

Abstract
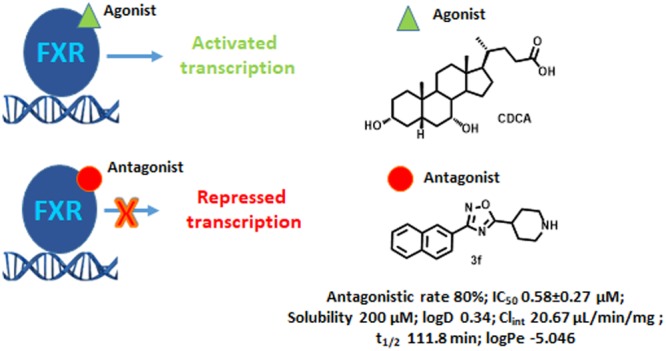
Recent findings have shown that Farnesoid X Receptor (FXR) antagonists might be useful in the treatment of cholestasis and related metabolic disorders. In this paper, we report the discovery of a new chemotype of FXR antagonists featured by a 3,5-disubstituted oxadiazole core. In total, 35 new derivatives were designed and synthesized, and notably, compounds 3f and 13, containing a piperidine ring, displayed the best antagonistic activity against FXR with promising cellular potency (IC50 = 0.58 ± 0.27 and 0.127 ± 0.02 μM, respectively). The excellent pharmacokinetic properties make compound 3f the most promising lead identified in this study.
Keywords: FXR antagonists; bile acid receptor; 1,2,4-oxadiazole core; piperidine ring; pyrrolidine ring
Farnesoid X Receptor (FXR, also known as BAR, NR1H4), a ligand-dependent transcription factor, belongs to the superfamily of metabolic nuclear receptors. FXR is highly expressed in the liver, intestine, kidney, and adrenals, and it responds to bile acids1 regulating their homeostasis and governing lipid and glucose metabolism in the liver.2−6 For all these reasons, FXR represents a promising pharmacological target in the management of metabolic disorders.
The most potent endogenous ligand of FXR is chenodeoxycholic acid (CDCA), a primary bile acid found in human bile. FXR agonists bind to the FXR ligand binding activation function 2 (AF-2) domain (LBD) inducing a receptor conformational change responsible for the release of corepressor molecules (such as N-Cor) and the recruitment of coactivators (such as SRC-1). The FXR/coactivator complex assisted by the RXR protein binds to specific regions of DNA enhancing the expression of target genes. On the other hand, the binding of an antagonist molecule to the FXR-LBD induces a shift of AF-2 helix which assumes an orientation unable to recruit the coactivators, thus leading to transcriptional silencing.7
The strong interest toward FXR agonists is linked to the ability of these compounds in regulating bile acid homeostasis in cholestasis, lowering plasma lipogenesis, repressing very low density lipoprotein production, increasing plasma triglyceride clearance, improving insulin sensitivity, and promoting the storage of glycogen.8,9
In recent years, several FXR agonists, belonging to steroidal and nonsteroidal scaffolds, have been identified as potential leads for the treatment of metabolic disorders with some of them progressed in advanced clinical trials for the treatment of patients with primary biliary cirrhosis (PBC) and nonalcoholic steatohepatitis (NASH).10 Unfortunately, the side effects resulted from the phase III clinical trials of 6-ECDCA, the most promising steroidal FXR agonist,10 raised concerns about the use of FXR agonists in the treatment of metabolic liver diseases. In addition, the recent finding that the FXR gene ablation protects from liver injury caused by bile duct ligation (BDL) has moved the attention toward the FXR antagonism in the treatment of obstructive cholestasis.11
As in the case of the agonists, both steroidal and nonsteroidal compounds12−14 have been identified as FXR antagonists. Among the steroidal scaffolds, the most promising are natural compounds with glyco-β-muricholic acid (GβMCA) currently in advanced preclinical trials for the treatment of obesity, NAFLD, and insulin resistance.15 In this work, starting from the recent discovery of Flesch et al.16 who showed that structural fragmentation on GW4064, a highly potent and selective nonsteroidal FXR agonist, can lead to the shift of the receptor modulation from the agonism to the antagonism, we aimed at developing novel nonsteroidal FXR antagonists. In particular, Flesch et al.16 reported that the removal of the bulky 4-substituent from the isoxazole ring of GW4064 can be a valuable starting point for the synthesis of FXR antagonists. In detail, the replacement of the isoxazole ring in GW4064 with a 3,5-disubsituted oxadiazole led to compounds with different activity profiles, from agonism to partial agonism and antagonism.
The 1,2,4-oxadiazole ring is interesting from the chemical synthesis point of view, being an accessible and highly diversifiable scaffold that can be exploited to improve the pharmacokinetics, pharmacodynamics, and the selectivity profile of the drug candidates.
This is the case of compound 1 in Figure 1, a highly simplified GW4064 derivative endowed with an antagonistic profile toward FXR.
Figure 1.
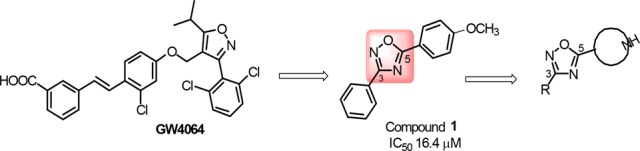
Despite the promising pharmacological profile of 1, a strong rationale of its activity, supported by the elucidation of the ligand binding mode, is lacking. Therefore, we have decided to explore further this scaffold, modifying both substituents at C-3 and C-5 of the oxadiazole moiety developing a small library of derivatives listed in Table 1. In particular, the lipophilic p-methoxyphenyl at C-5 in 1 was replaced by different protonable N-bearing heterocyclic substituents, such as a piperidine or a pyrrolidine ring. It is worth noting that a similar substituent has been recently reported by Teno et al.,13 in a new chemotype of FXR antagonists with the evidence that the replacement of a cyclohexyl component with an ionizable piperidine ring increases the binding activity to FXR. The research of Teno et al. led finally to the identification of N-acylated electrically neutral potent FXR antagonists. At variance with the Teno’s compounds, in our derivatives, the N-bearing ring is not acetylated and, hence, protonable, thus endowing basic properties to our compounds and allowing to explore the piperidine moiety as an available H-bond donor group in the FXR-LBD.
Table 1. FXR Antagonistic Activity of Substituted 1,2,4-Oxadiazole Derivatives.
![]()

Efficacy has been calculated in transactivation assay on HepG2 cells as percent of inhibition of transactivation caused by CDCA at 10 μM, set as 100%.
IC50 has been determined for efficacy ≥50%. IC50 values (μM) were calculated from at least three experiments. Results are expressed as mean ± SEM.
Here, we report the pharmacological evaluation, the assessment of the in vitro pharmacokinetic properties, and the ligand binding mode to FXR of the newly synthesized compounds that allowed the identification of novel potent FXR antagonists with promising pharmacokinetic properties.
The structure of the synthesized compounds was confirmed by spectroscopic and analytical techniques, and the structural characterization is available in the Supporting Information.
Synthesis started with oxadiazole derivatives 3a–f prepared from the nitriles 1a–f and N-Boc-isonipecotic acid (N-Boc-Inp-OH), as starting materials (Scheme 1).
Scheme 1. General Synthetic Procedure To Prepare 1,2,4-Oxadiazole Derivatives 3a–f.

Reagents and conditions: (a) NH2OH HCl, K2CO3 in CH3OH, reflux, 100–90% yield; (b) N-Boc-Inp-OH, DIPEA, HBTU in DMF dry, 80 °C, 60–80% yield; (c) TFA/CH2Cl2 1:1, 2 h, quantitative yield.
Briefly, treatment of nitriles 1a–f with hydroxylamine hydrochloride afforded the intermediate amidoximes 2a–f in quantitative yield.
Reaction with N-Boc-isonipecotic acid (N-Boc-Inp-OH), using HBTU as coupling agent, followed by deprotection of the Boc protecting group in acidic conditions, led to compounds 3a–f (Scheme 1). Compounds 3a–f were evaluated in a luciferase reporter assay on HepG2 cells transiently transfected with human FXR (Table 1). None of the above resulting compounds were able to transactivate FXR in agonistic mode at 10 μM concentration (Figure S3) whereas cell costimulation in the presence of CDCA (10 μM) reveals that, among this series, derivative 3f was relatively effective (80% antagonistic rate) in inhibiting FXR activation caused by CDCA, and the above efficacy data was accompanied by a promising potency (IC50 0.58 ± 0.27 μM). Having identified the 3-naphtyl-1,2,4-oxadiazole as a promising core in FXR antagonism, substitutions preserving the naphtyl at C-3 and the protonable core at C-5 have been explored. First, compounds 13 and 23 have been prepared reacting the amidoxima 2f with N-Boc-l-pipecolic acid (N-Boc-l-Pip-OH) and N-Boc-l-proline (N-Boc-l-Pro-OH) (Scheme 2), in order to explore the influence of different size and different position of the nitrogen in the heterocyclic moiety at C-5. Compound 13 was also demonstrated to be a high potent FXR antagonist (IC50 0.127 ± 0.02 μM), whereas a dramatic loss of potency was observed for the corresponding proline derivative 23, thus identifying the N-bearing six membered heterocycle as a key structural element at C-5 position on the central 1,2,4-oxadiazole moiety. Therefore, the effect of the introduction of N-alkyl side chain with differentiated lengths and functionalization has been the focus, using compounds 3f, 13, and 23 as cornerstone intermediates. N-Alkylation with β, γ, or δ-bromo esters, as depicted in Scheme 2, allowed the introduction of C-3/C-4 or C-5 side chains with the methyl ester end-group. Basic hydrolysis with LiOH and reduction with DIBAL-H afforded the corresponding acids and alcohols (Scheme 2).
Scheme 2. Synthesis of Analogues 4–32.

Reagents and conditions: (a) N-Boc-Inp-OH, DIPEA, HBTU in DMF, 80 °C, 80% yield; (b) TFA/CH2Cl2 1:1, 2 h, quantitative yield; (c) N-Boc-l-Pip-OH, DIPEA, HBTU in DMF dry, 80 °C, 78% yield; (d) N-Boc-l-Pro-OH, DIPEA, HBTU in DMF dry, 80 °C, 83% yield; (e) methyl 3-bromopropanoate, methyl 4-bromobutanoate, or methyl 5-bromopentanoate, DIPEA, in CH3CN dry, 60 °C, overnight, 65–92% yield; (f) LiOH in THF/H2O (2:1), 0 °C then room temperature, 24 h, 40–60% yield; (g) DIBAL-H (1 M in toluene) in THF dry, 0 °C, 48 h, 57–82% yield.
Alkylation at the nitrogen affects FXR antagonism. As shown in Table 1, the effects in terms of efficacy/potency are highly dependent on the position of the nitrogen in the cycle, the length of the alkyl chain, and the nature of the terminal functional group (COOH or CH2OH). Foremost, the introduction of alkyl side chains bearing a carboxylic group produces substantial changes in FXR antagonistic activity compared to nonalkylated counterparts, with a dramatic increase in IC50 values. Importantly, the above loss in potency is independent of the length of the N-alkyl chain and the position of the nitrogen on the heterocycle at C-5.
Of interest, the replacement of the carboxylic end group with a neutral alcoholic functional group preserves the antagonistic activity toward FXR. Furthermore, the biological activity indicates that the efficacy in antagonizing FXR correlates with the size of the installed side chain, with the best match found for the C-4 side chain as in compounds 9 and 19 (IC50 value of 1.17 and 7.0 μM, respectively). The comparison between the IC50 values of the N-alkylated piperidine derivatives with those of the N-free piperidine 3f and 13 shows that the modification at the nitrogen atom alters the ligand potency, prompting the nonsubstituted piperidine ring as a key structural element in the FXR antagonism, with compounds 3f and 13 being the best hits identified in this study.
All the synthesized compounds showed no agonistic activity toward FXR (Figure S3).
The efficacy of compounds 3f and 13 on FXR was then evaluated in a cell-free SRC-1 recruitment Alpha Screen experiment. In this assay, FXR-SRC-1 recruitment is measured in the presence of CDCA at 500 nM with or without different concentrations of 3f and 13. As reported in Figure 2A, both compounds show high efficacy in counteracting SRC-1 peptide recruitment prompted by CDCA when tested at 500 and 1000 nM. Afterward, we attempted to see whether 3f and 13 interacts with common off-targets, such as the nuclear receptors liver X receptor α and β (LXRα and -β), the peroxisome proliferator-activated receptor γ (PPARγ), and pregnane X receptor (PXR) (Figure 2B,C). Compounds 3f and 13 showed no agonistic and antagonistic effects toward any of these receptors.
Figure 2.

(A) Effect of 3f and 13 at 500 and 1000 nM in antagonizing SRC-1 recruitment on FXR by CDCA at 500 nM. Each measurement has been repeated in triplicate and SD < 5%. (B, C) Off-target activity. (B) HepG2 cells were stimulated with GW3965 (GW, 10 μM) or Rosiglitazone (ROSI, 500 nM), or Rifaximin (RIF, 10 μM) as positive controls and compounds 3f and 13 (10 μM); (C) cells were stimulated with 5 μM of compounds 3f and 13 in combination with the relative positive control. Results are expressed as mean ± SEM. *p < 0.05 versus not treated cells (NT).
In order to evaluate the physicochemical parameters of the above-mentioned analogs, LC-MS analysis was assessed (Table 2). Both compounds exhibited good aqueous solubility offering also the possibility to improve this property as well as their polarity by a salt formation via the amine group. Instead, large differences were observed in their metabolic stability, as resulted by in vitro incubation with rat liver microsomes. Compound 13 showed a very low metabolic stability in vitro, with only 9.4% of unmodified molecule remaining after 40 min, and these data rule out its pharmacological potential.
Table 2. In Vitro Pharmacokinetics for Compounds 3f and 13.
| compound | solubilitya (μM) | log D | Clintb | t1/2 (min) | %c |
|---|---|---|---|---|---|
| 3f | 200 | 0.34 | 20.67 | 111.8 | 76.2 |
| 13 | 110.4 | 0.58 | 201 | 11.5 | 9.4 |
Aqueous solubility at pH 7.4.
Reported as μL/min/mg protein.
Percentage of compound remaining in solution after 40 min of incubation. Each measurement has been repeated in triplicate and SD < 5%.
Collectively, compound 3f combines the high potency/efficacy in antagonizing FXR (Table 1) with excellent metabolic stability (about 76% remaining after 40 min of in vitro incubation, Table 2), thus representing a new promising chemotype of FXR antagonist. Furthermore, we also tested its behavior in the parallel artificial membrane permeability assay (PAMPA) to measure its effective permeability (expressed as log Pe) through an artificial lipid membrane.17 Propanolol and furosemide at 250 μM were used as positive and negative control molecules giving a log Pe of −4.875 for propanolol and −6.503 for furosemide. In this assay, 3f displayed an average propensity to cross the membrane in vitro (logPe −5.046).
To gain insights into the antagonism of compound 3f, we then investigated the downstream genes of FXR by real-time PCR (Figure 3). The HepG2 (1 × 106) were plated and, after 24 hours of starvation, were stimulated with CDCA (10 μM) and compound 3f at 0.1 and 1 μM concentration.
Figure 3.

Compound 3f modulates FXR target genes. Real-time PCR analysis of mRNA expression on FXR target genes in HepG2 cells primed with CDCA (10 μM) alone or in combination with increasing concentrations of 3f (0.1 and 1 μM) or with vehicle (DMSO) alone. Values are normalized to GAPDH. The relative mRNA expression is analyzed according to the CT method. Data were calculated from at least three experiments. Results are expressed as actual mean between the two concentrations ± SEM. *p < 0.05 versus not treated cells (NT); #p < 0.05 versus CDCA stimulated cells.
As shown in Figure 3, CDCA induces the expression of SHP, OSTα, PEPCK, and GC6P whereas a decrease of CYP7A1 relative to mRNA expression and 3f reverts the effect of CDCA on the above FXR targeted genes. These results demonstrate that compound 3f is a potent, effective, and selective FXR antagonist.
In order to elucidate the binding mode of the newly synthesized FXR antagonists, we performed docking calculations, which is a widely used computational technique to generate and rank ligand/protein complexes based on empirical scoring functions.18−20
In particular, we performed molecular docking calculations on the most potent derivatives 3f and 13 (Figure 4) in the crystal structure of the FXR-ligand binding domain (LBD) in the antagonistic conformation (PDB code: 4OIV).21
Figure 4.
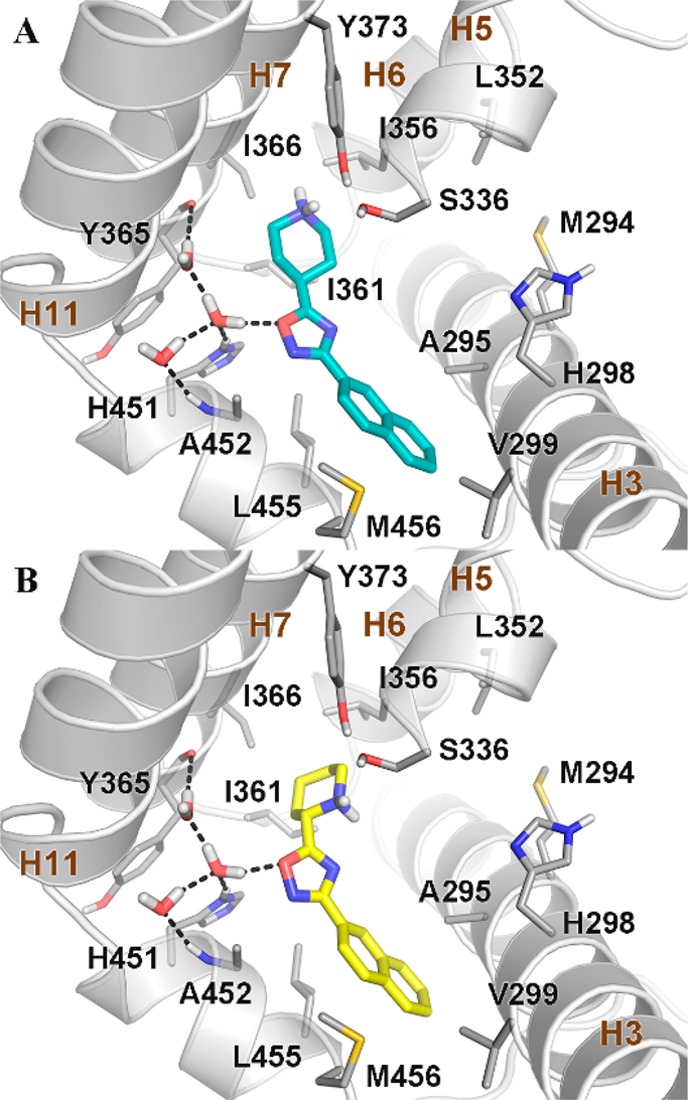
Docking pose of (A) 3f (cyan sticks) and (B) 13 (yellow sticks) in the crystal structure of the FXR-LBD (PDB code: 4OIV).21 FXR is shown as gray cartoons, with amino acids important for ligand binding depicted as sticks. Nonpolar hydrogens are omitted for clarity. Hydrogen bonds are shown as dashed black lines.
We note that the structure of the FXR-LBD in this conformation is rather different if compared with that of the FXR-LBD in the agonistic form. In particular, in the agonistic state, the ligand binding site is defined by helices 3, 5, 6, 7, 11, and 12, with the latter forming the recognition site for the coactivator peptides.22 At variance with the agonistic form, in the antagonistic state, the hydrophobic C-terminal part of H11 is bent filling part of the ligand binding pocket and sending H12 to the binding site of the coactivators peptides of the other FXR monomer. This conformational change blocks the receptor in a not activable state.21
Docking of 3f and 13 (lowest energy conformation, Figure 4) in the FXR-LBD antagonistic conformation shows that the ligand oxadiazole ring engages both a water-mediated hydrogen bond and a T-shape interaction with the side chain of His451 on H11. Additionally, the naphtyl ring of 3f and 13 establishes a number of hydrophobic contacts with residues such as Ala295, Val299, Leu455, and Met456. On the other side, the piperidine ring of 3f and 13 can establish favorable interactions with Ile356 and Ile361 and might form water-mediated hydrogen bonds with Ser336 and Tyr373. The overlap in the FXR-LBD of the binding mode of 3f and 13 with that of the FXR cocrystallized antagonist N-benzyl-N-(3-(tert-butyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide (NDB) shows that the ligands occupy similarly the ligand binding site, although NDB can form additional interactions with the receptor through its 3-(tert-butyl)-4-hydroxyphenyl moiety (Figure S1). Interestingly, additional docking calculations on 9 show that also the binding mode of this compound is superimposable to that of NBD (Figure S2). This finding explains why compounds endowed with an alcoholic side chain on the piperidine nucleus (i.e., 9, 19, 22, and 26) have an FXR antagonistic profile.
In conclusion, we report the discovery of a new chemotype of FXR antagonists, featuring a central oxadiazole ring with a protonable piperidine nucleus at C-5 and a naphtyl group at C-3. We note that the piperidine moiety plays a different role in our compounds if compared with the compounds reported by Teno et al.13 In particular, in the Teno’s compounds, the piperidine works as a spacer to place the H-bond acceptor group (the carbonyl CO) in the proper position to interact with His298, while in our compounds, the piperidine is not acetylated and therefore available as H-bond donor group in the FXR-LBD. Furthermore, the presence of a free piperidine makes our compounds basic with the piperidine protonated and an overall net charge equal to +1.
Our study identified compounds 3f and 13 as potent and selective FXR antagonists. The ability in reverting the effect of CDCA on the expression of several FXR targeted genes combined with the excellent pharmacokinetic properties makes compound 3f the most promising lead identified in this study and suitable for further development.
Glossary
Abbreviations
- AF-2
activation function 2 (domain)
- Boc
tert-butyloxycarbonyl
- CDCA
chenodeoxycholic acid
- Clint
intrinsic clearance
- CYP7A1
cholesterol 7 α-hydroxylase
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- 6-ECDCA
6-ethyl chenodeoxycholic acid
- FXR-LBD
Farnesoid X Receptor–Ligand Binding Domain
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- NAFLD
nonalcoholic fatty liver disease
- GC6P
glucose-6-phosphatase
- NDB
N-benzyl-N-(3-(tert-butyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide
- OSTα
organic solute transporter α
- PAMPA
parallel artificial membrane permeability assay
- PCR
polymerase chain reaction
- PDB
protein data bank
- PEPCK
phosphoenolpyruvate carboxykinase
- SHP
small heterodimer partner
- SRC-1
steroid receptor coactivator-1
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00534.
Experimental procedures; docking poses of 3f and 13 with the crystallographic pose of NBD at the FXR-LBD; docking pose of 9 and its superposition with the crystallographic pose of NBD at the FXR-LBD; FXR transactivation on HepG2 cells; 1H NMR of compounds 3a–3f and 4–32 (PDF)
This work was supported by University of Naples Federico II “Finanziamento della Ricerca in Ateneo (DR/2016/341, February 2016)”, by MIUR-ITALY PRIN2015 “Top-down and Bottom-up approach in the development of new bioactive chemical entities inspired on natural products scaffolds” (Project N. 2015MSCKCE_003), and by the Swiss National Science Foundation (Project N. 200021_163281). V.L. also thanks the COST action CA15135 (Multitarget paradigm for innovative ligand identification in the drug discovery process MuTaLig) for the support.
The authors declare no competing financial interest.
Supplementary Material
References
- Forman B. M.; Goode E.; Chen J.; Oro A. E.; Bradley D. J.; Perlmann T.; Noonan D. J.; Burka L. T.; McMorris T.; Lamph W. W.; Evans R. M.; Weinberger C. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 1995, 81, 687–693. 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- Makishima M.; Okamoto A. Y.; Repa J. J.; Tu H.; Learned R. M.; Luk A.; Hull M. V.; Lustig K. D.; Mangelsdorf D. J.; Shan B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Parks D. J.; Blanchard S. G.; Bledsoe R. K.; Chandra G.; Consler T. G.; Kliewer S. A.; Stimmel J. B.; Willson T. M.; Zavacki A. M.; Moore D. D.; Lehmann J. M. Bile acids: natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lee F. Y.; Barrera G.; Lee H.; Vales C.; Gonzalez F. J.; Willson T. M.; Edwards P. A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1006–1011. 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudel T.; Staels B.; Kuipers F. The farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler., Thromb., Vasc. Biol. 2005, 25, 2020–2030. 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- Cipriani S.; Mencarelli A.; Palladino G.; Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. 10.1194/jlr.M001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowski A. M.; Pike A. C.; Dauter Z.; Hubbard R. E.; Bonn T.; Engström O.; Ohman L.; Greene G. L.; Gustafsson J. A.; Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Prawitt J.; Caron S.; Staels B. How to modulate FXR activity to treat the metabolic syndrome. Drug Discovery Today: Dis. Mech. 2009, 6, e55–e64. 10.1016/j.ddmec.2010.05.002. [DOI] [Google Scholar]
- Cariou B.; Staels B. FXR: a promising target for the metabolic syndrome?. Trends Pharmacol. Sci. 2007, 28, 236–243. 10.1016/j.tips.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Sepe V.; Distrutti E.; Fiorucci S.; Zampella A. Farnesoid X receptor modulators 2014-present: a patent review. Expert Opin. Ther. Pat. 2018, 28, 351–364. 10.1080/13543776.2018.1459569. [DOI] [PubMed] [Google Scholar]
- Stedman C.; Liddle C.; Coulter S.; Sonoda J.; Alvarez J. G.; Evans R. M.; Downes M. Benefit of farnesoid X receptor inhibition in obstructive cholestasis. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11323–11328. 10.1073/pnas.0604772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. Recent progress on bile acid receptor modulators for treatment of metabolic diseases. J. Med. Chem. 2016, 59, 6553–6579. 10.1021/acs.jmedchem.5b00342. [DOI] [PubMed] [Google Scholar]
- Teno N.; Yamashita Y.; Iguchi Y.; Fujimori K.; Une M.; Nishimaki-Mogami T.; Hiramoto T.; Gohda K. Nonacidic chemotype possessing N-acylated piperidine moiety as potent farnesoid X receptor (FXR) antagonists. ACS Med. Chem. Lett. 2018, 9, 78–83. 10.1021/acsmedchemlett.7b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J.; Schierle S.; Gellrich L.; Kaiser A.; Merk D. Structural optimization and in vitro profiling of N-phenylbenzamide-based farnesoid X receptor antagonists. Bioorg. Med. Chem. 2018, 26, 4240–4253. 10.1016/j.bmc.2018.07.017. [DOI] [PubMed] [Google Scholar]
- Gonzalez F. J.; Jiang C.; Patterson A. D. An intestinal microbiota-farnesoid X receptor axis modulates metabolic disease. Gastroenterology 2016, 151, 845–59. 10.1053/j.gastro.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesch D.; Gabler M.; Lill A.; Carrasco Gomez R.; Steri R.; Schneider G.; Stark H.; Schubert-Zsilavecz M.; Merk D. Fragmentation of GW4064 led to a highly potent partial farnesoid X receptor agonist with improved drug-like properties. Bioorg. Med. Chem. 2015, 23, 3490–3498. 10.1016/j.bmc.2015.04.035. [DOI] [PubMed] [Google Scholar]
- Miele E.; Valente S.; Alfano V.; Silvano M.; Mellini P.; Borovika D.; Marrocco B.; Po A.; Besharat Z.; Catanzaro G.; Battaglia G.; Abballe L.; Zwergel C.; Stazi G.; Milite C.; Castellano S.; Tafani M.; Trapencieris P.; Mai A.; Ferretti E. The histone methyltransferase EZH2 as a druggable target in SHH medulloblastoma cancer stem cells. Oncotarget 2017, 8, 68557–68570. 10.18632/oncotarget.19782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzini M.; Braile C.; Valenti S.; Cappelli A.; Vomero S.; Marinelli L.; Limongelli V.; Novellino E.; Betti L.; Giannaccini G.; Lucacchini A.; Ghelardini C.; Norcini M.; Makovec F.; Giorgi G.; Ian Fryer R. Ethyl 8-Fluoro-6-(3-nitrophenyl)-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylate as novel, highly potent, and safe antianxiety agent. J. Med. Chem. 2008, 51, 4730–4743. 10.1021/jm8002944. [DOI] [PubMed] [Google Scholar]
- Famiglini V.; La Regina G.; Coluccia A.; Pelliccia S.; Brancale A.; Maga G.; Crespan E.; Badia R.; Riveira-Muñoz E.; Esté J. A.; Ferretti R.; Cirilli R.; Zamperini C.; Botta M.; Schols D.; Limongelli V.; Agostino B.; Novellino E.; Silvestri R. Indolylarylsulfones carrying a heterocyclic tail as very potent and broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 2014, 57, 9945–9957. 10.1021/jm5011622. [DOI] [PubMed] [Google Scholar]
- Nuti E.; Casalini F.; Avramova S. I.; Santamaria S.; Fabbi M.; Ferrini S.; Marinelli L.; La Pietra V.; Limongelli V.; Novellino E.; Cercignani G.; Orlandini E.; Nencetti S.; Rossello A. Potent arylsulfonamide inhibitors of tumor necrosis factor-α converting enzyme able to reduce activated leukocyte cell adhesion molecule shedding in cancer cell models. J. Med. Chem. 2010, 53, 2622–2635. 10.1021/jm901868z. [DOI] [PubMed] [Google Scholar]
- Xu X.; Xu X.; Liu P.; Zhu Z. Y.; Chen J.; Fu H. A.; Chen L. L.; Hu L. H.; Shen X. Structural basis for small molecule NDB (N-Benzyl-N-(3-(tert-butyl)-4-hydroxyphenyl)-2,6-dichloro-4-(dimethylamino) benzamide) as a selective antagonist of farnesoid X receptor α (FXR α) in stabilizing the homodimerization of the receptor. J. Biol. Chem. 2015, 290, 19888–19899. 10.1074/jbc.M114.630475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi L. Z.; Devarakonda S.; Harp J. M.; Han Q.; Pellicciari R.; Willson T. M.; Khorasanizadeh S.; Rastinejad F. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol. Cell 2003, 11, 1093–1100. 10.1016/S1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.