

Abstract

We previously described the discovery of GSK5852 (1), a non-nucleoside polymerase (NS5B) inhibitor of hepatitis C virus (HCV), in which an N-benzyl boronic acid was essential for potent antiviral activity. Unfortunately, facile benzylic oxidation resulted in a short plasma half-life (5 h) in human volunteers, and a backup program was initiated to remove metabolic liabilities associated with 1. Herein, we describe second-generation NS5B inhibitors including GSK8175 (49), a sulfonamide-N-benzoxaborole analog with low in vivo clearance across preclinical species and broad-spectrum activity against HCV replicons. An X-ray structure of NS5B protein cocrystallized with 49 revealed unique protein-inhibitor interactions mediated by an extensive network of ordered water molecules and the first evidence of boronate complex formation within the binding pocket. In clinical studies, 49 displayed a 60–63 h half-life and a robust decrease in viral RNA levels in HCV-infected patients, thereby validating our hypothesis that reducing benzylic oxidation would improve human pharmacokinetics and lower efficacious doses relative to 1.
1. Introduction
The remarkable progress made since the identification of hepatitis C virus (HCV) in 1989 represents one of the success stories of drug discovery.1 The last decade marked an epoch of rapid advancement culminating in development of all-oral therapies with cure rates >90%.2 The impressive efficacy and convenience afforded by all-oral regimens have dramatically increased the number of patients seeking treatment.3,4 Unfortunately, the majority of the estimated 70 million people infected with HCV remain untreated due in part to the high cost of therapy, especially in many upper-middle and high-income countries where generics may not be available.5 The WHO has set an ambitious target of diagnosing 90% of the HCV-infected population and treating 80% of those diagnosed by 2030.6 The development of new direct-acting antivirals may serve to increase market competition and lower costs, thereby enabling more equitable access to safe and effective therapy options.
In our effort to identify therapies to treat HCV, we selected GSK5852 (1) as a candidate for clinical development (Figure 1).7 Compound 1 binds to the palm site II region of HCV RNA-dependent RNA polymerase (NS5B) and inhibits the initiation step of the polymerase RNA replication cycle.8 A benzyl boronic acid was a critical pharmacophore for broad spectrum antiviral activity against HCV genotype (GT) 1–6 replicons,9 as well as clinically relevant polymorph replicons resistant to the structurally related compound 2.10,11 Unfortunately, the 5 h plasma half-life (t1/2) of 1 in human subjects was shorter than predicted by preclinical allometric scaling, resulting in a high anticipated daily dose for efficacy (420 mg b.i.d).12 Despite the disappointing human pharmacokinetic (PK) profile, a single 420 mg oral dose of 1 produced a statistically significant reduction in plasma HCV RNA levels (1.33 log10 IU/mL) in HCV-infected subjects.12
Figure 1.

Metabolites 3 and 4 identified in clinical plasma samples from human volunteers dosed with 1 (GSK5852). Comparison of 1 with a structurally related clinical asset 2 (HCV-796).
Analyses of the plasma samples from human volunteers dosed with 1 identified a major circulating metabolite (3) with a long human half-life (45 h) (Figure 1).12 The exposure (AUC0–t) of 3 was ∼50% greater than the parent drug and exceeded safety margins established in 3-month toxicology studies (rat and dog).12 Additionally, compound 3 is a reported human metabolite of HCV-796 (2), a clinical asset associated with liver toxicity in a Phase-2 study.13 Although there is no direct evidence that 3 causes drug-induced liver injury, avoiding its formation seemed prudent.
A plausible mechanism for in vivo formation of 3 involves oxidation of the benzylic carbon with subsequent C–N bond cleavage.14 The detection of significant concentrations of benzoic acid 4 in plasma samples from humans dosed with 1 further supports this pathway (Figure 1). Importantly, the boronic acid moiety was found intact in metabolite 4 suggesting that high clearance of 1 occurs predominantly via debenzylative, not deborylative,15 metabolism. This distinction inspired the hypothesis that the half-life of parent drug could be extended by addressing benzylic oxidation while retaining the boron pharmacophore necessary for robust antiviral activity.
With a successful demonstration of clinical efficacy and a working hypothesis to guide discovery efforts, a backup program was initiated to remove liabilities associated with 1. Specifically, we aimed to identify a compound that met four criteria: (a) antiviral activity against clinically relevant NS5B C316N/Y replicons resistant to 2; (b) no potential to form metabolite 3; (c) low in vivo metabolism across preclinical species; and (d) a predicted human dose <100 mg/day. An asset achieving these objectives was anticipated to have an increased probability of delivering clinical efficacy for a broad patient population while minimizing safety risks inherent with high doses and long-lived metabolites.
2. Design Strategy
With goals of the backup effort clearly defined, strategies were devised to reduce/eliminate benzylic oxidation and prevent the in vivo formation of 3. Given the favorable antiviral profile of 1 across multiple wildtype (WT) and polymorph replicons, including the resistant mutations at amino acid 316 of NS5B, we preferred designs that maintained a similar binding mode, especially with respect to the boronic acid’s position in the binding site. From this perspective, increasing the steric bulk at the benzylic carbon appeared the simplest way of reducing oxidation while maintaining boron alignment. Unfortunately, adding a methyl group to the benzylic carbon reduced potency ∼50-fold against GT 1b C316N replicon (compare analogs 5 and 6, Table 1). We speculated that the Me-benzyl substituent introduces an unfavorable twist in the phenyl ring, which might be eliminated with constrained bonds that preorganize the desired conformation.
Table 1. Inhibition of HCV Replicons for Approach A (9–11) and Comparator Compoundsa.
![]()

See Supporting Information for assay details, number of replicates, 95% confidence intervals, and cellular toxicity data (CC50). Confidence intervals were derived from a pooled standard deviation (SD) calculated for each assay in log space (i.e., pEC50), where replicates are normally distributed; pooled assay SDs are supplied parenthetically in the following notes.
GT 1b WT (SD = 0.21 log units) and 316N (SD = 0.24 log units) assays used a stable replicon system, whereas GT 1a WT (SD = 0.23 log units) and 316Y (SD = 0.20 log units) were transiently transfected.
Data originally reported in ref (8).
Data generated from a closely related assay system.
Not tested.
Two approaches were therefore envisioned in which the benzylic substituent was cyclized to reduce rotational freedom (Figure 2): (a) onto the boron-containing phenyl ring (approach A) or (b) in the direction of the sulfonamide moiety (approach B). Exemplars of each design strategy were virtually constructed in Schrodinger’s modeling software, Maestro, and manually overlaid with the bound conformation of 1.16 Ideas were visually inspected for high-energy conformations, and energy minimization using a molecular mechanics force field and implicit solvent model was performed to confirm constrained conformational energetics and visualize the theoretical, local conformational minima.16 Modeling in this manner suggested that approach A would limit the twist of the phenyl ring; however, a sterically hindered conformation may be necessary to maintain alignment of the boronic acid. For example, both enantiomers of the indane analog of 1 appeared to require a partially eclipsed conformation of the sulfonamide-indane torsion to maximize overlap of the boronic acid with the bound conformation of 1 (Figure 3a). In contrast, minimized conformations of representative target molecules from approach B appeared to bias the desired bioactive conformation (Figure 3b).
Figure 2.

Strategic approaches to reduce the propensity for benzylic oxidation associated with N-benzyl boronic acids.
Figure 3.

Models of representative target molecules from approaches A–C overlaid with the conformation of 1 (grey) when bound to HCV NS5B GT 1b C316N polymerase (PDB code: 4KAI). Only proposed changes to 1 are shown for clarity. (a) Approach A: the (1R)-isomer of an indane boronic acid (magenta) in a partially eclipsed (nonminimum) conformation (fC2–C1–N–S ≈ −10°). (b) Approach B: the molecular-mechanics-minimized16 conformation of an oxazolidinone-linked phenyl boronic acid (cyan). (c) Approach C: the molecular-mechanics-minimized16 conformation of an N-phenyl boronic acid (green) that necessarily displaces the position of the boronic acid relative to 1 (d ≈ 3.7 Å).17
A third approach (C) was undertaken to excise the benzylic methylene group and construct N-phenyl sulfonamide analogs (Figure 2). Although approaches A and B could achieve conformations that align with the unhindered benzyl boronic acid when bound to NS5B (Figure 3a,b), the removal of the methylene would necessarily alter its position significantly (∼3.7 Å as shown in Figure 3c). Therefore, the opportunity in approach C to guarantee the elimination of benzylic oxidation is offset by the challenge of establishing new binding interactions that can drive potency against 316 polymorphs.
3. Chemistry and Antiviral Results
3.1. Approach A Cyclization: Indanes and Tetrahydronaphthalenes (Compounds 9–11)
Constrained analogs defined by approach A were supported by the observation that aryl substitution adjacent to the benzylic carbon with small lipophilic groups (Me, F, and CF3) retained antiviral activity (compare compounds 7 and 8, Table 1) while reducing in vivo clearance in rat (3.5 vs 36 mL/min/kg for 8 and 7, respectively, see the Supporting Information). We speculated that tethering the ortho-substituent to the benzylic carbon might further improve the clearance. A small set of bicyclic analogs was designed to vary both the ring size and position of the boronic acid on the phenyl ring. Analogs 9–11 were prepared via Fukuyama–Mitsunobu displacement of the bicyclic 2°-alcohol with the N-sulfonamide of 3. The six- and the five-membered ring derivatives (9 and 10) showed reduced activity against GT 1a and 1b WT replicons (>10-fold) and a more significant ∼1000-fold shift in potency versus the 316Y mutation as compared to 1 (GT 1a). Transferring the boronic acid from the para- to the meta-position (10 and 11, respectively) did not rescue the activity.
3.2. Approach B Cyclization: Saturated Heterocycles with Carbonyl Groups (Compounds 19–26)
X-ray crystal structures of compounds in the benzofuran series bound to NS5B highlighted an interaction between the sulfonyl oxygen of the inhibitor and residue Arg200.8 We sought to enable a similar interaction for the constrained analogs envisioned in approach B by incorporating structural motifs capable of hydrogen bonding to Arg200. As only one of the sulfonamide oxygens appeared to make meaningful contact with the protein, we reasoned that the sulfonamide could be replaced with carbonyl-containing groups such as lactams, ureas, and carbamates.
A representative synthesis of oxazolidinone 18 is shown in Scheme 1. Bromination of readily available benzylic ester 12 occurred in 71% yield to afford the benzylic bromide 13. Displacement of the bromide by aniline 14 required heating to 85 °C in DMF and gave a 47% yield of the α-amino ester 15. The ester moiety was reduced to the corresponding alcohol 16, and subsequent treatment with triphosgene afforded the oxazolidinone 17 as a racemate. The aryl bromide was then converted to the boronic acid (18) via a Pd-catalyzed borylation/hydrolysis procedure described previously.8 Other analogs in this series were synthesized in an analogous manner (procedures are provided in the Supporting Information).
Scheme 1. Representative Synthesis of Oxazolidinone 18.

Conditions: (i) NBS, CCl4, benzoyl peroxide, reflux, 4 h, 71%; (ii) aniline 14, DMF, 85 °C, 47%; (iii) LiBH4, THF, 84%; (iv) triphosgene, Et3N, DCM, 50%; (v) (BPin)2, PdCl2(dppf)·CH2Cl2, KOAc, dioxane, 100 °C; and (vi) PS-BBA, HCl, THF, 30% for 2 steps.
Results with lactam 19 and cyclic urea 20 were disappointing as even WT activity was diminished (EC50 > 60 nM); however, a marked improvement was noted for the cyclic carbamate 21 (Table 2). There was a preference for five- versus six-membered ring size with both the oxazolidinone (compound 21 and 22, respectively) and imidazolinone motifs (SI-1, see the Supporting Information). The substitution of the phenyl ring with the boronic acid is favored at the para position (21) relative to the meta position (23). The strong preference for para-substitution and five-membered ring size is not surprising as both help orient the boron pharmacophore in a manner consistent to 1.
Table 2. Inhibition of HCV Replicons for Approach B Compounds 19–26a.
![]()

See the Supporting Information for assay details, number of replicates, 95% confidence intervals, and cellular toxicity data (CC50). Confidence intervals were derived from a pooled SD calculated for each assay in log space (i.e., pEC50), where replicates are normally distributed; pooled assay SDs are supplied parenthetically in the following notes.
GT 1b WT (SD = 0.21 log units) and 316N (SD = 0.24 log units) assays used a stable replicon system, whereas GT 1a WT (SD = 0.23 log units) and 316Y (SD = 0.20 log units) were transiently transfected.
Not tested.
SAR developed within the benzyl boronic acid series demonstrates that electron-withdrawing groups (EWGs) adjacent to boron improve the antiviral activity, especially with respect to replicons containing mutations at amino acid 316 of NS5B. The incorporation of an ortho-fluoro substituent led to potent compound 18 (Scheme 1) which was resolved via chiral supercritical fluid chromatography to provide enantiomers 24 and 25 (Table 2).18 The in-silico docking of the two enantiomers predicted the (S)-isomer to be favored, and this was verified in the replicon assay where (S)-isomer 25 was >50-fold more potent than (R)-isomer 24. With respect to the 316N/Y polymorph activity, the positive impact of an EWG adjacent to boron was evident, as analog 25 displayed low nanomolar activity in the GT 1b 316N and GT 1a 316Y replicon assays (EC50 of 5.0 and 24 nM, respectively). This potency is in the range of the most active benzyl boronic acid compounds. The critical role of the boronic acid was again noted because its removal resulted in a >100-fold loss in activity against the 4 replicons tested (compound 26).
3.3. Approach B Cyclization: Aromatic Heterocycles (Compounds 31–37)
Encouraged by the antiviral profile of compound 25 and the prospect of replacing the sulfonamide as a means of avoiding metabolite 3, we considered other structural motifs capable of interacting with the conserved Arg200 residue. Five-membered heteroaromatic systems (Table 3) should be inherently more stable to metabolism than the nonaromatic oxazolidinone group, as well as eliminate the need for chiral synthesis or separation. However, retraction of the lone pair from the position of exocyclic carbamate oxygen to a heteroatom in an aromatized ring posed the risk of losing direct contact with Arg200.
Table 3. Inhibition of HCV Replicons for Approach B Heteroaromatic Compounds 31–37a.


See the Supporting Information for assay details, number of replicates, 95% confidence intervals, and cellular toxicity data (CC50). Confidence intervals were derived from a pooled SD calculated for each assay in log space (i.e., pEC50), where replicates are normally distributed; pooled assay SDs are supplied parenthetically in the following notes.
GT 1b WT (SD = 0.21 log units) and 316N (SD = 0.24 log units) assays used a stable replicon system, whereas GT 1a WT (SD = 0.23 log units) and 316Y (SD = 0.20 log units) were transiently transfected.
Heterocycles bearing a phenyl boronic acid were synthesized, as shown in Scheme 2. Hydrazine 27 was prepared via in situ reduction of the diazonium salt of the corresponding aniline. Acid-catalyzed cyclocondensation between 27 and (dimethylamino)propanone 28 afforded triazole 29 in a regioselective manner, whereby triazole formation proceeds via initial displacement of dimethylamine followed by cyclization onto the carbonyl group. The methyl amide analog 30 was prepared from the ethyl ester (29); then, the aryl bromide was subsequently converted to the boronic acid (31) using the standard Pd-catalyzed borylation/hydrolysis procedure described herein.
Scheme 2. Representative Synthesis of Heteroaromatic Analog (Compound 31).

Conditions: (i) AcOH, 100 °C, 67%; (ii) NaOH, THF, MeOH, H2O, 100 °C, 100%; (iii) MeNH2·HCl, HATU, DIEA, DMF, 95%; (iv) (BPin)2, PdCl2(dppf)·CH2Cl2, KOAc, dioxane, 80 °C; and (v) NaIO4, NH4OAc, H2O, THF, 24% over 2 steps.
We were pleased to find that the N-linked triazole 31 was active and essentially equipotent with oxazolidinone 25 (Table 3). A nitrogen atom adjacent to the connecting benzofuran core (position 2 as defined in Table 3) appeared critical as a replacement with a group incapable of hydrogen-bonding dramatically eroded potency (compound 32). Heteroaromatic systems with various nitrogen and oxygen substitutions were well-tolerated, provided a nitrogen resided in position 2 (examples 33–37). There was no difference in the antiviral activity between C- or N-linked phenyl boronic acid analogs (e.g., 31 vs 34), and position 4 was tolerant of nitrogen (31), carbon (33), and oxygen (35) substitution. Position 3 was also amenable to N- for C-substitution with respect to HCV WT activity but a >10-fold loss in potency was noted against the more sensitive 316Y replicon (compare analogs 31 and 36). A larger change at position 3, for instance, adding a methyl group (compound 37), adversely impacted activity which is consistent with steric constraints in this portion of the binding pocket.
3.4. Approach C: N-Phenyl Sulfonamides (Compounds 44–49)19
In addition to cyclization approaches A and B, we attempted to circumvent oxidative debenzylation of 1 by directly eliminating the culprit, the benzylic carbon. The desired compounds required appendage of the phenyl ring to the sulfonamide nitrogen which was attempted via Chan–Lam coupling of 3 with aryl boronic acids (Scheme 3).20 Although there are few reports describing the N-arylation of sulfonamides,21 both meta- and para-bromophenyl boronic acids underwent Cu-mediated C–N bond formation with the 2°-sulfonamide of 3 to provide the desired N-phenyl derivatives 38 and 39 in low yields (26 and 39%, respectively).22 Chan–Lam couplings with 3 were generally slow, often requiring several days of stirring, and little product was observed with more electron-deficient phenyl boronic acids.23 However, this approach provided access to target molecules 44 and 45 in sufficient quantity for biological testing.
Scheme 3. Synthesis of N-Phenyl Boronic Acid Analogs (44–49).

Conditions: (i) Cu(OAc)2·H2O, TEA, 4 Å mol. sieves, DCM, 26–39%; (ii) K2CO3, DME, H2O, 80–100 °C or K2CO3, HMPA, 60 °C, 29–93%; (iii) SnCl2 or H2 with Pd/C or sulfided Pt/C, 50–100%; (iv) NaNO2, CuBr, HBr, MeCN, H2O, 0–60 °C, 73–76%; (v) NCS, DMF, 60 °C, 99%; (vi) LiBH4, THF, MeOH, 5 °C, 100%; (vii) MOMCl, DIEA, THF, 50 °C, 68%; (viii) (BPin)2, PdCl2(dppf)-CH2Cl2 or Pd(dppb)Cl2, KOAc, dioxane, 80 °C; (ix) PS-BBA, HCl, H2O, THF or NaIO4, HCl, H2O, THF, 15–74% (2 steps); and (x) HCl, H2O, THF, MeOH, 71% (2 steps).
Our desire to incorporate additional EWGs (ideally adjacent to the boron pharmacophore) guided us toward nucleophilic aromatic substitution (SNAr) as a method for N-arylation (Scheme 3). The poor nucleophilicity of aryl sulfonamide 3 necessitated the use of highly activated electrophilic aromatic partners and in practice was limited to fluoronitrobenzenes. Importantly, substrates with EWGs adjacent to the nitro-facilitated fluoro-displacement thereby serving to compliment the substrate scope observed for the Chan–Lam coupling. Substrates with 3-F, -Cl, -CHF2, -CO2Me, -CN, or -CF3 substituents afforded the SNAr products with increasing yields (29, 64, 76, 83, 93, and 97%, respectively; see the Supporting Information). Conversely, 3-OMe, -Me, and -CHO fluoro-4-nitrobenzenes failed to provide the product.
The nitro group served a dual role—it increased the SNAr reactivity of the fluorobenzene substrate and provided a convenient handle for elaboration to the boronic acid. The nitro group was selectively reduced to aniline (41) and subsequently converted to the boronic acid (46–48) using a two-step procedure: a Sandmeyer reaction to produce the bromide, followed by standard Pd-catalyzed borylation/hydrolysis.
In the case of the 3-CO2Me derivative 41, chlorination ortho- to the aniline was carried out with N-chlorosuccinimide prior to the Sandmeyer reaction. Subsequent reduction of the methyl ester with LiBH4 and protection as the MOM ether afforded 43, which was converted to the pinacol boronic ester. Acid cleavage of the MOM group liberated the benzyl alcohol which rapidly cyclized on the boronic ester, displacing the pinacol group, and forming the benzoxaborole analog 49. It is worth noting that the synthetic route depicted in Scheme 3 was used to prepare up to 5 kg of 49 to supply the 3-month safety and first-time-in-human studies, after some improvements made by process chemists. A more efficient, convergent synthesis of 49 was ultimately developed to support Phase-2 studies.24
In removing the benzylic carbon, we recognized that the boron pharmacophore would shift significantly within the binding pocket relative to 1; therefore, both meta- and para-disposed boronic acids were studied (44 and 45). The para-substituted boronic acid 45 proved superior with a ∼10-fold advantage in potency against the GT 1b 316N polymorph; however, poor potency was apparent in the GT 1a 316Y assay (Table 4). Despite the shift in boronic acid position in this series, we again observed that the replicon activity was improved with the addition of an ortho-fluoro group (compound 46). In general, all analogs with small EWGs adjacent to the boron performed well against the 316N/Y replicons (compounds 46–48) with chloro-derivative 47 being one of the most potent analogs identified in any series.
Table 4. Inhibition of HCV Replicons for N-Phenyl Boronic Acids 44–49a.
![]()

See the Supporting Information for assay details, number of replicates, 95% confidence intervals, and cellular toxicity data (CC50). Confidence intervals were derived from a pooled SD calculated for each assay in log space (i.e., pEC50), where replicates are normally distributed; pooled assay SDs are supplied parenthetically in the following notes.
GT 1b WT (SD = 0.21 log units) and 316N (SD = 0.24 log units) assays used a stable replicon system, whereas GT 1a WT (SD = 0.23 log units) and 316Y (SD = 0.20 log units) were transiently transfected.
The promising antiviral profile and novelty of the N-phenyl series prompted further expansion of the SAR. A benzoxaborole group was introduced as a structurally unique pharmacophore that offered diversity away from the boronic acid, as well as potential for enhanced chemical and metabolic stability.25 It was gratifying that compound 49 displayed a similar activity to 47 in the GT 1 WT and C316 polymorphic replicon assays.
4. DMPK Results
The PK profiles of the most active compounds from each approach were obtained in rats to assess our progress toward the backup program objectives (Table 5). Compound 11 (approach A) is rapidly cleared in vivo (rat Cl = 69 mL/min/kg), which is consistent with the short in vitro half-life in rat and human hepatocytes (t1/2 ≈ 45 and <15 min, resp.). Disappointingly, the rat clearance of oxazolidinone 25 (approach B) was not improved relative to 1, and the oral exposure and bioavailability (% F) were worse. Fortunately, the heteroaryl analogs (31 and 33) exhibited excellent rat PK profiles and a >10-fold higher AUC from oral dosing than 1. The two heterocycles were indistinguishable from a PK perspective, demonstrating low clearance and moderate oral bioavailability from solution dosing. The N-phenyl analogs also showed an improvement in rat PK parameters compared to 1. A low clearance in line with the heteroaryl series was observed in rat for 47 and 49, whereas the benzoxaborole 49 had slightly higher oral bioavailability than its boronic acid counterpart.
Table 5. In Vivo Rat PK Profiles of Second-Generation NS5B Inhibitors.
| i.v.a |
p.o.b |
|||||
|---|---|---|---|---|---|---|
| Cmpd | series | Cl (mL/min/kg) | Vdss (L/kg) | t1/2 (h) | AUC (ng h/mL) | F (%) |
| 1c | N-benzyl | 29 | 1.4 | 1.0 | 700 | 23 |
| 11 | indane | 69 | 3.5 | 1.1 | 40 | 4 |
| 25 | oxazolidinone | 22 | 1.3 | 1.2 | 280 | 8 |
| 31 | heteroaryl | 4.5 | 0.9 | 2.6 | 7600 | 40 |
| 33 | 5.4 | 1.3 | 3.4 | 10000 | 64 | |
| 47 | N-phenyl | 5.1 | 1.1 | 3.7 | 4800 | 27 |
| 49 | 6.9 | 0.9 | 2.8 | 5400 | 43 | |
Dosed i.v. at 1 mg/kg in DMSO/20% HP-β-CD (10/90).
Dosed p.o. at 5 mg/kg in DMSO/20% HP-β-CD (10/90).
Dosed i.v. at 1 mg/kg and P.O. at 5 mg/kg in DMSO/Solutol/4%HP-β-CD with 0.05% acetic acid (5/10/85) to CD rats (n = 3). Further details are provided in the Supporting Information.
Having identified potent antiviral compounds with improved rat PK relative to 1, the top leads were examined for cytochrome P450 (CYP) inhibition (Table 6). The heteroaryl analogs 31 and 33 had low micromolar activity versus CYP2C9, raising a potential risk for development. Interestingly, the two N-phenyl derivatives displayed different profiles. The boronic acid analog 47 was a low micromolar inhibitor of CYP2C9, whereas the benzoxaborole derivative 49 posed significantly less risk of directly inhibiting the major CYPs, including CYP1A2 and CYP2D6 (data not shown).
Table 6. Cytochrome P450 (CYP) Inhibition Data for the Second-Generation NS5B Inhibitorsa.
| IC50 (μM) (substrate probe) |
||||||
|---|---|---|---|---|---|---|
| Cmpd | series | CYP2C9 (diclofenac) | CYP2C19 (mephenytoin) | CYP3A4 (atorvastatin) | CYP3A4 (midazolam) | CYP3A4 (nifedipine) |
| 1 | N-benzyl | 4.9 | 18 | 7.1 | >33 | 28 |
| 31 | heteroaryl | 2.7 | 1.2 | 8.9 | >33 | >33 |
| 33 | 1.5 | 23 | 2.0 | >33 | >33 | |
| 47 | N-phenyl | 2.6 | 7.4 | 4.2 | >33 | 8.1 |
| 49 | 22 | >33 | >33 | >33 | >33 | |
Experimental details are provided in the Supporting Information.
As anticipated, eliminating the benzylic carbon translated to low clearance for 49 across all preclinical species studied (Table 7). Relative to 1, compound 49 showed a significant improvement in clearance that did not exceed 13% of hepatic blood flow in any of the species tested.26 Bioavailability (%F) from solution dose (5 mg/kg) ranges from 43 to 82% in mouse, rat, and dog (data not shown).
Table 7. In Vivo PK Plasma or Blood Clearance (Cl) in Mouse, Rat, Dog, and Cynomolgus Monkey.
| in vivo
clearance [mL/min/kg], (% liver blood
flow of species)a |
||||
|---|---|---|---|---|
| Cmpd | mouse | ratb | dogb | Cyno |
| 1c | 1.4 (1.1%) | 29 (37%) | 11 (19%) | 48 (∼100%) |
| 49b,d | 0.7 (0.6%) | 6.9 (8.9%) | 1.2 (2.1%) | 5.7 (13%) |
Cl as a percentage of liver blood flow is calculated as compound Cl (blood or plasma)/species hepatic blood flow. Hepatic blood flow estimates of 126, 77.6, 56.1, and 44 mL/min/kg were used for mouse (0.02 kg), rat (0.25 kg), dog (10 kg), and cynomolgus monkey (5 kg), respectively.26
Blood clearance was used.
Dosed i.v. at 1 mg/kg in 5–10% DMSO/85–95% HP-β-CD (20% in sterile water) with or without solutol or DMSO/30% sulfobutylether- β-cyclodextrin (5/95).
Dosed i.v. at 1 mg/kg in DMSO/20% hydroxypropyl-β-cyclodextrin (10/90, pH adjusted).
5. Discussion
Our backup effort was predicated on the hypothesis that reducing or eliminating benzylic oxidation associated with benzyl boronic acid 1 would decrease preclinical in vivo clearance, prevent formation of metabolite 3, and ultimately translate into improved human PK and therapeutic dose. Our main concern, however, was the ability to alter benzylic metabolism while maintaining a favorable antiviral profile, especially against the clinically relevant polymorphs at amino acid 316 that confer resistance to inhibitors in this compound class. At the time of initiating the backup effort, over 600 analogs had been prepared of which only 14 displayed an EC50 < 50 nM versus GT 1a 316Y—all 14 compounds contained a benzyl boronic acid. It was therefore an acknowledged risk that designs violating this established pharmacophore might not meet the target antiviral profile.
Robust synthetic chemistry and commercially available starting materials allowed rapid preparation of analogs cyclized between the benzylic carbon and the boron-substituted phenyl ring (approach A). Unfortunately, derivatives 9–11 were >sixfold less active than the untethered benzylic analog 7 against WT GT 1 replicons. The loss in activity substantiated our concerns that an energetic penalty is incurred in adopting the bioactive confirmation, which requires a partially eclipsed conformation of the sulfonamide-indane torsion (Figure 3a). More disappointing, compound 11 has high in vivo clearance (69 mL/min/kg) that approximates the rate of hepatic blood flow in rat and greatly exceeds the modest clearance associated with compound 8 (3.5 mL/min/kg, see the Supporting Information).
The high degree of structural overlap between 1 and five-membered heterocycles inspired pursuit of approach B. Recognizing that only one of the sulfonamide oxygens of 1 interacts with NS5B (Figure 4a), carbonyl-containing heterocycles were considered reasonable replacements. X-ray crystallography of 25 bound to NS5B GT 1b 316N protein (Figure 4b) revealed that the carbonyl group of the oxazolidinone is well-positioned to directly replace the sulfonyl oxygen in hydrogen bonding with the Arg200 residue. As expected, the aryl boronic acids of 1 and 25 are closely aligned, rationalizing the analogous SAR in this portion of the molecule and the strong preference for the (S)-stereoisomer. Whereas an energy-minimized conformation16 of the five-membered ring overlays well with the bound conformations of 1 and 25 (Figure 3b), which was confirmed by crystallography (Figure 4b), the larger six-membered oxazolidinone 22 minimizes to a conformation that significantly displaces the boronic acid (data not shown). This shift likely accounts for the strong preference for five- versus six-membered rings with respect to the replicon activity (25 vs 22, respectively; Table 2).
Figure 4.
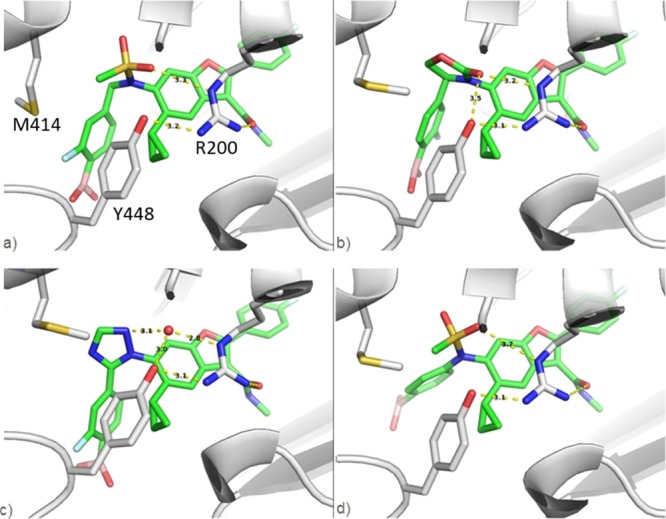
X-ray crystal structures of NS5B GT 1b 316N bound with (a) benzyl boronic acid 1 (PDB ID 4KAI); (b) oxazolidinone 25 (PDB ID 6MVK); (c) triazole 31 (PDB ID 6MVQ); and (d) N-phenyl 45 (PDB ID 6MVP). Highlighted are interactions with Arg200 and Tyr448 [residues labeled in panel (a) only for clarity].
A cocrystal structure of NS5B and heteroaryl analog 31 revealed the critical role of nitrogen at position 2 (Figure 4c). The triazole N(2) interacts with an ordered water molecule that hydrogen bonds to both Arg200 and Tyr448. These same through–water interactions are observed in all cocrystal structures obtained with heteroaryl inhibitors containing nitrogen in the 2-position regardless of the position of the boronic acid (data not shown). Therefore, it is not surprising that replacing the nitrogen with an atom unable to hydrogen bond erodes antiviral activity (compound 32). The phenyl boronic acid of 31 establishes an analogous, highly structured, water-mediated H-bond to the protein as observed with 1, therefore supporting the translation of SAR and the low-nanomolar activity versus GT 1a 316Y replicon.
X-ray structures obtained for our N-phenyl series again show that only one of the S=O groups on 45 hydrogen-bonds to Arg200 (Figure 4d), consistent with the X-ray data obtained for all sulfonamide analogs. As expected, eliminating the benzylic carbon causes significant movement of the boron pharmacophore. On the basis of crystallographic measurements, the boron atom shifts 3.6 Å between compounds 1 and 45 in the binding pocket. Regardless of this, para- (45–49) and meta- (44) boronic acid analogs displayed EC50’s < 7 nM against GT1 WT replicons. Considering the impact that boronic acid orientation has had on activity, it is gratifying to find the N-phenyl analogs represent a second robust pharmacophore for obtaining replicon potency.
The low nanomolar activity of analogs 46–49 in the GT 1a 316Y assay is noteworthy as this level of potency had been difficult to achieve within the benzofuran class of inhibitors. A cocrystal structure of 49 bound to GT 1a 316Y protein reveals an extensive network of interactions, mediated by highly ordered water molecules that had not been observed with other series (Figure 5). Six water-mediated hydrogen bonds are established in total, none of which contact Tyr316, in contrast to the benzyl and heteroaryl boronic acid analogs that form through–water interactions with Tyr316. The oxaborole moiety interacts directly with two water molecules—one contacts the backbone N–H of residue Gly449 and the second H-bonds to another ordered water molecule bridging Arg386 and Asn411. The X-ray data suggests that the boronic acid contributes to binding NS5B by establishing multiple H-bonds without covalent interaction to the protein. Improved binding affinities in the absence of covalent protein interactions have also been described with other enzyme inhibitors that incorporate boronic acids.27
Figure 5.
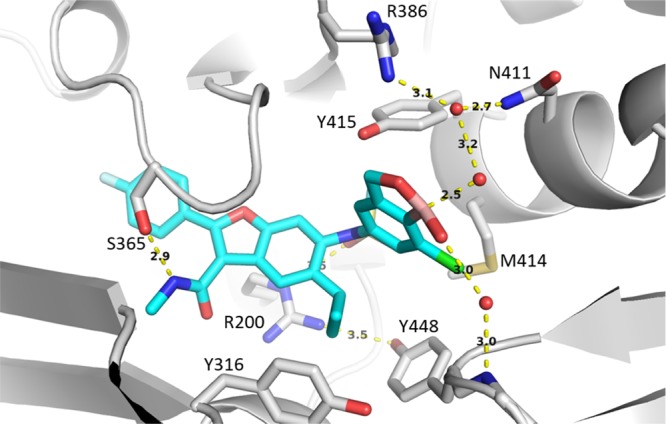
X-ray crystal structure of benzoxaborole analog 49 highlighting three ordered water molecules and key interactions to NS5B GT 1a 316Y protein (PDB ID 6MVO).
It is worth noting the distorted trigonal planar geometry of the boron observed in the crystal structure of 49 (Figure 5). A water molecule is well-positioned to occupy the empty p-orbital of the Lewis acidic boron and induce a tetrahedral configuration. The crystal structure reveals a hybrid geometry, somewhere between trigonal planar and tetrahedral, perhaps indicating an equilibrium between the bound and unbound water states. Further evidence for an equilibrating water complex is the distance between the boron and the water oxygen, which at 2.5 Å is close enough for a strong interaction but not as short as predicted for a covalent bond. Despite the unique dependence of antiviral activity on the presence of a boronic acid which could not be replicated with conventional acid isosteres, this is the first evidence of formation of a boronate complex within this series.
At physiological pH, equilibrium favors the boronate complex of 49 over the uncharged boronic acid based on its acidic pKa of 5.4.28 The relatively low pKa, which is consistent with pH-solubility data and pKa’s for related boron heterocycles,29 is due to the adjacent electron-withdrawing halogen and strain introduced by the five-membered oxaboracycle (see the Supporting Information for additional pKa data). Formation of a tetrahedral boronate relieves ring strain as the boron bond angles transition from ∼120° to ∼109°, close to the ideal internal angle of 108° for a five-membered ring, thereby giving rise to a more Lewis acidic boron. The resulting ionic species can establish strong hydrogen bonds to neighboring water molecules and the protein. Interestingly, formation of the boronate complex and establishment of the extensive water–protein binding network appear specific to the binding orientation of the N-phenyl series as addition of the benzoxaborole group to active analogs in the other series (i.e., 1, 21, 31) resulted in a substantial loss in potency versus the GT 1a 316Y replicon (SI-2-4, see the Supporting Information).
Two of the three approaches to eliminating benzylic oxidation were successful in retaining the antiviral activity, and the most active leads were evaluated in PK. Although the oxazolidinone series (e.g., 25) in general displays poor rat PK, both the heteroaryl and N-phenyl series show improvement (Table 5). The heteroaryl analogs 31 and 33 have lower clearance and >10-fold higher oral exposure in rats relative to 1, whereas the N-phenyl derivatives 47 and 49 show ∼sevenfold improvements in oral AUC. The lowered clearance in both series is particularly gratifying because it serves to validate our hypothesis for removing benzylic oxidation. Additionally, there is no in vivo or in vitro evidence of metabolite 3 being formed from any of the backup compounds. The major route of metabolism appears to proceed via oxidative-deborylation to form the corresponding phenol metabolite (data not shown).
There is a tendency for both backup series from approaches B and C to inhibit CYP isoform 2C9 which poses a potential safety issue involving medicines with narrow therapeutic indices such as warfarin or phenytoin.30 The drug–drug interaction risk is highest with the heteroaryl derivatives although opportunities to attenuate this activity may exist given the tolerance for N-, O-, and C-substitution, and the diversity of heteroaromatic systems available. Benzoxaborole 49 is fortunately inactive for CYP inhibition and shows a >10-fold improvement in hERG activity31 versus the free boronic acid 47 for reasons that are not clear.
The predicted human PK for 1 was complicated by significant differences in IV clearance between species (high in monkey, low in mouse). As a result, allometric scaling was conducted with 2 species (rat and dog) which ultimately underpredicted human metabolism (predicted t1/2 ≈ 15 h vs observed t1/2 ≈ 5 h).12 We sought to avoid this risk by identifying a candidate with low blood clearance across preclinical species. As expected, removal of the benzylic carbon improves in vivo clearance in mouse, rat, dog, and monkey for 49 versus 1. We were particularly pleased that the monkey clearance of 49 is >eightfold lower than 1 because post hoc analysis had highlighted that monkey was the most accurate of the preclinical species in predicting the poor human PK of 1. Benzoxaborole 49 is also stable in human cryopreserved hepatocytes showing little metabolism after 2 h of incubation while 1 was rapidly metabolized (t1/2 ≈ 1 h, see the Supporting Information). Allometric scaling for 49 was completed using four preclinical species, and a dose of 65 mg q.d. was predicted to maintain steady-state trough plasma concentrations (Cτ) above the protein-adjusted EC90 for the GT 1b 316N replicon (Cτ > 117 ng/mL) over 24 h. In contrast, a dose of 1100 mg q.d. was predicted for compound 1 using the same criterion.
6. Conclusions
Identification of metabolites produced from 1 in human subjects exposed the metabolic liability responsible for poor PK that formed the basis of the backup strategy. Guided by SAR and structure-based design, we devised three synthetic approaches to prevent metabolism that occurred via benzylic oxidation and subsequent C–N bond cleavage. Two of the approaches delivered structurally distinct chemotypes with low in vivo rat clearance and robust anti-HCV profiles. The potent inhibition of GT1a 316Y replicon with N-phenyl boronic acids was particularly exciting as one analog, benzoxaborole 49, successfully met our criteria for a clinical candidate.
Our hypothesis assumed that reducing benzylic oxidation would improve human PK and lower doses required for efficacy relative to 1. The first-time-in-human study with 49 validated our proposal, as the plasma half-life ranged from 60 to 63 h in HCV-infected subjects, exceeding predictions.32 No formation of metabolite 3 was observed and >90% of parent compound was recovered unchanged in feces. To the best of our knowledge, 49 has the longest plasma half-life of any boron-containing compound reported in human subjects, including the marketed drug bortezomib.33 Compound 49 has completed phase 2 clinical trials for treating HCV infection, the results of which will be reported in due course. We hope that our experience encourages medicinal chemists to consider incorporating boron within drug design, an area that we believe remains underdeveloped.34
7. Experimental Section
7.1. HCV Stable and Transient Replicon Assays
Experimental protocols and method for EC50 determination are provided in the Supporting Information.
7.2. Animals
All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed.
7.3. Humans
The human biological samples were sourced ethically, and their research use was in accordance with the terms of the informed consents under an IRB/EC-approved protocol.
7.4. Compound Purity and Identity
The purity of the final compounds was determined to be ≥95% by 1H NMR and LCMS (unless otherwise noted). Structural assignments were consistent with the spectroscopic data. All final compounds underwent the Immediate Processing Quality Control (IPQC) protocol upon submission to GSK’s centralized compound depository and prior to biological testing as an additional confirmation of identity and purity (listed in the Supporting Information). 1H NMR spectra were taken on a Varian (Agilent) Inova 400 NMR spectrometer. Mass spectrometric analyses and compound purity determinations were conducted on a Waters Acquity UPLC system (Phenomenex Kinetex column at 40 °C, mobile phase of water with 0.2% v/v formic acid and acetonitrile with 0.15% v/v formic acid) and Waters Acquity SQD with alternating positive/negative electrospray ionization scanning from 125 to 1000 amu, with a scan time of 105 ms and an interscan delay of 20 ms. High-resolution mass spectrometric analysis was performed on a Waters qTOF Premiere mass spectrometer using flow injection operating in W mode. Chiral analytical HPLC was performed on an Agilent 1100 analytical system.
7.5. Synthesis of Benzoxaborole (49)19
7.5.1. Methyl 5-(N-(5-Cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)-2-nitrobenzoate (40)
A mixture of 5-cyclopropyl-2-(4-fluorophenyl)-N-methyl-6-(methylsulfonamido)benzofuran-3-carboxamide (3,7 55.0 g, 123 mmol), methyl 5-fluoro-2-nitrobenzoate (36.7 g, 184 mmol), and Na2CO3 (39.1 g, 369 mmol) in DMF (400 mL) was heated to 70 °C with vigorous stirring. After 72 h, LCMS indicated nearly a complete reaction. The mixture was cooled to RT, diluted with EtOAc (300 mL), and filtered through a bed of Celite to remove solids. The filter cake was washed with EtOAc until the filtrate ran colorless which gave a total filtrate volume of 1.2 L. The filtrate was transferred to a separatory funnel, partitioned with 1.4 L of 5% aqueous NaCl, and the phases were separated. The aqueous solution was extracted with two additional 300 mL portions of EtOAc. The combined EtOAc solutions were washed with 5% aqueous NaCl (2×), saturated aqueous NaCl (1×), dried over Na2SO4, and concentrated to approximately 200 mL by rotary evaporation. At this point, a solid began to crystallize. The suspension was diluted with 200 mL of DCM and the suspension stirred overnight. The suspension was then cooled in an ice water bath for 2 h, and the solid was collected by vacuum filtration. The filter cake was washed twice with cold 1:1 EtOAc/DCM, suction air dried for 30 min, and then dried in vacuo overnight to afford the title compound (59.6 g, 83%) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.52 (q, J = 4.42 Hz, 1H), 8.13 (d, J = 9.07 Hz, 1H), 8.08 (s, 1H), 7.93–8.01 (m, 2H), 7.57 (d, J = 2.63 Hz, 1H), 7.53 (dd, J = 9.07, 2.73 Hz, 1H), 7.38–7.46 (m, 2H), 7.29 (s, 1H), 3.83 (s, 3H), 3.60 (s, 3H), 2.85 (d, J = 4.59 Hz, 3H), 1.87–2.04 (m, 1H), 0.87 (m, 2H), 0.73 (m, 1H), 0.46 (m, 1H). ES-LCMS m/z: 582 (M + 1).
7.5.2. Methyl 2-Amino-5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)benzoate (41)
A stirred suspension of methyl 5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)-2-nitrobenzoate (40, 59.5 g, 102 mmol) and 10% Pd/C (6.00 g) in 2:1 THF/EtOH (1.2 L) was saturated with hydrogen by bubbling hydrogen gas through for 10 min and then subjected to hydrogenation (1 atm) at RT. After 24 h, the mixture was purged with nitrogen, the catalyst was removed by filtration through Celite, and the filtrate was concentrated to dryness at reduced pressure to give the title compound (55.2 g, 98%) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.46 (q, J = 4.42 Hz, 1H), 8.16 (s, 1H), 7.91–8.00 (m, 3H), 7.61 (dd, J = 8.98, 2.73 Hz, 1H), 7.41 (t, J = 8.88 Hz, 2H), 7.15 (s, 1H), 6.76–6.86 (m, 3H), 3.79 (s, 3H), 3.28 (s, 3H), 2.83 (d, J = 4.68 Hz, 3H), 2.25–2.38 (m, 1H), 0.82–1.10 (m, 3H), 0.42 (m, 1H). ES-LCMS m/z: 552 (M + 1).
7.5.3. Methyl 2-Amino-3-chloro-5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)benzoate
A suspension of methyl 2-amino-5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)benzoate (41, 55.0 g, 100 mmol) in DMF (320 mL) was heated to 60 °C. The resulting yellow solution was treated with NCS (14.0 g, 105 mmol) in one portion. The solution quickly darkened. After 5 min, LCMS indicated complete conversion of the starting material to the desired chlorinated compound. The solution was cooled to RT and diluted with EtOAc (1 L). The resulting solution was washed with 5% aqueous NaCl (1 × 1 L), 5% aqueous sodium bisulfite (2 × 200 mL), saturated aqueous NaHCO3 (3 × 200 mL), and saturated aqueous NaCl (1 × 200 mL). The solution was dried over Na2SO4 and concentrated to dryness at reduced pressure to afford the title compound (58.0 g, 99%) as a brown solid. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (q, J = 4.42 Hz, 1H), 8.25 (s, 1H), 7.94–8.04 (m, 3H), 7.90 (d, J = 2.63 Hz, 1H), 7.34–7.46 (m, 2H), 7.18 (s, 1H), 6.93 (br s, 2H), 3.83 (s, 3H), 3.33 (s, 3H), 2.84 (d, J = 4.59 Hz, 3H), 2.26–2.36 (m, 1H), 0.84–1.11 (m, 3H), 0.40 (br s, 1H). ES-LCMS m/z: 586 (M + 1).
7.5.4. Methyl 2-Bromo-3-chloro-5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)benzoate
To a 1 L 3-necked flask equipped with a magnetic stirrer was added methyl 2-amino-3-chloro-5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)benzoate (58.0 g, 99.0 mmol) followed by MeCN (500 mL) and 48% aqueous HBr (500 mL). The dark brown solution was cooled to 0 °C in an ice water/brine bath and treated with a solution of NaNO2 (8.20 g, 119 mmol) in water (50 mL) over a 5 min period. After 30 min, LCMS indicated complete consumption of the starting material. The solution was then treated with CuBr (18.5 g, 129 mmol) over 2 min and warmed to 50 °C. After 30 min at 50 °C, LCMS indicated complete conversion to the desired bromo compound. The solution was cooled to RT and partitioned between EtOAc (1 L) and water (1.5 L). The phases were separated, and the aqueous solution was extracted with EtOAc (2 × 200 mL). The combined EtOAc solutions were washed with 5% aqueous NaCl (1 × 1 L), 5% aqueous sodium bisulfite (2 × 300 mL), saturated aqueous NaHCO3 (2 × 300 mL), saturated aqueous NaCl (1 × 300 mL), and dried over Na2SO4. The drying agent was removed by filtration through a pad of silica gel, and the filtrate was concentrated to dryness at reduced pressure to afford the title compound (64.3 g, quantitative) as a reddish-brown foam. The crude product was carried forward without further purification. 1H NMR (400 MHz, DMSO-d6): δ 8.50 (q, J = 4.29 Hz, 1H), 8.16 (s, 1H), 7.92–8.03 (m, 2H), 7.79 (d, J = 2.73 Hz, 1H), 7.65 (d, J = 2.73 Hz, 1H), 7.41 (t, J = 8.93 Hz, 2H), 7.24 (s, 1H), 3.81–3.89 (m, 3H), 3.45–3.53 (m, 3H), 2.85 (d, J = 4.59 Hz, 3H), 2.04–2.16 (m, 1H), 0.71–1.07 (m, 3H), 0.40 (br s, 1H). ES-LCMS m/z: 649 (M + 1).
7.5.5. 6-(N-(4-Bromo-3-chloro-5-(hydroxymethyl)phenyl)methylsulfonamido)-5-cyclopropyl-2-(4-fluorophenyl)-N-methylbenzofuran-3-carboxamide
A solution of methyl 2-bromo-3-chloro-5-(N-(5-cyclopropyl-2-(4-fluorophenyl)-3-(methylcarbamoyl)benzofuran-6-yl)methylsulfonamido)benzoate (54.3 g, 84.0 mmol) in anhydrous THF (351 mL) and anhydrous MeOH (39 mL) was cooled in an ice water/brine bath to −5 °C (internal temperature). To the stirred solution was added 2 M LiBH4/THF (125 mL, 250 mmol) via addition funnel at a rate so as to maintain the temperature below 5 °C. The addition required 35 min. The brine bath was then replaced with an ice water bath, and the stirring of the solution continued. After another 1.5 h, LCMS indicated the complete reaction. The solution was diluted with saturated aqueous NaHCO3 (200 mL) followed by water (400 mL) and then EtOAc (600 mL). The mixture was stirred vigorously for 30 min and then transferred to a separatory funnel. After the phases separated, the solid remained in the aqueous phase; therefore, an additional 200 mL portion of water was added and the mixture was shaken, and the phases again separated. The aqueous solution was extracted with EtOAc (2 × 200 mL). The combined EtOAc solutions were washed with 5% aqueous NaCl (1×), saturated aqueous NaCl (1×), dried over Na2SO4, and concentrated to dryness at a reduced pressure to afford the title compound (54.5 g, quantitative) as an orange-brown foam. The crude material was carried forward to the next step without purification. 1H NMR (400 MHz, DMSO-d6): δ 8.45–8.54 (m, 1H), 8.12 (s, 1H), 7.98 (dd, J = 8.93, 5.41 Hz, 2H), 7.59 (d, J = 2.73 Hz, 1H), 7.54 (d, J = 2.63 Hz, 1H), 7.41 (t, J = 8.88 Hz, 2H), 7.22 (s, 1H), 5.64 (t, J = 5.56 Hz, 1H), 4.48 (d, J = 5.56 Hz, 2H), 3.45 (s, 3H), 2.84 (d, J = 4.59 Hz, 3H), 2.06–2.17 (m, 1H), 0.75–1.06 (m, 3H), 0.47 (br s, 1H). ES-LCMS m/z: 623 (M + 1).
7.5.6. 6-(N-(4-Bromo-3-chloro-5-((methoxymethoxy)methyl)phenyl)methylsulfonamido)-5-cyclopropyl-2-(4-fluorophenyl)-N-methylbenzofuran-3-carboxamide (43)
A solution of 6-(N-(4-bromo-3-chloro-5-(hydroxymethyl)phenyl)methylsulfonamido)-5-cyclopropyl-2-(4-fluorophenyl)-N-methylbenzofuran-3-carboxamide (50.0 g, 80.0 mmol) in THF (500 mL) was treated with DIEA (42.1 mL, 241 mmol) followed by MOM-Cl (15.3 mL, 201 mmol). The resulting solution was heated to 50 °C under stirring. After 18h, LCMS indicated the complete reaction. The solution was cooled to RT and diluted with EtOAc (600 mL) followed by water (600 mL). After stirring vigorously for 10 min, the mixture was transferred to a separatory funnel, and the phases were separated. The aqueous phase was extracted with EtOAc (1 × 200 mL). The combined EtOAc solutions were washed with an aqueous solution of 5% citric acid and 5% NaCl (3 × 300 mL), saturated aqueous NaHCO3 (2 × 300 mL), saturated brine (1 × 300 mL), and dried over Na2SO4. The drying agent was removed by filtration, and the filtrate concentrated to dryness at a reduced pressure to give an orange-brown foam. This material was dissolved in EtOAc (250 mL), and the solution was heated to reflux with stirring. To the solution was added hexane (375 mL) over a 5 min period maintaining reflux temperature. The solution was then allowed to cool to RT under stirring during which time a light tan solid was crystallized. After 2 h, the solution was cooled in an ice water bath and stirred for an additional 2 h. The solid was collected by filtration in a medium-fritted funnel. The filter cake was washed with cold 3:2 hexane/EtOAc (250 mL), dried by suction filtration for 30 min, and then dried in vacuo to afford the title compound (36.4 g, 68% over 3 steps) as a light tan solid. 1H NMR (400 MHz, DMSO-d6): δ 8.49 (q, J = 4.30 Hz, 1H), 8.14 (s, 1H), 7.93–8.00 (m, 2H), 7.62 (d, J = 2.74 Hz, 1H), 7.52 (d, J = 2.74 Hz, 1H), 7.40 (t, J = 8.94 Hz, 2H), 7.21 (s, 1H), 4.68 (s, 2H), 4.57 (s, 2H), 3.44 (s, 3H), 3.24 (s, 3H), 2.83 (d, J = 4.59 Hz, 3H), 2.06–2.19 (m, 1H), 0.74–1.05 (m, 3H), 0.44 (br s, 1H). ES-LCMS m/z: 665 (M + 1).
7.5.7. 6-(N-(7-Chloro-1-hydroxy-1,3-dihydrobenzo[c][1,2]oxaborol-5-yl)methylsulfonamido)-5-cyclopropyl-2-(4-fluorophenyl)-N-methylbenzofuran-3-carboxamide (49)
A mixture of 6-(N-(4-bromo-3-chloro-5-((methoxymethoxy)methyl)phenyl)methylsulfonamido)-5-cyclopropyl-2-(4-fluorophenyl)-N-methylbenzofuran-3-carboxamide (46.4 g, 69.7 mmol), bis(pinacolato)diboron (44.2 g, 174 mmol), KOAc (27.4 g, 279 mmol), and Pd(dppb)Cl2 (2.10 g, 3.48 mmol) in 1,4-dioxane (350 mL) was sparged with nitrogen for 15 min and then heated to 108 °C under nitrogen. After 22 h, LCMS showed the complete conversion of starting material to an 84:16 mixture of the desired product/protio by-product (replacement of bromine by hydrogen). The mixture was cooled to RT, combined with the crude reaction mixture from a 1 g scale pilot reaction and diluted with 500 mL of EtOAc. The mixture was filtered through a pad of silica gel to remove solids, and the filtrate was concentrated at reduced pressure to give a light tan solid. This material was dissolved in THF (437 mL), and the solution was diluted with MeOH (87 mL) followed by 1 M aqueous HCl (440 mL). A tan solid rapidly precipitated. The mixture was heated to 70 °C. The solid slowly dissolved affording a yellow solution. After 18 h, LCMS indicated the complete reaction. The solution was cooled to approximately 40 °C and poured into a rapidly stirred mixture of water (1 L) and MTBE (1 L). To the mixture was added 1 M aqueous NaOH (440 mL). The pH was then adjusted to approximately 12 by addition of 3 M aqueous NaOH. The mixture was transferred to a separatory funnel, and the phases were separated. The aqueous phase was washed with MTBE (3 × 250 mL) and then treated with concentrated HCl to a pH of approximately 2. The resulting cloudy solution was extracted with EtOAc (4 × 500 mL). The combined EtOAc extracts were washed with 5% aqueous NaCl (1×), saturated aqueous NaCl (1×), and dried over Na2SO4. The drying agent was removed by filtration through a pad of Celite. The light-yellow filtrate was concentrated to dryness at reduced pressure to give a tan foam (39.0 g). This material was dissolved in MeCN (400 mL). The solution was stirred with a slow addition of 0.1 M aqueous HCl (800 mL) via an addition funnel over a 40 min period. Early in the addition, the solution was seeded with a small amount of previously prepared, crystalline 49 which induced vigorous crystallization. The suspension was stirred overnight at RT. The solid was collected by vacuum filtration rinsing with 2:1 MeCN/water. The material was suction air dried for 1h and then dried to constant weight in vacuo to afford 49 (28.8 g, 71%) as an off-white powder. 1H NMR (400 MHz, DMSO-d6): δ 9.18 (s, 1H), 8.50 (q, J = 4.42 Hz, 1H), 8.09 (s, 1H), 7.93–8.02 (m, 2H), 7.37–7.46 (m, 3H), 7.28 (d, J = 1.56 Hz, 1H), 7.22 (s, 1 H), 4.97 (s, 2H), 3.48 (s, 3H), 2.85 (d, J = 4.59 Hz, 3H), 2.02–2.14 (m, 1H), 0.92–1.04 (m, 1H), 0.82 (br s, 2H), 0.49 (br s, 1H). ES-LCMS m/z: 569 (M + 1), 100% purity. HRMS m/z: calcd for C27H23BClFN2O6S, 569.1121; found, 569.1121.
Acknowledgments
The author would like to thank GlaxoSmithKline scientists Iain Reid for pKa measurements and Dr. Katja Remlinger for the statistical input associated with this manuscript.
Glossary
Abbreviations
- HCV
hepatitis C virus
- NS5B
nonstructural protein 5B
- WHO
World Health Organization
- DAA
direct acting antiviral
- GT
genotype
- DILI
drug-induced liver injury
- WT
wild type
- PDB
Protein Data Bank
- PS-BBA
polymer-supported benzene boronic acid
- SFC
supercritical fluid chromatography
- VCD
vibrational circular dichroism
- SNAr
nucleophilic aromatic substitution
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01719.
Purities of final compounds; chemical syntheses of final compounds; EC50/pEC50 determination methods and replicon assay protocols; replicon data for final compounds and SI-1-4; in vivo/in vitro DMPK profiling; VCD analysis; crystal structures and RCSB ID/PDB ID numbers; potentiometric pKa determination protocol; and pH solubility profile of compound 49 (PDF)
Molecular formula strings for final compounds (CSV)
Author Present Address
∥ Abbvie, 1 North Waukegan Road, North Chicago, IL, 60064.
Author Present Address
⊥ Open Eye Scientific Software, Inc., 9 Brisbee Ct, Santa Fe, NM, 87507.
Author Present Address
# Gilead Sciences, 333 Lakeview Drive, Foster City, CA, 94404.
Author Present Address
¶ BioCryst Pharmaceuticals Inc., 4505 Emperor Blvd., Durham, NC 27703.
Author Present Address
∇ PRA Health Sciences, 4130 Parklake Ave, Raleigh, NC, 27612.
Author Present Address
○ KBI Biopharma, 1101 Hamlin Road, Durham, NC, 27704.
The authors declare the following competing financial interest(s): All authors are current or former employees of GlaxoSmithKline.
Notes
The coordinates for the corresponding X-ray crystal structures reported in this paper have been deposited with RCSB. Authors will release the atomic coordinates and experimental data upon article publication. The PDB codes listed in figure legends are as follows: Figures 3 and 4a (4KAI); Figure 4b (6MVK), 4c (6MVQ), 4d (6MVP), and 5 (6MVO).
Supplementary Material
References
- Choo Q.; Kuo G.; Weiner A.; Overby L.; Bradley D.; Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- Kwong A. D. The HCV revolution did not happen overnight. ACS Med. Chem. Lett. 2014, 5, 214–220. 10.1021/ml500070q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating G. M. Ledipasvir/Sofosbuvir: a review of its use in chronic hepatitis C. Drugs 2015, 75, 675–685. 10.1007/s40265-015-0381-2. [DOI] [PubMed] [Google Scholar]
- Łucejko M.; Parfieniuk-Kowerda A.; Flisiak R. Ombitasvir/paritaprevir/ritonavir plus dasabuvir combination in the treatment of chronic HCV infection. Expert Opin. Pharmacother. 2016, 17, 1153–1164. 10.1080/14656566.2016.1176143. [DOI] [PubMed] [Google Scholar]
- WHO Progress report on access to hepatitis C treatment: focus on overcoming barriers in low- and middle-income countries. 2018 (WHO/CDS/HIV/18.4) http://www.who.int/hepatitis/publications/hep-c-access-report-2018/en/ (accessed Oct 2018).
- WHO Combating hepatitis B and C to reach elimination by 2030. 2016 http://apps.who.int/iris/bitstream/handle/10665/206453/WHO_HIV_2016.04_eng.pdf;jsessionid=E4E17821B997F1608838F6E6AA64498E?sequence=1 (accessed Oct, 2018).
- a Johns B. A.; Shotwell J. B.. Therapeutic Compounds. WO 2011103063 A1, 2011.; b Johns B. A.; Shotwell J. B.; Haigh D.. Compounds. WO 2012067663 A1, 2012.
- Maynard A.; Crosby R. M.; Ellis B.; Hamatake R.; Hong Z.; Johns B. A.; Kahler K. M.; Koble C.; Leivers A.; Leivers M. R.; Mathis A.; Peat A. J.; Pouliot J. J.; Roberts C. D.; Samano V.; Schmidt R. M.; Smith G. K.; Spaltenstein A.; Stewart E. L.; Thommes P.; Turner E. M.; Voitenleitner C.; Walker J. T.; Waitt G.; Weatherhead J.; Weaver K.; Williams S.; Wright L.; Xiong Z. Z.; Haigh D.; Shotwell J. B. Discovery of a potent boronic acid derived inhibitor of the HCV RNA-dependent RNA polymerase. J. Med. Chem. 2014, 57, 1902–1913. 10.1021/jm400317w. [DOI] [PubMed] [Google Scholar]
- Voitenleitner C.; Crosby R.; Walker J.; Remlinger K.; Vamathevan J.; Wang A.; You S.; Johnson J.; Woldu E.; Van Horn S.; Horton J.; Creech K.; Shotwell J. B.; Hong Z.; Hamatake R. In VitroCharacterization of GSK2485852, a Novel Hepatitis C Virus Polymerase Inhibitor. Antimicrob. Agents Chemother. 2013, 57, 5216–5224. 10.1128/aac.00874-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneteman N. M.; Howe A. Y. M.; Gao T.; Lewis J.; Pevear D.; Lund G.; Douglas D.; Mercer D. F.; Tyrrell D. L. J.; Immermann F.; Chaudhary I.; Speth J.; Villano S. A.; O’Connell J.; Collett M. HCV796: A selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 2009, 49, 745–752. 10.1002/hep.22717. [DOI] [PubMed] [Google Scholar]
- Yeung K.-S.; Beno B. R.; Parcella K.; Bender J. A.; Grant-Young K. A.; Nickel A.; Gunaga P.; Anjanappa P.; Bora R. O.; Selvakumar K.; Rigat K.; Wang Y.-K.; Liu M.; Lemm J.; Mosure K.; Sheriff S.; Wan C.; Witmer M.; Kish K.; Hanumegowda U.; Zhuo X.; Shu Y.-Z.; Parker D.; Haskell R.; Ng A.; Gao Q.; Colston E.; Raybon J.; Grasela D. M.; Santone K.; Gao M.; Meanwell N. A.; Sinz M.; Soars M. G.; Knipe J. O.; Roberts S. B.; Kadow J. F. Discovery of a hepatitis C virus NS5B replicase palm site allosteric inhibitor (BMS-929075) advanced to phase 1 clinical studies. J. Med. Chem. 2017, 60, 4369–4385. 10.1021/acs.jmedchem.7b00328. [DOI] [PubMed] [Google Scholar]
- Wilfret D. A.; Walker J.; Voitenleitner C.; Baptiste-Brown S.; Lovern M.; Kim J.; Adkison K.; Shotwell B.; Mathis A.; Moss L.; Lee D.; Yu L.; Gan J.; Spaltenstein A. A randomized, double blind, dose escalation, first time in human study to assess the safety, tolerability, pharmacokinetics, and antiviral activity of single doses of GSK2485852 in chronically infected hepatitis C subjects. Clin. Pharmacol. Drug Dev. 2014, 3, 439–448. 10.1002/cpdd.142. [DOI] [PubMed] [Google Scholar]
- Feldstein A.; Kleiner D.; Kravetz D.; Buck M. Severe hepatocellular injury with apoptosis induced by a hepatitis C polymerase inhibitor. J. Clin. Gastroenterol. 2009, 43, 374–381. 10.1097/mcg.0b013e318178d91f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. S.; Lin G.; Yang J. W. Oxidative N-debenzylation of N-benzyl-N-substituted benzylamines catalyzed by cytochrome P450. Bull. Korean Chem. Soc. 2004, 25, 249–252. 10.5012/bkcs.2004.25.2.249. [DOI] [Google Scholar]
- Labutti J.; Parsons I.; Huang R.; Miwa G.; Gan L.-S.; Daniels J. S. Oxidative deboronation of the peptide boronic acid proteasome inhibitor bortezomib: contributions from reactive oxygen species in this novel cytochrome P450 reaction. Chem. Res. Toxicol. 2006, 19, 539–546. 10.1021/tx050313d. [DOI] [PubMed] [Google Scholar]
- Virtual compounds were constructed in Maestro and manually overlaid with the conformation of 1 (coordinates obtained from the cocrystal structure of HCV NS5B GT 1b C316N with 1, PDB Code: 4KAI). Illustrated minimized conformations represent local minima optimized from this overlaid conformation using OPLS3, an implicit aqueous solvent model, and default minimization parameters in Macromodel. Maestro and Macromodel (release 2017-4) are products of Schrödinger, LLC.
- Images Generated with the PyMol Molecular Graphics System, version 1.7.6.6, Schrödinger, LLC, 2002.
- Chiral separation via SFC was performed on intermediate 17 to avoid issues of deborylation associated with 18. See Supporting Information for details related to the instrument, chiral column, and solvent conditions that afforded enantiomers 25 and 26 in >98% enantiomeric excess (ee), respectively.
- Chong P. Y.; Miller J. F.; Peat A. J.; Shotwell J. B.. Benzofuran compounds for the treatment of hepatitis c virus infections. WO 2013028371 A1, 2013.
- For review, see:Lam P. Y. S.Chan–Lam coupling reaction: copper-promoted C-element bond oxidative coupling reaction with boronic acids. In Synthetic Methods in Drug Discovery; Blakemore D. C., Doyle P. M., Fobian Y. M., Eds.; Royal Society of Chemistry: Cambridge, 2016; pp 242–273. [Google Scholar]
- Demont E. H.; Redshaw S.; Walter D. S.. Hydroxyethylamine Compounds Having asp2 Inhibitory Activity for the Treatment of Alzheimer’s Disease. WO 2004080376 A2, 2004.; b Eickmeier C.; Fuchs K.; Peter S.; Dorner-Ciossek C.; Heine N.; Handschuh S.; Klinder K.; Kostka M.. Substituted 1,2-Ethylendiamines, Medicaments Comprising Said Compound; Their Use and Their Method of Manufacture. WO2006103038 A1, 2006.
- Chan D. M. T.; Monaco K. L.; Wang R.-P.; Winters M. P. New N- and O-arylations with phenylboronic acids and cupric acetate. Tetrahedron Lett. 1998, 39, 2933–2936. 10.1016/s0040-4039(98)00503-6. [DOI] [Google Scholar]
- Reaction optimization during clinical development identified conditions for Chan-Lam N-arylation coupling of an electron-deficient aryl boronic ester with the 2°-sulfonamide of a derivative of 3 (carboxylic acid in place of N-Me carboxamide).Vantourout J. C.; Li L.; Bendito-Moll E.; Chabbra S.; Arrington K.; Bode B. E.; Isidro-Llobet A.; Kowalski J. A.; Nilson M. G.; Whellhouse K. M. P.; Woodard J. L.; Xie S.; Leitch D. C.; Watson A. J. B. Mechanistic insight enables practical, scalable, room temperature Chan-Lam N-arylation of N-aryl sulfonamides. ACS Catal. 2018, 8, 9560–9566. 10.1021/acscatal.8b03238. [DOI] [Google Scholar]
- Kowalski J. A.; Leitch D. C.; Allen C. L.; Arrington K.; Barcan G. A.; Calandra N.; Erickson G. A.; Li L.; Liu L.; Nilson M. G.; Strambeanu I. I.; VanGelder K.; Woodard J. L.; Xie S.. Convergent synthesis of the NS5B inhibitor GSK8175 enabled by transition metal catalysis. J. Org. Chem. 10.1021/acs.joc.8b02269 [DOI] [PubMed] [Google Scholar]
- Experience working with boron-containing compounds in our laboratories suggests a trend towards greater metabolic stability and chemical stability, with respect to protodeborylation in solid and solution states, for benzoxaborole versus phenylboronic acid derivatives.
- Hepatic blood flow values for various species used in calculation of % Liver Blood Flow (126, 77.6, 56.1 and 44 mL/min/kg) for mouse (0.02 kg), rat (0.25 kg), dog (10 kg) and cynomolgus monkey (5 kg) are the values accepted internally by GlaxoSmithKline. For cynomolgus monkey, the parameter was taken fromDavies B.; Morris T. Physiological parameters in laboratory animals and humans. Pharm. Res. 1993, 10, 1093–1095. 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- a Windsor I. W.; Palte M. J.; Lukesh J. C.; Gold B.; Forest K. T.; Raines R. T. Sub-picomolar inhibition of HIV-1 protease with a boronic acid. J. Am. Chem. Soc. 2018, 140, 14015–14018. 10.1021/jacs.8b07366. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Freund Y. R.; Akama T.; Alley M. R. K.; Antunes J.; Dong C.; Jarnagin K.; Kimura R.; Nieman J. A.; Maples K. R.; Plattner J. J.; Rock F.; Sharma R.; Singh R.; Sanders V.; Zhou Y. Boron-based phosphodiesterase inhibitors show novel binding of boron to PDE4 bimetal center. FEBS Lett. 2012, 19, 3410–3414. 10.1016/j.febslet.2012.07.058. [DOI] [PubMed] [Google Scholar]
- pKa was determined by acid/base titration in a water/MeOH mixture using potentiometric measurements. Details provided in Supporting Information.
- Tomsho J. W.; Pal A.; Hall D. G.; Benkovic S. J. Ring structure and aromatic substituent effects on the pKa of the benzoxaborole pharmacophore. ACS Med. Chem. Lett. 2012, 3, 48–52. 10.1021/ml200215j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Food & Drug Administration . Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm (accessed Oct 2018).
- Activity versus the human ether-a-go-go (hERG) ion channel was assessed in a patch clamp electrophysiology assay that measures the reduction in current induced by the test compound. The IC50 for compounds 47 and 49 were determined to be 1.6 μM and 16 μM, respectively. For additional information see:Webster R.; Leishman D.; Walker D. Towards a drug concentration effect relationship for QT prolongation and torsade de pointes. Curr. Opin. Drug Discovery Dev. 2002, 5, 116–126. [PubMed] [Google Scholar]
- Gardner S. D.; Kim J.; Baptiste-Brown S.; Lopez V.; Hamatake R.; Gan J.; Edwards S.; Elko-Simms L.; Dumont E. F.; Leivers M.; Hong Z.; Paff M. T. GSK2878175, A pan-genotypic non-nucleoside NS5B polymerase inhibitor, in healthy and treatment-naïve chronic hepatitis C subjects. J. Viral Hepatitis 2018, 25, 19–27. 10.1111/jvh.12753. [DOI] [PubMed] [Google Scholar]
- Papandreou C. N.; Daliani D. D.; Nix D.; Yang Y.; Madden T.; Wang X.; Pien C. S.; Millikan R. E.; Tu S.-M.; Pagliaro L.; Kim J.; Adams J.; Elliott P.; Esseltine D.; Petrusich A.; Dieringer P.; Perez C.; Logothetis C. J. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J. Clin. Oncol. 2004, 22, 2108–2121. 10.1200/jco.2004.02.106. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Supuran C. T.; Winum J.-Y. Benzoxaborole compounds for therapeutic uses: a patent review (2010-2018). Expert Opin. Ther. Pat. 2018, 28, 493–504. 10.1080/13543776.2018.1473379. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.