

Abstract
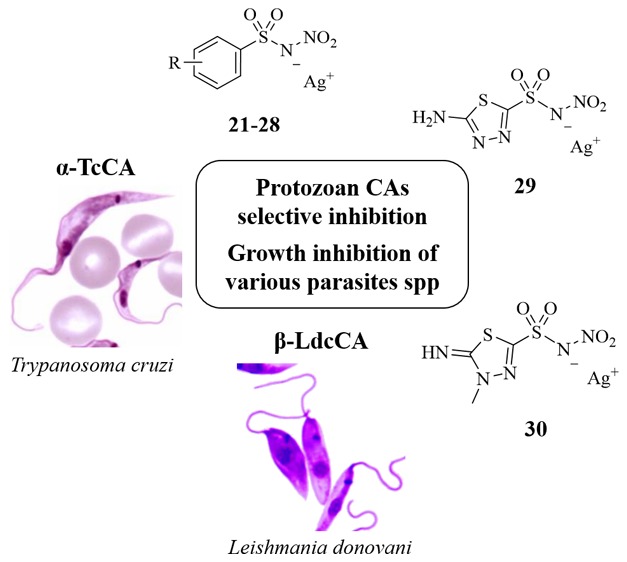
Trypanosoma cruzi and Leishmania spp. are protozoa of the Trypanosomatidae family, respectively, responsible of the neglected tropical disorders (NTDs) Chagas disease and leishmaniasis. The present pharmacotherapy is often ineffective and exhibits serious side effects. The metalloenzyme carbonic anhydrases (CAs, EC 4.2.1.1) recently identified in these protozoans (α-TcCA and β-LdcCA) are novel promising targets for chemotherapeutic interventions. Herein, we report a series of N-nitrosulfonamides, as a novel chemotype to yield the target CA isoform selective inhibition over ubiquitous human isozymes. Two derivatives selected among the most active and selective ones for TcCA/LdcCA over off-target CAs were progressed as silver salts to in vitro studies with various developmental forms and spp of Trypanosoma cruzi and leishmania. Excellent values of parasites growth inhibition (IC50) were observed, with some selectivity index (over cytotoxicity for macrophages and Vero cells) being comparable or better than reference drugs. These findings make N-nitrosulfonamides and their salts promising lead compounds for a rational optimization of innovative agents for the treatment of Chagas disease and leishmaniasis based on CA inhibition.
Keywords: Chagas disease, Trypanosoma cruzi, leishmania, carbonic anhydrase, zinc-binding group, inhibition, silver, antiparasitic
World Health Organization (WHO) included Chagas disease (American trypanosomiasis) and leishmaniasis in the list of neglected tropical diseases (NTDs). Parasites of the kinetoplastidae family are responsible for these infections, both belonging to the vector-borne diseases affecting 20 million people and killing more than 50,000 every year.1
Trypanosoma cruzi is naturally transmitted by kissing bugs (mainly belonging to the genera Triatoma and Rhodnius), which primarily diffuse in Latin America. The disease evolves producing potentially fatal lesions to organs in the cardiac, digestive, or neurological systems.1
Leishmaniasis is transmitted by the bite of an infected phlebotomine and works out skin or visceral aches that could turn out to be fatal if untreated. Among the NTDs, leishmaniasis is the first-in-class in terms of mortality and morbidity.1
Available pharmacological treatments for the majority of NTDs are limited in terms of cost and toxicity and ineffective, and resistance phenomena constantly increase throughout the world.2−4 Pharmaceutical industry shows poor interest in searching new effective drugs for the treatment of NTDs due to high costs and expected low financial return. It is urgent to find new therapeutic targets for these parasitosis, which WHO classifies as priority infections.2,5 Novel targets have been identified driven by large-scale analysis on the completely known genome sequence of both protozoans. Indeed, endeavors to enrich the therapeutic arsenal against Chagas disease and leishmaniasis based on enzymatic inhibition have been starting in many laboratories with synthetic drugs representing a valuable source for new treatments.6,7
The metalloenzyme carbonic anhydrases (CAs, EC 4.2.1.1) recently identified in these protozoans are novel promising targets for chemotherapeutic interventions.6,8,9 CAs catalyze the reversible hydration of CO2 to bicarbonate and proton, a pivotal reaction for all cells and complex organisms, which is also basic in the growth and virulence of pathogenic microorganisms.9 CAs from Trypanosoma cruzi (TcCA) and Leishmania donovani chagasi (LdcCA) were cloned and characterized in 2013,10−12 resulting in the design of novel antiprotozoal agents that act by a totally new mechanism of action and lack cross-resistance to existing drugs. The α-CA TcCA is endowed with a very high catalytic activity for the CO2 hydration reaction and was shown to be inhibited in the nanomolar range by many types of CA inhibitors (CAIs) such as aromatic/heterocyclic sulfonamides,10,13,14 sulfamates,10 thiols,10 anions,15 dithiocarbamates,15 hydroxamates,16 and benzoxaboroles.17 Thiols and hydroxamates exhibited in vitro antitrypanosomal activity, inhibiting the three phases of the pathogen’s life cycle.10,16 The β-CA LdcCA also features an effective catalytic activity and was shown to be efficiently inhibited by sulfonamides and heterocyclic thiols with nanomolar inhibition constants.12,18 Some such thiol derivatives displayed in vitro antileishmania activity in preliminary assays being able to reduce parasites’ growth and causing their death.12 Identification of new protozoans CAIs with effective antitrypasonomal or antileishmania activities is more than ever worth the endeavor due to such targets’ remarkable druggability.
Nifurtimox and benznidazole have been the first effective drugs for treating acute-phase human Chagas infection, with the first being no longer available on the market because of undesirable side effects.19 They feature heteroaromatic nitro moieties that are pivotal for the antiprotozoa mechanism of action. Parasite resistance arisen with benznidazole drove the development of alternative therapies. Indeed, combined treatment of benznidazole with drugs with different mechanisms of action such as azoles, nitric oxide, or clomipramine could be a strategy to improve the pharmacotherapy efficacy.6
Noteworthy, a new chemotype able to afford α- and β-CAs inhibition was reported by us in 2016, namely, N-nitrosulfonamides.20 Interestingly, these latter were shown to inhibit ubiquitous, off-targets isoforms, such as CA II, feebler than lead sulfonamides though holding remarkable submicromolar inhibition of the human (h) tumor-associated CA IX (α-CA) and the β-CA from the pathogen fungus Malassezia Globosa.
Considering the above, we have herein extended the set of N-nitrosulfonamides and screened them on a wider pattern of human and pathogen (from protozoa and fungi) CAs, among which are the targets α-TcCA and β-LdcCA.
Furthermore, we produced silver salts of all such derivatives based on their marked effects against viruses, bacteria, fungi, and protozoa.20 The antimicrobial behaviors of silver, silver ions, and silver-containing compounds have long been investigated with various antimicrobial mechanisms of action having been proposed to date.21−24 The biologically active silver ion (Ag+) irreversibly damages key enzyme systems in the cell membranes of pathogens. Conversely, silver exhibits low toxicity in the human body and minimal risk is expected due to clinical exposure.21 Recently, silver nanoparticles (Ag-NPs) were demonstrated to produce reactive oxygen species to which Leishmania parasites are very sensitive.24 Moreover, the commercially available antibiotic silver sulfadiazine shares a wealth of features with silver N-nitrosulfonamides. These latter derivatives are thus endowed with multiple potential antiprotozoa entities to be synergistically exploited to overcome resistance issues displayed by single-targeted therapy.
The general synthetic strategy proposed by Minksztym24 for the chemoselective mononitration of aminosulfonamides was applied to a set of ten starting compounds being commercially available (1–3, 7, 8) or yielded by methylation (4–6) or deacetylation (17, 19) reactions (Schemes 1 and 2).
Scheme 1. Synthesis of Aromatic N-Nitrosulfonamides: (a) H2O; (b) NH4OH(aq).

Scheme 2. Synthesis of Heteroaromatic N-Nitrosulfonamides.

Quenching and workup of the NH4NO3/H2SO4 based nitration reaction was switched for the most unstable derivatives 16, 18, and 20 from water to NH4OH(aq) to generate the stable ammonium salts instead of the zwitterion forms of N-nitrosulfonamides (Schemes 1 and 2).25,26
The production of the silver salts of the derivatives was achieved by different methods depending on the nature of the compound or the form it was produced as in the previous step (Scheme 3). Silver carbonate was used as the base (to remove the proton) and the source of silver ion in an aqueous phase in case of zwitterion of amino aromatic compounds. NaOH/AgNO3 was used for the zwitterion of the amino aliphatic compound 15 and for ammonium salts 16, 18, and 20. All compounds silver salts precipitated in aqueous phases and were therefore recrystallized by the same solvent. All the obtained derivatives were properly characterized by means of 1H NMR, 13C NMR, and MS (see Supporting Information).
Scheme 3. Synthesis of Silver Salts of N-Nitrosulfonamides: (c) Ag2CO3, H2O; (d) NaOH, AgNO3, H2O.

The inhibition profiles of the N-nitrosulfonamide derivatives were evaluated against six α-CAs and three β-CA isoforms in addition to acetazolamide (AAZ) as standard inhibitor, by a stopped flow CO2 hydrase assay.27
Five human CA isoforms, among which the ubiquitous CA I and II (involved in many physiopathological processes), the membrane-associated CA IV (involved in ocular aches, stroke and arthritis), and IX and XII (overexpressed in hypoxic tumors) were included in the kinetic study to work out thorough structure–activity relationships (SARs) and selectivity profiles.28 Along with the target TcCA and LdcCA, the activity of two additional β-CA isoforms from pathogenic fungi was studied with the reported inhibitors, namely, MgCA from Malassezia globosa (responsible of the production of dandruff)29 and Can2 from Cryptococcus neoformans (that can cause fungal meningitis and encephalitis).30 The inhibitory efficacy against nine such isoforms was also assessed with the silver salts of all derivatives to verify whether the monovalent metal ion affects the enzymatic activities. The inhibition constants (KIs) of these latter do not show significant variations out of the error ranges, witnessing no significant action of the Ag+ ion to each CA activity. Tables 1 (hCAs) and 2 (pathogens CAs) gather the inhibition data of silver salts 21–30 (inhibition data of 9–20 are not shown; comparisons can be made with data previously reported).20
Table 1. Inhibition Data of CA I, II, IV, IX, and XII with N-Nitrosulfonamides 21–30 and the Standard Sulfonamide Inhibitor Acetazolamide (AAZ) by a Stopped Flow CO2 Hydrase Assay27.

|
KI (μM)a |
||||||
|---|---|---|---|---|---|---|
| compd | R | CA I | CA II | CA IV | CA IX | CA XII |
| 21 | 2-NH2 | 29.0 | 60.9 | 39.2 | 0.52 | 0.75 |
| 22 | 3-NH2 | 54.7 | 7.7 | 4.3 | 5.4 | 2.6 |
| 23 | 4-NH2 | 67.4 | 53.4 | 23.6 | 9.4 | 2.0 |
| 24 | 2-N(CH3)2 | 80.6 | 6.2 | 32.2 | 8.0 | 3.6 |
| 25 | 3-N(CH3)2 | 45.9 | 18.1 | 4.5 | 6.8 | 5.7 |
| 26 | 4-N(CH3)2 | 58.3 | 64.2 | 11.0 | 4.5 | 3.9 |
| 27 | CH2NH2 | 39.6 | 55.8 | 3.1 | 5.4 | 0.65 |
| 28 | 3-NH2, 4-OH, 5-NO2 | 19.8 | 45.0 | 1.9 | 5.2 | 0.55 |
| 29 | 7.3 | 2.9 | 1.4 | 0.84 | 0.92 | |
| 30 | 4.9 | 2.2 | 4.8 | 0.23 | 0.76 | |
| AAZ | 0.25 | 0.012 | 0.075 | 0.025 | 0.006 | |
Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values).
Table 2. Inhibition Data of TcCA, LdcCA, MgCA, and Can2 with N-Nitrosulfonamides and AAZ.
| KI (μM)a |
|||||
|---|---|---|---|---|---|
| compd | R | TcCA | LdcCA | MgCA | Can2 |
| 21 | 2-NH2 | 3.2 | 4.7 | 0.52 | 7.4 |
| 22 | 3-NH2 | 0.15 | 0.49 | 1.7 | 0.25 |
| 23 | 4-NH2 | 0.10 | 0.23 | 0.76 | 0.40 |
| 24 | 2-N(CH3)2 | 5.0 | 4.8 | 32.2 | 4.3 |
| 25 | 3-N(CH3)2 | 1.4 | 0.50 | 4.5 | 1.1 |
| 26 | 4-N(CH3)2 | 0.43 | 0.65 | 0.30 | 0.42 |
| 27 | CH2NH2 | 0.47 | 0.71 | 7.1 | 1.0 |
| 28 | 3-NH2, 4-OH, 5-NO2 | 0.85 | 1.0 | 0.57 | 0.35 |
| 29 | 0.35 | 0.52 | 4.1 | 0.76 | |
| 30 | 0.32 | 0.44 | 2.7 | 2.3 | |
| AAZ | 0.06 | 0.09 | 0.076 | 0.01 | |
Mean from three different assays, by a stopped flow technique (errors were in the range of ±5–10% of the reported values).
The following structure–activity relationships (SAR) can be drawn from the inhibition data reported in Table 1 and 2.
According to preliminary data previously reported,20N-nitro aromatic sulfonamides exhibited low CA I and II inhibitory effectiveness, with KIs spanning in a low to medium micromolar range (2.2–80.6 μM). Heteroaromatic derivatives 29 and 30 turned out as the most potent inhibitors against these ubiquitous hCAs. Whereas CA IV was targeted by all derivatives in a low micromolar range (1.4–39.2 μM), a wealth of submicromolar KI values against CA IX and XII (0.23–9.4 μM) confirmed the favorite efficacy of N-nitrosulfonamides against the tumor-associated isoforms. CA XII was the most affected isozyme among the considered cluster, though the greatest inhibition was measured with the thiadiazole derivative 30 with CA IX (KI of 0.23 μM). It should be noted that CA XII features more Thr and Ser residues in the active site than other hCAs. Likely extended H-bond networks between N-nitrosulfonamide moieties and such hydrophilic residues could justify the reported low KI values. Noteworthy, TcCA turned out to be the most affected α-CA among those herein studied (Table 2). Most derivatives inhibited TcCA in a medium nanomolar range (0.10–0.85 μM), except for compounds bearing 2-NH2, 2-N(CH3)2, or 3-N(CH3)2 moieties at the phenyl ring (KIs in the range 1.4–5.0 μM). The incorporation of primary amino groups at the meta or para position of the phenyl ring confers to 22 and 23, the greatest TcCA inhibitory efficacies as well as the strongest CA inhibition properties of the study. In agreement with the inhibition data previously reported,20 β-CAs were generally more efficiently inhibited by N-nitrosulfonamides than α-CAs. Indeed, most KIs shown in Table 2 for LdcCA, MgCA, and Can2 lie into a submicromolar range. While the first isozyme is undoubtedly the most affected one among the three (KIs in the range 0.23–4.8 μM), equally efficient inhibitions were measured against the fungal MgCA and Can2. The 4-NH2-phenyl derivative 23 arose again as the most potent one against the target LdcCA (KI of 0.23 μM). Unlike against TcCA, 22 inhibited the isozyme comparably with the heterocyclic derivatives 29 and 30 (KIs of 0.49, 0.52, and 0.44 μM, respectively).
Striking target/off-target CAs selectivity profiles can be ascribed to many N-nitrosulfonamide derivatives. As a general trend, the designed compounds acted one to more than two orders of magnitude more potently against TcCA and LdcCA than ubiquitous h-isoforms CA I and II; e.g., derivative 23 showed TcCA/CA II and LdcCA/CA II inhibition ratios of 540 and 230, respectively.
The noticeable in vitro inhibition results of N-nitrosulfonamides against TcCA and LdcCA isoforms led us to study the inhibitory activity of some such inhibitors against various Trypanosoma cruzi and Leishmania forms. Compounds 22 and 23 demonstrated the most potent and selective inhibition against the target CAs and were progressed in the study in the silver salt forms.
The percentages of inhibition of the Trypanosoma cruzi epimastigote forms at different concentrations of synthetic compounds are shown in Table 3. The experiments showed that compounds 22 and 23 possess better activity than the reference drug benznidazole (Bnz) against the epimastigotes forms of Trypanosoma cruzi in both Dm28c clone and Y strain. At the concentration of 5.03 ± 0.95 μM compound 22 inhibited by 50% (IC50) the proliferation of T. cruzi Dm28c. For T. cruzi Y, IC50 values were reached at the concentrations of 12.00 ± 1.06 and 2.51 ± 0.40 μM for 22 and 23, respectively. Anyhow, the two derivatives possess higher toxicity than Bnz for Raw 267.4 macrophages cells (Table 3). As a result, only 22 shows a better SI (5.87 ± 1.14) than benznidazole (4.77 ± 0.91) for T. cruzi Dm28c, whereas uniquely 23 display a higher SI for the Y strain of the parasite 11.58 ± 2.72 with respect to the standard (8.09 ± 0.40). Compounds 22 and 23 were also screened against both T. cruzi forms relevant to human infection. Table 4 summarizes the trypanocidal activity against the nonreplicative (trypomastigotes) and replicative (amastigotes) stages of T. cruzi, Dm28c-Luc clone. Both inhibitors 22 and 23 showed a potent activity against trypomastigotes, reaching IC50 values of 4- to 19.5-fold better than Bnz, respectively. For intracellular amastigotes, 22 (IC50 = 5.2 ± 1.1) and 23 (IC50 = 8.3 ± 1.5) showed lower efficacy than Bnz (IC50 = 1.7 ± 0.3). The higher toxicity than Bnz against Vero cells found for compounds 22 and 23 resulted in reduced selectivity index with respect to the reference drug (Table 4).
Table 3. Minimum Inhibitory Concentration (MIC) and Concentration That Reduced the Proliferation of Epimastigotes by 50% (IC50) Values Derived from Growth Inhibition Assays of T. cruzi Dm28c Clone and Y Straina.
| MIC
(μM) |
IC50 (μM) |
selectivity
index (SI) |
|||||
|---|---|---|---|---|---|---|---|
| compd | epimastigotes forms T. cruzi Dm28c | epimastigotes forms T. cruzi Y | epimastigotes forms T. cruzi Dm28c | epimastigotes forms T. cruzi Y | raw 267.4 cells toxicity CC50 (μM) | epimastigotes forms T. cruzi Dm28c | epimastigotes forms T. cruzi Y |
| benznidazole | 32 | 32 | 29.12 ± 3.03 | 17.00 ± 0.64 | 137.54 ± 12.05 | 4.77 ± 0.91 | 8.09 ± 0.40 |
| 22 | 16 | 32 | 5.03 ± 0.95 | 12.00 ± 1.06 | 29.28 ± 0.38 | 5.87 ± 1.14 | 2.47 ± 0.41 |
| 23 | 32 | 8 | 11.99 ± 0.14 | 2.51 ± 0.40 | 34.89 ± 3.47 | 2.32 ± 0.79 | 11.58 ± 2.72 |
Determination of cytotoxicity (CC50) and the selectivity index (SI50) of 22 and 23 was done using RAW 264.7 macrophages. Average values of three independent experiments ± standard deviations. SI3 = IC50 Raw 267.4 cells/IC50 epimastigote forms of T. cruzi Dm28c and T. cruzi Y.
Table 4. Analysis of Cytotoxicity and Trypanocidal Effect of Compoundsa.
| IC50 (μM) |
selectivity
index (SI) |
||||
|---|---|---|---|---|---|
| compd | trypomastigotes | intracellular amastigotes | Vero cells toxicity CC50 (μM) | trypomastigotes | intracellular amastigotes |
| benznidazole | 15.6 ± 1.9 | 1.7 ± 0.3 | >500 | >32 | >294.1 |
| 22 | 0.8 ± 0.3 | 5.2 ± 1.1 | 21.1 ± 2.8 | 26.4 | 4.1 |
| 23 | 3.9 ± 1.1 | 8.3 ± 1.5 | 24.1 ± 3.6 | 6.2 | 2.9 |
Average values of three independent experiments ± standard deviations. SI = IC50 Vero cells/IC50 trypomastigote and intracellular amastigote forms of T. cruzi, Dm28c-Luc clone.
Minimum inhibitory concentrations (MIC) of 22 and 23 against Leishamnia infantum and L. amazonensis are shown in Table 5, whereas IC50, CC50, and SI values are represented in Figure 1. While compound 22 shows MIC values of 25 against both Leishmania spp, 23 exhibits a 2-fold greater activity against L.amazonensis than L. infantum (Table 5). These values are higher than the reference drug amphotericin B (AMP). Derivatives 22 and 23, respectively, display IC50 of 16.61 and 8.43 μM against the promastigote forms of L.amazonensis and 16.64 and 17.67 μM for L. infantum. The standard AMP is more effective with IC50 values of 1.65 and 1.77 μM against the two Leishmania sp. Nonetheless, the concentration of AMP, which reduced 50% of RAW 267.4 macrophage cells viability (CC50), is very low (1 μM), whereas inhibitors 22 and 23 possess lower toxicity (CC50 values of 29.28 and 34.89 μM, respectively;Figure 1B). As a result of the latter, whereas the SI of the reference compound AMP was more than 2-fold higher than 22 against both Leishmanias, 23 showed a better selectivity profile than AMP against L. amazonensis (Figure 1C).
Table 5. Minimum Inhibitory Concentration (MIC) Assays of L. amazonensis and L. infantuma.
| MIC
(μM) |
||
|---|---|---|
| compd | promastigotes forms L. amazonensis | promastigotes forms L. infantum |
| amphotericin B | 8 | 8 |
| 22 | 25 | 25 |
| 23 | 12.5 | 25 |
Average values of three independent experiments ± standard deviations.
Figure 1.

(A) Concentration that reduced the proliferation of promastigotes by 50% (IC50 μM). (B) Cytotoxic concentration that reduced 50% of RAW 267.4 cells (CC50 μM). (C) Selectivity index: RAW 267.4 cells (CC50)/IC50 against L. amazonensis and L. infantum.
We proposed herein an innovative approach to afford agents for the treatment of Chagas disease and leishmaniasis based on a new CA inhibitory chemotype and on the antimicrobial properties of silver. A set of N-nitrosulfonamides and silver salts thereof were screened against a panel of nine human and pathogens isoforms, among which the targets are TcCA and LdcCA. Most such derivatives showed selective (nanomolar) inhibition of the parasite CAs over human ubiquitous ones. The best inhibitors 22 and 23 showed potent inhibition activity against various developmental forms and spp of Trypanosoma cruzi and Leishmania. The two compounds showed to be more effective than the reference drug benznidazole in inhibiting epimastigote proliferation of both T. cruzi stocks belonging to TcI (Dm28c clone) and TcII (Y strain) lineage. Anyhow, their higher toxicity than Bnz against macrophage cells led to SI comparable to the reference drug. Moreover, both tested compounds displayed 4- to 19.5-fold greater efficacy against T. cruzi forms relevant to human infection, but rather low SI values were calculated owing to low micromolar inhibition of Vero cells in comparison to Bnz.
Both L. amazonensis and L. infantum were inhibited by 22 and 23 in a low micromolar range, but less efficiently than the reference drug amphotericin B. Nevertheless, comparable SI values with the standard were calculated based on a rather less toxic effect of 22 and 23 against RAW 264.7 macrophages. The SI of 23 against L.infantum is even better than amphotericin B.
The present results make N-nitrosulfonamides innovative chemotypes to yield the selective inhibition of the target pathogens CAs over human isoforms. The reported in vitro assays against various strains of T. cruzi and L. donovani are of remarkable interest in the field of NTDs and represent a new interesting starting point for the rational CAI optimization for the treatment of Chagas disease and leishmaniasis.
Glossary
ABBREVIATIONS
- CA
carbonic anhydrase
- CAI
carbonic anhydrase inhibitor
- KI
inhibition constant
- Bzn
benznidazole
- AMP
amphotericin B
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00430.
Synthetic procedures, characterization of compounds, in vitro kinetic procedure, and antiparasitic and cytotoxicity assays (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was financed in part by the Coordenação de Aperfeiçoamento Pessoal de Nível Superior–Brasil (CAPES), Finance code 001, by grants from Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) and Fundação Oswaldo Cruz (FIOCRUZ), and Platform of Bioassay of Instituto Oswaldo Cruz (FIOCRUZ) for use of their facilities, Conselho Nacional de Desenvolvimento Científico e Tecnológico (MCTI-CNPq). Ente Cassa di Risparmio di Firenze, Italy, is gratefully acknowledged for a grant to A.N. (ECR 2016.0774).
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Available from: http://www.who.int/chagas/en/ and http://www.who.int/leishmaniasis/en/.
- Mackey T. K.; Liang B. A.; Cuomo R.; Hafen R.; Brouwer K. C.; Lee D. E. Emerging and reemerging neglected tropical diseases: a review of key characteristics, risk factors, and the policy and innovation environment. Clin. Microbiol. Rev. 2014, 27, 949–979. 10.1128/CMR.00045-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett M. P.; Croft S. L. Management of trypanosomiasis and leishmaniasis. Br. Med. Bull. 2012, 104, 175–196. 10.1093/bmb/lds031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes P. M.; Silva G. K.; Gutierrez F. R.; Silva J. S. Current status of Chagas disease chemotherapy. Expert Rev. Anti-Infect. Ther. 2011, 9, 609–620. 10.1586/eri.11.31. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Sustaining the drive to overcome the global impact of neglected tropical diseases. Second WHO report on neglected tropical diseases; 2013. Available from: http://www.who.int/neglected_diseases/9789241564540/en/.
- Vermelho A. B.; Capaci G. R.; Rodrigues I. A.; Cardoso V. S.; Mazotto A. M.; Supuran C. T. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg. Med. Chem. 2017, 25, 1543–1555. 10.1016/j.bmc.2017.01.034. [DOI] [PubMed] [Google Scholar]
- Ortiz C.; Moraca F.; Medeiros A.; Botta M.; Hamilton N.; Comini M. A. Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules 2016, 21, 368. 10.3390/molecules21030368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Inhibition of carbonic anhydrase from Trypanosoma cruzi for the management of Chagas disease: an underexplored therapeutic opportunity. Future Med. Chem. 2016, 8, 311–324. 10.4155/fmc.15.185. [DOI] [PubMed] [Google Scholar]
- Capasso C.; Supuran C. T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets 2015, 19, 1689–1704. 10.1517/14728222.2015.1067685. [DOI] [PubMed] [Google Scholar]
- Pan P.; Vermelho A. B.; Capaci G. R.; Scozzafava A.; Tolvanen M. E. E.; Parkkila S.; Capasso C.; Supuran C. T. Cloning, characterization, and sulfonamide and thiol inhibition studies of an a-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J. Med. Chem. 2013, 56, 1761–1771. 10.1021/jm4000616. [DOI] [PubMed] [Google Scholar]
- de Menezes D. R.; Calvet C. M.; Rodrigues G. C.; de Souza Pereira M. C.; Almeida I. R.; de Aguiar A. P.; Supuran C. T.; Vermelho A. B. Hydroxamic acid derivatives: a promising scaffold for rational compound optimization in Chagas disease. J. Enzyme Inhib. Med. Chem. 2016, 31, 964–973. 10.3109/14756366.2015.1077330. [DOI] [PubMed] [Google Scholar]
- Syrjanen L.; Vermelho A. B.; Rodrigues I. A.; Corte-Real S.; Salonen T.; Pan P.; Vullo D.; Parkkila S.; Capasso C.; Supuran C. T. Cloning, characterization, and inhibition studies of a b-carbonic anhydrase from Leishmania donovani chagasi, the protozoan parasite responsible for leishmaniasis. J. Med. Chem. 2013, 56, 7372–7381. 10.1021/jm400939k. [DOI] [PubMed] [Google Scholar]
- Guzel-Akdemir O.; Akdemir A.; Pan P.; Vermelho A. B.; Parkkila S.; Scozzafava A.; Capasso C.; Supuran C. T. A class of sulfonamides with strong inhibitory action against the a-carbonic anhydrase from Trypanosoma cruzi. J. Med. Chem. 2013, 56, 5773–5781. 10.1021/jm400418p. [DOI] [PubMed] [Google Scholar]
- Alafeefy A. M.; Ceruso M.; Al-Jaber N. A.; Parkkila S.; Vermelho A. B.; Supuran C. T. A new class of quinazoline-sulfonamides acting as efficient inhibitors against the a-carbonic anhydrase from Trypanosoma cruzi. J. Enzyme Inhib. Med. Chem. 2015, 30, 581–585. 10.3109/14756366.2014.956309. [DOI] [PubMed] [Google Scholar]
- Pan P.; Vermelho A. B.; Scozzafava A.; Parkkila S.; Capasso C.; Supuran C. T. Anion inhibition studies of the a-carbonic anhydrase from the protozoan pathogen Trypanosoma cruzi, the causative agent of Chagas disease. Bioorg. Med. Chem. 2013, 21, 4472–4476. 10.1016/j.bmc.2013.05.058. [DOI] [PubMed] [Google Scholar]
- Rodrigues G. C.; Feijo D. F.; Bozza M. T.; Pan P.; Vullo D.; Parkkila S.; Supuran C. T.; Capasso C.; Aguiar A. P.; Vermelho A. B. Design, synthesis,and evaluation of hydroxamic acid derivatives as promising agents for the management of Chagas disease. J. Med. Chem. 2014, 57, 298–308. 10.1021/jm400902y. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Cadoni R.; Dumy P.; Supuran C. T.; Winum J. Y. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. J. Enzyme Inhib. Med. Chem. 2018, 33, 286–289. 10.1080/14756366.2017.1414808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruso M.; Carta F.; Osman S. M.; Alothman Z.; Monti S. M.; Supuran C. T. Inhibition studies of bacterial, fungal and protozoan b-class carbonic anhydrases with Schiff bases incorporating sulfonamide moieties. Bioorg. Med. Chem. 2015, 23, 4181–4187. 10.1016/j.bmc.2015.06.050. [DOI] [PubMed] [Google Scholar]
- Crespillo-Andújar C.; Chamorro-Tojeiro S.; Norman F.; Monge-Maillo B.; López-Velez R.; Pérez-Molina J. A. Toxicity of nifurtimox as second-line treatment after benznidazole intolerance in patients with chronic Chagas disease: When available options fail. Clin. Microbiol. Infect. 2018, 24, 1344. 10.1016/j.cmi.2018.06.006. [DOI] [PubMed] [Google Scholar]
- Nocentini A.; Vullo D.; Bartolucci G.; Supuran C. T. N-Nitrosulfonamides: A new chemotype for carbonic anhydrase inhibition. Bioorg. Med. Chem. 2016, 24, 3612–3617. 10.1016/j.bmc.2016.05.072. [DOI] [PubMed] [Google Scholar]
- Lansdown A. B. Silver in health care: antimicrobial effects and safety in use. Curr. Probl. Dermatol. 2006, 33, 17–34. 10.1159/000093928. [DOI] [PubMed] [Google Scholar]
- Lok C. N.; Ho C. M.; Chen R.; He Q. Y.; Yu W. Y.; Sun H.; Tam P. K.; Chiu J. F.; Che C. M. Silver nanoparticles: partial oxidation and antibacterial activities. JBIC, J. Biol. Inorg. Chem. 2007, 12, 527–534. 10.1007/s00775-007-0208-z. [DOI] [PubMed] [Google Scholar]
- Rai M.; Yadav A.; Gade A. Silver nanoparticles as a new generation of antimicrobials. Biotechnol. Adv. 2009, 27, 76–83. 10.1016/j.biotechadv.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Allahverdiyev A. M.; Abamor E. S.; Bagirova M.; Ustundag C. B.; Kaya C.; Kaya F.; Rafailovich M. Antileishmanial effect of silver nanoparticles and their enhanced antiparasitic activity under ultraviolet light. Int. J. Nanomed. 2011, 6, 2705–2714. 10.2147/IJN.S23883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minksztym K. Synthesis of Aromatic Aminosulfonic Acid Nitroamides Synthesis. Synthesis 2007, 12, 1819. 10.1055/s-2007-983715. [DOI] [Google Scholar]
- Mathews B. R. Benzene Sulfonnitramide, Toluene-4-sulfonnitramide, 2-nitroluene-4-sulfonnitrani ide and Some of their Salts. J. Phys. Chem. 1920, 24, 108. 10.1021/j150200a003. [DOI] [Google Scholar]
- Khalifah R. G. The carbon dioxide hydration activity of carbonic anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Hewitson K. S.; Vullo D.; Scozzafava A.; Mastrolorenzo A.; Supuran C. T. Molecular cloning, characterization, and inhibition studies of a beta-carbonic anhydrase from Malassezia globosa, a potential antidandruff target. J. Med. Chem. 2012, 55, 3513–3520. 10.1021/jm300203r. [DOI] [PubMed] [Google Scholar]
- Schlicker C.; Hall R. A.; Vullo D.; Middelhaufe S.; Gertz M.; Supuran C. T.; Muehlschlegel F. A.; Steegborn C. Structure and Inhibition of the CO2-Sensing Carbonic Anhydrase Can2 from the Pathogenic Fungus Cryptococcus neoformans. J. Mol. Biol. 2009, 385, 1207–1220. 10.1016/j.jmb.2008.11.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.