

Abstract
STARD3 is a cellular protein that represents an attractive target for cancer therapy, being overexpressed in breast cancer and implied in the development of colorectal, gastric, and prostate cancers. Unfortunately, no STARD3 inhibitor has been identified yet. In this work, an in silico strategy was applied to predict a reliable binding mode of cholesterol into STARD3 and to develop a pharmacophore-based virtual screening protocol that allowed the identification of the first STARD3 inhibitor ever reported. The identified compound VS1 binds STARD3 with micromolar affinity (IC50 = 35 μM) and shows antiproliferative activity in breast (MCF7 and MDA- MB-231) and colon (HCT-116) cancer cell lines in the same concentration range (IC50 = 49.7–105.5 μM). Although VS1 has a moderate potency, we demonstrated that it specifically targets STARD3 in the cells and induces its degradation. Overall, the results confirm the reliability of the computational strategies herein applied and the identification of the first hit compound for the development of novel potent STARD3 inhibitors.
Keywords: STARD3 inhibitor, virtual screening, breast and colon cancers
Although many advances in the cancer treatment have been made, scientists are continuously looking for new approaches in order to provide the best possible outcome for all patients. An emerging challenge is the identification of new genes involved in cancer development and progression in order to develop new therapeutic molecules to be used alone or in combination with current therapies. In the last years, some research groups have focused their attention on a protein initially found to be overexpressed in breast cancer: the StAR-related lipid transfer domain containing 3 (STARD3).1 STARD3 is a member of a subfamily of lipid trafficking proteins characterized by a C-terminal steroidogenic acute regulatory domain, which shares a 35% of homology with the domain of the StAR protein STARD1, a transporter of cholesterol in mitochondria. They both belong to the StAR-related lipid-transfer (START) domain proteins superfamily, involved in the nonvesicular transport of lipids in membranes.2 In humans, 15 different proteins contain the START domain, and based on their sequence homology, they are categorized into six different subfamilies, each one sharing similar ligand binding specificities.
Although the functions and structures of the members of this family are not totally clarified, the importance of several START proteins in cancer is well reported.3,4 Even if its role has not been clarified yet, STARD3 was found to be located in the late endosomal membranes and specifically involved in cholesterol transport, transferring such sterol from the endoplasmic reticulum to the endosomes, or vice versa.5 It was demonstrated that STARD3 is overexpressed in different human cancer cell lines, particularly in HER2-overexpressing breast cancer.6 In fact, STARD3 and HER2 are coamplified and co-overexpressed in about 25% of breast cancers.7 The molecular mechanism by which STARD3 cooperates with HER2 is still unclear. Furthermore, this gene is implicated in therapy resistance of breast cancer, and patients with a high level of STARD3 expression had metastasis, local recurrence, and shorter overall survival.8,9 This feature has been supported by STARD3 silencing in HER2 positive breast cancer cell lines, which induces the reduction of cell viability and increases cell death,10 reinforcing the idea that STARD3 can be implicated in the intratumoral steroidogenesis and cancer progression. Recently, new evidence suggest a possible involvement of STARD3 also in colorectal, prostate, and gastric cancers.11−13 High levels of STARD3 were found in tubular and papillary adenocarcinoma cells, suggesting that STARD3 affects cholesterol metabolism in gastric cancer tissues by increasing cholesterol transport to mitochondria and consequently activating steroidogenesis.13 In neoplastic prostate tissue, STARD3 is coexpressed with CYT7, another gene involved in androgen synthesis. Even if the link is not clear, STARD3 expression seems to be correlated with high stage, high Gleason score and short relapse-free time in the prostate cancer patients.
Due to its involvement in cancer, STARD3 is an attractive candidate as a target for cancer therapy, and the identification of selective STARD3 inhibitors is an interesting but yet unexplored field of study. For this reason, we aimed at developing a virtual screening (VS) study focused on the identification of new inhibitors of the STARD3 mediated cholesterol transport. In 2000, Tsujishita and co-workers reported the first X-ray structure of the START domain of STARD3 protein solved in the apo form. The crystal structure revealed the presence of a wide hydrophobic pocket able to accommodate only one molecule of cholesterol. Furthermore, by performing titration experiments, they also proved that the STARD3 domain binds cholesterol at 1:0.8 ratio.14 Recently, a new high-resolution X-ray structure of the apo domain has been solved by Horvarth and co-workers;15 nevertheless, no clear data about the binding disposition of cholesterol within STARD3 binding site were reported in literature until now. In this study, a computational protocol including consensus docking, molecular dynamic simulations, and binding free energy evaluations was employed to predict a reliable binding mode of cholesterol into STARD3. The results allowed the development of a receptor-based pharmacophore screening that led to the identification of a hit compound (VS1) endowed with an interesting inhibitory activity. The specificity of the inhibitor was evaluated by measuring the stability of STARD3 and its target protein, the focal adhesion kinase (FAK). Furthermore, the effects of VS1 were evaluated in breast and colorectal cancer cell lines, demonstrating its antiproliferative activity.
As a first step, we performed an in-depth docking evaluation to predict a reliable binding mode for cholesterol into the STARD3 binding site. A consensus docking (CD) procedure was thus employed for this task since our previous studies highlighted the capability of the CD approach to predict ligand binding poses better than single docking evaluations.16,17 Through this approach, a ligand is docked into the protein target by multiple docking methods, and the best-ranked poses generated by the different docking procedures are clustered together to search for common binding modes. The docking pose that shows the highest consensus level represents the final predicted binding mode, as it is considered more reliable than the other poses. Based on these considerations, 12 available docking procedures were employed to dock cholesterol into the STARD3 binding site, and the 12 obtained docking poses were then clustered together using an RMSD cutoff of 2.0 Å. As shown in Figure S1 in the Supporting Information, the CD approach identified four clusters of poses with a comparable consensus level, each corresponding to a different hypothetical binding mode. In this case, the CD protocol seemed not able to identify a particularly reliable binding mode among those predicted by the different docking methods. This can be probably due to the fact that the protocol was applied on the X-ray structure of an apo protein, instead of using a receptor extracted from a cocrystal structure as in our previous analyses and prospective VS studies.17,18 This could affect the quality of the CD protocol, making the identification of the most reliable binding mode more challenging. However, it is worth noting that the approach was still able to identify a preferential disposition of cholesterol within STARD3 since in three out of the four different clusters of poses (10 out of 12 docking poses) the ligand shared the same orientation into the protein binding site, with the hydroxyl group pointing toward the inner side of the cavity. Nevertheless, all predicted binding modes were analyzed through molecular dynamic (MD) simulations. A representative docking pose belonging to each cluster of poses obtained by CD (poses C1–C4) was subjected to a 20 ns MD simulation with explicit water. By analyzing the RMSD of cholesterol’s position during the simulation with respect to the starting pose C1, we observed an average RMSD of 1.2 Å, suggesting a remarkably stable binding mode. A different behavior was observed for the ligand in poses C2 and C3 since it underwent a certain deviation from its initial conformation, as shown by the RMSD plots in Figure S2 (see Supporting Information). A detailed analysis revealed that, in both cases, cholesterol moved from its starting position and unexpectedly converged on pose C1. Notably, this movement allowed the formation of an H-bond between the hydroxyl group of the molecule and residue S362. This specific H-bond appeared to be important for cholesterol binding to the STARD3 domain, as suggested by previous studies.19 The H-bond analysis of the MD trajectories confirmed the strength of this interaction since the hydrogen bond was kept for 89% and 72% of the whole simulation by cholesterol in poses C2 and C3, respectively. Considering that in C1 the same H-bond was maintained for 93% of the MD simulation, this analysis further evidenced the fast convergence of poses C2 and C3 into C1, probably guided by the formation of the ligand–protein interaction with S362. On the contrary, this H-bond was not observed for the ligand in pose C4 since the hydroxyl group of the molecule was placed in the opposite side of the binding pocket with respect to S362 (Figure S1D). The C1 and C4 binding modes were thus further analyzed through the molecular mechanics and Poisson–Boltzmann surface area (MM-PBSA) method, which has shown to reliably estimate the ligand–protein interaction energy.20,21 This approach averages the contribution of solvation free energy and gas phase energy for snapshots of the ligand–protein complex and the unbound components extracted from MD trajectories. The results of the MM-PBSA analysis suggested pose C1 as the most favorable binding mode since it showed an interaction energy (ΔPBSA = −48.6 kcal/mol) that was about 8 kcal/mol lower than that estimated for the binding mode C4 (Table S1 in the Supporting Information).
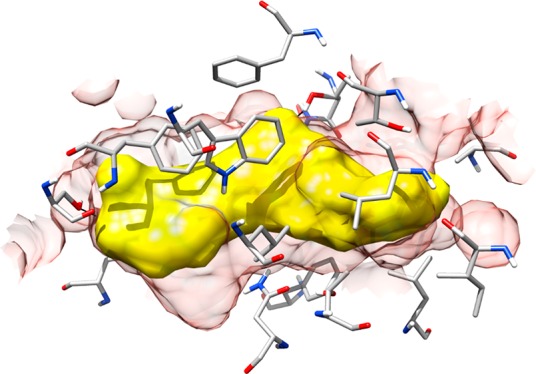
The results obtained from these analyses suggested the MD-refined C1 pose as the most reliable binding disposition of cholesterol within STARD3 binding site. As shown in Figure 1A, cholesterol is localized in the L-shaped binding pocket of STARD3 and forms a large number of hydrophobic interactions. In particular, the phenanthrene core of the molecule interacts with the residues located in the central portion of the binding site, mainly delimited by V314, N349, F388, W404, and L406, whereas the terminal aliphatic chain interacts with the hydrophobic residues at the top of the cavity, i.e., A337, F347, and L410. Finally, the ligand forms the H-bond with S362, maintained for almost the whole MD simulation. The obtained binding mode seemed to be in agreement with the one proposed by Murcia and co-workers in 2006 as the most likely cholesterol orientation biologically admitted into the STARD3 binding domain.19 Therefore, a VS protocol based on the key cholesterol–STARD3 interactions was developed. A structure-based pharmacophore model was built taking into account the main interactions established by cholesterol in the predicted binding mode into the STARD3 binding cavity. In particular, we considered (a) the H-bond between the hydroxyl group and S362, (b) the lipophilic interactions of the phenanthrene core, and (c) the hydrophobic interactions of the terminal aliphatic chain (Figure 1B). The pharmacophore model was generated by means of the software LigandScout22 and included a double H-bond donor/acceptor feature (1) representing the cholesterol hydroxyl group interacting with S362, two hydrophobic features (2, 3) describing the interactions of cholesterol phenanthrene core, and other two hydrophobic features (4, 5) representing the terminal aliphatic chain of the molecule (Figure 2B). Additionally, the model was refined by adding excluded volume spheres mimicking the steric hindrance of the STARD3 binding site. The pharmacophore model was used to filter the Enamine database, comprising 1 782 240 commercially available compounds. Only compounds matching at least four out of five pharmacophore features and respecting the imposed volume constraints were retrieved. The double H-bond donor/acceptor feature was set as obligatory in order to identify molecules that could form H-bonds with S362, while the four hydrophobic features were set as optional. Applying these filters, 5456 compounds were selected and subjected to docking studies. Recent results suggested that CD could be profitably applied in VS studies. In fact, this approach not only performed as well as the best available docking methods found in literature, but it also allowed us to experimentally identify new active compounds.18,23
Figure 1.
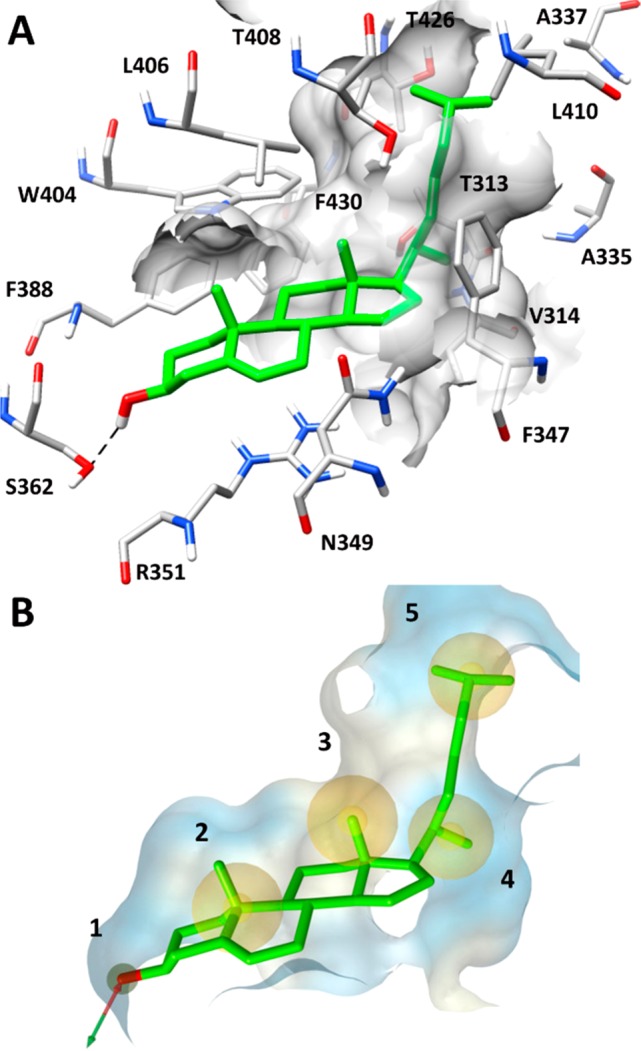
(A) Minimized average structure of cholesterol into the STARD3 binding site obtained from MD simulation and (B) the corresponding structure-based pharmacophore model used for the VS study.
Figure 2.
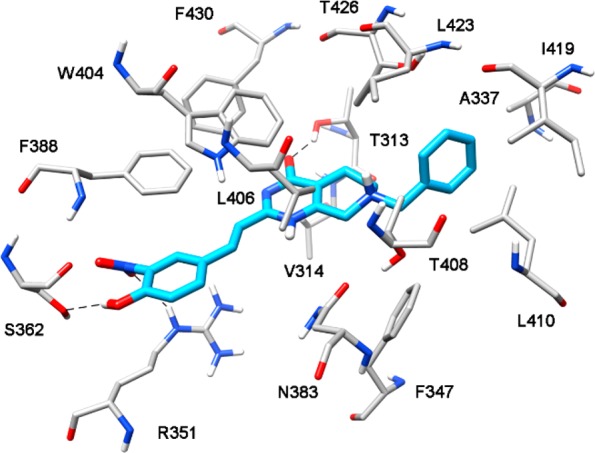
Minimized average structure of compound VS1 bound to STARD3 binding site.
On these bases, the 5456 molecules selected through the pharmacophore search were subjected to a CD protocol including the 12 docking procedures already used in this study. For each molecule, the 12 docking poses predicted by the 12 docking methods were clustered together to search for common binding modes. As reported in Table S2, none of the docked compounds reached a consensus level above 8; in fact, only two compounds achieved consensus level 8, ten compounds showed consensus 7, and 30 molecules obtained a consensus level of 6. However, these results are in agreement with previous CD studies reported in literature, which highlighted the high level of selectivity of this procedure.17 Based on the CD results, only the 42 compounds with a minimum consensus level of 6 were considered as potential active ligands and further analyzed. The visual analysis of the binding mode predicted for the 42 potential STARD3 inhibitors identified by the CD procedure revealed that only 12 compounds formed the H-bond interaction with S362 in their docking pose. Therefore, these 12 compounds were subjected to MD simulations to verify the stability of their predicted binding mode. The same MD protocol applied to cholesterol was employed in this step; therefore, the 12 different ligand–protein complexes were subjected to 20 ns of MD simulation, which were then analyzed in terms of H-bond stability and ligand RMSD with respect to the starting coordinates.
Only compounds showing an average RMSD value lower than 2.0 Å and maintaining the H-bond interaction with S362 for more than 70% of the whole simulation were retained. We applied the RMSD threshold of 2.0 Å because it was already used in previous VS studies;23−25 differently, the threshold of 70% for the H-bond with S362 was used because in the three MD simulations in which cholesterol assumed the binding mode corresponding to the pharmacophore model (see above), the H-bond with SS36 was maintained for at least 70% of the simulations. Moreover, the same threshold was recently applied for identifying new LDH inhibitors.26 By applying these filters, eight compounds were rejected, while the remaining four potential ligands were purchased and tested for STARD3 inhibitory activity through a competition binding fluorescent assay. Taking into consideration the binding of cholesterol to STARD3 with a stoichiometry of 0.8:1 and the concentration of protein and substrate utilized in the assay developed by Tsujishita and Hurley,14 we carried out a binding titration curve of STARD3 (Figures S3 and S4). Then, a concentration of 5 μM of STARD3 was selected (55.9% of completely bound state, which represented the optimal assay conditions). The IC50 values obtained for the four tested compounds are reported in Table 1. Among all compounds, VS1 was the only one able to inhibit the interaction of cholesterol with an IC50 of 35.0 ± 4.7 μM.
Table 1. Structure and Activity of the Tested Compounds.

Figure 2 shows the binding mode of compound VS1 into the STARD3 binding site. The para-hydroxyl group on the styryl moiety of the ligand forms a stable hydrogen bond with S362, whereas the nitro group in meta position on the same ring shows an H-bond interaction with R351. The central bicyclic portion of the ligand interacts with the residues delimiting the binding cavity on the two sides, i.e., V314, W404, L406, and F430, while the carbonyl group at C-4 forms an H-bond with T313 side chain. Interestingly, this additional interaction was found to be quite stable since it was maintained for almost 70% of the whole MD simulation. Finally, the benzyl moiety of the ligand mimics the terminal aliphatic chain of cholesterol, filling the small hydrophobic cavity mainly defined by A337, T408, L410, I419, L423, and T426.
Interestingly, the comparison between the interaction of VS1 and that of VS2-VS4 highlights that only VS1 forms H-bonds with T313, R351, and S362. In fact, compound VS2 does not form any hydrogen bond with R351, whereas compounds VS3 and VS4 do not show the interaction with T313 (see Figure S11 in the Supporting Information). These evidence suggest a possible connection between the triple H-bond network and the inhibitory activity observed for VS1.
To examine the potential therapeutic effects of STARD3 inhibition, VS1 was tested in cancer cells. Breast (MCF7 and MDA-MB-231) and colon (HCT-116) cancer cell lines were selected as models.6,8,11 As reported in Table 2, VS1 showed an interesting activity, demonstrating to inhibit cell viability of all cell lines in the micromolar range.
Table 2. Antiproliferative Activity of VS1 on Representative Breast and Colon Cancer Cell Lines.
| cell line | IC50 (μM) |
|---|---|
| MCF7 | 105.5 ± 10.4 |
| MDA-MB-231 | 49.7 ± 6.8 |
| HCT-116 | 70.7 ± 4.5 |
To determine the effectiveness of cytotoxic agents, the survival of a single cancer cell could be tested through a clonogenic assay.27 For this purpose, MCF7, MDA-MB-231, and HCT-116 cell lines were treated with different concentrations of VS1 and grown until colonies were visible. Results shown in Figure 3A demonstrated that VS1 could impair the growth of cancer cells in a dose-dependent manner. To confirm the specificity of this result, STARD3 was knocked down with two shRNAs. Both shRNAs inhibited the growth of cancer cells, confirming that VS1 is a valid inhibitor of STARD3 (Figure 3B).
Figure 3.

(A) Cells were treated with different doses of VS1 or (B) with two specific shRNAs for STARD3. Colony forming efficiency was evaluated by visual inspection.
It has been reported that high affinity or covalent inhibitors induce degradation of target protein.28,29 Since NIH3T3 cell line tolerated well the protein synthesis inhibitor cycloheximide (CHX),29VS1 was used in combination with CHX. This assay allows to follow the kinetics of protein degradation. Compared to control, VS1 accelerated the degradation of STARD3 protein in CHX-treated cells starting after 12 h (Figure 4A). Moreover, the mRNA level of STARD3 was unaffected by the treatment (Figure 4B).
Figure 4.

Compound VS1 decreases STARD3 protein stability. (A) NIH3T3 fibroblasts were treated with 100 μM of compound VS1 for 24 h followed by 10 μg/mL of CHX for the indicated time (hours). (B) STARD3 mRNA levels: cells were treated for 48 h at two different concentrations of VS1. The inhibitor does not affect the mRNA levels of target. (C) MDA-MB-231 cell line was treated with 100 μM of compound VS1 for 24 and 48 h. Western blot analysis shows that the level of FAK and pTyr397-FAK increase in treated cells.
Finally, since FAK was demonstrated to be regulated by STARD3 in MDA-MB-231 cells,8 we evaluated the levels of FAK and pTyr397-FAK in VS1-treated MDA-MB-231 cells. The results confirmed that inhibition of STARD3 by VS1 increased the level of FAK and pTyr397-FAK (Figure 4C).
In conclusion, we developed a VS study aimed at the identification of potential STARD3 inhibitors. A receptor-based pharmacophore screening combined with CD and MD simulations allowed us to identify four promising compounds predicted to interact within the STARD3 binding domain through a strong H-bond with S362. A competition binding fluorescent assay performed for testing the inhibitory activity of the selected compounds revealed an IC50 of 35.0 μM for compound VS1. The biological activity of VS1 was thus evaluated by treating two breast cancer cell lines (MCF7 and MDA- MB-231) and a colon cancer cell line (HCT-116) with different concentrations of the inhibitor. Results showed a promising antitumoral activity of VS1 on all cell lines, with IC50 values ranging from 49.7 to 105.5 μM. The reduction of cancer cell proliferation induced by VS1 was also confirmed through clonogenic assays. Even if further investigations are needed to test the efficacy and the safety of this molecule, compound VS1 represents the first available STARD3 inhibitor endowed with a relevant biological activity in cancer cell lines. Although the IC50 of VS1 on STARD3 is in the micromolar range, the induction of STARD3 degradation produced by the ligand suggests a potent and specific activity of VS1 at cellular level. Pretreatment of NIH3T3 fibroblast cells with VS1, followed by treatment with protein synthesis inhibitor CHX, induced the reduction of STARD3 levels over time, suggesting a potent activity of VS1 on STARD3. Moreover, in agreement with literature data, Western blot analysis confirmed the implication of STARD3 in FAK pathway, as shown by the higher levels of FAK and pTyr397-FAK in VS1-treated MDA-MB-231 cells, with respect to untreated cells. In this study, only one compound was identified as a STARD3 inhibitor, and the obtained results were not corroborated by any close analogs of this compound. However, due to the absence of similar commercially available compounds, to better explore the SAR of this potential class of inhibitors the synthesis of new derivatives is ongoing, and they will be reported in due course.
Acknowledgments
F.R. and I.C. are thankful to Associazione Italiana per la Ricerca sul Cancro (AIRC).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00509.
Supplementary figures, tables, and experimental details (PDF)
Author Contributions
The manuscript was written through contributions of all authors.
This work was funded by MyFirst AIRC (15639).
The authors declare no competing financial interest.
Supplementary Material
References
- Tomasetto C.; Régnier C.; Moog-Lutz C.; Mattei M. G.; Chenard M. P.; Lidereau R.; Basset P.; Rio M. C. Identification of Four Novel Human Genes Amplified and Overexpressed in Breast Carcinoma and Localized to the Q11-Q21.3 Region of Chromosome 17. Genomics 1995, 28 (3), 367–376. 10.1006/geno.1995.1163. [DOI] [PubMed] [Google Scholar]
- Alpy F. Give Lipids a START: The StAR-Related Lipid Transfer (START) Domain in Mammals. J. Cell Sci. 2005, 118 (13), 2791–2801. 10.1242/jcs.02485. [DOI] [PubMed] [Google Scholar]
- Yuan B. Z.; Miller M. J.; Keck C. L.; Zimonjic D. B.; Thorgeirsson S. S.; Popescu N. C. Cloning, Characterization, and Chromosomal Localization of a Gene Frequently Deleted in Human Liver Cancer (DLC-1) Homologous to Rat RhoGAP. Cancer Res. 1998, 58 (10), 2196–2199. [PubMed] [Google Scholar]
- King S. R.; Bhangoo A.; Stocco D. M. Functional and Physiological Consequences of StAR Deficiency: Role in Lipoid Congenital Adrenal Hyperplasia. Endocr. Dev. 2011, 20, 47–53. 10.1159/000321214. [DOI] [PubMed] [Google Scholar]
- Wilhelm L. P.; Wendling C.; Védie B.; Kobayashi T.; Chenard M.; Tomasetto C.; Drin G.; Alpy F. STARD3Mediates Endoplasmic Reticulum-to-endosome Cholesterol Transport at Membrane Contact Sites. EMBO J. 2017, 36 (10), 1412–1433. 10.15252/embj.201695917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev B.; Sihto H.; Li S.; Hölttä-Vuori M.; Ilola J.; Lundin J.; Isola J.; Kellokumpu-Lehtinen P.-L.; Joensuu H.; Ikonen E. Elevated Levels of StAR-Related Lipid Transfer Protein 3 Alter Cholesterol Balance and Adhesiveness of Breast Cancer Cells: Potential Mechanisms Contributing to Progression of HER2-Positive Breast Cancers. Am. J. Pathol. 2015, 185 (4), 987–1000. 10.1016/j.ajpath.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Dressman M. A.; Baras A.; Malinowski R.; Alvis L. B.; Kwon I.; Walz T. M.; Polymeropoulos M. H. Gene Expression Profiling Detects Gene Amplification and Differentiates Tumor Types in Breast Cancer. Cancer Res. 2003, 63 (9), 2194–2199. [PubMed] [Google Scholar]
- Cai W.; Ye L.; Sun J.; Mansel R. E.; Jiang W. G. Expression of MLN64 Influences Cellular Matrix Adhesion of Breast Cancer Cells, the Role for Focal Adhesion Kinase. Int. J. Mol. Med. 2010, 25 (4), 573–580. 10.3892/ijmm_00000379. [DOI] [PubMed] [Google Scholar]
- Vinatzer U.; Dampier B.; Streubel B.; Pacher M.; Seewald M. J.; Stratowa C.; Kaserer K.; Schreiber M. Expression of HER2 and the Coamplified Genes GRB7 and MLN64 in Human Breast Cancer: Quantitative Real-Time Reverse Transcription-PCR as a Diagnostic Alternative to Immunohistochemistry and Fluorescence in Situ Hybridization. Clin. Cancer Res. 2005, 11 (23), 8348–8357. 10.1158/1078-0432.CCR-05-0841. [DOI] [PubMed] [Google Scholar]
- Kao J.; Pollack J. R. RNA Interference-Based Functional Dissection of the 17q12 Amplicon in Breast Cancer Reveals Contribution of Coamplified Genes. Genes, Chromosomes Cancer 2006, 45 (8), 761–769. 10.1002/gcc.20339. [DOI] [PubMed] [Google Scholar]
- Uhlen M.; Zhang C.; Lee S.; Sjöstedt E.; Fagerberg L.; Bidkhori G.; Benfeitas R.; Arif M.; Liu Z.; Edfors F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science (Washington, DC, U. S.) 2017, 357 (6352), eaan2507 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Stigliano A.; Gandini O.; Cerquetti L.; Gazzaniga P.; Misiti S.; Monti S.; Gradilone A.; Falasca P.; Poggi M.; Brunetti E.; et al. Increased Metastatic Lymph Node 64 and CYP17 Expression Are Associated with High Stage Prostate Cancer. J. Endocrinol. 2007, 194 (1), 55–61. 10.1677/JOE-07-0131. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Zhang Z.-Y.; Du W.-D.; Ye L.; Xu S.; Zuo X.-B.; Zhou F.-S.; Chen G.; Ma X.-L.; Schneider M. E.; et al. Association Analysis of ERBB2 Amplicon Genetic Polymorphisms and STARD3 Expression with Risk of Gastric Cancer in the Chinese Population. Gene 2014, 535 (2), 225–232. 10.1016/j.gene.2013.11.030. [DOI] [PubMed] [Google Scholar]
- Tsujishita Y.; Hurley J. H. Structure and Lipid Transport Mechanism of a StAR-Related Domain. Nat. Struct. Biol. 2000, 7 (5), 408–414. 10.1038/75192. [DOI] [PubMed] [Google Scholar]
- Horvath M. P.; George E. W.; Tran Q. T.; Baumgardner K.; Zharov G.; Lee S.; Sharifzadeh H.; Shihab S.; Mattinson T.; Li B.; et al. Structure of the Lutein-Binding Domain of Human StARD3 at 1.74 Å Resolution and Model of a Complex with Lutein. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72 (8), 609–618. 10.1107/S2053230X16010694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuccinardi T.; Poli G.; Romboli V.; Giordano A.; Martinelli A. Extensive Consensus Docking Evaluation for Ligand Pose Prediction and Virtual Screening Studies. J. Chem. Inf. Model. 2014, 54 (10), 2980–2986. 10.1021/ci500424n. [DOI] [PubMed] [Google Scholar]
- Poli G.; Martinelli A.; Tuccinardi T. Reliability Analysis and Optimization of the Consensus Docking Approach for the Development of Virtual Screening Studies. J. Enzyme Inhib. Med. Chem. 2016, 31, 167–173. 10.1080/14756366.2016.1193736. [DOI] [PubMed] [Google Scholar]
- Chiarelli L. R.; Mori M.; Barlocco D.; Beretta G.; Gelain A.; Pini E.; Porcino M.; Mori G.; Stelitano G.; Costantino L.; et al. Discovery and Development of Novel Salicylate Synthase (MbtI) Furanic Inhibitors as Antitubercular Agents. Eur. J. Med. Chem. 2018, 155, 754–763. 10.1016/j.ejmech.2018.06.033. [DOI] [PubMed] [Google Scholar]
- Murcia M.; Faráldo-Gómez J. D.; Maxfield F. R.; Roux B. Modeling the Structure of the StART Domains of MLN64 and StAR Proteins in Complex with Cholesterol. J. Lipid Res. 2006, 47 (12), 2614–2630. 10.1194/jlr.M600232-JLR200. [DOI] [PubMed] [Google Scholar]
- Milella L.; Milazzo S.; De Leo M.; Vera Saltos M. B.; Faraone I.; Tuccinardi T.; Lapillo M.; De Tommasi N.; Braca A. α-Glucosidase and α-Amylase Inhibitors from Arcytophyllum Thymifolium. J. Nat. Prod. 2016, 79 (8), 2104–2112. 10.1021/acs.jnatprod.6b00484. [DOI] [PubMed] [Google Scholar]
- Poli G.; Lapillo M.; Granchi C.; Caciolla J.; Mouawad N.; Caligiuri I.; Rizzolio F.; Langer T.; Minutolo F.; Tuccinardi T. Binding Investigation and Preliminary Optimisation of the 3-Amino-1,2,4-Triazin-5(2 H)-One Core for the Development of New Fyn Inhibitors. J. Enzyme Inhib. Med. Chem. 2018, 33 (1), 956–961. 10.1080/14756366.2018.1469017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45 (1), 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- Poli G.; Giuntini N.; Martinelli A.; Tuccinardi T. Application of a FLAP-Consensus Docking Mixed Strategy for the Identification of New Fatty Acid Amide Hydrolase Inhibitors. J. Chem. Inf. Model. 2015, 55 (3), 667–675. 10.1021/ci5006806. [DOI] [PubMed] [Google Scholar]
- Poli G.; Gelain A.; Porta F.; Asai A.; Martinelli A.; Tuccinardi T. Identification of a New STAT3 Dimerization Inhibitor through a Pharmacophore-Based Virtual Screening Approach. J. Enzyme Inhib. Med. Chem. 2016, 31 (6), 1011–1017. 10.3109/14756366.2015.1079184. [DOI] [PubMed] [Google Scholar]
- Tuccinardi T.; Poli G.; Corchia I.; Granchi C.; Lapillo M.; Macchia M.; Minutolo F.; Ortore G.; Martinelli A. A Virtual Screening Study for Lactate Dehydrogenase 5 Inhibitors by Using a Pharmacophore-Based Approach. Mol. Inf. 2016, 35 (8–9), 434–439. 10.1002/minf.201501026. [DOI] [PubMed] [Google Scholar]
- Poli G.; Scarpino A.; Aissaoui M.; Granchi C.; Minutolo F.; Martinelli A.; Tuccinardi T. Identification of Lactate Dehydrogenase 5 Inhibitors Using Pharmacophore- Driven Consensus Docking. Curr. Bioact. Compd. 2018, 14 (2), 197–204. 10.2174/1573407213666170208102317. [DOI] [Google Scholar]
- Rizzolio F.; Lucchetti C.; Caligiuri I.; Marchesi I.; Caputo M.; Klein-Szanto A. J.; Bagella L.; Castronovo M.; Giordano A. Retinoblastoma Tumor-Suppressor Protein Phosphorylation and Inactivation Depend on Direct Interaction with Pin1. Cell Death Differ. 2012, 19 (7), 1152–1161. 10.1038/cdd.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J. C.; Gollapalli D. R.; Hedstrom L. Inhibitor Mediated Protein Degradation. Chem. Biol. 2012, 19 (5), 629–637. 10.1016/j.chembiol.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo Spena C.; De Stefano L.; Palazzolo S.; Salis B.; Granchi C.; Minutolo F.; Tuccinardi T.; Fratamico R.; Crotti S.; D’Aronco S.; et al. Liposomal Delivery of a Pin1 Inhibitor Complexed with Cyclodextrins as New Therapy for High-Grade Serous Ovarian Cancer. J. Controlled Release 2018, 281, 1–10. 10.1016/j.jconrel.2018.04.055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.