

Abstract
The potent N-methyl-d-aspartate (NMDA) receptor antagonists 1–3 have been demonstrated to show antiproliferative and cytotoxic effects in MCF-7 and SKBR3 breast cancer cell lines. To improve the knowledge about the role played by the NMDA receptor in the antitumor activity of these compounds, the enantiomers of 1 were prepared and evaluated for their affinity for the phencyclidine (PCP) site of the NMDA receptor and for their cytotoxic effect in MCF-7 and SKBR3 cell lines, both expressing the NMDA receptor. The (S)-1 enantiomer, showing negligible affinity for the PCP site, exhibited antiproliferative activity higher than that of (R)-1, which instead bound the PCP site. The downregulation of NMDA GluN1 expression resulted in a decreased (S)-1-induced cytotoxicity and apoptotic cell death, unequivocally demonstrating the involvement of the NMDA receptor in the antitumor effect of this compound. Due to its interesting biological profile, (S)-1 represents a lead compound to develop novel antitumor agents for breast cancer treatment.
Keywords: NMDA receptor; 1,4-dioxane compounds; breast cancer; apoptosis; silencing of NMDA GluN1 subunit
Breast cancer is the second most common type of cancer worldwide after lung cancer and one of the leading causes of death in women.1 It comprises several biologically different entities with distinct functional features and clinical implications, and its heterogeneity makes it a challenging solid tumor from both diagnostic and therapeutic points of view.2,3 Different therapeutic strategies could be applied, including surgery, radiation therapy, therapy with selective estrogen receptor modulators, monoclonal antibodies, as well as aromatase, tyrosine kinase, cyclin-dependent kinase, mammalian target of rapamycin (mTOR) and poly(ADP-ribose) polymerase (PARP) inhibitors.4
Functional N-methyl-d-aspartate (NMDA) receptors have been demonstrated to be expressed in breast cancer cells and might have a role in maintaining cell growth and viability.5 These receptors are hetero-tetrameric integral membrane proteins belonging to the ionotropic glutamate receptor (iGluR) family, together with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and 2-carboxy-3-carboxymethyl-4-isopropenylpyrrolidine (kainate, KA) receptors.6 NMDA receptors are ion channels with high calcium permeability, whose opening is gated by the binding and cooperative interaction of l-glutamate and the obligatory coagonist glycine.7,8 In addition, channel opening is regulated by several other ligands interacting with different receptor binding sites, including those for polyamines, Zn2+, Mg2+, as well as phencyclidine (PCP). All the compounds interacting with the PCP site, which is located within the cation channel, behave as noncompetitive NMDA receptor antagonists by inhibiting the Ca2+ influx through the cation channel blockade.9 NMDA receptors are assembled by combination of seven subunits, namely, GluN1, GluN2A/B/C/D, and GluN3A/B. In particular, they mainly consist of two obligatory GluN1 subunits and two out of four types of regulatory subunits GluN2A, B, C, or D, which assemble as a dimer of dimers. The resulting complex so formed can also combine with either GluN3A or 3B by replacing one of the GluN2 subunits.10,11 GluN1 subunit is essential for calcium conductivity of the channel whereas GluN2 and GluN3 subunits determine electrophysiological and pharmacological properties of the receptor.6 NMDA receptors are distributed in different areas of the central nervous system and are involved in the regulation of processes underlying learning, memory, and neuron maturation.10,11 Moreover, several studies report the expression of different NMDA receptor subunits in various types of neoplastic cells.12,13 In particular NMDA GluN1 and GluN2B subunits have been demonstrated to be expressed in MCF-7 and SKBR3 breast cancer cells. Moreover, the noncompetitive NMDA receptor antagonist MK-801 reduced cell viability on both cell lines, and also inhibited tumor growth of MCF-7 tumor xenografts in nu/nu mice, suggesting the active role played by NMDA receptor in breast cancer.5 The study of the effects of other potent NMDA receptor ligands on these breast cancer cell lines might help to better investigate such a role.
In the past decade we have demonstrated that the 1,4-dioxane ring is a bioversatile scaffold of ligands selectively targeting different receptor systems,14−18 including the PCP site of NMDA receptor.19 In particular, derivatives 1-3 proved to be potent noncompetitive NMDA receptor antagonists with affinity and functional profiles similar to those of the dissociative anesthetic (S)-(+)-ketamine and different from those of MK-801.19 Moreover, compound 1 has recently been conjugated with bifunctionalizable species to form novel copper(II) complexes which showed antiproliferative activity against a panel of human tumor cell lines, including MCF-7 and SKBR3. The most interesting final complex exerted its cytotoxic activity by causing changes characteristic of paraptosis cell-death.20
Based on these observations, to deeply investigate the involvement of NMDA receptor in breast cancer, in the present study, the cytotoxic effect of 1–3 in MCF-7 and SKBR3 breast cancer cell lines was evaluated using the sulforhodamine B (SRB) assay, according to the National Cancer Institute protocol.21 The functionally related compounds (S)-(+)-ketamine and MK-801 were also included in the study for useful comparison. Interestingly, the results reported in Table 1 show that the 1,4-dioxanes 1–3 inhibit the growth of MCF-7 and SKBR3 cells with similar GI50 values, which are remarkably lower than those of the reference compounds (S)-(+)-ketamine and MK-801. This result indicates that there seems to be no clear relationship between affinity for the PCP binding site of NMDA receptor and cytotoxic activity in these breast cancer cell lines.
Table 1. Affinity (Ki) for PCP Binding Site of NMDA Receptora and Cytotoxic Activity against MCF-7 and SKBR3 Breast Cancer Cell Linesb of 1–3 and Reference Compounds (S)-(+)-Ketamine and MK-801.
| PCP site of the NMDA receptora | MCF-7
(SKBR3) |
|||
|---|---|---|---|---|
| compd | Ki, nM | GI50, μM | TGI, μM | LC50, μM |
| 1 | 712 ± 99a | 74.4 ± 3.4 | 137.3 ± 5.6 | 253.1 ± 5.1 |
| (31.2 ± 5.3) | (200 ± 7.1) | (>300) | ||
| 2 (trans) | 413 ± 20a | 65.3 ± 2.3 | 107.2 ± 5.1 | 176.0 ± 6.1 |
| (23.8 ± 1.9) | (191.8 ± 4.3) | (>300) | ||
| 3 (cis) | 893 ± 40a | 89.7 ± 1.9 | 148 ± 4.7 | 244.2 ± 5.3 |
| (25.8 ± 2.1) | (180.3 ± 8.9) | (>300) | ||
| (S)-(+)-ketamine | 419 ± 4a | >300 | >300 | >300 |
| (>300) | (>300) | (>300) | ||
| MK-801 | 1.5 ± 0.1 | >300 | >300 | >300 |
| (>300) | (>300) | (>300) | ||
From ref (19).
In vitro cytotoxic activity in human MCF-7 and SKBR3 cells was carried out using SRB assay. Growth Inhibition 50 (GI50) represents the drug concentration (μM) required to inhibit 50% net of cell growth. Total growth inhibition (TGI) represents the drug concentration (μM) required to inhibit 100% of cell growth. Lethal concentration 50 (LC50) represents the drug concentration (μM) required to kill 50% of the initial cell number. Each quoted value represents the mean of quadruplicate determinations ± standard error (SE) (n = 5).
Considering that the interaction with NMDA receptor is well-known to be highly stereospecific and stereochemistry can influence quantitatively and qualitatively the biological profile of the ligands,22 to improve the knowledge about the role played by NMDA receptor in the antiproliferative activity of the 1,4-dioxane derivatives of the present study, the enantiomers of 1 were prepared and evaluated for their affinity for the PCP site of the NMDA receptor and for their cytotoxic effect in MCF-7 and SKBR3 breast cancer cell lines. Since 1–3 show similar NMDA receptor affinity and activity19 as well as comparable antitumor profile, compound 1 was selected on the basis that only one center of chirality is present. In contrast to the diastereoisomeric cyclohexyl derivatives 2 and 3, which contain two centers of chirality, only two enantiomers had to be separated.
The enantiomers of 1 were prepared following a procedure different from that used for the corresponding racemic mixture,19 to improve the overall yield (Scheme 1). Treatment of the tosyl derivatives (S)-(+)- and (R)-(−)-423 with NaN3 afforded the azides (R)-(+)- and (S)-(−)-5, which were reduced with LiAlH4 to give the desired primary amines (R)-(+)- and (S)-(−)-1, respectively, in overall yield of 78–79%.
Scheme 1. Synthesis of the Enantiomers (R)-(+)-1 and (S)-(−)-1.

Reagents: (a) NaN3, DMF; (b) LiAlH4, Et2O.
The enantiomeric purity, determined by 1H NMR spectroscopy in the presence of the chiral shift reagent (R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid [(+)-MTPA],24 was found to be >98% (detection limit) for both enantiomers. Indeed, the 1H NMR spectrum of racemic compound (±)-1 showed two doublets at 4.57 and 4.60 ppm for one of the protons in the 5-position of the 1,4-dioxane ring, whereas only one doublet was observed for (R)-(+)-1 and (S)-(−)-1 at 4.60 and 4.57 ppm, respectively.
The affinity of the novel enantiomers (R)-1 and (S)-1 for the PCP binding site of the NMDA receptor was determined in competition experiments with the radioligand [3H]-(+)-MK-801 on membrane preparations from pig brain cortex.19 The results reveal that (R)-1 binds to the PCP binding site of the NMDA receptor with a Ki value of 497 ± 18 nM, which is comparable with the Ki values of the racemic compound (±)-1 (712 ± 99 nM) and (S)-(+)-ketamine (419 ± 4 nM). Instead (S)-1 shows negligible affinity for the PCP site (Ki > 10000 nM).
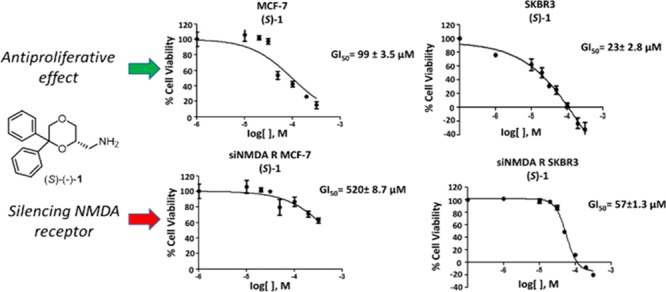
After confirming the expression of NMDA receptor mRNA in MCF-7 and SKBR3 cell lines by quantitative RT-PCR, cytofluorimetric, and Western blot analysis (see Supporting Information, Figure S1), the antiproliferative effects of the enantiomers (R)-1 and (S)-1 were evaluated in both cell lines by the SRB assay, according to the National Cancer Institute protocol.21 Both the enantiomers demonstrated to reduce dose-dependently the growth of both cell lines, with GI50 of 250 μM (MCF-7) and 74 μM (SKBR3) for (R)-1 (Figure 1A,B), and 99 μM (MCF-7) and 23 μM (SKBR3) for (S)-1 (Figure 1C,D), after 48 h of treatment. The GI50 values of (S)-1 are not significantly different from those of the racemic 1 and are lower than those of (R)-1 in both cell lines. The observation that the enantiomer (S)-1, endowed with negligible affinity for the PCP binding site of the NMDA receptor, shows cytotoxic activity in breast cancer cell lines higher than that of (R)-1, which instead binds to the PCP site with good affinity, strengthens the hypothesis that the affinity for PCP binding site is not correlated with the cytotoxic effect.
Figure 1.

MCF-7 and SKBR3 cells were treated for 48 h with different doses of (R)-1 and (S)-1. Cell viability was determined by SRB assay. Data shown are expressed as mean ± SE of three separate experiments.
These results prompted us to investigate mechanisms responsible for the cytotoxic effect of (S)-1, selected for its significantly higher antitumor activity in both cell lines. For the following experiments GI50 doses of 100 μM and 25 μM of enantiomer (S)-1 were used for MCF-7 and SKBR3, respectively. Both MCF-7 and SKBR3 cells were treated with the enantiomer (S)-1 for up to 72 h and cell death process was analyzed by Annexin V-FITC and propidium iodide (PI) staining followed by biparametric FACS analysis. As shown in Figure 2A, results indicated that (S)-1 treatment causes an increased percentage of cells undergoing apoptotic cell death (Annexin V+/PI–, early apoptosis; Annexin V+/PI+, late apoptosis) in both cell lines 72 h after treatment. These results were strengthened by Western blot analysis showing the cleavage of procaspase-3 in the active caspase-3 after 72 h of treatment with (S)-1 in both cell lines (Figure 2B). Overall these data demonstrated that (S)-1 treatment induces apoptotic cell death in MCF-7 and SKBR3 breast cancer cells.
Figure 2.

(A) MCF-7 and SKBR3 cells were cultured with (S)-1 (100 μM and 25 μM, respectively) for 48 and 72 h. Cells were double stained with Annexin V-FITC/PI and analyzed by flow cytometry. Data, representative of one of three separate experiments, are the percentage of Annexin V and/or PI positive cells. (B) Representative immunoblots of caspase-3 in MCF-7 and SKBR3 cells treated for 72 h as described above. Densitometric values were normalized to GAPDH used as loading control. (C,D) ROS generation was evaluated by DCFDA staining and FACS analysis in MCF-7 and SKBR3 cells treated for 24, 48, and 72 h as described above, alone or in combination with NAC. (C) *p < 0.01 vs untreated cells; (D) *p < 0.01 vs untreated cells; #p < 0.01 vs (S)-1 treated cells.
To elucidate the molecular mechanism by which (S)-1 induces apoptosis, the mitochondrial depolarization (Δψm) was analyzed by JC-1 staining in untreated or (S)-1-treated MCF-7 and SKBR3 cells. Treatment with (S)-1 did not influence mitochondrial membrane potential (data not shown), suggesting that mitochondria are not involved in the apoptotic cell death.
In addition, reactive oxygen species (ROS) production in MCF-7 and SKBR3 breast cancer cells treated with (S)-1 at different times (0–72 h) was measured through 2′,7′-dichloro fluorescein diacetate (DCFDA) staining and cytofluorimetric analysis. A significant increase in ROS concentration was detected after 72 h of treatment (Figure 2C). Moreover, pretreatment of cells with the ROS scavenger N-acetylcysteine (NAC, 10 mM) completely inhibited ROS production, indicating that (S)-1 stimulates peroxide accumulation in breast cancer cells (Figure 2D). This result suggests that the NMDA receptor might mediate the cytotoxic activity of (S)-1, as such receptor has been reported to be involved in ROS production.25
To unequivocally evaluate the involvement of NMDA receptor in the (S)-1- and (R)-1-mediated cytotoxic activity against breast cancer cells, the GluN1 subunit present in all NMDA receptors was knocked down in both MCF-7 and SKBR3 cell lines. As evaluated by qRT-PCR and Western blot analysis, transfection with siNMDA receptor (siNMDA R) significantly reduced the mRNA and protein levels respect to untransfected cells (see Supporting Information, Figure S2). The effect of NMDA receptor gene knockdown on cytotoxicity induced by (S)-1 and (R)-1 treatment was evaluated by SRB assay in siNMDA R MCF-7 and SKBR3 cell lines, compared with untransfected cells. For (S)-1 the IC50 value shifted from 99 and 23 μM to 520 and 57 μM for MCF-7 and SKBR3 cells, respectively. On the contrary, no significant changes were observed after treatment with (R)-1 (Table 2).
Table 2. Antiproliferative Activity of the Enantiomers (R)-1 and (S)-1 in siNMDA R MCF-7 and SKBR3 Cell Lines, Compared with Untransfected Cellsa.
| GI50 (μM) |
||
|---|---|---|
| cell line | (R)-1 | (S)-1 |
| MCF-7 untrasfected | 250 ± 6.5 | 99 ± 3.5 |
| MCF-7 siNMDA R | 210 ± 2.3 | 520 ± 8.7 |
| SKBR3 untrasfected | 74 ± 4.8 | 23 ± 2.8 |
| SKBR3 siNMDA R | 66 ± 1.9 | 57 ± 1.3 |
Cells were treated for 48 h with different doses of (R)-1 and (S)-1. Cell viability was determined by SRB assay. Data are expressed as mean ± SE of three separate experiments.
Since, as demonstrated above, (S)-1 treatment induces apoptotic cell death in MCF-7 and SKBR3 breast cancer cells, untransfected and siNMDA R transfected cells treated with (S)-1 were stained with Annexin V and PI and analyzed by cytofluorimetry, to evaluate whether the silencing of NMDA GluN1 subunit also affected the cell death apoptotic process (Figure 3). Data evidenced a marked decrease in the percentage of double positive cells (Annexin V+/PI+) in siNMDA R respect to untransfected cells, suggesting the involvement of NMDA receptor in the cytotoxic response induced by (S)-1 (Figure 3A). The activation of procaspase-3 in siNMDA R cells, 72 h after treatment with (S)-1, was also analyzed. Densitometric analysis of the immunoblots revealed that siNMDA R transfection increased the basal levels of activated caspase-3 (the cleaved form) respect to untrasfected MCF-7 and SKBR3 cells (Figure 3B,C). However, the increase of active caspase-3 form, observed after drug treatment, was lower in siNMDA-transfected cells with respect to untransfected ones (Figure 3B,D), confirming the results obtained by cytofluorimetric analysis.
Figure 3.

(A) Untransfected or siNMDA R transfected MCF-7 and SKBR3 cells were treated for 72 h with (S)-1 (100 μM and 25 μM, respectively). Cells were double stained with Annexin V-FITC/PI and analyzed by flow cytometry. Data, representative of one of three separate experiments, are the percentage of Annexin V and/or PI positive cells. (B) Representative immunoblots of caspase-3 in MCF-7 and SKBR3 cells treated for 72 h as above-described. (C,D) Densitometric values were normalized to GAPDH used as loading control. (C) *p < 0.01 siNMDA R transfected vs untransfected cells. (D) *p < 0.01 (S)-1 treated vs untreated cells.
Overall, these data indicated that the downregulation of NMDA receptor mRNA expression resulted in a reduction in (S)-1-induced cytotoxicity and apoptotic cell death, demonstrating the involvement of NMDA receptor in the antitumor activity of this compound. No significant difference in (R)-1-mediated cytotoxicity was observed in siNMDA R transfected with respect to untransfected cells, confirming once more that the interaction with the PCP site of NMDA receptor might have a secondary or even no role in the anticancer activity of this class of compounds. Probably, (S)-1 binds NMDA receptor in a site different from that of PCP.
The enantiomers (R)-1 and (S)-1 were also evaluated for their in vitro ADME profile. For both compounds, hepatic in vitro intrinsic clearance, determined in rat and human liver microsomes, and permeability studies in Caco-2 cell line were assessed according to previously reported procedures.26−28 The results, reported in the Supporting Information (Table S1), revealed that both enantiomers showed high in vitro metabolic stability in human liver microsomes and good permeability in Caco-2 cell line with no impact from P-gp efflux transporter, thus suggesting a potential for good oral bioavailability in human.
In summary, the noncompetitive NMDA receptor antagonists 1–3 showed antiproliferative and cytotoxic effects in MCF-7 and SKBR3 breast cancer cells. The effects were significantly higher than those of (S)-(+)-ketamine and MK-801, indicating that the affinity of these compounds for the PCP binding site of the NMDA receptor might not correlate with their cytotoxic activity. This hypothesis was strengthened by the study of enantiomers (R)-1 and (S)-1 that highlighted an intriguing result. Indeed, the enantiomer (S)-1, showing negligible affinity for the PCP site of the NMDA receptor, displayed antiproliferative activity in MCF-7 and SKBR3 cells. This activity was considerably higher than that of (R)-1, which instead interacted with the PCP site with moderate affinity. The investigation of mechanisms responsible for the antitumor activity revealed that (S)-1 induced apoptosis and ROS production in a time- and dose-dependent, but in a mitochondria-independent manner in both MCF-7 and SKBR3 cell lines. The downregulation of NMDA GluN1 expression resulted in a decrease in the (S)-1-induced cytotoxicity and apoptotic cell death in MCF-7 and SKBR3 cells, unequivocally demonstrating the involvement of NMDA receptor in the antitumor effect of this compound. Moreover, the observation that no significant differences in (R)-1-mediated cytotoxicity were observed in siNMDA R transfected with respect to untransfected cells confirmed that the interaction with the PCP site of the NMDA receptor might have a secondary or even no role in the anticancer activity of this class of compounds. Due to its interesting biological profile, enantiomer (S)-1 may represent a useful tool to investigate the physiological functions in which NMDA receptor is involved and a lead compound for the development of novel agents for the treatment of breast cancer. Several studies report that NMDA receptor inhibition reduces cancer cell growth, whereas others describe the potential antiproliferative effect of NMDA receptor activation.14 According to evidence from literature,25 (S)-1-induced ROS production might be due to NMDA receptor activation. Therefore, future studies will be devoted to evaluating the functional activity of (S)-1 at the NMDA receptor and its affinity for binding sites different from the PCP site.
Acknowledgments
This work was supported by grants from the University of Camerino (Fondo di Ateneo per la Ricerca 2018) and the Fondazione Umberto Veronesi (Postdoctoral Fellowship 2018 to M.B.M.).
Glossary
Abbreviations
- mTOR
mammalian target of rapamycin
- PARP
poly(ADP-ribose) polymerase
- iGluRs
ionotropic glutamate receptors
- NMDA
N-methyl-d-aspartate
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- KA
2-carboxy-3-carboxymethyl-4-isopropenylpyrrolidine
- PCP
phencyclidine
- SRB
sulforhodamine B
- (+)-MTPA
(R)-(+)-α-methoxy-α-(trifluoromethyl)phenylacetic acid
- SE
standard error
- ROS
reactive oxygen species
- NAC
N-acetylcysteine
- DCFDA
2′,7′-dichlorofluorescein diacetate
- PI
propidium iodide.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00536.
Experimental details of synthesis and biological assays for (R)-1 and (S)-1; results of in vitro ADME studies; NMDA receptor expression in MCF-7 and SKBR3 cells; reduction of NMDA mRNA and protein levels in MCF-7 and SKBR3 cells transfected with siNMDA R with respect to untransfected cells; data of SRB assay performed on MCF-7 and SKBR3 siNMDA R transfected cells (PDF)
Author Contributions
∇ M.B.M. and C.A. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Howell A.; Sims A. H.; Ong K. R.; Harvie M. N.; Evans D. G. R.; Clarke R. B. Mechanisms of disease: prediction and prevention of breast cancer-cellular and molecular interactions. Nat. Clin. Pract. Oncol. 2005, 2, 635–646. 10.1038/ncponc0361. [DOI] [PubMed] [Google Scholar]
- Spitale A.; Mazzola P.; Soldini D.; Mazzucchelli L.; Bordoni A. Breast cancer classification according to immunohistochemical markers: clinicopathologic features and short-term survival analysis in a population-based study from the South of Switzerland. Ann. Oncol. 2009, 20, 628–635. 10.1093/annonc/mdn675. [DOI] [PubMed] [Google Scholar]
- Weigelt B.; Baehner F. L.; Reis-Filho J. S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: a retrospective of the last decade. J. Pathol. 2010, 220, 263–280. 10.1002/path.2648. [DOI] [PubMed] [Google Scholar]
- Makhoul I.Therapeutic Strategies for Breast Cancer. In The Breast, Comprehensive Management of Benign and Malignant Diseases, 5th ed.; Bland K. I., Copeland E. M., Klimberg V. S., Gradishar W. J., Eds.; Elsevier: 2018; pp 315–330. [Google Scholar]
- North W. G.; Gao G.; Memoli V. A.; Pang R. H.; Lynch L. Breast cancer expresses functional NMDA receptors. Breast Cancer Res. Treat. 2010, 122, 307–314. 10.1007/s10549-009-0556-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynelis S. F.; Wollmuth L. P.; McBain C. J.; Menniti F. S.; Vance K. M.; Ogden K. K.; Hansen K. B.; Yuan H.; Myers S. J.; Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R.; Kleckner N. W.; McBain C. J. The glycine coagonist site of the NMDAR. Adv. Exp. Med. Biol. 1990, 268, 17–26. 10.1007/978-1-4684-5769-8_3. [DOI] [PubMed] [Google Scholar]
- Blanke M. L.; VanDongen A. M. J.. Activation mechanisms of the NMDA receptor. In Biology of the NMDA Receptor; Van Dongen A. M., Ed.; CRC Press: Boca Raton, FL, 2009; pp 283–312. [PubMed] [Google Scholar]
- Tomek S. E.; LaCrosse A. L.; Nemirovsky N. E.; Olive M. F. NMDA receptor modulators in treatment of drug addiction. Pharmaceuticals 2013, 6, 251–268. 10.3390/ph6020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hallaq R. A.; Conrads T. P.; Veenstra T. D.; Wenthold R. J. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J. Neurosci. 2007, 27, 8334–8343. 10.1523/JNEUROSCI.2155-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothwell S. L. C.; Barber J. L.; Monaghan D. T.; Jane D. E. A.; Gibb J.; Jones S. NR2B- and NR2D-containing synaptic NMDA receptors in developing rat substantia nigra pars compacta dopaminergic neurons. J. Physiol. 2008, 586, 739–750. 10.1113/jphysiol.2007.144618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch S. I.; Tang A. H.; Burket J. A.; Benson A. D. NMDA receptors on the surface of cancer cells: target for chemotherapy?. Biomed. Pharmacother. 2014, 68, 493–496. 10.1016/j.biopha.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra A.; Koiri R. K. N-Methyl-D-aspartate (NMDA) receptors: therapeutic target against cancer. Int. J. Immunother. Cancer Res. 2015, 1, 13–17. 10.17352/2455-8591.000004. [DOI] [Google Scholar]
- Piergentili A.; Quaglia W.; Del Bello F.; Giannella M.; Pigini M.; Barocelli E.; Bertoni S.; Matucci R.; Nesi M.; Bruni B.; Di Vaira M. Properly substituted 1,4-dioxane nucleus favours the selective M3 muscarinic receptor activation. Bioorg. Med. Chem. 2009, 17, 8174–8185. 10.1016/j.bmc.2009.10.027. [DOI] [PubMed] [Google Scholar]
- Mammoli V.; Bonifazi A.; Del Bello F.; Diamanti E.; Giannella M.; Hudson A. L.; Mattioli L.; Perfumi M.; Piergentili A.; Quaglia W.; Titomanlio F.; Pigini M. Favourable involvement of α2A-adrenoreceptor antagonism in the I2-imidazoline binding sites-mediated morphine analgesia enhancement. Bioorg. Med. Chem. 2012, 20, 2259–2265. 10.1016/j.bmc.2012.02.016. [DOI] [PubMed] [Google Scholar]
- Bonifazi A.; Piergentili A.; Del Bello F.; Farande Y.; Giannella M.; Pigini M.; Amantini C.; Nabissi M.; Farfariello V.; Santoni G.; Poggesi E.; Leonardi A.; Menegon S.; Quaglia W. Structure–activity relationships in 1,4-benzodioxan-related compounds. 11. Reversed enantioselectivity of 1,4-dioxane derivatives in α1-adrenergic and 5-HT1A receptor binding sites recognition. J. Med. Chem. 2013, 56, 584–588. 10.1021/jm301525w. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Bonifazi A.; Giannella M.; Giorgioni G.; Piergentili A.; Petrelli R.; Cifani C.; Micioni Di Bonaventura M. V.; Keck T. M.; Mazzolari A.; Vistoli G.; Cilia A.; Poggesi E.; Matucci R.; Quaglia W. The replacement of the 2-methoxy substituent of N-((6,6-diphenyl-1,4-dioxan-2-yl)methyl)-2-(2-methoxyphenoxy)ethan-1-amine improves the selectivity for 5-HT1A receptor over α1-adrenoceptor and D2-like receptor subtypes. Eur. J. Med. Chem. 2017, 125, 233–244. 10.1016/j.ejmech.2016.09.026. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Bonifazi A.; Giorgioni G.; Petrelli R.; Quaglia W.; Altomare A.; Falcicchio A.; Matucci R.; Vistoli G.; Piergentili A. Novel muscarinic acetylcholine receptor hybrid ligands embedding quinuclidine and 1,4-dioxane fragments. Eur. J. Med. Chem. 2017, 137, 327–337. 10.1016/j.ejmech.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Bonifazi A.; Del Bello F.; Mammoli V.; Piergentili A.; Petrelli R.; Cimarelli C.; Pellei M.; Schepmann D.; Wünsch B.; Barocelli E.; Bertoni S.; Flammini L.; Amantini C.; Nabissi M.; Santoni G.; Vistoli G.; Quaglia W. Novel potent N-methyl-D-aspartate (NMDA) receptor antagonists or σ1 receptor ligands based on properly substituted 1,4-dioxane ring. J. Med. Chem. 2015, 58, 8601–8615. 10.1021/acs.jmedchem.5b01214. [DOI] [PubMed] [Google Scholar]
- Morelli M. B.; Amantini C.; Santoni G.; Pellei M.; Santini C.; Cimarelli C.; Marcantoni E.; Petrini M.; Del Bello F.; Giorgioni G.; Piergentili A.; Quaglia W. Novel antitumor copper(II) complexes designed to act through synergistic mechanisms of action, due to the presence of an NMDA receptor ligand and copper in the same chemical entity. New J. Chem. 2018, 42, 11878–11887. 10.1039/C8NJ01763H. [DOI] [Google Scholar]
- Grever M. R.; Shepartz S. A.; Chabner B. A. The National Cancer Institute: cancer drug discovery and development program. Semin. Oncol. 1992, 19, 622–638. [PubMed] [Google Scholar]
- Köhler J.; Bergander K.; Fabian J.; Schepmann D.; Wünsch B. Enantiomerically pure 1,3-dioxanes as highly selective NMDA and σ1 receptor ligands. J. Med. Chem. 2012, 55, 8953–8957. 10.1021/jm301166m. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Barocelli E.; Bertoni S.; Bonifazi A.; Camalli M.; Campi G.; Giannella M.; Matucci R.; Nesi M.; Pigini M.; Quaglia W.; Piergentili A. 1,4-Dioxane, a suitable scaffold for the development of novel M3 muscarinic receptor antagonists. J. Med. Chem. 2012, 55, 1783–1787. 10.1021/jm2013216. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Mattioli L.; Ghelfi F.; Giannella M.; Piergentili A.; Quaglia W.; Cardinaletti C.; Perfumi M.; Thomas R. J.; Zanelli U.; Marchioro C.; Dal Cin M.; Pigini M. Fruitful adrenergic α2C-agonism/α2A-antagonism combination to prevent and contrast morphine tolerance and dependence. J. Med. Chem. 2010, 53, 7825–7835. 10.1021/jm100977d. [DOI] [PubMed] [Google Scholar]
- Girouard H.; Wang G.; Gallo E. F.; Anrather J.; Zhou P.; Pickel V. M.; Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J. Neurosci. 2009, 29, 2545–2552. 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F.; Bacchi A.; Braggio S.; Castelletti L.; Cavallini P.; Cavanni P.; Cremonesi S.; Dal Cin M.; Feriani A.; Gehanne S.; Kajbaf M.; Marchió L.; Nola S.; Oliosi B.; Pellacani A.; Perdonà E.; Sava A.; Semeraro T.; Tarsi L.; Tomelleri S.; Wong A.; Visentini F.; Zonzini L.; Heidbreder C. 1,2,4-Triazolyl-5-Azaspiro[2.4]heptanes: Lead identification and early lead optimization of a new series of potent and selective dopamine D3 receptor antagonist. J. Med. Chem. 2016, 59, 8549–8576. 10.1021/acs.jmedchem.6b00972. [DOI] [PubMed] [Google Scholar]
- Skolnik S.; Lin X.; Wang J.; Chen X. H.; He T.; Zhang B. Towards prediction of in vivo intestinal absorption using a 96-well Caco-2 assay. J. Pharm. Sci. 2010, 99, 3246–3265. 10.1002/jps.22080. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Bonifazi A.; Giorgioni G.; Cifani C.; Micioni Di Bonaventura M. V.; Petrelli R.; Piergentili A.; Fontana S.; Mammoli V.; Yano H.; Matucci R.; Vistoli G.; Quaglia W. 1-[3-(4-Butylpiperidin-1-yl)propyl]-1,2,3,4-tetrahydroquinolin-2-one (77-LH-28–1) as a model for the rational design of a novel class of brain penetrant ligands with high affinity and selectivity for dopamine D4 receptor. J. Med. Chem. 2018, 61, 3712–3725. 10.1021/acs.jmedchem.8b00265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.