

SUMMARY
The repertoire and abundance of proteins displayed on the surface of neuronal dendrites are tuned by regulated fusion of recycling endosomes (REs) with the dendritic plasma membrane. While this process is critical for neuronal function and plasticity, how synaptic activity drives RE fusion remains unexplored. We demonstrate a multistep fusion mechanism that requires Ca2+ from distinct sources. NMDA receptor Ca2+ initiates RE fusion with the plasma membrane, while L-type voltage-gated Ca2+ channels (L-VGCCs) regulate whether fused REs collapse into the membrane or reform without transferring their cargo to the cell surface. Accordingly, NMDA receptor activation triggered AMPA-type glutamate receptor trafficking to the dendritic surface in an L-VGCC-dependent manner. Conversely, potentiating L-VGCCs enhanced AMPA receptor surface expression only when NMDA receptors were also active. Thus L-VGCCs play a role in tuning activity-triggered surface expression of key synaptic proteins by gating the mode of RE fusion.
In Brief
Recycling endosomes (REs) in neuronal dendrites regulate the abundance of important synaptic molecules by regulated fusion with the dendritic plasma membrane. Hiester et al. describe a multistep mechanism for RE fusion that requires coincident Ca2+ entry from distinct sources: NMDA receptors and L-type voltage-gated Ca2+ channels.
Graphical Abstract
INTRODUCTION
Neurons have evolved the ability to couple synaptic activity to membrane trafficking pathways that regulate the abundance of important receptors and channels displayed on the plasma membrane (PM) of dendrites and dendritic spines, the primary postsynaptic sites of excitatory connectivity in the CNS. For example, following strong synaptic stimulation, AMPA-type excitatory neurotransmitter receptors (AMPA receptors) are mobilized to the dendritic PM through regulated fusion of vesicular organelles known as recycling endosomes (REs) (Park et al., 2004, 2006; Wang et al., 2008; Kennedy et al., 2010). REs reside throughout the dendritic shaft and within ~50% of dendritic spines, making them ideally poised for local delivery of cargo molecules to synaptic sites and dendritic branches (Cooney et al., 2002; Park et al., 2006; Wang et al., 2008; Kennedy et al., 2010). Disrupting dendritic membrane trafficking, either by inhibiting postsynaptic vesicle fusion machinery (Lledo et al., 1998; Lu et al., 2001; Yang et al., 2008a, 2008b; Kennedy et al., 2010; Ahmad et al., 2012; Heimer-McGinn et al., 2013; Jurado et al., 2013; Arendt et al., 2015) or by specifically inhibiting RE trafficking (Gerges et al., 2004; Park et al., 2004, 2006; Brown et al., 2007), blocks important forms of synaptic plasticity. Furthermore, accumulating evidence indicates that impaired neuronal RE function is associated with neurodegenerative and neurodevelopmental disorders (Li and DiFiglia, 2012; Das et al., 2013).
Despite the critical role of regulated dendritic RE trafficking in neurons, little is known about how synaptic activity is coupled to RE fusion and insertion of integral membrane cargo into the PM. While NMDA receptor activity is required for RE fusion, it is unknown whether Ca2+ is the second messenger that triggers RE fusion and, if so, whether NMDA receptors are the sole Ca2+ source. In addition to NMDA receptors, other sources of dendritic Ca2+ in hippocampal neurons include L-, T-, and R-type voltage-gated Ca2+ channels (VGCCs), all of which have been shown to contribute to Ca2+ signaling in neuronal dendrites and spines (for a review, see Higley and Sabatini, 2012).
Here, we investigated how synaptic activity is coupled to RE fusion and surface expression of AMPA receptors. As expected, NMDA receptor activation and Ca2+ influx was required. Surprisingly, blocking L-type voltage-gated Ca2+ channels (L-VGCCs) severely impaired activity-triggered surface delivery of RE cargoes, including AMPA receptors. Blocking L-VGCCs did not prevent REs from fusing with the PM but instead limited transfer of RE cargo to the cell surface by preventing them from fully collapsing into the PM. Conversely, potentiating L-VGCC function was sufficient to enhance RE fusion and AMPA receptor surface trafficking if NMDA receptors were also active. Thus, NMDA receptors and L-VGCCs cooperatively regulate the transfer of RE cargo molecules, including AMPA receptors, to the dendritic PM. More broadly, our data reveal a multistep membrane fusion mechanism with at least two distinct Ca2+ sources: NMDA receptor Ca2+ for initiating RE docking/fusion and L-VGCC Ca2+ for driving subsequent collapse and cargo transfer to the PM.
RESULTS
REs in Dendrites and Spines Contain AMPA Receptors
Several previous studies indicate that a substantial portion of AMPA receptors are housed in REs and inserted into the neuronal PM in response to activity (Ehlers, 2000; Park et al., 2004; Kennedy et al., 2010). We confirmed and extended previous co-localization studies with a quantitative analysis of endogenous AMPA receptor trafficking through dendritic shaft and spine REs using an antibody feeding approach. Neurons were transfected with a construct that expresses the RE marker transferrin receptor (TfR) fused to mCherry (TfR-mCh) to identify REs and incubated with antibodies directed against GluA1. The remaining primary antibodies bound to surface receptors were blocked with unlabeled secondary antibodies. Cells were then permeabilized and incubated with fluorescent secondary antibodies to label the internalized pool of receptors. Using this method, we observed that a large fraction of REs within dendrites and spines housed internalized GluA1 (Figures 1A and S1A). Figure 1B plots the intensity of internalized GluA1 signal in individual shaft and spine REs. The same analysis was performed on non-permeabilized cells to measure background signal from residual surface anti-GluA1. We used the intensity distribution of non-permeabilized cells to set a conservative threshold for classifying REs as AMPA receptor positive or negative (Figure 1B, see Experimental Procedures for details). We observed that the majority of dendritic shaft (83.1% ± 8.5%) and spine REs (77.7% ± 7.6%) contain detectable internalized GluA1 (Figures 1B and S1C). Similar results were obtained using an antibody against GluA2 (Figures S1B and S1D). To confirm that internalized signal was not a result of nonspecific fluid-phase uptake of antibody, we performed separate experiments in which we preincubated anti-GluAI with a 10-fold molar excess of the peptide antigen used to generate the antibody, which eliminated the internal anti-GluA1 signal (Figure S1E). Together, these data indicate that endogenous AMPA receptors populate REs in dendritic shafts and spines.
Figure 1. REs in Dendrites and Spines Harbor AMPA Receptors and Require Ca2+ for Fusion.

(A) Antibody-labeled GluA1 internalized during a 1-hr feeding period was selectively visualized (see Experimental Procedures) following fixation and permeabilization (top panels). Non-permeabilized neurons served as a control (bottom panels). Note the strong co-localization of internalized GluA1 and TfR-mCh in dendritic shaft and spine REs (arrowheads). Scale bar, 2 μm.
(B) Quantification of internalized GluA1 signal within shaft (black) and spine (red) REs for non-per-meabilized (n = 920 shaft REs from 10 neurons and 560 spine Res from 10 neurons) and permeabilized (n = 787 shaft REs from 11 neurons and 669 spine REs from 13 neurons) neurons.
(C) NH4Cl treatment of neurons co-expressing TfR-mCh and SEP-GluA1 reveals RE localization of SEP-tagged GluA1 in a small subset of dendritic shaft and dendritic spine REs (arrowheads). Scale bar, 2 μm.
(D) Quantification of the percentage of shaft and spine REs that contain detectable endogenous GluA1 (black bars) or expressed SEP-GluA1 (gray bars). ***p < 0.001 (one-way ANOVA, Bonferroni multiple comparisons test).
(E) Schematic of the dual-fluorescent reporter of RE fusion TfR-mCh-SEP. The SEP signal, but not the mCh signal, is quenched within the acidic RE lumen. Fusion events are observed as an abrupt increase in the SEP signal.
(F) Representative uncaging-induced spine exocytosis event. The asterisk marks the location of the uncaging pulses, the red arrowhead marks the mCh-positive RE within the dendritic spine prior to stimulation, and the yellow arrowhead marks the burst of SEP fluorescence indicative of a RE fusion event.
(G) Quantification of the number of fusion events before and after local stimulation of individual dendritic spines by uncaging (indicated by the dotted red line) plotted as the number of RE fusion events over time (normalized to control [left panel]) for, —MNI-Glu (MNI-Glu was excluded from the bath solution),AP5, BAPTA-AM, and 0 mM extracellular(Ex.) Ca2+-treated neurons(control, 181 spines from 29 neurons, — MNI-Glu 135spinesfrom 21 neurons, AP5 162 spines from 24 neurons, BAPTA-AM 158 spines from 15 neurons, 0 mM Ex. Ca2+ from 106 spines from 24 neurons).
(H) Quantification of the probability of a stimulated dendritic spine undergoing an RE fusion event within a 5-min window following glutamate uncaging, normalized to the —MNI-Glu control. ***p < 0.001, **p < 0.01, *p < 0.05 (n = number of neurons, one-way ANOVA, Bonferroni multiple comparisons test).
All error bars represent the SEM.
Ca2+ Influx Is Coupled to RE Fusion in Dendrites and Spines
We next investigated how activity-triggered RE fusion is regulated. Numerous previous studies have used superecliptic pHluorin (SEP)-tagged AMPA receptors to investigate trafficking and surface delivery (Yudowski et al., 2007; Lin et al., 2009; Kopec et al., 2006; Makino and Malinow, 2009; Araki et al., 2010; Mao et al., 2010; Widagdo et al., 2015). However, in agreement with our previous work (Kennedy et al., 2010), we could only observe internal SEP-GluA1 in a very sparse subset of dendritic shaft and spine REs (Figures 1C and 1D; see also Figure S2C for comparison). Lack of SEP-GluA1 signal in REs was not due to a distinct trafficking compartment for GluA1, because nearly all internal SEP-GluA1 (revealed by NH4Cl treatment) strongly co-localized with TfR-mCh (Figure 1C, arrowheads). The discrepancy between our antibody feeding approach and expressed SEP-GluA1 could be the result of a trafficking defect of the expressed receptor. Indeed, recent reports demonstrated that fusion of fluorescent proteins to the N terminus of GluA1 impairs its synaptic localization (Díaz-Alonso et al., 2017; Watson et al., 2017). Alternatively, the discrepancy could be the result of the greatly enhanced signal amplification obtained using antibody-based detection. In either case, to further investigate RE trafficking, we used the RE marker TfR, which has been validated in numerous previous studies and allows us to easily track RE movement and fusion (Park et al., 2006; Wang et al., 2008; Kennedy et al., 2010; Jullié et al., 2014; Roman-Vendrell et al., 2014). Importantly, TfR-based fluorescent reporters localize to REs throughout the dendritic compartment and strongly co-localize with an independent RE marker (Rab11) in spine and shaft REs (Figures S1F and S1G).
RE Fusion Depends on Ca2+ Influx
A rise in cytosolic Ca2+ is the trigger for many, but not all, forms of regulated membrane fusion (Hille et al., 1999). Whether regulated RE fusion in dendrites and spines requires Ca2+ has not been directly addressed, because standard synaptic stimulation protocols rely on presynaptic neurotransmitter release, which itself requires Ca2+. To address this issue, we bypassed the requirement for neurotransmitter secretion by using 4-methoxy-7-nitro-indolinyl (MNI)-glutamate uncaging to directly stimulate individual dendritic spines. We uncaged MNI-glutamate immediately adjacent to spine heads of dissociated hippocampal neurons in an extracellular solution containing reduced (0.5 mM) Mg2+. Laser intensity was tuned such that the uncaging-evoked postsynaptic Ca2+ transient was spatially restricted to the stimulated spine, and the amplitude and duration resembled a spontaneous quantal NMDA-receptor-dependent Ca2+ transient (Figures S1H and S1I). Uncaging-induced spine Ca2+ transients were blocked by AP5 or BAPTA-AM or by performing the experiment in Ca2+-free artificial cerebrospinal fluid (ACSF) (Figures S1J and S1K). We stimulated individual RE-containing dendritic spines of neurons expressing TfR-mCh-SEP (Figure 1E) by delivering 30 uncaging pulses at a rate of 0.5 Hz (Matsuzaki et al., 2004; Patterson et al., 2010), which was sufficient to induce local NMDA-receptor-dependent RE fusion in 15.3% ± 2.7% of dendritic spines within 5 min of the stimulus (Figures 1F–1H). Chelating intracellular Ca2+ with BAPTA-AM blocked uncaging-induced RE fusion confirming a requirement for Ca2+ (Figures 1G and 1H). Fusion was also impaired when we stimulated spines in Ca2+-free ACSF, demonstrating that Ca2+ influx is required for driving RE fusion (Figures 1G and 1H).
L-VGCCs Regulate RE Fusion and AMPA Receptor Surface Delivery
While NMDA receptors are a major source of dendritic Ca2+ (Berdichevsky et al., 1983), other ion channels also contribute to postsynaptic Ca2+ influx. Namely, voltage-gated calcium channels (VGCCs), specifically L-VGCCs, are important sources of postsynaptic Ca2+ (Murphy et al., 1991; Magee and Johnston, 1997; Dolmetsch et al., 2001; Golding et al., 2002; Wheeler et al., 2008; Lee et al., 2009; Kim et al., 2015). To determine whether L-VGCCs influence activity-triggered RE fusion, we stimulated neurons expressing TfR-mCh-SEP using a solution lacking Mg2+ and containing the NMDA receptor co-agonist glycine (“cLTP” solution), which was previously demonstrated to trigger RE fusion and AMPA receptor surface delivery (Lu et al., 2001; Park et al., 2004, 2006; Kennedy et al., 2010). For these experiments, we measured total dendritic surface SEP signal during a 10-min baseline period and then exposed the neurons to the cLTP stimulus. Under control conditions, this treatment led to a robust increase in the SEP signal as TfR-mCh-SEP was inserted into the dendritic PM (Figures 2A and 2B; Movie S1). We confirmed that the increased SEP signal following synaptic stimulation was due to surface delivery and not intracellular pH changes caused by synaptic stimulation (Rathje et al., 2013). We first used a fast-stepping motor to rapidly position a multibarrel pipette flowing pH 6.0 extracellular solution over the imaged neurons. Exposure to pH 6.0 solution nearly instantaneously (within 1 s) quenched ~80% of the signal, confirming that the majority of the SEP signal originates from TfR-SEP at the PM (Figure S2A). We then imaged neurons as they were briefly exposed to extracellular solution at pH 6.0 to quench surface TfR-SEP fluorescence prior to and following cLTP stimulation. The increased TfR-SEP signal following stimulation was rapidly quenched to pre-stimulation levels, confirming the increased SEP signal originated from surface-localized TfR-SEP (Figure S2B).
Figure 2. L-VGCCs Are Required for Activity-Dependent RE Fusion and AMPA Receptor Surface Delivery.

(A) Representative dendritic segments from TfR-SEP-expressing hippocampal neurons before and 10 min after cLTP stimulation for control, AP5-treated (50 μM), and MK801-treated (10 μM) neurons. Scale bar, 5μm.
(B) Quantification of RE cargo delivery over time for control (n = 8 neurons), AP5-treated (n = 10 neurons), and MK801-treated (n = 11 neurons) neurons. Grey box indicates the duration of the cLTP stimulus. AP5 and MK801 were present only during the cLTP stimulation. ***p < 0.001 (one-way repeated-measures ANOVA, Bonferroni multiple comparisons test).
(C) Representative segments of hippocampal neurons showing surface TfR-SEP signal before and 10 min after cLTP stimulation for control, nimodipine-treated (10 μM), verapamil-treated (25 μM), and diltiazem-treated (50 μM) neurons. Scale bar, 5 μm.
(D) Quantification of RE cargo delivery overtime for control (n = 11 neurons), nimodipine-treated (n = 12 neurons), verapamil-treated (n = 12 neurons), and diltiazem-treated (n = 13 neurons) neurons. Grey box indicates the duration of the cLTP stimulus. Nimodipine, verapamil, and diltiazem were present only during the cLTP stimulation. ***p < 0.001 (one-way repeated-measures ANOVA, Bonferroni multiple comparisons test).
(E) Representative segments of neurons stained for total surface GluA1 (total anti-GluA1, left column) and for newly inserted surface GluA1 (anti-GluA1-biotin, right column) for the following conditions: unstimulated negative control (—cLTP), cLTP-stimulated positive control (Cont.), cLTP-stimulated plus AP5 (100 μM, cLTP+AP5), cLTP stimulated plus nimodipine (10 μM, cLTP+Nim), and cLTP stimulated plus verapamil (25 μM, cLTP+Verap). Scale bar, 5 μm.
(F) Quantification of the normalized (to unstimulated control neurons) anti-GluA1-biotin signal for unstimulated negative control neurons (—cLTP, n = 32), positive control cLTP-stimulated neurons (Cont., n = 35), and cLTP-stimulated neuronstreated with AP5 (+AP5, n = 33), nimodipine (+Nim, n = 29), orverapamil (+Verap, n = 33). ***p < 0.001, **p < 0.01 (n = number of dendritic segments, one-way ANOVA, Bonferroni multiple comparisons test).
All error bars represent the SEM.
Activity-triggered TfR-SEP surface delivery was blocked by AP5 and MK801, confirming a requirement for NMDA receptors (Figures 2A and 2B). Importantly, activity-triggered RE trafficking was also robustly impaired by three L-VGCC antagonists: nimodipine (10 μM), verapamil (25 μM), and diltiazem (50 μM) (Figures 2C and 2D). We performed this assay using a range of concentrations for one of the antagonists (nimodipine) and found that impairment of TfR-SEP insertion was saturated even at low (1 μM) doses (Figure S2D). Given the differential nimodipine sensitivity of the two predominant forms of L-VGCCs (CaV1.2 and CaV1.3), these data suggest that dendritic RE fusion is primarily driven by CaV1.2-type L-VGCCs (Xu and Lipscombe, 2001).
Based on our antibody feeding experiments localizing AMPA receptors to REs, we predicted that activity-triggered AMPA receptor surface trafficking would be similarly regulated by L-VGCCs. To assay activity-triggered AMPA receptor surface expression, we first tried using two independent approaches for labeling the total surface pool of GluA1-containing AMPA receptors before and after cLTP stimulation. Even though this stimulation protocol drives robust RE fusion, we did not observe a reliable change in the total steady-state level of surface GluA1-containing AMPA receptors using either fluorescence-based antibody surface labeling (Figures S2E and S2F) or surface biotinylation (Figure S2G). Because AMPA receptors are rapidly internalized following agonist binding (Ehlers, 2000; Lin et al., 2000; Passafaro et al., 2001), we wondered if GluA1 surface delivery mediated by RE fusion was offset by stimulation-induced internalization, leaving the steady-state surface level unperturbed. Thus, we devised a method to selectively label recently inserted AMPA receptors. We first blocked the preexisting pool of surface GluA1 with an unlabeled primary antibody against GluA1 (Figure S2H). We then stimulated neurons using our cLTP simulation protocol. Following stimulation, we applied a biotinylated version of the same antibody (anti-GluA1-biotin) that we used to block preexisting surface receptors. Using labeled streptavidin, we visualized recently inserted GluA1-containing AMPA receptors. We quantified the streptavidin signal over randomly selected dendritic regions defined by total surface GluA1 detected with a fluorescent secondary antibody, which labels both anti-GluA1 and anti-GluA1-biotin. Using this approach, we detected reliable NMDA receptor-dependent GluA1 surface delivery following cLTP (Figures 2E and 2F; no cLTP 100.0% ± 3.2%, cLTP 118.2% ± 3.2% [p = 0.011 versus no stimulation], and cLTP + D-2-amino-5-posphonovalerate (AP5) 99.4% ± 3.4% [p = 0.0006 versus cLTP]). While this increase is modest, we note that the size of the effect that we identified is consistent with the degree of synaptic potentiation that requires postsynaptic insertion of GluA1-containing AMPA receptors (Penn et al., 2017). Consistent with our previous results demonstrating a critical role for L-VGCCs in regulating RE cargo delivery to the PM, we found that pharmacologically blocking L-VGCCs with either nimodipine (10 μM) or verapamil (25 μM) also prevented activity-triggered membrane insertion of GluA1-containing AMPA receptors (Figures 2E and 2F; cLTP 118.2 ± 3.2%, cLTP + nimodipine [Nim] 99.24 ± 1.6 [p = 0.0003 versus cLTP], and cLTP + verapamil [Verap] 95.8 ± 2.2 [p < 0.0001 versus cLTP]).
L-VGCCs Cooperate with NMDA Receptors to Regulate RE Fusion and AMPA Receptor Surface Delivery
To further characterize the functional relationship between NMDA receptors and L-VGCCs, we asked whether potentiating L-VGCC function would enhance NMDA-receptor-dependent RE fusion. Since cLTP stimulation is likely to maximally stimulate dendritic L-VGCC activity through synaptic depolarization and back-propagating action potentials (bAPs), we utilized a weaker stimulation paradigm for this experiment in which we wash on ACSF containing 0 mM Mg2+, 200 μM glycine, and 2 μM tetrodotoxin (TTX) to block evoked synaptic activity and bAPs. This protocol relies on synaptic NMDA receptor activation by spontaneous quantal glutamate release (Lu et al., 2001). We confirmed that NMDA receptors are activated under these conditions using GCaMP6s to monitor intracellular Ca2+ dynamics (Chen et al., 2013; Sinnen et al., 2016). Upon removal of Mg2+ from the extracellular solution, we observed robust Ca2+ transients originating in dendritic spines (Figure 3A). Enhancing L-VGCC function with the agonist Bay K8644 had no effect on Ca2+ levels prior to Mg2+ washout but robustly potentiated postsynaptic Ca2+ transients when cells were exposed to 0 Mg2+/glycine/TTX solution, often resulting in large dendritic Ca2+ spikes (Figures 3B and 3D; Movie S2). These potentiated Ca2+ signals were blocked by AP5, indicating that quantal NMDA receptor activation is coupled to L-VGCC Ca2+ entry under these conditions (Figures 3C and 3E). While we observed modest TfR-SEP surface delivery using this protocol in the absence of Bay K8644, neurons treated with Bay K8644 during stimulation displayed robust TfR-SEP delivery to the PM (Figures 3F and 3G). To provide further evidence for L-VGCC triggered insertion of AMPA receptors through RE fusion, we used our GluA1 surface-labeling strategy. We observed that Bay K8644 treatment increased GluA1-containing AMPA receptor insertion in an NMDA-receptor-dependent manner (Figures 3H and 3I; no stimulation [No Stim] 100.0% ± 3.3%, positive control stimulated [Stim. Cont.] 98.9% ± 3.3%, stimulation [Stim] + Bay K8644 116% ± 3.1% [p = 0.0018 versus Stim. Cont.], Stim + Bay K8644/AP5 92.7% ± 4.0% [p < 0.0001 versus Stim + Bay K8644]).
Figure 3. Potentiation of L-VGCCs Stimulates RE Fusion and AMPA Receptor Surface Delivery in an NMDAR-Dependent Manner.

(A) Representative segment of a hippocampal neuron expressing mCh and GCaMP6s showing mCh expression (left panel). Kymograph of the GCaMP6s signal generated from the white line in (A) before and after exposing the neuron to 0 Mg2+/glycine/TTX solution (Stim., right panel).
(B) The same analysis as in (A) except with the addition of Bay K8644 (10 μM) in both the baseline imaging solution and stimulation solution.
(C) AP5 completely blocks Ca2+ transients from a neuron incubated in Bay K8644 stimulation solution. Kymograph analysis is shown to the right. Note that the timescale shown at the bottom of the right panel of the kymograph in (C) applies to both (A) and (B).
(D) Quantification of the integrated Ca2+ signal after stimulation within spines and shafts for control neurons and neurons treated with Bay K8644. **p < 0.01, *p < 0.05 (n = 5 neurons per treatment, two-tailed Student’s t test).
(E) Quantification of the total (spine plus shaft) integrated Ca2+ signal after stimulation with Bay K8644 before and after treatment with AP5. *p < 0.05 (n = 8 neurons, paired Student’s t test).
(F) Representative segments of hippocampal neurons showing surface RE cargo levels before and 10 min after stimulation for control and Bay-K8644-treated neurons.
(G) Quantification of RE cargo delivery overtime for control (n = 12 neurons) and Bay-K8644-treated (n = 8 neurons) neurons. Gray box indicates the duration of the stimulus, while the blue bar indicates the presence of Bay K8644. ***p < 0.001 (one-way repeated-measures ANOVA, Bonferroni multiple comparisons test).
(H) Representative dendritic segments of neurons stained for total surface GluAI (total anti-GluA1, left column) and newly inserted surface GluAI (GluA1-biotin, right column) for the following conditions: unstimulated negative control (−Stim.), control stimulated (+Stim Cont.), stimulated plus Bay K8644 (10 μM, Stim+Bay K), and stimulated plus Bay K8644 and AP5 (10 μM/100 μM, cLTP+Bay K/AP5). Scale bar, 5 μm.
(I) Quantification of the normalized (to unstimulated control neurons) GluA1-Biotin signal for unstimulated control neurons (−Stim, n = 39), control stimulated neurons (Cont, n = 39), stimulated neurons treated with Bay K8644 (+Bay K,n = 57), and stimulated neurons treated with Bay K8644 and AP5 (+Bay K/AP5, n = 42). ***p < 0.001, **p < 0.01 (n = number of dendritic segments, one-way ANOVA, Bonferroni multiple comparisons test).
All error bars represent the SEM.
Thus far, our data support a model where NMDA receptors cooperate with L-VGCCs to regulate activity-triggered RE cargo delivery, including AMPA receptors, to the dendritic PM. We also wanted to test whether L-VGCC activation in the absence of NMDA receptor activation was sufficient to drive RE fusion. To directly activate L-VGCCs, we depolarized the neuronal cell membrane by increasing extracellular KCl (50 μM, osmolarity adjusted) in the presence of AP5 (50 μM) to block NMDA receptors. KCl depolarization stimulated significant L-VGCC-dependent dendritic Ca2+ influx in the presence of AP5 but did not drive robust RE fusion with the PM (Figures S3A and S3B), indicating that Ca2+ influx through L-VGCCs alone is insufficient. In contrast, when AP5 was omitted, KCl triggered RE fusion that was comparable to cLTP, confirming a critical role for NMDA receptors (Figure S3B).
L-VGCCs Regulate the Mode of RE Fusion in the Dendritic Shaft
To gain a better understanding for how L-VGCCs regulate activity-triggered RE fusion and surface delivery of AMPA receptors, we performed fast imaging of neurons expressing TfR-mCh-SEP and quantified the frequency of TfR-SEP fusion events before and after cLTP stimulation in the presence and absence of L-VGCC antagonists. In agreement with previous studies, cLTP stimulation robustly potentiated the frequency of RE fusion in both dendrites and spines (Kennedy et al., 2010; Keith et al., 2012; Woolfrey et al., 2015). Representative examples of discrete RE fusion events reported by TfR-mCh-SEP are shown in Figures 4A and 4B (see also Figure S1G). Surprisingly, blocking L-VGCCs had no effect on activity-stimulated RE fusion frequency in the dendritic shaft and did not significantly affect the probability of RE fusion following glutamate uncaging in dendritic spines (Figures 4C–4F). Thus, REs can dock and fuse with the PM even when L-VGCCs are blocked.
Figure 4. NMDA Receptor Activation, but Not L-VGCC Activation, Is Required for Initiation of RE Fusion.

(A and B) RE fusion events reported by TfR-mCh-SEP occur within dendritic shafts (A) and dendritic spines (B) during cLTP stimulation. Red arrowheads indicate the presence of the mCh-positive RE prior to the fusion event, while green and yellow arrowheads indicate the appearance of the SEP signal. Kymographs for each channel (generated from the line displayed in the bottom left panel of the color merge) and a plot of the relative intensity for each channel at the site of fusion (circle 1) or at an adjacent site (circle 2) are shown on the bottom right panel of the color merge. Scale bars, 2μm.
(C) Quantification of the rate of dendritic shaft RE fusion events before and after cLTP stimulation for control (n = 12), AP5-treated (n = 10), or nimodipine-treated (n = 10 neurons) neurons. **p < 0.01 (paired Student’s t test).
(D) Quantification of the pooled normalized rate of dendritic spine RE fusion events before and during cLTP stimulation for control (82 total events from 5 neurons), AP5-treated (55 total events from 7 neurons), or nimodipine-treated (96 total events from 7 neurons) neurons.
(E) Histograms showing the normalized distribution of dendritic spine RE fusion events before and after local stimulation of individual dendritic spines by MNI-glutamate uncaging (timing of the uncaging stimulus is indicated by the dotted vertical red line) for control (left) and nimodipine-treated (right) neurons (control, 181 spines from 29 neurons; nimodipine, 130 spines from 25 neurons). Note that the control dataset shown here is the same shown in Figure 2F.
(F) Quantification of the probability of spine RE fusion within a 5-min window following stimulation, normalized tothe –MNI-Glu control (see Figures 2F and 2G). N.S., not significant (two-tailed Student’s t test).
All error bars represent the SEM.
Since blocking L-VGCCs had little effect on the number of stimulated RE fusion events but robustly impaired surface delivery of RE cargo, we hypothesized that L-VGCCs regulate the mode of RE fusion. Intriguingly, previous reports demonstrated that under basal conditions, dendritic REs can undergo full fusion, where the entire endosome merges with the PM, resulting in complete cargo delivery to the cell surface, or “display”-mode fusion, where REs dock and open a fusion pore but then pinch away from the PM with little orno cargo transfer to the cell surface (Yudowski et al., 2007; Jullié et al., 2014; Roman-Vendrell et al., 2014). To ascertain whether synaptic activity regulates the mode of RE fusion and whether L-VGCCs play a role in this process, we categorized fusion events reported by TfR-SEP as either full or display-mode fusion before and immediately following synaptic stimulation. A hallmark of display-mode fusion is an abrupt burst of SEP signal, followed by a stable, delayed period where the SEP fluorescence remains elevated at the fusion site since cargo remains trapped in the RE. In contrast, full fusion events are typified by the abrupt appearance of SEP signal followed by a rapid, monotonic decay as cargo diffuses away from the fusion site. Representative examples of full and display-mode fusion events in dendritic shafts are shown in Figures 5A–5D. For these experiments, we enhanced event detection and quantification by photobleaching existing surface SEP fluorescence immediately before imaging to reduce background signal (Movie S3). The same stretch of dendrite was imaged before and after cLTP stimulation, allowing us to make direct, pairwise comparisons (Figures 5E and 5F). Under basal conditions, display mode events made up 18.8% ± 1.7% of the total events, in agreement with previous experiments (Jullié et al., 2014). Upon stimulation, we found that display-mode events still occurred, but the overall fraction was significantly reduced (8.9% ± 2.4% of total events), indicating that synaptic activity promotes full fusion events in dendritic shafts (Figure 5F). Blocking L-VGCCs with either nimodipine or verapamil led to a striking increase in the raw number and relative fraction of display-mode events following stimulation (Figures 5E and 5F; Movie S3). Thus, L-VGCCs potently regulate the mode of individual RE fusion events in dendritic shafts, explaining the dramatic reduction in RE cargo delivery to the PM when L-VGCCs are blocked.
Figure 5. L-VGCCs Regulate the Mode of RE Fusion in Dendritic Shafts.

(A and B) TfR-SEP was expressed in hippocampal neurons to visualize fusion events. Representative image series showing a full fusion (A) and display fusion event (B) occurring in the dendritic shaft following cLTP stimulation. In both (A) and (B), the panel on the left shows the surface TfR-SEP signal before photobleaching. Note the difference in timescales between full (A, top) and display-mode (B, bottom) fusion.
(C and D) Traces plotting the TfR-SEP signal as a function of time at the site of RE fusion for the fusion events shown in (A) (full fusion) and (B) (display fusion).
(E) Representative traces for all fusion events detected in a segment of dendrite from a control neuron (left), a neuron treated with nimodipine (10 μM, middle), and a neuron treated with verapamil (25 μM, right). Asterisks mark events classified as display fusion events.
(F) Quantification of the number of display fusion events expressed as a fraction of the total number of events before and after cLTP stimulation for control (n = 5 neurons), nimodipine-treated (n = 4 neurons), or verapamil-treated (n = 4 neurons) neurons. ***p < 0.001, *p < 0.05 (paired Student’s t test).
L-VGCCs Regulate the Extent of RE Fusion, but Not Fusion Mode, in Dendritic Spines
Interestingly, when we analyzed RE fusion in dendritic spines, we observed few display-mode events under control conditions or when we inhibited L-VGCCs (control, 0 out of 21 events; nimodipine, 2 out of 18 events), suggesting that spine and shaft RE fusion are regulated by distinct mechanisms. To further investigate a possible role for L-VGCC function in spine RE fusion, we used an independent method based on our dual-color TfR-mCh-SEP reporter. By tracking the ratio of SEP to mCh signals (SEP/mCh) before, during, and following RE fusion and comparing these ratios to the intrinsic SEP/mCh ratio (SEPneut/mCh) determined by neutralizing endosomes with NH4Cl, we can determine the extent of cargo delivery (Kennedy et al., 2010). If all the REs in a spine completely fuse with the PM, then SEP/mCh will abruptly increase to SEPneut/mCh and remain at this value following fusion, even as cargo diffuses away from the site of insertion. If RE fusion occurs in display mode, with no cargo delivery to the surface, then SEP/mCh will abruptly increase to SEPneut/mCh but then revert to its original value as cargo is recaptured and SEP signal is quenched. Finally, if a spine contains multiple REs (Cooney et al., 2002; Park et al., 2006), we can determine whether all the REs within a spine simultaneously fuse (SEP/mCh = SEPneut/mCh at the time of fusion) or only a fraction (SEP/mCh < SEPneut/mCh at the time of fusion) of REs fuse with the PM. Representative examples and plots of SEP/mCh for full and partial spine fusion events are shown in Figures 6A and 6B. Intriguingly, blocking L-VGCCs with nimodipine led to significantly more partial fusion events where only a fraction of the total RE pool was mobilized to fuse (SEP/mCh normalized to SEPneut/mCh immediately after fusion control, 0.91 ± 0.05; nimodipine, 0.70 ± 0.06 [p = 0.006]) (Figures 6C and 6D). Figure 6E shows the cumulative distribution of normalized SEP/mCh values at the time of fusion for control and nimodipine-treated neurons. Note that in both nimodipine and control conditions, SEP/mCh remained nearly constant following fusion, ruling out a major contribution of display-mode fusion in spines (Figure 6C). Thus, in contrast to RE fusion in dendritic shafts, L-VGCCs do not appear to influence fusion mode in spines but instead play a role in mobilization and fusion for a subset of spine REs. One prediction from this observation is that a significant fraction of spines contain multiple REs. Since confocal microscopy can only resolve signals to ~250 nm, REs in spines nearly always appear as a single organelle. Thus, we used structured illumination microscopy (SIM), which offers a substantial increase in lateral resolution compared to confocal microscopy, to more precisely quantify spine RE structure (Gustafsson, 2005). We used SIM to image neurons expressing soluble mCh to visualize dendritic spines and a HALO-tagged version of TfR (TfR-HT) to label REs with the HALO-tag dye ligand JF646 (Figure 6F) (Grimm et al., 2016). Using this method, we readily detected dendritic spines that contained multiple REs (Figure 6F, arrows; Figure 6G). Under basal conditions, nearly 20% of RE-positive dendritic spines contained more than one resolvable RE (Figure 6H). Intriguingly, blocking L-VGCCs during cLTP led to a robust decrease in the fraction of dendritic spines that lacked REs and an increase in the fraction of dendritic spines that contained one or more REs compared to unstimulated control neurons (Figure 6H). Most of this difference could be accounted for by spines that contained multiple REs (Figure 6H). This “backup” of spine REs following synaptic stimulation in the presence of nimodipine is consistent with our previous data demonstrating an increased fraction of spine fusion events in which only a fraction of the total pool of REs within a given spine fuse with the PM. Combined, these data suggest that L-VGCCs initiate fusion of a subset of spine REs.
Figure 6. Blocking L-VGCCs Partially Limits RE Fusion in Dendritic Spines.

(A and B) Representative full (A) and partial (B) spine fusion events reported with TfR-mCh-SEP following cLTP stimulation. Red arrowheads indicate the presence of a spine endosome(s) prior to fusion, while green and yellow arrowheads denote the appearance of the fusion event. Note the persistent mCh signal within the spine 5 s following fusion in (B) indicating the presence of additional endosome(s) that did not fuse with the PM (red arrow in upper right panel in B). Scale bars, 2μm. Panel (ii) shows kymographs for each channel (generated from the line displayed in the bottom left panel of the color merge) and a plot of the relative intensity for each channel at the site of fusion (generated from the circular ROI shown in the bottom right panel of the color merge). Panel (iii) plots the SEP/mCh ratio over time, normalized to SEPneut/mCh (i.e., a value of 1 indicates all the TfR-mCh-SEP is at the PM of the spine).
(C) Plot showing the average normalized SEP/mCh ratio over time for spine RE fusion events occurring during cLTP stimulation for control (n = 21 events from 8 neurons) and nimodipine-treated (n = 18 events from 7 neurons) neurons.
(D) The average normalized SEP/mCh before fusion (i. pre-fusion) and immediately following fusion (ii. fusion) for control (black) and nimodipine-treated (orange) neurons. **p < 0.01 (two-tailed Student’s t test).
(E) Cumulative frequency distribution of normalized SEP/mCh ratios at the time of RE fusion for all events in control and nimodipine-treated neurons. **p < 0.01 (Kolmogorov-Smirnov test).
(F) Representative dendritic segment of a neuron expressing mCh (top panel) and TfR-HT labeled with JF646 (middle panel) imaged using SIM. Arrowheads denote spines containing a single RE, while arrows denote spines containing multiple REs. Scale bar, 2 μm.
(G) Representative images of dendritic spines that contain multiple discrete REs. Scale bar, 500 nm.
(H) Quantification of the proportion of total dendritic spines that contain zero, one, or multiple REs in unstimulated neurons (no cLTP, 530 spines from 12 neurons), stimulated neurons (cLTP Cont., 522 spines from 12 neurons), and stimulated neurons treated with nimodipine (cLTP+Nim, 567 spines from 11 neurons). n = number of neurons; ***p < 0.001, **p < 0.01, *p < 0.05; N.S., not significant (two-way ANOVA, Bonferroni’s multiple comparisons test).
All error bars represent the SEM.
DISCUSSION
In the present study, we investigated how synaptic activity is coupled to RE fusion in neuronal dendrites and spines and whether these same regulatory mechanisms affect the delivery of AMPA receptors to the dendritic PM. Our data support a multistep mechanism requiring NMDA receptor activation to initiate RE docking and fusion with the PM and coincident L-VGCC activation regulating the transfer of integral membrane cargo to the cell surface. Further, we show that L-VGCCs regulate activity-triggered surface delivery of AMPA receptors.
Multistep RE Fusion Regulated by Different Ca2+Sources
Several previous studies demonstrated that NMDA receptor activation is required for regulated RE exocytosis (Park et al., 2004, 2006; Wang et al., 2008; Kennedy et al., 2010), leading to the assumption that Ca2+ entry through NMDA receptors is sufficient to drive the entire process of RE fusion and surface delivery of RE cargo. Our experiments confirm that Ca2+ influx and NMDA receptors are required but reveal an unexpected role for L-VGCCs. We found that L-VGCC blockers potently impaired RE cargo delivery to the PM following synaptic activity. Surprisingly, blocking L-VGCCs had little effect on mobilization and fusion of REs with the PM. However, blocking L-VGCCs drastically increased the incidence of display-mode events that transfer less RE cargo to the PM in dendritic shafts (Jullié et al., 2014). In contrast, blocking NMDA receptors completely prevented all forms of activity-triggered RE fusion. Taken together, these observations support a model in which NMDA receptor signaling initiates RE docking and fusion with the PM, while L-VGCCs gate the collapse of the RE into the PM and subsequent transfer of integral membrane RE cargo (Figure 7). This mechanism, which relies on at least two different Ca2+ sources, contrasts with regulated fusion of other secretory organelles where L-VGCC activity is sufficient (Kolarow et al., 2007; Matsuda et al., 2009; Xia et al., 2009).
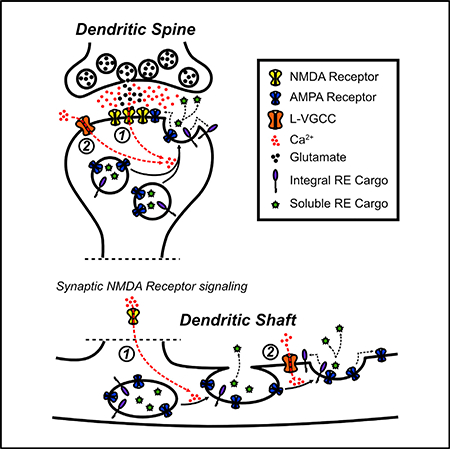
Figure 7. NMDA Receptors and L-VGCCs Cooperatively Regulate Activity-Triggered RE Fusion and AMPA Receptor Delivery in Dendrites and Spines.

(A) In dendritic spines, both NMDA receptor activation (1) and L-VGCC activation (2) contribute to mobilizing the entire pool of REs to fuse with the PM in response to synaptic stimulation.
(B) In dendritic shafts, synaptic NMDA receptor activation (1) drives the initial mobilization of REs to the PM and fusion pore opening, while subsequent L-VGCC activation (2) promotes full RE collapse and fusion into the PM, which delivers integral membrane proteins to the dendritic PM.
The dual requirement for NMDA receptors and L-VGCCs significantly expands the degree to which neural activity can regulate dendritic RE trafficking. A secondary role for L-VGCC activation provides a mechanism that could allow for selective trafficking of different classes of RE cargo proteins. For example, only soluble factors within the RE lumen would be released during display mode fusion, while integral membrane proteins would be inserted into the PM (along with release of soluble factors) during full fusion. Given that L-VGCCs are potently regulated by neuromodulators and hormones (Davare et al., 2001; Hoogland and Saggau, 2004; Sarkar et al., 2008), we speculate that L-VGCCs could serve as a state-dependent switch that defines the functional outcome of plasticity-related RE fusion events.
The role of L-VGCCs appears to be different in dendritic spines. Our SIM data are consistent with previous serial electron microscopy reconstruction studies of hippocampal pyramidal cell dendrites (Cooney et al., 2002; Park et al., 2006), and show that a subset of spines house multiple endosomal organelles. Instead of regulating the mode of RE fusion in dendritic spines, L-VGCCs appear to regulate whether the entire spine RE pool is mobilized to fuse with the PM (Figure 7). Blocking L-VGCCs did not reduce the probability of spine fusion, but led to a backup of REs within spines, with significantly more spines housing multiple endosomes following synaptic stimulation. The simplest interpretation is that synaptic stimulation drives formation of new vesicles within spines (either by local endocytosis or by mobilization of REs into the spine head from the dendritic shaft) and that blocking L-VGCCs prevents these endosomes from fusing. The molecules and mechanisms conferring sensitivity of a subset of spine REs to L-VGCC activation will await further investigation, but this heterogeneity may allow for L-VGCC regulation of specific cargo molecules near excitatory synapses.
Functional Consequences
The composition of channels and receptors displayed on the dendritic PM is not homogeneous but can vary from branch to branch or from proximal to distal dendritic regions (Hoffman et al., 1997; Lörincz et al., 2002; Kerti et al., 2012). Recent studies demonstrated that dendritic branches can act as fundamental units for plasticity (Frick et al., 2004; Losonczy and Magee, 2006; Govindarajan et al., 2011; Makino and Malinow, 2011; Cichon and Gan, 2015) and that regulation of the functional properties and abundance of postsynaptic ion channels is a critical component of dendritic branch plasticity (Frick et al., 2004; Losonczy et al., 2008; Nestor and Hoffman, 2012). More specifically, spatially restricted AMPA receptor surface delivery in response to local synaptic activity supports synaptic plasticity (Makino and Malinow, 2009; Patterson et al., 2010). Numerous studies have reported that L-VGCCs are required for various forms of long-term potentiation (LTP) (Grover and Teyler, 1990; Huang and Malenka, 1993; Magee and Johnston, 1997; Morgan and Teyler, 2001). More recently, L-VGCC activation was demonstrated to play a critical role in forms of LTP that depend on locally generated postsynaptic dendritic spikes (Golding et al., 2002; Remy and Spruston, 2007; Kim et al., 2015). REs are ideally poised to contribute to these activity-dependent changes in dendritic and synaptic properties, because they contain AMPA receptors and other important cargoes and are distributed throughout the dendritic arbor and within a large fraction of dendritic spines. A requirement for coincident NMDA receptor and L-VGCC activation could localize RE fusion near stimulated synapses, providing a mechanism for spatial constraint of modifications to activated regions of the cell. Whether the L-VGCC requirement for these forms of neural plasticity is a result of RE fusion, established transcriptional regulatory mechanisms, or as-yet-unidentified mechanisms awaits further investigation.
Finally, numerous brain disorders have been linked to L-VGCCs. Gain-of-function alleles in CACNA1C, the gene that encodes the L-VGCC subunit CaV1.2, lead to autism-like brain disorders associated with Timothy syndrome, while numerous polymorphisms associated with CACNA1C have been implicated in bipolar disorder, schizophrenia, autism, and depression (Splawski et al., 2004; Bhat et al., 2012; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). How loss or gain of L-VGCC function contributes to brain disorders is not well understood, but most previous work has focused on L-VGCC-regulated gene transcription and neurotrophin secretion. Our study reveals a new and important role for L-VGCCs in regulating trafficking of dendritic REs and AMPA receptors, expanding the repertoire of mechanisms linking L-VGCCs to synaptic function in health and disease.
EXPERIMENTAL PROCEDURES
Experimental Model and Subject Details
All animal procedures were carried out in accordance with a protocol approved by the University of Colorado Denver Institutional Animal Care and Use Committee. All dissociated cultures were prepared from Sprague-Dawley rats. Timed pregnant dams (typically embryonic day 16) were obtained from Charles River Laboratories and housed under standard conditions. This study used dissociated cultures from both male and female pups.
Cell Culture
Dissociated hippocampal cultures were prepared from neonatal rat pups as previously described (Beaudoin et al., 2012) and grown on 18-mm poly-D-lysine-coated (Sigma) coverslips in 12-well cell culture dishes in Neurobasal-A medium (Invitrogen) supplemented with B27 (Invitrogen) and Glutamax (Invitrogen) at an approximate density of 100,000 cells per well. Neurons were maintained at 37°C in a humidified incubator at 5% CO2. All neuronswere between 16days in vitro (DIV16) and DIV21 at the time of experiment. A detailed description of the expression constructs and pharmacological reagents used for experiments performed in this study can be found in Supplemental Experimental Procedures.
Imaging
Live and fixed cell imaging of dissociated neurons was carried out at 32°C on an Olympus IX71 equipped with a spinning-disc scan head (Yokogawa). Excitation illumination was delivered from an acousto-optic tunable filter (AOTF) controlled laser launch (Andor). Images were acquired using a 60x Plan-Apochromat 1.4 numerical aperture (NA) objective and collected on a 1,024 × 1,024 pixel Andor iXon EM-CCD camera. Forall imaging experiments in this study, the apical portion of the dendritic arbor extending 25–100 μm from the cell soma was imaged. Data acquisition and analysis were performed with Metamorph (Molecular Devices), Andor IQ, and ImageJ software. Some images were low-pass filtered and interpolated for display. Only raw, unprocessed data were used for quantification. A detailed description of the methods used to assay glutamate receptor internalization, stimulate neural activity, image RE and Ca2+ dynamics, and quantify surface GluA1 levels, as well as the methods used for SIM imaging, can be found in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Recycling endosomes (REs) in dendrites harbor AMPA-type glutamate receptors
Fusion of REs with the PM requires Ca2+ entry from different sources
NMDAR Ca2+ initiates RE docking, and L-VGCCs regulate the mode of membrane fusion
L-VGCCs regulate activity-triggered surface expression of AMPA receptors
ACKNOWLEDGMENTS
We would like to thank Tim Benke, Mark Dell’Acqua, William Sather, and Michael Tadross for critical discussions and Luke Lavis (Janelia Farm Research Campus) for generously providing us with HaloTag ligand. This work was supported by National Institute of General Medical Sciences (NIGMS) grant GM007635-37 and National Institute of Neurological Diseases and Stroke (NINDS) grant F31NS100403 (B.L.S.); National Science Foundation grant HRD-1201885 and the HHMI Gilliam Scholars Program (A.M.B.); and NINDS grant NS082271, the Brain Research Foundation, the Boettcher Foundation, and the Pew Charitable Trusts (M.J.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, three figures, and three movies and can be found with this article online at https://doi.org/10.10167j.celrep.2017.10.105.
REFERENCES
- Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Südhof TC, and Malenka RC (2012). Postsynaptic complex in controls AMPA receptor exocytosis during LTP. Neuron 73, 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki Y, Lin DT, and Huganir RL (2010). Plasma membrane insertion of the AMPA receptor GluA2 subunit is regulated by NSF binding and Q/R editing of the ion pore. Proc. Natl. Acad. Sci. USA 107, 11080–11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Zhang Y, Jurado S, Malenka RC, Südhof TC, and Chen L (2015). Retinoic acid and LTP recruit postsynaptic AMPA receptors using distinct SNARE-dependent mechanisms. Neuron 86, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin GMJ, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, and Arikkath J (2012). Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat. Protoc 7, 1741–1754. [DOI] [PubMed] [Google Scholar]
- Berdichevsky E, Riveros N, Sánchez-Armáss S, and Orrego F (1983). Kainate, N-methylaspartate and other excitatory amino acids increase calcium influx into rat brain cortex cells in vitro. Neurosci. Lett 36, 75–80. [DOI] [PubMed] [Google Scholar]
- Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, and Gould TD (2012). CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol 99, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TC, Correia SS, Petrok CN, and Esteban JA (2007). Functional compartmentalization of endosomal trafficking for the synaptic delivery of AMPA receptors during long-term potentiation. J. Neurosci 27,13311–13315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon J, and Gan WB (2015). Branch-specific dendritic Ca(2+) spikes cause persistent synaptic plasticity. Nature 520, 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney JR, Hurlburt JL, Selig DK, Harris KM, and Fiala JC (2002). Endosomal compartments serve multiple hippocampal dendritic spines from a widespread rather than a local store of recycling membrane. J. Neurosci 22, 2215–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Scott DA, Ganguly A, Koo EH, Tang Y, and Roy S (2013). Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, and Hell JW (2001). A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293, 98–101. [DOI] [PubMed] [Google Scholar]
- Díaz-Alonso J, Sun YJ, Granger AJ, Levy JM, Blankenship SM, and Nicoll RA (2017). Subunit-specific role for the amino-terminal domain of AMPA receptors in synaptic targeting. Proc. Natl. Acad. Sci. USA 114, 7136–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, and Greenberg ME (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2000). Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, and Johnston D (2004). LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci 7, 126–135. [DOI] [PubMed] [Google Scholar]
- Gerges NZ, Backos DS, and Esteban JA (2004). Local control of AMPA receptor trafficking at the postsynaptic terminal by a small GTPase of the Rab family. J. Biol. Chem 279, 43870–43878. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, and Spruston N (2002). Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418, 326–331. [DOI] [PubMed] [Google Scholar]
- Govindarajan A, Israely I, Huang SY, and Tonegawa S (2011). The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron 69, 132–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott-Schwartz J, Liu Z, Lionnet T, and Lavis LD (2016). Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 13, 985–988. [DOI] [PubMed] [Google Scholar]
- Grover LM, and Teyler TJ (1990). Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347, 477–479. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL (2005). Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. Proc. Natl. Acad. Sci. USA 102, 13081–13086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer-McGinn V, Murphy ACH, Kim JC, Dymecki SM, and Young PW (2013). Decreased dendritic spine density as a consequence of tetanus toxin light chain expression in single neurons in vivo. Neurosci. Lett 555, 36–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, and Sabatini BL (2012). Calcium signaling in dendritic spines. Cold Spring Harb. Perspect. Biol 4, a005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, Billiard J, Babcock DF, Nguyen T, and Koh DS (1999). Stimulation of exocytosis without a calcium signal. J. Physiol 520, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, and Johnston D (1997). K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387, 869–875. [DOI] [PubMed] [Google Scholar]
- Hoogland TM, and Saggau P (2004). Facilitation of L-type Ca2+ channels in dendritic spines by activation of beta2 adrenergic receptors. J. Neurosci 24, 8416–8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, and Malenka RC (1993). Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J. Neurosci 13, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jullié D, Choquet D, and Perrais D (2014). Recycling endosomes undergo rapid closure of a fusion pore on exocytosis in neuronal dendrites. J. Neurosci 34, 11106–11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Goswami D, Zhang Y, Molina AJM, Südhof TC, and Malenka RC (2013). LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El-Husseini A, and Dell’acqua ML (2012). Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J. Neurosci 32, 7119–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, and Ehlers MD (2010). Syntaxin-4 defines a domain for activity-dependent exocytosis in dendritic spines. Cell 141, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerti K, Lorincz A, and Nusser Z (2012). Unique somato-dendritic distribution pattern of Kv4.2 channels on hippocampal CA1 pyramidal cells. Eur. J. Neurosci 35, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Hsu C-L, Cembrowski MS, Mensh BD, and Spruston N (2015). Dendritic sodium spikes are required for long-term potentiation at distal synapses on hippocampal pyramidal neurons. eLife 4, e06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, and Lessmann V (2007). Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J. Neurosci 27, 10350–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, and Malinow R (2006). Glutamate receptor exocytosis and spine enlargement during chemically induced longterm potentiation. J. Neurosci 26, 2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJR, Escobedo-Lozoya Y, Szatmari EM, and Yasuda R (2009). Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, and DiFiglia M (2012). The recycling endosome and its role in neurological disorders. Prog. Neurobiol 97, 127–141. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, and Sheng M (2000). Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat. Neurosci 3, 1282–1290. [DOI] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, and Huganir RL (2009). Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat. Neurosci 12, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Südhof TC, Malenka RC, and Nicoll RA (1998). Postsynaptic membrane fusion and long-term potentiation. Science 279, 399–403. [DOI] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Támils G, Shigemoto R, and Nusser Z (2002). Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat. Neurosci 5, 1185–1193. [DOI] [PubMed] [Google Scholar]
- Losonczy A, and Magee JC (2006). Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron 50, 291–307. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Makara JK, and Magee JC (2008). Compartmentalized dendritic plasticity and input feature storage in neurons. Nature 452, 436–441. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, and Wang YT (2001). Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29, 243–254. [DOI] [PubMed] [Google Scholar]
- Magee JC, and Johnston D (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213. [DOI] [PubMed] [Google Scholar]
- Makino H, and Malinow R (2009). AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron 64, 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, and Malinow R (2011). Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron 72, 1001–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Takamiya K, Thomas G, Lin DT, and Huganir RL (2010). GRIP1 and 2 regulate activity-dependent AMPA receptor recycling via exocyst complex interactions. Proc. Natl. Acad. Sci. USA 107, 19038–19043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, and Poo MM (2009). Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J. Neurosci 29, 14185–14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GCR, and Kasai H (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan SL, and Teyler TJ (2001). Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J. Neurophysiol 86, 1289–1296. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, and Baraban JM (1991). L-typevoltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7, 625–635. [DOI] [PubMed] [Google Scholar]
- Nestor MW, and Hoffman DA (2012). Differential cycling rates of Kv4.2 channels in proximal and distal dendrites of hippocampal CA1 pyramidal neurons. Hippocampus 22, 969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, and Ehlers MD (2004). Recycling endosomes supply AMPA receptors for LTP. Science 805,1972–1975. [DOI] [PubMed] [Google Scholar]
- Park M, Salgado JM, Ostroff L, Helton TD, Robinson CG, Harris KM, and Ehlers MD (2006). Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52, 817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Piech V, and Sheng M (2001). Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat. Neurosci 4, 917–926. [DOI] [PubMed] [Google Scholar]
- Patterson MA, Szatmari EM, and Yasuda R (2010). AMPA receptors are exocytosed in stimulated spines and adjacent dendrites in a Ras-ERK-dependent manner during long-term potentiation. Proc. Natl. Acad. Sci. USA 107, 15951–15956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn AC, Zhang CL, Georges F, Royer L, Breillat C, Hosy E, Petersen JD, Humeau Y, and Choquet D (2017). Hippocampal LTP and contextual learning require surface diffusion of AMPA receptors. Nature 549, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathje M, Fang H, Bachman JL, Anggono V, Gether U, Huganir RL, and Madsen KL (2013). AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc. Natl. Acad. Sci. USA 110, 14426–14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy S, and Spruston N (2007). Dendritic spikes induce single-burst long-term potentiation. Proc. Natl. Acad. Sci. USA 104, 17192–17197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman-Vendrell C, Chevalier M, Acevedo-Canabal AM, Delgado-Peraza F, Flores-Otero J, and Yudowski GA (2014). Imaging of kiss-and- run exocytosis of surface receptors in neuronal cultures. Front. Cell. Neurosci 8, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar SN, Huang RQ, Logan SM, Yi KD, Dillon GH, and Simpkins JW (2008). Estrogens directly potentiate neuronal L-type Ca2+ channels. Proc. Natl. Acad. Sci. USA 105, 15148–15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Gibson ES, and Kennedy MJ (2016). Local and use-dependent effects of β-amyloid oligomers on NMDA receptor function revealed by optical quantal analysis. J. Neurosci 36, 11532–11543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Wang Z, Edwards JG, Riley N, Provance DW Jr., Karcher R, Li XD, Davison IG, Ikebe M, Mercer JA, Kauer JA, and Ehlers MD (2008). Myosin Vb mobilizes recycling endosomes and AMPA receptors for postsynaptic plasticity. Cell 135, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JF, Ho H, and Greger IH (2017). Synaptic transmission and plasticity require AMPA receptor anchoring via its N-terminal domain. eLife 6,1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Barrett CF, Groth RD, Safa P, and Tsien RW (2008). CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol 183, 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Chai YJ, Ridder MC, Chau YQ, Johnson RC, Sah P, Huganir RL, and Anggono V (2015). Activity-dependent ubiquitination ofGluA1 and GluA2 regulates AMPA receptor intracellular sorting and degradation. Cell Rep. 10, 783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Sanderson JL, and Dell’Acqua ML (2015). The palmitoyl acyltransferase DHHC2 regulates recycling endosome exocytosis and synaptic potentiation through palmitoylation of AKAP79/150. J. Neurosci 35, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Lessmann V, and Martin TFJ (2009). Imaging of evoked dense-core-vesicle exocytosis in hippocampal neurons reveals long latencies and kiss-and-run fusion events. J. Cell Sci 122, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, and Lipscombe D (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci 21, 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, and Zhou Q (2008a). Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc. Natl. Acad. Sci. USA 105, 11388–11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Frerking M, and Zhou Q (2008b). Spine expansion and stabilization associated with long-term potentiation. J. Neurosci 28, 5740–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudowski GA, Puthenveedu MA, Leonoudakis D, Panicker S, Thorn KS, Beattie EC, and von Zastrow M (2007). Real-time imaging of discrete exocytic events mediating surface delivery of AMPA receptors. J. Neurosci 27, 11112–11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.