J = 7.2 Hz, 3H). MS: [ESI + H]+: 532.3. HPLC: 8.21 min.
N-(3-(8-Methoxy-2,4-dioxo-3-propyl-3,4-dihydropyrido[2,1-f]purin-1(2H)-yl)propyl)-4-(methylsulfonyl)benzamide (19)
To a solution of EDC (0.061 g, 0.32 mmol, 1.2 equiv) in CHCl3 (5 mL) was added 4-(methylsulfonyl)benzoic acid (0.060 g, 0.30 mmol, 1.1 equiv). Amine 16b (0.090 g, 0.27 mmol, 1 equiv) was taken up in CHCl3 (5 mL) and was subsequently added dropwise via an automatic syringe at a rate of 0.15 mL min–1. The reaction was stirred at room temperature and monitored by TLC (4% MeOH in CH2Cl2). After 3 h, the reaction was completed and CHCl3 (50 mL) was added. The organic layer was washed with 1 M HCl (60 mL), H2O (60 mL), and brine (60 mL), dried over MgSO4, and concentrated under vacuum. The product was purified by column chromatography using 2% MeOH in CH2Cl2 to afford the title compound (0.075 g, 0.14 mmol, 54%). 1H NMR (400 MHz, CDCl3): δ 8.86 (d, J = 7.2 Hz, 1H), 8.36 (t, J = 5.6 Hz, 1H), 8.19 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 8.4 Hz, 2H), 6.90–6.71 (m, 2H), 4.45–4.28 (m, 2H), 4.13–3.99 (m, 2H), 3.91 (s, 3H), 3.55–3.41 (m, 2H), 3.11 (s, 3H), 2.27–2.09 (m, 2H), 1.83–1.61 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (600 MHz, DMSO-d6, 318 K): δ 164.7, 161.1, 153.5, 150.7, 150.6, 149.3, 142.8, 138.9, 127.9, 127.5, 126.7, 107.4, 99.9, 95.4, 56.0, 43.2, 41.6, 40.8, 36.8, 27.4, 20.7, 10.9. MS: [ESI + H]+: 514.2. HRMS-ESI+: [M + H]+ calcd: 518.1760 found: 518.1791, C24H28O6N5S. HPLC: 6.89 min.
Computational Studies
All calculations were performed using the Schrodinger Suite.31 Since compound 17b shares high similarity with the ligands on which we previously published,7 the same homology model based on the high-resolution antagonist-bound crystal structure of the adenosine A2A receptor (PDB: 4EIY(27)) was used for the docking studies performed here. Based on these proposed docking poses, we used induced fit docking32 with core constraints on the pyridopurinedione to dock the different ligands.
Biology
[3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-5-one ([3H]PSB-11, specific activity 56 Ci mmol–1) was a gift from Prof. C. E. Müller (University of Bonn, Germany). Unlabeled PSB-11, 1-deoxy-1-[6-[3- and 2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide]] (2-Cl-IB-MECA) were purchased from Tocris Ltd. (Abingdon, U.K.). 5′-N-Ethylcarboxamidoadenosine (NECA) was purchased from Sigma-Aldrich (Steinheim, Germany). Adenosine deaminase was purchased from Boehringer Mannheim (Mannheim, Germany). Bicinchoninic acid (BCA) and BCA protein assay reagents were purchased from Pierce Chemical Company (Rockford, IL). Chinese hamster ovary (CHO) cells stably expressing the human A3 adenosine receptor (CHOhA3) were a gift from Dr. K.-N. Klotz (University of Würzburg, Germany). All other chemicals were obtained from standard commercial sources and were of analytical grade.
Cell Culture and Membrane Preparation
Chinese hamster ovary (CHO) cells, stably expressing the human A3 adenosine receptor (CHOhA3), were cultured and membranes were prepared and stored as previously reported.7,33 Protein determination was performed based on the bicinchoninic acid (BCA) method.34
Y265F7.36 Site-Directed Mutagenesis
The single tyrosine mutation introduced in TM7 of the hA3AR was performed with the QuickChange II Site-Directed Mutagenesis system (Stratagene, Huizen, The Netherlands). The wild-type pcDNA3.1(+)-A3AR plasmid DNA with N-terminal 3 × HA-tag was used as a template for polymerase chain reaction (PCR) mutagenesis. Mutant primers for directional PCR product cloning were designed using the online Quickchange primer design program (Agilent Technologies, Santa Clara, CA) and obtained from Eurogentec (Maastricht, The Netherlands). Forward primer used for this procedure was 5′-cacagcttgtgctgttcatgggcatcctgct-3′ and the reverse primer was 5′-agcaggatgcccatgaacagcacaagctgtg-3′. All DNA sequences were verified by Sanger sequencing at LGTC (Leiden, The Netherlands).
Transient Expression of Wild Type (WT) and Mutant Receptors in CHO-K1 Cells
CHO-K1 cells were seeded into 150 mm culture dishes to achieve 60% confluence in the presence of 20 mL culture medium consisting of Dulbecco’s modified Eagle’s medium/F12 (1:1) supplemented with 10% (v/v) newborn calf serum, streptomycin (50 μg mL–1), and penicillin (50 IU mL–1). The cells were transfected approximately 24 h later with plasmid DNA (20 μg of DNA/dish) by the PEI method35 (PEI/DNA = 3:1) and left for 24 h. Subsequently, the medium was removed and fresh medium containing 5 mM sodium butyrate was added (to enhance the receptor expression level36), and cells were grown for an additional 24 h at 37 °C and 5% CO2. Membrane preparation followed the procedure described above for the CHO cell membranes stably expressing the hA3AR.7,33
Radioligand Displacement Assay
Radioligand displacement experiments were performed as in previously published methods.7 Membrane aliquots containing 15 μg of protein were incubated in a total volume of 100 μL assay buffer (50 mM Tris–HCl, 5 mM MgCl2, supplemented with 0.01% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate and 1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.4) at 25 °C for 120 min. Displacement experiments were performed using six concentrations of competing antagonist in the presence of ∼10 nM [3H]PSB-11. Nonspecific binding was determined in the presence of 100 μm NECA and represented less than 10% of total binding. Incubation was terminated by rapid filtration performed on 96-well GF/B filter plates (PerkinElmer, Groningen, the Netherlands) in a PerkinElmer Filtermate-harvester (PerkinElmer, Groningen, the Netherlands). After the filter plate was dried at 55 °C for 30 min, the filter-bound radioactivity was determined by scintillation spectrometry using a 2450 MicroBeta2 Plate Counter (PerkinElmer, Boston, MA).
Radioligand Competition Association Assay
The competition association assay was performed by incubation of ∼10 nM [3H]PSB-11 in the absence or presence of the competing hA3AR antagonist at its IC50 concentration with membrane aliquots. The amount of receptor-bound radioligand was determined at different time points up to 240 min. Incubations were terminated and samples were obtained as described under the Radioligand Displacement Assay.
[35S] GTPγS Binding Assay
The assays were started by adding 15 μg of homogenized CHOhA3 membranes in an ice-cold assay buffer to a total volume of 80 μL containing 50 mM Tris–HCl buffer, 5 mM MgCl2, 1 mM EDTA, 0.05% bovine serum albumin and 1 mM dithiothreitol, 100 mM NaCl, pH 7.4, supplemented with 1 μm GDP and 5 μg saponin. The assays were performed in a 96-well plate format, where stock solutions of the compounds were added to a total volume of 100 μL using an HP D300 Digital Dispenser (Tecan, Männedorf, Switzerland). The final concentration of dimethyl sulfoxide (DMSO) per assay point was ≤0.1%. The basal level of [35S] GTPγS binding was determined in the absence of the ligand, whereas the maximal level of [35S] GTPγS binding was determined in the presence of 10 μm 2-Cl-IB-MECA. For the insurmountability experiments, membrane preparations were preincubated with or without antagonists (0.1-,1-, 3-, 10-fold Ki values) for 60 min at 25 °C, prior to the addition of 2-Cl-IB-MECA (10 μm to 0.1 nM) and 20 μL [35S] GTPγS (final concentration ∼0.3 nM), after which incubation continued for another 30 min at 25 °C. For the surmountability (control) experiments, antagonists (1-, 3-, 10-fold Ki values) and 2-Cl-IB-MECA (10 μm to 0.1 nM) were co-incubated with [35S] GTPγS for 30 min at 25 °C. For all experiments, incubations were terminated and samples were obtained as described under the Radioligand Displacement Assay, using GF/B filters (Whatman International, Maidstone, U.K.).
Data Analysis
All experimental data were analyzed using the nonlinear regression curve fitting program GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA). Data from the radioligand displacement assays were fit into one-site binding mode, and the obtained IC50 values were converted into Ki values using the Cheng–Prusoff equation to determine the affinity of the ligands.37 The observed association rate constants (kobs) derived from both assays were obtained by fitting association data using one-phase exponential association. The dissociation rate constants were obtained by fitting dissociation data to a one phase exponential decay model. The kobs values were converted into association rate constants (kon) using the equation kon = (kobs – koff)/[L], where [L] is the amount of radioligand used for the association experiments. Association and dissociation rate constants for unlabeled compounds were calculated by fitting the data into the competition association model using “kinetics of competitive binding”.22
 |
where X is the time (min), Y is the specific [3H]PSB-11 binding (DPM), k1 and k2 are the kon (M–1 min–1) and koff (min–1) of [3H]PSB-11, respectively, Bmax is the total binding (DPM), L is the radioligand concentration (nM), and I is the concentration of the unlabeled competitor (nM). Association and dissociation rate constants for [3H]PSB-11 (k1 = 0.281 ± 0.04 × 108 M–1 min–1 and k2 = 0.3992 ± 0.02 min–1) were obtained from Xia et al.7 With that, the k3, k4, and Bmax were calculated, where k3 represents the kon (M–1 min–1) of the unlabeled ligand, k4 stands for the koff (min–1) of the unlabeled ligand and Bmax equals the total binding (DPM). All competition association data were globally fitted. The residence time (RT, in min) was calculated using the equation RT = 1/koff, as koff values are expressed in min–1. [35S] GTPγS binding curves were analyzed by nonlinear regression using “log (agonist) vs response–variable slope” to obtain potency, inhibitory potency, or efficacy values of agonists and antagonists (EC50 and Emax, respectively). In the (in)surmountability assays, Schild EC50 shift equations were used to obtain Schild-slopes and pA2 values. All experimental values obtained are means of three independent experiments performed in duplicate.
Acknowledgments
We thank Prof C. E. Müller (Bonn University, Germany) for her kind help in obtaining [3H]PSB-11, the radiolabeled probe used in this study. We are also thankful to our colleague, Lindsey Burggraaff, for her assistance with computational modeling studies and Ing. G.K. Spijksma for assistance with HRMS. X.Y. is supported by a grant from the Chinese Scholarship Council.
Glossary
Abbreviations
- BCA
bicinchoninic acid
- CHO
Chinese hamster ovary
- CHO-K1
a subclone from the parental CHO cell line
- 2-Cl-IB-MECA
1-[2-chloro-6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-1-deoxy-N-methyl-β-d-ribofuranuronamide
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- Emax
maximum response elicited by an unlabeled ligand in a functional assay (relatively to 2-Cl-IB-MECA) at membranes of CHO cells stably expressing the A3 adenosine receptor
- EtOAc
ethyl acetate
- G418
geneticin
- GTPγS
guanosine 5′-O-[γ-thio]triphosphate
- hA1AR
human A1 adenosine receptor
- hA3AR
human A3 adenosine receptor
- MeCN
acetonitrile
- NECA
5′-(N-ethylcarboxamide)adenosine
- PSB-11
8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H- imidazo[2,1-i]-purin-5-one
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b02026.
Author Contributions
X.Y., L.H.H., and A.P.I. conceived the study. L.H.H., D.v.d.E., and A.P.I. supervised the project. Chemical synthesis was designed and supervised by X.Y. and J.P.D.v.V. and performed by J.O., B.J.K., and J.P.D.v.V. The bioassays were supervised by L.H.H and performed by X.Y. The computational work was performed by E.B.L. The manuscript was written by X.Y., L.H.H., D.v.d.E., and A.P.I.
The authors declare no competing financial interest.
Notes
The homology model was based on the crystal structure of the adenosine A2A receptor (PDB: 4EIY). Authors will release the atomic coordinates upon article publication.
Supplementary Material
References
- Fredholm B. B.; IJzerman A. P.; Jacobson K. A.; Klotz K. N.; Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H.; Cunhamelo J. R.; Saul W. F.; Beaven M. A. Activation of phospholipase-C via adenosine receptors provides synergistic signals for secretion in antigen-stimulated Rbl-2h3 cells - evidence for a novel adenosine receptor. J. Biol. Chem. 1990, 265, 745–753. [PubMed] [Google Scholar]
- Borea P. A.; Varani K.; Vincenzi F.; Baraldi P. G.; Tabrizi M. A.; Merighi S.; Gessi S. The A3 adenosine receptor: history and perspectives. Pharmacol. Rev. 2015, 67, 74–102. 10.1124/pr.113.008540. [DOI] [PubMed] [Google Scholar]
- Yang H.; Avila M. Y.; Peterson-Yantorno K.; Coca-Prados M.; Stone R. A.; Jacobson K. A.; Civan M. M. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Curr. Eye Res. 2005, 30, 747–754. 10.1080/02713680590953147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. A.; Spina D.; Page C. P. Adenosine receptors and asthma. Br. J. Pharmacol. 2008, 153, S446–456. 10.1038/bjp.2008.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priego E. M.; Perez-Perez M. J.; von Frijtag Drabbe Kuenzel J. K.; de Vries H.; IJzerman A. P.; Camarasa M. J.; Martin-Santamaria S. Selective human adenosine A3 antagonists based on pyrido[2,1-f]purine-2,4-diones: novel features of hA3 antagonist binding. ChemMedChem 2008, 3, 111–119. 10.1002/cmdc.200700173. [DOI] [PubMed] [Google Scholar]
- Xia L.; Burger W. A. C.; van Veldhoven J. P. D.; Kuiper B. J.; van Duijl T. T.; Lenselink E. B.; Paasman E.; Heitman L. H.; IJzerman A. P. Structure-affinity relationships and structure-kinetics relationships of pyrido[2,1-f]purine-2,4-dione derivatives as human adenosine A3 receptor antagonists. J. Med. Chem. 2017, 60, 7555–7568. 10.1021/acs.jmedchem.7b00950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priego E. M.; Kuenzel J. V.; IJzerman A. P.; Camarasa M. J.; Perez-Perez M. J. Pyrido[2,1-f]purine-2,4-dione derivatives as a novel class of highly potent human A3 adenosine receptor antagonists. J. Med. Chem. 2002, 45, 3337–3344. 10.1021/jm0208469. [DOI] [PubMed] [Google Scholar]
- Weichert D.; Gmeiner P. Covalent molecular probes for class A G protein-coupled receptors: advances and applications. ACS Chem. Biol. 2015, 10, 1376–1386. 10.1021/acschembio.5b00070. [DOI] [PubMed] [Google Scholar]
- Murrison E. M.; Goodson S. J.; Edbrooke M. R.; Harris C. A. Cloning and characterisation of the human adenosine A3 receptor gene. FEBS Lett. 1996, 384, 243–246. 10.1016/0014-5793(96)00324-9. [DOI] [PubMed] [Google Scholar]
- Glukhova A.; Thal D. M.; Nguyen A. T.; Vecchio E. A.; Jorg M.; Scammells P. J.; May L. T.; Sexton P. M.; Christopoulos A. Structure of the adenosine A1 receptor reveals the basis for subtype selectivity. Cell 2017, 168, 867–877. 10.1016/j.cell.2017.01.042. [DOI] [PubMed] [Google Scholar]
- Li A. H.; Chang L.; Ji X. D.; Melman N.; Jacobson K. A. Functionalized congeners of 1,4-dihydropyridines as antagonist molecular probes for A3 adenosine receptors. Bioconjugate Chem. 1999, 10, 667–677. 10.1021/bc9900136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi P. G.; Cacciari B.; Moro S.; Romagnoli R.; Ji X. D.; Jacobson K. A.; Gessi S.; Borea P. A.; Spalluto G. Fluorosulfonyl- and bis-(beta-chloroethyl)amino-phenylamino functionalized pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives: irreversible antagonists at the human A3 adenosine receptor and molecular modeling studies. J. Med. Chem. 2001, 44, 2735–2742. 10.1021/jm010818a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X. D.; Gallorodriguez C.; Jacobson K. A. A selective agonist affinity label for A3 adenosine receptors. Biochem. Biophys. Res. Commun. 1994, 203, 570–576. 10.1006/bbrc.1994.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Michiels T. J. M.; de Jong C.; Soethoudt M.; Dekker N.; Gordon E.; van der Stelt M.; Heitman L. H.; van der Es D.; IJzerman A. P. An affinity-based probe for the human adenosine A2A receptor. J. Med. Chem. 2018, 61, 7892–7901. 10.1021/acs.jmedchem.8b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone R. P.; Fournier D. J.; Makriyannis A. Ligand based structural studies of the CB1 cannabinoid receptor. J. Pept. Res. 2002, 60, 348–356. 10.1034/j.1399-3011.2002.21069.x. [DOI] [PubMed] [Google Scholar]
- Narayanan A.; Jones L. H. Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci. 2015, 6, 2650–2659. 10.1039/C5SC00408J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimster N. P.; Connelly S.; Baranczak A.; Dong J. J.; Krasnova L. B.; Sharpless K. B.; Powers E. T.; Wilson I. A.; Kelly J. W. Aromatic sulfonyl fluorides covalently kinetically stabilize transthyretin to prevent amyloidogenesis while affording a fluorescent conjugate. J. Am. Chem. Soc. 2013, 135, 5656–5668. 10.1021/ja311729d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller C. E.; Diekmann M.; Thorand M.; Ozola V. [3H]8-Ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-f]-purin-5-one ([3H]PSB-11), a novel high-affinity antagonist radioligand for human A3 adenosine receptors. Bioorg. Med. Chem. Lett. 2002, 12, 501–503. 10.1016/S0960-894X(01)00785-5. [DOI] [PubMed] [Google Scholar]
- Weichert D.; Kruse A. C.; Manglik A.; Hiller C.; Zhang C.; Hubner H.; Kobilka B. K.; Gmeiner P. Covalent agonists for studying G protein-coupled receptor activation. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 10744–10748. 10.1073/pnas.1410415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Dong G.; Michiels T. J. M.; Lenselink E. B.; Heitman L.; Louvel J.; IJzerman A. P. A covalent antagonist for the human adenosine A2A receptor. Purinergic Signalling 2017, 13, 191–201. 10.1007/s11302-016-9549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky H. J.; Mahan L. C. The kinetics of competitive radioligand binding predicted by the law of mass-action. Mol. Pharmacol. 1984, 25, 1–9. [PubMed] [Google Scholar]
- Doornbos M. L. J.; Wang X.; Vermond S. C.; Peeters L.; Perez-Benito L.; Trabanco A. A.; Lavreysen H.; Cid J. M.; Heitman L. H.; Tresadern G.; IJzerman A. P. Covalent allosteric probe for the metabotropic glutamate receptor 2: design, synthesis, and pharmacological characterization. J. Med. Chem. 2018, 223–233. 10.1021/acs.jmedchem.8b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jörg M.; Glukhova A.; Abdul-Ridha A.; Vecchio E. A.; Nguyen A. T. N.; Sexton P. M.; White P. J.; May L. T.; Christopoulos A.; Scammells P. J. Novel irreversible agonists acting at the A1 adenosine receptor. J. Med. Chem. 2016, 59, 11182–11194. 10.1021/acs.jmedchem.6b01561. [DOI] [PubMed] [Google Scholar]
- van Muijlwijk-Koezen J. E.; Timmerman H.; van der Sluis R. P.; van de Stolpe A. C.; Menge W. M. P. B.; Beukers M. W.; van der Graaf P. H.; de Groote M.; IJzerman A. P. Synthesis and use of FSCPX, an irreversible adenosine A1 antagonist, as a ‘receptor knock-down’ tool. Bioorg. Med. Chem. Lett. 2001, 11, 815–818. 10.1016/S0960-894X(01)00069-5. [DOI] [PubMed] [Google Scholar]
- Strange P. G. Use of the GTP gamma S ([35S]GTP gamma S and Eu-GTP gamma S) binding assay for analysis of ligand potency and efficacy at G protein-coupled receptors. Br. J. Pharmacol. 2010, 161, 1238–1249. 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; Cherezov V.; Stevens R. C. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone R. P.; Khanolkar A. D.; Xu W.; Ayotte L. A.; Thakur G. A.; Hurst D. P.; Abood M. E.; Reggio P. H.; Fournier D. J.; Makriyannis A. (-)-7′-Isothiocyanato-11-hydroxy-1 ‘,1′-dimethylheptylhexahydrocannabinol (AM841), a high-affinity electrophilic ligand, interacts covalently with a cysteine in helix six and activates the CB1 cannabinoid receptor. Mol. Pharmacol. 2005, 68, 1623–1635. 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- Singh J.; Petter R. C.; Baillie T. A.; Whitty A. The resurgence of covalent drugs. Nat. Rev. Drug Discovery 2011, 10, 307–317. 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Soethoudt M.; Stolze S. C.; Westphal M. V.; van Stralen L.; Martella A.; van Rooden E. J.; Guba W.; Varga Z. V.; Deng H.; van Kasteren S. I.; Grether U.; IJzerman A. P.; Pacher P.; Carreira E. M.; Overkleeft H. S.; Ioan-Facsinay A.; Heitman L. H.; van der Stelt M. Selective photoaffinity probe that enables assessment of cannabinoid CB2 receptor expression and ligand engagement in human cells. J. Am. Chem. Soc. 2018, 140, 6067–6075. 10.1021/jacs.7b11281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel U.; Siepe M.; Schwer C.; Schlensak C.; Loop T. Sevoflurane-induced preconditioning in the isolated mouse heart is mediated by A1-and A3-adenosine receptors. Eur. J. Anaesthesiol. 2010, 27, 72. 10.1097/00003643-201006121-00229. [DOI] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Heitman L. H.; Göblyös A.; Zweemer A. M.; Bakker R.; Mulder-Krieger T.; van Veldhoven J. P. D.; de Vries H.; Brussee J.; IJzerman A. P. A series of 2,4-disubstituted quinolines as a new class of allosteric enhancers of the adenosine A3 receptor. J. Med. Chem. 2009, 52, 926–931. 10.1021/jm8014052. [DOI] [PubMed] [Google Scholar]
- Smith P. K.; Krohn R. I.; Hermanson G. T.; Mallia A. K.; Gartner F. H.; Provenzano M. D.; Fujimoto E. K.; Goeke N. M.; Olson B. J.; Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Boussif O.; Lezoualch F.; Zanta M. A.; Mergny M. D.; Scherman D.; Demeneix B.; Behr J. P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in-vivo - polyethylenimine. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 7297–7301. 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman C. M.; Howard B. H.; Reeves R. Expression of recombinant plasmids in mammalian-cells is enhanced by sodium-butyrate. Nucleic Acids Res. 1983, 11, 7631–7648. 10.1093/nar/11.21.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung-Chi C.; Prusoff W. H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.