

Abstract
The application of class I HDAC inhibitors as cancer therapies is well established, but more recently their development for nononcological indications has increased. We report here on the generation of improved class I selective human HDAC inhibitors based on an ethylketone zinc binding group (ZBG) in place of the hydroxamic acid that features the majority of HDAC inhibitors. We also describe a novel set of HDAC3 isoform selective inhibitors that show stronger potency and selectivity than the most commonly used HDAC3 selective tool compound RGFP966. These compounds are again based on an alternative ZBG with respect to the ortho-anilide that is featured in HDAC3 selective compounds reported to date.
Keywords: Histone deacetylase, HDAC3 selective, nonhydroxamate, nonbenzamide
Cellular phenotypes are ultimately controlled by both the cell’s genomic DNA sequences and epigenetic factors that have no effect on gene sequence but that do impact gene expression. Epigenetic regulation mechanisms are driven by chemical transformations that structurally modify either DNA itself or the histone proteins around which DNA is packaged in the nucleus of eukaryotic cells.1 Two enzymes strongly involved in post-translational modification of proteins are histone deacetylases (HDACs) and histone acetyl transferase (HATs). Of the numerous substrates that these enzymes process, their interplay in controlling the acetylation level of lysine residues in the N-terminal tail of histones is of particular importance.2 Histone acetylation diminishes the charge–charge interactions between nucleic acids in DNA and the positively charged lysine residues in histones and is a key requisite for relaxation of chromatin structure and gene transcription. Favoring histone acetylation, as is expected by inhibition of HDACs, therefore, promotes gene-expression, and HDAC inhibitors continue to nurture extensive interest in drug discovery due to their broad impact on cellular events.
Class I HDACs are predominantly located in the nucleus and are key players in regulating gene expression and cellular differentiation. Class I HDAC inhibitors were originally developed as anticancer agents,3 and to date four HDACis have been approved for the treatment of cutaneous and peripheral T-cell lymphomas and multiple myeloma.4 However, the finding that many cancers and most normal cells are relatively resistant to HDACi induced protein hyperacetylation has resulted in a trend toward exploration of HDACis for noncancer indications. While clinical trials initiated prior to 2015 were almost exclusively focused on their use as antiproliferative agents in cancer, ca.70% of post-2015 clinical studies aim to exploit positive pharmacological effects induced by HDACis rather than their (ultimately) cytotoxic properties.5 At tolerated doses, HDAC inhibitors can still induce gene-expression changes making their use in cases where therapeutic benefit may come from altering a target gene’s expression of interest. One example is the use of HDACi for the treatment of Duchenne muscular dystrophy (DMD)6 where HDAC inhibition is thought to limit the fibro-fatty differentiation of fibro-adipocytic precursors (FAPs) and stimulate the regeneration of muscles by acting on muscle resident satellite cells. Immunological diseases7 such as encephalomyelitis and rheumatoid arthritis, where HDAC enzymes play a key role in T-cell function, are further examples where inhibitors may prove beneficial.
Despite the therapeutic potential of HDACis, concerns about their use (particularly for class I HDACis) as chronic treatments persist due to known on- and off-target undesired effects. Consequently the development of isoform selective HDAC inhibitors has become the focus of intense recent activity. Subtype-selective HDACis offer the potential for selective pharmacological effects while managing the general mechanism based toxicity associated with HDAC inhibition.8 Among class I enzymes the development of HDAC3 selective inhibitors9 is an attractive goal. It is known that the accessory proteins required to direct HDAC3 activity are different to those used by HDACs 1–2,10 suggesting that its functions may differ and be associated with specific effects. HDAC3 has been associated with circadian cycle mediated metabolism,11 inflammation,12 and CNS diseases.13 Moreover, in addition to its nuclear function HDAC3 is also found in the cytoplasm14 suggesting additional roles in post-translationally modifying cytoplasmic targets. HDAC3 activity (though not the entire protein) was shown to be dispensable in HDAC3 inactive mice15 suggesting that on-target toxicity might be of limited relevance.
We have previously16 reported on the discovery of what we believe to be the first series of bona fide selective Plasmodium falciparum HDAC inhibitors (e.g., compound 1, Figure 1) that effectively impeded the growth of malaria parasites in erythrocytes. In tandem with this work (which aimed to avoid human HDAC inhibition), we also focused on developing improved human class I selective and isoform selective inhibitors, predominantly with a view to their use as therapeutic agents for nononcological indications. Here, we describe how SAR from our work toward PfHDAC inhibitors was used in guiding these efforts. We report an evolution of class I HDAC inhibitors (e.g., compound 2, Figure 1) to provide a new set of potent and highly bioavailable inhibitors of human HDACs 1–3. Our profiling of compound 6, a preclinical development candidate that demonstrated proof-of-concept in an mdx mouse model for Duchenne muscular dystrophy,17 is included. In addition, efforts employing information from our PfHDAC (and pan-class I HDACi) work to generate isoform selective inhibitors is described. This led to the discovery of new HDAC3 inhibitors with improved potency and isoform selectivity with respect to benzamide analogs such as RGFP966.18
Figure 1.

PfHDAC selective and human class I isoform selective HDAC inhibitors.
Our past work to generate selective PfHDACis began by screening a structurally divergent subset of our human HDAC inhibitor collection on a P. falciparum parasite proliferation assay in erythrocytes. An early hit was compound 3, an oxazole analog of known19 alkyl-ketone substituted imidazoles (e.g., 4) that were themselves designed in our laboratories from apicidin.20 Replacement of the central imidazole ring in compounds like 4 with alternative five-membered aromatic heterocycles (data not shown) reduced human HDAC1 activity by 5–10-fold for 5-aryl-2-alkyl-1,3,4-oxadiazoles, ca. 10-fold for 5-aryl-2-alkyloxazoles and by 50-fold or more for other heterocycles (triazole, N-alkylimidazole, or 1,2,4-oxadiazoles). The oxazole based compound 3 was therefore considered an appealing starting point with a view to favoring PfHDAC vs human HDAC inhibition, and two additional oxazole based analogs (5–6) were prepared using known SAR from the imidazole series as a guide. Changes to the naphthalene ring (Ar = A in Table 1) and/or to the amide functionality (R in Table 1) were immediately introduced to avoid the high metabolic turnover in liver microsomes that had been a feature of early imidazole compounds.19
Table 1. HDAC1 Inhibition and Cellular Class I HDAC Inhibition for Oxazole Based HDACis.

HDAC1 IC50 (see Supporting Information for full data with standard deviations).
HeLa cellular class I HDAC EC50.
See ref (19).
Compounds 5 and 6 ultimately failed to show relevant selectivity in the P. falciparum parasite growth assay (with respect to human HDAC assays) and were not further pursued as an avenue toward antiparasitic agents. Intriguingly, however, we observed that 2-methoxyquinoline compound 6 showed stronger than anticipated human HDAC inhibition. While the relative potency of the naphthyl substituted oxazole-imidazole matched pair 5 vs 7 followed the expected trend (around 10-fold weaker potency for the oxazole analog), this was reversed for the 2-methoxyquinoline analogs 6 vs 2. Testing of further 2-methoxyquinoline substituted oxazole analogs (e.g., compounds 8 and 9) confirmed that, in the presence of the 2-methoxyquinoline substituent as the aromatic group, the oxazole series was robust in generating strong human class I HDAC inhibition (Table 1). Compounds 6, 8, and 9 had IC50s below 3.5 nM against HDAC isoforms 1–3 and were potent inhibitors of class I HDAC’s in our HeLa cellular assay. Notably the cellular potency of 9 is almost an order of magnitude stronger than previously reported inhibitors from the related imidazole class (e.g., 2).
Compound 6 was chosen for further profiling by virtue of its structural analogy with the optimized imidazole 2. Biochemical studies demonstrated that 6 showed potent inhibition of the class I enzymes HDAC1, 2, and 3 (IC50 of 1.7, 2.8, and 1.1 nM, respectively). Strong selectivity against class II HDAC isoforms was measured with 6 having an IC50 against HDAC6 of 177 nM and proving inactive at 5 μM against HDACs 4–7. In vitro studies showed that 6 caused no inhibition of metabolic enzymes from the cytochrome P450 (CYP) family with IC50s against CYP isoforms 3A4, 2D6, 2C9, and 1A2, all being >20 μM. Since hydroxamic acid HDACis are typically positive in Ames tests, compound 6 was tested against two strains of bacteria (Salmonella typhimurium and Escherichia coli). No reverse mutations were detected, highlighting an attractive Ames-negative genotoxicity profile for 6. Interestingly although the MPO2 score21 (a recent molecular property based predictor for identifying CNS permeable compounds) for 6 is 4.1 (slightly lower than the preferred value (of 5) for probable brain penetrant compounds), it showed medium-high permeability in a porcine brain endothelial cell blood–brain barrier (BBB) assay (P = 10.5 × 10–6 cm/s).22 The apparent BBB permeability for 6 suggests that it may provide a path to higher CNS levels than hydroxamic-acid based class I HDACis. The in vivo pharmacokinetic profile for compound 6 is summarized in Table 2. In rat 6 showed 100% bioavailability with a 3.3 h plasma half-life and moderate (20 mL/min/kg) plasma clearance. In line with its structure, 6 was found to be metabolized by a number of oxidative and dealkylative transformations, but no metabolites were found circulating in rat plasma after sampling 4 h postdose. High oral bioavailability (62%) was also measured in mouse where plasma half-life and clearance were 3.3 h and 13 mL/min/kg, respectively. Modest mouse exposure was a characteristic of previously reported imidazole based compounds, meaning i.p. dosing proved necessary as the administration route for compound 2 in a human HCT116 colon carcinoma mouse xenograft study.19 The mouse profile for 6 was clearly significantly improved, with a 5 mg/kg oral dose generating an AUC of 9.3 μM·h, 10-fold higher than 2 (AUC 0.9 μM·h). Changing the central scaffold from an imidazole to an oxazole (which replaces a hydrogen bond donor with a hydrogen bond acceptor) improves the PK profile likely by increasing absorption/membrane permeability. Oral bioavailability was retained in larger animals (minipig) where bioavailability was 30% in spite of a potentially significant liver first pass.
Table 2. In Vivoa PK Parameters for Compound 6.
| species | F (%) | Clp (mL/min/kg) | Vdss (L/kg) | t1/2 (h) | AUCpob (μM–h) |
|---|---|---|---|---|---|
| mouse | 62 | 13 | 3.2 | 3.3 | 9.3 |
| rat | 100 | 20 | 5.3 | 3.3 | 21.5 |
| minipig | 30 | 20 | 0.64 | 3.7 | 2.6 |
Compound 6 was dosed as a tartrate salt.
Plasma exposure after oral administration of 20, 10, and 5 mg/kg doses in mouse, rat, and minipig, respectively. Data are normalized linearly downward to 5 mg/kg.
An off-target activity for imidazole based compounds such as 2 was hERG potassium ion channel binding (IC50 of ca. 3 μM) and in our hERG assay 6 showed a similar IC50 of 3.6 μM. In a functional (patch-clamp) assay the IC50 for 6 was 10 μM. In contrast, no significant binding (IC50 > 16 μM or >30 μM) was measured on a panel of additional ion channels including the sodium NaV1.5, calcium CaV1.2, and four further K+ channels. The presence of hERG activity for 6 was not viewed as a hurdle for the advancement of this compound. Based on 6’s free fraction in human plasma (1.8%), therapeutic doses should result in unbound compound concentrations significantly below the hERG IC50. Nonetheless, the observation of hERG binding for 6 (as well as for a number of further oxazole based analogs on our program) led us to explore approaches to dial out hERG binding from this compound series (Table 3). Chemistry to modulate the moderately basic and lipophilic properties of 6 (calculated23 pKa 7.65, calculated logD 3.09) were explored since the combination of these properties are known to be associated with hERG activity.24 To allow the optimal N-methylazetidine amide to be retained, ways to reduce lipophilicity by making structural changes at the oxazole’s 2-heteroaryl substituent were explored. Compound 10, which has the same amine pKa as 6 but that is less lipophilic (calculated log D 2.63 vs 3.09, respectively), showed that this approach could be successful. Compound 10 had no hERG binding (IC50 > 30 μM) but retained cellular class I HDAC potency within 2-fold of 6. The impact of the basic amine pKa was clear from strong hERG channel binding (0.19 μM) that was measured for compound 8 (whose amine pKa is around a log unit higher than 6, 8.81 vs 7.65, respectively). A marginal improvement in hERG binding came from reducing the pKa of the azetidine nitrogen in 6 by introducing a fluorine atom on the β-carbon. The lower amine pKa of 11 vs 6 (4.66 vs 7.65) led to 3–4-fold reduced hERG binding but also impacted HDAC biochemical and cellular potency that were both reduced by an order of magnitude. More success was achieved from SAR to explore compounds containing (alternative) electroneutral amide functionality. A number of compounds of this type did not bind to the hERG channel, with compounds 12–14 (all of which have hERG IC50s above 30 μM) providing examples. While compound 12 was somewhat less active against HDACs than compound 6, the final two compounds in Table 3 showed broadly similar inhibition profiles as 6, with cellular class I HDAC inhibition within 2–4-fold.
Table 3. Oxazole Based HDACis.

HDAC1 IC50 (see Supporting Information for full data with standard deviations).
HeLa cellular class I HDAC EC50.
See ref (23).
Throughout the course of work toward both PfHDACis16 and human class I HDACis,19 our compounds were routinely tested on human HDAC isoforms 1–3 (as well as on isoforms from class II HDACs). These data sets were explored in the search for SAR that could lead to the development of isoform selective inhibitors. Figure 2 highlights SAR trends that were the basis for the discovery of HDAC3 selective inhibitors. A consistent profile emerged for compounds that contained an ethyl ketone as their zinc binding group, with essentially equipotent activity being found across HDACs 1–3. Of the 675 ethyl ketone analogs in our collection that were tested on both HDAC1 and HDAC3, only a handful (1.6%) showed 10-fold or higher selectivity (i.e., IC50 HDAC1/IC50 HDAC3) for HDAC3 (Figure 2A). The lack of robust selective starting points as well as the absence of any clear SAR trends that we could identify (or that stood up to testing) made ethyl ketone analogs a challenging starting point toward selective HDAC3 inhibitors. Similar potencies on HDACs 1–2 were typical for compounds from our work targeting PfHDAC (that were based on an imidazole central core together with a methylamide zinc binding group). However, for these compounds a trend toward stronger inhibition of HDAC3 was evident from a set of 84 analogs that were prepared with a thiazole-5-carboxamide adjacent to the imidazole (Figure 2B). A much larger proportion of this compound set (>20%) was at least 10-fold selective. Higher HDAC3 selectivity than had previously been achieved was observed for a number of compounds, with the 4-N,N-dimethylmethanaminophenyl compound (15, Table 4) proving the best in this set, with 32-fold selectivity. While the amide ZBG appears to favor HDAC3 selectivity, the SAR depicted in Figure 2B illustrates that the nature of the aromatic functionality attached to the central imidazole ring is also important. The evident selectivity measured for 15 was in contrast to the lack of selectivity found for the 2-naphthyl analog 16 (for which the HDAC3 IC50 was comparable with HDAC1). Although marginally more selectivity was found for compounds with Ar = 2-methoxy-3-quinoline (e.g., 17), again these analogs consistently failed to generate highly selective profiles. The origin of selectivity for HDAC3 appears to lie in (stronger) attenuation of HDAC1 activity rather than improved inhibition of HDAC3. Although compounds 15, 16, and 18 are similar in terms of their HDAC3 potency, compounds 15 and 18 are less efficient HDAC1 inhibitors. It is perhaps unsurprising therefore that the Ar fragments that feature our most potent HDAC1 inhibitors (such as 2-naphthyl or 2-methoxy-3-quinoline) proved consistently suboptimal with respect to selectivity (see Supporting Information). To explore whether further improved HDAC3 selectivity and improved HDAC3 potency could be achieved, as well as to improve our understanding of SAR, a 2D-array of compounds containing structurally diverse amides (R in Table 4) and either a “highly” selective (cf. compound 15) or a “moderately” selective (cf. compound 18) Ar group was prepared. Given the dangers of comparing activity ratios (that are especially sensitive to assay variability) compounds were in all cases tested in at least triplicate. Compounds 19–21 illustrate some of the findings from this set of 60 compounds, more than a third of which generated HDAC3 selectivity above 10-fold. In virtually all cases the 4-N,N-dimethylmethanaminophenyl analogs generated improved selectivity with respect to the corresponding 4-pyrazolophenyl, though in many cases the improvement was marginal. It is clear from the work we have conducted that achieving HDAC3 selectivity requires the correct zinc-binding group, aryl substituent, and pendant amide. However, the SAR was not without trends that could be followed. While the presence of a basic amine in the aryl fragment was associated with high selectivity for compound 15, basic groups in the amide R fragment consistently failed to generate selective profiles. Amides R that did generate high selectivity (arbitrarily defined as >15-fold) in the presence of the 4-pyrazolophenyl Ar-group always generated high selectivity in the presence of the 4-N,N-dimethylmethanaminophenyl Ar-fragment (see Supporting Information). Examples included compounds 19–20 that were 20-fold and 39-fold selective, respectively.
Figure 2.
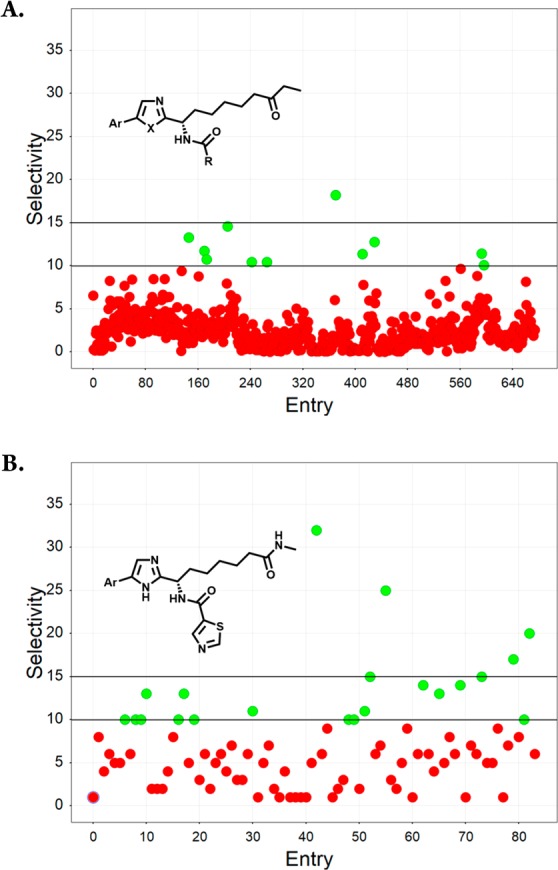
HDAC3 selectivity trends for (A) compounds based on an ethyl ketone ZBGand prepared as human class I HDACis, X = N, O; (B) compounds based on a methyl amide ZBG prepared as PfHDACis.
Table 4. HDAC3 Selective Inhibitors.

IC50 values are the mean of at least three independent measurements. See Supporting Information for full IC50 profiles and standard deviations.
Selectivity for HDAC3 (IC50 HDAC1/IC50 HDAC3).
Compound 21 provided a further example of a 50-fold selective HDAC3 inhibitor that emerged from this work, and it was encouraging that the improved selectivity for analogs 20 and 21 also aligned with improved potency against HDAC3 with respect to 15 (26 and 28 nM vs 162 nM, respectively). In view of the benefits we have described herein for replacement of the central imidazole core with an oxazole, a final compound that was made was analog 22. In this case significantly poorer HDAC3 potency and selectivity were found for 22, suggesting that the imidazole core is preferred for selective profiles.
Compound 21 showed weak inhibition of HDAC2 (IC50 of 4.1 μM, 160-fold selectivity over HDAC3) and had IC50 > 15 μM against HDAC isoforms 4–7. In line with its weak potency against HDAC1–2 in biochemical assays, low activity was also measured for 21 in our cellular class I HDAC assay (IC50 12 μM). The lower concentration of HDAC3 with respect to isoforms 1 and 2 in the HeLa cells in which the assay is run is expected to desensitize the assay to HDAC3-selective inhibitors. However, at concentrations of 21 that did not inhibit class I HDAC’s intracellularly (1 μM), changes in the expression of UCP1 (a gene whose transcription has been demonstrated to be dependent25 on HDAC3) were apparent, giving a first indication of HDAC3 selective effects in a cellular context. Compound 21 demonstrated good stability in vitro, proving stable (t1/2 > 4 h) in both plasma and hepatocytes from mouse and humans. In view of its strong selectivity and early profile, compound 21 represents a novel lead for the development of HDAC3 selective inhibitors.
The synthesis of oxazole analogs containing the ethyl ketone zinc binding group is outlined in Scheme 1. Amide coupling between Boc-protected l-Aoda amino acid 23 and the appropriate aryl α-aminoketone 24 was followed by ring closure of the β-ketoamide intermediate to furnish the 2,5-disubstituted 1,3-oxazole 26. Elaboration of the amino substituent by TFA-mediated deprotection followed by amide coupling with the appropriate carboxylic acid gave compounds 8–9 and 12–14 together with the azetidine precursors 28–29. The azetidine analogs were further elaborated by N-deprotection followed by reductive amination with formaldehyde to give compounds 6 and 11. Acid-mediated hydrolysis of 6 afforded the 2-quinolinone analog 10.
Scheme 1. Synthesis of Oxazole Compounds Containing an Ethyl Ketone ZBG.

Reagents and conditions: (a) HOBt, EDC, DIPEA, DMF, 20 °C; (b) Ph3P, C2Cl6, TEA, DCM, 20 °C; (c) TFA/DCM (9:1), 0 to 20 °C; (d) HCHO, NaOAc, NaCNBH3, MeOH, 0 to 20 °C; (e) HCl 4 N, DCM, 20 °C.
Modification of the above route by employing (S)-2-((benzyloxy)carbonyl)amino-8-(tert-butoxy)-8-oxooctanoic acid in place of 23 was used for the synthesis of oxazole analog 22 (see Supporting Information). The methylamide based compounds 15–21 reported in Table 4 were prepared as we have previously described.16
In summary, we have reported herein work that led to an advanced set of nonhydroxamic acid class I HDAC inhibitors (exemplified by compound 6) that show improved pharmacokinetics and cellular activity with respect to previously reported compounds from this class. We found that hERG binding, an undesired off-target biological activity, could be dialed out from this compound series without a strong compromise in terms of HDAC activity profiles. Overall, the profile of the potential clinical candidate 6 compares favorably with current clinical HDAC inhibitors, including Panobinostat.4 We also reported how a change to the zinc-binding group discovered originally in the context of work toward PfHDAC inhibitors could be employed as a key first step toward biasing SAR for HDAC3 selective profiles. Although SAR in this direction was capricious (with apparently small structural changes having a large impact on activity profiles) following SAR trends allowed us to identify robustly HDAC3 selective compounds for further optimization. Notably, compound 21 was a 26 nM inhibitor of HDAC3 and was 50-fold selective, data that compare favorably with perhaps the most common HDAC3-selective tool compound RGFP9669,18 (that when tested head-to-head in our assays showed an HDAC3 IC50 of 118 nM and was 20-fold selective).
Acknowledgments
We acknowledge Fabrizio Colaceci, Maria Vittoria Orsale, and Roberto Speziale (in vivo work and bioanalysis); Ivan Fini and Odalys Gonzalez (BBB assay); Maria Rosaria Battista (CYP1A2 assay); Nadia Gennari, Ottavia Cecchetti, and Daniela Natale (HDAC assays/HDAC3 biology); Costanza Iaccarino and Letizia Lazzaro (compound purification and QC); and Sergio Altamura, whose science and inspiration were universally appreciated and are missed.
Glossary
ABBREVIATIONS
- HDAC
histone deacetylase
- hERG
human ether-a-go-go related gene ion channel
- Pf
Plasmodium falciparum
- ZBG
zinc binding group
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00517.
Synthetic details, biological protocols, IC50 data, SAR graphics (PDF)
Author Contributions
† A.B. and J.M.O. assisted manuscript preparation, data interpretation, and project overview. These authors contributed equally. A.F., HDAC3 selectivity. A.D.M. and A.Ce., in vitro profiling and hERG. E.M. and M.V., DMPK studies. I.B., I.R., A.Ci., S.P., and F.F., Compound synthesis. S.M., E.N., and P.P., compound synthesis, supervision, and target design. V.S., project overview. S.H., manuscript preparation, data interpretation, and project overview. All authors have approved the final version of the manuscript.
Parts of this work received financial support from the Italian National Compound and Screening Collection (CNCCS consortium) and through the RESEARCH project (Life2020, POR FESR 2014/2020 regione Lazio).
The authors declare no competing financial interest.
Supplementary Material
References
- Goldberg A. D.; Allis C. D.; Bernstein E. Epigenetics: a landscape takes shape. Cell 2007, 128, 635–638. 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Allis C. D.; Jenuwein T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009, 280, 168–176. 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- Tandon N.; Ramakrishnan V.; Kumar S. K. Clinical use and applications of histone deacetylase inhibitors in multiple myeloma. Clin. Pharmacol.: Adv. Appl. 2016, 8, 35–44. 10.2147/CPAA.S94021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abend A.; Kehat I. Histone deacetylases as therapeutic targets--from cancer to cardiac disease. Pharmacol. Ther. 2015, 147, 55–62. 10.1016/j.pharmthera.2014.11.003. [DOI] [PubMed] [Google Scholar]
- Mozzetta C.; Consalvi S.; Saccone V.; Tierney M.; Diamantini A.; Mitchell K. J.; Marazzi G.; Borsellino G.; Battistini L.; Sassoon D.; Sacco A.; Puri P. L. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol. Med. 2013, 5, 626–639. 10.1002/emmm.201202096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellmeier W.; Seiser C. Histone deacetylase function in CD4+ T cells. Nat. Rev. Immunol. 2018, 18, 617–634. 10.1038/s41577-018-0037-z. [DOI] [PubMed] [Google Scholar]
- Marx-Blümel L.; Marx C.; Kühne M.; Sonnemann J. Assessment of HDACi-Induced Cytotoxicity. Methods Mol. Biol. 2017, 1510, 23–45. 10.1007/978-1-4939-6527-4_3. [DOI] [PubMed] [Google Scholar]
- Cao F.; Zwinderman M. R. H.; Dekker F. J. The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 2018, 23, 551–564. 10.3390/molecules23030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P. J.; Fairall L.; Santos G. M.; Schwabe J. W. R. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 2012, 481, 335–340. 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery R. L.; Potthoff M. J.; Haberland M.; Qi X.; Matsuzaki S.; Humphries K. M.; Richardson J. A.; Bassel-Duby R.; Olson E. N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 2008, 118, 3588–3597. 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetze K. B.; McKinsey T. A.; Long C. S. Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J. Mol. Cell. Cardiol. 2014, 70, 100–107. 10.1016/j.yjmcc.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvaez M.; McQuown S. C.; Rogge G. A.; Astarabadi M.; Jacques V.; Carreiro S.; Rusche J. R.; Wood M. A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 2647–2652. 10.1073/pnas.1213364110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagianni P.; Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene 2007, 26, 5439–5449. 10.1038/sj.onc.1210612. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Feng D.; Fang B.; Mullican S. E.; You S.-H.; Lim H.-W.; Everett L. J.; Nabel C. S.; Li Y.; Selvakumaran V.; et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol. Cell 2013, 52, 769–782. 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ontoria J. M.; Paonessa G.; Ponzi S.; Ferrigno F.; Nizi E.; Biancofiore I.; Malancona S.; Graziani R.; Roberts D.; Willis P.; Bresciani A.; Gennari N.; Cecchetti O.; Monteagudo E.; Orsale M. V.; Veneziano M.; Di Marco A.; Cellucci A.; Laufer R.; Altamura S.; Summa V.; Harper S. Discovery of a Selective Series of Inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016, 7, 454–459. 10.1021/acsmedchemlett.5b00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Full details of this study will be reported elsewhere.
- Chou C. J.; Herman D.; Gottesfeld J. M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. 10.1074/jbc.M807045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzel O.; Llauger-Bufi L.; Pescatore G.; Rowley M.; Schultz-Fademrecht C.; Monteagudo E.; Fonsi M.; Gonzalez Paz O.; Fiore F.; Steinkühler C.; Jones P. Discovery of a potent class I selective ketone histone deacetylase inhibitor with antitumor activity in vivo and optimized pharmacokinetic properties. J. Med. Chem. 2009, 52, 3453–3456. 10.1021/jm9004303. [DOI] [PubMed] [Google Scholar]
- Jones P.; Altamura S.; Francesco R. de; Paz O. G.; Kinzel O.; Mesiti G.; Monteagudo E.; Pescatore G.; Rowley M.; Verdirame M.; Steinkühler C. A novel series of potent and selective ketone histone deacetylase inhibitors with antitumor activity in vivo. J. Med. Chem. 2008, 51, 2350–2353. 10.1021/jm800079s. [DOI] [PubMed] [Google Scholar]
- Rankovic Z. CNS Physicochemical Property Space Shaped by a Diverse Set of Molecules with Experimentally Determined Exposure in the Mouse Brain. J. Med. Chem. 2017, 60, 5943–5954. 10.1021/acs.jmedchem.6b01469. [DOI] [PubMed] [Google Scholar]
- For preliminary details on the BBB assay see:; Elbaum D.; Beconi M. G.; Monteagudo E.; Di Marco A.; Quinton M. S.; Lyons K. A.; Vaino A.; Harper S. Fosmetpantotenate (RE-024), a phosphopantothenate replacement therapy for pantothenate kinase-associated neurodegeneration: Mechanism of action and efficacy in nonclinical models. PLoS One 2018, 13, e0192028 10.1371/journal.pone.0192028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calculated using ChemAxon software version 15.
- Shamovsky I.; Connolly S.; David L.; Ivanova S.; Nordén B.; Springthorpe B.; Urbahns K. Overcoming undesirable HERG potency of chemokine receptor antagonists using baseline lipophilicity relationships. J. Med. Chem. 2008, 51, 1162–1178. 10.1021/jm070543k. [DOI] [PubMed] [Google Scholar]
- Emmett M. J.; Lim H.-W.; Jager J.; Richter H. J.; Adlanmerini M.; Peed L. C.; Briggs E. R.; Steger D. J.; Ma T.; Sims C. A.; et al. Histone deacetylase 3 prepares brown adipose tissue for acute thermogenic challenge. Nature 2017, 546, 544–548. 10.1038/nature22819. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.